Изобретение относится к области органической химии, а именно к новым индивидуальным биологически активным химическим веществам, конкретно к новому производному 1,3,5-оксадиазина, содержащему в своей молекуле фрагмент гуанидина и фторированные радикалы и может быть использовано в экспериментальной и клинической биохимии, в фармакологии для создания лекарственных средств, в частности, онкологического профиля.
С медико-биологической точки зрения новое соединение содержит две важные функциональные группировки: гуанидиновый фрагмент и фторированные радикалы.
Развитие исследований биологически активных веществ (БАВ), содержащих гуанидиновый фрагмент (HN2)2C=N-, тесно смыкается с успехами химии природных соединений.
Класс биологически активных соединений гуанидинового ряда очень большой. Среди них следует упомянуть ряд известных веществ (препаратов).
1. Вещества, являющиеся конкурентными антагонистами фолиевой кислоты, например бисептол (действующее начало триметоприм), сульфадимезин и бигуанидин, проявляющие антибактериальную активность.
2. Антибиотики стрептомицинового ряда, антибактериальная активность которых основана на другом механизме действия и заключается в подавлении синтеза белка за счет связывания антибиотика с рибосомами и нарушения считывания генетической информации с матричной РНК, что приводит к образованию неполноценных белков.
3. Наконец, природные противоопухолевые антибиотики блеомицин В2 и флеомицин Д1, обладающие способностью вызывать интенсивный распад ДНК (но не РНК) вирусов, бактерий и клеток эукариот. При этом они обладают некоторой специфичностью к ДНК в зависимости от фазы клеточного цикла.
Биомишенью подавляющего числа этих БАВ являются важные биогенные вещества и биополимеры клеток, имеющие высокое сродство к пиримидиновым и пуриновым основаниям, пиридинам, ксантинам. Взаимодействие этих соединений с биомишенями осуществляется либо по механизму подмены метаболита, либо по пути конкурентного антагонизма с нормальными метаболитами. Повышенная гидрофобность БАВ обеспечивает то, что они труднее, чем нормальные метаболиты гидратируются, легче перемещаются в клетке и более сильно взаимодействуют с биомишенью, а повышенная электрофильность способствует их более прочному связыванию валентными связями с активным центром биомишени. Показательным при этом является подобие механизмов биоактивности как циклических, так и ациклических соединений рассматриваемого ряда.
Интерес к фторсодеражащим БАВ обусловлен тем, что атомы фтора и фторсодержащие группировки в молекуле БАВ способны изменять его физико-химические свойства и влиять на характер взаимодействия с различными биологическими мишенями. В ряде случаев такое введение способствует получению более устойчивых к внешним воздействиям соединений (за счет более прочной связи C-F по сравнению с С-Н), что в свою очередь может быть причиной ингибирования ферментов, которые катализируют реакции окисления субстратов с разрывом связи С-Н в интактном субстрате.
С другой стороны, такое введение атомов фтора и фторсодержащих группировок может приводить к увеличению гидрофобности молекул БАВ, что может стать причиной изменения путей и скоростей метаболизма БАВ в организме как на токсикинетической, так и на токсидинамической стадиях действия таких соединений в организме. При этом организм, как правило, не может отличить фторсодержащее соединение от соединений, содержащих С-Н связи (так называемый "эффект маскировки").
Известен фторсодержащий 4Н-1,3,5-оксадиазин формулы (I):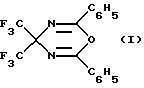
и различные способы его получения:
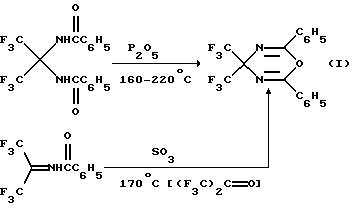
1. Путем дегидратации 3,3-бис(бензоил)-2,2-диаминогексафторпропана под действием пентаоксида фосфора при температуре 160-220oС.
2. Вследствие циклизации N-бензоилимина гексафторацетона (ГФА) при его нагревании до 170oС с триоксидом серы (Гамбарян Н.П., Зейфман Ю.В. Некоторые новые реакции циклоприсоединения к бензоилимину гексафторацетона. Изв. АН СССР, сер.хим., 1969, с.20-59) по схеме 1.
Описаны фторсодержащие 4Н-1,3,5-оксадиазины формулы (II):
и способ их получения, заключающийся в 2,4-циклоприсоединении по Дильсу-Альдеру N-полифторациламинов ГФА и нитрилов карбоновых кислот по схеме 2: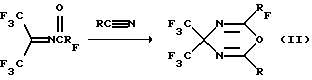
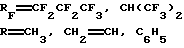
Способ получения отличается мягкими условиями процесса (20-30oС) и количественным выходом продуктов (Крюков Л.Н., Крюкова Л.Ю., Стерлин Р.Н., Кнунянц И. Л. Способ получения фторсодержащих 1,3,5-4Н-оксадиазинов. Авторское свидетельство СССР 743997, С 07 D 234/04, 1978, БИ 24 (1980); Крюков Л. Н., Крюкова Л.Ю., Курыкин М.А., Стерлин Р.Н., Кнунянц И.Л. Взаимодействие N-полифторацилиминов гексафторацетона с нитрилами карбоновых кислот. ЖВХО им. Д.И. Менделеева, 1979. т.24, 4, с.393-395.
Наиболее близким аналогом предлагаемого соединения является фторсодержащий 4Н-1,3,5-оксадиазин, содержащий в цикле гуанидиновый фрагмент, а именно: 2,2,6,6-тетракис(трифторметил)-4-амино-5,6-дигидро-1,3,5-оксадиазин формулы (III):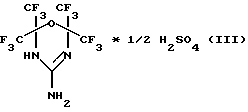
Способ его получения заключается в том, что смесь сульфата гуанидина и гексафторацетона в соотношении 1:4 в N,N-диметилформамиде подвергают циклизации при 70-100oС в течение 5-6 часов, затем ее последовательно обрабатывают водой, концентрированной серной кислотой и еще раз водой до отрицательной реакции на сульфат-анион по схеме 3 (Давыдов А.В., Кнунянц И.Л. Конденсация гуанидина и его солей с гексафторацетоном. ЖВХО им. Д.И. Менделеева. 1977, т.22, 3, с.358-360).
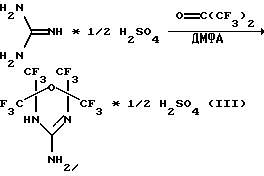
Вещество охарактеризовано физико-химическими константами, однако никаких сведений о биологических свойствах соединения не приводится. Отметим также, что заявителю не известны сведения о гомологах этого соединения.
Изобретательской задачей являлось получение новых перспективных биологически активных соединений, содержащих гуанидиновый фрагмент в молекуле, химическая структура которых (компактность, цикличность, наличие перфторированных заместителей) позволяла бы рассчитывать на выявление у них высокой специфической активности, (прежде всего специфической противоопухолевой активности), требуемой для современных лекарственных средств.
Задача была решена тем, что было получено новое соединение, а именно: 2,2,6,6-тетракис(трифторметил)-4-этиламино-5,6-дигидро-1,3,5-оксадиазин формулы (IV):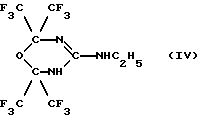
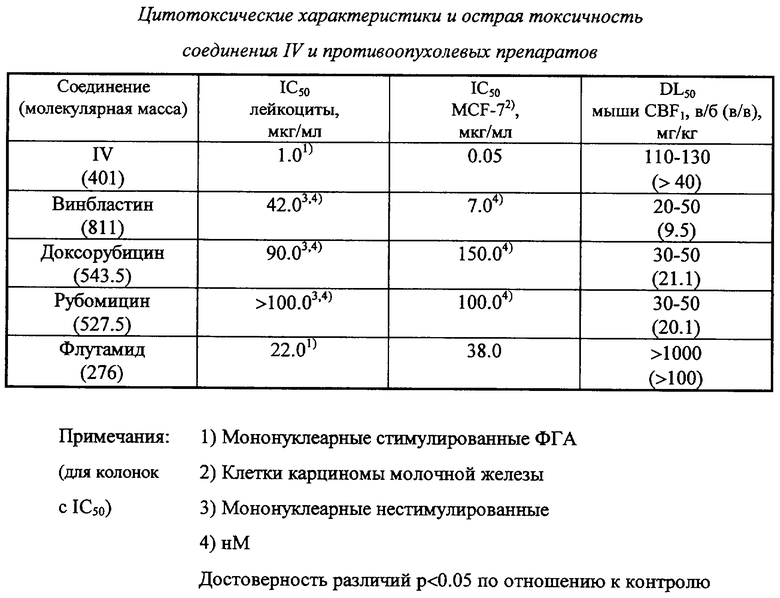
у которого было выявлено требуемое сочетание свойств для современного противоопухолевого средства. Оно в 2 раза эффективнее эталонных противоопухолевых средств (таких как доксорубицин), и при этом оно почти на 2 порядка безопаснее для здоровых клеток, чем для опухолевых.
Заявителем были получены и протестированы более 30 различных веществ, гомологичных указанному (в том числе и вещество по способу прототипа), которые не показали требуемого сочетания свойств.
В дальнейшем планируется тестирование этого соединения на другие виды активности.
Предлагается также способ получения 2,2,6,6-тетракис(трифторметил)-4-этиламино-5,6-дигидро-1,3,5-оксадиазина путем реакции циклизации гексафторацетона с этилгуанидином.
Предлагается также фармацевтическая композиция, обладающая цитотоксическим действием в отношении клеток карциномы молочной железы, содержащая действующее начало и подходящий фармацевтический носитель, отличающаяся тем, что в качестве действующего начала она содержит 2,2,6,6-тетракис(трифторметил)-4-этиламино-5,6-дигидро-1,3,5-оксадиазина в эффективном количестве. Если композиция используется для введения in vivo, эффективное количество составляет 10-70% от общего веса композиции.
Изобретение иллюстрируется следующими примерами.
Пример 1. Получение 2,2,6,6-тетракис(трифторметил)-4-этиламино-5,6-дигидро-1,3,5-оксадиазина ["синтазин"] (IV).
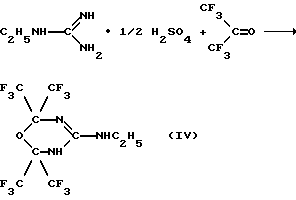
В охлажденный до -78oС автоклав загружают 2.6 г (1.9•10-2 моль) сульфата этилгуанидина в 15 мл N,N-диметилформамида и 14.8 г (8.9•10-2 моль) гексафторацетона. Реакционную смесь выдерживают при 70-100oС в течение 5-6 часов, охлаждают до -78oС и вскрывают автоклав. При отогревании массы до 15-25oС отгоняется непрореагировавший легколетучий гексафторацетон. Остаток промывают водой (25 мл • 4), органический слой отделяют и затем к нему при охлаждении до 0-5oС и перемешивании в течение 30-45 мин добавляют 9.0 г концентрированной серной кислоты. Смесь нагревают до 60-70oС 2-2.5 часа. Образовавшуюся при этом кристаллическую массу промывают теплой (30-40oС) водой до отрицательной реакции фильтрата на сульфат-анион (качественная реакция с раствором хлорида бария) и сушат. После перекристаллизации из бензола получают 6.7 г продукта (IV) с т.пл. 78-79oС. Выход 80.9%.
Найдено, %: С 27.25; Н 1.86; F 56.64; N 10.66. C9H7F12N3O.
Вычислено, %: С 26.94; Н 1.75; F 56.83; N 10.47.
Спектр 1Н-ЯМР: 1.05 т (3Н, СН3); 2.37 дк (2Н, CH2); 5.95 уш.с (1Н, NH); 6.66 уш. с (1Н, NH). Спектр 19F-ЯМР: 2.90 м (3F, CF3); 3.50 м (3F, CF3). ИК-спектр, см-1: 1608 (С=N), 2986 и 2992 (NH). Масс-спектр, м/z: 401 [М]+.
Спектр 1Н-ЯМР получен на приборе "Broker AC-300P" (ФРГ) при комнатной температуре в DMSO-d6. Химические сдвиги (в м.д.) групп протонов измерялись относительно остаточного сигнала растворителя.
Спектр 19F-ЯМР в растворе DMSO-d6 записывали на приборе "Bruker AC-200P" (ФРГ). Химические сдвиги приведены в δ-шкале (в м.д.) относительно трифторуксусной кислоты (внешний эталон).
ИК-спектр в таблетке с Kbr регистрировали на спектрометре "Hitachi 270-30" (Япония) в области 400-3400 см-1. Стандарт - полистирол.
Масс-спектр получали на приборе GC-MS M-80B "Hitachi" (Япония) при энергии ионизирующих электронов 70 эВ, прямом вводе вещества и температуре испарения 250oС.
Количественный элементный анализ соединения осуществляли на CHNX-анализаторе "Carlo Erba 1106" (Италия).
Контроль за протеканием процесса и гомогенностью продукта проводили методом хроматографии в тонком слое на пластинах "Kieselgel 60 F254" (ФРГ).
Пример 2. Композиции 2,2,6,6-тетракис(трифторметил)-4-этиламино-5,6-дигидро-1,3,5-оксадиазина, используемые для биологических испытаний in vitro или in vivo
а) Соединение IV растворяли в ДМСО (по 10 мг/мл), а затем разводили культуральной средой до концентрации 200 мкг/мл. Композицию в последовательных разбавлениях использовали для изучения неспецифической и специфической цитотоксичности, а также острой токсичности.
б) Соединение IV растворяли в 0,1%-ном водном растворе крахмального клейстера в концентрации 10-70 вес.%.
Композиции могут быть использованы для введения препарата по схеме in vivo.
Пример 3. Изучение цитотоксичности и острой токсичности соединения IV в сравнении с известными противоопухолевыми препаратами
Изучение цитотоксичности проводили на лимфоцитах периферической крови человека и клетках аденокарциномы молочной железы человека линии MCF-7.
Лимфоциты периферической крови человека культивировали в пластиковых флаконах фирмы "Costar" (Нидерланды) в среде RPMI 1640, а клетки аденокарциномы молочной железы человека линии MCF-7 - в среде DMEM с добавлением 10% фетальной сыворотки крупного рогатого скота. 100 ЕД/мл пенициллина и 100 мкг/мл стрептомицина при 37oС в СО2-инкубаторе (содержание СО2 5%,. влажность 95%).
Для проведения теста на цитотоксичность клетки в логарифмической фазе роста собирали с помощью раствора Версена, переносили их в культуральную среду, подсчитывали количество в камере Горяева и суспендировали в культуральной среде в концентрации 50 тыс/мл.
Соединение IV растворяли в ДМСО (по 10 мг/мл), а затем его аликвоты разводили культуральной средой до концентрации 200 мкг/мл. В крайний ряд 96-луночных плат вносили по 200 мкл готовых растворов исследуемого соединения, а в остальные лунки - по 100 мкл культуральной среды. Растворы препаратов последовательно титровали, перенося из одного ряда в другой по 100 мкл смеси. Клетки высевали, добавляя в лунки по 100 мкл приготовленной суспензии клеток. Контрольные клетки высевали в лунки без добавления растворов тестируемых препаратов. Продолжительность инкубации в стандартных условиях для куьтивирования клеток составляла 72 ч.
Количественное определение выживших после инкубации клеток проводили, используя МТТ (бромид 3-(4,5-диметилтиазол-2-ил)-2,5-дифенил-тетразолия) по методике Mosmann (Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytoxicity assaya, J.Immunol. Meth. , 1983, v.65, p.55-63). Исходный раствор МТТ готовили, растворяя этот реактив в 0,01 М фосфатно-солевом буфере (натрий-фосфатный буфер, содержащий 0,15 М NaCl, pH 7,4 при 20oС) в концентрации 5 мг/мл с последующим фильтрованием через фильтр с размером пор 0,45 мкм. Хранили при 4oС до 1 мес. Рабочий раствор МТТ получали перед употреблением, смешивая 4 мл культуральной среды и 1 мл исходного раствора МТТ.
После окончания инкубации во все лунки планшеты добавляли по 50 мкл рабочего раствора МТТ и продолжали инкубацию в тех же условиях еще 4 ч. Среду культивирования удаляли с помощью насоса. Добавляли во все лунки по 150 мкл ДМСО (для растворения образовавшихся там кристаллов формазана) и измеряли поглощение растворов в каждой лунке при длине волны 540 нм.
Выживаемость клеток (в процентах) для каждого разведения испытуемого соединения представляли как отношение поглощения полученного раствора к поглощению раствора в контрольном опыте.
На основании полученных данных по концентрационным кривым для каждого изучаемого препарата определяли величину IC50 (концентрацию соединения в cpeде культивирования, вызывающую ингибирование пролиферации клеток на 50% по сравнению с контролем) и оценивали максимальную его концентрацию (минимальное разведение), не влияющую на интенсивность пролиферации клеток. Результаты приведены в таблице.
В качестве объектов для исследования острой токсичности соединения IV использовались здоровые мыши BDF2, самцы массой 18-22 г в возрасте 2-2.5 месяца, на каждую точку по 6 особей. Препараты вводились в растворах ДМСО. Оценка токсичности строго соответствовала общепринятой методике (Принципы и методы оценки токсичности химических веществ. 4.1. Совместное издание программы ООН по окружающей среде и Всемирной организации здравоохранения, ВОЗ, Женева, 1981). Установлено, что среднесмертельная доза (DL50) соединения IV превышает 100 мг/кг, то есть оно является малотоксичным (4 класс токсичности). При этом значимых патологических реакций у подопытных животных не было зафиксировано.
Аналогичные токсикологические испытания были проведены для веществ, используемых для сравнения. Эти результаты также представлены в таблице, из которой следует, что соединение IV обладает выраженной специфической цитотоксической активностью, причем соотношение специфическая/общая токсичность принципиально лучше, чем у известных противоопухолевых препаратов. Кроме того, это соединение обладает относительно низкой острой токсичностью, особенно в сравнении с препаратами, применяемыми для химиотерапии карциомы молочной железы и перечисленными в таблице.
| название | год | авторы | номер документа |
|---|---|---|---|
| 4-НИТРО-3-ТРИФТОРМЕТИЛ-ПЕРФТОРНОНАНОИЛАНИЛИД (ФЛУСТАТ), СПОСОБ ЕГО ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ | 2001 |
|
RU2186057C1 |
| КОНЪЮГАТЫ БИОЛОГИЧЕСКИ АКТИВНЫХ ВЕЩЕСТВ С α -ФЕТОПРОТЕИНОМ, ОБЛАДАЮЩИЕ ИЗБИРАТЕЛЬНЫМ ДЕЙСТВИЕМ ПО ОТНОШЕНИЮ К РАКОВЫМ ОПУХОЛЯМ, СПОСОБ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ) И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1996 |
|
RU2071351C1 |
| ПЕПТИД, ЯВЛЯЮЩИЙСЯ АНАЛОГОМ ФРАГМЕНТА АЛЬФА-ФЕТОПРОТЕИНА, КОНЪЮГАТ ПЕПТИДА С ДОКСОРУБИЦИНОМ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ ДЛЯ ЛЕЧЕНИЯ ОНКОЛОГИЧЕСКИХ ЗАБОЛЕВАНИЙ | 2006 |
|
RU2317102C1 |
| ПОЛИМЕРОСОДЕРЖАЩЕЕ ЛЕКАРСТВЕННОЕ СРЕДСТВО НА ОСНОВЕ ПРОТИВООПУХОЛЕВОГО ПРЕПАРАТА ЭТОПОЗИДА | 2015 |
|
RU2595859C1 |
| МОДУЛЯТОР КЛЕТОЧНОГО ИММУНИТЕТА | 1997 |
|
RU2146528C1 |
| БИЦИКЛИЧЕСКИЕ ГЕТЕРОЦИКЛИЛЬНЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЯ | 2020 |
|
RU2811612C2 |
| ПРОИЗВОДНОЕ АКРИЛАНИЛИДА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЯ В ФАРМАКОЛОГИИ | 2016 |
|
RU2742372C2 |
| ИМИДАЗОПИРИДИНОВЫЕ ИНГИБИТОРЫ IAP | 2007 |
|
RU2466131C2 |
| ПРОИЗВОДНЫЕ АНТРАНИЛОВОЙ КИСЛОТЫ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1997 |
|
RU2195454C2 |
| АНТАГОНИСТЫ ЕР4 | 2016 |
|
RU2761341C2 |
Изобретение относится к органической химии, конкретно к новому фторпроизводному 1,3,5-оксадиазина, обладающему выраженным противоопухолевым действием в отношении клеток аденокарциномы молочной железы. Описывается 2,2,6,6-тетракис(трифторметил)-4-этиламино-5,6-дигидро-1,3,5-оксадиазин формулы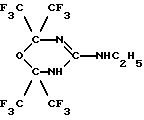
Также предлагается способ его получения и фармацевтическая композиция на его основе. Технический результат - получено новое соединение, обладающее полезными биологическими свойствами. 3 с.п.ф-лы, 1 табл.
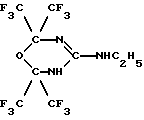
2. Способ получения 2,2,6,6-тетракис(трифторметил)-4-этиламино-5,6-дигидро-1,3,5-оксадиазина, отличающийся тем, что гексафторацетон подвергают реакции циклизации с этилгуанидином.
| Давыдов А.В | |||
| и др | |||
| Конденсация гуанидина и его солей с гексафторацетоном | |||
| ЖВХО им | |||
| Д.И | |||
| Менделеева, 1977, т.22, №3, с.358-360 | |||
| Способ получения фторсодержащих 1,3,5-4н-оксадиазинов | 1978 |
|
SU743997A1 |
| Способ получения 5-арилиденамино -6-арил-2-тригалогенметил-2,3-дегидро1,3,5-оксадиазин-4-онов | 1978 |
|
SU697513A1 |
| АРИЛАЛКИЛ-ДИАЗИНОНЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ, СОДЕРЖАЩИЙ ИХ ФАРМАЦЕВТИЧЕСКИЙ СОСТАВ, СПОСОБ ЕГО ПОЛУЧЕНИЯ | 1996 |
|
RU2167159C2 |

