Предпосылки изобретения
С-нуклеозиды являются вызывающими интерес соединениями, которые имеют высокую активность как фармацевтические агенты. Одно из этих соединений, Тиазофурин, [6,2-(β-D-рибофуранозил)тиазол-4-карбоксамид)] , обладает значительной активностью как против линии лимфоидных клеток человека (F.Earle and R. I. GIaser, Cancer Res., 1983, 43, 133), линии клеток опухоли легкого (D. N.Carnex, G.S. Abluwalia, H.N.Jayaram, D.A.Cooney and D.G.Johns, J.CIin. Invest., 1985, 75, 175), так и против клеток рака яичника человека, имплантированных мыши (J. P. Micha, P.R.Kuchera, C.N.Preve, M.A.Rettenmaier, J.A. Stratton, P.J.DiSaia, Gynecol.Oncol., 1985, 21, 351). Тиазофурин также эффективен при лечении острой миелоидной лейкемии (G.T.Tricot, H.N.Jasyaram, C. R. NichoIs, K. Pennington, E.Lapis, G.Weber и R.Hoffman, Cancer Res., 1997, 47, 4988). Кроме того, недавние исследования вызвали интерес к Тиазофурину как возможному средству для лечения пациентов с хронической миелоидной лейкемией (ХМЛ) при бластном кризисе (G.Weber, патент США, 5405837, 1995). В клетках Тиазофурин превращается в свой активный метаболит, тиазол-4-карбоксамидадениновый динуклеотид (ТАД), который ингибирует IMP дегидрогеназу и в результате истощает пулы гуанозинового нуклеотида (E.OIah, Y.Natusmeda, T. lkegami, Z.Kote, M.Horanyi, I.Szeleney, E.Paulik, T.Kremmer, S.R.Hollan, J. Sugar and G.Weber, Proc.Natl. Acad.Sci.USA, 1988, 85, 6533).
Хотя Тиазофурин известен в течение более 15 лет, и в настоящее время испытания находятся в фазе II/III на людях, не существует подходящего способа синтеза для крупномасштабного производства. Тиазофурин впервые был синтезирован независимо M.Fuertes et al. (J.Org.Chem., 1976, 41, 4076) и Srivastava et al. (J.Med.Chem., 1977, 20, 256) с низким выходом. В обоих способах авторы получали побочные продукты (то есть соединение 12) и использовали колоночную хроматографию на каждой стадии для очистки продуктов. Основным недостатком этих способов является образование производного фурана, а также использование высокотоксичного газа сульфида водорода.
W. J. Hannon et al. (J.Org.Chem., 1985, 50, 1741) разработали несколько иной способ получения Тиазофурина с выходом 19%. Способ Ханнона (Hannon) также имеет недостатки из-за низкого выхода, использования газа H2S и хроматографических очисток. Недавно P. Vogel et al. (Helv.Chem.Acta., 1989, 72, 1825) синтезировал Тиазофурин в девять стадий с выходом 25%. Позднее D.C. Humber et al. (J. Chem.Soc.Perkin Trans. 1, 1990, 283) разработали синтез Тиазофурина, исходя из бензил(2,3,5-три- -бензиоил-β-D-рибофуранозил)пенициллината.
-бензиоил-β-D-рибофуранозил)пенициллината.
Единственным известным способом, который является совершенно пригодным для крупномасштабного производства, является способ Парсонса (США 4451684). К сожалению, в способе Парсонса используют как цианид ртути, так и сульфид водорода, причем оба они создают проблемы безопасности и окружающей среды. Кроме того, способ Парсонса также приводит к получению смеси продуктов.
Рассмотренные выше проблемы, которые сопутствуют крупномасштабному производству Тиазофурина, характерны и для крупномасштабного производства других С-нуклеозидов. В производстве тиокарбоксамидов, например, в большинстве известных способов используют газообразный сульфид водорода как реагент для превращения цианогруппы в соответствующую тиокарбоксамидную группу. Такие способы создают проблемы для окружающей среды. В производстве С-нуклеозидов вообще большинство или все известные синтезы приводят к получению смеси продуктов в ходе стадии замыкания кольца. Таким образом, существует насущная потребность в новой процедуре для крупномасштабного производства Тиазофурина и других С-нуклеозидов.
Сущность изобретения
Настоящее изобретение относится к новому способу синтеза С-нуклеозидов, при котором получают производное сахара по положению C1 в одну стадию с получением гетероцикла, а затем этот гетероцикл ароматизируют в ходе другой одной стадии.
В одной группе предпочтительных воплощений цианосахар превращают в тиокарбоксамид и после этого конденсируют с образованием азольного кольца. Во второй группе предпочтительных воплощений цианосахар конденсируют с аминокислотой с получением азольного кольца. В третьей группе предпочтительных воплощений галогеносахар конденсируют с предварительно образованным гетероциклом с получением азольного кольца.
У способа по настоящему изобретению имеется много преимуществ. Одним преимуществом является то, что данный способ исключает необходимость в использовании газообразного сульфида водорода, который небезопасен для окружающей среды. Другим преимуществом является то, что выход значительно выше по сравнению с предыдущими способами. Третьим преимуществом является то, что способ по настоящему изобретению исключает необходимость процедуры хроматографической очистки, что дает возможность снизить издержки производства.
Эти и различные другие задачи, признаки, аспекты и преимущества настоящего изобретения станут более понятны из последующего подробного описания предпочтительных воплощений изобретения вместе с сопровождающими графическими материалами, в которых одинаковые цифры представляют одинаковые соединения.
Краткое описание графических материалов
Фиг. 1 - серия реакционных схем, демонстрирующих различные воплощения настоящего изобретения.
Фиг.2 - другая серия реакционных схем, демонстрирующих различные воплощения настоящего изобретения.
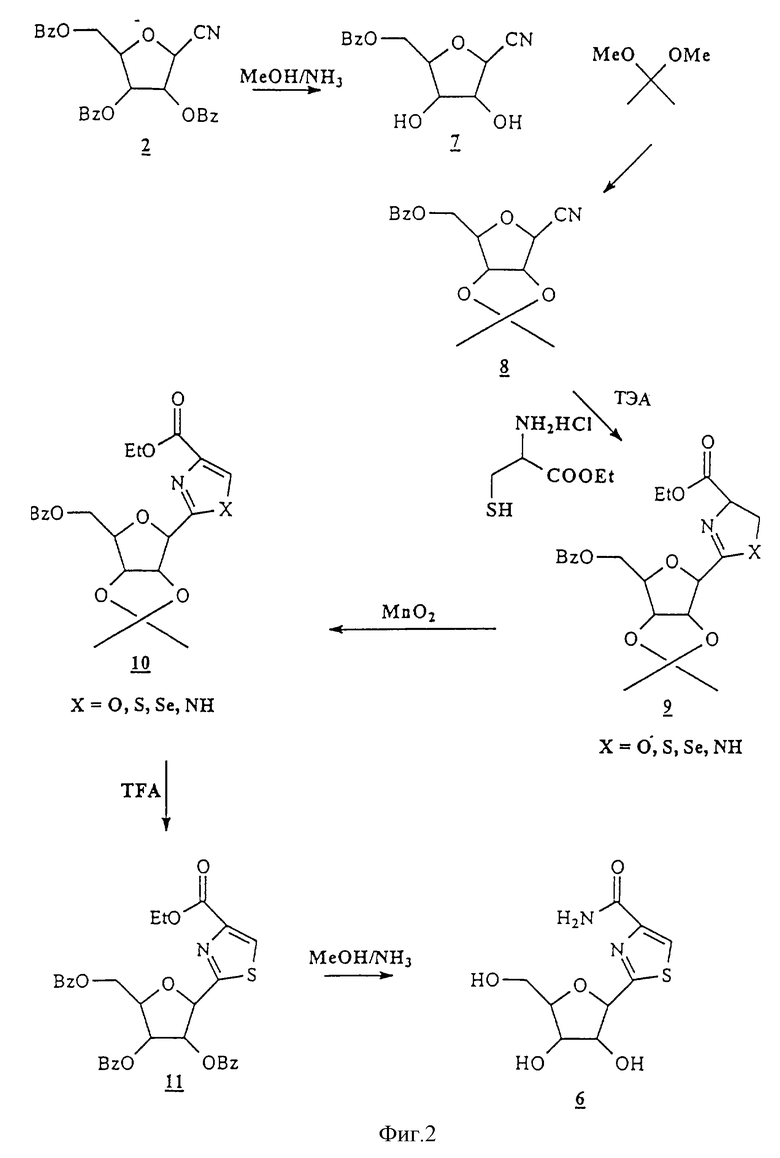
Фиг.3 - другая серия реакционных схем, демонстрирующих различные воплощения настоящего изобретения.
Подробное описание
Существует три предпочтительные группы способов для осуществления настоящего изобретения, каждая из которых проиллюстрирована относительно производства Тиазофурина на фиг.1 - 3.
В первой предпочтительной группе воплощений цианосахар конвертируют в тиокарбоксамид и после этого конденсируют с получением азольного кольца. В конкретном примере, показанном на фиг.1, блокированный цианосахар 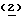 конвертируют в тиокарбоксамид
конвертируют в тиокарбоксамид 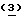 и после этого конденсируют с этилбромпируватом с получением промежуточного соединения Тиазофурина
и после этого конденсируют с этилбромпируватом с получением промежуточного соединения Тиазофурина  . Показанный способ обеспечивает получение Тиазофурина с количественным выходом без каких-либо побочных продуктов
. Показанный способ обеспечивает получение Тиазофурина с количественным выходом без каких-либо побочных продуктов 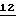 или α-аномера
или α-аномера  ).
).
Во второй группе предпочтительных воплощений цианосахар конденсируют с аминокислотой с получением азольного кольца. В конкретном примере, показанном на фиг.2, известный циано 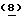 конденсируют с гидрохлоридом этилового эфира цистеина с получением продукта с замкнутым кольцом
конденсируют с гидрохлоридом этилового эфира цистеина с получением продукта с замкнутым кольцом  , который затем ароматизируют с использованием активированного диоксида марганца с получением промежуточного соединения Тиазофурина
, который затем ароматизируют с использованием активированного диоксида марганца с получением промежуточного соединения Тиазофурина 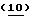 . Это ключевое промежуточное соединение
. Это ключевое промежуточное соединение  подходящим способом трансформируют в Тиазофурин с хорошим выходом.
подходящим способом трансформируют в Тиазофурин с хорошим выходом.
В третьей группе предпочтительных воплощений галогеносахар конденсируют с предварительно образованным гетероциклом с получением азольного кольца. В конкретном примере, показанном на фиг.1, предварительно образованный гетероцикл  конденсируют с известным галогеносахаром
конденсируют с известным галогеносахаром  с получением ключевого промежуточного соединения
с получением ключевого промежуточного соединения  , из которого легко можно получить Тиазофурин.
, из которого легко можно получить Тиазофурин.
Разумеется, способы по изобретению, описанные здесь, не ограничены только производством Тиазофурина, и они без труда могут быть распространены, в том числе особенно вторая и третья группа способов, на практически все С-нуклеозиды. Вообще С-нуклеозид согласно настоящему изобретению подпадает под общую структуру А, где А представляет собой О, S, СН2 или NR, где R представляет собой Н или блокирующую группу; Х представляет собой О, S, Se или NH; R1, R2, R3 и R4 независимо представляют собой Н или низший алкил; и Z1, Z2 и Z3 независимо представляют собой Н или не Н.
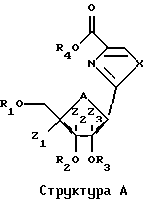
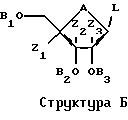
Для различных соединений, охваченных структурой А, может иметь место значительная вариабельность в сахарной части молекулы. Среди прочих обстоятельств, сахар сам по себе не должен быть простым фураном. Например, кислород может быть заменен серой с получением тиосахара или азотом с получением аминосахара. Кроме того, сахар может быть замещен в положениях C'2, С'3 и С'4 группой, иной чем водород. К тому же сахар может иметь D- или L-конфигурацию и может быть альфа- или бета-аномером. К тому же сахар может иметь блокирующие группы на различных стадиях синтеза. Все эти варианты охвачены Структурой Б, где А представляет собой О, S, CH2 или NR, где R представляет собой Н или блокирующую группу; B1, B2 и В3 независимо представляют собой блокирующие группы или низший алкил; и Z1, Z2 и Z3 независимо представляют собой Н или не Н. Группа L является реакционноспособной функциональной группой, такой как CN, галоген или СНО.
Снова обращаясь ко второй группе предпочтительных воплощений, использование гидрохлорида этилового эфира цистеина можно распространить и на использование соединения согласно Структуре В, где Х представляет собой О, S, Se или NH; Y представляет собой Н или низший алкил; и R4 представляет собой Н или низший алкил.

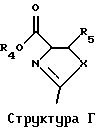
Точно так же в третьей группе предпочтительных воплощений использование предварительно полученного гетероцикла можно распространить на использование соединения согласно Структуре Г, где R4 представляет собой Н или низший алкил, a R5 представляет собой Н.
Существуют, разумеется, многочисленные подходящие блокирующие группы. Среди прочих можно использовать бензоил, бензил, силил или изопропилиден. Кроме того, предполагается, что блокирующие группы в положениях C'2 и С'3 на сахаре могут образовывать изопропилиденовую группу, как показано на Структуре Д.
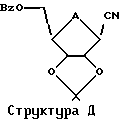
Эта изопропилиденовая группа может быть удалена многими способами, в том числе путем обработки реагентом, выбранным из группы, состоящей из трифторуксусной кислоты, муравьиной кислоты, уксусной кислоты, Н+-смолы в органическом растворителе или иода в метаноле. Применяя настоящий способ к Структуре Д, можно затем получить соединение согласно Структуре Е, где R5 представляет собой Н.
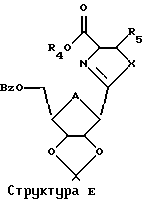
Это воплощение также включает в себя ароматизацию структуры Е с использованием активированного диоксида марганца или других реагентов и с последующим деблокированием защитных групп с получением Тиазофурина или родственных С-нуклеозидов.
Особенно предпочтительные воплощения по изобретению включают в себя реакцию А или реакцию Б, показанные ниже.


Эти и другие признаки станут понятными из следующих рабочих примеров, которые должны расцениваться как иллюстрация различных аспектов заявленного предмета изобретения, а не ограничение объема заявленного предмета изобретения.
Экспериментальная часть
Пример 1
2,3,5-Три-O-бензоил-β-D-рибофуранозил-1-карбонитрил (2)
Смесь 1-O-ацетил-2,3,5-три-O-бензоил-b-D-рибофуранозы (высушенной при 60oС, 1 мм рт.ст., 12 ч) (630 г, 1,249 моль), триметилсилилцианида (высушенного над молекулярными ситами, 24 ч) (250 мл, 1,875 моль) и дихлорметана (высушенного над сульфатом магния и хранившегося над молекулярным ситом) (1,25 л) перемешивали и охлаждали до 0-2oС. Медленно добавляли хлорид олова (50 мл, 0,425 моль) (1,5 часа), поддерживая температуру реакции 0-2oС, и полученную смесь перемешивали и поддерживали температуру от -5 до 0oС в течение дополнительных 1,5 часов. Реакционную смесь медленно (30 минут) и при энергичном перемешивании добавляли к холодному (5oС) 10% раствору гидроксида натрия (1,5 л), который поддерживали при 5-8oС во время добавления. Слои разделяли, и органический слой промывали водой (3•500 мл) до нейтральной реакции и сушили над безводным сульфатом магния (приблизительно 150 г). Смесь фильтровали и высушивающий агент промывали дихлорметаном (3•500 мл). Фильтрат и промывные воды объединяли, и раствор концентрировали (<30oС, 20 мм) до объема 2-2,5 л. Оставшийся раствор фильтровали через фильтр (13,5 см внутренний диаметр • 6,5 см) из Силикагеля и фильтр далее элюировали дихлорметаном (2,5 л). Растворы дихлорметана объединяли и выпаривали (<30oС, 20 мм) до сиропа (приблизительно 750 мл). Этот сироп смешивали с этанолом (1,5 л), и смесь нагревали (приблизительно 60oС) с получением гомогенного раствора. Добавляли затравочные кристаллы 2,3-ангидро-3,4,6-три-O-бензоил-β-D-аллононитрила, и раствор перемешивали при температуре окружающей среды в течение 2 часов, а затем медленно охлаждали до 0oС в течение 2-часового периода. Кристаллическое твердое вещество собирали, промывали холодным (-5oС) этанолом (3•600 мл), промывали гексанами (600 мл) и сушили при 45oС и 1 мм рт. ст. в течение 12 час; 452 г (0,959 моль, 76%), т.пл. 73-75oС (справочная т. пл. 78-80oС). 1H ЯМР (ДМСО-d6): d 4,61 (m, 2), 4,80 (q, 1), 5,88 (t, 1), 6,05 (t, 1), 7,45-7,57 (m, 6), 7,64-7.71 (m, 4), 7,88-7,94 (m, 4) и 8.07 (d, 2).
Пример 2
2,5-Ангидро-3,4,6-три-O-бензоил-D-аллонтиоамид (3)
Способ А: Сульфид водорода пропускали через холодную (5oС) перемешиваемую суспензию 2', 3', 5'-три- -бензоил-β-D-рибофуранозилцианида (
-бензоил-β-D-рибофуранозилцианида (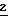 , 50 г, 106,16 моль) в сухом EtOH (900 мл) в течение 5 минут, затем одной порцией добавляли N, N-диметиламинопиридин (1,2 г, 10 ммоль). Сульфид водорода медленно пропускали через перемешиваемую реакционную смесь в течение 5 часов (выходную трубку из реакционной колбы опускали в раствор хлорной извести, приготовленный на 5% NaOH). Через 5 часов колбу герметически закупоривали, и перемешивание продолжали при температуре ниже 25oС в течение 16 часов. Через реакционную смесь пропускали в течение 1 часа аргон для удаления последних следов H2S. Суспензию перемешивали при 0oС в течение 2 часов, и твердое вещество отфильтровывали, промывали холодным сухим EtOH и сушили над P2O5 под вакуумом. Выход 52 г (97%); т.пл. 133-135oС. 1H ЯМР (CDCl3): δ 4,72 (m, 2H), 4,74 (m, 1Н), 5,12 (d, 1H), 5,71 (t, 1H), 5,98 (t, 1H), 7,30-7,60 (m, 10Н), 7,86 (d, 2H), 8,14 (m, 4H) и 8,46 (bs, 1H).
, 50 г, 106,16 моль) в сухом EtOH (900 мл) в течение 5 минут, затем одной порцией добавляли N, N-диметиламинопиридин (1,2 г, 10 ммоль). Сульфид водорода медленно пропускали через перемешиваемую реакционную смесь в течение 5 часов (выходную трубку из реакционной колбы опускали в раствор хлорной извести, приготовленный на 5% NaOH). Через 5 часов колбу герметически закупоривали, и перемешивание продолжали при температуре ниже 25oС в течение 16 часов. Через реакционную смесь пропускали в течение 1 часа аргон для удаления последних следов H2S. Суспензию перемешивали при 0oС в течение 2 часов, и твердое вещество отфильтровывали, промывали холодным сухим EtOH и сушили над P2O5 под вакуумом. Выход 52 г (97%); т.пл. 133-135oС. 1H ЯМР (CDCl3): δ 4,72 (m, 2H), 4,74 (m, 1Н), 5,12 (d, 1H), 5,71 (t, 1H), 5,98 (t, 1H), 7,30-7,60 (m, 10Н), 7,86 (d, 2H), 8,14 (m, 4H) и 8,46 (bs, 1H).
Способ Б: Раствор 2',3',5'-три- -бензоил-β-D-рибофуранозилцианида (
-бензоил-β-D-рибофуранозилцианида ( , 4,71 г, 10,0 ммоль) и тиоацетамида (1,50 г, 20,00 ммоль) в сухом ДМФ (50 мл) насыщали безводным хлороводородом и нагревали при 70-60oС в течение 2 часов. Реакционную смесь охлаждали и выпаривали до сухого состояния. Остаток растворяли в метиленхлориде (150 мл), промывали насыщенным раствором NаНСО3 (100 мл), водой (100 мл) и рассолом (70 мл). Органический экстракт сушили над безводным MgSO4, фильтровали и промывали СН2Cl2 (50 мл). Объединенный фильтрат упаривали до сухого состояния. Остаток растворяли в минимальном количестве сухого этанола, что при охлаждении привело к получению чистого продукта. Выход 4,20 г (83%). Т.пл. и спектральные характеристики этого продукта согласуются с характеристиками продукта, полученного предыдущим Способом А.
, 4,71 г, 10,0 ммоль) и тиоацетамида (1,50 г, 20,00 ммоль) в сухом ДМФ (50 мл) насыщали безводным хлороводородом и нагревали при 70-60oС в течение 2 часов. Реакционную смесь охлаждали и выпаривали до сухого состояния. Остаток растворяли в метиленхлориде (150 мл), промывали насыщенным раствором NаНСО3 (100 мл), водой (100 мл) и рассолом (70 мл). Органический экстракт сушили над безводным MgSO4, фильтровали и промывали СН2Cl2 (50 мл). Объединенный фильтрат упаривали до сухого состояния. Остаток растворяли в минимальном количестве сухого этанола, что при охлаждении привело к получению чистого продукта. Выход 4,20 г (83%). Т.пл. и спектральные характеристики этого продукта согласуются с характеристиками продукта, полученного предыдущим Способом А.
Пример 3
Этиловый эфир 2-(2', 3',5'-три-O-бензоил-β-D-рибофуранозил)тиазол-4-карбоновой кислоты (4)
К перемешиваемой смеси 2,5-ангидро-3',4',6'-три- -бензоил-D-аллонтиоамида (
-бензоил-D-аллонтиоамида ( , 10,12 г, 20,00 ммоль) и твердого NаНСО3 (16,8 г, 200 ммоль) в сухом 1,2-диметоксиэтане (60 мл) при 0oС в атмосфере аргона добавляли этилбромпируват (7,8 г, 40,0 ммоль) в течение 10 минут. После добавления реакционную смесь перемешивали при 0oС в атмосфере аргона в течение 6 часов. Тонкослойная хроматография (ТСХ) показала полную конверсию исходного вещества в единственный продукт (Гексан:ЕtOАс, 7:3). Реакционную смесь охлаждали до -15oС в смеси сухой лед/ССl4 в атмосфере аргона. В течение 15 минут медленно добавляли раствор трифторуксусного ангидрида (12,6 г, 60,00 ммоль) и 2,6-лутидина (12,84 г, 120 ммоль), растворенного в сухом 1,2-диметоксиэтане (20 мл). После добавления реакционную смесь перемешивали при -15oС в течение 2 часов в атмосфере аргона. Реакционную смесь фильтровали, промывали сухим метиленхлоридом (100 мл). Объединенный фильтрат выпаривали до сухого состояния при пониженном давлении. Остаток растворяли в СН2Сl2 (200 мл), и pH доводили до 7 насыщенным раствором NaHCO3. Органический экстракт промывали 1 н. HCl (100 мл), насыщенным NаНСО3 (200 мл) и рассолом (100 мл). Органический слой сушили над безводным Na2SO4, фильтровали, промывали СН2Сl2 (100 мл) и выпаривали до сухого состояния. Неочищенное вещество использовали как есть для следующей реакции. Небольшое количество очищали флэш-хроматографией над силикагелем, используя смесь гексан-этилацетат в качестве элюента. 1H ЯМР (CDCl3): δ 1,36 (t, 3H), 4,40 (m, 2H), 4,62 (dd, 1H), 4,74 (m, 1H), 4,86 (dd, 1H), 5,74 (d, 1H), 5,84 (m, 2H), 7,30-7,60 (m, 9H), 7,91 (d, 2H), 7,98 (d, 2H), 8,08 (m, 2H) и 8,12 (s, 1H).
, 10,12 г, 20,00 ммоль) и твердого NаНСО3 (16,8 г, 200 ммоль) в сухом 1,2-диметоксиэтане (60 мл) при 0oС в атмосфере аргона добавляли этилбромпируват (7,8 г, 40,0 ммоль) в течение 10 минут. После добавления реакционную смесь перемешивали при 0oС в атмосфере аргона в течение 6 часов. Тонкослойная хроматография (ТСХ) показала полную конверсию исходного вещества в единственный продукт (Гексан:ЕtOАс, 7:3). Реакционную смесь охлаждали до -15oС в смеси сухой лед/ССl4 в атмосфере аргона. В течение 15 минут медленно добавляли раствор трифторуксусного ангидрида (12,6 г, 60,00 ммоль) и 2,6-лутидина (12,84 г, 120 ммоль), растворенного в сухом 1,2-диметоксиэтане (20 мл). После добавления реакционную смесь перемешивали при -15oС в течение 2 часов в атмосфере аргона. Реакционную смесь фильтровали, промывали сухим метиленхлоридом (100 мл). Объединенный фильтрат выпаривали до сухого состояния при пониженном давлении. Остаток растворяли в СН2Сl2 (200 мл), и pH доводили до 7 насыщенным раствором NaHCO3. Органический экстракт промывали 1 н. HCl (100 мл), насыщенным NаНСО3 (200 мл) и рассолом (100 мл). Органический слой сушили над безводным Na2SO4, фильтровали, промывали СН2Сl2 (100 мл) и выпаривали до сухого состояния. Неочищенное вещество использовали как есть для следующей реакции. Небольшое количество очищали флэш-хроматографией над силикагелем, используя смесь гексан-этилацетат в качестве элюента. 1H ЯМР (CDCl3): δ 1,36 (t, 3H), 4,40 (m, 2H), 4,62 (dd, 1H), 4,74 (m, 1H), 4,86 (dd, 1H), 5,74 (d, 1H), 5,84 (m, 2H), 7,30-7,60 (m, 9H), 7,91 (d, 2H), 7,98 (d, 2H), 8,08 (m, 2H) и 8,12 (s, 1H).
Пример 4
Этиловый эфир 2-(β-D-рибофуранозил)тиазол-4-карбоновой кислоты (5)
Неочищенный этиловый эфир (2',3',5'-три- -бензоил-β-D-рибофуранозил)тиазол-4-карбоновой кислоты (4, 15,00 г) растворяли в сухом этаноле (100 мл) и обрабатывали порошком этилата натрия (1,36 г, 20 ммоль) в атмосфере аргона. Реакционную смесь перемешивали при комнатной температуре в течение 12 часов в атмосфере аргона. Раствор нейтрализовали Н+ -смолой Dowex 50W-X8, фильтровали и промывали метанолом (100 мл). Фильтрат выпаривали до сухого состояния. Остаток распределяли между водой (100 мл) и хлороформом (150 мл). Водный слой промывали хлороформом (100 мл) и выпаривали до сухого состояния. Остаток растворяли в метаноле (100 мл), добавляли силикагель (15 г) и выпаривали до сухого состояния. Высушенный силикагель с адсорбированным на нем соединением помещали в верхнюю часть колонки, упакованной силикагелем (5•20 см) в CH2Cl2. Колонку элюировали смесью СН2Сl2/ацетон (7:3, 500 мл) с последующим элюированием смесью СН2Сl2/метанол (95:5:1000 мл). СН2Сl2/метанольные фракции собирали вместе и выпаривали с получением чистого соединения
-бензоил-β-D-рибофуранозил)тиазол-4-карбоновой кислоты (4, 15,00 г) растворяли в сухом этаноле (100 мл) и обрабатывали порошком этилата натрия (1,36 г, 20 ммоль) в атмосфере аргона. Реакционную смесь перемешивали при комнатной температуре в течение 12 часов в атмосфере аргона. Раствор нейтрализовали Н+ -смолой Dowex 50W-X8, фильтровали и промывали метанолом (100 мл). Фильтрат выпаривали до сухого состояния. Остаток распределяли между водой (100 мл) и хлороформом (150 мл). Водный слой промывали хлороформом (100 мл) и выпаривали до сухого состояния. Остаток растворяли в метаноле (100 мл), добавляли силикагель (15 г) и выпаривали до сухого состояния. Высушенный силикагель с адсорбированным на нем соединением помещали в верхнюю часть колонки, упакованной силикагелем (5•20 см) в CH2Cl2. Колонку элюировали смесью СН2Сl2/ацетон (7:3, 500 мл) с последующим элюированием смесью СН2Сl2/метанол (95:5:1000 мл). СН2Сl2/метанольные фракции собирали вместе и выпаривали с получением чистого соединения  . Небольшое количество кристаллизовали из смеси 2-пропанол/эфир в виде бесцветного продукта. Выход 4,8 г (83%); т.пл. 62-64oС. 1Н ЯМР (DMSO-d6): δ 1,36 (t, 3H), 3,52 (m, 2H), 3,84 (m, 2H), 4,06 (m, 1H), 4,28 (m, 2H), 4,94 (t, 1H), 4,98 (d, 1H), 5.08 (d, 1H),5,46(d, 1Н) и 8,52(s, 1H).
. Небольшое количество кристаллизовали из смеси 2-пропанол/эфир в виде бесцветного продукта. Выход 4,8 г (83%); т.пл. 62-64oС. 1Н ЯМР (DMSO-d6): δ 1,36 (t, 3H), 3,52 (m, 2H), 3,84 (m, 2H), 4,06 (m, 1H), 4,28 (m, 2H), 4,94 (t, 1H), 4,98 (d, 1H), 5.08 (d, 1H),5,46(d, 1Н) и 8,52(s, 1H).
Пример 5
2-β-D-Рибофуранозилтиазол-4-карбоксамид (Тиазофурин) (6)
Неочищенный этиловый эфир 2-(β-рибофуранозил)тиазол-4-карбоновой кислоты ( , 4,6 г, 15,92 ммоль) помещали в стальную бомбу и смешивали со свежеприготовленным метанольным аммиаком (насыщенным при 0oС, 70 мл). Реакционную смесь перемешивали при комнатной температуре в течение 12 часов. Стальную бомбу охлаждали, осторожно открывали и содержимое выпаривали до сухого состояния. Остаток растирали с сухим этанолом (60 мл) и выпаривали до сухого состояния. Остаток обрабатывали сухим этанолом (60 мл), что после растирания привело к получению светло-желтого твердого вещества. Твердое вещество фильтровали, промывали этилацетатом и сушили. Это твердое вещество кристаллизовали из смеси этанол/этилацетат с получением чистого продукта. Выход 3,6 г (87%); т. пл. 142-144oС, 1H ЯМР (DMSO-d6): δ 3,57 (m, 2H), 3,89 (bs, 2H), 4,06 (m, 1H), 4,84 (t, 1H), 4,93 (d, 1H), 5,06 (m, 1H), 5,37 (d, 1H), 7,57 (s, 1H), 7,69 (s, 1H) и 8,21 (s, 1H).
, 4,6 г, 15,92 ммоль) помещали в стальную бомбу и смешивали со свежеприготовленным метанольным аммиаком (насыщенным при 0oС, 70 мл). Реакционную смесь перемешивали при комнатной температуре в течение 12 часов. Стальную бомбу охлаждали, осторожно открывали и содержимое выпаривали до сухого состояния. Остаток растирали с сухим этанолом (60 мл) и выпаривали до сухого состояния. Остаток обрабатывали сухим этанолом (60 мл), что после растирания привело к получению светло-желтого твердого вещества. Твердое вещество фильтровали, промывали этилацетатом и сушили. Это твердое вещество кристаллизовали из смеси этанол/этилацетат с получением чистого продукта. Выход 3,6 г (87%); т. пл. 142-144oС, 1H ЯМР (DMSO-d6): δ 3,57 (m, 2H), 3,89 (bs, 2H), 4,06 (m, 1H), 4,84 (t, 1H), 4,93 (d, 1H), 5,06 (m, 1H), 5,37 (d, 1H), 7,57 (s, 1H), 7,69 (s, 1H) и 8,21 (s, 1H).
Пример 6
5-O-Бензоил-β-D-рибофуранозил-1-карбонитрил (7)
Раствор 2', 3',5'-три- -бензоил-β-D-рибофуранозил-1-карбонитрила (
-бензоил-β-D-рибофуранозил-1-карбонитрила (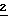 , 61 г, 129,40 ммоль) в хлороформе (200 мл) добавляли при перемешивании в охлажденный во льду насыщенный сухой метанольный аммиак (500 мл) в атмосфере аргона. Реакционную смесь перемешивали при 0oС в течение 4,5 часов. ТСХ показала полную конверсию исходного материала. Реакционную смесь выпаривали до сухого состояния. Остаток растворяли в этилацетате (500 мл), промывали насыщенным раствором NаНСО3 (100 мл), водой (300 мл) и рассолом (150 мл). Органический экстракт сушили над безводным MgSO4, фильтровали, промывали этилацетатом (100 мл), и фильтраты объединяли и выпаривали до сухого состояния с получением темно-коричневой жидкости. Жидкость растворяли в бензоле (100 мл), разбавляли гексаном (50 мл) и ацетоном (15 мл). Раствор при стоянии при комнатной температуре в течение ночи давал кристаллы. Твердое вещество отфильтровывали, промывали гексаном и сушили. Выход 29 г (85%); т.пл. 116-117oС.
, 61 г, 129,40 ммоль) в хлороформе (200 мл) добавляли при перемешивании в охлажденный во льду насыщенный сухой метанольный аммиак (500 мл) в атмосфере аргона. Реакционную смесь перемешивали при 0oС в течение 4,5 часов. ТСХ показала полную конверсию исходного материала. Реакционную смесь выпаривали до сухого состояния. Остаток растворяли в этилацетате (500 мл), промывали насыщенным раствором NаНСО3 (100 мл), водой (300 мл) и рассолом (150 мл). Органический экстракт сушили над безводным MgSO4, фильтровали, промывали этилацетатом (100 мл), и фильтраты объединяли и выпаривали до сухого состояния с получением темно-коричневой жидкости. Жидкость растворяли в бензоле (100 мл), разбавляли гексаном (50 мл) и ацетоном (15 мл). Раствор при стоянии при комнатной температуре в течение ночи давал кристаллы. Твердое вещество отфильтровывали, промывали гексаном и сушили. Выход 29 г (85%); т.пл. 116-117oС.
Пример 7
5-O-Бензоил-2,3-O-изопропилиден-β-D-рибофуранозил-1-карбонитрил (8)
Твердый 5'- -бензоил-β-D-рибофуранозил-1-карбонитрил (
-бензоил-β-D-рибофуранозил-1-карбонитрил ( , 26,3 г, 100 ммоль) добавляли к перемешиваемому раствору 72% перхлорной кислоты (4 мл) в 2,2-диметоксипропане (30 мл) и сухого ацетона (150 мл) в атмосфере аргона одной порцией. Реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Раствор нейтрализовали гидроксидом аммония и выпаривали до сухого состояния. Остаток растворяли в хлороформе (250 мл) и промывали водой (2•200 мл) и рассолом (100 мл). Органическую фазу сушили над безводным MgSО4, фильтровали, промывали хлороформом (50 мл) и фильтрат выпаривали до сухого состояния. Остаток после кристаллизации из смеси эфир/гексан дал бесцветные кристаллы. Выход 28,5 г (95%); т.пл. 62-63oС. 1H ЯМР (СDСl3): δ 1,35 (s, 3Н), 1,52 (s, 3H), 4,51 (m, 2H), 4,59 (m, 2H), 4,77 (d, 1Н), 4,87 (d, 1H), 5,10 (m, 1H), 7,46 (m, 2H), 7,57 (m, 1H) и 8,07 (m, 2H).
, 26,3 г, 100 ммоль) добавляли к перемешиваемому раствору 72% перхлорной кислоты (4 мл) в 2,2-диметоксипропане (30 мл) и сухого ацетона (150 мл) в атмосфере аргона одной порцией. Реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Раствор нейтрализовали гидроксидом аммония и выпаривали до сухого состояния. Остаток растворяли в хлороформе (250 мл) и промывали водой (2•200 мл) и рассолом (100 мл). Органическую фазу сушили над безводным MgSО4, фильтровали, промывали хлороформом (50 мл) и фильтрат выпаривали до сухого состояния. Остаток после кристаллизации из смеси эфир/гексан дал бесцветные кристаллы. Выход 28,5 г (95%); т.пл. 62-63oС. 1H ЯМР (СDСl3): δ 1,35 (s, 3Н), 1,52 (s, 3H), 4,51 (m, 2H), 4,59 (m, 2H), 4,77 (d, 1Н), 4,87 (d, 1H), 5,10 (m, 1H), 7,46 (m, 2H), 7,57 (m, 1H) и 8,07 (m, 2H).
Пример 8
Этиловый эфир 2-(5'-O-бензоил-2',3'-O-изопропилиден-β-D-рибофуранозил)тиазолин-4-карбоновой кислоты (9)
К перемешиваемому раствору 5'- -бензоил-2',3'-
-бензоил-2',3'- -изопропилиден-β-D-рибофуранозил-1-карбонитрила (
-изопропилиден-β-D-рибофуранозил-1-карбонитрила ( , 4,71 г, 15,55 ммоль) в сухом метиленхлориде (150 мл) при комнатной температуре в атмосфере аргона добавляли гидрохлорид этилового эфира цистеина (1,49 г, 8 ммоль) и (0,81 г, 8 ммоль) в час 0, через 2 часа, через 4 часа и через 6 часов. Реакционную смесь перемешивали при комнатной температуре в атмосфере аргона в течение 24 часов. Реакционную смесь разбавляли метиленхлоридом (100 мл), промывали водой (200 мл) и рассолом (150 мл). СН2Сl2 экстракт сушили над безводным MgSO4, фильтровали, промывали СН2Сl2 (50 мл) и фильтрат выпаривали до сухого состояния. Остаток использовали как есть для следующей реакции. Небольшое количество неочищенного продукта очищали флэш-хроматографией над силикагелем, используя смесь гексан/этилацетат в качестве элюента и характеризовали посредством протонной спектроскопии. 1Н ЯМР (CDCl3): δ 1,24 (t, 3Н), 1,35 (s, 3Н), 1,52 (s, 3Н), 3,40 (m, 2H), 4,20 (m, 2H), 4,42 (m, 3Н), 4,80 (m, 2H), 5,12 (m, 2H), 7,42 (m, 2H), 7,58 (m, 1H) и 8,08 (m, 2H).
, 4,71 г, 15,55 ммоль) в сухом метиленхлориде (150 мл) при комнатной температуре в атмосфере аргона добавляли гидрохлорид этилового эфира цистеина (1,49 г, 8 ммоль) и (0,81 г, 8 ммоль) в час 0, через 2 часа, через 4 часа и через 6 часов. Реакционную смесь перемешивали при комнатной температуре в атмосфере аргона в течение 24 часов. Реакционную смесь разбавляли метиленхлоридом (100 мл), промывали водой (200 мл) и рассолом (150 мл). СН2Сl2 экстракт сушили над безводным MgSO4, фильтровали, промывали СН2Сl2 (50 мл) и фильтрат выпаривали до сухого состояния. Остаток использовали как есть для следующей реакции. Небольшое количество неочищенного продукта очищали флэш-хроматографией над силикагелем, используя смесь гексан/этилацетат в качестве элюента и характеризовали посредством протонной спектроскопии. 1Н ЯМР (CDCl3): δ 1,24 (t, 3Н), 1,35 (s, 3Н), 1,52 (s, 3Н), 3,40 (m, 2H), 4,20 (m, 2H), 4,42 (m, 3Н), 4,80 (m, 2H), 5,12 (m, 2H), 7,42 (m, 2H), 7,58 (m, 1H) и 8,08 (m, 2H).
Пример 9
Этиловый эфир 2-(5'-O-бензоил-2',3'-O-изопропилиден-β-D-рибофуранозил)тиазол-4-карбоновой кислоты (10)
Способ А: К энергично перемешиваемому раствору неочищенного этилового эфира 2-(5'- -бензоил-2', 3'-
-бензоил-2', 3'- -изопропилиден-β-D-рибофуранозил)тиазолин-4-карбоновой кислоты (
-изопропилиден-β-D-рибофуранозил)тиазолин-4-карбоновой кислоты ( , 7,0 г) в метиленхлориде (300 мл) добавляли активированный диоксид марганца (27,8 г) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 24 часов, фильтровали через целит и промывали ацетоном (200 мл). Фильтраты объединяли и выпаривали до сухого состояния с получением маслянистого остатка. Выход 5,9 г (88% от цианосахара
, 7,0 г) в метиленхлориде (300 мл) добавляли активированный диоксид марганца (27,8 г) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 24 часов, фильтровали через целит и промывали ацетоном (200 мл). Фильтраты объединяли и выпаривали до сухого состояния с получением маслянистого остатка. Выход 5,9 г (88% от цианосахара  ). Небольшое количество неочищенного продукта очищали флэш-хроматографией над силикагелем, используя смесь СН2Сl2-этилацетат в качестве элюента и характеризовали посредством протонной спектроскопии. 1H ЯМР (CDCl3): δ 1,39 (t, 6H), 1,63 (s, 3Н), 4,39 (m, 3Н), 4,60 (m, 2Н), 4,84 (m, 1H), 5,26 (m, 1H), 5,40 (d, 1H), 7,40 (m, 2Н), 7,52 (m, 1H), 7,89 (m, 2H) и 8,02 (s, 1H).
). Небольшое количество неочищенного продукта очищали флэш-хроматографией над силикагелем, используя смесь СН2Сl2-этилацетат в качестве элюента и характеризовали посредством протонной спектроскопии. 1H ЯМР (CDCl3): δ 1,39 (t, 6H), 1,63 (s, 3Н), 4,39 (m, 3Н), 4,60 (m, 2Н), 4,84 (m, 1H), 5,26 (m, 1H), 5,40 (d, 1H), 7,40 (m, 2Н), 7,52 (m, 1H), 7,89 (m, 2H) и 8,02 (s, 1H).
Способ Б: Смесь неочищенного этилового эфира 2-(5'- -бензоил-2',3'-
-бензоил-2',3'- -изопропилиден-β-D-рибофуранозил)тиазолин-4-карбоновой кислоты (
-изопропилиден-β-D-рибофуранозил)тиазолин-4-карбоновой кислоты ( , 7,0 г) и активированного диоксида марганца (27,8 г) в сухом бензоле (150 мл) нагревали при 80oС в течение 2 часов. Реакционную смесь фильтровали через целит и промывали ацетоном (200 мл). Фильтраты объединяли и выпаривали до сухого состояния с получением маслянистого остатка. Выход 6,0 г (89% от цианосахара
, 7,0 г) и активированного диоксида марганца (27,8 г) в сухом бензоле (150 мл) нагревали при 80oС в течение 2 часов. Реакционную смесь фильтровали через целит и промывали ацетоном (200 мл). Фильтраты объединяли и выпаривали до сухого состояния с получением маслянистого остатка. Выход 6,0 г (89% от цианосахара  ). Небольшое количество неочищенного продукта очищали флэш-хроматографией на силикагеле, используя смесь СН2Сl2-этилацетат в качестве элюента, и посредством протонной спектроскопии было обнаружено, что продукты, полученные обоими способами, идентичны во всех отношениях.
). Небольшое количество неочищенного продукта очищали флэш-хроматографией на силикагеле, используя смесь СН2Сl2-этилацетат в качестве элюента, и посредством протонной спектроскопии было обнаружено, что продукты, полученные обоими способами, идентичны во всех отношениях.
Способ В: К энергично перемешиваемому раствору неочищенного этилового эфира 2-(5'- -бензоил-2', 3'-
-бензоил-2', 3'- -изопропилиден-β-D-рибофуранозил)тиазолин-4-карбоновой кислоты (
-изопропилиден-β-D-рибофуранозил)тиазолин-4-карбоновой кислоты ( , 2,0 г) в метиленхлориде (100 мл) добавляли пероксид никеля (10,0 г) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 24 часов, фильтровали через целит и промывали ацетоном (200 мл). Фильтраты объединяли и выпаривали до сухого состояния с получением маслянистого остатка. Выход 5,9 г (88% от цианосахара
, 2,0 г) в метиленхлориде (100 мл) добавляли пероксид никеля (10,0 г) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 24 часов, фильтровали через целит и промывали ацетоном (200 мл). Фильтраты объединяли и выпаривали до сухого состояния с получением маслянистого остатка. Выход 5,9 г (88% от цианосахара  ). Было установлено, что продукт, полученный этим способом, идентичен продуктам, полученным способами А и Б, во всех отношениях.
). Было установлено, что продукт, полученный этим способом, идентичен продуктам, полученным способами А и Б, во всех отношениях.
Пример 10
Этиловый эфир 2-(5'-O-бензоил-β-D-рибофуранозил)тиазол-4-карбоновой кислоты (11)
Раствор неочищенного этилового эфира 2-(5'- -бензоил-2',3'-
-бензоил-2',3'- -изопропилиден-β-D-рибофуранозил)тиазол-4-карбоновой кислоты (
-изопропилиден-β-D-рибофуранозил)тиазол-4-карбоновой кислоты (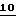 , 4,5 г, 10,39 ммоль) в смеси трифторуксусная кислота/тетрагидрофуран/вода (30:20:6 мл) перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь выпаривали до сухого состояния. Остаток суспендировали в метиленхлориде (100 мл), охлаждали и нейтрализовали насыщенным раствором NaHCO2. Водный раствор экстрагировали CH2Cl2 (2•100 мл), промывали насыщенным раствором NаНСО3 (100 мл), водой (100 мл) и рассолом (100 мл). Органический экстракт сушили над МgSO4, фильтровали, промывали СН2Cl2 (100 мл) и фильтрат выпаривали до сухого состояния. Остаток кристаллизовали из смеси этанол/вода (1:1) с получением бесцветных кристаллов. Твердое вещество фильтровали и сушили над P2O5 под вакуумом. Выход 4,0 г (98%); т.пл. 82-85oС. 1H ЯМР (CDCl3): δ 1,33 (t, 3H), 4,31 (m, 4H), 4,45 (m, 3H), 4,55 (m, 1Н), 4,74 (m, 1Н), 5,32 (d, 1H), 7,37 (m, 2H), 7,51 (m, 1H) и 7,99 (m, 3H).
, 4,5 г, 10,39 ммоль) в смеси трифторуксусная кислота/тетрагидрофуран/вода (30:20:6 мл) перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь выпаривали до сухого состояния. Остаток суспендировали в метиленхлориде (100 мл), охлаждали и нейтрализовали насыщенным раствором NaHCO2. Водный раствор экстрагировали CH2Cl2 (2•100 мл), промывали насыщенным раствором NаНСО3 (100 мл), водой (100 мл) и рассолом (100 мл). Органический экстракт сушили над МgSO4, фильтровали, промывали СН2Cl2 (100 мл) и фильтрат выпаривали до сухого состояния. Остаток кристаллизовали из смеси этанол/вода (1:1) с получением бесцветных кристаллов. Твердое вещество фильтровали и сушили над P2O5 под вакуумом. Выход 4,0 г (98%); т.пл. 82-85oС. 1H ЯМР (CDCl3): δ 1,33 (t, 3H), 4,31 (m, 4H), 4,45 (m, 3H), 4,55 (m, 1Н), 4,74 (m, 1Н), 5,32 (d, 1H), 7,37 (m, 2H), 7,51 (m, 1H) и 7,99 (m, 3H).
Пример 11
2-β-D-Рибофуранозилтиазол-4-карбоксамид (Тиазофурин) (6)
Этиловый эфир 2-(5'- -бензоил-β-D-рибофуранозил)тиазол-4-карбоновой кислоты (
-бензоил-β-D-рибофуранозил)тиазол-4-карбоновой кислоты ( , 3,7 г, 942 ммоль) помещали в стальную бомбу и смешивали со свежеприготовленным холодным метанольным аммиаком (70 мл, насыщенный при 0oС). Смесь защищали от влаги и перемешивали при комнатной температуре в течение 12 часов. Стальную бомбу охлаждали до 0oС, осторожно открывали и выпаривали до образования клейкой пены. Остаток растирали с сухим толуолом (3•50 мл) и толуольный слой отбрасывали. Полученный остаток обрабатывали безводным этанолом (60 мл) и растирали с получением светло-желтого твердого вещества. Твердое вещество фильтровали, промывали этилацетатом и сушили. Твердое вещество кристаллизовали из смеси этанол/пропилацетат с получением 2,25 г (90%) чистого продукта: т. пл. 145-147oС. 1H ЯМР (DMSO-d6): δ 3,57 (М, 2Н), 3,89 (s, 2Н), 4,07 (m, 1H), 4,83 (t, 1Н), 4,92 (d, 1H), 5,05 (d, 1Н), 5,36 (d, 1H), 7,56 (s, 1), 7,69 (s, 1) и 8,20 (s, 1).
, 3,7 г, 942 ммоль) помещали в стальную бомбу и смешивали со свежеприготовленным холодным метанольным аммиаком (70 мл, насыщенный при 0oС). Смесь защищали от влаги и перемешивали при комнатной температуре в течение 12 часов. Стальную бомбу охлаждали до 0oС, осторожно открывали и выпаривали до образования клейкой пены. Остаток растирали с сухим толуолом (3•50 мл) и толуольный слой отбрасывали. Полученный остаток обрабатывали безводным этанолом (60 мл) и растирали с получением светло-желтого твердого вещества. Твердое вещество фильтровали, промывали этилацетатом и сушили. Твердое вещество кристаллизовали из смеси этанол/пропилацетат с получением 2,25 г (90%) чистого продукта: т. пл. 145-147oС. 1H ЯМР (DMSO-d6): δ 3,57 (М, 2Н), 3,89 (s, 2Н), 4,07 (m, 1H), 4,83 (t, 1Н), 4,92 (d, 1H), 5,05 (d, 1Н), 5,36 (d, 1H), 7,56 (s, 1), 7,69 (s, 1) и 8,20 (s, 1).
Таким образом, описаны конкретные воплощения и применения способа производства Тиазофурина и других С-Нуклеозидов. Однако специалистам должно быть очевидно, что возможны многие другие модификации, кроме тех, которые уже описаны, не выходящие за пределы изложенной здесь концепции изобретения. Следовательно, предмет изобретения ограничен не иначе, как только объемом прилагаемой формулы изобретения.

Предлагается способ синтеза нуклеозида, соответствующего структуре А, где А = 0; Х = S; Z1, Z2, Z3, R1, R2 и R3 представляют собой Н; R4 представляет собой Н или низший алкил. Соединение структуры Б, где В1, В2 и В3 представляют собой блокирующие группы или низший алкил, а L - -СN, подвергают взаимодействию со структурой В, где ХY = SH, а R4 - низший алкил. В результате L структуры Б превращается в структуру Г, содержащую гетероциклическое кольцо, которое затем ароматизируют в одну стадию. Когда L=С(S) NH2, соединение структуры Б вводят в реакцию с этилбромпируватом / NaHCO3. Полученный нуклеозид далее подвергают реакции с образованием тиазофурина. Технический результат - повышение выхода продукта, сокращение продолжительности процесса. 2 с. и 9 з.п. ф-лы, 3 ил.


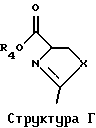

предоставляют соединение, соответствующее Структуре Б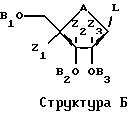
L структуры Б подвергают реакции с образованием Структуры Г, имеющей гетероциклическое кольцо, в одну стадию и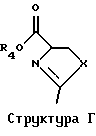
ароматизируют это гетероциклическое кольцо в одну стадию, где А представляет собой О, где L представляет собой -CN; X представляет собой S; R1, R2 и R3 представляют собой Н; R4 представляет собой Н или низший алкил; В1, В2 и В3 независимо представляют собой блокирующие группы или низший алкил и Z1, Z2 и Z3 представляют собой Н и где стадия реакции L Структуры Б с образованием Структуры Г включает в себя взаимодействие Структуры Б со Структурой В, где XY представляет собой SH и R4 представляет собой низший алкил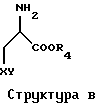
2. Способ по п. 1, где соединение, соответствующее Структуре Б, представляет собой Структуру Д
3. Способ по п. 2, где стадия реакции L с образованием Структуры Г включает в себя взаимодействие Структуры Д со Структурой В, где Y представляет собой Н или низший алкил
4. Способ по п. 1, при котором далее соединение Структуры Е ароматизируют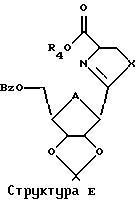
5. Способ по п. 4, где стадия ароматизирования включает в себя обработку соединения Структуры Е активированным диоксидом марганца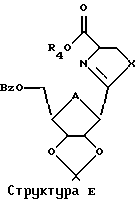
6. Способ по п. 5, где указанное соединение содержит изопропилиденовую группу и эту изопропилиденовую группу удаляют путем обработки реагентом, выбранным из группы, состоящей из трифторуксусной кислоты, муравьиной кислоты, уксусной кислоты, Н+ смолы в органическом растворителе и иода в метаноле.
10. Способ по п. 9, где указанный нуклеозид далее подвергают реакции с образованием Тиазофурина.
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| US 4451648 А, 29.05.1984 | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| HENNEN, WJ | |||
| et al | |||
| Journal of Organic Chemistry, 1985, V | |||
| Устройство для выпрямления многофазного тока | 1923 |
|
SU50A1 |
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| SRIVASTAVA, P.C | |||
| et al | |||
| Journal of Medicinal Chemistry, 1977, V.20, №2, pp.256-262 | |||
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| Общая органическая химия, т.9 | |||
| Кислородсодержащие, серосодержащие и другие гетероциклы/Под ред | |||
| Н.К | |||
| Кочеткова | |||
| - М.: Химия, 1985, с.472-473. | |||

