Настоящее изобретение относится к окислению аммиака. Для производства азотной кислоты и цианистого водорода широко применяют окисление аммиака. Для получения азотной кислоты подвергают окислению воздухом аммиак до образования окиси азота, а для получения цианистого водорода смесь аммиака и метана (обычно в качестве природного газа) подвергают окислению воздухом. В обоих способах для достижения окисления аммиака над катализатором направляют газовую смесь при повышенной температуре. В этом случае нежелательны такие побочные реакции, как, например, образование азота или закиси азота. Поэтому желательно иметь катализатор, который помимо хорошей его активности обладает хорошей избирательностью.
На протяжении многих лет для катализаторов применяли обычно платину, причем иногда в смеси с другими благородными металлами, в виде сеток или тканей, изготовленных из металлической проволоки. Такие катализаторы имеют хорошую активность и избирательность, однако они имеют также и недостатки: катализаторы не только очень дорогостоящие, но их металлы демонстрируют значительную летучесть при температурах, с которыми они сталкиваются во время их применениями, поэтому они теряются в потоке газов. Хотя достаточно известно, что применяют средство в форме ловушки, устанавливаемой вниз по потоку, для улавливания летучего металла для последующего его извлечения, однако срок службы катализатора из-за постоянного улетучивания металла уменьшается, и его необходимо часто заменять. К тому же, требуются значительные капитальные затраты на извлечение металлов из ловушки, установленной вниз по потоку, а также на изготовление новых сеток или тканей для катализаторов.
Таким образом, такие катализаторы из благородных металлов желательно заменить другими.
Известно, что оксид кобальта проявляет активность для окисления аммиака. С целью улучшения активности и избирательности катализатора из оксида кобальта предлагают включать в него различные промоторы, например, из редкоземельных металлов.
Например, в качестве катализаторов для окисления аммиака предлагают применять композиции на основе оксида лантана/оксида церия (3)/оксида Кобальта, имеющие общую формулу LaI-хСехСоОз (где: х=0-1) и полученные специальным способом совместного осаждения, как описано, например, в патенте CN-A-86108985. Указано, что, как показали испытания, проведенные в небольшом масштабе, такие материалы имеют хорошую активность и избирательность, однако существует предположение, что их активность и/или избирательность снижаются при рабочих температурах на верхнем конце температурного интервала, обычно применяемого для окисления аммиака (интервал 800-1000oC).
Было установлено нами, что в этом типе катализатора важно, чтобы масса кобальта присутствовала в качестве фазы смешанных оксидов, например, в качестве Перовскитовой структуры RЕСоО3 (RE = редкоземельный металл) или в его форме, в которой кислород не является стехиометрическим и он не присутствует в виде свободных оксидов кобальта, например оксидов кобальта (II,III) Со3O4 или моноксида кобальта СоО. Полагаем, что если значительное количество кобальта присутствует в качестве свободных оксидов во время применения при высоких температурах, например, свыше примерно 850oC, то свободные оксиды кобальта способны катализировать окисление во время побочных реакций, например, азот или закись азота, тогда как если масса кобальта "блокирована" в фазе смешанных оксидов, например такой? как Перовскитовая структура, способность к окислению более ограничена требуемым окислением.
Изготовление катализатора просто путем совместного осаждения оксидов компонентов (или соединений, которые легко разлагаются в нем) либо испарением раствора смеси термически разлагаемых солей, например нитратов, требуемых металлов с последующим прокаливанием при умеренных температурах, например 600-900oC, необязательно блокирует массу кобальта в фазе смешанных оксидов, например Перовскитовая структура, даже если компоненты присутствуют в требуемых пропорциях. Для получения требуемой структуры необходима термообработка продукта. Согласно вышеупомянутой заявке CN-A-86108985 катализаторы подвергали прокаливанию при температуре 900oC в течение 5 часов до их применения, что приводит к получению катализатора, содержащего более 30 ат.% кобальта в виде свободных оксидов. Считаем, что такая термообработка не является соответствующей и для уменьшения количества присутствующего свободного оксида кобальта требуется термообработка при более высоких температурах и/или в течение более продолжительного периода времени. Однако нагрев при очень высокой температуре, свыше примерно 1150oC, может привести к разложению фаз смешанных оксидов, выделению свободных оксидов кобальта. Альтернативно, или дополнительно, для удаления свободных оксидов кобальта из композиции можно применять стадии, например композицию можно промыть аммиачным раствором или другим раствором, содержащим комплексообразующий агент для кобальта. Примером такого комплексообразующего агента является этилендиаминтетрауксусная кислота.
Таким образом, настоящее изобретение обеспечивает получение катализатора окисления, содержащего оксиды (а), по меньшей мере, одного элемента А, выбранного из редкоземельных металлов и иттрия, и (в) кобальт, причем кобальт и элемент А присутствуют в таких пропорциях, что атомное отношение содержания элемента А к кобальту находится в интервале 0,8-1,2; по меньшей мере, некоторые оксиды кобальта и элемента А присутствуют в качестве фазы смешанных оксидов, при этом, по меньшей мере, 30%, предпочтительно менее 25% кобальта (по атомам), присутствуют в форме свободных оксидов кобальта.
Итак, настоящий катализатор содержит, по меньшей мере, одну фазу смешанных оксидов, содержащую кобальт и, по меньшей мере, один элемент А. Катализатор может также содержать свободные оксиды элемента А и/или одну или более фаз смешанных оксидов, включающих два или более элементов А. Атомное соотношение элемента А и кобальта составляет 0,8-1,2, в частности 1,0-1,2. В качестве свободных оксидов кобальта присутствуют предпочтительно менее 25% (по атомам) кобальта, а в частности предпочтительно, чтобы в качестве моноокиси кобальта, СоО, присутствовало менее 15% (по атомам) кобальта. Содержание различных фаз можно определить рентгенографией (XRF) или термогравиметрическим анализом (TGA), позволяющим применять в последнем случае потери в массе, связанные с характерным термическим разложением Со3О4, которое происходит при температуре примерно 930oC на воздухе. Предпочтительно менее 10%, в частности менее 5% по массе, композиции составляет свободный оксид кобальта (II,III), а менее 2% по массе - свободный моноксид кобальта.
В качестве части или всего элемента А применяют предпочтительно, по меньшей мере, один элемент, выбранный из иттрия, церия, лантана, неодимия и празеодимия. Элемент А может содержать смесь, по меньшей мере, одного элемента Vv с переменной валентностью, выбранного из церия и празеодима, и, по меньшей мере, одного элемента Vn с непеременной валентностью, выбранного из иттрия и редкоземельных элементов с непеременной валентностью, например лантан или неодим. В частности предпочтительно, чтобы атомные соотношения элемента Vv с переменной валентностью к элементу Vn с непеременной валентностью находилось в интервале 0-1, особенно 0-0,3. Предпочтительно большинство кобальта присутствует в качестве Перовскитовой фазы АСоО3, но когда элемент А содержит два или более элементов, например Vv и Vn, необязательно, чтобы присутствовала смешанная Перовскитовая фаза, например Vvx Vn1-x СоО3, где х находится между 0 и 1. Таким образом может присутствовать Перовскитовая фаза, например VnCoO3 или VvСоО3, смешанная с другими фазами, например Vv2O, Vn2О3(VvxVn1-x)2O3 или VvxVn1-xO2.
Как было указано, катализатор может быть в форме, в которой содержание кислорода не стехиометрическое. Это является результатом переменной валентности кобальта, а также любой переменной валентности редкоземельного элемента, присутствующего как часть или всего элемента А.
Настоящий катализатор мажет быть образован путем нагрева композиции, содержащей оксиды кобальта и элемента А, предпочтительно на воздухе, до температуры в интервале 900-1200oC для получения материала, в котором присутствует в качестве свободных оксидов только небольшая часть кобальта.
Композиции можно приготовить путем осаждения, например путем добавки раствора растворимых солей соответствующих металлов в раствор основания, например карбонат аммония или гидроокись для осаждения соответствующих металлов в качестве (основных) карбонатов, гидроокисей или оксидов с последующим прокаливанием для превращения осажденных соединений в оксиды. Применение соединений щелочных металлов в качестве основания для осуществления осаждения менее предпочтительно, поскольку они вызывают неизбежно некоторое загрязнение продукта натрием, который может действовать как яд катализатора. Осаждение можно осуществлять альтернативно, но менее предпочтительно, путем добавки основания в раствор смешанных солей. Либо композицию можно получить путем формирования раствора термически разлагаемых солей, например нитратов или солей органических кислот, например оксалаты или цитраты, металлов в соответствующих пропорциях и испарения раствора до сухости с последующим прокаливанием для осуществления разложения до соответствующих оксидов. Менее предпочтительно композицию можно приготовить путем смешения предварительно формованных оксидов металлов в соответствующих пропорциях.
В другом альтернативном варианте можно использовать часть или весь материал элемента А в качестве носителя, на который наносят покрытие из кобальта и любого оставшегося элемента А. Таким образом тонкоизмельченный оксид элемента А, например оксид церия (3), можно пропитать раствором, содержащим соль кобальта и возможно также соль элемента А, например соль лантана, с последующим разложением солей кобальта и любого элемента А. Или же такой материал на носителе можно получить путем осаждения кобальта и возможно некоторого элемента А в качестве термически разлагаемых соединений на тонкоизмельченную, например осажденную, окись элемента А или соединения, способного разлагаться на нем.
Какой бы ни применяли способ для приготовления композиции оксидов, настоящую композицию необходимо подвергнуть прокаливанию, например, на воздухе при достаточно высокой температуре и в течение достаточно продолжительного периода времени для получения достаточного материала со смешанной структурой оксидов, например Перовскитовой структурой, для объединения большинства, если по существу не всех, свободных оксидов кобальта в одной или более фазах смешанных оксидов. Как было указано, температура прокаливания находится предпочтительно в интервала 900-1200oC. Продолжительность необходимого нагрева будет зависеть от применяемой температуры и применяемого способа приготовления композиции. В том случае, если температура нагрева ниже 1100oC, предпочтителен нагрев, по меньшей мере, в течение 6 часов. С другой стороны, предпочтительна продолжительность нагрева при температуре свыше 1150oC менее 6 часов для уменьшения разложения фаз, содержащих оксид кобальта, на свободный моноксид кобальта. Однако катализаторы, полученные путем испарения раствора, содержащего смесь органических солей, например цитраты соответствующих металлов, до сухости с последующим прокаливанием, могут потребовать термообработки в течение более непродолжительных периодов времени и/или при температурах на 200-300oC ниже температур, необходимых для композиций, полученных, например, осаждением. С другой стороны, если катализатор получают путем прокаливания смеси предварительно формованных оксидов, могут потребоваться более продолжительные периоды времени и/или более высокие температуры для получения материала, в котором присутствует в качестве свободных оксидов только небольшая часть кобальта.
Согласно заявке CN-A-86108985 катализаторы подвергли испытанию в небольшом масштабе с катализаторами в форме слоя крупнозернистого порошка.
По практическим причинам нежелательно применять слой порошкообразного катализатора в установке натурального размера для окисления аммиака, поскольку желательно, чтобы катализатор был в такой форме, чтобы он мог прямо заменить обычно применяемые сетки из благородного металла.
В патенте Чехии, CS 266106 предлагают применять катализатор в форме сетки из нержавеющей стали, имеющей покрытие из смеси оксида кобальта, промотированной небольшим количеством оксида церия (3), окиси хрома (3) и/или окиси алюминия. Однако такие катализаторы, которые содержат значительно больше кобальта, чем это требуется для Перовскитовой структуры, будут неизбежно содержать значительное количество свободных оксидов кобальта.
Для получения соответствующей площади поверхности катализатора при использовании проволочного носителя необходимо снабдить носитель керамическим покрытием, тонким слоем, а затем осадить на этот тонкий слой активный материал. В качестве таких тонких слоев обычно применяют композиции из окиси алюминия или окиси лантана. Однако с обычными высокотемпературными стальными носителями существует риск, что материал тонкого слоя или примеси, оставшиеся в нем, например щелочь, образующаяся в результате применения щелочных растворов алюмината для образования тонкого слоя, могут постепенно диффундировать во время применения в активный материал, нарушать требуемую структуру и мешать каталитическому процессу.
Однако было установлено нами, что при использовании первичных носителей, изготовленных из жаропрочного ферритового сплава, содержащего алюминий, можно получить хорошее сцепление тонкого слоя с первичным носителем без применения щелочных растворов и тем самым можно также устранить проблему миграции щелочных примесей в активные катализаторы.
Каталитические процессы с использованием слоя неупорядоченно упакованных элементов носителя катализатора, имеющих множество сквозных каналов и на которых поддерживается катализатор и в которых элементы носителя можно изготовить из таких сплавов, предложены в патенте Великобритании -А-2077136. В этой ссылке описано окисление аммиака в качестве примера каталитического процесса, для которого можно применять такие элементы. Дана также ссылка на патент Великобритании -А-1568861, в котором описаны способы применения подходящего тонкого слоя, в которых не используют щелочные растворы.
Соответствующие сплавы железа/алюминия являются теми, которые описаны в патенте Великобритании, 2077136, а в частности теми, которые имеют следующий состав по массе, %:
Хром - 10-25
Алюминий - 3-6
Иттрий и/или церий - 0-1
Кобальт - 0-5
Углерод - 0-0,5
Железо (и обычные примеси) - Недостающее до баланса количество
Присутствие иттрия и/или церия предпочтительно, поскольку они оказывают стабилизирующий эффект на оксид алюминия, образованный после обжига сплава или конечного катализатора. Для уменьшения миграции компонентов из сплава или тонкого слоя в активный катализатор возможно также желательно присутствие кобальта; предпочтительные сплавы содержат 15-25% хрома, 4-6% алюминия, 0,3-1% иттрия, церия и/или 1-3% кобальта, 0-0,5% углерода, остальное железо и обычные примеси.
Согласно настоящему изобретению катализатор изготавливают предпочтительно путем образования металлической ткани, сетки или подкладки из проволоки сплава железа/алюминия, нанесения тонкого слоя из оксида алюминия, оксида церия (3), двуокиси циркония или оксида лантана, например, как описано в патенте Великобритании -А-1568861, а затем путем нанесения дисперсии, содержащей композицию активных оксидов, или раствор соединений, способных разлагаться на активные оксиды. Тонкий слой наносят на сплав предпочтительно после осуществления поверхностного окисления сплава путем обжига сплава на воздухе при температуре, например, 1000oC. Тонкий слой наносят предпочтительно в виде золя, а когда используют тонкий слой из оксида алюминия, то он предпочтительно содержит также оксид иттрия и/или оксид церия (3). Покрытую металлическую ткань, сетку или подкладку подвергают затем прокаливанию на воздухе при высокой температуре для уменьшения количества свободных оксидов кобальта. В то же время это прокаливание будет обеспечивать некоторое спекание между смежными проволоками с нанесенным покрытием для связи металлической сетки или подкладки в прочную структуру в токах, где смежные нити проволоки контактируют друг с другом.
Установлено, что во время такого обжига конечной композиции при высокой температуре для образования требуемой структуры из смешанных оксидов с минимальным содержанием свободных оксидов кобальта присутствуют оксид алюминия и оксид лантана, если их используют в качестве тонкого слоя, в виде диффузионных слоев, преходящих в смежные компоненты, но с этого времени они становятся относительно устойчивыми, таким образом во время применения происходит в дальнейшем мало миграции. Вместо применения металлического первичного носителя можно использовать подкладку, или сетку, или ткань, изготовленную из керамики, например альфа-окиси алюминия, волокон или нитей, например, путем ткачества; такой керамический первичный носитель может иметь вторичный носитель в виде тонкого слоя согласно вышеупомянутому.
Альтернативно, вместо применения металлической сетки или подкладки можно применять монолитный носитель в форме сотового или вспененного керамического материала, например оксида алюминия или двуокиси циркония, либо монолитной конструкции, изготовленной из сплава железа/алюминия, например, как предложено в патенте Великобритании -А-2077136, но однако необязательно применять его в форме неупорядочно упакованного слоя элементов согласно патенту Великобритании -А-2077136. Таким образом, можно применять монолитные конструкции с их каналами, ориентированными под заданным углом к направлению потока газа. Как было описано, такие монолитные носители могут также иметь тонкий слой вторичного носителя.
Таким образом, согласно настоящему изобретению предложен катализатор для окисления, содержащий первичный носитель в форме сетки, ткани, подкладки или монолита, образованного из жаропрочного сплава железа/алюминия, или в форме сетки, ткани, подкладки, монолита или пенопласта из керамического материала, вторичный носитель в виде свободного от щелочи тонкого слоя из оксида алюминия или оксида лантана на первичном носителе; а на вторичном носителе активное покрытие из оксидов (а), по меньшей мере, одного элемента А, выбранного из редкоземельных металлов и иттрия, а также из (в) кобальта, причем содержание кобальта и элемента А находится в таких пропорциях, что атомное отношение элемента А к кобальту находится в интервале 0,8-1,2, по меньшей мере, некоторое количестве оксидов кобальта и элемента А присутствует в виде смешанной фазы оксидов, при этом менее 30%, предпочтительно менее 25%, кобальта (по атомам) присутствует в форме свободных оксидов кобальта.
Когда применяют керамическую сотовую конструкцию или пенопласт, то его можно получить из каталитической композиции, таким образом исключается необходимость в отдельном материале для носителя.
Катализаторы в соответствии с настоящим изобретением, особенно те, которые в форме тканей, сеток или подкладок, можно применять в качестве прямой замены обычных катализаторов из благородных металлов по существу без модификации процесса окисления аммиака, за исключением, конечно, того, что можно исключить устройства с обычными ловушками из благородного металла. Во время окисления аммиака до оксида азота для получения азотной кислоты процесс окисления можно осуществлять при температурах 800-1000oC, в частности 850-950oC, давлении 1-15 бар абс. с концентрацией аммиака в воздухе 5-15%, часто примерно 10% по объему.
Кроме их применения для реакций окисления аммиака катализаторы можно также использовать для других окислений.
Настоящее изобретение проиллюстрировано следующими примерами.
Пример 1
Путем смешения раствора нитратов лантана, церия и кобальта в таких пропорциях, чтобы на один атом церия было 3 атома лантана и 4 атома кобальта, приготовили катализатор. Раствор подвергли испарению до сухости, а полученный порошок прокалили на воздухе при температуре 1100oC в течение 8 часов для получения структуры из смешанных оксидов. Термогравиметрический анализ (TGA) показал, что 5,8% атомов кобальта присутствовали в качестве свободного оксида кобальта.
Тем же способом, но исключив нитрат церия и применяя такие пропорции, что на один атом кобальта был один атом лантана, приготовили второй катализатор. Термогравиметрический анализ показал, что 13,3% атомов кобальта присутствовали в виде свободного оксида кобальта.
Катализаторы подвергли испытанию, поместив примерно 0,1 г полученного порошкообразного катализатора в трубку микрореактора и направив через трубку микрореактора смесь гелия, содержащую 5% по объему аммиака и 10% по объему кислорода, с линейной скоростью 5000 м/час. Это соответствует объемной скорости 1,8•106 час-1. Температуру повышали затем от 100oC до 1000oC со скоростью 30oC/мин и анализировали выходящий газ при различных температурах.
Для целей сравнения испытали при тех же условиях подкладку (0,13 г) из 5 слоев металлической ткани и платины/родия (которая, как было установлено, обеспечивает оптимальную избирательность для окисления аммиака до окиси азота).
Избирательность, определенная как [NOJ/([NO] + 2[N2]), где [NO] и [N2] соответственно представляют объемные соотношения окиси азота и азота в выходящем газе при различных температурах, показана в табл. 1.
Пример 2
Смесь соединений лантана, церия и кобальта подвергли осаждению путем постепенной добавки раствора, содержащего нитраты лантана, церия и кобальта в соотношениях атомов La: Ce:Co 4:1:5, в раствор осадителя, содержащий смесь карбоната аммония и щавелевой кислоты. На всем протяжении осаждения смесь непрерывно перемешивали, значение рН поддерживали между 6 и 7, а температуру поддерживали между 48 и 57oC. Суспензии затем дали отстояться, при этом было отмечено образование флоккулированного осадка. Всплывающая жидкость была темно-розового цвета, который указывал на то, что не весь кобальт осадился. Осадок отфильтровали, высушили на воздухе при температуре 120oC в течение 6 часов, а затем прокалили на воздухе при температуре 600oC в течение дополнительных 6 часов. Прокаленный материал разрезали на множество частей, каждая примерно 10 г.
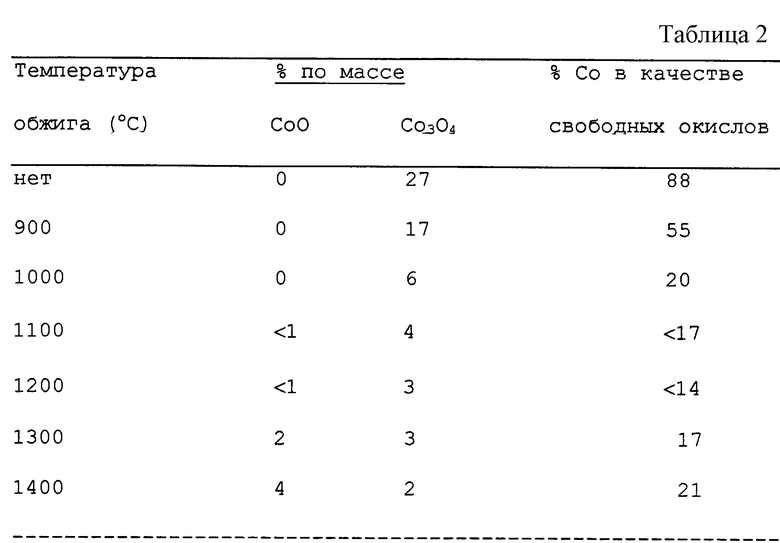
Одну часть прокаливали на воздухе при температуре 900oC в течение 6 часов. Другие части прокаливали на воздухе при температурах 1000oC, 1200oC, 1300oC и 1400oC соответственно. Химический анализ образца, прокаленного при температуре 900oC, показал, что атомные отношения металлов были La:Се:Со 4,6: 1,06: 5, то есть он имел атомное отношение редкоземельного металла к кобальту примерно 1/13, которое согласуется с наблюдением, что не весь кобальт осадился. Рентгеновский флуоресцентный анализ (XRF) показал, что присутствовало 22,6% кобальта по массе катализатора.
Рентгеноструктурный анализ (ХRД) частей, прокаленных при различных температурах, был проведен с использованием двуокиси кремния в качестве внутреннего эталона для определения относительного содержания присутствующих оксидов кобальта (II, III) и моноксида кобальта. По этим данным вычислили атомное относительное содержание кобальта, присутствующего в качестве свободных оксидов кобальта. Результаты представлены в таблице 2.
Исследования электронной микроскопией показывают, что ни один из образцов не содержал Перовскитовую смешанную фазу лантана/церия/кобальта, хотя возможно, что Перовскитовая фаза лантана/кобальта, LаСоО3, присутствующая в образцах, прокаленных при температуре 900oC и выше, содержала небольшое количество (менее примерно 2%) церия. Однако электронная микроскопия показала, что многие частицы имеют Перовскитовую фазу лантана/кобальта, присоединенную или нанесенную на частицы окиси церия и/или окиси церия, легированной лантаном.
Из этих данных можно увидеть, что моноксид кобальта, обнаруженный в образцах, прокаленных при высоких температурах, может образоваться в результате разложения Перовскитовой фазы лантан/кобальт и/или оксиды кобальта (II,III); другие исследования показали, что оксиды кобальта (II,III) обратимо разлагаются, образуя моноксид кобальта при температуре примерно 930oC.
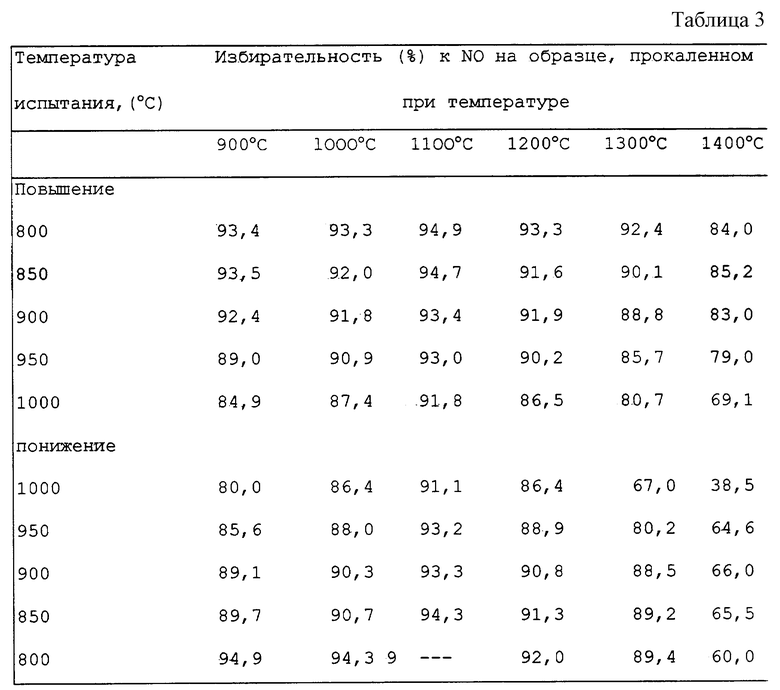
Прокаленные образцы подвергли испытанию на их избирательность для окисления аммиака способом, описанным в Примере 1, за исключением, что после повышения температуры до 1000oC температуру поддерживали на этом уровне в течение 10 минут, а затем ее уменьшали со скоростью примерно 30oC в минуту. Во время повышения и понижения температуры осуществляли анализы выходящего газа. Результаты представлены в таблице 3.
Из этих данных видно, что при высоких рабочих температурах (свыше примерно 900oC) отмечается снижение избирательности для тех образцов, которые имеют большое содержание кобальта в виде свободных оксидов кобальта. Более плохая избирательность образцов во время цикла охлаждения в противоположность циклу нагрева возможно является следствием разложения свободных оксидов кобальта (II,III) до менее избирательного моноксида кобальта при высоких рабочих температурах; это различие между данными цикла нагрева и охлаждения менее очевидное при более низких рабочих температурах возможно в результате образования во время высокотемпературной части операции испытания свободного моноксида кобальта, претерпевающего обратимый переход обратно в оксиды (II, III) кобальта при понижении температуры.
Пример 3
Приготовили примерно 20 кг катализатора способом, описанным в Примере 2, с окончательным его прокаливанием при температуре 900oC в течение 6 часов. Рентгеноструктурный анализ показал, что атомные отношения металлов были La: Се: Со 8,54:2,08:10. Термогравиметрический анализ показал, что 23,8% атомов кобальта присутствовали в форме свободных окислов кобальта.
Из катализатора образовали небольшие цилиндрические гранулы, а образец гранул подвергли испытанию на избирательность способом, описанным в Примере 2. При температуре испытания 900oC избирательность составила 92%.
Остальные гранулы катализатора поддерживали затем на проволочной сетке в качестве катализатора в реакторе для окисления аммиака в промышленной установке для производства азотной кислоты, которая работала в дальнейшем при типичных рабочих условиях установки для производства азотной кислоты (11-12% аммиака в воздухе; рабочее давление =1,1 бар; температура на входе = 200oC; а температура на выходе = 910-925oC) в течение 6 месяцев. Затем взяли для анализа образец катализатора и подвергли его испытанию на избирательность способом из Примера 2. Термогравиметрический анализ показал, что только 5,7% атомов кобальта присутствуют в виде свободных окислов кобальта, а избирательность при температуре испытания 900oC составила 96%.
Эти данные показывают, что уровень свободных оксидов кобальта, присутствующих в катализаторе, значительно уменьшился во время первых 6 месяцев работы при повышенной температуре, а это сопровождалось повышением избирательности. Эксплуатационные качества катализатора после 6 месяцев работы такие же, как у свежего катализатора из платиновой/родиевой сетки.
Операцию по осуществлению способа окисления аммиака продолжили затем в течение добавочных 6 месяцев при тех же условиях, как и прежде, а после этого подвергли анализу другой образец, который показал, что 5,5% атомов кобальта присутствовали в виде свободных оксидов кобальта. Это указывает на то, что содержание свободных окислов кобальта стало стабильным только с очень незначительным дальнейшим снижением, происходящим во время второго 6-месячного периода работы.

Предложен способ окисления аммиака, при котором аммиак и воздух вступают в реакцию в присутствии катализатора окисления, содержащего оксиды (а) церия и лантана и (б) кобальта, причем кобальт, церий и лантан присутствуют в таких пропорциях, что атомное отношение церия плюс лантан к кобальту находится в пределах от 0,8 до 1,2, при этом указанные оксиды присутствуют в качестве фазы смешанного оксида, причем менее 30 ат.% кобальта присутствуют в виде свободных оксидов кобальта. Технический результат - повышение активности и избирательности катализатора, используемого в способе окисления аммиака. 3 табл.
Способ окисления аммиака, при котором аммиак и воздух вступают в реакцию в присутствии катализатора окисления, содержащего оксиды (а) церия и лантана и (б) кобальта, причем кобальт, церий и лантан присутствуют в таких пропорциях, что атомное отношение церия плюс лантан к кобальту находится в пределах от 0,8-1,2, отличающийся тем, что указанные оксиды присутствуют в качестве фазы смешанного оксида, причем менее 30 ат. % кобальта присутствует в виде свободных оксидов кобальта.
| CN 86108985 A, 20.04.1988 | |||
| СПОСОБ ОКИСЛЕНИЯ АММИАКА | 1992 |
|
RU2009995C1 |
| УСТРОЙСТВО для КОНТРОЛЯ ГЕОМЕТРИЧЕСКОЙ ФОРМЫ ЦИЛИНДРИЧЕСКИХ ИЗДЕЛИЙ | 0 |
|
SU384563A1 |
| Устройство для управления процессом термообезмасливания парафина | 1972 |
|
SU562567A1 |

