РОДСТВЕННЫЕ ЗАЯВКИ
По настоящей заявке испрашивается приоритет согласно предварительной заявке на патент США № 60/934201, поданной 12 июня 2007 года, и предварительной заявки на патент США № 61/067627, поданной 29 февраля 2008 года. Описания указанных заявок во всей полноте включены в настоящее описание посредством ссылки.
УРОВЕНЬ ТЕХНИКИ
Атазанавира сульфат, известный также как сульфат диметилового эфира (3S,8S,9S,12S)-3,12-бис(1,1-диметилэтил)-8-гидрокси-4,11-диоксо-9-(фенилметил)-6-[[4-(2-пиридинил)фенил]метил]-2,5,6,10,13-пентаазатетрадекандионовой кислоты, предотвращает образование зрелых вирионов ВИЧ в клетках, инфицированных ВИЧ-1, селективным ингибированием вирус-специфического процессинга некоторых полипротеинов (вирусных полипротеинов Gag и Gag-Pol). В настоящее время атазанавира сульфат утвержден в качестве лекарственного средства для лечения ВИЧ инфекции.
Противопоказанием для применения атазанавира является совместное применение с лекарственными средствами, которые являются высокозависимыми от CYP3A для клиренса и для которых повышенные концентрации в плазме связаны с серьезными и/или опасными для жизни событиями. Вследствие ингибиторного действия атазанавира на CYP3A, CYP2C8 и UGT1A1 для пациентов, принимающих атазанавир, предписывается осторожность при назначении лекарственных средств, метаболизируемых, главным образом, CYP3A, CYP2C8 и UGT1A1. Обычно неблагоприятные эффекты, связанные с атазанавиром, включают гипербилирубинемию, быстропроходящую сыпь, тошноту, головную боль и желтуху/склеральную желтуху. Неблагоприятные последствия, которые испытывают некоторые пациенты и причинная взаимосвязь которых не была установлена, включают сахарный диабет/гипергликемию, увеличение продолжительности PR интервала, гемофилию и перераспределение жира.
Несмотря на полезные активные свойства атазанавира существует потребность в новых соединениях для лечения указанных выше заболеваний и состояний.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к новым соединениям, которые представляют собой азапептиды, и их фармацевтически приемлемым солям. Точнее, изобретение относится к новым азапептидам, представляющим собой производные атазанавира сульфата, который является ингибитором ВИЧ протеазы. Данное изобретение предоставляет также апирогенные композиции, содержащие одно или несколько соединений согласно изобретению и носитель, и применение заявленных соединений и композиций в способах лечения заболеваний и состояний, которые лечатся введением ингибиторов ВИЧ протеазы. Изобретение также относится к применению одного или нескольких заявленных соединений в качестве реагентов в аналитических исследованиях, которые включают применение атазанавира.
Соединения согласно настоящему изобретению представляют собой соединения формулы А:
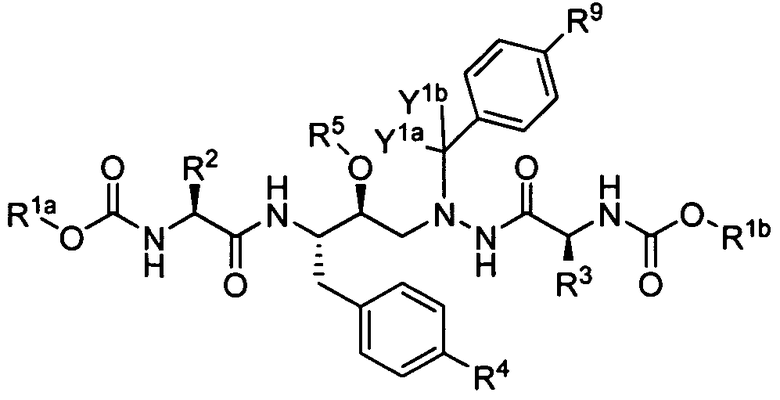
(А)
или их соли, гидраты или сольваты, где
каждый из R1a и R1b независимо выбран из С1-С3 алкила, где один или несколько атомов водорода в алкиле необязательно заменены атомом дейтерия;
каждый из R2 и R3 независимо выбран из изопропила, втор-бутила и трет-бутила, где один или несколько атомов водорода в изопропиле, втор-бутиле или трет-бутиле необязательно заменен(ы) атомом дейтерия;
R4 выбран из Н, ОН и -О-(СR6R7-O)n-R8;
R5 выбран из Н и -(СR6R7-O)n-R8, где:
каждый R6 и R7 независимо выбран из Н, С1-С6 алкила, С2-С6 алкенила, С2-С6 алкинила и С3-С7 циклоалкила; или
R6 и R7 вместе с атомом углерода, к которому они присоединены, образуют 3-7-членный циклоалкил;
каждый R8 независимо выбран из -С(О)Н, -С(О)-(С1-С7 алкила), -Р(О)-(ОН)2, -S(O)-OH, -S(O)2-OH и A-R11, где
А представляет собой остаток α-аминокислоты; и
R11 выбран из Н, С1-С6 алкила, -С(О)-(С1-С7 алкила), А-R12,
где R12 выбран из Н, С1-С6 алкила и -С(О)-(С1-С7 алкила);
n равно 0 или 1;
где любой алкил в R5 является необязательно замещенным;
каждый из Y1a и Y1b независимо выбран из Н и D;
R9 выбран из 2-тиенила, 3-тиенила, тиазол-5-ила, тиазол-2-ила, пиридин-2-ила, пиридин-3-ила, пиридин-4-ила, пиразин-2-ила, 2-метил-2Н-тетразол-5-ила, 2-(d 3-метил)-2Н-тетразол-5-ила, 1-метил-1Н-тетразол-5-ила и 1-(d 3-метил)-1Н-тетразол-5-ила; и
по меньшей мере, одна из переменных R1a, R1b, R2, R3 или Y включает атом дейтерия.
Соединения, их фармацевтически приемлемые соли и композиции согласно изобретению могут применяться для лечения заболеваний, которые эффективно лечатся соединением, являющимся ингибитором ВИЧ протеазы. Само по себе настоящее изобретение включает способ лечения заболевания, чувствительного к лечению соединением, которое является ингибитором ВИЧ протеазы, причем способ включает введение субъекту, нуждающемуся в таком лечении, эффективного количества: (i) соединения или его фармацевтически приемлемой соли; или (ii) апирогенной композиции (например, фармацевтической композиции) согласно настоящему изобретению.
Заболевания или состояния, чувствительные к лечению соединением, обладающим ингибиторной активностью в отношении ВИЧ протеазы, включают, но без ограничения, ВИЧ инфекцию.
Соединения или композиции согласно настоящему изобретению могут также применяться в качестве реагентов в способах определения концентрации атазанавира сульфата в растворе, исследованиях метаболизма атазанавира сульфата и других аналитических исследованиях. Дополнительная возможность применения соединений любой из приведенных в описании формул включает их применение в качестве внутренних стандартов для определения точной концентрации атазанавира сульфата в биологических матрицах, таких как плазма.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
ФИГ.1 представляет собой график, показывающий стабильность соединений согласно данному изобретению в микросомах печени человека по сравнению с атазанавиром.
ФИГ.2 представляет собой график, показывающий стабильность соединений согласно настоящему изобретению в микросомах печени человека по сравнению с атазанавиром.
ФИГ.3 представляет собой график, показывающий стабильность соединений согласно настоящему изобретению в микросомах печени человека по сравнению с атазанавиром.
ФИГ.4 представляет собой график, показывающий уровни содержания в плазме соединений согласно изобретению после перорального введения шимпанзе по сравнению с атазанавиром.
ФИГ.5 представляет собой график, показывающий уровни содержания в плазме соединений согласно настоящему изобретению после перорального введения шимпанзе по сравнению с атазанавиром.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Термины «улучшать» и «лечить» используются взаимозаменяемо и включают как терапевтическое лечение, так и профилактическое лечение (снижение вероятности развития). Оба термина означают снижение, подавление, ослабление, уменьшение, приостановку или стабилизацию развития или прогрессирования заболевания (например, заболевания или расстройства, определенного в описании), снижение тяжести заболевания или ослабление симптомов, связанных с заболеванием.
Термин «заболевание» означает любое состояние или расстройство, которое наносит ущерб или причиняет вред нормальной функции клетки, ткани или органа.
Следует представлять, что некоторое изменение природной изотопной распространенности имеет место в синтезированном соединении в зависимости от источника химических веществ, используемых в синтезе. Таким образом, препарат атазанавира будет по существу содержать небольшие количества дейтерированных изотопологов. Концентрация стабильных изотопов водорода, которая имеет место согласно природной распространенности, несмотря на такое отклонение, является небольшой и незначительной по сравнению со степенью стабильного изотопного замещения соединений согласно настоящему изобретению (см., например, Wada E. et al., Seikagaku 1994, 66:15; Ganes L.Z. et al., Comp. Biochem. Physiol. Mol. Integr. Physiol. 1998, 119:725).
Если не определено иначе, когда положение обозначено конкретно как «Н» или как «водород», следует представлять, что это положение содержит водород согласно природной распространенности его изотопного состава. Также если не определено иначе, когда положение точно определено как «D» или как «дейтерий», следует представлять, что положение содержит дейтерий в количестве, которое, по меньшей мере, в 3500 раз превышает природное содержание дейтерия, равное 0,015% (т.е., по меньшей мере, 52,5% введения дейтерия).
Термин «фактор изотопного обогащения», когда используется в данном описании, означает соотношение между изотопным избытком D в конкретном положении в соединении согласно настоящему изобретению и природным содержанием данного изотопа. Природное содержание дейтерия составляет 0,015%.
В других вариантах осуществления изобретения соединение согласно настоящему изобретению имеет фактор изотопного обогащения для каждого дейтерия, присутствующего в сайте, указанном в качестве возможного сайта дейтеризации на соединение, равный, по меньшей мере, 4000 (60% введения дейтерия), по меньшей мере, 4500 (67,5% введения дейтерия), по меньшей мере, 5000 (75% введения дейтерия), по меньшей мере, 5500 (82,5% введения дейтерия), по меньшей мере, 6000 (90% введения дейтерия), по меньшей мере, 6333,3 (95% введения дейтерия), по меньшей мере, 6466,7 (97% введения дейтерия), по меньшей мере, 6600 (99% введения дейтерия) или, по меньшей мере, 6633,3 (99,5% введения дейтерия). Следует представлять, что фактор изотопного обогащения каждого дейтерия, присутствующего в сайте, обозначенном в качестве сайта дейтеризации, не зависит от других дейтерированных сайтов. Например, если в соединении имеются два сайта дейтеризации, один сайт может быть дейтерированным на 52,5%, в то время как другой может быть дейтерированным на 75%. Тогда полученное соединение рассматривается как соединение, в котором фактор изотопного обогащения равен, по меньшей мере, 3500 (52,5%).
Термин «изотополог» относится к соединению, которое отличается от конкретного соединения согласно изобретению только изотопным составом. Изотопологи могут отличаться уровнем обогащения изотопов в одном или нескольких положениях и/или положением(ями) изотопного обогащения.
Следует представлять, что термин «соединение», когда относится к соединениям согласно данному изобретению, относится к совокупности молекул, имеющих идентичную химическую структуру, за исключением того, что может иметь место изменение изотопного состава атомов, образующих молекулу. Таким образом, специалисту в данной области техники должно быть понятно, что соединение, представленное конкретной химической структурой и включающее указанные атомы дейтерия, будет также содержать меньшие количества изотопологов, содержащих атомы водорода в одном или нескольких указанных положениях дейтерия в данной структуре. Относительное количество таких изотопологов в соединении согласно настоящему изобретению будет зависеть от ряда факторов, включая изотопную чистоту дейтерированных реагентов, использованных для получения соединения, и эффективности введения дейтерия на различных стадиях синтеза, используемых для получения соединения. Однако, как показано выше, относительное количество таких изотопологов будет составлять менее 47,5% соединения.
Подразумевается также, что термин «соединение» включает любые сольваты или гидраты данного соединения.
Соль соединения согласно настоящему изобретению образуется между кислотной и оснóвной группой соединения, такой как функциональная аминогруппа, или основанием и кислотной группой соединения, такой как функциональная карбоксильная группа. В соответствии с другим вариантом осуществления изобретения соединение представляет собой фармацевтически приемлемую кислотно-аддитивную соль.
Термин «фармацевтически приемлемый», когда используется в данном описании, относится к компоненту, который в пределах области действия стандартной медицинской оценки подходит для использования в контакте с тканями людей и других млекопитающих без избыточной токсичности, раздражения, аллергической реакции и т.п. и обладает приемлемым соотношением «польза/риск». Термин «фармацевтически приемлемая соль» означает любую нетоксичную соль, которая при введении реципиенту способна предоставлять непосредственно или опосредованно соединение согласно настоящему изобретению. Термин «фармацевтически приемлемый противоион» относится к ионной части соли, которая является нетоксичной, когда высвобождается из соли при введении реципиенту.
Кислоты, обычно используемые для получения фармацевтически приемлемых солей, включают неорганические кислоты, такие как сероводородная кислота, хлористоводородная кислота, бромистоводородная кислота, йодистоводородная кислота, серная кислота и фосфорная кислота, а также органические кислоты, такие как пара-толуолсульфоновая кислота, салициловая кислота, винная кислота, аскорбиновая кислота, малеиновая кислота, бензолсульфоновая кислота, фумаровая кислота, глюконовая кислота, глюкуроновая кислота, муравьиная кислота, глутаминовая кислота, метансульфоновая кислота, этансульфоновая кислота, бензолсульфоновая кислота, молочная кислота, щавелевая кислота, пара-бромфенилсульфоновая кислота, угольная кислота, янтарная кислота, лимонная кислота, бензойная кислота и уксусная кислота, а также родственные неорганические и органические кислоты. Следовательно, такие фармацевтически приемлемые соли включают сульфат, пиросульфат, бисульфат, сульфит, бисульфит, фосфат, моногидрофосфат, дигидрофосфат, метафосфат, пирофосфат, хлорид, бромид, йодид, ацетат, пропионат, деканоат, каприлат, акрилат, формиат, изобутират, капрат, гептаноат, пропиолат, оксалат, малонат, сукцинат, суберат, себакат, фумарат, малеат, бутин-1,4-диоат, гексин-1,6-диоат, бензоат, хлорбензоат, метилбензоат, динитробензоат, гидроксибензоат, метоксибензоат, фталат, терефталат, сульфонат, ксилолсульфонат, фенилацетат, фенилпропионат, фенилбутират, цитрат, лактат, β-гидроксибутират, гликолят, малеат, тартрат, метансульфонат, пропансульфонат, нафталин-1-сульфонат, нафталин-2-сульфонат, манделат и другие соли. В одном варианте осуществления изобретения фармацевтически приемлемые кислотно-аддитивные соли включают соли, полученные с минеральными кислотами, такими как хлористоводородная кислота и бромистоводородная кислота, в частности соли, полученные с органическими кислотами, такими как малеиновая кислота.
Для соединений согласно изобретению, включающих -P(O)-(OH)2, -S(O)-OH, -S(O)2-OH, подходящие катионные фрагменты для образования фармацевтически приемлемых солей включают, но без ограничения, щелочные металлы, такие как натрий, калий и литий; щелочноземельные металлы, такие как кальций и магний; другие металлы, такие как алюминий и цинк; аммиак и органические амины, такие как моно-, ди- или триалкиламины; дициклогексиламин; трибутиламин; пиридин; N-метил-, N-этиламин; диэтиламин; триэтиламин; моно-, бис- или трис-(2-гидрокси-низший алкил)амины, такие как моно-, бис или трис-(2-гидроксиэтил)амин, 2-гидрокси-трет-бутиламин или трис-(гидроксиметил)метиламин, N,N-ди-низший алкил-N-(гидрокси низший алкил)амины, такие как N,N-диметил-N-(2-гидроксиэтил)амин или три-(2-гидроксиэтил)амин; N-метил-D-глюкамин; аминокислоты, такие как аргинин, лизин и т.п., и цвиттерионы, такие как глицин и т.п.
Термин «гидрат», когда используется в данном описании, относится к соединению, которое дополнительно включает стехиометрическое или нестехиометрическое количество воды, связанной нековалентными внутримолекулярными силами.
Термин «сольват», когда используется в данном описании, относится к соединению, которое дополнительно включает стехиометрическое или нестехиометрическое количество растворителя, такого как вода, ацетон, этанол, метанол, дихлорметан, 2-пропанол и т.п., связанного нековалентными внутримолекулярными связями.
Заявленные соединения могут существовать в различных стехиометрических формах. Стереоизомеры представляют собой соединения, которые различаются только расположением их атомов в пространстве. Энантиомеры представляют собой пары стереоизомеров, «зеркальные изображения» которых не могут совмещаться при наложении, в большинстве случаев ввиду того, что они содержат асимметрически замещенный атом углерода, который действует как хиральный центр. Термин «энантиомер» относится к одной из пары молекул, которые являются зеркальными изображениями друг друга и не совмещаются при наложении. Диастереомеры представляют собой стереоизомеры, которые не являются «зеркальными изображениями», главным образом, так как они содержат два или несколько асимметрически замещенных атомов углерода. Символы «R» и «S» означают конфигурацию заместителей относительно одного или нескольких хиральных атомов углерода.
Когда стереохимия заявленных соединений названа или представлена структурой, названный или изображенный стереоизомер составляет, по меньшей мере, 60%, 70%, 80%, 90%, 99% или 99,99% из расчета на чистую массу относительно других стереоизомеров. Когда единственный энантиомер назван или представлен структурой, представленный структурой или названный энантиомер является, по меньшей мере, на 60%, 70%, 80%, 90%, 99% или 99,9% оптически чистым. Оптическая чистота, выраженная в процентах по массе, представляет собой отношение массы энантиомера к суммарной массе энантиомера и его оптического изомера.
Когда заявленное соединение названо или представлено структурой без указания стереохимии и содержит, по меньшей мере, один хиральный центр, следует представлять, что название или структура включает один энантиомер соединения, свободный от соответствующего оптического изомера, рацемическую смесь соединения и смеси, обогащенные одним энантиомером относительно соответствующего оптического изомера («скалемические смеси»).
Когда заявленное соединение названо или представлено структурой без указания стереохимии и содержит, по меньшей мере, два хиральных центра, следует представлять, что название или структура включает диастереомер, свободный от других диастереомеров, пару диастереомеров, свободную от других диастереомерных пар, смеси диастереомеров, смеси диастереомерных пар, смеси диастереомеров, в которых один диастереомер обогащен относительно другого(их) диастереомера(ов), и смеси диастереомерных пар, в которых одна диастереомерная пара обогащена относительно другой(их) диастереомерной(ных) пары (пар).
Выражение «по существу свободен от других стереоизомеров», когда используется в описании, означает, что присутствует менее 25% других стереоизомеров, предпочтительно менее 10% других стереоизомеров, более предпочтительно менее 5% других стереоизомеров и наиболее предпочтительно менее 2% других стереоизомеров или менее «Х»% других стереоизомеров (где Х представляет собой число в интервале от 0 до 100).
Термин «стабильные соединения», когда используется в данном описании, относится к соединениям, которые обладают стабильностью, достаточной для их производства, и которые сохраняют целостность в течение достаточного периода времени для того, чтобы применяться для целей, подробно описанных далее (например, для введения в терапевтические препараты, получения промежуточных продуктов для применения при получении соединений, обладающих терапевтическими свойствами, для выделения или хранения промежуточных продуктов, для лечения заболевания или состояния, чувствительного к терапевтическим средствам).
Символ «D» относится к дейтерию. Термин «стереоизомер» относится как к энантиомерам, так и диастереомерам. Приставка «трет»-, символы «t» и «t-» означают «третичный». Аббревиатура «США» означает Соединенные Штаты Америки. Аббревиатура “FDA” означает Управление по контролю за продуктами и лекарствами. Аббревиатура “NDA” означает Заявку на новое лекарственное средство.
Термин «необязательно замещенный» относится к возможной замене одного или нескольких атомов водорода другим фрагментом. За исключением особо оговоренных случаев, любой атом водорода, включая концевые атомы водорода, может быть необязательно заменен.
Термин «галоген» относится к любому элементу из -Cl, -F, -Br или -I.
Термин «оксо» относится к группе =О.
Термин «алкокси» относится к -О-алкилу.
Термин «алкиламино» относится к -NH-алкилу.
Термин «диалкиламино» относится к N(алкил)алкилу, где два алкильных фрагмента являются одинаковыми или различающимися.
Термин «алкил» относится к прямым или разветвленным алкильным цепям, содержащим от 1 до 12 атомов углерода, предпочтительно от 1 до 8 атомов углерода, более предпочтительно от 1 до 4 атомов углерода, если не указано иного. Примеры алкильных групп с прямой или разветвленной цепью включают метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, трет-бутил, пентил, гексил, гептил и октил. Алкил может быть необязательно замещенным.
Алкильная или арильная группы, которые являются необязательно замещенными, обычно будут содержать от одного до четырех заместителей, которые выбраны независимо. Примеры необязательных заместителей включают С1-7 алкил, галоген, цианогруппу, гидроксильную группу, карбоксигруппу, алкоксигруппу, оксогруппу, аминогруппу, алкиламиногруппу, диалкиламиногруппу, гетероциклоалкил, алкилгетероциклоалкил, арил, алкиларил, гетероарил и алкилгетероарил.
Термин «гетероциклоалкил» относится к неароматической моноциклической, бициклической, трициклической, спироциклической или тетрациклической кольцевой системе, которая включает один или несколько гетероатомов, таких как атом азота, атом кислорода или атом серы, по меньшей мере, в одном из циклов. Каждый цикл может быть четырех-, пяти-, шести-, семи- или восьмичленным. Примеры таких гетероциклоалкилов включают тетрагидрофурил, тетрагидротиофенил, морфолино, тиоморфолино, пирролидинил, пиперазинил, пиперидинил и тиазолидинил, а также циклические формы сахаров.
Термин «алкилгетероциклоалкил» относится к гетероциклоалкильной группе, включающей алкильный заместитель. Примеры алкилгетероциклоалкилов включают 4-метилпиперазин-1-ил и 4-метилпиперидин-1-ил.
Термин «арил» относится к карбоциклическим ароматическим группам, таким как фенил и нафтил.
Термин «алкиларил» относится к арильной группе, связанной с основной частью молекулы через алкильную цепь.
Термин «гетероарил» относится к моноциклическим ароматическим группам, содержащим в цикле один или несколько гетероатомов, таких как атом азота, атом кислорода или атом серы, и включает имидазолил, тиенил, фурил, пиридил, пиримидил, пиранил, пиразолил, пирролил, пиразинил, тиазолил, оксазолил и тетразолил. Гетероарильные группы включают также конденсированные полициклические ароматические кольцевые системы, в которых, по меньшей мере, один цикл включает один или несколько гетероатомов, таких как атом азота, атом кислорода или атом серы. Примеры гетероарильных групп включают бензотиенил, бензофурил, индолил, хинолинил, бензотиазол, бензоксазол, бензимидазол, хинолинил, изохинолинил и изоиндолил.
Термин «алкилгетероарил» относится к гетероарильной группе, связанной с основной частью молекулы через алкильную цепь.
Термин «остаток α-аминокислоты» относится к группе общей формулы -С(О)-CHR-NH- и включает остаток природных и синтетических аминоксилот с D- или L-конфигурацией.
За исключением особо оговоренных случаев термин «α-аминокислота» включает α-аминокислоты с (D)-, (L)- или рацемической (D,L)-конфигурацией. Следует представлять, что когда переменная R8 представляет собой α-аминокислоту, она связана с остальной частью молекулы через атом углерода карбонильной группы, непосредственно связанный с α-углеродом аминокислоты. Согласно структуре формулы I такая связь приводит к образованию сложного эфира.
В данном описании переменная может обозначаться общим символом (например, «каждый R») или может обозначаться более точно (например, R1, R2, R3 и т.д.). За исключением особо оговоренных случаев считается, что указанное обозначение включает все специфические варианты осуществления данной конкретной переменной.
Соединения согласно изобретению представляют собой соединения, представленные формулой А:
(А)
или их соль, гидрат или сольват,
где
каждый из R1a и R1b независимо выбран из С1-С3 алкила, в котором один или несколько атомов водорода необязательно заменен(ы) атомом дейтерия;
каждый из R2 и R3 независимо выбран из изопропила, втор-бутила и трет-бутила, где один или несколько атомов водорода в изопропиле, втор-бутиле или трет-бутиле заменен(ы) атомом дейтерия;
R4 выбран из Н, ОН и -О-(CR6R7-O)n-R8;
R5 выбран из Н и -О-(CR6R7-O)n-R8, где
каждый из R6 и R7 независимо выбран из Н, С1-С6 алкила, С2-С6 алкенила, С2-С6 алкинила и С3-С7 циклоалкила, или
R6 и R7 вместе с атомом углерода, к которому они присоединены, образуют 3-7-членный циклоалкил;
каждый R8 независимо выбран из -C(O)H, -C(O)-(C1-C7 алкила), -P(O)-(OH)2, -S(O)-OH, -S(O)2-OH и A-R11, где
А представляет собой остаток α-аминокислоты; и
R11 выбран из С1-С6 алкила, -С(О)-(С1-С7 алкила), А-R12,
где R12 выбран из Н, С1-С6 алкила и -С(О)-(С1-С7 алкила); и
n равно 0 или 1;
где любой алкил в R5 является необязательно замещенным;
каждый Y1a и Y1b независимо выбран из Н и D;
R9 выбран из 2-тиенила, 3-тиенила, тиазол-5-ила, тиазол-2-ила, пиридин-2-ила, пиридин-3-ила, пиридин-4-ила, пиразин-2-ила, 2-метил-2Н-тетразол-5-ила, 2-(d 3-метил)-2Н-тетразол-5-ила, 1-метил-1Н-тетразол-5-ила и 1-(d 3-метил)-1Н-тетразол-5-ила; и
по меньшей мере, одна из переменных R1a, R1b, R2, R3 или Y содержит атом дейтерия.
Конкретные варианты осуществления формулы А включают соединения, где
а) один или оба из R2 и R3 содержат атом дейтерия;
b) каждый из R2 и R3 независимо выбран из -C(CH3)3, -C(CD3)3, -CH(CH3)2, -CD(CD3)2, -CH2CH2(CH3)2 и -CD2CD2(CD3);
с) один или оба из R1a и R1b содержат атом дейтерия;
d) каждый из R1a и R1b независимо выбран из -CH3, -CD3, -CH2CH3, -CD2CD3, -CD2CD2CD3 и -CH2CH2CH3;
е) R5 представляет собой H, P(O)-(OH)2, -CH2-O-P(O)-(OH)2 или их фармацевтически приемлемую соль;
f) R2 выбран из -C(CD3)3, -CD(CD3)2 и -CD2CD2(CD3)2; или
g) имеют место одновременно два или несколько из параметров, описанных в пунктах а)-f), представленных выше.
В одном варианте осуществления изобретения соединения согласно изобретению представляют собой соединения формулы I:
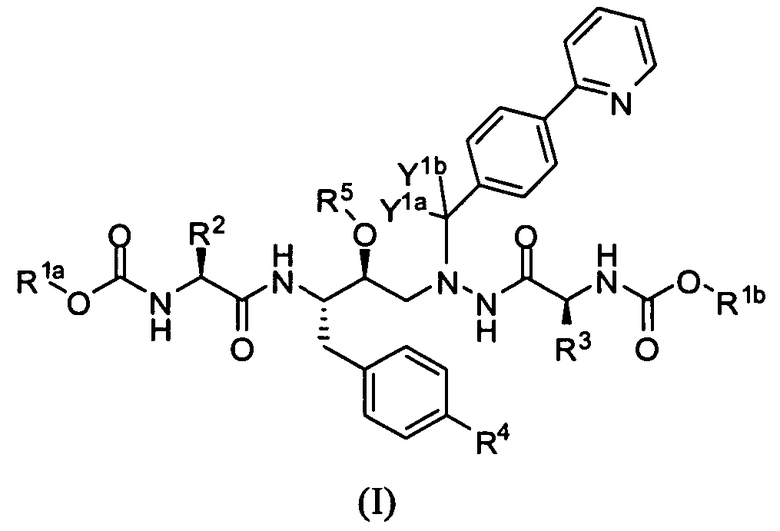
или их фармацевтически приемлемую соль, где:
каждый из R1a и R1b независимо выбран из CH3, CH2D, CHD2 и CD3;
каждый из R2 и R3 независимо представляет собой -С(СН3)3, где от 1 до 9 атомов водорода необязательно заменены атомом дейтерия;
R4 выбран из Н, ОН и -О-(CR6R7-O)n-R8;
R5 выбран из Н и -(CR6R7-O)n-R8, где:
R6 и R7 независимо выбраны из Н и С1-С3 алкила;
каждый R8 независимо выбран из α-аминокислоты, -C(O)H, -C(O)-(C1-C7 алкила), где указанный C1-C7 алкил является необязательно замещенным, -P(O)-(OH)2 и -S(O)-OH;
n равно 0 или 1;
Y1a и Y1b независимо выбраны из Н и D; и
по меньшей мере, одна из переменных R1a, R1b, R2, R3 или Y содержит атом дейтерия.
Конкретные варианты соединений формулы I включают соединение, где:
i. каждый из R1a и R1b независимо выбран из СН3 и CD3;
ii. каждый из R2 и R3 независимо выбран из -С(СН3)3 и -C(CD3)3;
iii. R2 представляет собой -С(СD3)3;
iv. Y1a и Y1b являются одинаковыми;
v. каждый из Y1a и Y1b представляет собой дейтерий;
vi. R4 выбран из Н и -O-(CR6R7-O)n-R8;
vii. R4 и R5 одновременно представляют собой Н;
viii. каждый R6 и каждый R7 представляет собой Н;
ix. каждый R8 независимо выбран из α-аминокислоты с (L)-конфигурацией; -С(О)Н; -С(О)-(С1-С3 алкила), где указанный С1-С3 алкил является необязательно замещенным цианогруппой, гидроксильной группой, карбоксигруппой, алкоксигруппой, аминогруппой, алкиламиногруппой, диалкиламиногруппой, гетероциклоалкилом, алкилгетероциклоалкилом, арилом, алкиларилом, гетероарилом и алкилгетероарилом; -Р(О)-(ОН)2; или соли -Р(О)-(ОН)2, где катион выбран из Na+, Mg2+ или аммония; -S(O)-OH; и соли -S(O)-OH, где катион выбран из Na+, Mg2+ или аммония;
х. каждый R8 независимо выбран из L-серина, L-лизина, L-тирозина, L-валина, L-глутаминовой кислоты, L-аспаргиновой кислоты, L-3-пиридилаланина, L-гистидина, -С(О)Н, -С(О)-(С1-С3 алкила), -C(O)CH2OCH3; -C(O)CH2CH2OCH3; -C(O)CH2CH2C(O)OH; -C(O)CH2CH2NH2; -С(О)СН2СН2NHCH3; -C(O)CH2CH2N(CH3);  ;
;  ;
; 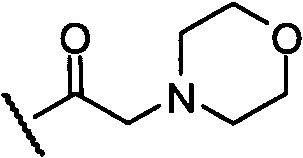 ; -Р(О)-(ОН)2; соли -Р(О)-(ОН)2, где катион выбран из Na+, К+ или Са2+; -S(O)-OH; и соли -S(O)-OH, где катион выбран из Na+, К+ или Са2+; или
; -Р(О)-(ОН)2; соли -Р(О)-(ОН)2, где катион выбран из Na+, К+ или Са2+; -S(O)-OH; и соли -S(O)-OH, где катион выбран из Na+, К+ или Са2+; или
xi. в соединении присутствуют два или несколько параметров i.-x.
Примеры вариантов осуществления изобретения, где встречаются два или несколько из указанных выше параметров, включают, но без ограничения, следующие конкретные варианты осуществления изобретения.
В одном конкретном варианте осуществления R2 и R3 независимо выбраны из -С(СН3)3 и -C(CD3)3, и R1a и R1b независимо выбраны из СН3 и CD3.
В другом конкретном варианте осуществления изобретения R2 представляет собой -C(CD3)3, и R1a представляет собой CD3.
В еще одном конкретном варианте осуществления изобретения R2 представляет собой -C(CD3)3, R1a представляет собой CD3, и R1b представляет собой CD3.
В еще одном варианте осуществления изобретения Y1a и Y1b являются одинаковыми (т.е. оба одновременно представляют собой дейтерий или одновременно представляют собой Н), и либо каждый из R1a и R1b независимо выбран из СН3 и CD3, либо R2 и R3 независимо выбраны из -С(СН3)3 и -C(CD3)3. В более точно определенном варианте осуществления Y1a и Y1b являются одинаковыми (например, оба представляют собой дейтерий), R1a и R1b независимо выбраны из СН3 и CD3, и R2 и R3 независимо выбраны из -С(СН3)3 и -C(CD3)3.
В еще одном более точно определенном варианте осуществления изобретения R4 выбран из Н и -O-(CR6R7-O)n-R8; и либо R1a и R1b независимо выбраны из СН3 и CD3, либо R2 и R3 независимо выбраны из -С(СН3)3 и -C(CD3)3. В более точно определенном варианте осуществления R4 выбран из Н и -O-(CR6R7-O)n-R8; R1a и R1b независимо выбраны из СН3 и CD3, R2 и R3 независимо выбраны из -С(СН3)3 и -C(CD3)3.
В еще одном конкретном варианте осуществления изобретения R4 выбран из Н и -O-(CR6R7-O)n-R8; и Y1a и Y1b являются одинаковыми. В более точно определенном варианте осуществления изобретения R4 выбран из Н и -O-(CR6R7-O)n-R8; и Y1a и Y1b представляют собой дейтерий. В еще одном более точно определенном варианте осуществления изобретения R4 выбран из Н и -O-(CR6R7-O)n-R8, Y1a и Y1b являются одинаковыми (т.е. оба одновременно представляют собой дейтерий или одновременно представляют собой Н), и либо R1a и R1b независимо выбраны из СН3 и CD3, либо R2 и R3 независимо выбраны из -С(СН3)3 и -C(CD3)3. В наиболее точно определенном варианте осуществления изобретения R4 выбран из Н и -O-(CR6R7-O)n-R8, Y1a и Y1b являются одинаковыми (т.е. оба одновременно представляют собой дейтерий или одновременно представляют собой Н), R1a и R1b независимо выбраны из СН3 и CD3, и R2 и R3 независимо выбраны из -С(СН3)3 и -C(CD3)3.
В еще одном конкретном варианте осуществления изобретения каждый из R6 и R7 представляет собой Н, и либо R1a и R1b независимо выбраны из СН3 и CD3, либо R2 и R3 независимо выбраны из -С(СН3)3 и -C(CD3)3. В более точно определенном варианте осуществления изобретения каждый R6 и R7 представляет собой Н, R1a и R1b независимо выбраны из СН3 и CD3, и R2 и R3 независимо выбраны из -С(СН3)3 и -C(CD3)3.
В еще одном конкретном варианте осуществления изобретения каждый из R6 и R7 представляет собой Н, и Y1a и Y1b являются одинаковыми. В более точно определенном варианте осуществления изобретения каждый из R6 и R7 представляет собой Н, и Y1a и Y1b представляют собой дейтерий. В еще более точно определенном варианте осуществления изобретения каждый из R6 и R7 представляет собой Н, Y1a и Y1b являются одинаковыми (т.е. оба одновременно представляют собой дейтерий или одновременно представляют собой Н), и либо R1a и R1b независимо выбраны из СН3 и CD3, либо R2 и R3 независимо выбраны из -С(СН3)3 и -C(CD3)3. В наиболее точно определенном варианте осуществления изобретения каждый из R6 и R7 представляет собой Н, Y1a и Y1b являются одинаковыми (например, оба представляют собой дейтерий), R1a и R1b независимо выбраны из СН3 и CD3, R2 и R3 независимо выбраны из -С(СН3)3 и -C(CD3)3.
В еще одном конкретном варианте осуществления изобретения каждый из R6 и R7 представляет собой Н, и R4 выбран из Н и -O-(CR6R7-O)n-R8. В более точно определенном варианте осуществления изобретения каждый из R6 и R7 представляет собой Н, R4 выбран из Н и -O-(CR6R7-O)n-R8, и либо R1a и R1b независимо выбраны из СН3 и CD3, либо R2 и R3 независимо выбраны из -С(СН3)3 и -C(CD3)3. В наиболее точно определенном варианте осуществления изобретения каждый из R6 и R7 представляет собой Н, R4 выбран из Н и -O-(CR6R7-O)n-R8, R1a и R1b независимо выбраны из СН3 и CD3, и R2 и R3 независимо выбраны из -С(СН3)3 и -C(CD3)3.
В еще одном конкретном варианте осуществления изобретения каждый из R6 и R7 представляет собой Н, R4 выбран из Н и -O-(CR6R7-O)n-R8, Y1a и Y1b являются одинаковыми. В более точно определенном варианте осуществления изобретения каждый из R6 и R7 представляет собой Н, R4 выбран из Н и -O-(CR6R7-O)n-R8, Y1a и Y1b представляют собой дейтерий. В еще более точно определенном варианте осуществления изобретения каждый из R6 и R7 представляет собой Н, R4 выбран из Н и -O-(CR6R7-O)n-R8, Y1a и Y1b являются одинаковыми (т.е. оба одновременно представляют собой дейтерий или оба одновременно представляют собой Н), и либо R1a и R1b независимо выбраны из СН3 и CD3, либо R2 и R3 независимо выбраны из -С(СН3)3 и -C(CD3)3. В наиболее точно определенном варианте осуществления изобретения каждый из R6 и R7 представляет собой Н, R4 выбран из Н и -O-(CR6R7-O)n-R8, Y1a и Y1b являются одинаковыми (т.е. оба одновременно представляют собой дейтерий или оба одновременно представляют собой Н), R1a и R1b независимо выбраны из СН3 и CD3, и R2 и R3 независимо выбраны из -С(СН3)3 и -C(CD3)3.
В еще одном ряду вариантов осуществления изобретения для любого одного из перечисленных выше вариантов осуществления изобретения R8 независимо выбран из α-аминокислоты с (L)-конфигурацией; -С(ОН); -С(О)-(С1-С3 алкила), где указанный С1-С3 алкил является необязательно замещенным цианогруппой, гидроксильной группой, карбоксигруппой, алкоксигруппой, аминогруппой, алкиламиногруппой, диалкиламиногруппой, циклогетероалкилом, алкилгетероциклоалкилом, арилом, алкиларилом, гетероарилом и алкилгетероарилом; -Р(О)-(ОН)2; соли -Р(О)-(ОН)2, где катион выбран из Na+, К+ или Са2+; -S(O)-OH; и соли -S(O)-OH, где катион выбран из Na+, К+ или Са2+.
В еще одном ряду вариантов осуществления изобретения для любого одного из перечисленных выше вариантов осуществления изобретения R8 независимо выбран из L-серина, L-лизина, L-тирозина, L-валина, L-глутаминовой кислоты, L-аспаргиновой кислоты, L-3-пиридилаланина, L-гистидина, -С(О)Н, -С(О)-(С1-С3 алкила), -C(O)CH2OCH3; -C(O)CH2CH2OCH3; -C(O)CH2CH2C(O)OH; -C(O)CH2CH2NH2; -С(О)СН2СН2NHCH3; -C(O)CH2CH2N(CH3)2; ; ; ; -Р(О)-(ОН)2; соли -Р(О)-(ОН)2, где катион выбран из Na+, Mg2+ или аммония; S(O)-OH; и соли -S(O)-OH, где катион выбран из Na+, Mg2+ или аммония.
В еще одном конкретном варианте осуществления изобретения R4 и R5 одновременно представляют собой Н, и либо R1a и R1b независимо выбраны из СН3 и CD3, либо R2 и R3 независимо выбраны из -С(СН3)3 и -C(CD3)3. В еще более точно определенном варианте осуществления изобретения R4 и R5 одновременно представляют собой Н, R1a и R1b независимо выбраны из СН3 и CD3, и R2 и R3 независимо выбраны из -С(СН3)3 и -C(CD3)3.
В еще одном конкретном варианте осуществления изобретения R4 и R5 одновременно представляют собой Н, и Y1a и Y1b являются одинаковыми. В более точно определенном варианте осуществления изобретения R4 и R5 одновременно представляют собой Н, и Y1a и Y1b одновременно представляют собой дейтерий. В еще более точно определенном варианте осуществления изобретения R4 и R5 одновременно представляют собой Н, Y1a и Y1b являются одинаковыми (т.е оба одновременно представляют собой дейтерий или одновременно представляют собой Н), и либо R1a и R1b независимо выбраны из СН3 и CD3, либо R2 и R3 независимо выбраны из -С(СН3)3 и -C(CD3)3. В наиболее точно определенном варианте осуществления изобретения R4 и R5 одновременно представляют собой Н, Y1a и Y1b являются одинаковыми (т.е. оба одновременно представляют собой дейтерий или одновременно представляют собой Н), R1a и R1b независимо выбраны из СН3 и CD3, и R2 и R3 независимо выбраны из -С(СН3)3 и -C(CD3)3.
В еще одном варианте осуществления изобретения соединение представляет собой соединение формулы Ia:
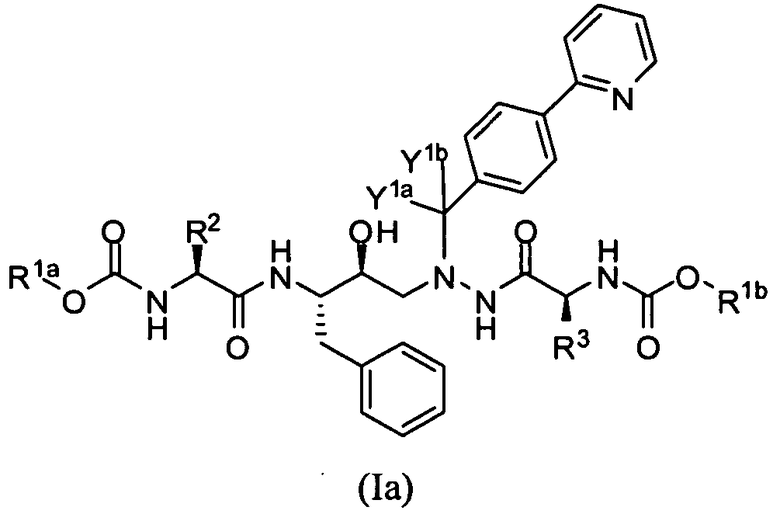
или его фармацевтически приемлемую соль и выбрано из любого одного из соединений, представленных в таблице 1 ниже.
Примеры вариантов осуществления соединений формулы Ia
В еще одном варианте осуществления изобретения соединение представляет собой соединение формулы Ib:
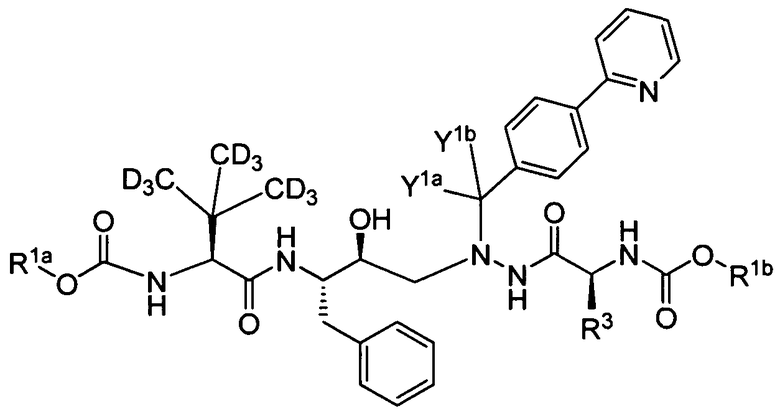
(Ib)
или его фармацевтически приемлемую соль, где:
каждый из R1a и R1b независимо выбран из -CD3 и -CH3;
R3 выбран из -C(CD3)3 и -C(CH3)3; и
Y1a и Y1b являются одинаковыми и выбраны из Н и D.
В еще одном варианте осуществления изобретения соединение представляет собой соединение формулы Ic:
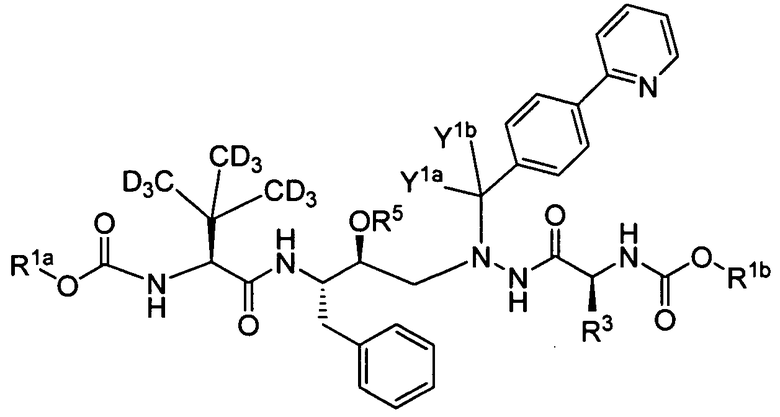
(Ic)
или его фармацевтически приемлемую соль, где:
каждый из R1a и R1b независимо выбран из -CD3 и -CH3;
R3 выбран из -C(CD3)3 и C(CH3)3;
R5 представляет собой -Р(О)-(ОН)2, -СН2-Р(О)-(ОН)2 или его фармацевтически приемлемую соль из вышеуказанных фрагментов; и
Y1a и Y1b являются одинаковыми и выбраны из Н и D.
В еще одном варианте осуществления изобретения соединение согласно изобретению выбрано из следующих соединений;
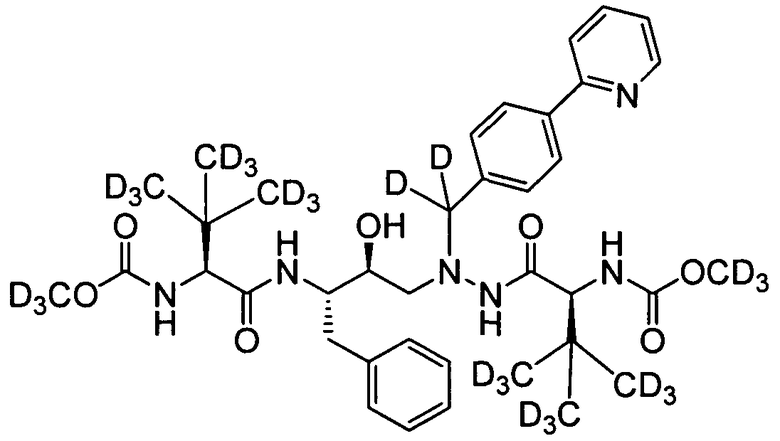 соединение 131;
соединение 131;
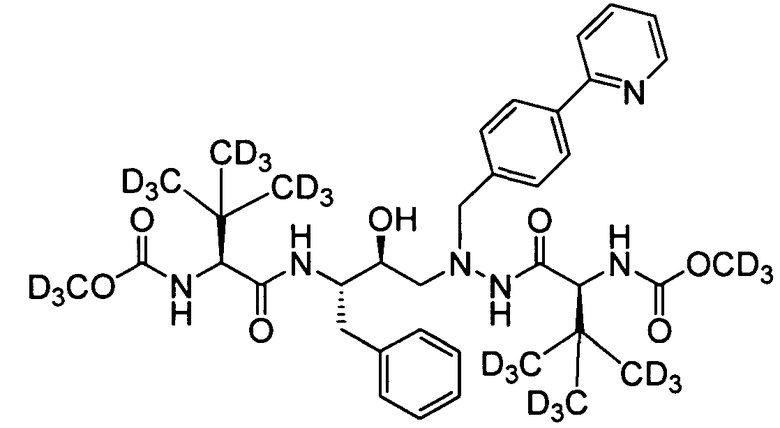 соединение 122;
соединение 122;
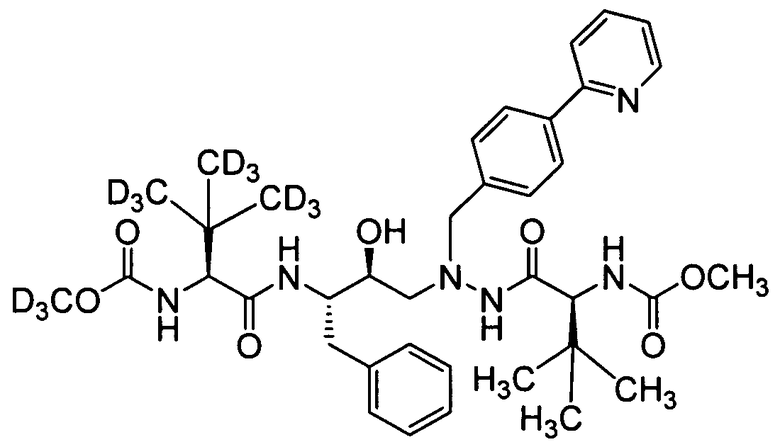 соединение 114;
соединение 114;
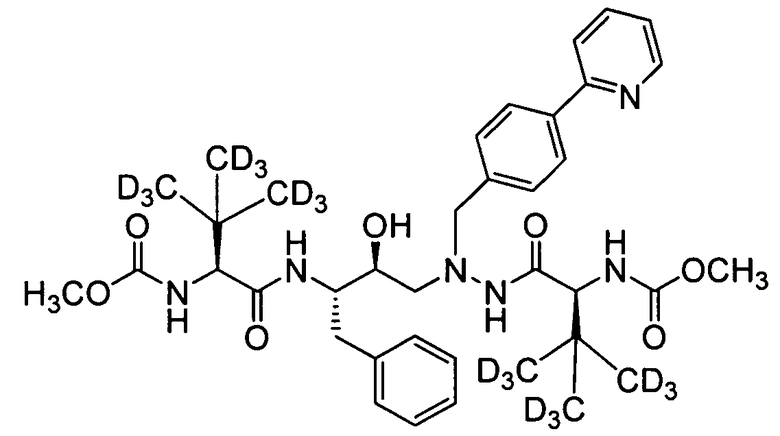 соединение 106;
соединение 106;
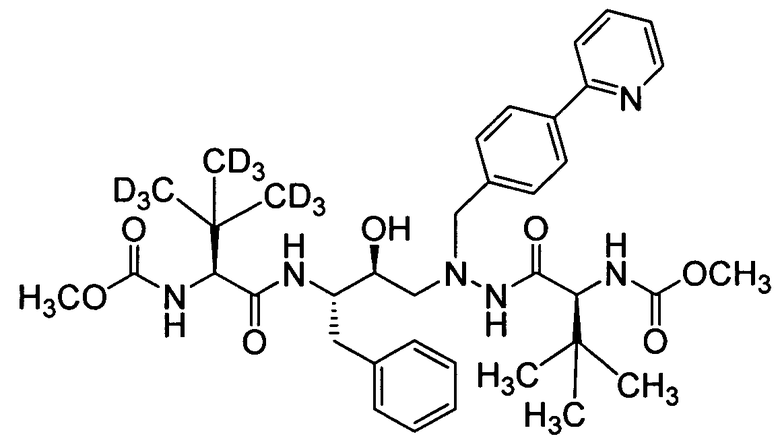 соединение 104;
соединение 104;
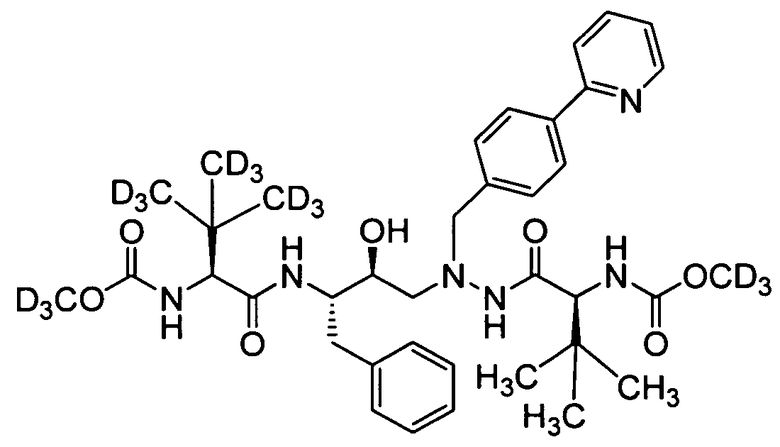 соединение 120; и
соединение 120; и
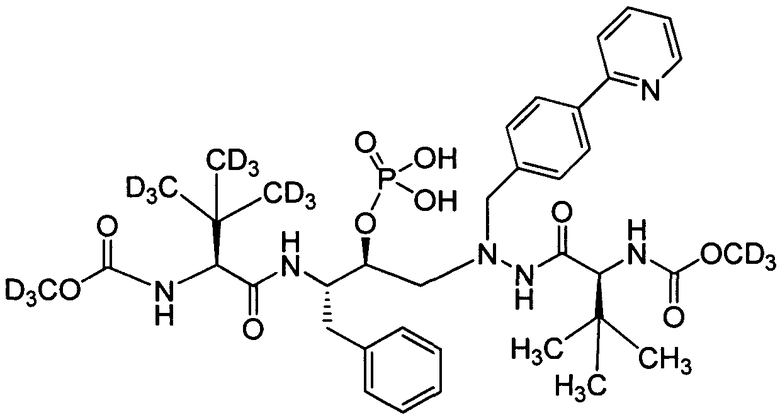
 соединение 123
соединение 123
или фармацевтически приемлемой соли любого из вышеуказанных соединений.
В еще одном варианте осуществления изобретения соединение согласно изобретению выбрано из следующих соединений:
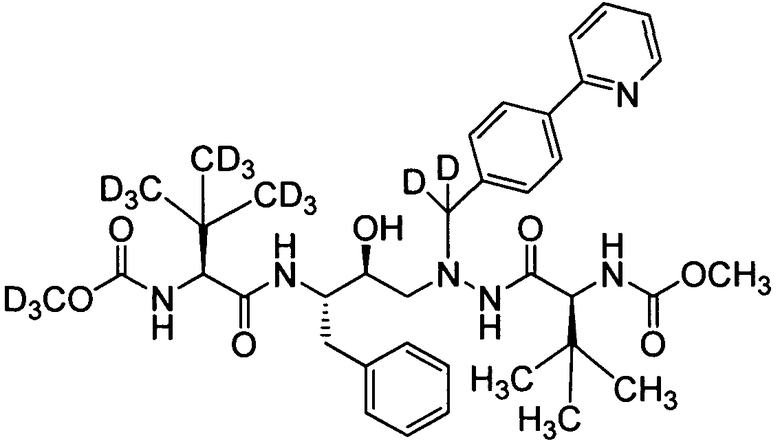
 соединение 176; и
соединение 176; и
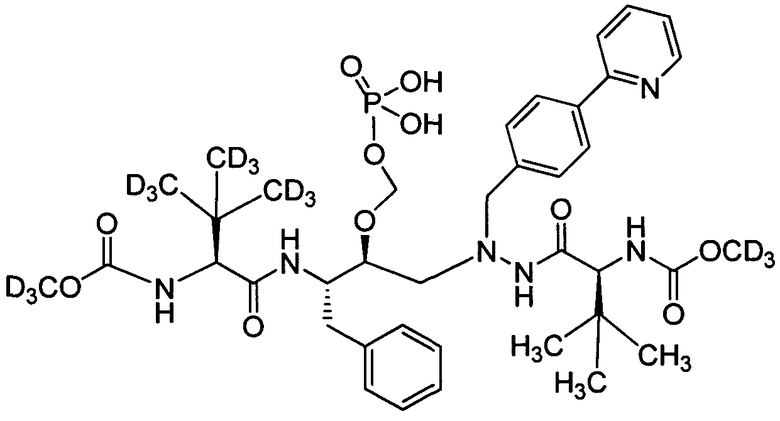 соединение 177
соединение 177
или фармацевтически приемлемой соли любого из вышеуказанных соединений.
В еще более точно описанном варианте осуществления изобретения соединение выбрано из соединения 114, соединения 120, соединения 122 и соединения 131.
В еще одной серии вариантов осуществления изобретения любой атом, не обозначенный как дейтерий, в любой из серий вариантов осуществления, представленных выше, присутствует в его природном изотопном содержании.
Синтез соединений формулы I может быть легко осуществлен способами, известными специалисту в данной области техники. Подходящие методики и промежуточные продукты описаны, например, в патенте США № 5849911; РСТ WO 97/46514; Bold G. et al., J. Med. Chem. 1998, 41:3387; Xu, Z. et al., Org. Process Res. Dev. 2002, 6:323; PCT WO 2006/014282.
Такие способы могут быть осуществлены с использованием соответствующих дейтерированных и необязательно содержащих другие изотопы реагентов и/или промежуточных продуктов синтеза соединений, описанных здесь, или с использованием стандартных методик синтеза, известных в данной области техники для введения изотопных атомов в химическую структуру. Некоторые промежуточные продукты могут использоваться после очистки (например, фильтрации, дистилляции, сублимации, кристаллизации, растирания, твердофазной экстракции и хроматографии) или без очистки.
ТИПИЧНЫЙ ПРИМЕР СИНТЕЗА
Удобный способ синтеза соединений формулы Ia представлен на схеме 1
Схема 1. Общий способ получения соединений формулы Ia, где R 1a =R 1b , R 2 =R 3 .
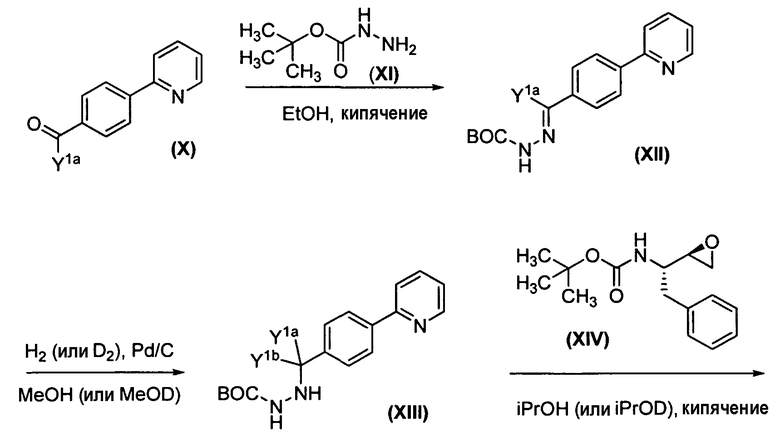
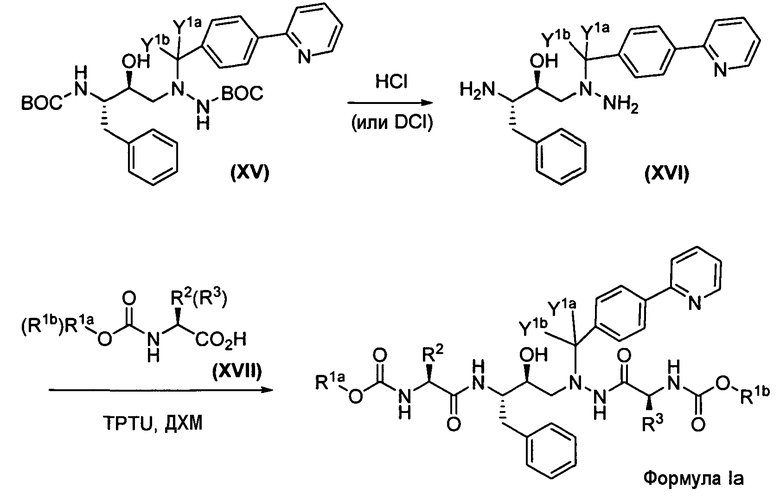
Альдегид Х подвергается обработке коммерчески доступным трет-бутоксикарбонилгидразидом (XI) для получения ВОС-защищенного промежуточного гидразон-производного XII, которое затем восстанавливается с использованием либо газообразного водорода, либо газообразного дейтерия для получения соответствующего ВОС-защищенного гидразида XIII. BOC-защищенный гидразид XIII затем подвергается обработке коммерчески доступным эпоксидом (XIV) для получения XV, из которого затем удаляется защита с помощью хлористоводородной кислоты для получения XVI. Соответствующее карбамат-производное трет-лейцина XVII подвергается обработке XVI в присутствии тетрафторбората О-(1,2-дигидро-2-оксо-1-пиридил)-N,N,N',N'-тетраметилурония (TPTU) с получением соединения формулы Ia.
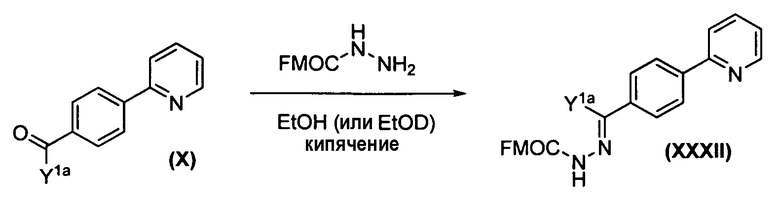
Применение различных защитных групп либо на XI, либо XIV вместе с подходящим способом удаления защиты, как описано в публикации Zhang, H. et al., J. Labelled Compounds Radiopharm. 2005, 48:1041-1047, дает возможность синтезировать соединения формулы Ia, которые не являются симметрично замещенными. В этом способе различные схемы дейтерирования для R1a и R1b и/или R2 и R3 могут быть достигнуты, как показано на схемах 1b и 1с.
Схема 1b. Общий способ, где R 1a ≠R 1b , R 2 ≠R 3
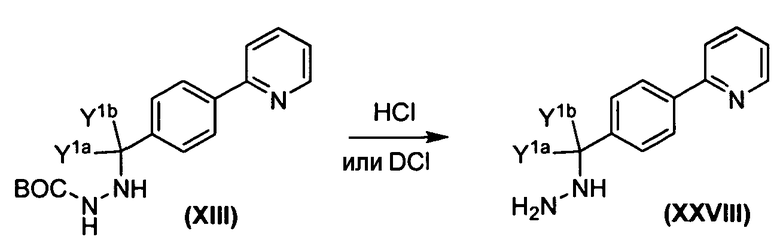
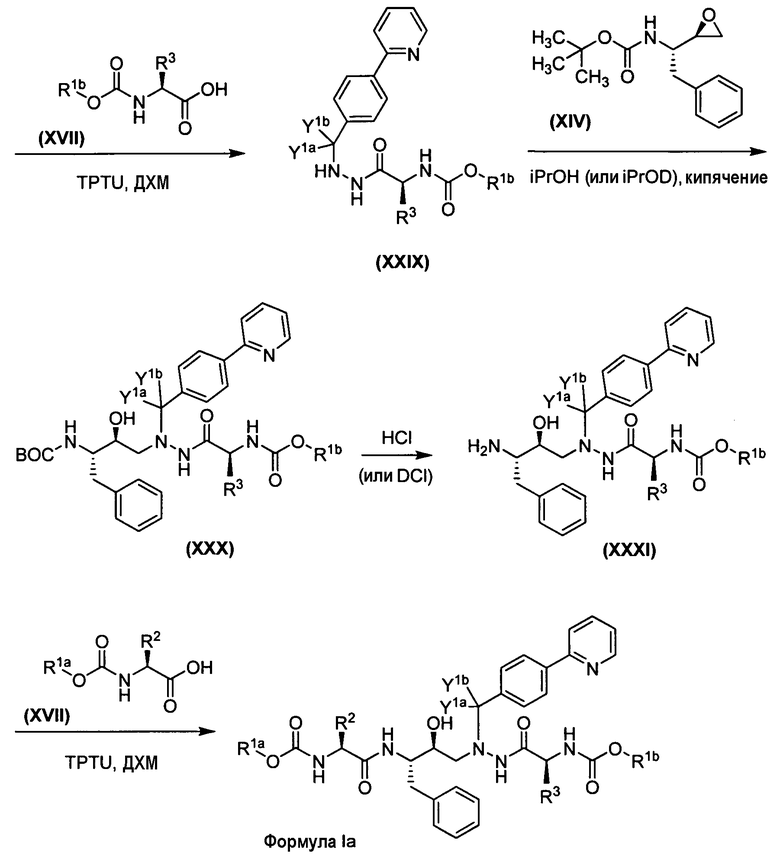
Схема 1с. Общий способ введения различных R и Y групп
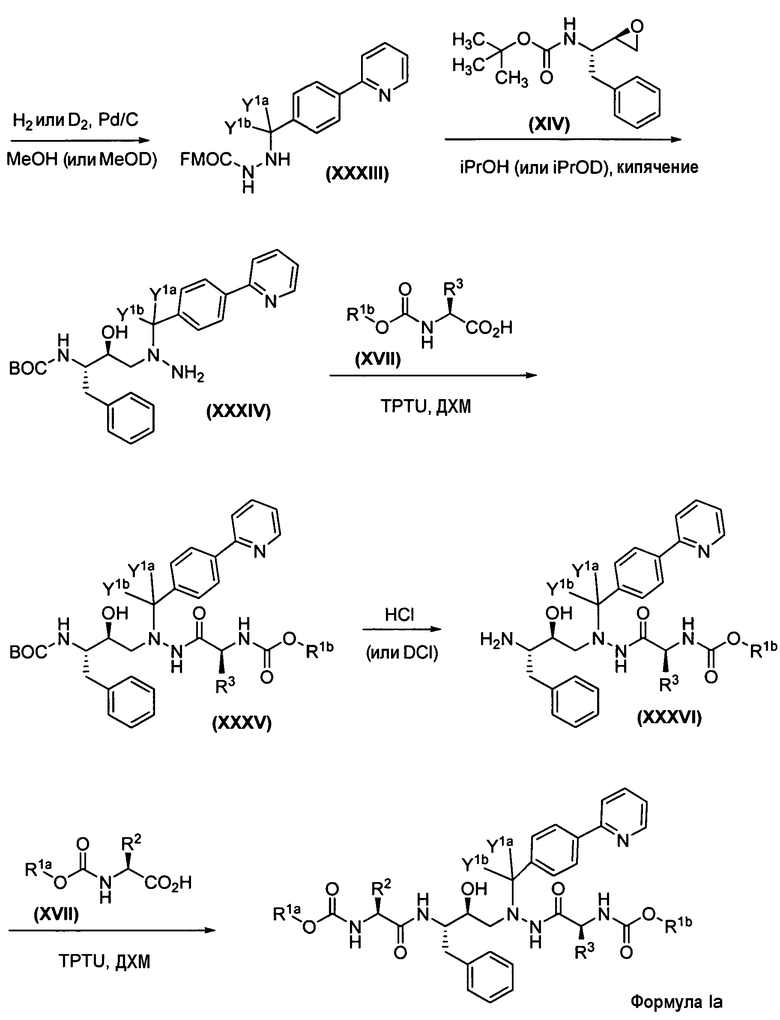
Недейтерированный альдегид Х, применяемый в схемах 1 и 1с, представленных выше, является коммерчески доступным. Дейтерированный вариант альдегида Х синтезируется в соответствии с методикой, описанной в публикациях Thompson, A.F. et al., JACS 1939, 61:1374-1376; Scott, CA et al., Syn Comm 1976, 6:135-139, как показано на схеме 2 ниже.
Схема 2. Получение дейтерированного промежуточного продукта Х
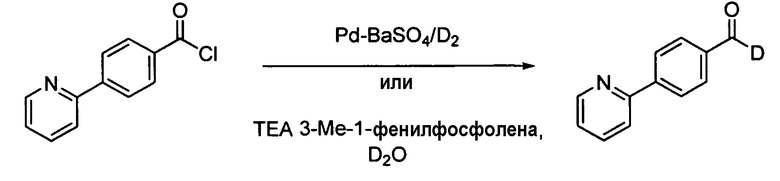
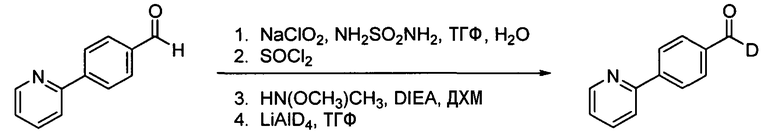
Альтернативно, недейтерированный альдегид Х может подвергаться окислению до карбоновой кислоты, превращаться в амид Вейнреба через ацилхлорид и восстанавливаться с помощью LiAlD4 с получением целевого дейтерированного альдегида, как представлено ниже на схеме 2b.
Схема 2b. Альтернативное получение дейтерированного промежуточного продукта Х
Дейтерированные варианты карбамат-производного трет-лейцина XVII получают в соответствии со схемами 3-5.
Схема 3. Способ получения дейтерированного трет-лейцина (XIII)
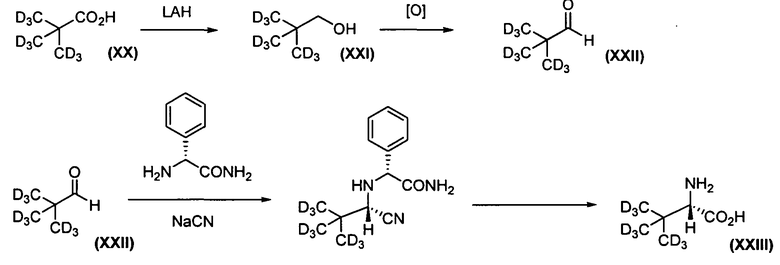
Как показано на схеме 3, трет-лейцин XXIII, где R2 и/или R3 представляют собой -С(СD3)3, может быть получен исходя из коммерчески доступной d 9-пивалиновой кислоты (ХХ). ХХ подвергается восстановлению до спирта XXI алюмогидридом лития, как описано в публикации Brainard, R.L. et al., Organometallics 1986, 5:1481-1490. Данный спирт ХХI подвергается окислению до альдегида XXII в любых мягких условиях (см., например, Herrerias, C.I. et al., Tet. Lett. 2005, 47: 13-17). Альдегид XXII подвергается превращению в трет-лейцин XXIII с использованием асимметричного синтеза Стреккера, как описано в публикации Boesten, W.H.J. et al., Org. Lett. 2001, 3:1121-1124. Альтернативный асимметричный синтез Стрекера описан в публикации Davis, F.A. et al., J. Org. Chem. 1996, 61:440-441.
Схема 4. Превращение дейтерированного трет-лейцина в соответствующий карбамат
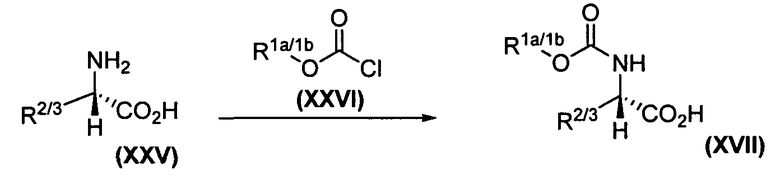
Как показано на схеме 4, дейтерированный трет-лейцин XXV подвергается взаимодействию с подходящим хлорметилформиатом XXVI, как описано в публикации заявки на патент США 2005131017, с получением целевого карбамат-производного трет-лейцина XVII, который использовался в синтезе, представленном на схеме 1.
Схема 5. Превращение дейтерированного трет-бутилхлорида в соответствующий пивалальдегид (XXII)
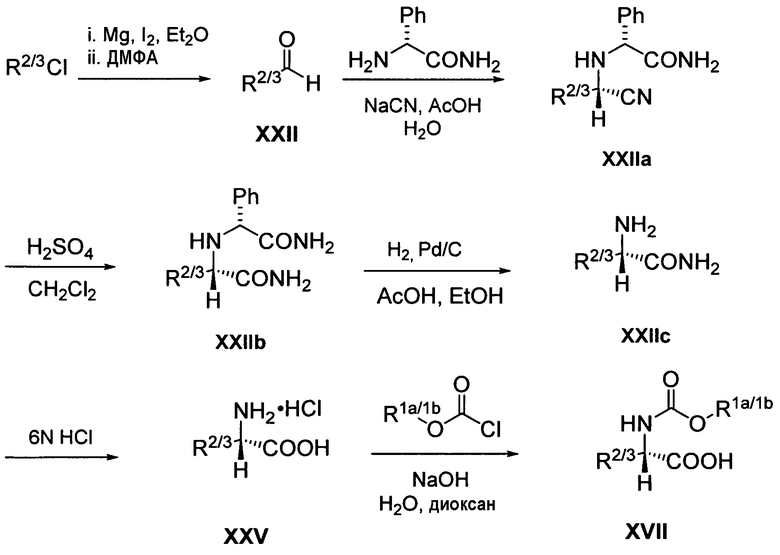
Как видно из схемы 5, дейтерированный трет-бутилхлорид подвергается превращению в соответствующий пивалальдегид (XXII) при кипячении с обратным холодильником в безводном простом эфире в присутствии магния и йода с последующим добавлением безводного диметилформамида (ДМФА). Пивалальдегид (XII) подвергается взаимодействию с (R)-фенилглицинамидом и NaCN в водной уксусной кислоте с получением нитрила (XXIIa). Нитрил (XXIIa) подвергается гидролизу серной кислотой с получением амида (XXIIb), который затем подвергается гидрированию над палладием на углероде с получением амида (XXIIc). Амид (XXIIc) подвергается гидролизу хлористоводородной кислотой с получением соответствующей карбоновой кислоты (XXV), которая затем подвергается взаимодействию с дейтерированным метилхлорформиатом в присутствии NaOH с получением дейтерированного промежуточного продукта XVII.
Для получения соединений формулы А может использоваться ряд новых промежуточных продуктов. Таким образом, изобретение предоставляет также соединение, которое выбрано из следующей группы соединений:
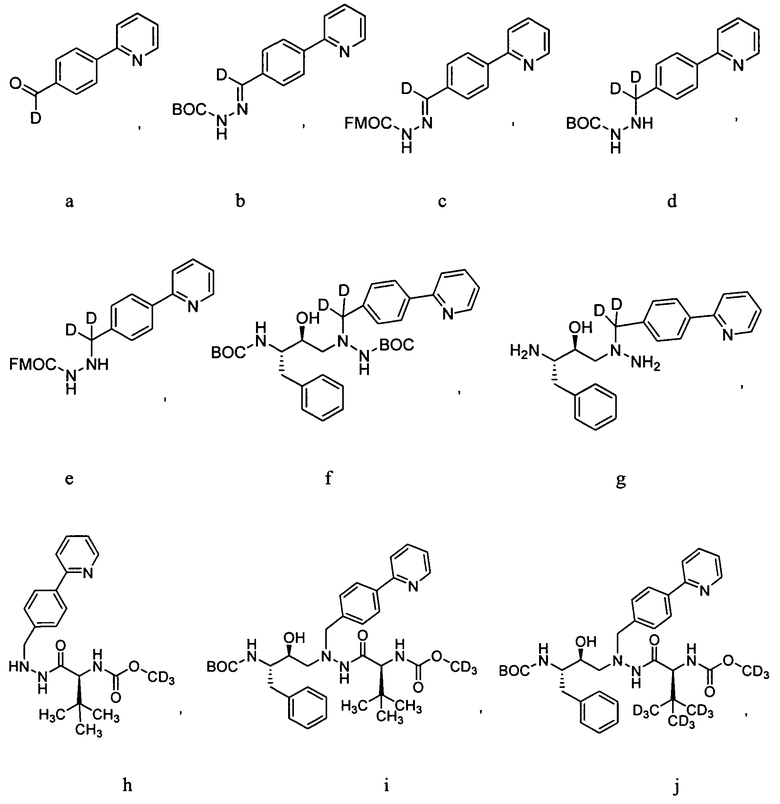
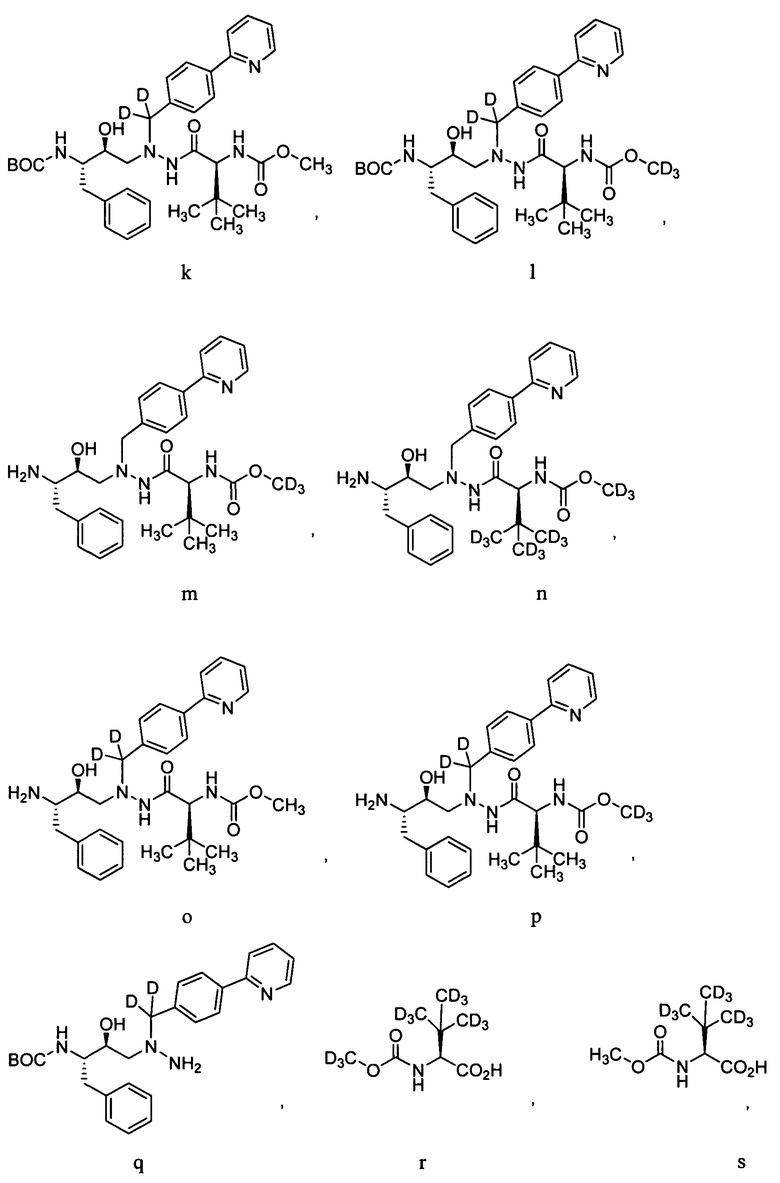
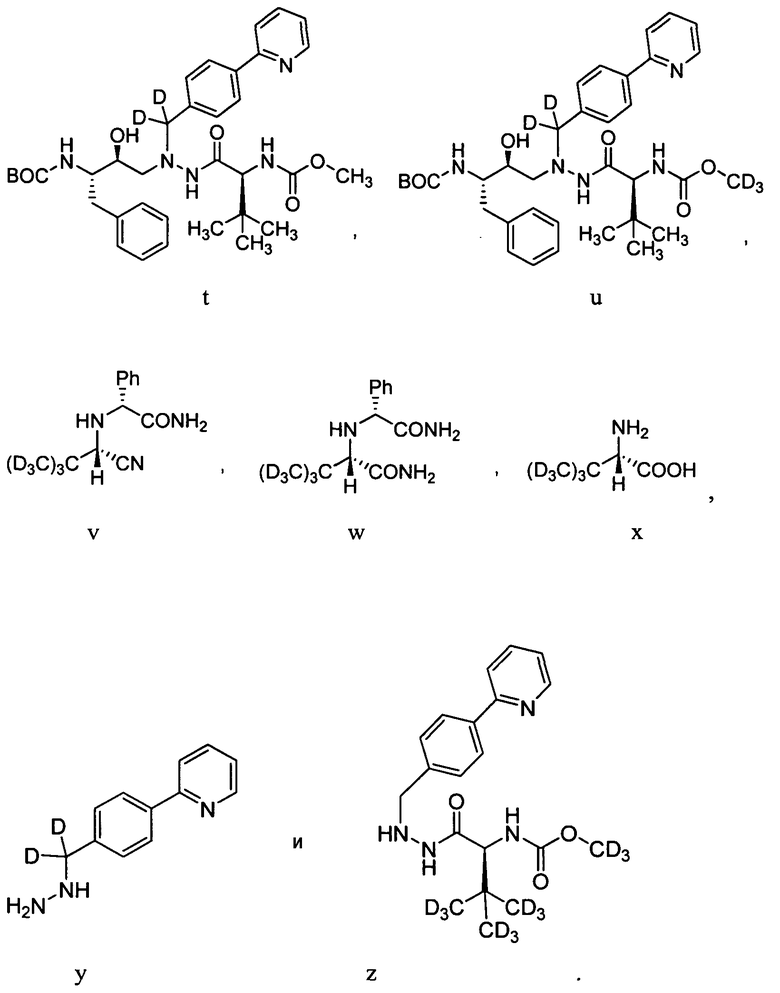
В некоторых условиях синтеза соединения 103, 104, 106, 111, 113, 114, 120, 121, 122, 123, 129 и 131 были получены с относительным содержанием изотопа в каждом положении, обозначенном как «D», по меньшей мере, примерно 75%. В других условиях синтеза соединения 103, 104, 106, 111, 113, 114, 120, 121, 122, 123, 129 и 131 были получены с относительным содержанием изотопа в каждом положении, обозначенном как «D», примерно более 95%.
Пролекарства соединений согласно изобретению, представленных формулой А, где R5 представляет собой -Р(О)-(ОН)2 или его соль, могут быть получены в соответствии с методикой, описанной в WO 2001 000635А. Пролекарства согласно изобретению, представленные формулой А, где R5 представляет собой -(CR6R7-O)n-R8, где R6 и R7 представляют собой Н и каждый R8 представляет собой -Р(О)-(ОН)2 или его соль, могут быть получены в соответствии с методиками, описанными в публикации Safadi, M. et al., Pharmaceutical Research, 1993, 10(9): 1350. Другие подходящие способы получения пролекарств соединений согласно изобретению можно найти в публикации РСТ WO 2006/014282.
В других вариантах осуществления изобретения соединение согласно изобретению содержит, по меньшей мере, 52,5% введенного дейтерия, по меньшей мере, 60% введенного дейтерия, по меньшей мере, 67,5% введенного дейтерия, по меньшей мере, 75% введенного дейтерия, по меньшей мере, 82,5% введенного дейтерия, по меньшей мере, 90% введенного дейтерия или, по меньшей мере, 95% введенного дейтерия в каждом положении, обозначенном как дейтерий, в соединении согласно изобретению. Соединение согласно изобретению может быть в количестве, например, по меньшей мере, 100 мг, например, по меньшей мере, 200 мг, предпочтительно, по меньшей мере, 400 мг, более предпочтительно, по меньшей мере, 500 мг и необязательно до 10 кг.
Подразумевается, что конкретные способы и соединения, представленные выше, не являются ограничивающими объем данного изобретения. Химические структуры в схемах, где указаны переменные, соответственно являются определениями химических групп (фрагментов, атомов и т.д.) соответствующего положения в формуле соединений независимо от того, одинаковы обозначения переменных (т.е. R1, R2, R3 и т.д.) или нет. Определение приемлемости химической группы в структуре соединения для применения в синтезе другого соединения находится в компетенции специалиста в данной области техники. Дополнительные способы синтеза соединений формулы I и их синтетических предшественников, включая пути синтеза, не представленные точно на схемах, известны специалисту в данной области техники. Способы оптимизации условий реакции и, если необходимо, снижения образования конкурентных побочных продуктов известны в данной области техники. Помимо приведенных в описании ссылок на публикации, в которых описан синтез, специалист в данной области техники может определить схемы реакций и методики при применении программных средств баз данных коммерчески доступных структур, например SciFinder® (CAS отделение Американского Химического Общества), STN® (CAS отделение Американского Химического общества), CrossFire Beilstein® (Elsevier MDL), или поисковых систем Интернета, таких как Google®, или баз данных с поиском по ключевому слову, таких как текстовая база данных US Patent and Trademark Office.
Способы, представленные в данном описании, также могут дополнительно включать стадии, до или после конкретно описанных стадий, для введения или удаления подходящих защитных групп в порядке, позволяющем в итоге синтезировать соединения согласно изобретению. Кроме того, различные стадии синтеза могут осуществляться в альтернативной последовательности или в порядке, позволяющем получить целевые соединения. Химия синтетических превращений и методики использования защитных групп (введение защиты и удаление защиты), применимые в синтезе заявленных соединений, известны в данной области техники и включают, например, превращения и методы, описанные в следующих публикациях: Larock R., Comprehensive Organic Transformations, VCH Publishers (1989); Greene T.W. et al., Protective Groups in Organic Synthesis, 3rd Ed., John Wiley and Sons (1999); Fieser L. et al., Fieser and Fieser's Reagents for Organic Synthesis, John Wiley and Sons (1994); Paquette L., ed., Encyclopedia of Reagents for Organic Synthesis, John Wiley and Sons (1995) и последующие издания.
Комбинациями заместителей и переменных, предполагаемыми согласно настоящему изобретению, являются только такие комбинации и переменные, которые приводят к образованию стабильных соединений.
КОМПОЗИЦИИ
Настоящее изобретение предоставляет также апирогенные композиции, содержащие эффективное количество соединения любой из формул А, I, Ia, Ib или Ic (например, включая любую из приведенных в описании формул) или фармацевтически приемлемую соль указанного соединения; и приемлемый носитель. Предпочтительно композиция согласно настоящему изобретению изготавливается по рецептуре, предназначенной для фармацевтического применения (по рецептуре «фармацевтическая композиция»), где носитель представляет собой фармацевтически приемлемый носитель. Носитель(и) называется(ются) «приемлемым(и)», когда является(ются) совместимым(и) с другими ингредиентами композиции и в случае фармацевтически приемлемого носителя не вреден(ны) для его реципиента в количестве, используемом в лекарственном средстве.
Фармацевтически приемлемые носители, адъюванты и наполнители, которые могут применяться в фармацевтических композициях согласно данному изобретению, включают, но без ограничения, ионно-обменные вещества, оксид алюминия, стеарат алюминия, лецитин, сывороточные белки, такие как альбумин сыворотки человека, буферные вещества, такие как фосфаты, глицин, сорбиновую кислоту, сорбат натрия, смеси частичных глицеридов растительных насыщенных жирных кислот, воду, соли или электролиты, такие как протаминсульфат, гидрофосфат натрия, гидрофосфат калия, хлорид натрия, соли цинка, коллоидный диоксид кремния, трисиликат магния, поливинилпирролидон, производные целлюлозы, полиэтиленгликоль, натрийкарбоксиметилцеллюлоза, полиакрилаты, воски, блок-сополимеры полиэтилена и полиоксипропилена, полиэтиленгликоль и ланолин.
Если необходимо, растворимость и биодоступность соединений согласно настоящему изобретению в фармацевтических композициях могут повышаться способами, известными в данной области техники. Один способ включает применение липидных эксципиентов в композиции (см. “Oral Lipid-Based Formulations: Enhancing the Bioavailability of Poorly Water-Soluble Drugs (Drugs and the Pharmaceutical Sciences)”, David J. Hauss, ed. Informa Healthcare, 2007; “Role of Lipid Excipients in Modifying Oral and Parenteral Drug Delivery: Basic Principles and Biological Examples”, Kishor M. Wasan, ed. Wiley-Interscience, 2006).
Другим известным способом повышения биодоступности является применение аморфной формы соединения согласно изобретению, необязательно введенной в композицию с полоксамером, таким как LUTROL™ и PLURONIC™ (BASF Corporation), или блок-сополимерами этиленоксида и пропиленоксида (см. патент США № 7014866, публикации патентов США 20060094744 и 20060079502).
Фармацевтические композиции согласно изобретению включают композиции, подходящие для перорального, ректального, назального, местного (включая трансбуккальное и подъязычное), пульмонального, вагинального или парентерального (включая подкожное, внутримышечное, внутривенное и внутрикожное) введения. В некоторых вариантах осуществления изобретения соединение приведенных в описании формул вводится чрескожным способом (например, с использованием чрескожного пластыря или ионтофореза). Другие композиции могут быть удобно представлены в единичной дозированной форме, например в форме таблеток, капсул замедленного высвобождения и в липосомах, и могут быть получены любым из способов, которые хорошо известны в фармацевтической области (см., например, Remington's Pharmaceutical Sciences, Mack Publishing Company, Philadelphia, PA (17th ed. 1985).
Такие препаративные способы включают стадию объединения соединения, подлежащего введению, с такими ингредиентами, как носитель, который состоит из одного или нескольких вспомогательных ингредиентов. Обычно композиции получают тщательным смешением активных ингредиентов с жидкими носителями, липосомами или тонко измельченными твердыми носителями или с теми и другими с получением однородной смеси и затем, если нужно, формованием продукта.
В некоторых вариантах осуществления соединение вводится перорально. Композиции согласно настоящему изобретению, подходящие для перорального введения, могут быть представлены в форме дискретных единиц, таких как капсулы, саше или таблетки, каждая из которых содержит предопределенное количество активного ингредиента; в форме порошков или гранул; раствора или суспензии в водной жидкости или неводной жидкости; в форме жидкой эмульсии типа «масло в воде»; жидкой эмульсии типа «вода в масле»; включенными в липосомы; или в виде болюсов и т.д. Мягкие желатиновые капсулы могут применяться для включения в них таких суспензий, что может полезно повышать скорость абсорбции соединений.
В случае таблеток для перорального применения традиционно используемые носители ключают лактозу и кукурузный крахмал. Лубриканты, такие как стеарат магния, также обычно добавляются. При пероральном введении в форме капсул применимые разбавители включают лактозу и высушенный кукурузный крахмал. Когда водные суспензии вводятся перорально, активный ингредиент объединяется с эмульгирующим и суспендирующим агентами. Если нужно, могут добавляться некоторые подсластители, и/или вкусовые добавки, и/или красители.
Композиции, подходящие для перорального введения, включают леденцы, содержащие ингредиенты во вкусовой основе, обычно сахарозу и гуммиарабик или трагакант; и пастилки, содержащие активный ингредиент в инертной основе, такой как желатин и глицерин или сахароза и гуммиарабик.
Композиции, подходящие для парентерального введения, включают стерильные водные и неводные растворы для инъекции, которые могут содержать антиоксиданты, буферные добавки, бактериостаты и растворенные вещества, которые придают композиции изотоничность с кровью предполагаемого реципиента; и водные и неводные стерильные суспензии, которые могут включать суспендирующие агенты и загустители. Композиции могут быть представлены в контейнерах с единичной дозой или множественной дозой, например, в запаянных ампулах и пузырьках и могут храниться в условиях сушки вымораживанием (лиофилизованными), при которых требуется только добавление стерильного жидкого носителя, например воды для инъекций, непосредственно перед применением. Приготовленные для немедленного введения растворы и суспензии для инъекций могут быть получены из стерильных порошков, гранул и таблеток.
Такие растворы для инъекций могут быть представлены, например, в форме стерильной водной или масляной суспензии для инъекций. Такая суспензия может быть изготовлена по рецептуре и в соответствии с методиками, известными в данной области техники, с использованием подходящих диспергирующих или смачивающих агентов (таких как, например, Tween 80) и суспендирующих агентов. Стерильный препарат для инъекции также может представлять собой стерильный раствор или суспензию для инъекций в нетоксичном разбавителе или растворителе, подходящем для парентерального введения, например в виде раствора в 1,3-бутандиоле. Примеры приемлемых разбавителей и растворителей, которые могут использоваться, включают маннит, воду, раствор Рингера и изотонический раствор хлорида натрия. Кроме того, стерильные нелетучие масла удобно используются в качестве растворителя или суспензионной среды. Для этой цели может применяться любое успокаивающее нелетучее масло, включая синтетические моно- или диглицериды. Жирные кислоты, такие как олеиновая кислота и ее глицеридные производные, могут применяться для получения препарата для инъекций в виде природных фармацевтически приемлемых масел, таких как оливковое масло или касторовое масло, особенно в их полиоксиэтилированных вариантах. Такие масляные растворы или суспензии могут также содержать спиртовой разбавитель или диспергирующее средство с длинной цепью.
Фармацевтические композиции согласно настоящему изобретению могут применяться в форме суппозиториев для ректального введения. Такие композиции могут быть получены смешением соединения согласно настоящему изобретению с подходящим не вызывающим раздражения эксципиентом, который является твердым веществом при комнатной температуре, но жидким при ректальной температуре и, следовательно, будет плавиться в прямой кишке с высвобождением активных компонентов. Такие материалы включают, но без ограничения, масло какао, пчелиный воск и полиэтиленгликоли.
Фармацевтические композиции согласно настоящему изобретению могут вводиться с помощью назального аэрозоля или ингаляцией. Такие композиции получают в соответствии с методиками, хорошо известными в области получения фармацевтических препаратов, и могут быть получены в виде растворов в физиологическом растворе с применением бензилового спирта или других подходящих консервантов, ускорителей абсорбции для повышения биодоступности, фторуглеродов и/или других солюбилизирующих или диспергирующих агентов, известных в данной области техники (см., например, Rabinowitz J.D. and Zaffaroni A.C. Патент США № 6803031 (Alexza Molecular Delivery Corporation)).
Местное введение фармацевтических композиций согласно настоящему изобретению является особенно полезным, когда желательное лечение включает области или органы, для которых приемлемо местное применение. Для местного применения на кожу фармацевтические композиции должны изготавливаться по рецептуре, подходящей для мази, содержащей активные компоненты, суспендированные или растворенные в носителе. Носители для местного введения соединения согласно данному изобретению включают, но без ограничения, минеральное масло, вазелиновое масло, обесцвеченное вазелиновое масло, пропиленгликоль, полиоксиэтиленполиоксипропиленовое соединение, эмульгирующийся воск и воду. Альтернативно, фармацевтическая композиция может приготавливаться по рецептуре лосьона или крема, содержащего активное соединение, суспендированное или растворенное в носителе. Подходящие носители включают, но без ограничения, минеральное масло, сорбитанмоностеарат, полисорбат 60, воск сложных цетиловых эфиров, цетеариловый спирт, 2-октилдодеканол, бензиловый спирт и воду. Фармацевтические композиции согласно настоящему изобретению могут также применяться местно в толстом кишечнике посредством композиции в форме ректальных суппозиториев или в форме подходящего препарата для клизмы. Местное ведение с помощью чрескожных пластырей или лекарственного электрофореза также включено в данное изобретение.
Применение пациентом терапевтических средств может представлять собой местное введение терапевтических средств в заданную область. Для доставки пациенту композиций могут использоваться различные способы, такие как инъекция, применение катетеров, трокаров, струйных инъекторов высокого давления, плуроник-геля, стентов, полимеров для замедленного высвобождения лекарственных средств или другого устройства, которое обеспечивает внутренний доступ.
Таким образом, в соответствии с еще одним вариантом осуществления изобретения соединения данного изобретения могут вводиться в композиции, предназначенные для нанесения в виде покрывающего слоя на медицинское устройство, которое может имплантироваться, такого как протезы, искусственные клапаны, сосудистые трансплантаты, стенты или катетеры. Подходящие покрытия и общие способы получения покрытых имплантируемых устройств известны в данной области техники, и их примеры описаны в патентах США № 6099562; 5886026 и 5304121. Покрытия обычно представляют собой биологически совместимые полимерные материалы, такие как гидрогелевый полимер, полиметилдисилоксан, поликапролактон, полиэтиленгликоль, полимолочная кислота, этиленвинилацетат и их смеси. Покрытия могут необязательно дополнительно покрываться подходящим верхним покрытием из фторсиликона, полисахаридов, полиэтиленгликоля, фосфолипидов или их сочетания для придания композиции свойств контролируемого высвобождения действующего вещества. Покрытия для инвазивных устройств должны быть включены в определение фармацевтически приемлемого носителя, адъюванта или разбавителя, которые используются в данном описании.
В соответствии с другим вариантом осуществления изобретения изобретение предоставляет способ покрытия медицинского устройства, которое может имплантироваться, включающий стадию контактирования указанного устройства с композицией покрытия, описанной выше. Специалисту в данной области техники будет понятно, что нанесение покрытия на устройство будет осуществляться перед его имплантацией млекопитающему.
В соответствии с другим вариантом осуществления изобретение предоставляет способ импрегнирования имплантируемого устройства для высвобождения лекарственного средства, включающий стадию контактирования указанного устройства для высвобождения лекарственного средства с соединением или композицией настоящего изобретения. Имплантируемые устройства для высвобождения лекарственного средства включают, но без ограничения, биодеградируемые полимерные капсулы или ядра, не подвергающиеся разложению, способные диффундировать полимерные капсулы и биоразлагаемые полимерные вафли.
В соответствии с еще одним вариантом осуществления изобретение предоставляет имплантируемое медицинское устройство, покрытое соединением или композицией, включающей соединение согласно настоящему изобретению, таким образом, что указанное соединение является терапевтически активным.
В соответствии с еще одним вариантом осуществления изобретение предоставляет имплантируемое устройство для высвобождения лекарственного средства, импрегнированное соединением или композицией, содержащей соединение данного изобретения, так что указанное соединение высвобождается из указанного устройства и является терапевтически активным.
Когда орган или ткань является доступным(ой) ввиду удаления из пациента, такой орган или такая ткань может промываться в среде, содержащей композицию согласно настоящему изобретению, причем композиция согласно изобретению может окрашиваться на органе, или композиция согласно изобретению может применяться любым другим удобным способом.
В другом варианте осуществления изобретения композиция согласно настоящему изобретению включает второе терапевтическое средство. В одном варианте осуществления изобретения второе терапевтическое средство представляет собой одно или несколько дополнительных соединений согласно изобретению. В конкретном варианте осуществления изобретения каждое из двух или нескольких соединений согласно изобретению, присутствующих в таких композициях, различаются положениями изотопного обогащения. Обычно такая композиция включает три, четыре, пять или большее количество различных соединений согласно настоящему изобретению.
В еще одном варианте осуществления изобретения второе терапевтическое средство может быть выбрано из любого соединения или терапевтического средства, которое, как известно, обладает полезными свойствами или демонстрирует полезные свойства при введении с соединением, обладающим таким же механизмом действия, что и атазанавир. Такие лекарственные средства включают лекарственные средства, которые указаны как применимые в сочетании с атазанавиром, в том числе, но без ограничения, соединения, описанные в РСТ публикациях WO 2003020206, WO 2005058248, WO 2006060731 и WO 20050277855.
Предпочтительно второе терапевтическое средство представляет собой лекарственное средство, применимое в лечении или профилактике ВИЧ инфекции (то есть противоретровирусное средство).
В одном варианте осуществления изобретения второе терапевтическое средство выбрано из других противоретровирусных средств, включая, но без ограничения, второй ингибитор ВИЧ протеазы (например, ампренавир, фосампренавир, типранавир, индинавир, саквинавир, лопинавир, ритонавир, дарунавир или нелфинавир), ненуклеозидный обратный ингибитор транскриптазы (NNRTI) (например, этравирин, делавирдин, эфавиренз, невирапин или рилпивирин), нуклеозидный/нуклеотидный обратный ингибитор транскриптазы (NRTI) (например, зидовудин, ламивудин, эмтрицитабин, фумарат тенофовирдисопроксила, диданозин, ставудин, абакавир, рацивир, амдоксовир, априцитабин, энтекавир, адефовир или элвуцитабин), ингибитор входа вируса (например, энфувиртид, маравирок, викривирок, PRO 140 или TNX-355), ингибитор интегразы (например, ралтегравир или элвитегравир), иммунное противоретровирусное средство (например, иммунитин, пролейкин, ремун, BAY 50-4798 или IR103), ингибитор созревания вируса (например, бевиримат), клеточный ингибитор (например, дроксия или гидроксимочевина) или сочетания двух или нескольких указанных выше соединений.
В более точно определенном варианте осуществления изобретения второе терапевтическое средство выбрано из ритонавира, эфавиренза, диданозина, тенофовирдисопроксила, нелфинавирмезилата, ампренавира, ралтегравира, саквинавира, лопинавира, невирапина, эмтрицитабина, абакавира, ламивудина, зидовудина, маравирока, ставудина, дарунавира, фосампренавира, викривирока, фармацевтически приемлемых солей любого их вышеуказанных соединений и их сочетаний.
В еще более точно определенном варианте осуществления изобретения второе терапевтическое средство выбрано из ритонавира, эфавиренза, диданозина, ралтегравира, тенофовирдисопроксила, ламивудина, абакавира, зидовудина, эмтрицитабина, эфавиренза, фармацевтически приемлемых солей любого из вышеуказанных соединений и их сочетаний. В другом конкретно определенном варианте осуществления изобретения композиции согласно настоящему изобретению включают соединение любой из формул А, I, Ia, Ib или Ic и два-три вторых терапевтических средства, указанных выше в данном абзаце. В еще более точно определенном варианте осуществления изобретения композиции согласно настоящему изобретению включают соединение любой из формул А, I, Ia, Ib или Ic и два вторых терапевтических средства, представленных выше в данном абзаце.
В другом варианте осуществления изобретение предоставляет отдельные дозированные формы соединения согласно данному изобретению и любого одного или нескольких из описанных выше вторых терапевтических средств, где соединение и второе терапевтическое средство объединены друг с другом. Термин «объединены друг с другом», когда используется в данном описании, означает, что отдельные дозированные формы упакованы вместе или иным образом связаны друг с другом, что подразумевает, что раздельные дозированные формы предназначены для продажи и введения вместе (в пределах 24 часов, последовательно или одновременно).
В еще одном варианте осуществления изобретение предоставляет фармацевтическую композицию, содержащую эффективное количество соединения любой из формул А, I, Ia, Ib или Ic, введение которого объекту исследования приводит к периоду полувыведения соединения из сыворотки, превышающему период полувыведения из сыворотки атазанавира, когда атазанавир вводится эквивалентному объекту исследования в фармацевтической композиции, включающей молярно эквивалентное количество атазанавира, и когда вводится в таком же режиме дозирования, что и соединение любой из формул А, I, Ia, Ib или Ic. В других вариантах осуществления изобретения период полувыведения из сыворотки соединения любой из формул А, I, Ia, Ib или Ic составляет по меньшей мере 110%, 120%, 130%, 140%, 150% или 160% или более периода полувыведения из сыворотки крови атазанавира, достигнутого при использовании молярно эквивалентной композиции атазанавира, введенной в таком же режиме дозирования. В более точно определенном варианте осуществления изобретения соединение любой из формул А, I, Ia, Ib или Ic вводится в единичной дозе.
В родственном варианте осуществления изобретение предоставляет фармацевтическую композицию, содержащую эффективное количество соединения любой из формул А, I, Ia, Ib или Ic или его фармацевтически приемлемой соли, где период полувыведения из сыворотки крови соединения после введения единичной дозы композиции объекту исследования составляет более 5,0 часов, более 6,0 часов, более 7,0 часов или более 8,0 часов.
В еще одном варианте осуществления изобретение предоставляет фармацевтическую композицию, содержащую эффективное количество соединения любой из формул А, I, Ia, Ib или Ic, введение которой объекту исследования приводит к значению AUC0-τ (где τ = интервал дозирования) соединения, большему, чем значение AUC0-τ атазанавира, когда атазанавир вводится эквивалентному объекту исследования в молярно эквивалентной фармацевтической композиции и вводится в таком же режиме дозирования, что и соединение любой из формул А, I, Ia, Ib или Ic. В других вариантах осуществления изобретения значение AUC0-τ, достигнутое при введении композиции согласно изобретению, составляет, по меньшей мере, 120%, 130%, 140%, 150%, 160% или более значения AUC0-τ, достигнутого при введении молярно эквивалентной композиции атазанавира в таком же режиме дозирования. В более точно определенном варианте осуществления соединение по любой из формул А, I, Ia, Ib или Ic вводится один раз в сутки.
В еще одном варианте осуществления изобретение предоставляет фармацевтическую композицию, содержащую эффективное количество соединения любой из формул А, I, Ia, Ib или Ic, пероральное введение которой объекту исследования приводит к максимальной концентрации соединения в сыворотке (Сmax), которая больше максимальной концентрации соединения в сыворотке атазанавира, когда атазанавир вводится перорально эквивалентному объекту исследования в молярно эквивалентной фармацевтической композиции и в таком же режиме дозирования, что и соединение любой из формул А, I, Ia, Ib или Ic. В родственном варианте осуществления изобретения максимальная концентрация в сыворотке соединения любой из формул А, I, Ia, Ib или Ic, достигнутая в результате перорального введения соединения согласно настоящему изобретению, составляет, по меньшей мере, 120%, 125%, 130%, 135% или более максимальной концентрации в сыворотке атазанавира, достигнутой в результате перорального введения молярно эквивалентной композиции атазанавира, вводимой в таком же режиме дозирования. В более точно определенном варианте осуществления изобретения соединение любой из формул А, I, Ia, Ib или Ic вводится один раз в сутки.
В еще одном варианте осуществления изобретение предоставляет фармацевтическую композицию, содержащую эффективное количество соединения любой из формул А, I, Ia, Ib или Ic, пероральное введение которой объекту исследования приводит к минимальной концентрации в сыворотке соединения (Сmin), которая больше минимальной концентрации в сыворотке атазанавира при пероральном введении атазанавира эквивалентному объекту исследования в молярно эквивалентной фармацевтической композиции в таком же режиме дозирования, что и соединение любой из формул А, I, Ia, Ib или Ic. В родственном варианте осуществления изобретения минимальная концентрация в сыворотке соединения любой из формул А, I, Ia, Ib или Ic, достигнутая при пероральном введении композиции согласно настоящему изобретению, составляет, по меньшей мере, 125%, 150%, 175%, 200% или более минимальной концентрации в сыворотке атазанавира, достигнутой при пероральном введении молярно эквивалентной композиции атазанавира в таком же режиме дозирования. В более точно определенном варианте осуществления изобретения соединение любой из формул А, I, Ia, Ib или Ic вводится раз в сутки.
Соединения согласно настоящему изобретению также демонстрируют большую резистентность к определенному метаболизму по сравнению с атазанавиром. Таким образом, в еще одном варианте осуществления изобретение предоставляет фармацевтическую композицию, содержащую эффективное количество соединения любой из формул А, I, Ia, Ib или Ic, пероральное введение которой объекту исследования приводит к показателю клиренса сыворотки, который меньше показателя клиренса в сыворотке атазанавира после перорального введения атазанавира эквивалентному объекту исследования в молярно эквивалентной фармацевтической композиции и в таком же режиме дозирования, что и соединение любой из формул А, I, Ia, Ib или Ic. В других вариантах осуществления показатель клиренса сыворотки соединения после перорального введения композиции согласно настоящему изобретению составляет менее 90%, менее 80%, менее 70% или менее 60% показателя клиренса сыворотки атазанавира после перорального введения молярно эквивалентной композиции атазанавира в таком же режиме дозирования. В более точно определенном варианте осуществления изобретения соединение любой из формул А, I, Ia, Ib или Ic вводится раз в сутки.
В родственном варианте осуществления изобретение предоставляет фармацевтическую композицию, содержащую 150 мг соединения любой из формул А, I, Ia, Ib или Ic или его фармацевтически приемлемой соли, где показатель клиренса сыворотки соединения после перорального введения единичной дозы соединения шимпанзе составляет менее 90 мл/час/кг, менее 80 мл/час/кг, менее 75 мл/час/кг или менее 70 мл/час/кг.
В еще одном родственном варианте осуществления изобретение предоставляет фармацевтическую композицию, содержащую 50 мг соединения любой из формул А, I, Ia, Ib или Ic или его фармацевтически приемлемой соли, где скорость клиренса сыворотки соединения после перорального введения в единичной дозе композиции шимпанзе составляет менее 350 мл/час/кг, менее 325 мл/час/кг, менее 300 мл/час/кг, менее 275 мл/час/кг.
В еще одном родственном варианте осуществления изобретение предоставляет фармацевтическую композицию, содержащую эффективное количество соединения любой из формул А, I, Ia, Ib или Ic, пероральное введение которой объекту исследования приводит к количеству соединения, выведенного интактным в течение 24 часов после перорального введения, которое больше количества атазанавира, выведенного интактным в течение 24 часов после перорального введения атазанавира эквивалентному объекту исследования в молярно эквивалентной фармацевтической композиции и в таком же режиме дозирования, что и соединение любой из формул А, I, Ia, Ib или Ic. В других вариантах осуществления изобретения количество соединения любой из формул А, I, Ia, Ib или Ic, выведенного интактным в течение 24 часов после перорального введения композиции согласно настоящему изобретению, составляет более 140%, более 160%, более 180%, более 200%, более 250% или более относительно количества атазанавира, выводимого интактным в течение 24 часов после перорального введения молекулярно эквивалентной композиции атазанавира, введенной в таком же режиме дозирования. В более точно определенном варианте осуществления изобретения соединение любой из формул А, I, Ia, Ib или Ic вводится один раз в сутки.
В еще одном варианте осуществления изобретение предоставляет фармацевтическую композицию, содержащую эффективное количество соединения любой из формул А, I, Ia, Ib или Ic, введение которой объекту исследования приводит к а) AUC0-12, b) Cmax или с) Сmin (минимальной концентрации в промежутке между дозировками), аналогичным этим показателя у атазанавира, когда атазанавир вводится эквивалентному объекту исследования в фармацевтической композиции, содержащей количество атазанавира, большее, чем количество соединения любой из формул А, I, Ia, Ib или Ic из расчета на моль активного ингредиента при введении в таком же режиме дозирования, что и соединение любой из формул А, I, Ia, Ib или Ic. В других вариантах осуществления изобретения эффективное количество соединения любой из формул А, I, Ia, Ib или Ic составляет не более 80%, 70%, 60%, 50%, 40% или менее количества атазанавира, необходимого для получения аналогичной AUC0-12, аналогичной Сmin и/или аналогичной Сmax при введении в таком же режиме дозирования, что и соединение любой из формул А, I, Ia, Ib или Ic. В более точно определенном варианте осуществления изобретения соединение любой из формул А, I, Ia, Ib или Ic вводится раз в сутки.
В еще одном варианте осуществления изобретение предоставляет фармацевтическую композицию, содержащую от 250 мг до 275 мг соединения любой из формул А, I, Ia, Ib или Ic, введение которой раз в сутки объекту исследования в отсутствии совместного введения ритонавира приводит к Сmin в интервале от 275 до 625 нг/мл в плазме и/или среднему значению концентрации в плазме в стационарном состоянии («Сss», также определяемая как AUC0-τ, где τ представляет собой интервал времени между дозировками) в интервале от 925 до 1425 нг/мл плазмы.
В еще одном варианте осуществления изобретение предоставляет фармацевтическую композицию, содержащую от 275 мг до 300 мг соединения любой из формул А, I, Ia, Ib или Ic, введение которой раз в сутки объекту исследования приводит к Сmin в плазме в интервале от 300 до 625 нг/мл плазмы и/или Сss в интервале от 1000 до 1550 нг/мл плазмы.
В еще одном варианте осуществления изобретение предоставляет фармацевтическую композицию, содержащую от 300 мг до 325 мг соединения любой из формул А, I, Ia, Ib или Iс, введение которой раз в сутки объекту исследования приводит к Сmin в плазме в интервале от 350 до 750 нг/мл плазмы и/или Сss в интервале от 1100 до 1675 нг/мл плазмы.
В еще одном варианте осуществления изобретение предоставляет фармацевтическую композицию, содержащую от 325 мг до 350 мг соединения любой из формул А, I, Ia, Ib или Ic, введение которой раз в сутки объекту исследования без совместного введения ритонавира приводит к Сmin в плазме в интервале от 375 до 800 нг/мл плазмы и/или Сss в интервале от 1200 до 1800 нг/мл плазмы.
В еще одном варианте осуществления изобретение предоставляет фармацевтическую композицию, содержащую от 350 мг до 375 мг соединения любой из формул А, I, Ia, Ib или Ic, введение которой раз в сутки объекту исследования без совместного введения ритонавира приводит к Сmin в плазме в интервале от 400 до 850 нг/мл плазмы и/или Сss в интервале от 1300 до 1925 нг/мл плазмы.
В еще одном варианте осуществления изобретение предоставляет фармацевтическую композицию, содержащую от 375 мг до 400 мг соединения любой из формул А, I, Ia, Ib или Ic, введение которой раз в сутки объекту исследования приводит к Сmin в плазме в интервале от 425 до 900 нг/мл плазмы и/или Сss в интервале от 1400 до 2050 нг/мл плазмы.
В еще одном варианте осуществления изобретение предоставляет фармацевтическую композицию, содержащую от 400 мг до 425 мг соединения любой из формул А, I, Ia, Ib или Ic, введение которой раз в сутки объекту исследования без совместного введения ритонавира приводит к Сmin в плазме в интервале от 450 до 975 нг/мл плазмы и/или Сss в интервале от 1500 до 2175 нг/мл плазмы.
В еще одном варианте осуществления изобретение предоставляет фармацевтическую композицию, содержащую от 425 мг до 450 мг соединения любой из формул А, I, Ia, Ib или Ic, введение которой раз в сутки объекту исследования без совместного введения ритонавира приводит к Сmin в плазме в интервале от 500 до 1025 нг/мл плазмы и/или Сss в интервале от 1575 до 2300 нг/мл плазмы.
В каждом из перечисленных выше вариантов осуществления изобретения фармацевтически приемлемая соль соединения любой из формул А, I, Ia, Ib или Ic и/или атазанавира может использоваться вместо соединения в форме свободного основания.
В более точно определенном варианте осуществления изобретения в каждой из композиций, упомянутых выше, соединение представляет собой соединение формулы I. В еще более точно определенном варианте осуществления изобретения в каждой из композиций, упомянутых выше, соединение представляет собой соединение формулы Ib. В еще более точно определенном варианте осуществления изобретения в каждой из композиций, упомянутых выше, соединение выбрано из соединения 114, соединения 120, соединения 122 и соединения 131.
Термин «молярно эквивалентное количество», когда используется в данном описании, означает, что количество, присутствующее в первой композиции, равно количеству, присутствующему во второй композиции, из расчета на мольное количество активного ингредиента.
Термин «объект исследования» означает любое млекопитающее, предпочтительно шимпанзе или человека.
Термин «эквивалентный объект исследования», когда используется в данном описании, означает объект исследования такого же вида и пола, что и объект исследования, который показывает не более 10% изменчивости по сравнению с объектом исследования в фармакокинетическом параметре, испытанном после введения равного количества атазанавира объекту исследования и эквивалентному объекту исследования. Специалисту в данной области техники будет понятно, что одним из способов снижения изменчивости является совместное введение соединения согласно изобретению и атазанавира.
В фармацевтических композициях согласно изобретению соединение согласно изобретению присутствует в эффективном количестве. Термин «эффективное количество», когда используется в данном описании, относится к количеству, которое при введении в походящем режиме дозирования является достаточным для лечения (терапевтического или профилактического) целевого расстройства. Например, эффективное количество является достаточным для снижения или облегчения тяжести, продолжительности или развития расстройства, подлежащего лечению, предотвращения развития расстройства, подлежащего лечению, побуждения регрессии расстройства, подлежащего лечению, или повышения или улучшения терапевтического(их) действия(й) других терапевтических средств. Предпочтительно соединение присутствует в композиции в количестве от 0,1 до 50% мас., более предпочтительно от 1 до 30% мас., наиболее предпочтительно от 5 до 20% мас.
Соотношение доз для животных и доз для людей (в миллиграммах на квадратный метр поверхности тела) описано в публикации Freireich et al., (1966) Cancer Chemother. Rep. 50: 219. Площадь поверхности тела может быть приблизительно определена исходя из роста и массы пациента (см. Scientific Tables, Geigy Pharmaceuticals, Ardsley, N Y, 1970, 537).
В одном варианте осуществления изобретения эффективное количество соединения согласно настоящему изобретению может находиться в интервале от примерно 200 до примерно 800 мг на лечение. В более точно определенных вариантах осуществления изобретения интервал эффективного количества составляет от примерно 250 мг до примерно 600 мг, от примерно 250 мг до примерно 400 мг, от примерно 300 до примерно 500 мг или наиболее точно от примерно 325 мг до примерно 450 мг. Лечение обычно представляет собой введение от одного до двух раз в сутки. Эффективные дозы будут также изменяться, что понятно специалисту в данной области техники, в зависимости от заболеваний, подлежащих лечению, тяжести заболевания, способа введения, пола, возраста и общего состояния здоровья пациента, используемого наполнителя, возможности сочетания с другими терапевтическими лечениями, например применения других лекарственных средств и заключения лечащего врача. Например, руководство по выбору эффективной дозы может определяться обращением к ранее описанной информации для атазанавира.
Для фармацевтических композиций, которые содержат второе терапевтическое средство, эффективное количество второго терапевтического средства находится в интервале от примерно 20% до 100% дозы, обычно используемой в монотерапевтическом режиме с применением только данного средства. Предпочтительно, эффективное количество находится в интервале от примерно 70% до 100% обычной монотерапевтической дозы. Обычные монотерапевтические дозы вторых терапевтических средств хорошо известны в данной области техники (см., например, Wells et al., eds., Pharmacotherapy Handbook, 2nd Edition, Appleton and Lange, Stamford, Conn. (2000); PDR Pharmacopoeia, Tarascon Pocket Pharmacopoeia 2000, Deluxe Edition, Tarascon Publishing, Loma Linda, Calif. (2000), содержание которых введено в данное описание во всей полноте посредством ссылки).
Ожидается, что некоторые из вторых терапевтических средств, упомянутых выше, будут действовать синергически с соединениями согласно настоящему изобретению. Когда имеет место эффект синергизма, он позволяет снизить эффективную дозу второго терапевтического средства и/или соединения согласно настоящему изобретению относительно дозы, необходимой при монотерапии. Это дает преимущество снижения до минимума токсического побочного действия второго терапевтического средства или соединения согласно настоящему изобретению, преимущество синергического повышения эффективности, преимущество повышенной простоты введения или применения и/или снижения общей стоимости препарата или рецептуры соединения.
СПОСОБЫ ЛЕЧЕНИЯ
В другом варианте осуществления изобретение предоставляет способ лечения ВИЧ инфекции у пациента, нуждающегося в таком лечении, включающий стадию введения пациенту эффективного количества соединения любой из формул А, I, Ia, Ib или Ic или фармацевтически приемлемой композиции, содержащей соединение любой из формул А, I, Ia, Ib или Ic.
Способы, представленные в данном описании, включают также способы, где пациент определен как нуждающийся в конкретно заявленном лечении. Идентификация пациента как нуждающегося в таком лечении может быть осуществлена решением пациента или специалиста в области медицины и может быть субъективной (например, субъективным мнением) или объективной (например, количественно определена опытным путем или диагностическим методом).
В еще одном варианте осуществления изобретения любой из упомянутых выше способов лечения включает дополнительную стадию совместного введения указанному пациенту одного или нескольких вторых терапевтических средств. Выбор второго терапевтического средства может быть осуществлен из любого второго терапевтического средства, которое, как известно, может применяться для совместного введения с атазанавиром. Выбор второго терапевтического средства также зависит от конкретного заболевания или состояния, подлежащего лечению. Примеры вторых терапевтических лекарственных средств, которые могут использоваться в способах согласно данному изобретению, включают терапевтические лекарственные средства, перечисленные выше для применения в комбинированных композициях, содержащих соединение согласно настоящему изобретению и второе терапевтическое лекарственное средство.
В частности, способы комбинированного терапевтического лечения согласно настоящему изобретению включают совместное введение соединения любой из формул А, I, Ia, Ib или Ic и второго ингибитора ВИЧ протеазы (например, ампренавира, фосампренавира, типранавира, индинавира, саквинавира, лопинавира, ритонавира, дарунавира или нелфинавира), ненуклеозидного обратного ингибитора транскриптазы (NNRTI) (например, этравирина, делавирдина, эфавиренза, невирапина или рилпивирина), нуклеозидного/нуклеотидного обратного ингибитора транскриптазы (NRTI) (например, зидовудина, ламивудина, эмтрицитабина, зидовудина, тенофовирдисопроксила фумарата, диданозина, ставудина, абакавира, рацивира, амдоксовира, априцитабина или элвуцитабина), ингибитора входа вируса (например, энфувиртида, маравирока, викривирока, PRO 140 или TNX-355), ингибитора интегразы (например, ралтегравира или элвитегравира), иммунного противоретровирусного средства (например, иммунитина, пролейкина, ремуна, BAY 50-4798 или IR103), ингибитора созревания вируса (например, бевиримата), клеточного ингибитора (например, дроксии или гидроксимочевины) или сочетания двух или нескольких указанных выше соединений.
В более точно описанном варианте осуществления изобретения способы комбинированного терапевтического лечения включают совместное введение соединения любой из формул А, I, Ia, Ib или Ic и второго терапевтического средства, выбранного из группы, включающей ритонавир, эфавиренз, диданозин, тенофовирдисопроксил, нелфинавирмезилат, ампренавир, ралтегравир, саквинавир, лопинавир, невирапин, эмтрицитабин, абакавир, ламивудин, зидовудин, маравирок, ставудин, дарунавир, фосампренавир, викривирок, фармацевтически приемлемые соли любого их вышеуказанных соединений и их сочетания для лечения ВИЧ инфекции у пациента, нуждающегося в таком лечении.
В еще более точно определенном варианте осуществления изобретения второе терапевтическое средство выбрано из группы, включающей ритонавир, эфавиренз, диданозин, ралтегравир, тенофовирдисопроксил, ламивудин, абакавир, зидовудин, эмтрицитабин, эфавиренз, фармацевтически приемлемые соли любого из вышеуказанных соединений и их сочетания. В другом конкретно определенном варианте осуществления изобретения способ включает совместное введение соединения любой из формул А, I, Ia, Ib или Ic и от двух до трех вторых терапевтических средств, упомянутых выше в данном абзаце. В еще более точно определенном варианте осуществления изобретения способ включает совместное введение соединения любой из формул А, I, Ia, Ib или Ic и два вторых терапевтических средства, упомянутых выше в данном абзаце.
Термин «совместно введенный», когда используется в данном описании, означает, что второе терапевтическое средство может вводиться вместе с соединением согласно данному изобретению как часть единичной дозированной формы (такой, как композиция согласно данному изобретению, содержащая соединение согласно данному изобретению, и второе терапевтическое средство, которое описано выше) или как раздельные дозированные формы. Альтернативно, дополнительное лекарственное средство может вводиться до введения, совместно или после введения соединения согласно данному изобретению. В таком комбинированном терапевтическом лечении оба соединения согласно данному изобретению и второе(ые) терапевтическое(ие) средство(а) вводятся удобными способами. Введение пациенту композиции согласно данному изобретению, содержащей соединение согласно данному изобретению и второе терапевтическое средство, не исключает раздельного введения такого же терапевтического средства, любого другого второго терапевтического средства или любого соединения согласно данному изобретению указанному пациенту в другое время в течение курса лечения.
Эффективные количества этих вторых терапевтических средств хорошо известны специалисту в данной области техники, и указание по дозировке можно найти в патентах и публикациях заявок на патенты, указанных в описании в качестве ссылок, в публикациях Wells et al., eds., Pharmacotherapy Handbook, 2nd Edition, Appleton and Lange, Stamford, Conn. (2000); PDR Pharmacopoeia, Tarascon Pocket Pharmacopoeia 2000, Deluxe Edition, Tarascon Publishing, Loma Linda, Calif. (2000), а также других медицинских публикациях. Однако квалифицированный специалист в данной области техники сможет определить оптимальный интервал дозировки второго терапевтического средства.
При простом лечении пациентов рекомендованная доза Reyataz® (атазанавира сульфат) для лечения ВИЧ-1 инфекции составляет 400 мг раз в сутки с пищей. При совместном введении с тенофовиром рекомендованная доза Reyataz® составляет 300 мг, и рекомендованная доза ритонавира составляет 100 мг. При длительном лечении пациентов рекомендованная доза Reyataz для лечения ВИЧ-1 инфекции составляет 300 мг с ритонавиром 100 мг один раз в сутки с пищей. Исходя из данных, полученных при испытании на животных и представленных в данном описании, ожидается, что некоторые соединения согласно данному изобретению после приема дозы в интервале от 325 мг до 450 мг в сутки обладают полезным действием для людей, которое состоит в достижении Сmin и/или AUC, которая сравнима с Сmin и/или AUC, достигаемой при введении дозы атазанавира 300 мг раз в сутки с 100 мг ритонавира. Соответственно, один вариант осуществления изобретения предоставляет способ лечения ВИЧ инфекции введением субъекту, нуждающемуся в таком лечении, раз в сутки композиции, включающей соединение согласно данному изобретению в дозе в интервале от 325 мг до 450 мг. В еще одном варианте осуществления изобретения такая композиция вводится без совместного введения ритонавира.
Еще один вариант осуществления изобретения относится к способу лечения ВИЧ инфекции введением композиции, содержащей соединение согласно настоящему изобретению, один раз в сутки в дозе в интервале от 250 мг до 400 мг.
В еще одном варианте осуществления изобретения, где второе терапевтическое средство вводится субъекту, эффективное количество соединения согласно настоящему изобретению меньше его эффективного количества без введения второго терапевтического средства. В другом варианте осуществления изобретения эффективное количество второго терапевтического средства меньше его эффективного количества без введения соединения согласно настоящему изобретению. В этом случае нежелательные побочные эффекты, связанные с высокими дозами любого агента, могут быть снижены до минимума. Другие возможные преимущества (включая, но без ограничения, улучшенные режимы дозирования и/или снижение стоимости лекарственного средства) будут понятны специалисту в данной области техники.
В соответствии с еще одним аспектом изобретение предоставляет применение соединения формулы I, одного или в комбинациии с одним или несколькими перечисленными выше вторыми терапевтическими средствами, в производстве лекарственного средства в форме единой композиции или в форме отдельных дозированных форм для лечения или профилактики у пациента заболевания, расстройства или его симптома, упомянутых выше. Другим аспектом настоящего изобретения является соединение любой из формул А, I, Ia, Ib или Ic для применения в лечении или профилактики у пациента заболевания, расстройства или его симптома, упомянутого в описании. В дополнительном аспекте соединения согласно настоящему изобретению могут применяться в медицине, например в терапии. В любом из этих применений соединение предпочтительно вводится без совместного введения ритонавира.
МЕТОДЫ ДИАГНОСТИКИ И НАБОРЫ
Соединения и композиции согласно настоящему изобретению также могут применяться в качестве реагентов в методах определения концентрации атазанавира в растворе или биологическом образце, таком как плазма, в исследованиях метаболизма атазанавира, а также в других аналитических исследованиях.
В соответствии с одним вариантом осуществления изобретение предоставляет способ определения концентрации атазанавира в растворе или биологическом образце, где способ включает следующие стадии:
a) добавление к раствору биологического образца соединения формулы А в известной концентрации;
b) внесение раствора или биологического образца в измерительное устройство, которое отличает атазанавир от соединения формулы А;
с) калибровка измерительного устройства для корреляции определения количества соединения формулы А с известной концентрацией соединения формулы А, добавленного к биологическому образцу или раствору;
d) определение количества атазанавира в биологическом образце с помощью указанного калиброванного измерительного устройства;
е) определение концентрации атазанавира в растворе образца с использованием корреляции между определенным количеством и концентрацией, полученной для соединения формулы А.
Измерительные устройства, которые могут отличить атазанавир от соответствующего соединения формулы А, включают любое измерительное устройство, которое может различить два соединения, которые отличаются друг от друга только содержанием изотопов. Примеры измерительных устройств включают масс-спектрометр, ЯМР-спектрометр или ИК-спектрометр.
В другом варианте осуществления изобретение предоставляет способ оценки метаболической стабильности соединения формулы А, включающий стадии контактирования соединения формулы А с источником метаболизирующего фермента в течение некоторого периода времени и сравнение количества соединения формулы А с продуктами метаболизма соединения формулы I после указанного периода времени.
В родственном варианте осуществления изобретение предоставляет способ оценки метаболической стабильности соединения формулы А у пациента после введения соединения формулы А. Данный способ включает стадии отбора образцов сыворотки, мочи или фекалий у пациента после введения соединения формулы А указанному пациенту; и сравнение количества соединения формулы А с продуктами метаболизма соединения формулы А в образце сыворотки, мочи или фекалий.
Настоящее изобретение предоставляет также наборы для применения в лечении ВИЧ инфекции. Эти наборы включают (а) фармацевтическую композицию, содержащую соединение любой из формул А, I, Ia, Ib или Ic или его соль, где указанная фармацевтическая композиция находится в контейнере; и (b) инструкции, описывающие способ применения фармацевтической композиции для лечения ВИЧ инфекции.
Контейнером может быть любой сосуд, или другой герметичный аппарат, или аппарат, который может герметично закрываться, где может находиться указанная фармацевтическая композиция. Примеры включают бутыли, ампулы, разделенные или многокамерные держатели ампул, где каждое отделение или камера включает единичную дозу указанной композиции, или распределительное устройство, которое высвобождает единичные дозы указанной композиции. Контейнер может иметь любую удобную форму, известную в данной области техники, и изготавливаться из фармацевтически приемлемого материала, например, в форме бумажной или картонной коробки, стеклянной или пластиковой бутыли или банки, сумки, которая может герметично закрываться (например, удерживать «сменный блок» таблеток для внесения их в другой контейнер), или в форме блистерной упаковки с дозами для выдавливания их из модуля согласно терапевтическому графику. Вид применяемого контейнера может зависеть от конкретной дозированной формы, которую он включает, например традиционная картонная коробка не должна использоваться для содержания жидкой суспензии. Допустимо применение в одной упаковке более одного контейнера для продажи единичной дозированной формы. Например, таблетки могут находиться в бутыли, которая, в свою очередь, размещена внутри коробки. В одном варианте осуществления изобретения контейнер представляет собой блистерную упаковку.
Наборы согласно настоящему изобретению могут включать устройство для введения или для дозирования единичной дозы фармацевтической композиции. Такое устройство может включать аппарат для ингаляционной терапии, если указанная композиция представляет собой композицию, которая может вводиться ингаляцией; шприц и иглу, если указанная композиция представляет собой композицию для инъекций; шприц, ложку, нагнетающее устройство или емкость с маркировкой объема или без маркировки объема, если указанная композиция представляет собой жидкую композицию для перорального введения; или любое другое отмеряющее или доставляющее устройство, подходящее для дозированной формы композиции, присутствующей в наборе.
В некотором варианте осуществления изобретения наборы согласно настоящему изобретению могут включать отдельную емкость контейнера фармацевтической композиции, содержащей второе терапевтическое средство, например одно из упомянутых выше, для совместного введения с соединением согласно настоящему изобретению.
Изобретение, описанное в общих чертах, далее будет более подробно описано с помощью примеров, которые представлены только для иллюстрации некоторых аспектов и вариантов осуществления настоящего изобретения, но не предназначены для ограничения изобретения каким бы то ни было образом.
Примеры
Пример 1. Синтез 1,14-ди(метил-d 3 )-(3S,8S,9S,12S)-3,12-бис[(1,1-диметилэтил)-d 9 ]-8-гидрокси-4,11-диоксо-9-(фенилметил)-6-[[4-(2-пиридинил)фенил]метил]-2,5,6,10,13-пентаазатетрадекандиоата (соединение 122)
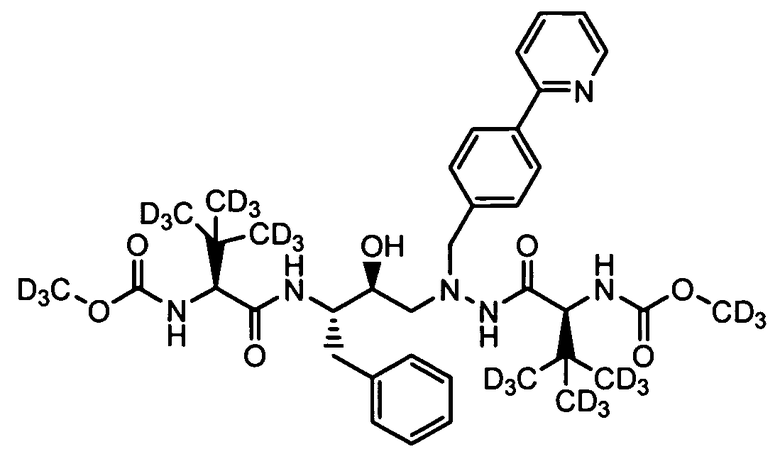
Соединение 122
Соединение 122 получают в соответствии со схемой 1, представленной выше. Подробности каждой стадии синтеза описаны ниже и относятся к общему способу А.
Синтез трет-бутил-2-(4-(пиридин-2-ил)бензилиден)гидразинкарбоксилата (XII, Y 1a =H)
Смесь 4-(пиридин-2-ил)бензальдегида X (17,7 г, 96,6 ммоль) и трет-бутилкарбазата (12,2 г, 92,3 ммоль) в этаноле (125 мл) кипятят с обратным холодильником в атмосфере азота в течение 4 часов (час). Реакционную смесь охлаждают до 40°C и добавляют лед (60 г). Полученную смесь перемешивают в течение 20 минут (мин). Осадок собирают фильтрацией, промывают водой и сушат в вакуумной печи (60°C), получая продукт XII, где Y1a=H (25,0 г, 91,1%).
Синтез трет-бутил-2-(4-(пиридин-2-ил)бензил)гидразинкарбоксилата (XIII, Y 1a =Y 1b =H)
Раствор XII, Y1a=H (23,15 г, 77,85 ммоль) в метаноле (350 мл) обрабатывают 20% палладием на активированном угле (2,3 г, 50% мас.) и гидрируют при давлении 10 фунтов на кв. дюйм (689,475 Па) в течение 4 часов. Реакционную смесь фильтруют через целит, осадок на фильтре промывают метанолом и растворитель удаляют на роторном испарителе. Остаток перекристаллизовывают из гептана и сушат в вакуумной печи (40°С), получая XIII, где Y1a=Y1b=H (22,48 г, 96,5%).
Синтез трет-бутил-2-((2S,3S)-3-(трет-бутоксикарбониламино)-2-гидрокси-4-фенилбутил)-2-(4-(пиридин-2-ил)бензил)гидразинкарбоксилата (XV, Y 1а =Y 1b =H)
Смесь трет-бутил-(S)-1-((R)-оксиран-2-ил)-2-фенилэтилкарбамата XIV (1,18 г, 4,48 ммоль), XIII, Y1a=Y1b=H (1,23 г, 4,11 ммоль) и изопропанола (15 мл) кипятят с обратным холодильником в атмосфере азота в течение ночи. Растворитель удаляют на роторном испарителе и остаток очищают хроматографией на диоксиде кремния (100 г) (элюирование: дихлорметан/этилацетат 8:2), получая продукт XV, где Y1a=Y1b=H (1,74 г, 75%).
Синтез (2S,3S)-3-амино-4-фенил-1-(1-(4-(пиридин-2-ил)бензил)гидразинил)бутан-2-ола (XVI, Y 1a =Y 1b =H)
Раствор XV, Y1a=Y1b=H (2,84 г, 5,05 ммоль) в дихлорметане (30 мл) перемешивают в атмосфере азота при комнатной температуре и обрабатывают 4 н. HCl в диоксане (60 мл). Перемешивание продолжают при комнатной температуре в течение 20 минут. Добавляют метанол в количестве, достаточном для растворения образовавшегося осадка, и перемешивание продолжают при комнатной температуре в течение 2 часов. Растворители удаляют с помощью роторного испарителя и остаток сушат в вакуумной печи (60°C), получая XVI, где Y1a=Y1b=H (3,27 г, 5,05 ммоль, предполагая полную конверсию) в форме сложной гидрохлоридной соли.
Синтез 1,14-ди(метил-d 3 )-(3S,8S,9S,12S)-3,12-бис[(1,1-диметилэтил)-d 9 ]-8-гидрокси-4,11-диоксо-9-(фенилметил)-6-[[4-(2-пиридинил)фенил]метил]-2,5,6,10,13-пентаазатетрадекандиоата (122)
Смесь (S)-2-(метоксикарбониламино)-3,3-диметилбутановой кислоты XVII-d 12 (R1a=R1b=CD3, R2=R3=C(CD3)3; 0,90 г, 4,44 ммоль; получена в соответствии со схемой 5 и примером 13) и тетрафторбората O-(1,2-дигидро-2-оксо-1-пиридил)-N,N,N',N'-тетраметилурония (TPTU) (1,32 г, 4,44 ммоль) в дихлорметане (40 мл) обрабатывают диизопропилэтиламином (1,16 г, 8,88 ммоль) и перемешивают в атмосфере азота при комнатной температуре в течение 30 минут. Полученный раствор добавляют к охлажденной на ледяной бане суспензии гидрохлорида XVI, Yla=Ylb=H (1,15 г, 1,78 ммоль) и полученную смесь перемешивают при комнатной температуре в течение ночи. Реакционную смесь разбавляют дихлорметаном (140 мл), промывают водой (2×100 мл) и насыщенным раствором бикарбоната натрия (150 мл), сушат над сульфатом натрия и фильтруют. Растворитель удаляют на роторном испарителе и сырой продукт очищают хроматографией на диоксиде кремния (120 г) (элюирование: 2% этанола в смеси гептан/этилацетат (4,5 л)). Растворитель удаляют из чистых фракций и остаток (0,57 г) переносят в этилацетат (10 мл), перемешивают при 60°С в течение 20 минут и разбавляют MTBE (60 мл). После охлаждения осадок собирают фильтрацией, промывают MTBE и сушат в вакуумной печи (55°C), получая соединение 122 (0,40 г). Менее чистые фракции, полученные в результате хроматографии, дают дополнительные 0,57 г сырого продукта.
1H-ЯМР (300 МГц, CDCl3): δ 2,54 (д, 1H), 2,87-2,95 (м, 3H), 3,57 (д, 2H), 3,75 (д, 1H), 3,91-4,08 (м, 3H), 4,81 (уш., 1H), 5,15-5,30 (м, 2H), 6,38-6,43 (м, 2H), 7,14-7,23 (м, 6H, частично приглушенный CDCl3), 7,41 (д, 2H), 7,68-7,76 (м, 2H), 7,94 (д, 2H), 8,68 (д, 1H). ВЭЖХ (метод: колонка С18-RP 20 мм; элюирование с градиентом: 2-95% ацетонитрила + 0,1% муравьиной кислоты в течение 3,3 минут с 1,7 мин удержанием при 95% ацетонитрила; длина волны: 254 нм): время удержания: 3,22 минуты. МС (M+H+): 729,6.
Пример 2. Синтез 1,14-диметил-(3S,8S,9S,12S)-3,12-бис[(1,1-диметилэтил)-d 9 ]-8-гидрокси-4,11-диоксо-9-(фенилметил)-6-[[4-(2-пиридинил)фенил]метил]-2,5,6,10,13-пентаазатетрадекандиоата (соединение 106)
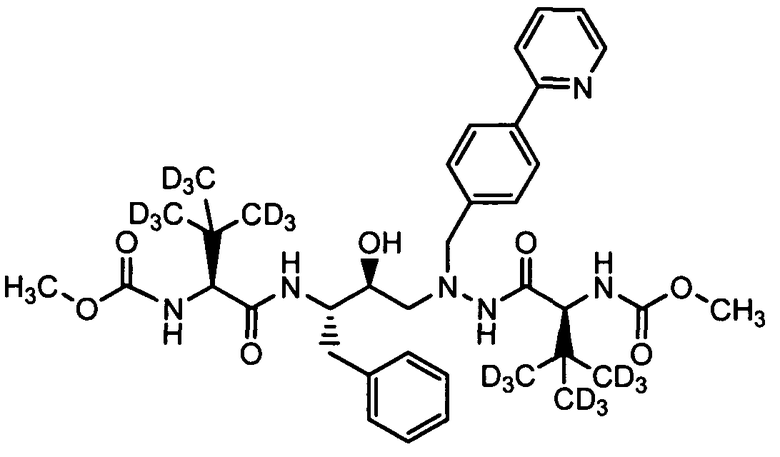
Соединение 106
Соединение 106 получают в соответствии со схемой 1, представленной выше, и следуя общему способу А, описанному выше.
Синтез 1,14-диметил-(3S,8S,9S,12S)-3,12-бис[(1,1-диметилэтил)-d 9 ]-8-гидрокси-4,11-диоксо-9-(фенилметил)-6-[[4-(2-пиридинил)фенил]метил]-2,5,6,10,13-пентаазатетрадекандиоата (106)
Соединение 106 получают общим способом A, описанным выше, из (2S,3S)-3-амино-4-фенил-1-(1-(4-(пиридин-2-ил)бензил)гидразинил)бутан-2-ола (XVI, Y1a=Y1b=H, гидрохлорид) и (S)-2-(метоксикарбониламино)-3,3-диметилбутановой кислоты-d 9 (XVII- d 9, R1a=Rlb=CH3, R2=R3=C(CD3)3; получена в соответствии со схемой 5).
1H-ЯМР (300 МГц, CDCl3): δ 2,54 (д, 1H), 2,84-2,89 (м, 1H), 2,93 (д, 2H), 3,57 (д, 2H), 3,63 (с, 3H), 3,66 (с, 3H), 3,75 (д, 1H), 3,91-4,08 (м, 3H), 4,81 (уш., 1H), 5,15-5,32 (м, 2H), 6,36-6,45 (м, 2H), 7,18-7,24 (м, 6H, частично затененный CDCl3), 7,41 (д, 2H), 7,68-7,76 (м, 2H), 7,94 (д, 2H), 8,68 (д, 1H). ВЭЖХ (метод: колонка C18-RP 20 мм; элюирование с градиентом: 2-95% ACN + 0,1% муравьиная кислота в течение 3,3 мин с удержанием в течение 1,7 мин при 95% ACN; длина волны: 254 нм): время удержания: 3,23 мин. МС (M+H+): 723,6.
Пример 3 . Синтез 1,14-ди(метил-d 3 )-(3S,8S,9S,12S)-3,12-бис(1,1-диметилэтил)-8-гидрокси-4,11-диоксо-9-(фенилметил)-6-[[4-(2-пиридинил)фенил]метил]-2,5,6,10,13-пентаазатетрадекандиоата (соединение 103)
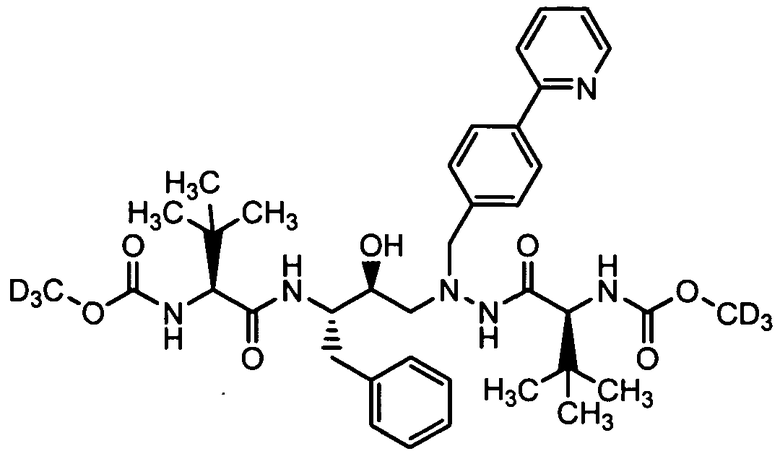
Соединение 103
Соединение 103 получают в соответствии со схемой 1, представленной выше, и следуя общему способу A, описанному выше.
Синтез 1,14-ди(метил-d 3 )-(3S,8S,95,12S)-3,12-бис(1,1-диметилэтил)-8-гидрокси-4,11-диоксо-9-(фенилметил)-6-[[4-(2-пиридинил)фенил]метил]-2,5,6,10,13-пентаазатетрадекандиоата (103)
Соединение 103 получают в соответствии с общим способом A из (2S,3S)-3-амино-4-фенил-1-(1-(4-(пиридин-2-ил)бензил)гидразинил)бутан-2-ола (XVI, Y1a=Y1b=H, гидрохлорид) и известного соединения (S)-2-(метоксикарбониламино)-3,3-диметилбутановой кислоты-d3 (XVII-d 3, R1a=R1b=CH3, R2=R3=C(CD3)3) (Zhang, Huiping et al., Journal of Labelled Compounds & Radiopharmaceuticals, 2005, 48(14), 1041-1047).
1H-ЯМР (300 МГц, CDCl3): δ 0,79 (c, 9H), 0,87 (с, 9H), 2,52 (д, 1H), 2,82-2,95 (м, 3H), 3,58 (д, 2H), 3,77 (д, 1H), 3,91-4,08 (м, 3H), 4,81 (с, 1H), 5,15-5,32 (м, 2H), 6,35-6,45 (м, 2H), 7,16-7,24 (м, 6H, частично затененный CDCl3), 7,41 (д, 2H), 7,68-7,76 (м, 2H), 7,94 (д, 2H), 8,68 (д, 1H). ВЭЖХ (метод: колонка C18-RP 20 мм; элюирование с градиентом: 2-95% ACN + 0,1% муравьиная кислота в течение 3,3 мин с удержанием в течение 1,7 мин при 95% ACN; длина волны: 254 нм): время удержания: 3,24 мин. МС (M+H+): 711,3.
Пример 4. Синтез 1,14-ди(метил-d 3 )-(3S,8S,9S,12S)-3,12-бис[(1,1-диметилэтил)-d 9 ]-8-гидрокси-4,11-диоксо-9-(фенилметил)-6-[[4-(2-пиридинил)фенил]метил-d 2 ]-2,5,6,10,13-пентаазатетрадекандиоата (соединение 131)
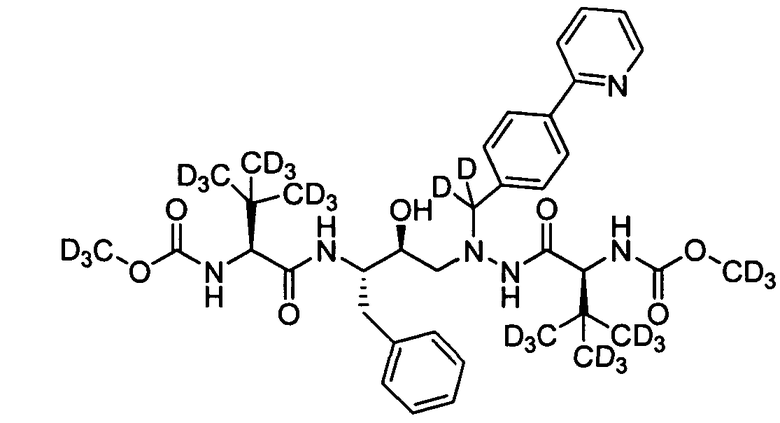
Соединение 131
Соединение 131 получают в соответствии со схемой 1, представленной выше, и следуя общему способу A, описанному выше. В данном синтезе используют газообразный дейтерий (Cambridge Isotopes, 99,8% атом. D), MeOD (Aldrich, 99,5% атом. D), iPrOD (Aldrich, 98% атом. D) и хлорид дейтерия (Aldrich, 99% атом. D). Дейтерированный альдегид X получают в соответствии со схемой 2b с использованием LiAlD4 (Cambridge Isotopes, 98% атом. D).
1H-ЯМР (300 МГц, CDCl3): δ 2,71 (дд, 2H), 2,94 (д, 2H), 3,56 (д, 2H), 3,77 (д, 1H), 4,02-4,05 (м, 1H), 4,83 (с, 1H), 5,19-5,29 (м, 2H), 6,40-6,47 (м, 2H), 7,20-7,23 (м, 6H, частично затененный CDCl3), 7,41 (д, 2H), 7,69-7,76 (м, 2H), 7,95 (д, 2H), 8,69 (д, 1H). ВЭЖХ (метод: колонка C18-RP 20 мм; элюирование с градиентом: 2-95% ACN + 0,1% муравьиная кислота на 3,3 мин с 1,7 мин удержанием при 95% ACN; длина волны: 254 нм): время удержания: 3,22 мин; чистота: 99,2%. МС (M+H+): 731,7.
Пример 5. Синтез 1,14-ди(метил-d 3 )-(3S,8S,9S,12S)-3-(1,1-диметилэтил)-12-[(1,1-диметилэтил)-d 9 ]-8-гидрокси-4,11-диоксо-9-(фенилметил)-6-[[4-(2-пиридинил)фенил]метил]-2,5,6,10,13-пентаазатетрадекандиоата (соединение 120)
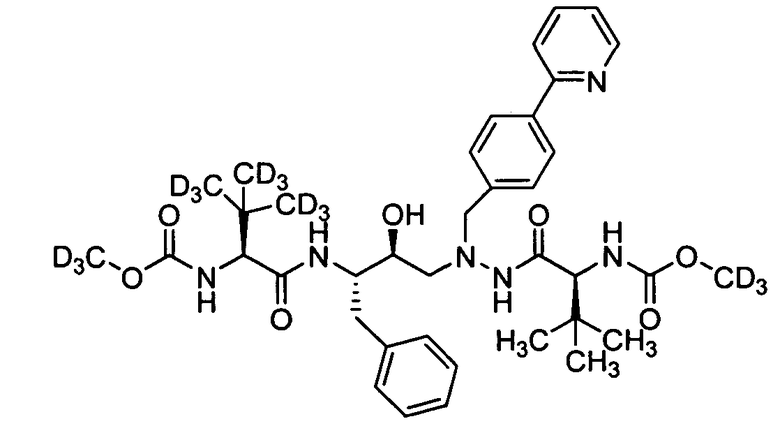
Соединение 120
Соединение 120 получают в соответствии со схемой 1b, представленной выше.
1H-ЯМР (300 МГц, CDCl3): δ 0,78 (с, 9H), 2,72 (дд, 2H), 2,94 (д, 2H), 3,58-3,63 (м, 2H), 3,78 (д, 1Н), 3,92-4,09 (м, 3H), 4,88 (с, 1Н), 5,28 (дд, 2H), 6,46 (д, 1Н), 6,73 (с, 1Н), 7,14-7,25 (м, 6H, частично затененный CDCl3), 7,42 (д, 2H), 7,68-7,78 (м, 2H), 7,94 (д, 2H), 8,68 (д, 1Н). ВЭЖХ (метод: колонка C18-RP 20 мм; элюирование с градиентом: 2-95% ACN + 0,1% муравьиная кислота на 3,3 мин с 1,7 мин удерживанием при 95% ACN; длина волны: 254 нм): время удержания: 3,23 мин; чистота: 99,6%. МС (M+H+): 720,6.
Пример 6 . Синтез 1,14-ди(метил-d 3 )-(3S,8S,9S,12S)-3-[(1,1-диметилэтил-d 9 ]-12-(1,1-диметилэтил-8-гидрокси-4,11-диоксо-9-(фенилметил)-6-[[4-(2-пиридинил)фенил]метил]-2,5,6,10,13-пентаазатетрадекандиоата (соединение 121)
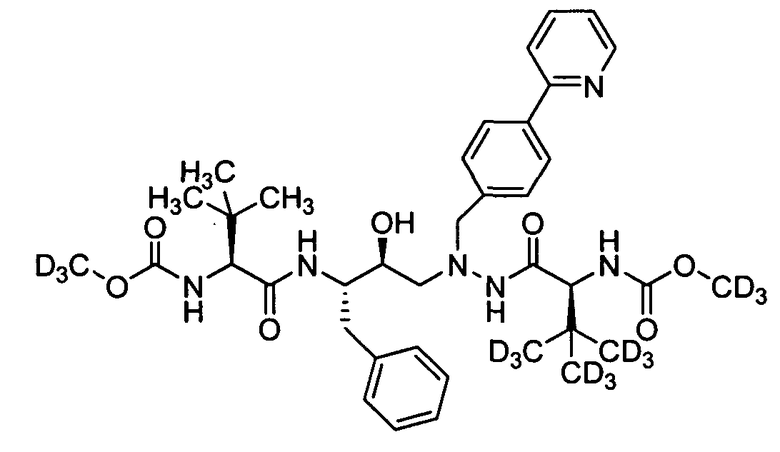
Соединение 121
Соединение 121 получают в соответствии со схемой 1c, представленной выше. 1H-ЯМР (300 МГц, CDCl3): δ 0,86 (с, 9H), 2,72 (дд, 2H), 2,94 (д, 2H), 3,60-3,63 (м, 2H), 3,80 (д, 1Н), 3,92-4,09 (м, 3H), 4,89 (с, 1Н), 5,30 (дд, 2H), 6,43 (д, 1Н), 6,74 (с, 1Н), 7,14-7,26 (м, 6H, частично затененный CDCl3), 7,42 (д, 2H), 7,68-7,79 (м, 2H), 7,93 (д, 2H), 8,68 (д, 1Н). ВЭЖХ (метод: колонка C18-RP 20 мм; элюирование с градиентом: 2-95% ACN + 0,1% муравьиная кислота на 3,3 мин с 1,7 мин удерживанием при 95% ACN; длина волны: 254 нм): время удержания: 3,22 мин; чистота: 99,4%. МС (M+H+): 720,6.
Пример 7. Синтез 1,14-диметил-(3S,8S,9S,12S)-3-(1,1-диметилэтил)-12-[(1,1-диметилэтил)-d 9 ]-8-гидрокси-4,11-диоксо-9-(фенилметил)-6-[[4-(2-пиридинил)фенил]метил]-2,5,6,10,13-пентаазатетрадекандиоата (соединение 104)
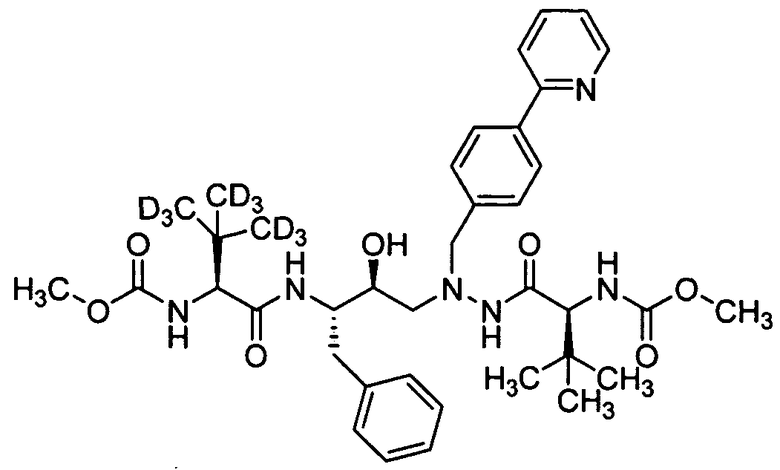
Соединение 104
Соединение 104 получают в соответствии со схемой 1c, представленной выше.
1H-ЯМР (300 МГц, CDCl3): δ 0,78 (с, 9H), 2,70 (дд, 2H), 2,94 (д, 2H), 3,59-3,66 (м, 8H), 3,78 (д, 1Н), 3,92-4,09 (м, 3H), 4,86 (с, 1Н), 5,27 (дд, 2H), 6,44 (д, 1Н), 6,63 (с, 1Н), 7,14-7,26 (м, 6H, частично затененный CDCl3), 7,42 (д, 2H), 7,68-7,79 (м, 2H), 7,94 (д, 2H), 8,69 (д, 1Н). ВЭЖХ (метод: колонка C18-RP 20 мм; элюирование с градиентом: 2-95% ACN + 0,1% муравьиная кислота на 3,3 мин с 1,7 мин удерживанием при 95% ACN; длина волны: 254 нм): время удержания: 3,23 мин; чистота: 99,8%. МС (M+H+): 714,6.
Пример 8. Синтез 1,14-диметил-(3S,8S,9S,12S)-3,12-бис[(1,1-диметилэтил)-d 9 ]-8-гидрокси-4,11-диоксо-9-(фенилметил)-6-[[4-(2-пиридинил)фенил]метил-d 2 ]-2,5,6,10,13-пентаазатетрадекандиоата (соединение 113)
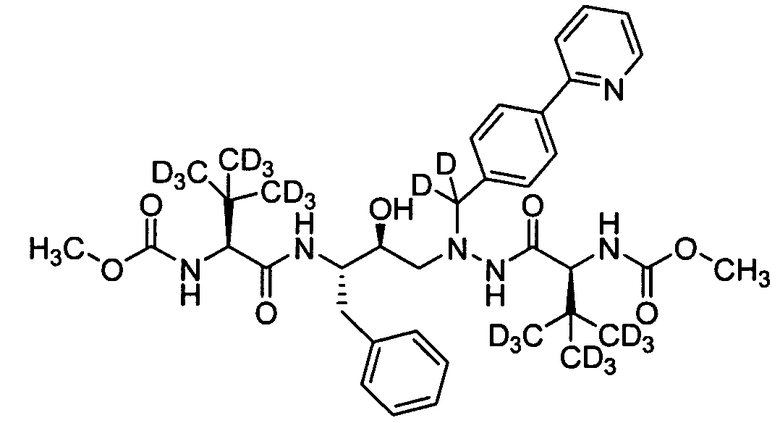
Соединение 113
Соединение 113 получают в соответствии со схемой 1, представленной выше, и следуя общему способу A, описанному выше. В данном синтезе используют газообразный дейтерий (Cambridge Isotopes, 99,8% атом. D), MeOD (Aldrich, 99,5% атом. D), iPrOD (Aldrich, 98% атом. D) и хлорид дейтерия (Aldrich, 99% атом. D). Дейтерированный альдегид X получают в соответствии со схемой 2b с использованием LiAlD4 (Cambridge Isotopes, 98% атом. D).
1H-ЯМР (300 МГц, CDCl3): δ 2,69 (дд, 2H), 2,94 (д, 2H), 3,56-3,59 (м, 2H), 3,64 (с, 3H), 3,67 (с, 3H), 3,77 (д, 1Н), 4,02-4,05 (м, 1Н), 4,84 (с, 1Н), 5,18-5,32 (м, 2H), 6,40-6,45 (м, 2H), 7,14-7,26 (м, 6H, частично затененный CDCl3), 7,41 (д, 2H), 7,61-7,80 (м, 2H), 7,95 (д, 2H), 8,69 (д, 1Н). ВЭЖХ (метод: колонка C18-RP 20 мм; элюирование с градиентом: 2-95% ACN + 0,1% муравьиная кислота на 3,3 мин с 1,7 мин удерживанием при 95% ACN; длина волны: 254 нм): время удержания: 3,25 мин; чистота: 99,4%. МС (M+H+): 725,4.
Пример 9. Синтез 1-метил-14-(метил-d 3 ]-3S,8S,9S,12S)-3-(1,1-диметилэтил)-12-[(1,1-диметилэтил)-d 9 ]-8-гидрокси-4,11-диоксо-9-(фенилметил)-6-[[4-(2-пиридинил)фенил]метил]-2,5,6,10,13-пентаазатетрадекандиоата (соединение 114)
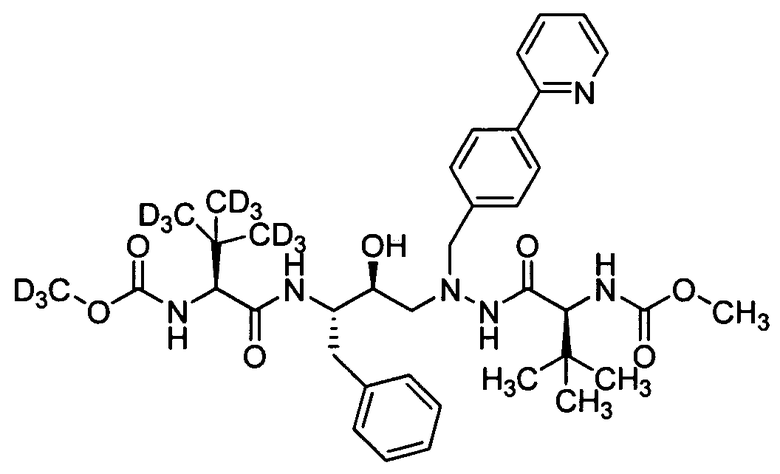
Соединение 114
Соединение 114 получают в соответствии со схемой 1c, представленной выше. Pd(OH)2 используют вместо Pd/C для превращения XXXII в XXXIII.
1H-ЯМР (300 МГц, CDCl3): δ 0,78 (с, 9H), 2,70 (дд, 2H), 2,93 (д, 2H), 3,59-3,63 (м, 5H), 3,78 (д, 1Н), 3,92-4,04 (м, 3H), 4,84 (с, 1Н), 5,30 (дд, 2H), 6,44 (д, 1Н), 6,60 (с, 1Н), 7,20-7,26 (м, 6H, частично затененный CDCl3), 7,41 (д, 2H), 7,70-7,79 (м, 2H), 7,94 (д, 2H), 8,68 (д, 1Н). МС (M+H+): 717,4.
Пример 10. Синтез 1-метил-14-(метил-d 3 )-(3S,8S,9S,12S)-3-(1,1-диметилэтил)-12-[(1,1-диметилэтил)-d 9 ]-8-гидрокси-4,11-диоксо-9-(фенилметил)-6-[[4-(2-пиридинил)фенил]метил-d 2 ]-2,5,6,10,13-пентаазатетрадекандиоата (соединение 123)
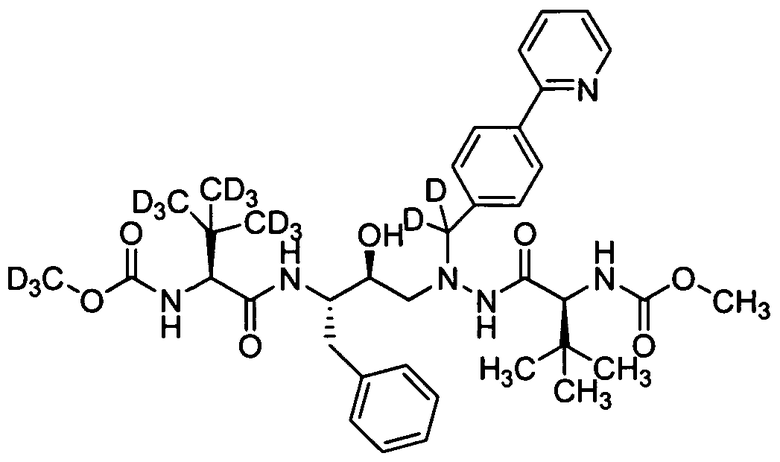
Соединение 123
Соединение 123 получают в соответствии со схемой 1c, представленной выше. В данном синтезе используют газообразный дейтерий (Med-Tech, 98% атом. D), EtOD (Aldrich, 99,5% атом. D), MeOD (Aldrich, 99,5% атом. D), iPrOD (CDN, 99,1% атом. D) и хлорид дейтерия (Aldrich, 99% атом. D). Pd(OH)2 используют вместо Pd/C для превращения XXXII в XXXIII.
1H-ЯМР (300 МГц, CDCl3): δ 0,79 (с, 9H), 2,72 (дд, 2H), 2,93 (д, 2H), 3,56-3,63 (м, 5H), 3,77 (д, 1Н), 4,04 (д, 1Н), 4,81 (с, 1Н), 5,30 (дд, 2H), 6,41 (д, 1Н), 6,51 (с, 1Н), 7,14-7,26 (м, 6H, частично затененный CDCl3), 7,41 (д, 2H), 7,69-7,76 (м, 2H), 7,94 (д, 2H), 8,68 (д, 1Н). МС (MH-H+): 719,5.
Пример 11. Синтез 1,14-диметил-(3S,8S,9S,12S)-3-(1,1-диметилэтил)-12-[(1,1-диметилэтил)-d 9 ]-8-гидрокси-4,11-диоксо-9-(фенилметил)-6-[[4-(2-пиридинил)фенил]метил-d 2 ]-2,5,6,10,13-пентаaзaтетрадекандиоата (соединение 111)
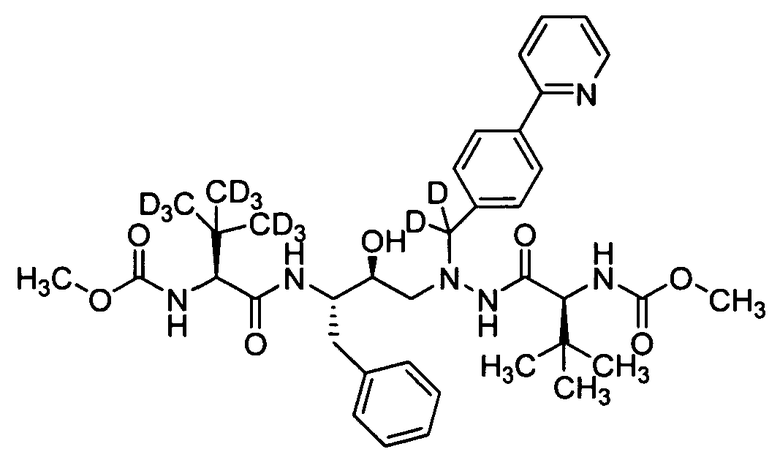
Соединение 111
Соединение 111 получают в соответствии со схемой 1c, представленной выше. В данном синтезе используют газообразный дейтерий (Med-Tech, 98% атом. D), EtOD (Aldrich, 99,5% атом. D), MeOD (Aldrich, 99,5% атом. D), iPrOD (CDN, 99,1% атом. D) и хлорид дейтерия (Aldrich, 99% атом. D). Pd(OH)2 используют вместо Pd/C для превращения XXXII в XXXIII.
1H-ЯМР (300 МГц, CDCl3): δ 0,79 (с, 9H), 2,74 (дд, 2H), 2,93 (д, 2H), 3,58-3,66 (м, 8H), 3,77 (д, 1Н), 4,03 (д, 1Н), 4,82 (с, 1Н), 5,30 (дд, 2H), 6,41 (д, 1Н), 6,51 (с, 1Н), 7,20-7,26 (м, 6H, частично затененный CDCl3), 7,41 (д, 2H), 7,70-7,76 (м, 2H), 7,94 (д, 2H), 8,68 (д, 1Н). МС (M+H+): 716,5.
Пример 12. Синтез 1,14-ди(метил-d 3 )-(3S,8S,9S,12S)-3-(1,1-диметилэтил)-12-[(1,1-диметилэтил)-d 9 ]-8-гидрокси-11-диоксо-9-(фенилметил)-6-[[4-(2-пиридинил)фенил]метил-d 2 ]-2,5,6,10,13-пентаазатетрадекандиоата (соединение 129)
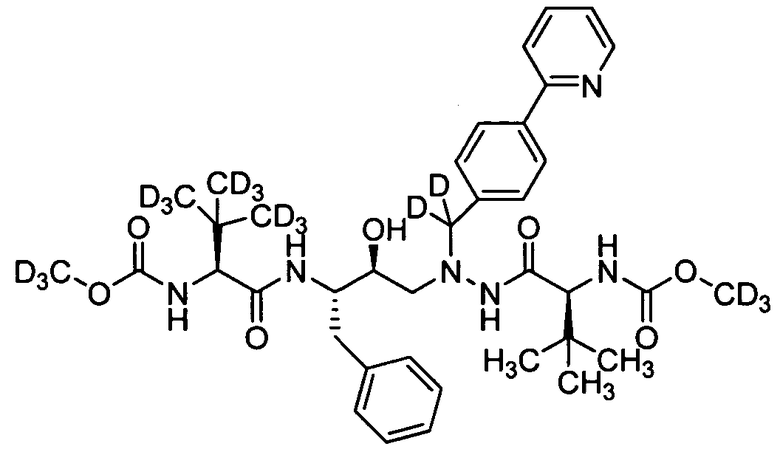
Соединение 129
Соединение 129 получают в соответствии со схемой 1c, представленной выше. В данном синтезе используют газообразный дейтерий (Med-Tech, 98% атом. D), EtOD (Aldrich, 99,5% атом. D), MeOD (Aldrich, 99,5% атом. D), iPrOD (CDN, 99,1% атом. D) и хлорид дейтерия (Aldrich, 99% атом. D). Pd(OH)2 используют вместо Pd/C для превращения XXXII в XXXIII.
1H-ЯМР (300 МГц, CDCl3): δ 0,79 (с, 9H), 2,71 (дд, 2H), 2,93 (д, 2H), 3,52-3,61 (м, 2H), 3,76 (д, 1Н), 3,99-4,05 (м, 1Н), 4,82 (с, 1Н), 5,19-5,21 (м, 2H), 6,40-6,47 (м, 2H), 7,20-7,26 (м, 6H, частично затененный CDCl3), 7,42 (д, 2H), 7,69-7,76 (м, 2H), 7,95 (д, 2H), 8,69 (д, 1Н). МС (M+H+): 722,5.
Пример 13. Синтез (S)-2-(d 3 -метоксикарбониламино)-3,3-d 9 -диметилбутановой кислоты (XVII-d 12 )
Промежуточный продукт XVII- d 12 (R2=R3=C(CD3)3; R1a=R1b=CD3) получают в соответствии со схемой 5, представленной выше. Подробное описание синтеза представлено ниже.
Синтез d 9 -пивалальдегида (XXII, R 2 =R 3 =C(CD 3 ) 3 )
В 4-горлую круглодонную колбу объемом 3 л, снабженную механической мешалкой, обратным холодильником, капельной воронкой и термометром, загружают несколько небольших кристаллов йода и затем магниевую стружку (24,7 г, 1,029 моль). Дно колбы нагревают феном до тех пор, пока йод не начнет испаряться, затем дают возможность раствору охладиться и в капельную воронку загружают раствор трет-бутилхлорида-d 9 (100,0 г, 1,029 моль, Cambridge Isotopes, 99% атом. D) в безводном эфире. Раствор трет-бутилхлорида-d9 в эфире (3-5 мл) добавляют непосредственно на сухой магний. Добавляют безводный эфир (1 л) и несколько небольших кристаллов йода и полученную смесь нагревают в течение 0,5 часа для инициирования реакции. Оставшуюся часть раствора трет-бутилхлорида-d9 в эфире при перемешивании добавляют со скоростью не более чем одна капля в секунду. Смесь кипятят с обратным холодильником во время добавления галогенид-эфирной смеси и не применяют внешнего охлаждения. После этого реакционную смесь кипятят с обратным холодильником в течение нескольких часов до тех пор, пока не исчезнет весь магний. Смесь охлаждают до -20°С и к смеси добавляют раствор безводного ДМФА (73,0 г, 1,0 моль) в эфире (100 мл) в течение 35 минут таким образом, чтобы температура реакции не превышала -15°C. Затем быстро добавляют вторую порцию безводного ДМФА (73,0 г, 1,0 моль) при -8°С. После перемешивания в течение дополнительных 5 минут добавляют гидрохинон (0,5 г), перемешивание прекращают, охлаждающую баню удаляют и смесь оставляют на ночь в атмосфере азота при температуре окружающей среды. Смесь охлаждают до 5°С и в реакционную смесь небольшими порциями добавляют водную 4M HCl (600 мл) для гашения реакции. Смесь разбавляют водой (400 мл) и слои делят. Водный слой экстрагируют эфиром (3×200 мл) и объединенные органические слои сушат и фильтруют. Фильтрат подвергают фракционной перегонке при атмосферном давлении азота для удаления большей части эфира. Остаток переносят в небольшую колбу и продолжают фракционную перегонку для получения целевого соединения XXII (R2=R3=C(CD3)3) (39,5 г, 40% выход) в виде бесцветного масла при 65-75°С. Соединение XXII (R2=R3=C(CD3)3) хранят в атмосфере азота в холодильнике.
Синтез (R)-2-((S)-1-циано-2,2-d 9 -диметилпропиламино)-2-фенилацетамида (XXIIa, R 2 =R 3 =C(CD 3 ) 3 )
К суспензии (R)-фенилглицинамида (60,7 г, 400 ммоль) в воде (400 мл) при перемешивании добавляют соединение XXII (R2=R3=C(CD3)3) (39,5 г, 415 ммоль) при комнатной температуре. Одновременно в течение 30 минут добавляют 30% водный раствор NaCN (68,8 г, 420 ммоль) и ледяную уксусную кислоту (25,4 г, 423 ммоль), в результате чего температура реакционной смеси возрастает до 34°С. Смесь перемешивают в течение 2 часов при 30°C, затем перемешивают при 70°С в течение 20 часов. После охлаждения до 30°С продукт выделяют фильтрацией. Твердый продукт промывают водой (500 мл) и сушат в вакууме при 50°С, получая целевое соединение XXIIa (R2=R3=C(CD3)3) (90,0 г, 88% выход) в виде твердого вещества желтовато-коричневого цвета [α]D=-298° (c=1,0, CHCl3).
Синтез (S)-2-((R)-2-амино-2-оксо-1-фенилэтиламино)-3,3-d 9 -диметилбутанамида (XXIIb, R 2/3 =C(CD 3 ) 3 )
Раствор соединения XXIIa (R2=R3=C(CD3)3) (64,2 г, 252,4 ммоль) в дихлорметане (500 мл) добавляют к концентрированной серной кислоте (96%, 350 мл) при 15-20°С через дополнительную воронку при охлаждении на ледяной бане. Полученную смесь перемешивают при комнатной температуре в течение 1 часа. Смесь выливают в лед и осторожно нейтрализуют раствором NH4OH до pH 9. Смесь экстрагируют дихлорметаном и объединенные органические слои промывают водой, сушат, фильтруют и концентрируют в вакууме, получая целевое соединение XXIIb (R2=R3=C(CD3)3) (55,0 г, 80% выход) в виде пены желтого цвета [α]D=-140° (c=1,0, CHCl3).
Синтез (S)-2-амино-3,3-d 9 -диметилбутанамида (XXIIc, R 2/3 =C(CD 3 ) 3 )
Смесь соединения XXIIb (R2=R3=C(CD3)3) (77,0 г, 282,7 ммоль), 10% Pd/C (~50% воды, 20 г) и уксусной кислоты (50 мл) в этаноле (1,2 л) гидрируют при давлении 30 фунтов на квадратный дюйм (206,843 кПа) при комнатной температуре в течение нескольких дней до тех пор, пока ЖХМС не показывает, что реакция завершена. Смесь фильтруют через целит и промывают EtOAc. Фильтрат концентрируют в вакууме, остаток разбавляют водой (1 л) и подщелачивают 1M раствором NaOH до pH 9. Смесь экстрагируют дихлорметаном и водный слой упаривают в вакууме до половины объема, насыщают твердым NaCl и экстрагируют ТГФ. Объединенные экстракты сушат, фильтруют и концентрируют в вакууме. Остаток обрабатывают толуолом для удаления остатков воды, затем растирают с дихлорметаном, получая целевое соединение XXIIc (R2=R3=C(CD3)3) (38,0 г, 96% выход) в виде твердого белого вещества.
Синтез гидрохлорида (S)-2-амино-3,3-d 9 -диметилбутановой кислоты (XXV, R 2/3 =C(CD 3 ) 3 )
Смесь соединения XXIIc (R2=R3=C(CD3)3) (31,0 г, 222,6 ммоль) в 6M водном растворе HCl (1,5 л) кипятят с обратным холодильником в течение 24 часов. Смесь концентрируют в вакууме, получая сырой продукт. Твердый продукт снова растворяют в воде (500 мл) и промывают EtOAc (2×200 мл) для удаления загрязняющих примесей предыдущих стадий. Водный слой затем концентрируют в вакууме, обрабатывают толуолом и сушат в вакууме при 50°С, получая HCl соль целевого соединения: d 9-гидрохлорид (S)-2-амино-3,3-диметилбутановой кислоты (XXV, R2=R3=C(CD3)3) (33,6 г, 85% выход) в виде твердого белого вещества.
Синтез (S)-2-(d 3 -метоксикарбониламино)-3,3-d 9 -диметилбутановой кислоты (XVII-d 12 )
К раствору соединения XXV (R2=R3=C(CD3)3) (4,42 г, 25,0 ммоль) в смеси диоксана (12,5 мл) и 2M раствора NaOH (60 мл) по каплям добавляют метилхлорформиат-d 3 (5,0 г, 50,0 ммоль, Cambridge Isotopes, 99% атом. D), поддерживая температуру смеси ниже 50°C. Полученную смесь нагревают до 60°С и перемешивают в течение ночи, после чего охлаждают до комнатной температуры. Смесь промывают дихлорметаном и водный слой подкисляют конц. HCl до pH 2 и экстрагируют EtOAc. Объединенные экстракты сушат, фильтруют и концентрируют в вакууме, получая целевое соединение (S)-2-(метоксикарбониламино)-3,3-диметилбутановую кислоту-d12 (XXII-d 12) (3,8 г) в виде масла желтого цвета.
Пример 14 . Синтез (S)-2-(метоксикарбониламино)-3,3-d 9 -диметилбутановой кислоты (XVII-d 9 )
Промежуточный продукт XVII-d 9 (R2=R3=C(CD3)3; R1a=R1b=CH3) получают в соответствии со схемой 5 и способом, описанным для синтеза XVII- d 12, заменяя метилхлорформиат на метилхлорформиат-d3 в конечной стадии.
Пример 15. Синтез (S)-2-(d 3 -метоксикарбониламино)-3,3-диметилбутановой кислоты (XVII-d 3 )
Промежуточный продукт XVII-d 3 (R2=R3=C(CH3)3; R1a=R1b=CD3), описанный в литературе (Zhang, H. et al., J. Label. Comp. Radiopharm. 2005, 48(14): 1041-1047), получают из метилхлорформиата-d3 (Cambridge Isotopes, 99% атом. D).
Пример 16 . Оценка метаболической стабильности
Некоторые исследования метаболизма в печени в условиях in vitro были описаны ранее в следующих публикациях, которые во всей полноте введены в данное описание посредством ссылки: Obach, RS, Drug Metab Disp, 1999, 27:1350; Houston, J.B. et al., Drug Metab. Rev., 1997, 29:891; Houston, J.B., Biochem. Pharmacol., 1994, 47:1469; Iwatsubo, T. et al., Pharmacol. Ther., 1997, 73:147; и Lave, T., et al., Pharm. Res., 1997, 14:152.
Микросомальные биологические испытания. Микросомы печени человека (20 мг/мл, пул от 50 индивидуумов) получают от Xenotech LLC (Lenexa, KS). Инкубационные смеси получают следующим образом. Приготавливают исходные растворы (10 мM), растворы исследуемых соединений 103, 106, 122 и атазанавира в ДМСО. Исходные растворы с концентрацией 10 мМ разбавляют до 1 мМ ацетонитрилом (ACN). Препараты печеночных микросом 20 мг/мл разбавляют до 0,625 мг/мл 0,1 M фосфатно-калиевым буфером (pH 7,4), содержащим 3 мМ MgCl2. 1 мМ испытываемого соединения добавляют к разбавленным микросомам для получения смеси, содержащей 1,25 мкM испытываемого соединения. Смеси «микросома-испытываемое соединение» добавляют в лунки глубиной 2 мл 96-луночного полипропилeнового планшета (опыт проводят в трех экземплярах). Планшет нагревают до 37°С перед инициированием реакций добавлением предварительно нагретого NADPH в 0,1M фосфатно-калиевом буфере (pH 7,4), содержащем 3 мМ MgCl2. Конечная реакционная смесь содержит:
Реакционные смеси выдерживают при 37°С, отбирают аликвоты 50 мкл на 0, 3, 7, 12, 20 и 30 минуте и добавляют в 96-луночный микропланшет с неглубокими лунками, которые содержат 50 мкл охлажденного льдом ACN с внутренним стандартом для остановки реакций. Планшеты хранят при -20°С в течение 30 минут, после чего в лунки планшета добавляют 100 мкл воды перед центрифугированием с получением пеллета осажденных белков. Супернатант переносят в другой 96-луночный микропланшет и анализируют для определения остаточных количеств исходных веществ с помощью ЖХ-МС/МС при использовании масс-спектрометра Applied Biosystems API 4000.
Значения t1/2 для испытываемых соединений in vitro вычисляют из тангенсов угла наклонов графика линейной регрессии остаточного содержания в % исходных веществ (ln) по времени инкубирования с использованием формулы: in vitro t1/2=0,693/k, где k = -[тангенс угла наклона графика зависимости остаточного содержания (%) исходных веществ (ln) от времени инкубирования]. Анализ полученных данных выполняют с использованием Microsoft Excel Software.
Результаты представлены на фигуре 1 и в таблице 2 ниже.
Стабильность испытываемых соединений в микросомах печени человека
В условиях опыта испытываемые соединения 103, 106 и 122 показывают более продолжительный период полураспада по сравнению с атазанавиром. Соединения 106 и 122 показывают наибольшие отличия от атазанавира, показывая приблизительно 40% и 67% повышение периода полураспада соответственно.
Описанный выше опыт повторяют с использованием атазанавира и соединений 103, 104, 106, 111, 114, 120, 121, 122, 123 и 131. Результаты представлены на фигурах 2 и 3 и в таблице 3 ниже:
Стабильность испытываемых соединений в микросомах печени человека
В условиях опыта соединения 104, 106, 111, 114, 120, 122, 123 и 131 демонстрируют увеличение периода полураспада ≥ 24% по сравнению с атазанавиром.
Пример 17 . Фармакокинетические свойства
Фармакокинетические свойства соединений согласно изобретению исследуют на крысах и на шимпанзе, используя пероральное и внутривенное введение.
Фармакокинетика в опыте на крысах. Соединение 122 и атазанавир растворяют в 5% растворе глюкозы с 10% DMI, 15% EtOH и 35% PG соответственно до 2 мг/мл. Затем получают комбинированную дозу смешением обоих растворов в соотношении 1:1 для получения конечных концентраций 1 мг/мл для каждого соединения (рН 5-6) для внутривенного и перорального введения.
В данном исследовании используют самцов крыс Sprague-Dawley (масса тела: от 170 г до 235 г). Крысам вводят либо перорально, либо внутривенно соединение 122 (2 мг/мг) атазанавир (2 мг/мг) либо комбинацию (1:1) соединения 122 и атазанавира (1 мг/кг каждого). Образцы крови (300 мкл) отбирают из ретробульбарной вены перед введением и через 0,083, 0,25, 0,5, 1, 2, 4, 6, 8, 10, 12 и 24 часа после введения. Образцы крови помещают в гепаринизованные пробирки Эппендорфа (с использованием испарения) и затем центрифугируют со скоростью 8000 об/мин в течение 6 минут. 100 мкл аликвоты плазмы переносят в чистые пробирки Эппендорфа и хранят с лекарственным препаратом при -20°С до биологического анализа. Для биологического анализа плазму оттаивают и добавляют к ней 20 мкл метанола и 500 мкл раствора внутреннего стандарта с концентрацией 50 нг/мл (кветиапин в метаноле). Образец подвергают вихревому встряхиванию, центрифугируют со скоростью 15000 об/мин в течение 5 минут и супернатант переносят в стеклянные ампулы автоматического пробоотборника.
Анализы образцов плазмы проводят с использованием метода высокоэффективной жидкостной хроматографии/масс-спектрометрии (ВЭЖХ/МС). ЖХ система включает жидкостной хроматограф Agilent (Agilent Technologies Inc. USA), снабженный изократическим насосом (1100 серий), автоматическим пробоотборником (1100 серий) и дегазатором (1100 серий). Масс-спектрометрический анализ проводят с использованием аппарата API3000 (тройной квадруполь) от AB Inc. (Канада) с ESI интерфейсом. Данные собирают и создают контрольную систему, используя программное обеспечение Analyst 1.4 от ABI Inc. После совместного внутривенного введения соединения 122 и атазанавира атазанавир выводится из крови более быстро. Ускоренное снижение содержания атазанавира по сравнению с соединением 122 начинается в интервале между 1 и 2 часами после внутривенного введения.
Период полувыведения и AUC после внутривенной инъекции представлены в таблице 4 ниже. Соединение 122 показывает 10,7% увеличение периода полувыведения и 6,0% повышение значения AUC после внутривенной инъекции.
Период полувыведения соединения 122 по сравнению с атазанавиром после совместного внутривенного введения крысам
Совместное пероральное введение соединения 122 и атазанавира дает еще более ярко выраженное различие в фармакокинетических свойствах этих двух соединений. Как показано в таблице 5, соединение 122 показывает значительное повышение Сmax по сравнению с атазанавиром после совместного перорального введения. Значения Сmax, периода полувыведения и AUC этих двух соединений после совместного перорального введения представлены в таблице ниже. Соединение 122 показывает 43% повышение периода полувыведения, 67% повышение Сmax и 81% повышение AUC по сравнению с атазанавиром после совместного перорального введения двух соединений крысам.
Период полувыведения, Сmax, Cmin и AUC соединения 122 в сравнении с атазанавиром после совместного перорального введения крысам
Фармакокинетические свойства при испытании на шимпанзе
Серия опытов А: Приготавливают раствор атазанавира с концентрацией 4 мг/мл и каждого из соединений 114, 120 и 122 в 10% DMI (диметилизосорбид), 15% EtOH, 35% PG в D5W. В частности, для приготовления раствора каждого соединения 240 мг соединения растворяют в растворе, включающем 6 мл DMI, 9 мл EtOH и 21 мл PG. Когда соединение полностью растворяется, добавляют 24 мл D5W и раствор тщательно перемешивают. Таким образом получают 60 мл раствора каждого соединения с концентрацией 4 мг/мл.
Затем пятьдесят пять мл раствора каждого лекарственного средства объединяют и смесь фильтруют со стерилизацией с использованием 0,2 мкм фильтра. Таким образом получают 220 мл 1:1:1:1 смеси атазанавир:соединение 114:соединение 120:соединение 122. Конечная концентрация каждого лекарственного средства в растворе равна 1 мг/мг. Каждому животному вводят 50 мл данного средства внутривенным или пероральным способами.
В данном исследовании используют четырех шимпанзе (двух самцов и двух самок) и перед введением раствора соединения их выдерживают в течение ночи без кормления. Перед введением соединения животным вводят седативное средство кетамин и/или телазол. Внутривенное введение осуществляют внутривенным вливанием в течение 30 минут.
Приблизительно 4,5 мл крови отбирают в вакуумированные пробирки с гепарином натрия в качестве антикоагулянта на 0 (перед вливанием), 15 мин, 29,5 мин (сразу после завершения вливания) и затем на 6, 15, 30 и 45 минутах и через 1, 2, 4, 6, 8, 10, 12, 24 часа после завершения вливания. Аналогичную методику используют для забора крови после перорального введения с отбором образцов на 0 (перед введением), 15 и 30 минутах и через 1, 1,5, 2, 4, 6, 8, 10, 12, 24 часа после введения. После отбора образцов вакуумированные пробирки встряхивают вручную несколько раз для обеспечения адекватного смешения. Образцы крови сразу помещают на влажный лед и центрифугируют в пределах 1 часа от времени забора. После центрифугирования полученную плазму хранят замороженной при -70°С до проведения анализа. Результаты представлены на фигурах 4 и 5 и в таблицах 6 и 7.
Увеличение периода полувыведения соединений согласно настоящему изобретению относительно атазанавира, выраженное в процентах, после совместного внутривенного введения представлено в таблице 6 ниже. Соединения 120, 122 и 114 показывают значительно более длительные периоды полувыведения, чем атазанавир, при совместном введении шимпанзе.
Увеличение периода полувыведения относительно атазанавира, выраженное в процентах, после совместного внутривенного введения шимпанзе
Концентрации (нг/мл) соединений 120, 122 и 144, обнаруженных интактными в моче через 24 часа после внутривенного или перорального введения, представлены в таблице 7. В таблице 7 представлено также соотношение содержания каждого испытанного соединения согласно данному изобретению по сравнению с атазанавиром. Более высокие концентрации неметаболизированных исследуемых соединений в моче по сравнению с атазанавиром показывают более медленную скорость метаболизма испытываемых соединений по сравнению с атазанавиром.
Повышение концентраций испытываемых соединений в моче по сравнению с атазанавиром при совместном введении шимпанзе
Серия опытов В : Так же как и в серии опытов А, но с тем отличием, что пероральная доза каждого из атазанавира и соединений 114 и 120 составляет 150 мг, и разбавитель включает 10% этанола, 40% полипропиленгликоля в 2,5% лимонной кислоте. Сmax, Cmin, период полувыведения, AUC и клиренс (CL, мл/мин/кг) соединений после совместного введения представлены в таблице 8, таблице 9 и на фигурах 4 и 5. При совместном введении шимпанзе соединения 114 и 120 показывают в значительной степени более продолжительный период полувыведения, более высокие Сmax, Cmin и AUC и обладают более медленными скоростями клиренса, чем атазанавир.
Серия опытов В: разности t1/2, Сmax, Cmin, AUC и клиренса испытываемых соединений после совместного перорального введения шимпанзе
Концентрации в нг/мл вводимых соединений, обнаруженных интактными в моче через 24 часа после перорального введения, представлены в таблице 9. В моче обнаружены более высокие концентрации неметаболизированных соединений 120 и 114 по сравнению с атазанавиром, включая более медленную скорость метаболизма испытываемых соединений.
Серия опытов В: Повышение концентраций испытываемых соединений по сравнению с атазанавиром при совместном введении шимпанзе
Пример 18. Противовирусная активность в отношении ВИЧ
Противовирусную активность в отношении ВИЧ соединения согласно данному изобретению испытывают на CEM-SS клетках, инфицированных ВИЧ-1. Перед применением в количественном определении противовирусной активности CEM-SS клетки высевают в Т-75 колбы в RPMI 1640 среду, снабженную 10% термоинактивированной фетальной телячьей сывороткой, 2 ммоль/л L-глютамина, 100 Ед./мл пенициллина и 100 мкг/мл стрептомицина. За день до испытания клетки разделяют в соотношении 1:2 для гарантии, что они находятся в фазе экспоненциального роста во время заражения. Подсчет общего количества клеток и жизнеспособных клеток проводят с использованием гемоцитометра и удаления окраски трипанового синего красителя. Жизнеспособность клеток составляет более 95% для клеток, предназначенных для применения в биологическом испытании. Клетки повторно суспендируют с концентрацией 5×104 клеток на мл в среде культуры ткани и добавляют в планшеты микротитратора, содержащие лекарственные средства, в объеме 50 мкл.
Вирус, используемый в биологическом испытании, представляет собой вирус HIV-IRF лимфоцит-тропического вирусного штамма. Вирус получают от NIH AIDS Research and Reference Reagent Program и исходные вирусные пулы получают в CEM-SS клетках. Предварительно титрованную аликвоту вируса удаляют из холодильника (-80°С) и дают возможность медленно оттаять до комнатной температуры в биологически безопасном ящике. Вирус снова суспендируют и разбавляют в среде культуры ткани, так чтобы количество вируса, добавленное в каждую лунку в объеме 50 мкл, представляло собой количество, которое, как определено, приводит к 85-95% поражению клеток на 6 день после инфицирования.
Каждый планшет содержит лунки с контрольными клетками (только клетки), лунки контроля вируса (клетки плюс вирус), лунки токсичности соединения (только клетки плюс соединение), лунки колориметрического контроля соединения (только соединение), а также экспериментальные лунки (соединение плюс клетки плюс вирус). Образцы испытывают в тех повторах с одиннадцатью полулогарифмическими разбавлениями на соединение исходя из 0,1 мкМ соединения. Соединения 104, 120 и 122 испытывают, как атазанавир и AZT. Все соединения также испытывают в присутствии 2 мг/мл гликопротеина α1-кислоты (AAGP), 10 мг/мл альбумина сыворотки человека (HSA) или смеси AAGP плюс HAS.
После инкубирования при 37°С в инкубаторе с 5% СО2 опытные планшеты окрашивают тетразолиевым красителем ХТТ (гидроксид 2,3-бис(2-метокси-4-нитро-5-сульфофенил)-5-[(фениламино)карбонил]-2Н-тетразолия). ХТТ-тетразолий метаболизируется митохондриальными ферментами метаболически активных клеток до растворимого формазан-продукта, что дает возможность провести быстрый количественный анализ ингибирования ВИЧ, индуцированного поражением клеток испытываемыми веществами, обладающими противовирусной активностью в отношении ВИЧ. ХТТ раствор приготавливают ежедневно как исходный раствор с концентрацией 1 мг/мл в RPMI 1640. Приготавливают раствор феназинметосульфата (PMS) с концентрацией 0,15 мг/мл в PBS и хранят в темноте при -20°С. Исходную смесь ХТТ/PMS приготавливают непосредственно перед применением, добавляя 40 мкл PMS на мл раствора ХТТ. В каждую лунку микропланшета добавляют пятьдесят микролитров ХТТ/PMS и микропланшет снова инкубируют в течение 4 часов при 37°С. Микропланшеты герметично закрывают с помощью приспособлений для заклеивания микропланшетов и осторожно встряхивают или переворачивают вверх дном несколько раз для смешивания растворимого формазанового продукта, и планшеты считывают спектрофотометрически при 450/650 нм с помощью планшет-ридера Molecular Devices Vmax.
Необработанные данные получают с помощью программного обеспечения Softmax Pro 4.6 и переносят в крупноформатную таблицу Microsoft Excel 2003 для анализа с помощью соответствующих вычислений линейного графика функции. Результаты испытания представлены в таблице 10 ниже.
Противовирусная активность в отношении ВИЧ в CEM-SS клетках, инфицированных ВИЧ-1
Значения ЕС50 соединений 122 и 120 составляют менее 0,4 и 0,5 нМ соответственно в среде культуры клеток и в 5-6 раз возрастают до 2 и 3 нМ соответственно в присутствии 0,5 мг/мл AAGP. Значение ЕС50 соединения 104 составляет менее 0,3 нМ в среде культуры клеток и более чем в 7 раз возрастает до 2 нМ в присутствии AAGP. Значения ЕС50 соединений 104, 120 и 122 в присутствии 10 мг/мл HSA составляет 0,8, 1 и 0,9 нМ соответственно, то есть являются от двух до более чем в три раза менее сильнодействующими, чем только в среде культуры клеток. Противовирусная активность снижается от 8 до 15 раз для соединения 122 и 120 в присутствии AAGP плюс HSA со значениями EC50, равными 6 и 4 нМ соответственно. Значение ЕС50 соединения 104 равно 4 нМ в присутствии AAGP плюс HSA, что более чем в 13 раз является менее сильнодействующим, чем только в среде культуры клеток. Присутствие AAGP, одного или в сочетании с HSA, приводит к наиболее значительному связыванию белка и потере противовирусной активности соединений 104, 120 и 122. Каждый из этих белков сыворотки подвергается воздействию, аналогичному воздействию на атазанавир. Каждое из соединений согласно данному изобретению, испытанных в данном опыте, обладает эффективностью, по меньшей мере, равной эффективности атазанавира.
Пример 19. Синтез d3-метил-(S)-1-(2-((2S,3S)-3-((S)-2-метоксикарбамоиламино-3,3-диметилбутанамидо)-2-гидрокси-4-фенилбутил)-2-(4-(пиридин-2-ил)бензил)гидразинил)-3,3-диметил-1-оксобутан-2-илкарбамата (соединение 102)
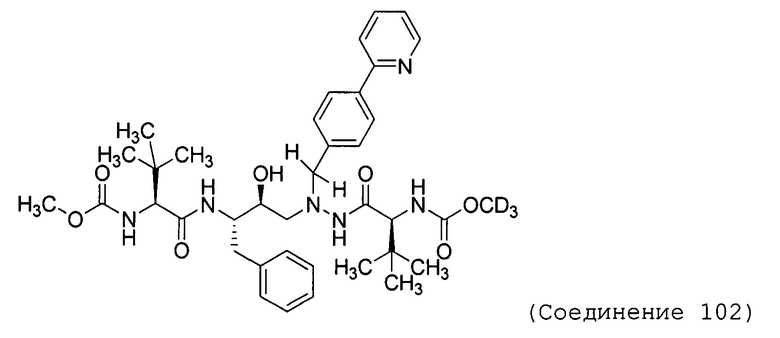
Соединение 102 получают, как изложено ниже.
Синтез бензил 2-((2S,3S)-3-((трет-бутоксикарбонил)амино)-2-гидрокси-4-фенилбутил)-2-(4-(пиридин-2-ил)бензил)гидразин-карбоксилата (12). К смеси коммерчески доступного эпоксида 11 (869 г, 3,3 мол.) и коммерчески доступного гидразида 10а (Y1a=Y1b=H, 1000 г, 3,0 мол.) в изопропаноле (10 л) добавляют уксусную кислоту (198 г, 3,3 мол.). Реакционную смесь нагревают до 55°C и перемешивают в течение 28 часов. Реакционную смесь охлаждают до ~25°C в течение 40 минут и полученную суспензию перемешивают в течение 1 часа. Осадок собирают фильтрованием, промывают изопропанолом и сушат в вакуум-сушильном шкафу (60°C) с получением указанного в заголовке соединения 12 (1,44 кг, 78%).
Синтез трет-бутил ((2S,3S)-3-гидрокси-1-фенил-4-(1-(4-(пиридил-2-ил)бензил)гидразинил)бутан-2-ил)карбамата (13). Смесь соединения 12 (120 г, 201,0 мол.) и 5% Pd/C (17,12 г, 4,02 мол.) в изопропилацетате (1,44 л) в присутствии пиридина (32,4 мл, 402 ммоль) гидрируют при 60 фунтов на кв. дюйм водорода при 25-30°C в течение 3 часов. Реакционную смесь разбавляют метанолом, фильтруют и затем заменяют растворитель на гептан для осаждения требуемого продукта, который затем выделяют фильтрованием. Сушка в вакуум-сушильном шкафу (25-30°C) дает указанное в заголовке соединение 13 (78,5 г, 84%).
Синтез d3-метил ((S)-1-(2-((2S,3S)-3-трет-бутоксикарбониламино-2-гидрокси-4-фенилбутил)-2-(4-(пиридин-2-ил)бензил)гидразинил)-3,3-диметил-1-оксобутан-2-ил)карбамата (15). Смесь соединения 13 (131,38 г, 283,97 ммоль), (S)-2-(d3-метоксикарбониламино)-3,3-диметилбутановой кислоты (XVII-d3; полученной, как описано в Примере 15) и гидрата HOBt (32,62 г, 212,98 ммоль) растворяют в 2-Me-THF (920 мл, не содержащий ингибитора). Добавляют через делительную воронку раствор EDC·HCl (65,33 г, 340,76 ммоль) в воде (920 мл). Двухфазную смесь встряхивают в течение 4 часов. Слои разделяют и органический слой промывают 5% водным раствором NaCl, 5% водным раствором NaHCO3, водой и затем 5% водным раствором NaCl. Затем органический слой концентрируют в вакууме. Неочищенный продукт суспендируют в t-BuOMe (1,0 л) и полученную суспензию встряхивают в течение 15 часов. Суспензию фильтруют и осадок на фильтре промывают t-BuOMe и сушат в вакууме с получением указанного в заголовке соединения 15 в виде белого твердого вещества (161,23 г, 89%).
Синтез бисгидрохлорида d3-метил ((S)-1-(2-((2S,3S)-3-амино-2-гидрокси-4-фенилбутил)-2-(4-(пиридин-2-ил)бензил)гидразинил)-3,3-диметил-1-оксобутан-2-ил)карбамата (16). Смесь соединения 15 (100 г, 157,2 ммоль) в изопропаноле (700 мл) нагревают до 40-45°C. Добавляют раствор 4,95 н. HCl в изопропаноле (Acros, 95,2 мл, 471,7 ммоль) в течение примерно 1 часа. Реакционную смесь нагревают при 48-52°C в течение примерно 22 часов. Затем смесь охлаждают до комнатной температуры и перемешивают в течение 3 часов. Смесь фильтруют, промывают смесью 1:1 н-гептан/изопропанол и осадок на фильтре сушат в вакуум-сушильном шкафу (50°C) с получением соединения 16 в виде белого твердого вещества (95,2 г, 99,4%).
Синтез d3-метил-(S)-1-(2-((2S,3S)-3-((S)-2-метоксикарбамоиламино-3,3-диметилбутанамидо)-2-гидрокси-4-фенилбутил)-2-(4-(пиридин-2-ил)бензил)гидразинил)-3,3-диметил-1-оксобутан-2-илкарбамат (Соединение 102). К смеси соединения 16 (10,01 г, 15,0 ммоль), коммерчески доступного (S)-2-метоксикарбониламино-3,3-диметилбутановой кислоты XVII (Excelsyn, 3,41 г, 18,0 ммоль), гидрата HOBt (1,72 г, 11,25 ммоль) в 2-Me-THF (151 мл) добавляют N-метилморфолин (5,77 мл, 52,5 ммоль) с последующим добавлением раствора гидрохлорида EDC (3,45 г, 18,0 ммоль) в воде (200 мл). Реакционную смесь перемешивают в течение 4,8 часов, после чего слои разделяют и органический слой промывают водой (3х). Органический слой концентрируют в вакууме и полученный маслянистый остаток суспедируют в толуоле (300 мл). Полученную суспензию перемешивают при 50°C в течение 90 минут и затем при комнатной температуре в течение ночи. Затем суспезию фильтруют, осадок на фильтре промывают толуолом и сушат в вакуум-сушильном шкафу (50°C) в течение ночи с получением Соединения 102 в виде белого твердого вещества (11,16 г, 98%). 1H-ЯМР (400 МГц, d6-DMSO): δ 0,62 (с, 9Н), 0,77 (с, 9Н), 2,59-2,69 (м, 1Н), 2,69-2,87 (м, 3Н), 3,53 (с, 3Н), 3,61 (шир. д, J=6,3, 1Н), 3,65 (д, J=9,9, 1Н), 3,85 (д, J=9,1, 1Н), 3,97 (прибл. т, J=14,4, 2H), 4,02-4,11 (м, 1Н), 5,04 (с, 1Н), 6,90 (д, J=9,3, 1Н), 7,01 (д, J=9,1, 1Н), 7,10-7,17 (м, 1Н), 7,20 (шир. с, 4Н), 7,25 (кв, J=7,3, 1Н), 7,34 (прибл. т, J=6,3, 1Н), 7,44 (дд, J=7,1, 2H), 7,56 (д, J=8,6, 1Н), 7,83-7,95 (м, 2Н), 7,99 (д, J=7,8, 2H), 8,66 (д, J=4,0, 1Н), 9,13 (с, 1Н). ВЭЖХ (метод: колонка 150 мм × 4,6 мм C18-RP - метод элюирования с градиентом: 85-30% ACN (+0,1% муравьиная кислота) + 0,025% фосфорная кислота в воде в течение 30,0 минут, затем 35-85% ACN (+0,1% муравьиная кислота) в течение 0,1 мин с 4,9-минутным удерживанием при 85% ACN; длина волны: 220 нм): время удерживания: 18,74 мин. МС (M+H+): 708,376.
Примечание: свободное основание соединения 102 может быть превращено в моносульфатную соль путем растворения в теплой смеси ацетон/NMP), обработки концентрированной серной кислотой, охлаждения и фильтрования.
Пример 20. Испытание дополнительных соединений на противовирусную активность
Соединение 102 испытывают на противовирусную активность в испытании, аналогичном описанному в примере 18, со следующими изменениями. Лимфоцит-трофический вирусный штамм представляет собой HIV-1IIIB. Образцы испытывают в тройном повторе в диапазоне концентраций с 5 полулогарифмическими разбавлениями, изменяющемся от 0,032 нМ до 10 нМ или 0,32 нМ до 100 нМ. В таком же испытании атазанавир испытывают при концентрации в диапазоне между 0,156 нМ и 50 нМ. Образцы испытывают в присутствии изменяющихся количеств сыворотки человека (0, 10, 20 и 40% сыворотки человека) или AAGP (0,5 и 1,0 мг/мл). Результаты представлены в Таблице ниже.
Соединение 102 демонстрирует эффективность, аналогичную эффективности атазанавира. Соединение 102 не проявляло значительного повышения или снижения эффективности по сравнению с атазанавиром в присутствии повышающихся концентраций сыворотки человека. Эффект AAGP является немного более значимым на EC50 соединения 102, чем на EC50 атазанавира. Результаты этого испытания подтверждают, что соединение 102 обладает противовирусной активностью в отношении ВИЧ на уровне, аналогичном активности атазанавира.
Предполагается, что специалист в данной области техники без дополнительного описания может, используя приведенное описание и иллюстративные примеры, синтезировать и применить соединения согласно настоящему изобретению, а также осуществить на практике заявленные способы. Следует представлять, что приведенное выше обсуждение и примеры лишь представляют подробное описание некоторых предпочтительных вариантов осуществления изобретения. Специалисту в данной области техники понятно, что различные модификации и эквиваленты могут быть выполнены без выделения из общего представления и объема настоящего изобретения. Все патенты, статьи из научных журналов и другие документы, обсужденные или цитированные выше, введены в описание посредством ссылки.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ РЕЗИКВИМОДА | 2018 |
|
RU2808163C2 |
| ДВУХЗАМЕЩЕННЫЕ СОЕДИНЕНИЯ ПИРАЗОЛА ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ | 2017 |
|
RU2772212C2 |
| СОЕДИНЕНИЯ ДЛЯ ЛЕЧЕНИЯ PD-L1 ЗАБОЛЕВАНИЙ | 2020 |
|
RU2838028C2 |
| ИНГИБИТОРЫ MMPL3, КОМПОЗИЦИИ НА ИХ ОСНОВЕ И ПУТИ ИХ ПРИМЕНЕНИЯ | 2019 |
|
RU2795229C2 |
| ИНГИБИТОРЫ MEK И ИХ ТЕРАПЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2021 |
|
RU2812929C1 |
| ПРОИЗВОДНОЕ ХИНАЗОЛИНОНА, СПОСОБ ЕГО ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ПРИМЕНЕНИЯ | 2017 |
|
RU2730500C2 |
| ТРИЦИКЛИЧЕСКОЕ ТЕТРАГИДРОИЗОХИНОЛИНОВОЕ ПРОИЗВОДНОЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ В МЕДИЦИНЕ | 2021 |
|
RU2830109C1 |
| Ингибиторы альдостеронсинтазы (CYP11B2) человека | 2022 |
|
RU2811745C1 |
| КЛАСС КОНДЕНСИРОВАННЫХ КОЛЬЦЕВЫХ СОЕДИНЕНИЙ И ИХ ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ | 2021 |
|
RU2831125C1 |
| (АЛЬФА-ЗАМЕЩЕННЫЕ АРАЛКИЛАМИНО- И ГЕТЕРОАРИЛАЛКИЛАМИНО)ПИРИМИДИНИЛ- И 1, 3, 5-ТРИАЗИНИЛБЕНЗИМИДАЗОЛЫ, ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ И ИХ ПРИМЕНЕНИЕ В ЛЕЧЕНИИ ПРОЛИФЕРАТИВНЫХ ЗАБОЛЕВАНИЙ | 2012 |
|
RU2608742C2 |
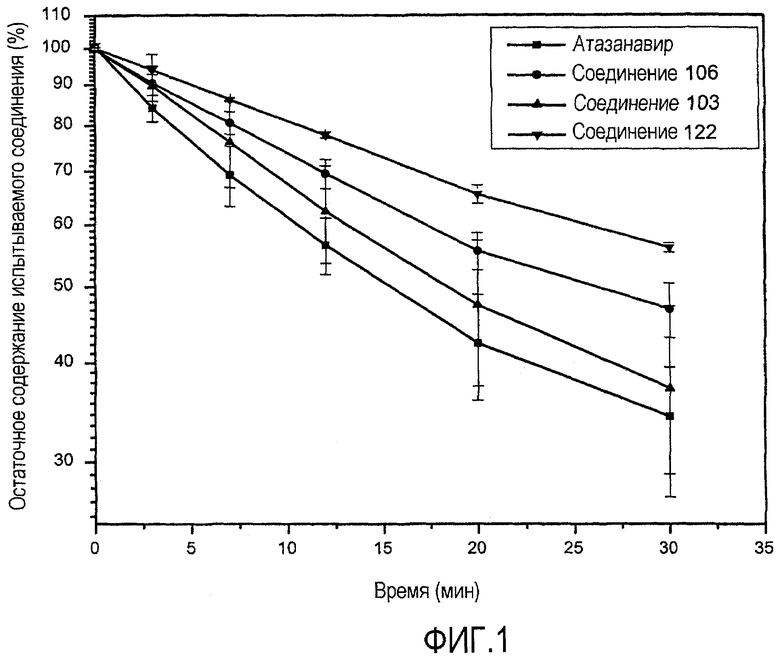
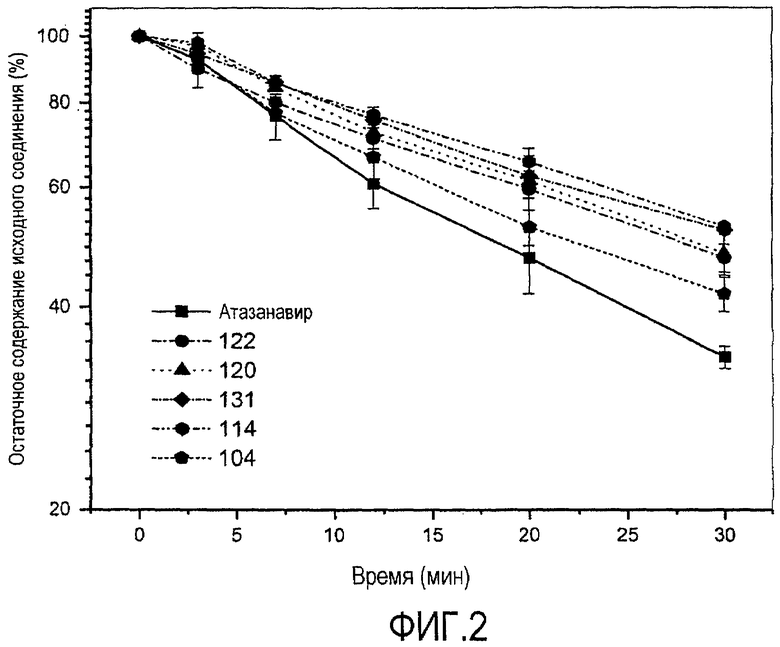
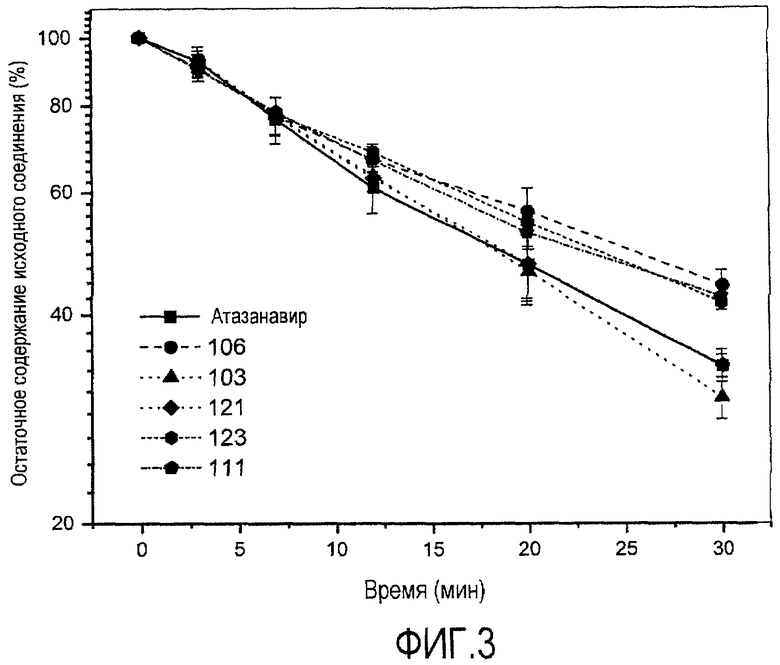
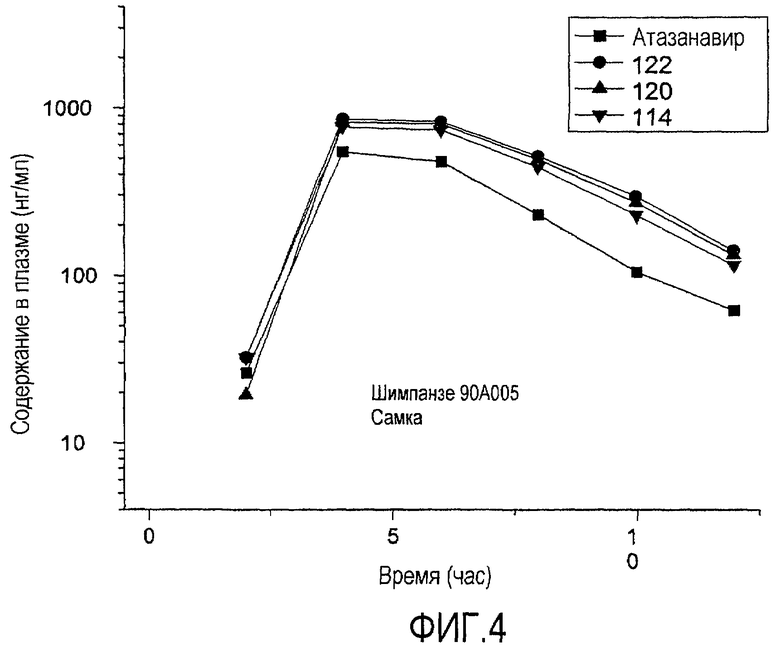
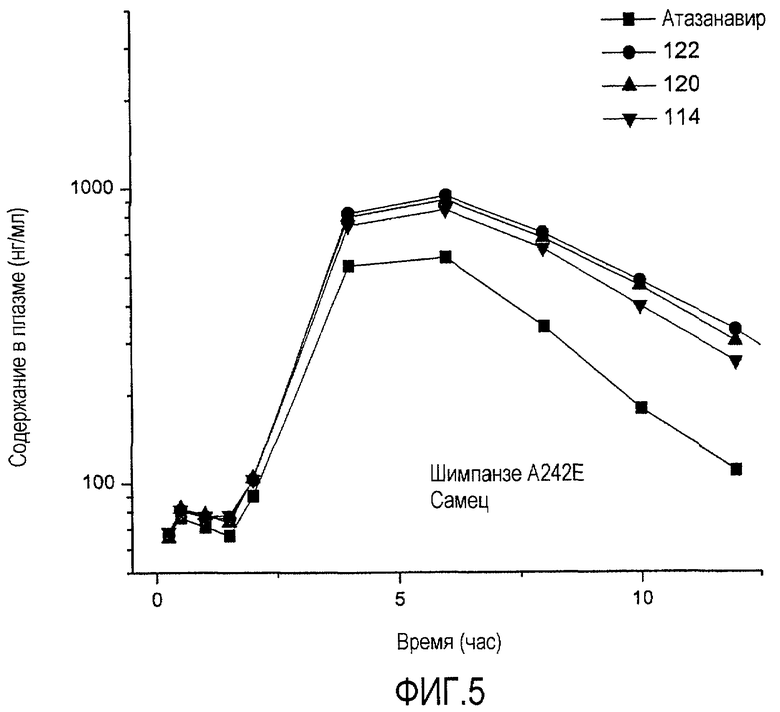
Изобретение относится к новым производным сульфата атазанавира - ингибитора ВИЧ протеазы. Данное изобретение также относится к фармацевтическим композициям, обладающим противовирусной активностью в отношении ВИЧ. 2 н. и 17 з.п. ф-лы, 5 ил., 11 табл., 20 пр.
1. Соединение формулы:
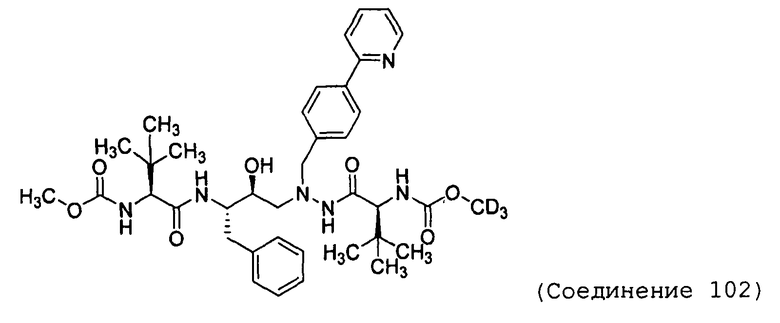
или его фармацевтически приемлемая соль, где любой атом, не обозначенный как дейтерий, представлен в его природной изотопной распространенности (в его природном изотопном содержании).
2. Фармацевтическая композиция, обладающая противовирусной активностью в отношении ВИЧ, содержащая эффективное количество соединения по п.1 и фармацевтически приемлемый носитель.
3. Композиция по п.2, дополнительно содержащая эффективное количество второго терапевтического средства, выбранного из второго ингибитора ВИЧ-протеазы, ненуклеозидного обратного ингибитора транскриптазы, нуклеозидного/нуклеотидного обратного ингибитора транскриптазы, ингибитора входа вируса, ингибитора интегразы, иммунного противоретровирусного средства, ингибитора созревания вируса, клеточного ингибитора или сочетаний двух или большего количества вышеуказанных терапевтических средств.
4. Композиция по п.3, где второе терапевтическое средство выбрано из ритонавира, эфавиренза, диданозина, тенофовирдисопроксилфумарата, нелфинавирмезилата, ампренавира, калия ралтегравира, саквинавира, лопинавира, невирапина, фамотидина, эмтрицитабина, абакавира, ламивудина, зидовудина, маравирока, ставудина, дарунавира, фосампренавира, викривирока, фармацевтически приемлемой соли любого из вышеуказанных терапевтических средств и их сочетаний.
5. Композиция по п.4, где второе терапевтическое средство выбрано из ритонавира, эфавиренза, диданозина, тенофовирдисопроксилфумарата, фармацевтически приемлемой соли любого из вышеуказанных терапевтических средств и их сочетаний.
6. Композиция по п.5, содержащая эффективное количество двух или трех дополнительных вторых терапевтических средств, независимо выбранных из ритонавира, диданозина, ралтегравира, тенофовирдисопроксила, ламивудина, абакавира, зидовудина, эмтрицитабина, эфавиренза и фармацевтически приемлемой соли любого из вышеуказанных терапевтических средств и их сочетаний.
7. Композиция по п.6, содержащая два дополнительных вторых терапевтических средства, независимо выбранных из ритонавира, диданозина, ралтегравира, тенофовирдисопроксила, ламивудина, абакавира, зидовудина, эмтрицитабина, эфавиренза, фармацевтически приемлемой соли любого из вышеуказанных терапевтических средств и их сочетаний.
8. Соединение по п.1 для применения в лечении ВИЧ-инфекции у пациента.
9. Соединение по п.8 для введения с эффективным количеством второго терапевтического средства, выбранного из второго ингибитора ВИЧ-протеазы, ненуклеозидного обратного ингибитора транскриптазы, нуклеозидного/нуклеотидного обратного ингибитора транскриптазы, ингибитора входа вируса, ингибитора интегразы, иммунного противоретровирусного средства, ингибитора созревания вируса, клеточного ингибитора и сочетаний двух или нескольких из вышеуказанных терапевтических средств.
10. Соединение по п.9 для введения с эффективным количеством второго терапевтического средства, выбранного из ритонавира, эфавиренза, диданозина, тенофовирдисопроксилфумарата, нелфинавирмезилата, ампренавира, калия ралтегравира, саквинавира, лопинавира, невирапина, фамотидина, эмтрицитабина, абакавира, ламивудина, зидовудина, маравирока, ставудина, дарунавира, фосампренавира, викривирока, фармацевтически приемлемой соли любого из вышеуказанных терапевтических средств и их сочетаний.
11. Соединение по п.10, где второе терапевтическое средство выбрано из ритонавира, эфавиренза, диданозина, ралтегравира, тенофовирдисопроксилфумарата, ламивудина, абакавира, зидовудина, эмтрицитабина, эфавиренза, фармацевтически приемлемой соли любого из вышеуказанных терапевтических средств и их сочетаний.
12. Соединение по п.11 для введения с эффективным количеством двух-трех дополнительных вторых терапевтических средств, независимо выбранных из ритонавира, эфавиренза, диданозина, ралтегравира, тенофовирдисопроксила, ламивудина, абакавира, зидовудина, эмтрицитабина, эфавиренза, фармацевтически приемлемой соли любого из вышеуказанных терапевтических средств и их сочетаний.
13. Соединение по п.12 для введения с эффективным количеством двух дополнительных вторых терапевтических средств, независимо выбранных из ритонавира, эфавиренза, диданозина, ралтегравира, тенофовирдисопроксила, ламивудина, абакавира, зидовудина, эмтрицитабина, эфавиренза, фармацевтически приемлемой соли любого из вышеуказанных терапевтических средств и их сочетаний.
14. Композиция по п.2 для применения в лечении ВИЧ-инфекции у пациента.
15. Композиция по п.14 для введения с эффективным количеством второго терапевтического средства, выбранного из второго ингибитора ВИЧ-протеазы, ненуклеозидного обратного ингибитора транскриптазы, нуклеозидного/нуклеотидного обратного ингибитора транскриптазы, ингибитора входа вируса, ингибитора интегразы, иммунного противоретровирусного средства, ингибитора созревания вируса, клеточного ингибитора или сочетаний двух или нескольких из вышеуказанных терапевтических средств.
16. Композиция по п.15, где второе терапевтическое средство выбрано из ритонавира, эфавиренза, диданозина, тенофовирдисопроксилфумарата, нелфинавирмезилата, ампренавира, калия ралтегравира, саквинавира, лопинавира, невирапина, фамотидина, эмтрицитабина, абакавира, ламивудина, зидовудина, маравирока, ставудина, дарунавира, фосампренавира, викривирока, фармацевтически приемлемой соли любого из вышеуказанных терапевтических средств и их сочетаний.
17. Композиция по п.16, где второе терапевтическое средство выбрано из ритонавира, эфавиренза, диданозина, ралтегравира, тенофовирдисопроксилфумарата, фармацевтически приемлемой соли любого из вышеуказанных терапевтических средств и их сочетаний.
18. Композиция по п.17 для введения с эффективным количеством двух-трех дополнительных вторых терапевтических средств, независимо выбранных из ритонавира, эфавиренза, диданозина, ралтегравира, тенофовирдисопроксила, ламивудина, абакавира, зидовудина, эмтрицитабина, эфавиренза, фармацевтически приемлемой соли любого из вышеуказанных терапевтических средств и их сочетаний.
19. Композиция по п.18 для введения с эффективным количеством двух дополнительных вторых терапевтических средств, независимо выбранных из ритонавира, эфавиренза, диданозина, ралтегравира, тенофовирдисопроксила, ламивудина, абакавира, зидовудина, эмтрицитабина, эфавиренза, фармацевтически приемлемой соли любого из вышеуказанных терапевтических средств и их сочетаний.
| RU 2004106787 A, 10.12.2004 | |||
| WO 2006014282 A2, 09.02.2006 | |||
| XU ZHONGMIN et al | |||
| "Process Research and Development for an Efficient Synthesis of the HIV Protease Inhibitor BMS-232632" ORGANIC PROCESS RESEARCH AND DEVELOPMENT, 2002, vol.6, no.3, 2002, pages 323-328 | |||
| ZHANG HUIPING et al | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |

