Предметом данного изобретения являются фармацевтические составы, содержащие полимерные матрицы, в частности составы, содержащие ИЛ-6, предназначенные для применения при лечении заболеваний, опосредованных ИЛ-1 и/или TNFα, например хронических воспалительных состояний. Показано, однако, что специфические полимеры по изобретению, особенно поли(этиленкарбонат)ные полимеры, описанные ниже, более широко применяются в качестве матричных материалов в составах пролонгированного выделения, содержащих фармакологически активные соединения, и показано, в частности, что они обладают новым, неожиданным и очень желательным свойством подвергаться негидролитической поверхностной эрозии in vivo. Поэтому также предлагают и приводят в качестве примера матрицы, содержащие другие лекарственные средства, совместно со способом получения полимеров и фармацевтическими составами, содержащими их. Кроме того, новым и неожиданным является применение ИЛ-6 для лечения состояний, опосредованных ИЛ-1 и/или TNFα, (ранее считали, что многие подобные состояния обостряются под воздействием ИЛ-6), поэтому в изобретении предлагают также новое применение ИЛ-6 для лечения, например, хронических воспалительных состояний, вызываемых патогенами, демиелинизирующих заболеваний и острых и сверхострых воспалительных состояний, таких как септический шок.
Лечение заболеваний, опосредованных ИЛ-1 и/или TNFα
Многочисленные спонтанно проявляющиеся хронические воспалительные состояния имеют неизвестную (возможно автоиммунную) этиологию, при этом полагают, что они опосредованы ИЛ-1 и/или TNFα. Например, рассеянный склероз (PC), калечащее нервное расстройство, отличительным признаком которого являются рассеянные бляшки демиелинизации в спинном и головном мозге, привлекает внимание исследовательских организаций в течение многих лет. Хотя точная этиология рассеянного склероза еще не установлена окончательно, считают, что она имеет сильную автоиммунную компоненту, что подтверждается, например, расширенной сферой действия определенных антигенов главного комплекса гистосовместимости человека у пациентов, страдающих этим заболеванием. Существующие в настоящее время противовоспалительные лекарственные препараты, такие как АКТГ (адренокортикотропический гормон) или кортикостероиды, например преднизон, по-видимому, ускоряют восстановление после острых приступов, особенно при введении на ранней стадии, но не влияют на основную этиологию заболевания. Длительное введение кортикостероидов или иммунодепрессантов приводит к возникновению опасности серьезных побочных эффектов. Недавно было показано, что рекомбинантная форма ИФН-β1 уменьшает краткосрочное образование бляшек, но не удалось показать, что он влияет на процесс длительного развития заболевания. Изучение эффективности лечения осложняется тем фактом, что естественное развитие заболевания проявляется в виде чередования спонтанной ремиссии и хронического рецидива. Коротко говоря, несмотря на многолетние интенсивные исследования, до сих пор не существует общепринятой конкретной терапии для этого очень серьезного заболевания.
Считают, что другие хронические воспалительные состояния индуцируются внешними агентами, например патогенами. Например, болезнь Лайма является серьезным хроническим состоянием, причиной которого является инфицирование переносимой клещами спирохетой Borrelia burgdorferi. После начальной острой стадии, характеризующейся поражениями кожи и гриппоподобными симптомами, заболевание переходит в хроническую стадию, которая может быть охарактеризована артритом и хроническими невралгическими отклонениями. Обычно заболевание лечат антибиотиками и нестероидными противовоспалительными средствами, но оптимальная терапия, в частности для установленного заболевания, еще не разработана.
Острые или сверхострые неконтролируемые воспалительные состояния могут также вызываться внешними причинами, например тяжелыми ожогами или тяжелыми инфекциями. Например, септический шок, и в частности респираторный дистресс-синдром у взрослых (РДСВ), является опасным для жизни состоянием, для которого в настоящее время не существует эффективного лечения. Начало заболевания быстрое, а смертность, как правило, превышает 50%. Септический шок обычно является результатом тяжелой бактериальной инфекции и, как правило, характеризуется лихорадкой, за которой на поздних стадиях часто следует гипотермия, колеблющееся кровяное давление (гипердинамический синдром) с последующей гипотензией на поздних стадиях, метаболическим ацидозом, нарушением умственных способностей и обширной дисфункцией органов и, наконец, во многих случаях заканчивается смертью. Наиболее часто септический шок является результатом грамм-отрицательной бактериальной инфекции (эндотоксический шок), но также он может являться и результатом грамм-положительных бактериальных инфекций или других инфекций. Термин "септический шок", используемый в тексте, должен поэтому интерпретироваться широко как термин для обозначения шокового состояния, включая РДСВ, являющегося результатом микробной инфекции, особенно бактериальной инфекции, главным образом грамм-отрицательной бактериальной инфекции.
ИЛ-6 является известным цитокином. Известно, что его применяют для лечения различных состояний, например тромбоцитопении и некоторых раков. Обычно он продуцируется организмом в ответ на бактериальные инфекции и участвует в медиации воспаления, лихорадки и септического шока. Он является сильным иммуностимулятором, а ряд литературных данных подтверждает, что управляемые ИЛ-6 механизмы вызывают некоторые автоиммунные и воспалительные заболевания, включая системную красную волчанку, рассеянный склероз и ревматоидный артрит, а также септический шок.
Было большой неожиданностью обнаружение того факта, что ИЛ-6 пригоден для лечения хронических воспалительных заболеваний (не являющихся гломерулонефритом), например рассеянного склероза, и для лечения острых и сверхострых воспалительных состояний, например септического шока. Механизм его действия неизвестен, но, не подразумевая никакую частную теорию, мы считаем, что посредством механизма обратной связи ИЛ-6 может подавлять или ингибировать экспрессию, выделение или функционирование других цитокинов, в частности TNFα и/или ИЛ-1, возможно путем положительной регуляции выделения антагониста растворимого TNFα рецептора и/или ИЛ-1 рецептора, подавляя тем самым активность результирующего автоиммунного, воспалительного или шокового состояний, которые опосредованы главным образом этими цитокинами. Однако показано, что в случае состояний, характеризующихся ИЛ-6 опосредованными комплемент-активирующими антиген-антитело (IgG) комплексами, в частности гломерулонефрита, (который обычно вызывается накоплением таких комплексов в почке), ИЛ-6 обостряет это состояние. Таким образом, мы показали, что ИЛ-6 оказывает лечащее действие в животных моделях, например, для PC и артрита Лайма, о которых полагают, что они первично вызываются ИЛ-1 и/или TNFα, но обостряет гломерулонефрит у мышей с волчанкой, которая, как полагают, непосредственно вызывается ИЛ-6. Мы показали также, что ИЛ-6 обладает лечебным действием в мышиных моделях эндотоксического шока, который, как предполагают аналогичным образом, вызывается главным образом ИЛ-1 и/или TNFα.
Поэтому мы считаем, что ИЛ-6 пригоден в качестве агента для подавления или ингибирования экспрессии, выделения или функционирования TNFα и/или ИЛ-1 и особенно для лечения воспалительных состояний, не являющихся гломерулонефритом, и для лечения септического шока. Воспалительные состояния, которые поддаются излечению с помощью ИЛ-6, включают, например, артритные состояния, в частности вызываемые патогенами артритные состояния, например артрит болезни Лайма, вызываемый бактериями артрит и полиартрит; рассеянный склероз и другие демиелинизирующие состояния (в частности, заболевания, характеризующиеся демиелинизацией нервов, мозга и/или спинного мозга, включая, в частности, рассеянный склероз, острый рассеянный энцефаломиелит или послеинфекционный энцефалит, оптический нейромиелит, шум в ушах, диффузный церебральный склероз, болезнь Шилдера, андренолейкодистрофию, третичную болезнь Лайма, тропический спастический парапоэз и другие заболевания, при которых демиелинизация, особенно автоиммунная демиелинизация, является основным симптомом); острые тяжелые воспалительные состояния, такие как ожоги, септический шок, менингит и пневмония, и автоиммунные заболевания, включая полихондриты, склеродому, грануломатоз Вегенера, дерматомиозит, хронический активный гепатит, миастению беременных, псориаз, псориазный артрит, синдром Стевена-Джонсона, миастенический синдром мальабсорбции, автоиммунное воспалительное заболевание кишечника (включая, например, язвенный колит и болезнь Крохна), эндокринную офтальмопатию, болезнь Гравеса, саркоидоз, первичный билиарный цирроз печени, юношеский диабет (диабет mellitus тип I), увеит (передний или задний), кератоконъюнктивит сухой и весенний кератоконъюнктивит и интерстициальный фиброз легких.
Таким образом, согласно изобретению предложены:
I) Способ
ингибирования экспрессии, выделения или функционирования TNFα и/или ИЛ-1;
лечения или профилактики воспалительного состояния, не являющегося гломерулонефритом;
лечения или профилактики состояния, опосредованного ИЛ-1 и/или TNFα;
лечения или профилактики любых описанных выше состояний;
лечения или профилактики демиелинизирующего заболевания, в частности рассеянного склероза;
лечения или профилактики вызываемого внешними причинами воспалительного состояния;
лечения или профилактики воспалительной реакции на тяжелую острую инфекцию, например септический шок, менингит или пневмонию;
лечения ожогов;
лечения или профилактики хронического вызываемого патогеном воспалительного состояния, например болезни Лайма;
указанный способ включает в себя введение терапевтически или профилактически эффективного количества ИЛ-6, например ингибирующего TNFα и/или ИЛ-1 количества ИЛ-6, в частности чрИЛ-6 (в частности, особенно в тех случаях, когда ИЛ-6 вводят как единственный терапевтический или профилактический агент или возможно вводят в сочетании с антимикробными или вазоактивными агентами, в частности, не в сочетании с TNFα агонистами или антагонистами или с анти-TNFα антителом), возможно в медленно выделяемой или депонированной форме, в частности в сочетании с полимерной матрицей, например поли(этиленкарбонат)ной матрицей, описываемой ниже, субъекту, например млекопитающему, в частности человеку, в случае необходимости в таком лечении или профилактике;
II) Применение ИЛ-6, в частности чрИЛ-6, при изготовлении лекарства для использования в способе (I), например, для лечения или профилактики любого из вышеперечисленных под (I) состояний, причем лекарство может находиться в лекарственной форме с пролонгированным выделением, может содержать полимерную матрицу, в частности поли(этиленкарбонат)ную матрицу, описываемую далее;
III) Применение ИЛ-6, в частности чрИЛ-6, для лечения или профилактики любого из перечисленных в п.(I) состояний и
IV) Фармацевтический состав, содержащий ИЛ-6, в частности чрИЛ-6, для применения по способу (I), например для лечения или профилактики любого из состояний, описанных выше в п.(I), возможно в форме с пролонгированным выделением, возможно содержащей, кроме того, полимерную матрицу, в частности поли(этиленкарбонат)ную матрицу, как описано далее; например состав пролонгированного выделения (в частности, состав, который претерпевает биологический распад in vivo в течение нескольких дней, недель или месяцев), содержащий ИЛ-6 в полимерной матрице, например, в форме микрочастицы или запаса, в частности, если полимер обнаруживает негидролитическую поверхностную эрозию in vivo, особенно любая из систем для введения лекарственных средств, описанных здесь, для применения при лечении любого из описанных выше состояний, например для лечения хронического воспалительного состояния.
Под ИЛ-6 понимают любое соединение, соответствующее известным разновидностям интерлейкина-6 (известного также как интерферон бета-2 (ИФН-βII), В-клеточный стимуляторный фактор 2 (ВСФ-2), интерлейкин HP-1 (HR l), гепатоцитный стимулирующий фактор (ГСФ), фактор роста гибридомной плазмацитомы (ГПРФ) и 26кД фактор). Предпочтителен рекомбинантный ИЛ-6, хотя может быть использован также и нерекомбинантный ИЛ-6, например продуцируемый ИЛ-6-секретирующими раковыми клеточными линиями. ИЛ-6 производится в коммерческих масштабах или может быть получен известными способами: ЕР 220574, 257406, 326120, WO 88/00206, GB 2063882, 2217327, содержания этих источников включены здесь по сноске. ИЛ-6 может быть гликозилирован, как, например, продуцируемый эукариотическими клетками, в частности СНО клетками, либо негликозилированным, как, например, продуцируемый прокариотическими клетками, в частности Е.coli. Предпочтителен рекомбинантный человеческий ИЛ-6 (рчИЛ-6), хотя известно, что ИЛ-6 остается активным при межвидовом переносе, поэтому ИЛ-6, полученный от продуцентов, не являющихся человеком, также может быть использован и подпадает под приводимое здесь определение ИЛ-6. Считают, что белки, имеющие минорные вариации в последовательности ИЛ-6, например вставки, делеции или мутации 1, 2, 3 или более аминокислот; белки слияния, содержащие ИЛ-6 и другой белок; активные фрагменты ИЛ-6 и/или другие подобные варианты, процессированные или мутантные формы ИЛ-6, которые обладают ИЛ-6 активностью, подпадают под определение ИЛ-6.
Известны приемлемые фармацевтические составы, содержащие ИЛ-6 совместно с фармацевтически приемлемым разбавителем или носителем. ИЛ-6 может быть введен парентерально, например в форме раствора для инъекций или суспензии, в частности, согласно или аналогично описанию Remington's Rharmac. Science, 1980. К числу пригодных носителей относятся такие водные носители, как солевой раствор, раствор Рингера, декстрозный раствор, раствор Ханка, а также неводные носители, такие как фиксированные масла или этилолеат. Для обычного парентерального введения пригоден ИЛ-6 в лиофилизированной форме в количествах, соответствующих унифицированной дозе, которые могут быть смешаны с носителем для получения подходящего раствора или суспензии для инъекции.
С другой стороны, ИЛ-6 может быть введен с использованием имплантируемой системы введения лекарственных средств или системы введения лекарственных средств с пролонгированным выделением лекарства, например, в форме микрочастиц или депонированной форме в сочетании с полимером для образования полимерной матрицы, посредством чего лекарство медленно выделяется из матрицы. Это предпочтительно в тех случаях, когда, например, состояние, которое должно быть излечено, является хроническим, в частности хроническим воспалительным состоянием, и требуемое лечение продолжается недели или месяцы. Под полимером понимают любую пригодную (например, фармакологически приемлемую) линейную молекулу с высоким молекулярным весом, образованную из повторяющихся звеньев (включая гомополимеры, сополимеры и гетерополимеры), возможно разветвленную или перекрестно сшитую, которая может быть получена, в частности, путем полимеризации простой молекулы или путем сополимеризации более чем одной молекулы (например, получение поли(этиленкарбонат)а из этиленоксида и диоксида углерода, как описано ниже), и возможно содержащую включения других звеньев в полимерной цепи. Предпочтительно, чтобы полимер был линейным и состоял из углерода, кислорода и водорода, например поли-DL-лактид-со-гликолид, полиэтиленгликоль или поли(этиленкарбонат). Предпочтительно, чтобы полимер обнаруживал негидролитическую поверхностную эрозию подобно описанному далее поли(этиленкарбонат)у.
Дозировка будет изменяться в зависимости от точного типа применяемого ИЛ-6, реципиента, способа введения и природы и степени тяжести состояния, подлежащего лечению. ИЛ-6 вводят крупным млекопитающим, например человеку, путем подкожной инъекции или в виде формы с пролонгированным выделением лекарства таким образом, чтобы обеспечить дозу от 0,5 мкг/кг/день до 30 мкг/кг/день, предпочтительно от 2,5 мкг/кг/день до 10 мкг/кг/день или в любой другой дозировке, которая является безопасной и эффективной для in vivo активности в известных терапевтических способах применения ИЛ-6, в частности в дозировке, повышающей содержание тромбоцитов. В случае тяжелых острых воспалительных состояний, например септического шока, допустимо введение более высоких доз внутривенно с тем, чтобы добиться быстрой и сильной реакции. Частота введения ИЛ-6 может быть уменьшена от ежедневной до введения через день или каждую неделю, либо еще реже в случае форм пролонгированного выделения, которые предпочтительны в тех случаях, когда лечение проводят в течение длительных периодов времени. Лечение ИЛ-6 может приводить к появлению лихорадок, жара и гриппоподобных симптомов, которые в норме могут быть излечены или предотвращены путем совместного введения ненаркотических анальгетиков, таких как аспирин, ацетаминофен или индометацин. Другие серьезные побочные эффекты обычно появляются только при более высоких дозах, например свыше 10 мкг/кг/день, и могут, как правило, быть устранены понижением дозы.
Полимерные матрицы для пролонгированного выделения
Далее в изобретении предлагают фармацевтические составы, пригодные для пролонгированного выделения лекарственных средств, которые пригодны, например, для введения ИЛ-6, в частности, при вышеописанных показаниях, а также для введения других лекарственных средств. Фармацевтическими составами являются главным образом те составы, которые содержат полимеры поли(этиленкарбонат)а, иногда именуемые как поли(этиленкарбонат)ы и ПЭКы.
Несмотря на то, что из уровня техники известен ряд примеров применения поли(этиленкарбонат)ов в системах для введения лекарств, в них не описаны специфические полимеры по изобретению и не описаны полимеры, способные испытывать негидролитическую поверхностную эрозию in vivo. Из уровня техники неизвестно также о таких системах для ввода ряда специфических лекарственных средств, описанных здесь, в частности ИЛ-6, и нет подтверждений тому, что для введения таких лекарств необходима система пролонгированного выделения.
Наиболее неожиданными являются показатели распада полимеров по изобретению. На основании общих химических представлений можно ожидать, что карбонатные эфирные связи являются в принципе расщепляемыми. Однако было высказано предположение, что поликарбонаты должны быть стабильными в умеренных условиях in vitro.
Согласно Chem. Pharm. Bull. 31(4), 400-1403 (1983), поли(этиленкарбонат)ы распадаются in vivo, однако исследованный полимер не был полностью идентифицирован, в частности, с помощью современных спектроскопических методов. Согласно стр. 1402, распад in vivo может быть объяснен только действием гидролитических ферментов.
Согласно Chem. Pharm. Bull. 32(7), 2795-2802 (1984), микрочастицы делали из поли(этиленкарбонат)а, содержащего дибукаин. Хотя описание относится к первоначально процитированному достижению, оказалось, что выделение дибукаина было связано не с in vitro или in vivo способами распада полимера, а с диффузией через полимер. Физические и химические свойства исследованного поли(этиленкарбонат)а также не были установлены в достаточной степени.
Согласно Makromol, Chem. 183, 2085-2092 (1982), главным образом стр. 2086, полагают, что эпоксидные полимеры диоксида углерода способны к биологическому распаду, и отмечено, что предварительные результаты подтвердили возможность биологического распада полимеров диоксид углерода-этиленоксид, а отсюда и возможность их применения в контролируемом выделении лекарств. Для подтверждения утверждения относительно возможности биологического распада ссылались на Jinko Zoki 3 (Suppl)- -(1974). В этой публикации было указано, что поли(этиленкарбонат) принадлежит к группе соединений, которые наиболее легко гидролизуются, и даже фермент проназа без труда их разрушает. Это означает, что может быть возможен ферментативный гидролиз in vitro и in vivo, поскольку проназа представляет собой смесь гидролитических ферментов. Однако это утверждение кажется очень сомнительным. Мы подвергали поли(этиленкарбонат)ы по нашему изобретению в форме спрессованных дисков диаметром 5 мм с весом 25 мг действию 10 мг/мл проназы и 5 мМ CaCl2•2H2O в забуференном фосфатом физиологическом растворе (ЗФР) с рН 7,4 и 10 мг/мл проназы Е и 5 мМ СаСl2•2Н2O в забуференном фосфатом физиологическом растворе c рН 7,4 (при 37oС) и при этом не наблюдали никакого распада (см. Фиг.1). Раствор проназы обновляли каждый день.
Было неожиданностью установление того факта, что ряд поли(этиленкарбонат)ов с определенным содержанием этиленкарбоната, вязкостью и температурным интервалом стеклования, которые не распадаются посредством гидролиза (например, в присутствии гидролитических ферментов, в частности проназы, или в основных условиях), тем не менее распадаются in vitro и in vivo, а именно (и исключительно) из-за поверхностной эрозии. Выражение "поверхностная эрозия" применяют в литературе, особенно в отношении гидролитического разложения полиангидридов и полиортоэфиров, но его никогда точно не определяли.
Поверхностная эрозия имеет место, если есть распад массы только на поверхности полимерных частиц, без снижения молекулярного веса сохраняющегося полимерного остатка. В тех случаях, когда в литературе голословно утверждали, что наблюдали поверхностную эрозию, определений молекулярного веса остаточной массы параллельно с определениями массовых потерь никогда не проводили и поэтому в действительности поверхностная эрозия никогда не имела места.
В действительности же почти у всех исследовавшихся когда-либо полимеров наблюдали полимерную объемную эрозию. Системы, подверженные полимерной объемной эрозии, имеют существенный недостаток, заключающийся в том, что если полимер насыщают лекарственным соединением, например пептидом, который относительно нестабилен при воздействии биологической среды, в которую он будет выделяться, то лекарственное соединение уже контактирует со средой в объемной части и может терять свою активность задолго до того, как оно выделится из полимера. Если же полимер будет испытывать поверхностную эрозию, в частности, при отсутствии объемной эрозии, то введенное лекарственное соединение, например пептид, будет оставаться защищенным от пагубного воздействия биологической среды вплоть до того момента, когда развивающаяся поверхностная эрозия не достигнет частиц лекарства и частица лекарства не выделится из поверхности остаточной полимерной массы. В случае систем введения лекарств на основе полимерных матриц, подверженных поверхностной эрозии в противоположность объемной эрозии, частица лекарства подвергается пагубному воздействию биологической среды в течение короткого периода времени, что обеспечивает более длительное, сильное и стабильное выделение фармакологически активного лекарства из полимерной матрицы.
Для полиангидридов в последних публикациях Proc. Nat. Acad. Sci. USA 90, 552-556 и 4176-4180 (1993) описаны некоторые характеристики процесса, подобного поверхностной эрозии. Однако в этих случаях задействован, по-видимому, весь объем, а определений молекулярного веса не проводили. Кроме того, такая эрозия является эрозией гидролитического типа. А сейчас было установлено, что выбранная группа поли(этиленкарбонат)ов, описанная ниже, показывает, как in vitro, так и in vivo, исключительно негидролитическую поверхностную эрозию.
В изобретении предлагают полимер, распадающийся in vivo и in vitro посредством поверхностной эрозии, которая происходит по негидролитическому механизму, и имеющий этиленкарбонатные звенья формулы А
-(-C(O)-O-CH2-CH2-O-)-
имеющий содержание этиленкарбоната от 70 до 100 мол.%, внутреннюю вязкость от 0,4 до 4,0 дл/г при измерении в хлороформе при 20oС и температуру стеклования от 15 до 50oС.
Содержание этиленкарбоната в полимере согласно изобретению составляет от 70 до 100 мол.%, в частности 80-100%, предпочтительно от 90 до 99,9%, например от 94 до 99,9%. Внутренняя вязкость полимера составляет от 0,4 до 4,0 дл/г при измерении в хлороформе при 20oС. Предпочтительно, чтобы полимер имел внутреннюю вязкость, измеренную при 20oС и концентрации 1 г/дл в хлороформе, равную от 0,4 до 3,0 дл/г.
Его температура стеклования составляет от 15 до 50oС, предпочтительно от 18 до 50oС.
В литературе описаны поли(этиленкарбонат)ы, имеющие температуру стеклования от 5 до 17oС.
Полимеры по изобретению получают предпочтительно посредством сополимеризации этиленоксида с диоксидом углерода, способ получения также является частью изобретения. Как результат реализации этого способа получения, полимер содержит в большинстве случаев в качестве со-единицы звено этиленоксида формулы Б
-(-CH2-CH2-O-)-
Если полимеры по изобретению выдерживают в водной среде, например в забуференном фосфатом физиологическом растворе с рН 7,4, то среда практически не попадает в их объемную часть, как это видно, например, из Фиг.2. Поэтому никакой объемной эрозии не происходит и оставшаяся масса будет сохраняться постоянной (100%) в течение периода времени свыше 28 дней, как это показано на правом графике Фиг.3.
В настоящее время поли-DL-лактид-со-гликолиды являются наиболее широко применяемыми матричными материалами для систем пролонгированного выделения лекарственных средств. Такие полимеры, однако, в отличие от полимеров по изобретению, распадаются посредством гидролиза. Например, распад массы в ЗФР, как показано в левой части Фиг.3 для одного из наиболее сложных типов поли-DL-лактид-со-гликолидов, а именно инициированного глюкозой поли-DL-лактид-со-гликолида (DL-ПЛГГЛЮ), описан в GB 2145422.
Различие в течение распада между поли(этиленкарбонат)ами по изобретению и поли-DL-лактид-со-гликолидами (DL-ПГЛ) из уровня техники in vivo представлены на Фиг.3. В то время, как полилактид-со-гликолиды испытывают объемную эрозию, о чем свидетельствует понижение молекулярного веса остаточной массы DL-ПЛГГЛЮ, молекулярный вес остаточной массы поли(этиленкарбонат)ов остается постоянным (100%).
Остаточная масса всего имплантанта уменьшается in vivo в обоих случаях до нуля в течение 1 месяца, что означает, что поли(этиленкарбонат) претерпевает поверхностную эрозию, а не объемную эрозию. Вследствие отсутствия объемной эрозии заполненный полимер хранится длительное время, например до его введения, он непроницаем для влаги и сохраняется в том же сухом состоянии, в котором он был получен. Имплантированное в него лекарство, если оно чувствительно к влажности, остается стабильным.
В изобретении также предлагают способ получения полимера, в котором этиленоксид и СО2 полимеризуют в молярном соотношении от 1:4 до 1:5 в присутствии катализатора. Очевидно, что в рамках этой реакции возможно введение звеньев этиленоксида в полимерную цепь, если две эпоксидные молекулы реагируют друг с другом без участия СО2 молекулы, например если окси-анионный промежуточный продукт атакует другую молекулу этиленоксида перед его карбоксилированием посредством СО2. Поэтому вероятно, что полимер содержит несколько звеньев этиленоксида. Полимер по изобретению, если он содержит звенья этиленоксида, имеет случайное распределение звеньев этиленкарбоната и этиленоксида согласно суммарной формуле Аm-Вn=
-(С(O)-O-СН2-СН2-O-)-m-(-СН2-СН2-O-)-n,
в которой 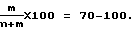
Однако большинство звеньев этиленоксида в полимерах по изобретению имеют статистически смежные звенья этиленкарбоната, особенно в тех случаях, когда молекулярное соотношение звеньев этиленоксида невелико. Это означает, что в таких случаях большинство результирующих эфирных групп распределено случайным образом между карбонатными группами вдоль полимерной цепи. 1Н-ЯМР спектры продуктов по изобретению в СDСl3 подтверждают это предположение. Они дают сигналы при δ= прибл. 4,37 ррm (Пик Iа) звеньев этиленкарбоната (этиленовые звенья между двумя карбонатными группами), при прибл. 4,29 и 3,73 ppm (Пики Iб и Iв) этиленовых звеньев между одной карбонатной и одной эфирной группой и прибл. 3,65 ppm (Пик Iг) этиленовых звеньев между двумя эфирными группами. Затем в пределах точности ЯМР вычисляют долю звеньев этиленкарбоната (А) согласно формуле
В качестве структурной особенности поли(этиленкарбонат)ов в литературе часто вместо содержания в них этиленкарбоната приводят содержание в них эфирных групп. Соотношение эфирных групп (Э) в полимерах по изобретению может быть вычислено по формуле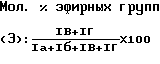
Согласно WO 92/2260 получают поли(этиленкарбонат)ы, которые содержат звенья этиленоксида и звенья этиленкарбоната в молярном соотношении от 2 до 400: 2, это означает, что полимер содержит по меньшей мере 50 мол.% этиленоксида и менее 50 мол.% звеньев этиленкарбоната. В заявке WO 92/2260 упомянуты биологический распад полимеров и их применение в качестве способных к биологической эрозии матриц для пролонгированного выделения фармакологически активных соединений. Однако не представлено никаких данных о том, что полимеры действительно способны к биологическому распаду. В целом же поли(этиленкарбонат)ы, имеющие такое большое количество эфирных групп, слабо поддаются биологическому распаду. В WO 92/2260 не содержится никаких указаний на возможность поверхностной эрозии полимеров.
В примерах US 3248415 описывают поли(этиленкарбонат)ы с низким молекулярным весом, составляющим Мв=700-5000, имеющие менее чем 70 мол.% звеньев этиленкарбоната, отличающиеся от полимеров по изобретению, однако ничего не сообщается об их биологическом распаде.
Согласно WO 89/05664 описаны поли(этиленкарбонат)ы, которые содержат в описанной структуре II звенья этиленоксида и этиленкарбоната в молярном соотношении от 1 до 8:1, это означает, что полимер содержит по меньшей мере 50 мол. % звеньев этиленоксида и отсюда самое большее 50 мол.% звеньев этиленкарбоната, отличаясь от полимеров по изобретению. Несмотря на то, что полимеры описывают как применимые для медикаментозных средств, способных к биологическому распаду, например имплантантов, которые могут содержать лекарственное соединение, не приведено никакой информации о поверхностной эрозии.
В способе по изобретению содержание звеньев этиленоксида, а отсюда и содержание эфирных групп, которое снижает или ингибирует скорость биологического распада полимера, значительно понижено путем создания таких реакционных условий, как описанное молярное соотношение реакционных компонентов, температура реакции, а также путем выбора соответствующего катализатора, полученного, например, из Zn(C2H5)2 и воды или ацетона, либо ди- или трифенола, в частности флороглюцина, в молярном соотношении от 0,9:1 до 1:0,9 или 2: 1 до 1:2 соответственно либо предпочтительно полученного из Zn(C2H5)2 и диола, особенно этиленгликоля, в молярном соотношении от 0,9:1 до 1:0,9.
Способ предпочтительно реализуют в растворяющей или диспергирующей системе органического растворителя, например диоксана и СO2. СО2 предпочтительно вводят в жидкой форме, и он присутствует в избытке. Давление предпочтительно от 20 до 70 бар, а температура предпочтительно от 10 до 80oС, особенно от 20 до 70oС.
Полученные таким образом полимеры по изобретению содержат обычно менее 15% эфирных групп, предпочтительно менее 10%, в частности менее 5%, например менее 3%. Поли(этиленкарбонат)ы по изобретению, если они получены с использованием катализатора из этиленгликоля или ацетона и диэтилцинка, обладают низкими полидисперсностями (Мв/Мн), обычно менее чем 5, в частности менее 2,5.
В способе согласно изобретению считают, что катализатор или его часть является цепным инициатором (со)-полимера. Когда реакция заканчивается и цепь завершена, заключительной концевой группой является гидроксильная группа. Противоположная сторона цепи, где цепь начиналась, может быть занята катализаторной группой или ее фрагментом. Если катализатор получают из этиленгликоля и диэтилцинка или воды и диэтилцинка, то предполагают, что оба конца полимерной цепи идентичны. Однако если катализатор получают из ди- или трифенола и диэтилцинка, то в тот конец цепи, где она начинается, будет включена ароматическая группа, в то время как другой конец цепи будет гидроксильной группой. Из Фиг.4 видно, что биологический распад поли(этиленкарбонат)а, в случае если одна из его концевых групп блокирована, например, ароматическим инициатором, в частности флороглюцином, протекает медленнее.
По этой причине предполагают, что распад полимерной цепи начинается с концевой гидроксильной группы. С другой стороны, может быть принята во внимание более поздняя модификация концевой гидроксильной группы, например, путем этерификации для блокировки концевых гидроксильных групп и для регуляции биологического распада поли(этиленкарбонат)ов по изобретению. Подходящими концевыми эфирными группами являются биологически совместимые эфирные группы, подобные эфирным группам жирных кислот (C1-48), предпочтительно (С1-30), особенно эфирные группы жирных кислот (C1-18), например эфирные группы уксусной кислоты и стеариновой кислоты либо эфирная группа угольной кислоты, в частности группа этиленкарбоната, либо эфирная группа памоевой кислоты либо эфирная группа молочной или гликолевой, или полимолочной, или полигликолевой, либо полимолочной-со-гликолевой кислоты.
Поли(этиленкарбонат)ы по изобретению стабильны в течение нескольких часов в горячей воде (90-100oС) без существенного снижения молекулярного веса. Значительное увеличение температуры стеклования, например до примерно 18oС или 20oС, наблюдают после выдерживания в кипящей бидистиллированной воде в течение 5 часов. Путем проведения этой реакционной стадии получают полимер большей чистоты. Мы обнаружили, что обработанные таким образом полимеры лучше обрабатываются.
Поли(этиленкарбонат)ная часть полимеров по изобретению является, как уже говорилось ранее, негидролизуемой, поэтому они стабильны по меньшей мере в течение 1 месяца при воздействии гидролитических ферментов в физиологических условиях или воды при рН 12 и 37oС (см. Фиг.1 и 8). Однако было установлено, что полимеры по изобретению распадаются in vivo и in vitro посредством поверхностной эрозии под воздействием аниона супероксидного радикала O2 -. Анионы супероксидного радикала O2 - вырабатываются в воспалительных клетках in vivo и ех vivo в присутствии поли(этиленкарбонат)ов по изобретению, как это видно из Фиг. 5. Полилактид-со-гликолиды, наиболее широко применяемые матричные материалы для систем пролонгированного выделения лекарств в наше время, распадающиеся посредством объемного гидролиза, не индуцируют продуцирование анионов супероксидного радикала O2 -, что показано на том же рисунке для инициированного глюкозой поли-DL-лактид-со-гликолида, который был использован и для Фиг.3.
In vitro была создана водная система, содержащая супероксид калия в качестве источника O2 - и демонстрирующая поверхностную эрозию поли(этиленкарбонат)ов по изобретению (см. Фиг.8). In vitro выбрали рН 12, поскольку радикал O2 - достаточно стабилен при этом значении рН.
Интересно, что поли(пропиленкарбонат), отличающийся от поли(этиленкарбонат)а замещением водорода этиленового звена метильной группой, вообще не способен к биологическому распаду.
Используя суспензии микрочастиц поли(этиленкарбонат)ов по изобретению, было проведено токсикологическое исследование на 48-и крысах в течение 21-х суток и на 24-х собаках в течение 35-и суток. Каждому виду было сделано два введения на 1-е и 17-е сутки. После подкожного и внутримышечного введения 10 и 40 мг полимерных микрочастиц/кг веса тела не наблюдали ни клинических симптомов системной токсичности, ни соответствующих воздействий на гематологические параметры, на клинические параметры крови, на вес тела и на потребление пищи. Места введения тестировали на гистопатологические изменения через 4 и 21 день после введения у крыс и через 18 и 35 дней после введения у собак. Кроме ожидавшейся воспалительной реакции не было выявлено никаких необычных гистопатологических изменений.
Время распада полимеров по изобретению может регулироваться в широких пределах в зависимости от их молекулярного веса, содержания этиленоксида, идентичности концевых групп, например биосовместимых эфирных групп, и присутствия O2 - радикальных нейтрализаторов, например витамина С, и может длиться от 6 дней до 6 месяцев и дольше, в частности до 1 года. Радикальный нейтрализатор предпочтительно может быть введен в полимер в качестве добавки.
Молекулярный вес Мв (со)-полимеров по изобретению составляет от 80 000, предпочтительно от 100 000, в частности от 200 000 до 2 000 000 Дальтон согласно определению с помощью гелевой проникающей хроматографии с метиленхлоридом в качестве элюента и полистиролом в качестве эталона.
В Chem. Pharm. Bull. 32(7), 2795-2802 (1984), обсуждавшейся ранее, упоминают, что использовали поли(этиленкарбонат)ы, имеющие молекулярный вес от 50 000 до 150 000 Дальтон. Мы обнаружили, что in vitro и in vivo распад полимера может иметь место в достаточной степени только в том случае, если молекулярный вес составляет более 80 000, предпочтительно более 100 000 (Фиг.6); это является предпочтительным аспектом изобретения.
Полимеры по изобретению могут быть применены в фармацевтических составах, особенно в качестве матричных материалов для связывания фармакологически активных соединений. Поскольку в условиях in vivo и in vitro не наблюдается объемной эрозии и активное соединение защищено полимером, то активное соединение выделяется как только (но не раньше) оно появится на матричной поверхности из-за поверхностной эрозии матрицы. В водной системе in vitro pH 7,4, не содержащей O2 -, выделялись только следовые количества активного соединения (см. Фиг.9).
Еще одним преимуществом поверхностной эрозии является то, что размер молекулы фармакологически активного соединения не влияет на скорость выделения.
Таким образом, в изобретении предлагают фармацевтический состав фармакологически активного соединения в полимере, способном к негидролитической поверхностной эрозии, особенно с линейной, а именно 1:1 линейной корреляцией выделения активного соединения и негидролитического распада массы полимера и защитой активного соединения в полимерной матрице.
Составы предпочтительно применяют в форме микрочастиц или имплантантов.
Получение фармацевтических форм согласно изобретению можно осуществлять уже известными способами, микрочастиц - путем соответствующей воздушной сушки или методами эмульгирования, имплантантов - путем смешивания лекарственного соединения и поли(этиленкарбонат)ов в виде твердых частиц при более высоких температурах, при которых поли(этиленкарбонат)ы становятся мягкими и легко обрабатываемыми, возможно с последующим охлаждением смеси до твердого состояния и придания ему подходящей формы. Возможно также смешивание лекарственного соединения в растворенном или диспергированном состоянии с раствором поли(этиленкарбонат)а с последующим испарением растворителя, после чего твердый остаток формируют в удобные имплантируемые формы.
Фармацевтические составы, содержащие микрочастицы, могут быть получены путем обработки их соответствующими галеновыми эксципиентами и пропускания через соответствующие дозировочные устройства.
В зависимости от свойств лекарственного препарата и способа производства содержание введенного лекарства может варьировать в широких пределах, порядка от 0,001 до приблизительно 70%, в частности от 0,001 до 20%, предпочтительно от 0,001 до 5% по весу. Следует избегать проникновения среды в полимер из-за высокой лекарственной нагрузки и ограничивать верхние значения содержания нагрузки.
В медицинской практике введения лекарственных соединений может применяться любой тип фармакологически активного соединения в сочетании с поли(этиленкарбонат)ом по изобретению. В случае микрочастиц предпочтительно применять те типы лекарственных соединений, которые фармакологически активны в небольших количествах и должны поддерживаться на неизменном уровне в крови в течение длительных периодов времени, например гормонов, пептидов или белков, в частности соматостатинов, интерферонов или интерлейкинов, особенно те соединения, которые являются нестабильными и разлагаются после перорального применения в желудочно-кишечной системе и поэтому предпочтительно их парентеральное введение.
Депонированные формы согласно изобретению могут быть использованы для введения различных классов активных агентов, например таких фармакологически активных агентов, как контрацептивы, седативы, стероиды, сульфонамиды, вакцины, витамины, препараты от мигрени, ферменты, бронхолитические средства, кардиоваскулярные препараты, анальгетики, антибиотики, антигены, противоконвульсивные препараты, противовоспалительные препараты, препараты против болезни Паркинсона, ингибиторы секреции пролактина, противоастматические препараты, гериартрические и противомалярийные препараты. Активный агент может быть выбран из широкого круга химических соединений, например липофильных и/или гидрофильных активных агентов, включая пептиды, такие как октреотид (GB 2234896).
Активные белки или пептиды предпочтительно являются цитокинами, например интерлейкинами, гранулоцит-макрофаг-колониестимулирующий фактор (G-CSF), макрофаг-колониестимулирующий фактор (M-CSF), гранулоцит-колониестимулирующий фактор (GM-CSF) или фактор, ингибирующий лейкемию, LIF, интерферонами, эритропоэтинами, циклоспоринами либо гормонами или их аналогами, в частности октреотидом.
Фармацевтические составы могут быть применены для
иммуномодуляции, когда активный ингредиент содержит цитокин, например интерлейкин (IL-3, IL-6), либо факторы стимуляции гемопоэтических колоний (G-CSP, например филграстим, GM-CSF, например молграмостим, сарграмостим, M-CSF), в частности, как адьюванта для вакцин,
восстановления кроветворения после миелосупрессивной терапии или после пересадки костного мозга, когда активный ингредиент содержит гемопоэтический ростовой фактор, например GM-CSF, G-CSF, ИЛ-3, ИЛ-6, LIF, фактор столовых клеток (SCF) или их сочетания,
создания высокой локальной концентрации активного ингредиента, например, когда активный ингредиент содержит лекарственное средство или цитокин, GM-CSF, IL-6, IL-2, IL-4 или их сочетания для стимуляции защитного иммунного ответа, например, при совместном введении с облученными опухолевыми клетками или вакционными антигенами (аналог облученных опухолевых клеток, "трансфецированных" соответствующими цитокиновыми генами),
индукции сильных иммунных ответов, когда активный ингредиент содержит, например, GM-CSF, введенный в сочетании с антигенами, особенно опухолевыми антигенами, вирусными антигенами или бактериальными антигенами,
индуцирования заживления ран путем локального инъецирования составов, например, когда активный ингредиент содержит GM-CSF,
индуцирования антиген-специфической иммунной устойчивости, когда активный ингредиент является, например, GM-CSF в сочетании с ингибиторами побочных молекул (сорецепторов), особенно ингибиторами CD28-B7 взаимодействия, CD40-CD40 лигандного взаимодействия, для адгезии факторных взаимодействий,
сопутствующей терапии при цитостатическом лечении или как вакцинный адъювант, когда активный ингредиент является, например, цитокином, особенно интерлейкином (IL-3, IL-6), или индуктором секреции цитокинов, например липидным производным, в частности соединением, описанным ЕР 309411, особенно в примере 1, известным также, как MRL 953,
специфической иммунной суппрессии, например, когда активный ингредиент является иммунофилинсвязывающим иммунодепрессантом, в частности циклоспорином (например, циклоспорином А), аскомицином (например, FK506) или рапамицином (например, рапамицином или его производным, как описано в WO 94/09010, в частности 40-O-гидроксиэтил-рапамицином),
лечения или профилактики автоиммунных заболеваний и воспалительных состояний путем медленного выделения противоспалительных цитокинов, например IL-6, IL-10 или TGFβ; либо интерферонов, например INF-β1 или бетасерона; либо растворимых рецепторов цитокинов или антагонистов цитокиновых рецепторов, например IL-1, TNFα или IL-4,
лечения или профилактики аллергических заболеваний путем медленного выделения растворимой α-цепи рецептора с высоким сродством к IgE (FcE RI),
лечения раков, например, с помощью октреотида, цитокинов, особенно интерлейкинов,
селективного лечения, например для лечения лейшманиоза, грибных инфекций, заболеваний накопления ферментов (Тау Sachs, Gauckerillness),
терапии СПИДа и ARC,
вакцинации, например, вакциной на основе столбнячного токсина,
гематопоэза, например, когда активным ингредиентом является эритропоэтин,
внутрисуставного введения в воспаленные суставы, когда активный ингредиент является противоспалительным лекарственным препаратом, особенно если он является препаратом, не биодоступным при пероральном введении или имеющим очень короткий период полураспада, например ингибиторами IL-1β конвертирующих ферментов, ингибиторами металлопротеазы.
Известен из WO 94/01133 способ усиления иммунного ответа млекопитающего на вакцину, отличающийся тем, что он включает введение млекопитающему в случае необходимости в вакцинации эффективного количества GM-CSF, сконъюгированного с вакциной. Однако введение GM-CSF не было аккуратно замедленным способом согласно текущему изобретению, что обеспечивает практически постоянное выделение активного соединения в течение длительного периода времени, посредством чего сроки повторного введения GM-CSF могут быть уменьшены.
В изобретении предлагают, в частности, фармацевтический состав фармакологически активного соединения в полимере, способном к негидролитической поверхностной эрозии, для парентерального введения интерлейкина или CSF, в частности, в полимере, как описано ранее.
В изобретении также предлагают способ введения такого состава субъекту, который включает в себя его введение парентерально субъекту, нуждающемуся в подобном лечении.
Депонированные препараты согласно изобретению могут применяться по известным показаниям для конкретного лекарственного соединения, включенного в них.
Точные количества лекарственного соединения или депонированного препарата, которые должны быть введены, зависят от ряда факторов, например состояния, подлежащего лечению, требуемой продолжительности лечения, скорости выделения лекарственного соединения и способности поли(этиленкарбонат)а к распаду.
Требуемые препараты могут быть получены известным способом.
Требуемое количество фармакологически активного агента и скорость его выделения могут быть определены на основании известных методов in vivo и in vitro, например насколько долго концентрация конкретного активного агента в кровяной плазме сохраняется на необходимом уровне. Способность к распаду матрицы также можно изучить с помощью методов in vitro и, особенно, in vivo, например путем определения количества матричных материалов в подкожной ткани через определенные промежутки времени.
Депонированные препараты по изобретению могут быть введены, например, в форме микрочастиц посредством перорального, носового или легочного, предпочтительно подкожного, внутримышечного или внутривенного введения главным образом в виде суспензии в подходящем жидком носителе или в форме имплантантов, например подкожно.
Повторное введение депонированных препаратов по изобретению может быть проведено тогда, когда происходит достаточно полный распад полимерной матрицы, например через 1, 2 или 3 недели либо 1 месяц.
Преимуществом поли(этиленкарбонат)ных матриц по изобретению является то, что в ходе выделения лекарственного соединения полимерные цепи распадаются на части с небольшим молекулярным размером, которые переносятся жидкостями организма от места введения.
Примерами лекарственных нагрузок для предпочтительного соединения октреотида являются акромегалии в парентеральной жидкой депонированной форме, имеющей микрочастицы, которые содержат пептид в количестве от по меньшей мере 0,1, предпочтительно 0,5 до 20 процентов по весу относительно (со)-полимерной матрицы, предпочтительно от 2,0 до 10, в частности от 3 до 6% по весу. Общая доза октреотида составляет от 20 до 30 мг при акромегалии и до 100-200 мг при раке мозга, в частности, на 1 месяц лечения.
Время выделения пептида из микрочастиц может составлять от 5 дней до приблизительно 2 недель или более.
Удобно, когда препарат пролонгированного введения содержит октреотид в (со)-полимерном носителе, который при подкожном введении кролику или крысе в дозировке 2 мг октреотида на кг веса тела животного создает концентрацию октреотида в плазме крови по меньшей мере 0,3 нг/мл и предпочтительно менее чем 20 нг/мл в течение длительного периода.
Фармацевтические составы по изобретению могут содержать дополнительные добавки, предпочтительно также введенные в (со)-полимер, например нейтрализатор радикалов, особенно нейтрализатор супероксидного радикального аниона O2 -. Присутствие такого нейтрализатора, например менадиона или витамина С, понижает скорость распада поли(этиленкарбонат)а (Фиг.7).
Другим типом добавки является нейтрализатор гидроксильного радикала, получающегося возможно под влиянием супероксидного радикального аниона O2 -, например полиол, особенно сахарный спирт, в частности маннитол. Было обнаружено, что эта добавка также оказывает благоприятное влияние на увеличение веса тела опытных животных, которым вводят микроинкапсулированный ИЛ-3. Без этой добавки увеличение веса тела задерживается. Когда состав находится в форме микрочастиц, та же самая или другая добавка может быть присоединена наружным образом к имеющимся микрочастицам, поскольку это оказывает положительное влияние на стабильность суспензии микрочастиц - против флокуляции и осаждения.
Если имеется добавка, то ее количество составляет предпочтительно от 1 до 90% по весу по отношению к общему весу препарата.
Значительный in vitro и in vivo распад массы под влиянием супероксидного радикального аниона O2 - может быть виден из Фиг.8. Кривые распада для остаточной массы являются приблизительно линейными и имеют различный наклон, поскольку условия распада in vivo и in vitro различны. Количество распадающейся массы в единицу времени является практически постоянным.
Кривые для in vivo выделения фармакологически активного соединения, например человеческого ИЛ-3, под влиянием супероксидного радикального аниона O2 - являются, как и кривые распада, практически линейными (Фиг.10), что означает, что количество выделяющегося лекарственного соединения в единицу времени также практически постоянно.
Сочетание in vivo выделения человеческого ИЛ-3 и in vivo распада массы представлено на Фиг.11, показывая наличие 1:1 корреляции между in vivo распадом массы и выделением лекарственного соединения.
ПРИМЕРЫ 1-6: Общий способ синтеза поли(этиленкарбонат)ов с катализатором, получаемым из диэтилцинка и воды.
Количества реагентов, растворителя, катализатора и т.д. для каждого эксперимента приведены в таблице 1.
200 мл сухого диоксана и 19,5 г (158 ммоль) Zn(C2H5)2 помещали в колбу емкостью 750 мл в атмосфере азота. Колба была снабжена механической мешалкой, капельной воронкой, термометром и входным клапаном для N2. Капельная воронка была снабжена СаСl2-трубкой. Раствор охлаждали до 10oС на ледяной бане и медленно добавляли раствор 2,7 мл Н2О в диоксане (см. таблицу 1) таким образом, чтобы температура сохранялась в интервале 10-15oС. Реакционную смесь дополнительно перемешивали в течение 45 минут при комнатной температуре до тех пор, пока изначально бесцветный раствор не становился бледно-желтым. Этот катализаторный раствор переносили в автоклав, обрабатывали его 40 г СО2 и прогревали при 125oС в течение времени, указанного в таблице 1. Затем смесь охлаждали до комнатной температуры и добавляли 560 г (12,7 моль) СО2 с последующим добавлением 132 г (3 моль) этиленоксида в течение 1 часа. Реакцию проводили в течение промежутка времени, указанного в таблице 1. После этого в течение нескольких часов снижали давление. Продукт, липкую суспензию, разбавляли диоксаном и осаждали путем вливания диоксанового
раствора в 0,25 М водную НС1. Осадок растворяли в соответствующем количестве СН2Сl2 (2-4 литра), промывали водной 0,5 М НСl (2х) и Н2О (1х). Раствор сушили над безводным Na2SO4 и упаривали до конечного объема от 0,5 до 1,5 литров, в зависимости от вязкости раствора. Продукт осаждали посредством приливания CH2Cl2 раствора в 4-кратном объеме метанола. Белый осадок отфильтровывали и сушили в течение ночи при 0,5 мбар/50oС. Сырой продукт переосаждали из ацетона для дальнейшей очистки, см. таблицу 2. Все продукты показывали идентичные 1Н-ЯМР спектры, за исключением относительных интенсивностей сигналов при 3,65, 3,73, 4,29 и 4,37 ppm из-за различий в содержании этиленкарбонатного звена.
Все эксперименты проводили в автоклаве NB2 емкостью 1,0 литр. Молярное соотношение Н2О: Zn(C2H5)2=0,95 для всех экспериментов. Катализатор предварительно обрабатывали 40 г СО2 при 125oС в течение 1 часа, за исключением Примера 1 (10 часов).
ПРИМЕРЫ 7-11: Основной способ синтеза поли(этиленкарбонат)ов с катализатором, полученным из диэтилцинка и диола.
1. Получение катализатора.
200 мл сухого диоксана помещали в сухую четырехгорлую колбу объемом 750 мл в атмосфере азота. С помощью стеклянного шприца добавляли 19,50 г (158 ммоль) диэтилцинка. Колба была снабжена механической мешалкой, капельной воронкой, термометром и входным клапаном для аргона. Капельную воронку заполняли 100 мл сухого диоксана и снабжали трубкой с хлоридом кальция. Затем аппарат помещали в поток аргона. К диоксану добавляли 9,00 г (145 ммоль, 0,92 моль-экв.) свежего дистиллированного сухого этиленгликоля (хранят на молекулярных ситах) посредством капельной воронки под током аргона. Колбу при механическом перемешивании охлаждали до 10oС на ледяной бане в атмосфере аргона. Раствор этиленгликоля в диоксане добавляли по каплям к перемешиваемому раствору диэтилцинка в диоксане в течение периода времени, равного 30 минутам, в течение этого времени температуру поддерживали в диапазоне 10-14oС. Выделение газообразного этана и осаждение наблюдали одновременно при добавлении раствора этиленгликоля. После того, как добавление было закончено, охлаждающую баню удаляли, а смесь перемешивали дополнительно в течение 60 минут, позволяя ей нагреваться до комнатной температуры. Гетерогенную смесь затем переносили в автоклав (автоклав NB2 емкостью 1 л) под аргоном. Автоклав заполняли приблизительно 40 г (0,9 моль) диоксида углерода и прогревали при 125oС в течение 1 часа при перемешивании для предварительной обработки катализатора диоксидом углерода.
2. Полимеризация.
Автоклав с предварительно обработанным катализатором охлаждали до комнатной температуры и заполняли дополнительно 560 г (12,7 моль) диоксида углерода. Затем к перемешиваемой в автоклаве смеси добавляли 132 г (3 моль) этиленоксида (99,8%-ного) путем медленного введения в течение 1 часа. После окончания добавления автоклав нагревали до температуры, указанной в таблице 3, и смесь перемешивали в течение указанного времени при этой температуре.
3. Обработка.
Автоклав охлаждали до комнатной температуры, а давление медленно понижали до атмосферного. Продукт, белую липкую суспензию, переносили в 7 л дихлорметана, добавляли 1035 мл 0,4 М раствора НСl, смесь перемешивали в течение 3 часов при комнатной температуре. Фазы разделяли, органический слой дважды промывали 3 литрами 0,5 М НСl и дважды - 4,5 литрами воды. Раствор дихлорметана затем сушили над 120 г сульфата натрия и концентрировали до конечного объема приблизительно 2 литра. Продукт осаждали путем медленного добавления этого раствора к 6 литрам метанола. Осадок сушили в течение 16 часов в вакууме при 40oС, получая в результате сырой полимер, который очищали затем следующим образом.
Сырой продукт растворяли в дихлорметане, а раствор вливали в 5-кратный объем ацетона в течение 15 минут для осаждения продукта. Осадок сушили в течение 16 часов в вакууме при 40oС с образованием соответствующего поли(этиленкарбонат)а. Физические свойства продуктов представлены в таблице 4. Все продукты показали сильные ИК-поглощения при 1750 и 1225 см-1. 1H-ЯМР сигнал этиленкарбонатных звеньев регистрировали при 4,37 ppm.
ПРИМЕР 12: Экспериментальный способ синтеза поли(этиленкарбонат)а с катализатором, полученным из диэтилцинка и флороглюцина.
1. Получение катализатора.
200 мл сухого диоксана помещали в сухую четырехгорлую колбу емкостью 750 мл в атмосфере азота. Посредством стеклянного шприца добавляли 19,60 г (158,7 ммоль) диэтилцинка. Колба была снабжена механической мешалкой, капельной воронкой, термометром и входным клапаном для аргона. Капельную воронку заполняли 100 мл сухого диоксана и снабжали трубкой с хлоридом кальция. Установку помещали под ток аргона. Под током аргона к диоксану через капельную воронку добавляли 13,34 г (105,8 ммоль, 0,92 моль-экв.) сухого флороглюцина. Колбу с механическим перемешиванием охлаждали до 10oС на ледяной бане под аргоном. Раствор флороглюцина в диоксане добавляли по каплям к перемешиваемому раствору диэтилцинка в диоксане в течение периода времени, равного 30 минутам, в течение этого периода температуру поддерживали в диапазоне 10-14oС. Выделение газообразного этана и образования осадка наблюдали одновременно при добавлении раствора флороглюцина. После того, как добавление было завершено, охлаждающую баню удаляли, а смесь перемешивали дополнительно в течение 30 минут, позволяя ей нагреваться до комнатной температуры. Гетерогенную смесь переносили затем в автоклав (автоклав NB2 емкостью 1 л) под аргоном. Автоклав заполняли приблизительно 40 г (0,9 моль) диоксида углерода и прогревали в течение 1 часа при 125oС при перемешивании для предварительной обработки катализатора диоксидом углерода.
2. Полимеризация.
Автоклав с предварительно обработанным катализатором охлаждали до комнатной температуры и заполняли дополнительно 560 г (12,7 моль) диоксида углерода. Затем к перемешиваемой в автоклаве смеси добавляли 132 г (3 моль) этиленоксида (99,8%-ного) путем медленного введения в течение 1 часа. После окончания добавления этиленоксида автоклав выдерживали при перемешивании в течение 260 часов при 21oС.
3. Обработка.
Давление в автоклаве медленно понижали до атмосферного давления. Продукт переносили в 4 литра дихлорметана, добавляли 1035 мл 0,4 М НСl и полученную смесь перемешивали в течение 3 часов при комнатной температуре. Фазы разделяли, а органический слой промывали дважды 1,5 литрами 0,5 М HCl и дважды - двумя литрами воды. Затем раствор дихлорметана сушили над 120 г сульфата натрия и концентрировали до конечного объема приблизительно 1 литр. Продукт осаждали путем медленного вливания этого раствора в 3 литра метанола. Осадок сушили в течение 16 часов в вакууме при 40oС, получая в результате сырой полимер, который очищали далее следующим образом.
Сырой продукт растворяли в дихлорметане, а полученный раствор вливали в 5-кратный объем ацетона в течение 15 минут для осаждения продукта. Осадок снова растворяли в дихлорметане, переосаждали из метанола и сушили в течение 16 часов в вакууме при 40oС с получением соответствующего поли(этиленкарбонат)а.
Физические свойства продукта:
Мв=258000 Д, Мн=35600 Д, Тст=15,4oС.
ИК: Сильные поглощения при 1751 и 1225 см-1.
Согласно 1Н-ЯМР, продукт содержит приблизительно 96% этиленкарбоната.
ПРИМЕР 13: Экспериментальный способ синтеза поли(этиленкарбонат)а с катализатором, полученным из диэтилцинка и ацетона.
132 г (3 моль) этиленоксида подвергали сополимеризации с 600 г (13,6 моль) СО2 при 50oС в течение 96 часов в присутствии катализатора, полученного из 8,43 г (145,16 ммоль) ацетона и 19,62 г (159 ммоль) диэтилцинка.
Получение катализатора и его полимеризацию проводили аналогично процессу, описанному для примеров 7-11, с тем исключением, что для получения катализатора вместо диола применяли ацетон.
Полученный таким образом поли(этиленкарбонат) содержал 93% этиленкарбоната и имел следующие свойства: Мв=233 кД, Мн=109 кД, Мв/Мн=2,14, Тст= 22,4oС.
ПРИМЕР 14: Синтез стеароилированного по концевой группе поли(этиленкарбонат)а.
1 г поли(этиленкарбонат)а, имевшего Мв=153000 Д, Мн=68900 Д, Тg=29,1oС, растворяли в 30 мл сухого дихлорметана. Раствор обрабатывали затем 0,98 г (12,38 ммоль) пиридина и 10 г (33,0 ммоль) стеароилхлорида. Реакционную смесь перемешивали при комнатной температуре в течение 48 часов, затем разбавляли 50 мл дихлорметана и промывали последовательно 2 х 150 мл насыщенного бикарбоната натрия и воды. Органический слой сушили над безводным сульфатом натрия, а продукт осаждали путем капельного вливания дихлорметанового раствора в 300 мл н-гексана. Полученный в результате сырой продукт очищали далее путем растворения в дихлорметане и осаждения из 3-кратного объема диэтилового эфира. И наконец, продукт сушили в вакууме при 40oС в течение 16 часов, получая в результате стеароилированный по концевой группе поли(этиленкарбонат).
Мв=144000 Д. Мн=71000 Д, Тст=25,6oС.
ПРИМЕР 15: Синтез ацетилированного по концевой группе поли(этиленкарбонат)а.
1 г поли(этиленкарбонат)а (имевшего Мв=153000 Д, Мн=68900 Д, Тст=29,1oС) растворяли в 10 мл сухого дихлорметана. Добавляли 0,98 г (12,38 ммоль) пиридина с последующим добавлением 10,08 г (98,7 ммоль) уксусного ангидрида. Реакционную смесь перемешивали при комнатной температуре в течение 120 часов. Затем ее разводили 50 мл дихлорметана и медленно вливали в 200 мл насыщенного бикарбоната натрия. Смесь перемешивали в течение 30 минут, а затем разделяли слои. Органический снова промывали 150 мл насыщенного карбоната натрия, а затем - водой. Дихлорметановый раствор сушили над безводным сульфатом натрия, а продукт осаждали капельным вливанием этого раствора в 300 мл диэтилового эфира. Осадок снова растворяли в дихлорметане и переосаждали из диэтилового эфира. Продукт сушили в течение 16 часов при 40oС в вакууме с получением поли(этиленкарбонат)а с концевой группой, этерифицированной ацетатом.
Мв=150000 Д, Мн=69100 Д, Тст=26,8oС.
ПРИМЕР 16: Очистка поли(этиленкарбонат)а путем обработки кипящей водой.
1 г поли(этиленкарбонат)а (из Примера 8, имевшего Мв=328000 Д, Мн=149000 Д, Тст= 16,4oС) измельчали на мелкие кусочки и перемешивали в 50 мл кипящей воды в течение 2 часов. Воду удаляли и заменяли свежей водой, которую снова нагревали до температуры кипения. Спустя еще 3 часа кусочки полимера отделяли и сушили в вакууме при 40oС в течение 16 часов. Полученный продукт имел следующие физические свойства: Мв=340000 Д, Мн=148000 Д, Тст=28,3oС. Таким образом, наблюдали разительное увеличение температуры стеклования, которое не было связано с изменением молекулярного веса полимера.
ПРИМЕР 17: Состав (микрочастицы) с 1%-ным чИЛ-3 лекарственным наполнением.
1. Приготовление микрочастиц, содержащих лекарство.
1 г поли(этиленкарбонат)а Мв=328000 Д из Примера 8 (ПЭК) растворяли в 10 мл метиленхлорида при перемешивании с последующим добавлением 12,1 мг человеческого интерлейкина 3 (чИЛ-3), растворенного в 0,6 мл воды. Смесь интенсивно перемешивали с помощью Ultra-Turax в течение одной минуты при скорости 20000 об./мин (внутренняя W/O-фаза). 1 г желатина А растворяли в 2000 мл деионизированной воды при 50oС, раствор охлаждали до 20oС (внешняя W-фаза). W/O-фазу и W-фазу интенсивно смешивали. Посредством этого внутреннюю W/O-фазу гомогенно диспергировали во внешней W-фазе в виде правильных капелек. Полученную тройную эмульсию медленно перемешивали в течение 1 часа. Этим метиленхлорид упаривали, а из капелек внутренней фазы генерировались и затвердевали микрочастицы.
После седиментации микрочастиц супернатант отсасывали, а микрочастицы регенерировали путем фильтрации под вакуумом или центрифугирования и промывали водой для удаления желатина. И наконец, микрочастицы либо сушили замораживанием, используя в качестве агента для увеличения объема маннит, либо сушили в вакуумном сушильном шкафу (свободные составы на основе маннита) в течение 72 часов и сортировали (0,125 мм размер сита) для получения конечного продукта.
2. Технология приготовления плацебо.
1 г ПЭК Мв=328000 Д из Примера 8 растворили в 10 мл метиленхлорида при перемешивании (внутренняя O-фаза). 1 г желатина А растворяли в 2000 мл деионизированной воды при 50oС, а раствор охлаждали до 20oС (внешняя W-фаза). О- и W-фазы интенсивно смешивали. Посредством этого O-фазу гомогенно диспергировали в виде правильных капелек во внешней W-фазе. Полученную эмульсию медленно перемешивали в течение 1 часа, а далее обрабатывали способом, описанным выше.
ПРИМЕРЫ 18-26.
Все галеновые препараты, описанные здесь, готовили, используя ПЭКы, синтезированные согласно Примеру 8 в Таблице 3, а далее очищали способом, аналогичным описанному в Примере 16. Все они имели Мв от 300000 до 450000 Д, содержание этиленкарбоната свыше 94%, а Тg в интервале от 18 до 50oС.
ПРИМЕР 18: Составы (микрочастицы), имеющие 0,2%-ное чИЛ-2 наполнение.
2,9 мг человеческого интерлейкина 2 (чИЛ-2) растворяли в 1,5 мл воды и готовили содержащие ИЛ-2 микрочастицы согласно описанию из Примера 17.
Микрочастицы сушили замораживанием с применением маннита в качестве агента для увеличения объема и сортировали (размер сита 0,125 мм) для получения конечного продукта.
ПРИМЕР 19: Состав (микрочастицы), имеющий 0,2%-ное чИЛ-2 наполнение (безводный).
Препарат готовили как описано в Примере 18, но 2,9 мг человеческого интерлейкина диспергировали непосредственно в органической фазе (ПЭК, растворенный в метиленхлориде).
ПРИМЕР 20: Состав (имплантанты), имеющий 0,8%-ное чИЛ-3 наполнение.
1. Литье под давлением.
25 мг микрочастиц, состоящих из 100%-ного (в/в) поли(этиленкарбонат)а (плацебо), 99% (в/в) поли(этиленкарбонат)а и 1% (в/в) человеческого интерлейкина-3 или 79,2% (в/в) поли(этиленкарбонат)а, 20% (в/в) маннита и 0,8% (в/в) человеческого интерлейкина-3, формовались сжатием в течение 3 минут при 60-70oС и 160 бар в имплантанты (таблетки) диаметром 5 мм. Таблетки хранили при 4oС в закрытых стеклянных флаконах перед применением для экспериментов по выделению лекарств in vitro и in vivo.
2. Эксперименты по выделению лекарств in vitro.
Три таблетки, не содержащие маннит, содержащие маннит препараты человеческого интерлейкина-3 и препараты плацебо качались при 37oС в синтетической культуральной среде, содержащей 2,5% (об./об.) N-[2-гидроксиэтил]-пиперазин-N'-[2-этансульфоновой кислоты] (1 М), 10% (об./об.) фетальной сыворотки теленка и 2% (об./об.) раствора пенициллин/стрептомицин. Образцы вынимали из среды через 0,5, 1, 2, 5 часов и на 1, 2, 3, 7, 14, 20-е сутки, а среду обновляли. Содержание человеческого интерлейкина-3 в образцах измеряли с помощью твердофазного иммуноферментного анализа.
3. Эксперимент по выделению лекарства in vivo
Самцов крыс, содержавшихся в оптимальных условиях, анестезировали путем ингаляции наркотика и каждой крысе имплантировали в подкожный кожный карман одну таблетку препаратов интерлейкина-3 и препарата плацебо. Через 1, 4, 7, 14, 21-ые сутки крыс умерщвляли путем передозировки наркотической ингаляции. Сохранившиеся таблетки извлекали, освобождали от прилипшей ткани и сушили. Потерю массы таблеток определяли гравиметрически. Затем определяли содержание человеческого интерлейкина-3 сохранившихся таблеток путем ЖХВР и твердофазного иммуноферментного анализа.
ПРИМЕР 21: Состав (w/o/w микрочастицы), имеющий 0,0002%-ное чИЛ-2 наполнение.
4 г ПЭК растворяли в 80 мл метиленхлорида с помощью магнитной мешалки. К этому раствору добавляли соответствующее количество ИЛ-2 (113,2 мг для 2%, 11,32 мг для 0,2% и т.д.), растворенное в 6 мл дистиллированной воды или воды с несколькими каплями этанола. Смесь интенсивно перемешивали с помощью Ultra-Turax для диспергирования ИЛ-2 раствора в полимерной фазе (внутренняя W/O-фаза). 1 г желатина А растворяли в 200 мл 1/15 М фосфатного буфера (рН 7,4) при 50oС, раствор охлаждали до 20oС (внешняя W- фаза). W/O- и W-фазы интенсивно перемешивали. Посредством этого внутреннюю W/O-фазу разбивали на мелкие капельки, которые были гомогенно диспергированы во внешней W-фазе. Полученную тройную эмульсию медленно перемешивали в течение 1 часа. При этом метиленхлорид испарялся, а из капелек внутренней фазы формировались при затвердевании микрочастицы.
После седиментации (или центрифугирования) микрочастиц супернатант отсасывали, а частицы отделяли путем фильтрации под вакуумом и промывали водой для удаления желатина. И наконец, микрочастицы сушили в вакууме в течение 24 часов и просеивали для получения конечного продукта.
Эффективность инкапсуляции, определенная с помощью ЖХВР и биооценки, составляла от 10 до 100%.
ПРИМЕР 22: Состав (s/o/w микрочастицы), имеющий 0,0002%-2%-ное ИЛ-2 наполнение.
Препараты готовили как описано в Примере 21, за исключением того, что ИЛ-2 не растворяли в воде. Вместо растворения ИЛ-2 лекарство диспергировали непосредственно в полимерной фазе (О-фаза). Эффективность инкапсуляции, определенная с помощью ЖХВР и биооценки, составляла от 10 до 100%.
Замечание: Количество полимера, метиленхлорида, воды и лекарства варьируют в широком диапазоне, что не изменяло качества продукта. Получают большие лекарственные наполнения до 20%. Во внешней фазе желатин заменяют другими эмульгаторами, такими как поливиниловый спирт и т. д., и/или изменяют концентрацию эмульгатора/буфера. Описанные процессы сепарации и сушки заменяют другими хорошо известными фармацевтическими технологиями, например фильтрацией, лиофилизацией или распылительной сушкой.
ПРИМЕР 23: Состав (w/o/w и s/o/w микрочастицы), имеющий 1%-ное чGM-СSF наполнение.
Получение проводили согласно способу, описанному в Примерах 21 и 22. Согласно этим описаниям готовят S/O/W- и W/O/W-композиции. Однако эффективность инкапсуляции W/O/W-композиций составляла 60%, в то время как S/O/W-композиции имели более низкие эффективности инкапсуляции.
ПРИМЕР 24: Состав (w/o/w и s/o/w микрочастицы), имеющий от 1 до 10%-ного наполнения октреотид-памоатом (SMS-PA).
Получение проводили согласно способу, описанному в Примерах 19 и 20. Однако SMS-PA не является водорастворимым веществом. Поэтому лекарство не растворяли, а диспергировали в воде для получения W/O/W-препаратов. Эффективность инкапсуляции, которую определяли с помощью ЖХВР, составляла от 20 до 100%.
ПРИМЕР 25: Состав (w/o/w и s/o/w микрочастицы), имеющий от 1% до 10%-ного наполнения октреотид-ацетатом.
Получение проводили согласно способу, описанному в Примерах 21 и 22. Эффективность инкапсуляции определяли посредством ЖХВР, и она составляла от 2 до 40%, что ощутимо меньше по сравнению с липофильным SMS-PA.
Более высокие значения были получены в S/О/W-препаратах после применения материала лиофилизированного активного соединения (меньшая частица лекарства).
ПРИМЕР 26: Выделение октреотид-памоата (SMS-PA) из микрочастиц у кроликов и имплантанты у кроликов и крыс.
Осуществляли подкожную имплантацию поли(этиленкарбонат)овых дисков или инъекцию микрочастиц поли(этиленкарбонат)а (лекарственное пополнение 1,95%) в количестве приблизительно 2 мг лекарственного вещества/кг веса тела самцам кроликов (гибрид шиншиллы, вес тела около 3 кг) и подкожную имплантацию дисков самцам крыс (Wistar, вес тела около 375 г). Каждой крысе или кролику вводили количества, равные 40 и соответственно 300 мг содержащего лекарство полимера в форме микрочастиц, соответственно спрессованного в виде имплантанта или в виде суспензии.
Имплантационные диски для крыс и кроликов имели диаметр, равный 0,5 и 1 см соответственно, и изготовлялись как описано в Примере 20.
Для определения выделения лекарства отбирали образцы крови на 14-е и 21-е сутки у крыс и кроликов соответственно и измеряли остаточные количества лекарства в имплантантах с помощью радиоиммуноанализа и ЖХВР.
Может быть выявлена линейная корреляция потери массы поли(этиленкарбонат)а и выделения SMS-PA (Фиг.13), как это продемонстрировано для чИЛ-3 с высокой молекулярной массой (Фиг.11). В течение 3 недель после введения у кроликов разлагалось максимум 75% имплантированного материала, у крыс в течение 2 недель после введения разлагалось максимум 95% имплантированного материала. Для биологической деградации поли(этиленкарбонат)а необходима предварительная воспалительная реакция (включая инвазию нейтрофильных лейкоцитов и других клеток). Можно ожидать, что течение воспалительной реакции будет являться видоспецифичным, обеспечивая видоспецифичные профили содержания лекарства в плазме. Это было установлено для SMS-PA (Фиг.12). У крыс биодеградация поли(этиленкарбонат)а происходит намного быстрее, чем у кроликов. У кроликов уровни SMS-PA в плазме возрастают медленно, достигая фазы постоянного выделения на 9-е сутки, и эта фаза продолжается по меньшей мере до 21-х суток.
ПРИМЕР 27: Состав (w/o/w микрочастицы), имеющий 0,0002%-2% рчИЛ-6 наполнение.
4 г ПЭК растворяют в 80 мл метиленхлорида путем перемешивания с помощью магнитной мешалки. К этому раствору добавляют соответствующее количество рчИЛ-6 (113,2 мг для 2%-ного, 11,32 мг для 0,2%-ного и т. д.), растворенного в 6 мл дистиллированной воды или воды с несколькими каплями этанола. Смесь интенсивно перемешивают с помощью Ultra-Turax для диспергирования раствора ИЛ-6 в полимерной фазе (внутренняя W/O-фаза). 1 г желатина А растворяют в 200 мл 1/15 М фосфатного буфера (рН 7,4) при 50oС и охлаждают раствор до 20oС (внешняя W-фаза). W/O- и W-фазы интенсивно смешивают. Посредством этого внутреннюю W/O-фазу дробят на мелкие капельки, которые были диспергированы гомогенно во внешней W-фазе. Полученную тройную эмульсию медленно перемешивают в течение 1 часа, метиленхлорид испаряется, а капельки внутренней фазы затвердевают в микрочастицы.
После седиментации (или центрифугирования) микрочастиц супернатант отсасывают, а микрочастицы выделяют путем фильтрации под вакуумом и промывают водой для удаления желатина. И наконец, микрочастицы сушат в вакууме в течение 24 часов и просеивают для получения конечного продукта.
Эффективность инкапсуляции, тестированная с помощью ЖХВР и биооценки, составляет от 10 до 100%.
ПРИМЕР 28: Состав (s/о/w микрочастицы), имеющий 0,0002%-2% рчИЛ-6 наполнение.
Препараты получают как описано в Примере 27, с тем исключением, что ИЛ-6 не растворяют в воде. Вместо растворения ИЛ-6 лекарство диспергируют непосредственно в полимерной фазе (О-фаза). Эффективность инкапсуляции, тестированная с помощью ЖХВР и биооценки, составляет от 10 до 100%.
Замечание: количество полимера, метиленхлорида, воды и лекарства варьируют в широком диапазоне, что не сказывается на качестве продукта. Получают более высокие наполнения лекарством - до 20%. Во внешней фазе желатин заменяют другими эмульгаторами, такими как поливиниловый спирт и т. д., и/или изменяют концентрацию эмульгатора/буфера. Описанные процессы сепарации и сушки заменяют другими хорошо известными фармацевтическими технологиями, такими как фильтрация, лиофилизация или распылительная сушка.
ПРИМЕРЫ 29-31: Применение ИЛ-6 для лечения состояний, обусловленных TNFα и/или ИЛ-1.
ПРИМЕР 29: Животная модель рассеянного склероза: модель хронического рецидива экспериментально индуцируемого аллергического энцефаломиелита на крысе Lewis (ХР-ЭАЭ).
Экспериментально индуцируемый аллергический энцефаломиелит (ЭАЭ) у крысы представляет собой хорошо изученную экспериментальную модель рассеянного склероза у людей (IMMUNOL. 5 (1966) 131-208, 113 (1974) 712,13 (1981) 3. J. PATH 47 (1965) 61). Крысам вводят нервную ткань других видов вместе с адъювантом, а полученная аллергическая реакция приводит к поражениям крысиных нервов, которые имитируют автоиммунные поражения, вызываемые рассеянным склерозом. Крысы становятся частично или полностью парализованными, а тяжесть заболевания оценивают при введении тестируемых лекарств и без него. Ряд лекарств, таких как стероиды и иммунодепрессанты, активно ослабляют развитие заболевания, но не способны предотвращать рецидивы после того, как болезнь установлена.
Модель хронического рецидива экспериментально индуцируемого аллергического энцефаломиелита (ХР-ЭАЭ) J. NEUROIMMUNOL: 10 (1985) 159-166) считают наиболее актуальной моделью, которая близко имитирует актуальные трудности лечения пациентов, страдающих рассеянным склерозом, у которых установлено это заболевание. В этой модели заболевание индуцируют путем введения смеси спинного мозга морской свинки и полного адъюванта Фройнда, обогащенного Mycobacterium tuberculosis. Обычно у 75-80% сенсибилизированных крыс развивается ХР-ЭАЭ, дающий 2-3 клинических рецидива в течение первых 40 суток. Спустя 60-80 суток приблизительно у 50% крыс с ХР-ЭАЭ имеют место следующие рецидивы, за которыми следует полное выздоровление только в 35% всех случаев. У остальных 65% этих животных наблюдают прогрессирующее состояние заболевания. Лекарственное лечение начинают на 16-е сутки после выздоровления от первого приступа заболевания.
Рекомбинантный человеческий интерлейкин-6 (рчИЛ-6, Sandoz), растворенный в рассоле, вводили внутрибрюшинно каждые 2-е сутки начиная с 16-х суток, используя 10 мкг ИЛ-6 на крысу (приблизительно 50 мкг/кг). Контрольные животные и животные в ИЛ-6 группе перенесли обычный тяжелый приступ заболевания (острый) на 11-14-е сутки. По шкале тяжести (от 0 - отсутствие заболевания до 4 - полный паралич животного) контрольная группа имела средний балл 3,0, а ИЛ-6 группа - 3,2. Введение ИЛ-6 каждые вторые сутки с 16-х по 30-е сутки (всего 7 введений) приводило к практически полному подавлению заболевания. Только у одной из 5-и ИЛ-6 обработанных крыс наблюдали слабый повторный приступ заболевания (тяжесть 0,4). У пяти из 5-и контрольных животных наблюдали повторный приступ заболевания со средним баллом тяжести, составлявшим порядка 1,8, на 16-е сутки и третий приступ заболевания на 22-29-е сутки. В ИЛ-6 обработанной группе следующих рецидивов не наблюдали.
ПРИМЕР 30: Животная модель артрита: вызываемый Borrelia артрит у мышей с тяжелым комбинированным иммунодефицитом (ТКИД).
Артрит Лайма (или болезнь артрита Лайма) представляет собой уникальную форму хронического артрита, поскольку для него с уверенностью можно назвать стимулирующее событие. Заболевание является одной из известных характерных особенностей, индуцируемых инфицированием спирохетой Borrelia burgdorferi, переносчиком которой является клещ. Особенности синовиальных поражений у пациентов с артритом Лайма имеют большое сходство с симптомами поражений в синовиальной оболочке у пациентов с ревматоидным артритом. У обеих групп пациентов можно наблюдать гипертрофию клеток синовиальной выстилки, гиперплазию синовиальных клеток, васкулярную пролиферацию и инфильтрацию одноядерных клеток в областях субсиновиальной оболочки. Многие плазматические клетки, высокоэндотелиальные венулы, рассеянные макрофаги и некоторые дендритные клетки имеют интенсивное представление антигена МНС класса II. Кроме того, такие цитокины, как ИЛ-1, ИЛ-6 и TNF-альфа, были обнаружены в синовиальной жидкости пациентов с различными артритами, подтверждая тем самым тот факт, что эти цитокины могут способствовать патогенезу разрушения суставов. Недавно была создана мышиная модель артрита Лайма на ТКИД мышах, которые недостаточны по функциональным Т и В клеткам (Immunology Today, 12: 11 (1991). Инфицирование иммунодефицитных мышей Borrelia burgdorferi приводит к возникновению рельефного и устойчивого олигоартрита. Вызываемый Borrelia артрит у ТКИД мышей чувствителен к кортикостероидам (преднизолон 30 мг/кг подкожно) и не чувствителен к иммунодепрессированным агентам, подобным SIM (циклоспорин А), в дозах, не превышающих 30 мг/кг подкожно. Ее рассматривают как хорошую модель индуцируемого цитокином артрита, включая другие типы артрита, для которых стимулирующее событие с определенностью не известно.
Шестинедельных С. В.-17 ТКИД мышей (гомозиготные по ТКИД мутации, получены из Bomholtgard, Denmark, 5-6 животных/группу) инокулировали 100 mio. Bоrrelia burgdorferi организмов путем подкожной инъекции в основание хвоста. В качестве контрольных животных использовали С.В.-17 иммунокомпетентных мышей (из того же источника). У них при инъецировании Bоrrelia burgdorferi заболевание не развивается. Рекомбинантный человеческий ИЛ-6 (рчИЛ-6, Sandoz, раствор для хранения 5 мг/мл) разводили физиологическим раствором и вводили 5 раз в неделю за 17 инъекций в дозировке 10 микрограмм /мышь внутрибрюшинно. Мышей осматривали ежедневно произвольным образом на клинические симптомы артрита в тибиотарсальных и запястно-локтевых суставах. Клинический артрит регистрировали согласно следующим параметрам:
- отсутствие симптомов
? симптомы сомнительные
(+) покраснение суставов
+ слабая припухлость
++ умеренная припухлость
+++ сильная припухлость тибиотарсальных и запястно-локтевых суставов.
В момент максимального развития клинического артрита мышей умерщвляли, а суставы фиксировали в растворе Schaffer'a, залитом в пластичные 9100, и окрашивали гематоксилинэозином (см. табл.5).
У ТКИД мышей, которые не подвергались обработке ИЛ-6, развивается тяжелый артрит в результате инфицирования Borrelia burgdorferi, начиная примерно с 13-х суток после инъекции антигена. Низкая доза рчИЛ-6 понижает тяжесть артрита в пределах 60-75% у всех пораженных животных.
ПРИМЕР 31: Муриновая модель септического шока.
Было решено исследовать влияние ИЛ-6 в модели мышиного эндотоксичного шока с использованием мышей, чувствительных к d-галактозамину, поскольку ее широко применяют как модель человеческого септического шока. Наши методы и результаты имеют следующий вид.
Самкам OF1 мышей с весом 18-22 г вводили инъекции по 0,2 мл внутрибрюшинно ЗФР раствора, содержавшего 0,15 мг/кг липополисахаридного эндотоксина (ЛПС) и 500 мг/кг d-галактозамина. Мышей делили на группы по 10 мышей в каждой и обрабатывали следующим образом:
Эксперимент 1 (см. табл.6),
Эксперимент 2 (см. табл.7).
рчИЛ-6 (ИЛS 969, Sandoz), рчИЛ-2 (Sandoz) и рчИЛ-4 (Sandoz) растворяли в ЗФР. Все инъекции (объем 0,2 мл) вводили внутрибрюшинно. В группе 3 (эксперимент 1) и группах 3-8 (эксперимент 2) ИЛ-6 и ИЛ-2 разводили в ЛПС/d-ГАЛ растворе и таким образом мыши получали одну инъекцию объемом 0,2 мл. Числа в скобках показывают дозу интерлейкина, вводимую каждой мыши. Множественное дозирование ЗФР требовалось для контроля внутригрупповой вариабельности из-за вызываемой стрессом реакции на управление в разное время до или после ЛПС введения.
За выживанием мышей наблюдали в течение 48 часов. Для статистических вычислений мы применяли Chi-квадратичный тест. Как показано на Фиг.1, спустя 24 часа после введения ЛПС 9 из 10 контрольных мышей погибали. Обработка ИЛ-6 за 3 часа перед введением ЛПС или 2 часа спустя после ЛПС введения понижала смертность соответственно на 60% (р=0,12) и 70% (р=0,26). С другой стороны, введение ИЛ-6 одновременно с введением ЛПС понижало групповую смертность на 10% (р<0,01). Защитные эффекты имели большую длительность, так, спустя 48 часов смертность в группе 3 медленно возрастала, например, до 30%, отражая значительное защитное действие по сравнению с контрольной группой (р<0,01). Смертность группы 4 изменялась с 70% до 80%, в то время как в группах 1 и 2 не наблюдали никаких изменений.
Основываясь на этих результатах, мы протестировали действие ИЛ-6 в различных дозах. Мы давали ИЛ-6 во время инъекций ЛПС, поскольку согласно первому эксперименту это было оптимальное время. Мы исследовали также влияние ИЛ-3 и ИЛ-4, введенных одновременно с ЛПС, как путь исключения возможных артефактов из-за применения рекомбинантных белков в ЛПС/d-ГАЛ препаратах. Мы тестировали также, был ли ИЛ-6 эффективен в защите мышей от смерти от эндотоксина в дозе 100 мкг/мышь, введенной до или после ЛПС.
Результаты эксперимента 2 (Фиг.2) соответствуют результатам эксперимента 1. Также в этом эксперименте обработка ИЛ-6 защищала мышей от смерти от эндотоксина. Когда ИЛ-6 вводили вместе с ЛПС, результирующая защита 24 часа спустя после ЛПС была дозозависимой при дозе 20 (30% смертей, р=0,03), 4 (50% смертей, р=0,16) и 0,8 (70% смертей, р=0,61) мкг/мышь, в то время как при дозе 100 мкг/мышь (60% смертей, р=33) мыши были защищены менее эффективно, чем при 20 мкг/мышь. Предварительная или последующая обработка 100 мкг ИЛ-6/мышь обеспечивала защиту, сравнимую с защитой, наблюдаемой в тех случаях, когда ту же самую дозу ИЛ-6 вводили вместе с ЛПС. Аналогичные результаты выживания мышей были получены спустя 48 часов после ЛПС.
ИЛ-4, введенный одновременно с ЛПС, не был эффективен в защите мышей от смерти от эндотоксина, в то время как ИЛ-2 понижал выживание мышей.
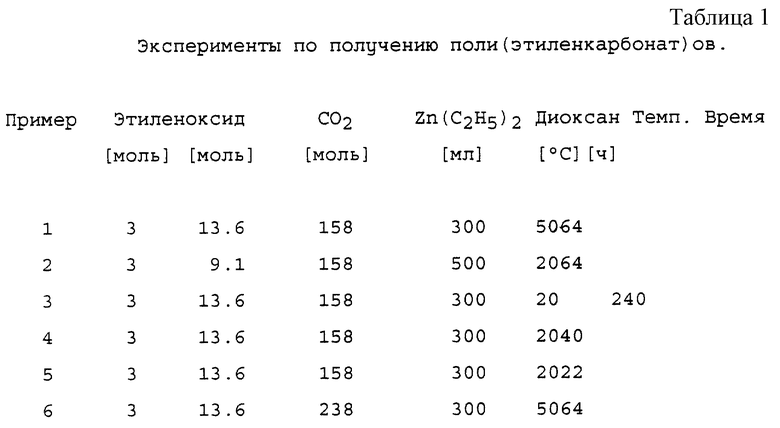
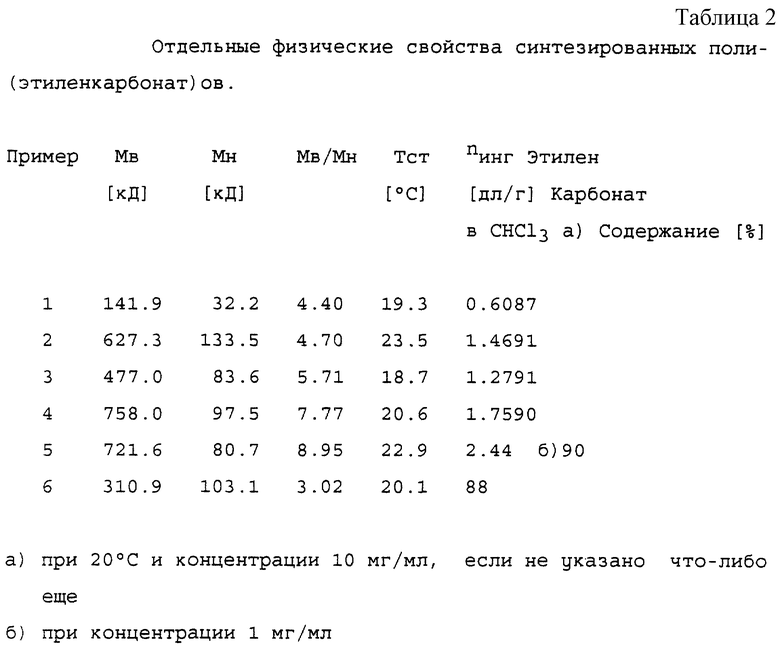
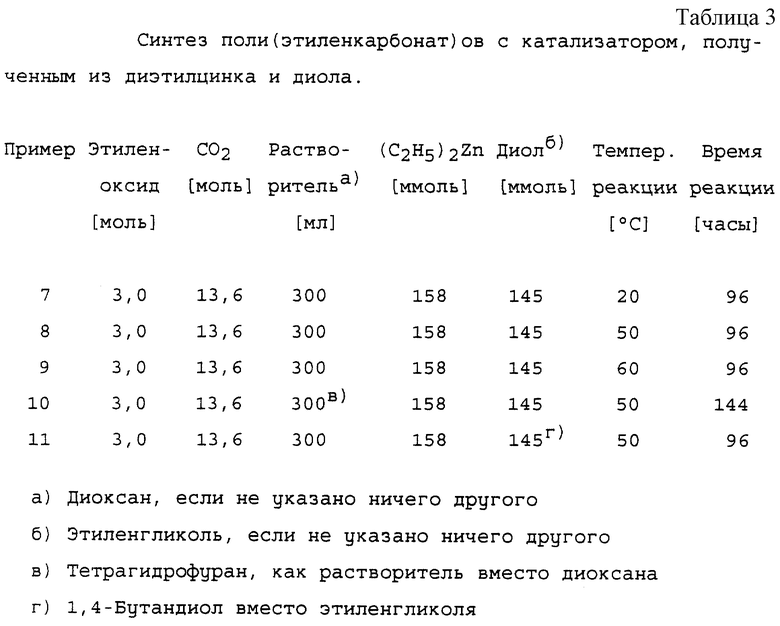
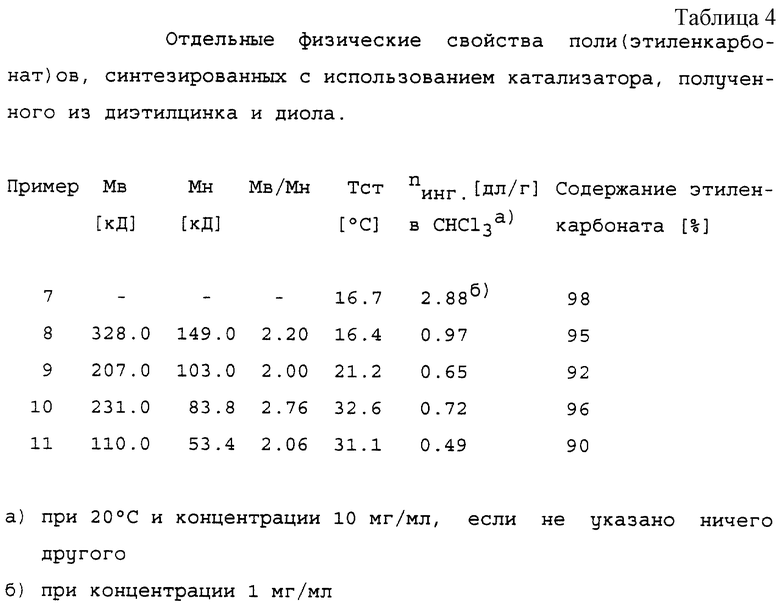
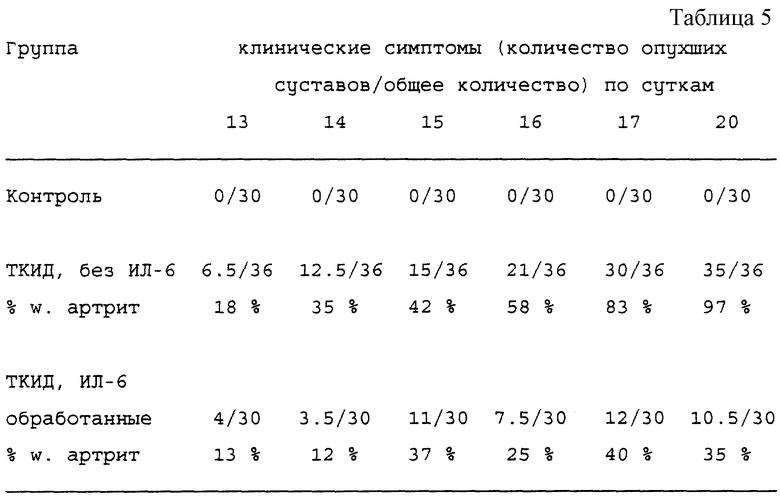


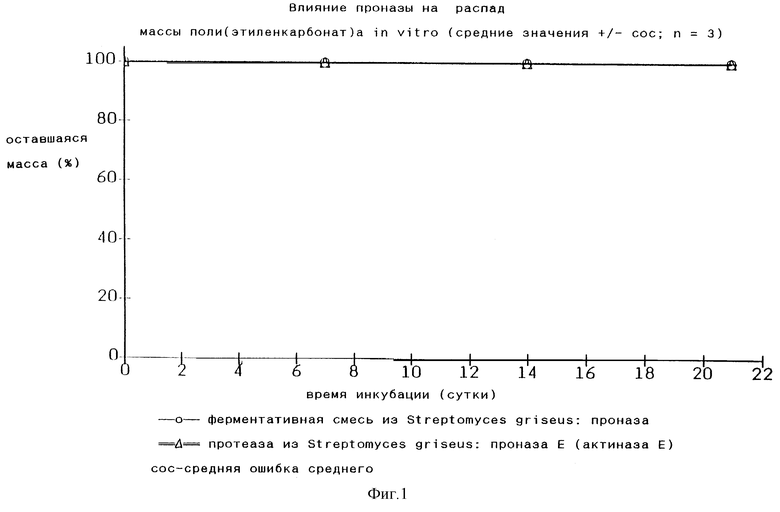
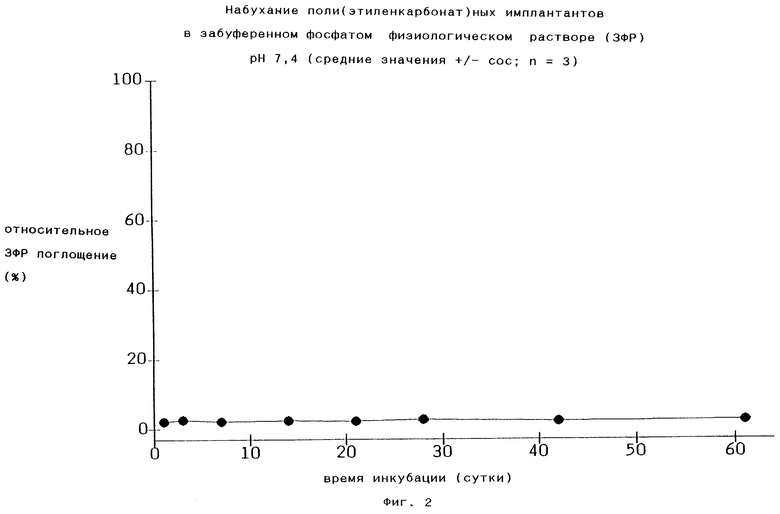
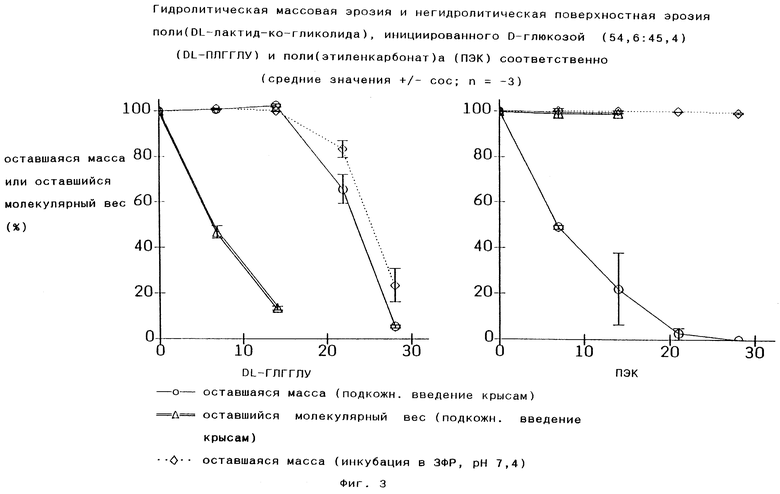
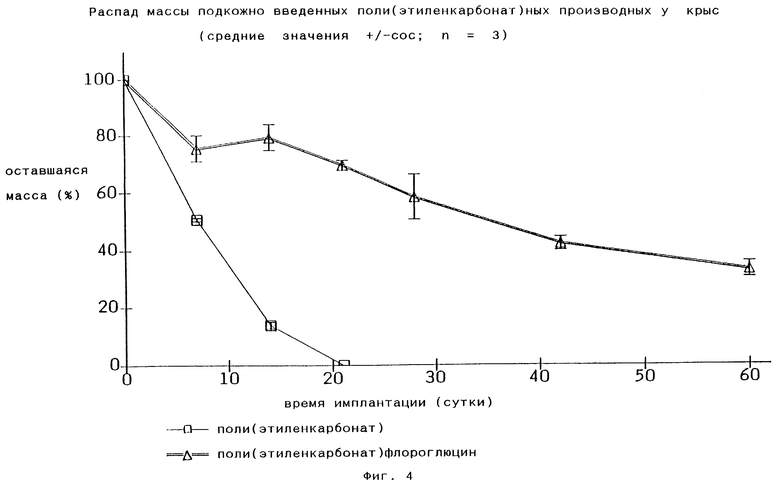
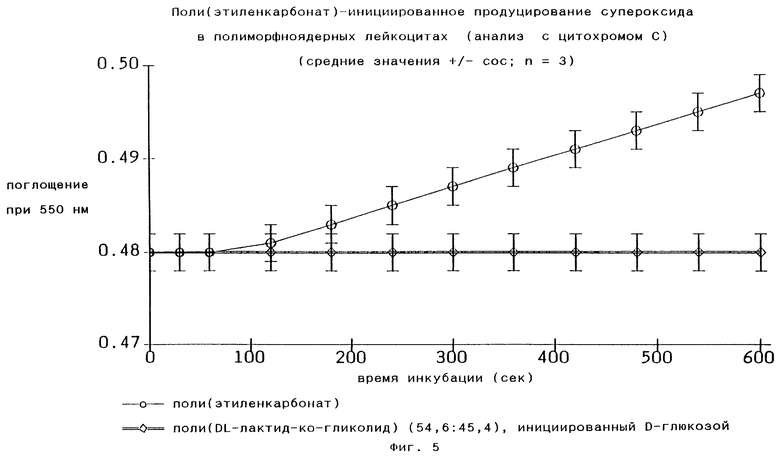
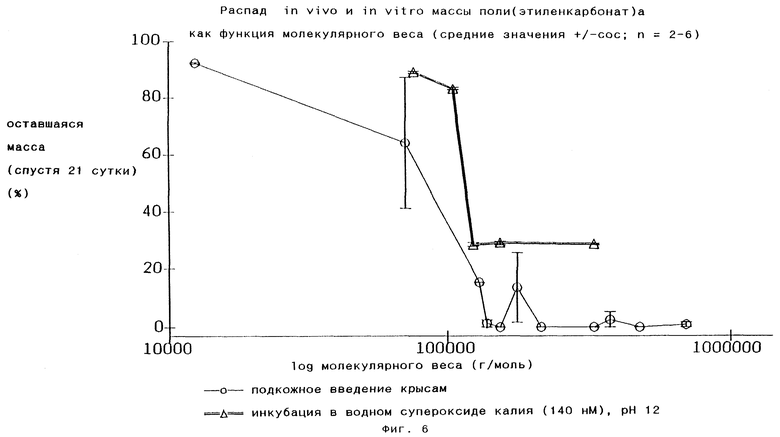
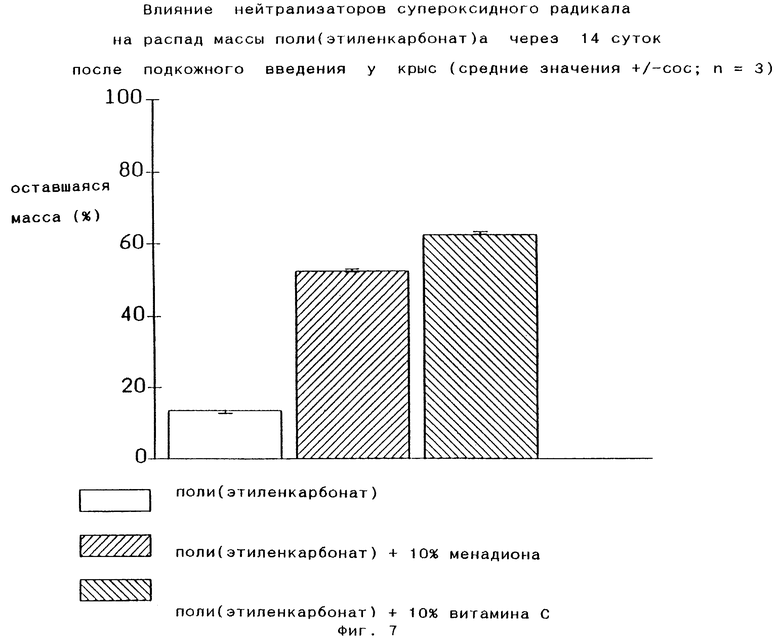


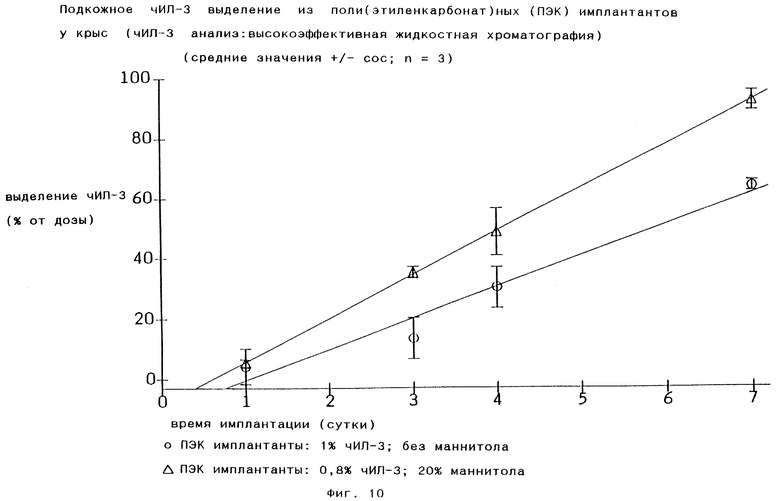
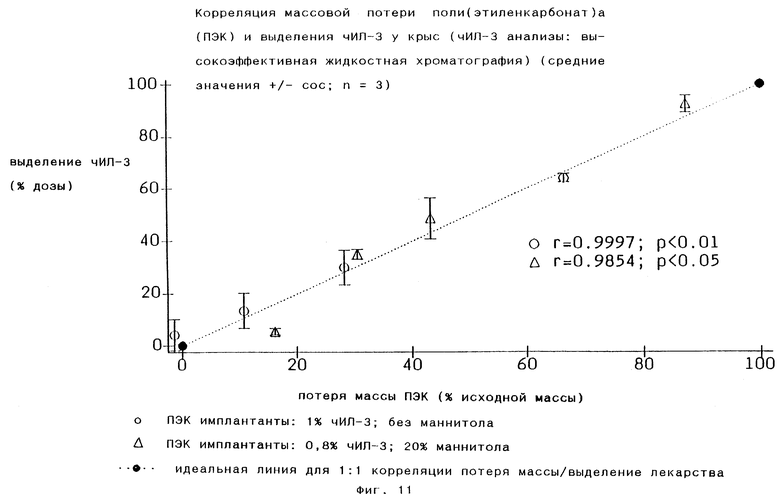
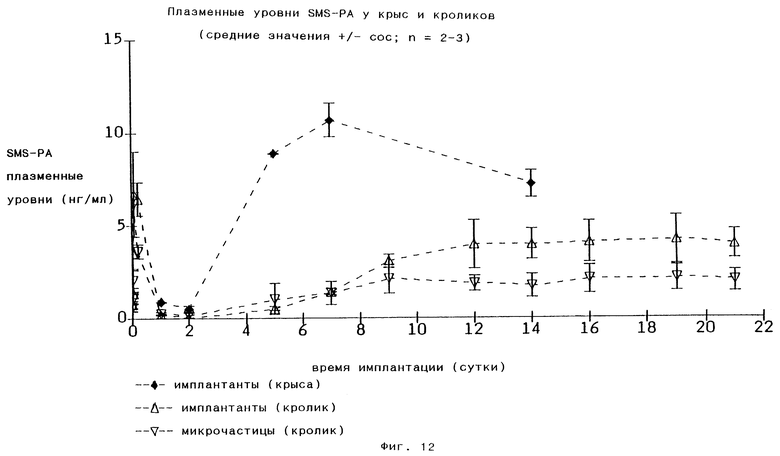
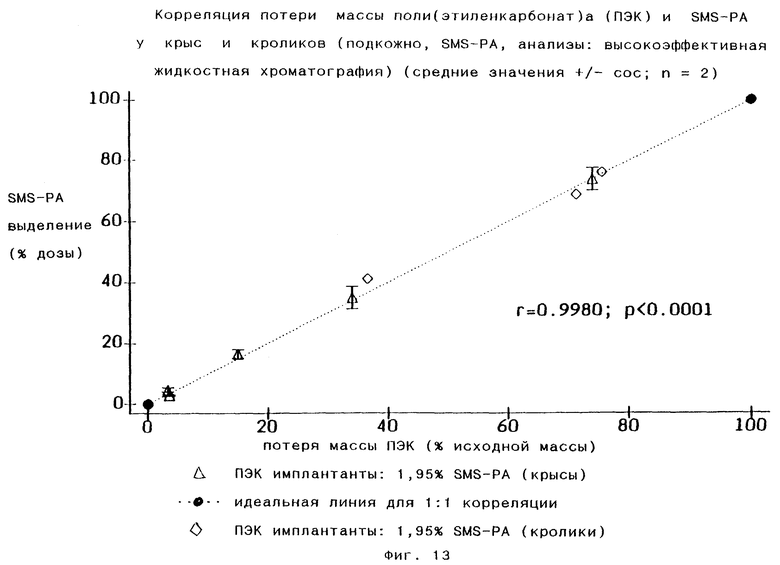
Изобретение относится к специфическим полимерным матрицам - поли(этиленкарбонат)ам, способу их получения, фармацевтическим составам на их основе, предназначенным для лечения заболеваний, таких как хронические воспалительные состояния. Поли(этиленкарбонат), способный к биологическому распаду, содержит звенья формулы -(С(O)-О-СН2-СН2-О-)- в количестве от 70 до 100 мол. %, имеет внутреннюю вязкость 0,4-4,0 дл/г, измеренную в хлороформе при 20oС и концентрации 1 г/дл, и температуру стеклования 15-50oС. Полимер получают полимеризацией этиленоксида и диоксида углерода при их мольном соотношении от 1:4 до 1:5 при 10-80oС в присутствии катализатора, полученного реакцией диэтилцинка и растворителя, выбранного из группы: вода, ацетон, диол, ди- или трифенол. Фармацевтическая композиция содержит поли(этиленкарбонат) и фармацевтически активное соединение. Полимер по изобретению обнаруживает негидролитическую поверхностную эрозию in vivo, он стабилен в течение нескольких часов в горячей воде без существенного снижения молекулярного веса. Фармакологические композиции могут использоваться для лечения хронических воспалительных состояний, вызываемых патогенами, демиелинизирующих заболеваний и острых воспалительных состояний. 4 с. и 21 з.п. ф-лы, 13 ил., 7 табл.
-(C(O)-O-CH2-CH2-O-)- (A)
и имеющий содержание этиленкарбоната от 70 до 100 мол.%, внутреннюю вязкость от 0,4 до 4,0 дл/г, измеренную в хлороформе при температуре 20oС при концентрации 1 г/дл, и температуру стеклования от 15 до 50oС.
-(CH2-CH2-O-)- (B)
9. Полимер по пп.1-7, отличающийся тем, что он содержит концевую гидроксильную группу.
| US 4665136 А, 12.05.1987 | |||
| US 3953383 А, 27.04.1976 | |||
| Способ получения пектина | 1975 |
|
SU535937A1 |
| Реферативный журнал Химия, №7С238, 1985 | |||
| Энциклопедия полимеров | |||
| Советская энциклопедия, 1974, т.2, с.933-935. | |||

