Область изобретения
Настоящая заявка относится к области L-нуклеозидов.
Предпосылки изобретения
В последние несколько десятилетий значительные усилия были направлены на исследование возможных применений аналогов D-нуклеозидов в качестве противовирусных агентов. Некоторые из этих работ принесли результаты, и в настоящее время на рынок в качестве противовирусных лекарств поступило множество аналогов нуклеозидов, включая ингибиторы обратной транскриптазы ВИЧ (AZT, ddl, ddC, d4T и 3ТС).
Разнообразные аналоги пуриновых D-нуклеозидов были также исследованы в поисках иммуномодуляторов. Например, было показано, что аналоги гуанозина с заместителями в положении 7 и/или 8 стимулируют иммунную систему (обзоры см. в Weigle, W. O. CRC Crit. Rev. Immunol. 1987, 7, 285; Lin et al., J. Med. Chem. 1985, 28, 1194-1198; Reitz et al., J. Med. Chem. 1994, 37, 3561-3578, Michael et al., J. Med. Chem. 1993. 36, 3431-3436). Некоторые 3-β-D-рибофуранозилтиазоло[4,5-d]пиримидины также продемонстрировали значительную активность в отношении иммунных реакций, включая пролиферацию клеток селезенки мыши и активность in vivo против вируса Semliki Forest (Nagahara et al., J. Med. Chem 1990, 33, 407-415; Robins et al., патент США 5041426). В других исследованиях было показано, что 7-деазагуанозин и аналоги проявляют противовирусную активность у мышей против множества разных РНК-содержащих вирусов, хотя это соединение не проявляет противовирусных свойств в культуре клеток. 3-Деазагуаниновые нуклеозиды и нуклеотиды также продемонстрировали широкий спектр противовирусной активности против некоторых ДНК- и РНК-содержащих вирусов (Revankar et al., J. Med. Chem 1984, 27, 1389-1396). Определенные 7- и 9-деазагуаниновые С-нуклеозиды проявляют способность защищать мышей от летального инфицирования вирусом Semliki Forest (Girgis et al., J. Med. Chem. 1990, 33, 2750-2755). Определенные 6-сульфенамидные и 6-сульфинамидные пуриновые нуклеозиды продемонстрировали значительную противоопухолевую активность (Robins et al. Патент США 4328336). Некоторые пиримидо[5,4-D] пиримидиновые нуклеозиды были эффективны при лечении лимфомы L1210 у мышей BDF1 (Robins et al. Патент США 5041542), и было выдвинуто предположение, что противовирусная и противоопухолевая активность вышеуказанных нуклеозидов является результатом их роли в качестве иммуномодуляторов (Bonnet et al., J. Med. Chem. 1993, 36, 635-653).
Одна из возможных мишеней иммуномодуляции включает в себя стимуляцию или подавление Тh1- и Тh2-лимфокинов. Клетки типа I (Тh1) продуцируют интерлейкин 2 (IL-2), фактор некроза опухолей (TNFα) и гамма-интерферон (INFγ) и они отвечают главным образом за клеточные иммунные реакции, такие как гиперчувствительность замедленного типа и противовирусный иммунитет. Клетки типа II (Тh2) продуцируют интерлейкины IL-4, IL-5, IL-6, IL-9, IL-10 и IL-13 и вовлечены главным образом в ассистирование гуморальным иммунным реакциям, таким как наблюдаемые при реакциях на аллергены, например, переключение изотипов антител IgE и IgG4 (Mosmann, 1989, Annu Rev Immunol, 7:145-173). Показано, что аналоги D-гуанозина производят разные эффекты на лимфокины IL-1, IL-6, INFα и TNFα (не прямо) in vitro (Goodman, 1988, Int J Immunopharmacol, 10, 579-88) и in vivo (Smee et al., 1991, Antiviral Res 15:229). Однако способность аналогов D-гуанозина, таких как 7-тио-8-оксогуанозин, модулировать цитокины типа I и типа II непосредственно в Т-клетках была слабая или не была описана.
Таким образом, сохраняется необходимость в новых L-нуклеозидных аналогах, включая новые пуриновые L-нуклеозидные аналоги. Имеется особая потребность в новых пуриновых L-нуклеозидах, которые обладают иммуномодулирующей активностью, и особенно в новых пуриновых L-нуклеозидах, которые модулируют Тh1- и Тh2-активность.
Настоящее изобретение направлено на новые соединения пуриновых L-нуклеозидов, их терапевтические применения и синтез.
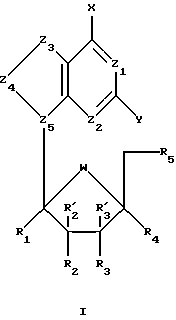
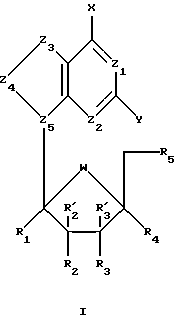
По одному из аспектов данного изобретения предложены пуриновые L-нуклеозидные аналоги формулы I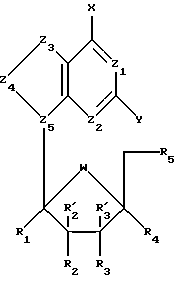 , (I)
, (I)
где R1, R'2, R'3 и R4 представляют собой Н;
R2, R3 и R5 представляют собой ОН;
Z1 представляет собой N;
Z2 выбран из группы, содержащей N и СН;
Z3 выбран из группы, содержащей -NR-, -C(R)2-, -S-, где R, одинаковые или разные, выбраны из группы, содержащей Н, Вr, NH2, алкил и алкенил;
Z4 выбран из группы, содержащей -С=О, -NR-, -C(R)2-, где R, одинаковые или разные, выбраны из группы, содержащей Н и Вr;
Z5 представляет собой N;
химическая связь между Z3 и Z4 выбрана из группы, содержащей С-С, С=С, C-N, C=N и C-S;
химическая связь между Z4 и Z5 выбрана из группы, содержащей C-N, C=N и N-N;
Х выбран из группы, содержащей Н, ОН, SH, -SNH2, -S(О)NH2, -S(O)2NH2;
Y выбран из группы, содержащей Н и NH2;
W представляет собой О; и
когда Y представляет собой NH2, тогда Z3 нe представляет собой -S-.
По другому аспекту данного изобретения фармацевтическая композиция включает в себя терапевтически эффективное количество соединения формулы I или его фармацевтически приемлемого эфира или соли в смеси по меньшей мере с одним фармацевтически приемлемым носителем.
По еще одному аспекту данного изобретения соединение в соответствии с формулами I применяют при лечении любого состояния, которое положительно реагирует на введение этого соединения, в соответствии с любым составом или протоколом, который приводит к достижению положительной реакции. Среди прочего предполагается, что соединения формулы I можно применять для лечения инфекции, инвазии (заражения паразитами), рака, опухоли или другой неоплазмы или аутоиммунного заболевания.
Фиг. 1-6 (схемы 1-6) изображают стадии химического синтеза, которые можно использовать, чтобы синтезировать соединения по настоящему изобретению. Схемы, относящиеся к синтезу определенной композиции, указаны в примерах, приведенных здесь далее.
Фиг. 7 представляет собой графическое изображение действия приведенных для примера аналогов L-гуанозина на Тh1 и Th2.
При использовании в данном описании нижеследующих терминов, они используются так, как определено ниже.
Термин "нуклеозид" относится к соединению, образованному любой пентозной или модифицированной пентозной группировкой, которая присоединена в определенном положении гетероцикла или в естественном положении пурина (положение 9) или пиримидина (положение 1) или в эквивалентном положении в аналоге.
Термин "нуклеотид" относится к фосфатному эфиру, замещающему нуклеозид в положении 5'.
Термин "гетероцикл" относится к моновалентному насыщенному или ненасыщенному карбоциклическому радикалу, имеющему по меньшей мере один гетероатом, такой как N, О или S, в кольце, каждое доступное положение которого могут возможно и независимо замещать гидрокси, оксо, амино, имино, низший алкил, бромо, хлоро и/или циано. В этот класс заместителей включены пурины, пиримидины.
Термин "пурин" относится к азотсодержащим бициклическим гетероциклам.
Термин "пиримидин" относится к азотсодержащим моноциклическим гетероциклам.
Термин "D-нуклеозиды" при использовании в настоящем изобретении описывает нуклеозидные соединения, которые имеют D-рибозную сахарную группировку (например, аденозин).
Термин "L-нуклеозиды" при использовании в настоящем изобретении описывает нуклеозидные соединения, которые имеют L-рибозную сахарную группировку.
Термин "L-конфигурация" используется в настоящем изобретении, чтобы описывать химическую конфигурацию рибофуранозильной группировки соединений, которая присоединена к нуклеиновым основаниям. L-конфигурация сахарной группировки соединений по данному изобретению отличается от D-конфигурации рибозных сахарных группировок нуклеозидов, встречающихся в природе, таких как цитидин, аденозин, тимидин, гуанозин и уридин.
Термин "С-нуклеозиды" используется в данном описании, чтобы обозначать тип связи, которая образована между рибозной сахарной группировкой и гетероциклическим основанием. В С-нуклеозидах эта связь идет от положения С-1 рибозной сахарной группировки к углероду гетероциклического основания. Связь, которая образуется в N-нуклеозидах, относится к типу углерод-углерод.
Термин "N-нуклеозиды" используется в данном описании, чтобы обозначать тип связи, которая образована между рибозной сахарной группировкой и гетероциклическим основанием. В N-нуклеозидах эта связь идет от положения С-1 рибозной сахарной группировки к азоту гетероциклического основания. Связь, которая образуется в N-нуклеозидах, относится к типу углерод-азот.
Термин "защитная группа" относится к химической группе, которая присоединена к атому кислорода или азота, чтобы предотвращать его дальнейшее реагирование в ходе дериватизации других группировок в молекуле, где расположен этот углерод или азот. Специалистам по органическому синтезу известно широкое разнообразие защитный групп для кислорода и азота.
Термин "низший алкил" относится к метилу, этилу, н-пропилу, изопропилу, н-бутилу, трет-бутилу, изобутилу или н-гексилу. Этот термин также применим к циклическим, разветвленным или простым цепям, содержащим от одного до шести атомов углерода.
Термин "арил" относится к моновалентному ненасыщенному ароматическому карбоциклическому радикалу, имеющему одно кольцо (например, фенил) или два конденсированных кольца (например, нафтил), которые могут быть возможно замещены гидроксилом, низшим алкилом, хлоро и/или циано.
Термин "гетероцикл" относится к моновалентному насыщенному или ненасыщенному карбоциклическому радикалу, имеющему по меньшей мере один гетероатом, такой как N, О, S, Se или Р, в кольце, каждое доступное положение которого могут возможно и независимо замещать или не замещать, например, гидрокси, оксо, амино, имино, низший алкил, бромо, хлоро и/или циано.
Термин "моноциклический" относится к моновалентному насыщенному карбоциклическому радикалу, имеющему по меньшей мере один гетероатом, такой как О, N, S, Se или Р, в кольце, каждое доступное положение которого могут возможно и независимо замещать сахарная группировка или любые другие группы, как то, бромо, хлоро и/или циано, так что эта моноциклическая кольцевая система в конечном счете ароматизируется [например, тимидин, 1-(2'-деокси-β-D-эритро-пентофуранозил)-тимин].
Термин "иммуномодуляторы" относится к природным или синтетическим продуктам, способным модифицировать нормальную или нарушенную иммунную систему посредством стимуляции или подавления.
Термин "эффективное количество" относится к количеству соединения формулы (I), которое восстанавливает иммунную функцию до нормальных уровней или усиливает иммунную функцию сверх нормальных уровней, чтобы ликвидировать инфекцию.
Соединения формул I и от I-A до I-Е могут иметь по несколько центров асимметрии. Соответственно, их можно получать в любой оптически активной форме или в виде рацемической смеси. Объем данного изобретения, как оно описано и заявлено, охватывает индивидуальные оптические изомеры и их нерацемические смеси, а также рацемические формы соединений формулы I.
Термин "α" или "β" указывает специфическую стереохимическую конфигурацию заместителя при асимметричном атоме углерода в химической структуре, как она выведена. Соединения, описанные здесь, все находятся в L-фуранозильной конфигурации.
Термин "энантиомеры" относится к паре стереоизомеров, которые представляют собой несовместимые зеркальные образы друг друга. Смесь пары энантиомеров в соотношении 1:1 является "рацемической" смесью.
Термин "изомеры" относится к разным соединениям, имеющим одну формулу. "Стереоизомеры" представляют собой изомеры, которые различаются только пространственным расположением атомов.
"Фармацевтически приемлемые соли" могут быть любыми солями, образованными неорганическими или органическими кислотами или основаниями.
Соединения
Соединения по настоящему изобретению в общем описаны формулой I. Однако имеется несколько подгрупп таких соединений, которые представляют особый интерес, включая соединения, соответствующие нижеприведенным формулам от I-A до I-Е.
Соединения формулы I-A представляют собой 8-замещенные L-гуанозиновые аналоги, имеющие структуру в соответствии с формулой: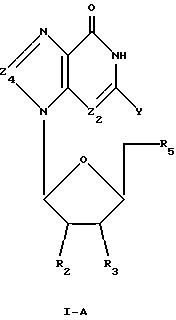
где R2, R3 и R5 представляют собой ОН;
Z2 выбран из группы, содержащей N и СН;
Z4 выбран из группы, содержащей -С=О, -NR-, -C(R)2-, где R, одинаковые или разные, выбраны из группы, содержащей Н и Вr; и
Y выбран из группы, содержащей Н и NH2.
Соединения формулы I-Б представляют собой 7-замещенные 8-оксо-L-гуанозиновые аналоги, имеющие структуру в соответствии с формулой: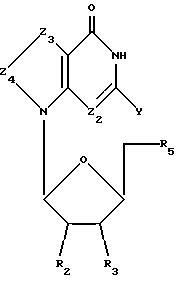 , (I-Б)
, (I-Б)
где R2, R3 и R5 представляют собой ОН;
Z2 выбран из группы, содержащей N и СН;
Z3 представляет собой -NR-, где R, одинаковые или разные, выбраны из группы, содержащей Н, Вr, NH2, алкил и алкенил;
Z4 представляет собой -С=О;
химическая связь между Z3 и Z4 представляет собой C-N; и
Y выбран из группы, содержащей Н и NH2.
Соединения формулы I-В представляют собой 7-деаза-7,8-моно- или дизамещенные L-гуанозиновые аналоги, имеющие структуру в соответствии с формулой: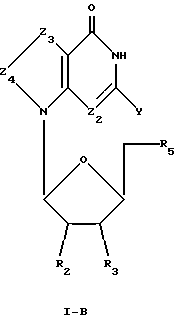
где R2, R3 и R5 представляют собой ОН;
Z2 выбран из группы, содержащей N и СН;
Z3 представляет собой -C(R)2-, где R, одинаковые или разные, выбраны из группы, содержащей Н, Br, NH2, алкил и алкенил;
Z4 представляет собой -C(R)2-, где R, одинаковые или разные, выбраны из группы, содержащей Н и Br;
химическая связь между Z3 и Z4 представляет собой С=С; и
Y выбран из группы, содержащей Н и NH2.
Соединения формулы I-Г представляют собой 7-деаза-8-аза-7-замещенные L-гуанозиновые аналоги, имеющие структуру в соответствии с формулой: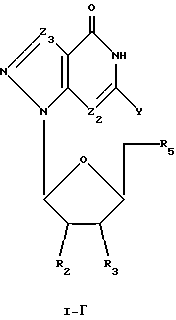
где R2, R3 и R5 представляют собой ОН;
Z2 выбран из группы, содержащей N и СН;
Z3 представляет собой -C(R)2-, где R, одинаковые или разные, выбраны из группы, содержащей Н, Вr, NH2, алкил и алкенил; и
Y выбран из группы, содержащей Н и NH2.
Соединения формулы I-Д представляют собой тиазоло[4,5-d]пиримидиновые L-нуклеозиды, имеющие структуру в соответствии с формулой: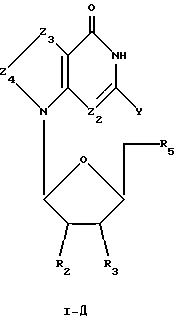
где R2, R3 и R5 представляют собой ОН;
Z2 выбран из группы, содержащей N и СН;
Z3 представляет собой -S-;
Z4 представляет собой -С=О;
химическая связь между Z3 и Z4 представляет собой C-S; и
Y выбран из группы, содержащей Н и NH2.
Соединения формулы I-Е представляют собой β-L-пуриновые нуклеозиды, имеющие структуру в соответствии с формулой: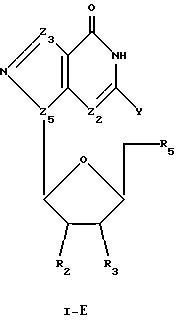
где R2, R3 и R5 представляют собой ОН;
Z2 выбран из группы, содержащей N и СН;
Z3 представляет собой -C(R)2-, где R, одинаковые или разные, выбраны из группы, содержащей Н, Вr, NH2, алкил и алкенил;
Z5 представляет собой N; и
Y выбран из группы, содержащей Н и NH2.
Применения
Предполагается, что соединения формул I, I-А, I-Б, I-В, I-Г, I-Д и I-Е, соединения по настоящему изобретению, будут применяться для лечения разнообразных состояний и, фактически, любого состояния, которое будет положительно реагировать на введение одного или более чем одного из этих соединений. Среди прочего в особенности предполагается, что соединения по данному изобретению можно применять для лечения инфекции, инвазии, рака или опухоли или аутоиммунного заболевания.
Предполагается, что инфекции, которые можно лечить соединениями по настоящему изобретению, включают в себя вызываемые респираторно-синтициальным вирусом (RSV), вирусом гепатита В (HBV), вирусом гепатита С (HCV), вирусами простого герпеса типа 1 и 2, генитального герпеса, генетического кератита, герпетического энцефалита, опоясывающего лишая, вирусом иммунодефицита человека (HIV), вирусом гриппа А, хантавирусом (геморрагическая лихорадка), вирусом папилломы человека (HPV), вирусом кори, а также грибками.
Предполагается, что инвазии, которые можно лечить соединениями по данному изобретению, включают в себя протозойные инвазии, а также заражения гельминтами и другими паразитами.
Предполагается, что рак и опухоли, которые можно лечить, включают в себя вызываемые каким-либо вирусом, а эффект может включать в себя ингибирование трансформации инфицированных вирусом клеток в неопластическое состояние, ингибирование распространения вирусов от трансформированных клеток к другим, нормальным, клеткам и/или остановку роста трансформированных вирусом клеток.
Предполагается, что аутоиммунные и другие болезни, которые можно лечить, включают в себя артрит, псориаз, кишечное заболевание, юношеский диабет, волчанку, рассеянный склероз, подагру и подагрический артрит, ревматоидный артрит, отторжение трансплантата, аллергию и астму.
Другие предположительные применения соединений по настоящему изобретению включают в себя применение в качестве промежуточных соединений химического синтеза других нуклеозидных и нуклеотидных аналогов, которые в свою очередь полезны в качестве терапевтических агентов или для других целей.
По еще одному аспекту способ лечения млекопитающего включает в себя введение терапевтически и/или профилактически эффективного количества фармацевтического препарата, содержащего соединение по настоящему изобретению. В этом аспекте эффект может относиться к модуляции некоторой части иммунной системы млекопитающего, особенно к модуляции профилей Тh1- и Тh2-лимфокинов. Когда происходит модуляция Th1- и Тh2-лимфокинов, предполагается, что такая модуляция может включать в себя стимуляцию как Тh1, так и Th2, подавление как Тh1, так и Th2, стимуляцию одного, либо Тh1, либо Th2 и подавление другого или бимодальную модуляцию, при которой одно воздействие на уровни Тh1/Th2 (такое как генерализованное подавление) происходит при низкой концентрации, тогда как другое воздействие (такое как стимуляция одного, либо Тh1, либо Th2, и подавление другого) происходит при более высокой концентрации.
Вообще, наиболее предпочтительными применениями по настоящему изобретению являются такие, при которых активные соединения соответственно менее цитотоксичны для клеток хозяина, не являющихся клетками-мишенями, и соответственно более активны в отношении мишени. В этом отношении преимущество может состоять в том, что L-нуклеозиды могут быть более стабильными, чем D-нуклеозиды, что может приводить к лучшей фармакокинетике. Этого результата можно достичь, потому что L-нуклеозиды могут не распознаваться ферментами и поэтому могут иметь более длительные периоды полужизни.
Предполагается, что соединения по настоящему изобретению можно будет вводить в любом подходящем фармацевтическом составе и по любому подходящему протоколу. Так, введение может происходить перорально, парентерально (включая методики подкожной, внутривенной, внутримышечной, интрастернальной инъекции или вливания), с помощью ингаляционного аэрозоля, или ректальным или местным образом и так далее, а также в составах стандартной дозировки, содержащих общепринятые нетоксичные фармацевтически приемлемые носители, адъюванты и растворители для введения.
Например, можно предполагать, что соединения по настоящему изобретению можно приготавливать в виде состава в смеси с фармацевтически приемлемым носителем. Например, соединения по настоящему изобретению можно вводить перорально в виде фармакологически приемлемых солей. Поскольку соединения по настоящему изобретению по большей части растворимы в воде, их можно вводить внутривенно в физиологическом солевом растворе (например, забуференном до рН около 7,2-7,5). Для этой цели можно использовать общепринятые буферы, такие как фосфаты, бикарбонаты или цитраты. Разумеется, специалист может модифицировать эти составы в соответствии с требованиями, чтобы получать многочисленные составы для определенного пути введения без того, чтобы делать композиции по настоящему изобретению нестабильными или нарушать их терапевтическую активность. В частности, модификацию данных соединений с целью сделать их более растворимыми в воде или другом растворителе для введения, можно, например, легко осуществить путем минимальных модификаций (образование соли, этерификация и т.д.), что вполне находится в пределах навыков, обычных для данной области. Также не выходя за пределы навыков, обычных для данной области, можно модифицировать пути введения и режимы дозировки определенного соединения, чтобы контролировать фармакокинетику данных соединений для получения максимально полезного эффекта у пациентов.
В определенных фармацевтических лекарственных формах предпочтительны про-лекарственные формы соединений, особенно такие, которые включают в себя ацилированные (ацетилированные и другие) производные, пиридиновые эфиры и различные солевые формы данных соединений. Специалист в данной области может определить, как с легкостью модифицировать данные соединения до про-лекарственных форм, чтобы способствовать доставке активных соединений в целевые участки организма-хозяина или пациента. При наличии навыков, обычных в данной области, можно пользоваться преимуществами благоприятных фармакокинетических параметров про-лекарственных форм, в случаях их применимости, для доставки данных соединений в целевые участки в организме-хозяине или в пациенте, чтобы максимизировать желаемые эффекты соединения.
В дополнение, соединения па данному изобретению можно вводить отдельно или в комбинации с другими агентами для лечения вышеуказанных инфекций или состояний. Способы комбинированной терапии по данному изобретению включают в себя введение по меньшей мере одного соединения по настоящему изобретению или функционального производного такого соединения и по меньшей мере одного другого фармацевтически активного ингредиента. Активный(е) ингиредиент(ы) и фармацевтически активные агенты можно вводить по отдельности или совместно и при введении по отдельности это может происходить одновременно или порознь в любом порядке. Количество активных(ого) ингредиентов(а) и фармацевтически активных(ого) агентов(а) и относительный временной порядок введения выбирают, чтобы достичь желаемого комбинированного терапевтического эффекта. Комбинированная терапия предпочтительно включает в себя введение одного соединения по настоящему изобретению или физиологически активного производного этого соединения и одного из агентов, упомянутых здесь ниже.
Примеры таких дополнительных терапевтических агентов включают в себя агенты, которые эффективны для модулирования иммунной системы или связанных состояний, такие как AZT, 3ТС, аналоги 8-замещенного гуанозина, 2',3'-дидеоксинуклеозиды, интерлейкин II, интерфероны, такие как α-интерферон, тукаресол, левамизол, изопринозин и циклолигнаны. Определенные соединения по настоящему изобретению могут быть эффективными для усиления биологической активности определенных агентов по настоящему изобретению путем снижения метаболизма или инактивации других соединений и как таковые они подлежат введению совместно с последними для достижения этого желаемого эффекта.
Что касается дозировки, то обычному специалисту в данной области понятно, что терапевтически эффективное количество варьирует в зависимости от инфекции или состояния, которые подлежат лечению, от тяжести заболевания, от применяемого режима лечения, от фармакокинетики используемого агента, также как от пациента (животного или человека), который подлежит лечению. Эффективные дозировки могут быть в пределах от 1 мг/кг веса тела или меньше до 25 мг/кг веса тела или больше. В общем, терапевтически эффективное количество данного соединения в лекарственной форме обычно находится в пределах от чуть меньше примерно 1 мг/кг до примерно 25 мг/кг веса пациента, в зависимости от используемого соединения, состояния или инфекции, которое лечится, и пути введения. Такие пределы дозировки обычно создают эффективные концентрации активного компонента в крови, находящиеся в диапазоне от примерно 0,04 до примерно 100 мкг/с3 крови пациента. Однако предполагается, что будет разработан эффективный режим, предусматривающий введение небольшого количества с последующим увеличением количества до тех пор, пока либо побочные эффекты не станут чрезмерно нежелательными, либо не будет достигнут желаемый эффект.
Введение активного соединения можно варьировать в диапазоне от непрерывного (внутривенная капельница) до нескольких пероральных введений в день (например, 4 раза в день), и оно может включать в себя пероральное, местное, парентеральное, внутримышечное, внутривенное, подкожное, чрезкожное (что может включать в себя применение агента, способствующего проникновению), трансбуккальное и суппозиторное введение, наряду с другими путями введения.
Для приготовления фармацевтических композиций по настоящему изобретению терапевтически эффективное количество одного или более чем одного соединения по настоящему изобретению предпочтительно смешивать до однородности с фармацевтически приемлемым носителем в соответствии с общепринятыми фармацевтическими методиками для смешивания, чтобы получить дозу. Носитель может принимать широкое разнообразие форм в зависимости от формы препарата, желательной для ведения, например перорального или парентерального. При приготовлении фармацевтических композиций в пероральной лекарственной форме можно использовать любую из обычных фармацевтических сред. Так, для жидких пероральных препаратов, таких как суспензии, эликсиры и растворы, можно использовать подходящие носители и добавки, включая воду, гликоли, масла, спирты, корригенты, консерванты, красители и тому подобное. Для твердых пероральных препаратов, таких как порошки, таблетки, капсулы, и для твердых препаратов, таких как суппозитории, можно использовать подходящие носители и добавки, включая крахмалы, сахарные носители, такие как декстроза, маннит, лактоза и родственные носители, разбавители, гранулирующие агенты, смазывающие вещества, связывающие вещества, разрыхлители и тому подобное. При желании таблетки или капсулы можно с помощью стандартных методик покрывать энтеросолюбильной оболочкой или изготавливать как препараты для непрерывного высвобождения.
Составы для парентерального введения обычно включают в себя стерильную воду или водный раствор хлорида натрия, хотя можно включать в него другие ингредиенты, в том числе такие, которые помогают диспергированию. Разумеется, когда надо использовать стерильную воду и поддерживать ее стерильность, композиции и носители также надо стерилизовать. Также можно готовить инъецируемые суспензии, в каковом случае можно применять подходящие жидкие носители, суспендирующие агенты и тому подобное.
Результаты тестирования
Тесты in vitro проводили с девятью L-гуанозиновыми соединениями, результаты описаны ниже. Этими девятью соединениями были следующие:
17316 8-меркапто-L-гуанозин
17317 2-амино-9-β-L-рибофуранозилпурин-6-сульфенамид
17318 2-амино-9-β-L-рибофуранозилпурин-6-сульфинамид
17319 2-амино-9-β-L-рибофуранозилпурин-6-сульфонамид
17320 7-деаза-8-аза-β-L-гуанозин
17321 7-деаза-8-аза-7-амино-β-L-гуанозин
17322 7-деаза-8-аза-7-бром-β-L-гуанозин
17323 5-амино-3-β-L-рибофуранозилтиазоло[4,5-d]пиримидин-2,7(6Н)-дион 34
17324 8-аллилокси-β-L-гуанозин
Мононуклеарные клетки периферической крови (МКПК) выделяли из светлого слоя после центрифугирования 60 мл крови здоровых доноров в градиенте плотности Ficoll-Hypaque. Затем из МКПК выделяли Т-клетки, используя Lymphokwik, реагент для выделения лимфоцитов, специфичный для Т-клеток (LK-25T, One Lambda, Canoga Park CA). Затем полученные при среднем выходе 40-60•106 Т-клетки инкубировали в течение ночи при 37oC в 20-30 мл среды RPMI-AP5 (среда RPMI-1640, ICN, Costa Mesa, Ca), содержащей 20 мМ буфер HEPES (рН 7,4), 5% аутологичной плазмы, 1% L-глутамина, 1% пенициллина/стрептомицина и 0,05% 2-меркаптоэтанола для удаления любых примесных прилипающих клеток. Во всех экспериментах промывали Т-клетки средой RPMI-AP5, а затем высевали их на микротитровальные планшеты с 96 лунками при концентрации клеток 1•106 клеток/мл.
Эти Т-клетки активировали добавлением 500 нг иономицина и 10 нг 12-миристата, 13-ацетата форбола (МАФ) (Calbiochem, La Jolla, Ca) и инкубировали в течение 48-72 ч при 37oС. Т-клетки, активированные иономицином и МАФ, обрабатывали тестируемым L-гуанозином в концентрации 0,5-50 мкМ или контрольным противовирусным интерфероном-альфа (Accurate, Westbury, NY) в концентрации 250-10000 ед./мл немедленно после активации и еще раз через 24 ч. Т-клетки с каждого планшета использовали для анализа методом иммунофлюоресценции, а супернатант использовали для определения внеклеточных цитокинов. После активации 900 мкл супернатанта от клеток с каждого микропланшета переносили на другой микропланшет для анализа продукции произведенных клетками цитокинов. Клетки затем использовали для иммунофлюоресцентного анализа уровней внутриклеточных цитокинов и экспрессии рецепторов к цитокинам.
Концентрации произведенных клетками цитокинов определяли в супернатантах от клеток с каждого микропланшета. Вызванные активацией изменения уровней интерлейкина-2 (IL-2) определяли с использованием имеющихся в продаже наборов для твердофазного иммуноферментного анализа (R&D systems Quantikine kit, Minneapolis, MN) или биологическим методом с использованием зависимой от IL-2 клеточной линии CTLL-2 (АТСС, Rockville, MD). Вызванные активацией изменения уровней интерлейкина-4 (IL-4), фактора некроза опухолей (TNFα), интерлейкина-8 (IL-8) (R&D systems Quantikine kit, Minneapolis, MN) и интерферона-гамма (INF-γ) (Endogen, Cambridge, MA) определяли с использованием наборов для иммуноферментного анализа ELISA. Все результаты иммуноферментного анализа выражали в пг/мл, а результаты биологического определения с CTLL-2 выражали в импульсах в минуту, что отражало зависимое от IL-2 включение 3H-тимидина (ICN, Costa Mesa, CA) клетками CTLL-2.
Результаты для каждого из девяти L-гуанозиновых аналогов по уровням IL-2, TNFα,INF-γ, IL-4 и IL-5 представлены на фиг. 7.
Синтез
Соединения по настоящему изобретению можно производить в соответствии со способами синтеза, сведения о каждом из которых могут быть легко получены обычными специалистами в данной области. В общем, соединения по настоящему изобретению синтезируют конденсацией подходящего нуклеозидного основания с необходимым сахарным синтоном с получением защищенного L-нуклеозида, что при дальнейших манипуляциях и снятии защиты с защищенных гидроксильных групп сахара в конечном счете дает нуклеозидный аналог, имеющий желаемую рибофуранозильную группировку в L-конфигурации.
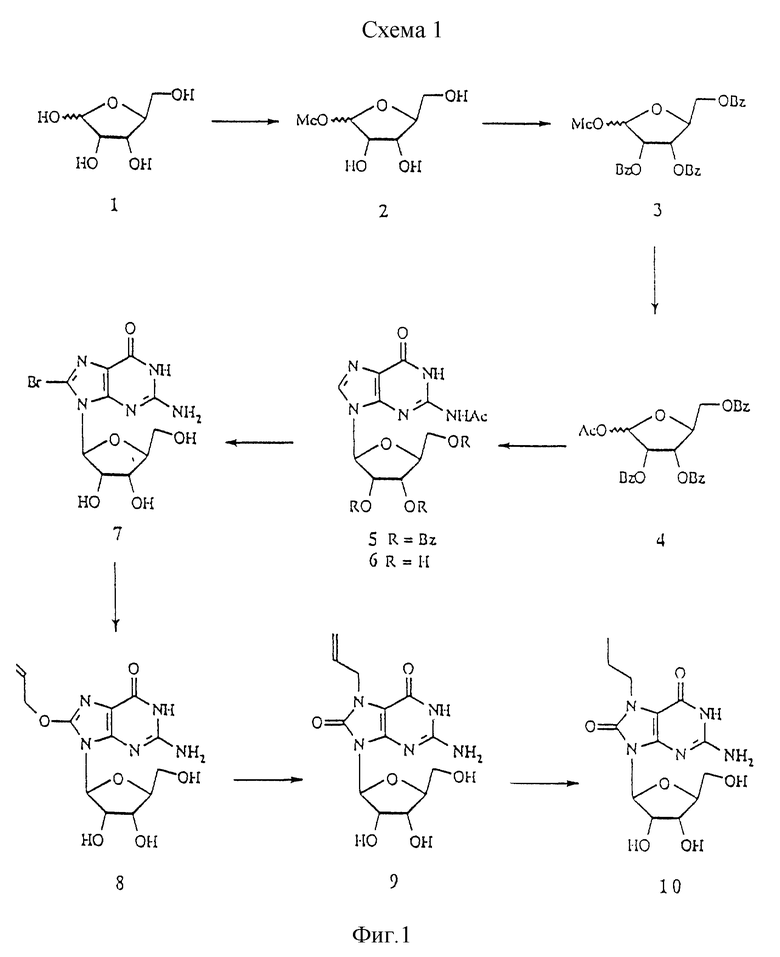
Схема 1 показывает синтез определенных 7- и 8-замещенных L-гуанозиновых аналогов L-рибозу 1 метилировали по С-1 и полученный продукт 2 бензоилировали с получением соединения 3, которое превращали в 4 обработкой уксусным ангидридом в присутствии серной кислоты. Взаимодействие 4 и силилированного N2-ацетилгуанина в присутствии триметилсилилтрифлата дало соединение 5 в соответствии с обычно используемой процедурой (Vorbruggen et al. Chem. Ber., 1981, 14, 1234). 5 превращали в 6 с помощью аммиака в метаноле. Бромирование 5 дало 8-бром-производное 7, которое превращали в 8-аллилокси-производное 8 обработкой аллиловым спиртом и гидридом натрия. 8 нагревали в смеси воды с метанолом, что приводило к получению 7-аллил-8-оксо-производного 9, которое гидрогенизировали с получением 7-пропил-8-оксо-L-гуанозина 10.
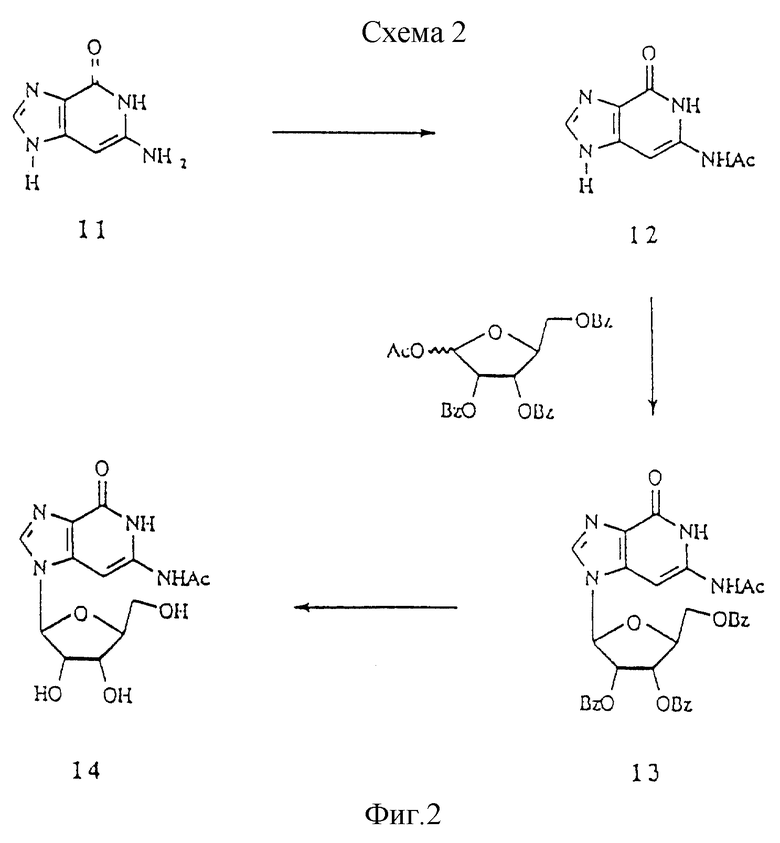
Схема 2 показывает синтез N2-ацетил-3-деаза-L-гуанозина. 3-Деазагуанозин 11 (Cook et at. J. Med. Chem. 1976, 27, 1389) обрабатывали уксусным ангидридом в пиридине с получением N2-ацетил-3-деазагуанина 12, который силилировали и сочетали с 1-ацетил-2,3,5-O-трибензоил-L-рибозой с получением соединения 13. Удаление бензоильной группы с помощью аммиака в метаноле дало N2-ацетил-3-деаза-L-гуанозин 14.
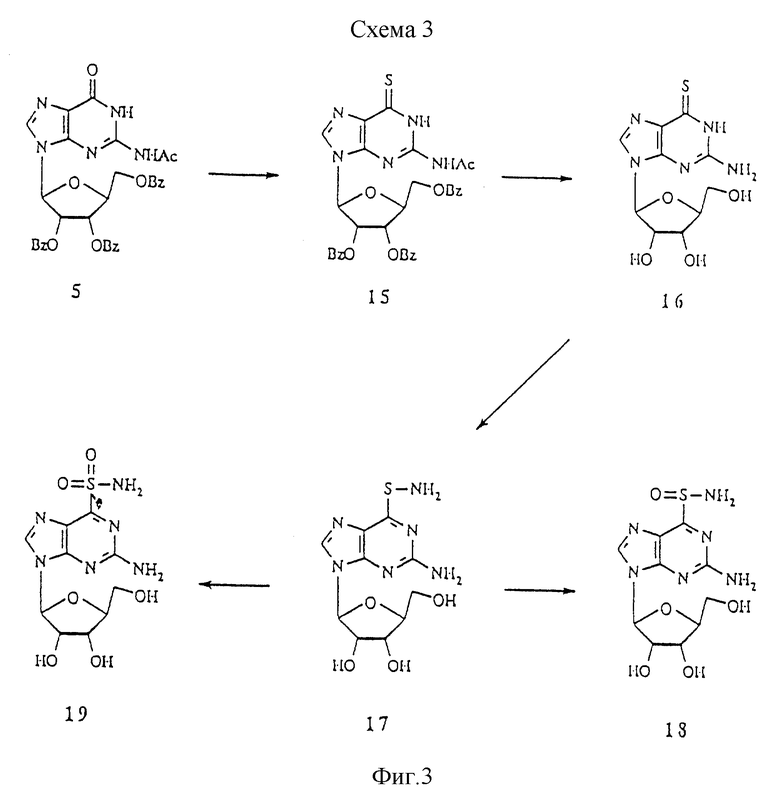
Схема 3 показывает синтез 6-меркапто-L-гуанозина и его производных. N2-Ацетил-2', 3', 5'-О-трибензоил-β-L-гуанозин 5 обработкой пентасульфидом фосфора (Fox et al., J. Am. Chem. Soc. 1958, 80, 1669) превращали в 6-меркапто-производное 15, с которого снимали защиту с получением 6-меркапто-β-L-гуанозина 16. Сульфенамидное производное 17 получали в реакции 16 с NH2-Cl, генерируемым in situ. Сульфенамид 17 окисляли с помощью МСРВА (4-(2-метил-4-хлорфенокси)-масляной кислоты) до сульфинамида 18 и сульфонамида 19, контролируя количество реагента (Revankar et al., J. Med. Chem. 1990, 33, 121).
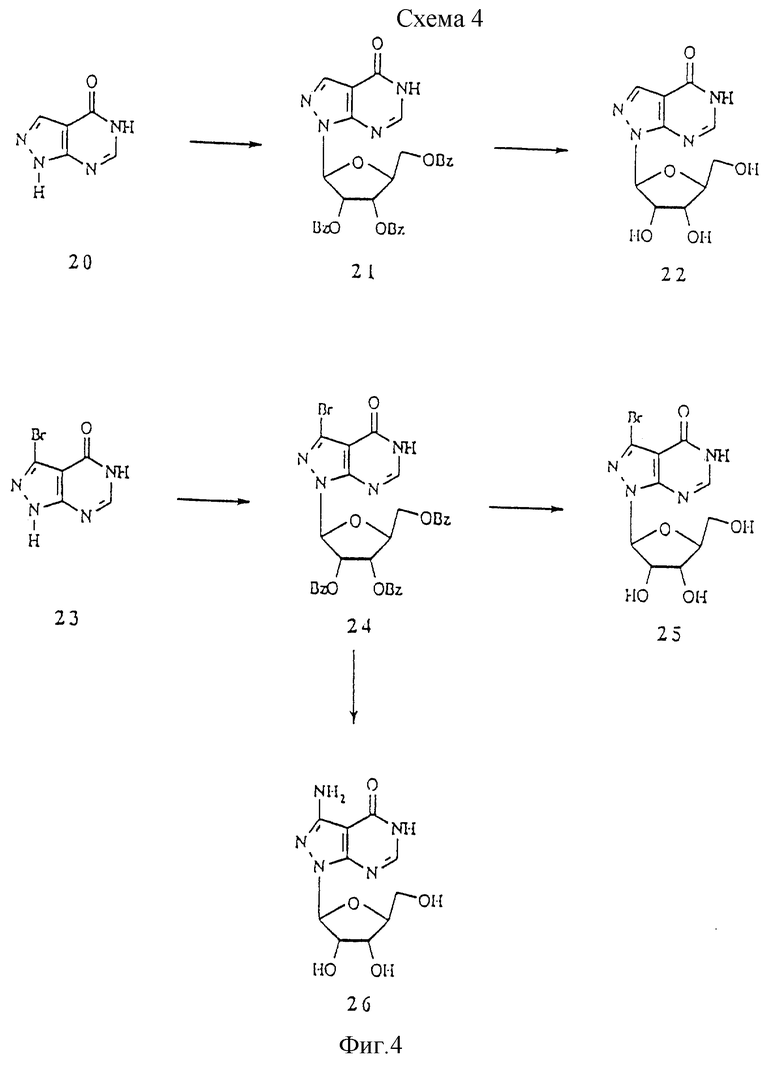
Схема 4 показывает синтез 1-β-L-рибофуранозилпиразоло[3,4-d]пиримидин-4(5Н)-она и производных. Имеющийся в продаже 4-гидроксипиразоло[3,4-d]пиримидин 20 сочетали с защищенной L-рибозой, получая защищенный нуклеозид 21, с которого снимали защиту с получением 1-β-L-рибофуранозилпиразоло[3,4-d]пиримидин-4(5Н)-она 22. Аналогичным образом 3-бром-4-гидроксипиразоло[3,4-d]пиримидин 23 (Cottam et al., J. Med. Chem. 1984, 27, 1119) сочетали с L-рибозой, получая защищенный нуклеозид 24, с которого снимали защиту с получением 3-бром-1-β-L-рибофуранозилпиразоло[3,4-d] пиримидин-4(5Н)-она 25. Обработка 24 аммиаком в присутствии меди и хлорида меди при 100oС дала 3-амино-производное 26.
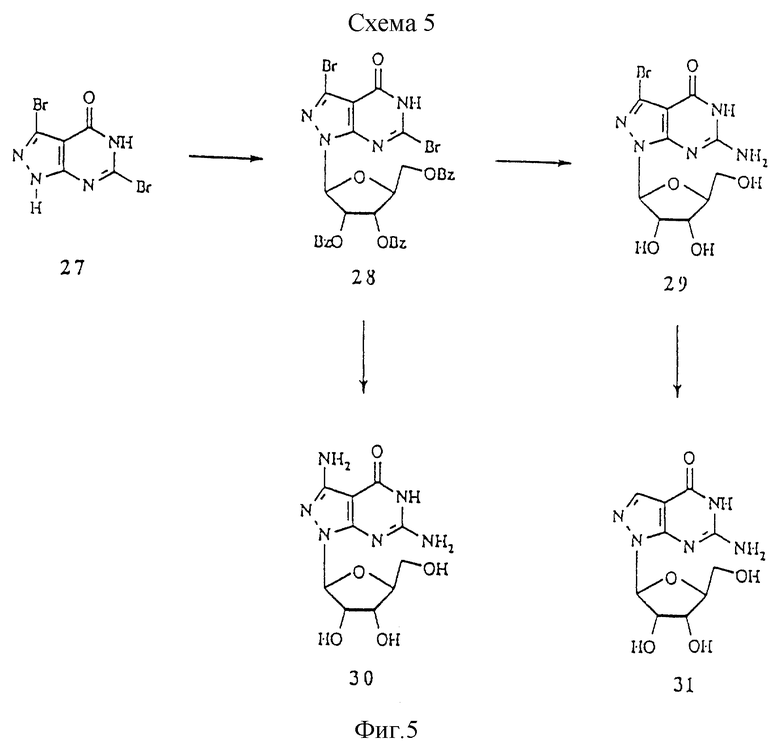
Схема 5 показывает синтез 8-аза-7-деаза-L-гуанозиновых аналогов. 3,6-Дибромпиразоло[3,4-d]пиримидин-4(5Н)-он 27 (Petrie III et al., J. Med. Chem. 1985, 28, 1010) сочетали с защищенной L-рибозой с получением нуклеозида 28, который обрабатывали аммиаком с получением 8-аза-3-бром-7-деаза-β-L-гуанозина 29. Обработка 28 аммиаком при 120oС дала 3-амино-производное 30. Гидрогенизирование 29 над Pd/C дало 8-аза-7-деаза-β-L-гуанозин 31.
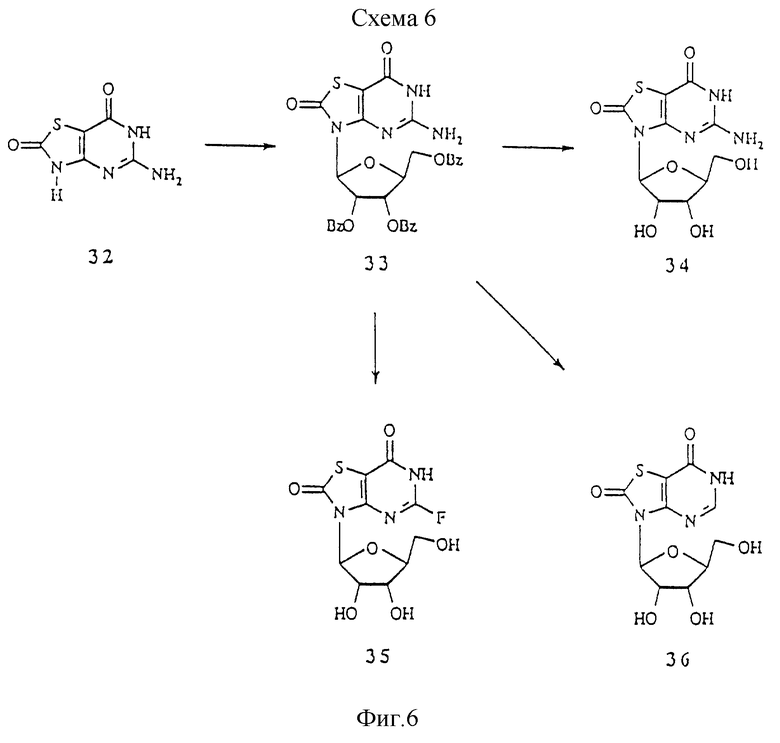
Схема 6 показывает синтез 5-амино-3-β-L-рибофуранозилтиазоло[4,5-d]пиримидин-2,7(6Н)-диона и его аналогов. 5-Аминотиазоло[4,5-d]пиримидин-2,7(3H, 6Н)-дион 32 (Baker et al., J. Chem. Soc. С 1970, 2478) сочетали с незащищенной рибозой, получая нуклеозид 33, с которого снимали защиту с получением 5-амино-3-β-L-рибофуранозилтиазоло[4,5-d] пиримидин-2,7(6Н)-диона 34. Соединение 33 можно защитить нитроэтоксифенильной группой, а затем обработать бутилнитритом и фтористым водородом в пиридине с получением фторпроизводного 35. Обработка 33 трет-бутилнитритом (Nagahara et al., J. Med. Chem. 1990, 33, 407) в тетрагидрофуране может заменить аминогруппу водородом с получением 36.
3-Деаза-L-гуанозина может быть получен посредством силилирования метил-5-цианометилимидазол-4-карбоксилата и его взаимодействия с 1-О-ацетил-2', 3',5'-О-трибензоил-L-рибофуранозой 4, с последующей циклизацией и бромированием.
Соединения, описанные в схемах 1-6, представляют собой β-L-гуанозиновые аналоги. Соответствующие α-L-аналоги можно получить аналогичным образом, но используя L-рибозу, имеющую другие защитные группы. 1-Ацетил-2,3,5-О-трибензоил-L-рибофуранозу в качестве реагента можно заменить производными 1-бром-β-L-рибозы, что должно дать α-L-нуклеозиды в качестве основных продуктов.
Примеры
Нижеследующий раздел дает экспериментальные примеры, выполненные в лаборатории заявителей. Примеры по возможности широкие, но не исчерпывающие. Выполненная работа включает в себя все описанные ниже примеры, но не ограничена этими примерами.
Пример 1
1-О-Метил-L-рибофураноза 2
Холодный раствор сухого хлористого водорода (4,4 г, 0,12 моль) в метаноле (100 мл) медленно добавляли к раствору L-(+)-рибозы 1 (50 г; 0,33 моль в 1000 мл метанола) при комнатной температуре. После добавления этот раствор перемешивали 2,5 ч и тушили реакцию пиридином (100 мл). Смесь перемешивали 10 минут и растворитель выпаривали. Остаток растворяли в пиридине (100 мл) и полученный раствор концентрировали досуха с получением 1-О-Метил-L-рибофуранозы 2 в виде бледно-желтого сиропа.
Пример 2
1-О-Метил-2',3',5'-О-трибензоил-L-рибофураноза 3
Бензоилхлорид (154,5 г; 1,1 моль) добавляли по каплям в течение 10 мин к раствору 1-метил-L-рибофуранозы 2 (0,33 моль) в пиридине (350 мл) при 0oС. После добавления этот раствор стоял при комнатной температуре в течение 14 ч и реакцию тушили его перемешиванием с водой (50 мл) при 0oС в течение 1 ч. Водный слой экстрагировали с помощью CH2Cl2 (2•100 мл) и объединенный органический слой концентрировали. Остаток растворяли в CH2Cl2 (500 мл), промывали последовательно насыщенным NaHCO3 (3•100 мл), водой (200 мл), соляным раствором (200 мл), сушили над Na2SO4, фильтровали и упаривали с толуолом (2•300 мл). Дальнейшая сушка под вакуумом дала 1-О-Метил-2',3',5'-О-трибензоил-L-рибофуранозу 3 в виде желтого сиропа (80 г; 0,17 моль).
Пример 3
1-О-Ацетил-2',3',5'-О-трибензоил-1-рибофураноза 4
1-О-Метил-2', 3', 5'-О-трибензоил-L-рибофуранозу 3 (80 г, 0,17 моль) растворяли при комнатной температуре в смеси уксусной кислоты (354 мл) и уксусного ангидрида (36 мл). Полученный раствор охлаждали до 0oС и к нему по каплям добавляли серную кислоту (96%; 8,23 г, 0,084 моль). После добавления эту реакционную смесь держали при комнатной температуре 18 ч, выливали в лед (500 г) и перемешивали, пока лед не растаивал. Добавляли ЕtOАс (1,2 л), а затем воду (1 л). Органический слой промывали смесью воды и соляного раствора (в соотношении 1:4), насыщенным NаНСО3 (500 мл), соляным раствором (500 мл), фильтровали через слой силикагеля и концентрировали до неочищенного продукта в виде желтого твердого вещества. Перекристаллизация из смеси гексанов с ЕtOАс (в соотношении 300 мл/100 мл) дала 1-О-Ацетил-2',3',5'-О-трибензоил-L-рибофуранозу 4 в виде белых игл (50 г; общий выход из L-рибозы 59,6%).
Пример 4
N2-Ацетил-2',3',5'-О-трибензоил-β-L-гуанозин 5
N2-Ацетилгуанин (4,125 г; 21,35 ммоль) суспензировали в пиридине (50 мл) при 80oС в течение 25 минут, а затем выпаривали пиридин под высоким вакуумом. Эту же процедуру повторяли еще раз. Полученный материал сушили под вакуумом в течение ночи и силилировали нагреванием с избытком HMDS (50 мл), пиридина (10 мл) и TMSCI (150 мкл) под аргоном в течение 2,5 ч. После охлаждения реакционной смеси до комнатной температуры выпаривали растворители под вакуумом. Остатки HMDS и пиридина выпаривали вместе с ксилолом (2•40 мл). Полученную суспензию перемешивали под аргоном при температуре образования флегмы в течение 10 мин и по каплям в течение 20 минут добавляли раствор тетраметилсилантрифлата (4,50 мл; 23,276 моль) в дихлорэтане (35 мл). Полученную реакционную смесь промывали холодным NаНСО3 (5% водный раствор, 2•150 мл), соляным раствором (150 мл), сушили (Na2SО4) и упаривали досуха. Эту реакционную смесь чистили с помощью тонкослойной хроматографии (400 г силикагеля; элюент: 28% ЕtOАс, 2% ЕtOН в CH2Cl2, объем/объем) с получением 5,60 г (46%) N2-Ацетил-2',3',5'-О-трибензоил-β-L-гуанозина 5.
Пример 5
β-L-Гуанозин 6
Раствор N2-Ацетил-2',3',5'-О-трибензоил-β-L-гуанозина 5 в насыщенном аммиаком метаноле стоял при комнатной температуре в течение двух дней. Аммиак и метанол выпаривали и неочищенный продукт растворяли в воде и хлороформе (два слоя). Водный слой трижды промывали хлороформом и концентрировали. Неочищенный продукт чистили кристаллизацией из смеси воды и метанола с получением β-L-гуанозина 6 в виде бесцветного твердого вещества.
Пример 6
8-Бром-β-L-гуанозин 7
К суспензии L-гуанозина 6 (1,24 г) в воде (7,5 мл) добавляли порциями 35 мл насыщенного раствора брома в воде, содержащего 0,35 мл брома. Твердые вещества отфильтровывали, последовательно промывали холодной водой, холодным ацетоном и сушили. Кристаллизация из воды дала чистый 8-бром-β-L-гуанозин 7 в виде бесцветного твердого вещества.
Пример 7
8-Аллилокси-β-L-гуанозин 8
К перемешиваемой смеси NaH (984 мг) в безводном диметилсульфоксиде (30 мл) по каплям добавляли аллиловый спирт (10 мл) с последующим добавлением 8-бром-L-гуанозина 7 (1,78 г; 4,92 ммоль) в диметилсульфоксиде (10 мл). Полученную реакционную смесь перемешивали при 60oС в течение ночи, охлаждали до комнатной температуры и разбавляли этиловым эфиром (350 мл). Образовавшиеся преципитаты отфильтровывали, растворяли в воде (18 мл) и нейтрализовали уксусной кислотой. Образовавшиеся преципитаты отфильтровывали и перекристаллизовывали из смеси воды и метанола с получением 836 мг 8-аллилокси-L-гуанозина в виде желтоватого твердого вещества.
Пример 8
7-Аллил-8-оксо-β-L-гуанозин 9
Смесь 8-аллилоксигуанозина 8 (560 мг) в смеси метанола и воды (50 мл; 1: 1 объем/объем) перемешивали при температуре образования флегмы и через 2 ч получали прозрачный раствор. Этот раствор выдерживали при температуре образования флегмы еще 5 ч и охлаждали до комнатной температуры. Коричневый преципитат (побочный продукт) отфильтровывали и фильтрат концентрировали с получением неочищенного продукта. Кристаллизация из смеси воды и этанола дала 83 г целевого соединения в виде коричневатого твердого вещества. Фильтрат концентрировали и остаток хроматографировали на кремнеземе, используя 5% Et3N и 20% МеОН в хлориде метилена, с получением 260 мг 7-аллил-8-оксо-β-L-гуанонзина 9 в виде бесцветного твердого вещества.
Пример 9
8-Оксо-7-пропил-β-L-гуанозин 10
Суспензию 120 мг 7-аллил-8-оксо-β-L-гуанонзина 9 и 80 мг 10% палладия на углероде встряхивали в аппарате для гидрогенизации при комнатной температуре и давлении водорода 55 фунтов на квадратный дюйм в течение 2 ч. Палладиевый катализатор отфильтровывали и фильтрат концентрировали. Неочищенный продукт кристаллизовали из смеси воды и этанола с получением 75 мг 8-оксо-7-пропил-β-L-гуанозина 10 в виде желтоватого твердого вещества.
Пример 10
N2-Ацетил-3-деазагуанин 12
К суспензии 3-деазагуанина 11 (2,0 г) в безводном пиридине (30 мл) добавляли уксусный ангидрид (5 мл) и полученную реакционную смесь нагревали до 90oС. Твердые вещества постепенно растворялись с образованием коричневого раствора. Через 10 минут снова появлялись преципитаты. Эту смесь перемешивали при 90oС еще 90 минут и охлаждали до 50oС. Преципитаты отфильтровывали и промывали ацетонитрилом, водой и снова ацетонитрилом с получением 1,79 г N2-ацетил-3-деазагуанина 12 в виде светло-коричневого твердого вещества.
Пример 11
N2-Ацетил-3-деаза-β-L-гуанозин 14
Суспензию N2-ацетил-3-деазагуанина 12 (576 мг, 3,0 ммоль), гексаметилдисилазана (HMDS, 15 мл), пиридина (2 мл) и сульфата аммония (10 мг) перемешивали при температуре образования флегмы в условиях, исключающих увлажнение, в течение 2,5 ч. Растворители выпаривали и остаток сушили под вакуумом в течение 2 ч с получением пенистого сиропа. Этот остаток растворяли в хлориде метилена (безводный, 30 мл) и добавляли 1-ацетил-2,3,5-трибензоилрибозу (1,51 г, 3,0 ммоль) с последующим медленным добавлением триметилсилилтрифлата (4,5 ммоль; 0,81 мл). Полученный раствор держали при температуре образования флегмы в течение 20 ч. Растворители выпаривали и остаток растворяли в этилацетате, промывали с помощью 5% NаНСО3, сушили (Na2SO4) и концентрировали.
Хроматография на кремнеземе с 5% Et3N и 2-10% этанолом в хлориде метилена дала три основных продукта: 340 мг продукта с более высоким Rf, 368 мг продукта со средним Rf и 335 мг продукта с более низким Rf, все в виде желтоватого твердого вещества.
Раствор продукта со средним Rf 13 (350 мг) в насыщенном аммиаком метаноле держали при комнатной температуре два дня. Аммиак и метанол выпаривали и остаток хроматографировали на кремнеземе, используя 5% Et3N и 20% этанол в хлориде метилена, с получением 114 мг N2-ацетил-3-деаза-β-L-гуанозина 14 в виде белого твердого вещества.
Пример 12
N2-Ацетил-6-меркапто-2',3',5'-О-трибензоил-β-L-гуанозин 15
К перемешиваемой суспензии N2-ацетил-2',3',5'-О-трибензоил-L-гуанозина 5 (5,60 г; 8,78 ммоль) и пентасульфида фосфора (8,0 г, 36,0 ммоль) в пиридине (210 мл) по каплям добавляли воду (590 мкл) и полученную реакционную смесь грели при температуре образования флегмы в течение 8 ч. Каждый раз, как только раствор начинал терять мутность, к нему добавляли несколько капель воды. В конце нагревания при температуре образования флегмы пиридин выпаривали с получением сиропа, который медленно добавляли к интенсивно перемешиваемой кипящей воде (1000 мл). Полученную смесь перемешивали 45 минут и экстрагировали с помощью ЕtOАс (3•250 мл). Органический слой экстрагировали соляным раствором (2•200 мл), водой (2•100 мл), сушили (Na2SО4) и упаривали досуха. Хроматография на силикагеле (400 г) с 23% ЕtOН в CH2Cl2 (объем/объем) дала 3,53 г (61,5%) N2-ацетил-6-меркапто-2',3',5'-О-трибензоил-β-L-гуанозина 15 в виде бесцветного твердого вещества.
Пример 13
6-Меркапто-β-L-гуанозин 16
Раствор N2-ацетил-6-меркапто-2,3',5'-О-трибензоил-β-L-гуанозина 15 (3,53 г; 4,40 ммоль) в насыщенном аммиаком метаноле (200 мл) перемешивали при комнатной температуре в течение 62 ч. Аммиак и метанол выпаривали и остаток перетирали с хлороформом. Осадок отфильтровывали и промывали теплым хлороформом (50 мл), снова растворяли в разбавленном водном растворе аммиака и подкисляли уксусной кислотой. Полученный осадок фильтровали и сушили под вакуумом с получением 1,48 г (91,6%) 6-меркапто-β-L-гуанозина 16 в виде бесцветного твердого вещества.
Пример 14
2-Амино-9-(-β-L-рибофуранозил)-пурин-6-сульфенамид 17
К перемешиваемому водному раствору гипохлорита натрия (5,25%; 2,25 мл; 1,725 ммоль), охлаждаемому до 0oС в ледяной бане, добавляли гидроксид аммония (1,4 М; 6 мл; 8,4 ммоль), охлажденный до 0oС. Полученную смесь перемешивали при 0oС 15 минут и добавляли к ней холодный (0oС) раствор 6-меркапто-L-гуанозина 16 (450 мг; 1,5 ммоль) в 2 М КОН (750 мл). Эту реакционную смесь перемешивали 2 ч, пока она не нагревалась до комнатной температуры. Образовавшиеся преципитаты отфильтровывали, промывали холодным ЕtOН и сушили с получением 240 мг (51%) 2-амино-9-(β-L-рибофуранозил)-пурин-6-сульфенамида 17 в виде бесцветного твердого вещества.
Пример 15
2-Амино-9-(β-L-рибофуранозил)-пурин-6-сульфинамид 18
Смесь 2-амино-9-(β-L-рибофуранозил)-пурин-6-сульфенамида 17 (200 мг, 0,637 ммоль), этанола (90 мл) и воды (6,4 мл) интенсивно перемешивали при -10oС в соляно-ледяной бане. По каплям в течение 15 минут добавляли раствор MCPBA (80%; 0,637 ммоль) в этаноле (5,5 мл). Этой смеси давали перемешиваться и нагреваться по мере того, как таял лед (8 ч), и перемешивали при температуре окружающей среды еще 14 ч. Небольшое количество преципитатов отфильтровывали и фильтрат упаривали при 23oС досуха. Остаток перетирали с этиловым эфиром (30 мл) и твердое вещество собирали фильтрацией и промывали этиловым эфиром (10 мл). Твердое вещество снова суспензировали в этиловом эфире (25 мл), отфильтровывали и сушили с получением 182 мг (87%) 2-амино-9-(β-L-рибофуранозил)-пурин-6-сульфинамида 18 в виде бесцветного твердого вещества.
Пример 16
2-Амино-9-(β-L-рибофуранозил)-пурин-6-сульфонамид 19
К перемешиваемой суспензии 2-амино-9-(β-L-рибофуранозил)-пурин-6-сульфенамида 17 (150 мг; 0,478 ммоль) в этаноле (28,5 мл) и воде (2,8 мл) при комнатной температуре добавляли порциями в течение 1 ч раствор МСРВА (80%; 412 мг; 1,91 ммоль) в этаноле (2,8 мл). Эта реакционная смесь становилась прозрачной через 3 ч. Раствор перемешивали еще 15 ч при комнатной температуре, и он становился мутным. Эту реакционную смесь концентрировали при комнатной температуре досуха. Остаток перетирали с этиловым эфиром (30 мл) и твердое вещество собирали фильтрацией. Неочищенный продукт растворяли в смеси метанола и воды и сорбировали силикагелем (2,0 г). Растворитель выпаривали и сухой силикагель, содержащий продукт, наносили на колонку с кремнеземом (100 г), набитую в хлориде метилена. Колонку элюировали 20% метанолом в CH2Cl2 (объем/объем) с получением 87 мг (56,2%) 2-амино-9-(β-L-рибофуранозил)-пурин-6-сульфонамида 19 в виде бесцветного твердого вещества.
Пример 17
1-(2', 3', 5'-О-трибензоил-β-L-рибофуранозил)-пиразоло[3,4-d]пиримидин-4(5Н)-он 21
Смесь 4-гидроксипиразоло[3,4-d] пиримидина 200 (100 мг; 0,74 ммоль), 1,1,1,3,3,3-гексаметилдисилазана (HMDS, 10 мл) и (NH4)2SО4 (10 мг; 0,076 ммоль) нагревали при температуре образования флегмы в течение 3 ч с образованием прозрачного раствора. Избыток HMDS выпаривали с получением желтого твердого вещества, которое сушили под вакуумом в течение 15 минут. Добавляли 1-О-ацетил-2', 3', 5'-О-трибензоил-L-рибофуранозу с последующим добавлением ацетонитрила (безводный, 5 мл). К вышеуказанной взвеси при
комнатной температуре по каплям добавляли триметилсилиловый эфир трифторметансульфоновой кислоты (245 мг, 1,1 ммоль). После добавления прозрачный раствор стоял при комнатной температуре в течение 14 ч. Растворитель выпаривали и желтый остаток растворяли в ЕtOАс (50 мл), промывали насыщенным NаНСО3 (2•20 мл), водой (3•20 мл), сушили над Na2SО4 и концентрировали. Тонкослойная хроматография на силикагеле (5% метанол в хлориде метилена) дала 1-(2',3',5'-О-трибензоил-β-L-рибофуранозил)-пиразоло[3,4-d]пиримидин-4(5Н)-она 21 в виде белого твердого вещества (117 мг, 41,4%).
Пример 18
1-β-L-рибофуранозилпиразоло[3,4-d]пиримидин-4(5Н)-он 22
1-(2', 3', 5'-О-Трибензоил-β-L-рибофуранозил)-пиразоло[3,4-d]пиримидин-4(5Н)-он 21 (152 мг; 0,26 моль) растворяли в насыщенном растворе NН3 в МеОН (75 мл) при 0oС. Полученный раствор 24 ч стоял при комнатной температуре, после чего его концентрировали. Остаток растворяли в воде (30 мл) и промывали с помощью ЕtOАс (3•15 мл). После выпаривания воды кристаллическое твердое вещество замачивали в ацетонитриле (2 мл), фильтровали и сушили под вакуумом с получением 1-β-L-рибофуранозилпиразоло[3,4-d]пиримидин-4(5Н)-она 22 в виде белого кристаллического твердого вещества (70 мг, 99%).
Пример 19
3-Бром-1-(2',3',5'-О-трибензоил-β-L-рибофуранозил)-пиразоло[3,4-d]пиримидин-4(5Н)-он 24
Ацетонитрил (30 мл) добавляли к смеси 3-бромпиразоло[3,4-d]пиримидин-4(5Н)-она 23 (1,08 г, 4,0 моль) и 1-О-ацетил-2',3',5'-О-трибензоил-β-L-рибофуранозы (3,02 г, 6,0 ммоль). Полученную взвесь нагревали до температуры образования флегмы и по каплям добавляли этерат трифторборана. Полученный раствор в течение ночи грели при температуре образования флегмы. Растворитель выпаривали, остаток растворяли в ЕtOАс (100 мл), полученный раствор промывали насыщенным NaHCO3, водой, сушили над Na2SO3 и концентрировали. Тонкослойная хроматография на кремнеземе (5% ацетон в хлориде метилена) дала 3-бром-1-(2',3',5'-О-трибензоил-β-L-рибофуранозил)-пиразоло[3,4-d]пиримидин-4(5Н)-он 24 в виде бледно-желтого твердого вещества (1,1 г; 41,7%).
Пример 20
3-Бром-1β-L-рибофуранозилпиразоло[3,4-d]пиримидин-4(5Н)-он 25
3-Бром-1-(2', 3', 5'-О-трибензоил-β-L-рибофуранозил)-пиразоло[3,4-d] пиримидин-4(5Н)-он 24 (280 мг, 0,43 ммоль) растворяли в насыщенном растворе NН3 в МеОН (25 мл) при 0oС. Этот раствор в герметически закрытом баллоне из нержавеющей стали грели при 100oС в течение 6 ч. После охлаждения выпаривали аммиак и метанол. Остаток растворяли в воде (40 мл), промывали с помощью ЕtOАс (4•20 мл) и концентрировали. Остаток замачивали в ацетонитриле и полученное твердое вещество отфильтровывали и сушили под вакуумом с получением 3-бром-1β-L-рибофуранозилпиразоло[3,4-d]пиримидин-4(5Н)-она 25 в виде белого твердого вещества (140 мг; 95%).
Пример 21
3-Амино-1-β-L-рибофуранозилпиразоло[3,4-d]пиримидин-4(5Н)-он 26
3-Бром-1-(2', 3',5'-О-трибензоил-β-L-рибофуранозил)-пиразоло[3,4-d]пиримидин-4(5Н)-он 24 (714 мг; 1,08 ммоль) растворяли в насыщенном растворе NН3 в МеОН (25 мл) при 0oС. Добавляли тонкую медную проволоку (21 мг; 0,33 ммоль) и хлорид меди (33 мг; 0,33 ммоль). Эту смесь в герметически закрытом баллоне из нержавеющей стали грели при 100oС в течение 16 ч. После охлаждения к реакционной смеси добавляли силикагель (2 г) и растворитель выпаривали. Силикагель с сорбированным неочищенным продуктом наносили на колонку с кременеземом и проводили элюцию посредством 5% Еt3N, 17% МеОН в CH2Cl2. Продукт чистили дальше перекристаллизацией (95% EtOH) с получением 3-амино-1-β-L-рибофуранозилпиранозоло[3,4-d] пиримидин-4(5Н)-она 26 в виде белых игл (110 мг; 36%).
Пример 22
3,6-Дибром-1-(2', 3', 5'-О-трибензоил-β-L-рибофуранозил)-пиразоло[3,4-d] пиримидин-4(5Н)-он 28
Ацетонитрил (80 мл) добавляли к смеси 3,6-дибромпиразоло[3,4-d]пиримидин-4(5Н)-она 27 (1,18 г; 4,0 ммоль) и 1-О-ацетил-2',3',5'-О-трибензоил-L-рибофуранозы (3,02 мг; 6,0 ммоль). Эту взвесь нагревали до температуры образования флегмы с последующим медленным добавлением этерата трифторборана (851 мг; 6,0 ммоль). Эту реакционную смесь грели при температуре образования флегмы в течение 6 ч. После удаления растворителя остаток растворяли в EtOAc (200 мл), промывали насыщенным NаНСО3 (2•50 мл), водой (2•50 мл), сушили (Nа2SО3) и концентрировали. Неочищенный продукт чистили тонкослойной хроматографией на кремнеземе (5% ацетон в хлориде метилена) с получением 1,49 г (50,5%) 3,6-дибром-1-(2', 3', 5'-О-трибензоил-β-L-рибофуранозил)-пиразоло[3,4-d]пиримидин-4(5Н)-она 28 в виде желтой пены.
Пример 23
3-Бром-7-деаза-8-аза-β-L-гуанозин 24
3,6-Дибром-1-(2', 3', 5'-О-трибензоил-β-L-рибофуранозил)-пиразоло[3,4-d] пиримидин-4(5Н)-он (260 мг; 0,35 ммоль) 28 растворяли в насыщенном растворе NН3 в МеОН (20 мл) при 0oС. Этот раствор в герметически закрытом баллоне из нержавеющей стали грели при 120oС в течение 16 ч. После охлаждения и удаления растворителя остаток растворяли в воде (100 мл), промывали посредством CH2Cl2 (5•15 мл) и концентрировали с получением желтого твердого вещества. Это твердое вещество растворяли в смеси метанола и хлорида метилена (1:1) и пропускали через слой силикагеля. Фильтрат концентрировали и твердый остаток растворяли в МеОН (5 мл) с последующим медленным добавлением диэтилового эфира (40 мл). Полученный преципитат отфильтровывали, промывали диэтиловым эфиром (2•2 мл) и сушили под вакуумом с получением 3-бром-7-деаза-8-аза-β-L-гуанозина 29 в виде беловатого твердого вещества (102,2 мг; 80,2%).
Пример 24
3-Амино-7-деаза-8-аза-L-гуанозин 25
3,6-Дибром-1-(2', 3', 5'-О-трибензоил-β-L-рибофуранозил)-пиразоло[3,4-d] пиримидин-4(5Н)-он 28 (500 мг; 0,68 ммоль) растворяли в насыщенном растворе NH2 в МеОН (50 мл) при 0oС с последующим добавлением тонкой медной проволоки (21,5 мг; 0,34 ммоль) и хлорида меди (19,8 мг; 0,20 ммоль). Эту смесь в герметически закрытом баллоне из нержавеющей стали грели при 120oС в течение 16 ч. После охлаждения и удаления растворителя остаток растворяли в МеОН, твердое вещество отфильтровывали и фильтрат концентрировали. Очистка остатка тонкослойной хроматографией на кремнеземе (20% МеОН в CH2Cl2) дала 3-амино-7-деаза-8-аза-L-гуанозин 30 в виде белого твердого вещества (62 мг; 30,9%).
Пример 25
7-Деаза-8-аза-L-гуанозин 26
3-Бром-7-деаза-8-аза-β-L-гуанозин 29 (246 мг, 0,68 ммоль) растворяли в ЕtOН (50%, 60 мл) с последующим добавлением 10% Pd/C (67 мг). Эту смесь встряхивали при давлении водорода 50 фунтов на кв. дюйм и при комнатной температуре в течение 6 ч. Палладиевый катализатор отфильтровывали и фильтрат концентрировали. Неочищенный продукт растворяли в МеОН с последующим добавлением силикагеля (2 г). После удаления метанола сухой силикагель с сорбированным неочищенным продуктом наносили на колонку с кремнеземом и проводили элюцию посредством 17% МеОН в СН2С12 с получением 7-деаза-8-аза-β-L-гуанозина 31 в виде белого твердого вещества (102,4 мг; 53,2%).
Пример 26
5-Амино-3-β-L-рибофуранозилтиазоло[4,5-d]пиримидин-2,7(6Н)-дион 34
5-Аминотиазоло[4,5-d] пиримидин-2,7(6Н)-дион 32 (400 мг; 2,71 ммоль) суспензировали в ацетонитриле (16 мл) и добавляли гексаметилдисилазан (0,96 мл), триметилхлорсилан (0,55 мл) и триметилсилилтрифлат (0,45 мл). Эту смесь перемешивали при температуре образования флегмы в течение 3,5 ч. По каплям добавляли раствор триметилсилилтрифлата (0,45 мл) в ацетонитриле (1,0 мл) и перемешивание с нагреванием продолжали еще 30 минут. Добавляли взвесь 1-О-ацетил-2,3,5-О-трибензоил-L-рибофуранозы (1,22 г; 2,28 ммоль) в ацетонитриле и смесь перемешивали при температуре образования флегмы в течение 30 минут. Реакционную смесь охлаждали и медленно вливали в интенсивно перемешиваемую смесь бикарбоната натрия (2,81 г) и воды (96 мл), что приводило к образованию липкого твердого вещества. Добавляли этилацетат и смесь перемешивали, пока это твердое вещество не растворялось. Водный слой дважды экстрагировали этилацетатом и объединенный органический слой промывали бикарбонатом натрия, сушили (Na2SО4) и концентрировали. Неочищенный продукт чистили хроматографией на кремнеземе, используя 5% Et3N и 5% атанол в хлориде метилена, с получением 1,10 г 5-амино-3-(2',3',5'-О-трибензоил-β-L-рибофуранозил)-тиазоло[4,5-d]-пиримидин-2,7(6Н)-диона 33 в виде белого твердого вещества.
5-Амино-3-(2', 3', 5'-О-трибензоил-β-L-рибофуранозил)-тиазоло[4,5-d]-пиримидин-2,7(6Н)-дион 33 (1,09 г; 1,717 ммоль) растворяли в метаноле (25 мл) и добавляли метоксид натрия (5,4 М в метаноле; 0,64 мл). Этот раствор стоял при комнатной температуре в течение 64 ч. Большую часть метанола выпаривали и добавляли воду (20 мл) и Амберлит в Н-форме (1 г). Эту суспензию осторожно перемешивали 20 минут и смолу отфильтровывали отсасыванием и промывали водой (2•10 мл). Фильтрат концентрировали и неочищенный продукт чистили кристаллизацией из метанола с получением 368 мг 5-амино-3-β-L-рибофуранозилтиазоло[4,5-d] пиримидин-2,7(6Н)-диона 34 в виде бесцветного твердого вещества.
Пример 27
3-β-L-Рибофуранозилтиазоло[4,5-d]пиримидин-2,7(6Н)-дион 36
К раствору 5-амино-3-β-L-рибофуранозилтиазоло[4,5-d]пиримидин-2,7(6Н)-диона 34 (1,40 г; 2,22 ммоль) в безводном тетрагидрофуране (50 мл) при комнатной температуре добавляли по каплям трет-бутилнитрит (15,05 ммоль; 1,72 мл). Полученный раствор перемешивали при комнатной температуре в течение 1 ч и при 50oС в течение 14 ч. Растворитель выпаривали и остаток хроматографировали на кремнеземе, используя 20-30% этилацетат в хлориде метилена, с получением 612 мг деаминированного продукта в виде пены.
500 мг деаминированного продукта растворяли в метаноле (15 мл) и добавляли 30% гироксид аммония (75 мл). Полученный раствор стоял при комнатной температуре в течение ночи. Растворитель выпаривали и остаток хроматографировапи на кремнеземе, используя 10-20% метанол в хлориде метилена, с получением 184 мг 3-β-L-рибофуранозилтиазоло[4,5-d]пиримидин-2,7(6Н)-диона 36 в виде бесцветного твердого вещества.
Пример 28
Метиловый эфир 5-цианометил-1-(2,3,5-О-трибензоил-L-рибофуранозил)-имидазол-4-карбоновой кислоты 38
Метиловый эфир 5-цианометилимидазол-4-карбоксикислоты 37 (Robins et al. J. Org. Chem. 1963, 28, 3041; 500 мг; 3,02 ммоль) грели при температуре дефлегмации в безводных условиях в течение 12 ч с HMDS (8 мл) и сульфатом аммония (30 мг). Избыток HMDS удаляли отгонкой под пониженным давлением с получением триметилсилилового производного в виде желтовато-коричневого масла. Это масло растворяли в сухом 1,2-дихлорэтане (20 мл) и добавляли 1-О-ацетил-2,3,5-трибензоилрибофуранозу (1,53 г; 3,03 ммоль) с последующим добавлением хлорида олова (516 мкг; 4,39 ммоль). Эту реакционную смесь перемешивали при температуре окружающей среды в течение 18 ч и затем вливали в 50 мл холодного водного 5% раствора NаНСО3. Преципитат отфильтровывали через целит и фильтрат экстрагировали хлороформом (3•50 мл). Экстракты сушили (Nа2SО4) и упаривали под пониженным давлением с получением светло-бежевой пены (1,8 г). Этот материал чистили хроматографией на кремнеземе, используя смесь гексанов с этилацетатом (1: 1), с получением 1,65 г (89%) метилового эфира 5-цианометил-1-(2,3,5-О-трибензоил-L-рибофуранозил)имидазол-4-карбоксикислоты 38 в виде бесцветного твердого вещества.
Пример 29
3-Деаза-β-L-гуанозин 39
Метиловый эфир 5-цианометил-1-(2,3,5-О-трибензоил-L-рибофуранозил)имидазол-4-карбоксикислоты 38 (1,03 г; 1,69 ммоль) растворяли в метаноле (60 мл) и насыщали безводным аммиаком при 0oС. Эту реакционную смесь помещали в герметически закрытый стальной баллон и держали при 100oС в течение 18 ч. Смесь охлаждали до комнатной температуры и упаривали досуха. Остаток суспензировали в теплом хлороформе и оставшиеся твердыми вещества отфильтровывали, промывали хлороформом (5•10 мл) и сушили. Неочищенный продукт перекристаллизовывали из воды с получением 320 мг (70%) 3-деаза-β-L-гуанозина 39 в виде бесцветного твердого вещества.
Пример 30
3-Бром-3-деаза-β-L-гуанозин 40
К перемешиваемому раствору 3-деаза-β-L-гуанозина 39 (200 мг; 0,708 ммоль) в 8 мл смеси воды и метанола (1:1) при 0oС добавляли бром (20 мкл; 0,39 ммоль). После перемешивания в течение 15 минут эту реакционную смесь упаривали досуха. Неочищенный материал суспензировали в хлороформе, фильтровали и сушили с получением 210 мг (82%) 3-бром-3-деаза-β-L-гуанозина 40 в виде бесцветного твердого вещества.

Изобретение относится к производному пуринового L-нуклеозида формулы (I), где R1, R2', R3' и R4 - Н; R2, R3 и R5 - ОН; Z1 - N; Z2 выбран из N и СН; Z3 - из -NR-, -С(R)2, -S-, где R, одинаковые или разные, выбраны из Н, Br, NH2, алкила и алкенила; Z4 выбран из -С=O, -NR-, -C(R)2-, где R, одинаковые или разные, выбраны из Н и Br; Z5 - N; Х выбран из Н, ОН, SH, -SNH2, -S(O)NH2, -S(O)2NH2; Y - из Н и NН2; W - О, и когда Y представляет собой NH2, тогда Z3 не представляет собой -S-. Также описана фармацевтическая композиция, содержащая соединения по изобретению. Предложенные соединения и композиции на их основе обладают иммуномодулирующей активностью. 2 с. и 6 з. п. ф-лы, 7 ил.
где R1, R2', R3' и R4 - Н;
R2, R3 и R5 - ОН;
Z1 - N;
Z2 выбран из группы, содержащей N и СН;
Z3 - из группы, содержащей -NR-, -C(R)2-, -S-, где R, одинаковые или разные, выбраны из группы, содержащей Н, Вr, NH2, алкил и алкенил;
Z4 - из группы, содержащей -С= O, -NR-, -C(R)2-, где R, одинаковые или разные, выбраны из группы, содержащей Н и Вr;
Z5 - N;
химическая связь между Z3 и Z4 выбрана из группы, содержащей С-С, С= С, C-N, C= N и C-S;
химическая связь между Z4 и Z5 выбрана из группы, содержащей C-N, C= N и N-N;
Х выбран из группы, содержащей Н, ОН, SH, -SNH2, -S(O)NH2, -S(O)2NH2;
Y - из группы, содержащей Н и NH2;
W - О
и когда Y представляет собой NH2, тогда Z3 нe представляет собой -S-.
где R2, R3 и R5 - ОН;
Z2 выбран из группы, содержащей N и СН;
Z4 - из группы, содержащей -С= O, -NR-, -C(R)2-, где R, одинаковые или разные, выбраны из группы, содержащей Н и Вr;
Y - из группы, содержащей Н и NH2.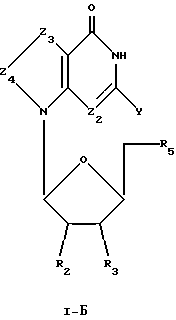
где R2, R3 и R5 - ОН;
Z2 выбран из группы, содержащей N и СН;
Z3 представляет собой -NR-, где R, одинаковые или разные, выбраны из группы, содержащей Н, Вr, NH2, алкил и алкенил;
Z4 представляет собой -С= O;
химическая связь между Z3 и Z4 представляет собой C-N;
Y выбран из группы, содержащей Н и NH2.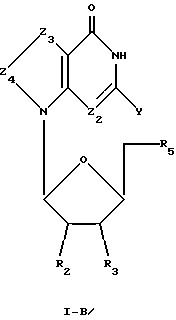
где R2, R3 и R5 - ОН;
Z2 выбран из группы, содержащей N и СН;
Z3 представляет собой -C(R)2-, где R, одинаковые или разные, выбраны из группы, содержащей Н, Br, NH2, алкил и алкенил;
Z4 представляет собой -C(R)2-, где R, одинаковые или разные, выбраны из группы, содержащей Н и Br;
химическая связь между Z3 и Z4 представляет собой С= С;
Y выбран из группы, содержащей Н и NH2.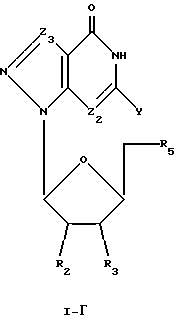
где R2, R3 и R5 - ОН;
Z2 выбран из группы, содержащей N и СН;
Z3 представляет собой -C(R)2-, где R, одинаковые или разные, выбраны из группы, содержащей Н, Вr, NH2, алкил и алкенил; и
Y выбран из группы, содержащей Н и NH2.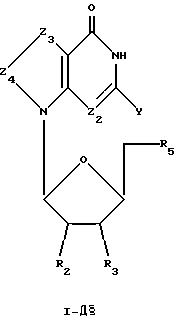
где R2, R3 и R5 представляют собой ОН;
Z2 выбран из группы, содержащей N и СН;
Z3 представляет собой -S-;
Z4 представляет собой -С= O,
химическая связь между Z3 и Z4 представляет собой C-S;
Y выбран из группы, содержащей Н и NH2.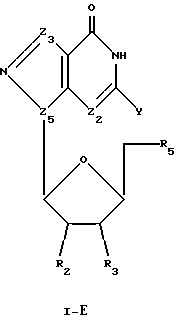
где R2, R3 и R5 - ОН;
Z2 выбран из группы, содержащей N и СН;
Z3 представляет собой -C(R)2-, где R, одинаковые или разные, выбраны из группы, содержащей Н, Вr, NH2, алкил и алкенил;
Z5 представляет собой N;
Y выбран из группы, содержащей Н и NH2.
Приоритет по пунктам:
16.10.1996 по пп. 1-7;
12.08.1997 по п. 8.

