Изобретение относится к фармацевтической композиции, имеющей повышенную активирующую остеогенез остеогенную активность, содержащей непептидное остеогенное активирующее оптически активное вещество и биоразлагаемый сополимер, которая является агентом, полезным для лечения и/или предотвращения заболеваний костей.
Предпосылки изобретения
Заболевания костей (например, переломы костей) могут иметь место у всех категорий людей вследствие различных причин в спорте и при дорожно-транспортных авариях. И, поскольку требуется длительное время для их излечения, заболевания костей вносят значительные затруднения в повседневную нормальную жизнь пациента. В последние годы число больных остеопорозом увеличилось в связи со старением населения. Так, частота переломов костей конечностей, связанных с остеопорозом, заметно увеличилась пропорционально этому. В частности, перелом кости шейки бедра делает необходимой длительную госпитализацию и часто приводит к внутренним осложнениям, в том числе к деменции, обусловленной длительной госпитализацией, и создает большие социальные и экономические проблемы. Неотъемлемой задачей является обеспечение возможности ранней выписки из больницы пациентов с переломом костей.
Излечивание перелома кости является формой заживления ран, характеризующейся местным проявлением и прогрессированием. Обычно различные местные факторы действуют хорошо в месте перелома для активирования заживления in vivo. Такие факторы включают в себя биоактивные вещества пептидного типа, такие как костные морфогенетические белки (BMP) и трансформирующие факторы роста (TGF), которые, как сообщалось, активируют остеогенез на моделях животных "Proceedings of the National Acagemy of Sciences, USA, vol.87, pp. 2220-2224 (1990), and Endocrinology, vol.124, pp.2991-2993 (1989)".
Что касается непептидного остеогенного активирующего вещества, сообщалось, например, о производных простагландина A1, производных витамина D3, производных бензилфосфоновой кислоты, производных фенолсульфофталеновой кислоты.
Вышеуказанные биоактивные вещества пептидного типа являются пептидами или белками с молекулярными массами более 5000 и они быстро метаболизируются in vivo и не являются стабильными. С учетом этого были получены некоторые препараты в попытке получения удовлетворительной стабильности, но все они не достигали достаточной активирующей остеогенез активности, и не существует препаратов, удовлетворительных в отношении качества и т.д. "Clinical Orthopaedics and Related Research, vol.178, pp.274-285". Также вышеуказанные непептидные активирующие остеогенез вещества не являются клинически эффективными в отношении активирования остеогенеза активности для излечивания переломов костей.
По этим причинам существует большая потребность в агенте высокого качества для лечения костного заболевания, который является высокостабильным, безопасным и активным, клинически эффективным при продолжительном лечении переломов костей.
Описание изобретения
Было проведено обширное исследование для решения указанных проблем и обнаружено, что агент для лечения заболевания костей, содержащий непептидное активирующее остеогенез оптически активное вещество и биоразлагаемый сополимер, неожиданно хорошо может служить для активации заживления переломов костей с более сильной остеогенной стимулирующей активностью непептидного остеогенного активирующего вещества, чем при его введении отдельно. Было проведено дальнейшее исследование на основании данного открытия и создано настоящее изобретение.
Таким образом, данное изобретение относится к:
(1) фармацевтической композиции, содержащей непептидное остеогенез активирующее оптически активное вещество формулы: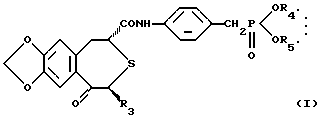
где R3 представляет низшую алкильную группу; и R4 и R5 независимо представляют низшую алкильную группу или связаны вместе с образованием низшей алкиленовой группы и сополимер молочной кислоты и гликолевой кислоты,
(2) фармацевтической композиции по п.(1), в которой R3, R4 и R5 независимо представляют C1-6-алкильную группу,
(3) фармацевтической композиции по п.(1), в которой соединение представляет собой (2R, 4S)-(-)-N-[4-(диэтоксифосфорилметил)фенил]-1,2,4,5-тетрагидро-4-метил-7,8-метилендиокси-5-оксо-3-бензотиепин-2-карбоксамид,
(4) фармацевтической композиции по п.(1), в которой весовое отношение указанного сополимера относительно непептидного остеогенного активирующего оптически активного вещества является приблизительно 1-100-кратным,
(5) фармацевтической композиции по п.(2), которая дополнительно содержит фосфорную кислоту или ее соль,
(6) фармацевтической композиции по п.(5), в которой фосфорная кислота или ее соль представляет собой фосфат натрия,
(7) фармацевтической композиции по п.(2), в которой относительное содержание (2R, 4S)-(-)-N-[4-(диэтоксифосфорилметил)фенил] -1,2,4,5-тетрагидро-4-метил-7,8-метилендиокси-5-оксо-3-бензотиепин-2-карбоксамида в расчете на указанный сополимер равно приблизительно 5-30% (масс.) и относительное содержание фосфата натрия в расчете на (2R,4S)-(-)-N-[4-(диэтоксифосфорилметил)фенил] -1,2,4,5-тетрагидро-4-метил-7,8-метилендиокси-5-оксо-3-бензотиепин-2-карбоксамид и данный сополимер равно приблизительно 0,1-20% (масс.).
(8) фармацевтической композиции по п.(1), в которой отношение молочная кислота/гликолевая кислота равно приблизительно 90/10-50/50 мас.% и средневесовая молекулярная масса равна приблизительно 8000-50000,
(9) фармацевтической композиции по п.(1) в форме суспензии,
(10) фармацевтической композиции по п.(1), используемой для инъекции, и т.д.
Применимые непептидные остеогенные активирующие оптически активные вещества данного изобретения включают серосодержащие гетероциклические соединения, такие как (2R,4S)-(-)-N-[4-(диэтоксифосфорилметил)фенил]-1,2,4,5-тетрагидро-4-метил-7,8-метилендиокси-5-оксо-3-бензотиепин-2-карбоксамид или его соли, описанные в US5071841, US5158943 и JP529460, производные бензопирана, такие как N-(4-диметоксифосфорилметилфенил)-4-оксо-4Н-1-бензопиран-2-карбоксамид или его соли, описанные в ЕР625522, производные фосфоновой кислоты, такие как диэтил-4-(7-циклогексил-3,4-дигидро-2-нафталинкарбоксамид)бензилфосфонат или его соли, описанные в W096/01267, производные простагландина A1, описанные в Journal of Pharmacology and Experimental Therapeutics, vol.258, pp.1120-1126 (1991), производные витамина D3, описанные в Bioorganic and Medicinal Chemistry Letters, vol.3, pp.1815-1819 (1993), производные бензилфосфоновой кислоты, описанные в ЕР524023, производные бисфосфоновых кислот, описанные в Bone, vol.13, pp.249-255 (1992), и производные витамина К2, описанные в Biochemical and Biophysical Research Communicatios, vol.187, pp.814-820 (1992).
Фармацевтическая композиция данного изобретения может содержать одно или несколько непептидных остеогенных активирующих веществ, описанных выше, в качестве активного ингредиента.
В вышеуказанных непептидных остеогенез активирующих оптически активных веществах предпочтительно используют для данного изобретения соединение, представленное формулой (I), или его соль.
Оптически активное соединение, представлено формулой (I):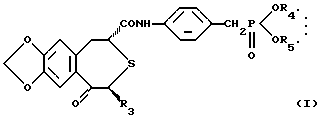
где R3 представляет низшую алкильную группу; R4 и R5 независимо представляют низшую алкильную группу или связываются вместе с образованием низшей алкиленовой группы.
Примерами низшей алкильной группы, представленной R3, R4 или R5 в формуле (I), являются алкильные группы, имеющие 1-6 (предпочтительно 1-4) атомов углерода, такие как метил, этил, пропил, изопропил, бутил, изобутил, втор-бутил, трет-бутил, пентил, изопентил, неопентил и гексил. R4 и R5 могут связываться вместе с образованием низшей алкиленовой группы. В этом случае часть молекулы: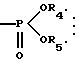
может обозначать группу: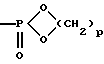
где р равно целому числу от 2 до 4.
Предпочтительные группы для R3, R4 и R5 включают алкильные группы, имеющие 1-4 атома углерода, такие как метил и этил. Соединение (I) является оптически активным соединением (2R,4S)конфигурации и по существу не содержит соединения (2S, 4R)конфигурации. Предпочтительным является соединение (I) с оптической чистотой, близкой к 100%.
Наиболее предпочтительным в качестве соединения (I) является, например, соединение (2R, 4S)-(-)-N-[4-(диэтоксифосфорилметил)фенил] -1,2,4,5-тетрагидро-4-метил-7,8-метилендиокси-5-оксо-3-бензотиепин-2-карбоксамид (далее называемое также соединением А).
Солью непептидного остеогенного активирующего соединения согласно данному изобретению является предпочтительно фармацевтически приемлемая соль. Фармацевтически приемлемые соли включают соли с неорганическими основаниями, соли с органическими основаниями и соли с основными или кислотными аминокислотами. Неорганические основания, способные к образованию таких солей, включают гидроксиды щелочных металлов (например, натрия, калия) и щелочноземельных металлов (например, кальция, магния), такие органические основания включают триметиламин, триэтиламин, пиридин, пиколин, N,N-дибензилэтилендиамин и диэтаноламин, такие неорганические кислоты включают хлористоводородную кислоту, бромистоводородную кислоту, иодистоводородную кислоту, фосфорную кислоту, азотную кислоту и серную кислоту, такие органические кислоты включают муравьиную кислоту, уксусную кислоту, трифторуксусную кислоту, щавелевую кислоту, винную кислоту, фумаровую кислоту, малеиновую кислоту, метансульфоновую кислоту, бензолсульфоновую кислоту, п-толуолсульфокислоту и лимонную кислоту, и такие основные или кислотные аминокислоты включают аргинин, лизин, аспарагиновую кислоту и глутаминовую кислоту.
Непептидное остеогенное активирующее оптически активное вещество данного изобретения может быть получено, например, по общеизвестной методике (например, согласно US5071841, US5158943, описанным выше) и по способу, описанному ниже, или его модификации. Например, соединение (I) или его соль может быть получено взаимодействием оптически активного соединения формулы (II):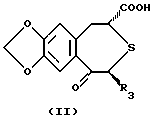
где R3 имеет значения, приведенные выше, или реакционноспособного производного по карбоксильной группе или его соли с соединением формулы (III):
где R4 и R5 имеют значения, приведенные выше, его реакционноспособным производным по аминогруппе или его солью.
Предпочтительные реакционноспособные производные аминогруппы на соединении (III) включают таутомерные изомеры имино- или енамино-формы оснований Шиффа, образующиеся в результате взаимодействия соединения (III) и карбонильного соединения, такого как альдегид (например, ацетальдегид) или кетон (например, ацетон); силильные производные, образующиеся в результате взаимодействия соединения (III) и силильного соединения, такого как бис(триметилсилил)ацетамид, моно(триметилсилил)ацетамид или бис(триметилсилил)мочевина; и производные, получающиеся при взаимодействии соединения (III) и трихлорида фосфора или фосгена.
Предпочтительные реакционноспособные производные карбоксильной группы на соединении (II) включают галогенангидриды, ангидриды кислот, активированные амиды и активированные сложные эфиры, которые получают по общепринятым методикам. Предпочтительные реакционноспособные производные включают хлорангидриды; азиды кислот; смешанные ангидриды кислот с замещенной фосфорной кислотой, такой как диалкилфосфорная кислота, фенилфосфорная кислота, дифенилфосфорная кислота, дибензилфосфорная кислота или галогенированная фосфорная кислота, диалкилфосфорная кислота, сернистой кислотой, тиосерной кислотой, серной кислотой, сульфоновой кислотой, такой как метансульфоновая кислота, алифатической карбоновой кислотой, такой как уксусная кислота, пропионовая кислота, масляная кислота, изомасляная кислота, пивалиновая кислота, пентановая кислота, изопентановая кислота или трихлоруксусная кислота, или ароматической карбоновой кислотой, такой как бензойная кислота; симметричные ангидриды кислот; активированные амиды с имидазолом, 4-замещенным имидазолом, диметилпиразолом, триазолом или тетразолом; активированные сложные эфиры, такие как цианометиловый эфир, метоксиметиловый эфир, диметилиминометиловый эфир, виниловый эфир, пропаргиловый эфир, п-нитрофениловый эфир, трихлорфениловый эфир, пентахлорфениловый эфир, мезилфениловый эфир, фенилазофениловый эфир, фенилтиоэфир, п-нитрофениловый эфир, п-крезилтиоэфир, карбоксиметилтиоэфир, пираниловый эфир, пиридиловый эфир, пиперидиловый эфир или 8-хинолилтиоэфир; и сложные эфиры N-гидроксисоединений, такие как N, N-диметилгидроксиламин, 1-гидрокси-2-(1Н)-пиридон, N-гидроксисукцинимид, N-гидроксифталимид, 1-гидрокси-1Н-бензо-триазол и N-гидрокси-5-норборнен-2,3-дикарбоксиимид. Эти реакционноспособные производные могут быть необязательно выбраны в соответствии с типом соединения (II).
Предпочтительные соли реакционноспособных производных соединения (II) или (III) включают основные соли, например соли щелочных металлов, такие как соль натрия и соль калия, соли щелочноземельных металлов, такие как соль кальция и соль магния, соль аммония, и соли органических оснований, таких как соль триметиламина, соль триэтиламина, соль пиридина, соль пиколина, соль дициклогексиламина и соль дибензилэтилендиамина.
Указанную реакцию, как правило, проводят в обычном растворителе, таком как вода, спирт (например, метанол, этанол), ацетон, диоксан, ацетонитрил, хлороформ, метиленхлорид, этиленхлорид, тетрагидрофуран, этилацетат, N,N-диметилформамид или пиридин, но ее можно проводить в любом другом органическом растворителе, если он не мешает протеканию реакции. Указанные обычные растворители могут быть использованы в смеси с водой. В данной реакции при применении соединения (II) или (III) в форме свободной кислоты или ее соли реакцию предпочтительно осуществляют в присутствии обычного конденсирующего агента, такого как N,N'-дициклогексилкарбодиимид; N-циклогексил-N'-морфолиноэтилкарбодиимид; N-циклогексил-N'-(4- диэтиламиноциклогексил)карбодиимид; N,N'-диэтилкарбодиимид; N,N'-диизопропилкарбодиимид; N-этил-N'-(3-диметиламинопропил)карбодиимид; N,N'-карбонилбис(2-метилимидазол); пентаметиленкетен-N-циклогексилимин; дифенилкетен-N-циклогексилимин; этоксиацетилен; 1-алкокси-1-хлорэтилен; триалкилфосфит; этилполифосфат; изопропилполифосфат; оксихлорид фосфора; дифенилфосфорилазид; тионилхлорид; оксалилхлорид; низший алкилгалогенформиат, такой как этилхлорформиат или изопропилхлорформиат; трифенилфосфин; соль 2-этил-7-гидроксибензизооксазолия; внутримолекулярная соль гидроксида 2-этил-5-(м-сульфофенил)изоксазолия; N-гидрокси-бензотриазол; 1-(п-хлорбензолсульфонилокси)-6-хлор-1Н-бензотриазол или реагент Вилсмейера, полученный взаимодействием N,N'-диметилформамида с тионилхлоридом, трихлорметилхлорформиатом или оксихлоридом фосфора. Также желательной является методика с использованием конденсирующего агента, такого как N,N'-дициклогексилкарбодиимид, в присутствии N-гидроксибензотриазола или N-гидрокси-5-норборнен-эндо-2,3-дикарбоксиимида. Данную реакцию можно также проводить в присутствии неорганического или органического основания, такого как бикарбонат щелочного металла, три(низший)алкиламин, пиридин, N-(низший)алкилморфолин или N, N-ди(низший)алкилбензиламин. Хотя температура реакции не ограничивается, данную реакцию обычно проводят в условиях охлаждения - нагревания (-10 -120oС). Время реакции равно обычно приблизительно 0,5-100 часам, предпочтительно приблизительно 1-50 часам.
Соединение (I), полученное таким образом, можно выделить и очистить известными способами разделения и очистки, такими как концентрирование, концентрирование при пониженном давлении, экстракция растворителем, кристаллизация, перекристаллизация, повторное растворение и хроматография.
Исходное соединение (II) может быть получено, например, оптическим разделением рацемата соединения (II), описанным в US5158943. В частности, оптически активное соединение получают приготовлением соли рацемата соединения (II) и оптически активного основания (например, α-метилбензиламина, бруцина, хинина, цинхонина), повторения фракционной кристаллизации на основе различия растворимости между образующимися диастереоизомерами с получением слаборастворимой соли в чистом виде и с последующей обработкой кислотой. Другое оптически активное соединение можно получить этерификацией рацемата соединения (II) оптически активным спиртом (например, оптически активным метиллактатом, метилманделатом), получением другого сложного эфира в чистом виде на основе различия физических свойств между образующимися диастереоизомерами с последующим проведением гидролиза.
Сополимер молочной кислоты и гликолевой кислоты согласно данному изобретению представляет собой сополимер, который растворим или нерастворим в воде и является разлагаемым in vivo.
Полимеризация может быть произвольной полимеризацией, блок-сополимеризацией или графт-полимеризацией.
Среднюю молекулярную массу указанных сополимеров данного изобретения выбирают предпочтительно из диапазона приблизительно от 2000 до 800000, более предпочтительно приблизительно от 5000 до 200000.
Относительное содержание молочной кислоты и гликолевой кислоты в сополимере молочной кислоты-гликолевой кислоты равно предпочтительно приблизительно 100/0-50/50 по массе и более предпочтительно приблизительно 90/10-50/50 по массе. Средневесовая молекулярная масса сополимера молочной кислоты-гликолевой кислоты равна предпочтительно приблизительно от 5000 до 100000, более предпочтительно приблизительно 8000-50000. Сополимер молочной кислоты-гликолевой кислоты можно синтезировать по общеизвестной методике получения, такой как описано в ЕР172636. Данный сополимер предпочтительно синтезируют дегидрогенизационной поликонденсацией в отсутствии катализатора.
В данной заявке средневесовую молекулярную массу определяют как средневесовую массу в расчете на полистирол, измеренную при помощи гель-проникающей хроматографии (ГПХ). Измерения проводили с применением колонки ГПХ KF804L•2 (производимой Showa Denko) и монитора RI L-3300 (производимого Hitachi Ltd.) с хлороформом в качестве подвижной фазы.
Количество данного сополимера меняется в соответствии с величиной фармакологической активности непептидного остеогенез активирующего вещества, скорости и продолжительности высвобождения лекарственного средства из данного сополимера и так далее, пока не будет достигнута желаемая цель. Например, сополимер используют в приблизительно 0,2-10000-кратных количествах (соотношение по массе), предпочтительно приблизительно 1-1000-кратных количествах более предпочтительно приблизительно 1-100-кратных количествах, относительно количества биоактивного вещества.
Фармацевтическая композиция данного изобретения может быть получена обычными способами получения фармацевтической композиции, например, она может быть получена диспергированием непептидного активирующего вещества в данном сополимере или помещением непептидного остеогенного активирующего вещества в предварительно сформованный полый сополимер. В частности, используемые способы включают способ сушки в воде, способ разделения фаз, способ сушки распылением и их модификации.
Фармацевтическая композиция данного изобретения, полученная по способу, указанному выше, может быть в форме, например, мелких частиц, сфер, палочек, игл, гранул, пленок или кремов, но форма ее не ограничивается только указанными формами для достижения желаемой цели.
В данной заявке фармацевтическую композицию мелких частиц называют также микрокапсулами или микросферами.
Ниже описаны примеры способов получения микрокапсул.
(1) Способ сушки в воде (м/в, т.е. способ получения эмульсии типа "масло в воде").
В данном способе сначала готовят раствор в органическом растворителе, содержащий сополимер. Органический растворитель, используемый для получения фармацевтической композиции данного изобретения, предпочтительно имеет точку кипения не выше 120oС. Такие органические растворители включают галогенированные углеводороды (например, дихлорметан, хлороформ, хлорэтан, дихлорэтан, трихлорэтан, четыреххлористый углерод), алифатические сложные эфиры (например, этиловый эфир, бутиловый эфир), простые эфиры (например, этиловый простой эфир, изопропиловый простой эфир) и ароматические углеводороды (например, бензол, толуол, ксилол). Данные растворители можно использовать в сочетании из двух или нескольких типов растворителей в подходящих соотношениях. Предпочтительным растворителем является дихлорметан или ацетонитрил, более предпочтительно дихлорметан. Концентрацию сополимера в растворе органического растворителя обычно выбирают в диапазоне приблизительно от 0,01 до 80 мас. %, предпочтительно приблизительно 0,1-70 мас.% и более предпочтительно приблизительно 1-60 мас.%, хотя она изменяется в зависимости от молекулярной массы данного сополимера и от типа растворителя, и т.д.
Непептидное остеогенное активирующее вещество добавляют и растворяют в полученном таким образом растворе в органическом растворителе, содержащем данный сополимер, если необходимо, после лиофилизации или сушки в вакууме. Количество непептидного остеогенного активирующего вещества составляет приблизительно от 0,001 до 90 мас.%, предпочтительно приблизительно 0,1-80 мас.% и более предпочтительно приблизительно 0,1-50 мас.% в расчете на концентрацию сополимера в растворе органического растворителя, хотя количество изменяется в зависимости от типа лекарственного средства, механизма его действия на остеогенез, продолжительности действия и т.д.
Полученный таким образом раствор в органическом растворителе добавляют затем к водной фазе с получением эмульсии типа масло в воде при помощи приводной мешалки турбинного типа или т.п. Объем водной фазы обычно выбирают из диапазона приблизительно от 1 до 10000-кратного, предпочтительно приблизительно от 2 до 5000-кратного и более предпочтительно приблизительно от 5 до 2000-кратного относительно объема масляной фазы.
К водной фазе может быть добавлен эмульгатор. Это может быть любой эмульгатор, если он способен образовывать стабильную эмульсию типа масло в воде. Примеры таких эмульгаторов включают анионогенные поверхностно-активные вещества, неионогенные поверхностно-активные вещества, полиоксиэтиленовые производные касторового масла, поливинилпирролидон, поливиниловый спирт, карбоксиметилцеллюлозу, лецитин, желатин и гиалуроновую кислоту. Они могут быть использованы в подходящем сочетании. Концентрация эмульгатора в водной фазе равна предпочтительно приблизительно 0,001-20 мас.%, более предпочтительно приблизительно 0,01-10 мас. % и еще более предпочтительно приблизительно 0,05-5 мас.%.
Выпаривание растворителя из масляной фазы может быть выполнено общепринятыми способами, в том числе по методике, в которой растворитель выпаривают при нормальном или постепенно уменьшающемся давлении во время перемешивания пропеллерной мешалкой или магнитной мешалкой и т.д., и по методике, в которой растворитель выпаривают при доведении вакуума до нужной величины с использованием роторного испарителя и т.д. Полученные микрокапсулы разделяют центрифугированием или фильтрованием, после чего их промывают, например, водой или гептаном, несколько раз для удаления свободных непептидных остеогенных активирующих веществ, эмульгатора и т.д., прилипших к поверхности микрокапсул. Затем микрокапсулы опять диспергируют в дистиллированной воде и т.д. и лиофилизуют.
В описанном выше м/в-способе микрокапсулы могут быть получены по р/м/в-способу (растворитель/масло/вода), в котором непептидное остеогенное активирующее вещество диспергируют в растворе органического растворителя, содержащем данный сополимер.
(2) Способ сушки в воде (в/м/в способ)
В данном способе непептидное остеогенное активирующее вещество или его соль сначала растворяют или диспергируют в воде с получением указанной выше концентрации для получения внутренней водной фазы, если необходимо, с растворением или суспендированием, добавляя удерживающее лекарственное средство вещество, такое как белок (например, желатин), морская водоросль (например, агар), полисахарид (например, альгиновая кислота), синтетическое высокомолекулярное вещество (например, поливиниловый спирт), основная аминокислота (например, аргинин, лизин) или т.п. Внутренняя водная фаза может быть дополнена органической кислотой, такой как уксусная кислота, щавелевая кислота или лимонная кислота, неорганической кислотой, такой как угольная кислота или фосфорная кислота, гидроксидом щелочного металла, таким как гидроксид натрия, основной аминокислотой, такой как аргинин или лизин, или их солью (например, солями с органическими кислотами, такими как уксусная кислота, щавелевая кислота, лимонная кислота, или солями с неорганическими кислотами, такими как угольная кислота, фосфорная кислота и соляная кислота), в качестве регулятора pН для сохранения стабильности и растворимости непептидного остеогенного активирующего оптически активного вещества или его соли. В качестве стабилизатора непептидного остеогенного активирующего оптически активного вещества могут быть добавлены белок (например, альбумин, желатин), производное крахмала (например, декстрин, пуллулан и т.д.), органическая кислота (например, лимонная кислота), соль щелочного металла этилендиаминтетрауксусной кислоты (например, этилендиаминтетраацетат натрия), кислая соль щелочного металла сернистой кислоты (например, бисульфит натрия), синтетическое высокомолекулярное вещество (например, полиэтиленгликоль) или т. п. Могут быть также добавлены обычные консерванты, такие как п-оксибензоаты (например, метилпарабен, пропилпарабен), бензиловый спирт, хлорбутанол и тимеросал. Дополнительное количество непептидного остеогенного активирующего вещества равно приблизительно 0,001-90 мас.%, предпочтительно приблизительно 0,01-80 мас. %, более предпочтительно приблизительно 0,1-50 мас. %, хотя это количество меняется в зависимости от типа лекарственного средства, механизма действия его на остеогенез или продолжительности действия и т.д.
Полученную внутреннюю водную фазу добавляют к раствору (масляная фаза), содержащему данный сополимер, с последующей эмульгирующей обработкой с получением эмульсии типа вода в масле. Эмульгирование достигается известными способами диспергирования, которые включают в себя способ прерывистого встряхивания, способ с применением смесителя, такого как пропеллерный смеситель или турбинный смеситель, способ с применением коллоидной мельницы, способ с применением гомогенизатора и способ обработки ультразвуком. Описанный выше раствор (масляная фаза), содержащий данный сополимер, представляет собой раствор, полученный растворением сополимера в органическом растворителе. Данный растворитель может быть любым растворителем, если его точка кипения не превышает приблизительно 120oС и если он не смешивается с водой. Такие растворители включают галогенированные углеводороды (например, дихлорметан, хлороформ, хлорэтан, дихлорэтан, трихлорэтан, четыреххлористый углерод), алифатические сложные эфиры (например, этилацетат, бутилацетат), простые эфиры (например, этиловый простой эфир, изопропиловый простой эфир) и ароматические углеводороды (например, бензол, толуол, ксилол). Указанные растворители можно применять в сочетании из двух или более типов в подходящих соотношениях.
Затем полученную эмульсию типа вода в масле добавляют к водной фазе с получением эмульсии типа вода/масло/вода, из которой растворитель масляной фазы выпаривают с получением микрокапсул. Специфический способ для данного получения идентичен способу, описанному в (1) выше.
(3) Способ разделения фаз
В данном способе к вышеописанной эмульсии вода/масло постепенно добавляют коацерватирующий агент при перемешивании для осаждения и отверждения данного сополимера. В качестве коацерватирующего агента могут быть использованы силиконовое масло, растительные масла и жиры (например, кунжутное масло, соевое масло, кукурузное масло, хлопковое масло, льняное масло), минеральные масла, углеводороды (например, н-гексан, н-гептан), если они представляют собой полимерные соединения минерального масла или растительного масла, которые могут смешиваться с растворителем данного сополимера и которые не растворяют данный сополимер, для инкапсулирования. Они могут быть использованы в сочетании из двух или нескольких типов коацерватирующих агентов.
Полученные микрокапсулы после их фильтрования и отделения повторно промывают гептаном и т. д. для удаления коацерватирующего агента. Затем свободное лекарственное средство и растворитель удаляют с использованием того же способа сушки в воде. Для предотвращения флокуляции частиц во время промывания могут быть добавлены антифлокулянты: водорастворимые сахара, такие как маннит, лактол, глюкоза, и крахмалы (например, кукурузный крахмал), аминокислоты, такие как глицин и аланин, и белки, такие как желатин, фибрин и коллаген.
(4) Способ сушки распылением
Для получения микрокапсул по данному способу вышеописанную эмульсию типа вода в масле распыляют через распылительное сопло в сушильную камеру распылительной сушки для испарения органического растворителя и воды в виде мелких капелек за короткое время и получают микрокапсулы. Примерами распылительного сопла являются двухкомпонентная форсунка, сопло высокого давления и сопло с вращающимся диском. Для предотвращения флокуляции микрокапсул через другое сопло может распыляться водный раствор описанного выше антикоагулянта одновременно с распылением эмульсии типа вода в масле. Полученные таким образом микрокапсулы могут быть нагреты при пониженном давлении для облегчения удаления содержащихся в них воды и растворителя.
Кроме получения вышеописанных микрокапсул, фармацевтическая композиция согласно данному изобретению может быть получена путем растворения диспергированного в данном сополимере непептидного остеогенного активирующего оптически активного вещества и формования данного раствора в сферы, иглы, гранулы, пленки или т.п. при помощи подходящего для этого способа. Диспергированное в сополимере непептидное остеогенное активирующее оптически активное вещество получают, например, согласно способу, описанному в US3773919.
Кроме того, фармацевтическая композиция данного изобретения также может быть получена распылением до подходящего размера частиц диспергированного в данном сополимере непептидного остеогенного активирующего оптически активного вещества согласно способу, такому как описано в JP62234656, в котором используют разбрызгиватель струйной мельницы турбинного типа или ультразвуковой струйный разбрызгиватель. В частности, непептидное остеогенное активирующее оптически активное вещество добавляют к органическому растворителю, содержащему сополимер, и растворяют в нем. Затем твердый раствор, полученный вакуумной сушкой, грубо измельчают и просеивают с последующим удалением растворителя, после чего грубые частицы измельчают до контролируемого размера частиц при помощи ультразвукового струйного пульверизатора с получением фармацевтической композиции данного изобретения.
Фармацевтическая композиция данного изобретения может быть использована в форме микрокапсул как таковых или сформулирована в различные дозированные формы с микрокапсулами, сферами, палочками, иглами, шариками, пленками или кремами в качестве исходного материала. Фармацевтическая композиция данного изобретения может содержать фосфорную кислоту или ее соль (например, фосфат натрия, фосфат калия) в количестве приблизительно 0-30%. Фармацевтическая композиция данного изобретения может также вводиться в виде парентерального средства для местного введения (например, в виде инъецируемых препаратов для внутримышечного, подкожного введения, введения в органы или суставы и т.д., твердых препаратов, таких как вводимые надолго препараты, гранулы и порошки, жидких препаратов, таких как суспензии и мази).
Инъецируемый препарат может быть приготовлен в виде водной суспензии суспендированием микрокапсул в воде вместе с диспергирующим агентом (например, поверхностно-активными веществами, такими как Твин 80 и НСО-60, полисихаридами, такими как карбоксиметилцеллюлоза, альгинат натрия и гиалуроновая кислота и полисорбат), консервантом (например, метилпарабен, пропилпарабен), изотоническим агентом (например, хлорид натрия, маннит, сорбит, глюкоза), буфером (например, карбонат кальция), агентом для доведения рН (например, фосфат натрия, фосфат калия) и т.д., и он может быть приготовлен в виде масляной суспензии диспергированием микрокапсул в растительном масле, таком как кунжутное масло или кукурузное масло, с фосфолипидом, таким как лецитин, или без него или триглицеридом жирной кислоты с цепью умеренной длины (например, MIGLYOL 812).
Фосфат может повышать остеогенную активирующую активность фармацевтической композиции согласно данному изобретению.
Концентрация фосфата натрия или фосфата калия в таком инъецируемом препарате равна приблизительно от 0,1 мМ до 500 мМ, предпочтительно приблизительно 1 мМ-100 мМ.
Предпочтительным вариантом данного изобретения является следующий вариант.
(A) сополимер молочной кислоты-гликолевой кислоты, в котором соотношение молочной кислоты/гликолевой кислоты равно приблизительно 90/10-50/50 по массе и средневесовая молекулярная масса равна приблизительно 8000-50000,
(B) (2R,4S)-(-)-N-[4-(диэтоксифосфорилметил)фенил]-1,2,4,5-тетрагидро-4-метил-7,8-метилендиокси-5-оксо-3-бензотиепин-2-карбоксамид и (С) фосфат натрия.
Относительное содержание (В) в расчете на (А) равно приблизительно 30 мас.%.
Относительное содержание (С) в расчете на (А) и (В) равно приблизительно 0,1-20 мас.%.
При использовании микрокапсул, например, в виде инъецируемой суспензии размер их частиц выбирают в диапазоне приблизительно 0,1-300 мкм (средний диаметр частиц), если они удовлетворяют требованиям, касающимся степени диспергирования и способности прохождения через иглу шприца. Предпочтительно размер частиц равен приблизительно 1-150 мкм, более предпочтительно приблизительно 2-100 мкм.
Фармацевтическая композиция данного изобретения представляет собой предпочтительно суспензию, как описано выше.
Фармацевтическая композиция данного изобретения предпочтительно находится в форме мелких частиц. Это связано с тем, что такая композиция будет вызывать меньшую боль у пациента при введении через иглу для инъекции для подкожной или внутримышечной инъекции.
Фармацевтическая композиция данного изобретения предпочтительно представляет собой препарат для инъекции.
Способы получения микрокапсул в виде стерильного препарата включают (но не ограничиваются ими) способ, в котором весь процесс получения является стерильным, способ, в котором в качестве стерилизующего агента используют гамма-излучение, и способ, в котором добавляют антисептическое средство.
Фармацевтическая композиция данного изобретения может быть использована для предотвращения и лечения заболеваний костей (например, переломов костей, рефрактур, остеопороза, остеомаляции, синдрома Бехчета (тройного симптомокомплекса), анкилозирующего спондилоартрита, ревматоидного артрита и деструкции ткани сустава, вызываемой деформирующим гонартритом, и родственных заболеваний), для восстановления костной ткани после хирургии по поводу миеломной болезни (болезни Калера), рака легких, рака молочной железы и т.д. и для регенерации периодонтальной ткани в парадонтопатии, поскольку данная композиция обнаруживает свойство пролонгированного (замедленного) высвобождения с повышенной активностью непептидного остеогенного активирующего вещества и имеет время пролонгированного высвобождения от 1 недели до 3 месяцев в зависимости от типа и содержания сополимера и т.д. Фармацевтическая композиция данного изобретения особенно эффективна в случае пациентов с переломами костей, поскольку указанные пациенты обычно имеют фиксированную пораженную часть, покрытую гипсовой повязкой, и желают охотнее ускорить выздоровление посредством однократного введения, чем посредством многочисленных введений. Препарат пролонгированного действия, состоящий из фармацевтической композиции данного изобретения, можно использовать в сочетании с другими активными агентами. Например, в случае соединения, представленного формулой (I), в качестве остеогенного активирующего вещества для сочетания с ним можно использовать препараты кальция (например, карбонат кальция), кальцитонин, витамин D (например, альфакальцидол), половой гормон (например, эстроген, эстрадиол), простагландин A1, бисфосфоновую кислоту, иприфлавон, соединение фтора (например, фторид натрия), витамин K2, BMP (морфогенетический белок костей), FGF (фибробластный фактор роста), PDGF (тромбоцитарный фактор роста), TGF-β (трансформирующий фактор роста-β), IGF-1 (инсулин-подобный фактор роста-1), IGF-2 (инсулин-подобный фактор роста-2), РТН (паратиреоидный гормон) и так далее.
Обладая низкой токсичностью, фармацевтическая композиция данного изобретения может безопасно использоваться для млекопитающих (например, для людей, крупного рогатого скота, лошадей, свиней, кошек, мышей, крыс, кроликов).
Ожидается, что фармацевтическая композиция данного изобретения может служить в качестве безопасного препарата с высокой эффективностью, обеспечивая постоянное действие лекарственного средства с низкой токсичностью и удовлетворяя требованиям предотвращения и лечения заболеваний костей, восстановления поврежденной костной ткани и регенерации периодонтальной ткани в случае периодонтита и т. д., поскольку данная композиция постоянно высвобождает лекарственное средство на протяжении длительного периода времени. Например, при применении фармацевтической композиции данного изобретения для лечения переломов костей (например, перелома кости шейки бедра) она может эффективно проявлять ее остеогенное активирующее действие местно и значительно сокращать время выздоровления, которое обычно составляет 2-6 месяцев после перелома кости. Поэтому пациенты быстро возвращаются к нормальной общественной жизни и могут быть также предотвращены различные осложнения, вызываемые переломами костей в старческом возрасте.
Доза фармацевтической композиции данного изобретения может быть эффективным количеством непептидного остеогенного активирующего оптически активного вещества, находящимся, однако, в зависимости от типа и содержания данного непептидного остеогенного активирующего оптически активного вещества, времени высвобождения лекарственного средства, конкретного животного и т. д. Например, при применении фармацевтической композиции данного изобретения в форме микрокапсул для лечения участка кости с переломом ее можно вводить в дозе приблизительно от 0,01 до 500 мг, предпочтительно приблизительно 5-50 мг в расчете на содержание активного ингредиента (например, соединения (I)) на взрослого больного (с весом 50 кг) на одну дозу, от одного раза в неделю до одного раза в месяц.
Краткое описание рисунков
Фиг. 1 - графическое изображение минерального содержания (мг) малоберцовой кости через 2 недели после введения соединения А в виде микрокапсул и микрокапсул плацебо (контроль) крысам.
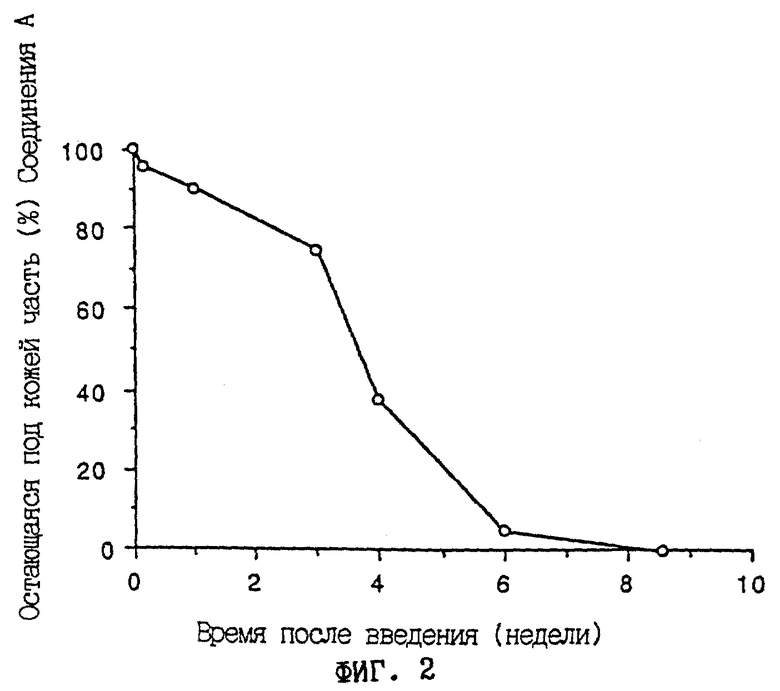
Фиг. 2 - графическое изображение временного изменения скорости удерживания соединения А в месте введения у крыс, получивших подкожную имплантацию таблетки, содержащей соединение А в виде микрокапсул в спину. Абсцисса указывает время (недели) после введения. Ордината показывает остающуюся под кожей дозу (%) соединения А.
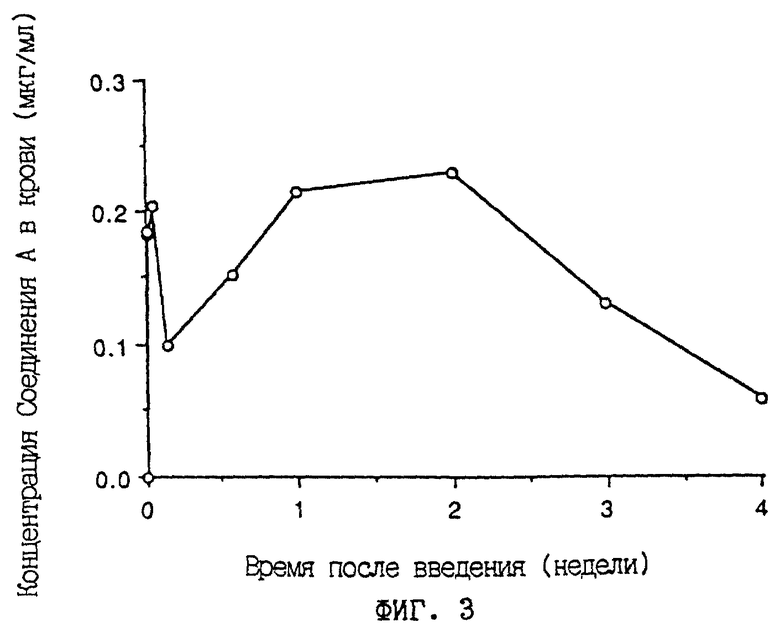
Фиг. 3 - графическое изображение изменения во времени концентрации соединения А в крови крыс, получивших подкожную имплантацию в спинки находящегося в виде микрокапсул соединения А.
Абсцисса указывает время (недели) после введения. Ордината показывает концентрацию (мкг/мл) соединения А в крови.
Наилучший способ воплощения изобретения
Данное изобретение описано далее более подробно при помощи следующих приводимых в качестве справочных примеров, рабочих примеров и примеров испытаний, которые не предназначены для ограничения данного изобретения. В рабочих примерах и примерах испытаний комнатной температурой называют температуру между приблизительно 0 и 30oС.
Примеры
Справочный пример 1
Получение (R)-α-метоксикарбонилбензилового эфира (2R,4S)-(-)-1,2,4,5-тетрагидро-4-метил-7,8-метилендиокси-5-оксо-3-бензотиепин-2-карбоновой кислоты
Раствор гидрохлорида 1-этил-3-(3-диметиламинопропил) карбодиимида (12,59 г) в дихлорметане (200 мл) добавляли по каплям к раствору (±)-т-1,2,4,5-тетрагидро-4-метил-7,8-метилендиокси-5-оксо-3-бензотиепин-2-карбоновой кислоты (15,34 г) и метил-(R)-(-)-миндальной кислоте (18,19 г) в N,N-диметилформамиде (ДМФ) (200 мл) при 0oС с последующим добавлением 4-диметиламинопиридина (ДМАР) (3,34 г). После перемешивания при 0oС в течение 1 часа и при комнатной температуре в течение 15 часов смесь выливали на воду и экстрагировали этилацетатом. Слой этилацетата промывали водой и сушили (MgSO4), после чего растворитель выпаривали. Оставшиеся кристаллы собирали фильтрованием, промывали смесью эфир-гексан и дважды перекристаллизовывали из смеси этилацетат-гексан с получением соединения заголовка (4,09 г, выход 17%).
Точка плавления: 140-141oС.
Оптическое вращение [α]D (23oС): -244,2oС (с=0,50, СНСl3).
Справочный пример 2
Получение (2R, 4S)-{-)-1,2,4,5-тетрагидро-4-метил-7,8-метилендиокси-5-оксо-3-бензотиепин-2-карбоновой кислоты
Смесь (R)-α-метоксикарбонилбензилового эфира (2R,4S)-(-)-1,2,4,5-тетрагидро-4-метил-7,8-метилендиокси-5-оксо-3-бензотиепин-2-карбоновой кислоты, полученного в справочном примере 1 (4,18 г), уксусной кислоты (45 мл) и концентрированной соляной кислоты (30 мл) перемешивали в течение 30 минут при нагревании с обратным холодильником. Реакционную смесь выливали в воду (800 мл). Полученное кристаллическое вещество собирали фильтрованием и растворяли в этилацетате (150 мл). Слой этилацетата промывали водой и сушили (MgS04), после чего растворитель выпаривали. Оставшиеся кристаллы собирали фильтрованием, промывали гексаном и перекристаллизовывали из смеси этилацетат-гексан с получением соединения заголовка (1,62 г, выход 59%) в форме бесцветных игольчатых кристаллов.
Точка плавления: 194-195oС.
Оптическое вращение [α]D (23oС): -210,8o (с=0,50, СН3ОН).
Справочный пример 3
Получение (2R, 4S)-(-)-N-[4-(диэтоксифосфорилметил)фенил] -1,2,4,5-тетрагидро-4-метил-7,8-метилендиокси-5-оксо-3-бензотиепин-2-карбоксамида (соединения А)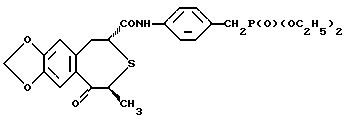
Раствор гидрохлорида 1-этил-3-(3-диметиламинопропил)карбодиимида (0,39 г) в дихлорметане (7 мл) добавляли по каплям к раствору (2R,4S)-(-)-1,2,4,5-тетрагидро-4-метил-7,8-метилендиокси-5-оксо-3-бензотиепин-2-карбоновой кислоты (0,47 г), полученной в справочном примере 2, и диэтил 4-аминобензилфосфоната (0,41 г) в N,N-диметилформамиде (ДМФ) (7 мл) при 0oС с последующим добавлением 1-гидроксибензотриазола (HOBt) (0,28 г). После перемешивания при 0oС в течение 1 часа и при комнатной температуре в течение 15 часов смесь выливали на воду и экстрагировали этилацетатом. Слой этилацетата промывали водой и сушили (MgSO4), после чего растворитель выпаривали. Оставшиеся кристаллы собирали фильтрованием и перекристаллизовывали из смеси этилацетат-гексан и смеси метанол-гексан с получением соединения А (0,37 г, выход 44%) в форме бесцветных призм.
Точка плавления: 181-182oС.
Оптическое вращение [α]D (23oС): -187,4o (с=0,50, СНСl).
Пример 1
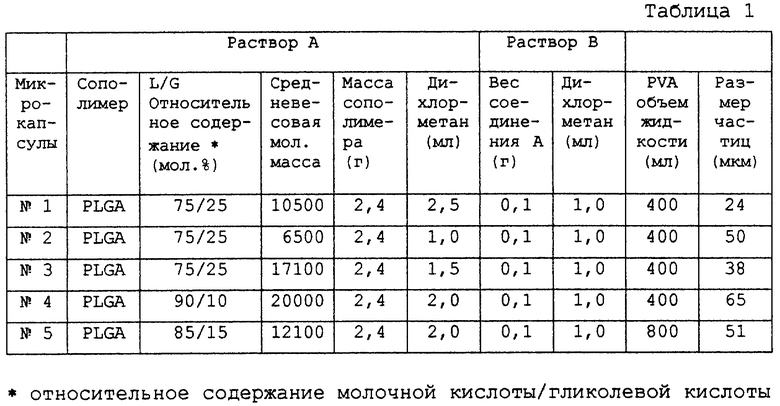
Готовили раствор в дихлорметане сополимера молочной кислоты-гликолевой кислоты, далее называемого ПМГК; относительное содержание молочной кислоты-гликолевой кислоты (мол. %) и средневесовая молекулярная масса согласно ГПХ показаны в таблице 1; производимого Wako Pure Chemical Industry. С применением формулы, показанной в таблице 1 (раствор далее называют раствором А). Подобным образом раствор в дихлорметане соединения А также готовили с применением формулы, показанной в таблице 1 (раствор далее называют раствором В). Растворы А и В однородно смешивали
вместе. Данную смесь вводили в 0,1% водный раствор поливинилового спирта (EG-40, производимого Nippon Synthetic Chemical Industry) (далее называемый раствором ПВС), охлажденный до 15oС заранее, в объеме, указанном в таблице 1, и эмульгировали с применением гомосмесителя турбинного типа при 7000 об/мин с получением эмульсии типа масло в воде, которую затем перемешивали при комнатной температуре в течение 3 часов для выпаривания дихлорметана. После отверждения масляную фазу центрифугировали при 2000 об/мин, используя центрифугу (05PR-22, Hitachi Ltd. ). Собранную фракцию микрокапсул опять диспергировали в дистиллированной воде, затем центрифугировали с последующим промыванием высвобожденного лекарственного средства и т.д. Собранные микрокапсулы опять диспергировали в небольшом количестве дистиллированной воды и затем лиофилизовали. Получали микрокапсулы с размерами частиц, показанными в таблице 1. Содержание микрокапсул, включавших в себя соединение А, было 100%.
Пример 2
500 мг микрокапсул 1, полученных в примере 1, однородно диспергировали в двух тест-пробирках раствора фибриногена для Tisseel (производимого Nippon Zoki Pharmaceutical). Постепенно добавляли в две тест-пробирки раствор тромбина для Tisseel. Затем каждую смесь сразу же набирали в пластиковый шприц. Шприцу давали стоять при 37oС в течение 30 минут для отверждения содержимого. После отверждения содержимое выдавливали из кончика шприца и разрезали бритвой на гранулы с объемом приблизительно 200 мкл.
Пример 3
К микрокапсулам 3, полученным в примере 1, которые содержат соединение А (относительное содержание 4%), добавляли 20% измельченного желатина (производимого Nitta Gelatin) с получением содержащего микрокапсулы препарата в виде таблетки диаметром 5,5 мм и весом 125 мг.
Пример 4
Микрокапсулы 7, содержащие соединение А (относительное содержание 10%), получали так же, как в примере 1, за исключением того, что PLGA (ПМГК) имела относительное содержание молочной кислоты-гликолевой кислоты 85/15 (мол.%) и средневесовую молекулярную массу 14900 (Wako Pure Chemical Industry). Средний размер частиц был 31 мкм.
Пример 5
Раствор в дихлорметане, содержащий 2,4 г PLGA (ПМГК) (Wako Pure Chemical Industry), относительное содержание молочной кислоты/гликолевой кислоты которого равно 85/15 и средневесовая молекулярная масса равна 14900, и 0,1 г соединения А, получали так же, как описано в примере 1. В раствор добавляли 0,2 г эстрадиола. Затем раствор PVA добавляли в данный раствор для получения эмульсии типа масло в воде. Получали микрокапсулы 8, содержащие соединение А и эстрадиол.
Средний размер частиц был 27 мкм.
Пример испытаний 1
500 мг микрокапсул 1, полученных в примере 1, точно взвешивали и переносили в стеклянную центрифужную пробирку и давали стоять при 37oС в виде порошка. После определенного времени микрокапсулы растворяли в небольшом количестве ацетонитрила и количественно анализировали при помощи ВЭЖХ. Испытания стабильности представлены в таблице 2.
Содержание лекарственного средства было более 95% даже после 4 недель.
Пример испытаний 2
Микрокапсулы 2 и 3, полученные в примере 1, по 5 мг каждая, точно взвешивали и переносили в стеклянный сосуд с последующим встряхиванием при 37oС на водяной бане (TAITEC, термостат М-100, 115 толчков/мин) в присутствии 10 мл тест-раствора высвобождения (фосфатный буфер с добавлением 10% бычьего сывороточного альбумина, pН 7,0). К каждой пробе 100 мкл, взятой после определенного времени, добавляли 100 мкл ацетонитрила. После встряхивания смесь центрифугировали. Полученный супернатант анализировали при помощи ВЭЖХ для определения количества высвободившегося соединения А. Испытания на высвобождение представлены в таблице 3.
Были получены три вида различных распределений высвобождения.
Пример испытаний 3
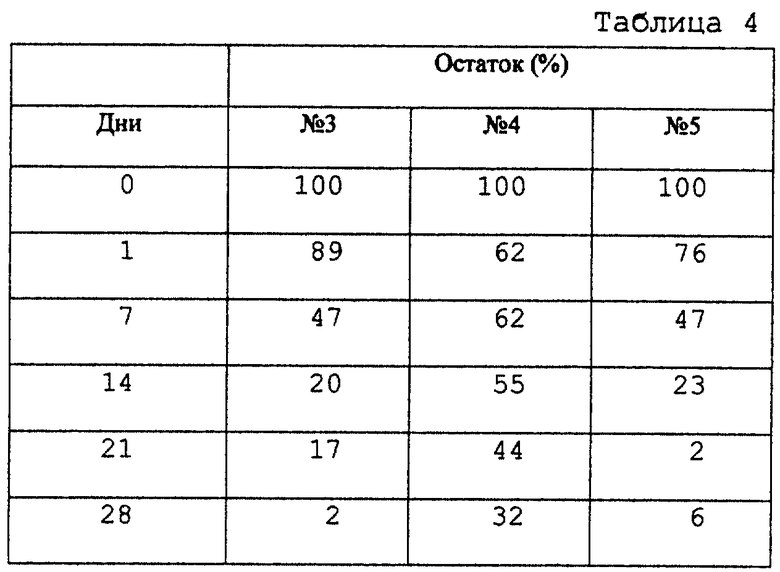
Микрокапсулы 3-5, полученные в примере 1, по 25 мг каждая, диспергировали в 0,3 мл диспергирующего раствора (раствора 1,5 мг карбоксиметилцеллюлозы, 0,3 мг полисорбата 20 и 15 мг маннита в дистиллированной воде) и инъецировали подкожно в головы самцов крыс SD (n=5) в возрасте 5 недель при анестезии эфиром при помощи иглы для инъекции 22G. Крыс умерщвляли при заданных интервалах времени после введения. Микрокапсулы, остающиеся в месте введения, вынимали и анализировали ВЭЖХ для определения количества соединения А в микрокапсулах. Результаты представлены в таблице 4.
Также и in vivo были получены различные типы распределения высвобождения микрокапсул. В частности, было обнаружено, что микрокапсулы 3 имеют пролонгированное высвобождение в течение 1 месяца, микрокапсулы 4 более 1 месяца и микрокапсулы 5 в течение 3 недель.
Пример испытаний 4
При анестезии пентобарбиталом головы самцов крыс SD в возрасте 6 недель надрезали и отделяли надкостницу (периост). Затем делали отверстие диаметром 4 мм в левом своде черепа при помощи стоматологической бормашины. Спустя 1 неделю после наложения швов 25 мг микрокапсул 3, полученных в примере 1, диспергировали в 0,3 мл диспергирующего агента (раствора 1,5 мг карбоксиметилцеллюлозы, 0,3 мг полисорбата 20 и 15 мг маннита в дистиллированной воде) и вводили подкожно в правый висок при помощи иглы для инъекции 22G. Для контроля использовали крыс, получавших 0,3 мл указанного выше раствора диспергирующего агента. Спустя три недели крыс умерщвляли. Свод черепа разрезали и подвергали мягкому рентгенографическому анализу. При помощи полученной фотографии зону новообразованной костной ткани в поврежденном участке кости определяли анализом изображений.
Значительное увеличение зоны вновь образованной костной ткани отмечали в группе с введением микрокапсул, что свидетельствует о превосходной остеогенез активирующей способности фармацевтической композиции согласно данному изобретению.
Пример испытаний 5
При анестезии пентобарбиталом левые конечности самцов крыс SD (n=8) в возрасте 6 недель надрезали и центральную часть левой малоберцовой кости обнажали и затем разрезали режущим инструментом. 25 мг микрокапсул 1, полученных в примере 1, погружали в участок разреза малоберцовой кости с последующим наложением швов. Спустя две недели крыс убивали. Малоберцовую кость вырезали и анализировали с применением анализатора минерального содержимого костей (DSC-600, Aloca Co., Ltd., Tokyo) для определения минерального содержимого кости. Также определяли минеральное содержимое кости в необработанной правой малоберцовой кости. Измерение для правой малоберцовой кости вычитали из измерения для левой малоберцовой кости для получения содержимого минеральных компонентов в каллусе. Для контроля микрокапсулы плацебо без соединения А готовили по способу, использованному для получения микрокапсул 1, и сравнивали с капсулами, содержащими А так же, как в примере испытаний 4. Эти результаты показаны на фиг.1.
Введение содержащих лекарственное средство микрокапсул приводило к значительно повышенному минеральному содержимому кости, демонстрируя превосходную остеогенную активирующую способность фармацевтической композиции согласно данному изобретению.
Пример испытаний 6
25 мг микрокапсул 1, содержащих соединение А (относительное содержание 4%), полученных в примере 1, вводили местно модели крыс с переломом малоберцовой кости согласно способу, описанному в примере испытаний 5. После 2 и 3 недель определяли минеральное содержимое кости. Для контроля минеральное содержимое кости определяли в группе, получившей микрокапсулы с плацебо (не содержащие лекарственного средства), и в самопроизвольно выздоравливающей группе. Эти результаты показаны в таблице 5.
Было обнаружено, что группа, получавшая микрокапсулы, содержащие лекарственное средство, имела значительно более высокое содержание минеральных веществ в сравнении с контрольными группами, что свидетельствовало о превосходной остеогенной активирующей способности фармацевтической композиции согласно данному изобретению.
Пример испытаний 7
Содержащий микрокапсулы препарат в виде таблетки, полученный в примере 3, имплантировали подкожно в спинки самцов крыс SD (n=8) в возрасте 5 недель под анестезией эфиром. Количество лекарственного средства, остающегося в месте введения, определяли при помощи ВЭЖХ во времени. Результаты представлены на фиг.2. Исследуемая таблетка обнаружила свойство пролонгированного действия в течение 6 недель.
Пример испытаний 8
В соответствии со способом Miyamoto et al. "Shinpei Miyamoto, Hideki Yoshikava and Kunio Takaoka: The Bone, 7, 85-96 (1983)" большеберцовые кости кроликов (весом 3-4 кг) разрезали с образованием костного повреждения длиной 5 мм, которое затем заполняли введением содержащего микрокапсулы препарата в виде таблетки, полученного в примере 3, и затем рану фиксировали снаружи. Для контроля использовали препарат в виде таблетки, содержащий микрокапсулы с плацебо, которые не содержали соединения А. Мягкий рентгеновский радиографический анализ выявил заживление кости у кроликов, которым вводили таблетку, содержащую соединение А, спустя 2 месяца после хирургии, тогда как у кроликов, получавших таблетку с плацебо, не было замечено заживление.
Пример испытаний 9
Микрокапсулы 7, содержащие соединение А (относительное содержание 10%), полученные в примере 4, вводили подкожно в спинки самцов крыс SD (n=5) в возрасте 5 недель под эфирной анестезией согласно способу, описанному в примере испытаний 2 (100 мг/кг веса тела крысы в расчете на соединение А). Концентрацию лекарственного средства в крови определяли при помощи ВЭЖХ во времени. Результаты показаны на фиг.3. Даже при 4 неделях после введения получали концентрацию лекарственного средства в крови 0,05-0,1 мкг/мл.
Пример испытаний 10
Крысиную модель перелома малоберцовой кости получали согласно способу, описанному в примере испытаний 5. Этой модели перелома вводили местно микрокапсулы 7, содержащие соединение А (относительное содержание 10%), полученные в примере 4, в форме лиофилизированного порошка (1 мг/крысу) или суспензии в диспергирующем агенте, описанном в примере испытаний 4 (5 мг/0,25 мл на крысу). Спустя две недели определяли минеральное содержимое кости согласно способу, описанному в примере испытаний 5, и сравнивали с контрольной группой, получавшей микрокапсулы плацебо. Результаты представлены в таблице 7.
Было обнаружено, что независимо от того, было ли лекарственное средство в форме лиофилизованного порошка или в форме суспензии, группа с микрокапсулами, содержащими лекарственное средство, имела значительно большее содержание минеральных веществ кости в сравнении с контрольными группами, что свидетельствовало о превосходной остеогенной активирующей способности фармацевтической композиции согласно данному изобретению.
Пример испытаний 11
Крысиную модель перелома малоберцовой кости получали согласно способу, описанному в примере испытаний 5. Микрокапсулы 7 суспендировали в диспергирующей среде, содержащей различные концентрации фосфата натрия, которую готовили с использованием раствора D-сорбита (2,5 г), хлорида натрия (0,9 г) и карбоксиметилцеллюлозы (0,5 г) в дистиллированной воде.
Суспензию вводили локально (5 мг/0,25 мл на крысу). Спустя две недели минеральное содержимое кости определяли согласно способу, описанному в примере испытаний 5. Результаты показаны в таблице 7.
Результаты изобретения
Обнаруживая повышенную остеогенез усиливающую активность при низкой токсичности на протяжении продолжительного периода времени, фармацевтическая композиция согласно данному изобретению может безопасно использоваться в качестве профилактического/терапевтического средства для различных заболеваний костей у млекопитающих, например, переломов костей, остеопороза, остеомаляции, синдрома Бехчета кости, анкилозного спондилоартрита, ревматоидного артрита, гонартрита (gonarthritis deformans) и деструкции тканей суставов, в качестве средства восстановления костной ткани после хирургии по поводу множественной миеломы, рака легких, рака молочной железы и т.д. и в качестве активатора регенерации периодонтальной ткани в случае периодонтопатии и т.д.
Промышленная применимость
Фармацевтическая композиция, содержащая непептидное остегенное оптически активное вещество и сополимер молочной кислоты/гликолевой кислоты представляет собой ценное средство для лечения и/или предотвращения заболеваний костей.





Изобретение относится к области фармакологии и обеспечивает фармацевтическую композицию, содержащую непептидное остеогенез активирующее оптически активное вещество и сополимер молочной кислоты и гликолевой кислоты, которая может быть безопасно использована в качестве более эффективного профилактического терапевтического средства для различных заболеваний костей (например, переломов костей). 10 з.п.ф-лы, 3 ил., 7 табл.
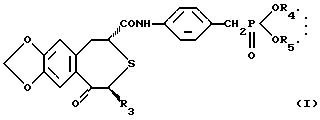
где R3 представляет низшую алкильную группу;
R4 и R5 независимо представляют низшую алкильную группу или связаны вместе с образованием низшей алкиленовой группы,
и сополимер молочной кислоты и гликолевой кислоты.
Приоритет по пунктам:
05.06.1995 по пп. 1-4, 8-11;
04.06.1996 по пп. 5-7.
| Способ испытания абразивного инструмента | 1973 |
|
SU460488A1 |
| УСТРОЙСТВО для ШОВНОЙ СВАРКИ ДАВЛЕНИЕМ | 0 |
|
SU376197A1 |
| Способ получения производных дифосфоновой кислоты или их натриевых солей | 1985 |
|
SU1475487A3 |
| NELSAI at ab "Evaluation and Comparisone of biodegrecdable substance as osteogenic Agents" | |||
| B: "Oral surgery, oral medicin, oral pathology", 1977, v | |||
| Зубчатое колесо со сменным зубчатым ободом | 1922 |
|
SU43A1 |
| Polylactic acid-polyethylene glycol block polymer B: "Clin orth | |||
| and Rel | |||
| Research", 1993, v | |||
| ДИФФЕРЕНЦИАЛЬНАЯ ТЕРМИОННАЯ ЛАМПА | 1920 |
|
SU294A1 |

