Данное изобретение относится к лекарственному средству, в частности к амидному производному, обладающему способностью ингибировать Са2+ каналы, активируемые высвобождением Са2+, и к фармацевтической композиции, содержащей его в качестве активного ингредиента, в частности ингибитора Са2+ каналов, активируемых высвобождением или выделением Са2+.
Предпосылки создания изобретения
Давно известно, что ионы кальция (Са2+) важны для внутриклеточного вторичного медиатора при активации различных клеток. Внутриклеточные Са2+ действуют также как важный регуляторный фактор в воспалительных клетках. Однако было высказано предположение, что блокаторы потенциалзависимых Са2+ каналов (далее называемых здесь ПЗКК), такие как нифедипин, не проявляют ингибирующей активности в отношении активации воспалительных клеток и что в воспалительных клетках имеется другой, а не через ПЗКК, механизм всасывания Са2+.
Hoth et al. сообщают, что Са2+ селективные и активируемые истощением запасов Са2+ кальциевые каналы, а именно активируемые выделением Са2+ каналы (здесь далее называемые "АВККК", также называемые зависимыми от накопления Са2+ каналами), присутствуют в тучных клетках и лимфоцитах, и эти клетки не чувствительны к мембранному потенциалу (Pflugers Arch., 430, pp. 315-322 (1955)). Известно, что АВККК есть у нескольких видов клеток, участвующих в воспалительном процессе, таких как тучные клетки, лимфоциты, астроциты (J. Biol. Chem., 270, pp. 29-32 (1995)) и тому подобные, и что это глубоко связано, например, с продукцией цитокинов и выделением липидного медиатора (J. Immunol. , 155, pp. 285-296 (1995) и Br. J. Pharmacol., 114, pp. 598-601 (1995)).
Недавно было обнаружено, что противоартритное средство, тенидап, обладает активностью блокатора АВККК (Cell Calcium, 14, pp. 1-16 (1933)). Следовательно, существует возможность терапевтического действия блокатора АВККК при хронических воспалительных заболевания, включая ревматоидный артрит.
Известно, что АВККК присутствуют также и у эндотелиальных клеток (Am. J. Physiol. , 269, с. 733-738 (1995)) и у эпителиальных клеток (J. Biol. Chem., pp. 29169-29175 (1995)). Так как сообщалось, что непрерывное всасывание кальция играет роль в радикальном воздействии на эндотелиальные клетки (Am. J. Physiol. , 261, с. 889-896 (1991), предполагается, что блокатор АВККК должен обладать защитным (профилактическим) действием в отношении связанного с эндотелиальными клетками поражения тканей.
Кроме того, сообщалось, что блокада всасывания кальция подавляет пролиферацию клеток и продукцию интерлейкина 2 (ИЛ-2) (Br. J. Pharmacol., 113, pp. 861-868 (1994)). Следовательно, ингибитор АВККК может использоваться в качестве средства для профилактики и лечения пролиферативных или прогрессирующих заболеваний (например, злокачественных опухолей и тому подобного) и аутоиммунных заболеваний, а также в качестве супрессора отторжения при трансплантации.
С другой стороны, известно, что в чувствительных к раздражению клетках, таких как клетки гладких мышц и нервные клетки, внутриклеточное содержание кальция в основном регулируется с помощью ПЗКК, а не с помощью АВККК. Поэтому ожидается, что блокатор кальциевых каналов, обладающий селективностью в отношении АВККК по сравнению с ПЗКК, может использоваться в качестве средства для профилактики или лечения различных воспалительных заболеваний, аллергических заболеваний, аутоиммунных заболеваний, повреждения тканей, пролиферативных заболеваний и т.п. без нежелательного действия на сердечно-сосудистую и центральную нервную системы.
Недавно было сообщено о некоторых соединениях, проявляющих ингибирующую активность АВККК, таких как производные циклоалкилпиперазинилэтанола, описанные в опубликованном описании патента Германии 4404249, и 2-(3,4-дигидро-1-изохинолил)ацетамидное производное, описанное в WO 94/00435. Также сообщалось, что 5-амино-1-[3,5-дихлор-4-(4-хлорбензоил)фенил] метил] -1Н-1,2,3-триазол-4-карбоксамид ингибирует АВККК (J. Pharm. Exp. Ther., 257, pp. 967-971 (1991)). Однако не описаны соединения, чья селективность в отношении АВККК по сравнению с ПЗКК была бы подтверждена.
С другой стороны, в опубликованном описании патента Германии 2525024 описано 5-(гетероциклоиламинофенил)-1-фенилпиразольное производное, которое проявляет противовоспалительное действие. Однако в этом патенте не описывается и не предполагается его ингибирующая активность в отношении АВККК и продуцирования ИЛ-2.
В WO 95/18097 описывается производное антраниловой кислоты, представленное следующей формулой (I), которое ингибирует циклическую ГМФ-фосфодиэстеразу. В формуле R1-R4 представляют Н, галоген,..., пиразолил, который может быть замещен,...; n равно 0-6, W представляет N или СН, Y представляет О или S,... (подробнее см. указанную опубликованную патентную заявку).
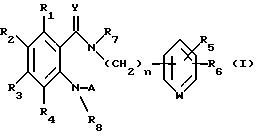
В не прошедшей экспертизу, опубликованной Японской патентной заявке 9-59236 описано R1, R2-дизамещенное бензамидное производное, представленное следующей формулой (1), которое может использоваться для профилактики и лечения ревматических, аллергических и других воспалительных заболеваний. В формуле R1 представляет замещенное или незамещенное ароматическое гетероциклическое кольцо,..., R2 представляет галоген, нитро, -NR5R6,..., А представляет -C(=Z)NR3R4 или -NR4C(=Z)R3, R3 представляет замещенное или незамещенное ароматическое углеводородное кольцо, замещенное или незамещенное ароматическое гетероциклическое кольцо (подробнее см. указанную опубликованную патентную заявку). Однако не существует иллюстративного описания пиразолильной группы в качестве ароматического гетероциклического кольца. Кроме того, не существует описание ингибирующей активности в отношении АВККК и/или продуцирования ИЛ-2.
Описание изобретения
Заявители по данному изобретению провели обширные исследования по отбору соединений, обладающих превосходной ингибирующей АВККК активностью. В результате этих усилий было обнаружено, что некоторые амидные производные, которые обладают совершенно другими структурами, чем те, о которых сообщалось как об ингибиторах АВККК, проявляют превосходную ингибирующую активность АВККК. Данное изобретение было завершено дополнительным обнаружением того, что эти соединения обладают высокой селективностью в отношении АВККК по сравнению с ПЗКК.
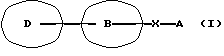
Соответственно данное изобретение относится к новому амидному производному, представленному следующей общей формулой (I)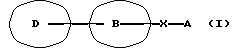
[символы в формуле имеют следующие значения:
D представляет пиразолильную группу, которая может содержать от 1 до 3 галогенозамещенных или незамещенных низших алкильных групп в качестве заместителя(ей),
В представляет фениленовую или тиофендиильную группу,
Х представляет группу формулы -NH-CO- или -CO-NH-,
А представляет фенильную группу, которая может быть замещена одним или нескольким атомами галогена, или пяти- или шестичленную моноциклическую гетероарильную группу, которая может быть замещена одной или несколькими низшими алкильными группами, при условии, что исключены 4-метил-4'-[3,5-бис(трифторметил)-1Н-пиразол-1-ил] -1,2,3-тиадиазол-5-карбоксианилид (далее указан здесь как "соединение А") и 4'-хлор-5-(1-метил-5-трифторметил-1Н-пиразол-3-ил)тиофен-2-карбоксианилид (далее указан здесь как "соединение Б"); и далее так же] или его фармацевтически приемлемой соли.
В связи с этим соединения А и Б являются известными соединениями, описанными как SEW 04225 и KM 02904 в каталоге реагентов, опубликованном MAYBRIDGE (UK, Cornwall, August, 1995). Однако сообщения об их фармацевтическом использовании и других применениях отсутствуют.
Предпочтительное соединение общей формулы (I) по настоящему изобретению является амидным производным или его фармацевтически приемлемой солью, в котором D представляет пиразолильную группу, замещенную по меньшей мере одной трифторметильной группой, или D представляет 1-метил-3-трифторметил-1Н-пиразол-5-ил или 3,5-бис(трифторметил)-1Н-пиразол-1-ил, и А представляет фенильную группу, которая может быть замещена одним или несколькими атомами галогена, или пяти- или шестичленную моноциклическую гетероарильную группу, выбранную из группы, включающей тиазолил, тиадиазолил, тиенил и пиридил, которые могут быть замещены одной или несколькими алкильными группами.
Данное изобретение относится также к фармацевтической композиции, которая содержит амидное производное, представленное следующей общей формулой (I'), включающей соединения А и Б, или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель, в частности к фармацевтической композиции для использования при ингибировании высвобождения Са2+, активируемого Са2+ каналами.
Предпочтительно оно является ингибитором продуцирования ИЛ-2, средством для использования при профилактике и лечении аллергических, хронических воспалительных или аутоиммунных заболеваний или средством для использования при профилактике и лечении бронхиальной астмы или ревматоидного артрита.
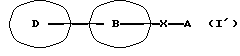
[символы в формуле имеют следующие значения:
D представляет пиразолильную группу, которая может содержать от 1 до 3 галогенозамещенных или незамещенных низших алкильных групп в качестве заместителя(ей),
В представляет фениленовую или тиофениленовую группу,
Х представляет группу формулы -NH-CO или -CO-NH-,
А представляет фенильную группу, которая может быть замещена одним или нескольким атомами галогена, или пяти- или шестичленную моноциклическую гетероарильную группу, которая может быть замещена одной или несколькими низшими алкильными группами; и далее так же].
Если не указано иного, термин "низший", как он использован здесь, означает прямую или разветвленную углеродную цепь, содержащую от 1 до 6 атомов углерода. "Низшая алкильная группа" представляет предпочтительно метил, этил или пропил. "Пяти- или шестичленная моноциклическая гетероарильная группа" представляет пяти- или шестичленную гетероарильную группу, содержащую от 1 до 4 гетероатомов, выбранных из атома азота, атома серы и атома кислорода, и является предпочтительно тиенилом, тиазолилом, тиадиазолилом или пиридилом. Предпочтительно "фениленовая группа" представляет 1,4-фенилен, и "тиофендиильная группа" является 2,5-тиофендиилом.
"Галоген" предпочтительно является F или Сl. "Галогенозамещенная низшая алкильная группа" является предпочтительно трифторметилом.
Соединение по данному изобретению может существовать в виде геометрических изомеров или таутомеров в зависимости от видов групп заместителей, и эти изомеры в разделенной форме или в смеси включены в настоящее изобретение. К тому же соединение по настоящему изобретению может иметь асимметрические атомы углерода, то есть может существовать в виде (R) и (S) оптического изомера на основе таких углеродных атомов. Все эти смеси и отдельные виды этих оптических изомеров включены в настоящее изобретение.
Соединение (I) или (I') по данному изобретению может образовывать соль присоединения кислоты или в зависимости от вида групп заместителей соль с основанием. Такие соли являются фармацевтически приемлемыми солями и их предпочтительные примеры включают соли присоединения неорганических кислот (например, хлороводородной кислоты, бромоводородной кислоты, йодоводородной кислоты, серной кислоты, фосфорной кислоты и т.п.) или органических кислот (например, муравьиной кислотой, уксусной кислотой, пропионовой кислотой, щавелевой кислотой, малоновой кислотой, янтарной кислотой, фумаровой кислотой, малеиновой кислотой, молочной кислотой, яблочной кислотой, винной кислотой, лимонной кислотой, метансульфоновой кислотой, этансульфоновой кислотой, п-толуолсульфоновой кислотой, аспарагиновой кислотой, глютаминовой кислотой и т. п. ) и соли с неорганическими основаниями (например, натрия, калия, магния, кальция, алюминия и т. п.) или органическими основаниями (например, метиламином, этиламином, этаноламином, лизином, орнитином и т.п. ), а также аммониевые соли.
Кроме того, в это изобретение включены различные гидраты и соль ваты и полиморфизм соединения (I) или (I') или их солей.
(Способ получения)
Соединение по настоящему изобретению и его фармацевтически приемлемая соль могут быть получены путем использования характерных признаков его основной структуры или определенных видов его заместителей и путем применения различных известных методов синтеза. В этом случае в зависимости от вида каждой функциональной группы иногда может быть эффективно с точки зрения методики получения заменить указанную функциональную группу соответствующей защитной группой, а именно группой, которая может быть легко преобразована в указанную функциональную группу на стадии исходного продукта или промежуточных соединений. Затем интересующее может быть получено путем удаления защищающей группы, как требуется. Примеры таких функциональных групп включают гидроксильную группу, карбоксильную группу и т.п., и примеры их защитных групп включают те, которые описаны в "Protective Groups in Organic Synthesis", 2nd edition, изданное Greene and Wuts, которые могут необязательно использоваться в зависимости от условий реакции.
В нижеследующем описаны типичные способы получения соединения по настоящему изобретению.
Способ получения 1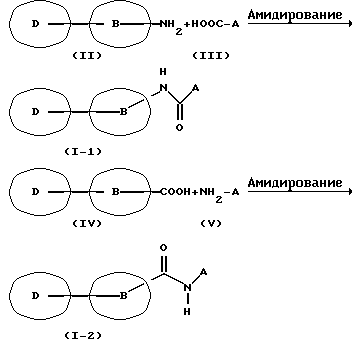
По этому способу, как показано на вышеприведенной схеме реакции, соединение (I-1) или (I-2) по настоящему изобретению получается путем осуществления реакции амидирования производного амина, представленного общей формулой (II) или (V) с производным карбоновой кислоты, представленным общей формулой (III) или (IV).
Производное карбоновой кислоты (III) или (IV), которое может использоваться по способу получения 1, является свободной карбоновой кислотой или ее реакционноспособным производным, и примеры реакционноспособного производного включают галогенангидриды кислот, такие как хлорангидриды кислот, бромангидриды кислот и т.п.; азиды кислот; активные сложные эфиры, которые могут быть получены с использованием метанола, этанола, бензилового спирта, фенола, которые могут быть замещены, 1-гидроксибензотриазола, N-гидроксисукцинимида и т.п.; симметричных ангидридов кислот, и смешанных ангидридов кислот с этоксикарбонилхлоридом, изобутилкарбонилхлоридом, алкилкарбоновой кислотой, п-толуолсульфоновой кислотой и т.п. Эти реакционноспособные производные коммерчески доступны или могут быть получены обычными методами.
Реакция амидирования может проводиться обычными методами.
Когда реакцию проводят с использованием свободной карбоновой кислоты, необходимо использовать конденсирующий агент, такой как N,N'-дициклогексилкарбодиимид (ДЦК), 1-(3-диметиламинопропил)-3-этилкарбодиимид (WSCD) или т. п., или активирующий карбоновую кислоту агент, такой как 1,1'-карбонилдиимидазол, N, N'-дисукцинимидилкарбонат, дифенилфосфорилазид, оксихлорид фосфора, трихлоридфосфор, трифенилфосфин/N-бромсукцинимид или т.п.
Реакцию проводят с использованием аминного производного, представленного общей формулой (II) или (V), и производного карбоновой кислоты, представленного общей формулой (III) или (IV), в эквимолярном количестве, или одного из них в избыточном количестве, в реакционноинертном органическом растворителе, таком как пиридин, тетрагидрофуран (ТГФ), диоксан, эфир, бензол, толуол, дихлорметан, 1,2-дихлорэтан (ДХЭ), хлороформ, диметилформамид (ДМФ), этилацетат, ацетонитрил или т.п. Температура реакции выбирается произвольно в зависимости от типа реакционных производных.
В зависимости от вида производных реакции в некоторых случаях может быть благоприятным добавление основания, такого как триэтиламин, пиридин, пиколин, N, N-диметиланилин, карбонат калия, гидроксид натрия или т.п., с точки зрения ускорения реакции. Также возможно использование пиридина в качестве растворителя.
Способ получения 2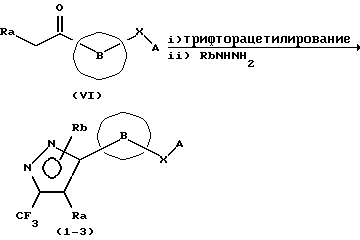
(В вышеприведенной схеме реакции каждый из Ra и Rb представляет Н или низшую алкильную группу).
При этом способе получения соединение (1-3) по данному изобретению получают путем трифторацетилирования соседнего с кетоном атома углерода у соединения, представленного общей формулой (VI), и затем циклизации путем взаимодействия его с производным гидразина.
Первая стадия трифторацетилирования может быть проведена путем взаимодействия соединения с трифторацетилирующим агентом (например, этилтрифторацетатом, ангидридом трифторуксусной кислоты или т.п.) при температуре от -78oС до температуры кипения (с обратным холодильником) в растворителе, таком как метанол, этанол, 1,3-диметилимидазолидин-2-он (ДМИ), ТГФ, ДМФ или т. п. , в присутствии основания, такого как метоксид натрия, этоксид натрия, гексаметилдизилазид щелочного металла, гидрид натрия, алкиллитий, триэтиламин или т.п.
Вторая стадия, реакция циклизации, может быть проведена путем взаимодействия соединения, полученного на первой стадии, с производным гидразина в растворителе, таком как метанол, этанол или т.п., или без растворителя в присутствии или в отсутствии кислоты, такой как уксусная кислота, хлороводородная кислота или т.п., или кислоты Льюиса, такой как изопропоксид титана (IV), хлорид титана (IV), комплекс трифторида бора-диэтилового эфира или т. п. Эту реакцию можно проводить при температуре от температуры охлаждения до температуры кипения с обратным холодильником.
(Способ получения исходных соединений)
Исходные соединения для вышеизложенных способов получения коммерчески доступны или могут быть легко получены хорошо известными специалистам методами.
Каждый из продуктов реакции, полученных с помощью вышеизложенных способов получения, выделяют и очищают, получая в виде свободного соединения, его соли, гидрата или сольвата. Соль может быть получена с помощью обычного способа получения соли. Выделение и очистку осуществляют путем использования обычно применяемых химических методов, таких как экстракция, концентрирование, выпаривание, кристаллизация, фильтрация, перекристаллизация, различные виды хроматографии и т.п. Различные виды изомеров могут быть выделены обычными методами с использованием физико-химических различий между изомерами. Например, оптические изомеры могут быть разделены обычным методом разделения рацематов, такого как фракционная кристаллизация или хроматография. Кроме того, оптический изомер может быть также синтезирован из соответствующего оптически активного исходного соединения.
ПРОМЫШЛЕННАЯ ПРИМЕНИМОСТЬ
Соединение по настоящему изобретению может быть использовано в качестве активного компонента фармацевтических композиций. Поскольку оно обладает ингибирующей АВККК и продуцирования ИЛ-2 активностью, оно особенно полезно в качестве ингибитора АВККК или продуцирования ИЛ-2.
Оно также, в частности, может использоваться в качестве средства для использования при профилактике и лечении аллергических, хронических воспалительных или аутоиммунных заболеваний, которые связаны с АВККК и/или продуцированием ИЛ-2. В этой связи примеры аллергических, хронических воспалительных или аутоиммунных заболеваний включают различные заболевания, которые связаны с АВККК и/или продуцированием ИЛ-2, такие как бронхиальная астма, псориаз, атопические заболевания, включая атопический дерматит, воспалительные заболевания кишечника, включая болезнь Крона, пептическая язва, гломерулонефрит, гепатит, панкреатит, коллагеноз, ревматоидный артрит, остеоартрит, отторжение трансплантата и т.п.
Применимость соединения по настоящему изобретению при вышеуказанных заболеваниях видна из результатов испытаний in vitro по ингибированию АВККК и продукцирования ИЛ-2, которые будут описаны далее, а также из результатов различных испытаний, проведенных с использованием животных моделей для заболеваний, таких как вызываемая антигеном эозинофилия дыхательных путей в качестве типичной модели бронхиальной астмы, некоторые модели заболеваний, зависящих от Т-клеток и вызываемый коллагеном артрит у мышей. Кроме того, поскольку соединения по данному изобретению обладают также ингибирующим эффектом в отношении продуцирования ИЛ-4, ИЛ-5, ММР-1 и TNFα, такие результаты также подтверждают его применимость при вышеуказанных заболеваниях.
С другой стороны, антипролиферативное действие ингибитора АВККК предполагает, что он должен быть применим при профилактике или лечении пролиферативных и прогрессирующих заболеваний, таких как злокачественные опухоли, артериосклероз, множественный склероз органов, различные типы фиброза, ожоговые рубцы и т.п. Также, поскольку блокатор АВККК ингибирует активацию воспалительных клеток, таких как тучные клетки, лейкоциты и астроциты, которые участвуют в воспалительном процессе в некоторых периферических тканях и тканях мозга, можно ожидать его действия по защите тканей от их поражения, такого как поражение при ишемии-реперфузии, травмах головы, церебральном инфаркте и инфаркте миокарда.
В частности, соединение по настоящему изобретению, которое обладает селективной по отношению ПЗКК ингибирующей активностью АВККК, является полезным, поскольку оно может вызывать ингибирование АВККК без активируемых с помощью ПЗКК нежелательных реакций в центральной нервной системе и сердечно-сосудистой системе и т.п.
Далее представлены некоторые испытания и их результаты для подтверждения фармакологического действия соединения по настоящему изобретению.
(1) АВККК ингибирующая активность
Суспензию клеток Тюрка (Jurkat) (6•106/мл), насыщенную индикаторной на кальций флуоресцентной краской фура-2 (fura-2) (1 мкМ) распределяли порциями по 100 мкл в ячейки 96-ячеечной микроплаты. Увеличение внутриклеточного содержания кальция, стимулированное ингибитором кальциевого насоса тапсигарджином (thapsigargin), вызывали путем добавления в каждую ячейку 100 мкл сбалансированного солевого раствора Хэнкса, содержащее исследуемое лекарство, в концентрации в два раза выше конечной концентрации и 2 мкМ тапсигарджина (конечная концентрация 1мкМ), и через 30 минут после добавления количественно оценивали отношение (R) интенсивности флуоресценции, получаемой при возбуждении светом с длиной волны 340 нм/500 нм и 380 нм/500 нм соответственно. При расчетах R собственную флуоресценцию исследуемого лекарственного вещества измеряли в бесклеточной системе и корректировали влияние собственной флюоресценции на флюоресценцию фура-2.
Внутриклеточную концентрацию кальция определяли с помощью следующей формулы расчета на основе максимальной реакции R (Rmax), получаемой при стимуляции 25 мкМ иономицина, минимальной реакции R (Rmin), получаемой при стимуляции 5 мкМ иономицина + 1 мМ ЭГТА, эффективности флюоресценции (Sb2) связывающего кальций красителя при длине возбуждающей волны 380 нм/500 нм и эффективности флюоресценции (Sf2) красителя диссоциации кальция при длине волны возбуждения 380 нм/500 нм.
Формула расчета: Внутриклеточная концентрация кальция (нМ)=224•[(R-Rmin)/(Rmax-R)]•[Sf2/Sb2].
Используя рассчитанную таким образом внутриклеточную концентрацию кальция в присутствии предварительно определенной концентрации каждого из лекарств и показатели для контрольного растворителя, получали отношение блокады всасывания кальция (блокады АВККК) для расчета его концентрации для 50% подавления АВККК (значение IC50).
Значение IC50 для соединений по примерам с 1 по 6 находилось в интервале 0,51 до 0,050 мкМ.
(2) Селективность ингибирования ДВККК в сравнении с ПЗКК
Суспензию нейробластов крыс PC12-h5 (2•106/мл), насыщенную флюоресцентным красителем - индикатором кальция fura-2 (1мкМ), распределяли порциями по 100 мкл в ячейки 96-ячеечной микроплаты. Повышение внутриклеточного содержания кальция, стимулированное высокой концентрацией хлорида калия, вызывали добавлением в каждую ячейку 100 мкл сбалансированного солевого раствора Хэнкса, содержащего исследуемое лекарственное вещество в концентрации в два раза выше конечной концентрации и 100 мМ КСl (конечная концентрация 50 мМ) и через 30 минут после добавления рассчитывали отношение интенсивности флюоресценции (R) по двум интенсивностям флюоресценции, полученным при возбуждении при длинах волн 340 нм/500 нм и 380 нм/500 нм соответственно. При расчете R собственную флюоресценцию лекарственного вещества измеряли в бесклеточной системе и корректировали влияние собственной флюоресценции на флюоресценцию фура-2.
Значение IC50 ингибирования ПЗКК рассчитывали таким же образом, как и в случае вышеописанного ингибирования АВККК, и сравнивали с ингибированием АВККК.
Ингибирование ПЗКК соединениями по примерам с 1 по 6 было в 16 раз или более слабым, чем их ингибирование АВККК.
(3) Ингибирующее действие на продуцирование ИЛ-2
Ингибирующее действие соединения по изобретению на продуцирование ИЛ-2 клетками Тюрка (Jurkat) испытывали в соответствии с методом, описанным S. Clare Chung et al. in Br. J. Pharmacol., 113: 861-868, 1994, и рассчитывали его значение IC50.
Соединения по данному изобретению показывают значения IC50, равные 1 мкМ или менее.
(4) Действие на модели контактной гиперчувствительности, вызываемой TNCB
Действие соединения этого изобретения на вызываемую TNCB контактную гиперчувствительность у пятинедельных мышей-самцов ICR определяли почти таким же образом, что и методом, описанным в Current Protocols in Immunology (John Wiley & Sons, Inc., 1994). Соединения по данному изобретению ингибировали вызываемую TNCB контактную гиперчувствительность в степени, зависящей от дозы.
(5) Ингибирующее действие на вызванный конканавалином А (Кон-А) гепатит у мышей
На четырех пятинедельных мышах-самках Balb/c (SLC) это испытание осуществляли методом, подобным методу, описанному G. Tiegs et al. in J. Clin. Invest., 1992, 90: 196-203. Соединения по изобретению ингибировали вызванный Кон-А гепатит в степени, зависящей от дозы.
(6) Ингибирующее действие на вызываемый коллагеном артрит у мышей
Подавляющее действие на артрит у пятинедельных мышей-самцов DBA/1J (Charles River, Japan) определяли методом, подобным методам, описанным Fumio Nishikaku and Yoshihiko Koda in Immunopharmacology, 25, 65-74 (1993) и Fuminori Kato, Manasao Nomura and Kyoto Nakamura in Annals of the Rheumatic Disease, 55, 535-539 (1996). Соединения по данному изобретению показывают значительное ингибирование артрита.
(7) Ингибирующее действие на вызываемую антигеном эозинофилию дыхательных путей у крыс
Подавляющее действие на вызываемую антигеном эозинофилию дыхательных путей у четырехнедельных крыс-самцов BN определяли почти таким же образом, что и по методу, описанному W. Tlwood et al. in Inflamm. Res., 44, 83-86, 1995. В этой связи лекарственное средство вводили за 30 минут перед воздействием антигена в случае внутривенной инъекции или за 1 час до и через 3 часа после воздействия антигена в случае перорального введения.
На этой модели соединения по данному изобретению уменьшали число инфильтрированных лейкоцитов в целом и число инфильтрированных в дыхательные пути эозинофилов.
Фармацевтическая композиция, которая содержит соединение (I') по данному изобретению или его соль и фармацевтически приемлемый носитель, может быть получена обычным методом с использованием по меньшей мере одного из соединений, представленных общей формулой (I'), или его солей и носителя для медицинского использования, наполнителя и других добавок, обычно используемых в фармацевтических препаратах. Его введение может осуществляться или путем перорального введения в виде таблеток, пилюль, капсул, гранул, порошков, растворов и т. п., или путем парентерального введения в виде внутривенных, внутримышечных и тому подобных инъекций, суппозиториев, препаратов для чрескожного всасывания и т.п.
Твердая композиция для использования при пероральном введении в соответствии с данным изобретением применяется в виде таблеток, порошков, гранул и т.п. В такой твердой композиции одно или несколько активных веществ смешиваются с по меньшей мере одним инертным разбавителем, таким как лактоза, маннит, глюкоза, гидроксипропилцеллюлоза, микрокристаллическая целлюлоза, крахмал, поливинилпирролидон или силикат алюминия-магния. По обычным технологическим процессам композиция может содержать другие добавки помимо инертного разбавителя, такие как смазывающее средство (например, стеарат магния или т. п. ), дезинтегрирующее средство (например, целлюлозогликолят кальция или т. п.), стабилизирующее средство (например, лактоза и т.п.) и способствующее солюбилизации средство (например, глютаминовую кислоту, аспарагиновую кислоту или т. п.) Если необходимо, таблетки или пилюли могут быть покрыты пленками сахара или веществом, растворимым в желудке или кишечнике, таким как сахароза, желатин, гидроксипропилцеллюлоза, фталат гидроксипропилметилцеллюлозы или т.п.
Жидкая композиция для перорального введения включает фармацевтически приемлемые эмульсии, растворы, суспензии, сиропы, эликсиры и т.п. и содержит обычно используемый инертный разбавитель, такой как очищенная вода или этанол. В дополнение к инертному растворителю эта композиция может также содержать вспомогательные вещества, такие как увлажняющее средство, суспендирующее средство и т. п. , а также подсластители, улучшающее вкус и запах средство, ароматизаторы и антисептики.
Инъекционные препараты для парентерального введения включают асептические водные или неводные растворы, суспензии и эмульсии. Примеры растворителя для использования в водных растворах и суспензиях включают дистиллированную воду для инъекций и физиологический раствор. Примеры растворителя для использования в неводных растворах и суспензиях включают пропиленгликоль, полиэтиленгликоль, растительное масло (например, оливковое масло или т.п.), спирт (например, этанол и т.п.) и полисорбат 80. Такая композиция может дополнительно содержать вспомогательные средства, такие как антисептическое, увлажняющее средство, эмульгирующее средство, диспергирующее средство, стабилизирующее средство (например, лактозу) и способствующее солюбилизации средство (например, глютаминовую кислоту или аспарагиновую кислоту). Эти композиции стерилизуются путем фильтрации через задерживающий бактерии фильтр, добавления бактерицидного вещества или облучения. Альтернативно они могут использоваться путем первоначального изготовления твердых стерильных композиций и затем их растворения в стерильной воде или стерильном растворителе для использования для инъекций перед их применением.
В случае перорального введения соответствующая суточная доза обычно составляет от примерно 0,001 до 10 мг/кг веса тела, и суточная доза вводится один раз в сутки или разделена на от 2 до 4 приемов в сутки. В случае внутривенной инъекции соответствующая суточная доза обычно составляет от примерно 0,0001 до 1 мг/кг веса тела и суточная доза вводится один раз в сутки или делится на некоторое число введений в сутки. Решение о дозе принимается произвольно, принимая во внимание симптомы, возраст, пол и т.п. каждого пациента, нуждающегося в лечении.
НАИЛУЧШИЙ СПОСОБ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
Данное изобретение подробно описано далее на следующих примерах. Должно быть понятно, однако, что соединения данного изобретения не ограничиваются соединениями, описанными в следующих примерах.
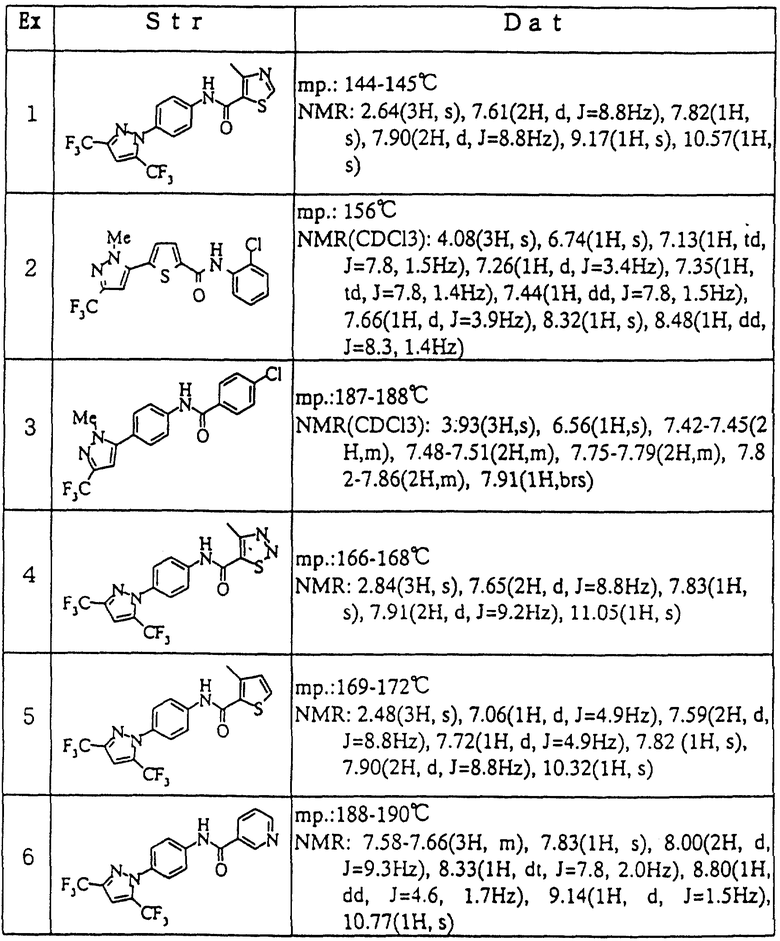
Пример 1
Смесь 4-метилтиазол-5-карбоновой кислоты (108 мг), 4-[3,5-бис(трифторметил)-1Н-пиразол-1-ил] анилина (223 мг), гидрохлорида WSCD (152 мг) и DCE (5 мл) перемешивали в течение ночи при комнатной температуре. К реакционной смеси добавляли воду (10 мл) и полученный таким образом продукт экстрагировали смешанным растворителем из диэтилового эфира (5 мл) и этилацетата (10 мл). Экстракт промывали 1н соляной кислотой, насыщенным водным раствором гидрокарбоната натрия и насыщенным раствором соли, в этом порядке. Полученный органический слой сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Полученный таким образом остаток очищали с помощью колоночной хроматографии на силикагеле (элюент; н-гексан: этилацетат= 2: 1) и затем перекристаллизовывали из смешанного растворителя, состоящего из этилацетата и н-гексана, с получением 4-метил-4'-[3,5-бис(трифторметил)-1Н-пиразол-1-ил] тиазол-5-карбоксианилида (143 мг) в виде бесцветных игл.
Пример 2
Смесь 5-(1-метил-3-трифторметил-1Н-пиразол-5-ил)тиофен-2-карбонилхлорида (150 мг) с дихлорметаном (1,5 мл) добавляли к смеси 2-хлоранилина (68 мг), пиридина (42 мг) и дихлорметана (2 мл) и перемешивали в течение 30 минут при комнатной температуре. К реакционной смеси добавляли насыщенный водный раствор гидрокарбоната натрия, и полученный таким образом продукт экстрагировали этилацетатом и затем экстракт промывали насыщенным раствором соли. Полученный органический слой сушили над безводным сульфатом магния и затем концентрировали при пониженном давлении. Полученный остаток перекристаллизовывали из этанола с получением 2'-хлор-5-(1-метил-3-трифторметил-1Н-пиразол-5-ил)тиофен-2-карбоксианилида (80 мг) в виде бесцветных кристаллов. В этом случае вышеуказанное исходное соединение 5-(1-метил-3-трифторметил-1Н-пиразол-5-ил)тиофен-2-карбонилхлорид получали в виде коричневого твердого вещества путем обработки 5-(1-метил-3-трифторметил-1Н-пиразол-5-ил)тиофен-2-карбоновой кислоты оксалилхлоридом.
Пример 3
Метоксид натрия (257 мг) добавляли к смеси 4'-ацетил-4-хлорбензанилида (1,00 г) и ДМИ (10 мл) при 0oС и полученную таким образом смесь перемешивали в течение 2 часов при комнатной температуре. К реакционному раствору добавляли этилтрифторацетат (0,522 мл) и перемешивали в течение 2 дней при 60oС. К реакционной смеси добавляли воду (50 мл) и 1н соляную кислоту (10 мл) и полученный таким образом продукт экстрагировали этилацетатом и затем экстракт промывали водой и насыщенным раствором соли, в этом порядке. Полученный таким образом органический слой сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении. Смесь полученного таким образом остатка с метилгидразином (0,206 мл), уксусной кислотой (2 мл) и этанолом (20 мл) перемешивали в течение 21 часа при комнатной температуре. После концентрирования реакционной смеси при пониженном давлении полученный таким образом остаток добавляли к этилацетату (100 мл) и промывали насыщенным водным раствором гидрокарбоната натрия и насыщенным раствором соли. Полученный таким образом органический слой сушили над безводным сульфатом натрия и затем концентрировали при пониженном давлении. Полученный таким образом остаток очищали с помощью колоночной хроматографии на силикагеле (элгоент; н-гексан: этилацетат = 3:1) и затем перекристаллизовывали из смешанного растворителя, состоящего из этилацетата и н-гексана, с получением 4-хлор-4'-(1-метил-3-трифторметил-1Н-пиразол-5-ил) бензанилида (440 мг) в виде бесцветного кристаллического порошка.
Соединения по примеру 4: (4-метил-4'[3,5-бис-(трифторметил)-1Н-пиразол-1-ил] -1,2,3-тиадиазол-5-карбоксианилид), примеру 5: (3-метил-4'-[3,5-бис-(трифторметил)-1Н-пиразол-1-ил] тиофен-2-карбоксианилид) и примеру 6: (4'-[3,5-бис(трифторметил)-1Н-пиразол-1-ил] никотинанилид), представленные в таблице, были соответственно получены таким же образом, который описан в примере 1.
Структуры и физико-химические свойства соединений по примерам представлены в таблице. В таблице Ех означает примера, Str означает структурная формула, Dat означает физико-химические свойства, mр. означает температуру плавления, NMR означает спектр ядерно-магнитного резонанса (ДМСО-d6, ТМС внутренний стандарт) δ м.д. и NMR (СDСl3) означает спектр ядерно-магнитного резонанса (CDCl3, ТМС внутренний стандарт) δ м.д., Hz=Гц, s=с, d=д, td=тд, dd=дд, m=м, brs=шир. с., dt=дт.
| название | год | авторы | номер документа |
|---|---|---|---|
| ТРИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ ПИРАЗОЛА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1998 |
|
RU2191176C2 |
| ПРОИЗВОДНЫЕ АЗОЛА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ | 1995 |
|
RU2161612C2 |
| ПРОИЗВОДНОЕ БЕНЗАЗЕПИНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ПРОИЗВОДНОЕ ДИФТОРБЕНЗАЗЕПИНА И ПРОИЗВОДНОЕ (ЗАМЕЩЕННОГО) АМИНОБЕНЗОИЛДИФТОРБЕНЗАЗЕПИНА | 1994 |
|
RU2137760C1 |
| АРИЛЭТЕНСУЛЬФОНАМИДНЫЕ ПРОИЗВОДНЫЕ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1996 |
|
RU2172735C2 |
| ПРОИЗВОДНОЕ 4,4-ДИФТОР-1,2,3,4-ТЕТРАГИДРО-5Н-БЕНЗАЗЕПИНА, ЕГО СОЛЬ И СОДЕРЖАЩАЯ ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2002 |
|
RU2268882C1 |
| НОВЫЕ ПРОИЗВОДНЫЕ ХИНУКЛИДИНА И МЕДИЦИНСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 1995 |
|
RU2143432C1 |
| КОНДЕНСИРОВАННОЕ ПРОИЗВОДНОЕ ТИАЗОЛА ИЛИ ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМАЯ СОЛЬ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ПРОЯВЛЯЮЩАЯ АКТИВНОСТЬ АГОНИСТА 5-НТ*003-РЕЦЕПТОРА НА ЕГО ОСНОВЕ | 1995 |
|
RU2098418C1 |
| 1,2,3,4-ТЕТРАГИДРОХИНОКСАЛИНДИОНОВЫЕ ПРОИЗВОДНЫЕ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1995 |
|
RU2149873C1 |
| ПРОИЗВОДНОЕ ТИОФЕНА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ | 1997 |
|
RU2172737C2 |
| ПРОИЗВОДНЫЕ АМИДИНОНАФТИЛА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1995 |
|
RU2154633C2 |
Изобретение относится к амидному производному общей формулы I, символы в формуле имеют следующие значения: D представляет пиразолильную группу, которая может иметь 1-3 галогенозамещенных или незамещенных низших алкильных группы в качестве заместител(я)ей, В представляет фениленовую или тиофендиильную группу, Х представляет группу формулы -NH-CO- или -CO-NH- и А представляет фенильную группу, которая может быть замещенной одним или несколькими атомами галогена, или пяти- или шестичленную моноциклическую гетероарильную группу, которая может быть замещенной одной или более из низших алкильных групп. Описана также фармацевтическая композиция на основе заявленных соединений, которая предназначена для ингибирования выделения Са2+ каналов, активируемых выделением Са2+. Соединения могут быть использованы в качестве средства для профилактики или лечения различных воспалительных заболеваний и аллергических заболеваний. 2 c. и 7 з.п. ф-лы, 1 табл.
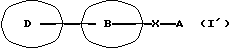
символы в формуле имеют следующие значения:
D представляет пиразолильную группу, которая может иметь 1-3 галогенозамещенных или незамещенных низших алкильных группы в качестве заместител(я)ей;
В представляет фениленовую или тиофендиильную группу;
Х представляет группу формулы -NH-CO- или -CO-NH-;
А представляет фенильную группу, которая может быть замещена одним или несколькими атомами галогена, или пяти- или шестичленную моноциклическую гетероарильную группу, содержащую 1-4 гетероатома, выбранных из атома азота, атома серы и атома кислорода, которая может быть замещена одной или несколькими низшими алкильными группами, при условии, что исключены 4-метил-4'-[3,5-бис(трифторметил)-1Н-пиразол-1-ил] -1,2,3-тиадиазол-5-карбоксианилид и 4'-хлор-5-(1-метил-5-трифторметил-1Н-пиразол-3-ил)тиофен-2-карбоксианилид,
или его фармацевтически приемлемая соль.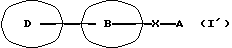
символы в формуле имеют следующие значения:
D представляет пиразолильную группу, которая содержит от 1 до 3 галогенозамещенных или незамещенных низших алкильных групп в качестве заместител(я)ей;
В представляет фениленовую или тиофендиильную группу;
Х представляет группу формулы -NH-CO- или -CO-NH-;
А представляет фенильную группу, которая может быть замещена одним или несколькими атомами галогена, или пяти- или шестичленную моноциклическую гетероарильную группу, содержащую 1-4 гетероатома, выбранных из атома азота, атома серы и атома кислорода, которая может быть замещена одной или несколькими низшими алкильными группами.
| Прибор для очистки паром от сажи дымогарных трубок в паровозных котлах | 1913 |
|
SU95A1 |
| Приспособление в пере для письма с целью увеличения на нем запаса чернил и уменьшения скорости их высыхания | 1917 |
|
SU96A1 |
| Способ получения 1-арил-2/1-имидазолил/алкильных эфиров,тиоэфиров или их солей | 1976 |
|
SU622405A3 |

