Настоящее изобретение касается макролидов, обладающих противовоспалительной активностью, а более конкретно оно касается дез-диметиламинопроизводных макролидов с противовоспалительной активностью, их фармацевтически приемлемых солей, а также фармацевтических составов, содержащих эти соединения в качестве активных ингредиентов.
Известно, что многие антибиотики, в частности антибиотики класса макролидов с 14 атомами, представляющие собой производные эритромицина, помимо антибактериальной активности обладают и противовоспалительными свойствами [Clin. Immunother., (1996), 6, 454-464].
Эритромицин - это природный макролид (The Merck Index, XII edition, n°3720, page 625), обладающий очень широким спектром клинического применения при лечении инфекций, вызываемых грамположительными бактериями, некоторыми грамотрицательными бактериями или микоплазмой.
В последнее время интерес научного сообщества сфокусировался на противовоспалительном и иммуномодулирующем компоненте эритромицина и его производных [Journal of Antimicrobial Chemotherapy, (1998), 41, Suppl.B, 37-46].
Эта активность была хорошо подтверждена материалами клинических испытаний как in vivo, так и in vitro.
Например, доказана эффективность макролидов в терапии воспалительных процессов, таких как панбронхиолит [Thorax, (1997), 52, 915-918], бронхиальная астма [Chest, (1991), 99, 670-673] и кистозный фиброз [The Lancet, (1998), 351, 420]; или при моделировании у животных воспалительных процессов, таких как перитонит мышей, вызванный зимозаном [Journal of Antimicrobial Chemotherapy, (1992), 30, 339-348] и нейтрофильный рекрутмент трахеи крыс, вызванный эндотоксином [Antimicrobial Agents and Chemotherapy, трахеи крыс, вызванный эндотоксином [Antimicrobial Agents and Chemotherapy, (1994), 38, 1641-1643]; или в in vitro изучении таких клеток иммунной системы, как нейтрофилы [The journal of Immunology, (1997), 159, 3395-4005] и Т-лимфоциты [Life Sciences, (1992), 51, PL 231-236]; или при модулировании цитокинов, таких как интерлейкин 8 (IL-8) [Am. J. Respir. Crit. Care Med., (1997). 156, 266-271] или интерлейкин 5 (IL-5) (EP 0775489 и ЕР 0771564, Taisho Pharmaceutical Co., Ltd).
Специфическая терапевтическая эффективность макролидов при заболеваниях, в которых обычные противовоспалительные лекарственные средства (такие, как, например кортикостероиды) показали свою неэффективность [Thorax, (1997), 52, 915-918, цитируемый выше], объясняет высокий интерес, адресованный этому новому потенциальному классу противовоспалительных средств.
Тем не менее высокая антибактериальная активность общеизвестных макролидов не допускает их расширенное применение для постоянного лечения воспалительных процессов, не обусловленных патогенными микроорганизмами, из-за быстрого появления устойчивых штаммов.
Поэтому было бы желательно располагать новыми веществами с макролидной структурой, которые обладают противовоспалительной активностью и в то же время лишены свойств антибиотиков. Для большей ясности мы даем формулу эритромицина, в которой приведена нумерация, принятая в описании настоящей заявки.

Некоторые классы производных эритромицина, обладающие противовоспалительной активностью, описаны в литературе.
Например, в уже цитированных заявках на ЕП заявлены права на Тайшо (Таisho)-производные эритромицина по 3-, 9-, 11и 12-позициям в качестве сильных ингибиторов синтеза интерлейкин 5.
В заявке ЕП 0283055 (Sour Pliva) N-алкил производные азитромицина без кладиноза и дезозамина формулы
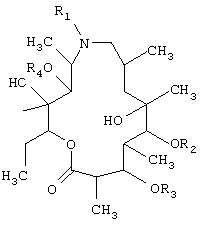
в которой
R1 представляет собой водород, низший алкил или низший алканоил;
R2, R3 и R4, одинаковые или различные, представляют собой водород или низший алканоил, описаны как противовоспалительные средства.
В заявке WO 92/16226 от имени Smith-Kline Beecham Corporation заявлено использование эритромицина как противовоспалительного средства, которое за счет подавления гликопротеина млекопитающих mdr-P снижает высвобождение интерлейкин 1.
Среди макролидных производных, описываемых в литературе, есть несколько 3-дездиметиламино-9-оксиимино производных. Ограниченный интерес к этому классу соединений подтвержден тем фактом, что уже известна существенная роль диметиламино-группы в активности рибосомного связывания типичных макролидов [Tetrahedron Letters, (1994), 35, 3837-3840].
В заявке США 3928387 (Hoffmann-La Roche Inc.) описан оксим 3’-дездиметиламино-3’, 4’-дегидроэритромицина А как промежуточное соединение, пригодное для получения антибиотика 1745 А/Х.
В заявке ЕП 0254534 (Robinson, William S.) предъявлены права на очень широкий класс макролидов с антивирусной активностью. Среди них описано соединение формулы

правильное химическое название которого 9-O-метилоксим 3’-дездиметиламино-3’, 4’-дегидроэритромицина А вопреки тексту самой заявки ЕП 0254534 ошибочно приведено как 9-O-метилоксим дездиметиламиноэритромицина (с.10, строка 46).
В настоящее время обнаружено, что при удалении диметиламино-группы из 3’-положения дезозамина 9-оксииминовых макролидов образуются соединения, обладающие противовоспалительной активностью и в значительной степени лишенные свойств антибиотиков.
Следовательно, предметом настоящего изобретения являются соединения формулы
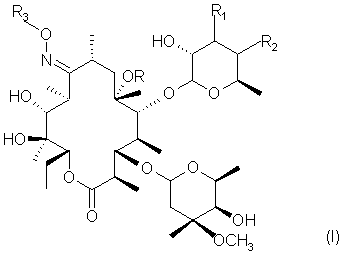
в которой R представляет собой водород или метил;
R1 и R2 оба представляют собой водород, или они вместе образуют химическую связь;
R3 представляет собой водород, линейную или разветвленную C1-C5 алильную группу, или цепь формулы

где А представляет собой водород или фенильную группу, или 5 или 6-членный гетероцикл, насыщенный или ненасыщенный, содержащий от 1 до 3 гетероатомов, выбранных из азота, кислорода и серы, необязательно замещенных одним или двумя заместителями, выбранными из C1-C5 алкильных групп или фенильных групп;
Х и Y, одинаковые или различные, представляют собой О, или NR4, где R4 -водород, линейная или разветвленная C1-C5 алкильная группа, бензилоксикарбонильная группа;
r - целое число от 1 до 6;
m - целое число от 1 до 8;
n - целое число от 0 до 2;
а также их фармацевтически приемлемые соли;
при этом исключаются соединения оксим 3'-дездиметиламино-3',4'-дегидроэритромицина А и 9-O-метилоксим 3'-дездиметиламино-3'4'-дегидроэритромицина А.
Следующим предметом настоящего изобретения является применение оксима 3'-дездиметиламино-3',4'-дегидроэритромицина А и 9-O-метилоксима 3'-дездиметиламино-3',4'-дегидроэритромицина А в качестве противовоспалительных средств.
Указанные соединения формулы I представляют собой макролиды противовоспалительного действия, лишенные антибиотической активности, и поэтому они пригодны для лечения воспалительных процессов.
Под термином линейные или разветвленные C1-C5 алкильные группы подразумевается группа, выбранная из метильной, этильной, н-пропильной, изопропильной, н-бутильной, изобутильной, вторичной бутильной, трет-бутильной, н-пентильной и изопентильной групп.
Под термином 5- или 6-членный гетероцикл, насыщенный или ненасыщенный с 1-3 гетероатомами, выбранными из азота, кислорода и серы, понимают такие гетероциклы, как пиррол, тиофен, фуран, имидазол, пиразол, тиазол, изотиазол, изоксазол, оксазол, пиридин, пиразин, пиримидин, пиридазин, триазол, тиадиазол или их частично или полностью насыщенные формы.
Предпочтительными соединениями формулы I являются такие соединения, в которых R, R1 и R2 представляет собой водород.
В классе этих соединений предпочтительны, в частности, такие соединения, в которых R3 представляет собой цепь с формулой
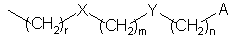
в которой
X, Y, А, r, m и n имеют упомянутые выше значения.
Еще более предпочтительны такие соединения, в которых R3 представляет собой цепь формулы

в которой
r равен 2, m равен 2 или 6, n равен 1, Y представляет собой NR4, Х представляет собой О или NR4, R4 представляет собой водород, а А представляет собой фенил или тиазолил.
Примерами фармацевтически приемлемых солей формулы (I) являются соли органических или неорганических кислот, таких как соляная, бромистоводородная, иодистоводородная, азотная, серная, фосфорная, уксусная, винная, лимонная, бензойная, янтарная и глутаровая кислоты.
Соединения формулы I, являющиеся предметом настоящего изобретения, получают, используя схему синтеза, которая включает: (а) удаление диметиламино-группы с 3’-позиции, и (6) необязательное введение функциональной группы указанного оксима.
Указанное удаление диметиламино-группы проводят путем окисления, пиролиза и необязательного восстановления, которые осуществляют известными методами. Людям, квалифицированным в данной области, очевидно, что для того, чтобы избежать препятствий со стороны функциональных групп, которые необязательно присутствуют при заместителе R3, предпочтительно, чтобы это удаление диметиламино-группы проводилось исходя из промежуточных соединений с формулой

в которой
R имеет значения, указанные ранее, a R3’ представляет собой водород, или линейную или разветвленную С1-С5 алкильную группу.
Их окислением получают соответствующие N-оксимы с формулой

в которой
R и R3’ имеют указанные ранее значения;
А их пиролизом, при необязательном последующем восстановлении, получают соединения с формулами

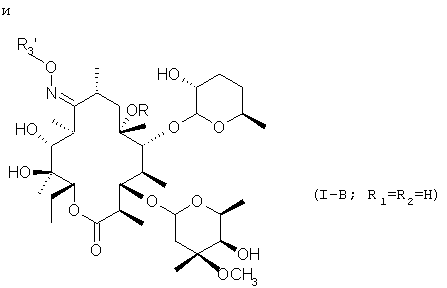
являющиеся предметом настоящего изобретения (I-R3=водород или C1-C5 алкил).
Соединения формулы I, в которой R3 отличен от водорода, можно получить согласно общепринятой методике, исходя из соединений формул I-A и I-B, в которых R3’ представляет собой водород, путем введения функциональной группы указанного оксима.
Обычно указанное введение функциональной группы проводят взаимодействием с соединением формулы
R3”-W (IV)
в которой R3" имеет все значения R2, кроме водорода, a W - уходящая группа (предпочтительно атом хлора или брома, или мезильная группа). Другой путь синтеза, особенно подходящий для получения соединений формулы I, в которой R3 представляет собой цепь формулы

где
X, Y, А, r, m и n имеют указанные ранее значения;
включает взаимодействие соединения формулы I, в которой R3 представляет собой водород, с промежуточным соединением формулы

где W, X, Y, m и n имеют значения, указанные ранее, a Z представляет собой защитную группу; с получением промежуточного соединения формулы
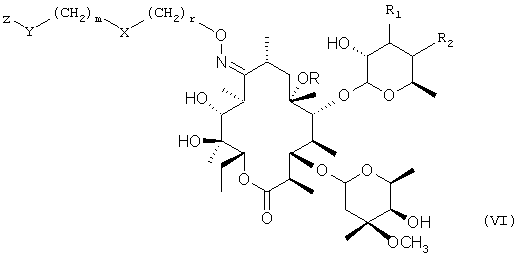
в которой R, R1, R2, X, Y, Z, r и m имеют значения, указанные ранее;
после удаления указанной защитной группы Z это соединение взаимодействует с производным формулы

где А, W и n имеют значения, указанные ранее; с получением соединения формулы I.
Указанные соединения формулы I, в которой Y представляет собой NR4, можно получить, следуя изложенной выше схемы синтеза, а также при использовании альдегида формулы
А-СНО (VIII),
в которой А имеет значения, указанные ранее,
а вместо соединения формулы VII произвести удаление указанной защитной группы Z из промежуточного соединения формулы VI.
Кроме того, соединения формулы I, в которой R1=R2=H, могут быть получены восстановлением соответствующих соединений формулы I, в которой R1 и R2 образуют химическую связь.
Указанные соединения формулы I, являющиеся предметом настоящего изобретения, обладают противовоспалительной активностью и лишены антибиотической активности.
Фармакологическую активность соединений формулы I оценивали в тестах in vitro и in vivo, сравнивая ее с активностью известных макролидов, таких как эритромицин, кларитромицин и рокситромицин, которые обладают как противовоспалительной активностью, так и антибиотической активностью.
Указанная противовоспалительная активность in vitro оценивалась по подавлению высвобождения интерлейкин 8 и высвобождения аниона пероксида (пример 11), a in vivo - по подавлению нейтрофилии, вызываемой повторными введениями LPS (пример 12).
Во всех этих экспериментах соединения, являющиеся предметом настоящего изобретения, показали свою очень высокую активность как противовоспалительные средства, и их противовоспалительная активность эквивалентна активности одного из названных соединений или выше ее.
Для терапевтического применения соединение формулы I можно использовать как фармацевтическую форму, пригодную для орального или парентерального введения.
Таким образом, предметом настоящего изобретения являются фармацевтические соединения, содержащие терапевтически активное количество соединения формулы I или его соль в смеси с фармацевтически приемлемым носителем.
Для лучшей иллюстрации настоящего изобретения предназначены следующие далее примеры.
Пример 1
Получение бензилового эфира [6-(2-гидроксиэтиламино)-гексил]-карбаминовой кислоты
К раствору бензилового эфира (6-гидроксигексил)-карбаминовой кислоты (25 г; 99,47 ммоль), полученного так, как это описано в заявке WO 96/18633, в CH2Cl2 (350 мл) и охлажденному льдом до температуры приблизительно в 10°С сначала добавили раствор КВr (1,18 г; 9,94 ммоль) в воде (20 мл) и TEMPO (0,155 г; 0,994 ммоль). Затем по каплям, выдерживая температуру в 10-12°С, в течение приблизительно 15-20 мин добавляли раствор, полученный из NаНСО3 (7,5 г; 89,28 ммоль) и NaClO (4,5% водный раствор; 197 мл; 125 ммоль).
Спустя 15 мин после окончания процесса прикапывания получившиеся фазы разделили и водную фазу однократно экстрагировали СH2Cl2 (100 мл). Собранные органические экстракты дважды промыли соляным раствором (20% NaCl) и высушили на сульфате натрия.
К полученному раствору (приблизительно 800 мл) добавили 3  -молекулярные сита (30 г), а затем при быстром прикапывании и при охлаждении водой и льдом раствор 2-аминоэтанола (35,9 мл; 0,597 мол) в этаноле (600 мл).
-молекулярные сита (30 г), а затем при быстром прикапывании и при охлаждении водой и льдом раствор 2-аминоэтанола (35,9 мл; 0,597 мол) в этаноле (600 мл).
По окончании прикапывания полученную смесь выдерживали с перемешиванием при комнатной температуре в течение 2 ч, а затем отфильтровали.
К полученному раствору по каплям добавляли NaBH4 (4,54 г; 120 ммоль), охладили водой и льдом при перемешивании в атмосфере азота.
По окончании этого процесса реакционную смесь выдерживали с перемешиванием при комнатной температуре в течение 2 ч, после чего растворитель выпарили.
Остаток собрали, добавили воду и этилацетат и образовавшиеся фазы разделили, затем дважды провели экстракцию водной фазы этилацетатом.
Собранные органические экстракты промыли соляным раствором (20% NaCl, высушили на сульфате натрия и сгущали до получения маслянистого остатка, который затем довели до загустевания.
Остаток растирали в порошок в гексане, отфильтровали и промыли смесью гексана и этилового эфира, получив при этом в виде белого твердого вещества бензиловый эфир [6-(2-гидроксиэтиламино)-гексил]-карбаминовой кислоты (26,22 г; выход 89%).
1Н-ЯМР (200 МГц, CDCl3) δ(ppm): 7.33-7.25 (m, 5H, Аr); 5.05 (s. 2H, СООСН2); 4.96 (уширенный-t, 1H, NH); 3.63-3.58 (m, 2H. *CH2-OH); 3.19-3.09 (m, 2H, CH2NCO); 2.72-2.67 (m, N-*CH2-CH2O); 2.59-2.52 (m, 4H, ОН and СН3); 1.53-1.23 (m, 8H, 4CH2).
Пример 2
Получение бензилового эфира 6-(бензилоксикарбониламиногексил)-(2-гидроксиэтил)-карбаминовой кислоты
Раствор бензилового эфира бензилхлорформиата (50% в толуоле; 42,5 мл; 0,128 моль) в этилацетате (85,5 мл) и 1N NaOH (128 мл; 0,128 моль) одновременно по каплям добавляли к раствору бензилового эфира [6-(2-гидроксиэтиламино)-гексил]-карбаминовой кислоты (31,5 г; 0,107 моль), полученного так, как это описано в примере 1, в смеси воды (87 мл), 1N NaOH (17 мл) и этилацетата (180 мл). Охлаждали при 0-5°С, контролируя температуру и рН (приблизительно равный 8).
По окончании прикапывания реакционную смесь выдерживали при 0-5°С с перемешиванием в течение 30 мин, после чего охлаждение прекратили и добавили еще 1N NaOH (15 мл), чтобы снова вернуться к прежнему значению рН 8. После этого реакционную смесь оставили на ночь при перемешивании при комнатной температуре.
Образовавшиеся фазы разделили и водную фазу снова экстрагировали этилацетатом. Собранные органические экстракты промыли соляным раствором, высушили на сульфате натрия и сгустили под вакуумом с получением маслянистого остатка.
Хроматографической очисткой (элюант этилацетат:петролатум при соотношении от 60:40 до 70:30) был получен в виде масла бензиловый эфир 6-(бензилоксикарбониламиногексил)-(2-гидроксиэтил)-карбаминовой кислоты (42,5 г; выход 92%).
1Н-ЯМР (200 МГц, СDСl3) δ(ppm): 7.39-7.25 (m, 10Н, Аr); 5.10 и 5.07 (2s, 4H, 2COOCH2); 3.71 (уширенный сигнал, 2Н, *СН2-ОН); 3.43-3.01 (m, 4H, 2CH2NCO); 1.57-1.19 (m, 8H, 4СН2).
Действуя аналогично, получили следующие соединения:
Бензиловый эфир (2-бензилоксикарбониламиноэтил)-(2-гидрокси-этил)-карбаминовой кислоты
исходя из 2-(2-аминоэтиламино)-этанола
(выход 32%)
1Н-ЯМР (200 МГц, СDCl3) δ(ppm): 7.33-7.28 (m, 10Н, 2Ph); 5.06 and 5.04 (2s, 4H, 2CH2-Ph); 3.73-3.34 (уширенный-m, 8H, 4СН2);
Бензиловый эфир [2-(бензил-бензилоксикарбониламино)-этил]-(2-гидроксиэтил)-карбаминовой кислоты
исходя из 2-[2-(бензиламино)-этиламино]-этанола, полученного так, как это описано в заявке WO 96/18633.
(выход 50%)
1H-ЯМР (200 МГц, СDСl3) δ(ppm): (сильно уширенные сигналы) 7.42-7.13 (m, 15H, 3Аr); 5.12 и 5.09 (2s, 4H, СООCH2*); 4.55 (s, 2H, N-*CH2-Ph);
Бензиловый эфир [2-(2-гидроксиэтокси)-этил]-карбаминовой кислоты
исходя из 2-(2-аминоэтокси)-этанола
(выход 85%)
1Н-ЯМР (200 МГц, СDСl3) δ(ppm): 7.36-7.28 (m, 5H, Аr); 5.18 (уширенный сигнал, 1Н, NH); 5.08 (s, 2Н, Ph-*CHO); 3.74-3.34 (m, 8H, *CH2-*СН2-O-*СН2-*СН2-ОН); 2.13 (уширенный-t, 1H, ОН).
Пример 3
Получение___этилового___эфира___2-[бензилоксикарбонил-(6-бензилоксикарбониламиногексил)-амино]-метансульфоновой кислоты
Триэтиламин (8,95 мл; 64,31 ммоль) добавили к раствору бензилового эфира 6-(бензилоксикарбониламиногексил)-(2-гидрокси-этил)-карбаминовой кислоты (13,78 г; 32,15 ммоль), полученного так, как это описано в примере 2, в СН2Сl2 (140 мл). Полученную смесь охлаждали при 0-5°С, а затем по каплям добавляли к раствору метансульфонилхлорида (3,36 мл; 43,41 ммоль) в СН2Сl2 (20 мл).
По окончании этого процесса смесь выдерживали при комнатной температуре с перемешиванием в течение 60 мин, промывали сначала 5% водной лимонной кислотой, затем соляным раствором (20% NaCl), 5% водным NaHCO3 и в завершение снова соляным раствором. После высушивания на сульфате натрия и упаривания в вакууме в виде коричневого масла был получен этиловый эфир 2-[бензилоксикарбонил-(6-бензилоксикарбониламиногексил)-амино]-метансульфоновой кислоты (16,37 г; выход 100%).
1H-ЯМР (200 МГц, CDCl3) δ(ppm): 7.35-7.27 (m, 10Н, Аr); 5.11 и 5.07 (2s, 4Н, 2СООСН2); 4.36-4.19 (m, 2H, CH2OSO2); 3.57-3.51 (m, 2H, SO-CH2-*CH2N); 3.32-3.07 (m, 4H, 2CH2N); 2.91 и 2.85 (2s-конформационные варианты структуры, 3Н, СН3); 1.50-1.20 (m, 8H, 4СН2).
Действуя аналогично, получили следующие соединения:
Этиловый____эфир 2[2-(бензилоксикарбониламино)-этокси]-метансульфоновой кислоты, исходя из бензилового эфира [2-(2-гидрокси-этокси)]-карбаминовой кислоты, полученной так, как это описано в примере 2.
(выход 98%)
1Н-ЯМР (200 МГц, CDCl3) δ(ppm): 7.36-7.28 (m, 5H, Аr); 5.16 (уширенный сигнал, 1Н, NH); 5.08 (s, 2Н, СООСН2); 4.34-4.30 (m, 2H, SO3CH2); 3.72-3.67 (m, 2H, SО3-CH2-*CH2);
3.60-3.34 (m, 4H, N-*CH2-*CH2); 2.98 (s, 3Н, SО3СН3);
этиловый___эфир___2-[2-бензил-бензилоксикарбониламино)-этил]-бензилоксикарбониламино]-метансульфоновой кислоты
исходя из бензилового эфира [2-(бензил-бензилоксикарбониламино)-этил]-(2-гидроксиэтил)-карбаминовой кислоты, полученного так, как это описано в примере 2.
(выход 72%)
1Н-ЯМР (200 МГц, СDСl3) δ(ppm): 7.40-7.00 (m, 15Н. 3Аr); 5.13-5.01 (уширенный сигнал, 4H, 2СООСН2); 4.51-3.30 (уширенный-m, 10Н, 4CH2N и CH2SO3); 2.92-2.76 (уширенный сигнал, 3Н, СН3);
этиловый_________эфир_________2-[бензилоксикарбонил-(2-бензилоксикарбониламиноэтил)-амино]-метаносульфоновой кислоты
исходя из бензилового эфира (2-бензилоксикарбониламиноэтил)-(2-гидроксиэтил)-карбаминовой кислоты, полученного так, как это описано в примере 2.
(выход 100%)
1Н-ЯМР (200 МГц, СDCl3) δ(ppm): 7.35-7.27 (m, 10Н. 2Аr); 5.10 и 5.04 (2s, 4H, 2СООСН2); 4.41-4.15 (m, 2H. *CH2-MeSO2); 3.63-3.23 (m, 6H, N-*CH2-*CH2-N-*CH2); 2.90 (уширенный-s, 3Н, MeSO2).
Пример 4
Получение N-оксида оксима эритромицина А
Раствор Н2O2 (72,00 г; титр 34% вес/объем; 0,72 моль) в воде (780 мл) по каплям в течение 1 ч добавляли к раствору оксима эритромицина А (35,00 г; 0,0467 моль) в метаноле (1400 мл) при механическом перемешивании, сохраняя температуру в 20-25°С. По окончании этого процесса добавления полученную реакционную смесь выдерживали при перемешивании в течение 24 ч при комнатной температуре. После дополнительного добавления Н2O2 (8 мл) полученную смесь выдерживали при перемешивании еще в течение 6 ч.
Метанол выпарили в вакууме при температуре приблизительно в 40°С, поддерживая при этом постоянным объем воды (примерно 700 мл).
После фильтрования, промывки водой и высушивания был получен N-оксид оксима эритромицина А в виде белого кристаллического твердого вещества (36,3 г; выход 99%).
1Н-ЯМР (200 МГц, ДМСО-d6) δ(ppm): 10.71-10.19 (уширенный сигнал, 2Н, сдвиг Н); 5.14-5.08 (m, 1Н, Н-13); 4.72 (d, 1H, Jнн=4,4 Гц, Н-1"); 4.45 (d, 1H, Jнн=7,0 Гц, Н-1').
Пример 5
Получение оксима 3'-де(диметиламино)-3',4'-дегидроэритромицина А (соединение 1)
Раствор N-оксида оксима эритромицина А (30,00 г; 38,3 ммоль), полученного так, как это описано в примере 4, в диметилформамиде (235 мл) нагревали при 150°С на предварительно подогретой масляной бане (175-180°С), а затем оставили при этой температуре и при механическом перемешивании на 15-20 мин. После охлаждения и выпаривания диметилформамида масляный остаток собрали с деминерализованной водой, нагрели и охладили. Отфильтрованное твердое вещество растирали в порошок под вакуумом при 40-45°С, получив при этом сырой продукт (25,5 г).
Этот сырой продукт вначале перекристаллизовывали из ацетонитрила (110 мл), отфильтровали, промыли водой под вакуумом при 50°С, получив при этом кристаллический продукт (20 г), которой снова перекристаллизовывали из смеси метанол/вода=65/35 (400 мл), отфильтровали и высушили под вакуумом при 40-50°С. Таким образом получили соединение 1 в виде кристаллического продукта (10,3 г; выход 38,2%).
Из кристаллизационного раствора потом извлекли дополнительное количество продукта (3,7 г), что дало общий выход, составляющий 51,8%.
1Н-ЯМР (200 МГц, CDCl3) δ(ppm): 5.67-5.55 (m. 2H, *СН=*СН); 4.44 (d, 1Н, JHH=7,0 Гц, Н-1’); 4.33-4.22 (m, 1Н, Н-5’); 4.13-4.04 (m, 1H, H-2’); 3.84-3.73 (m, 1H, Н-8); 3.69 (s, lH, H-11).
13С-ЯМР (200 МГц, CDCl3) δ(ppm): 171.11 (s, C-9); 132.2 и 126.1 (2s, C-3’ и С-4’).
Пример 6
Получение оксима 3’-де(диметиламино)-эритромицина А (соединение 2)
Оксид платины (0,615 г) добавляли при комнатной температуре к раствору соединения 1 (20,00 г; 28,4 ммоль), полученного так, как это описано в примере 5, в этаноле (850 мл) (полное растворение было получено после слабого нагревания).
Полученную смесь гидрировали в аппарате Парра (1,36 атм), и поглощение происходило без промежуточных стадий.
После того как отфильтровали катализатор и в вакууме удалили растворитель, полученный белый кристаллический остаток растирали в порошок с петролатумом. Потом его отфильтровали и высушили под вакуумом при 50°С, получив при этом соединение 2 (19,8 г; выход 99%).
1Н-ЯМР (200 МГц, CDCl3) δ(ppm): 5.05-4.98 (m. 1H, Н-13); 4.94 (d, 1Н, JHH=4,4 Гц, Н-1’’); 4.23 (d, 1Н, JHН=7,4 Гц, H-1’); 3.44-3.30 (m, 1H, H-2’); 2.07-1.21 (m, 2H, Н-3’); 1.65-1.45 (m, 2H, H-4’).
13С-ЯМР (200 МГц, CDCl3) δ(ppm): 171.2 (s, C-9); 104.7 (s, C-1'); 31.8(s, C-3’); 29.5 (s, С-4’).
Пример 7
Получение Е-9-[О-[2-[бензилоксикарбонил-(6-бензилоксикарбониламиногексил)-амино]-этил]-оксима 3’-де(диметиламино)-эритромицина А (соединение 3).
Соединение 2 (12,51 г; 17,73 ммоль), полученное так, как это описано в примере 6, добавили к 95% раствору трет-бутилата кальция (2,45 г; 19,48 ммоль) в безводном тетрагидрофуране (120 мл) при перемешивании в среде азота, поддерживая при этом температуру приблизительно в 25°С. Полученную реакционную смесь выдерживали при перемешивании в течение 30 мин при комнатной температуре. Затем добавили 18-краун-6 эфир (4,69 г; 17,73 ммоль) и раствор этилового эфира 2-[бензилоксикарбонил-(6-бензилоксикарбониламиногексил)-амино]-метансульфоновой кислоты (8,98 г; 17,73 ммоль), полученного так, как это описано в примере 3, в безводном тетрагидрофуране (60 мл); при этом перемешивание продолжали при комнатной температуре в течение ночи.
После выпаривания в вакууме растворителя остаток соединили со смесью этилацетата и соляного раствора (20% NaCl) и полученные фазы разделили. Водную фазу еще раз экстрагировали этилацетатом. Собранные и высушенные органические экстракты сгустили в вакууме, получив при этом сырой продукт (23,4 г).
При хроматографической очистке (элюант СН2Сl2:СН3ОН=97:3) было получено слегка загрязненное соединение 3 (15,42 г), которое использовали без последующей очистки.
1Н-ЯМР (200 МГц, СDCl3) δ (ppm): 7.35-7.28 (m, 10Н, Аr); 5.14-5.06 (m, 1H, H-13); 5.11 и 5.07 (2s, 4H, 2СООСН2); 4.84 (d, 1H, JHH=4,4 Гц, Н-1’’); 4.28 (d, 1H, JHH=7.4 Гц, H-l’).
Действуя аналогичным образом, получили следующие соединения:
(Е)-9-[O-[2-[2-бензилоксикарбониламино)-этокси]-этил]-оксим____3'-де(диметиламино)-эритромицина А (соединение 4), исходя из этилового эфира 2-[2-(бензилоксикарбониламино)-этокси]-метансульфоновой кислоты.
(выход 62%)
1Н-ЯМР (200 МГц, CDCl3) δ(ppm): 7.34-7.21 (m, 5H, Аr); 6.09 (уширенный сигнал, 1Н, NH); 5.13-5.05 (m, 1H, Н-13); 5.06 (s, 2H, СООСН2); 4.79 (d, 1H, JHH=4,4 Гц, Н-1’’); 4.26 (d, 1H, JHH=7,4 Гц, Н-1’);
(Е)-9-[O-[2-метоксиэтокси1-метил]-оксим_____3’-де(диметиламино)-эритромицина А (соединение 5), исходя из метоксиэтоксиметилхлорида
(выход 32,5%)
1Н-ЯМР (200 МГц, СDСl3) δ(ppm): 5.21-5.12 (m, 2H, О-CH2-О); 5.12-5.05 (m, 1H, Н-13); 4.85 (d, 1H, JHH=5,4 Гц, Н-1’’); 4.39 (d. 1H, JHH=7,5 Гц, Н-1’); 3.40 (s. 3Н, СН2-O-*СН3); 3.27 (s, 3Н, Н-3”).
13С-ЯМР (200 МГц, CDCl3) δ(ppm): 172.5 (s, С-9); 97.44 (s. NO-C-0); 32.12 (s,C-31); 29.84 (s, C-41);
(Е)-9-[O-[2-[[2-(бензил-бензилоксикарбонил-амино)-этил]-бензилоксикарбониламино]-этил]-оксим 3’-де(диметиламино-эритромицина А (соединение 12), исходя из этилового эфира 2-[[2-(бензил-бензилоксикарбониламино)-этил]-бензилоксикарбониламино]-метансульфоновой кислоты
1Н-ЯМР (200 МГц, CDCl3) δ(ppm): 7.40-7.16 (уширенный-m, 15Н, 3Ph); 5.17-5.00 (m. 5H, 2*CH2-Ph и Н-13).
(Е)-9-[О-[2-[[2-(бензилоксикарбониламино)-этил-бензилоксикарбонил-амино]-этил]-оксим 3’-де(диметиламино)-эритромицина А (соединение 13), исходя из этилового эфира 2-[бензилоксикарбонил-(2-бензилоксикарбониламиноэтил)-амино]-метансульфоновой кислоты
(выход 42%)
1Н-ЯМР (200 МГц, CDCl3) δ(ppm): 7.38-7.25 (m, 10H, 2Ph); 5.11 и 5.05 (2s, 4H, 2СООСН2); 5.14-5.00 (m, 1H, Н-13); 4.89-4.79 (уширенный-m, 1H, H1’’); 2.26 (d, 1Н, JHH=7,4 Гц, Н1’).
Пример 8
Поучение (Е)-9-[O-[2-[(6-аминогексил)-амино]-этил]-оксима 3’-де(диметиламино)-эритромицина А (соединение 6)
10% Pd/C (1,6 г) добавили к раствору соединения 3 (15.42 г; 13,8 ммоль), полученного так, как это описано в примере 7, в этаноле (160 мл).
Полученную смесь гидрировали в аппарате Парра (1,02 атм). Спустя 2 ч катализатор отфильтровали, а растворитель выпарили.
Остаток очищали тонкослойной хроматографией (элюант СН2Сl2:СН3ОН:NН3=85:15:1,5; затем 80:20:2), получив в результате соединение 5 (8,48 г) в виде аморфного твердого вещества белого цвета.
1Н-ЯМР (200 МГц, СОСl3) δ(ppm): 5.07-5.00 (m, 1H, H-13); 4.79 (d, 1H, JHH=4,4 Гц, Н-1’’); 4.21 (d, 1H. JHH=7,4 Гц, Н-1’).
13С-ЯМР (200 МГц, CDCl3) δ(ppm): 171.8 (s, C-9); 71.6 (s,=N-O-C); 49.43 и 49.0 (2s,=N-O-C-*C-N-*C); 41.1 (s, С-NH2)
Действуя аналогичным образом, получили следующие соединения:
(Е)-9-[О-[2-(2-аминоэтокси)-этил]-оксим______3’-де(диметиламино)-эритромицина А (соединение 7), исходя из соединения 4
(выход 85%)
1Н-ЯМР (200 МГц, СDСl3) δ(ppm): 5.12-5.05 (m, 1H, H-13); 4.83 (d, 1H, JHH=4,4 Гц, H-l’’); 4.26 (d, 1H,JHH=7,5 Гц, Н-1’).
(Е)-9-[O-[2-[(2-бензиламиноэтил)-амино]-этил]-оксим 3’-де(диметиламино)-эритромицина А (соединение 14), исходя из соединения 12
(выход 48%)
1H-ЯМР (200 МГц, СОСl3) δ(ppm): 7.32-7.17 (m, 5Н, Ph); 5.08-5.00 (m, 1H. H-13); 4.74 (d, 1H, JHH=4,6 Гц, Н-1’’); 4.22 (d, 1H, JHH=7,4 Гц, Н-1’); АВ система: Va=3.80, Vb=3.76, Jab=13,7 Гц, *CH2Ph.
13С-ЯМР (200 МГц, CDCl3) δ(ppm): 171.3 (s, C-9); 105.0 (s, С-1’); 32.2 (s, C-3’); 29.8 (s, C-4’).
(Е)-9-[О-[2-[(2-аминоэтил)-амино]-этил]-оксим 3’-де(диметиламино)-эритромицина А (соединение 15), исходя из соединения 13
(выход 70%)
1Н-ЯМР (200 МГц, CDCl3) δ(ppm): 5.08-5.00 (m, 1H, H-13); 4.76 (d, 1H, JHH=4,6 Гц, Н-1’’); 4.23 (d, 1H, JHH=7,4 Гц, Н-1’).
Пример 9
Получение (Е)-9[О-[2-[6-[(тиазол-2-ил-метил)-амино]-гексиламино)-этил]-оксима 3’-де(диметиламино)-эритромицина А (соединение 8)
Суспензию соединения 6 (3 г; 3,53 ммоль), полученного так, как это описано в примере 8, а также 97% 2-тиазолкарбальдегида (0,412 г; 3,53 ммоль) и молекулярных сит (3 , 6,75 г) в этаноле (60 мл) выдерживали при перемешивании в течение 3 ч.
, 6,75 г) в этаноле (60 мл) выдерживали при перемешивании в течение 3 ч.
После того как молекулярные сита отфильтровали на целите, добавили 10% Pd/C (0,3 г) и полученную смесь гидрировали в гидрогенизаторе Парра (1,02 атм). Спустя 20 ч катализатор отфильтровали, а растворитель выпарили.
Хроматографической очисткой остатка (элюант СНС13:петролатум:триэтиламин=90:10:10) в виде аморфного твердого вещества было получено соединение 8 (1,66 г; выход 49,8%).
1Н-ЯМР (200 МГц, CDCl3) δ(ppm): 7.66 (d, 1H, JHH=3,2 Гц, CHN); 7.22 (d, 1H, CHS); 5.08-5.02 (m, 1H, H-13); 4.78 (d, 1H, JHH=4,4 Гц, Н-1’’); 4.21 (d, 1H, JHH=7.4 Гц, Н-1’); 4.07 (s, 2H, *CH2-thiaz).
13С-ЯМР (200 МГц, CDCl3) δ(ppm): 172.0 (s, S-C=N); 171.7 (s, C-8); 142.4 (s, CHN); 118.7 (s, CHS); 104.9 (s, С-1’’); 96.3 (s, С-1’’); 71.7 (s, N-O-C).
Действуя аналогичным способом, были получены следующие соединения:
(Е)-9-[O-[2-[6-(бензиламино)-гексиламино]-этил]-оксим 3’-де(диметиламино)-эритромицина А (соединение 9), исходя из соединения 6 и бензальдегида.
1Н-ЯМР (200 МГц, CDCl3) δ(ppm): 7.27-7.17 (m, 5H, Аr); 5.07-5.01 (m, 1H, H-13); 4.75 (d, 1H, Jнн=4,4 Гц, Н-1’’); 4.19 (d, 1H, Jнн=7.4 Гц, Н-1’); 3.73 (s, 2H, *CH2-Ph).
13С-ЯМР (200 МГц, СDCl3) δ(ppm): 171.8 (s, C-9); 104.9 (s, С-1’); 96.3 (s, С-1”); 71.8 (s,=N-O-C); 53.8 (s, N-*C-Ph); 49.7, 49.2 и 49.1 (3s, 3N-C).
(Е)-9-[О-[2-[2-[(тиазол-2-ил-метил)-амино]-этокси]-этил]-оксим 3’-де(диметиламино)-эритромицина А (соединение 10), исходя из соединения 7 и 2-тиазолкарбальдегида.
1Н-ЯМР (200 МГц, CDCl3) δ(ppm): 7.64 (d, 1H, Jнн=3.2 Гц, CHN); 7.19 (d. 1H, CHS); 5.10-5.02 (m, 1H, H-13); 4.78 (d, 1H, Jнн=4.4 Гц, Н-1’’); 4.21 (d, 1H. JHH=7,4 Гц, Н-1’); 4.13 (s, 2H,*CH2-thiaz).
13С-ЯМР (200 МГц, СDСl3) δ(ppm): 172.7 (s, SC=N); 171.5 (s, C-9); 142.4 (s. CHN’); 118.6 (s, CHS); 104.7 (s, С-1’); 96.4 (s, С-1’’); 50.7 (s, N-*C-thiaz).
(Е)-9-[О-[2-[2-(бензиламино)-этокси]-этил]-оксим 3'-де(диметиламино)-эритромицина А (соединение 11), исходя из соединения 7 и бензальдегида.
1Н-ЯМР (200 МГц, CDCl3) δ(ppm): 7.33-7.10 (m, 5H, AT); 5.12-5.04 (m, 1H, H-13); 4.78 (d, 1H, JНН=4,4 Гц, Н-Н; 4.72 (d, 1H, JHH=7,4 Гц, Н-1'); 3.80 (s, 2H, NCH2).
13С-ЯМР (200 МГц, CDCl3) δ(ppm): 171.5 (s, C-9); 104.8 (s, С-1'); 96.5 (s, C-1"); 69.4, 70.8 и 72.4 (3s, 3ОСH2); 53.6 (s, *C-Ph); 48.2 (s, O-C-*C-N).
(E)-9-[О-[2-[2-[(тиазол-2-ил-метил)-амино]-этиламино]-этил]-оксим 3'-де(диметиламино)-эритромицина А (соединение 16), исходя из соединения 15 и 2-тиазолкарбальдегида.
1Н-ЯМР (200 МГц, CDCl3) δ(ppm): 7.62 (d, 1H, JHH=3,0 Гц, N-*CH=CH); 7.18 (d, 1H, S-*CH=CH); 4.18 (d, 1H, JHH=7,4 Гц, Н1').
13С-ЯМР (200 МГц, CDCl3) δ(ppm): 172.6 (s, SC=N); 171.3 (s, C-9); 142.4 (s, CHN); 118.6 (s, CHS); 104.9 (s, С-1'); 96.4 (s, C-1"); 32.17 (s, C-3'); 29.8 (s, C-4').
(E)-9-[O-[2-[6-[(2-фенил-1Н-имидазол-4-ил-метил)-амино]-гексиламино]-этил]-оксим 3'-де(диметиламино)-эритромицина А (соединение 17), исходя из соединения 6 и 2-фенил-1H-имидазол-4-карбальдегида.
1Н-ЯМР (200 МГц, CDCl3) δ(ppm): 7.86-7.21 (m, 5H, Ar); 6.91 (s, 1H, CH-Imid.); 5.11-5.02 (m, 1H, H13); 4.76 (d, 1H, Jнн=4,2 Гц, H1"); 4.21 (d, 1H, JHH=7,4 Гц, Н1'); 3.76 (s, 2H, *CH2-Imid); 3.23 (s, 3H, ОМе).
13С-ЯМР (200 МГц, CDCl3) δ(ppm): 171.7 (s, C-9); 104.8 (s, С-1'); 96.3 (s, C-1"); 32.1 (s, C-3'); 29.9 (s, C-4').
(Е)-9-[O-[2-[6-[(1-метил-2-фенил-1Н-имидазол-4-ил-метил)-амино]-гексиламино]-этил]-оксим 3’-де(диметиламино)-эритромицина А (соединение 18), исходя из соединения 6 и 1-метил-2-фенил-1Н-имидазол-4-карбальдегида.
1Н-ЯМР (200 МГц, СОСl3) δ(ррm): 7.57-7.31 (m, 5Н, Аr); 6.87 (s, 1H, CH-Imid); 5.09-5.00 (m, 1H, Н13); 4.76 (d, 1H, JHH=4,2 Гц, H1”); 4.20 (d, 1H, JHH=7,4 Гц, H1’); 3.71 (s, 2H,*CH2-Imid); 3.62 (s, 3H, NMe); 3.21 (s, 3Н, ОМе).
13С-ЯМР (200 МГц, СОСl3) δ(ррm): 171.8 (s, C-9); 104.9 (s. С-1’); 96.3 (s, С-1”); 32.1 (s, С-31); 29.7 (s, С-4’).
Пример 10
Получение (Е)-9-[О-[2-[2-[(тиазол-2-ил-метил)-(мегиламино)-этокси]-этил]-оксима 3’-де(диметиламино)-эритромицина А (соединение 19)
Соединение 10 (0,23 г; 0,258 ммоль), формальдегид (37% вес/объем; 42 мкл; 0,516 ммоль), 10% Pd на древесном угле (25 мг) и смесь этанола и воды при их соотношении 4:1 (10 мл) загрузили в аппарат Парра при 1,02 атм. Спустя 2 ч и 3 ч добавляли дополнительное количество формальдегида (42 мкл + 21 мкл). По окончании реакции (занявшей в общей сложности 6 ч) катализатор отфильтровали, а растворитель выпарили. Получившийся в результате остаток очищали тонкослойной хроматографией (элюант СН2Сl2:СН3ОН=95:5), получив при этом соединение 19 в виде аморфного твердого вещества.
1Н-ЯМР (200 МГц, СОСl3) δ(ррm): 7.64 (d, 1H, JHH=3,6 Гц, N-*CH=CH); 7.21 (d, 1H, S-*CH=CH); 5.10-5.00 (m, 1H, Н13); 4.80 (d, 1H, JHH=4,6 На, H1”); 4.23 (d, 1H, JHH=7,4 Гц, Н1’); 3.94 (s, 2H,*CH2-Thiaz.).
13С-ЯМР (200 МГц, CDCl3) δ(ррm): 172.0 (s, SC=N); 171.8 (s, C-9); 142.2 (s, CHN); 119.3 (s, CHS); 104.9 (s, С-1’); 96.4 (s, С-1’’); 32.2 (s, C-3’); 29.9 (s, C-4’).
Пример 11
Фармакологическая активность in vitro
А) Высвобождение интерлейкин 8
Иммортализованная эндотелиальная клеточная линия человека (ECV304) была получена от Американской коллекции тканевых культур (Rockville, Md). Ее выращивали в среде (Medium 199, mod. Earie's salts) (GIBCO, Life Technologies, Grand Island, N.Y.) с добавками 20% фетальной телячьей сыворотки (GIBCO), пенициллина в количестве 100 единиц/мл и стрептомицина (SIGMA, St. Louis, МО) в количестве 100  г/мл; процесс проводили во влажной атмосфере, содержащей 5% СО2 при 37°С.
г/мл; процесс проводили во влажной атмосфере, содержащей 5% СО2 при 37°С.
Клетки культивировали на пластинах с 96 ячейками до получения конфлюентного монослоя. Соединения, оценку которых следовало произвести, растворили в диметилсульфоксиде (ДМСО) при концентрации 10-2 М и разбавили культуральной средой.
Эти соединения перед проведением провокационной пробы были предварительно инкубированы в течение 1 ч с указанными клетками.
Высвобождение интерлейкин 8 вызвали при добавлении 0,66  г/мл липополисахарида В (Е. coli 055:B5, Difco, Detroit, Mi) до конечного объема в 200
г/мл липополисахарида В (Е. coli 055:B5, Difco, Detroit, Mi) до конечного объема в 200  л.
л.
Спустя ночь получившийся супернатант был собран для проведения теста в отношении интерлейкин 8.
Специфическую иммунореактивность указанного культурального супернатанта в отношении интерлейкин 8 измеряли с использованием набора ELISA kit (Amersham, UK).
Результаты измерений выражены величинами достигаемого самого высокого ингибирования (эффективности), и, там, где это возможно, как концентрация, при которой достигают 50% этого эффекта (IС50).
Б) Высвобождение анионов пероксида
Нейтрофилы были выделены центрифугированием (на центрифуге Ficoll-Hypaque) венозной крови здоровых волонтеров при последующем осаждении на 6% декстране и осмотическом лизисе эритроцитов. Затем нейтрофилы промыли и повторно суспендировали в среде, составленной из RPMI-1640 с добавкой 5% фетальной телячьей сыворотки и 1,34 ммоль/л двунатриевый дигидрат этилендиаминтетрауксусной кислоты (ЭДТК). Клетки выдерживали в течение 24 ч при 4°С, а перед проведением теста суспензию центрифугировали и повторно суспендировали в сбалансированном солевом растворе Хенкса. Жизнеспособность и чистота полученного препарата нейтрофилов была проверена по окрашиванию трипановым синим и ализариновым синим.
Анионы супероксида определяли хемилюминесценцентным способом, усиленным за счет применения Lucigenin (нитрат бис-N-метилакридина).
Нейтрофилы (2×106 клеток/мл) предварительно в течение 30 мин инкубировали при 37°С в 900  л сбалансированного солевого раствора Хенкса в присутствии испытуемого соединения и в его отсутствии (система считывания).
л сбалансированного солевого раствора Хенкса в присутствии испытуемого соединения и в его отсутствии (система считывания).
Продуцирование анионов пероксида определяли с помощью биологического счетчика Lumac/ЗМ; процесс проводили после введения в систему считывания 100  л сбалансированного нитратом бис-N-метилакридина солевого растора Хенкса (2 ммоль/л) и N-формил-L-метионил-L-лейцил-L-фенилаланина в качестве стимулирующего агента при концентрации 1×10-5 М.
л сбалансированного нитратом бис-N-метилакридина солевого растора Хенкса (2 ммоль/л) и N-формил-L-метионил-L-лейцил-L-фенилаланина в качестве стимулирующего агента при концентрации 1×10-5 М.
N-формил-L-метионил-L-лейцил-L-фенилаланин растворили в ДМСО (1×10-2 М), а потом разбавили в сбалансированном солевом растворе Хенкса. Соединение, предназначенное для испытания, растворили в ДМСО при концентрации 10-2 М. Более низкие концентрации относительно ДМСО были получены из этого раствора и испытаны. Количество ДМСО, присутствующее в указанной системе считывания, было ниже 1%.
Пиковые значения яркости свечения, полученные для каждой из исследуемых концентраций соединения, преобразовали в величины процентного ингибирования по сравнению с эталонными соединениями.
Концентрации, способные ингибировать продуцирование анионов пероксида на 50% (IС50), вычисляли с помощью полученной кривой “доза-реакция”.
В табл.1 приведены результаты, полученные для некоторых соединений формулы I, являющихся представителями всего этого класса; и эти результаты сравнивают с данными для эритромицина, кларитромицина и рокситромицина.

Пример 12
Фармакологическая активность in vivo
- Животные
Использовались мужские особи крыс Sprague-Dawley, которые весили 200 300 г.
Крысы, используемые в экспериментах, не имели признаков очевидного инфицирования. Указанные животные перед их умерщвлением выдерживали в течение 7 суток в стандартных условиях.
- Введение эндотокосина
LPS (Е. coli липосахарид) (эндотоксин; тип сыворотки 055:В5; Sigma Chemical Co., St. Louis, МО) растворили в стерильном соляном растворе и ввели методом интраперитонеальной инъекции в дозировке, составляющей 6 мг/кг (1 мл/кг). Аналогично контрольным животным путем инъекции был введен соляной раствор и/или носитель (соляной раствор +0,5% Твин 20).
- Введение соединений (профилактическая обработка)
Каждое из соединений вводили методом интраперитонеальной инъекции дважды в сутки на протяжении 6 дней, а на 7 день - за 1 ч до введения LPS и 5 ч спустя после его введения. Указанные соединения суспендировали в соляном растворе вместе с 0,5% Твин 20.
- Бронхоальвеолярное орошение
Спустя 24 ч после инъекции LPS крыс умерщвили путем передозировки нембутала (100 мг/кг, интраперитонеальная инъекция). Провели катетеризацию трахеи, а язык промыли инсталляцией при 37°С двух 5 мл аликвот фосфатно-солевого буфера и эту жидкость немедленно удалили. Жидкость снова ввели инъекционным способом и для каждой аликвоты всю процедуру повторили трижды.
- Подсчет клеток и их дифференциация
200 мкл жидкости бронхоальвеолярного орошения (BALF) развели 1 мл холодной воды и 19 мл Isoton. Общее количество клеток подсчитывали дважды при использовании Contraves autolyser 800. Если общее их количество было меньше 2000, то жидкости бронхоальвеолярного орошения центрифугировали в течение 10 мин при скорости вращения 800 об/мин для отделения клеток от супернатанта. Полученные супернатанты слили, а клетки повторно суспендировали в небольшом количестве фосфатно-солевого буфера. Для проведения цитологического исследования 50 мкл полученного раствора и прошедшие повторное суспендирование клетки центрифугировали на Shandon Cytospin центрифуге в течение 1 мин при скорости вращения 1300 об/мин. Предметные стекла были зафиксированы в ацетоне и высушены с помощью DiffQuick. Дифференциальный подсчет клеток проводили на каждом предметном стекле путем подсчета выбранных наугад 200 клеток; согласно стандартным морфологическим критериям клетки были классифицированы как нейтрофилы, эозинофилы и мононуклеарные клетки.
В табл.2 изложены результаты, полученные для некоторых соединений формулы I, являющихся представителями всего класса в целом.

| название | год | авторы | номер документа |
|---|---|---|---|
| БИЦИКЛИЧЕСКИЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СПИРОСОЕДИНЕНИЯ | 2010 |
|
RU2506266C2 |
| ПРОИЗВОДНЫЕ ПИРРОЛИДИНА В КАЧЕСТВЕ АГОНИСТОВ PPAR | 2017 |
|
RU2711991C1 |
| ТИЕНОДИАЗЕПИНОВЫЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ | 2018 |
|
RU2795005C2 |
| СИНТЕЗ ЛАКТОНОВ РЕЗОРЦИЛОВОЙ КИСЛОТЫ, ИСПОЛЬЗУЕМЫХ В КАЧЕСТВЕ ТЕРАПЕВТИЧЕСКИХ АГЕНТОВ | 2009 |
|
RU2534527C2 |
| ЛИГАНДЫ ДЛЯ АГРЕГИРОВАННЫХ МОЛЕКУЛ ТАУ-БЕЛКА | 2009 |
|
RU2518892C2 |
| ИНДАЗОЛЬНЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ FGFR, ИХ ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ | 2015 |
|
RU2719428C2 |
| Азопроизводные аминофенолов, обладающие способностью ингибировать образование конечных продуктов гликирования | 2024 |
|
RU2839138C1 |
| ХИНОЛИНОВЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ SMO | 2015 |
|
RU2695815C2 |
| 2-ЦИАНОПИРИМИДИН-4-ИЛКАРБАМАТ, ИЛИ ПРОИЗВОДНОЕ МОЧЕВИНЫ, ИЛИ ЕГО СОЛЬ И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2019 |
|
RU2788740C2 |
| ПЕСТИЦИДНЫЕ КОМПОЗИЦИИ И СВЯЗАННЫЕ С НИМИ СПОСОБЫ | 2012 |
|
RU2614976C2 |
Изобретение относится к 3’-Дездиметиламино-9-оксиимино макролидам формулы (I): 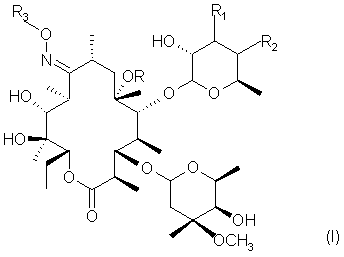
в которой R представляет собой водород или метил; R1 и R2 оба представляют собой водород или вместе образуют химическую связь; R3 представляет собой водород или линейную, или разветвленную C1-C5 алильную группу, или цепь формулы

где А представляет собой водород или фенильную группу, или 5 или 6-членный гетероцикл, насыщенный или ненасыщенный и содержащий от 1 до 3 гетероатомов, выбранных из азота, кислорода и серы, необязательно замещенный одним или двумя заместителями, выбранными из C1-C5 алкильных групп или фенильных групп, Х и Y, одинаковые или различные, представляют собой О или NR4, где R4 – водород, линейная или разветвленная C1-C5 алкильная группа, бензилоксикарбонильная группа; r - целое число от 1 до 6; m - целое число от 1 до 8; n - целое число от 0 до 2; и их фармацевтически приемлемые соли; за исключением соединений оксима 3’-дездиметиламино-3’,4’-дегидроэритромицина А и 9-O-метилоксим 3’-дездиметиламино-3’4’-дегидроэритромицина А. Изобретение также относится к фармацевтической композиции на основе соединений формулы 1, обладающей противовоспалительным действием, и применению оксима 3’-дездиметиламино-3’,4’-дегидроэритромицина А в качестве противовоспалительного средства. Технический результат – получение противовоспалительных средств. 3 н. и 3 з.п. ф-лы, 2 табл.

в которой R представляет собой водород или метил;
R1 и R2 оба представляют собой водород, или они вместе образуют химическую связь;
R3 представляет собой водород, линейную или разветвленную C1-C5 алильную группу, или цепь формулы

где А представляет собой водород или фенильную группу, или 5 или 6-членный гетероцикл, насыщенный или ненасыщенный, содержащий от 1 до 3 гетероатомов, выбранных из азота, кислорода и серы, необязательно замещенный одним или двумя заместителями, выбранными из C1-C5 алкильных групп или фенильных групп;
Х и Y, одинаковые или различные, представляют собой О, или NR4, где R4 – водород, линейная или разветвленная C1-C5 алкильная группа, бензилоксикарбонильная группа;
r – целое число от 1 до 6;
m – целое число от 1 до 8;
n – целое число от 0 до 2;
а также их фармацевтически приемлемые соли; при этом исключаются соединения: оксим 3’-дездиметиламино-3’,4’-дегидроэритромицина А и 9-O-метилоксим 3’-дездиметиламино-3’4’-дегидроэритромицина А.
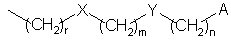
в которой X, Y, А, r, m и n имеют значения, указанные в п.1.

в которой r равно 2;
m равно 2 или 6;
n равно 1;
Y представляет собой NR4;
Х представляет собой О или NR4;
R4 представляет собой водород;
А представляет собой фенил или тиазолил.
| ПАРОЭЖЕКТОРНАЯ ФРЕОНОВАЯ ХОЛОДИЛЬНАЯ МАШИНА | 0 |
|
SU254534A1 |
| US 3928387 A, 23.12.1975 | |||
| Сигнальное устройство к дисковой резательной машине для изготовления вельвета | 1951 |
|
SU96013A1 |
| ПРОИЗВОДНЫЕ ЭРИТРОМИЦИНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ЭРИТРОМИЦИНА | 1993 |
|
RU2114859C1 |
| Lemahien Ronald A | |||
| et al., J | |||
| Med | |||
| Chem, 1974, 17(9), p | |||
| Инжектор отработанного пара для паровозов | 1924 |
|
SU953A1 |

