Данное изобретение относится к молекулам нуклеиновых кислот, включающим индуцируемый теплом промотор, а также к экспрессирующим векторам и клеткам-хозяевам, содержащим, по меньшей мере, одну молекулу нуклеиновой кислоты согласно изобретению. Кроме того, данное изобретение относится к наборам и способам получения одного или большего количества белков, используя молекулы нуклеиновых кислот согласно изобретению, и к различным применениям вышеупомянутого.
Микроорганизмы способны отвечать на ряд стрессовых ситуаций, таких как тепловой или холодовой шок, этанол, ионы тяжелых металлов, кислородная депривация или пищевая депривация, в частности глюкозная депривация. Известно, что дрожжи и другие грибы накапливают трегалозу в течение фаз пониженного роста. Обычно это стадии развития, которые, например, толерантны к водной депривации и теплу, такие как споры, конидии, склероции или клетки в стационарной фазе роста. Также уже известно, что клетки Saccharomyces cerevisiae накапливают трегалозу во время одночасового теплового шока от 27°С до 40°С и что накопление трегалозы коррелирует с повышенной термотолерантностью. Были использованы селективные мутации для того, чтобы показать, что трегалоза действительно является необходимым фактором для индукции термотолерантности.
HSE (элементы теплового шока) и STRE (элементы, отвечающие на стресс) присутствуют в промоторных областях генов, индуцируемых при стрессе, таких как гены S. cerevisiae, ответственные за синтез трегалозы. По-видимому, данные элементы опосредуют активацию стресс-генов при индукции стресса, включая индукцию тепловым шоком. В настоящее время общепризнанным является то, что фосфорилирование Msn2p и Msn4p посредством пути Ras/цАМФ ингибирует факторы транскрипции Msn2p и Msn4p. В отсутствие такого ингибирования (например, в условиях стресса) Msn2p и Msn4p становятся активными. STRE с последовательностью ССССТ приписывают роль в ответе на условия стресса.
Благодаря своей способности осуществлять котрансляционные и посттрансляционные модификации, которые сходны с модификациями у человека, грибы и, в частности, дрожжи являются привлекательными системами для продукции рекомбинантных белков. Для продукции рекомбинантных белков кодирующая последовательность гена, который кодирует интересующий белок, часто экспрессируется под контролем подходящего гетерологичного промотора. Так называемые индуцируемые промоторы, которые можно индуцировать конкретными условиями окружающей среды, оказались особенно выгодными для данной цели. Например, промоторы генов, которые кодируют ключевые ферменты в метаболизме у метилотрофов, такие как промотор МОХ (метанолоксидаза) или FMD (формиатдегидрогеназа), предоставляют возможности широкого применения для экспрессии гетерологичных генов, которая строго регулируется источником углерода.
Для исследования в молекулярной биологии были созданы экспрессирующие векторы, которые включают промотор, индуцируемый теплом, например промотор гена hsp70 Drosophila. Промоторы, используемые в прошлом для индукции теплового шока в клетках грибов и, в частности, у дрожжей, имеют тот недостаток, что они не отвечают избирательно на тепловой шок. Поэтому нельзя достаточно хорошо контролировать механизм их активации и дезактивации, что может, в частности, вызывать проблемы во время продукции белков, которые являются повреждающими по отношению к клеткам. Например, в промоторе TPS1 S. cerevisiae обнаруживаются несколько последовательностей, известных как общие элементы стресса (STRE-элементы), а именно ССССТ и AGGGG, но не более чем одна последовательность, действующая как элемент теплового шока (HSE), а именно GGAACAGAACAATCG. Кроме того, вследствие своего широкого стрессорного ответа известные в настоящее время промоторы активируются фактором стресса в степени, которая не достаточна для многих применений.
Поэтому целью изобретения является предоставление промотора, характеристика индукции теплом которого настолько избирательна, насколько возможно, в частности промотор, который активен в грибах и, в частности, в дрожжах и который пригоден для экспрессии белков при высоких температурах.
Согласно изобретению данная цель достигается посредством молекулы нуклеиновой кислоты, включающей индуцируемый теплом промотор и которая выбрана из следующих нуклеиновых кислот:
(a) нуклеиновая кислота, последовательность которой включает последовательность промотора гена Hansenula polymorpha, кодирующего белок с активностью трегалоза-6-фосфатсинтазы;
(b) нуклеиновая кислота с последовательностью, указанной в SEQ ID NO:1;
(c) нуклеиновая кислота с последовательностью, которая проявляет, по меньшей мере, 40% идентичность на протяжении длины в 300 п.н. с одной из последовательностей, указанных в (а) или (b);
(d) нуклеиновая кислота, которая гибридизуется с комплементарной цепью одной из нуклеиновых кислот, указанных в (а), (b) или (с);
(e) производная одной из нуклеиновых кислот, указанных в (а), (b) или (с), полученная заменой, присоединением и/или делецией одного или большего количества нуклеотидов;
(f) фрагмент одной из нуклеиновых кислот, указанных в (а)-(е), который сохраняет функцию индуцируемого теплом промотора;
(g) комбинация нескольких нуклеиновых кислот, указанных в (а)-(f), где последовательности нуклеиновых кислот могут отличаться или быть одинаковыми;
или посредством молекулы нуклеиновой кислоты, последовательность которой комплементарна последовательности одной из нуклеиновых кислот, указанных в (а)-(g).
Термин “индуцируемый теплом промотор” в используемом в данном контексте виде относится к последовательности нуклеиновой кислоты, которая при повышении температуры в культуральной среде от 25°С до, по меньшей мере, 37°С, предпочтительно до 47°С, приводит к увеличению, по меньшей мере, примерно на 50% в транскрипции (синтезе РНК) гена, находящегося под транскрипционным контролем промотора.
“Активность трегалоза-6-фосфатсинтазы” относится к превращению глюкоза-6-фосфата (Glu6P) и UDP-глюкозы (UDPG) в трегалоза-6-фосфат и UDP, которое катализируется ферментом трегалоза-6-фосфатсинтазой (TPS). Трегалоза-6-фосфатсинтазную активность белка или полипептида можно измерить, например, способом, описанным ниже в “Материалах и способах”.
Признак “последовательность, которая гибридизуется с комплементарной цепью одной из нуклеиновых кислот, указанных в (а), (b) или (с)”, относится к последовательности, которая гибридизуется в жестких условиях с комплементарной цепью нуклеиновой кислоты, имеющей признаки, указанные в (а), (b) или (с). Например, гибридизацию можно проводить при 68°С в 2×SSC или согласно протоколу набора для мечения диоксигенином производства Boehringer (Mannheim). Следующим примером жестких условий гибридизации является инкубация при 65°С в течение ночи в 7% SDS, 1% БСА, 1 мМ ЭДТА, 250 мМ натрий-фосфатном буфере (рН 7,2) с последующей промывкой при 65°С в 2×SSC, 0,1% SDS.
Термин “% идентичности”, как известно в данной области, относится к степени сходства между последовательностями двух или более молекул ДНК или двух или более молекул полипептидов, которая выявляется при сравнении последовательностей. Процент “идентичности” является результатом, получаемым на основании процента идентичных областей в двух или более последовательностях при рассмотрении пробелов или других конкретных особенностей последовательности.
Идентичность соотносимых молекул ДНК или полипептидов можно определить посредством известных процедур. В основном применяют специализированные компьютерные программы, используя алгоритмы, которые вводят поправку на конкретные требования. Предпочтительные способы определения идентичности сначала генерируют наибольшие совпадения между изучаемыми последовательностями. Компьютерные программы для определения идентичности между двумя последовательностями включают, но не ограничены этим, пакет программ GCG, включая GAP (Devereux J., et al., Nucleic Acids Research 12 (12): 387 (1984); Genetics Computer Group University of Wisconsin, Madison, (WI)); BLASTP, BLASTN и FASTA (Altschul S., et al., J. Molec Biol 215: 403/410 (1990)). Программу BLAST X можно приобрести в Национальном центре биотехнологической информации (NCBI) и из других источников (BLAST Manual, Altschul S., et al., NCB NLM NIH Bethesda MD 20894; Altschul S., et al., J. Mol. Biol. 215: 403/410 (1990)). Хорошо известный алгоритм Smith Waterman также можно использовать для определения идентичности.
Предпочтительные параметры сравнения последовательностей включают в себя следующее:
Алгоритм: Needleman and Wunsch, J. Mol. Biol. 48: 443-453 (1970)
Матрикс сравнения: Совпадения = +10
Несоответствия = 0
Штраф пробела: 50
Штраф длины пробела: 3
Программа GAP также пригодна для применения с указанными выше параметрами. Указанные выше параметры представляют собой параметры, устанавливаемые по умолчанию для сравнений последовательностей нуклеиновых кислот.
Могут применяться другие алгоритмы, штрафы открытия пробела, штрафы удлинения пробела, матрицы сравнения, включая те, которые представлены в руководстве по программированию Program Manual, Wisconsin Package, Version 9, September 1997. Сделанные выборы будут зависеть от конкретного сравнения, которое будет проводиться, и, кроме того, от того, проводится ли сравнение между парами последовательностей, и в этом случае предпочтительны GAP или Best Fit, или между одной последовательностью и большой базой данных последовательностей, и в этом случае предпочтительны FASTA или BLAST.
Неожиданно в настоящее время обнаружено, что молекулы нуклеиновых кислот согласно изобретению и, в частности, промотор гена трегалоза-6-фосфатсинтазы (TSP1) Hansenula polymorpha не содержат, по меньшей мере, в первых 300 п.н. выше кодирующей последовательности ни одного STRE-элемента, которые были обнаружены в S. cerevisiae и для которых было сделано предположение, что они являются первично отвечающими элементами для стрессорного ответа, включая индукцию данного гена тепловым шоком. Затем было обнаружено, что данный промотор хорошо и очень избирательно отвечает на тепло.
Молекулы нуклеиновых кислот согласно изобретению могут быть либо получены синтетически стандартными способами, либо выделены из подходящих библиотек ДНК и затем мутированы, как это необходимо. Получение таких библиотек также известно в данной области. Выделение предпочтительно выполняют посредством подготовки зонда длиной, по меньшей мере, 200-400 п.н. из кодирующей последовательности гена TPS1 Н. polymorpha (см. фиг.6), которая используется для скрининга библиотеки ДНК, в частности библиотеки геномной ДНК. Зонд такого вида можно получить посредством ПЦР (полимеразной цепной реакции), используя подходящие праймеры, каждый из которых предпочтительно должен быть длиной, по меньшей мере, 20-21 п.н. и иметь подходящие последовательности согласно фиг.6 (или соответствующие комплементарные последовательности), и геномную ДНК или кДНК Н. polymorpha в качестве “матрицы”.
Таким образом, способность нуклеиновой кислоты согласно изобретению отвечать на тепло приписывается исключительно двум элементам, отвечающим на тепло, которые присутствуют в последовательности промотора, а именно элементу NGAANNNNNNNGAAN (SEQ ID №2), включающему нуклеотиды 627-641 в последовательности промотора (SEQ ID №l), и элементу TGAATATAAAGGAAA (SEQ ID №5), включающему нуклеотиды 698-713 в указанной последовательности промотора. Таким образом, способность отвечать на тепло проявляется двумя элементами, состоящими из 15 п.н., которые разделены 57 п.н. Следовательно, для гетерологической экспрессии гена может быть сконструирован промотор, содержащий один (или гомологичный) элемент или два элемента в любой комбинации, разделенных участком нуклеотидов со схожей последовательностью. Могут быть также задействованы элементы STRE с последовательностями ССССТ и AGGGG, что имеет место в генах теплового шока S.cerevisiae, но которые не являются обязательными. Возможные варианты таких промоторов представлены в примерах.
Зонды могут быть либо синтезированы, либо получены фрагментацией имеющейся в распоряжении ДНК TPS1, где это применимо. Конечно, также можно прямо проводить скрининг посредством зондов, которые соответствуют частям последовательности промотора; однако эта процедура менее предпочтительна из-за в лучшем случае неполной консервативности последовательности в пределах некодирующих частей.
В варианте молекул нуклеиновых кислот согласно изобретению последовательность нуклеиновой кислоты проявляет, по меньшей мере, 60%, предпочтительно, по меньшей мере, 80% идентичности на участке из 300 п.н. с одной из последовательностей, указанных выше в (а) или (b).
Молекулы нуклеиновых кислот, которые включают индуцируемый теплом промотор и которые проявляют, по меньшей мере, 90% идентичности на участке из 300 п.н. с одной из последовательностей, указанных выше в пунктах (а) или (b), особенно предпочтительны. Однако наиболее предпочтительны молекулы нуклеиновых кислот, которые проявляют, по меньшей мере, 95% идентичности на участке из 300 п.н. с одной из последовательностей, указанных выше в пунктах (а) или (b).
Молекулы нуклеиновых кислот, предпочтительные для выполнения изобретения, представляют, по меньшей мере, один элемент теплового шока с последовательностью NGAANNNNNNNGAAN (SEQ ID № 2) или комплементарной ей последовательностью, где нуклеотидами, обозначенными N, независимо друг от друга могут быть А, Т, С или G. Молекулы нуклеиновых кислот согласно изобретению предпочтительно представляют, по меньшей мере, один элемент теплового шока с последовательностью NGAANNBWMNNGAAN (SEQ ID №3) или комплементарной ей последовательностью, где В представляет собой G, С или Т, W означает А или Т, и М означает С или А.
В особо предпочтительном варианте изобретения элемент теплового шока выбран из TGAAGCCTCTTGAAA (SEQ ID №4) и/или TGAATATAAAGGAAA (SEQ ID №5) и/или комплементарных им последовательностей, где два или более элементов теплового шока там, где они присутствуют, могут представлять одну и ту же или различные последовательности. Предпочтительная молекула нуклеиновой кислоты согласно изобретению представляет, по меньшей мере, два различных элемента теплового шока.
В предпочтительном варианте изобретения молекулы нуклеиновых кислот согласно изобретению не содержат STRE-элемента, имеющего последовательность ССССТ или AGGGG.
Изобретение также предоставляет фрагменты молекул нуклеиновых кислот согласно изобретению, как указано выше, которые сохраняют функцию индуцируемого теплом промотора. Фрагмент, включающий последовательность от нуклеотида 228 до нуклеотида 792 в SEQ ID №l, особенно предпочтителен. Следующий предпочтительный фрагмент включает последовательность от нуклеотида 493 до нуклеотида 792 в SEQ ID №l. Также можно использовать фрагмент, содержащий последовательность от нуклеотида 627 до нуклеотида 713 в SEQ ID №l.
Молекулы нуклеиновых кислот согласно изобретению могут, кроме того, содержать, по меньшей мере, одну последовательность нуклеиновой кислоты гетерологичного гена под транскрипционным контролем индуцируемого теплом промотора.
“Гетерологичный ген” следует относить к кодирующей части структурного гена, который либо не экспрессируется под контролем своего собственного (гомологичного) промотора, либо не экспрессируется в организме, из которого ген получен, или не экспрессируется ни под контролем исходного промотора, ни в исходном организме.
В следующем варианте изобретения молекулы нуклеиновых кислот согласно изобретению включают последовательность нуклеиновой кислоты под транскрипционным контролем индуцируемого теплом промотора, которая выбрана из следующих последовательностей:
i) последовательность нуклеиновой кислоты, которая кодирует полипептид с аминокислотной последовательностью трегалоза-6-фосфатсинтазы Hansenula polymorpha;
(ii) последовательность нуклеиновой кислоты, которая указана в SEQ ID №6;
(iii) последовательность нуклеиновой кислоты, которая проявляет, по меньшей мере, 80% идентичность с последовательностью, указанной в SEQ ID №6;
(iv) последовательность нуклеиновой кислоты, которая кодирует полипептид с аминокислотной последовательностью, указанной в SEQ ID №7 или с ее частичной последовательностью, где полипептид проявляет трегалоза-6-фосфатсинтазную активность;
(v) последовательность нуклеиновой кислоты, которая, принимая во внимание вырожденность генетического кода, кодировала бы полипептид с аминокислотной последовательностью, указанной в SEQ ID №7, или с ее частичной последовательностью, где полипептид проявляет трегалоза-6-фосфатсинтазную активность;
(vi) последовательность нуклеиновой кислоты, которая кодирует полипептид, аминокислотная последовательность которого проявляет, по меньшей мере, 80% идентичности с аминокислотной последовательностью, указанной в SEQ ID №7.
Последовательность нуклеиновой кислоты, указанная в (iii), предпочтительно проявляет, по меньшей мере, 90% идентичности с последовательностью, указанной в SEQ ID №6. В альтернативной форме молекул нуклеиновых кислот согласно изобретению последовательность нуклеиновой кислоты, указанная в (vi), кодирует полипептид, аминокислотная последовательность которого проявляет, по меньшей мере, 90% идентичности с аминокислотной последовательностью, указанной в SEQ ID №7.
Молекула нуклеиновой кислоты согласно изобретению может, кроме того, включать последовательность, кодирующую сигнальный пептид, который обеспечивает перенос экспрессированного белка, при этом последовательность нуклеиновой кислоты, кодирующая сигнальный пептид, предпочтительно непосредственно связана с экспрессируемым гетерологичным геном. Секреция и модификация многих эукариотических белков требует, чтобы N-конец последовательности белка был слит с сигнальной последовательностью, для того чтобы направлять полипептиды в секреторный аппарат. Компоненты гена GAM1 S. occidentalis и гормонального гена краба Carcinus maenas, которые были успешно использованы для секреции гирудина (Weydemann et al., 1995), могут быть здесь рассмотрены для примера. Молекула нуклеиновой кислоты согласно изобретению может, кроме того, включать элемент терминатора, содержащий сигнальные структуры для РНК-полимеразы, которые приводят к терминации транскрипции. Примерами терминирующих элементов, которые могут быть использованы, являются терминаторы МОХ или РНО1 Н. polymorpha.
Следующим объектом изобретения является клетка-хозяин, содержащая, по меньшей мере, одну молекулу нуклеиновой кислоты согласно изобретению, при этом клеткой-хозяином является прокариотическая или эукариотическая клетка. Эукариотической клеткой может быть, например, растительная клетка. Эукариотической клеткой предпочтительно является клетка гриба, особенно предпочтительна клетка дрожжей. Особо рассматриваются грибы в качестве клеток-хозяев для выполнения данного изобретения, например гифомицеты, такие как Aspergillus, Neurospora, Mucor, Trichoderma, Acremonium, Sordaria и Penicillium, или дрожжи, такие как Saccharomyces, Hansenula, Pichia, Kluyveromyces, Schwanniomyces, Yarrowia, Arxula, Trichosporon и Candida.
В наиболее предпочтительном варианте изобретения дрожжевая клетка является клеткой факультативных метилотрофных дрожжей Hansenula, предпочтительно Hansenula polymorpha. H. polymorpha является термотолерантной дрожжевой клеткой и относится к небольшой группе так называемых метилотрофных дрожжей, которые способны использовать метанол в качестве источника углерода и энергии. Н. polymorpha выделяли из почвенных образцов при инкубации при 37°С (Levine and Cooney, 1973). Высокая температура, при которой Н. polymorpha продолжает расти и продуцировать белок, дает возможность удалить другие нежелательные организмы. Основанием для этого является то, что было показано, что Н. polymorpha не только имеет очень высокую оптимальную температуру роста, в районе 37°С, но также способна переживать невредимой температуры примерно в 50°С (смотри фиг.1). Жизнеспособность Н. polymorpha после вхождения в стационарную фазу не падает в течение примерно 50 часов даже при 47°С (фиг.2).
Следующим предметом данного изобретения является экспрессирующий вектор, включающий, по меньшей мере, одну молекулу нуклеиновой кислоты согласно изобретению. Такой экспрессирующий вектор также может содержать другие последовательности нуклеиновых кислот в дополнение к индуцируемому теплом промотору, например последовательность, которая кодирует полипептид, ген селектируемого маркера, начало репликации Е. coli и т.д.
Данное изобретение также предоставляет набор, включающий:
(a) экспрессирующий вектор согласно изобретению, который пригоден для включения в него клонированной нуклеиновой кислоты, которая кодирует рекомбинантный белок, и
(b) подходящую клетку-хозяина для индукции индуцируемого теплом промотора и для продукции рекомбинантного белка.
Термин “клонирование” должен включать все способы клонирования, известные в данной области, которые могут применяться для этой цели. Не все эти способы описаны в данной работе в отдельности, будучи известными специалисту в данной области.
Кроме того, изобретение предоставляет набор, включающий:
(a) экспрессирующий вектор и
(b) клетку-хозяина, пригодную для индукции индуцируемого теплом промотора и для продукции белка, кодируемого кодирующей последовательностью под транскрипционным контролем индуцируемого теплом промотора.
Молекулы нуклеиновых кислот, клетки-хозяева, экспрессирующие векторы и наборы согласно изобретению могут быть использованы для рекомбинантной экспрессии гена под контролем индуцируемого теплом промотора или для продукции одного или большего количества белков.
“Рекомбинантную экспрессию в подходящей клетке-хозяине” следует относить ко всем способам экспрессии, известным на данной стадии в этой области в известных системах экспрессии, которые можно использовать для данной цели. Не все эти способы описаны здесь в отдельности, поскольку известны специалисту в данной области.
Следующим предметом изобретения является способ получения одного или большего количества белков, указанный способ включает:
(i) клонирование, по меньшей мере, одной нуклеиновой кислоты, кодирующей рекомбинантный белок, в экспрессирующем векторе согласно изобретению, так чтобы клонированная таким образом нуклеиновая кислота находилась под транскрипционным контролем индуцируемого теплом промотора;
(ii) введение экспрессирующего вектора, полученного на этапе (i) в клетку-хозяина, подходящую для индукции индуцируемого теплом промотора и для продукции рекомбинантного белка;
(iii) культивирование клетки-хозяина, полученной в (ii);
(iv) индукцию индуцируемого теплом промотора способами, по существу известными.
В случае, когда экспрессирующий вектор согласно изобретению содержит последовательность, кодирующую полипептид, и находящуюся под транскрипционным контролем индуцируемого теплом промотора, способ согласно изобретению для получения одного или большего количества белков включает следующие этапы:
(i) введение экспрессирующего вектора в клетку-хозяина, подходящую для индукции индуцируемого теплом промотора и для продукции рекомбинантного белка;
(ii) культивирование клетки-хозяина, полученной в (i);
(iii) индукцию индуцируемого теплом промотора способами, по существу известными.
Далее изобретение описывается с более подробными деталями, со ссылками на фигуры, на которых показано следующее:
Фиг.1 показывает кривые роста Н. polymorpha при 27°С, 37°С и 47°С.
Фиг.2 показывает жизнеспособность после вхождения в стационарную фазу при 27°С, 37°С и 47°С.
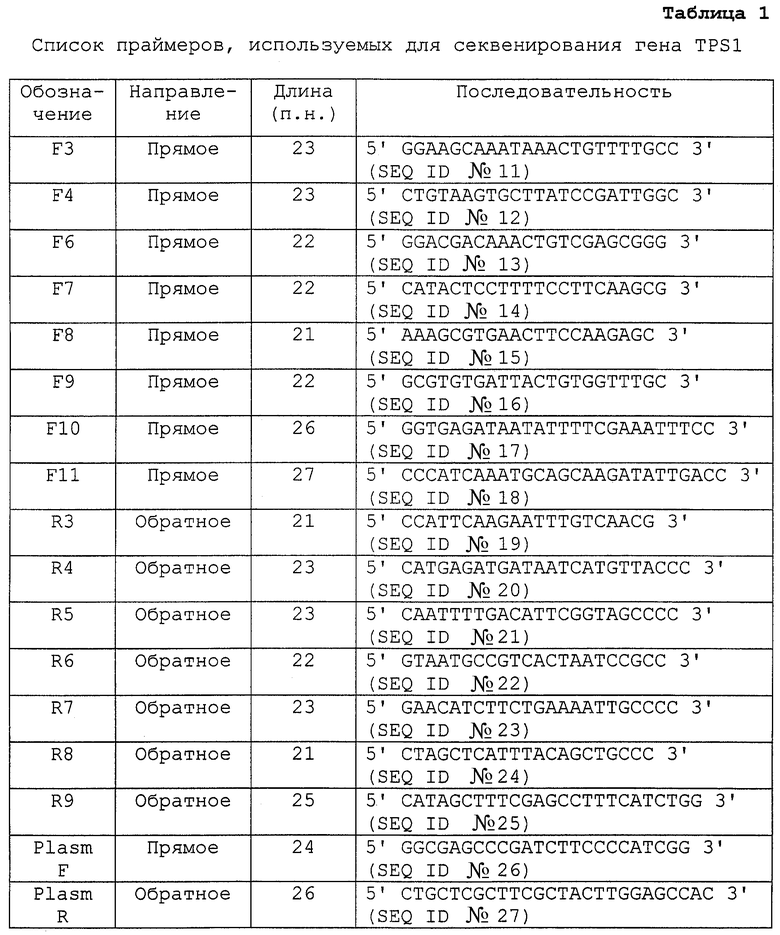
Фиг.3А показывает Нозерн-блот РНК Н. polymorpha дикого типа после теплового шока от 27°С до 47°С и последующего охлаждения до 27°С. Клетки культивировали в среде YDP при 27°С до ранней экспоненциальной фазы роста; затем температуру увеличивали до 47°С (нулевое время) и снова снижали до 27°С через 120 минут.
Фиг.3В показывает Вестерн-блот белка Tps1 (Tps1p) H. polymorpha после теплового шока от 27°С до 47°С и последующего охлаждения до 27°C (см. фиг.3А), из чего можно видеть корреляцию между увеличением TPSl-мРНК и увеличением белка Tps1 (Tps1p).
Фиг.3С показывает внутриклеточную концентрацию трегалозы и трегалоза-6-фосфатсинтазную активность, нанесенные против времени для Н. polymorpha после теплового шока от 27°С до 47°С и последующего охлаждения до 27°С (см. фиг.3А). Не закрашенными кружками изображена внутриклеточная концентрация трегалозы, закрашенными квадратами показана трегалоза-6-фосфатсинтазная активность. Из фигуры очевидна корреляция между увеличением TPSl-мРНК и увеличением трегалоза-6-фосфатсинтазной активности и внутриклеточной концентрацией трегалозы.
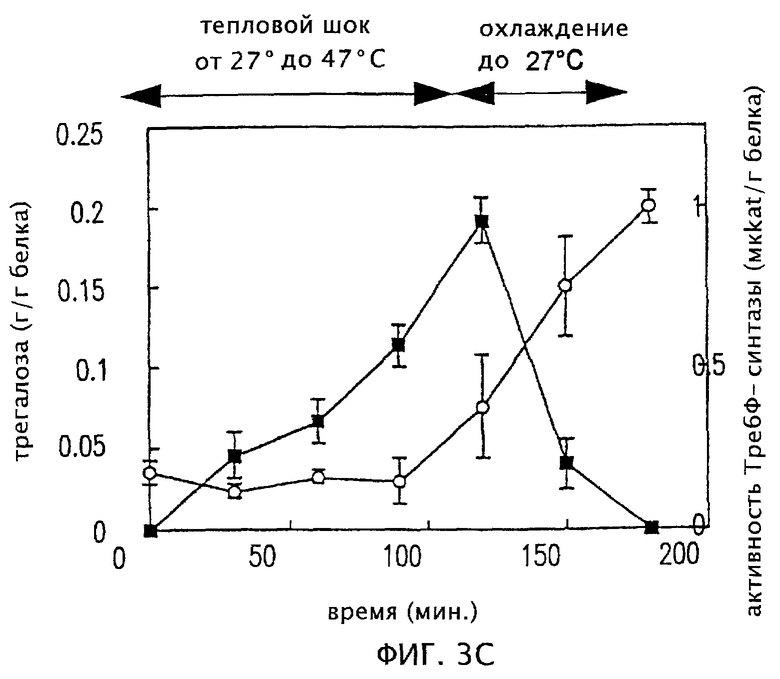
На фиг.4 показаны три гистограммы, изображающие трегалоза-6-фосфатсинтазную активность (белые столбики) и внутриклеточную концентрацию трегалозы (черные столбики) в клетках Hansenula polymorpha, культивируемых при 27°С (А), 37°С (В) и 47°С (С) и при глюкозной депривации спустя 7, 10, 17 и 36 часов. Накопление трегалозы коррелирует с увеличением трегалоза-6-фосфатсинтазной активности (фиг.4А), увеличением TPSl-мРНК (фиг.4 В) и увеличением белка Tps1 (Tps1p) (фиг.4 С).
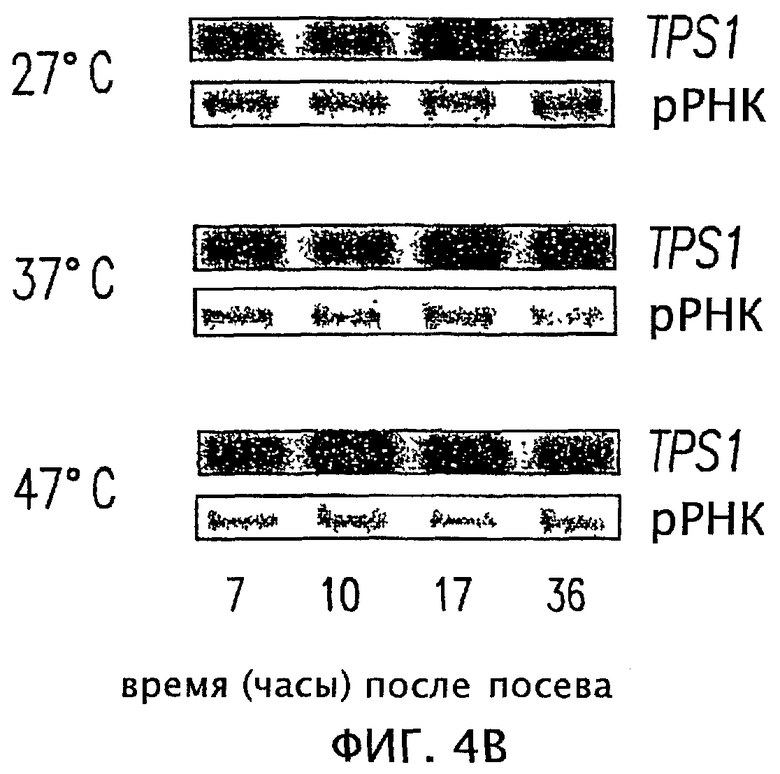
На фиг.5 показана гомология некоторых областей последовательности ДНК трегалоза-6-фосфатсинтазы ряда организмов.
На фиг.6 показана последовательность ДНК гена TPS1 Н. polymorpha (SEQ ID №8) и выведенная аминокислотная последовательность (SEQ ID №6). Элементы теплового шока в последовательности промотора подчеркнуты.
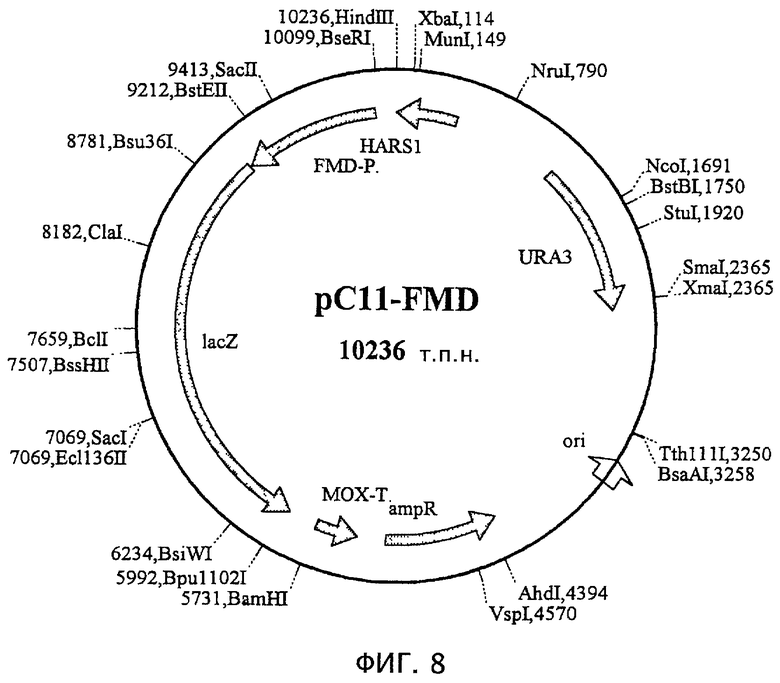
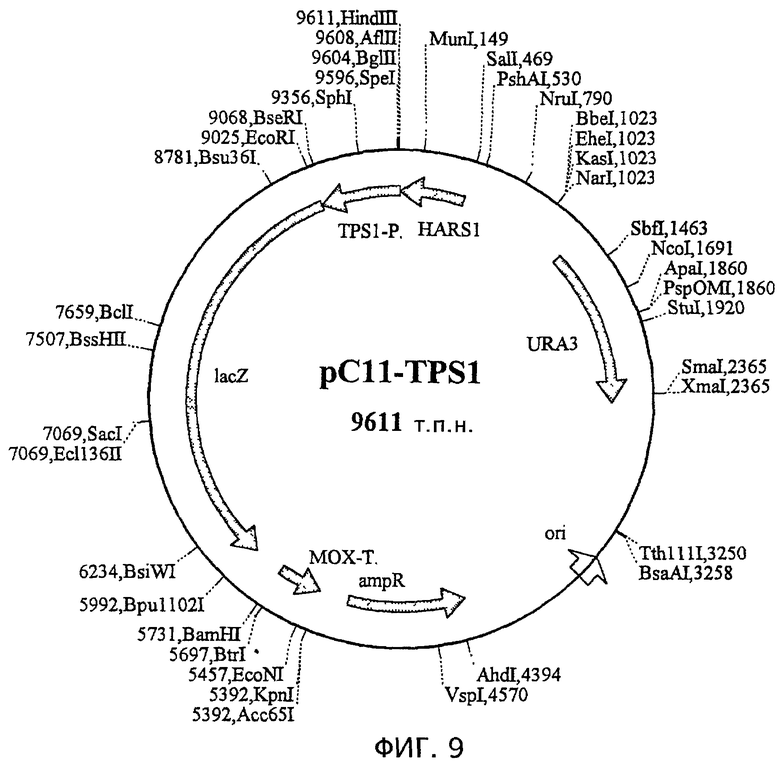
На фиг.7 показана плазмида рС11, производная рМ1 (М. Suckow, персональное сообщение), которая была получена встраиванием гена lacZ в полилинкер рМ1. Плазмида содержит последовательность HARS1 (автономно реплицирующиеся последовательности Н. polymorpha), ori (начало репликации) pBR322, ген устойчивости к ампициллину, ген URA3 для размножения и селекции в Н. polymorpha и Е. coli и терминатор МОХ после гена lacZ для терминации процесса транскрипции.
На фиг.8 показана плазмида pCll-FMD, полученная посредством встраивания промотора FMD перед репортерным геном lacZ плазмиды рС11.
На фиг.9 показана плазмида pCll-TPS1, полученная посредством встраивания промотора TPS1 перед репортерным геном lacZ плазмиды рС11.
На фиг.10 показано сравнение между активностью промоторов FMD (А) и TPS1 (В) при 30, 37 и 44°С в трех различных источниках углерода (2% глюкозы, 2% глицерина или 2% метанола).
На фиг.11 показана плазмида pTPSlConphysMT, используемая в примере 4. МОХ-Т = терминатор МОХ, Conphys = ген Соnрhys3, TPS1 = промотор TPS1 Hansenula polymorpha, HARS = автономно реплицирующиеся последовательности Н. polymorpha, tet = ген устойчивости к тетрациклину, URA3 = URA3 S. cerevisiae, amp = ген устойчивости к ампициллину.
ПРИМЕРЫ
Материалы и способы
Специальные реактивы и материалы
Bio 101, Vista, USA - набор Geneclean II
BioRad Lab., Munich, Germany - анализ белка BioRad (Bredford)
Boehringer, Mannheim, Germany - набор GOD/POD для измерения глюкозы, этанольный набор, таблетки для “полного” коктейля ингибиторов протеаз “COMPLETE”
Fluka Chemie AG, Buchs, Switzerland - циклогексимид (актидион), SDS, D+-тpeгaлoзa, PEP, трицин, НАДФ, фенольный реактив Фолина-Чиокалтеу
ICN Biochemicals, Ohio/USA - "Liquigel" 40% акриламид/N'N'-метиленбисакриламид (37,5:1)
Kodak, New York, USA - регистрирующая пленка для научных целей ВIOМАХ MR
Mediatech, Herndon, USA - сульфат генетицина G418 (антибиотик)
Perkin Elmer Applied Biosystems - набор для секвенирования ДНК Forest City, USA
Pharmacia Biotech, Sweden - колонки Nap-10 (с сефадексом G-25), все используемые рестриктазы, полимераза Taq
Qiagen GmbH, Germany - набор для плазмид Plasmide
Midi (50)
Schleicher + Schuell, Dassel, Germany - Protran BA 83 0,2 мкм/⊘ 82 мм
(круглый фильтр из нитрата целлюлозы), Protran BA 83 0,2 мкм (мембрана для переноса при блоттинге)
Sigma, St. Louis, USA - моноклональные антикроличьи иммуноглобулины козы (конъюгат с щелочной фосфатазой), трегалоза почек свиньи (№ в каталоге Т-8778), UDPG, глюкоза-6-Р, LDH, пируваткиназа
Stratagene, La Jolla, USA - набор Prime-It II (набор для мечения со случайным праймером), колонки NucTrap (колонки для очистки проб, включая защитное устройство от бета-излучения для колонок под давлением)
US Biological, Swampscott, USA - бактериологический агар, бульон YPD, дополненный композицией W/Peptone X, бульон LB Миллера
Используемая аппаратура
Прибор для электропорации - генератор импульсов для Е. coli, BioRad Laboratories, Hercules USA
ВЭЖХ - DIONEX DX-300, DIONEX, Sunnyvale, USA
Охлаждаемые центрифуги - Centricon H-401, Kontron Instr. AG, Zurich, Switzerland IEC Centra GP8R, Brouwer, Lucerne, Switzerland
Biofuge 17RS, Heraeus Sepatech, Germany
Аппаратура для ПЦР - Progene, Techne, Cambridge, United Kingdom
Визуализатор фосфора - GS 250 молекулярный визуализатор (включая связанное оборудование), BioRad Laboratories, Hercules, USA
Фотометр - Anthos 2001 (для планшетов для микротитрования), Anthos Labtec Instruments, Salzburg, Austria
Shimadzu UV-160A, Japan
Секвенатор - ABI PRISM 301 Genetic Analyzer, Perkin Elmer, Applied Biosystems, Foster City, USA
Бактериальный штамм и условия культивирования
Штамм DH5α E. coli (F' endAlhsdR17rkmk+supE44thi-lrecAlgyra relA(lacZYA-argF) U169 (φ80Δ(lacZ)M15)(Gibco BRL, Gaithersburg MD, USA) использовали для клонирования гена TPS1 Н. polymorpha, следуя стандартному протоколу (Sambrook et al., 1989). Среду для Е. coli также получали в соответствии со стандартными процедурами (Sambrook et. al.,1989).
Выделение плазмидной ДНК из Е. coli (STET prep)
Плазмидную ДНК выделяли в соответствии с модифицированным протоколом, согласно Sambrook et. al. (1989). Использовали шпатель для того, чтобы соскабливать клеточный материал с чашки. Затем полученный материал добавляли к 500 мкл STET (8% [мас./об.] сахарозы, 5% [об./об.] тритона Х-100, 50 мМ ЭДТА, 50 мМ трис-HCl, рН 8,0) с 35 мкл лизоцима (10 мг/мл) и перемешивали. Затем образцы кипятили в течение 1 мин 40 с при 100°С и центрифугировали в течение 10 минут при 20000 g и 4°С. Примерно 400 мкл надосадка отбирали пипеткой и осаждали ДНК, используя 400 мкл изопропанола. После центрифугирования в течение 10 минут при 20000 g и 4°С надосадок полностью удаляли и осадок ДНК один раз промывали ледяным 70% [об./об.] этанолом. Наконец ДНК сушили при комнатной температуре и суспендировали в 50-70 мкл ТЕ (10 мМ трис-HCl, рН 8,0, 1 мМ ЭДТА, рН 8,0).
Штамм дрожжей и условия культивирования
Используемым штаммом дрожжей был Hansenula polymorpha дикого типа (имеющийся в наличии благодаря Р. Piper, London (1994)). Исходные культуры выращивали на агаре YPD (2% [мас./об.] глюкозы, 2% [мас./об.] бактопептона, 1% [мас./об.] дрожжевого экстракта, 2% [мас./об.] агара), и исходные культуры обновляли каждые шесть недель. Данные культуры служили в качестве инокулята для жидких культур YPD (состав тот же, что и для агара YPD, но без 2% [мас./об.] агара).
Штамм RB11 Н. polymorpha (штамм Н. polymorpha odc1, без оротидин-5-фосфатдекарбоксилазы (ауксотрофный по урацилу) (Weydemann et al., 1995)) использовали для экспериментов в примерах 3 и 4. Используемая полная среда содержала 2% глюкозы или глицерина, 1% дрожжевого экстракта и 2% бактопептона; селективная среда содержала 0,17% азотистого основания дрожжей, 0,5% сульфата аммония, 2% глюкозы или глицерина, 38,4 мг/л аргинина, 57,6 мг/л изолейцина, 48 мг/л фенилаланина, 57,6 мг/л валина, 6 мг/мл треонина, 50 мг/л инозитола, 40 мг/л триптофана, 15 мг/л тирозина, 60 мг/л лейцина, 4 мг/л гистидина. Урацил не присутствовал в селективной среде.
Для культивирования клеточных культур в автоклавированную жидкую среду инокулировали исходную культуру и инкубировали в течение ночи в инкубаторах с качалками при 27, 37 или 47°С, в зависимости от эксперимента.
Определение оптической плотности культур клеток Н. polymorpha
Чтобы определить оптическую плотность (OD) 200 мкл культуры клеток (соответствующим образом разведенной YPD там, где это применимо) помещали в лунку планшета для микротитрования и измеряли при 620 нм, используя фотоспектрометр Anthos 2001. 200 мкл YPD использовали в качестве пустого контроля.
Эксперименты по росту и тепловому шоку с Н. polymorpha
Ночные культуры использовали для инокуляции в среду YPD в колбах Эрленмейера. Засев данной исходной культуры при той температуре, при которой потом начинали сам эксперимент (27°С для тепловых шоков, 27, 37 или 47°С для ростовых экспериментов) проводили особенно тщательно.
Культуры высевали при начальной плотности OD620 0,2 для каждого ростового эксперимента и непрерывно поддерживали в инкубаторах с качалками (Multitron). Напротив, для экспериментов по тепловому шоку культуры высевали при начальной OD 0,05. Культурам давали возможность расти при 27°С до OD620 0,4 (примерно 1-1,5×108 клеток в мл культуры) перед проведением теплового шока при 47°С в водяной бане с качалкой (Aquatron). Затем в течение следующих двух часов отбирали образцы. Затем культуру клеток охлаждали во второй водяной бане в течение одного часа до 27°С.
Трансформация Н. polymorpha путем электропорации
В 100 мл YPD высевали 5 мл плотно выросшей ночной культуры. Культуру встряхивали при 37°С примерно в течение трех часов до OD600 0,8-1,2. Клетки собирали центрифугированием при 3000 об/мин и снова суспендировали в 20 мл буфера Kpi (50 мМ/рН 7,5). После добавления 0,5 мл DTT и встряхивания в течение 15 мин при 37°С клетки осаждали центрифугированием при 2500 об/мин и дважды промывали буфером STM (270 мМ сахарозы, 10 мМ трисС1, 1 мМ MgCl2, pH 7,5). Затем их суспендировали в 0,25 мл буфера STM, и аликвоты объемом 60 мкл хранили при -70°С. Для трансформации векторами, интегрирующими в рДНК, плазмидную ДНК сначала линеаризовали XhoI или SacI. 0,1-1 мкг линеаризованной плазмидной ДНК смешивали со свежими компетентными клетками, размороженными на льду. Затем эти препараты помещали в 2 мм кюветы. Трансформацию выполняли в генераторе импульсов Gene Pulser (Bio-Rad, Munich) при 2,0 кВ, 25 мкФ и 200 Ом. Затем клетки инкубировали в 1 мл YPD в течение одного часа при 37°С для восстановления перед высеванием на селективную среду. Макроспоровые колонии наблюдали после инкубации от двух до четырех дней при 37°С.
Определение концентрации глюкозы в среде
Концентрацию глюкозы в среде определяли методом GOD (Набор GOD/POD, Bohringer). Образцы разводили 1:200 водой. 190 мкл 1% (мас./об.) раствора фермента GOD (поставляемого в наборе в форме порошка) добавляли к 10 мкл каждого образца, и смесь инкубировали приблизительно 25 минут при 27°С. В данном определении в качестве стандарта также используется 10 мкл раствора глюкозы (0,91 мкг глюкозы), поставляемого в наборе. Поглощение измеряли на спектрофотометре Anthos 2001 при 405 нм.
Экстракция и количественное определение трегалозы
Экстракция трегалозы
1-10 мл культуры клеток фильтровали через фильтр из стекловолокна (Whatman GF/C) и три раза промывали водой. Фильтр помещали в пробирку Эппендорф с 1 мл воды и встряхивали с перемешиванием в течение 30 секунд, перед тем как осторожно отжимали и удаляли. Затем суспензию клеток кипятили в течение 10 минут в водяной бане. Чтобы полностью отделить надосадок от клеточного материала, суспензию центрифугировали три раза при 20000 g.
Определение трегалозы ВЭЖХ
Экстрагированные сахара отделяли с помощью анионообменной колонки (колонка DIONEX CarboPac РА1, 4×250 мм) и определяли амперометрически на золотом электроде (PED = импульсный электрохимический детектор). Состав элюирующего градиента представляет собой следующее:

Следствием данных условий является время удержания трегалозы примерно 3,7 минуты. В каждом случае инъецировали 20 мкл образца. В качестве стандарта использовали раствор трегалозы в концентрации 0,1 мг/мл.
Определение трегалозы ферментативным анализом
В некоторых случаях в качестве альтернативы более дорогому методу ВЭЖХ использовали в равной степени надежный способ ферментативного анализа (Parrou and Francois, 1997, с модификациями): 25 мкл экстракта трегалозы смешивали с 12,5 мкл трегалозы (Sigma) и 37,5 мкл буферного раствора (0,2 М ацетат натрия, 0,03 М CaCl2, pH 5,7) и инкубировали в течение пяти часов при 37°С в водяной бане. Это приводило к полному распаду трегалозы на две единицы глюкозы. После краткого центрифугирования образцы три минуты инкубировали при 95°С и затем снова центрифугировали еще пять минут при 20000 g. Концентрацию трегалозы определяли косвенно посредством определения концентрации глюкозы (набор GOD/POD, смотри выше). Для этой цели использовали 10 мкл полученного надосадка.
Определение белка
Определение белка по Петерсону (незначительно модифицированное) (Peterson(1997))
Чтобы определить концентрацию суммарного белка в культуре клеток 1 мл суспензии клеток преципитировали в 1 мл 10% (мас./об.) ТХУ и центрифугировали в течение 10 минут при 3000 g. Надосадок отсасывали пастеровской пипеткой, соединенной с водоструйным насосом, и осадок промывали 1 мл 1N PCA. Затем осадок суспендировали в 5-12 мл (в зависимости от OD изучаемой клеточной культуры) раствора 0,8 N NaOH:10% (мас./об.) SDS (1:1) и инкубировали, по меньшей мере, в течение одного часа при 60°С. К 200 мкл суспензии добавляли 600 мкл 6-кратного разведения реагента СТС (10% Na2СО3, 0,1% CuSO4·5Н20, 0,2% тартрата KNa). Ровно через 10 минут добавляли 200 мкл 6-кратного разведения реагента Фолина-Чиокалтеу и недолго перемешивали. Образцы оставляли в темноте на 30 минут, после чего измеряли поглощение при 750 нм, в качестве стандарта служил БСА.
Определение белка по Брэдфорду (1976)
Чтобы определить концентрацию белка в бесклеточном экстракте, 100 мкл соответствующим образом разведенного экстракта смешивали с 700 мкл воды. Затем добавляли 200 мкл реактива для анализа белка BioRad (Bradford) и недолго встряхивали (Vortex). Поглощение измеряли при 595 нм, в качестве стандарта служил БСА.
Измерения ферментативной активности
Подготовка проницаемых клеток
Ферментативную активность трегалоза-6-фосфатсинтазы (Тре-6-Ф-синтазы) измеряли в проницаемых клетках (De Virgilio et al., 1991). С этой целью 1-6 мл клеток фильтровали (на фильтрах из стекловолокна GF/C, Whatman), дважды промывали, используя ледяную воду, и снова суспендировали при встряхивании и перемешивании в 1 мл лизирующего буфера (0,2 М трицин, рН 7,0, 0,5% [об./об.] тритон Х-100). Фильтры извлекали и пробирки Эппендорф замораживали в жидком азоте и хранили при -20°С. Перед проведением измерения клетки размораживали на водяной бане в течение трех минут при 30°С. Затем их дважды промывали в 0,2 М трицине (рН 7,0) и центрифугировали в течение 20 секунд при 4°С и 8000 об/мин (Biofuge 17RS) после каждой промывки. Наконец клетки снова суспендировали в 600 мкл 0,2 М трицина (рН 7,0).
Активность трегалоза-6-фосфатсинтазы
Активность Тре-6-Ф-синтазы определяли комбинированным ферментативным анализом согласно Hottiger et al. (1987) при 50°С, при этом всегда использовали 60 мкл проницаемых клеток. Как субстрат (без глюкоза-6-Ф), так и пустые контроли ферментов (без проницаемых клеток) анализировали в качестве контролей.
Вестерн-блот-анализы
Экстракция белка разрушением клеток
5-15 мл культуры клеток центрифугировали в течение 5 минут при 4°С и 3000 об/мин (IEC Centra GP8R) и затем надосадок декантировали. Осадок суспендировали в 1 мл воды и переносили в пробирку Сарстедта (с закручивающейся крышкой). После центрифугирования в течение 10 секунд надосадок отсасывали пипеткой и осадок взвешивали, вес пустой пробирки служил в качестве веса тары. 1 мкл 0,2 М трицин-буфера (рН 7,0; с ингибиторами протеиназ [2 табл./25 мл]) добавляли на мг осадка и осадок ресуспендировали. Добавляли стеклянные шарики непосредственно до нижнего уровня мениска жидкости, после чего пробирки Сарстедта осторожно фиксировали в гомогенизаторе клеток (Fastprep FP120) в холодильной камере. Гомогенизатор клеток запускали дважды по 30 секунд при установке 6,0, получая в результате разрушение более 90% клеток. Далее с этого момента строгое внимание обращали на содержание образцов хорошо охлажденными в течение всего времени. С помощью иглы в пробирке Сарстедта делали небольшое отверстие. Пробирку помещали в стеклянную пробирку и центрифугировали при 4°С и 100 g, отделяя, таким образом, экстракт от стеклянных шариков. Затем один раз добавляли трицин-буфер в количестве, используемом для разрушения клеток, в пробирки Сарстедта, которые снова центрифугировали. Затем мутный экстракт переносили в пробирки Эппендорф и центрифугировали три раза в течение 10 минут при 25000 g и 4°С (Biofuge 17RS), каждый раз потом использовали надосадок, содержащий растворимые белки (включая Тре6Ф-синтазу).
Получение образцов
Концентрацию белка в полученных экстрактах затем определяли методом Бредфорда (смотри выше). В соответствии с полученными значениями их разводили водой до 2,5 мкг белка/мкл, и один объем 5х буфера образца добавляли к четырем объемам данного раствора белка. Затем образцы денатурировали в течение пяти минут при 95°С и либо использовали сразу же для SDS гель-электрофореза, либо замораживали. Для анализа использовали 10 мкл, т.е. 20 мкг белка.
Буфер для образца: 1 мл 0,5 М трис-HCl, рН 6,8; 0,8 мл глицерина; 1,6 мл 10% [мас./об.] SDS; 0,2 мл 0,05% [мас./об.] бромфенолового синего, 4 мл воды. 19 объемов буфера для образца добавляли к одному объему 2-β-меркаптоэтанола непосредственно перед использованием.
Электрофорез в SDS-полиакриламидном геле (SDS-ПААГ)
Применяли систему в соответствии с Laemmli et al. (1970) для разделения белков по их молекулярной массе. Для использования в качестве разделяющего и концентрирующего геля соответственно получали 10% и 4% акриламидный гель (общие размеры 10×10 см) в следующем составе:
Разделяющий гель: 2,5 мл 40% (мас./об.) акриламида/бис-акриламида; 2,5 мл 1,5 М трис-HCl, рН 8,8; 100 мкл 10% (мас./об.) SDS; 4,95 мл воды; 50 мкл 10% (мас./об.) персульфата аммония, 5 мкл TEMED
Концентрирующий гель: 1 мл 40% (мас./об.) акриламида/бисакриламида; 2,5 мл 0,5 М трис-НС1, рН 6,8; 100 мкл 10% (мас./об.) SDS; 6,4 мл воды; 50 мкл 10% (мас./об.) персульфата аммония, 10 мкл TEMED;
5х рабочий буфер: 15 г трис, 72 г глицина, 5 г SDS, Н2О, добавленная до 1 л. Значение рН должно быть примерно 8,3, без последующего доведения.
На каждый гель наносили 20 мкг белка. В качестве стандарта использовали предварительно окрашенный стандарт "Kaleidoscope prestained standard" BioRad следующего состава: миозин (204 кД), β-галактозидаза (121 кД), БСА (78 кД), карбо-ангидраза (39 кД), ингибитор трипсина сои (30 кД). Гель-электофорез проводили примерно один час (но не долее того времени, когда фронт образца достигал нижнего края геля) при постоянном напряжении 200 В. Затем полученные гели красили 0,1% (мас./об.) Кумасси синим R250 в 10% (об./об.) уксусной кислоте/50% (об./об.) этаноле (и отмывали от краски примерно через один час 10% (об./об.) уксусной кислотой, 20% (об./об.) этанолом) или с помощью блоттинга переносили на нитроцеллюлозу (обратитесь к следующему разделу).
Иммуноблоттинг
Затем SDS-ПААГ-гели с помощью блоттинга переносили на нитроцеллюлозу в устройстве для блоттинга (Scieplas) в буфере для переноса (250 мМ трис, 1250 мМ глицин, 15% (об./об.) метанол) в течение 1 часа 15 минут при 40 В и 4°С.
Иммунное окрашивание
Нитроцеллюлозную мембрану сначала выдерживали, по меньшей мере, в течение одного часа в растворе для насыщения, включающем 3% (мас./об.) БСА в TBS (TBS: 20 мМ трис, 500 мМ NaCl, pH доводили до 7,5 с помощью НС1), после чего промывали в течение 5 минут, используя TTBS (TTBS: такой же, как TBS, но с 0,05% твина-20). Затем добавляли поликлональное анти-Tpslp антитело кролика (разведенное 1:50 в 1% [мас./об.] БСА в TTBS) (Euro-gentec, Belgium) в течение ночи при 4°С, с целью связывания с белком Tps1 (Tps1p) H. polymorpha, присутствующим на нитроцеллюлозе.
Затем нитроцеллюлозный блот дважды промывали в течение 5 минут TTBS и инкубировали 1 час 30 минут с моноклональным антикроличьим антителом, связанным с щелочной фосфатазой (разведенным 1:10000 в 1% [мас./об.] БСА в TTBS). После чего дважды промывали по 5 минут TTBS и один раз в течение 5 минут TBS. Для того чтобы проявить окраску полос 1 мл 10х буфера для проявления окраски (100 мМ трис-HCl, pH 9,5, 1 мМ MgCl2) разводили 1:10 водой и добавляли 45 мкл NBT (75 мг/мл 70% [об./об.] DMF) и 35 мкл Х-фосфата (50 мг 5-бром-4-хлор-3-индолилфосфат, соль толуидиния/мл DMF). Мембраны инкубировали в темноте в указанной смеси в течение 20 минут (или до того момента, когда полосы становились четко видимыми) перед промывкой водой, чтобы остановить реакцию.
ПЦР колоний с клетками H. polymorpha
ПЦР колоний выполняли в соответствии с протоколом Huxley et. al. (1990, модифицирован): индивидуальные колонии собирали желтым наконечником пипетки и соскребали в пробирку для ПЦР. Затем пробирки нагревали в течение 2 минут при полной мощности в микроволновой печи. Наконец в каждую пробирку добавляли 25 мкл смеси для ПЦР (0,2 мкл полимеразы Taq, 2,5 мкл 10х буфера для ПЦР, 2,5 мкл 25 мМ MgCl2, 0,5 мкл 10 мМ дНТФ, каждый из праймеров в конечной концентрации 0,5 мкМ и вода, добавленная, чтобы довести объем до 25 мкл), и ресуспендировали клетки. Затем пробирки сразу же помещали в устройство для ПЦР, которое предварительно нагревали до 92°С, и запускали программу.
Нозерн-блот-анализ
РНК экстрагировали из Н. polymorpha согласно протоколу Piper (1994, адаптированный). С этой целью собирали культуру клеток объемом 40 мл в логарифмической и 20 мл в стационарной фазе и (в экспериментах по тепловому шоку) сразу же охлаждали ледяной стерильной водой DEPC. Затем клетки осаждали центрифугированием и снова промывали стерильной водой DEPC. Осадок, полученный после центрифугирования и удаления надосадка, хранили при -20°С. После размораживания к осадку добавляли 1-2 г стеклянных шариков, 2 мл буфера для экстракции РНК (20 мМ трис-HCl, рН 8,5, 10 мМ Na2-ЭДTA, 1% [мас./об.] SDS) и 2 мл фенола. Затем полученную смесь непрерывно встряхивали с перемешиванием в течение 5 минут при комнатной температуре, после чего центрифугировали в течение 5 минут при 3500 об/мин (IEC Centra GP8R). Верхнюю водную фазу переносили в новую пробирку, содержащую равный объем фенола/хлороформа (1:1). Суспензию встряхивали с перемешиванием в течение 1 минуты и центрифугировали 5 минут при 3500 об/мин и надосадок помещали в новую пробирку, содержащую равный объем хлороформа. Повторяли встряхивание с перемешиванием в течение 1 минуты, центрифугировали при 3500 об/мин 2 минуты и надосадок переносили в пробирки Соrех, объемом 15 мл. Добавляли 6М ацетат аммония до конечной концентрации 1М ацетата аммония, затем 2 объема этанола (ледяного) и пробирки выдерживали в морозильном отделении при -20°С, по меньшей мере, 20 минут. Затем РНК осаждали центрифугированием в течение 15 минут при 7500 g и 4°С. Надосадок сливали и пробирки сушили на промокательной бумаге. Затем осадок суспендировали в 1 мл ТЕ и РНК осаждали добавлением 3М ацетата натрия (до конечной концентрации 0,2 М) и 2,5 объемов ледяного этанола. После центрифугирования в течение 15 минут при 7500 g и 4°С осадок промывали ледяным 70% (об./об.) этанолом и сушили при комнатной температуре. Наконец РНК суспендировали в 400 мкл ТЕ.
Получение образца
50 мкг РНК на образец сушили в SpeedVac в течение 10-15 минут для Нозерн-блот-анализа (согласно Sambrook et al., 1989). Затем РНК ресуспендировали в 50 мкл буфера для образца (конечные концентрации: 20 мМ MOPS, pH 7,0, 0,5 мМ ацетат натрия, 1 мМ ЭДТА, pH 8,0, 2,2 М формальдегид, 50% [об./об. л] формамид) и нагревали в течение 10 минут при 55°С. Наконец в каждый образец добавляли 5,5 мкл буфера для загрузки (10х) и 1 мкл раствора бромистого этидия (1 мкл/мл).
Предварительный гель и основной гель
Предварительный гель (1% [мас./об.] агароза и 0,65 М формальдегид в буфере MOPS, содержащем 40 мМ MOPS, pH 7,0, 10 мМ ацетат натрия, 2 мМ ЭДТА, рН 8,0) использовали для проверки целостности экстрагированной РНК и визуальной калибровки нанесенного количества. Основной гель-электрофорез (состав, идентичный составу предварительного геля) проводили в течение 34 часов при 80 В в буфере MOPS, который служил в качестве рабочего буфера.
Блоттинг
Сначала гели дважды промывали в течение 20 минут в 10х SSC (1,5 М NaCl, 170 мМ цитрат натрия). Затем РНК блотировали в течение ночи посредством капиллярного переноса (с использованием в качестве буфера для переноса 20х SSC) на нитроцеллюлозную мембрану (ВА 83). Затем мембрану промывали в 6х SSC, помещали между фильтровальными бумагами 3ММ (Whatman) и помещали в вакуумную печь в течение 2 часов при 80°С, что давало возможность фиксировать РНК на нитроцеллюлозе.
Гибридизация
Нитроцеллюлозную мембрану предгибридизовали в специальной печи (Hybaid) в 10 мл раствора для РНК-гибридизации (0,5 М NaHPO4, рН 7,2, 1 мМ ЭДТА, 1% [мас./об.] БСА, 7% [мас./об.] SDS) в течение 5 часов при 60°С. Для этапа основной гибридизации 150 мкл радиоактивной пробы (в итоге примерно 1×107 имп/мин) добавляли к 10 мл раствора для гибридизации РНК и мембрану инкубировали в течение ночи при 60°С. Наконец избыток радиоактивности отмывали дважды по 15 минут при 60°С 300 мл буфера для промывки (1 мМ ЭДТА, 40 мМ Na2HPO4, рН 7,2, 1% [мас./об.] SDS). Нитроцеллюлозную мембрану экспонировали на пленке BioMax.
Определение фитазы
Клетки Н. polymorpha собирали из 3 мл ночных культур и суспендировали в 200 мкл среды YNB и 1 мл 5% глицерина. После выращивания в течение 1-2 дней сначала определяли OD600. Затем клетки осаждали центрифугированием и затем использовали 25 мкл надосадка. К этой аликвоте добавляли 25 мкл 5 М NaAc и 50 мкл 4-нитрофенилфосфата. Смесь инкубировали в течение 30 минут при 37°С. Ферментативное превращение субстрата прекращали добавлением 100 мкл 15% трихлоруксусной кислоты. После добавления 100 мкл 1 М NaOH образцы надосадка позитивных культур окрашивались в насыщенный желтый цвет. Желтую окраску количественно оценивали измерением OD405 на фотометре.
Анализ при наслаивании Х-gаl - определение β-галаксозидазы
Тестируемые штаммы культивировали в селективной среде в течение 4-6 часов при 37°С. Каплю объемом 4 мкл из каждой культуры помещали на селективную чашку и инкубировали в течение ночи при 37°С. Чашку покрывали свежим верхним слоем агара (0,5% агароза, 0,5 М Na2HPO4/NaH2PO4 (рН 7); 0,2% SDS; 2% DMF (диметилформамид) 2 мг/мл X-gal (о-нитрофенил-β-D-галакто-пиранозид) при 70°С. Через несколько минут клоны с экспрессией lacZ проявляли синюю окраску.
ПРИМЕР 1
Клонирование гена TPS1 Н. polymorpha
Получение радиоактивного TPSl-зонда
На основании сравнения последовательностей известных генов TPS1 S. cerevisiae, S. pombe, К. lactis, Candida albicans и A. niger (см. фиг.6) можно было получить два вырожденных праймера из двух высоко консервативных областей, которые в ходе ПЦР (состоящей из 30 циклов, каждый из которых включал в себя 1 минуту при 92°С, 30 секунд при 52°С, 1 минуту при 72°С) с геномной ДНК Н. polymorpha амплифицировали фрагмент длиной примерно 650 п.н. Последовательности двух праймеров были следующими:
F1 (прямой): 5' TGGCCVYTNTTCCAYTACCATCCYGG 3' (SEQ ID NO:9)
Rl (обратный): 5' GGCRTGBAAYTTYTGHGGHACACC 3' (SEQ ID NO:10)
В=С, G, Т Н=А, С, Т R=А, G
V=А, С, G N=А, С, G, T Y=С, Т
Затем продукт ПЦР наносили на препаративный 1% (мас./об.) агарозный гель и разделяли электрофоретически. Вырезали полосу 650 п.н., экстрагировали, используя набор Geneclean II (Bio 101, Vista, USA) и метили радиоактивным [α-32Р]-dCTP. Для этой цели использовали набор Prime-It II и колонки NucTrap для очистки. Указанный радиоактивный зонд использовали для скрининга TPS1 Н. polymorpha и Нозерн-блот-анализа.
Библиотека геномной ДНК Н. polymorpha
Библиотека геномной ДНК была доступна благодаря R. Hibrands (University of Groningen, Netherlands). Получение библиотеки геномной ДНК не является существенным моментом при условии, что фрагменты имеют длину ≥ 2 т.п.н. Фрагменты геномной ДНК Н. polymorpha длиной 2-5 т.п.н (возможно длина в несколько раз большая) клонировали в сайте рестрикции BamHI плазмиды pHRP2 (7813 п.н.). Данная плазмида (Faber et al., 1992) содержит ori (начало репликации) и ген устойчивости к ампициллину для репликации и селекции в Е. coli. Для трансформации Н. polymorpha присутствует последовательность HARS1 (автономно реплицирующаяся последовательность Н. polymorpha) и ген LEU2 S. cerevisiae, действующий как маркер, который также функционирует в Н. polymorpha. Данная библиотека содержит около 20000 различных клонов.
Трансформация Е. coli
Трансформацию Е. coli с использованием библиотеки геномной ДНК осуществляли путем электропорации (Sambrook et al., 1989) и клетки высевали на чашки 50LB+Amp (75 мг/л) (2000-4000 колоний на чашку). Чашки инкубировали в течение ночи при 37°С.
Скрининг гена TPS1 Н. polymorpha
Чтобы провести анализ ДНК индивидуальных колоний нитроцеллюлозные мембраны осторожно помещали на чашки (согласно Sambrook et al., 1989). Для того чтобы получить четыре асимметрично расположенных отверстия в мембране и геле использовали иглу. Эти отверстия выполняли функцию маркеров, для того чтобы провести ориентацию мембран на чашках, воспроизводимую на более поздних этапах. Когда наносили мембраны, получали реплики колоний, присутствующих на чашке.
Затем раскладывали четыре пластиковые чашки, содержащие промокательную бумагу 3ММ (Whatman), и каждую чашку смачивали одним из четырех различных растворов. Лишнюю жидкость удаляли. Нитроцеллюлозные мембраны сначала помещали (при этом колонии помещали лицевой стороной вверх) на промокательную бумагу, смоченную в 10% (мас./об.) SDS в течение 3 минут. Затем мембраны помещали во вторую чашку, содержащую денатурирующий раствор (0,5 N NaOH, 1,5 M NaCl) и выдерживали в ней в течение 5 минут. Затем их последовательно выдерживали на промокательной бумаге с нейтрализующим раствором (1,5 М NaCl, 0,5 М трис-HCl, рН 7,4) и с 2×SSC (10×SSC 1,5 М NaCl, 170 мМ цитрат натрия) по 5 минут в каждом случае. Для того чтобы фиксировать ДНК на нитроцеллюлозе, каждую мембрану помещали между двумя промокательными бумагами 3ММ и помещали в вакуумную печь при 80°С в течение 2 часов. Затем мембраны смачивали в течение 5 минут в 2×SSC, перед тем как обмакнуть на 30 минут в раствор для предварительной промывки при 50°С (5×SSC, 0,5% [маc./об.] SDS, 1 мМ ЭДТА, рН 8,0). Влажный бумажный платок Kleenex использовали, чтобы вытереть избыточный бактериальный материал перед тем, как мембраны помещали на 2 часа в раствор для предгибридизации (6×SSC, 0,25% [маc./об.] порошкового снятого молока) при 68°С. Для процесса основной гибридизации радиоактивный TPSl-зонд (см. раздел “подготовка радиоактивного TPSl-зонда) с активностью примерно 1×107 имп/мин помещали в 40 мл раствора для предгибридизации и мембраны инкубировали в нем в течение ночи при 68°С. После коротких полосканий три раза в 2×SSC, 0,1% (маc./об.) SDS и промывки в течение 1 часа при 68°С в 1×SSC, 0,1% (маc./об.) SDS мембраны сушили и экспонировали на пленке BioMax. Сигналы на проявленных пленках позволили отобрать 8 позитивных колоний на чашках и создать из них исходные культуры. Из этих колоний экстрагировали плазмиды. Чтобы проверить, присутствует ли в действительности фрагмент длиной 650 п.н., выполняли ПЦР.
ПРИМЕР 2
Секвенирование гена TPS1 Н. polymorpha
Выделение плазмид
Для секвенирования отобрали две колонии, которые при ПЦР с праймерами, направленными от внутренней части фрагмента в 650 п.н. наружу (F4 и R4, см. таблицу 1) и от плазмиды по направлению к вставке (Plasm. F и Plasm. R, см. таблицу 1) давали наибольшие возможные полосы. Чистые экстракты плазмид получали из указанных двух колоний (No. 20.1 и 21.3) с помощью набора Plasmid Midi Kit (Qiagen).
Секвенирование
Последовательности получали посредством циклической программы секвенирования (аппаратура для ПЦР: Progene) и автоматического секвенатора ABI 301 (Perkin Elmer). Для этой цели использовали 0,5 мкл (0,5 мкг) плазмидной ДНК, 1 мкл праймера (конечная концентрация 0,5 мкМ), 4 мкл реакционной смеси (набор для секвенирования ДНК) и 4 мкл воды. Применяемая программа секвенирования состояла из 27 циклов, включающих 30 секунд при 96°С, 15 секунд при 50°С и 4 минуты при 60°С. При завершении программы к реакционной смеси добавляли 10 мкл воды и ДНК осаждали ацетатом натрия и этанолом. Осадок дважды промывали, используя 1 мл ледяного 70% (об./об.) этанола. Затем ДНК недолго сушили и ресуспендировали в 25 мкл TSR (реактив, подавляющий матрицу, набор для секвенирования ДНК). После инкубации в течение двух минут образцы были готовы далее для секвенирования в ABI 301.
Праймеры, используемые для секвенирования плазмиды клона No. 21.3 перечислены в таблице 1. Они были получены при FMI на оборудовании для синтеза нуклеиновых кислот "ExpediteТМ Nucleic Acid Synthesis". Последовательности анализировали с помощью программы GCG (Devereux et al., 1984).
Промотор, выделенный из Н. polymorpha и способ его действия более подробно описаны ниже. Указанный промотор, который контролирует экспрессию TPS1, исследовали, измеряя увеличение TPSl-мРНК при определенных условиях. Было обнаружено, что в то время как при температурах, очень низких для Н. polymorpha, этот промотор экспрессировал небольшие количества TPS1, экспрессия очень сильно возрастала при высоких температурах, а именно намного сильнее, чем в случае с описанными ранее индуцируемыми тепловым шоком промоторами (см. фигуру 3А, Нозерн-блот при тепловом шоке). Индуцируемое теплом увеличение TPS1-мРНК коррелирует с увеличением Tps1 белка (фигура 3В), с увеличением активности трегалоза-6-фосфатсинтазы и с увеличением внутриклеточной концентрации трегалозы (фигура 3С). Для того чтобы оптимизировать влияние температуры, можно, например, избирательно укоротить промотор и соединить с дополнительными участками, содержащими HSE.
Кроме индукции теплом также наблюдали накопление трегалозы, зависимое от глюкозной депривации, предположительно вследствие близкой биологической связи между этими двумя факторами стресса (см. фигуру 4А). Указанное накопление трегалозы коррелирует с увеличением активности трегалоза-6-фосфат-синтазы, увеличением TPSl-мРНК (фигура 4В), и возрастание накопления трегалозы наблюдали вместе с увеличением белка Tps1 во время глюкозной депривации (фигура 4 С).
Чрезвычайно высокое накопление TPSl-мРНК свидетельствует о том, что TPSl-мРНК высоко стабильна, что делает ее (и кДНК на ее основе или на основе полученной от нее информации) не только полезным средством для выделения промотора, но также особенно ценным способом защиты других организмов против ряда стрессовых условий, таких как жара или засуха. ТРS1-ДНК, предоставленную с подходящими промоторами и векторами (например, как описано в WO 93/17093 и WO 96/00789), можно, например, применять для защиты растений от недостатка воды, таким образом давая возможность культивировать их в более теплых районах и районах с более низким количеством осадков. Конечно, для этой цели также можно использовать не только TPSl-ДНК, но также близкую ей ДНК.
ПРИМЕР 3
Сравнительная экспрессия бактериального гена lacZ под контролем промоторов EMD и TPS1
На основе интегрирующего вектора pCll H. polymorpha (фиг.7) сконструировали два производных, которые отличались только соответствующим промотором перед репортерным геном lacZ. В случае pCll-FMD (фиг.8) ген lacZ находится под контролем промотора FMD, который уже хорошо охарактеризован. В случае pC11-TPS1 (фиг.9) указанный ген находится под контролем тестируемого индуцируемого теплом промотора. В целях данного эксперимента в качестве индуцируемого теплом промотора использовали фрагмент между нуклеотидами 228 и 792 последовательности, указанной под номером SEQ ID №l (называемый ниже TPSl-промотором).
H. polymorpha RB11 трансформировали pCll-FMD и pCll-TPSI (см. раздел “Материалы и способы”). Стабильные штаммы, в которых соответствующая плазмида присутствовала в стабильно интегрированном в геном состоянии, получали отдельно примерно из 1000 клонов прототрофных по урацилу клеток для каждой трансформации. В указанном случае процедура представляла собой следующее: после трансформации клетки высевали на чашки, содержащие селективную среду. Через три дня были видны макроскопические дискретные колонии. В обоих случаях 1000 дискретных отдельных колоний переносили в стерильных условиях на новые селективные чашки, которые затем инкубировали в течение двух дней при 37°С. Далее указанную процедуру повторяли два раза (пассирование). Затем колонии клеток переносили на чашки с полной средой и снова инкубировали в течение двух дней при 37°С (стабилизация). Наконец колонии клеток снова переносили на селективные чашки для того, чтобы исключить любые оставшиеся свободные плазмиды. После инкубации указанных чашек в течение двух дней при 37°С получение штаммов было завершено. Точное количество копий и локус интеграции плазмид в индивидуальных штаммах не определяли; однако в этом отношении согласно Gatzke et al. (1995) различные получаемые штаммы, несомненно, должны отличаться друг от друга.
Так как и количество копий, и геномное окружение оказывают большое влияние на степень транскрипции гена, следовало предположить, что индивидуальные клоны клеток будут также значительно отличаться друг от друга в отношении активности β-галактозидазы. Это было подтверждено экспериментально (данные не показаны). Поэтому было невозможно сравнить силу промоторов непосредственно с помощью индивидуальных штаммов. Несмотря на это, чтобы сделать возможным проведение объективных исследований промоторов, объединили 500 индивидуальных штаммов, которые были получены отдельно, имея целью создать репрезентативные смеси штаммов в отношении числа копий и локуса интеграции. Так как используемые для получения штаммов плазмиды pCll-FMD и pCll-TPS1 идентичны, за исключением соответствующего промотора, локализованного перед геном lacZ, можно предположить, что они интегрируются в геном хозяина гомологичным образом. Это предположение было подтверждено наблюдением того, что различные смеси штаммов при идентичной трансформации только незначительно отличаются друг от друга по своей β-галактозидазной активности (данные не показаны). Поэтому определение β-галактозидазной активности смесей штаммов, полученных при трансформации плазмидами, которые в значительной степени идентичны, должно позволить объективные сравнения промоторов в Н. polymorpha.
Функционирование lacZ под контролем промоторов FMD или TPS1 осуществляли при трех различных температурах в трех различных источниках углерода (см. фигуру 10). С этой целью смеси штаммов, описанные выше, культивировали до OD600, равным 5 в 10 мл селективной среды при указанных температурах и источниках углерода, после чего готовили экстракты клеток, активности β-галактозидазы в которых определяли посредством измерений ONPG в жидкой среде. Процедура представляла собой следующее: при достижении желаемой плотности культуры центрифугировали в течение 10 минут при 4°С, осадки клеток промывали в 10 мл lacZ-буфера (50 мМ натрий-фосфатный буфер, рН 7; 10 мМ КС1; 1 мМ MgSO4), ресуспендировали в 500 мкл lacZ-буфера и переносили в пробирки Эппендорф объемом 1,5 мл. Стеклянные шарики диаметром 0,45 мм добавляли к суспензиям (до мениска жидкости), после чего клетки разрушали в Vibrax (Janke & Kunkel; 6 минут; 4°С; 2200 об/мин). Лизаты клеток отбирали и центрифугировали (настольная центрифуга; 4°С; 10 минут). Растворимые фракции использовали как для определения β-галактозидазных активностей, так и для измерения суммарного содержания белка. Для измерений β-галактозидазной активности 1 мл раствора ONPG (4 мг ONPG/мл lacZ-буфера) добавляли к различным разведениям растворимых фракций и затем каждую смесь переносили в пластиковые 1 см-кюветы. Затем измеряли OD420 с 30-секундными интервалами в течение 3-минутного периода, чтобы позволить провести измерение ΔЕ.
Чтобы определить суммарное содержание белка в клеточных экстрактах 790 мкл Н2О смешивали с 10 мкл соответствующей растворимой фракции (разведенной 1:10, 1:5, 1:2 или не разведенной, в соответствии с количеством белка) и добавляли 200 мкл реактива Брэдфорда (Biorad). После инкубации в течение 10 минут при комнатной температуре фотометрически определяли OD490 и корректировали по контрольному образцу, содержащему 1асZ-буфер вместо экстракта клеток. Затем определяли концентрацию белка в экстракте клеток на основании значений поглощения посредством калибровочной кривой для БСА. Специфические β-галактозидазные активности рассчитывали в соответствии со следующей формулой:
Объемная активность (мU/мл) = ΔЕ V/ε d v суммарного белка,
V: общий объем,
v: объем образца,
ε: коэффициент экстинкции
(0,0045 мМ см),
d: плотность слоя (1 см).
Известно, что промотор FMD главным образом контролируется типом источника углерода; температурная зависимость еще не описана (патент ЕР № 299108). Это было подтверждено выполненными в данной работе измерениями (см. фиг.10А). Показали, что β-галактозидазные активности были низкими в условиях с глюкозой (репрессия глюкозой), тогда как значительно более высокие значения были измерены в условиях глицерина или метанола (дерепрессия или индукция). Изменения температуры не приводили к разительным изменениям полученных при измерении значений (см. фиг.10А). Это также наблюдали в используемой в данной работе тест-системе. Активности β-галактозидазы были низкими при 30°С или 37°С, но существенно возрастали при 44°С (см. фиг.10В). Указанное зависимое от температуры повышение активности промотора не происходило в условиях метанола (фиг.10В), феномен, который еще не был описан. Неожиданно наибольшие β-галактозидазные активности, измеренные для промоторов TPS1, были значительно выше, чем таковые для промоторов FMD (см. фиг.10А, В).
ПРИМЕР 4
Сравнительная экспрессия гена фитазы под контролем промоторов FMD или TPS1
Рекомбинантные штаммы создавали посредством трансформации векторами pTPSlConphysMT и pFMTConphysMT согласно стандартным процедурам. За исключением промоторного элемента в экспрессирующей кассете два используемых для трансформации вектора идентичны. Индуцируемый теплом промотор, входящий в pTPSlConphysMT представляет собой фрагмент, соответствующий последовательности между нуклеотидами 228 и 792 в SEQ ID №l, 3'-конец которого имеет сайт рестрикции EcoRI (называемый ниже TPSl-промотором), тогда как pFMTConPhysMT содержит промотор FMD. Карта плазмиды и нуклеотидная последовательность вектора pTPSlConphysMT показаны на фиг.11. В качестве репортерного гена использовали мутеин фитазы.
После трансформации электропорацией получали рекомбинантные штаммы Н. polymorpha, выращивая полученные трансформацией прототрофные по урацилу штаммы на селективной среде в течение, по меньшей мере, 80 поколений (Gatzke et al., 1995). Представителей трансформантов двух полученных коллекций штаммов культивировали, сравнивая при различных условиях в жидких культурах объемом 3 мл. Культивирование проводили в среде YNB, забуференной 0,1 М фосфатным буфером рН 5,0 с добавлением 2% глюкозы или 5% глицерина. Через 48 часов количественно измеряли секретированную фитазу в аликвотах надосадка культуры с помощью способа, описанного в “Материалах и способах”.

В данном исследовании промотор TPS1 сравнивали с наиболее широко используемым в настоящее время промотором, промотором FMD. Применение промотора TPS1 в результате давало слабо увеличенные значения экспрессии при 37°С при сравнении с промотором FMD. При 40°С и 44°С наблюдали в два-три раза более высокую экспрессию в том случае, когда использовали промотор TPS1, чем наблюдали с промотором FMD.
ПРИМЕР 5
Сравнительная экспрессия гена фитазы под контролем вариантов промотора TPS1
На основании интегрирующего вектора pCll H.polymorpha (фиг.7), были сконструированы несколько производных векторов, которые отличались только последовательностью промотора перед репортерным геном фитазы.
В качестве примеров конструировали следующие различные конструкции на основании фрагмента между нуклеотидами 228 и 792 последовательности, представленной в SEQ ID №l (“исходный” промотор, индуцируемый теплом), и двух элементов, представленных в SEQ ID №2 и 5.
В первой серии промежуточная последовательность, состоящая из 52 п.н., между двумя элементами была модифицирована со 100% до 40%, при этом число в 52 п.н. не изменялось.
Во второй и третьей сериях элемент 1 и 2 был делегирован.
В сериях 4 и 5 эти конструкции модифицировали таким образом, что последовательности SEQ ID №4 и 3 использовали вместо последовательностей SEQ ID №2 и 5, производя замену элементов, способных отвечать на тепловой шок, указанными нуклеотидами.
В дополнительной серии использовали комбинацию исходных и замещенных последовательностей. Для полноразмерного мутагенеза использовали только пример с двумя исходными элементами, сравнивая 100%-ную и 40%-ную гомологию.
Различные последовательности получали индивидуально с помощью нуклеотидного синтеза, включая сайты рестрикции для правильного клонирования в вектор. Рекомбинантные штаммы получали трансформацией вышеуказанных векторов в соответствии со стандартными процедурами. Мутеин фитазы был использован в качестве репортерного гена. После осуществления трансформации электропорацией рекомбинантные штаммы H.polymorpha получали выращиванием урацил-фототрофных клонов, полученных трансформацией на селективной среде, на по меньшей мере 80 поколениях. Репрезентативные трансформанты, содержащие различные конструкции, культивировали и сравнивали в различных условиях в 3 мл жидкой культуры. Культивирование осуществляли в среде YNB, забуференной 0,1 М фосфатного буфера при рН 5,0, дополненной 2% глюкозой, и культивирование осуществляли при 40°С. Через 48 часов секретируемую фитазу подвергали количественному анализу в аликвотах культурального надосадка с помощью методов, описанных в разделе Материалы и Способы.
Исследование показало, что все варианты промотора TPS1 применимы для рекомбинантной экспрессии.
Библиография
Bradford, M.M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72: 248-254.
Devereux J., Haeberii P. and Smithies O. (1984) A comparative set of sequence analysis programs for the VAX. Nucl Acids Res 12: 387-395.
De Virgilio C., Burckert N., Boller T. and Wiemken A. (1991) A method to study the rapid phosphorylation-related modulation of neutral trehalase activity by temperature shifts in yeast. FEBS Lett 291: 355-358.
Faber K.N., Swaving G.J., Faber F., Ab G., Harder W., Veenhuis M. and Haima P. (1992) Chromosomal targeting of replicating plasmids in the yeast Hansenula polymorpha. J Gen Microbiol 138: 2405-2416.
Gatzke R., Weydemann U., Janowicz Z.A. & Hollenberg C.P. (1995) Stable multicopy integration of vector sequences in Hansenula polymorpha. Appl. Microbiol. Biotechnol 43, 844-849).
Hottiger Т., Schmutz P. and Wiemken A. (1987) Heat-induced accumulation and futile cycling of trehalose in Saccharomyces cerevisiae. J Bacteriol 169: 5518-5522.
Huxley С., Green E.D. and Dunham I. (1990) Rapid assessment of Saccharomyces cerevisiae mating type by PCR. Trends Genet 6 (8): р. 236.
Laemmli U.K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227: 680-685.
Levine D.W. and Cooney C.L. (1973) Isolation and characterization of a thermotolerant methanol-utilizing yeast. Appi Microbiol 26: 982-990.
Parrou J.L. and Francois J. (1997) A simplified procedure for a rapid and reliable assay of both glycogen and trehalose in whole yeast cells. Anal Biochem 248: 186-188.
Peterson G.C. (1977) A simplification of the protein assay method of Lowry et al. which is more generally applicable. Anal Biochem 83: 346-356.
Piper P.W. (1994) Measurement of transcription. In: Molecular Genetics of yeast. A practical approach, J.R. Johnston (ed.). IRL Press, Oxford.
Sambrook J., Fritsch E.F. and Maniatis T. (1989) Molecular Cloning: A Laboratory Manual. Second edition. Cold Spring Harbor Laboratory Press, Cold Spring Harbor.
Weydemann U, Keup P, Piontek M, Strasser AWM, Schweden J, Gellissen G, Janowicz ZA (1995) High-level secretion of hirudin by Hansenula polymorpha - authentic processing of three different preprohirudins. Appl Microbiol Biotechnol 44: 844-849.









| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБЫ УВЕЛИЧЕНИЯ УРОЖАЯ И СТРЕССОУСТОЙЧИВОСТИ У РАСТЕНИЯ | 2012 |
|
RU2632569C2 |
| КОНСТРУКЦИЯ И СПОСОБ КОНСТРУИРОВАНИЯ СИНТЕТИЧЕСКОГО ДВУНАПРАВЛЕННОГО РАСТИТЕЛЬНОГО ПРОМОТОРА UBI1 | 2012 |
|
RU2639538C2 |
| ОПТИМИЗИРОВАННАЯ ЭКСПРЕССИЯ L1 HPV45 В ДРОЖЖАХ | 2004 |
|
RU2360001C2 |
| ОПТИМИЗИРОВАННАЯ ЭКСПРЕССИЯ HPV 58 L1 В ДРОЖЖАХ | 2004 |
|
RU2370538C2 |
| МОЛЕКУЛЫ НУКЛЕИНОВОЙ КИСЛОТЫ, КОТОРЫЕ ВОЗДЕЙСТВУЮТ НА СУБЪЕДИНИЦУ С ВАКУОЛЯРНОЙ АТФАЗЫ И ПРИДАЮТ УСТОЙЧИВОСТЬ К ЖЕСТКОКРЫЛЫМ НАСЕКОМЫМ-ВРЕДИТЕЛЯМ | 2011 |
|
RU2644669C2 |
| ЭНХАНСЕР ПАЛОЧКОВИДНОГО ВИРУСА САХАРНОГО ТРОСТНИКА (SCBV) И ЕГО ПРИМЕНЕНИЕ В ФУНКЦИОНАЛЬНОЙ ГЕНОМИКЕ РАСТЕНИЙ | 2013 |
|
RU2639517C2 |
| ИНТЕРФЕРОН-ПОДОБНЫЙ БЕЛОК ZCYTO21 | 2001 |
|
RU2292394C2 |
| СЛИТЫЕ БЕЛКИ КАРЦИНОЭМБРИОНАЛЬНОГО АНТИГЕНА | 2005 |
|
RU2380375C2 |
| СПОСОБ ПОЛУЧЕНИЯ ГИАЛУРОНАНА В РЕКОМБИНАНТНОЙ КЛЕТКЕ-ХОЗЯИНЕ | 2002 |
|
RU2346049C2 |
| ЛОКУСЫ ФУНКЦИОНАЛЬНОСТИ FAD2 И СООТВЕТСТВУЮЩИЕ СПЕЦИФИЧНЫЕ К УЧАСТКУ-МИШЕНИ СВЯЗЫВАЮЩИЕ БЕЛКИ, СПОСОБНЫЕ ИНДУЦИРОВАТЬ НАПРАВЛЕННЫЕ РАЗРЫВЫ | 2013 |
|
RU2656159C2 |











Изобретение относится к биотехнологии, а именно к индуцируемым теплом промоторам, наборам и способам получения одного или большего количества белков с использованием индуцируемого теплом промотора. Индуцируемый теплом промотор выбирают из следующих нуклеиновых кислот: (а) нуклеиновой кислоты с последовательностью, указанной в описании; (b) нуклеиновой кислоты с последовательностью, которая проявляет по меньшей мере 40% идентичности на участке длиной в 300 п.н. с последовательностью, указанной в (а); (с) нуклеиновой кислоты, которая гибридизуется в жестких условиях с комплементарной цепью одной из нуклеиновых кислот, указанных в (а) или (b); (d) фрагмента одной из нуклеиновых кислот, указанных в (а)-(с), который сохраняет функцию индуцируемого теплом промотора; (е) комбинации нескольких нуклеиновых кислот, указанных в (а)-(d), где последовательности нуклеиновых кислот могут отличаться или быть одинаковыми; или нуклеиновой кислоты, имеющей последовательность, комплементарную последовательности одной из нуклеиновых кислот, указанных в (а)-(е). Представлен вектор, используемый для экспрессии белков, несущий индуцируемый теплом промотор. Вектор входит в набор для экспрессии белков наряду с клеткой-хозяином, подходящей для индукции индуцируемого теплом промотора и для продуцирования рекомбинантного белка. Изобретение позволяет получить промотор, параметр тепловой индукции которого избирателен, насколько это возможно, в частности промотор, который активен у дрожжей и который подходит для экспрессии белка при высоких температурах. 5 с. и 27 з.п. ф-лы, 16 ил., 2 табл.

(vi) последовательности нуклеиновой кислоты, которая кодирует полипептид, аминокислотная последовательность которого обладает по меньшей мере 80% идентичностью с аминокислотной последовательностью, указанной в SEQ ID NO:7.
| US 5792921 А, 11.08.1998 | |||
| BLAZOUEZ MIGUEL et al | |||
| J | |||
| Bacteriol, vol | |||
| Приспособление для удаления таянием снега с железнодорожных путей | 1920 |
|
SU176A1 |
| ЩЕЛКУНОВ С.Н | |||
| Генетическая инженерия | |||
| - Новосибирск: Из-во Новосибирского университета, 1997, с.192 и 193. | |||

