Изобретение относится к способу получения промежуточных соединений ароматических производных амидинов, которые обладают противосвертывающим действием, основанным на превосходном ингибировании активированного коагуляционного фактора Х (далее сокращенно как Fxa), и описаны в выложенной заявке (kokai) 5-208946 на патент Японии.
Предпосылки создания изобретения
В качестве промежуточных соединений ароматических производных амидинов, описанных в выложенной заявке (kokai) 5-208946 на патент Японии, известны соединения следующих формул (V), (Va) и (Vb) и их соли: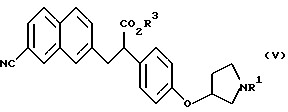
(где R1 представляет защитную группу для атома азота и R3 представляет атом водорода, аралкильную группу или алкильную группу, имеющую 1-6 углеродных атомов);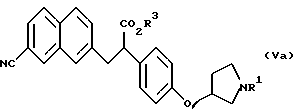
(где R1 и R3 имеют такие же значения, как описанные выше); и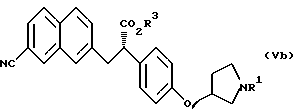
(где R1 и R3 имеют такие же значения, как описанные выше).
В упомянутой публикации описаны также способы получения указанных выше соединений.
Типичный способ получения указанных промежуточных соединений включает следующие стадии:
бромирование 7-метил-2-нафталинкарбонитрила с образованием 7-бромметил-2-нафталинкарбонитрила (первая стадия);
дальнейшее превращение 7-бромметил-2-нафталинкарбонитрила в фосфониевую соль и [(7-циано)-2-нафтил)метил]трифенилфосфонийбромид (вторая стадия);
синтез этил 2-[4-[[(3S)-1-(трет-бутоксикарбонил)-3-гидроксипирролидинил] окси] фенил] -2-оксоацетата с применением реакции Мицунобу (Mitsunobu) между этил-2-(4-гидроксифенил)-2-оксоацетатом и (3R)-1-(трет-бутоксикарбонил)-3-гидроксипирролидином (третья стадия);
введение полученных этил 2-[4-[[3S)-1-(третбутоксикарбонил)-3-гидроксипирролидинил] окси] фенил]-2-оксоацетата и [(7-циано)-2-нафтил)метил] трифенилфосфонийбромида в реакцию Виттига (четвертая стадия);
дальнейшее проведение каталитического гидрирования с образованием соединений, представленных формулой (V) или (Va) (пятая стадия); и
растворение соединений, представленных формулой (Va), в этаноле при нагревании, добавление к полученному раствору небольшого количества гидрида натрия и кристаллизация при перемешивании смеси при комнатной температуре с получением соединений, представленных формулой (Vb) (шестая стадия).
Однако вышеописанный известный способ имеет следующие недостатки:
1) бромирование на первой стадии проводят в тетрахлорметане, который предположительно является канцерогеном;
2) продукт первой стадии, т.е. 7-бромметил-2-нафталинкарбонитрил, вызывает раздражение кожи при выделении в виде кристаллов;
3) используются сравнительно дорогие реагенты диэтил азодикарбоксилат и 1,8-диазабицикло[5.4.0]-7-ундецен;
4) побочные продукты, образующиеся на третьей и четвертой стадиях, ведут себя как каталитические яды при каталитическом гидрировании на пятой стадии и, чтобы удалить побочные продукты, требуется очистка путем колоночной хроматографии на силикагеле;
5) моногидрат оксида палладия - сульфат бария, который является катализатором каталитического гидрирования, должен быть приготовлен непосредственно при использовании; и
6) выход шестой стадии низок, к тому же используется гидрид натрия, что вызывает проблему, связанную с безопасностью.
Короче, известный способ неудовлетворителен как промышленный способ.
Таким образом, задачей настоящего изобретения является создание промышленно применимого способа получения соединений, представленных формулами (V), (Va) и (Vb), и их солей путем применения безопасного недорогого и легко доступного исходного материала(ов) и вспомогательного материала(ов) и без стадии хроматографической очистки на силикагеле, а также создание промышленного способа получения промежуточных соединений производных ароматических амидинов, описанных в выложенной заявке (kokai) 5-208946 на патент Японии.
Раскрытие сущности изобретения
Учитывая вышесказанное, были проведены серьезные исследования и было найдено
что галогенирование на первой стадии может быть эффективно осуществлено в алкилнитрильном растворителе, что позволяет проводить реакцию на следующей стадии без выделения продукта;
что применение производного пирролидинилоксифенилуксусной кислоты, полученного конденсацией 4-гидроксифенилуксусных кислот и сульфонилоксипирролидинов, в качестве исходного материала дает соединение, представленное формулой (V) или (Va), без необходимости реакции образования фосфониевой соли и каталитического гидрирования, а также без необходимости получения дорогого реагента или реагента, которое требуется в момент использования; и
что добавление основания к диастереомерной смеси соединений, представленных формулой (Va), облегчает образование соединений, представленных формулой (Vb). На основании этих открытий и было создано настоящее изобретение.
Настоящее изобретение в общем представляют схемы реакций I и II (см. в конце описания).
Таким образом, в соответствии с настоящим изобретением предположен способ получения соединения, представленного формулой (III) или (IIIa), или его соли реакцией соединения, представленного формулой (I) или (Iа), и соединения, представленного формулой (II), в присутствии основания.
В соответствии с настоящим изобретением предложен также способ получения соединения, представленного формулой (V) (или (Va)), или его солей реакцией соединения, представленного формулой (III) (или (IIIa)), или его солью и соединения, представленного формулой (IV), в присутствии основания.
В соответствии с настоящим изобретением также предложен способ получения соединения, представленного формулой (Vb), реакцией соединения, представленного формулой (Va), и основанием.
Далее, в соответствии с настоящим изобретением предложен способ получения соединения, представленного формулой (IVa):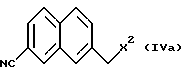
(где X2 представляет атом галогена), галогенированием соединения, представленного формулой (VII) в алкилнитрильном растворителе.
Из соединений, показанных на вышеописанных схемах, некоторые являются новыми соединениями, которые впервые получены в настоящем изобретении. Поэтому настоящее изобретение касается также таких новых соединений, полезных в качестве промежуточных соединений в синтезе.
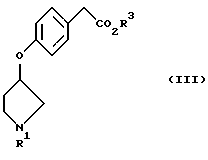
Кроме того, в соответствии с настоящим изобретением предложены соединения, представленные формулой (III):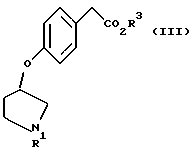
(где R1 и R3 имеют такие же значения, как описанные выше), и их соли.
В соответствии с настоящим изобретением предложены также соединения, представленные формулой (IIIa):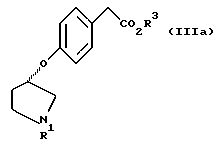
(где R1 и R3 имеют такие же значения, как описанные выше), и их соли.
Еще в соответствии с настоящим изобретением предлагаются соединения, представленные формулой (Vc):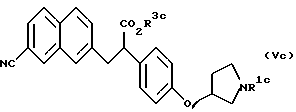
(где R1c представляет трет-бутоксикарбонильную группу и R3c представляет атом водорода, аралкильную группу или алкильную группу, имеющую 1-6 углеродных атомов (отличную от этильной группы)), и их соли.
Далее, в соответствии с настоящим изобретением предложены соединения, представленные формулой (Vd):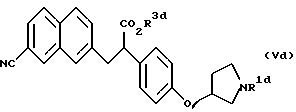
(где R1d представляет бензильную группу и R3d представляет атом водорода, аралкильную группу или алкильную группу, имеющую 1-6 углеродных атомов), и их соли.
Предпочтительный способ осуществления изобретения
Далее настоящее изобретение описано более подробно. В первую очередь описаны заместители соединений по настоящему изобретению.
R1 представляет защитную группу для атома азота. В качестве защитной группы могут быть использованы обычно применяемые защитные группы. Примеры включают трет-бутоксикарбонильную группу, бензилоксикарбонильную группу, п-нитробензилоксикарбонильную группу, бензильную группу, формильную группу, ацетильную группу и трифенилметильную группу. В настоящем изобретении предпочтительной является трет-бутоксикарбонильная группа или бензильная группа.
R2 представляет метансульфонильную группу или п-толуолсульфонильную группу.
Алкильная группа, имеющая 1-6 углеродных атомов, представленная символом R3, может быть неразветвленной, разветвленной или циклической и ее примеры включают метильную группу, этильную группу, пропильную группу, изопропильную группу, бутильную группу, изобутильную группу, трет-бутильную группу, пентильную группу, гексильную группу, циклопропильную группу, циклобутильную группу, циклопентильную группу и циклогексильную группу. Аралкильная группа представляет собой группу, образованную алкильной группой, имеющей 1-6 углеродных атомов, и арильной группой, и ее примеры включают бензильную группу и нафтилметильную группу. Группа R3 в настоящем изобретении предпочтительно представляет собой алкильную группу, имеющую 1-6 углеродных атомов, а более предпочтительно метильную группу или этильную группу.
X1 представляет уходящую группу. В качестве уходящей группы может быть использована любая такая группа, которую обычно применяют, и ее примеры включают атом галогена, метансульфонилоксигруппу и п-толуолсульфонилоксигруппу. При использовании в настоящем изобретении примеры атома галогена включают атом фтора, атом хлора, атом брома и атом иода. Из них предпочтительным является атом брома.
X2 представляет атом галогена. Примеры атома галогена включают атом фтора, атом хлора, атом брома и атом иода. Из них предпочтительным является атом брома.
Соединения, представленные формулой (I) или формулой (Iа), соединения, представленные формулой (II), и соединения, представленные формулой (VII), используемые в настоящем изобретении, являются легко доступными известными соединениями или соединениями, которые можно легко получить описанными в литературе способами.
Соединения, представленные формулой (I), являются известными соединениями, и оптически активный (3R)-1-(трет-бутоксикарбонил)-3-метансульфонилоксипирролидин (смотри выложенную заявку (kokai) 2-28180 на патент Японии) и оптически активный (3R)-1-(трет-бутоксикарбонил)-3-п-толуолсульфонилоксипирролидин (смотри WO 9200295), представленные формулой (Iа), также являются известными соединениями.
Из соединений, представленных формулой (II), известными являются метил п-гидроксифенилацетат и этил п-гидроксифенилацетат. Другие сложные алкиловые эфиры п-гидроксифенилуксусной кислоты могут быть легко получены конденсацией соответствующего спирта и легко доступной п-гидроксифенилуксусной кислоты.
7-Галогенметил-2-нафтилинкарбонитрил как пример соединения формулы (IV) также является известным соединением (выложенные заявки (kokai) 5-20008946 и 7-17937 на патент Японии).
Далее способ получения по настоящему изобретению описан более подробно.
(Стадия А) Способ получения соединения, представленного формулой (III) или формулой (IIIa), или его солей.
Для получения соединения, представленного формулой (III) или формулой (IIIa), или его солей соединение, представленное формулой (I) или формулой (Iа), подвергают взаимодействию с соединением, представленным формулой (II), в присутствии основания и, необязательно, катализатора.
Нет особого ограничения в отношении используемого на этой стадии растворителя, пока он не оказывает вредного влияния на реакцию. Примеры растворителя включают органические растворители, такие, как апротонные полярные растворители, простые эфиры, ароматические углеводороды и спирты, смеси органических растворителей и смеси органических растворителей и воды.
Примеры апротонных полярных растворителей включают N,N-диметилформамид, N,N-диметилацетамид, диметилсульфоксид и ацетонитрил. Примеры простых эфиров включают тетрагидрофуран, диоксан, диметоксиэтан, диметиловый эфир диэтиленгликоля и диметиловый эфир триэтиленгликоля. Примеры ароматических углеводородов включают бензол, толуол и ксилол. Примеры спиртов включают метанол и этанол. Из указанных растворителей предпочтительно используют апротонные полярные растворители или ароматические углеводороды, а более предпочтительно -N,N-диметилформамид или толуол.
Нет особого ограничения в отношении основания, пока оно не оказывает вредного влияния на реакцию, причем можно использовать слабое или сильное основание. Примеры сильного основания включают гидрид щелочного металла, такой, как гидрид натрия или гидрид лития; гидрид щелочноземельного металла, такой, как гидрид кальция; алкоксид щелочного металла, такой, как метоксид натрия, метоксид лития, этоксид натрия, этоксид лития, трет-бутоксид натрия или трет-бутоксид калия; гидроксид щелочного металла, такой, как гидроксид натрия или гидроксид калия; и карбонат щелочного металла, такой, как карбонат натрия или карбонат калия. Сильные основания являются предпочтительными. В частности, предпочтительным является гидрид щелочного металла, а более предпочтительным гидрид натрия.
Примеры катализатора, который используют в настоящем изобретении, включают катализаторы межфазного переноса и молекулярные сита. Примеры катализаторов межфазного переноса включают олеофильные соли четвертичного аммония, такие, как тетра(н-бутил)аммонийбромид, тетра(н-бутил)аммонийхлорид, тетраэтиламмонийбромид, тетра(н-бутил)аммонийсероводород, триэтилбензиламмонийбромид или триэтилбензиламмонийхлорид, и краун-эфиры, такие, как 18-краун-6,15-краун-5. Катализаторы межфазного переноса являются предпочтительными. В частности, являются предпочтительными олеофильные соли четвертичного аммония, а более предпочтительным является тетра(н-бутил)аммонийбромид. Введение катализаторов увеличивает выход соединений, представленных формулой (III) или формулой (IIIa) или их солей.
Нет особого ограничения в отношении температуры реакции, пока она не превышает температуру кипения растворителя. Реакцию обычно осуществляют в температурном интервале от 0oС до примерно температуры кипения используемого растворителя, предпочтительно при 60oС-110oС. Время реакции изменяется в соответствии с температурой реакции и обычно реакцию проводят в течение от 15 минут до одного дня, предпочтительно 4 часов или менее.
(Стадия В) Способ получения соединения, представленного формулой (IV).
Соединение, представленное формулой (IV), может быть получено известным способом и предпочтительно его получают галогенированием соединения, представленного формулой (VII), в алкилнитрильном растворителе. В процессе галогенирования может быть введен инициатор радикалов.
Нет особого ограничения в отношении алкилнитрильного растворителя, пока он не оказывает вредного влияния на реакцию, и можно использовать неразветвленные или разветвленные C2-C7 алкилнитрилы. Примеры неразветвленных или разветвленных С2-С7 алкилнитрилов включают ацетонитрил, пропионитрил, н-бутиронитрил, изобутиронитрил, валеронитрил, гексаннитрил и гептаннитрил. Из них предпочтительными являются неразветвленные или разветвленные алкилнитрилы, имеющие 2-4 углеродных атома, такие, как ацетонитрил, пропионитрил, н-бутиронитрил или изобутиронитрил, а более предпочтительным является ацетонитрил.
Нет особого ограничения в отношении инициатора радикалов, пока он не оказывает вредного влияния на реакцию, и его примеры включают пероксиды, такие, как дибензоилпероксид, или азосоединения, такие, как азабисизобутиронитрил. Вместо введения инициатора радикалов, могут быть осуществлены такие операции, как облучение светом или нагревание. Из разнообразных инициаторов радикалов предпочтительными являются азосоединения, причем особенно предпочтительным является 2,2'-азобисизобутиронитрил.
Галогенирование может быть осуществлено введением галогенирующего агента. Нет особого ограничения в отношении галогенирующего агента, пока он не оказывает вредного влияния на реакцию. Его примеры включают сульфурилгалогениды и N-галогеноимиды. Из них предпочтительными являются N-галогеноимиды, причем более предпочтительным является N-бромсукцинимид.
Нет особого ограничения в отношении температуры реакции, пока она не превышает температуру кипения растворителя. Реакцию обычно осуществляют при 40oС-120oС, предпочтительно при примерно 80oС. Время реакции зависит от температуры реакции и обычно реакцию проводят в течение от одного часа до одного дня, предпочтительно 1-4 часа.
(Стадия С) Способ получения соединения, представленного формулой (V) или формулой (Va), или его солей.
Для получения соединения, представленного формулой (V) или формулой (Va), или его соли соединение, представленное формулой (III) или формулой (IIIa), подвергают взаимодействию с соединением, представленным формулой (IV), в присутствии основания.
Нет особого ограничения в отношении используемого на этой стадии растворителя, пока он не оказывает вредного влияния на реакцию. Примеры растворителя включают органические растворители, такие, как апротонные полярные растворители, простые эфиры, сложные эфиры, ароматические углеводороды и спирты, смеси органических растворителей и смеси органических растворителей и воды.
Примеры апротонных полярных растворителей включают N,N-диметилформамид, N,N-диметилацетамид, диметилсульфоксид и ацетонитрил. Примеры простых эфиров включают тетрагидрофуран, диоксан, диметоксиэтан, диметиловый эфир диэтиленгликоля и диметиловый эфир триэтиленгликоля. Примеры сложных эфиров включают метилацетат, этилацетат, метилпропионат и этилпропионат. Примеры ароматических углеводородов включают бензол, толуол и ксилол. Примеры спиртов включают метанол и этанол. Предпочтительно используют смеси органических растворителей. Из них предпочтительными являются смеси апротонного полярного растворителя и ароматического углеводорода, причем более предпочтительной является смесь, содержащая N, N-диметилформамид и толуол.
Нет особого ограничения в отношении основания, пока оно не оказывает вредного влияния на реакцию, причем можно использовать слабое или сильное основание. Примеры сильного основания включают гидрид щелочного металла, такой, как гидрид натрия или гидрид лития; гидрид щелочноземельного металла, такой, как гидрид кальция; алкоксид щелочного металла, такой, как метоксид натрия, метоксид лития, этоксид натрия, этоксид лития, трет-бутоксид натрия или трет-бутоксид калия; гидроксид щелочного металла, такой, как гидроксид натрия или гидроксид калия; и карбонат щелочного металла, такой, как карбонат натрия или карбонат калия. Сильные основания являются предпочтительными. В частности, предпочтительным является гидрид щелочного металла, а более предпочтительным гидрид натрия.
Нет особого ограничения в отношении температуры реакции, пока она не превышает температуру кипения растворителя, однако реакцию предпочтительно проводят при сравнительно низкой температуре, чтобы подавить побочную реакцию. Реакцию обычно осуществляют в температурном интервале от -10oС до комнатной температуры, предпочтительно при 60o-110oС. Время реакции изменяется в соответствии с температурой реакции и обычно реакцию проводят в течение от одного часа до одного дня, предпочтительно 3-12 часов.
Полученные описанным образом производные 2-фенил-3-нафтил-пропионовой кислоты, которые являются соединениями, представленными формулой (V) или (Va) являются важными промежуточными соединениями производных ароматических амидинов, описанными в выложенной заявке (kokai) 5-208946 на патент Японии.
Стадии А и С, стадии В и С или стадии А, В и С могут быть проведены непрерывно. Коротко говоря, соединение, представленное формулой (III) или формулой (IIIa), или его соль, которое(ую) получают на стадии А, и соединение, представленное формулой (IV), или его соль, которое(ую) получают на стадии В, можно использовать на последующей стадии С без выделения соединений на соответствующих стадиях.
Далее описан пример осуществления последовательных стадий. Сначала, на стадии А, получают соединение, представленное формулой (III) или формулой (IIIa), или его соль, реакцией в ароматическом углеводородном растворителе в присутствии сильного основания при использовании катализатора межфазного переноса. Затем, на стадии В, соединение, представленное формулой (IV), полученное реакцией в алкилнитриле, экстрагируют ароматическим углеводородом. Далее соединение, представленное формулой (III) или формулой (IIIa), или его соль и соединение, представленное формулой (IV), взаимодействуют друг с другом, без выделения этих соединений, в смеси растворителей, содержащей ароматический углеводородный растворитель и апротонный полярный растворитель, в присутствии сильного основания и в результате получают соединение, представленное формулой (V) или формулой (Va), или его соль.
Последовательные стадии, не требующие такой операции, как выделение, являются предпочтительными в промышленном способе. В частности, поскольку выделенные кристаллы 7-бромметил-2-нафталинкарбонитрила вызывают раздражение кожи, стадию В и стадию С предпочтительно выполняют последовательно.
(Стадия D) Способ получения оптически активного соединения, представленного формулой (Vb).
Для получения соединения, представленного формулой (Vb), из соединения, представленного формулой (Va), соединение, представленное формулой (Va), может быть подвергнуто взаимодействию с основанием.
Коротко, соединение, представленное формулой (Va), которое является смесью R-диастереомера и S-диастереомера, подвергают взаимодействию с основанием и в результате получают соединение, представленное формулой (Vb), которое является S-диастереомером.
В частности, R-диастереомер растворяют в растворителе, подходящем для инициирования кристаллизации S-диастереомера, и в растворенном состоянии подвергают взаимодействию с основанием для осуществления превращения R-диастереомера в S-диастереомер. Затем целевой S-диастереомер кристаллизуют, используя разницу в растворимости между R-диастереомером и S-диастереомером.
В этом случае нет особого ограничения в отношении растворителя, пока он не оказывал вредного влияния на реакцию, и может быть использован растворитель, позволяющий проводить кристаллизацию соединений, представленных формулой (Vb). В частности, можно использовать протонные растворители, включающие воду и спирты. Эти растворители можно использовать в отдельности или в сочетании. Протонные растворители можно смешивать с апротонными полярными растворителями, простыми эфирами, углеводородами или их смесями.
Примеры спиртов включают метанол и этанол. Примеры апротонных полярных растворителей включают N,N-диметилформамид, диметилсульфоксид и ацетонитрил. Примеры простых эфиров включают изопропиловый эфир, тетрагидрофуран, диоксан, диметоксиэтан, диметиловый эфир диэтиленгликоля и диметиловый эфир триэтиленгликоля. Примеры углеводородов включают бензол, толуол, ксилол, н-гексан и н-пентан.
Нет особого ограничения в отношении основания, пока оно не оказывает вредного влияния на реакцию. Примеры сильного основания включают алкоксиды щелочных металлов, такие, как метоксид натрия, метоксид лития, этоксид натрия, этоксид лития, трет-бутоксид натрия или трет-бутоксид калия; амиды щелочных металлов, такие, как амиды натрия; гидроксиды щелочных металллов, такие, как гидроксид натрия или гидроксид калия; и карбонаты щелочных металлов, такие как карбонат натрия или карбонат калия. Предпочтительно используют сильные основания. Из них предпочтительными являются алкоксиды щелочных металлов, причем более предпочтительным является этоксид натрия.
Количество основания не ограничено, пока оно не оказывает вредного влияния на реакцию, и оно предпочтительно составляет 10-30% мол.-эквивалент от соединения, представленного формулой (Va).
Нет особого ограничения в отношении температуры реакции, пока она не превышает температуру кипения растворителя. Реакцию обычно осуществляют в температурном интервале от -10oС до комнатной температуры, предпочтительно в интервале от 10oС до комнатной температуры. Время реакции изменяется в соответствии с температурой реакции и обычно реакцию проводят в течение от 30 минут до нескольких дней, предпочтительно 20 часов или менее.
Полученные описанным образом оптически активные производные 2-фенил-3-нафталинпропионовой кислоты, которые являются соединениями, представленными формулой (Vb), являются важными промежуточными соединениями производных ароматических амидинов, описанных в указанном ссылочном материале.
Производные ароматических амидинов или их соли могут быть получены из соединений, представленных формулой (V), (Va) или (Vb), способом, описанным в выложенной заявке (kokai) 5-208946 на патент Японии.
ПРИМЕРЫ
Далее настоящее изобретение подробно описано в примерах, которые не следует считать как ограничивающие изобретение.
Пример 1. Способ получения (3R)-1-бензил-3-метансульфонилоксипирролидина
(3R)-1-Бензил-3-пирролидинол (10,0 г, 56 ммоль) и триэтиламин (6,6 г, 65 ммоль) растворяли в толуоле (100 мл) и полученный раствор охлаждали до 5oС. В этот раствор добавляли метансульфонилхлорид (7,1 г, 62 ммоль) по каплям в течение 10 минут при 5oС. После перемешивания смеси в течение 30 минут добавляли к ней толуол (100 мл), после чего полученный раствор нагревали до комнатной температуры и перемешивали в течение 1,5 часов. Реакционную смесь промывали насыщенным водным раствором бикарбоната натрия (200 мл) и затем два раза водой (100 мл). После концентрирования полученного органического слоя при пониженном давлении остаток подвергали колоночной хроматографии с получением 9,8 г указанного в заголовке соединения (выход: 68%).
Спектр ядерного магнитного резонанса (СDСl3) δ:
2,19 (1Н, м), 2,30 (1Н, м), 2,49 (1Н, м), 2,76-2,89 (3Н, м), 2,99 (3Н, с), 3,64 (2Н, дд, J=12,9, 12,9 Гц), 5,20 (1Н, м), 7,25-7,35 (5Н, м)
Элементный анализ C12H17NSO3:
Вычислено: С, 56,45; Н, 6,71; N, 5,49.
Найдено: С, 55,70; Н, 6,33; N, 5,48.
FAB MC (m/z): 256(M++1).
Спектр поглощения в инфракрасной области vmax (KBr) см-1: 3028, 2944, 2804, 1496, 1454, 1356, 1170, 1146, 966.
Угол вращения [α]
Пример 2. Способ получения этилового эфира 2-[4-[(3S)-1-(трет-бутоксикарбонил)-3-пирролидинил]окси]фенилуксусной кислоты
Этиловый эфир п-гидроксифенилуксусной кислоты (39,6 г, 0,22 моль) растворяли в диметилформамиде (500 мл). К смеси при комнатной температуре прибавляли раствор 60%-ного гидрида натрия (8,8 г, 0,22 моль). Через сорок минут прибавляли (3R)-1-(трет-бутоксикарбонил)-3-метансульфонилоксипирролидин (53,1 г, 0,2 моль) и смесь нагревали в течение 15 минут при внутренней температуре 110oС. Полученную смесь охлаждали до комнатной температуры и концентрировали выпариванием растворителя при пониженном давлении. Для растворения остатка к нему добавляли этиловый эфир уксусной кислоты (500 мл). Полученный раствор промывали 4 раза 10%-ным водным раствором гидроксида калия (100 мл). Органический слой концентрировали при пониженном давлении и остаток подвергали колоночной хроматографии на силикагеле с получением 39,8 г указанного в заголовке соединения (выход: 57%).
Температура плавления: 39-40oС.
Спектр ядерного магнитного резонанса (СDСl3) δ:
1,25 (3Н, т, J=6,9 Гц), 1,47 (9Н, с), 2,08-2,17 (2Н, м), 3,46-3,63 (4Н, м), 3,54 (2Н, с), 4,14 (2Н, kв, J=6,9 Гц), 4,86 (1Н, м), 6,83 (2Н, д, J=8,3 Гц), 7,24 (2Н, д, J=8,3 Гц).
Элементный анализ C19H26NO5:
Вычислено: С, 65,31; Н, 7,79; N, 4,01.
Найдено: С, 65,20; Н, 7,59; N, 3,76.
MC(m/z): 349 (М+).
Спектр поглощения в инфракрасной области vmax(КВr)см-1:
2984, 1736, 1694, 1514, 1482, 1410, 1370, 1168, 1116.
Угол вращения [α]
Пример 3. Способ получения этилового эфира 2-[4-[(3S)-1-(трет-бутоксикарбонил)-3-пирролидинил]окси]фенилуксусной кислоты
Этиловый эфир п-гидроксифенилуксусной кислоты (39,6 г, 0,22 моль) растворяли в диметилформамиде (500 мл) и к смеси прибавляли при комнатной температуре раствор 60%-ного гидрида натрия (8,8 г, 0,22 моль). Через сорок минут прибавляли (3R)-1-(трет-бутоксикарбонил)-3-метансульфонилоксипирролидин (53,1 г, 0,2 моль) и смесь нагревали в течение 15 минут при внутренней температуре 110oС. Полученную смесь охлаждали до комнатной температуры и концентрировали выпариванием растворителя при пониженном давлении. Для растворения остатка к нему добавляли этиловый эфир уксусной кислоты (500 мл) и полученный раствор промывали 4 раза 10%-ным водным раствором гидроксида калия (100 мл). Анализ путем обращенно-фазовой хроматографии с использованием продукта, полученного в примере 2, в качестве стандартной пробы показал, что получили 43,7 г указанного в заголовке соединения, имеющего чистоту 70% (выход: 62%).
Пример 4. Способ получения этилового эфира 2-[4-[(3S)-1-(трет-бутоксикарбонил)-3-пирролидинил]окси]фенилуксусной кислоты
Повторяли процедуры примера 3 в диметилсульфоксиде (25 мл), за исключением того, что использовали этиловый эфир п-гидроксифенилуксусной кислоты (1,8 г, 10 ммоль), (3R)-1-(трет-бутоксикарбонил)-3-метансульфонилоксипирролидин (2,5 г, 10 ммоль) и 60%-ный гидрид натрия (440 мг, 10 ммоль). Анализ путем обращенно-фазовой хроматографии с использованием продукта, полученного в примере 2, в качестве стандартной пробы показал, что получили 1,8 г указанного в заголовке соединения (выход: 52%).
Пример 5. Способ получения этилового эфира 2-[4-[(3S)-1-(трет-бутоксикарбонил)-3-пирролидинил]окси]фенилуксусной кислоты
Повторяли процедуры примера 3 в диметилформамиде (25 мл), за исключением того, что использовали этиловый эфир п-гидроксифенилуксусной кислоты (1,8 г, 10 ммоль), (3R)-1-(трет-бутоксикарбонил)-3-метансульфонилоксипирролидин (2,5 г, 10 ммоль) и этоксид натрия (0,7 г, 10 ммоль). Анализ путем обращенно-фазовой хроматографии с использованием продукта, полученного в примере 2, в качестве стандартной пробы показал, что получили 1,7 г указанного в заголовке соединения (выход: 50%).
Пример 6. Способ получения этилового эфира 2-[4-[(3S)-1-(трет-бутоксикарбонил)-3-пирролидинил]окси]фенилуксусной кислоты
Повторяли процедуры примера 3 в диметилформамиде (25 мл), за исключением того, что использовали этиловый эфир п-гидроксифенилуксусной кислоты (2,0 г, 11 ммоль), (3R)-1-(трет-бутоксикарбонил)-3-метансульфонилоксипирролидин (2,5 г, 10 ммоль) и трет-бутоксид калия (1,2 г, 11 ммоль). Анализ путем обращенно-фазовой хроматографии с использованием продукта, полученного в примере 2, в качестве стандартной пробы показал, что получили 2,0 г указанного в заголовках соединения (выход: 58%).
Пример 7. Способ получения этилового эфира 2-[4-[(3S)-1-(трет-бутоксикарбонил)-3-пирролидинил]окси]фенилуксусной кислоты
Повторяли процедуры примера 3 в диметилформамиде (68 мл), за исключением того, что использовали этиловый эфир п-гидроксифенилуксусной кислоты (4,0 г, 22 ммоль), (3R)-1-(трет-бутоксикарбонил)-3-п-толуолсульфонилоксипирролидин (6,8 г, 20 ммоль) и 60%-ный гидрид натрия (880 мг, 22 ммоль). Анализ путем обращенно-фазовой хроматографии с использованием продукта, полученного в примере 2, в качестве стандартной пробы показал, что получили 4,4 г указанного в заголовке соединения (выход: 64%).
Пример 8. Способ получения этилового эфира 2-[4-[(3S)-1-(трет-бутоксикарбонил)-3-пирролидинил]окси]фенилуксусной кислоты
Этиловый эфир п-гидроксифенилуксусной кислоты (1,8 г, 10 ммоль) растворяли в этаноле (25 мл) и к смеси добавляли при комнатной температуре этоксид натрия (0,7 г, 10 ммоль). Спустя сорок минут прибавляли (3R)-1-(трет-бутоксикарбонил)-3-метансульфонилоксипирролидин (2,5 г, 10 ммоль) и смесь кипятили с обратным холодильником в течение 3 часов. Полученную смесь охлаждали до комнатной температуры и затем концентрировали для удаления растворителя при пониженном давлении. Для растворения остатка к нему добавляли этиловый эфир уксусной кислоты (30 мл) и полученный раствор промывали 4 раза 10%-ным водным раствором гидроксида калия (20 мл). Анализ путем обращенно-фазовой хроматографии с использованием продукта, полученного в примере 2, в качестве стандартной пробы показал, что получили 0,9 г указанного в заголовке соединения (выход: 25%).
Пример 9. Способ получения этилового эфира 2-[4-[(3S)-1-(трет-бутоксикарбонил)-3-пирролидинил]окси]фенилуксусной кислоты
Повторяли процедуры примера 8 в ацетонитриле (26 мл), за исключением того, что использовали этиловый эфир п-гидроксифенилуксусной кислоты (2,0 г, 11 ммоль), (3R)-1-(трет-бутоксикарбонил)-3-метансульфонилоксипирролидин (2,5 г, 10 ммоль) и 60%-ный гидрид натрия (440 мг, 11 ммоль). Анализ путем обращенно-фазовой хроматографии с использованием продукта, полученного в примере 2, в качестве стандартной пробы показал, что получили 2,2 г указанного в заголовке соединения (выход: 64%).
Пример 10. Способ получения этилового эфира 2-[4-[(3S)-1-(трет-бутоксикарбонил)-3-пирролидинил]окси]фенилуксусной кислоты
Этиловый эфир п-гидроксифенилуксусной кислоты (39,6 г, 0,22 моль) растворяли в толуоле (530 мл) и затем к смеси добавляли при комнатной температуре 60%-ный гидрид натрия (8,8 г, 0,22 моль). После нагревания смеси при внутренней температуре 45oС в течение одного часа, добавляли (3R)-1-(трет-бутоксикарбонил)-3-метансульфонилоксипирролидин (53,1 г, 0,2 моль) и четырехнормальный бутиламмонийбромид (19,3 г, 60 ммоль). Полученную смесь нагревали при внутренней температуре 80oС в течение 3 часов и затем охлаждали до комнатной температуре. Смесь промывали 3 раза 10%-ным водным раствором гидроксида калия (106 мл). Анализ путем обращенно-фазовой хроматографии с использованием продукта, полученного в примере 2, в качестве стандартной пробы показал, что получили 49,0 г указанного в заголовке соединения, имеющего чистоту 79% (выход: 70%).
Пример 11. Способ получения 7-бромметил-2-нафталинкарбонитрила
К 7-метил-2-нафталинкарбонитрилу (10,0 г, 60 ммоль) прибавляли при комнатной температуре 2,2'-азобисизобутиронитрил (982 мг, 6 ммоль) и ацетонитрил (100 мл). К полученной смеси прибавляли N-бромсукцинимид (10,6, 60 ммоль) и смесь нагревали с обратным холодильником в течение 2 часов. Полученную смесь охлаждали до комнатной температуры. Для экстрагирования к смеси добавляли воду (100 мл) и толуол (100 мл). Полученный органический слой промывали два раза водой (100 мл). После концентрирования органического слоя при пониженном давлении, остаток подвергали колоночной хроматографии на силикагеле с получением 11,4 г указанного в заголовке соединения (выход: 78%). Данные измерения приборами соответствовали данным, описанным в J. Med. Chem., 1991, 3105.
Пример 12. Способ получения 7-бромметил-2-нафталинкарбонитрила
К 7-метил-2-нафталинкарбонитрилу (10,0 г, 60 ммоль) добавляли при комнатной температуре 2,2'-азобисизобутиронитрил (982 мг, 6 ммоль) и ацетонитрил (100 мл). К полученной смеси добавляли N-бромсукцинимид (10,6 г, 60 ммоль) и смесь кипятили с обратным холодильником в течение 2 часов. Полученную смесь охлаждали до комнатной температуры. Для экстрагирования к смеси добавляли воду (100 мл) и толуол (100 мл). Полученный органический слой промывали два раза водой (100 мл). После концентрирования органического слоя при пониженном давлении, анализ путем обращенно-фазовой хроматографии с использованием продукта, полученного в примере 11, в качестве стандартной пробы показал, что получили 12,1 г указанного в заголовке соединения (выход: 82%).
Пример 13. Способ получения 7-бромметил-2-нафталинкарбонитрила
К 7-метил-2-нафталинкарбонитрилу (5,0 г, 30 ммоль) добавляли при комнатной температуре 2,2'-азобисизобутиронитрил (491 мг, 3 ммоль) и пропионитрил (50 мл). К полученной смеси добавляли N-бромсукцинимид (5,3 г, 30 ммоль) и смесь нагревали при внутренней температуре 80oС в течение 4 часов. После этого эксперимент проводили так, как в примере 12. Анализ путем обращенно-фазовой хроматографии с использованием продукта, полученного в примере 11, в качестве стандартной пробы показал, что получили 1,9 г указанного в заголовке соединения (выход: 82%).
Пример 14. Способ получения 7-бромметил-2-нафталинкарбонитрила
К 7-метил-2-нафталинкарбонитрилу (5,0 г, 30 ммоль) добавляли при комнатной температуре 2,2'-азобисизобутиронитрил (491 мг, 3 ммоль) и н-бутиронитрил (50 мл). К полученной смеси добавляли N-бромсукцинимид (5,3 г, 30 ммоль) и смесь нагревали при внутренней температуре 80oС в течение 2 часов. После этого эксперимент проводили так, как в примере 12. Анализ путем обращенно-фазовой хроматографии с использованием продукта, полученного в примере 11, в качестве стандартной пробы показал, что получили 5,4 г указанного в заголовке соединения 9 выход: 74%).
Пример 15. Способ получения 7-бромметил-2-нафталинкарбонитрила
К 7-метил-2-нафталинкарбонитрилу (5,0 г, 30 ммоль) добавляли при комнатной температуре 2,2'-азобисизобутиронитрил (491 мг, 3 ммоль) и изобутиронитрил (50 мл). К полученной смеси добавляли N-бромсукцинимид (5,3 г, 30 ммоль) и смесь нагревали при внутренней температуре 80oС в течение 2 часов. После этого эксперимент проводили так, как в примере 12. Анализ путем обращенно-фазовой хроматографии с использованием продукта, полученного в примере 11, в качестве
стандартной пробы показал, что получили 4,3 г указанного в заголовке соединения (выход: 58%).
Пример 16. Способ получения этилового эфира 2-[4-[[(3S)-1-(трет-бутоксикарбонил)-3-пирролидинил] окси] фенил] -3-(7-циано-2-нафтил)пропионовой кислоты
Этиловый эфир 2-[4-[(3S)-1-(трет-бутоксикарбонил)-3-пирролидинил]окси] фенилуксусной кислоты (1,8 г, 5 ммоль), полученный в примере 2, и 7-бромметил-2-нафталинкарбонитрил (1,5 г, 6 ммоль) растворяли в диметилформамиде (15 мл) и смесь охлаждали до 0oС. Затем добавляли 60%-ный гидрид натрия (0,2 г, 5,5 ммоль) и смесь перемешивали при той же температуре в течение 2,5 часов. Полученную смесь разбавляли этиловым эфиром уксусной кислоты (30 мл) и промывали три раза водой (10 мл). После концентрирования полученного органического слоя при пониженном давлении, остаток подвергали колоночной хроматографии на силикагеле с получением 2,37 г указанного в заголовке соединения (выход: 89%). Данные измерения приборами соответствовали данным ссылочного примера 35, описанного в выложенной заявке (kokai) 5-208946 на патент Японии.
Пример 17. Способ получения этилового эфира 2-[4-[[(3S)-1-(трет-бутоксикарбонил)-3-пирролидинил] окси] фенил] -3-(7-циано-2-нафтил)пропионовой кислоты
Этиловый эфир 2-[4-[(3S)-1-(трет-бутоксикарбонил)-3-пирролидинил]окси] фенилуксусной кислоты (40,0 г, 0,12 моль, чистота 79%), полученный в примере 3, и 7-бромметил-2-нафталинкарбонитрил (33,8 г, 0,14 моль) растворяли в диметилформамиде (340 мл) и смесь охлаждали до 0oС. Затем добавляли 60%-ный гидрид натрия (5,0 г, 0,13 моль) и смесь перемешивали при той же температуре в течение 3 часов. Полученную смесь разбавляли этиловым эфиром уксусной кислоты (680 мл) и промывали три раза водой (170 мл). Анализ путем обращенно-фазовой хроматографии с использованием продукта, полученного в примере 16, в качестве стандартной пробы показал, что получили 54,1 г указанного в заголовке соединения, имеющего чистоту 66% (выход: 92%).
Пример 18. Способ получения этилового эфира 2-[4-[[(3S)-1-(трет-бутоксикарбонил)-3-пирролидинил] окси] фенил] -3-(7-циано-2-нафтил)пропионовой кислоты
Этиловый эфир 2-[4-[(3S)-1-(трет-бутоксикарбонил)-3-пирролидинил]окси] фенилуксусной кислоты (40,0 г, 0,12 моль, чистота 79%), полученный в примере 10, и 7-бромметил-2-нафталинкарбонитрил (33,8 г, 0,14 моль) растворяли в диметилформамиде (400 мл) и смесь охлаждали до 0oС. Затем прибавляли 60%-ный гидрид натрия (5,0 г, 0,13 моль) и смесь перемешивали при той же температуре в течение 3 часов. Полученную смесь разбавляли этиловым эфиром уксусной кислоты (800 мл) и промывали три раза водой (200 мл). Анализ путем обращенно-фазовой хроматографии с использованием продукта, полученного в примере 16, в качестве стандартной пробы показал, что получили 53,5 г указанного в заголовке соединения, имеющего чистоту 67% (выход: 91%).
Пример 19. Способ получения этилового эфира 2-[4-[[(3S)-1-(трет-бутоксикарбонил)-3-пирролидинил] окси] фенил] -3-(7-циано-2-нафтил)пропионовой кислоты
Повторяли процедуры примера 17 в диметилформамиде, имеющем 0,2% воды (10 мл), за исключением того, что использовали этиловый эфир 2-[4-[(3S)-1-(трет-бутоксикарбонил)3-пирролидинил] окси]фенилуксусной кислоты (1,0 г, 2,8 ммоль), 7-бромметил-2-нафталинкарбонитрил (0,7 г, 2,8 моль) 60%-ный гидрид натрия (0,1 г, 3,1 ммоль). Анализ путем обращенно-фазовой хроматографии с использованием продукта, полученного в примере 16, в качестве стандартной пробы показал, что получили 1,3 г указанного в заголовке соединения (выход: 90%).
Пример 20. Способ получения этилового эфира 2-[4-[[(3S)-1-(трет-бутоксикарбонил)-3-пирролидинил] окси] фенил] -3-(7-циано-2-нафтил)пропионовой кислоты
Повторяли процедуры примера 17 в смеси, состоявшей из диметилформамида (7,5 мл) и толуола (7,5 мл), за исключением того, что использовали этиловый эфир 2-[4-[(3S)-1-(трет-бутоксикарбонил)-3-пирролидинил] окси]фенилуксусной кислоты (1,8 г, 5 ммоль), 7-бромметил-2-нафталинкарбонитрил (1,5 г, 6 ммоль) 60%-ный гидрид натрия (0,2 г, 5,5 ммоль). Анализ путем обращенно-фазовой хроматографии с использованием продукта, полученного в примере 16, в качестве стандартной пробы показал, что получили 2,7 г указанного в заголовке соединения (выход: 92%).
Пример 21. Способ получения этилового эфира 2-[4-[[(3S)-1-(трет-бутоксикарбонил)-3-пирролидинил] окси] фенил] -3-(7-циано-2-нафтил)пропионовой кислоты
Повторяли процедуры примера 17 в смеси, состоявшей из диметилового эфира триэтиленгликоля (9 мл) и толуола (9 мл), за исключением того, что использовали этиловый эфир 2-[4-[(3S)-1-(трет-бутоксикарбонил)-3-пирролидинил]окси]фенилуксусной кислоты (1,8 г, 5 ммоль), 7-бромметил-2-нафталинкарбонитрил (1,5 г, 6 ммоль) и 60%-ный гидрид натрия (0,2 г, 5,5 ммоль). Анализ путем обращенно-фазовой хроматографии с использованием продукта, полученного в примере 16, в качестве стандартной пробы показал, что получили 1,8 г указанного в заголовке соединения (выход: 71%).
Пример 22. Способ получения этилового эфира 2-[4-[[(3S)-1-(трет-бутоксикарбонил)-3-пирролидинил] окси] atybkъ-3-(7-циано-2-нафтил)пропионовой кислоты
Повторяли процедуры примера 17 в диметиловом эфире триэтиленгликоля (46 мл), за исключением того, что использовали этиловый эфир 2-[4-[(3S)-1-(трет-бутоксикарбонил) -3-пирролидинил]окси]фенилуксусной кислоты (3,5 г, 10 ммоль), 7-бромметил-2-нафталинкарбонитрил (3,0 г, 12 ммоль) и 60%-ный гидрид натрия (0,4 г, 11 ммоль). Анализ путем обращенно-фазовой хроматографии с использованием продукта, полученного в примере 16, в качестве стандартной пробы показал, что получили 4,4 г указанного в заголовке соединения (выход: 86%).
Пример 23. Способ получения этилового эфира 2-[4-[[(3S)-1-(трет-бутоксикарбонил)-3-пирролидинил] окси] фенил] -3-(7-циано-2-нафтил)пропионовой кислоты
Повторяли процедуры примера 17 в диметилформамиде (15 мл), за исключением того, что использовали этиловый эфир 2-[4-[(3S)-1-(трет-бутоксикарбонил)-3-пирролидинил] окси] фенилуксусной кислоты (1,7 г, 5 ммоль), 7-бромметил-2-нафталинкарбонитрил (1,5 г, 6 ммоль) и этоксид натрия (0,4 г, 5,5 ммоль). Анализ путем обращенно-фазовой хроматографии с использованием продукта, полученного в примере 16, в качестве стандартной пробы показал, что получили 1,6 г указанного в заголовке соединения (выход: 65%).
Пример 24. Способ получения этилового эфира 2-[4-[[(3S)-1-(трет-бутоксикарбонил)-3-пирролидинил] окси] фенил] -3-(7-циано-2-нафтил)пропионовой кислоты
Повторяли процедуры примера 17 в диметилформамиде (15 мл), за исключением того, что использовали этиловый эфир 2-[4-[(3S)-1-(трет-бутоксикарбонил)-3-пирролидинил] окси] фенилуксусной кислоты (1,7 г, 5 ммоль), 7-бромметил-2-нафталинкарбонитрил (1,5 г, 6 ммоль) и трет-бутоксид калия (0,6 г, 5,5 ммоль). Анализ путем обращенно-фазовой хроматографии с использованием продукта, полученного в примере 16, в качестве стандартной пробы показал, что получили 1,0 г указанного в заголовке соединения (выход: 38%).
Пример 25. Способ получения этилового эфира 2-[4-[[(3S)-1-(трет-бутоксикарбонил)-3-пирролидинил] окси] фенил] -3-(7-циано-2-нафтил)пропионовой кислоты
Повторяли процедуры примера 17 в диметилформамиде (10 мл), за исключением того, что использовали этиловый эфир 2-[4-[(3S)-1-(трет-бутоксикарбонил)-3-пирролидинил] окси] фенилуксусной кислоты (1,0 г, 2,8 ммоль), 7-хлорметил-2-нафталинкарбонитрил (0,6 г, 2,8 ммоль) и гидрид натрия (0,1 г, 3,1 ммоль). Анализ путем обращенно-фазовой хроматографии с использованием продукта, полученного в примере 16, в качестве стандартной пробы показал, что получили 0,9 г указанного в заголовке соединения (выход: 61%).
Пример 26. Способ получения этилового эфира 2-[4-[[(3S)-1-(трет-бутоксикарбонил)-3-пирролидинил] окси] фенил-3-(7-циано-2-нафтил)пропионовой кислоты
Повторяли процедуры примера 17 в диметилформамиде (10 мл), за исключением того, что использовали этиловый эфир 2-[4-[(3S)-1-(трет-бутоксикарбонил)-3-пирролидинил] окси] фенилуксусной кислоты (1,0 г, 2,8 ммоль), 7-п-толуолсульфонилоксиметил-2-нафталинкарбонитрил (0,9 г, 2,8 ммоль) и 60%-ный гидрид натрия (0,1 г, 3,1 ммоль). Анализ путем обращенно-фазовой хроматографии с использованием продукта, полученного в примере 16, в качестве стандартной пробы показал, что получили 0,9 г указанного в заголовке соединения (выход: 64%).
Пример 27. Способ получения этилового эфира 2-[4-[[(3S)-1-(трет-бутоксикарбонил)-3-пирролидинил] окси] фенил] -3-(7-циано-2-нафтил)пропионовой кислоты
Этиловый эфир п-гидроксифенилуксусной кислоты (1,4 г, 7,9 ммоль) растворяли в толуоле (20 мл) и затем к смеси добавляли при комнатной температуре 60%-ный гидрид натрия (310 мг, 7,9 ммоль). После нагревания смеси при внутренней температуре 45oС в течение одного часа, добавляли (3R)-1-(трет-бутоксикарбонил)-3-метансульфонилоксипирролидин (1,9 г, 7,1 ммоль) и четырехнормальный бутиламмонийбромид (690 мг, 2,4 ммоль). Полученную смесь нагревали при внутренней температуре 80oС в течение 3 часов и затем охлаждали до комнатной температуры. Смесь промывали 3 раза 10%-ным водным раствором гидроксида калия (4 мл). Анализ путем обращенно-фазовой хроматографии с использованием продукта, полученного в примере 2, в качестве стандартной пробы показал, что получили 1,7 г этилового эфира 2-[4-[(3S)-1-(трет-бутоксикарбонил)-3-пирролидинил]окси]фенилуксусной кислоты (выход: 70%).
Не концентрируя полученный органический слой, добавляли 7-бромметил-2-нафталинкарбонитрил (1,5 г, 6 ммоль) и диметилформамид (20 мл) и смесь охлаждали до 0oС. Затем добавляли 60%-ный гидрид натрия (220 мг, 5,5 ммоль) и полученную смесь перемешивали при той же температуре в течение 10 часов. Реакционную смесь промывали три раза водой (20 мл). Анализ путем обращенно-фазовой хроматографии с использованием продукта, полученного в примере 16, в качестве стандартной пробы показал, что получили 2,4 г указанного в заголовке соединения (выход: 92%). Общий для двух стадий выход составил 64%.
Пример 28. Способ получения этилового эфира 2-[4-[[(3S)-1-(трет-бутоксикарбонил)-3-пирролидинил] окси] фенил] -3-(7-циано-2-нафтил)пропионовой кислоты
Повторяли процедуры примера 27, за исключением того, что использовали этиловый эфир п-гидроксифенилуксусной кислоты (1,4 г, 7,9 ммоль), толуол (20 мл), 60%-ный гидрид натрия (310 мг, 7,9 ммоль, (3R)-1-(трет-бутоксикарбонил)-3-метансульфонилоксипирролидин (1,9 г, 7,1 ммоль) и четырехнормальный бутиламмонийбромид (690 мг, 2,4 ммоль). Анализ полученного органического слоя путем обращенно-фазовой хроматографии с использованием продукта, полученного в примере 2, в качестве стандартной пробы показал, что получили 1,7 г этилового эфира 2-[4-[(3S)-1-(трет-бутоксикарбонил)-3-пирролидинил]окси]фенилуксусной кислоты (выход: 70%).
Не концентрируя полученный органический слой, добавляли 7-бромметил-2-нафталинкарбонитрил (1,5 г, 6 ммоль) и диметилформамид (20 мл), а затем 60%-ный гидрид натрия (220 мг, 5,5 ммоль) и полученную смесь перемешивали при комнатной температуре в течение 8 часов. Реакционную смесь промывали три раза водой (20 мл). Анализ путем обращенно-фазовой хроматографии с использованием продукта, полученного в примере 16, в качестве стандартной пробы показал, что получили 2,3 г указанного в заголовке соединения (выход: 88%). Общий для двух стадий выход составил 61%.
Пример 29. Способ получения этилового эфира 2-[4-[[(3S)-1-(трет-бутоксикарбонил)-3-пирролидинил] окси] фенил] -3-(7-циано-2-нафтил)пропионовой кислоты
К 7-метил-2-нафталинкарбонитрилу (6,3 г, 38 ммоль) добавляли при комнатной температуре 2,2'-азобисизобутиронитрил (624 мг, 3,8 ммоль) и ацетонитрил (65 мл). К полученной смеси добавляли N-бромсукцинимид (6,8 г, 38 ммоль) и смесь кипятили с обратным холодильником в течение 2 часов. Полученную смесь охлаждали до комнатной температуры. Для экстрагирования к смеси прибавляли воду (65 мл) и толуол (65 мл). Полученный органический слой промывали два раза водой (65 мл). Анализ путем обращенно-фазовой хроматографии с использованием продукта, полученного в примере 11, в качестве стандартной пробы показал, что получили 7,4 г 7-бромметил-2-нафталинкарбонитрила (выход: 80%).
Не концентрируя этот органический слой, добавляли к нему этиловый эфир 2-[4-[(3S)-1-(трет-бутоксикарбонил)-3-пирролидинил]окси]фенилуксусной кислоты (8,7 г, 25 ммоль, чистота 79%), полученный в примере 10, и диметилформамид (81 мл) и полученную смесь охлаждали до 0oС. Затем добавляли 60%-ный гидрид натрия (1,1 г, 27,5 ммоль) и полученную смесь перемешивали при той же температуре в течение 5 часов. Полученный раствор разбавляли толуолом (40 мл) и промывали три раза водой (80 мл). Полученный органический слой концентрировали при пониженном давлении. Анализ остатка (19,3 г) путем обращенно-фазовой хроматографии с использованием продукта, полученного в примере 16, в качестве стандартной пробы показал, что получили 11,7 г указанного в заголовке соединения (выход: 91%).
Пример 30. Способ получения метилового эфира 2-[4-[[(3S)-1-(трет-бутоксикарбонил)-3-пирролидинил] окси] фенил] -3-(7-циано-2-нафтил)пропионовой кислоты
Метиловый эфир 2-[4-[[(3S)-1-(трет-бутоксикарбонил)-3-пирролидинил]окси] фенилуксусной кислоты (1,7 г, 5 ммоль и 7-бромметил-2-нафталинкарбонитрил (1,4 г, 5,5 ммоль) растворяли в диметилформамиде (15 мл) и смесь охлаждали до 0oС. Затем добавляли 60%-ный гидрид натрия (0,2 г, 5,5 ммоль) и полученную смесь перемешивали при той же температуре в течение 3 часов. Полученную смесь разбавляли этиловым эфиром уксусной кислоты (30 мл) и промывали три раза водой (10 мл). Полученный органический слой концентрировали при пониженном давлении и остаток подвергали колоночной хроматографии на силикагеле с получением 2,3 г указанного в заголовке соединения (выход: 93%).
Спектр ядерного магнитного резонанса (СDCl3) δ:
1,47 (9Н, с), 2,14 (2Н, м),
3,19 (1Н, дд, J=6,9, 13,9 Гц),
3,55 (5Н, м), 3,60 (3Н, с), 3,90 (1Н, ш),
4,86 (1Н, м), 6,83 (2Н, д, J=8,6 Гц),
7,23 (2Н, д, J=8,3 Гц),
7,41 (1Н, дд, J=1,5, 8,3 Гц),
7,56 (1Н, дд, J=1,5, 8,3 Гц),
7,61 (1Н, с), 7,78 (1Н, д, J=8,3 Гц),
7,85 (1Н, д, J=8,3 Гц), 8,13 (1Н, с).
Пример 31. Способ получения изопропилового эфира 2-[4-[[(3S)-1-(трет-бутоксикарбонил)-3-пирролидинил] окси]фенил]-3-(7-циано-2-нафтил)пропионовой кислоты
Изопропиловый эфир 2-[4-[[(3S)-1-(трет-бутоксикарбонил)-3-пирролидинил] окси] фенилуксусной кислоты (1,8 г, 5 ммоль) и 7-бромметил-2-нафталинкарбонитрил (1,5 г, 6 ммоль) растворяли в диметилформамиде (15 мл) и смесь охлаждали до 0oС. Затем добавляли 60%-ный гидрид натрия (0,2 г, 5,5 ммоль) и полученную смесь перемешивали при той же температуре в течение 3 часов. Полученную смесь разбавляли этиловым эфиром уксусной кислоты (30 мл) и промывали три раза водой (10 мл). Полученный органический слой концентрировали при пониженном давлении и остаток подвергали колоночной хроматографии на силикагеле с получением 2,4 г указанного в заголовке соединения (выход: 92%).
Спектр ядерного магнитного резонанса (СDСl3) δ:
1,05 (3Н, д, J=6,3 Гц),
1,08 (3Н, д, J=5,6 Гц),
2,14 (2Н, м), 3,17 (1Н, дд, J=6,6, 13,7 Гц,
3,55 (5Н, м), 3,85 (1Н, ш), 4,86 (1Н, м),
4,90 (1Н, м), 6,83 (2Н, д, J=8,6 Гц),
7,23 (2Н, ш), 7,42 (1Н, д, J=8,3 Гц),
7,55 (1Н, дд, J=1,6, 8,6 Гц),
7,62 (1Н, с), 7,79 (1Н, д, J=8,6 Гц),
7,84 (1Н, д, J=8,6 Гц), 8,113 (1Н, с)
Пример 32. Способ получения трет-бутилового эфира 2-[4-[[(3S)-1-(трет-бутоксикарбонил)-3-пирролидинил] окси]фенил]-3-(7-циано-2-нафтил)пропионовой кислоты
Трет-бутиловый эфир 2-[4-[(3S)-1-(трет-бутоксикарбонил)-3-пирролидинил] окси] фенилуксусной кислоты (1,9 г, 5 ммоль) и 7-бромметил-2-нафталинкарбонитрил (1,5 г, 6 ммоль) растворяли в диметилформамиде (15 мл) и смесь охлаждали до 0oС. Затем добавляли 60%-ный гидрид натрия (0,2 г, 5,5 ммоль) и полученную смесь перемешивали при той же температуре в течение 4 часов с последующим перемешиванием при комнатной температуре в течение 4 часов. Полученную смесь разбавляли этиловым эфиром уксусной кислоты (30 мл) и промывали три раза водой (10 мл). Полученный органический слой концентрировали при пониженном давлении и остаток подвергали колоночной хроматографии на силикагеле с получением 2,1 г указанного в заголовке соединения (выход: 79%).
Спектр ядерного магнитного резонанса (СDСl3) δ:
1,30 (9Н, с), 1,47 (9Н, с), 2,13 (2Н, м),
3,13 (1Н, дд, J=6,6, 13,9 Гц),
3,52 (5Н, м), 3,79 (1Н, ш), 4,87 (1Н, м),
6,83 (2Н, д, J=8,6 Гц),
7,21 (2Н, д, J=8,3 Гц),
7,43 (1Н, д, J=7,6 Гц),
7,55 (1Н, д, J=7,6 Гц), 7,62 (1Н, с),
7,77 (1Н, д, J=8,6 Гц),
7,86 (1Н, д, J=8,6 Гц), 8,13 (1Н, с)
Пример 33. Способ получения бензилового эфира 2-[4-[[(3S)-1-(трет-бутоксикарбонил)-3-пирролидинил] окси] фенил] -3-(7-циано-2-нафтил)пропионовой кислоты
Бензиловый эфир 2-[4-[(3S)-1-(трет-бутоксикарбонил)-3-пирролидинил]окси] фенилуксусной кислоты (2,1 г, 5 ммоль) и 7-бромметил-2-нафталинкарбонитрил (1,5 г, 6 ммоль) растворяли в диметилформамиде (15 мл) и смесь охлаждали до 0oС. Затем добавляли 60%-ный гидрид натрия (0,2 г, 5,5 ммоль) и полученную смесь перемешивали при той же температуре в течение 3 часов. Полученную смесь разбавляли этиловым эфиром уксусной кислоты (30 мл) и промывали три раза водой (10 мл). Полученный органический слой концентрировали при пониженном давлении и остаток подвергали колоночной хроматографии на силикагеле с получением 2,6 г указанного в заголовке соединения (выход: 90%).
Спектр ядерного магнитного резонанса (СDСl3) δ:
1,47 (9Н, с), 2,11 (2Н, м), 3,17 (1Н, дд),
3,51 (5Н, м), 3,93 (1Н, ш), 4,83 (1Н, м),
4,92 (1Н, д, J=13,2 Гц),
5,10 (1Н, д, J=9,9 Гц),
6,83 (2Н, д, J=8,6 Гц),
7,05 - 8,30 (7Н, м),
7,39 (1Н, д, J=8,2 Гц),
7,52 (1Н, д, J=9,9 Гц), 7,55 (1Н, с),
7,72 (1Н, д, J=8,6 Гц),
7,83 (1Н, д, J=8,6 Гц), 7,99 (1Н, с)
Пример 34. Способ получения этилового эфира 2-[4-[(3S)-1-бензил-3-пирролидинил]окси]фенилуксусной кислоты
Этиловый эфир п-гидроксифенилуксусной кислоты (9,1 г, 49,5 ммоль) растворяли в диметилформамиде (120 мл) и к смеси при комнатной температуре добавляли 60%-ный гидрид натрия (2,0 г, 49,5 ммоль). Спустя сорок минут добавляли (3R)-1-бензил-3-метансульфонилоксипирролидин (11,5 г, 45 ммоль) и полученную смесь сразу же нагревали на масляной бане с температурой 135oС. После нагревания при внутренней температуре 110oC в течение 15 минут, смесь охлаждали до комнатной температуры. Затем смесь концентрировали для удаления растворителя при пониженном давлении и добавляли этиловый эфир уксусной кислоты (120 мл) для растворения остатка. Полученную смесь промывали три раза 10%-ным водным раствором гидроксида калия (24 мл). Полученный органический слой концентрировали при пониженном давлении и остаток подвергали колоночной хроматографии на силикагеле с получением 10,9 г указанного в заголовке соединения (выход: 71%).
Спектр ядерного магнитного резонанса (СDCl3) δ:
1,24 (3Н, т, J=6,9 Гц), 1,98 (1H, м),
2,29 (1H, м), 2,60 (1H, м), 2,74 (2Н, м),
2,98 (1H, дд, 6,3, 6,3 Гц), 3,52 (2Н, с),
3,67 (2Н, дд, 12,9, 12,9 Гц),
4,13 (2Н, kв, J=6,9 Гц), 4,80 (1H, м),
6,78 (2Н, д, J=8,6 Гц),
7,15 (2Н, д, J=8,9 Гц),
7,24-7,35 (5Н, м)
Элементный анализ C21H25NO3:
Вычислено: С, 74,31; Н, 7,42; N, 4,13.
Найдено: С, 73,82; Н, 7,36; N, 4,01.
FAB MC (m/z): 340 (M++1).
Спектр поглощения в инфракрасной области vmax (KBr) см-1:
2984, 2800, 1732, 1614, 1512, 1296, 1244, 1148, 1028.
Угол вращения [α]
Пример 35. Способ получения этилового эфира 2-[4-[(3S)-1-бензил-3-пирролидинил]окси]фенил]уксусной кислоты
Этиловый эфир п-гидроксифенилуксусной кислоты (9,9 г, 55 ммоль) растворяли в диметилформамиде (120 мл) и к смеси при комнатной температуре добавляли 60%-ный гидрид натрия (2,2 г, 55 ммоль). Спустя 40 минут добавляли (3R)-1-бензил-3-метансульфонилоксипирролидин (12,8 г, 50 ммоль) и полученную смесь сразу же нагревали на масляной бане с температурой 135oС. После нагревания при внутренней температуре 110oС в течение 15 минут, смесь охлаждали до комнатной температуры. Затем смесь концентрировали для удаления растворителя при пониженном давлении и добавляли этиловый эфир уксусной кислоты (120 мл) для растворения остатка. Полученную смесь промывали три раза 10%-ным водным раствором гидроксида калия (24 мл). Полученный органический слой концентрировали при пониженном давлении и анализ остатка путем обращенно-фазовой хроматографии с использованием продукта, полученного в примере 34, в качестве стандартной пробы показал, что получили 12,2 г указанного в заголовке соединения (выход 72%, чистота 82%).
Пример 36. Способ получения этилового эфира 2-[4-[[(3S)-1-бензил-3-пирролидинил]окси]фенил]-3-(7-циано-2-нафтил)пропионовой кислоты
Этиловый эфир 2-[4-[(3S)-1-бензил-3-пирролидинил] окси] фенилуксусной кислоты (1,0 г, 3 ммоль), полученный в примере 34, и 7-бромметил-2-нафталинкарбонитрил (0,9 г, 3,6 ммоль) растворяли в диметилформамиде (10 мл) и смесь охлаждали до 0oС. Затем прибавляли 60%-ный гидрид натрия (0,1 г, 3,3 ммоль) и смесь перемешивали при той же температуре в течение 1,5 часов. Полученную смесь разбавляли этиловым эфиром уксусной кислоты (30 мл) и промывали три раза водой (10 мл). После концентрирования полученного органического слоя при пониженном давлении, остаток подвергали колоночной хроматографии на силикагеле с получением 0,3 г указанного в заголовке соединения (выход: 20%).
Спектр ядерного магнитного резонанса (СDCl3) δ:
1,10 (3Н, т, J=6,9 Гц), 1,97 (1H, м),
2,28 (1H, м), 2,60 (1H, м), 2,74 (2Н, м),
2,97 (1H, м),
3,16 (1H, дд, J=6,9, 13,5 Гц),
3,54 (1H, дд, J=8,9, 13,5 Гц),
3,67 (2Н, дд, J=12,9, 12,9 Гц),
3,85 (1H, дд, J=6,9, 8,6 Гц),
3,99-4,13 (2Н, м), 4,79 (1H, м),
6,76 (2Н, д, J=8,6 Гц),
7,20 (2Н, д, J=8,6 Гц), 7,23-7,34 (5Н, м),
8,41 (1Н, д, J=8,3 Гц),
7,54 (1Н, дд, J=8,6, 8,6 Гц), 7,60 (1Н, с),
7,76 (1Н, д, J=8,6 Гц),
7,85 (1Н, д, J=8,6 Гц), 8,11 (1Н, с)
Угол вращения [α]
Пример 37. Способ получения этилового эфира 2-[4-[[(3S)-1-бензил-3-пирролидинил]окси]фенил]-3-(7-циано-2-нафтил)пропионовой кислоты
Этиловый эфир 2-[4-[(3S)-1-бензил-3-пирролидинил] окси] -фенилуксусной кислоты (500 мг, 1,5 ммоль) растворяли в диметилформамиде (5 мл) и смесь охлаждали до -10oС. Затем добавляли 60%-ный гидрид натрия (60 мг, 1,5 ммоль) и смесь перемешивали при той же температуре в течение 0,5 часа. После этого к смеси постепенно добавляли 7-бромметил-2-нафталинкарбонитрил (370 мг, 1,5 ммоль) и полученную смесь перемешивали при той же температуре 4 часа. Полученную смесь разбавляли этиловым эфиром уксусной кислоты (15 мл) и промывали три раза водой (5 мл). После концентрирования полученного органического слоя при пониженном давлении анализ путем обращенно-фазовой хроматографии с использованием продукта, полученного в примере 36, в качестве стандартной пробы показал, что получили 480 мг указанного в заголовке соединения (выход 64%).
Пример 38. Способ получения этилового эфира 2-[4-[[(3S)-1-бензил-3-пирролидинил]окси]фенил-3-(7-циано-2-нафтил)-пропионовой кислоты
Этиловый эфир 2-[4-[(3S)-1-бензил-3-пирролидинил] окси] фенилуксусной кислоты (0,5 г, 1,5 ммоль) и 7-бромметил-2-нафталинкарбонитрил (440 мг, 1,8 ммоль) растворяли в диметоксиэтане (5 мл) и смесь охлаждали до 0oС. Затем добавляли 60%-ный гидрид натрия (66 мг, 1,65 ммоль) и смесь перемешивали при той же температуре в течение 5 часов. После этого повторяли процедуру примера 37. Анализ путем обращенно-фазовой хроматографии с использованием продукта, полученного в примере 36, в качестве стандартной пробы показал, что получили 158 мг указанного в заголовке соединения (выход 21%).
Пример 39. Способ получения этилового эфира 2-[4-[[(3S)-1-бензил-3-пирролидинил]окси]фенил]-3-(7-циано-2-нафтил)-пропионовой кислоты
Этиловый эфир 2-[4-[(3S)-1-бензил-3-пирролидинил] окси] -фенилуксусной кислоты (0,5 г, 1,5 ммоль) и 7-бромметил-2-нафталинкарбонитрил (440 мг, 1,8 ммоль) растворяли в диметиловом эфире диэтиленгликоля (5 мл) и смесь охлаждали до 0oС. Затем добавляли 60%-ный гидрид натрия (66 мг, 1,65 ммоль) и смесь перемешивали при той же температуре в течение 5 часов. После этого повторяли процедуру примера 37. Анализ путем обращенно-фазовой хроматографии с использованием продукта, полученного в примере 36, в качестве стандартной пробы показал, что получили 555 мг указанного в заголовке соединения (выход 73%).
Пример 40. Способ получения этилового эфира 2-[4-[[(3S)-1-бензил-3-пирролидинил]окси]фенил]-3-(7-циано-2-нафтил)-пропионовой кислоты
Этиловый эфир 2-[4-[(3S)-1-бензил-3-пирролидинил] окси] -фенилуксусной кислоты (0,5 г, 1,5 ммоль) и 7-бромметил-2-нафталинкарбонитрил (440 мг, 1,8 ммоль) растворяли в диметиловом эфире триэтиленгликоля (5 мл) и смесь охлаждали до 0oС. Затем добавляли 60%-ный гидрид натрия (66 мг, 1,65 ммоль) и смесь перемешивали при той же температуре в течение 6 часов. После этого повторяли процедуру примера 37. Анализ путем обращенно-фазовой хроматографии с использованием продукта, полученного в примере 36, в качестве стандартной пробы показал, что получили 647 мг указанного в заголовке соединения (выход 85%).
Пример 41. Способ получения этилового эфира 2-[4-[[(3S)-1-бензил-3-пирролидинил]окси]фенил]-3-(7-циано-2-нафтил)-пропионовой кислоты
Этиловый эфир 2-[4-[(3S)-1-бензил-3-пирролидинил] окси]-фенилуксусной кислоты (500 мг, 1,5 ммоль) растворяли в диметилформамиде (5 мл) и смесь охлаждали до -10oС. Затем добавляли этоксид натрия (102 мг, 1,5 ммоль) и смесь перемешивали при той же температуре в течение 0,5 часа. После этого к смеси постепенно прибавляли 7-бромметил-2-нафталинкарбонитрил (370 мг, 1,5 ммоль) и полученную смесь перемешивали при той же температуре 3 часа. Далее повторяли процедуры примера 37. Анализ путем обращенно-фазовой хроматографии с использованием продукта, полученного в примере 36, в качестве стандартной пробы показал, что получили 332 мг указанного в заголовке соединения (выход 44%).
Пример 42. Способ получения этилового эфира 2-[4-[[(3S)-1-бензил-3-пирролидинил]окси]фенил]-3-(7-циано-2-нафтил)-пропионовой кислоты
Этиловый эфир 2-[4-[(3S)-1-бензил-3-пирролидинил] окси] фенилуксусной кислоты (500 мг, 1,5 ммоль) растворяли в диметилформамиде (5 мл) и смесь охлаждали до -10oС. Затем добавляли амид натрия (60 мг, 1,5 ммоль) и смесь перемешивали при той же температуре в течение 0,5 часа. После этого к смеси постепенно добавляли 7-бромметил-2-нафталинкарбонитрил (370 мг, 1,5 ммоль) и полученную смесь перемешивали при той же температуре 2 часа. Далее повторяли процедуры примера 37. Анализ путем обращенно-фазовой хроматографии с использованием продукта, полученного в примере 36, в качестве стандартной пробы показал, что получили 158 мг указанного в заголовке соединения (выход 21%).
Пример 43. Способ получения этилового эфира 2-[4-[[(3S)-1-бензил-3-пирролидинил]окси]фенил]-3-(7-циано-2-нафтил)-пропионовой кислоты
Этиловый эфир 2-[4-[(3S)-1-бензил-3-пирролидинил] окси] фенилуксусной кислоты (500 мг, 1,5 ммоль) и 7-бромметил-2-нафталинкарбонитрил (363 мг, 1,8 ммоль) растворяли в диметилформамиде (5 мл) и смесь охлаждали до 0oС. Затем добавляли 60%-ный гидрид натрия (66 мг, 1,65 ммоль) и смесь перемешивали при той же температуре в течение 3 часов. После этого повторяли процедуру примера 37. Анализ путем обращенно-фазовой хроматографии с использованием продукта, полученного в примере 36, в качестве стандартной пробы показал, что получили 473 мг указанного в заголовке соединения (выход 62%).
Пример 44. Способ получения этилового эфира 2-[4-[[(3S)-1-бензил-3-пирролидинил]окси]фенил]-3-(7-циано-2-нафтил)-пропионовой кислоты
Этиловый эфир 2-[4-[(3S)-1-бензил-3-пирролидинил] окси] -фенилуксусной кислоты (6,4 г, 16 ммоль, чистота 82%) и 7-бромметил-2-нафталинкарбонитрил (4,7 г, 19,2 ммоль) растворяли в диметиловом эфире триэтиленгликоля (54 мл) и смесь охлаждали до 0oС. Затем добавляли 60%-ный гидрид натрия (704 мг, 17,6 ммоль) и смесь перемешивали при той же температуре в течение 8 часов. Полученную смесь разбавляли этиловым эфиром уксусной кислоты (108 мл) и промывали три раза водой (216 мл). После концентрирования полученного органического слоя при пониженном давлении анализ путем обращенно-фазовой хроматографии с использованием продукта, полученного в примере 36, в качестве стандартной пробы показал, что получили 6,4 г указанного в заголовке соединения (выход 79%).
Пример 45. Способ получения этилового эфира (2S)-2-[4-[[(3S)-1-(трет-бутоксикарбонил)-3-пирролидинил] окси] фенил-3-(7-циано-2-нафтил)пропионовой кислоты
Этиловый эфир 2-[4-[[(3S)-1-(трет-бутоксикарбонил)-3-пирролидинил]окси] фенил] -3-(7-циано-2-нафтил)пропионовой кислоты (54,1 г, 0,11 моль, чистота 66%), полученный в примере 17, нагревали и растворяли в этаноле (180 мл) и полученную смесь охлаждали до комнатной температуры при перемешивании с выпадением кристаллов. Затем добавляли этоксид натрия (1,1 г, 15,8 ммоль) с последующим перемешиванием при той же температуре в течение 30 минут, после чего прибавляли дополнительное количество этоксида натрия (1,1 г, 15,8 ммоль) с последующим перемешиванием в течение 18 часов. Кристаллы собирали фильтрованием и промывали этанолом (55 мл). Анализ методом ВЭЖХ (высокоэффективная жидкостная хроматография) в условиях ссылочного примера 49, описанного в выложенной заявке (kokai) 5-208946 на патент Японии, показал, что кристаллы имеют диастереомерную чистоту 94,7%. Кристаллы растворяли в этиловом эфире уксусной кислоты (1228 мл) и промывали три раза водой (250 мл). Полученный органический слой концентрировали при пониженном давлении и остаток перекристаллизовывали из этанола (550 мл) с получением 47,6 г указанного в заголовке соединения (выход 88%). Данные измерений приборами соответствовали данным ссылочного примера 49, описанного в выложенной заявке (kokai) 5-208946 на патент Японии. Диастереомерная чистота кристаллов составляла 99,5%.
Пример 46. Способ получения этилового эфира (2S)-2-[4-[[(3S)-1-(трет-бутоксикарбонил)-3-пирролидинил] окси]фенил]-3-(7-циано-2-нафтил)пропионовой кислоты
Этиловый эфир 2-[4-[[(3S)-1-(трет-бутоксикарбонил)-3-пирролидинил]окси] фенил] -3-(7-циано-2-нафтил)пропионовой кислоты (50,0 г, 0,10 моль, чистота 67%), полученный в примере 18, нагревали и растворяли в этаноле (165 мл) и полученный раствор охлаждали до комнатной температуры при перемешивании с выпадением кристаллов. Затем добавляли этоксид натрия (1,0 г, 14,6 моль) с последующим перемешиванием при той же температуре в течение 30 минут, после чего добавляли дополнительное количество этоксида натрия (1,0 г, 14,6 ммоль) с последующим перемешиванием в течение 18 часов. Кристаллы собирали фильтрованием и промывали этанолом (50 мл). Как было установлено, кристаллы имели диастереомерную чистоту 93,5%. Кристаллы растворяли в этиловом эфире уксусной кислоты (1900 мл) и промывали три раза водой (240 мл). Органический слой концентрировали при пониженном давлении и остаток перекристаллизовывали из этанола (500 мл) с получением 43,8 г указанного в заголовке соединения (выход 88%). Диастереомерная чистота кристаллов составляла 99,6%.
Пример 47. Способ получения этилового эфира (2S)-2-[4-[[(3S)-1-(трет-бутоксикарбонил)-3-пирролидинил] окси]фенил]-3-(7-циано-2-нафтил)пропионовой кислоты
Этиловый эфир 2-[4-[[(3S)-1-(трет-бутоксикарбонил)-3-пирролидинил]окси] фенил] -3-(7-циано-2-нафтил)пропионовой кислоты (11,3 г, 21,9 ммоль, чистота 61%), полученный в примере 29, нагревали и растворяли в этаноле (40 мл) и полученный раствор охлаждали до комнатной температуры при перемешивании с выпадением кристаллов. Затем добавляли этоксид натрия (235 мг, 3,28 ммоль) с последующим перемешиванием при той же температуре в течение 30 минут, после чего добавляли дополнительное количество этоксида натрия (235 мг, 3,28 ммоль) с последующим перемешиванием в течение 18 часов. Кристаллы собирали фильтрованием и промывали этанолом (10 мл). Полученные кристаллы имели диастереомерную чистоту 92,8%. Кристаллы растворяли в этиловом эфире уксусной кислоты (250 мл) и промывали три раза водой (50 мл). Полученный органический слой концентрировали при пониженном давлении и остаток перекристаллизовывали два раза из этанола (120 мл, 100 мл) с получением 9,23 г указанного в заголовке соединения (выход 82%). Диастереомерная чистота кристаллов составляла 99,8%.
Пример 48. Способ получения этилового эфира (2S)-2-[4-[[(3S)-1-(трет-бутоксикарбонил)-3-пирролидинил] окси]фенил]-3-(7-циано-2-нафтил)пропионовой кислоты
Этиловый эфир 2-[4-[[(3S)-1-(трет-бутоксикарбонил)-3-пирролидинил]окси] фенил]-3-(7-циано-2-нафтил)пропионовой кислоты (1,90 г, 3,7 ммоль) нагревали и растворяли в этаноле (6 мл) и полученный раствор охлаждали до комнатной температуры при перемешивании с выпадением кристаллов. Затем добавляли амид натрия (22 мг, 0,6 ммоль) с последующим перемешиванием при той же температуре в течение 30 минут, после чего добавляли дополнительное количество амида натрия (22 мг, 0,6 ммоль) с последующим перемешиванием в течение 18 часов. Кристаллы собирали фильтрованием с получением 1,48 г указанного в заголовке соединения (выход 78%). Диастереомерная чистота кристаллов составляла 92,3%.
Пример 49. Способ получения этилового эфира (2S)-2-[4-[[(3S)-1-бензил-3-пирролидинил]окси]фенил]-3-(7-циано-2-нафтил)пропионовой кислоты
Этиловый эфир 2-[4-[[(3S)-1-бензил-3-пирролидинил]окси]фенил-3-(7-циано-2-нафтил)пропионовой кислоты (5 г, 9,9 ммоль) нагревали и растворяли в этаноле (25 мл) и полученный раствор охлаждали до комнатной температуры при перемешивании с выпадением кристаллов. Затем добавляли этоксид натрия (100 мг, 1,5 ммоль) с последующим перемешиванием при той же температуре в течение часа, после чего добавляли дополнительное количество этоксида натрия (100 мг, 1,5 ммоль) с последующим перемешиванием в течение 15 часов. Кристаллы собирали фильтрованием с получением 3,3 г указанного в заголовке соединения (выход 66%). ВЭЖХ-анализ показал, что полученные кристаллы имели диастереомерную чистоту 82%. Кристаллы растворяли в этиловом эфире уксусной кислоты (75 мл) и промывали три раза водой (50 мл). Полученный органический слой концентрировали при пониженном давлении и остаток перекристаллизовывали два раза из этанола (31 мл, 23 мл) с получением 2,1 г указанного в заголовке соединения (выход 43%). Диастереомерная чистота кристаллов составляла 99,1%.
Температура плавления: 95,0-95,5oС.
Спектр ядерного магнитного резонанса (СDСl3) δ:
1,10 (3Н, т, J=6,9 Гц), 1,98 (1 Н, м),
2,28 (1Н, м), 2,60 (1Н, м), 2,74 (2Н, м),
2,98 (1Н, дд, J=6,9, 8,6 Гц),
3,16 (1Н, дд, J=6,9, 13,5 Гц),
3,54 (1Н, дд, J=8,6, 13,5 Гц),
3,67 (2Н, дд, J=12,9, 12,9 Гц),
3,85 (1Н, дд, J=6,9, 8,6 Гц),
3,996-4,12 (2Н, м), 4,79 (1Н, м),
6,76 (2Н, д, J=8,6 Гц),
7,19 (2Н, д, J=8,9 Гц), 7,24-7,35 (5Н, м),
7,41 (1Н, дд, J=8,3, 8,6 Гц),
7,54 (1Н, дд, J=8,3, 8,6 Гц), 7,60 (1Н, с),
7,71 (1Н, д, J=8,6 Гц),
7,84 (1Н, д, J=8,6 Гц), 8,11 (1Н, с)
Элементный анализ С33Н32N2О4:
Вычислено: С, 78,55; Н, 6,39; N, 5, 55.
Найдено: С, 78,40; Н, 6,48; N, 5,35.
МС (m/z): 504 (М+).
Спектр поглощения в инфракрасной области vmax (КВr) см-1: 2800, 2232, 1730, 1610, 1512, 1254, 1156, 850.
Угол вращения [α]
Пример 50. Способ получения этилового эфира (2S)-2-[4-[[(3S)-1-бензил-3-пирролидинил]окси]фенил]-3-(7-циано-2-нафтил)пропионовой кислоты
Этиловый эфир 2-[4-[[(3S)-1-бензил-3-пирролидинил] окси]-фенил]-3-(7-циано-2-нафтил)пропионовой кислоты (5 г, 9,9 ммоль) нагревали и растворяли в этаноле (50 мл) и полученный раствор охлаждали до комнатной температуры при перемешивании с выпадением кристаллов. Затем добавляли этоксид натрия (100 мг, 1,5 ммоль) с последующим перемешиванием при той же температуре в течение часа, после чего добавляли дополнительное количество этоксида натрия (100 мг, 1,5 ммоль) с последующим перемешиванием в течение 15 часов. Кристаллы собирали фильтрованием с получением 2,9 г указанного в заголовке соединения (выход 58%). ВЭЖХ-анализ показал, что полученные кристаллы имели диастереомерную чистоту 84%. Кристаллы растворяли в этиловом эфире уксусной кислоты (66 мл) и промывали три раза водой (50 мл). Полученный органический слой концентрировали при пониженном давлении и остаток перекристаллизовывали два раза из этанола (14,5 мл, 12 мл) с получением 2,0 г указанного в заголовке соединения (выход 40%). Диастереомерная чистота кристаллов составляла 93%.
Пример 51. Способ получения этилового эфира (2S)-2-[4-[[(3S)-1-бензил-3-пирролидинил]окси]фенил]-3-(7-циано-2-нафтил)пропионовой кислоты
Этиловый эфир 2-[4-[[(3S)-1-бензил-3-пирролидинил] окси]-фенил]-3-(7-циано-2-нафтил)пропионовой кислоты (5 г, 9,9 ммоль) нагревали и растворяли в этаноле (15 мл) и полученный раствор охлаждали до комнатной температуры при перемешивании с выпадением кристаллов. Затем добавляли этоксид натрия (100 мг, 1,5 ммоль) с последующим перемешиванием при той же температуре в течение часа, после чего добавляли дополнительное количество этоксида натрия (100 мг, 1,5 ммоль) с последующим перемешиванием в течение 15 часов. Кристаллы собирали путем фильтрования с получением 3,9 г указанного в заголовке соединения (выход 77%). ВЭЖХ-анализ показал, что полученные кристаллы имели диастереомерную чистоту 47%.
Промышленное применение
В соответствии с настоящим изобретением предложен способ получения производных 2-фенил-3-нафтилпропионовой кислоты и ее оптически активных соединений, которые играют важную роль в качестве промежуточных соединений для получения производных ароматических амидинов, которые обладают противосвертывающим действием, основанным на превосходной ингибирующей активности в отношении активированного коагуляционного фактора X. Способ по настоящему изобретению не только проще технологически, но и выгоден с точки зрения затрат. Таким образом, способ по настоящему изобретению является технологически и экономически подходящим способом для промышленного производства.
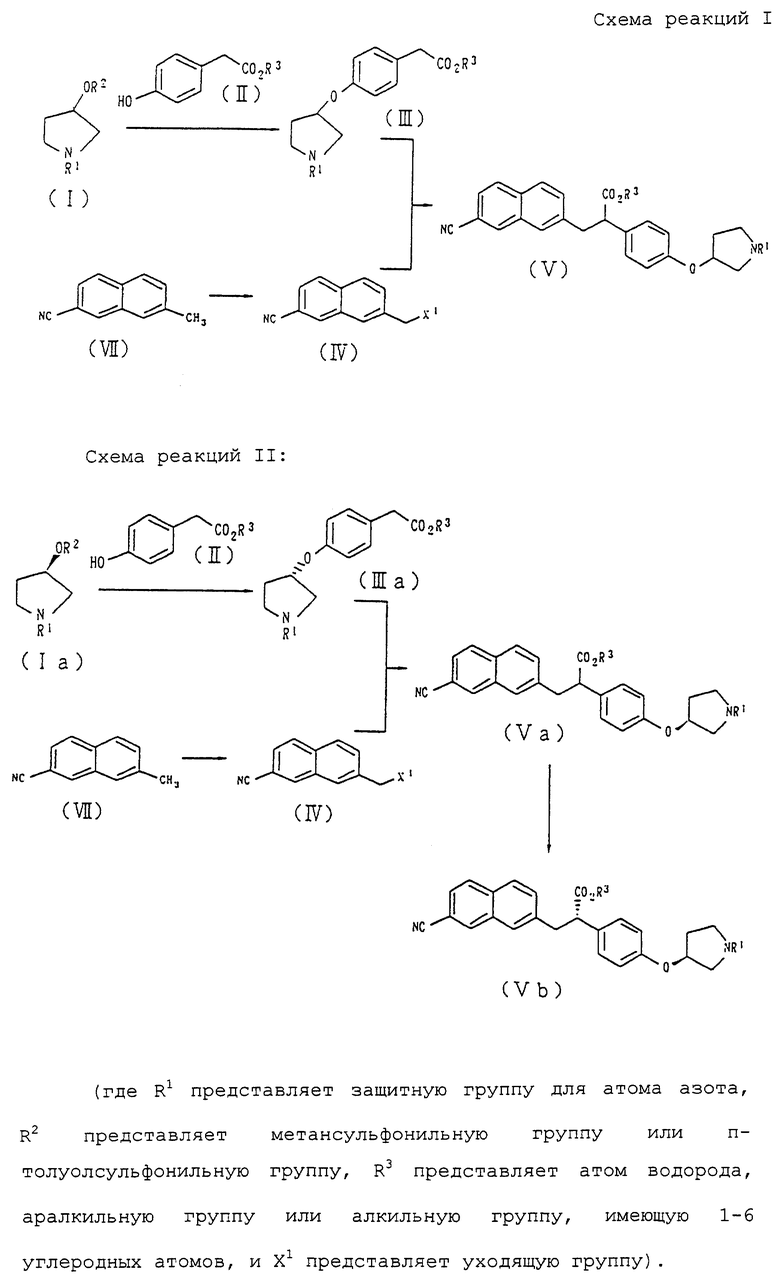
Изобретение относится к способу получения соединения формулы III или его соли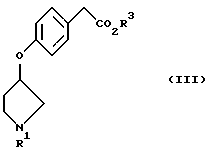
(где R1 представляет третбутоксикарбонил, бензил; R3 представляет C1-С6 алкил взаимодействием соединения формулы I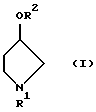
(где R1 имеет значение, определенное выше и R2 представляет метансульфонильную группу или п-толуолсульфонильную группу) с соединением формулы II
в присутствии основания. А также использованию соединения формулы III в получении соединения формулы V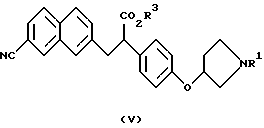
где R1 означает третбутоксикарбонил, бензил; R3 означает C1-С6 алкил, за счет взаимодействия соединения формулы III с соединением формулы IV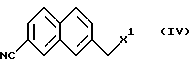
(где X -галоген, п-толуолсульфонилокси) в присутствии основания. 4 с. и 25 з.п. ф-лы.
(где R1 представляет третбутоксикарбонил, бензил и R3 представляет алкильную группу, имеющую 1-6 углеродных атомов),
отличающийся тем, что осуществляют взаимодействие соединения, представленного формулой I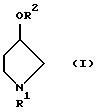
(где R1 имеет значение, определенное выше, и R2 представляет метансульфонильную группу или п-толуолсульфонильную группу)
с соединением, представленным формулой II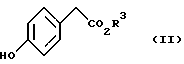
(где R3 имеет значение, определенное выше)
в присутствии основания.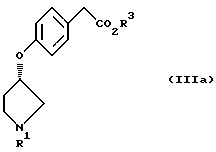
(где R1 представляет третбутоксикарбонил, бензил и R3 представляет алкильную группу, имеющую 1-6 углеродных атомов),
отличающийся тем, что осуществляют взаимодействие соединения, представленного формулой Ia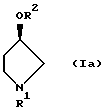
(где R1 имеет значение, определенное выше, и R2 представляет метансульфонильную группу или п-толуолсульфонильную группу)
с соединением, представленным формулой II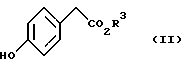
(где R3 имеет значение, определенное выше) в присутствии основания.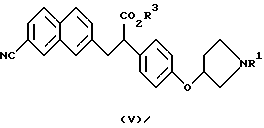
(где R1 представляет третбутоксикарбонил, бензил и R3 представляет алкильную группу, имеющую 1-6 углеродных атомов)
отличающийся тем, что осуществляют взаимодействие соединения, представленного формулой III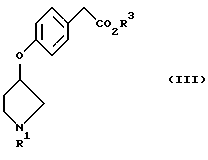
(где R1 и R3 имеют значения, определенные выше)
с соединением, представленным формулой IV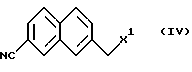
(где X1 представляет атом галогена, п-толуолсульфонилокси)
в присутствии основания.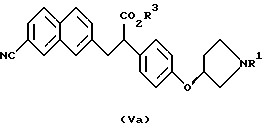
(где R1 представляет третбутоксикарбонил, бензил и R3 представляет алкильную группу, имеющую 1-6 атомов углерода)
отличающийся тем, что включает взаимодействие соединения, представленного формулой IIIa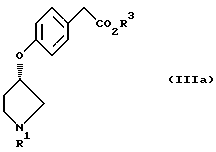
(где R1 и R3 имеют значение, определенные выше)
с соединением, представленным формулой IV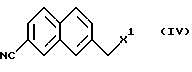
(где Х1 представляет атом галогена, п-толуолсульфонилокси)
в присутствии основания.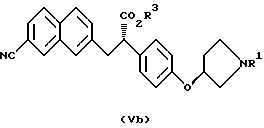
(где R1 представляет третбутоксикарбонил, бензил и R3 представляет аралкильную группу или алкильную группу, имеющую 1-6 углеродных атомов)
отличающийся тем, что соединение, представленное формулой Va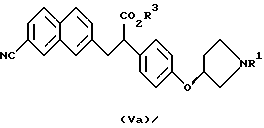
(где R1 и R3 имеют значения, определенные выше)
растворяют в протонном растворителе и подвергают взаимодействию с основанием.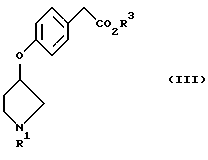
(где R1 представляет третбутоксикарбонил, бензил и R3 представляет алкильную группу, имеющую 1-6 углеродных атомов)
или его соль.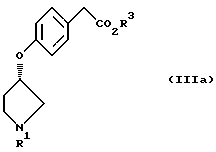
(где R1 представляет третбутоксикарбонил, бензил и R3 представляет алкильную группу, имеющую 1-6 углеродных атомов), или его соль.
Приоритет по пунктам:
06.09.1996, заявка 8/237013 по пп. 1-11, 12-20,28,29;
06.09.1996, заявка 8/237014 по пп. 21-26, 27.
| Кипятильник для воды | 1921 |
|
SU5A1 |
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| СПОСОБ ПОЛУЧЕНИЯ ХЛОРАЛЛЕНОВЫХ НИТРИЛОВ | 0 |
|
SU318569A1 |
| ЭЛЕКТРОННОЕ УСТРОЙСТВО ДЛЯ РАСЧЕТА МНОГОСЛОЙНЫХ ИНТЕРФЕРЕНЦИОННЫХ ПОКРЫТИЙ | 0 |
|
SU392515A1 |

