Введение
В последние годы для лечения депрессии и других заболеваний центральной нервной системы начали использовать селективные ингибиторы обратного захвата серотонина, среди которых заслуживают внимания флуоксетин, циталопрам, сертралин и пароксетин. Все они имеют различные химические структуры, что помогает объяснить их разные метаболические и фармакокинетические профили. Их эффективность действия в качестве антидепрессантов сравнима с эффективностью классических трициклических соединений, но их преимуществом является то, что они более безопасны и лучше переносимы пациентами. Настоящее изобретение относится к ряду новых 4-замещенных пиперидинов, имеющих арилокси функциональные группы и эффективно ингибирующих обратный захват серотонина и/или норадреналина в результате высокой аффинности к их нейрональным переносчикам. Данная характерная особенность придает им повышенные антидепрессивные возможности при лечении человека. Другим возможным терапевтическим применением этих соединений является лечение нервной булимии (чрезмерного аппетита), алкогольной зависимости, тревожных состояний, обсессивно-компульсивных расстройств (навязчивых состояний), паники, боли, предменструального синдрома и социофобии, а также профилактика мигрени. В литературе в качестве потенциальных антидепрессантов также описаны другие производные пиперидина с арилокси функциональными группами, хотя их химическая природа существенно отличается от таковой заявляемых в данном изобретении соединений, так как пиперидин замещен в 3-положении. То есть, например, это такие соединения, как 3-[(2-метоксифенокси)фенил]метил-пиперидин 1 (Melloni P., Carniel G., Della Torre A., Bonsignalari A., Buonamici M., Pozzi O., Ricciardi S., Rossi A.C. Eur.J.Med.Chem. Chim.Ther., 1984, 3, 235-242; Melloni P., Delia Torre A., De Munari S., Meroni M., Tonani R. Gazetta Chimica Italiana 1985, 115, 159-163) и 3-[(фенокси)фенил]метил-пиперидин 2 (патент Франции 2010615, Chem.Abstr.73: 66442j; патент Великобритании 1203149, Chem.Abstr.73: 120509b). В этих соединениях замещение пиперидинового кольца в 3-положении дает в результате дополнительный хиральный центр. Наличие двух хиральных центров приводит к диастереомерным смесям, в виде которых описано получение этих соединений
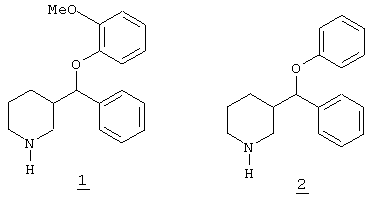
Ни в одном из случаев не описано получение и/или выделение чистых энантиомеров. Однако соединения, заявляемые в настоящем изобретении, имеют единственный хиральный центр, так как они имеют пиперидиновое кольцо, замещенное в 4-положении. Они были получены в виде рацемических смесей и в виде чистых энантиомеров при использовании синтетических способов, отличающихся от способов, используемых при получении соединений 1 и 2.
Кроме того, в качестве потенциальных антидепрессантов были описаны другие пиперидиновые производные, имеющие арилокси функциональные группы и пиперидиновое кольцо, замещенное в 4-положении (формулы 3 и 4)
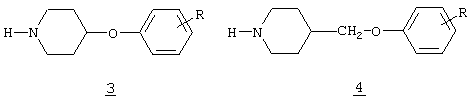
Так, в случае соединений типа 3 (патент Японии 9640999; Chem.Abstr.124:343333n) арилоксигруппа непосредственно связана с пиперидиновым кольцом, тогда как в соединениях типа 4 (патент Японии 9640999; Chem.Abstr.124:343333n) указанная группа связана с пиперидиновым кольцом через метиленовую группу, которая не имеет дополнительных замещений. Описанные в настоящей заявке соединения существенно отличаются от указанных выше, так как они содержат арилоксигруппу, связанную с пиперидиновым кольцом через метиленовую группу, где во всех случаях один из атомов водорода метиленовой группы замещен арильной группой, замещенной или незамещенной, как определено далее. Таким образом, эти соединения структурно отличаются от соединений типов 3 и 4, и методика синтеза, используемая для их получения, также является абсолютно иной.
Описание
Новые 4-замещенные пиперидины, описанные в настоящем изобретении, представлены общей формулой (I), в которой группы R1 и R2 представляют незамещенные арильные радикалы или арильные радикалы моно- или полизамещенные галогеном (фтором, хлором, бромом, иодом), алкилом, алкокси, циано, трифторметокси, трифторметилом, бензоилом, фенилом, нитро, амино, аминоалкилом, аминоарилом и карбониламино
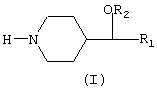
Соединения общей формулы (I) имеют асимметрический центр и были получены в виде рацемических смесей и в виде чистых энантиомеров. Настоящее изобретение включает все оптические изомеры соединений общей формулы (I) и их рацемические смеси. Настоящее изобретение также включает фармацевтически приемлемые соли этих соединений с неорганическими кислотами (такими как хлористоводородная, бромистоводородная, азотная, серная и фосфорная) и с органическими кислотами (такими как уксусная, фумаровая, винная, щавелевая, лимонная, п-толуолсульфоновая и метансульфоновая).
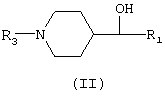
Рацемические соединения общей формулы (I) были получены с использованием хорошо известных способов синтеза, исходя из соединений общей формулы (II).
Образование группы алкиларилового простого эфира осуществляют с использованием реакции Мицунобу (Mitsunobu О., Synthesis 1981, 1; Hughes D.L., Organic Reactions 42, 335) с фeнолами R2-OH, где R2 представляет арильный радикал, замещенный или незамещенный, как описано для общей формулы (I) и соединениями общей формулы (II), в которой R1 представляет арильный радикал, замещенный или незамещенный, как описано для общей формулы (I), и R3 представляет водород или R4, который является алкоксикарбонильным радикалом, предпочтительно этоксикарбонилом и трет-бутоксикарбонилом
Группу алкиларилового простого эфира также получали с использованием реакции ароматического нуклеофильного замещения (Berglund R.A., Org.Proc.Res.Dev., 1997, 1, 328-330) с соединениями определенной выше общей формулы (II) и фторированными производными R2-F, где R2 представляет арильный радикал, моно- или полизамещенный галогеном (фтором, хлором, бромом, иодом), алкилом, алкокси, циано, трифторметокси, трифторметилом, бензоилом, фенилом, нитро, амино, аминоалкилом, аминоарилом и карбониламино. Соединения общей формулы (II) получали с использованием обычных синтетических способов, исходя из соединений общей формулы (III) (Duncan R.L., Helsley G.C., Welstead W.J., Da Vanzo J.P., Funderburk W.H., Lunsford C.D., J.Med.Chem., 1970, 13 (1), 1), в которой R5 представляет ацетильный радикал, этоксикрабонил, и R6 представляет циано или карбокси
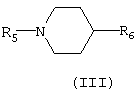
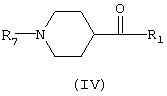
Соединения общей формулы (III), определенные выше, превращали в соединения общей формулы (IV), в которой R1 представляет арильный радикал, замещенный или незамещенный
 ,
,
как описано для соединений общей формулы (I), и R7 представляет водород, ацетил или R4, который является алкоксикарбонильным радикалом, предпочтительно этоксикарбонилом и трет-бутоксикарбонилом. Такое превращение проводили с использованием двух типов реакций: а) реакции Фриделя-Крафтса хлорангидридов кислот, производимых из соединений общей формулы (III), в которой R5 представляет ацетил или этоксикарбонил, и R6 представляет карбокси (Duncan R.L., Helsley G.C., Welstead W.J., Da Vanzo J.P., Funderburk W.H., Lunsford C.D., J.Med.Chem., 1970, 13 (1), 1) с бензолом или его подходящим образом функционализированными производными или b) реакции присоединения реактива Гриньяра, получаемого из подходящим образом функционализированных арилгалогенидов, к соединениям общей формулы (III), в которой R5 представляет ацетил, этоксикарбонил или трет-бутоксикарбонил, и R6 представляет циано (Duncan R.L., Helsley G.C., Welstead W.J., Da Vanzo J.P., Funderburk W.H., Lunsford C.D., J.Med.Chem., 1970, 13 (1), 1). Восстановление описанных соединений общей формулы (IV) дает определенные выше спирты общей формулы (II).
Энантиомеры, составляющие рацемические смеси общей формулы (I) получали двумя различными путями: а) разделением соответствующей рацемической смеси дробной кристаллизацией диастереомерных солей, полученных с хиральными кислотами (D или L-дибензоилвинной, D или L-винной, D или L-ди-п-толуолвинной и D или L-миндальной), и b) энантиоселективным синтезом. В последнем случае энантиомеры общей формулы (I) получали путем взаимодействия фенолов R2-OH или фторированных ароматических производных R2-F, определенных выше, с энантиомерами спиртов общей формулы (II), как это описано для рацемических смесей общей формулы (I). В энантиомерах спиртов общей формулы (II) R1 представляет арильный радикал, замещенный или незамещенный, как определено для соединений общей формулы (I), и R3 представляет водород или R4, который является алкоксикарбонильным радикалом, предпочтительно этоксикарбонилом и трет-бутоксикарбонилом. Энантиомеры спиртов общей формулы (II), определенные выше, получали энантиоселективным восстановлением (Ramachandran P.V., Teodorovic A.V., Rangaishenvi M.V., Brown H.C., J.Org.Chem., 1992, 57, 2379-2386) соединений общей формулы (IV) (Duncan R.L., Helsley G.C., Welstead W.J., Da Vanzo J.P., Funderburk W.H., Lunsford C.D., J.Med.Chem., 1970, 13 (1), 1), в которой R1 представляет арильный радикал, замещенный или незамещенный, как определено для соединений общей формулы (I), и R7 представляет водород или R4, определенный выше.
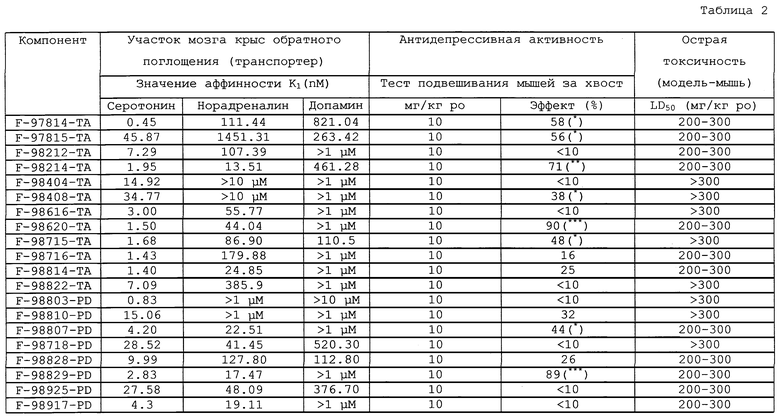
Фармакологическую активность соединений общей формулы (I) определяли с использованием хорошо разработанных фармакологических методик in vitro и in vivo. Аффинность соединений к серотониновым рецепторам обратного захвата (5НТ) оценивали на цельной коре головного мозга крысы с использованием [3Н]-пароксетина в качестве радиолиганда (Habert Е., Graham D., Tahraoui L., Claustre Y., Langer S.Z., Eur.J.Pharmacol., 1985, 118, 107-114), получая значения Ki в диапазоне между 0,5 и 500 нмоль/л. Аффинность соединений к рецепторам обратного захвата норадреналина (НА) оценивали на цельной коре головного мозга крысы с использованием [3Н]-низоксетина в качестве радиолиганда (Tejani-Butt S.M., J.Pharmacol.Exp.Ther., 1992, 260, 1, 427-436), получая значения Ki в диапазоне между 1 и 500 нмоль/л. Следующее использовали в качестве анализов, предсказывающих активность в качестве антидепрессанта: подвешивание мыши за хвост (Steru L., Chermat R., Thierry В., Mico J.A., Lenegre A., Steru M., Simon P., Porsolt R.D., Prog Neurophsychopharmacol. Biol. Psychiat., 1987, 11, 659-671), поведение крысы или мыши в безнадежном состоянии (Porsolt R.D., Anton G., Blavet N., Jalfre M. Eur. J.Pharmacol., 1978, 48, 379-394) и повышенную индуцированную иохимбином смертность крыс (Quinton R.M., Brit.J.Pharmacol., 1963, 21, 51-66). Соединения с Ki в диапазоне между 0,5 и 40 нмоль/л для одного из переносчиков или для обоих показали превосходную активность в качестве антидепрессанта на трех моделях при введении от 1 до 30 мг/кг перорально, внутриперитонеально или подкожно.
Следующие примеры иллюстрируют объем настоящего изобретения, который никоим образом не ограничивается такими примерами.
Пример 1
Фумарат (+/-)-4-[(4-трифторметоксифенокси)-2-(4-фторфенил)]метил-пиперидина
Смесь 1,1-диметил-этилового эфира (+/-)-4-[(4-трифторметоксифенил)гидрокси]метил-1-пиперидинкарбоновой кислоты (2,25 г, 7,27 ммоль), 2-пиридил-дифенилфосфина (1,90 г, 7,27 ммоль) и 1,3 г (7,4 ммоль) 4-трифторметоксифенола в 40 мл тетрагидрофурана (ТГФ) обрабатывали раствором диэтилазадикарбоксилата (ДЭАД) (1,15 мл) в 10 мл ТГФ. Реакционную смесь перемешивали при 20°С в течение 4-6 часов и концентрировали. Остаток растворяли в диэтиловом эфире, промывали водным раствором НСl (10%) и водным раствором NaOH (5%), сушили (безв. Na2SO4), фильтровали и концентрировали. Получали 2,4 г (71%) масла, которое растворяли в дихлорметане (50 мл) и обрабатывали трифторуксусной кислотой (2,1 мл) в 10 мл дихлорметана. Через 20 часов при 20°С смесь промывали водным раствором NaOH (5%) и насыщенным водным раствором NaCl. Сушка (безв. Na2SO4), фильтрование и концентрирование давали 1,3 г (71%) продукта, который суспендировали в безводном диэтиловом эфире (60 мл) и обрабатывали фумаровой кислотой (0,42 г), получая 1,0 г фумарата (выход 60%) с т.пл. 130-134°С. В спектре ЯМР-1H (DMSO-d6) наблюдался характерный сигнал при 4,31 м.д. (д, J=5,9 Гц, 1Н, 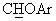 ), а в спектре ЯМР-13С (DMSO-d6) при 74,9 м.д. наблюдался сигнал, соответствующий
), а в спектре ЯМР-13С (DMSO-d6) при 74,9 м.д. наблюдался сигнал, соответствующий 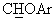 углероду.
углероду.
Пример 2
Гидрохлорид (+/-)-4-[(4-фторфенокси)(4-фторфенил)]метил-пиперидина
Смесь 1,1-диметил-этилового эфира (+/-)-4-[(4-фторфенил)гидрокси]метил-1-пиперидинкарбоновой кислоты (16,33 ммоль) и 1,9 г 4-фторфенола в 50 мл ТГФ обрабатывали 5,0 г трифенилфосфина и затем добавляли раствор ДЭАД (3,45 мл) в 10 мл ТГФ. Через 3 часа растворитель отгоняли и полученное масло обрабатывали гексаном, получая осадок, который отфильтровывали. Фильтрат концентрировали, остаток растворяли в дихлорметане (100 мл) и обрабатывали раствором трифторуксусной кислоты (8 мл) в 30 мл дихлорметана. Через 15 часов реакционную смесь обрабатывали обычным образом и получали гидрохлорид в ТГФ, получая 3,6 г его в виде аморфного и слегка гигроскопичного розоватого твердого вещества (выход 70%) с т.пл. 90°С (разл.). В спектре ЯМР-1H (CDCl3) гидрохлорида наблюдался характерный сигнал при 4,72 м.д. (д, J=5,8 Гц, 1Н, 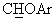 ), а в спектре ЯМР-13С (CDCl3) - сигнал при 83,1 м.д., соответствующий
), а в спектре ЯМР-13С (CDCl3) - сигнал при 83,1 м.д., соответствующий 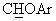 углероду.
углероду.
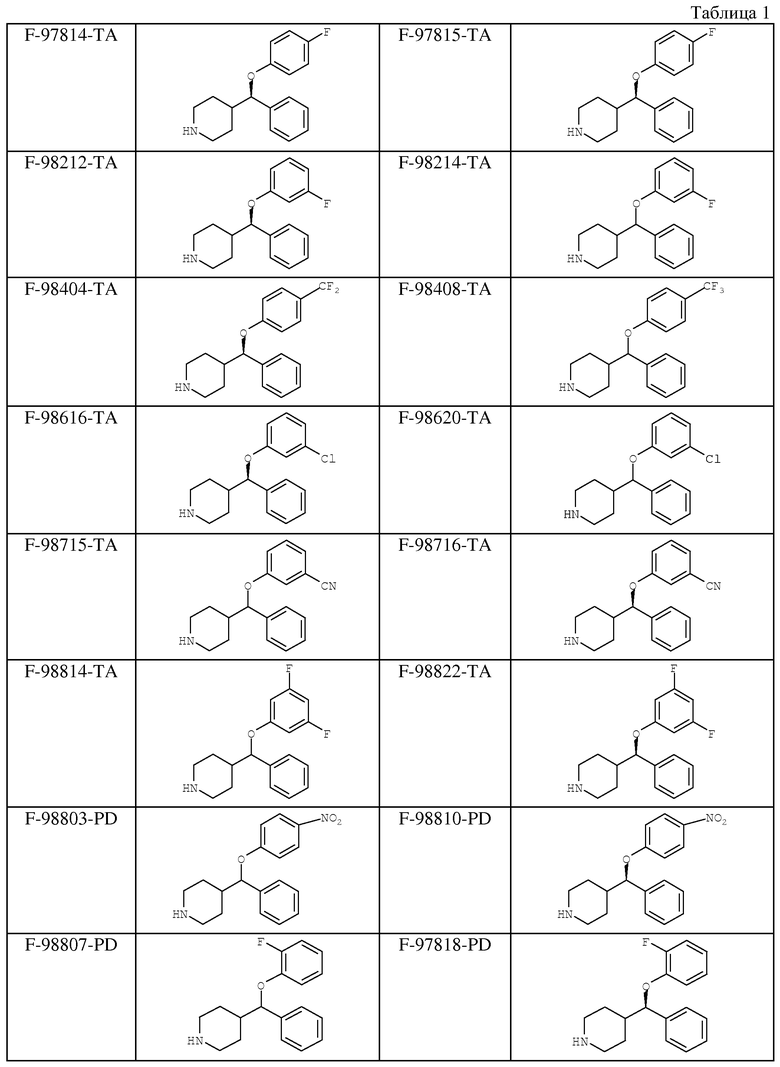
Аналогично были получены следующие соединения:
гидрохлорид (+/-)-4-[(4-фторфенокси)(4-хлорфенил)]метил-пиперидина (выход 54%, гигроскопичный),
фумарат (+/-)-4-[(4-метоксифенокси)(4-фторфенил)]метил-пиперидина (выход 60%, т.пл.=139-142°С),
гидрохлорид (+/-)-4-[(4-трифторметилфенокси)фенил]метил-пиперидина (выход 36%, гигроскопичный),
гидрохлорид (+/-)-4-[(фенокси)(4-хлорфенил)]метил-пиперидина (выход 72%, т.пл.=80°С (разл.)),
гидрохлорид (+/-)-4-[(4-бензоилфенокси)фенил]метил-пиперидина (выход 74%, т.пл. 70°С (разл.)), и
фумарат (+/-)-4-[(4-трифторметокси)фенил]метил-пиперидина (выход 58%, т.пл.=76°С (разл.)).
Пример 3
Сульфат (+/-)-4-[(4-фторфенокси)фенил]метил-пиперидина
Суспензию NaH (1,95 г, 60% в минеральном масле) в 20 мл диметилсульфоксида (ДМСО) обрабатывали раствором 1,1-диметил-этилового эфира (+/-)-4-(фенилгидрокси)метил-1-пиперидинкарбоновой кислоты (13,8 г, 47 ммоль) в 36 мл ДМСО. Добавляли бензоат калия (7,5 г, 47 ммоль)и 1,4-дифторбензол (6,1 мл, 56 ммоль), и реакционную смесь нагревали до 85°С до тех пор, пока не исчезало исходное вещество. Затем реакционную смесь обрабатывали насыщенным водным раствором NaCl и водой и экстрагировали этиловым эфиром. Остаток после упаривания органической фазы обрабатывали метанолом (200 мл) и водным раствором НСl (10%, 200 мл) и нагревали с обратным холодильником в течение часа. Продукт выделяли обычным способом, получая масло (9,6 г, 72% выход). В спектре ЯМР-1H (СDСl3) наблюдался сигнал при 4,70 м.д. (д, J=7,1 Гц, 1Н, 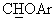 ), а в спектре ЯМР-13С (СDСl3) - сигнал при 85,0 м.д., соответствующий
), а в спектре ЯМР-13С (СDСl3) - сигнал при 85,0 м.д., соответствующий 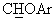 углероду. Масло обрабатывали раствором 1,85 мл конц. Н2SO4 в 90 мл воды, получая сульфат в виде твердого вещества с т.пл.=118-120°С (выход 75%).
углероду. Масло обрабатывали раствором 1,85 мл конц. Н2SO4 в 90 мл воды, получая сульфат в виде твердого вещества с т.пл.=118-120°С (выход 75%).
Пример 4
Сульфат (+/-)-4-[(3-фторфенокси)фенил]метил-пиперидина
Суспензию NaH (0,40 г, 60% в минеральном масле) в 6 мл ДМСО обрабатывали раствором 1,1-диметил-этилового эфира (+/-)-4-(фенилгидрокси)метил-1-пиперидинкарбоновой кислоты (2,55 г, 8,75 ммоль) в 6 мл ДМСО. Добавляли бензоат калия (1,35 г, 8,43 ммоль) и 1,3-дифторбензол (1,05 мл, 10,6 ммоль), и реакционную смесь нагревали до 85°С до тех пор, пока не исчезало исходное вещество. Затем реакционную смесь обрабатывали насыщенным водным раствором NaCl и водой и экстрагировали этиловым эфиром. Остаток после упаривания органической фазы обрабатывали метанолом (30 мл) и водным раствором НСl (10%, 30 мл) и нагревали с обратным холодильником в течение часа. Обычным способом обработки реакционной смеси получали 2,16 г янтарного масла (выход 88%). В спектре ЯМР-1H (CDCl3) наблюдался сигнал при 4,78 м.д. (д, J=6,4 Гц, 1Н, 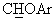 ), а в спектре ЯМР-13С (CDCl3) - сигнал при 84,6 м.д., соответствующий
), а в спектре ЯМР-13С (CDCl3) - сигнал при 84,6 м.д., соответствующий 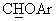 углероду. Масло обрабатывали раствором 0,20 мл конц. H2SO4 в 10 мл воды, получая сульфат в виде твердого вещества с т.пл.=72-76°С.
углероду. Масло обрабатывали раствором 0,20 мл конц. H2SO4 в 10 мл воды, получая сульфат в виде твердого вещества с т.пл.=72-76°С.
Следующие соединения были получены аналогичным образом:
гидрохлорид (+/-)-4-(феноксифенил)метил-пиперидина (выход 73%, гигроскопичен),
фумарат (+/-)-4-[(4-цианофенокси)фенил]метил-пиперидина (выход 81%, т.пл.=76°С (разл.)),
гидрохлорид (+/-)-4-[(3-трифторфенокси)фенил]метил-пиперидина (выход 72%, т.пл.=58°С (разл.)),
сульфат (+/-)-4-[(4-бромфенокси)фенил]метил-пиперидина (выход 70%, т.пл.=99-103°С),
гидрохлорид (+/-)-N,N-диметил-4-[[(4-пиперидинил)фенил]метил]окси-бензамида (выход 72%, т.пл.=45°С (гигроскопичный)),
гидрохлорид (+/-)-4-[(4-нитрофенилокси)фенил]метил-пиперидина (выход 80%, т.пл.=80°C (разл.)),
сульфат (+/-)-4-[(4-хлорфенил)(1-нафтилокси)]метил-пиперидина (выход 72%, т.пл.=186°С (разл.)),
сульфат (+/-)-4-[(1-нафтилокси)фенил]метил-пиперидина (выход 70%, т.пл.=152°С (разл.)),
сульфат (+/-)-4-[(2-фторфенокси)фенил]метил-пиперидина (выход 72%, т.пл.=76°С (разл.)),
гидрохлорид (+/-)-4-[(3-цианофенокси)фенил]метил-пиперидина (выход 80%, т.пл.=82°С (разл.)),
сульфат (+/-)-4-[(3-хлорфенокси)фенил]метил-пиперидина (выход 60%, т.пл.=101-104°С),
сульфат (+/-)-4-[(2-трифторметилфенокси)фенил]метил-пиперидина (выход 80%, т.пл.=110°С (разл.)),
оксалат (+/-)-4-[(2-цианофенокси)фенил]метил-пиперидина (выход 80%, т.пл.=105°С (разл.)),
гидрохлорид (+/-)-4-[[(2-бифенил)окси)фенил]метил-пиперидина (выход 84%, т.пл.=84-87°С),
гидрохлорид (+/-)-4-[[(4-бифенил)окси)фенил]метил-пиперидина (выход 82%, т.пл.=130°С),
сульфат (+/-)-4-[(3-бромфенокси)фенил]метил-пиперидина (выход 75%, т.пл.=98°С (разл.)),
сульфат (+/-)-4-[(2-иодфенокси)фенил]метил-пиперидина (выход 57%, т.пл.=105°С (разл.)),
сульфат (+/-)-4-[(3-иодфенокси)фенил]метил-пиперидина (выход 37%, т.пл.=127°С (разл.)),
сульфат (+/-)-4-[(3,5-дифторфенокси)фенил]метил-пиперидина (выход 86%, т.пл.=206-208°С),
сульфат (+/-)-4-[(3-фтор-2-метилфенокси)фенил]метил-пиперидина (выход 80%, т.пл.=125°С (разл.)),
гидрохлорид (+/-)-4-[(3-хлор-4-цианофенокси)фенил]метил-пиперидина (выход 70%, т.пл.=125°С (разл.)),
сульфат (+/-)-4-[(5-хлор-2-метилфенокси)фенил]метил-пиперидина (выход 75%, т.пл.=105°С (разл.)),
сульфат (+/-) -4-[(3-хлор-2-метилфенокси)фенил]метил-пиперидина (выход 89%, т.пл.=130°С (разл.)),
сульфат (+/-)-4-[(3,4-дихлорфенокси)фенил]метил-пиперидина (выход 91%, т.пл.=108°С (разл.)),
гидрохлорид (+/-)-4-[(3-метокси-5-фторфенокси)фенил]метил-пиперидина (выход 65%, т.пл.=200-203°С (разл.)) и
гидрохлорид (+/-)-4-[(3-фтор-5-цианофенокси)фенил]метил-пиперидина (выход 76%, т.пл.=70°С (разл.)).
Пример 5
Разделение (+/-)-4-[(3-фторфенокси)фенил]метил-пиперидина
К 7,1 г (25 ммоль) (+/-)-4-[(3-фторфенокси)фенил]метил-пиперидина, растворенного в 175 мл этанола (96%), добавляли 4,45 г L-(-)-дибензоилвинной кислоты. Получали белое твердое вещество (т.пл. 212°С (разл.)), которое обрабатывали водным раствором NaOH (5%) и экстрагировали хлороформом, получая левовращающий изомер (96% ее (ее=энантиомерный избыток), т.пл.=59-62°С, [α]546-11,4, с=0,576, СНСl3).
Полученный фильтрат концентрировали и свободное основание экстрагировали, обрабатывая водным раствором NaOH (5%) и хлороформом. Полученный продукт, растворенный в этаноле, обрабатывали D-(+)-дибензоилвинной кислотой, используя предшествующую методику. Получали белое твердое вещество (т.пл.=208°С (разл.)), который обрабатывали водным раствором NaOH (5%) и экстрагировали хлороформом, получая правовращающий изомер (98% ее, т.пл.=59-62°С, [α]546+11,4, с=0,618, CHCl3).
Аналогичным образом получали следующие соединения:
(+)-4-[(4-фторфенокси)фенил]метил-пиперидин (96% ее, т.пл.=100-102°С, [α]546+14, c=0,259, СНСl3),
(-)-4-[(4-фторфенокси)фенил]метил-пиперидин (96% ее, т.пл.=100-102°С, [α]546-14, с=0,237, СНСl3),
сульфат (+)-4-[(4-трифторметилфенокси)фенил]метил-пиперидина (96% ее, т.пл.=85°С (разл.), [α]265+17,8, с=0,556, СНСl3),
сульфат (-)-4-[(4-трифторметилфенокси)фенил]метил-пиперидина (96% ее, т.пл.=85°С (разл.), [α]365-15,5, с=0,508, СНСl3),
(+)-4-[(4-бромфенокси)фенил]метил-пиперидин (96% ее, т.пл.=129-131°С (разл.), [α]436+54, с=1,012, СНСl3),
(-)-4-[(4-бромфенокси)фенил]метил-пиперидин (95% ее, т.пл.=129-131°С (разл.), [α]436-54,1, с=1,048, СНСl3),
метансульфат (+)-4-[(3-хлорфенокси)фенил]метил-пиперидина (98% ее, т.пл.=200-202°С (разл.), [α]365+14,6, с=0,646, СНСl3),
метансульфат (-)-4-[(3-хлорфенокси)фенил]метил-пиперидина (99% ее, т.пл.=200-202°С (разл.), [α]365-13,6, с=0,690, СНСl3),
гидрохлорид (+)-4-[(3-цианофенокси)фенил]метил-пиперидина (95% ее, т.пл.=70°С (разл.), [α]436+26,5, с=0,600, СНСl3),
гидрохлорид (-)-4-[(3-цианофенокси)фенил]метил-пиперидина (98% ее, т.пл.=70°С (разл.), [α]365-27,1, с=0,680, СНСl3),
сульфат (+)-4-[(3,5-дифторфенокси)фенил]метил-пиперидин (96% ее, т.пл.=78°С (разл.), [α]436+19,4, с=0,80, СНСl3),
сульфат (-)-4-[(3,5-дифторфенокси)фенил]метил-пиперидин (98% ее, т.пл.=78°С (разл.), [α]436-19,8, c=0,724, СНСl3),
гидрохлорид (+)-4-[(3-фторфенокси)(3-фторфенил)]метил-пиперидина (96% ее, т.пл.=75°С (разл.), [α]546+15, с=0,183, СНСl3) и
гидрохлорид (-)-4-[(3-фторфенокси)(3-фторфенил)]метил-пиперидина (95,4% ее, т.пл.=75°С (разл.), [α]546-16, с=0,17, СНСl3).
Пример 6
(+)-4-[(4-фторфенокси)фенил]метил-пиперидин
К раствору 6,8 г (+)-В-хлордиизопиноканфеилборана ((+)-DIP-Cl) (21,25 ммоль) в дихлорметане (20 мл, сухой), охлажденному до 3-4°С, добавляли 4-бензоилпиперидин (2,0 г, 10,6 ммоль). После реакции в течение 72 часов добавляли 2,0 мл ацетальдегида (35,46 ммоль) и перемешивали при комнатной температуре в течение 3 часов. Добавляли 24 мл водного раствора NaOH (6н), дихлорметан и насыщенный водный раствор NaCl. Фазы разделяли и обычная обработка органической фазы давала (+)-α-фенил-4-пиперидинметанол в виде белого твердого вещества с т.пл.=64-66°С с выходом 90% (84% ее).
Растворяли 1,8 г аминоспирта - (+)-α-фенил-4-пиперидинметанола (9,6 ммоль), в метаноле (10 мл). Раствор охлаждали до 0°С и добавляли по каплям раствор ди-трет-бутилдикарбоната ((Вос)2O) (2,5 г, 11,27 ммоль) к 10 мл метанола. Смесь перемешивали в течение 24 часов при комнатной температуре, метанол концентрировали, добавляли воду и экстрагировали дихлорметаном. Обычная обработка органической фазы давала целевой спирт в виде слегка окрашенного масла с 93% выходом.
Полученный выше спирт (2,7 г, 9,3 ммоль), растворенный в ДМСО (25 мл), добавляли к суспензии NaH (60%, 0,6 г) в ДМСО (5 мл). Добавляли бензоат калия (1,53 г, 9,63 ммоль) и 1,4-дифторбензол (1,3 мл, 11,9 ммоль) и смесь нагревали (70-75°С) до исчезновения исходного вещества. Реакционную смесь выливали в воду и насыщенный водный раствор NaCl и экстрагировали диэтиловым эфиром. Полученное масло нагревали в условиях дефлегмации со смесью метанола (40 мл) и водного раствора соляной кислоты (40 мл) в течение 1 часа. Выделение продукта с использованием общепринятых способов дало (+)-4-[(4-фторфенокси)фенил]метил-пиперидин в виде масла с 54% выходом. Обработка 0,5 г (1,75 ммоль) данного масла D-дибензоилвинной кислотой в этаноле (96%, 30 мл) давала осадок, который отфильтровывали (т.пл.=198-199°С). Высвобождали аминоэфир, получая белое твердое вещество с 96% ее, т.пл.=102-104°С, и [α]546+15, с=0,105 (СНСl3).
Аналогично получали следующие соединения:
гидрохлорид (+)-4-[(4-нитрофенокси)фенил]метил-пиперидина (96% ее, т.пл.=55°С (разл.), [α]436+36, с=0,045, этанол),
гидрохлорид (-)-4-[(1-нафтилокси)фенил]метил-пиперидина (98% ее, т.пл.=65°С (разл.), [α]546-180, с=0,080, СНСl3) и
сульфат (+)-4-[(2-фторфенокси)фенил]метил-пиперидина (97,6% ее, т.пл.=105°С (разл.), [α]546+31, с=0,081, СНСl3).
Пример 7
(-)-4-[(4-фторфенокси)фенил]метил-пиперидин
4-Бензоил-пиперидин (7,35 г, 39,05 ммоль) добавляли к раствору 25 г (-)-DIP-Cl (78,125 ммоль) в дихлорметане (75 мл, сухой), охлажденному до 0-2°С. После протекания реакции в течение 72 часов добавляли 5,2 мл ацетальдегида (92,2 ммоль) и перемешивали при комнатной температуре в течение 3 часов. Добавляли 71 мл водного раствора NaOH (6н), дихлорметан и насыщенный водный раствор NaCl. Фазы разделяли, и обычная обработка органической фазы давала (-)-α-фенил-4-пиперидинметанол в виде белого твердого вещества с т.пл.=48-50°С с 85% выходом (86% ее).
В метаноле (10 мл) растворяли 2 г аминоспирта - (-)-α-фенил-4-пиперидинметанола (10,7 ммоль). Раствор охлаждали до 0°С и добавляли по каплям раствор (Вос)2О (2,6 г, 11,73 ммоль) в 7 мл метанола. Смесь перемешивали в течение 20 часов при комнатной температуре, метанол концентрировали, добавляли воду и экстрагировали дихлорметаном. Обычная обработка органической фазы давала желаемый спирт в виде слегка окрашенного масла с 90% выходом.
Полученный выше спирт (1,3 г, 4,5 ммоль), растворенный в ДМСО (10 мл), добавляли к суспензии NaH (60%, 210 г) в ДМСО (5 мл). Добавляли бензоат калия (715 г, 4,5 ммоль) и 1,4-дифторбензол (0,75 мл, 6,86 ммоль) и смесь нагревали (70-75°С) до исчезновения исходного соединения. Реакционную смесь выливали в воду и насыщенный водный раствор NaCl и экстрагировали эфиром. Полученное масло нагревали в условиях дефлегмации со смесью метанола (17 мл) и водного раствора соляной кислоты (17 мл) в течение 1 часа. Обычная обработка реакционной смеси давала (-)-4-[(4-фторфенокси)фенил]метил-пиперидин в виде масла с 64% выходом. Обработка данного масла L-дибензоилвинной кислотой в этаноле (96%, 35 мл) давала осадок, который отфильтровывали (т. пл.=193-194°C). Высвобождали аминоэфир, получая белое твердое вещество с 98% ее, т.пл.=100-102°С, [α]546-14, с=0,2, СНСl3).
Аналогичным образом получали следующие соединения:
гидрохлорид (-)-4-[(4-нитрофенокси)фенил]метил-пиперидина (98,7% ее, т.пл.=59°С (разл.), [α]436-31, с=0,042, этанол),
гидрохлорид (+)-4-[(1-нафтилокси)фенил]метил-пиперидина (94% ее, т.пл.=115°С (разл.), [α]546+156, с=0,128, СНСl3) и
сульфат (-)-4-[(2-фторфенокси)фенил]метил-пиперидин (97,6% ее, т.пл.=90°С (разл.), [α]546-31, с=0,140, CHCl3).
Пример 8
Сульфат (+/-)-4-[(3-фторфенокси)(3-фторфенил)]метил-пиперидина
Смесь 4-цианопиперидина (5 г, 40,92 ммоль), (Вос)2O (11,7 г, 53,7 ммоль), бикарбоната натрия (11,7 г, 139,3 ммоль) и воды (117 мл) перемешивали при комнатной температуре в течение 17 часов. Смесь экстрагировали дихлорметаном и органическую фазу сушили (безв. Na2SO4), фильтровали и концентрировали. Полученное масло очищали флэш-хроматографией (Still W.C., Kahn М., Mitra A. J.Org.Chem., 1978, 43, 2923), получая 1,1-диметил-этиловый эфир 4-циано-1-пиперидинкарбоновой кислоты в виде желтого масла с 43% выходом.
Суспензию Мg (0,5 г) в эфире (сухой, 22 мл) обрабатывали несколькими миллилитрами раствора (примерно 1/4 от общего количества) 1-бром-3-фторбензола (2,15 мл, 19,4 ммоль) в диэтиловом эфире (сухой, 16 мл) и кристаллом иода. Смесь нагревали до тех пор, пока не начиналось слабое кипение и исчезала окраска. Остаток раствора затем добавляли по каплям, поддерживая слабое кипение с обратным холодильником. После окончания добавления смесь нагревали с обратным холодильником в течение 1 часа 30 минут и оставляли охлаждаться до комнатной температуры. Добавляли по каплям к сухому диэтиловому эфиру (27 мл) 1,1-диметил-этиловый эфир 4-циано-1-пиперидинкарбоновой кислоты (2,7 г, 12,84 ммоль), и полученную смесь нагревали с обратным холодильником в течение 3 часов. Добавляли насыщенный водный раствор NH4Cl (50 мл) и экстрагировали диэтиловым эфиром. Обычная обработка органической фазы давала масло, которое очищали флэш-хроматографией (Still W.C., Kahn М., Mitra A. J.Org.Chem., 1978, 43, 2923), получая 2,4 г (61% выход) 1,1-диметил-этилового эфира 4-(3-фторбензоил)-1-пиперидинкарбоновой кислоты в виде желтоватого масла.
Полученный выше продукт (2,4 г, 7,8 ммоль) растворяли в метаноле (30 мл) и добавляли NaBH4 (0,2 г), растворенный в 3,5 мл воды. Смесь нагревали в течение 2 часов на масляной бане (50-60°С) и выделяли продукт обычным образом, получая 1,1-диметил-этиловый эфир (+/-)-4-[(3-фторфенил)гидрокси]метил-1-пиперидинкарбоновой кислоты в виде очень плотного желтоватого масла с количественным выходом.
Раствор полученного выше рацемического спирта (2,4 г, 7,8 ммоль) в ДМСО (25 мл) добавляли по каплям к суспензии NaH (60%) (0,62 г) в ДМСО (15 мл). Добавляли бензоат калия (1,53 г, 9,55 ммоль) и 1,3-дифторбензол (1,2 мл, 11,9 ммоль) и смесь нагревали на масляной бане (65-70°С) до исчезновения исходного вещества. Смесь выливали в смесь насыщенного раствора NaCl (50 мл) и воды (39 мл). Полученную смесь экстрагировали диэтиловым эфиром, и обычная обработка эфирной фазы давала масло, которое нагревали в условиях дефлегмации со смесью метанола (40 мл) и водного раствора НСl (10%, 40 мл) в течение 1 часа 30 минут. Целевой продукт (+/-)-4-[(3-фторфенокси)(3-фторфенил)]метил-пиперидин получали в виде янтарного масла с 50% выходом.
В спектре ЯМР-1H (CDCl3) данного продукта наблюдался сигнал при 4,55 м.д. (д, J=6,1 Гц, 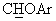 ), а в спектре ЯМР-13С (CDCl3) - сигнал при 83,9 м.д., соответствующий
), а в спектре ЯМР-13С (CDCl3) - сигнал при 83,9 м.д., соответствующий 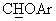 углероду. Полученное выше масло обрабатывали раствором 0,22 мл конц. Н2SO4 в 16,5 мл воды, получая сульфат в виде слегка окрашенного твердого вещества (т.пл.=158°С (разл.)).
углероду. Полученное выше масло обрабатывали раствором 0,22 мл конц. Н2SO4 в 16,5 мл воды, получая сульфат в виде слегка окрашенного твердого вещества (т.пл.=158°С (разл.)).
Аналогично были получены следующие соединения:
гидрохлорид (+/-)-4-[(2-фторфенокси)(3-фторфенил)]метил-пиперидин (выход 62%, т.пл.=90°С) и
гидрохлорид (+/-)-4-[(4-фторфенокси)(3-фторфенил)]метил-пиперидин (выход 30%, т.пл.=65°С).
Примеры, раскрывающие состав и способ приготовления композиций согласно настоящему изобретению.
Пример 1. Капсулы для перорального введения
Состав:
F-98214-TA 1 мг
Лактоза 90 мг
Микрокристаллическая целлюлоза 67 мг
Натриевая кроскармеллоза 3,5 мг
Стеарат магния 3,5 мг
Способ приготовления: Указанные ингредиенты смешиваются вместе и полученная в результате смесь упаковывается в желатиновые капсулы.
Доза активного ингредиента в других готовых формах, например таблетках, растворах, устанавливается в соответствии с активностью конкретного соединения общей формулы (I) и в зависимости от этого общий вес готовой формы регулируется микрокристаллической целлюлозой.
Пример 2. Таблетки
Состав:
Новые 4-замещенные пиперидины 0,05-10 мг
Микрокристаллическая целлюлоза 195 мг
Натриевая соль карбоксиметилкрахмала 2 мг
Колоидный диоксид кремния 1 мг
Стеарат магния 1 мг
Например, в случае использования в качестве активного ингредиента соединения F-98620-TA, таблетка содержит:
F-98620-TA 2 мг
Микрокристаллическая целлюлоза 195 мг
Натриевая соль карбоксиметилкрахмала 2 мг
Колоидный диоксид кремния 1 мг
Стеарат магния 1 мг
Способ приготовления
Таблетки могут быть изготовлены путем непосредственного прессования смеси или путем влажного гранулирования активного ингредиента и большей части наполнителя, которые высушиваются, смешиваются с оставшимся количеством ингредиентов и затем прессуются в таблетки.
Пример 3. Жидкие составы
100 мл такого состава содержат:
Новые 4-замещенные пиперидины 1-200 мг
Пропиленгликоль 5 г
Метилпарабен 200 мг
Пропилпарабен 100 мг
Ароматизирующий агент 130 мг
Сахарин натрия 10 мг
Вода, в количестве достаточном для 100 мл.
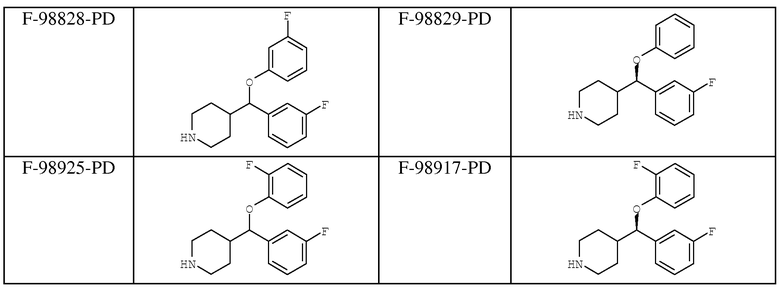
В таблице 1 указаны соединения, которые были протестированы на фармакологическую активность, и полученные в результате проведенных испытаний in vitro и in vivo данные приведены в таблице 2.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ АЗОТСОДЕРЖАЩИХ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБЫ ЛЕЧЕНИЯ ВОСПАЛИТЕЛЬНЫХ ЗАБОЛЕВАНИЙ И ЗАБОЛЕВАНИЙ ДЫХАТЕЛЬНЫХ ПУТЕЙ | 2001 |
|
RU2265011C2 |
| 2,7-ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ ОКТАГИДРО-1Н-ПИРИДО[1,2-А]ПИРАЗИНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ИНГИБИРОВАНИЯ ПРОЦЕССА СВЯЗЫВАНИЯ ДОПАМИНА С РЕЦЕПТОРОМ ДОПАМИНА D | 1995 |
|
RU2163239C2 |
| ФЕНИЛОКСАЗОЛИДИНОНЫ, ИМЕЮЩИЕ С-С-СВЯЗЬ С 4-8-ЧЛЕННЫМИ ГЕТЕРОЦИКЛИЧЕСКИМИ КОЛЬЦАМИ | 1996 |
|
RU2175324C2 |
| СОЕДИНЕНИЯ N, N-ЗАМЕЩЕННОГО ЦИКЛИЧЕСКОГО АМИНА | 1999 |
|
RU2232156C2 |
| Способ получения производных 2,3-дигидро-1,4-бензоксазина в виде смеси изомеров или в виде индивидуальных изомеров или их фармацевтически приемлемых солей | 1989 |
|
SU1826968A3 |
| 2,7-ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ ОКТАГИДРОПИРРОЛО[1,2-А]ПИРАЗИНА, СПОСОБ ЛЕЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ | 1996 |
|
RU2162470C2 |
| СОЕДИНЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ПРЕДОТВРАЩЕНИЯ ГИБЕЛИ НЕРВНЫХ КЛЕТОК, СПОСОБ ПРОФИЛАКТИКИ | 2001 |
|
RU2230060C2 |
| АЛЛОСТЕРИЧЕСКИЕ МОДУЛЯТОРЫ МЕТАБОТРОПНЫХ ГЛУТАМАТНЫХ РЕЦЕПТОРОВ | 2004 |
|
RU2360902C2 |
| 4-АРИЛОКСИ ИЛИ 4-АРИЛТИО-ПИПЕРИДИНОВЫЕ ПРОИЗВОДНЫЕ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 1994 |
|
RU2135495C1 |
| БОРСОДЕРЖАЩИЕ МАЛЫЕ МОЛЕКУЛЫ В КАЧЕСТВЕ ПРОТИВОВОСПАЛИТЕЛЬНЫХ СРЕДСТВ | 2007 |
|
RU2508113C2 |
Изобретение относится к новым 4-замещенным пиперидинам общей формулы (I), в которой R1 и R2 представляют арильные радикалы, замещенные или не замещенные, которые получаются в виде рацемических смесей или в виде чистых энантиомеров.
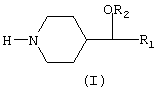
Эти соединения и их фармацевтически приемлемые соли ингибируют обратный захват серотонина и/или норадреналина и полезны в качестве антидепрессантов. Другими потенциальными терапевтическими применениями этих соединений являются лечение нервной булимии, обсессивно-компульсивных расстройств (навязчивых состояний), алкогольной зависимости, тревожных состояний, паники, боли, предменструального синдрома и социофобии, а также профилактика мигрени. 3 н. и 2 з.п.ф-лы, 2 табл.
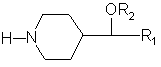
(I)
в которой R1 и R2 представляют незамещеные арильные радикалы или арильные радикалы, моно- или полизамещенные галогеном (фтором, хлором, бромом, иодом), алкилом, алкокси, циано, трифторметокси, трифторметилом, бензоилом, фенилом, нитро, амино, аминоалкилом, аминоарилом и карбониламино,
и их фармацевтически приемлемые соли с неорганическими кислотами и органическими кислотами.
4-(феноксифенил)метил-пиперидин,
4-[(4-фторфенокси)фенил]метил-пиперидин,
4-[(4-метоксифенокси)(4-фторфенил)]метил-пиперидин,
4-[(4-фторфенокси)(4-фторфенил)]метил-пиперидин,
4-[(4-фторфенокси)(4-хлорфенил)]метил-пиперидин,
4-[(4-трифторметилфенокси)фенил]метил-пиперидин,
4-[(4-трифторметоксифенокси)(4-фторфенил)]метил-пиперидин,
4-[фенокси(4-хлорфенил)]метил-пиперидин,
4-[(4-бензоилфенокси)фенил]метил-пиперидин,
4-[(4-трифторметоксифенокси)фенил]метил-пиперидин,
4-[(4-цианофенокси)фенил]метил-пиперидин,
4-[(3-трифторфенокси)фенил]метил-пиперидин,
4-[(3-фторфенокси)фенил]метил-пиперидин,
4-[(4-бромфенокси)фенил]метил-пиперидин,
N,N-диметил-4-[[(4-пиперидинил)фенил]метил]оксибензамид,
4-[(4-нитрофенилокси)фенил]метил-пиперидин,
4-[(4-хлорфенил)(1-нафтилокси)]метил-пиперидин,
4-[(1-нафтилокси)фенил]метил-пиперидин,
4-[(2-фторфенокси)фенил]метил-пиперидин,
4-[(3-цианофенокси)фенил]метил-пиперидин,
4-[(3-хлорфенокси)фенил]метил-пиперидин,
4-[(2-трифторметилфенокси)фенил]метил-пиперидин,
4-[(2-цианофенокси)фенил]метил-пиперидин,
4-[[(2-бифенил)окси]фенил]метил-пиперидин,
4-[(3-фторфенокси)(3-фторфенил)]метил-пиперидин,
4-[(2-фторфенокси)(3-фторфенил)]метил-пиперидин,
4-[(4-фторфенокси)(3-фторфенил)]метил-пиперидин,
4-[[(4-бифенил)окси)фенил]метил-пиперидин,
4-[(3-бромфенокси)фенил]метил-пиперидин,
4-[(4-иодфенокси)фенил]метил-пиперидин,
4-[(3-иодфенокси)фенил]метил-пиперидин,
4-[(3,5-дифторфенокси)фенил]метил-пиперидин,
4-[(3-фтор-2-метилфенокси)фенил]метил-пиперидин,
4-[(3-хлор-4-цианофенокси)фенил]метил-пиперидин,
4-[(5-хлор-2-метилфенокси)фенил]метил-пиперидин,
4-[(3-хлор-2-метилфенокси)фенил]метил-пиперидин,
4-[(3,4-дихлорфенокси)фенил]метил-пиперидин,
4-[(3-метокси-5-фторфенокси)фенил]метил-пиперидин, и
4-[(3-фтор-5-цианофенокси)фенил]метил-пиперидин.
| Способ получения производных пиперидина или их солей | 1977 |
|
SU867298A3 |

