Область изобретения
Изобретение относится к бициклическим и трициклическим нуклеозидным аналогам и синтезу таких нуклеозидных аналогов, которые применимы для получения синтетических олигонуклеотидов, способных образовывать специфичные по контексту дуплексы и триплексы с одноцепочечными и двухцепочечными нуклеиновыми кислотами. Такие комплексы проявляют более высокую термостабильность по сравнению с соответствующими комплексами, образуемыми обычными нуклеиновыми кислотами. Настоящее изобретение также относится к бициклическим и трициклическим нуклеозидным аналогам и синтезу таких нуклеозидов, которые бы могли быть использованы в качестве лекарственных средств и которые могли бы быть встроены в состав олигонуклеотидов с применением зависимых от матриц полимераз нуклеиновых кислот.
Предпосылки изобретения
Синтетические олигонуклеотиды являются соединениями, широко применяемыми в самых различных областях, таких как молекулярная биология и основанная на применении ДНК диагностика и терапия.
Лекарственные средства
В лекарственных средствах, например, успешно применяют олигонуклеотиды с целью блокирования трансляции in vivo конкретных мРНК, тем самым предотвращая синтез белков, которые являются нежелательными или вредными для клетки или организма. Данный подход опосредованного олигонуклеотидами блокирования трансляции известен как “антисмысловой метод”. С точки зрения кинетики процесса гибридизующийся олигонуклеотид, как считается, проявляет свой эффект либо путем индукции физического барьера на пути трансляции, либо путем активации клеточных ферментов, которые направленным образом разрушают мРНК-компонент дуплекса (таких как РНКаза-Н).
Недавно олигорибонуклеотиды и олигодезоксирибонуклеотиды, и их аналоги, которые сочетают РНКазную каталитическую активность со способностью к контекстно специфичному взаимодействию с комплементарной РНК-мишенью, (рибозимы) привлекли большое внимание в качестве антисмысловых зондов. Соответственно, для рибозимов была продемонстрирована эффективность и против вирусных мишеней, и против онкогенов при моделировании на культурах клеток.
Для полного подавления синтеза конкретного белка с помощью “антисмыслового метода” необходимым является блокирование/разрушение всех мРНК, которые кодируют такой белок, а во многих случаях количество таких мРНК очень велико. Обычно мРНК, которые кодируют конкретный белок, транскрибируются с одного или с нескольких генов. Следовательно, путем маркирования такого гена (“антигенный метод”), а не образующегося с него мРНК-продукта, будет возможным либо блокировать выработку кодируемого им белка с более высокой эффективностью, либо достичь существенного снижения количества олигонуклеотидов, необходимого для достижения желаемого эффекта. Для блокирования транскрипции олигонуклеотид должен быть способным гибридизоваться с двухцепочечной ДНК, проявляя специфичность в отношении контекста нуклеотидной последовательности. В 1953 году Уотсон и Крик показали, что дезоксирибонуклеиновая кислота (ДНК) состоит из двух цепей (Watson & Crick, 1953, Nature, 171, 737), которые удерживаются вместе в спиральной кон-формации за счет водородных связей, образуемых между расположенными напротив друг друга комплементарными основаниями двух таких цепей. Четыре основания, обычно находящиеся в составе ДНК, - это гуанин (G), аденин (А), тимин (Т) и цитозин (С): при этом основание G комплементарно С, а основание А - комплементарно Т. В РНК основание тимин заменено на основание урацил (U), который, как и Т, комплементарен основанию А. Химические группы оснований, которые участвуют в образовании стандартного дуплекса, представляют “поверхность” модели Уотсона-Крика. В 1959 г. Хугстен (Ноogseen) показал, что пуриновые основания (G и А) в дополнение к участию их поверхности в модели Уотсона-Крика, характеризуются еще и “поверхностью Хугстена”, которая может быть распознана со внешней стороны дуплекса, и использовал их для связывания пиримидиновых олигонуклеотидов по водородным связям, формируя тем самым тройную спираль. Хотя разработка “антигенного подхода” в принципе возможна с точки зрения практического применения в формировании трехцепочечных олигомеров, в настоящее время она ограничена рядом факторов, включая потребность в наличии гомопуриновых мотивов в составе последовательности гена-мишени и необходимость создания являющихся нефизиологичными высокой ионной силы и низкого значения рН с целью стабилизации трехцепочечного комплекса.
Использование олигонуклеотидов, известных как “аптамеры”, также активно исследуется. Этот перспективный для терапевтического применения новый класс олигонуклеотидов выбирают in vitro для специфического связывания на конкретной мишени с высоким уровнем аффинности к ней, таких как, например, рецепторы лигандов. Их характеристики связывания являются, по-видимому, отражением способности олигонуклеотидов формировать трехмерные структуры, поддерживаемые внутримолекулярным взаимодействием нуклеотидов.
Также нуклеозиды и нуклеозидные аналоги, как подтверждалось, эффективны в химиотерапии разнообразных вирусных инфекций и злокачественных опухолей.
Также различные типы двухцепочечных РНК, как было показано, эффективно подавляют рост некоторых типов злокачественных опухолей.
Диагностика
В молекулярной биологии олигонуклеотиды, как правило, используют в различных целях, таких как, например, (1) применение в качестве гибридизационных зондов при поиске, идентификации и количественном анализе нуклеиновых кислот-мишеней, (2) в качестве аффинных зондов в очистке нуклеиновых кислот-мишеней, (3) в качестве затравок для реакций секвенирования и процессах амплификации мишеней, таких как полимеразная цепная реакция (ПЦР), (4) для клонирования и мутирования нуклеиновых кислот и (5) в качестве строительных блоков для сборки макромолекулярных структур.
В диагностике применяются многие из упоминавшихся выше методик, основывающиеся на использовании олигонуклеотидов, в частности, те, которые поддаются простой автоматизации и обеспечивают воспроизводимые результаты при высокой чувствительности. Предметом данной области анализа является применение основанных на олигонуклеотидах методик, например, для (1) тестирования людей, животных и пищевых продуктов на присутствие патогенных микроорганизмов, (2) тестирования генетической предрасположенности к заболеванию, (3) идентификации наследственных и приобретенных генетических патологий, (4) поиска связи между биологическими объектами и подозреваемыми в следственной практике и (5) проверки присутствия микроорганизмов, участвующих в выработке пищевых продуктов и напитков.
Основные соображения
Для того чтобы их можно было применять для решения широкого круга задач, обозначенных выше, олигонуклеотиды должны удовлетворять значительному числу различных условий. В “антисмысловой терапии”, например, применяемый олигонуклеотид должен быть способен проникать внутрь клетки через ее мембрану, обладать существенной резистентностью к действию вне- и внутриклеточных ферментов и предпочтительно обладать способностью рекрутировать эндогенные ферменты, такие как РНКаза-Н. В диагностике и в разделах молекулярной биологии, основанных на использовании ДНК, важными оказываются иные свойства, такие как, например, способность олигонуклеотидов действовать в качестве эффективных субстратов для широкого круга различных ферментов, ассоциированных с природными нуклеиновыми кислотами, такими как, например, полимеразы, киназы, лигазы и фосфатазы. Однако фундаментальным свойством олигонуклеотидов, которое необходимо при всех их применениях, является способность распознавать и гибридизовать специфично по контексту последовательности с комплементарными одноцепочечными нуклеиновыми кислотами с участием либо водородных связей в соответствии с моделью Уотсона-Крика (А-Т и C-G), либо других вариантов водородных связей, таких как вариант Хугстена. Имеется два важных термина - “аффинность” и “специфичность”, - которые обычно используются для охарактеризования параметров гибридизуемости конкретного олигонуклеотида. Под аффинностью понимается мера силы связывания олигонуклеотида с комплементарной ему последовательностью-мишенью (выражается в величине термостабильности дуплекса - Тm). Каждая пара оснований в дуплексе вносит вклад в величину термостабильности и таким образом аффинность возрастает при увеличении размера (количества оснований) в составе олигонуклеотида. Под специфичностью понимается мера способности данного олигонуклеотида различать полностью комплементарную и неправильную последовательность-мишень. Другими словами, специфичность - это мера потери аффинности вследствие присутствия неправильных оснований в составе мишени. При постоянном размере олигонуклеотида специфичность возрастает параллельно увеличению количества несоответствий между олигонуклеотидом и его мишенями (т.е. при возрастании доли ошибок). С другой стороны, специфичность снижается тогда, когда размер данного олигонуклеотида увеличивается при постоянном числе несоответствий (т.е. доля ошибок уменьшается). Формулируя по-другому, увеличение аффинности олигонуклеотида происходит “за счет” специфичности, и наоборот.
Это свойство олигонуклеотидов создает ряд проблем при их практическом применении. В случае с очень длинными диагностическими процедурами, например, необходимо, чтобы олигонуклеотид характеризовался и высокой аффинностью с точки зрения обеспечения адекватной чувствительности на протяжении теста, и высоким уровнем специфичности с целью предотвращения получения ложно-позитивных результатов. Также олигонуклеотид, используемый в качестве антисмыслового зонда, должен характеризоваться и высокой аффинностью по отношению к соответствующей мРНК-мишени с целью эффективного подавления ее трансляции, и высоким уровнем специфичности с целью предотвращения непреднамеренного блокирования экспрессии других белков. При ферментативных процессах, таких как, например, амплификация с помощью ПЦР, аффинность олигонуклеотидной затравки должна быть достаточно высока для того, чтобы дуплекс “затравка-мишень” был стабильным в том температурном режиме, в котором активен данный фермент, а специфичность должна быть достаточно высокой для того, чтобы быть уверенным в амплификации именно последовательности-мишени.
Принимая во внимание ограничения, характерные для использования нативных олигонуклеотидов, новые подходы к увеличению уровней специфичности и аффинности должны разрабатываться в связи с применением основывающихся на ДНК методик в терапии, диагностике и молекулярной биологии в целом.
Конформационно ограниченные нуклеозиды
Известно, что олигонуклеотиды претерпевают конформационное превращение в процессе гибридизации с последовательностью-мишенью от относительно свободной спиральной структуры, характерной для одноцепочечного состояния, к упорядоченной структуре, характерной для двухцепочечного состояния.
Ряд конформационно ограниченных олигонуклеотидов, включающих бициклические и трициклические нуклеозидные аналоги (фиг.1А и 1В, на которых В = нуклеотидное основание), были синтезированы, встроены в олигонуклеотид и олигонуклеотидные аналоги и протестированы по их гибридизуемости и другим параметрам.
Бицикло[3.3.0]нуклеозиды (бцДНК) с дополнительными С-3’,С-5’-двухуглеродными “мостиками” (А и В) были синтезированы для всех пяти оснований (G, А, Т, С и U), в то время как (С) был синтезирован только с основаниями Т и А (M.Tarkoy, M.Bolli, B.Schweizer & C.Leumann, 1993, Helv. Chim. Acta, 76, 481; M.Tarkoy & C.Leumann, 1993, Angew. Chem. Int. Ed. Engl., 32, 1432; M.Egli, P.Lubini, M.Dobler & C.Leumann, 1993, J. Amer. Chem. Soc., 115, 5855; M.Tarkoy, M.Bolli & C.Leumann, 1994, Helv. Chim. Acta, 77, 716; M.Bolli & C.Leumann, 1995, Angew. Chem. Int. Ed. Engl., 34, 694; M.Bolli, P.Lubini & C.Leumann, 1995, Helv. Chim. Acta, 78, 2077; J.C.Litten, C.Epple & C.Leumann, 1995, Bioorg. Med. Chem. Lett., 5, 1231; J.C.Litten & C.Leumann, 1996, Helv. Chim. Acta, 79, 129; M.Bolli, J.C.Litten, R.Schultz & C.Leumann, 1996, Chem. Biol., 3, 197; M.Bolli, H.U.Trafelet & C.Leumann, 1996, Nucl. Acids Res., 24, 4660). ДНК-олигонуклеотиды, включающие несколько таких аналогов или полностью состоящие из них, в большинстве случаев способны формировать дуплексы согласно модели Уотсона-Крика с комплементарными им ДНК- и РНК-олигонуклеотидами. Термостабильность образующихся дуплексов, однако, либо существенно уступает (С), либо в некоторой степени ниже (А), либо сопоставима (В) с таковой у естественных вариантов ДНК и РНК. Все бцДНК-олигомеры характеризуются отчетливо более высокой чувствительностью к ионной силе среды гибридизации по сравнению с нативными вариантами. α -бц-ДНК (В) характеризуется большей стабильностью в отношении 3’-экзонуклеазной активности фосфодиэстеразы змеиного яда по сравнению с β -бцДНК, которая лишь ненамного превосходит по такой стабильности по сравнению с немодифицированными олигонуклеотидами.
Бикарбоцикло[3.1.0]нуклеозиды с дополнительным С-1’, С-6’- или С-6’, С-4’-одноуглеродным “мостиком” в циклопентановом кольце (соответственно, D и Е) были синтезированы для всех пяти оснований (Т, A, G, С и U). Однако только аналоги Т были включены в состав олигомеров. Внесение 1-10 мономеров D в смешанный полипиримидин-ДНК-олигонуклеотид обусловливало существенное снижение аффинности в отношении и ДНК-, и РНК-олигонуклеотидов по сравнению с немодифицированным контрольным олигонуклеотидом. Такое уменьшение было более выраженным в отношении одноцепочечной ДНК по сравнению с одноцепочечной РНК. Внесение одного мономера Е в два разных полипиримидин-ДНК-олигонуклеотида приводило к некоторому увеличению температуры плавления (на 0,8° С и 2,1° С) для дуплексов в отношении оцРНК по сравнению с немодифицированными контрольными дуплексами. Когда в состав 15-мерного олигонуклеотида, характеризующегося исключительно фосфотиоатными связями между нуклеозидами, были включены 10 аналогов Т величина Тm по отношению к комплементарному РНК-олигонуклеотиду увеличивалась приблизительно на 1,3° С на каждую модификацию по сравнению с такой же немодифицированной фосфотиоатной последовательностью. В отличие от контрольной последовательности, олигонуклеотид, включающий бициклический нуклеозид Е, не обеспечивал расщепление, опосредуемое РНКазой-Н. О параметрах гибридизации олигонуклеотидов, включающих аналоги G, А, С и U по формуле Е, ранее не сообщалось. Кроме того, собственно химия этих аналогов сама по себе не послужила поводом для дальнейших исследований полностью модифицированных олигонуклеотидов (К.-H.Altmann, R.Kesselring, E.Francotte & G.Rihs, 1994, Tetrahedron Lett., 35, 2331; K.-H.Altmann, R.Imwinkelried, R.Kesselring & G.Rihs, 1994, Tetrahedron Lett., 35, 7625; V.E.Marquez, M.A.Siddiqui, A.Ezzitouni, P.Russ, J.Wang, R.W.Wagner & M.D.Matteucci, 1996, J. Med. Chem., 39, 3739; A.Ezzitouni & V.E.Marquez, 1997, J. Chem. Soc., Perkin Trans., 1, 1073).
Бицикло[3.3.0]нуклеозид, включающий дополнительное С-2’, С-3’-диоксалановое кольцо, был синтезирован в виде димера с немодифицированным нуклеозидом, в котором дополнительное кольцо является частью межнуклеозидного “мостика”, заменяющего обычную фосфодиэфирную связь (F). Этот аналог был синтезирован только в вариантах димеров “тимин-тимин” или “тимин-5-метилцитозин”. 15-мерная полипиримидиновая последовательность, включающая семь таких димерных “блоков” и характеризующаяся перемежающимися фосфодиэфирными и рибоацетальными “мостиками”, проявляла существенно сниженную величину Тm по отношению к комплементарной одноцепочечной РНК по сравнению с контрольной последовательностью, характеризующейся исключительно нативными фосфодиэфирными связями между нуклеозидами (R.J.Jones, S.Swaminathan, J.F.Millagan, S.Wad-wani, B.S.Froehler & M.Matteucci, 1993, J. Amer. Chem. Soc., 115, 9816).
Два димера (G и Н) с дополнительными С-2’, С-3’-диоксановыми кольцами, образующими бицикло[4.3.0]системы в составе межнуклеозидных “мостиков” ацетального типа, были синтезированы в виде дитиминовых димеров и включены в среднюю часть 12-мерных полипиримидиновых олигонуклеотидов. Олигонуклеотиды, включавшие либо G, либо Н, образовывали существенно менее стабильные дуплексы с комплементарными им одноцепочечными РНК и ДНК по сравнению с немодифицированным контрольным олигонуклеотидом (J.Wang & М.D.Matteucci, 1997, Bioorg. Med. Chem. Lett., 7, 229).
Димеры, включающие бицикло[3.1.0]нуклеозиды с С-2’, С-3’-одноуглеродным “мостиком”, являющимся частью межнуклеозидных “мостиков” амидного и сульфонамидного типов (I и J), были синтезированы и включены в состав олигонуклеотидов. Олигонуклеотиды, включающие один или большее число таких аналогов, показали существенное снижение Тm по сравнению с немодифицированным нативным олигонуклеотидом (C.G.Yannopoulos, W.Q.Zhou, P.Nower, D.Peoch, Y.S.Sanghvi & G.Just, 1997, Synlett., 378).
Тример, имеющий формацетальные связи между нуклеозидами и включающий в середине бицикло[3.3.0]глюкозопроизводный аналог нуклеозида (К), был синтезирован и присоединен к 3’-концу олигонуклеотида. Величина Тm в отношении комплементарной одноцепочечной РНК уменьшалась на 4° С по сравнению с контрольной последовательностью и на 1,5° С по сравнению с последовательностью, включающей две 2’,5’-формацетальные связи в составе 3’-конца (C.G.Yannopoulos, W.Q.Zhou, P.Nower, D.Peoch, Y.S.Sanghvi & G.Just, 1997, Synlett., 378).
Совсем недавно было сообщено о том, что олигомеры, включающие трициклические аналоги нуклеозидов (L), обеспечивают более высокую стабильность дуплексов по сравнению с нативной ДНК (R.Steffens & C.Leumann [Poster SB-B4], 1997, Chimia, 51, 436).
Три бициклических ([4.3.0] и [3.3.0]) нуклеозида с дополнительными, присоединенными по С-2’, С-3’ шести- (М и N) или пятиатомными (O) кольцами, были синтезированы в виде аналогов тимина. Бициклические нуклеозиды М и N были включены по одному и по два в состав 14-мерных олиго-Т последовательностей. Величина Тm по отношению к одноцепочечным РНК и ДНК уменьшалась на 6-10° С на одну модификацию по сравнению с немодифицированными контрольными последовательностями. Полностью модифицированные олигонуклеотиды аналога О характеризовались увеличением Тm примерно на 1,0° С на одну модификацию по отношению к PНК-олигонуклеотиду по сравнению с контрольным ДНК-олигонуклеотидом. Также полностью модифицированная последовательность была существенно более резистентной к гидролизу фосфодиэстеразой змеиного яда по сравнению с немодифицированной контрольной последовательностью. Частично модифицированные олигонуклеотиды, в составе которых находится до четырех аналогов типа О включительно, однако, были менее термостабильны по сравнению с соответствующими немодифицированными олигонуклеотидами. Все олигонуклеотиды, включающие аналог О (как полностью, так и частично модифицированные), характеризовались существенно сниженной термостабильностью в отношении комплементарных ДНК-олигонуклеотидов по сравнению с немодифицированными олигонуклеотидами (P.Nielsen, H.M.Pfundheller & J.Wengel, 1997, Chem. Commun., 826; P.Nielsen, H.M.Pfundheller & J.Wengel, 1996, XII Intern. Roundtable: "Nucleosides, Nucleotides and Their Biological Applications", La Jolla, CA, Sept. 15-19, [Poster PPI-43]).
Попытка получить бициклические аналоги нуклеозида уридина (Q), которые по замыслу должны были включать дополнительное C-2’, С-4’-пятичленное кольцо, начинающееся с 4’-С-гидроксиметилнуклеозида (Р), оказалась неудачной (K.D.Nielsen, 1995, Specialerapport, Odense Univ., Denmark).
До сих пор работа с конформационно ограниченными нуклеозидами, применяемыми для конструирования синтетических олигонуклеотидов для придания им улучшенных гибридизационных характеристик, имела незначительный успех. В большинстве случаев олигонуклеотиды, включающие такие аналоги, образовывают менее стабильные дуплексы с комплементарными нуклеиновыми кислотами по сравнению с немодифицированными олигонуклеотидами. В других случаях, когда наблюдается некоторое улучшение стабильности дуплекса, это относится только либо к ДНК-, либо к РНК-мишени, или же это относится к полностью, но не частично модифицированным олигонуклеотидам, и наоборот. Дальнейший анализ большинства из описанных аналогов осложняется отсутствием данных об аналогах нуклеозидов G, А и С и отсутствием данных, характеризующих специфичность и тип гибридизации. Во многих случаях синтез описанных аналогов нуклеозидов очень сложен, в то время как в других случаях синтез полностью модифицированных олигонуклеотидов несовместим с широко применяемыми стандартными методами фосфорамидитного синтеза.
Сущность изобретения
С точки зрения преодоления ограничений, свойственных известным аналогам нуклеозидов, заявители представляют новые нуклеозидные аналоги (LNA) и олигонуклеотиды, в состав которых входят нуклеозиды LNA. Новые нуклеозидные аналоги LNA представляются для всех обычных оснований, что тем самым обеспечивает полный набор нуклеозидных аналогов, необходимых для включения в олигонуклеотиды. Как будет видно из нижеследующего, нуклеозидные аналоги LNA и LNA-модифицированные олигонуклеотиды открывают возможность для широкого круга вариантов улучшения олигонуклеотидов, применяемых в диагностике и терапии. Более того, нуклеозидные аналоги LNA и модифицированные ими олигонуклеотиды также представляют совершенно новые перспективы в диагностике и терапии, основывающихся на применении нуклеозидов и олигонуклеотидов.
Таким образом, настоящее изобретение касается олигомеров, включающих по крайней мере один нуклеозидный аналог
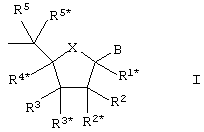
(здесь и далее обозначаемый как "LNA") основной формулы I:
где Х выбирают из -О-, -S-, -N(RN*)-, -C(R6R6*)-, -O-C(R7R7*)-, -С(R6R6*)-О-, -S-C(R7R7*)-, -C(R6R6*)-S-, -N(RN*)-C(R7R7*)-, -C(R6R6*)-N(RN*)- и –С(R6R6*)-C(R7R7*)-;
В выбирают из водорода, гидроксила, необязательно замещенной C1-4-алкоксигруппы, необязательно замещенного C1-4-алкила, необязательно замещенной C1-4-ацилоксигруппы, нуклеотидных оснований, ДНК-интеркалирующих групп, фотохимически активных групп, термохимически активных групп, хелатирующих групп, репортерных групп и лигандов;
Р обозначает положение радикала для формирования межнуклеозидного “мостика” со следующим мономером или 5’-концевую группу, при том, что такая связь или 5’-концевая группа необязательно включает заместитель R5;
один из заместителей R2, R2*, R3 и R3* является группой Р*, которая образует межнуклеозидный “мостик” с предшествующим мономером или 3’-концевую группу;
одну или две пары негеминальных заместителей (т.е. “несдвоенных” у одного углеродного атома) выбирают из имеющихся заместителей R1*, R4*, R5, R5*, R6, R6*, R7, R7*, RN*, а каждый из заместителей R2, R2*, R3 и R3*, не вовлеченных в Р*, образует бирадикал из 1-8 групп/атомов, выбираемых из –C(RaRb)-, -C(Ra)=C(Ra)-, -C(Ra)=N-, -О-, -Si(Ra)2-, -S-, -SO2-, -N(Ra) и >C=Z, где Z выбирают из -О-, -S- и –N(Ra)-, a Ra и Rb независимо друг от друга выбирают из водорода, необязательно замещенного C1-12-алкила, необязательно замещенного С2-12-алкенила, необязательно замещенного С2-12-алкинила, необязательно замещенного гидроксила, С1-12-алкоксигруппы, С2-12-алкенилоксигруппы, карбоксигруппы, С1-12-алкоксикарбонила, C1-12-алкилкарбонила, формила, арила, арилоксикарбонила, арилоксигруппы, арилкарбонила, гетероарила, гетероарилоксикарбонила, гетероарилоксигруппы, гетероарилкарбонила, аминогруппы, моно- и ди-(C1-6-амино)аминогруппы, карбамоила, моно- и ди-(C1-6-алкил)-аминокарбонила, амино-C1-6-алкил-аминокарбонила, моно- и ди-(C1-6-алкил)-амино-C1-6 -алкиламинокарбонила, C1-6-алкилкарбониламиногруппы, карбамидо, C1-6-алканоилоксигруппы, сульфоновой группы, C1-6-алкилсульфонилоксигруппы, нитрогруппы, азидогруппы, сульфанила, C1-6-алкилтиогруппы, галогена, ДНК-интеркалирующих групп, фотохимически активных групп, термохимически активных групп, хелатирующих групп, репортерных групп и лигандов, где арил и гетероарил могут быть необязательно замещенными и где два геминальных заместителя Ra и Rb вместе могут образовывать необязательно замещенный метилен (=CH2), и при том, что два геминальных или негеминальных заместителя, выбираемые из Ra, Rb и любого из заместителей R1*, R2, R2*, R3, R3*, R4*, R5, R5*, R6 и R6*, R7 и R7*, которые присутствуют и не вовлечены в Р, Р* или бирадикал (бирадикалы), вместе могут образовывать связанный бирадикал, выбираемый из бирадикалов того же типа, которые были. определены выше;
упомянутая пара (пары) негеминальных заместителей тем самым формируют моно- или бициклическую составляющую вместе с (i) атомами, к которым присоединены негеминальные заместители, и (ii) с любыми промежуточными атомами; и
каждый из заместителей R1*, R2, R2*, R3, R3*, R4*, R5, R5*, R6 и R6*, R7 и R7*, которые присутствуют и не вовлечены в Р, Р* или бирадикал (бирадикалы), независимо выбирают из водорода, необязательно замещенного C1-12-алкила, необязательно замещенного C2-12-алкенила, необязательно замещенного C2-12-алкинила, необязательно замещенного гидроксила, C1-12-алкоксигруппы, C2-12-алкенилоксигруппы, карбоксигруппы, C1-12-алкоксикарбонила, C1-12-алкилкарбонила, формила, арила, арилоксикарбонила, арилоксигруппы, арилкарбонила, гетероарила, гетероарилоксикарбонила, гетероарилоксигруппы, гетероарилкарбонила, аминогруппы, моно- и ди-(C1-6-амино)аминогруппы, карбамоила, моно- и ди-(C1-6-алкил)-аминокарбонила, амино-C1-6-алкиламинокарбонила, моно- и ди-(C1-6-алкил)-амино-С1-6-алкиламинокарбонила, С1-6-алкилкарбониламиногруппы, карбамидо, C1-6-алканоилоксигруппы, сульфоновой группы, C1-6-алкилсульфонилоксигруппы, нитрогруппы, азидогруппы, сульфанила, C1-6-алкилтиогруппы, галогена, ДНК-интеркалирующих групп, фотохимически активных групп, термохимически активных групп, хелатирующих групп, репортерных групп и лигандов, где арил и гетероарил могут быть необязательно замещенными и где два геминальных заместителя вместе могут определять оксогруппу, тиоксогруппу, иминогруппу или необязательно замещенный метилен или вместе могут формировать спиробирадикал, включающий 1-5-углеродную алкиленовую цепь, которая необязательно прерывается и(или) терминируется одним или несколькими гетероатомами/группами, выбираемыми из -О-, -S- и -(NRN)-, где RN выбирают из Н и C1-4-алкила и где два соседних (но геминальных) заместителя могут формировать дополнительную, двойную связь; и RN*, когда присутствует и не входит к бирадикал, выбирают из Н и C1-4-алкила;
и их основные соли и кислотно-аддитивные соли при условии что
(i) R2 и R3 одновременно не означают бирадикал, выбираемый из -O-CH2-CH2- и -O-СН2-СН2-СН2-, где LNA является бициклическим нуклеозидным аналогом;
(ii) R3 и R4 одновременно не означают бирадикал, выбираемый из -СН2-СН2- и -О-СН2-, где LNA является бициклическим нуклеозидным аналогом;
(iii) R3, R5 и R5* одновременно не означают трирадикал –СН2-СН(-)-СН2-, где LNA является трициклическим нуклеозидным аналогом;
(iv) R1* и R6* одновременно не означают бирадикал –СН2-, где LNA является бициклическим нуклеозидным аналогом; и
(v) R4* и R6* одновременно не означают бирадикал –CH2-, где LNA является бициклическим нуклеозидным аналогом.
Настоящее изобретение далее представляет нуклеозидные аналоги (здесь и далее LNA) общей формулы II:
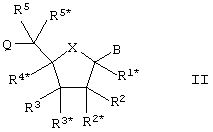
где заместитель В выбирают из нуклеотидных оснований, ДНК-интеркалирующих групп, фотохимически активных групп, термохимически активных групп, хелатирующих групп, репортерных групп и лигандов;
Х выбирают из -О-, -S-, -N(RN*)- и –C(R6R6*)-;
один из заместителей R2, R2*, R3 и R3* является группой Q*;
каждую из Q и Q* независимо выбирают из водорода, азидогруппы, галогена, цианогруппы, нитрогруппы, гидроксила, Prot-O-, Act-O-, меркаптогруппы, Prot-S-, Act-S-, C1-6-алкилтиогруппы, аминогруппы, Prot-N(RH)-, Act-N(RH)-, моно- или ди-(С1-6-алкил)аминогруппы, необязательно замещенной С1-6-алкоксигруппы, необязательно замещенного С1-6-алкила, необязательно замещенного C2-6-алкенила, необязательно замещенной C2-6-алкенилоксигруппы, необязательно замещенного C2-6-алкинила, необязательно замещенной C2-6-алкинилоксигруппы, монофосфата, дифосфата, трифосфата, ДНК-интеркалирующих групп, фотохимически активных групп, термохимически активных групп, хелатирующих групп, репортерных групп, лигандов, карбоксила, сульфоновой группы, гидроксиметила, Prot-O-CH2-, Act-O-СН2-, аминометила, Prot-N(RH)-CH2-, Act-N(RH)-CH2-, карбоксиметила, сульфонометила, где “Prot” обозначает защитную группу для -ОН, -SH и –NH(RH), ответственно, a “Act” обозначает активационную группу для -ОН, -SH и –NH(RH), ответственно, a RH выбирают из Н и C1-6-алкила;
(i) R2* и R4* вместе обозначают бирадикал, выбранный из -О-, -(CR*R*)R+S+1-, -(CR*R*)R-O-(CR*R*)S-, (CR*R*)R-S-(CR*R*)S-, -(CR*R*)R-N(R*)-(CR*R*)S-, -O-(CR*R*)R+S-O-, -S-O(CR*R*)R+S-O-, -O-(CR*R*)R+S-S-, -N(R*)-(CR*R*)R+S-O-, -O-(CR*R*)R+S-N(R*)-, -S-(CR*R*)R+S-S-, -N(R*)-(CR*R*)R+S-N(R*)-, -N(R*)-(CR*R*)R+S-S- и -S-(CR*R*)R+S-N(R*)-;
(ii) R2 и R3 одновременно означают бирадикал, выбираемый из -О-, -(CR*R*)r+s-, -(CR*R*)r-O-(CR*R*)s-, (CR*R*)r-S-(CR*R*)s- и -(CR*R*)r-N(R*)-(CR*R*)s-;
(iii) R2* и R3 вместе образуют бирадикал, выбираемый из -О-, -(CR*R*)r+s-, -(CR*R*)r-O-(CR*R*)s-, -(CR*R*)r-S-(CR*R*)s- и -(CR*R*)r-N(R*)-(CR*R*)s-;
(iv) R3 и R4* одновременно означают бирадикал, выбираемый из -(CR*R*)r-O-(CR*R*)s-, -(CR*R*)r-S-(CR*R*)s- и -(CR*R*)r-N(R*)-(CR*R*)s-;
(v) R3 и R5 одновременно означают бирадикал, выбираемый из -(CR*R*)r-O-(CR*R*)s-, -(CR*R*)r-S-(CR*R*)s- и -(CR*R*)r-N(R*)-(CR*R*)s-; или
(vi) R1* и R4* одновременно означают бирадикал, выбираемый из -(CR*R*)r-O-(CR*R*)s-, -(CR*R*)r-S-(CR*R*)s- и -(CR*R*)r-N(R*)-(CR*R*)s-;
(vii) R1* и R2* одновременно означают бирадикал, выбираемый из (CR*R*)r-O-(CR*R*)s-, (CR*R*)r-S-(CR*R*)s- и -(CR*R*)r-N(R*)-(CR*R*)s-;
при том, что каждый R* независимо друг от друга выбирают из водорода, галогена, азидогруппы, цианогруппы, нитрогруппы, гидроксила, меркаптогруппы, аминогруппы, моно- или ди-(С1-6-алкил) аминогруппы, произвольно замещенной C1-6-алкоксигруппы, произвольно замещенного C1-6-алкила, ДНК-интеркалирующих групп, фотохимически активных групп, термохимически активных групп, хелатирующих групп, репортерных групп и лигандов, а (или) два соседних (негеминальных) R* могут вместе образовывать двойную связь, а в каждом случае r и s составляют 0-3 при условии, что сумма r+s равна 1-4;
каждый из заместителей R1*, R2, R2*, R3, R4*, R5 и R5*, которые не вовлечены в Q, Q* или бирадикал, независимо выбирают из водорода, необязательно замещенного C1-12-алкила, необязательно замещенного С2-12-алкенила, необязательно замещенного С2-12-алкинила, необязательно замещенного гидроксила, С1-12-алкоксигруппы, С2-12-алкенилоксигруппы, карбоксигруппы, C1-12-алкоксикарбонила, C1-12-алкилкарбонила, формила, арила, арилоксикарбонила, арилоксигруппы, арилкарбонила, гетероарила, гетероарилоксикарбонила, гетероарилоксигруппы, гетероарилкарбонила, аминогруппы, моно- и ди-(C1-6-амино) аминогруппы, карбамоила, моно- и ди-(C1-6-алкил)-аминокарбонила, амино-С1-6-алкиламинокарбонила, моно- и ди(C1-6-алкил)-амино-C1-6-алкиламинокарбонила, C1-6-алкилкарбониламиногруппы, карбамидо, C1-6-алканоилоксигруппы, сульфоновой группы, C1-6-алкилсульфонилоксигруппы, нитрогруппы, азидогруппы, сульфанила, С1-6-алкилтиогруппы, галогена, ДНК-интеркалирующих групп, фотохимически активных групп, термохимически активных групп, хелатирующих групп, репортерных групп и лигандов, где арил и гетероарил могут быть необязательно замещенными и где два геминальных заместителя вместе могут определять оксогруппу, тиоксогруппу, иминогруппу или необязательно замещенный метилен или вместе могут формировать спиробирадикал, включающий 1-5-углеродную алкиленовую цепь, которая необязательно прерывается и(или) терминируется одним или несколькими гетероатомами/группами, выбираемыми из -О-, -S- и -(NRN)-, где RN выбирают из водорода и C1-4-алкила и где два соседних (но геминальных) заместителя могут формировать дополнительную, двойную связь; и RN*, когда присутствует и не входит к бирадикал, выбирают из водорода и C1-4-алкила; и их основные соли и кислотно-аддитивные соли при первом условии, что
(i) R2 и R3 вместе не образуют бирадикал, выбираемый из -О-СН2-СН2- и -О-СН2-СН2-СН2-, где LNA является бициклическим нуклеозидным аналогом;
(ii) R3 и R5 вместе не образуют бирадикал, выбираемый из -СН2-СН2-, -O-СН2- и -O-Si(iPr)2-O-Si(iPr)2-O-;
и при втором условии, что любая химическая группа (включая любые нуклеотидные основания), которая является реактивной в условиях, имеющих место в ходе синтеза олигонуклеотидов, необязательно должна быть защищена по своей функциональной составляющей.
Настоящее изобретение также представляет применение нуклеозидных аналогов (LNA) в конструировании олигомеров и применение таких олигомеров, равно как и нуклеозидных аналогов (LNA) в диагностике, молекулярно-биологических исследованиях и в терапии.
Краткое описание чертежей
На фигурах 1А и 1В показаны известные конформационно ограниченные нуклеотиды.
На фигуре 2 показаны нуклеотидные и нуклеозидные аналоги по настоящему изобретению.
На фигура 3 показан способ использования LNA-модифицированных олигонуклеотидов в специфичном по последовательности отборе ПЦР-ампликонов.
На фигурах 4А и 4В показано, что LNA-модифицированные олигонуклеотиды способны распознавать родственный им ПЦР-ампликон за счет инвазии цепи.
На фигуре 5 показано, что LNA-модифицированные олигонуклеотиды, иммобилизованные на твердую поверхность, активно функционируют в специфичном по последовательности отборе ПЦР-ампликона.
На фигуре 6 показано, что LNA-модифицированные олигонуклеотиды могут действовать в качестве субстратов для полинуклеотидкиназы Т4.
На фигуре 7 показано, что LNA-модифицированные олигонуклеотиды могут функционировать как затравки для полимераз нуклеиновых кислот.
На фигуре 8 показано, что LNA-модифицированные олигонуклеотиды могут функционировать как затравки в процессах амплификации мишеней.
На фигуре 9 показано, что LNA-модифицированные олигонуклеотиды, несущие 5’-антрахинон, могут быть ковалентно иммобилизованы на твердую подложку с помощью облучения и что иммобилизованный олигомер эффективен в отборе комплементарного ДНК-олигомера.
На фигуре 10 показано, что LNA-тимидин-5’-трифосфат (LNA-TTP) может действовать в качестве субстрата для терминальной дезоксинуклеотидилтрансферазы (TdT).
На фигуре 11 проиллюстрированы гибридизация и детекция мотива с помощью различных LNA-модифицированных Cγ 3-меченных октонуклеотидов.
На фигуры 12 и 13 проиллюстрированы гибридизация и детекция концевых ошибок в мотиве с помощью различных LNA-модифицированных Сγ 3-меченных октонуклеотидов.
На фигуре 14 проиллюстрировано блокирование с помощью LNA индуцированного [D-Ala2]-дельторфином обезболивания у “находящихся в сознании” крыс в тесте на раздражение теплой водой.
На фигурах 15А, 15В и 15С проиллюстрированы гибридизация и детекция концевых ошибок в мотиве с помощью AT и всех LNA-модифицированных Сγ 3-меченных октонуклеотидов.
На фигурах 16 и 17 показано, что LNA может быть внесен в живые клетки MCF-7 опухоли молочной железы человека.
На фигуры 18 и 19 показан способ использования меченных [α -33P]-ddNTP и реактивов ДНК-полимеразы ThermoSequenase™ в секвенировании матриц ДНК, включающих мономеры Т LNA-типа.
На фигурах 20 и 21 показано, что свободная от экзонуклеазной активности ДНК-полимераза I (фрагмент Кленова) может инкорпорировать аденозин-, цитозин-, гуанозин- и уридин-5’-трифосфаты LNA-типа в цепь ДНК.
На фигуре 22 показана способность терминальной дезокси-нуклеотидилтрансферазы (TdT) присоединять в концевом положении LNA-модифицированные олигонуклеотиды.
На фигурах 23А и 23В показано, что полностью перемешанные LNA-мономеры могут быть использованы для существенного увеличения вовлекаемости иммобилизованных, помеченных биотином олигомеров ДНК в специфичном по последовательности отборе ПЦР-ампликонов.
На фигурах 24-41 показаны возможные пути синтеза мономеров LNA по настоящему изобретению.
Подробное описание изобретения
По использованию в данном тексте термин “LNA” (Locked Nucleoside Analogues - замкнутые нуклеозидные аналоги) обозначает би- и трициклические нуклеозидные аналоги по настоящему изобретению, либо включенные в состав олигомера по настоящему изобретению (по базовой формуле I), либо существующего в виде дискретной химической единицы (по базовой формуле II). Термин “мономерный LNA” особенно применим к последнему случаю.
Олигомеры и аналоги нуклеозидов
Как отмечалось выше, настоящее изобретение, помимо прочего, представляет новые олигомеры (олигонуклеотиды), включающие один или большее число би-, три- или полициклических нуклеозидных аналогов (здесь и далее обозначаемых как “LNA”). Было установлено, что внесение таких LNA вместо и в дополнение, например, к известным нуклеозидам, придает получаемому в результате олигонуклеотиду интересные и многообещающие свойства. Би- и трициклические, а особенно бициклические LNA, по-видимому, особенно интересны в масштабе настоящего изобретения.
Каждый из вероятных LNA, включаемых в состав олигомера (олигонуклеотида), соответствует формуле I:
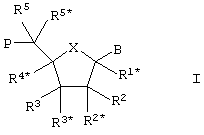
где Х выбирают из –О- (фуранозный мотив), -S-, -N(RN*)-, -C(R6R6*)-, -O-C(R7R7*)-, -C(R6R6*)-O-, -S-C(R7R7*)-, -C(R6R6*)-S-, -N(RN*)-C(R7R7*)-, -C(R6R6*)-N(RN*)- и -С(R6R6*)-С(R7R7*)-, где R6, R6*, R7, R7* и RN* соответствуют определенному далее.
Таким образом, вносимые в состав олигомера LNA могут включать либо пяти, либо шестиатомное кольцо в качестве существенной части би-, три- или полициклической структуры. Можно считать, что 5-атомные кольца (X представлен -О-, -S-, -N(RN*)-, -C(R6R6*)-) особенно интересны в связи с тем, что они способны занимать по сути те же самые конформации (однако при этом замкнутые введением одного или нескольких бирадикалов - см. ниже), что и нативное фуранозное кольцо в естественно встречающихся нуклеозидах. Среди вероятных 5-атомных колец наиболее интересными выглядят случаи, в которых Х представлен -О-, -S- и -N(RN*)-, а конкретно интересным считается вариант, в котором Х представлен -О-.
Заместитель В может представлять группу, которая, когда олигомер образует комплекс с ДНК или РНК, способна взаимодействовать (например, путем образования водородной связи или ковалентной связи, или за счет электронного взаимодействия) с ДНК или РНК, особенно нуклеотидами ДНК или РНК. С другой стороны, заместитель В может представлять группу, которая служит меткой или репортером, или же заместитель В может представлять группу (например, водород), которая, как ожидается, слабо или вообще не взаимодействует с ДНК или РНК. Таким образом, заместитель В предпочтительно выбирают из водорода, гидроксила, произвольно замещенной C1-4-алкоксигруппы, произвольно замещенного C1-4-алкила, произвольно замещенной C1-4-ацилоксигруппы, нуклеотидных оснований, ДНК-интеркалирующих групп, фотохимически активных групп, термохимически активных групп, хелатирующих групп, репортерных групп и лигандов.
В контексте настоящего изобретения термин “нуклеотидное основание” включает в себя нативные основания, равно как и неприродные основания. Для специалистов в данной области техники должно быть понятно, что различные основания, которые ранее были обозначены как “неприродными”, в последующем могут быть найдены в природе. Таким образом, понятие “нуклеотидное основание” включает не только известные пуриновые и пиримидиновые гетероциклы, но также и гетероциклические аналоги и их таутомеры. Иллюстрирующими примерами оснований являются аденин, гуанин, тимин, цитозин, урацил, пурин, ксантин, диаминопурин, 8-оксо-N6-метиладенин, 7-деазаксантин, 7-деазагуанин, N4,N4-этaнцитoзин, N6,N6-этан-2,6-диаминопурин, псевдоизоцитозин, 2-гидрокси-5-метил-4-тиазолопиридин, изоцитозин, изогуанин, инозин и “неприродными” основания, описанные Беннером с соавт. (Benner et al.: патент США №5432272). Подразумевается, что термин “нуклеотидное основание” охватывает каждый в отдельности и все вместе эти примеры, равно как и их аналоги и таутомеры. Особенно интересными основаниями являются аденин, гуанин, тимин, цитозин и урацил, которые рассматриваются как природные основания в связи с их терапевтическим и диагностическим применением в медицине.
По использованию в данном тексте термин “ДНК-интеркалирующая группа” обозначает группу, которая может интеркалировать спираль, дуплекс или триплекс ДНК или РНК. Примерами функциональных составляющих ДНК-интеркалирующих групп являются акридины, антрацен, хиноны, такие как антрахинон, индол, хинолин, изохинолин, дигидрохиноны, антрациклины, тетрациклины, метиленовый синий, антрациклинон, псоралены, кумарины, галогениды этидия, динемицин, комплексы металлов, такие как 1,10-фенантролин-медь, трис(4,7-дифенил-1,10-фенантролин)рутений-кобальт-ендиины, такие как кальхеамицин, порфирины, дистамицин, нетропцин, виологен, дауномицин. Особенно интересными примерами являются акридины, хиноны, такие как антрахинон, метиленовый синий, псоралены, кумарины и этидиумгалогениды.
В контексте данного изобретения термин “фотохимически активные группы” включает в себя соединения, которые способны претерпевать химические превращения при облучении видимым светом. Иллюстрирующими примерами таких функциональных групп являются хиноны, особенно 6-метил-1,4-нафтохинон, антрахинон, нафтохинон и 1,4-диметилантрахинон, диазирины, ароматические азиды, бензофеноны, псоралены, диазо-соединения и диазириновые соединения.
В контексте данного изобретения термин “термохимически реактивные группы” определяет функциональную группу, которая способна претерпевать индуцируемое термохимически образование ковалентной связи с другими группами. Иллюстративными примерами функциональных составляющих термохимически реактивных групп являются карбоновые кислоты, сложные эфиры карбоновых кислот, такие как активированные сложные эфиры, галогениды карбоновых кислот, такие как фториды, хлор-, бром и иодангидриды кислот, азиды карбоновых кислот, гидразиды-карбоновых кислот, сульфоновые кислоты, сложные эфиры сульфоновых кислот, галогениды сульфоновых кислот, семикарбазиды, тиосемикарбазиды, альдегиды, кетоны, первичные спирты, вторичные спирты, третичные спирты, фенолы, алкилгалогениды, тиолы дисульфиды, первичные амины, вторичные амины, третичные амины, гидразины, эпоксиды, имиды малеиновой кислоты и производные бороновых кислот.
В контексте данного изобретения термин “хелатирующая группа” обозначает молекулу, которая включает более одного сайта связывания и часто связывается с другой молекулой, атомом или ионом по более чем одному сайту связывания в одно и то же время. Примерами функциональных составляющих хелатирующих групп являются иминодиуксусная кислота, нитрилотриуксусная кислота, этилендиаминотетрауксусная кислота (EDTA), аминофосфоновая кислота и т.п.
В контексте данного изобретения термин “репортерная группа” обозначает группу, которая может быть детектирована сама по себе или в процессе детекционного анализа. Примерами функциональных репортерных групп являются биотин, дигоксигенин, флуоресцентные группы (группы, которые способны поглощать электромагнитное излучение, например, видимый свет или рентгеновское излучение, различных длин волн с последующей ремиссией поглощенной энергии в виде излучений с большей длиной волны: иллюстративными примерами являются DANSYL [(5-диметиламино)-1-нафталенсульфонил], DOXYL [N-оксил-4,4-диметилоксазолидин], PROXYL [N-оксил-2,2,5,5-тетраметилпирролидинил], TEMPO [М-оксил-2,2,6,6-тетраметилпиперидин], динитрофенол, акридины, кумарины, Сγ 3 и Сγ 5 (торговые марки систем детекции Biological Detection Systems Inc.), эритрозин, кумаровая кислота, умбеллиферон, техасский красный, родамин, тетраметилродамин, реактив Rox, 7-нитробензо-2-окса-1-диазол (NBD), пирен, флуоресцеин, европий, рутений, самарий и другие редкоземельные металлы), радиоизотопные метки, хемолюминесцирующие метки (метки, которые выявляются по эмиссии видимого света в ходе химической реакции), спиновые метки (свободный радикал, например, замещенный органический нитроксид, или другие парамагнитные зонды, например, Сu2+, Mg2+, связанные с биологической молекулой, которые могут быть детектированы с использованием электронной спиновой резонансной спектрометрии), ферменты (такие как пероксидазы, щелочные фосфатазы, β -галактозидазы, и гликозооксидазы), антигены, антитела, гаптены (группы, которые способны сочетаться с антителом, но которые не могут обусловливать иммунный ответ сами по себе, такие как пептиды и стероидные гормоны), системы-носители, предназначенные для проникновения через клеточные мембраны, такие как остатки жирных кислот, стероидные составляющие (холестерин), витамин А, витамин D, витамин Е, фолиевая кислота, лиганды конкретных рецепторов, опосредующие эндоцитоз группы, эпидермальный фактор роста (EGF), брадикинин и тромбоцитарный фактор роста (PDGF). Особенно интересными примерами являются биотин, флуоресцеин, техасский красный, родамин, динитрофенол, дигоксигенин, рутений, европий, Сγ 5, Сγ 3 и т.п.
В контексте данного изобретения термин “лиганд” обозначает нечто, что может связываться. Лигандами могут быть функциональные группы, как-то: ароматические группы (такие как бензол, пиридин, нафтален, антрацен и фенантрацен), гетероароматические группы (такие как тиофен, фуран, тетра-гидрофуран, пиридин, диоксан и пиримидин), карбоновые кислоты, сложные эфиры карбоновых кислот, галогениды карбоновых кислот, азиды карбоновых кислот, гидразиды карбоновых кислот, сульфоновые кислоты, сложные эфиры сульфоновых кислот, галогениды сульфоновых кислот, семикарбазиды, тиосемикарбазиды, альдегиды, кетоны, первичные спирты, вторичные спирты, третичные спирты, фенолы, алкилгалогениды, тиолы, дисульфиды, первичные амины, вторичные амины, третичные амины, гидразины, эпоксиды, имиды малеиновой кислоты, C1-20-алкильные группы, необязательно прерываемые или терминируемые одним или несколькими гетероатомами, такими как атомы кислорода атомы азота и(или) атомы серы, необязательно содержащие ароматические или моно/полиненасыщенные углеводороды, полиоксиэтилен, такой как полиэтиленгликоль, олиго/полиамиды, такие как поли-β -аланин, полиглицин, полилизин, пептиды, олиго/полисахариды, олиго/полифосфаты, токсины, антибиотики, клеточные яды и стероиды, а также “аффинные лиганды”, т.е. функциональные группы или биологические молекулы, характеризующиеся специфической аффинностью в отношении сайтов конкретных белков, антител, поли- и олигосахаридов и других биологических молекул.
Для специалистов в данной области техники должно быть ясно, что приведенные выше конкретные примеры ДНК-интеркалирующих групп, фотохимически активных групп, термохимически активных групп, хелатирующих групп, репортерных групп и лигандов соответствуют “активно-функциональной” части этих рассматриваемых групп. Для специалиста в данной области техники, кроме того, должно быть понятно, что ДНК-интеркалирующие группы, фотохимически активные группы, термохимически активные группы, хелатирующие группы, репортерные группы и лиганды обычно представлены в форме М-К-, где М - это “активно-функциональная” часть рассматриваемой группы, а К - спейсер, через который “активно-функциональная” часть присоединена к 5- или 6-атомному кольцу. Таким образом, должно быть понятно, что группа В, в случае когда В выбирают из ДНК-интеркалирующих групп, фотохимически активных групп, термохимически активных групп, хелатирующих групп, репортерных групп и лигандов, имеет форму М-К-, в которой М - это “активно-функциональная” часть ДНК-интеркалирующей группы, фотохимически активной группы, термохимически активной группы, хелатирующей группы, репортерной группы и лиганда, соответственно, а К - не являющийся обязательным спейсером, состоящим из 1-50 атомов, предпочтительно 1-30 атомов, в частности, 1-15 атомов, находящимся между 5- или 6-атомным кольцом и “активно-функциональной” частью.
В контексте настоящего изобретения термин “спейсер” обозначает термохимически и фотохимически неактивную, создающую “разрыв” группу, которая используется для соединения двух или большего числа различных составляющих, описанных выше. Спейсеры выбирают, исходя из ряда параметров, включая их гидрофобность, гидрофильность, молекулярную гибкость и размер (см., например, Hermanson et al., 1992, "Immobilized Affinity Ligand Techniques", Acad. Press, San Diego, CA, pp. 137-ff). В целом, длина спейсера составляет менее или примерно равна 400  , а в некоторых случаях предпочтительно составляет менее 100
, а в некоторых случаях предпочтительно составляет менее 100 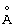 . Таким образом, спейсер включает цепочку атомов углерода, необязательно прерываемую или терминируемую одним или несколькими гетероатомами, такими как атомы кислорода, атомы азота и(или) атомы серы. Таким образом спейсер К может включать одну или несколько функциональных групп амидов, сложных эфиров, аминогрупп, простых эфиров и(или) тиоэфиров, а также необязательно ароматические или моно/полиненасыщенные углеводороды, полиоксиэтилен, такой как полиэтиленгликоль, олиго/полиамиды, такие как поли-β -аланин, полиглицин, полилизин, пептиды в целом, олигосахариды, олиго/полифосфаты. Более того, спейсер может включать их комбинированные сочетания. Длина спейсера может варьироваться, принимая во внимание желательное или являющееся необходимым расположение и пространственную ориентацию “активно-функциональной” части рассматриваемой группы по отношению к 5- или 6-атомному кольцу. В конкретных представляющих интерес вариантах спейсер включает химически отщепляемую группу. Примеры таких химически отщепляемых групп включают дисульфидные группы, отщепляемые в восстановительных условиях, пептидные фрагменты, отщепляемые с участием пептидаз, и т.п.
. Таким образом, спейсер включает цепочку атомов углерода, необязательно прерываемую или терминируемую одним или несколькими гетероатомами, такими как атомы кислорода, атомы азота и(или) атомы серы. Таким образом спейсер К может включать одну или несколько функциональных групп амидов, сложных эфиров, аминогрупп, простых эфиров и(или) тиоэфиров, а также необязательно ароматические или моно/полиненасыщенные углеводороды, полиоксиэтилен, такой как полиэтиленгликоль, олиго/полиамиды, такие как поли-β -аланин, полиглицин, полилизин, пептиды в целом, олигосахариды, олиго/полифосфаты. Более того, спейсер может включать их комбинированные сочетания. Длина спейсера может варьироваться, принимая во внимание желательное или являющееся необходимым расположение и пространственную ориентацию “активно-функциональной” части рассматриваемой группы по отношению к 5- или 6-атомному кольцу. В конкретных представляющих интерес вариантах спейсер включает химически отщепляемую группу. Примеры таких химически отщепляемых групп включают дисульфидные группы, отщепляемые в восстановительных условиях, пептидные фрагменты, отщепляемые с участием пептидаз, и т.п.
В одном из вариантов настоящего изобретения К представляет единственную связь таким образом, что “активно-функциональная” часть рассматриваемой группы напрямую присоединяемую в 5- или 6-атомному кольцу.
В предпочтительном варианте заместитель В базовых формул I и II предпочтительно выбирают из оснований нуклеотидов, в частности, аденина, гуанина, тимина, цитозина и урацила.
В составе олигомеров по настоящему изобретению (формула I) P обозначает положение радикала для межнуклеозидного “мостика” с последующим мономером или 5’-концевую группу. Первая из этих возможностей реализуется тогда, когда рассматриваемый LNA не является 5’-концевым мономером, в то время как вторая возможность соответствует тому, что рассматриваемый LNA таким мономером является. Должно быть понятно (что также с очевидностью вытекает из определения межнуклеозидного “мостика” и 5’-концевой группы, данных ниже), что такой межнуклеозидный “мостик” и 5’-концевая группа могут включать заместитель R5 (или в равной степени применим и заместитель R5*), в результате чего образуется двойная связь с группой Р. (Термин “5’-концевой” обозначает положение, соответствующее атому углерода 5’ в рибозной составляющей нуклеозида).
С другой стороны, межнуклеозидный “мостик” с предшествующим мономером или 3’-концевая группа (P*) могут образовываться от положений, определяемых заместителями R2, R2*, R3 и R3*, предпочтительно от положений, определяемых одним из заместителей R3 и R3*. Аналогичным образом, первая возможность реализуется тогда, когда рассматриваемый LNA не является “3’-концевым мономером”, в то время как последняя из этих возможностей соответствует тому, что рассматриваемый LNA является таким мономером. (Термин “3’-концевой” обозначает положение, соответствующее атому углерода 3’ в рибозной составляющей нуклеозида).
В контексте данного изобретения термин “мономер” обозначает природные нуклеозиды, не встречающиеся в естественных условиях нуклеозиды, РНК и т.п., равно как и LNA. Таким образом, термин “последующий мономер” относится к соседнему мономеру в направлении 5’, а термин “предшествующий мономер” обозначает соседний мономер в направлении 3’. Такие последующие и предшествующие мономеры, расположенные так по отношению к LNA-мономеру, могут быть природными или неприродными нуклеозидами и даже другими LNA.
Следовательно, в контексте настоящего изобретения (что может быть понято исходя из определенного выше) термин “олигомер” обозначает олигонуклеотид, модифицированный путем включения в него одного или нескольких LNA.
Ключевая часть настоящего изобретения связана с присутствием одного или большего числа колец, соединенных 5- или 6-атомным кольцом, что иллюстрируется базовой формулой I. Таким образом, одна или две пары негеминальных заместителей, выбираемых из R1*, R4*, R5, R5*, R6, R6*, R7, R7*, RH* и заместители R2, R2*, R3 и R3*, не образующие Р*, в каждом случае формируют бирадикал, состоящий из 1-8 групп/атомов, предпочтительно из 1-4 групп/атомов, независимо друг от друга выбираемых из –C(RaRb)-, -С(Ra)=С(Ra)-, -C(Ra)-N-, -О-, -Si(Ra)2-, -S-, -SO2-, -N(Ra)- и >C=Z. (Термин “присутствующий” указывает на то, что существование некоторых заместителей, например, R6, R6*, R7, R7*, RN*, зависит от того, включает ли Х такие заместители).
В группах, формирующих бирадикал (бирадикалы), Z выбирают из -О-, -S- и –N(Ra)-, а каждый из Ra и Rb независимо друг от друга выбирают из водорода, необязательно замещенного C1-12-алкила, необязательно замещенного С2-12-алкенила, необязательно замещенного С2-12-алкинила, необязательно замещенного гидроксила, C1-12-алкоксигруппы, С2-12-алкенилоксигруппы, карбоксигруппы, C1-12-алкоксикарбонила, C1-12-алкил-карбонила, формила, арила, арилоксикарбонила, арилоксигруппы, арилкарбонила, гетероарила, гетероарилоксикарбонила, гетероарилоксигруппы, гетероарилкарбонила, аминогруппы, моно- и ди-(C1-6-амино) аминогруппы, карбамоила, моно- и ди-(C1-6-алкил)-аминокарбонила, амино-C1-6-алкиламинокарбонила, моно- и ди(C1-6-алкил)-амино-С1-6-алкиламинокарбонила, C1-6-алкилкарбониламиногруппы, карбамидо, карбамидо, C1-6-алканоилоксигруппы, сульфоновой группы, C1-6-алкилсульфонилоксигруппы, нитрогруппы, азидогруппы, сульфанила, C1-6-алкилтиогруппы, галогена, ДНК-интеркалирующих групп, фотохимически активных групп, термохимически активных групп, хелатирующих групп, репортерных групп и лигандов (где последние группы могут включать спейсер в соответствии с определениями для заместителя В), где арил и гетероарил могут быть необязательно замещенными и где два геминальных заместителя Ra и Rb вместе могут обозначать необязательно замещенный метилен (=СН2, необязательно замещенный один или два раза заместителями в соответствии с определением в качестве произвольных заместителей для арила), и при том, что два геминальных или негеминальных заместителя, выбираемые из Ra, Rb и любого из заместителей R1*, R2, R2*, R3, R3*, R4*, R5, R5*, R6 и R6*, R7 и R7*, которые присутствуют и не вовлечены в Р, Р* или бирадикал (бирадикалы), вместе могут образовывать связанный бирадикал, выбираемый из бирадикалов того же типа, которые были определены выше. Должно быть понятно, что каждая из пар негеминальных заместителей тем самым образует моно- или бициклическую составляющую с участием (1) атомов, с которыми связаны негеминальные заместители, и (2) с любыми промежуточными атомами.
Можно считать, что бирадикалы, которые связаны с кольцевыми атомами в составе 5- или 6-атомных колец, являются предпочтительными в том смысле, что включение заместителей R5 и R5* может обусловливать нежелательные стерические взаимодействия с межнуклеозидным “мостиком”. Следовательно, предпочтительно, чтобы одна или две пары негеминальных заместителей, которые образуют один или два бирадикала, соответственно, выбирались из присутствующих заместителей R1*, R4*, R5, R5*, R6, R6*, R7, R7*, RN*, а заместители R2, R2*, R3 и R3* не образовывали Р*.
Предпочтительным является то, чтобы LNA, включенные в состав олигомеров, содержали только один бирадикал, образованный парой негеминальных заместителей. В частности, предпочтительно, чтобы R3* образовывал Р* и чтобы такой бирадикал образовывался между R2* и R4* или между R2 и R3.
Как уже говорилось, должно быть понятно (особенно при должном внимании к известным би- и трициклическим нуклеозидным аналогам см. раздел “Предпосылки изобретения”), что настоящее изобретение не касается олигомеров, включающих следующие би- или трициклические нуклеозидные аналоги:
(i) R2 и R3 одновременно означают бирадикал, выбираемый из -O-СН2-СН2- и -O-СН2-СН2-СН2-, где LNA является бициклическим нуклеозидным аналогом;
(ii) R3 и R4 одновременно означают бирадикал, выбираемый из -СН2-СН2- и -О-СН2-, где LNA является бициклическим нуклеозидным аналогом;
(iii) R3, R5 и R5* одновременно означают трирадикал –CH2-СН(-)-СН2-, где LNA является трициклическим нуклеозидным аналогом;
(iv) R1* и R6* одновременно означают бирадикал -СН2-, где LNA является бициклическим нуклеозидным аналогом; и
(v) R4* и R6* одновременно означают бирадикал -СН2-, где LNA является бициклическим нуклеозидным аналогом;
за исключением того, что такие би- или трициклические нуклеозидные аналоги сочетаются с одним или несколькими новыми LNA, определяемыми настоящим изобретением.
В контексте настоящего изобретения, т.е. в данном подробном описании и в формуле изобретения, ориентация бирадикалов такова, что расположенная по левую руку сторона занята заместителем с наименьшим номером, а сторона по правую руку занята заместителем с наивысшим номером: следовательно, когда R3 и R5 одновременно означают бирадикал “-O-CH2-”, то понятно, что атом кислорода соответствует заместителю R3, т.е. атом кислорода присоединен по положению заместителя R3, а метиленовая группа представляет заместитель R5.
Что касается многочисленных интересных структурных возможностей бирадикала (бирадикалов) во включаемых в состав олигомеров LNA по настоящему изобретению, то можно считать, что бирадикал (бирадикалы), образованный парой негеминальных заместителей предпочтительно выбирают из (CR*R*)r-(CR*R*)s-, (CR*R*)r-Y-(CR*R*)s-Y-, -Y-(CR*R*)r+s-Y-, -Y-(CR*R*)r-Y-(CR*R*)s-(CR*R*)r+s-Y-, -Y-Y-, где каждый Y независимо выбирают из -О-, -S-, -Si(R*)2-, -N(R*)-, >C=O, -C(=O) -N(R*)- и -N(R*)- С(=O)-, где каждый R* независимо выбирают из Н, галогена, азидогруппы, цианогруппы, нитрогруппы, гидроксила, меркаптогруппы, аминогруппы, моно- или ди(C1-6-алкил)аминогруппы, необязательно замещенной C1-6-алкоксигруппы, необязательно замещенного C1-6-алкила, ДНК-интеркалирующих групп, фотохимически активных групп, термохимически активных групп, хелатирующих групп, репортерных групп и лигандов, а (или) два соседних (негеминальных) R* могут формировать двойную связь; и каждый из r и s равен 0-4 при условии, что сумма r+s равна 1-5. Конкретно интересными вариантами являются те, в которых каждый бирадикал независимо выбирают из -Y-, -(CR*R*)r+s-, -(CR*R*)r-Y-(CR*R*)s- и –Y-(CR*R*)r+s-Y-, при том, что каждый из r и s равен 0-3 при условии, что сумма r+s равна 1-4.
Что касается положения бирадикала в LNA, то можно считать (основываясь на предварительных данных - см. примеры), что следующие варианты представляют наибольший интерес, как-то: R2* и R4* одновременно означают бирадикал, выбираемый из -Y-, -(CR*R*)r+s+1-, -(CR*R*)r-Y-(CR*R*)s- и –Y-(CR*R*)r+s-Y-; R2 и R3 одновременно означают бирадикал, выбираемый из -Y-, -(CR*R*)r+s-, -(CR*R*)r-Y-(CR*R*)s- и –Y-(CR*R*)r+s-Y-; R2* и R3 одновременно означают бирадикал, выбираемый из -Y-, (CR*R*)r+s-, -(CR*R*)r-Y-(CR*R*)s- и –Y-(CR*R*)r+s-Y-; R3 и R4* одновременно означают бирадикал, выбираемый из -Y-, -(CR*R*)r+s-, -(CR*R*)r-Y-(CR*R*)s- и –Y-(CR*R*)r+s-Y-; R3 и R5 одновременно означают бирадикал, выбираемый из -Y’-, -(CR*R*)r+s+1-, -(CR*R*)r-Y-(CR*R*)s- и –Y-(CR*R*)r+s-Y-; R1* и R4* одновременно означают бирадикал, выбираемый из -Y’-, -(CR*R*)r+s+1-, -(CR*R*)r-Y-(CR*R*)s- и –Y-(CR*R*)r+s-NR*-; R1* и R2* одновременно означают бирадикал, выбираемый из -Y-, -(CR*R*)r+s-, -(CR*R*)r-Y-(CR*R*)s- и –Y-(CR*R*)r+s-Y-; при том, что каждый r и s равен 0-3 при условии, что сумма r+s равна 1-4, Y соответствует определенному выше, а Y’ выбирают из -NR*-C(=O)- и -C(=O)-NR*-.
Представляющими конкретный интерес олигомерами являются те, у которых соблюдается один из следующих критериев по крайней мере для одного LNA, входящего в олигомер: R2* и R4* одновременно означают бирадикал, выбираемый из -О-, -S-, -N(R*)-, -(CR*R*)r+s+1-, -(CR*R*)r-O-(CR*R*)s-, -(CR*R*)r-S-(CR*R*)s-, -(CR*R*)r-N(R*)-(CR*R*)s-, -O-(CR*R*)r+s-O-, -S-(CR*R*)r+s-O-, -O-(CR*R*)r+s-S-, -N(R*)-(CR*R*)r+s-O-, -O-(CR*R*)r+s-N(R*)-, -S-(CR*R*)r+s-S-, -N(R*)-(CR*R*)r+s-N(R*)-, -N(R*)-(CR*R*)r+s-S- и -S-(CR*R*)r+s-N(R*), R2 и R3 одновременно означают бирадикал, выбираемый из -О-, (CR*R*)r+s-, -(CR*R*)r-O-(CR*R*)s-, -(CR*R*)r-S-(CR*R*)s- и (CR*R*)r-N(R*)-(CR*R*)s-; R2* и R3 одновременно означают бирадикал, выбираемый из -О-, -(CR*R*)r+s-, -(CR*R*)r-O-(CR*R*)s-, (CR*R*)r-S-(CR*R*)s- и -(CR*R*)r-N(R*)-(CR*R*)s-; R3 и R4* одновременно означают бирадикал, выбираемый из (CR*R*)r-O-(CR*R*)s-, -(CR*R*)r-S- (CR*R*)s- и -(CR*R*)r-N(R*)-(CR*R*)s-; R3 и R5 одновременно образуют бирадикал, выбираемый из (CR*R*)r-O-(CR*R*)s-, (CR*R*)r-S-(CR*R*)s- и -(CR*R*)r-N(R*)-(CR*R*)s-; R1* и R4* одновременно означают бирадикал, выбираемый из -(CR*R*)r-O-(CR*R*)s-, -(CR*R*)r-S-(CR*R*)s- и (CR*R*)r-N(R*)-(CR*R*)s-; или R1* и R2* одновременно означают бирадикал, выбираемый из -(CR*R*)r-O-(CR*R*)s-, -(CR*R*)r-S-(CR*R*)s- и -(CR*R*)r-N(R*)-(CR*R*)s-, при том, что каждый r и s равен 0-3 при условии, что сумма r+s равна 1-4, и при том, что RH представлен водородом или C1-4-алкилом.
Далее предпочтительным является то, чтобы один из R* выбирали из водорода, гидроксила, необязательно замещенной C1-6-алкоксигруппы, необязательно замещенного C1-6-алкила, ДНК-интеркалирующих групп, фотохимических активных групп, термохимически активных групп, хелатирующих групп, репортерных групп и лигандов, а все остальные R* были водородом.
В одном из предпочтительных вариантов одну группу R* в составе бирадикала по крайней мере одного из LNA выбирают из ДНК-интеркалирующих групп, фотохимических активных групп, термохимически активных групп, хелатирующих групп, репортерных групп и лигандов (где последние группы могут включать спейсер в соответствии с определенным для заместителя В).
Что касается заместителей R1*, R2, R2*, R3, R3*, R4*, R5, R5*, R6 и R6*, R7 и R7*, которые присутствуют и не вовлечены в образование Р, Р* или бирадикала (бирадикалов), то они независимо друг от друга выбираются из водорода, необязательно замещенного C1-12-алкила, необязательно замещенного С2-12-алкенила, необязательно замещенного С2-12-алкинила, гидроксила, C1-12-алкоксигруппы, С2-12-алкенилоксигруппы, карбоксигруппы, C1-12-алкоксикарбонила, C1-12-алкилкарбонила, формила, арила, арилоксикарбонила, арилоксигруппы, арилкарбонила, гетероарила, гетероарилоксикарбонила, гетероарилоксигруппы, гетероарилкарбонила, аминогруппы, моно- и ди-(C1-6-амино)-аминогруппы, карбамоила, моно- и ди-(C1-6-алкил)-аминокарбонила, амино-С1-6-алкиламинокарбонила, моно- и ди-(C1-6-алкил)-амино-C1-6-алкиламинокарбонила, C1-6-алкилкарбониламиногруппы, карбамидо, C1-6-алканоилоксигруппы, сульфоновой группы, C1-6-алкилсульфонилоксигруппы, нитрогруппы, азидогруппы, сульфанила, C1-6-алкилтиогруппы, галогена, ДНК-интеркалирующих групп, фотохимически активных групп, термохимически активных групп, хелатирующих групп, репортерных групп и лигандов (где последние группы могут включать спейсер в соответствии с определенным для заместителя В), где арил и гетероарил могут быть необязательно замещенными и где два геминальных заместителя вместе могут образовывать оксогруппу, тиоксогруппу, иминогруппу или необязательно замещенный метилен или вместе могут формировать спиробирадикал, включающий 1-5-углеродную алкиленовую цепь, которая необязательно прерывается и(или) терминируется одним или несколькими гетероатомамм/группами, выбираемыми из -О-, -S- и -(NRN)-, где RN выбирают из водорода и C1-4-алкила и где два соседних (но геминальных) заместителя могут формировать дополнительную, двойную связь; и RN*, когда присутствует и не входит к бирадикал, выбирают из водорода и C1-4-алкила.
Предпочтительно каждый из заместителей R1*, R2, R2*, R3, R3*, R4*, R5, R5*, R6 и R6*, R7 и R7* в составе LNA, которые присутствуют и не вовлечены в образование Р, Р* или бирадикала (бирадикалов), независимо выбирают из водорода, необязательно замещенного C1-6-алкила, необязательно замещенного C2-6-алкенила, гидроксила, C1-6-алкоксигруппы, C2-6-алкенилоксигруппы, карбоксила, C1-6-алкоксикарбонила, C1-6-алкилкарбонила, формила, аминогруппы, моно- и ди(C1-6-алкил) аминогруппы, карбамоила, моно- и ди(C1-6-алкил)аминокарбонила, C1-6-алкилкарбониламиногруппы, карбамидогруппы, азидогруппы, C1-6-алканоилоксигруппы, сульфоновой группы, сульфанила, C1-6-алкилтиогруппы, ДНК-интеркалирующих групп, фотохимически активных групп, термохимически активных групп, хелатирующих групп, репортерных групп и лигандов, и галогена, где два геминальных заместителя одновременно образуют оксогруппу и где RN*, в случае присутствия и неучастия в образовании бирадикала, выбирают из Н и C1-4-алкила.
В предпочтительном варианте настоящего изобретения Х выбирают из -О-, -S- и -(NRN)-, в частности, он является -О-, а каждый из заместителей R1*, R2, R2*, R3, R3*, R4*, R5, R5*, R6, R6*, R7 и R7* в составе LNA, которые присутствуют и не вовлечены в Р, Р* или бирадикал (бирадикалы), представлены водородом.
В еще более предпочтительном варианте настоящего изобретения R2* и R4* из LNA, входящего в состав олигомера, одновременно означают бирадикал. Предпочтительно Х представлен О, R2 выбирают из водорода, гидроксила и необязательно замещенной C1-6-алкоксигруппы, a R1*, R3, R5 и R5* представлены водородом, а, более конкретно, такой бирадикал выбирают из -О-, (СН2)0-1-О-(СН2)1-3-, (СН2)0-1-S-(СН2)1-3-, -(СН2)0-1-N(RN)-(CH2)1-3-, и -(СН2)2-4-, в частности, из -O-CH2-, -S-CH2- и –NRH-CH2. В целом, при должном внимании к полученным результатам, предпочтительным является то, чтобы бирадикал, образуемый заместителями R2* и R4*, формировали двухуглеродный “мостик”, т.е. чтобы этот бирадикал формировали 5-атомное фуранозное кольцо (X представлен О).
В другом варианте настоящего изобретения R2 и R3 в LNA, входящем в состав олигомера, одновременно означают бирадикал. Предпочтительно Х представлен О, R2* выбирают из галогена, гидроксила и необязательно замещенной C1-6-алкоксигруппы, R1*, R4*, R5 и R5* представлены водородом, а, более конкретно, такой бирадикал выбирают из -(CH2)0-1-O-(CH2)1-3-, -(CH2)0-1-S-(CH2)1-3-, -(CH2)0-1-N(RN)-(CH2)1-3-, и -(CH2)1-4-, в частности, из -O-СН2-, -S-CH2- и –NRH-CH2-. В последнем случае конкретный интерес представляют амино- и тиоварианты.
В другом варианте настоящего изобретения R2* и R3 в LNA, входящем в состав олигомера, одновременно означают бирадикал. Предпочтительно Х представлен О, R2 выбирают из галогена, гидроксила и необязательно замещенной C1-6-алкоксигруппы, R1*, R4*, R5 и R5* представлены водородом, а, более конкретно, такой бирадикал выбирают из (СН2)0-1-O-(СН2)1-3- и -(СН2)2-4-.
Еще в одном варианте настоящего изобретения R3 и R4* в LNA, входящем в состав олигомера, одновременно означают бирадикал. Предпочтительно Х представлен О, R2* выбирают из галогена, гидроксила и необязательно замещенной C1-6-алкоксигруппы, R1*, R2, R5 и R5* представлены водородом, а, более конкретно, такой бирадикал выбирают из (СН2)0-2-O-(СН2)0-2-.
В следующем варианте настоящего изобретения R3 и R5* в LNA, входящем в состав олигомера, одновременно означают бирадикал. Предпочтительно Х представлен О, R2* выбирают из галогена, гидроксила и необязательно замещенной C1-6-алкоксигруппы, R1*, R2, R4 и R5 представлены водородом, а, более конкретно, такой бирадикал выбирают из –О-(CHR*)2-3- и -(CHR*)1-3-O-(CHR*)0-3-.
Еще в следующем варианте настоящего изобретения R1* и R4* в LNA, входящем в состав олигомера, одновременно означают бирадикал. Предпочтительно Х представлен О, R2* выбирают из галогена, гидроксила и необязательно замещенной C1-6-алкоксигруппы, R2, R3, R5 и R5* представлены водородом, а, более конкретно, такой бирадикал выбирают из -(СН2)0-2-O-(СН2)0-2-.
В этих вариантах дополнительно предпочтительным является то, чтобы по крайней мере один LNA, входящий в состав олигомера, включал основание (заместитель В), выбираемое из аденина и гуанина. В частности, предпочтительно, чтобы олигомер имел включенный в его состав LNA, включающий и по крайней мере одно основание, выбираемое из тимина, урацила и цитозина, и по крайней мере одно основание, выбираемое из аденина и гуанина. Для мономеров LNA особенно предпочтительным является то, чтобы такое основание выбирали из аденина и гуанина.
Для этих представляющих интерес вариантов также предпочтительным является то, чтобы LNA соответствовал базовой формуле Iа (см. ниже).
В одном из ответвлений этих представляющих интерес вариантов все мономеры олигонуклеотида являются LNA-мономерами.
Как должно быть очевидно из формулы I (LNA в составе олигомера) (и базовой формулы II для мономерного LNA - см. ниже) и данных для нее определений, в олигомерах (и LNA-мономерах) могут присутствовать один или несколько асимметричных атомов углерода, что зависит от природы заместителей и вероятных бирадикалов, см. ниже. Олигомеры, конструируемые в соответствии со способом по настоящему изобретению, равно как и сами по себе олигомеры, должны включать все стереоизомеры, обусловливаемые присутствием любого из и всех вместе изомеров отдельных мономерных фрагментов, равно как и их смесей, включая смеси рацематов. Что касается 5- или 6-атомных колец, то можно, считать, что некоторые стереохимические конфигурации будут представлять особенный интерес, например, следующие:
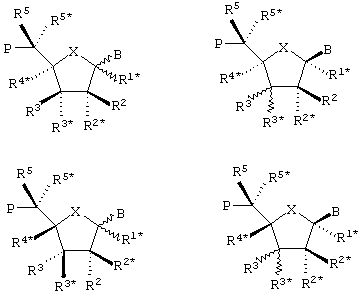
в которых волнистые линии обозначают возможность существования обоих диастереомеров, возникающих при взаимообмене двух рассматриваемых заместителей.
Особый интерес в связи со стереоизомерией представляет случай, в котором LNA характеризуется следующей формулой Iа:
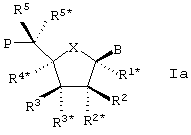
Также отдельным, представляющим интерес аспектом настоящего изобретения, является вариант формулы Iа, в котором В находится в α -конфигурации.
В этих случаях, равно как и в целом, R3* предпочтительно образует Р*.
Олигомеры в соответствии с настоящим изобретением обычно состоят из 1-10000 LNA базовой формулы I (или более детальной формулы Iа) и 0-10000 нуклеозидов, выбираемых из нативных нуклеозидов и нуклеозидных аналогов. Сумма количества нуклеозидов и количества LNA по крайней мере равна 2, предпочтительно по крайней мере 3, в частности, по крайней мере 5, особенно предпочтительно по крайней мере 7, и находится в диапазоне 2-15000, предпочтительнее в диапазоне 2-100, также 3-10, в частности, в диапазоне 2-50, а также 3-50 или 5-50, или 7-50.
Предпочтительно по крайней мере один LNA включает нуклеотидное основание в качестве В-заместителя.
В контексте настоящего изобретения термин “нуклеозид” обозначает гликозид гетероциклического основания. Термин “нуклеозид” широко применяется для обозначения не встречающихся в естественных условиях нуклеозидов, нативных нуклеозидов, а также и других нуклеозидных аналогов. Иллюстрирующими примерами нуклеозидов являются рибонуклеозиды, включающие остаток рибозы, а также дезоксирибонуклеозиды, включающие остаток дезоксирибозы. Что касается основания в таких нуклеозидах, то должно быть понятно, что оно может быть любым из встречающихся в естественных условиях оснований, например, аденин, гуанин, цитозин, тимин и урацил, равно как и любые их модифицированные варианты или любые другие ненативные основания.
При рассмотрении определений и известных нуклеозидов (нативных и не встречающихся в естественных условиях) и нуклеозидных аналогов (включая известные би- и трициклические аналоги), то ясно, что олигомер может включать один или большее число LNA (которые могут быть идентичными или дифференцированными как с точки зрения выбора заместителей, так и с точки зрения выбора бирадикала), а также один или большее число нуклеозидов и(или) нуклеозидных аналогов. В контексте настоящего изобретения термин “олигонуклеотид” обозначает последовательную цепочку нуклеозидов, соединенных с помощью межнуклеозидных “мостиков”, однако, должно быть понятно, что нуклеотидное основание в одном или нескольких единицах (мономерах) олигомера (олигонуклеотида) может быть модифицировано с помощью заместителя В в соответствии с определенным выше.
Олигомеры могут быть линейными, разветвленными или циклическими. В случае разветвленного олигомера точки ветвления могут быть расположены в нуклеозиде, в межнуклеозидном “мостике” или, как в одном из своеобразных вариантов, в LNA. Можно считать, что в последнем случае заместители R2, R2*, R3 и R3* могут образовывать две группы Р*, каждая из которых формирует межнуклеозидный “мостик” с предшествующим мономером: в частности, один из R2 и R2* образуют один Р*, а один из R3 и R3* образует следующий Р*.
Как отмечалось выше, LNA в олигомере соединяются с другими мономерами через межнуклеозидный “мостик”. В контексте настоящего изобретения термин “межнуклеозидный “мостик”” обозначает связь, состоящую из 2-4, предпочтительно 3, групп/атомов, выбираемых из –СН2-, -О-, -S-, -NRH-, >C=O, >C=NRH, >C=S, -S-i(R’’)2-, -SO-, -S(O)2-, -P(O)2-, -РО(ВН3)-, -P(O, S)-, -P(S)2-, -PO(R’’)-, -РО(ОСН3)-, и -PO(NHRH)-, где Rн выбирают из Н и C1-4-алкила, a R” выбирают из C1-6-алкила и фенила. Иллюстрирующими примерами таких межнуклеозидных “мостиков” являются -СН2-СН2-СН2-, -СН2-СО-СН2-, -СН2-СНОН-СН2-, -O-СН2-O-, -O-СН2-СН2-, -O-СН2-СН= (включая R5, когда он используется в качестве связи с последующим мономером), -СН2-СН2-O-, -NRH-CH2-CH2-, -CH2-CH2-NRH, -CH2-NRH-CH2-, -O-CH2-CH2-NRH-, -NRH-CO-O-, -NRH-CO-NRH-, -NRH-CS-NRH-, -NRH-C(=NRH)-NRH-, -NRH-CO-CH2-NRH-, -O-CO-O-, -O-CO-CH2-O-, -O-CH2-CO-O-, -CH2-CO-NRH-, -O-CO-NRH-, -NRH-CO-CH2-, -O-CH2-, -CO-NRH-, -O-CH2-CH2-NRH-, -CH=N-O-, -CH2-O-N= (включая R5, когда он используется в качестве связи с последующим мономером), -CH2-O-NRH-, -CO-NRH-CH2-, -CH2-NRH-O-, -CH2-NRH-CO-, -O-NRH-CH2-, -O-NRH-, -O-CH2-S-, -S-CH2-O-, -CH2-CH2-S-, -O-CH2-CH2-S-, -S-CH2-CH= (включая R5, когда он используется в качестве связи с последующим мономером), -S-СН2-СН2-, -S-CH2-CH2-O-, -S-CH2-CH2-S-, -CH2-S-CH2-, -CH2-SO-СН2-, -CH2-SO2-CH2-, -O-SO-O-, -O-S(O)2-O-, -O-S(O)2-CH2-, -O-S(O)2-NRH-, -NRH-S(O)2-CH2-, -O-S(О)2-СН2-, -O-Р(O)2-O-, -O-P(O, S)-O-, -O-P(S)2-O-, -S-P(O)2-O-, -S-P(O, S)-O-, -S-P(S)2-O-, -O-P(O)2-S-, -O-P(O, S)-S-, -O-P(S)2-S-, -S-P(O)2-S-, -S-P(O, S)-S-, -S-P(S)2-S-, -O-PO(R’’)-O-, -O-РО(ОСН3)-O-, -O-РО(ОСН2СН3)-O-, -O-PO(OCH2CH2S-R)-O-, -O-РО(ВН3)-O-, -O-PO(NHRN)-O-, -O-P(O)2-NRH-, -NRH-Р(O)2-O-, -O-P(О, NRH)-О-, -СН2-Р(O)2-O-, -O-Р(O)2-СН2- и -O-Si(R’’)2-O-; среди них -СН2-CO-NRH-, -CH2-NRH-O-, -S-CH2-O-, -O-P(О)2-O-, -O-P(О, S)-О-, -O-P(S)2-O-, -NRH-P(O)2-O-, -O-P(О, NRH)-О-, -O-РО(R’’)-О-, -O-РО(СН3)-O- и -O-PO(NHRN)-O-, где RH выбирают из Н и C1-4-алкила, a R’’ выбирают из C1-6-алкила и фенила, являются наиболее предпочтительными. Дальнейшие иллюстративные примеры приведены Месмейкером с соавт. (Mesmaeker et al., 1995, Cur. Opinion Struct. Biol., 5, 343-355). Сторона межнуклеозидного “мостика”, находящаяся по левую руку, связана с 5или 6-атомным кольцом в качестве заместителя Р*, в то время как ее сторона, находящаяся по правую руку, связана с 5’-сайтом предшествующего мономера.
Из вышеописанного также ясно, что группа Р также может составлять 5’-концевую группу в случае, когда рассматриваемый LNA является 5’-концевым мономером. Примерами таких 5’-концевых групп являются водород, гидроксил, необязательно замещенный C1-6-алкил, необязательно замещенная C1-6-алкоксигруппа, необязательно замещенная C1-6-алкилкарбонилоксигруппа, необязательно замещенная арилоксигруппа, монофосфат, дифосфат, трифосфат и группа -W-A’, где W выбирают из -О-, -S- и –N(RH)-, где RH выбирают из водорода и C1-6-алкила, и где А’ выбирают из ДНК-интеркалирующих групп, фотохимически активных групп, термохимически активных групп, хелатирующих групп, репортерных групп и лигандов (где последние группы могут включать спейсер в соответствии с ранее определенным для заместителя В).
В настоящем описании и формуле изобретения термины “монофосфат”, “дифосфат” и “трифосфат” обозначают группы, имеющие формулы -O-Р(O)2-O-, -O-Р(О)2-O-Р(O)2-O- и -O-Р(O)2-O-Р(О)2-O-Р(О)2-O-, соответственно.
В представляющем интерес конкретном варианте группа Р формирует 5’-концевые группы, выбираемые из монофосфата, дифосфата и трифосфата. Особенно привлекательным является случай, когда субстратом является вариант с трифосфатом.
Аналогичным образом группа Р* может образовывать 3’-концевую группу в случае, когда рассматриваемый LNA является 3’-концевым мономером. Примерами таких 3’-концевых групп являются водород, гидроксил, необязательно замещенная C1-6-алкоксигруппа, необязательно замещенная C1-6-алкилкарбонилоксигруппа, необязательно замещенная арилоксигруппа и группа -W-A’, где W выбирают из -О-, -S- и -N(RH)-, где RH выбирают из Н и C1-6-алкила, и где А’ выбирают из ДНК-интеркалирующих групп, фотохимически активных групп, термохимически активных групп, хелатирующих групп, репортерных групп и лигандов (где последние группы могут включать спейсер в соответствии с ранее определенным для заместителя В).
В предпочтительном варианте настоящего изобретения олигомер характеризуется следующей формулой V:
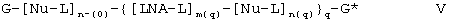
где q равно 1-50;
каждый из n(0), ....., n(q) независимо друг от друга равен 0-10000;
каждый из m(1), ....., m(q) независимо друг от друга равен 1-10000;
при условии, что сумма n(0), ....., n(q) и m(1), ....., m(q) равна 2-15000;
G обозначает 5’-концевую группу;
каждый Nu независимо обозначает нуклеозид, выбираемый из нативных нуклеозидов и нуклеозидных аналогов;
каждый LNA независимо является нуклеозидным аналогом;
каждая L обозначает межнуклеозидный “мостик” между двумя группами, выбираемыми из Nu и LNA, или L вместе с G* образует 3’-концевую группу; и каждая LNA-L независимо обозначает нуклеозидный аналог базовой формулы I в соответствии с определенным выше или, что предпочтительнее, базовой формулы Iа в соответствии с определенным выше.
В данном варианте, равно как и в целом в настоящем изобретении представляется перспективная возможность сочетания LNA с различными (т.е. пуриновыми и пиримидиновыми) основаниями: в частности, и основаниями, выбираемыми из тимина, цитозина и урацила, и с основаниями, выбираемыми из аденина и гуанина.
В другом варианте настоящего изобретения в состав олигомера дополнительно входит моно- или олигомерный PNA-сегмент формулы:
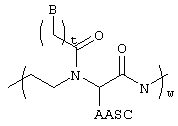
при том, что В соответствует предыдущему определению для формулы I, AASC обозначает водород или боковую цепь аминокислоты, t равна 1-5 и w равно 1-50.
В контексте настоящего изобретения термин “боковая цепь аминокислоты” обозначает группу, присоединенную к α -атому α -аминокислот, т.е. соответствующую рассматриваемой α -аминокислоте без глициновой составляющей, предпочтительно природной или легкодоступной α -аминокислоте. Иллюстрирующими примерами являются водород (сам по себе глицин), дейтерий (дейтеризованный глицин), метил (аланин), цианометил (β -цианоаланин), этил, 1-пропил (норвалин), 2-пропил (валин), 2-метил-1-пропил (лейцин), 2-гидрокси-2-метил-1-пропил (β -гидроксилейцин), 1-бутил (норлейцин), 2-бутил (изолейцин), метилтиоэтил (метионин), бензил (фенилаланин), n-аминобензил (n-аминофенилаланин), n-иодбензил (n-иодфенилаланин), n-фторбензил (n-фторфенилаланин), n-бромбензил (n-бромфенилаланин), n-хлорбензил (n-хлорфенилаланин), n-нитробензил (n-нитрофенилаланин), 3-пиридилметил (β -[3-пиридил]-аланин), 3,5-дииод-4-гидроксибензил (3,5-дийодтирозин), 3,5-дибром-4-гидроксибензил (3,5-дибромтирозин), 3,5-дихлор-4-гидроксибензил (3,5-дихлортирозин), 3,5-дифтор-4-гидроксибензил (3,5-дифтортирозин), 4-метоксибензил (O-метилтирозин), 2-нафтилметил (β -[2-нафтил]-аланин), 1-нафтилметил (β -[1-нафтил]-аланин), 3-индолилметил (триптофан), гидроксиметил (серин), 1-гидроксиэтил (треонин), меркаптометил (цистеин), 2-меркапто-2-пропил (пеницилламин), 4-гидроксибензил (тирозин), аминокарбонилметил (аспарагин), 2-аминокарбонилэтил (глутамин), карбоксилметил (аспарагиновая кислота), 2-карбоксиэтил (глутаминовая кислота), аминометил (α ,β -диаминопропионовая кислота), 2-аминоэтил (α ,γ -диаминобутировая кислота), 3-аминопропил (орнитин), 4-амино-1-бутил (лизин), 3-гуанидино-1-пропил (аргинин) и 4-имидазолилметил (гистидин).
Моно- и олигомерный PNA-сегмент может быть включен в состав олигомера в соответствии с описанным в заявке ЕР 0672677 А2.
Олигомеры по настоящему изобретению также призваны включить в себя химерные олигомеры. Термин “химерные олигомеры” обозначает два или большее число олигомеров, включающих мономеры различного происхождения, соединенные между собой напрямую или через спейсер. Иллюстрирующими примерами таких олигомеров, которые могут быть перекомбинированы, являются пептиды, РНК-олигомеры, содержащие LNA олигомеры и олигонуклеотидные олигомеры.
Помимо описанных выше олигомеров, настоящее изобретение также представляет мономерные LNA, применимые, например, для синтеза олигомеров, в качестве субстратов для, например, полимераз нуклеиновых кислот, полинуклеотидкиназ, терминальных трансфераз и в качестве терапевтических средств: см. ниже. Мономерные LNA по своей общей структуре (особенно в части возможных бирадикалов) соответствуют тем LNA, которые определяются как компоненты олигомеров, однако, что касается групп Р и Р*, то мономерные LNA в небольшой степени отличаются, что объясняется ниже. Кроме того, мономерные LNA могут быть защищены по своим функциональным составляющим, особенно в тех случаях, когда мономерные LNA должны включаться в состав олигомеров с использованием методов химического синтеза.
Представляющая интерес подгруппа мономерных LNA включает бициклические нуклеозидные аналоги (LNA) общей формулы II:
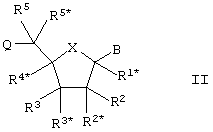
где заместитель В выбирают из оснований, ДНК-интеркалирующих групп, фотохимически активных групп, термохимически активных групп, хелатирующих групп, репортерных групп и лигандов; Х выбирают из -О-, -S-, -N(RH*)- и –C(R6R6*)-, предпочтительно из -О-, -S- и -N(RH*)-; один из заместителей R2, R2*, R3 и R3* является группой Q*;
каждую из Q и Q* независимо выбирают из водорода, азидогруппы, галогена, цианогруппы, нитрогруппы, гидроксила, Prot-O-, Act-O-, меркаптогруппы, Prot-S-, Act-S-, C1-6-алкилтиогруппы, аминогруппы, Prot-N(RH)-, Act-N(RH)-, моно- или ди(C1-6-алкил) аминогруппы, необязательно замещенной C1-6-алкоксигруппы, необязательно замещенного C1-6-алкила, необязательно замещенного С2-6-алкенила, необязательно замещенной С2-6-алкенилоксигруппы, необязательно замещенного С2-6-алкинила, необязательно замещенной С2-6-алкинилоксигруппы, монофосфата, дифосфата, трифосфата, ДНК-интеркалирующих групп, фотохимически активных групп, термохимически активных групп, хелатирующих групп, репортерных групп, лигандов, карбоксила, сульфоновой группы, гидроксиметила, Prot-O-CH2-, Act-O-CH2-, аминометила, Prot-N(RH)-CH2-, Act-N(RH)-CH2-, карбоксиметила, сульфонометила, где “Prot” обозначает защитную группу для -ОН, -SH и -NH(RH), соответственно, a “Act” обозначает активационную группу для -ОН, -SH и -NH(RH), соответственно, a RH выбирают из Н и C1-6-алкила;
R2* и R4* одновременно означают бирадикал, выбираемый из -О-, -S-, -N(R*)-, -(CR*R*)r+s+1-, -(CR*R*)r-O-(CR*R*)s-, (CR*R*)r-N-(CR*R*)s-, (CR*R*)r-N(R*)-(CR*R*)s-, -O-(CR*R*)r+s-O-, -S-(CR*R*)r+s-O-, -O-(CR*R*)r+s-S-, -N(R*)-(CR*R*)r+s-O-, -O-(CR*R*)r+s-N(R*)-, -S-(CR*R*)r+s-S-, -N(R*)-(CR*R*)r+s-N(R*)-, -N(R*)-(CR*R*)r+s-S- и -N-(CR*R*)r+s-N(R*)-; R2 и R3 одновременно означают бирадикал, выбираемый из -О-, (CR*R*)r+s, -(CR*R*)r-O-(CR*R*)s-, -(CR*R*)r-S-(CR*R*)s- и -(CR*R*)r-N(R*)-(CR*R*)s-; R2* и R3 одновременно означают бирадикал, выбираемый из -О-, -(CR*R*)r+s-, -(CR*R*)r-O-(CR*R*)s-; (CR*R*)r-S-(CR*R*)s- и (CR*R*)r-N(R*)-(CR*R*)s-; R3 и R4* одновременно означают бирадикал, выбираемый из (CR*R*)r-O-(CR*R*)s-, -(CR*R*)r-S-(CR*R*)s- и -(CR*R*)r-N(R*)-(CR*R*)s-; R3 и R5 одновременно означают бирадикал, выбираемый из -(CR*R*)r-O-(CR*R*)s-, -(CR*R*)r-S-(CR*R*)s- и -(CR*R*)r-N(R*)-(CR*R*)s-; R1* и R4* одновременно означают бирадикал, выбираемый из -(CR*R*)r-O-(CR*R*)s-, -(CR*R*)r-S-(CR*R*)s- и (CR*R*)r-N(R*)-(CR*R*)s-; R1* и R2* одновременно означают бирадикал, выбираемый из -(CR*R*)r-O-(CR*R*)s-, -(CR*R*)r-S-(CR*R*)s- и (CR*R*)r-N(R*)-(CR*R*)s-; где R* соответствует определенному выше для олигомеров и каждый из заместителей R1*, R2, R2*, R3, R4*, R5 и R5*, которые не вовлечены в образование Q, Q* или бирадикала, соответствуют тому, что было определено для олигомеров.
Также должно быть понятно, с учетом должного внимания к известным бициклическим нуклеозидным аналогам, что R2 и R3 вместе не образуют бирадикал, выбираемый из -О-СН2-СН2- и -О-СН2-СН2-СН2-; и R3 и R5 вместе не образуют бирадикал, выбираемый из -СН2-СН2-, -O-СН2- и -O-Si(iPr)2-O-Si(iPr)2-O-.
Мономерные LNA также включают в себя их основные соли и кислотно-аддитивные соли.
Более того, должно быть понятно, что любая химическая группа (включая любое основание), которая реактивна в тех условиях, которые имеют место в процессе химического синтеза олигонуклеотидов, необязательно может быть защищена в соответствии с известным в данной области техники. Это означает, что такие группы, как гидроксильная аминогруппа, сульфоновая группа, карбоксильная группа и меркаптогруппа, равно как и нуклеотидные основания, в составе мономерного LNA необязательно могут быть защищены по своим функциональным составляющим. Присоединение защитной группы (как и освобождение от нее) осуществляется с помощью методов, известных специалистам в данной области техники (см., например, T.W.Greene & P.G.M.Wuts, 1991, "Protective Groups in Organic Synthesis", 2nd ed., J.Wiley, NY и M.J.Gait, 1984, "Oligonucleotide Synthesis", IRL Press).
Иллюстрирующими примерами защитных групп для гидроксильной группы являются необязательно замещенный тритил, такой как 4,4’-диметокситритил (DMT), 4-монометокситритил (ММТ), и тритил, произвольно замещенный 9-(9-фенил)ксантенил (пиксил), необязательно замещенная этоксикарбонилоксигруппа, р-фенилазофенилоксикарбонилоксигруппа, тетрагидропиронил (ТНР), 9-флуоренилметоксикарбонил (Fmoc), метокситетрагидропиранил (МТНР), силилоксигруппа, такая как триметилсилил (TMS), три-изопропилсилил (TIPS), трет-бутилдиметилсилил (TBDMS), триэтилсилил и фенилдиметилсилил, бензилоксикарбонил или замещенные сложные эфиры бензилоксикарбонила, такие как 2-бромбензилоксикарбонил, трет-бутиловые эфиры, алкиловые эфиры, такие как метиловый эфир, ацетали (включающие две гидроксильные группы), ацилоксигруппа, такая как ацетил или галозамещенные ацетилы, например, хлорацетил или фторацетил, изобутирил, пивалоил, бензоил и замещенные бензоилы, метоксиметил (MOM), бензиловые эфиры или замещенные бензиловые эфиры, такие как 2,6-дихлорбензил (2,6-Cl2Bzl). С другой стороны, гидроксильная группа может быть защищена путем присоединения к твердой подложке, при этом необязательно через линкер.
Иллюстрирующими примерами защитных групп для аминогрупп являются Fmoc (флуоренилметоксикарбонил), ВОС (трет-бутил-оксикарбонил), трифторацетил, аллилоксикарбонил (АОС), бензилоксикарбонил (Cbz), замещенные бензилоксикарбонилы, такие как 2-хлорбензилоксикарбонил (2-ClZ), монометокситритил (ММТ), диметокситритил (DMT), фталоил и 9-(9-фенил)ксантенил (пиксил).
Иллюстрирующими примерами защитных групп для карбоксильной группы являются аллильные эфиры, метиловые эфиры, этиловые эфиры, 2-цианоэтиловые эфиры, триметилсилилэтиловые эфиры, бензиловые эфиры (Obzl), 2-адамантиловые эфиры (O-2-Ada), циклогексиловые эфиры (ОсНех), 1,3-оксазолины, оксазол, 1,3-оксазолидины, амиды или гидразиды.
Иллюстрирующими примерами защитных групп для меркаптогрупп являются тритил (Trt), ацетамидометил (АСМ), триметилацетамидометил (Таcm), 2,4,6-триметоксибензил (Tmob), трет-бутилсульфенил (StBu), 9-флуоренилметил (Fm), 3-нитро-2-пиридинсульфенил (Npys) и 4-метилбензил (Meb).
Далее необходимым или желательным может оказаться защитить любое из оснований, включенный в состав мономерного LNA, особенно тогда, когда такой мономерный LNA должен быть внесен в состав олигомера по настоящему изобретению. В контексте настоящего изобретения термин “защищенные основания” обозначает, что рассматриваемое основание несет защитную группу, выбираемую из групп, которые хорошо известны специалистам в данной области техники (см., например, "Protocols for Oligonucleotides and Analogs", vol. 20, ed. S.Agrawal, Humana Press, Totowa, NJ, 1993; S.L.Beaucage & R.P.Iyer, 1993, Tetrahedron, 49, 6123; S.L.Beaucage & R.P.Iyer, 1992, Tetrahedron, 48, 2223; и E.Uhlmann & A.Peyman, Chem. Rev., 90, 543). Иллюстрирующими примерами являются бензоил, изобутирил, грет-бутил, трет-бутилоксикарбонил, 4-хлорбензилоксикарбонил, 9-флуоренилметил, 9-флуоренилметилоксикарбонил, 4-метоксибензоил, 4-метокситрифенилметил, необязательно замещенная триазоловая группа, n-толуолсульфонил, необязательно замещенный сульфонил, изопропил, необязательно замещенные амидины, необязательно замещенный тритил, феноксиацетил, необязательно замещенный ацил, пиксил, тетрагидропиранил, необязательно замещенные силиловые эфиры и 4-метоксибензилоксикарбонил. Другие подходящие примеры представлены в Главе 1 монографии "Protocols for oligonucleotide conjugates", "Methods in Molecular Biology", vol. 26, ed. S.Agrawal, Humana Press, Totowa, NJ, 1993; S.L.Beaucage & R.P.Iyer, 1992, Tetrahedron, 48, 2223.
В предпочтительном варианте группа В мономерном LNA предпочтительно выбирают из нуклеотидных оснований и защищенных оснований.
В варианте настоящего изобретения с мономерным LNA одна из групп Q и Q*, предпочтительно Q*, образуют группу, выбираемую из Act-O-, Act-S-, Act-N(RH)-, Act-O-СН2-, Act-O-CH2-, Act-N(RH)-CH2-, а другие Q и Q*, предпочтительно Q*, образуют группу, выбираемую из водорода, азидогруппы, галогена, цианогруппы, нитрогруппы, гидроксила, Prot-O-, меркаптогруппы, Prot-S-, C1-6-алкилтиогруппы, аминогруппы, Prot-N(RH)-, моно- или ди(С1-6-алкил)аминогруппы, необязательно замещенной C1-6-алкоксигруппы, необязательно замещенного C1-6-алкила, необязательно замещенного С2-6-алкенила, необязательно замещенной С2-6-алкенилоксигруппы, необязательно замещенного С2-6-алкинила, необязательно замещенной С2-6-алкинилоксигруппы, монофосфата, дифосфата, трифосфата, ДНК-интеркалирующих групп, фотохимически активных групп, термохимически активных групп, хелатирующих групп, репортерных групп, лигандов, карбоксила, сульфоновой группы, гидроксиметила, Prot-O-CH2-, аминометила, Prot-N(RH)-СН2-, карбоксиметила, сульфонометила, a RH выбирают из водорода и C1-6-алкила.
В описанном выше случае группа “Prot” обозначает защитную группу для -ОН, -SH и -NH(RH), соответственно. Такие защитные группы выбирают из тех же групп, которые были определены выше для групп, защищающих гидроксильную группу, групп, защищающих меркаптогруппу и групп, защищающих аминогруппу, соответственно, принимая при этом во внимание необходимость обеспечения стабильности и ревертивности защитных групп. Однако предпочтительным является то, чтобы любая защитная группа для гидроксила выбиралась необязательно замещенного тритила, такого как диметокситритил (DMT), монометокситритил (ММТ) и тритил, и 9-(9-фенил)-ксантенила (пиксила), необязательно замещенного тетрагидропиранила (ТНР) (другие защитные группы для гидроксила, подходящие для проведения фосфорамидитного синтеза олигонуклеотидов, описаны у Agrawal ed., "Protocols for Oligonucleotide Conjugates", "Methods in Molecular Biology", vol. 26, Humana Press, Totowa, NJ, 1994 и "Protocols for Oligonucleotides and Analogs", vol. 20, ed. S.Agrawal, Humana Press, Totowa, NJ, 1993) или была защищена в виде ацеталя; чтобы любая защитная группа для -SH выбиралась из тритила, такого как диметокситритил (DMT), монометокситритил (ММТ) и тритил, и 9-(9-фенил)ксантенила (пиксила), необязательно замещенного тетрагидропиранила (ТНР) (другие защитные группы для меркаптогруппы, подходящие для проведения фосфорамидитного синтеза олигонуклеотидов, описаны у Agrawal - цит. выше); и чтобы любая защитная группа для группы –NH(RH) выбиралась из тритила, такого как диметокситритил (DMT), монометокситритил (ММТ) и тритил, и 9-(9-фенил)ксантенила (пиксила), необязательно замещенного тетрагидропиранила (ТНР) (другие защитные группы для аминогруппы, подходящие для проведения фосфорамидитного синтеза олигонуклеотидов, описаны у Agrawal цит. выше).
В описанном выше варианте, равно как и для всех вариантов мономерных LNA, описанных здесь, группа “Act” обозначает активационную группу для -ОН, -SH и –N(RH), соответственно. Такие активационные группы выбирают, например, из необязательно замещенного O-фосфорамидита, необязательно замещенного O-фосфорного триэфира, необязательно замещенного O-фосфорного диэфира, необязательно замещенного Р-фосфоната и необязательно замещенного O-фосфоната.
В контексте настоящего изобретения термин “фосфорамидит” обозначает группу формулы -Р(ORx)-N(Rv)2, где Rx обозначает необязательно замещенную алкильную группу, например, метил, 2-цианоэтил или бензил, а каждая группа Rv необязательно замещенные алкильные группы, например, этил или изопропил, или же группа –N(Rv)2 образует морфолиногруппу -N(CH2CH2)2O. Rx предпочтительно обозначает 2-цианоэтил, а две группы Rv предпочтительно идентичны и формируют изопропил. Таким образом, наиболее подходящим фосфорамидитом является N,N-диизопропил-O-(2-цианоэтил)фосфорамидит.
Должно быть понятно, что защитные группы, используемые в настоящем изобретении для отдельных мономеров LNA или нескольких мономерных LNA, могут быть выбраны таким образом, что тогда, когда LNA вносят в состав олигомера по настоящему изобретению, то имеется возможность осуществлять либо одновременного освобождения от защитной группы, либо последовательной депротекции защищенных функциональных групп. Последняя ситуация открывает возможность внесения избирательно по конкретному сайту одной или нескольких “активных/функциональных” групп, таких как ДНК-интеркалирующие группы, фотохимически активные группы, термохимически активные группы, хелатирующие группы, репортерные группы и лиганды, когда такие группы могут быть присоединены через спейсер в соответствии с описанным выше.
В предпочтительном варианте Q выбирают из водорода, азидогруппы, галогена, цианогруппы, нитрогруппы, гидроксила, Prot-O-, меркаптогруппы, Prot-S-, C1-6-алкилтиогруппы, аминогруппы, Prot-N(RH)-, моно- или ди(C1-6-алкил)аминогруппы, необязательно замещенной C1-6-алкоксигруппы, необязательно замещенного C1-6-алкила, необязательно замещенного С2-6-алкенила, необязательно замещенной С2-6-алкенилоксигруппы, необязательно замещенного С2-6-алкинила, необязательно замещенной С2-6-алкинилоксигруппы, монофосфата, дифосфата, трифосфата, ДНК-интеркалирующих групп, фотохимически активных групп, термохимически активных групп, хелатирующих групп, репортерных групп, лигандов, карбоксила, сульфоновой группы, гидроксиметила, Prot-O-CH2-, аминометила, Prot-N(RH)-CH2-, карбоксиметила, сульфонометила, где “Prot” является защитной группой для -ОН, -SH и -NH(RH), соответственно, a RH выбирают из Н и C1-6-алкила; и Q* выбирают из водорода, азидогруппы, галогена, цианогруппы, нитро-группы, гидроксила, Act-O-, меркаптогруппы, Act-S-, C1-6-алкилтиогруппы, аминогруппы, Act-N(RH)-, моно- или ди(С1-6-алкил)аминогруппы, необязательно замещенной C1-6-алкоксигруппы, необязательно замещенного C1-6-алкила, необязательно замещенного С2-6-алкенила, необязательно замещенной C2-6-алкенилоксигруппы, необязательно замещенного С2-6-алкинила, необязательно замещенной С2-6-алкинилоксигруппы, монофосфата, дифосфата, трифосфата, ДНК-интеркалирующих групп, фотохимически активных групп, термохимически активных групп, хелатирующих групп, репортерных групп, лигандов, карбоксила, сульфоновой группы, гидроксиметила, Prot-O-СН2-, аминометила, Prot-N(RH)-CH2-, карбоксиметила, сульфонометила, где “Act” является активационной группой для -ОН, -SH и -NH(RH), соответственно, a RH выбирают из водорода и C1-6-алкила.
Мономерные LNA базовой формулы II могут, при том, что они вносятся в состав олигомеров, представлять различные стереоизомеры. Следовательно, стереохимические варианты, описанные выше для LNA, внесенных в олигомеры, можно считать, в равной степени применимы в варианте с мономерными LNA (однако, необходимо заметить, что Р должно быть в этом случае заменено на Q).
В предпочтительном варианте настоящего изобретения мономерный LNA характеризуется базовой формулой IIа:
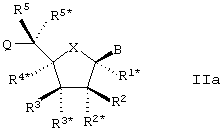
где заместители соответствуют данным выше определениям.
Более того, в связи с определениями заместителей, бирадикалов, R* и т.п., те же предпочтительные варианты, охарактеризованные выше для олигомера по настоящему изобретению, также применимы и для случая с мономерными LNA.
В конкретном предпочтительном варианте мономерные LNA по настоящему изобретению В соответствует основанию, а предпочтительно основание выбирают из тимина, цитозина, урацила, аденина и гуанина (в частности, из аденина и гуанина), Х представлен -О-, R2* и R4* одновременно означают бирадикал, выбираемый из (CH2)0-1-O-(CH2)1-3-, (СН2)0-1-S-(СН2)1-3- и -(СН2)0-1-N(RN)-(СН2)1-3-, в частности, -O-СН2-, -S-CH2- и –RN-СН2-, где RN выбирают из водорода и C1-4-алкила, Q представлен Prot-O-, R3* представлен Q*, который образует Act-OH, и каждый из R1*, R2, R3, R5 и R5* представляет водород. В этом варианте RN также может быть выбран из ДНК-интеркалирующих групп, фотохимически активных групп, термохимически активных групп, хелатирующих групп, репортерных групп и лигандов.
Еще в одном представляющем конкретный интерес варианте мономерных LNA по настоящему изобретению В представлен основанием, предпочтительно основанием, выбираемым из тимина, цитозина, урацила, аденина и гуанина (в частности, из аденина и гуанина), Х представлен -О-, R2* и R4* одновременно означают бирадикал, выбираемый из -(CH2)0-1-O-(CH2)1-3-, -(CH2)0-1-S-(CH2)1-3- и -(СН2)0-1-N(RN)-(СН2)1-3-, в частности, -О-СН2-, -S-CH2- и –RN-CH2-, где RN выбирают из Н и C1-4-алкила, Q выбирают из гидроксила, меркаптогруппы, C1-6-алкилтиогруппы, аминогруппы, моно- или ди(C1-6-алкил)аминогруппы, необязательно замещенной C1-6-алкоксигруппы, необязательно замещенной С2-6-алкенилоксигруппы, необязательно замещенной С2-6-алкинилоксигруппы, монофосфата, дифосфата и трифосфата, R3* - это Q*, выбираемая из водорода, азидогруппы, галогена, цианогруппы, нитрогруппы, гидроксила, меркаптогруппы, C1-6-алкилтиогруппы, аминогруппы, моно- или ди(C1-6-алкил)аминогруппы, необязательно замещенной C1-6-алкоксигруппы, необязательно замещенного C1-6-алкила, необязательно замещенного С2-6-алкенила, необязательно замещенной С2-6-алкенилоксигруппы, необязательно замещенного С2-6-алкинила и необязательно замещенной С2-6-алкинилоксигруппы, R3 выбирают из водорода, необязательно замещенного C1-6-алкила, необязательно замещенного С2-6-алкенила и необязательно замещенного С2-6-алкинила, а каждый из R1*, R2, R5 и R5* представлены водородом. Также в этом случае RN может быть выбран из ДНК-интеркалирующих групп, фотохимически активных групп, термохимически активных групп, хелатирующих групп, репортерных групп и лигандов.
Еще в одном конкретном представляющем интерес варианте мономерных LNA по настоящему изобретению В представлен основанием, Х представлен -О-, R2 и R3 одновременно означают бирадикал, выбираемый из (CH2)0-1-O-CH=CH-, (СН2)0-1-S-CH=CH- и (СН2)0-1-N(RN)-CH=CH-, где RN выбирают из Н и C1-4-алкила, Q выбирают из гидроксила, меркаптогруппы, C1-6-алкилтиогруппы, аминогруппы, моно- или ди(C1-6-алкил)аминогруппы, необязательно замещенной C1-6-алкоксигруппы, необязательно замещенной С2-6-алкенилоксигруппы, необязательно замещенной С2-6-алкинилоксигруппы, монофосфата, дифосфата и трифосфата, R3* - это Q*, выбираемая из водорода, азидогруппы, галогена, цианогруппы, нитрогруппы, гидроксила, меркаптогруппы, C1-6-алкилтиогруппы, аминогруппы, моно- или ди(C1-6-алкил) аминогруппы, необязательно замещенной C1-6-алкоксигруппы, необязательно замещенного C1-6-алкила, необязательно замещенного С2-6-алкенила, необязательно замещенной С2-6-алкенилоксигруппы, необязательно замещенного С2-6-алкинила и необязательно замещенной С2-6-алкинилоксигруппы, а каждый из R1*, R2*, R5 и R5* представлены водородом.
В одном из аспектов настоящего изобретения представляются различные производные LNA для твердофазного или жидкостного внесения в состав олигомера. Приводимые в качестве иллюстрирующих примеров мономеров, пригодных для внесения (1S,3R,4R,7S)-7-гидрокси-1-гидроксиметил-3-(тимин-1-ил)-2,5-диоксабицикло[2.2.1]гептана, (1S,3R, 4R,7S)-7-гидрокси-1-гидроксиметил-3-(цитозин-1-ил)2,5-диоксабицикло[2.2.1]гептана, (1S, 3R, 4R, 7S)-7-гидрокси-1-гидроксиметил-3-(урацил-1-ил)2,5-диоксабицикло[2.2.1]-гептана, (2S,3R,4R,7S)-7-гидрокси-1-гидроксиметил-3-(гуанин-1-ил)2,5-диоксабицикло[2.2.1]гептана, [1S,3R,4R,7S)-7-гидрокси-1-гидроксиметил-3-(аденин-1-ил)2,5-диоксабицикло[2.2.1]гептана с использованием фосфорамидитного метода, фосфотриэфирного метода и водородфосфонатного метода, соответственно, являются (1R,3R,4R,7S)-7-(2-цианоэтокси(диизопропиламино)-фосфинокси)-1-(4,4’-димeтoкcитpитилoкcимeтил)-3-(тимин-1-ил)-2,5-диоксабицикло[2.2.1]гептан, (1R,3R,4R,7S)-7-(4,4’-диметокситритилоксиметил)-3-(тимин-1-ил)2,5-диоксабицикло-[2.2.1]гептан-7-O-(2-хлорфенилфосфат) и [1R,3R,4R,7S)-7-(4,4’-диметокситритилоксиметил)-3-(тимин-1-ил)2,5-диоксабицикло[2.2.1]гептан-7-O-(водородфосфонат), а также их аналоги 3-(цитозин-1-ил), 3-(урацил-1-ил), 3-(аденин-1-ил) и 3-(гуанин-1-ил), соответственно. Кроме того, аналоги, в которых метиленоксибирадикалы мономеров заменены на метилентио-, метиленамино- или 1,2-этиленбирадикалы, также рассматриваются как представляющие интерес варианты настоящего изобретения. Метилентио- и метиленаминовые аналоги также считаются в равной степени применимыми, что и метиленоксианалоги: соответственно, конкретные реагенты, соответствующие внесению (1S,3R,4R,7S)-7-гидрокси-1-гидроксиметил-3-(тимин-1-ил)-2,5-диоксабицикло[2.2.1]гептана, (1S, 3R,4R,7S)-7-гидрокси-1-гидроксиметил-3-(цитозин-1-ил)2,5-диоксабицикло[2.2.1]-гептана, (1S,3R,4R,7S)-7-гидрокси-1-гидроксиметил-3-(урацил-1-ил)2,5-диоксабицикло[2.2.1]гептана, (1S,3R,4R,7S)-7-гидрокси-1-гидроксиметил-3-(гуанин-1-ил)2,5-диоксабицикло[2.2.1]гептана, (1S,3R,4R,7S)-7-гидрокси-1-гидроксиметил-3-(аденин-1-ил)2,5-диоксабицикло[2.2.1]гептана, также должны рассматриваться как конкретные, представляющие интерес реактивные мономеры по настоящему изобретению. Для метиленаминового аналога необходимо отметить, что вторичный амин может включать заместитель, выбираемый из необязательно замещенного C1-6-алкила, такого как метил и бензил, необязательно замещенного C1-6-алкилкарбонила, такого как трифторацетил, необязательно замещенного арилкарбонила и необязательно замещенного гетероарилкарбонила.
В конкретном представляющем интерес варианте настоящего изобретения представляется олигомер, включающий по крайней мере один LNA базовой формулы Iа:
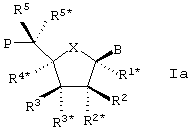
где X представлен -О-; В выбирают из оснований, ДНК-интеркалирующих групп, фотохимически активных групп, термохимически активных групп, хелатирующих групп, репортерных групп и лигандов; Р представляет положение радикала для межнуклеозидного “мостика” с последующим мономером или 5’-концевую группу, при том, что каждый межнуклеозидный “мостик” или 5’-концевая группа необязательно включают заместитель R5; R3* представлен группой Р*, которая образует межнуклеозидный “мостик” с предшествующим мономером или 3’-концевую группу; R2* и R4* одновременно образуют бирадикал, выбираемый из -О-, -S-, -N(R*)-, (CR*R*)r+s+1-, -(CR*R*)r-O-(CR*R*)s-, -(CR*R*)r-S-(CR*R*)s-, (CR*R*)r-N(R*)-(CR*R*)s-, -O-(CR*R*)r+s-O-, -S-(CR*R*)r+s-O-, -O-(CR*R*)r+s-S-, -N(R*)-(CR*R*)r+s-O-, -O-(CR*R*)r+s-N(R*)-, -S(CR*R*)r+s-S-, -N(R*)-(CR*R*)r+s-N(R*)-, -N(R*)-(CR*R*)s-S- и -S-(CR*R*)r+s-N(R*)-; где каждый R* независимо друг от друга выбирают из водорода, галогена, азидогруппы, цианогруппы, нитрогруппы, гидроксила, меркаптогруппы, аминогруппы, моно- или ди(C1-6-алкил)аминогруппы, необязательно замещенной C1-6-алкоксигруппы, необязательно замещенного C1-6-алкила, ДНК-интеркалирующих групп, фотохимически активных групп, термохимически активных групп, хелатирующих групп, репортерных групп и лигандов, а (или) два соседних (негеминальных) R* могут вместе образовывать двойную связь, и каждый из r и s равен 0-3 при условии, что сумма r+s равна 1-4; каждый из заместителей R1*, R2, R3, R5 и R5* независимо выбирают из водорода, необязательно замещенного C1-6-алкила, необязательно замещенного C2-6-алкенила, гидроксила, C1-6-алкоксигруппы, С2-6-алкенилалкоксигруппы, карбоксила, C1-6-алкоксикарбонила, C1-6-алкилкарбонила, формила, аминогруппы, моно- и ди(C1-6-алкил)аминогруппы, карбамоила, моно- и ди(C1-6-алкил)аминокарбонила, С2-6-алкилкарбониламиногруппы, карбамидо, азидогруппы, C1-6-алканоилоксигруппы, сульфоновой группы, сульфанила, C1-6-алкилтиогруппы, ДНК-интеркалирующих групп, фотохимически активных групп, термохимически активных групп, хелатирующих групп, репортерных групп и лигандов, и галогена, где два геминальных заместителя вместе могут образовывать оксогруппу; а также их соли (молекулярные соединения с кислотами и основаниями). В частности, R* выбирают из водорода, гидроксила, необязательно замещенной C1-6-алкоксигруппы, необязательно замещенного C1-6-алкила, ДНК-интеркалирующих групп, фотохимически активных групп, термохимически активных групп, хелатирующих групп, репортерных групп и лигандов, а любой из остальных заместителей R* представлен водородом. Конкретно бирадикал выбирают из -О-, -(CH2)0-1-O-(CH2)1-3-, (СН2)0-1-S-(CH2)1-3-, -(CH2)0-1-N(RN)-(CH2)1-3- и -(СН2)2-4-.
В другом конкретном представляющем интерес варианте настоящее изобретение представляет LNA базовой формулы IIа:
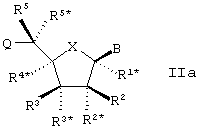
где X- -О-; В выбирают из оснований, ДНК-интеркалирующих групп, фотохимически активных групп, термохимически активных групп, хелатирующих групп, репортерных групп и лигандов; R3* представлен группой Q*; каждую из Q и Q* независимо выбирают из водорода, азидогруппы, галогена, цианогруппы, нитрогруппы, гидроксила, Prot-O-, Act-O-, меркаптогруппы, Prot-S-, Act-S-, C1-6-алкилтиогруппы, аминогруппы, Prot-N(RH)-, Act-NtRH)-, моно- или ди(C1-6-алкил)аминогруппы, необязательно замещенной C1-6-алкоксигруппы, необязательно замещенного C1-6-алкила, необязательно замещенного C2-6-алкенила, необязательно замещенной С2-6-алкенилоксигруппы, необязательно замещенного С2-6-алкинила, необязательно замещенной С2-6-алкинилоксигруппы, монофосфата, дифосфата, трифосфата, ДНК-интеркалирующих групп, фотохимически активных групп, термохимически активных групп, хелатирующих групп, репортерных групп, лигандов, карбоксила, сульфоновой группы, гидроксиметила, Prot-O-CH2-, Act-O-CH2-, аминометила, Prot-N(RH)-CH2-, Act-N(RH)-CH2-, карбоксиметила, сульфонометила, где “Prot” обозначает защитную группу для -ОН, -SH и -NH(RH), соответственно, a “Act” обозначает активационную группу для -ОН, -SH и -NH(RH), соответственно, a RH выбирают из водорода и C1-6-алкила; R2* и R4* одновременно означают бирадикал, выбираемый из -О-, -S-, -N(R*)-, (CR*R*)r+s+1-, -(CR*R*)r-O-(CR*R*)s-, -(CR*R*)r-S-(CR*R*)s-, (CR*R*)r-N(R*)-(CR*R*)s-, -O-(CR*R*)r+s-O-, -S-(CR*R*)r+s-O-, -O-(CR*R*)r+s-S-, -N(R*)-(CR*R*)r+s-O-, -O-(CR*R*)r+s-N(R*)-, -S(CR*R*)r+s-S-, -N(R*)-(CR*R*)r+s-N(R*)-, -N(R*)-(CR*R*)r+s-S- и -S-(CR*R*)r+s-N(R*)-; где каждый R* независимо друг от друга выбирают из водорода, галогена, азидогруппы, цианогруппы, нитрогруппы, гидроксила, меркаптогруппы, аминогруппы, моно- или ди(C1-6-алкил)аминогруппы, необязательно замещенной C1-6-алкоксигруппы, необязательно замещенного C1-6-алкила, ДНК-интеркалирующих групп, фотохимически активных групп, термохимически активных групп, хелатирующих групп, репортерных групп и лигандов, а (или) два соседних (негеминальных) R* могут вместе образовывать двойную связь, и каждый из r и s равен 0-3 при условии, что сумма r+s равна 1-4; каждый из заместителей R1*, R2, R3, R5 и R5* независимо выбирают из водорода, необязательно замещенного C1-6-алкила, необязательно замещенного С2-6-алкенила, гидроксила, C1-6-алкоксигруппы, С2-6-алкенилалкоксигруппы, карбоксила, C1-6-алкоксикарбонила, C1-6-алкилкарбонила, формила, аминогруппы, моно- и ди(C1-6-алкил)аминогруппы, карбамоила, моно- и ди(C1-6-алкил)аминокарбонила, C1-6-алкилкарбониламиногруппы, карбамидо, азидогруппы, C1-6-алканоилоксигруппы, сульфоновой группы, сульфанила, C1-6-алкилтиогруппы, ДНК-интеркалирующих групп, фотохимически активных групп, термохимически активных групп, хелатирующих групп, репортерных групп и лигандов, и галогена, где два геминальных заместителя вместе могут образовывать оксогруппу; а также их соли (молекулярные соединения с кислотами и основаниями); при условии, что любая химическая группа (включая любое из оснований), которая реактивна в условиях, при которых проводится синтез олигонуклеотида, может быть необязательно защищена по своим функциональным составляющим. Предпочтительно один из R* выбирают из водорода, гидроксила, необязательно замещенной C1-6-алкоксигруппы, необязательно замещенного C1-6-алкила, ДНК-интеркалирующих групп, фотохимически активных групп, термохимически активных групп, хелатирующих групп, репортерных групп и лигандов, а любой из остальных заместителей R* представлен водород. Конкретно бирадикал выбирают из -О-, -(СН2)0-1-O-(CH2)1-3-, -(CH2)0-1-S-(CH2)1-3-, -(СН2)0-1-N(RN)-(СН2)1-3- и -(СН2)2-4-.
В целом настоящее изобретение представляет олигомеры, характеризующиеся удивительно высокими гибридизационными параметрами с точки зрения уровней аффинности и специфичности. Следовательно, настоящее изобретение представляет олигомер, включающий по крайней мере один нуклеозидный аналог, который придает олигомеру Тm по отношению к комплементарному ДНК-олигонуклеотиду, которая по крайней мере на 2,5° С выше, предпочтительно по крайней мере на 3,5° С выше, конкретно по крайней мере на 4,0° С выше, особенно предпочтительно по крайней мере на 5,0° С выше, чем у соответствующего модифицированного контрольного олигонуклеотида, который не включает каких-либо нуклеозидных аналогов. В частности, Тm олигомера по крайней мере на 2,5× N° С выше, предпочтительно по крайней мере на 3,5× N° С выше, конкретно по крайней мере на 4,0× N° С выше, особенно предпочтительно по крайней мере на 5,0× N° С выше, где N - число нуклеозидных аналогов в олигомере.
В случае гибридизации с комплементарными РНК-олигонуклеотидами по крайней мере один нуклеозидный аналог придает олигомеру Тm по отношению к комплементарному ДНК-олигонуклеотиду, которая по крайней мере на 4,0° С выше, предпочтительно по крайней мере на 5,0° С выше, конкретно по крайней мере на 6,0° С выше, особенно предпочтительно по крайней мере на 7,0° С выше, чем у соответствующего модифицированного контрольного олигонуклеотида, который не включает каких-либо нуклеозидных аналогов. В частности, Тm олигомера по крайней мере на 4,0× N° С выше, предпочтительно по крайней мере на 5,0× N° С выше, конкретно по крайней мере на 6,0× N° С выше, особенно предпочтительно по крайней мере на 7,0× N° С выше, где N - число нуклеозидных аналогов в олигомере.
Термин “соответствующий немодифицированный контрольный олигонуклеотид” призван обозначить олигонуклеотид, полностью состоящий из природных нуклеотидов, которые представляют те же основания точно в таком же порядке (и в той же самой ориентации).
Значение температуры плавления (диссоциации дуплекса) Тm определяется в одном из вариантов условий (например, так, как это описано в примере 129):
1) 10 мМ Na2HPO4 - рН 7,0, 100 мМ NaCl, 0,1 мМ EDTA;
2) 10 мМ Na2HPO4 - рН 7,0, 0,1 мМ EDTA;
3) 3 М хлорид тетраметиламмония (ТМАС), 10 мМ Na2HPO4 - рН 7,0, 0,1 мМ EDTA;
предпочтительно в условиях (1), при эквимолярных количествах (обычно 1, мкМ) олигомера и комплементарного ДНК-олигонуклеотида.
Предпочтительно олигомер соответствует определенному выше, т.е. в нем по крайней мере один нуклеозидный аналог характеризуется формулой I, а В представлен нуклеотидным основанием. Конкретными представляющими интерес случаями являются те, когда по крайней мере один нуклеозидный аналог включает основание, выбираемое из аденина и гуанина.
Далее, что касается уровней специфичности и аффинности, олигомер при гибридизации с частично комплементарным ДНК-олигонуклеотидом или с частично комплементарным РНК-олигонуклеотидом, имеющими одно или большее число несоответствий с названным олигомером, должен проявлять снижение Тm вследствие таких несоответствий, которое окажется таким же или более значительным по сравнению с тем, что наблюдалось бы для соответствующего немодифицированного контрольного олигонуклеотида, который не включает каких-либо нуклеозидных аналогов. Также такой олигомер должен по сути иметь тот же уровень чувствительности величины Тm к ионной силе буфера для гибридизации, что и соответствующий немодифицированный контрольный олигонуклеотид.
Представляемые настоящим изобретением олигомеры обычно модифицированы по крайней мере на 30%, предпочтительнее модифицированы по крайней мере на 50%, в частности, модифицированы на 70% и в наиболее предпочтительных вариантах модифицированы на 100%.
Олигомеры по настоящему изобретению характеризуются существенно более высокой стабильностью по отношению к действию 3’-экзонуклеаз по сравнению с соответствующим немодифицированным контрольным олигонуклеотидом. Это важное свойство может быть протестировано в соответствии с описанным в примере 136.
Определения
В контексте настоящего изобретения термин “C1-12-алкил” обозначает линейную, циклическую или разветвленную углеводородную группу, включающую 1-12 атомов углерода, такую как метил, этил, пропил, изопропил, циклопропил, бутил, трет-бутил, изобутил, циклобутил, пентил, циклопентил, гексил, циклогексил и додецил. Аналогичным образом, термин “C1-6-алкил” обозначает линейную, циклическую или разветвленную углеводородную группу, включающую 1-6 атомов углерода, такую как метил, этил, пропил, изопропил, пентил, циклопентил, гексил, циклогексил, а термин “C1-4-алкил” обозначает линейную, циклическую или разветвленную углеводородную группу, включающую 1-4 атома углерода, такую как метил, этил, пропил, изопропил, циклопропил, бутил, изобутил, трет-бутил, циклобутил.
Предпочтительными примерами “C1-6-алкилов” являются метил, этил, пропил, изопропил, бутил, трет-бутил, изобутил, пентил, циклопентил, гексил, циклогексил, в частности, метил, этил, пропил, изопропил, трет-бутил, изобутил и циклогексил. Предпочтительными примерами “C1-4-алкилов” являются метил, этил, пропил, изопропил, бутил, трет-бутил и изобутил.
Сходным образом, термин “С2-12-алкенил” обозначает линейную, циклическую или разветвленную углеводородную группу, включающую 2-12 атомов углерода и имеющую одну ненасыщенную связь. Примерами алкенильных групп являются винил, аллил, бутенил, пентенил, гексенил, гептенил, октенил, додекаенил. Аналогичным образом, термин “С2-6-алкенил” призван обозначить линейную, циклическую или разветвленную углеводородную группу, включающую 2-6 атомов углерода и имеющую одну ненасыщенную связь. Предпочтительными примерами алкенилов являются винил, аллил, бутенил, а особенно аллил.
Сходным образом термин “С2-12-алкинил” обозначает линейную, циклическую или разветвленную углеводородную группу, включающую 2-12 атомов углерода и имеющую тройную связь. Их примерами являются этинил, пропинил, бутинил, октинил и додеканил.
В контексте настоящего изобретения, т.е. в связи с терминами “алкил”, “алкенил” и “алкинил”, термин “необязательно замещенный” обозначает то, что рассматриваемая группа может быть замещена одно- или многократно, предпочтительно 1-3 раза, группами, выбираемыми из гидроксила (который, в случае связи с ненасыщенным атомом углерода, может иметь таутомерную кетонную форму), C1-6-алкоксигруппы (например, C1-6-алкилокси), С2-6-алкенилоксигруппы, карбоксила, оксогруппы (образуя кетонную или альдегидную составляющую), C1-6-алкоксикарбонила, C1-6-алкилкарбонила, формила, арила, арилоксикарбонила, арилоксигруппы, арилкарбонила, гетероарила, гетероарилоксикарбонила, гетероарилоксигруппы, гетероарилкарбонила, аминогруппы, моно- и ди(C1-6-алкил)аминогруппы, карбамоила, моно- и ди(C1-6-алкил)аминокарбонила, амино-С1-6-алкиламинокарбонила, моно- и ди(C1-6-алкил) амино-C1-6-алкиламинокарбонила, C1-6-алкилкарбониламиногруппы, цианогруппы, гуанидиногруппы, карбамидогруппы, C1-6-алканоилоксигруппы, сульфоновой группы, C1-6-алкилсульфонилоксигруппы, нитрогруппы, сульфанила, C1-6-алкилтиогруппы, галогена, где любой арил и гетероарил может быть замещен так, как это описано ниже для “необязательно замещенных арила и гетероарила”.
Предпочтительно заместители выбирают из гидроксила, C1-6-алкоксигруппы, карбоксила, C1-6-алкоксикарбонила, C1-6-алкилкарбонила, формила, арила, арилоксикарбонила, арилкарбонила, гетероарила, аминогруппы, моно- и ди(C1-6-алкил)аминогруппы, карбамоила, моно- и ди(C1-6-алкил)аминокарбонила, амино-C1-6-алкиламинокарбонила, моно- и ди(C1-6-алкил)амино-C1-6-алкиламинокарбонила, C1-6-алкилкарбониламиногруппы, цианогруппы, карбамидогруппы, галогена, где арил и гетероарил могут быть замещены 1-5 раз, предпочтительно 1-3 раза, C1-4-алкилом, C1-4-алкоксигруппой, нитрогруппой, цианогруппой, аминогруппой или галогеном. Наиболее предпочтительными примерами являются гидроксил, C1-6-алкоксигруппа, карбоксил, арил, гетероарил, аминогруппа, моно- и ди(C1-6-алкил) аминогруппа и галоген, где арил и гетероарил могут быть 1-3 раза замещены C1-4-алкилом, C1-4-алкоксигруппой, нитрогруппой, цианогруппой, аминогруппой или галогеном.
В контексте настоящего изобретения термин “арил” обозначает полностью или частично ароматическое карбоциклическое кольцо или систему колец, такие как фенил, нафтил, 1,2,3,4-тетрагидронафтил, антрацил, фенантрацил, пиренил, бензопиренил, флуоренил и ксантенил, среди которых предпочтительным примером является фенил.
Термин “гетероарил” обозначает полностью или частично ароматическое карбоциклическое кольцо или систему колец, в которых один или большее число атомов углерода заменены на гетероатомы, например, на атомы азота (=N- или -NH), серы и(или) кислорода. Примерами таких гетероарильных групп являются оксазолил, изоксазолил, тиазолил, изотиозолил, пирролил, имидазолил, пиразолил, пиридинил, пиразинил, пиридазинил, пиперидинил, кумарил, фурил, хинолил, бензтиазолил, бензотриазолил, бензодиазолил, бензооксозолил, фталазинил, фталанил, триазолил, тетразолил, изохинолил, акридинил, карбазолил, дибензазепинил, индолил, бензопиразолил, феноксаpзонил.
В контексте настоящего изобретения, т.е. в связи с терминами “арил” и “гетероарил”, термин “необязательно замещенный” означает то, что такая рассматриваемая группа замещена одно- или многократно, предпочтительно 1-5 раз, в частности, 1-3 раза, группой (группами), выбираемой из гидроксила (который, в случае его присутствия в составе енольной системы, может иметь таутомерную кетонную форму), C1-6-алкила, C1-6-алкоксигруппы, оксогруппы (которая может быть представлена таутомерной енольной формой), карбоксила, C1-6-алкоксикарбонила, C1-6-алкилкарбонила, формила, арила, арилоксигруппы, арилоксикарбонила, арилкарбонила, гетероарила, аминогруппы, моно- и ди(C1-6-алкил)аминогруппы, карбамоила, моно- и ди(C1-6-алкил)-аминокарбонила, амино-C1-6-алкиламинокарбонила, моно- и ди(C1-6-алкил)-амино-C1-6-алкиламинокарбонила, C1-6-алкилкарбониламиногруппы, цианогруппы, гуанидиногруппы, карбамидогруппы, C1-6-алканоилоксигруппы, сульфоновой группы, C1-6-алкилсульфонилоксигруппы, нитрогруппы, сульфанила, дигалоген-С1-4-алкила, тригалоген-C1-4-алкила, галогена, где арильные и гетероарильные заместители могут быть 1-3 раза замещены C1-4-алкилом, C1-4-алкоксигруппой, нитрогруппой, цианогруппой, аминогруппой или галогеном. Предпочтительными примерами являются гидроксил, C1-6-алкил, C1-6-алкоксигруппа, карбоксил, C1-6-алкоксикарбонил, C1-6-алкилкарбонил, арил, аминогруппа, моно- и ди(С1-6-алкил) аминогруппа и галоген, где арил может быть 1-3 раза замещен C1-4-алкилом, C1-4-алкоксигруппой, нитрогруппой, цианогруппой, аминогруппой или галогеном.
Термин “галоген” включает фтор, хлор, бром и йод.
Должно быть понятно, что олигомеры (в состав которых включены LNA) и сами по себе LNA включают их возможные соли, среди которых наиболее предпочтительными являются фармацевтически приемлемые соли. Соли включают основные и кислотно-аддитивные соли. Примерами солей кислот являются гидрохлориды, соли натрия, соли кальция, соли калия и т.п. Примерами основных солей являются соли, в которых присоединяющийся ион выбирают из ионов щелочных металлов, таких как натрий и калий, щелочноземельных металлов, таких как кальций, и ионов аммония (+N(Rg)3Rh, где каждый из Rg и Rh независимо друг от друга обозначает необязательно замещенный C1-6-алкил, необязательно замещенный C2-6-алкенил, необязательно замещенный арил или необязательно замещенный гетероарил). Фармацевтически приемлемыми солями являются те соли, которые описаны в Remington’s Pharmaceutical Sciecnes, 17th edition, ed. A.R.Gennaro, Mack Publ. Co., Easton, PA, 1985 и в более поздних изданиях, а также в энциклопедии "Encyclopedia of Pharmaucetical Technology". Таким образом, термин “их соли (молекулярные соединения с кислотами и основаниями)” в данном тексте призван обозначить такие соли. Более того, олигомеры и LNA, равно как и любые промежуточные соединения или исходные материалы для них также могут находиться в форме гидратов.
Получение мономеров
В предпочтительном варианте нуклеозиды, включающие дополнительное 2’-О,4’-С-соединенное кольцо, были синтезированы с помощью следующей процедуры.
Синтез некоторых 4’-С-гидроксиметилнуклеозидов был описан ранее (R.D.Youssefyeh, J.P.H.Verheyden & J.G.Moffatt, 1979, J. Org Chem., 44, 1301; G.H.Jones, M.Taniguchi, D.Tegg & J.G.Moffatt, 1979, J. Org. Chem., 44, 1309; C.O-Yang, H.Y.Wu, E.B.Fraser-Smith & K.A.M.Walker, 1992, Tetrahedron Lett., 33, 37; H.Thrane, J.Fensholdt, M.Regner & J.Wengel, 1995, Tetrahedron, 51, 10389; K.D.Nielsen, F.Kirpekar, P.Roepstorff & J.Wengel, 1995, Bioorg. Med. Chem., 3, 1493). В качестве примера синтеза 2’-O,4’-С-соединенных бициклических нуклеозидов заявителями была выбрана стратегия, начинающаяся с 4’-С-гидроксиметилфуранозного производного (31). Бензилирование, ацетилирование и ацетолиз с последующим дополнительным ацетилированием приводили к получению фуранозы (33), являющейся ключевым промежуточным продуктом в нуклеозидном сочетании. В стереоселективной реакции с использованием силилированного тимина получали соединение 34, которое деацетилировали с получением нуклеозиддиола (35). Тозилирование с последующим замыканием кольца, индуцируемым основанием, привело к получению производного 2’-O,4’-С-соединенного бициклического нуклеозида (36). В результате дебензилирования получали освобожденный от защиты бициклический нуклеозидный аналог 37, который трансформировали в защищенный диметокситритилом 5’-O-4,4’-аналог 38 и затем в фосфорамидитное производное 39, необходимое для синтеза олигонуклеотида. Сходная процедура была применена для синтеза соответствующих урацилового, аденинового, цитозинового и гуанинового нуклеозидов, что рассматривается в разделе примеров. Такой метод сочетания является лишь одним из ряда возможных процедур, что должно быть очевидно для специалиста в данной области техники. Также возможным является применение стратегии, начинающейся с преобразованного нуклеозида. Таким образом, например, преобразование уридинового производного 62 в производное 44 была успешно осуществлена путем тозилирования, деизопропилидинирования и индуцированного основанием замыкания кольца. В другом примере конверсия нуклеозида 67 в нуклеозид 61В осуществляется в соответствии с фиг.34. Конверсия бициклического тиминового нуклеозида 37 в соответствующий 5-метилцитозиновый нуклеозид 65 осуществляли путем ряда известных реакций с использованием триазола и РОСl3 с последующим бензилированием и обработкой аммиаком. Сходная процедура применима для синтеза 57С и 44. Как пример других возможных стратегий синтеза можно привести сочетание предварительно циклизованных фуранозных дериватов, которые уже включают дополнительное кольцо, с нуклеотидными производными. Такая стратегия, кроме того, позволяет получить соответствующие α -нуклеозидные аналоги. При проведении реакции сочетания с защищенным метиленфуранозидом 4-С,2-O-метилен-D-рибофуранозы в основном был получен продукт с открытым кольцом. Однако сочетание 1-O-ацетилфуранозы (207) или тиофенилфуранозы (212) приводит к успешному получению LNA-нуклеозидов, а одним из получаемых продуктов являются α -аномеры. Инкорпорация таких (x-LNA-нуклеозидов будет возможной с использованием стандартных методик олигомеризации (применимых также в отношении LNA, включающих Z) с получением α -LNA-олигомеров. Кроме того, возможной является стратегия синтеза, осуществляемая путем нуклеозидного сочетания с использованием 4’-С-гидроксиметилфуранозы, которая уже активирована по замыканию кольца (например, вследствие наличия в ее составе мезильной или тозильной группы в составе 4’-С-гидроксиметильной группы), примером чему является конверсия фуранозы (78) в нуклеозид 79 с последующим освобождением от защиты и замыканием кольца с получением 36. Еще одной приемлемой стратегией является химическое или каталитическое трансгликозилирование или аномеризация подходящих фуранозных дериватов или нуклеозидов. Эти и другие стратегии обеспечивают синтез бициклических нуклеозидов, включающих другие нуклеотиды или их аналоги, за счет либо сочетания с этими нуклеотидами или аналогами, либо начиная с преформированных нуклеозидных производных соединений.
Описанные примеры призваны быть иллюстрирующими для процедур и примеров по настоящему изобретению. Для проверки структуры синтезированных соединений использовали одно- и двухмерный ЯМР, в частности, в эксперименте NOE.
Дополнительный вариант настоящего изобретения связан с представлением бициклических нуклеозидов, включающих дополнительные кольца различного размера и различной химической структуры. Для специалистов в области органического синтеза, исходя из приведенного выше методического описания, должно быть понятно, что циклизация других нуклеозидов является возможной с использованием сходных процедур, включая также нуклеозиды, имеющие различные С-ответвления. Для специалиста в данной области техники не составит труда отыскать в научной литературе руководства по получению таких промежуточных продуктов, как замещенных нуклеозидных аналогов: см., например, международную заявку WO 96/14329. Что касается колец различного химического состава, то ясно, что применение сходных процедур или процедур, вообще хорошо разработанных в области органической химии, синтез, например, тиоаналогов, приводимых в качестве примера оксоаналогов, возможен в той же степени, что и синтез соответствующих аминоаналогов (например, с применением реакций нуклеофильного замещения или реакций восстановительного алкилирования).
В разделе примеров описан синтез амино-LNА-аналогов (73-74F). Конверсия 74 и 74D стандартным образом выстроенные блоки для олигомеризации возможны с помощью 5’-O-DMT-защиты и 3’-O-фосфитилирования с последующими стандартными процедурами. В случае амино-LNА-аналогов защита 2’-аминофункциональной группы необходима из соображений контроля за линейной олигомеризацией. Такая защита может быть осуществлена с использованием стандартных защитных групп для аминогрупп, таких как Fmoc, трифторацетил или ВОС. С другой стороны, N-алкильная группа (например, бензил, метил, этил, пропил или функционализованный алкил) может сохраняться в ходе трансформаций нуклеозидов и олигомеризации. На фиг.35 и 36 показаны стратегии, основанные на использовании N-трифторацетильных и N-метильных производных. Как видно из фиг.37, конверсия нуклеозида 75 в 2’-тио-LNА-нуклеозидный аналог 76D была успешно осуществлена, равно как и последующий синтез его фосфорамидитного производного 76F. Соединение 76F характеризуется структурой, необходимой для автоматического синтеза 2’-тио-LNА-олигонуклеотидов. Синтон N-трифторацетил-2’-амино-LNA (74А) характеризуется структурой, необходимой для автоматического синтеза 2’-амино-LNA-олигонуклеотидов.
Синтез соответствующих цитозиновых, гуаниновых и адениновых производных 2’-тио- и 2’-амино-LNА-нуклеозидов может быть осуществлен с применением стратегий, аналогичных тем, которые показаны на фиг.35, 36 и 37. С другой стороны, стереохимия вокруг атома С-2’ может быть инвертирована перед проведением циклизации либо с использованием фуранозного синтона, имеющего стандартную конфигурацию (например, арабиноконфигурацию), либо путем инвертирования конфигурации вокруг атома углерода С-2’, начиная с нуклеозида/фуранозы, имеющих рибоконфигурацию. Последующая активация 2’-β -ОН, например, путем тозилирования, и двойное нуклеофильное замещение, как в описанном выше примере с урацилом/тимином, могут привести к получению желательных 2’-тио-LNA- или 2’-амино-LNА-нуклеозидов. Полученные таким образом защищенные аналоги цитозина, гуанина и аденина могут быть подготовлены для олигомеризации с применением стандартных реакций (DMT-протекция и фосфитилирование) в соответствии с описывавшимся выше для других примеров.
Получение олигомеров
Линейные, разветвленные (M.Groetli & B.S.Sproat, 1995, J. Chem. Soc., Chem. Commun., 495; R.H.E.Hudson & M.J.Damha, 1993, J. Amer. Chem. Soc., 115, 2119; M. Von Buren, G.V.Petersen, K.Rasmussen, G.Brandenburg, J.Wengel & F.Kirpekar, 1995, Tetrahedron, 51, 8491) и циклические (G.Prakash & E.T.Kool, 1992, J. Amer. Chem. Soc., 114, 3523) олиго- и полинуклеотиды по настоящему изобретению могут быть получены с использованием методов полимеризации, хорошо известных для химии нуклеиновых кислот специалистам в области органической химии. Для этой цели использовалась фосфорамидатная химия (S.L.Beaucage & R.P.Iyer, 1993, Tetrahedron, 49, 6123; S.L.Beaucage & R.P.Iyer, 1992, Tetrahedron, 48, 2223), однако также использовали методы, например, водород-фосфонатной химии, фосфотриэфирной химии или каталитического синтеза. В целом использовали стандартные условия сочетания и фосфорамидитный способ, хотя для некоторых мономеров по настоящему изобретению были использованы более продолжительные периоды реакции сочетания и(или) повторы реакции сочетания со свежими реактивами, и(или) применялись более концентрированные реактивы реакции сочетания. В качестве иной возможности также могут быть использованы более активные, чем 1H-тетразол, активаторы, позволяющие увеличивать скорость реакции сочетания. Все фосфорамидитные соединения 39, 46, 53, 57D, 61D и 66 обеспечивают сочетание с удовлетворительным выходом >95%. LNA-олигомер на основе только фосфотиоата (табл. 7) был синтезирован с применением стандартных процедур. Таким образом, фосфотиоатный LNA-олигомер был эффективно синтезирован (выход реакции сочетания ≥ 98%) в результате исключения нормального, например, с помощью йода/пиридина/пероксида водорода, окисления, используемого в синтезе фосфодиэфирных олигомеров, с окислением реактивом Бьюкажа (доступен на коммерческой основе). 2’-амино-LNA- и 2’-метиламино-LNА-олигонуклеотиды (табл. 9) были эффективно синтезированы (выход реакции сочетания ≥ 98%) с использованием амидитов 74А и 74F. 2’-тио-LNА-олигонуклеотиды (табл. 8) были эффективно синтезированы с использованием амидита 76F с применением стандартной фосфорамидитной процедуры в соответствии с тем, что было описано для LNA-олигонуклеотидов. После синтеза желательной последовательности проводилась ее обработка в стандартных условиях (отщепление от твердой подложки и удаление защитных групп с использованием 30%-ного аммиака при 55° С в течение 5 часов). Очистка LNA-олигонуклеотидов была осуществлена с использованием обратно-фазовых картриджей для очистки и(или) ВЭЖХ с обращенной фазой и(или) путем осаждения этанолом или бутанолом. Методы капиллярного гель-электрофореза, ВЭЖХ с обращенной фазой и MALDI-масс-спектрометрии были использованы для проверки чистоты синтезированных олигонуклеотидных аналогов и для проверки того, что желаемое число бициклических нуклеозидных аналогов по настоящему изобретению оказалось внесенным в состав.
Дополнительный аспект настоящего изобретения связан с представлением способов получения олигонуклеотидных аналогов, включающих LNA, в составе которых имеются неприродные межнуклеозидные “мостики”. Например, синтез соответствующих фосфотиоатных или фосфорамидатных аналогов возможен с использованием стратегий, хорошо известных в области химии олигонуклеотидов ("Protocols for Oligonucleotides and Analogs", vol. 20, ed. S.Agrawal, Humana Press, Totowa, NJ, 1993; S.L.Beaucage & R.P.Iyer, 1993, Tetrahedron, 49, 6123; S.L.Beaucage & R.P.Iyer, 1992, Tetrahedron, 48, 2223; E.Uhlmann & A.Peyman, Chem. Rev., 90, 543).
Таким образом, настоящее изобретение также представляет применение представляемых здесь LNA для получения LNA-модифицированных олигонуклеотидов. Должно быть понятно, что LNA-модифицированные олигонуклеотиды могут включать нормальные нуклеозиды (т.е. встречающиеся в естественных условиях, такие как рибонуклеозиды и/или дезоксирибонуклеозиды), равно как и модифицированные нуклеозиды, отличающиеся от тех, которые описываются общей формулой II. В конкретном варианте инкорпорация LNA обеспечивает модуляцию способности олигонуклеотида служить субстратом для ферментов, активных в отношении нуклеиновых кислот.
Более того, материалы твердых подложек, на которые иммобилизуют необязательно защищенный по нуклеотиду и необязательно защищенный по 5’-ОН LNA, особенно интересны в качестве материала для синтеза LNA-модифицированных олигонуклеотидов, в которых LNA-мономер включается по 3’-концу. В этом примере материалом твердой подложки предпочтительно является CPG, например, легко (на коммерческой основе) доступный материал CPG, на котором 3’-функционализованный, необязательно с защищенным нуклеотидом и необязательно защищенный по 5’-ОН LNA связывается с использованием условий, оговариваемых производителем конкретного материала. Может быть, например, использован материал BioGenex Universal CPG Support (BioGenex, США). Защитной группой для 5’-ОН. может быть, например, группа DMT. Функциональная 3’-группа должна выбираться с учетом условий, предлагаемых для рассматриваемого материала CPG.
Применение
Настоящее изобретение описывает неожиданную находку того, что различные новые производные бициклических нуклеозидных мономеров (LNA), в случае их внесения в состав олигонуклеотидов, в очень значительной степени увеличивают уровень аффинности таких модифицированных олигонуклеотидов в отношении и одноцепочечных ДНК, и одноцепочечных РНК по сравнению с немодифицированными олигонуклеотидами. Далее представляется неожиданное установление того, что и полностью, и частично LNA-модифицированные олигонуклеотиды проявляют резко повышенные гибридизационные свойства в отношении комплементарных им нуклеиновых последовательностей. В зависимости от конкретного применения, использование этих LNA предоставляет многообещающую возможность либо существенно повышать уровень аффинности стандартного олигонуклеотида без соответствующих потерь в специфичности (при постоянном размере олигонуклеотида), либо существенно повышать уровень специфичности без соответствующих потерь в аффинности (снижение размера олигонуклеотида). Также настоящее изобретение представляет неожиданное обнаружение того, что LNA-модифицированные олигонуклеотиды, в дополнение к существенному улучшению параметров гибридизуемости, проявляют ряд ценных физико-химических свойств, характерных для нормальных ДНК- и РНК-олигонуклеотидов. Приводимые здесь примеры включают отличную растворимость, реагирование LNA-модифицированных олигонуклеотидов на соли, такие как хлорид натрия и хлорид триметиламмония, напоминающее таковые немодифицированных олигонуклеотидов, способность LNA-модифицированных олигонуклеотидов работать в качестве затравок для различных полимераз, способность LNA-модифицированных олигонуклеотидов служить затравками в реакции амплификации мишеней с применением термостабильных ДНК-полимераз, способность LNA-модифицированных олигонуклеотидов выполнять роль субстрата для полинуклеотидкиназы Т4, способность биотинилированных LNA к специфичному по последовательности отбору ПЦР-ампликонов на покрытой стрептавидином твердой подложке, способность иммобилизованных LNA-модифицированных олигонуклеотидов к специфичному по последовательности отбору ампликонов и, что очень важно, способность LNA-модифицированных олигонуклеотидов к специфичному по последовательности распознаванию двухцепочечной ДНК по механизму инвазии цепи. Следовательно, для специалистов в данной области техники будет понятно, что новые нуклеозидные аналоги характеризуются исключительной применимостью в качестве средств для улучшения параметров и характеристик методик, основанных на использовании олигонуклеотидов и применяемых в терапии, диагностике и молекулярной биологии.
Объектом настоящего изобретения является представление мономерных LNA в соответствии с настоящим изобретением, которые могут быть инкорпорированы в состав олигонуклеотидов с применением методик и оборудования, хорошо известных специалистам в области синтеза олигонуклеотидов.
Другим объектом настоящего изобретения является представление полностью или частично LNA-модифицированных олигонуклеотидов (олигомеров), которые способны гибридизовать по специфичному по отношению к нуклеотидной последовательности типу с комплементарными олигонуклеотидами с образованием либо дуплексов, либо триплексов, проявляя существенно более высокий уровень аффинности по сравнению с соответствующими комплексами, образуемыми немодифицированными олигонуклеотидами.
Другим объектом настоящего изобретения является применение LNA для повышения уровня специфичности нормальных олигонуклеотидов без соответствующей потери в уровне аффинности. Это может быть достигнуто путем снижения длины (и тем самым аффинности) нормального олигонуклеотида до той степени, при которой будет достигнут тот же уровень аффинности, который обеспечивался включением LNA.
Еще одним объектом настоящего изобретения является представление полностью или частично модифицированных олигонуклеотидов, включающих и LNA, и нормальные нуклеозиды, а также иные нуклеозидные аналоги.
Следующим объектом настоящего изобретения является применение свойства высокой аффинности LNA для создания модифицированных олигонуклеотидов с исключительными свойствами аффинности, которые были бы способны связываться с соответствующими последовательностями-мишенями “в пределах” молекулы двухцепочечной ДНК по механизму “замещяющий цепь”.
Следующим объектом настоящего изобретения является представление различных классов LNA, которые, в случае их включения в состав олигонуклеотидов, будут различаться по своей аффинности в отношении соответствующих комплементарных нуклеозидов. В соответствии с настоящим изобретением это может быть достигнуто либо заменой нормальных нуклеотидов G, А, Т, С и U их производными, характеризующимися, например, измененной способностью к образованию водородных связей, либо использованием LNA, которые отличаются по своей базовой структуре. Доступность таких разных LNA обеспечивает возможность тонкой настройки уровней аффинности модифицированных олигонуклеотидов.
Другим объектом настоящего изобретения является представление LNA-модифицированных олигонуклеотидов, которые характеризуются более высокой устойчивостью к действию нуклеаз по сравнению с соответствующими немодифицированными олигонуклеотидами.
Еще одним объектом настоящего изобретения является представление LNA-модифицированных олигонуклеотидов, которые способны рекрутировать РНКазу-Н.
Дополнительным объектом настоящего изобретения является представление LNA, которые могут являться лекарственными средствами. Известны многочисленные примеры терапевтически применяемых нуклеозидных аналогов, и сходные производные представленных здесь нуклеозидных аналогов могут быть синтезированы с применением методик, известных из научной литературы (Е. De Clercq, 1995, J. Med. Chem., 38, 2491; P.Herdewijn & E.De Clercq, 1996, ″ Classical antiviral agents and design of new antiviral agents". In ″ A Textbook of Drug Design and Development", eds. P.Krogsgaard-Larsen, T.Liljefors & U.Madsen, Harwood Acad. Publ., Amsterdam, p. 425; I.K.Larsen, 1996, ″ Anticancer agents". In "A Textbook of Drug Design and Development", eds. P.Krogsgaard-Larsen, T.Liljefors & U.Madsen, Harwood Acad. Publ., Amsterdam, p. 460).
Как было показано, двухцепочечная РНК проявляет антивирусную активность и опухоль-супрессорную активность (Sharp et al., 1995, Eur. J. Biochem., 230, 97-103; Lengyel-P et al., 1993, Proc. Natl. Acad. Sci. USA, 90, 5893-5895; и Laurent-Crawford et al., 1992, AIDS Res. Human Retroviruses, 8, 285-290). Вероятно, что двухцепочечные LNA могут имитировать эффект терапевтически активных двухцепочечных РНК: соответственно, такие двухцепочечные LNA обладают потенциалом как лекарственные средства.
По использованию в данном тексте термин "природная нуклеиновая кислота” обозначает нуклеиновые кислоты в широком смысле этого слова, например, нуклеиновые кислоты интактных клеток любого происхождения или нуклеиновые кислоты, выделяемые из таких источников с применением химических или физических методов, или нуклеиновые кислоты, производные от таких первичных источников путем реакций амплификации. Нативные нуклеиновые кислоты могут быть одноцепочечными, двухцепочечными или частично двухцепочечными и могут быть относительно “чистыми” формами или являться смесью различных нуклеиновых кислот. Они также могут быть компонентами необработанного биологического образца, содержащего другие нуклеиновые кислоты и другие клеточные компоненты. С другой стороны, термин “синтетические нуклеиновые кислоты” обозначает любую нуклеиновую кислоту, получаемую методами химического синтеза.
Настоящее изобретение также представляет применение LNA-модифицированных олигонуклеотидов в методах терапии, диагностики и молекулярной биологии, основанных на нуклеиновых кислотах. LNA-модифицированные олигонуклеотиды могут быть использованы в детекции, идентификации, отборе, охарактеризовании, количественном анализе и фрагментировании природных или синтетических нуклеиновых кислот, а также в качестве агентов-блокаторов трансляции и транскрипции in vivo и in vitro. Во многих случаях может представлять интерес присоединение различных молекул к LNA-модифицированным олигонуклеотидам. Такие молекулы могут быть присоединены либо к концу олигонуклеотида, или же они могут быть присоединены по одной или нескольких внутренним позициям. С другой стороны, они могут быть присоединены к данному олигонуклеотиду опосредованно через спейсеры, присоединенные по 5’- или 3’-концу. Представительными группами таких молекул являются ДНК-интеркалирующие группы, фотохимически активные группы, термохимически активные группы, хелатирующие группы, репортерные группы и лиганды. Обычно все методы мечения немодифицированных ДНК- и РНК-олигонуклеотидов с помощью таких молекул также могут быть использованы для мечения LNA-модифицированных олигонуклеотидов. Все методы, применяемые для детекции помеченных олигонуклеотидов, в принципе также применимы и для соответствующим образом помеченных LNA-модифицированных олигонуклеотидов.
Терапия
Термин “замещение цепи” обозначает процесс, при котором олигонуклеотид связывается со своей комплементарной последовательностью-мишенью в составе двухцепочечных ДНК или РНК таким образом, чтобы заместить другую цепь на упомянутой цепи-мишени.
В одном из аспектов настоящего изобретения LNA-модифицированные олигонуклеотиды, способные осуществлять “вытеснение цепи”, используются в разработке новых лекарственных средств, применяемых в серии приемов “метода антигенов”. В противоположность олигонуклеотидам, способным формировать трехцепочечные спирали, такие олигонуклеотиды, “замещающие цепь”, способность взаимодействовать с любой последовательностью в составе двухцепочечной цепи ДНК при физиологических значениях ионной силы и рН.
Олигонуклеотиды “для заменяющий цепь” также могут быть высокоэффективно использованы в “антисмысловых методиках” - т.е. в случаях, когда последовательность-мишень РНК недоступна из-за наличия внутримолекулярных водородных связей. Такие внутримолекулярные структуры могут иметь место в мРНК и могут обусловливать существенные проблемы при попытках “прекратить” трансляцию такой мРНК “антисмысловыми методами”.
Другие классы клеточных РНК, такие как, например, тРНК, рРНК, “мяРНК” и мцРНК, несут внутримолекулярные структуры, которые являются необходимыми для их функционирования. Эти классы сложноустроенных РНК не кодируют белков, но играют существенную роль (в форме нуклеопротеиновых образований) участвует в обеспечении ряда клеточных функций, таких как сплайсинг мРНК, полиаденилирование, трансляция, “редактирование РНК”, поддержание стабильности концов хромосом и т.п. Благодаря их сложной пространственной структуре, которая не позволяют частично или даже полностью проявлять олигонуклеотидам их гибридизационные свойства, эти классы РНК до сих пор не привлекали внимания разработчиков “антисмысловых методов”.
Применение высокоаффинных LNA-мономеров должно обеспечивать конструирование антисмысловых зондов, характеризующихся существенной термостабильностью, эффективную гибридизуемость с такими РНК-мишенями. Следовательно, в предпочтительном варианте, LNA используются для обеспечения существенного уровня аффинности по отношению к олигонуклеотиду, что обеспечивает его гибридизуемость с РНК этих классов: тем самым может достигаться модулирование качественных и(или) количественных параметров частиц, в состав которых входят эти РНК.
В некоторых случаях важным может быть снижение уровня экспрессии некоего гена, в то время как в других случаях целесообразным может быть его активация. Как было показано Меллегардом с соавт. (N.E.Moellegaard, O.Buchardt, M.Egholm, P.E.Nielsen, 1994, Proc. Natl. Acad. Sci. USA, 91, 3892), олигомеры, способные обусловливать “замещение цепи”, могут функционировать в качестве РНК-транскрипционных активаторов. В одном из аспектов по настоящему изобретению LNA, способные обусловливать “замещение цепи”, применимы для активации генов, представляющих терапевтический интерес.
Для химиотерапии различных вирусных инфекций и злокачественных опухолей была подтверждена эффективность нуклеозидов и аналогов нуклеозидов. LNA-нуклеозиды потенциально могут быть использованы в качестве таких лекарственных средств, основанных на нуклеозидах.
Различные типы двухцепочечных РНК подавляют рост некоторых типов злокачественных опухолей. Дуплексы, в составе которых имеются один или большее число LNA-олигонуклеотидов, потенциально применимы в качестве таких двухцепочечных лекарественных средств.
Настоящее изобретение также представляет фармацевтическую композицию, включающую фармацевтически активный LNA-модифицированный олигонуклеотид или фармацевтически активный LNA-мономер в соответствии с определенным выше в сочетании с фармацевтически приемлемым носителем.
Такие композиции могут быть в форме, адаптированной для перорального, парентерального (внутривенного, внутрибрюшинного), внутримышечного, ректального, интраназального, кожного, вагинального, буккального, внутриглазного или внутрилегочного введения, предпочтительно в форме, пригодной для перорального введения, и такие композиции могут быть приготовлены с помощью приемов, хорошо известных специалистам в данной области техники, например, в соответствии с общими описаниями, имеющимися в "Remington’s Pharmaceutical Sciecnes", 17th ed., ed. A.R.Gennaro, Mark Publ. Co., Easton, PA, 1985 и в более поздних изданиях, а также в монографиях серии "Drugs and Pharmaceutical Sciences", выпускаемой издательством M.Dekker.
Диагностика
Некоторые диагностические и молекулярно-биологические процедуры были разработаны таким образом, чтобы утилизировать наборы различных олигонуклеотидов с целью одновременного анализа нуклеиновой кислоты-мишени на присутствие избытка возможных мутаций. Обычно олигонуклеотидные наборы иммобилизуют в заданном порядке на твердую подложку таким образом, чтобы присутствие конкретной мутации в нуклеиновой кислоте-мишени могло быть определено по расположению на твердой подложке после ее гибридизации. Одним из важных предварительных требований для успешного применения данного набора различных олигонуклеотидов в анализе нуклеиновых кислот связано с тем, чтобы все они были специфичными в отношении конкретной последовательности-мишени при одних и тех же применяемых в конкретном случае условиях гибридизации. Поскольку аффинность и специфичность стандартных олигонуклеотидов в отношении соответствующих комплементарных последовательностей-мишеней в значительной степени зависит от их нуклеотидной последовательности и общей длины, то эти критерии до сих пор трудно было полностью удовлетворить.
В предпочтительном варианте, следовательно, LNA используются в качестве средств для повышения аффинности и(или) специфичности зондов, а также в качестве средств выравнивания уровней аффинности различных олигонуклеотидов в отношении соответствующих им комплементарных последовательностей. Как представляется в настоящем изобретении, такое модулирование уровня аффинности может быть осуществлено, например, путем замены выбранных нуклеозидов в составе олигонуклеотида на LNA, несущий такое же основание. Как будет показано здесь далее, различные классы LNA проявляют разные уровни аффинности в отношении соответствующих им комплементарных нуклеозидов. Например, LNA с 2-3 “мостиками”, соединенный с основанием Т, проявляет меньшую аффинность по отношению к аденозину по сравнению с соответствующим LNA, имеющим 2-4 “мостика”. Следовательно, использование различных классов LNA обеспечивает дополнительную возможность тонкой настройки уровней аффинности олигонуклеотида.
В другом предпочтительном варианте высокие уровни аффинности и специфичности LNA-модифицированных олигонуклеотидов используется при специфичном по последовательности отборе и очистке природных или синтетических нуклеиновых кислот. В одном из аспектов природных и синтетические нуклеиновые кислоты контактируют с LNA-модифицированным олигонуклеотидом, иммобилизованным на твердой поверхности. В этом случае гибридизация и отбор происходят одновременно. Отобранные нуклеиновые кислоты могут быть, например, подвергнуты детекции, охарактеризованию, количественному анализу или амплификации прямо на поверхности с применением различных методов, хорошо известных в данной области техники, или же они могут быть сняты с этой поверхности до того, как будет проводиться такое охарактеризование или амплифицирование, путем помещения иммобилизованных модифицированного олигонуклеотида и отобранной с его помощью нуклеиновой кислоты в обезвоживающие условия, например, путем нагревания или используя буферы, характеризующиеся низкой ионной силой.
Твердая подложка может быть выбрана из широкого круга полимерных материалов, таких как, например, CPG (стекло с регулируемыми порами), полипропилен, полистирол, поликарбонат или полиэтилен, и она может иметь разнообразную форму, такую как, например, пробирка, микротитровальный планшет, палочка, шарик, фильтр и т.п. LNA-модифицированный олигонуклеотид может быть иммобилизован на твердую подложку с использованием его 5’- или 3’-конца (или по концевым составляющим линкера, присоединенного к 5’- или 3’-концу олигонуклеотида) с применением различных химических или фотохимических методов, обычно применяемых для иммобилизации олигонуклеотидов, или с помощью нековалентного сочетания, например, путем связывания биотинилированных LNA-модифицированных олигонуклеотидов на различных твердых подложках на иммобилизованном стрептавидине. Одним из предпочтительных методов иммобилизации LNA-модифицированных олигонуклеотидов на различных твердых подложках является фотохимический метод, в котором, используется фотохимически активный антрахинон, ковалентно присоединяемый к 5’- или 3’-концу модифицированного олигонуклеотида (необязательно через линкеры) в соответствии с описанным в международной заявке WO 96/31557. Таким образом, настоящее изобретение также представляет поверхность, несущую LNA-модифицированный олигонуклеотид.
В другом аспекте настоящего изобретения LNA-модифицированный олигонуклеотид несет лиганд, ковалентно присоединенный к его 5’- или 3’-концу. В этом случае LNA-модифицированный олигонуклеотид. контактируют с нативными или синтетическими нуклеиновыми кислотами в растворе, после чего сформировавшиеся гибридные молекулы отбираются на твердую подложку, несущую молекулы, которые могут специфически образом связывать конкретный лиганд.
Еще в одном аспекте LNA-модифицированные олигонуклеотиды, способные обеспечивать “замещение цепи”, используются для отбора нативных и синтетических нуклеиновых кислот без предварительной их денатурации. Такие модифицированные олигонуклеотиды, в частности, применимы в тех случаях, когда доступ к последовательности-мишени с помощью обычных олигонуклеотидов затруднен или вообще невозможен из-за быстрого образования стабильных внутримолекулярных структур. Примерами нуклеиновых кислот, включающих такие структуры, являются рРНК, тРНК, мяРНК и мцРНК.
В другом предпочтительном варианте LNA-модифицированные олигонуклеотиды, сконструированные с целью обеспечения высокого уровня специфичности, используются в качестве затравок при секвенировании нуклеиновых кислот и в качестве затравок в любой из ряда хорошо известных реакций амплификации, таких как реакция ПЦР. Как показано в данной заявке, дизайн LNA-модифицированных олигонуклеотидов определяет то, какой - экспоненциальный или линейный тип амплификации мишени они будут обеспечивать. Продукты амплификационной реакции могут быть проанализированы с помощью различных методов, применимых для анализа продуктов амплификации, полученных на обычных ДНК-затравках. В конкретном случае, когда затравки на основе LNA-модифицированных олигонуклеотидов конструируются таким образом, чтобы обеспечивать амплификацию линейного типа, получаемые в результате ампликоны будут иметь одноцепочечные концы, которые могут быть маркированы комплементарными зондами без денатурации. Такие концы могут быть использованы, например, для отбора ампликонов с помощью других комплементарных LNA-модифицированных олигонуклеотидов, присоединенных к твердой поверхности.
В другом аспекте LNA-модифицированные олигомеры, способные обеспечивать “замещение цепи”, используются в качестве затравок либо в экспоненциальных, либо в линейных реакциях амплификации. Применение таких олигомеров, как ожидается, обусловит повышение общего выхода ампликонов за счет эффективного конкурирования при регибридизации ампликонов на поздних этапах реакции амплификации. Было представлено (Demers et al., 1995, Nucl. Acids Res., 23, 3050-3055) использование высокоаффинных недостраиваемых олигомеров с целью увеличения общего выхода в ПЦР-реакции. Можно считать, что олигомеры обусловят такой эффект за счет недопущения регибридизации ампликонов на поздних этапах ПЦР-реакции. Можно ожидать, что LNA-модифицированные олигомеры, заблокированные по их 3’-концу, будут обеспечивать аналогичное преимущество. Заблокирование по 3’-концу может быть достигнуто разнообразными путями, как, например, с помощью замещения 3’-гидроксильной группы на водород или фосфат. Такие заблокированные по 3’-концу LNA-модифицированные олигомеры также могут быть использованы для избирательной амплификации близкородственных нуклеотидных последовательностей по способу, сходному с описанным ранее (Yu et al., 1997, Biotechniques, 23, 714-716).
В последние годы были представлены новые классы зондов, которые могут быть использованы, например, для выявления ампликонов, получаемых в реакциях амплификации мишеней в режиме “реального времени”. Один из классов таких зондов был назван "Molecular Beacons". Эти зонды синтезируются как частично автокомплементарные олигонуклеотиды, включающие флуорофор на одном из концов, а на другом конце - молекулу - квенчер (“тушитель флуоресценции”). Находясь в растворе в свободном виде, такой зонд упакован в структуру “шпильки” (за счет взаимодействия его автокомплементарных участков): в этом случае квенчер находится достаточно близко к флуорофору, чтобы тушить флуоресцентный сигнал. В условиях гибридизации с соответствующей нуклеиновой кислотой-мишенью происходит “раскрытие шпильки”, в результате чего происходит “разделение” флуорофора и квенчера и проявление флуоресцентного сигнала.
Другой класс зондов получил наименование “зондов Taqman”. Эти зонды также включают флуорофор и молекулу-квенчер. Однако в отличие от зондов "Molecular Beacons", способность квенчера подавлять флуоресцентный сигнал флуорофора поддерживается и после гибридизации зонда с его последовательностью-мишенью. Вместо этого флуоресцентный сигнал генерируется после гибридизации за счет физического удаления из состава зонда либо квенчера, либо флуорофора в результате 5’-экзонуклеазной активности полимеразы, которая инициирует синтез с затравки, расположенной с 5’-стороны от сайта связывания зонда Taqman.
Высокий уровень аффинности в отношении сайта-мишени является важным свойством обоих этих типов зондов: следовательно, такие зонды имеют тенденцию к слишком большому “накоплению” своего размера (обычно от 30 до 40 нуклеотидов). В результате возникают серьезные проблемы в получении высокоинформативных зондов. В предпочтительном варианте настоящего изобретения, следовательно, LNA используется для улучшения параметров и соответствующих характеристик применения зондов Taqman и зондов "Molecular Beacons" за счет уменьшения их размера при сохранении исходного уровня аффинности.
В следующем аспекте настоящего изобретения LNA используются для конструирования новых аффинных пар зондов (либо полностью, частично модифицированных олигонуклеотидов). Уровни аффинности могут быть легко выбраны в широком диапазоне и при этом очень большое число аффинных пар может быть определено и синтезировано. Один из компонентов аффинной пары может быть присоединен к представляющей интерес молекуле (таким как белки, ампликоны, ферменты, полисахариды, антитела, гаптены, пептиды, РНК и т.п.) с применением стандартных методов, в то время как другой компонент аффинной пары может быть присоединен, например, к твердой подложке, такой как шарики, мембраны, микротитровальные планшеты, палочки, пробирки и т.п. Твердая подложка может быть выбрана из широкого круга полимерных материалов, таких как, например, полипропилен, полистирол, поликарбонат или полиэтилен. Аффинные пары могут быть использованы в избирательном выделении, очистке, отборе и детекции разнообразных молекул-мишеней, обозначавшихся выше.
Принцип отбора LNA-помеченной молекулы путем взаимодействия с другим комплементарным LNA-олигонуклеотидом (либо полностью, либо частично модифицированным) может быть применен для создания ничем не ограниченного числа новых аффинных пар зондов.
В другом предпочтительном варианте высокие уровни аффинности и специфичности LNA-модифицированных олигонуклеотидов используются при конструировании зондов, применимых в гибридизации in situ. Например, LNA может быть использован для снижения размера традиционных ДНК-зондов при сохранении требуемого уровня аффинности, что тем самым обеспечит улучшение кинетических параметров данного зонда и его способности проникать в образец. Способность LNA-модифицированных олигонуклеотидов к “инвазии цепи” в двухцепочечных молекулах нуклеиновых кислот также обеспечивает значительные преимущества методу гибридизации in situ за счет того, что они обеспечивают гибридизацию без предварительной денатурации ДНК/РНК-мишеней.
В другом предпочтительном варианте LNA-модифицированные олигонуклеотиды, предполагаемые к использованию в “антисмысловой терапии”, конструируются таким образом, чтобы обладать и высоким уровнем аффинности, и способностью к инициации РНКазы-Н. Это может быть достигнуто, например, наличием LNA-сегментов, фланкирующих немодифицированный, расположенный в центре ДНК-сегмент.
Настоящее изобретение также обеспечивает набор, предназначенный для выделения, очистки, амплификации, детекции, идентификации, количественного анализа или отбора природных или синтетических нуклеиновых кислот, причем данный набор включает реакционное приспособление и один или несколько LNA-модифицированных олигонуклеотидов (олигомеров) в соответствии с описанным здесь. Предпочтительно LNA-модифицированные олигонуклеотиды иммобилизованы на поверхность упомянутого реакционного приспособления.
Настоящее изобретение также обеспечивает набор, предназначенный для выделения, очистки, амплификации, детекции, идентификации, количественного анализа или отбора природных или синтетических нуклеиновых кислот, при том, что этот набор включает реакционное приспособление и один или несколько. LNA в соответствии с описанным здесь. Предпочтительно LNA иммобилизованы на поверхность упомянутого реакционного приспособления (например, с использованием описанных выше методов иммобилизации).
Для наборов по настоящему изобретению реакционным приспособлением предпочтительно является материал твердой подложки, например, выбираемый из боросиликатного стекла, натрий-кальциевого стекла, полистирена, поликарбоната, полипропилена, полиэтилена, полиэтиленгликольтерефталата, поливинилацетата, поливинилпирролидинона, полиметилметакрилата и поливинилхлорида, а предпочтительно - полистирена и поликарбоната. Реакционное приспособление может иметь форму пробирки, ампулы, предметного стекла, пластины, пленки, шарика, гранулы, диска, пластинки, кольца, стержня, сетки, фильтра, лотка, микротитровального планшета, палочки или многолопастной палочки.
Наборы обычно сопровождаются письменной инструкцией, в которой описаны оптимальные условия применения данного набора.
Описанные выше диагностические и терапевтические аспекты настоящего изобретения далее иллюстрируются нижеследующими примерами.
Экспериментальная часть
Базовая информация
Все реактивы были получены из коммерческих источников и были использованы без дополнительной очистки. После высушивания органических фаз над Na2SO4 проводили фильтрование. Использовавшийся в хроматографии силикагель (поры 0,040-0,63 мм) был приобретен у фирмы "Merck". ЯМР-спектры записывали при 300 МГц или 250 МГц для 1H-ЯМР, или при 62,9 МГц для 13С-ЯМР, или при 202,33 МГц для 31Р-ЯМР. Величины δ указаны в м.д. по отношению к тетраметилсилану, являющемуся внутренним стандартом (1Н-ЯМР и 13С-ЯМР) и по отношению к 85%-ной Н3РO4, взятой в качестве внешнего стандарта (31Р-ЯМР). Обозначения пиков ЯМР даны в соответствии со стандартной номенклатурой нуклеозидов. Масс-спектры EI, масс-спектры FAB и масс-спектры PD (Plasma Desorption) были определены с целью получения информации о молекулярных массах синтезированных соединений. Олигонуклеотидные аналоги были синтезированы с применением фосфорамидитной технологии. Очистку 5’-O-DMT-включающих и 5’-O-DMT-освобожденных олигонуклеотидных аналогов осуществляли с использованием сменных картриджей для хроматографии с обращенной фазой, а при необходимости методом ВЭЖХ с обращенной фазой. Метод лазерной десорбционной масс-спектрометрии на матрице использовали для проверки молекулярной массы и состава мономеров в репрезентативных образцах олигонуклеотидов. Метод капиллярного гель-электрофореза использовали для проверки чистоты репрезентативных образцов олигонуклеотидов.
Конкретные нижеприведенные описания сопровождаются фигурами 2-41 и таблицами 1-10. За исключением специально оговариваемых случаев в нижеследующих примерах “LNA” обозначает вариант с 2’-4’-“мостиком”, проиллюстрированный формулой Z на фиг.2.
Получение LNA-мономеров
Пример 1
3-С-аллил-1,2-О-изопропилиден-α -D-рибофураноза (0А)
Метод 1. Раствор 5-O-трет-бутилдиметилсилил-1,2-O-изопропилиден-α -D-рибофуран-3-улозы (Y.Yoshimura, T.Sano, A. Matsuda & T.Ueda, 1988, Chem. Pharm. Bull., 36, 162) (17,8 г, 58,9 ммоль) в безводном THF (980 см3) перемешивали при 0° С и по каплям добавляли 1 М аллилбромид магния в безводном эфире (130 см3, 130 ммоль). После перемешивания в течение 2 часов добавляли насыщенный водный раствор хлорида аммония (800 см3) и полученную смесь экстрагировали дихлорметаном (3× 400 см3). Органическую фазу промывали соляным раствором (3× 450 см3) и высушивали над Na2SO4. Растворитель удаляли при пониженном давлении и остаток растворяли в безводном THF (700 см3). Добавляли 1,1 М раствор фторида тетрабутиламмония в THF (54,4 см3, 59,8 ммоль), полученную смесь перемешивали при комнатной температуре в течение 1 часа и упаривали досуха. Остаток растворяли в дихлорметане (1700 см3) и промывали насыщенным водным раствором кислого углекислого натрия (3× 500 см3) и высушивали над Na2SO4. Растворитель удаляли при пониженном давлении и остаток подвергали хроматографической очистки на силикагелевой колонке с использованием дихлорметана/метанола (объемное соотношение 98:2) в качестве элюента с получением фуранозы (0А) в виде твердого вещества белого цвета (9,42 г, 69%). Метод 2. Фуранозу (0А) аналогичным образом синтезировали из 5-O-третбутилдифенилсилил-1,2-O-изопропилиден-α -D-рибофуран-3-улозы (Т.F.Tam & B.Fraser-Reid, 1980, J. Chem. Soc., Chem. Commun., 556) (9,5 г, 22,2 ммоль) с использованием: безводного THF (425 см3), 1 М раствора аллилбромида магния в безводном эфире (130 см3, 130 ммоль), насыщенного водного раствора хлорида аммония (490 см3), эфира для экстрагирования (350 + 2× 160 см3), 1,1 М раствора фторида тетрабутиламмония в THF (22,3 см3, 24,6 ммоль), безводного THF (400 см3), дихлорметана (1400 см3), насыщенного водного раствора кислого углекислого натрия (3× 500 см3), соляного раствора (500 см3) и Na2SO4. δ H ((СD3)2SO) 5,84 (1Н, м, 2’-H), 5,65 (1Н, д, J 3,8, 1-Н), 5,12 (1Н, д, J 6,1, 3’-На), 5,06 (1Н, шс, 3’-Нb), 4,76 (1Н, с, 3-ОН), 4,64, (1Н, т, J 5,4, 5-ОН), 4,16 (1Н, д, J 3,8, 2-Н), 3,84 (1Н, дд, J 2,2, 8,1, 4-Н), 3,56 (1Н, ддд, J 2,3, 5,6, 11,8, 5-На), 3,42 (1Н, м, 5-Нb), 2,16 (1Н, дд, J 6,1, 14,3, 1’-На), 1,98 (1Н, дд, J 8,2, 14,3, 1’-Hb), 1,46 (3Н, с, СН3), 1,25 (3Н, с, СН3). δ C (СDСl3) 133,5 (С-2’), 117,9 (С-37), 110,8 (С(СН3)2), 102,9 (С-1), 82,6, 81,0, 77,7, (С-2, С-3, С-4), 59,4, (С-5), 36,4 (С-1’), 26,4, 26,3 (СН3). Получено: С-57,4, Н-8,0. Для С11Н18О5 рассчитано: С - 57,4, Н - 7,9%.
Пример 2
3-С-аллил-3,5-ди-О-бензил-1,2-O-изопропилиден-α -D-рибофураноза (0В)
60%-ную суспензию гидрида натрия (4,9 г, 123 ммоль) в безводном DMF (100 см3) перемешивали при 0° С и по каплям в течение 45 минут добавляли раствор фуранозы 0А (9,42 г, 40,9 ммоль) в безводном DMF (65 см3). Раствор перемешивали в течение 1 часа при 50° С и охлаждали до 0° С. По каплям добавляли смесь бензилбромида (14,5 см3, 121 ммоль) и безводного DMF (14,5 см3) и полученную смесь перемешивали при комнатной температуре в течение 18 часов. Реакционную смесь упаривали досуха и раствор остатка в дихлорметане (700 см3) промывали насыщенным водным раствором кислого углекислого натрия (3× 450 см3) и высушивали над Na2SO4. Растворитель удаляли при пониженном давлении и остаток подвергали хроматографической очистке на силикагелевой колонке с использованием петролейного эфира/этилацетата (о/о, 9:1) в качестве элюента с получением 0В в виде маслянистого вещества (14,5 г, 86%). δ H (CDCl3) 7,39-7,21 (10Н, м, Вn), 5,92 (1Н, м, 2’-Н), 5,71 (1Н, д, J 3,8, 1-Н), 5,17-5,09 (2Н, м, 3’-На, 3’-Hb), 4,67 (2Н, м, Вn), 4,60 (1Н, д, J 12,2, Bn), 4,52 (1Н, д, J 12,1, Bn), 4,43 (1Н, м, 4-Н), 4,42 (1Н, д, J 3,8, 2-H), 3,73 (1Н, дд, J 3,2, 10,8, 5-На), 3,66 (1Н, дд, J 7,4, 10,8, 5-Hb), 2,50 (1Н, дд, J 7,7, 14,9, 1’-Ha), 2,39 (1Н, дд, J 6,5, 14,9, 1’-Hb), 1,60 (3Н, с, СН3), 1,34 (3Н, с, СН3). δ C (СDСl3) 138,7, 138,1 (Bn), 132,6 (С-2’), 128,3, 128,2, 127,7, 127,5, 127,4, 127,4 (Bn), 118,5 (С-3'), 112,6 (С(СН3)2), 104,1 (С-1), 86,5, 82,1, 80,4 (С-2, С-3, С-4), 73,4, 68,6 (Bn), 67,0 (С-5), 35,8 (С-1’), 26,8, 26,6 (СН3). FAB-MS m/z 433 [M+Na]+. Получено: С - 73,4; Н - 7,4. Для С25Н30O5 рассчитано: С - 73,2; Н - 7,4%.
Пример 3
3-С-аллил-1,2-ди-O-ацетил-3,5-ди-O-бензил-D-рибофураноза (0С)
Раствор фуранозы 0В (12,42 г, 30,3 ммоль) в 80%-ной водной уксусной кислоты (150 см3) перемешивали при 90° С в течение 3 часов. Растворитель удаляли при пониженном давлении и остаток упаривали вместе с этанолом (3× 75 см3), толуолом (3× 75 см3) и безводным пиридином (2× 75 см3) и вновь растворяли в безводном пиридине (60 см3). Добавляли уксусный ангидрид (46 см3) и полученный раствор перемешивали при комнатной температуре в течение 48 часов. Добавляли смесь льда и воды (300 см3) и полученную смесь экстрагировали дихлорметаном (2× 300 см3). Объединенную органическую фазу промывали насыщенным водным раствором кислого углекислого натрия (3× 200 см3) и высушивали над Nа2SO4. Растворитель упаривали и остаток подвергали хроматографической очистке на силикагелевой колонке, используя петролейный эфир/этилацетат (о/о, 4:1) в качестве элюента с получением аномерной смеси 0С (β :α ≈ 2:1) в виде маслянистого вещества (13,3 г, 97%). δ C (CDCl3) 169,7, 169,6 (С=O), 138,7, 138,4, 137,7, 137,6 (Bn), 132,4, 132,2 (С-2’), 128,4 128,4, 128,2, 128,2, 127,8, 127,7, 127,7, 127,6, 127,3, 127,3, 126,9, 126,8 (Bn), 118,5 (С-3’), 99,4, 93,5 (С-1), 84,8, 83,7, 83,2, 82,0, 79,1, 75,5 (С-2, С-3, С-4), 73,7, 73,5, 69,3, 68,7 (Bn), 66,1 (С-5), 35,5, 34,9 (С-1), 21,1, 21,0, 20,7, 20,6 (СН3). Получено: С - 68,7; Н - 6,7. Для С26Н30O7 рассчитано: С - 68,8; Н - 6,6%.
Пример 4
1-(2-O-ацетил-3-С-аллил-3,5-ди-O-бензил-(β -D-рибофуранозил)тимин (1)
К перемешиваемому раствору аномерной смеси 0С (β :α ≈ 2:1, 11,8 г, 26,0 моль) (P.Nielsen, H.M.Pfundheller & J.Wengel, 1997, Chem. Commun., 825; P.Nielsen, H.M.Pfundheller, C.E.Olsen & J.Wengel, 1997, J. Chem. Soc., Perkin Trans. 1, в печати) и тимина (6,55 г, 52,0 ммоль) в безводном ацетонитриле (250 см3) добавляли N,O-бис (триметилсилил) ацетамид (44,9 см3, 182 ммоль). Реакционную смесь перемешивали в течение 1 часа с обратным холодильником и охлаждали до 0° С. По каплям добавляли триметилсилилтрифлат (8,00 cm3, 44,0 ммоль) и полученный раствор перемешивали при комнатной температуре в течение 12 часов. Добавляли охлажденный на льду насыщенный водный раствор кислого углекислого натрия (270 см3) и смесь экстрагировали дихлорметаном (3× 125 см3). Органическую фазу промывали насыщенным водным раствором кислого углекислого натрия (2× 125 см3) и соляным раствором (2× 125 см3) и высушивали над Na2SO4. Растворитель удаляли и остаток подвергали хроматографической очистке на силикагелевой колонке, используя дихлорметан/метанол (о/о, 98:2) в качестве элюента с получением нуклеозида 1 в виде твердого вещества белого цвета (11,6 г, 86%). δ H (СDСl3) 8,64 (1Н, шс, NH), 7,75 (1Н, д, J 1,1, 6-Н), 7,41-7,25 (10Н, м, Bn), 6,43 (1Н, д, J 8,2, 1’-Н), 5,88 (1Н, м, 2’’-Н), 5,66 (1Н, д, J 8,2, 2’-Н), 5,12 (1Н, с, 3’’-На), 5,07 (1Н, дд, J 1,5, 8,5, 3’’-Hb), 4,85 (1H, д, J 11,2, Bn), 4,64 (2Н, с, Bn), 4,63 (1Н, д, J 11,2, Bn), 4,33 (1Н, шс, 4’-Н), 3,81 (1Н, дд, J 2,7, 11,1, 5’-На), 3,65 (1Н, м, 5’-Hb), 2,81-2,65 (2Н, м, 1’’-Ha, 1’’-Hb), 2,08 (3Н, с, СOСН3), 1,52 (3Н, д, J 0,8, СН3). δ C (СDСl3) 170,1 (С=0), 163,6 (С-4), 150,9 (С-2), 138,1, 136,6 (Bn), 136,0 (С-6), 131,6 (С-2’’), 128,8, 128,4, 128,3, 127,6, 127,5, 127,1 (Bn), 118,5 (С-3’’), 111,1 (С-5), 84,2, 83,4, 83,1, 77,4 (С-1’, С-2’, С-3’, C-4’), 73,6, 69,2 (Bn), 65,6 (С-5’, 33,7 (С-1’’), 20,8 (СOСН3), 11,9 (СН3). Получено: С - 66,8; Н - 6,3; N - 5,1. Для C29H32N2O7 рассчитано: C - 66,9; Н - 6,2; N - 5,4%.
Пример 5
1-(3-С-аллил-3,5-ди-O-бензил-β -D-рибофуранозил)тимин (2)
К перемешиваемому раствору нуклеозида 1 (11,6 г, 22,3 ммоль) в метаноле (110 см3) добавляли метоксид натрия (3,03 г, 55,5 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов и нейтрализовывали разбавленной соляной кислотой. Растворитель частично выпаривали и остаток растворяли в дихлорметане (2× 400 см3). Органическую фазу промывали насыщенным водным раствором кислого углекислого натрия (2× 125 см3) и соляным раствором (2× 125 см3) и высушивали над Na2SO4. Растворитель удаляли при пониженном давлении с получением соединения 2 в виде твердого вещества белого цвета (10,1 г, 95%). δ H (СOСl3) 8,77 (1Н, шс, NH), 7,58 (1Н, д, J 1,2, 6-Н), 7,41-7,25 (10Н, м, Bn), 6,14 (1Н, м, 2’’-Н), 6,12 (1Н, д, J 7,8, 1’-Н), 5,23 (1Н, м, 3’’-На), 5,17 (1Н, шс, 3’’-Hb), 4,68 (1H, д, J 10,8, Bn), 4,59 (2Н, с, Bn), 4,55 (1H, д, J 10,9, Bn), 4,39 (1H, шс, 4’-H), 4,26 (1H, дд, J 7,8, 10,7, 2’-H), 3,84 (1H, дд, J 3,1, 11,0, 5’-На), 3,58 (1H, дд, J 1,4, 11,0, 5’-Hb), 3,04 (1H, д, J 10,8, 2’-OH), 2,82-2,78 (2Н, м, 1’’-Ha, 1’’-Hb), 1,51 (3Н, д, J 1,0, СН3). δ C (CDCl3) 163,5 (С-4), 151,1 (С-2), 137,3, 136,7 (Bn), 136,0 (С-6), 132,1 (С-2’’), 128,8, 128,5, 128,3, 127,9, 127,6 (Bn), 118,4 (С-3’’), 111,1 (С-5), 87,4, 82,6, 81,1, 79,3 (С-1’, С-2’, С-3’, С-4’), 73,7, 69,8 (Bn), 64,7 (С-5’), 35,1 (С-1’’), 11,9 (СН3). Получено: С – 67,8; Н – 6,1; N – 5,5. Для С27Н30Н2O6 рассчитано: С – 67,8; Н – 6,3; N – 5,9%.
Пример 6
1-(3-С-аллил-3,5-ди-O-бензил-2-O-метансульфонил-β -D-рибофуранозил)тимин (3)
К перемешиваемому раствору нуклеозида 2 (3,50 г, 7,31 ммоль) в безводном пиридине (23 см3) добавляли метансульфонилхлорид (1,69 см3, 21,89 ммоль) при 0° С. Реакционную смесь перемешивали при комнатной температуре в течение 1 часа, добавляли воду (100 см3) и проводили экстрагирование дихлорметаном (3× 150 см3). Органическую фазу промывали насыщенным водным раствором кислого углекислого натрия (3× 200 см3) и высушивали над Na2SO4. Растворитель удаляли при пониженном давлении и остаток подвергали хроматографической очистке на силикагелевой колонке с использованием дихлорметана/метанола (о/о 99:1) в качестве элюента с получением соединения 3 в виде твердого вещества белого цвета (3,64 г, 89%). δ н (СDСl3) 8,95 (1H, шс, NH), 7,71 (1Н, д, J 1,1, 6-Н), 7,39-7,25 (10Н, м, Bn), 6,52 (1Н, д, J 8,0, 1’-Н), 5,90 (1Н, м, 2’’-Н), 5,34 (1Н, д, J 7,9, 2’-Н), 5,20-5,09 (2Н, м, 3’’-На, 3’’-Hb), 4,91 (1Н, д, J 11,2, Bn), 4,68 (1Н, д, J 11,3, Bn), 4,64 (2Н, с, Bn), 4,33 (1Н, шс, 4’-Н), 3,81 (1Н, дд, J 2,5, 11,1, 5’-Ha), 3,73 (1Н, дд, J 1,1, 11,1, 5’-Hb), 3,08 (1Н, дд, J 5,5, 5,7, 1’’-Ha), 2,99 (3Н, с, СН3), 2,68 (1Н, м, 1’’-Hb), 1,51 (3Н, д, J 0,8, СН3). δ C (СDСl3) 163,4 (С-4), 150,8 (С-2), 137,9, 136,3 (Bn), 135,5 (С-6), 131,0 (C-2’’), 128,8, 128,3, 127,5, 127,2 (Bn), 119,3 (С-3’’), 111,6 (С-5), 84,1, 83,6, 82,4, 82,2 (С-1’, С-2’, С-3’, С-4’), 73,7, 68,9 (Bn), 66,2 (С-5’), 38,7 (СН3), 33,0 (С-1’’), 11,9 (СН3). Получено: С - 60,5; Н – 5,8; N - 4,9. Для C28H32N2O8S рассчитано: С - 60,4; Н - 5,8; N - 5,0%.
Пример 7
1-(3-С-аллил-3,5-ди-O-бензил-β -D-арабинофуранозил)тимин (4)
Раствор нуклеозида 3 (3,59 г, 6,45 ммоль) в этаноле (72 см3), воде (72 см3) и 1 М водном гидроксиде натрия (20,6 см3) перемешивали, нагревая с обратным холодильником, в течение 18 часов. После нейтрализации разбавленной соляной кислотой растворитель удаляли при пониженном давлении и остаток растворяли в дихлорметане (3× 150 см3). Органическую фазу промывали насыщенным водным раствором кислого углекислого натрия (3× 200 см3) и высушивали над Nа2SO4. Растворитель удаляли при пониженном давлении и остаток подвергали хроматографической очистке на силикагелевой колонке с использованием дихлорметана/метанола (о/о 99:1) в качестве элюента с получением соединения 4 в виде твердого вещества белого цвета (2,32 г, 74%). δ н (CDCl3) 7,60 (1H, д, J 1,2, 6-Н), 7,50-7,23 (10Н, м, Bn), 6,22 (1Н, д, J 2,9, 1’-Н), 5,80 (1H, м, 2’’-Н), 5,15-5,08 (2Н, м, 3’’-На, 3’’-Hb), 4,86-4,33 (6Н, м, 2× Bn, 2’-Н, 4’-Н), 3,82-3,71 (2Н, м, 5’-На, 5’-Hb), 2,72 (1H, м, 1’’-На), 2,52 (1H, дд, J 7,6, 16,1, 1’’-Hb), 1,70 (3Н, д, J 0,9, СН3). δ с (CDCl3) 165,1 (С-4), 150,4 (С-2), 138,4, 136,8 (Bn), 137,7 (С-6), 132,3 (С-2’’), 128,77, 128,4, 128,3, 128,0, 127,9, 127,6 (Bn), 118,5, (С-3’’), 107,8 (С-5), 88,0, 87,8, 83,7 (C-1’, C-3’, С-4’), 73,7, 72,9, 69,4 (Bn, С-2’), 64,7 (С-5’), 31,1 (С-1’’), 12,4 (СН3). Получено: С - 67,5; Н - 6,3; N - 5,3. Для С27Н30N2O6·0,25Н2O рассчитано С - 67,1; Н - 6,4; N - 5,8%.
Пример 8
1-(3,5-ди-О-бензил-3-С-(2-гидроксиэтил)-β -D-арабинофуранозил)тимин (5)
К перемешиваемому раствору нуклеозида 4 (2,26 г, 4,68 ммоль) в THF (12 см3) и воде (12 см3) добавляли перйодат натрия (3,04 г, 14,2 ммоль) и 2,5% раствор тетраоксида осмия в трет-бутаноле (в/о, 0,603 см3, 40 мкмоль). Раствор перемешивали при комнатной температуре в течение 45 минут. Добавляли воду (25 см3) и раствор экстрагировали дихлорметаном (2× 50 см3). Органическую фазу промывали насыщенным водным раствором кислого углекислого натрия (3× 30 см3) и высушивали над Nа2SO4. Растворитель удаляли при пониженном давлении и остаток вновь растворяли в THF (12 см3) и воде (12 см3). Полученную смесь перемешивали при комнатной температуре и добавляли боргидрид натрия (182 мг, 4,71 ммоль). После перемешивания в течение 1,5 часов добавляли воду (25 см3) и раствор экстрагировали дихлорметаном (2× 50 см3). Органическую фазу промывали насыщенным водным раствором кислого углекислого натрия (3× 30 см3) и высушивали над Na2SO4. Растворитель удаляли при пониженном давлении и остаток подвергали хроматографической очистке на силикагелевой колонке с использованием дихлорметана/метанола (о/о 98:2) в качестве элюента с получением соединения 5 в виде твердого вещества белого цвета (1,13 г, 49%). δ н (CDCl3) 9,29 (1Н, шс, NH), 7,47 (1Н, д, J 1,1, 6-Н), 7,38-7,25 (10Н, м, Bn), 6,22 (1Н, д, J 3,4, 1’-Н), 4,62 (2Н, с, Bn), 4,60 (1Н, м, 4’-Н), 4,46 (2Н, с, Bn), 4,35 (1Н, дд, J 3,4, 7,5, 2’-Н), 3,83-3,73 (4Н, м, 2× 5’-H, 2× 2’’-Н), 2,67 (1Н, шс, ОН), 2,07-2,01 (2Н, м, 2× 1’’-Н), 1,77 (3Н, д, J 0,5, СН3). δ с (СDСl3) 164,3 (С-4), 150,3 (С-2), 137,6, 137,4 (Bn, C-6), 136,7 (Bn), 128,6, 128,4, 128,2, 127,8, 127,6, 127,3, 127,1 (Bn), 108,4 (С-5), 88,0, 87,7, 81,6, 74,7 (C-1’, С-2’, С-3’, С-4’), 73,7, 69,6 (Bn), 64,6 (C-5’), 57,7 (C-2’’), 28,6 (С-1’’), 12,4 (СН3). FAB-MS m/z 483 [М+Н]+, 505 [M+Na]+. Получено: С - 63,6; Н - 6,2; N - 5,4. Для C26H30N2O7·0,5Н2O рассчитано С - 63,5; Н - 6,4; N - 5,7%.
Пример 9
(1S,5R,6R,8R)-5-гидрокси-6-(гидроксиметил)-8-(тимин-1-ил)-2,7-диоксабицикло[3.3.0]октан (6)
Раствор нуклеозида 5 (1,08 г, 2,20 ммоль) в безводном пиридине (5,0 см3) перемешивали при 0° С и по каплям добавляли раствор р-толуолсульфонилхлорида (462 мг, 2,47 ммоль) в безводном пиридине (2,0 см3). После перемешивания при комнатной температуре в течение 20 часов и добавления смеси воды и льда (70 см3) проводили экстрагирование дихлорметаном (2× 75 см3). Органическую фазу промывали насыщенным водным раствором кислого углекислого натрия (3× 50 см3) и высушивали над Na2SO4. Растворитель удаляли при пониженном давлении и остаток подвергали хроматографической очистке на силикагелевой колонке с использованием дихлорметана/метанола (о/о, 99:1) в качестве элюента с получением промежуточного соединения, которое после выпаривания растворяли в безводном DMF (4,0 см3). Этот раствор добавляли по каплям к перемешиваемой суспензии 60%-ного гидрида натрия (203 мг, 4,94 ммоль) в безводном DMF (4,0 см3) при 0° С. Полученную смесь перемешивали в течение 18 часов и добавляли воду (20 см3). После нейтрализации соляной кислотой добавляли дихлорметан (75 см3). Органическую фазу промывали насыщенным водным раствором кислого углекислого натрия (3× 50 см3) и высушивали над Na2SO4. Растворитель удаляли при пониженном давлении и остаток подвергали хроматографической очистке на силикагелевой колонке с использованием дихлорметана/метанола (о/о 98:2) в качестве элюента с получением твердого вещества белого цвета (858 мг). Раствор этого твердого вещества белого цвета в этаноле (10,0 см3) перемешивали при комнатной температуре и добавляли 20% гидроксид палладия на углероде (400 мг). Полученную смесь дегазировали аргоном и помещали в атмосферу водорода. После перемешивания в течение 2 часов смесь напрямую хроматографически очищали в силикагеле с использованием дихлорметана/метанола (о/о, 97:3) в качестве элюента с получением 6 в виде твердого вещества белого цвета (444 мг, 82%). δ н ((СD3)2SO) 11,3 (1Н, шс, NH), 7,36 (1Н, д, J 1,1, 6-Н), 5,80 (1Н, д, J 4,3, 1’-Н), 5,61 (1Н, с, ОН), 4,86 (1Н, м, 5’-Ha), 3,89 (1Н, д, J 4,2, 2’-Н), 3,85 (1Н, м, 2’’-Ha), 3,83-3,64 (3Н, м, 4’-Н, 5’-Hb, 2’’-Hb), 2,14 (1Н, м, 1’’-На), 1,81 (1Н, м, 1’’-Hb), 1,78 (3Н, д, J 1,0, СН3). δ c (CD3OD) 166,7 (С-4), 152,2 (С-2), 139,7 (С-6), 110,1 (С-5), 89,4, 89,1, 85,5, 85,2 (С-1’, С-2’, С-3’, С-4’), 71,4 (С-2’’), 61,6 (С-5’), 37,0 (C-1’’), 12,7 (СН3). Получено: С - 47,4; Н - 5,7; N - 9,0. Для C12H16N2O6·H2O рассчитано: С - 47,7; Н - 6,0; N - 9,3%.
Пример 10
(1S,5R,6R,8R)-6-(4,4’-диметокситритилоксиметил)-5-гидрокси-8-(тимин-1-ил)-2,7-диоксабицикло[3.3.0]нонан (7)
Раствор нуклеозида 6 (310 мг, 1,09 ммоль) в безводном пиридине (2,5 см3) перемешивали при комнатной температуре и добавляли 4,4’-диметокситритилхлорид (593 мг, 1,83 ммоль). После перемешивания в течение 3 дополнительно часов добавляли 4,4’-диметокситритилхлорид (100 мг, 0,310 ммоль). После перемешивания в течение еще 2 часов добавляли метанол (0,5 см3) и полученную смесь выпаривали. Остаток растворяли в дихлорметане (5 см3) и промывали насыщенным водным раствором кислого углекислого натрия (3× 5 см3). Органическую фазу высушивали над Na2SO4 и выпаривали при пониженном давлении. Остаток подвергали хроматографической очистке на силикагелевой колонке с использованием дихлорметана/метанола (о/о, 99:1) в качестве элюента с получением соединения 7 в виде твердого вещества белого цвета (618 мг, 97%). δ н (СDСl3) 9,04 (1Н, шс, NH), 7,47-7,16 (10Н, м, 6-Н, DMT), 6,86-6,82 (4Н, м, DMT), 6,06 (1Н, д, J 4,1, 1’-Н), 4,35 (1Н, д, J 4,1, 2’-Н), 4,03 (1Н, м, 4’-Н), 3,89 (1Н, м, 2’’-На), 3,79 (6Н, с, 2× ОСН3), 3,61 (1Н, м, 5’-На), 3,32-3,26 (2Н, м, 5’-Нb, 2’’-Hb), 1,94-1,69 (2Н, м, 1’’-На, 1’’-Hb), 1,89 (3Н, с, СН3). δ с (СDСl3) 163,4 (С-4), 158,6 (DMT), 150,1 (С-2), 144,3 (DMT), 137,2 (С-6), 135,6, 135,3, 129,9, 129,9, 128,9, 128,1, 127,9, 126,9, 125,2, 113,2 (DMT), 109,3 (С-5), 88,7, 87,3, 86,9, 83,5, 81,0 (DMT, С-1’, С-2’, С-3’, C-4’), 69,7 (С-2’’), 62,1 (С-5’), 55,1 (ОСН3), 36,5 (С-1’’), 12,5 (СН3).
Пример 11
(1S,5R,6R,8R)-5-(2-цианоэтокси(диизопропиламино)фосфинокси)-6-(4,4’-диметокситритилоксиметил)-8-(тимин-1-ил)-2,7-диоксабицикло[3.3.0]нонан (8)
Раствор нуклеозида 7 (436 мг, 0,743 ммоль) в безводном дихлорметане (2,2 см3) и диизопропилэтиламина (0,62 см3) перемешивали при комнатной температуре и добавляли 2-цианоэтил-N,N-диизопропилфосфоамидохлоридит (0,33 см3, 1,46 ммоль). После перемешивания в течение 1,5 часов добавляли метанол (0,4 см3) и этилацетат (5 см3) и полученную смесь промывали насыщенным водным раствором кислого углекислого натрия (3× 5 см3) и насыщенным соляным раствором (3× 5 см3). Органическую фазу высушивали над Na2SO4 и выпаривали при пониженном давлении. Остаток подвергали хроматографической очистке на силикагелевой колонке с использованием дихлорметана/метанола (о/о 97:3) в качестве элюента и выпаривали растворители с получением маслянистого вещества, которое растворяли в толуоле (1 см3) и осаждали гексаном при -30° С с получением соединения 8 в виде твердого вещества белого цвета (517 мг, 88%). δ р (CDCl3) 142,0, 141,9.
Пример 12
1-(3,5-ди-О-бензил-3-С(2-гидроксиэтил)-β -D-рибофуранозил)тимин (9)
К перемешиваемому раствору нуклеозида 2 (1,00 г, 2,09 ммоль) в THF (5,4 см3) и воде (5,4 см3) добавляли периодат натрия (1,34 г, 6,27 ммоль) и 2,5%-ный раствор тетраоксида осмия в грег-бутаноле (в/о, 0,265 см3, 19 мкмоль). Раствор перемешивали при комнатной температуре в течение 45 минут. Добавляли воду (25 см3) и раствор экстрагировали дихлорметаном (2× 50 см3). Органическую фазу промывали насыщенным водным раствором кислого углекислого натрия (3× 30 см3) и высушивали над Na2SO4. Растворитель удаляли при пониженном давлении и остаток вновь растворяли в THF (5,4 см3) и воде (5,4 см3). Смесь перемешивали при комнатной температуре и добавляли боргидрид натрия (79 мг, 2,08 ммоль). После перемешивания в течение 1,5 часов, добавляли воду (25 см3) и полученный раствор экстрагировали дихлорметаном (2× 50 см3). Органическую фазу промывали насыщенным водным раствором кислого углекислого натрия (3× 30 см3) и высушивали над Na2SO4. Растворитель удаляли при пониженном давлении и остаток подвергали хроматографической очистке на силикагелевой колонке с использованием дихлорметана/метанола (о/о, 98:2) в качестве элюента с получением соединения 9 в виде твердого вещества белого цвета (488 мг, 48%). δ н (СDСl3) 9,14 (1Н, шс, NH), 7,60 (1Н, д, J 1,1, 6-Н), 7,40-7,22 (10Н, м, Bn), 6,25 (1Н, д, J 7,7, 1’-Н), 4,59 (1Н, д, J 7,1, Bn), 4,49 (1Н, д, J 7,1 Bn), 4,39-3,30 (м, 8Н, 4’-Н, 2’-Н, Вn, 5’-Ha, 5’-Нb, 2’’-На, 2’’-Hb), 2,23-2,00 (2Н, м, 1’’-Ha, 1’’-Hb), 1,49 (3Н, д, J 0,7, СН3). δ с (CDCl3) 163,5 (С-4), 151,2 (С-2), 137,1, 136,5 (Bn), 135,7 (С-6), 128,7, 128,5, 128,2, 127,8, 127,6, 127,2 (Bn), 111,3 (С-5), 87,0, 82,7, 81,1, 78,3 (С-1’,С-2’, С-3’, С-4’), 73,7, 69,6 (Bn), 64,4 (С-5’), 57,0 (С-2’’), 32,4 (C-1’’), 11,8 (СН3). Получено: С - 63,9; Н - 6,3; N - 5,4. Для C26H30N2O7·0,25Н2О рассчитано: С - 64,1; Н - 6,3; N - 5,75%.
Пример 13
1-[3-С-(2-O-трет-бутилдиметилсилилоксиэтил)-3,5-ди-O-бензил-β -D-рибофуранозил]тимин (10)
Смесь нуклеозида 9 (1,80 г, 3,4 ммоль) и трет-бутилдиметилсилилхлорида (0,585 г, 3,9 ммоль) растворяли в безводном пиридине (20 см3). После выдерживания в течение 2 часов при комнатной температуре реакционную смесь упаривали досуха, дважды выпаривали вместе с толуолом (2× 10 см3) и вновь растворяли в дихлорметане (150 см3). Раствор промывали насыщенным водным раствором кислого углекислого натрия (2× 50 см3) и выпаривали с получением пены. Полученный материал очищали методом препаративной ВЭЖХ в силикагеле с использованием элюционного градиента (0-3% метанола в дихлорметане: о/о) с получением нуклеозида 10 в виде твердого вещества белого цвета (1,86 г, 92%). δ н (СDСl3) 7,61 (1Н, д, J 1,1, 6-Н), 7,35-7,20 (10Н, м, Bn), 6,27 (1Н, д, J 7,9, 1’-Н), 4,65-4,40 (4Н, м, Вn, 2’-H), 4,37 (1Н, с, Bn), 4,28 (1Н, т, J 7,9, 4’-Н), 4,35-3,55 (4Н, м, 2’’-На, 2’’-Нb, 5’-На, 5’-Hb), 2,30-2,05 (2Н, м, 1’’-На, 1’’-Hb), 1,46 (3Н, с, 5-СН3), 0,90 (9Н, м, СН3-C-Si), 0,08 (6Н, м, СН3-Si). δ с (СDСl3) 163,6 (С-6), 151,0 (С-2), 137,5, 136,6, 135,8 (С-5, Bn), 128,3, 128,1, 127,8, 127,2, 127,1, 126,8, 126,7 (Bn), 110,7 (С-4), 86,8, 82,5, 81,6, 78,3 (С-1’, С-2’, С-3’, С-4’), 73,3, 69,8 (Bn), 64,46 (С-5’), 58,2 (C-2’’), 32,9 (C-1’’), 25,6, 25,4, 17,9, -3,9, -5,7 (TBDMS), 11,6 (СН3). FAB+-MS: m/z 597,19 [М+Н]+, 619,18 [M+Na]+. Получено: С - 64,2; Н - 7,4; N - 4,2. Для С32Н44O7N2Si рассчитано: С - 64,4; Н - 7,4; N - 4,7%.
Пример 14
1-[3-С-(2-O-трет-бутилдиметилсилилоксиэтил)-3,5-ди-О-бензил-β -D-эритропентофуран-2-улозил]тимин (11)
Смесь нуклеозида 10 (2,14 г, 3,59 ммоль), дихромата пиридиния (1,48 г, 3,95 ммоль) и активированного порошкового молекулярного сита 3А (4 г) суспендировали в безводном дихлорметане (80 см3). После охлаждения смеси до -10° С при мощном перемешивании добавляли по каплям уксусный ангидрид (10 см3, 98 ммоль). Суспензию оставляли нагреваться до комнатной температуры и продолжали перемешивание в течение 1,5 часов до того момента, как реакцию останавливали добавлением триэтиламина (20 см3). Полученную смесь разводили дихлорметаном до 300 см3 и промывали водой (2× 200 см3). Органическую фазу выпаривали и полученный остаток хроматографически очищали в силикагеле с использованием градиента 1,0%, 1,2%, 1,3%, 1,4% и 1,5% метанола в дихлорметане (о/о, общий объем 250 см3 на каждом этапе градиента) с получением нуклеозида 11 (1,89 г, 84,4%) в виде твердого вещества белого цвета. δ н (СDСl3) 7,35-7,20 (11Н, м, Bn, 6-H), 6,40 (1Н, с, 1’-Н), 4,57 (1Н, с, Bn), 4,52 (1Н, с, Bn), 4,46 (1Н, д, J 11,0, Bn), 4,29 (1Н, д, J 11,0, Bn), 4,07 (1Н, дд, J 0,5, 2,2, 4’-Н), 3,95-3,70 (4Н, м, 2’’-На, 2’’-Нb, 5’-На, 5’-Hb), 2,05 (1Н, м, 1’’-На), 2,42 (1Н, м, 1’’-Hb), 1,42 (3Н, д, J 1,1, 5-СН3), 0,86 (9Н, с, СН3-C-Si), 0,01 (6Н, с, СН3-Si). δ с (СDСl3) 202,6 (С-2’), 163,7 (С-4), 151,2 (С-2), 137,7, 136,6, 136,5 (Bn, C-6), 128,7, 128,5, 128,2, 128,1, 127,7, 126,4, 126,3 (Bn), 110,9 (С-5), 84,5, 81,3, 80,2 (С-1’, С-3’, С-4’), 73,6, 70,4 (Bn), 66,0 (C-5’), 57,6 (C-2’’), 27,3 (С-1’’), 25,9, 25,7, 18,2, -5,8, -5,9 (TBDMS), 11,7 (СН3). FAB-MS m/z 595,14 [М+Н]+. Получено: С - 64,1; Н - 6,9; N - 4,5. Для C32H42N2Si рассчитано: С - 64,6; Н - 7,1; N - 4,7%.
Пример 15
(1S,5R,6R,8R)-1-гидрокси-5-бензилокси-6-бензилоксиметил-8-(тимин-1-ил)-2,7-диоксабицикло[3.3.0]октан (12)
Соединение 11 (1,80 г, 30,3 ммоль) растворяли в 0,5% HCl в метаноле (в/о, 20 см3) и полученную смесь перемешивали в течение 30 минут при комнатной температуре. После упаривания досуха остаток растворяли в дихлорметане (100 см3) и промывали насыщенным водным раствором кислого углекислого натрия (2× 40 см3). Органическую фазу выпаривали и остаток хроматографически очищали на силикагеле с использованием 2% метанола в дихлорметане (о/о) с выходом нуклеозида 12 (1,35 г, 93,5%) в виде твердого вещества белого цвета. δ н (CDCl3) 7,37-7,27 (11Н, м, Вn, 6-Н), 5,87 (1H, с, 1’-Н), 4,71 (2Н, с, Bn), 4,64 (1H, д, J 12,0, Bn), 4,56 (1H, д, J 12,0, Bn), 4,36 (1H, т, J 5,7, 4’-Н), 4,16 (1H, м, 2’’-На), 3,96 (1H, м, 2’’-Hb), 3,74 (2Н, м, 5’-На, 5’-Hb), 2,35-2,15 (2Н, м, 1’’-Ha, 1’’-Hb), 1,88 (3Н, с, СН3). δ с (CDCl3) 163,7 (С-4), 151,4 (С-2), 137,8, 137,3, 136,7 (Вn, С-6), 128,5, 128,4, 128,0, 127,8, 127,5 (Bn), 109,9 (С-5), 108,6 (С-2’), 88,8, 87,1, 80,9 (С-1’, С-3’, С-4’), 73,6, 68,5, 68,1, 67,9 (С-5’, C-2’’, Bn), 30,9 (С-1’’), 12,6 (СН3). FAB-MS: m/z 481,03 [М+Н]+, 503,02 [M+Na]+. Получено: С - 64,6; Н - 5,8; N - 5,7. Для C26H28O7N2 рассчитано: С - 65,0; Н - 5,9; N - 5,8%.
Пример 16
(1S,5R,6R,8R)-1,5-дигидрокси-6-гидроксиметил-8-(тимин-1-ил)-2,7-диоксабицикло[3.3.0]октан (13)
Соединение 13 было последовательно получено из соединения 12 путем каталитического отшепления защитной бензильной группы, выполненного таким же образом, как это было описано при получении соединения 6. Очистку 13 осуществляли хроматографически на силикагелевой колонке в градиенте метанола (от 6 до 14%) в дихлорметане. Аналитические количества соединения 13 (до 15 мг) дополнительно очищали методом ВЭЖХ с обращенной фазой на колонке (10× 250 мм), загруженной Nucleosil C18 (10 мкм). Скорость потока - 8 см3/мин. Элюент - 0-10% ацетонитрил. Время - 60 минут. Выход составил 82%. δ н (СD3ОD) 7,44 (1Н д, J 1,2, 6-Н), 5,83 (1Н, с, 1’-Н), 4,10-3,80 (5Н, м, 5’-На, 5’-Нb, 2’’-Ha, 2’’-Hb, 4’-Н), 2,39-2,25 (1Н, м, 1’’-На), 2,00-1,90 (1Н, м, 1’’-Hb), 1,87 (3Н, д, J 1,2, СН3). δ с (CD3OD) 166,3 (С-4), 152,7 (С-2), 139,8 (С-6), 110,0, 109,6 (С-2’, С-5), 87,8, 85,8, 84,6 (C-1’, С-3’, C-4’), 68,8, 61,6 (С-5’, С-2’’), 35,6 (С-1’’), 12,4 (СН3). FAB-MS: m/z 301,03 [М+Н]+. Получено: С - 46,6; Н - 5,7; N - 8,5. Для C12H16O7N2 рассчитано: С - 48,0; Н - 5,4; N - 9,3%.
Пример 17
(1S,5R,6R,8R)-5-бензилокси-6-бензилоксиметил-1-метокси-8-(3-N-метилтимин-1-ил)-2,7-диоксабицикло[3.3.0]октан (14), (1S,5R,6R,8R)-5-бензилокси-6-бензилоксиметил-1-гидрокси-8-(3-N-метилтимин-1-ил)-2,7-диоксабицикло[3.3.0]октан (15) и (1S,5R,6R,8R)-5-бензилокси-6-бензилоксиметил-1-метокси-8-(тимин-1-ил)-2,7-диоксабицикло[3.3.0]октан (16)
Смесь соединения 12 (1,04 г, 2,16 ммоль) и гидрида натрия (171 мг 60%-ной суспензии в минеральном масле, 4,30 ммоль) растворяли в безводном дихлорметане (4 см3) в течение 10 минут при перемешивании. Добавляли метилиодид (1 см3, 16 ммоль) и полученную реакционную смесь инкубировали при 36° С в течение 23 часов. После выпаривания остаток подвергали хроматографической очистке на силикагелевой колонке с использованием в качестве элюента градиента 0,4-2,4% метанола в дихлорметане (о/о) с получением продуктов 14, 15 и 16, а также исходного соединения 12 (212 мг, 20,5%). Соединение 14 (47 мг, 4,3%). δ н (CDCl3) 7,25-7,37 (11Н, м, Вn, 6-Н), 6,15 (1H, с, 1’-Н), 4,74 (1H, д, J 11,5, Bn), 4,67 (1H, д, J 11,3, Bn), 4,62 (1H, д, J 12,1, Bn), 4,55 (1H, д, J 11,9, Bn), 4,34 (1H, т, J 5,6, 4’-Н), 3,99 (1H, м, 2’’-На), 4,22 (1H, т, 2’’-Hb), 3,72 (2Н, м, 5’-На, 5’-На), 3,41 (3Н, с, СН3-O), 3,35 (3Н, с, СН3-N3), 2,27 (1H, м, 1’’-Ha), 2,41 (1H, м, 1’’-Hb), 1,93 (3Н, с, 5-СН3). δ с (CDCl3) 163,3 (С-4), 151,0 (С-2), 138,2, 137,3, 135,7 (Bn, C-6), 128,3, 128,2, 127,8, 127,6, 127,4, 126,9 (Bn), 111,8 (С-5), 108,5 (С-2’), 89,1, 84,8, 79,5 (C-1’, С-3’, С-4’), 73,5, 68,4, 68,2, 67,3 (Bn, 5 C-5’, C-2’’), 50,8 (СН3-O), 32,6 (С-1’’), 27,9 (СН3-N), 13,2 (СН3). FAB-MS: m/z 508,88 [M+H]+. Получено: С - 65,7; Н - 6,9; N - 4,8. Для С28H32O7N2 рассчитано: С - 66,1; Н - 6,3; N - 5,5%. Соединение 15 (97 мг, 9,1%). δ н (CDCl3) 7,37-7,28 (11Н, м, Bn, 6-Н), 5,86 (1H, с, 1’-Н), 4,72 (2Н, с, Bn), 4,64 (1H, д, J 11,9, Bn), 4,58 (1H, д, J 11,9, Bn), 4,37 (1H, т, J 5,6, 4’-Н), 4,13 (1H, м, 2’’-Ha), 3,93 (1H, м, 2’’-Hb), 3,75 (2Н, м, 5’-Ha, 5’-Hb), 3,34 (1H, с, СН3-N), 2,32-2,16 (2Н, м, 1’’-На, 1’’-Hb), 1,93 (3Н, с, СН3). δ с (СDСl3) 163,2 (С-4), 151,9 (С-2), 137,5, 137,1, 134,0 (Вn, С-6), 128,4, 128,3, 128,1, 127,9 127,7, 127,6, 127,3 (Bn), 108,8, 108,5 (С-2’, С-5), 88,7 (C-1’), 88,0, 81,0 (С-3’, С-4’), 73,5, 68,3, 67,9, 67,7 (Bn, С-5’, С-2’’), 30,6 (C-1’’), 27,8 (СН3-N), 13,2 (СН3). FAB-MS m/z 495,28 [М+Н]+, 517,24 [М+Nа]+. Соединение 16 (665 мг, 62,3%). δ н (СDСl3) 7,35-7,25 (11Н, м, Вn, 6-Н), 6,06 (1H, с, 1’-Н), 4,73 (1H, д, J 11,5, Bn), 4,6, (1H, д, J 11,3, Bn), 4,61 (1H, д, J 11,9, Bn), 4,55 (1H, д, J 12,0, Bn), 4,34 (1H, т, J 5,6, 4’-Н), 4,20 (1H, м, 2’’-На), 3,98 (1H, м, 2’’-Hb), 3,72 (2Н, м, 5’-На, 5’-Hb), 3,40 (3Н, c, СН3-O), 2,45-2,35 (1H, м 1’’-На), 2,30-2,20 (1H, м, 1’’-Hb), 1,90 (3Н, д, J 1,1, СН3). δ с (СDСl3) 163,2 (С-4), 150,1 (С-2), 138,2, 137,9, 137,3 (Вn, С-6), 128,4, 128,2, 127,8, 127,6 127,4, 127,1 (Bn), 110,8 (С-5), 109,3 (С-2’), 89,2, 84,2, 79,6 (C-1’, C-3’, C-4’), 73,6, 68,5, 68,3, 67,4 (Bn, C-5’, С-2’’), 50,8 (СН3-O), 32,6 (С-1’’), 12,5 (СН3). FAB-MS m/z 495,22 [М+Н]+, 517,23 [M+Na]+. Получено: С - 66,2; Н - 7,2; N - 4,4. Для С27Н30O7N2 рассчитано: С - 65,6; Н - 6,1; N - 5,7%.
Пример 18
[1S,5R,6R,8R)-5-гидрокси-6-гидроксиметил-1-метокси-8-(тимин-1-ил)-2,7-диоксабицикло[3.3.0]октан (17)
К раствору нуклеозида 16 (1,20 г, 2,43 ммоль) в метаноле (10 см3) добавляли 20% гидроксид палладия на угле (250 мг) и полученную смесь подвергали аккуратной дегазации при пониженном давлении. Применяли атмосферу водорода и перемешивали в течение 12 часов. После этого удаляли катализатор путем фильтрации реакционной смеси через стеклянную колонку (1× 8 см), загруженную силикагелем в метаноле. Эту колонку дополнительно промывали метанолом (20 см3). Все фракции собирали, упаривали досуха и выпаривали вместе с петролейным эфиром с выходом стекловидного твердого вещества. Этот остаток хроматографически очищали в силикагеле, используя для элюции градиент 5-10% метанола в дихлорметане (о/о). Эти фракции, включающие искомый продукт, отбирали, объединяли и упаривали досуха. Остаток растворяли в безводном метаноле (5 см3) и добавляли безводный бензол (100 см3). В результате лиофилизации получали нуклеозид 17 (0,61 г, 79%) в виде твердого вещества белого цвета. δ н (СD3ОD) 7,45 (1Н, с, 6-Н), 5,93 (1Н, с, 1’-Н), 4,15-3,81 (5Н, м, 5’-Ha, 5’-Нb, 2’’-Ha, 2’’-Нb, 4’-H), 3,43 (3Н, с, СН3-O), 2,47-2,40 (1Н, м, 1’’-Н), 2,03-1,93 (1Н, м, 1’’-Hb), 1,92 (3Н, с, СН3). δ C (СD3ОD) 164,1 (С-4), 150,1 (С-2), 138,3 (С-6), 109,6 (С-5), 108,3 (С-2’), 84,4, 84,1, 82,4 (С-1’, С-3’, С-4’), 68,0, 59,5 (С-5’, С-2’’), 49,6 (СН3-O), 34,0 (С-1’’), 10,5 (СН3). FAB-MS m/z 315,13 [М+Н]+, 337,09 [M+Na]+. Получено: С - 49,9; Н - 5,7; N - 8,2. Для C13H18O7N2 рассчитано: С - 49,7; Н - 5,8; N - 8,9%.
Пример 19
(1S,5R,6R,8R)-6-(4,4’-диметокситритилоксиметил)-5-гидрокси-1-метокси-8-(тимин-1-ил)-2,7-диоксабицикло[3.3.0]октан (18)
Смесь соединения 17 (0,95 г, 3,03 ммоль) и 4,4’-диметокситритилхлорида (1,54 г, 4,77 ммоль) растворяли в безводном пиридине (20 см3) и перемешивали в течение 4 часов при комнатной температуре. Реакционную смесь выпаривали с получением маслянистого остатка, который выпаривали вместе с толуолом (2× 20 см3). Добавляли дихлорметам (50 см3) и насыщенный водный раствор кислого углекислого натрия (50 см3), выделяли и выпаривали органическую фазу и полученный остаток очищали методом ВЭЖХ в силикагеле (остаток растворяли в минимальном количестве дихлорметана, содержащего 0,5% триэтиламина (о/о), который вносили на колонку, уравновешенную тем же растворителем). Колонку промывали этилацетатом/петролейным эфиром/триэтиламином (15:84,5:0,5, о/о/о, общий объем 1000 см3) и полученный продукт элюировали в градиенте метанола (0-2%) в дихлорметане, содержащем 0,5% триэтиламин (о/о) с получением соединения 18 (1,71 г, 92,8%) в виде твердого вещества белого цвета. δ н (CDCl3) 7,51-7,17 (10Н, м, DMT, 6-Н), 6,79-6,85 (4Н, м, DMТ), 6,04 (1Н, с, 1’-Н), 4,12-3,98 (3Н, м, 5’-На, 5’-Нb, 4’-Н), 3,77 (6Н, c, СН3-DMT), 3,49 (3Н, с, СН3-O), 3,45-3,32 (2Н, м, 2’’-На, 2’’-Hb), 2,11-2,01 (1Н, м, 1’’-На), 1,94-1,87 (1Н, м, 1’’-Hb), 1,93 (3Н, с, СН3). δ с (CDCl3) 164,2 (С-4), 158,6, 144,7, 135,7, 130,1, 128,2, 127,9, 126,8, 113,2 (DМТ), 150,7 (С-2), 137,7 (С-6), 109,8, 109,7 (С-5, С-2’), 86,5, 85,3, 85,0, 81,4 (DMТ, С-1’, C-3’, C-4’), 69,2, 62,4 (С-5’, С-2’’), 55,2 (СН3-DMT), 51,7 (СН3-O),35,5 (С-1’’), 12,7 (СН3). FAB-MS m/z 617,26 [М+Н]+, 639,23 [M+Na]+. Получено: С - 66,4; Р - 6,1; N - 4,2. Для С34Р36О9N2 рассчитано: С - 66,2; Р - 5,9; N - 4,5%.
Пример 20
(1S,5R,6R,8R)-5-(2-
цианоэтокси(диизопропиламимо)фосфинокси)-6-(4,4’-диметокситритилоксиметил)-1-метокси-8-(тимин-1-ил)-2,7-диоксабицикло[3.3.0]октан (19)
Соединение 18 (1,2 г, 1,95 ммоль) раcтворяли в безводном дихлорметане (10 см3). N,N-диизопропилэтиламин (1,35 см3, 7,8 ммоль) и 2-цианоэтил-N,N-диизопропилфосфорамидохлоридит (0,92 г, 3,9 ммоль) добавляли в условиях перемешивания при комнатной температуре. Через 72 часа полученную смесь разводили до 100 см3 в дихлорметане и промывали в насыщенном водном растворе гидрокарбоната (50 см3). Органическую фазу выпаривали и вносили на силикагелевую колонку ВЭЖХ, в которой использовали градиент элюента В (петролейный эфир/дихлорметан/этилацетат/пиридин - 45:45:10;0,5 - о/о) в элюенте А (петролейный эфир/дихлорметан/-пиридин - 50:50:0,5 - о/о). Фракции, содержащие искомый продукт, концентрировали, выпаривали вместе с толуолом (10 см3) и высушивали при пониженном давлении, Остаток растворяли в безводном бензоле (20 см3) и осаждали добавлением данного раствора в безводный петролейный эфир (400 см3) при перемешивании. Полученное в результате белое твердое вещество выделяли фильтреванием и высушивали с получением соединения 19 (0,96 г, 60,3%). δ p (СDСl3) 142,64, 142,52. FAB-MS m/z 817,26 [М+Н]+, 839,24 [М+Н]+. Получено: С – 62,8; Р - 6,4; N - 6,9. Для С43Р53О10N4Р рассчитано: С - 63,2; Н - 6,5; N - 6,9%.
Пример 21
1,2-O-изопропилиден-3-С-винил-α -D-рибофураноза (20)
Раствор 5-O-трет-бутилдиметилсилил-1,2-O-изопропилиден-α -D-эритропент-3-улофуранозы (Y.Yoshimura, T.Sano, A.Matsuda, T.Ueda, 1988, Chem. Pharm. Bull., 36, 162) (6,05 г, 0,020 моль) в безводном THF (250 см3) перемешивали при 0° С и по каплям добавляли 1 М раствор винилбромида магния в эфире (44 см3, 44 ммоль). Полученную реакционную смесь перемешивали при комнатной температуре в течение 2 часов, после чего добавляли насыщенный водный раствор хлорида аммония (200 см3) и осуществляли экстрагирование дихлорметамом (3× 300 см3). Объединенные экстракты промывали соляным раствором (3× 250 см3) и высушивали над Na2SO4. Растворитель удаляли и остаток повторно растворяли в безводном THF (225 см3). К этой смеси добавляли 1 М раствор фторида тетрабутиламмония в THF (22 см3, 22 ммоль), перемешивали при комнатной температуре проводили в течение 20 минут, после чего смесь выпаривали при пониженном давлении. Остаток растворяли в дихлорметане (500 см3) и промывали насыщенным раствором кислого углекислого натрия (2× 200 см3). Водную фазу экстрагировали с использованием непрерывной 12-часовой экстракции и объединенные экстракты высушивали над Na2SO4 и выпаривали. Остаток подвергали хроматографической очистке в силикагеле, используя дихлорметан/метанол (99:1, о/о) в качестве элюента с получением фуранозы 20 в виде твердого вещества белого цвета (3,24 г, 75%). δ н (CDCl3) 5,84 (1Н, д, J 3,7, 1-Н), 5,74 (1Н, дд, J 11,0, 17,2, 1’-Н), 5,52 (1Н, дд, J 1,6, 17,1, 2’-На), 5,29 (1Н, дд, J 1,3, 11,0, 2’-Hb), 4,21 (1Н, д, J 3,7, 2-Н), 3,98 (1Н, т, J 5,7, 4-Н), 3,68-3,64 (2Н, м, 5-На, 5-Hb), 2,88 (1Н, с, 3-ОН), 1,99 (1Н, т, J 6,3, 5-ОН), 1,60 (3Н, с, СН3), 1,35 (3Н, с, СН3). δ с (СDСl3) 133,6 (С-1’), 116,2 (С-2’), 113,0 (С(СН3)2), 103,8 (С-1), 83,4, 82,4 (С-4, С-2), 79,6 (С-3), 61,3 (С-5), 26,5, 26,4 (СН3).
Пример 22
3,5-ди-О-бензил-1,2-O-изопропилиден-3-С-винил-α -D-рибофураноза (21)
60%-ную суспензию гидрида натрия (в/о, 1,78 г, 44,5 ммоль) в безводном DMF (50 см3) перемешивали при 0° C и раствор фуранозы 20 (3,20 г, 14,8 ммоль) в безводном DMF (35 см3) добавляли по каплям в течение 30 минут. Смесь перемешивали при 50° С в течение 1 часа и затем охлаждали до 0° С. Раствор бензилбромида (5,3 мл, 44,5 моль) в безводном DMF (5,3 см3) добавляли по каплям и полученную смесь перемешивали при комнатной температуре в течение 20 часов. Реакционную смесь выпаривали и вновь растворяли в дихлорметане (300 см3), промывали насыщенным водным раствором кислого углекислого натрия (3× 200 см3) и высушивали над Na2SO4. Растворители удаляли при пониженном давлении и остаток подвергали хроматографической очистке петролейным эфиром/этилацетатом (9:1 о/о) в качестве элюента с получением фуранозы 21 в виде твердого вещества белого цвета (5,36 г, 91%). δ н (СDСl3) 7,40-7,26 (10Н, м, Bn), 5,90 (1Н, д, J 3,6, 1-Н), 5,72 (1Н, дд, J 11,1, 17,9, 1’-Н), 5,41 (1Н, дд, J 0,7, 11,1, 2’-На), 5,30 (1Н, дд, J 0,5, 17,8, 2’-Hb), 4,70-4,45 (6Н, м, Вn, 2-Н, 4-Н), 3,69 (1Н, дд, J 2,6, 10,8, 5-На), 3,50 (1Н, дд, J 7,9, 10,9, 5-Hb), 1,64 (3Н, с, СН3), 1,40 (3Н, с, СН3). δ с (СDСl3) 138,6, 138,3 (Bn), 134,5 (С-1’), 128,3-127,4 (Bn), 118,2 (С-2’), 112,9 (С(СН3)2), 104,7 (С-1), 84,7, 81,1, 81,0 (С-2, С-3, С-4), 73,3 (С-5), 69,4, 67,0 (Bn), 26,8, 26,6 (СН3).
Пример 23
3,5-ди-О-ацетил-3,5-ди-О-бензил-3-С-винил-α ,β -D-рибофураноза (22)
Раствор фуранозы 21 (4,40 г, 11,1 ммоль) в 80%-ной водной уксусной кислоте (50 см3) перемешивали при 90° С в течение 8 часов. Растворители удаляли и полученный остаток выпаривали вместе с 99%-ным этанолом (3× 25 см3), толуолом (3× 25 см3) и безводным пиридином (2× 25 см3) и вновь растворяли в безводном пиридине (20 см3). Добавляли уксусный ангидрид (17 см3) и полученный раствор перемешивали при комнатной температуре в течение 48 часов. Реакцию останавливали охлажденной льдом водой (10 см3) и экстрагировали дихлорметаном (2× 100 см3). Объединенные экстракты промывали насыщенным водным раствором кислого углекислого натрия (3× 100 см3) и высушивали над Na2SO4. Растворитель выпаривали и остаток подвергали хроматографической очистке в силикагеле с использованием петролейного эфира/этилацетата (4:1, о/о) в качестве элюента с получением фуранозы 22 в виде маслянистого вещества (4,27 г, 87%, α :β ≈ 1:1). δ с (CDCl3) 169,9, 169,8 (С=O), 139,0, 138,6, 138,0, 137,8 (Bn), 133,3, 132,4 (С-1’), 128,4-126,8 (Bn), 119,6, 119,5 (С-2’), 99,5, 94,0 (С-1), 85,4, 85,0, 84,3, 83,6, 77,7, 73,6, 73,5, 73,3, 70,0, 69,2, 67,5, 67,2 (С-2, С-3, С-4, С-5, Bn), 21,0, 20,9, 20,6, 20,4 (СН3).
Пример 24
1-(2-O-ацетил-3,5-ди-O-бензил-3-С-винил-β -D-рибофуранозил)тимин (23)
К перемешиваемому раствору соединения 22 (4,24 г, 9,6 ммоль) и тимина (2,43 г, 19,3 ммоль) в безводном ацетонитриле (100 см3) добавляли N,O-бис(триметилсилил)ацетамид (11,9 см3, 48,1 ммоль). Полученную реакционную смесь перемешивали с нагреванием с обратным холодильником в течение 30 минут. После охлаждения до 0° С добавляли по каплям триметил-силилтрифлат (3,2 см3, 16,4 ммоль) и полученный раствор перемешивали при комнатной температуре в течение 24 часов. Реакцию останавливали холодным насыщенным водным раствором кислого углекислого натрия (10 см3) и полученную в результате смесь экстрагировали дихлорметаном (3× 50 см3). Объединенные экстракты промывали насыщенным водным раствором кислого углекислого натрия (2× 50 см3) и соляным раствором (2× 50 см3) и высушивали над Na2SO4. Экстракт выпаривали при пониженном давлении и остаток подвергали хроматографической очистке в силикагеле с использованием дихлорметана/метанола (99:1, о/о) в качестве элюента с получением нуклеозида 23 в виде пены белого цвета (4,03 г, 83%). δ н (СDСl3) 8,78 (1Н, шс, NH), 7,75 (1Н, с, 6-Н), 7,38-7,26 (10Н, м, Bn), 6,49 (1Н, д, J 8,1, 1’-Н), 5,99-5,88 (2Н, м, 2’-Н и 1’’-H), 5,54-5,48 (2Н, м, 2’’-На, 2’’-Hb), 4,91-4,50 (4Н, м, Bn), 4,34 (1Н, с, 4’-Н), 3,80 (1Н, м, 5’-Ha), 3,54 (1Н, м, 5’-Hb), 2,11 (3Н, с, СОСН3), 1,48 (3Н, с, СН3). δ с (СDСl3) 170,1 (С=O), 163,8 (С-4), 151,0 (С-2), 138,9, 136,9 (Bn), 136,1 (С-6), 132,0 (C-1’’), 128,7, 128,5, 128,2, 127,8, 127,7, 127,5, 127,5, 127,1 (Bn), 120,7 (С-2’’), 111,3 (С-5), 85,4 (С-1’), 85,2 (С-3’), 84,3 (С-4’), 76,0 (С-2’), 73,7 (C-5’), 69,3, 67,6 (Bn), 20,6 (СОСН3), 11,7 (СН3). Получено: С - 66,3; Н - 6,0; N - 5,1. Для C28H30N2O7 рассчитано: С - 66,4; Н - 6,0; N - 5,5%.
Пример 25
1-(3,5-ди-O-бензил-3-С-винил-β -D-рибофуранозил)тимин (24)
К перемешиваемому раствору нуклеозида 23 (3,90 г, 1,1 ммоль) в безводном метаноле (40 см3) добавляли метоксид натрия (0,83 г, 15,4 ммоль). Смесь перемешивали при комнатной температуре в течение 42 часов и затем нейтрализовывали разбавленной водной соляной кислотой. Полученную смесь экстрагировали дихлорметаном (2× 150 см3) и объединенные экстракты промывали насыщенным водным раствором кислого углекислого натрия (3× 100 см3) и высушивали над Na2SO4. Растворитель удаляли при пониженном давлении с получением нуклеозида 24 в виде пены белого цвета (3,48 г, 97%). δ н (СDСl3) 8,89 (1Н, шс, NH), 7,60 (1Н, д, J 0,9, 6-Н), 7,36-7,26 (10Н, м, Bn), 6,23 (1Н, д, J 7,8, 1’-Н), 5,98 (1Н, дд, J 11,2, 17,7, 1’’-Н), 5,66 (1Н, д, J 17,7, 2’’-На), 5,55 (1Н, д, J 11,5, 2’’-Hb), 4,75-4,37 (6Н, м, 2’-H, 4’-H, Bn), 3,84 (1Н, дд, J 2,7, 10,8, 5’-Ha), 3,58 (1Н, д, J 11,2, 5’-Hb), 3,23 (1Н, д, J 10,6, 2’-ОН), 1,50 (3Н, с, СН3). δ с (CDCl3) 163,7 (С-4), 151,3 (С-2), 138,0, 136,9 (Bn), 136,0 (С-6), 131,2 (С-1’’), 128,8, 128,6, 128,3, 127,8, 127,7, 127,3 (Bn), 120,7 (С-2’’), 111,3 (С-5), 87,3 (C-1’), 84,6 (С-3’), 81,4 (С-4’), 78,0 (С-2’), 73,7 (C-5’), 70,0, 66,4 (Bn), 11,8 (СН3). Получено: С - 66,8; Н - 6,2; N - 5,9. Для С26Н28Н2O6 рассчитано: С - 67,2; Н - 6,1; N - 6,0%.
Пример 26
1-(3,5-ди-O-бензил-2-O-метансульфонил-3-С-винил-β -D-рибофуранозил)тимин (25)
Нуклеозид 24 (2,57 г, 5,53 ммоль) растворяли в безводном пиридине (18 см3) и охлаждали до 0° С. Метансульфонилхлорид (1,28 см3, 16,6 ммоль) добавляли по каплям и полученную смесь перемешивали при комнатной температуре в течение 30 минут. Реакцию останавливали водой (5 см3) и полученную в результате смесь экстрагировали дихлорметаном (3× 80 см3). Объединенные экстракты промывали насыщенным водным раствором кислого углекислого натрия (3× 120 см3) и высушивали над Na2SO4. Растворитель выпаривали при пониженном давлении и остаток подвергали хроматографической очистке в силикагеле с использованием дихлорметана/метанола (99:1, о/о) в качестве элюента с получением нуклеозида 25 в виде пены желтого цвета (2,53 г, 84%). δ н (СDСl3) 8,92 (1Н, шс, NH), 7,71 (1Н, д, J 1,4, 6-Н), 7,41-7,28 (10Н, м, Bn), 6,57 (1Н, д, J 7,8, 1’-Н), 5,99-5,61 (4Н, м, 2’-Н, 1’’-Н и 2’’-На, 2’’-Hb), 4,86-4,50 (4Н, м, Bn), 4,37 (1Н, дд, J 1,5, 2,4, 4’-Н), 8,82 (1Н, дд, J 2,6, 11,0, 5’-На), 3,55 (1Н, дд, J 1,2, 11,0, 5’-Hb), 3,02 (3Н, с, СН3), 1,47 (3Н, д, J 1,1, СН3). δ с (СDСl3) 163,7 (С-4), 151,5 (С-2), 138,7, 136,7 (Bn), 135,7 (С-6), 130,9 (С-1’’), 128,8, 128,5, 128,4, 127,6, 127,0 (Bn), 121,8 (С-2’’), 111,9 (С-5), 85,1 (С-1’), 84,5 (С-3’), 84,0 (С-4’), 80,7 (С-2’), 73,7 (С-5’), 69,2, 67,7 (Bn), 38,9 (СН3), 11,8 (СН3).
Пример 27
1-(3,5-ди-O-бензил-3-С-винил-β -D-арабинофуранозил)тимин (26)
Раствор нуклеозида 25 (2,53 г, 4,66 ммоль) в смеси этанола (50 см3), воды (50 см3) и 1 М водного раствора едкого натра (15 см3) перемешивали с нагреванием с обратным холодильником в течение 16 часов. Смесь нейтрализовывали разбавленной водной соляной кислотой, растворитель выпаривали при пониженном давлении, а полученный остаток экстрагировали дихлорметаном (3× 120 см3). Объединенные экстракты промывали насыщенным водным раствором кислого углекислого натрия (3× 150 см3) и высушивали над Na2SO4. Растворитель выпаривали при пониженном давлении и остаток подвергали хроматографической очистке на силикагелевой колонке с использованием дихлорметана/метанола (99:1) в качестве элюента с получением соединения 26 в виде пены белого цвета (1,61 г, 74%). δ н (CDCl3) 9,89 (1Н, шс, NH), 7,50 (1H, д, J 1,1, 6-Н), 7,41-7,26 (Bn), 6,28 (1Н, д, J 2,8, 1’-Н), 6,05 (1H, дд, J 11,1, 17,9, 1’’-Н), 5,58-5,50 (2Н, м, 2’’-Ha, 2’’-Hb), 4,98 (1H, д, J 9,0, 2’-OH), 4,64-4,31 (6Н, м, 2’-H, 4’-Н, Bn), 3,73 (2Н, м, 5’-На, 5’-Hb), 1,73 (1H, д, J 0,6, СН3). δ с (СDСl3) 165,1 (С-4), 150,5 (С-2), 138,4, 138,0, 136,7 (С-6, Bn), 130,4 (C-1’’), 128,8, 128,6, 128,5, 128,1, 128,0, 127,8 (Bn), 120,6 (С-2’’), 108,1 (С-5), 88,6 (С-1’), 87,9 (С-3’), 87,2 (С-4’), 73,7 (С-2’), 71,8 (С-5’), 69,7, 66,3 (Bn), 12,3 (СН3). Получено: С - 66,8; Н - 6,2; N - 5,9. Для C26H28N2O6 рассчитано: С - 67,2; Н - 6,1; N - 6,0.
Пример 28
1-(3,5-ди-O-бензил-3-С-гидроксиметил-β -D-арабинофуранозил)тимин (27)
К раствору нуклеозида 26 (2,00 г, 4,31 ммоль) в смеси THF (15 см3) и воды (15 см3) добавляли периодат натрия (2,76 г, 12,9 ммоль) и 2,5%-ный раствор тетраоксида осмия в трет-бутаноле (в/о, 0,54 см3, 43 мкмоль). Реакционную смесь перемешивали при комнатной температуре в течение 18 часов, останавливали реакцию водой (50 см3) и полученную смесь экстрагировали дихлорметаном (2× 100 см3). Объединенные экстракты промывали насыщенным водным раствором кислого углекислого натрия (3× 75 см3), высушивали над Na2SO4 и выпаривали при пониженном давлении. Остаток вновь растворяли в смеси THF (15 см3) и воды (15 см3) и добавляли борогидрид натрия (488 мг, 12,9 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа, добавляли воду (50 см3) и полученную смесь экстрагировали дихлорметаном (2× 100 см3). Объединенные экстракты промывали насыщенным водным раствором кислого углекислого натрия (3× 75 см3) и высушивали над Na2SO4. Растворитель удаляли и остаток подвергали хроматографической очистке на силикагелевой колонке с использованием дихлорметана/метанола (98:2) в качестве элюента с получением нуклеозида 27 в виде пены белого цвета (732 мг, 36%). δ н (СDСl3) 11,09 (1Н, шс, NH), 7,41 (1H, д, J 1,0, 6-Н), 7,38-7,26 (Bn), 6,16 (1H, д, J 2,6, 1’-Н), 5,12 (1H, д, J 5,4, 2’-ОН), 4,66-4,29 (6Н, м, 2’-Н, 4’-Н, Bn), 4,02-3,96 (2Н, м, 1’’-На, 1’’-Hb), 3,90 (1H, дд, J 7,2, 9,7, 5’-Ha), 3,79 (1H, дд, J 5,6, 9,7, 5’-Hb), 2,49 (1H, т, J 6,4, 1’’-ОН), 1,68 (3Н, д, J 0,6, СН3). δ с (СDСl3) 166,1 (С-4), 150,6 (С-2), 139,0, 137,9, 137,0 (C-6, Bn), 128,7, 128,6, 128,4, 128,3, 128,0 (Bn), 107,5 (С-5), 88,2 (С-1’), 88,1 (С-3’), 84,2 (C-4’), 73,7 (С-5’), 72,1 (С-2’), 69,3, 65,4 (Bn), 58,6 (C-1’’), 12,3 (СН3).
Пример 29
(1R,2R,4R,5S)-1-бензилокси-2-бензилоксиметил-4-(тимин-1-ил)-3,6-диоксабицикло[3.2.0]гептан (28)
Раствор соединения 27 (2,26 г, 4,83 ммоль) в безводном пиридине (20 см3) перемешивали при -40° С и добавляли раствор метансульфонилхлорид (0,482 см3, 4,83 ммоль) в безводном пиридине (10 см3). Реакционную смесь перемешивали при комнатной температуре в течение 17 часов, добавляли воду (50 см3) и полученную смесь экстрагировали дихлорметаном (2× 100 см3). Объединенные экстракты промывали насыщенным водным раствором кислого углекислого натрия (3× 100 см3), высушивали над Na2SO4 и выпаривали при пониженном давлении. Остаток подвергали хроматографической очистке на силикагелевой колонке с использованием дихлорметана/метанола (99:1 о/о) в качестве элюента с получением промежуточного соединения, которое после выпаривания растворителей растворяли в безводном DMF (15 см3). Полученный раствор добавляли по каплям в суспензию 60%-ного гидрида натрия (461 мг, 11,5 ммоль) в безводном DMF (15 см3) при 0° С. Реакционную смесь перемешивали при комнатной температуре в течение 30 минут, затем останавливали реакцию водой (60 см3). После нейтрализации разбавленной водной соляной кислотой смесь растворяли в дихлорметане (150 см3), промывали насыщенным водным раствором кислого углекислого натрия (3× 100 см3) и высушивали над Na2SO4. Растворители выпаривали и остаток подвергали хроматографической очистке на силикагелевой колонке с использованием дихлорметана/метанола (99:1) в качестве элюента с получением нуклеозида 28 в виде пены белого цвета (2,00 г, 93%). δ н (СDСl3) 9,13 (1H, шс, NH), 7,55 (1H, д, J 1,4, 6-Н), 7,40-7,26 (Bn), 5 5,99 (1Н, д, J 2,5, 1’-Н), 5,30 (1H, д, J 2,7, 2’-H), 4,88-4,57 (6Н, м, 1’’-На, 1’’-Нb, Bn), 4,22-4,19 (1H, м, 4’-Н), 3,92 (1H, дд, J 6,2, 10,8, 5’-На), 3,82 (1H, дд, J 3,7, 10,8, 5’-Hb), 1,91 (3Н, д, J 1,3, СН3). δ c (CDCl3) 163,8 (С-4), 150,3 (С-2), 137,6 (С-6), 137,5, 137,0 (Bn), 128,7, 128,6, 128,2, 128,0, 127,8, 127,3 (Bn), 109,8 (С-5), 85,7 (С-3’), 84,1 (С-1’), 83,5 (С-4’), 79,7 (С-1’’), 73,9 (С-2’), 73,6 (С-5’), 68,6, 10 67,8 (Bn), 12,4 (СН3). FAB m/z 451 [М+Н]4, 473 [M+Na]+. Получено: С - 66,3; Н - 5,9; N - 6,1. Для С25Н26Н2О6 рассчитано: С - 66,7; Н - 5,8; N - 6,2%.
Пример 30
[1R,2R,4R,5S)-1-гидрокси-2-гидроксиметил-4-(тимин-1-ил)-3,6-диоксабицикло[3.2.0]гептан (29)
К перемешиваемому раствору нуклеозида 28 (180 мг, 0,40 ммоль) в этаноле (3 см3) добавляли 10% гидроксид палладия на углероде (90 мг). Смесь несколько раз дегазировали аргоном и помещали в атмосферу водорода. Реакционную смесь перемешивали при комнатной температуре в течение 6 часов, затем отфильтровывали через целит. Фильтрат выпаривали при пониженном давлении и остаток подвергали хроматографической очистке на силикагелевой колонке с использованием дихлорметана/метанола (96:4) в качестве элюента с получением нуклеозида 29 в виде твердого вещества белого цвета (92 мг, 86%). δ н (CD3OD) 7,79 (1Н, д, J 1,2, 6-Н), 5,91 (1Н, д, J 2,5, 1’-Н), 4,96 (1Н, д, J 2,5, 2’-Н), 4,92 (1Н, д, J 7,4, 1’’-На), 4,58 (1Н, дд, J 0,9, 7,4, 1’’-Hb), 3,98 (1Н, дд, J 7,3, 12,8, 5’-На), 3,87-3,82 (2Н, м, 4’-Н, 5’-Hb), 3,34 (2Н, с, 3’-ОН, 5’-ОН), 1,87 (3Н, д, J 1,3, СН3). δ с (СD3ОD) 166,5 (С-4), 152,1 (С-2), 140,1 (С-6), 110,1 (С-5), 91,2 (C-2’), 85,1 (С-1’), 84,0 (С-4’), 79,6 (С-3’), 78,6 (С-1’’), 61,1 (С-5’), 12,3 (СН3).
Пример 31
(1R,2R,4R,5S)-1-(2-цианоэтокси(диизопропиламино)фосфинокси)-2-(4,4’-диметокситритилоксиметил)-4-(тимин-1-ил)-3,6-диоксабицикло[3.2.0]гептан (30)
К раствору диола 29 (250 мг, 0,925 ммоль) в безводном пиридине (4 см3) добавляли 4,4’-диметокситритилхлорид (376 мг, 1,11 ммоль) и полученную смесь перемешивали при комнатной температуре в течение 18 часов. Реакцию останавливали метанолом (1,5 см3) и смесь выпаривали при пониженном давлении. Раствор остатка в дихлорметане (30 см3) промывали насыщенным водным раствором кислого углекислого натрия (3× 20 см3), высушивали над Na2SO4 и выпаривали. Остаток подвергали хроматографической очистке на силикагелевой колонке с использованием дихлорметана/метанола (98:2, о/о) в качестве элюента с получением промежуточного соединения, которое растворяли в безводном дихлорметане (7,0 см3). Добавляли сначала N,N-диизопропилэтиламин (0,64 см3, 3,70 ммоль) и затем 2-цианоэтил-N,N-диизопропилфосфорамидохлоридит (0,41 см3, 1,85 ммоль) и полученную смесь перемешивали при комнатной температуре в течение 25 часов. Реакцию останавливали метанолом (3 см3) и полученную смесь растворяли в этилацетате (70 см3), промывали насыщенным водным раствором кислого углекислого натрия (3× 50 см3) и соляным раствором (3× 50 см3), высушивали над Na2SO4 и выпаривали при пониженном давлении. Остаток подвергали хроматографической очистке на силикагелевой колонке с использованием петролейного эфира/дихлорметана/этилацетата/триэтиламина (100:45:45:10 - о/о) в качестве элюента. Полученный остаток растворяли в толуоле (2 см3) и осаждали при перемешивании из петролейного эфира при -50° С. После выпаривания растворителей остаток выпаривали вместе с безводным ацетонитрилом (4× 5 см3) с получением соединения 30 в виде пены белого цвета (436 мг, 61%). 31Р-ЯМР (CDCl3) 146,6.
Пример 32
3,5-ди-O-бензил-4-С-гидроксиметил-1,2-O-изопропилиден-α -D-рибофураноза (31)
К раствору 3-О-бензил-4-С-гидроксиметил-1,2-O-изопропилиден-α -D-рибофуранозы (R.D.Youssefyeh, J.P.H.Verheyden & J.G.Moffatt, 1979, J. Org. Chem., 44, 1301) (20,1 г, 0,064 моль) в безводном DMF (10 см3) при -5° С добавляли суспензию гидрида натрия (60% в минеральном масле: в/о) четырьмя порциями в течение 1,5 часов, общее количество 2,85 г, 0,075 моль. Бензилбромид (8,9 см3, 0,075 моль) добавляли по каплям и перемешивание продолжали при комнатной температуре в течение 3 часов, после чего добавляли смесь льда и воды (50 см3). Полученную смесь экстрагировали EtOAc (4× 100 см3) и объединенную органическую фазу высушивали над Na2SO4. После выпаривания остаток подвергали хроматографической очистке на силикагелевой колонке с элюцией 5% EtOAc в петролейном эфире (о/о) с выходом соединения 31 (18,5 г, 71%). δ с (CDCl3) 138,0, 137,4, 128,5, 128,3, 128,0, 128,0, 127,8, 127,6 (Bn), 113,5 (С(СН3)2), 104,4 (С-1), 86,5 (С-4), 78,8, 78,6 (Bn), 73,6, 72,6, 71,6 (С-2, С-3, С-5), 63,2 (С-1’), 26,7, 26,1 (СН3).
Пример 33
4-С-(ацетоксиметил)-3,5-ди-О-бензил-1,2-O-изопропилиден-α -D-рибофураноза (32)
К раствору фуранозы 31 (913 мг, 2,28 ммоль) в безводном пиридине (4,5 см3) по каплям добавляли уксусный ангидрид (1,08 см3, 1,4 ммоль) и полученную реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Реакцию останавливали добавлением смеси льда и холодной воды (50 см3) и осуществляли экстракцию дихлорметаном (3× 50 см3). Объединенную органическую фазу промывали насыщенным водным раствором кислого углекислого натрия (2× 50 см3), высушивали над Na2SO4 и концентрировали при пониженном давлении. Остаток подвергали хроматографической очистке на силикагелевой колонке, используя в качестве элюента дихлорметан, с получением соединения 32 в виде прозрачного маслянистого вещества (911 мг, 90%). δ н (CDCl3) 7,34-7,25 (10Н, м, Bn), 5,77 (1Н, д, J 3,6, 1-Н), 4,78-4,27 (8Н, м, Вn, Н-5а, Н-5b, Н-3, Н-2), 3,58 (1Н, д, J 10,3, H-1’a), 3,48 (1H, д, J 10,5, Н-1’b), 2,04 (3Н, с, СОСН3), 1,64 (3Н, с, СH3), 1,34 (3Н, с, СН3). δ с (СDСl3) 171,1 (С=O), 138,2, 137,9, 128,6, 128,1, 128,0, 128,0, 127,8 (Bn), 114,0 (С(СН3)2), 104,5 (С-1), 85,4 (С-4), 79,3, 78,6 (С-2, С-3), 73,7, 72,7, 71,2 (Вn, С-5), 64,9 (С-1’), 26,7, 26,3 (С(СН3)2), 21,0 (СОСН3). Получено: С - 67,0; Н - 6,5. Для С25Н30O7·0,25Н2O рассчитано: С - 67,2; Н - 6,9%.
Пример 34
4-C-(ацетоксиметил)-1,2-ди-O-ацетил-3,5-ди-O-бензил-D-рибофураноза (33)
Раствор фуранозы 32 (830 мг, 1,88 ммоль) в 80%-ной уксусной кислоте (10 см3) перемешивали при 90° С в течение 4 часов. Растворитель удаляли при пониженном давлении и остаток выпаривали вместе с этанолом (3× 5 см3), толуолом (3× 5 см3) и безводным пиридином (3× 5 см3) и вновь растворяли в безводном пиридине (3,7 см3). Добавляли уксусный ангидрид (2,85 см3) и полученный раствор перемешивали в течение 72 часов при комнатной температуре. Раствор выливали в смесь льда и воды (20 см3) и полученную смесь экстрагировали дихлорметаном (2× 20 см3). Объединенную органическую фазу промывали насыщенным водным раствором кислого углекислого натрия (2× 20 см3), высушивали над Nа2SO4 и концентрировали при пониженном давлении. Остаток подвергали хроматографической очистке на силикагелевой колонке, используя в качестве элюента дихлорметан, с получением соединения 33 (β :α ≈ 1:3) в виде прозрачного маслянистого вещества (789 мг, 86%). δ с (СDСl3) 171,0, 170,3, 170,0, 169,3 (С=O), 138,1, 137,6, 136,3, 128,9, 128,6, 128,2, 128,0, 128,0, 127,9, 127,7, 124,0 (Bn), 97,8, 97,8 (С-1), 87,0, 85,0, 78,9, 74,5, 74,4, 73,8, 73,6, 72,0, 71,8, 71,0, 70,9, 64,6, 64,4 (С-2, C-3, C-4, Bn, C-5, C-1’), 21,0, 20,8, 20,6 (СОСН3). Получено: С - 64,2; Н - 6,3. Для С26Н30О9 рассчитано: С - 64,2; Н - 6,2%.
Пример 35
1-(4-С-(ацетоксиметил)-2-О-ацетил-3,5-ди-O-бензил-β -D-рибофуранозил)тимин (34)
К перемешиваемому раствору аномерной смеси 33 (736 мг, 1,51 ммоль) и тимина (381 мг, 3,03 ммоль) в безводном ацетонитриле (14,5 см3) добавляли N,О-бис-(триметилсилил) ацетамид (2,61 см3, 10,6 ммоль). Реакционную смесь перемешивали с нагреванием с обратным холодильником в течение 1 часа, затем охлаждали до 0° С. Триметилсилилтрифлат (0,47 см3, 2,56 ммоль) добавляли по каплям при перемешивании и полученный раствор перемешивали при 65° С в течение 2 часов. Реакцию останавливали холодным насыщенным водным раствором кислого углекислого натрия (15 см3) и проводили экстракцию дихлорметаном (3× 10 см3). Объединенную органическую фазу промывали насыщенным водным раствором кислого углекислого натрия (2× 20 см3) и соляным раствором (2× 20 см3) и высушивали над Na2SO4. Растворитель удаляли при пониженном давлении и остаток подвергали хроматографической очистке на силикагелевой колонке с использованием дихлорметана/метанола (98:2, о/о) в качестве элюента с получением нуклеозида 34 в виде твердого вещества белого цвета (639 мг, 76%). δ н (СDСl3) 8,98 (1Н, шс, NH), 7,39-7,26 (11Н, м, Вn, 6-Н), 6,22 (1Н, д, J 5,3, 1’-Н), 5,42 (1Н, т, J 5,4, 2’-H), 4,63-4,43 (5Н, м, 3’-Н, Bn), 4,41 (1Н, д, J 12,2, 5’-На), 4,17 (1Н, д, J 12,1, 5’-Hb), 3,76 (1Н, д, J 10,2, 1’’-На), 3,51 (1Н, д, J 10,4, 1’’-Hb), 2,09 (3Н, с, СОСН3), 2,03 (3Н, с, СОСН3), 1,53 (3Н, д, J 0,9, СН3). δ с (CDCl3) 170,8, 170,4 (С=O), 163,9 (С-4), 150,6 (С-2), 137,4 (С-6) 137,4, 136,1, 128,9, 128,8, 128,4, 128,2, 127,9 (Bn), 111,7 (С-5), 87,2, 87,2, 86,1 (С-1’, С-3’, C-4’), 77,6 (С-2’), 74,8, 73,9, 71,1, 63,8 (Вn, С-1’’, С-5’), 20,9, 20,8 (СОСН3), 12,0 (СН3). FAB-MS m/z 553 [М+Н]+. Получено: С - 62,7; Н - 5,9; N - 4,7. Для C29H32N2O9 рассчитано: С - 63,0; Н - 5,8; N - 5,1%.
Пример 36
1-(3,5-ди-O-бензил-4-С-(гидроксиметил)-β -D-рибофуранозил)тимин (35)
К перемешиваемому раствору нуклеозида 34 (553 мг, 1,05 ммоль) в метаноле (5,5 см3) добавляли метоксид натрия (287 мг, 5,25 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 10 минут, затем нейтрализовывали разбавленной соляной кислотой. Растворитель частично выпаривали и проводили экстракцию дихлорметаном (3× 20 см3). Объединенную органическую фазу промывали насыщенным водным раствором кислого углекислого натрия (3× 20 см3) и высушивали над Na2SO4. Растворитель удаляли при пониженном давлении с получением соединения 35 в виде твердого вещества белого цвета (476 мг, 97%). δ H (CDCl3) 7,47 (1Н, д, J 1,0 6-Н), 7,36-7,22 (10Н, м, Bn), 6,07 (1Н, д, J 3,8, 1’-Н), 4,87 (1H, д, J 11,7, Bn), 4,55 (1H, д, J 11,7, Bn), 4,50-4,32 (4Н, м, Bn, 2’-Н, 3’-Н), 3,84-53,53 (4Н, м, 5’-На, 5’-Нb, 1’’-На, 1’’-Hb), 1,50 (3Н, д, J 1,1, СН3). δ C (CDCl3) 164,3 (С-4), 151,3 (С-2), 137,6 (С-6) 136,4, 136,3, 128,8, 128,6, 128,4, 128,3, 127,9 (Bn), 111,1 (С-5), 91,1, 91,0, 88,1 (C-1’, С-3’, С-4’), 77,4 (С-2’), 74,8, 73,8, 71,4, 63,2 (Bn, C-5’, С-1’’), 12,0 (СН3). FAB-MS m/z 491 [M+Na]+. Получено: C - 63,4; Н - 6,0; N - 5,5. Для C25H28N2O7·0,25Н2О рассчитано: С - 63,5; Н - 6,1; N - 5,9%.
Пример 37
Промежуточное соединение 35А
Раствор нуклеозида 35 (225 мг, 0,48 ммоль) в безводном пиридине (1,3 см3) перемешивали при 0° С и добавляли небольшими порциями р-толуолсульфонилхлорид (118 мг, 0,62 ммоль). Раствор перемешивали при комнатной температуре в течение 16 часов и еще раз добавляли р-толуолсульфонилхлорид (36 мг, 0,19 ммоль). После перемешивания в течение еще 4 часов и добавления смеси льда и воды (15 см3) проводили экстракцию дихлорметаном (2× 15 см3). Объединенную органическую фазу промывали насыщенным водным раствором кислого углекислого натрия (3× 15 см3) и высушивали над Na2SO4. Растворитель удаляли при пониженном давлении и остаток подвергали хроматографической очистке на силикагелевой колонке с использованием дихлорметана/метанола (99:1, о/о) в качестве элюента с получением промежуточного соединения 35А (140 мг), которое использовали для дальнейшей очистки, выполнявшейся на следующем этапе.
Пример 38
(1S,3R,4R,7S)-7-бензилокси-1-бензилоксиметил-3-(тимин-1-ил)-2,5-диоксабицикло[2.2.1]гептан (36)
Промежуточное соединение 35А (1.59 мг) растворяли в безводном DMF (0,8 см3). Раствор добавляли по каплям к перемешиваемой суспензии 60%-ного гидрида натрия в минеральном масле (в/о, 32 мг, 0,80 ммоль) в безводном DMF (0,8 см3) при 0° С. Полученную смесь перемешивали при комнатной температуре в течение 72 часов и затем концентрировали при пониженном давлении. Остаток растворяли в дихлорметане (10 см3), промывали насыщенным водным раствором кислого углекислого натрия (3× 15 см3) и высушивали над Na2SO4. Растворитель удаляли при пониженном давлении и остаток подвергали хроматографической очистке на силикагелевой колонке с использованием дихлорметана/метанола (99:1, о/о) в качестве элюента с получением бициклического нуклеозида 36 в виде твердого вещества белого цвета (65,7 мг, 57%). δ H (СDСl3) 9,24 (1Н, шс, NH), 7,49 (1Н, с, 6-Н), 7,37-7,26 (10Н, м, Bn), 5,65 (1Н, с, 1’-Н), 4,70-4,71 (5Н, м, Вn, 2’-Н), 4,02-3,79 (5Н, м, 3’-Н, 5’-Ha, 5’-Нb, 1’’-На, 1’’-Hb), 1,63 (3H, с, СН3). δ C (СDСl3) 164,3 (С-4), 150,1 (С-2), 137,7, 137,1 (Bn), 135,0 (С-6), 128,8, 128,7, 128,4, 128,0, 127,9 (Bn), 110,4 (С-5), 87,5, 87,3 (С-1’, С-3’), 76,7, 75,8, 73,9, 72,3, 72,1 (Вn, C-5’, С-2’, С-4’), 64,5 (С-1’’), 12,3 (СН3). FAB-MS m/z 451 [М+Н]+.
Пример 39
(1S,3R,4R,7S)-7-гидрокси-1-гидроксиметил-3-(тимин-1-ил)-2,5-диоксабицикло[2.2.1]гептан (37)
Раствор нуклеозида 36 (97 мг, 0,215 ммоль) в этаноле (1,5 см3) перемешивали при комнатной температуре и добавляли 20% гидроксид палладия на углероде (50 мг). Смесь дегазировали несколько раз с использованием аргона и помещали в атмосферу водорода в баллоне. После перемешивания в течение 4 часов смесь подвергали хроматографической очистке на силикагелевой колонке с использованием дихлорметана/метанола (97:3, о/о) в качестве элюента с получением бициклического нуклеозида 37 в виде твердого вещества белого цвета (57 мг, 98%). δ H ((СD3)2SO) 11,33 (1Н, шс, NH), 7,62 (1Н, д, J 1,1 Гц, 6-Н), 5,65 (1Н, д, J 4,4 Гц, 3’-ОН), 5,41 (1Н, с, 1’-Н), 5,19 (1Н, т, J 5,6 Гц, 5’-ОН), 4,11 (1Н, с, 2’-Н), 3,91 (1Н, д, J 4,2 Гц, 3’-Н)-, 3,82 (1Н, д, J 7,7 Гц, 1’’-Н,,а), 3,73 (1Н, с, Н’-5а), 3,76 (1Н, g, 5’-Нb), 3,63 (1Н, д, J 7,7 Гц, 1’-Hb), 1,78 (3H, д, J 0,7 Гц, СН3). δ C (CDCl3) 166,7 (С-4), 152,1 (С-2), 137,0 (С-6), 110,9 (С-5), 90,5, 88,4 (С-1’, С-4’), 80,9, 72,5, 70,4 (С-2’, С-3’, С-5’), 57,7 (С-1’’), 12,6 (СН3). EI-MS m/z 270 [М]+.
Пример 40
(1R,3R,4R,7S)-1-(4,4’-диметокситритилоксиметил)-7-гидрокси-3-тимин-1-ил)-2,5-диоксабицикло[2.2.1]гептан (38)
К раствору нуклеозида 37 (1,2 г, 4,44 ммоль) в безводном пиридине (5 см3) добавляли 4,4’-диметокситритилхлорид (2,37 г, 7,0 ммоль) при 0° С. Раствор перемешивали при комнатной температуре в течение 2 часов, после чего реакцию останавливали смесью льда и воды (10 см3) и проводили экстракцию дихлорметаном. (3× 15 см3). Объединенную органическую фазу промывали насыщенным водным раствором кислого углекислого натрия (3× 10 см3) и соляным раствором (2× 10 см3) и высушивали над Nа2SO4. Растворитель удаляли при пониженном давлении и остаток подвергали хроматографической очистке на силикагелевой колонке с использованием дихлорметана/метанола (98:2, о/о) в качестве элюента с получением нуклеозида 38 в виде белого твердого вещества (2,35 г, 93%). δ H (CDCl3) 9,89 (1H, шс, NH), 7,64 (1H, с, 6-Н), 7,47-7,13 (9Н, м, DMT), 6,96-6,80 (4Н, м, DMT), 5,56 (1H, с, 1’-Н), 4,53 (1H, шс, 2’-H), 4,31 (1H, м, 3’-H), 4,04-3,75 (9Н, м, 1’’-На, 1’’-Нb, 3’-ОН, ОСН3), 3,50 (2Н, шс, 5’-На, 5’-Hb), 1,65 (3H, с, СН3). δ C (CDCl3) 164,47 (С-4), 158,66 (DMT), 150,13 (С-2), 144,56, 135,46, 135,35, 134,78, 130,10, 129,14, 128,03, 127,79, 127,05 (С-6, DMT), 113,32, 113,14 (DMT), 110,36 (С-5), 89,17, 88,16, 87,05 (С-1’, С-4’, DMT), 79,36, 71,81, 70,25, 58,38 (С-2’, С-3’, С-5’, С-1’’), 55,22 (ОСН3), 12,57 (СН3). FAB-MS m/z 595 [M+Na]+, 573 [М+Н]+.
Пример 41
(1R,3R,4R,7S)-7-(2-цианоэтокси(диизопропиламино)фосфинокси)-1-(4,4’-диметокситритилоксиметил)-3-(тимин-1-ил)-2,5-диоксабицикло[2.2.1]гептан (39)
К раствору нуклеозида 38 (2,21 г, 3,86 ммоль) в безводном дихлорметане (6 см3) при комнатной температуре добавляли N,N-диизопропилэтиламин (4 см3) и 2-цианоэтил-N,N-диизопропилфосфорамидохлоридит (1 см3, 4,48 ммоль) и перемешивание продолжали в течение 1 часа. Добавляли МеОН (2 см3) и полученную смесь разбавляли этилацетатом (10 см3) и последовательно промывали насыщенным водным раствором кислого углекислого натрия (3× 5 см3) и соляным раствором (2× 5 см3) и высушивали над Na2SO4. Растворитель выпаривали при пониженном давлении и остаток подвергали хроматографической очистке на щелочных глиноземных колонках с использованием дихлорметана/метанола (99:1, о/о) в качестве элюента с получением соединения 39 в виде пены белого цвета. Этот остаток растворяли в дихлорметане (2 см3) и полученный продукт осаждали из петролейного эфира (100 см3, охлажденный до -30° С) при мощном перемешивании. Преципитат отбирали фильтрованием и высушивали с получением нуклеозида 39 в виде твердого вещества белого цвета (2,1 г, 70%). δ P (CDCl3) 149,06, 148,74. FAB-MS m/z 795 [М+Na]+, 773 [М+Н]+.
Пример 42
1-(2-O-ацетил-4-С-ацетоксиметил-3,5-ди-O-бензил-β -D-рибофуранозил)урацил (40)
К перемешиваемому раствору аномерной смеси 33 (3,0 г, 6,17 ммоль) и урацила (1,04 г, 9,26 ммоль) в безводном ацетонитриле (65 см3) добавляли N,O-бис(триметилсилил)ацетамид (9,16 см3, 37,0 ммоль). Реакционную смесь перемешивали в течение 1 часа при комнатной температуре и охлаждали до 0° С. Триметилсилилтрифлат (1,8 см3, 10,0 ммоль) добавляли по каплям и полученный раствор перемешивали при 60° С в течение 2 часов. Реакцию останавливали добавлением насыщенного водного раствора кислого углекислого натрия (10 см3) при 0° С и проводили экстракцию дихлорметаном (3× 20 см3). Объединенную органическую фазу промывали соляным раствором (2× 20 см3) и высушивали над Na2SO4. Растворитель удаляли при пониженном давлении и остаток подвергали хроматографической очистке на силикагелевой колонке с использованием дихлорметана/метанола (99:1, о/о) в качестве элюента с получением нуклеозида 40 в виде белого твердого вещества (2,5 г,7S%). δ H (СDСl3) 9,57 (1H, шс, NH), 7,63 (1Н, д, J 8,2, 6-Н), 7,40-7,24 (10Н, м, Bn), 6,18 (1Н, д, J 4,5, 1’-Н), 5,39-5,32 (2Н, м, 2’-Н, 5-Н), 4,61 (1Н, д, J 11,6, Bn), 4,49-4,40 (5Н, м, 3’-H, Вn, 1’’-На), 4,37 (1Н, д, J 12,3, 1’’-Hb), 3,76 (1H, д, J 10,1, 5’-На), 3,49 (1H, д, J 10,1, 5’-Hb), 2,09 (3H, с, СОСН3), 2,04 (3H, с, СОСН3). δ C (CDCl3) 170,47, 169,94 (С=0), 163,32 (С-4), 150,30 (С-2), 140,24 (С-6), 137,15, 136,95, 128,65, 128,52, 128,32, 128,19, 128,02, 127,77 (Bn), 102,57 (С-5), 87,41, 86,14 (C-1’, С-4’), 77,09, 74,84, 74,51, 73,75, 70,60, 63,73 (С-2’, С-3’, С-5’, С-1’’, Bn), 20,79, 20,68 (СОСН3). FAB-MS m/z 539 [M]+.
Пример 43
1-(3,5-ди-O-бензил-4-С-гидроксиметил-β -D-рибофуранозил)урацил (41)
К перемешиваемому раствору нуклеозида 40 (2,0 г, 3,7 ммоль) в метаноле (25 см3) добавляли метоксид натрия (0,864 г, 95%, 16,0 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 10 минут и нейтрализовывали 20%-ной водной соляной кислотой. Растворитель частично выпаривали и остаток экстрагировали этилацетатом (3× 50 см3). Объединенную органическую фазу промывали насыщенным водным раствором кислого углекислого натрия (3х20 см3) и высушивали над Na0SO4. Растворитель удаляли при пониженном давлении и остаток подвергали хроматографической очистке на силикагелевой колонке с использованием дихлорметана/метанола (98,5:1,5, о/о) в качестве элюента с получением нуклеозида 41 в виде белого твердого вещества (1,58 г, 95%). δ H (СDСl3) 9,95 (1Н, шс, NH), 7,69 (д, J 8,1, 6-Н), 7,35-7,17 (10Н, м, Bn), 6,02 (1Н, д, J 2,3, 1’-Н), 5,26 (1Н, д, J 8,1, 5-Н), 4,80 (1Н, д, J 11,7, Bn), 4,47 (1Н, д, J 11,7, Bn), 4,45-4,24 (4Н, м, Вn, 2’-H, 3’-Н), 3,81 (1Н, д, J 11,9, 1’’-На), 3,69 (2Н, шс, 2’-ОН, 1’’-ОН), 3,67 (2Н, м, 5’-На, 1’’-Hb), 3,48 (1Н, д, J 10,3, 5’-Hb). δ C (СDСl3) 163,78 (С-4), 150,94 (С-2), 140,61 (С-6), 137,33, 137,22, 128,59, 128,18, 128,01 (Bn), 102,16 (С-5), 91,46, 88,36 (С-1’, С-4’), 76,73, 74,66, 73,71, 73,29, 70,81, 62,81 (C-2’, С-3’, С-5’, С-1’’, Bn). FAB-MS m/z 455 [M+H]+.
Пример 44
Промежуточное соединение 42
Раствор нуклеозида 41 (1,38 г, 3,0 ммоль) в безводном пиридине (2 см3) и безводном дихлорметане (6 см3) перемешивали при -10° С и небольшими порциями добавляли р-толуолсульфонилхлорид (0,648 г, 3,4 ммоль) в течение 1 часа. Полученный раствор перемешивали при -10° С в течение 3 часов. Реакцию останавливали добавлением смеси льда и воды (10 см3) и смесь экстрагировали дихлорметаном (3× 50 см3). Объединенную органическую фазу промывали насыщенным водным раствором кислого углекислого натрия (3× 20 см3) и высушивали над Na2SO4. Растворитель удаляли при пониженном давлении и остаток подвергали хроматографической очистке на силикагелевой колонке с использованием дихлорметана/метанола (99:1, о/о) в качестве элюента с получением промежуточного соединения 42 (0,9 г), которое на следующем этапе использовали без дополнительной очистки.
Пример 45
(1S,3R,4R,7S)-7-бензилокси-1-бензилоксиметил-3-(урацил-1-ил)-2,5-диоксабицикло[2.2.1]гептан (43)
Соединение 42 (0,7 г) растворяли в безводном DMF (3 см3), 60%-ную суспензию гидрида натрия (в/о, 0,096 г, 24 ммоль) добавляли четырьмя порциями в течение 10 минут при 0° С и реакционную смесь перемешивали при комнатной температуре в течение 12 часов. Реакцию останавливали метанолом (10 см3) и растворители удаляли при пониженном давлении. Остаток растворяли в дихлорметане (20 см3), промывали насыщенным водным раствором кислого углекислого натрия (3× 6 см3) и высушивали над Na2SO4. Растворитель удаляли при пониженном давлении и остаток подвергали хроматографической очистке на силикагелевой колонке с использованием дихлорметана/этанола (99:1, о/о) в качестве элюента с получением нуклеозида 43 (0,30 г, 60%). δ H (CDCl3) 9,21 (1H, шс, NH), 7,70 (1H, д, J 8,2, 6-H), 7,37-7,24 (10Н, м, Bn), 5,65 (1H, с, 1’-Н), 5,52 (1Н, д, J 8,2, 5-Н), 4,68-4,45 (5Н, м, 2’-Н, Bn), 4,02-3,55 (5Н, м, 3’-Н, 5’-На, 1’’-На, 5’-Нb, 1’’-Hb). δ C (СDСl3) 163,33 (С-4), 149,73 (С-2), 139,18 (С-6), 137,46, 136,81, 128,58, 128,54, 128,21, 128,10, 127,79, 127,53 (Bn), 101,66 (С-5), 87,49, 87,33 (С-1’, С-4’), 76,53, 75,71, 73,77, 72,33, 72,00, 64,35 (С-2’, С-3’, С-5’, C-1’’, Bn). FAB-MS m/z 459 [M+Ma]+.
Пример 46
(1S,3R,4R,7S)-7-гидрокси-1-гидроксиметил-3-(урацил-1-ил)-2,5-диоксабицикло[2.2.1]гептан (44)
К раствору соединения 43 (0,35 г, 0,8 ммоль) в абсолютном этаноле (2 см3) добавляли 20% гидроксид палладия на углероде (0,37 г) и полученную смесь несколько раз дегазировали водородом и перемешивали в атмосфере водорода в течение 4 часов. Растворитель удаляли при пониженном давлении и остаток подвергали хроматографической очистке на силикагелевой колонке с использованием дихлорметана/метанола (9:1, о/о) в качестве элюента с получением нуклеозида 44 в виде твердого вещества белого цвета (0,16 г, 78%). δ H (CD3OD) 7,88 (1Н, д, J 8,1, 6-Н), 5,69 (1Н, д, J 8,1, 5-Н), 5,55 (1Н, с, 1’-Н), 4,28 (1Н, с, 2’-Н), 4,04 (1Н, с, 3’-Н), 3,96 (1Н, д, J 7,9, 1’’-На), 3,91 (2Н, с, 5’-Н), 3,76 (1Н, д, J 7,9, 1’’-Hb). δ C (СD3OD) 172,95 (С-4), 151,82 (С-2), 141,17 (С-6), 101,97 (С-5), 90,52, 88,50 (С-1’, С-4’), 80,88, 72,51, 70,50, 57,77 (С-2’, С-3’, С-5’, С-1’’). FAB-MS m/z 257 [М+Н]+.
Пример 47
[1R,3R,4R,7S)-1-(4,4’-диметокситритилоксиметил)-7-гидрокси-3-(урацил-1-ил)-2,5-диоксабицикло[2.2.1]гептан (45)
К раствору соединения 44 (0,08 г, 0,31 ммоль) в безводном пиридине (0,5 см3) добавляли 4,4’-диметокситритилхлорид (0,203 г, 0,6 ммоль) при 0° С и полученную смесь перемешивали при комнатной температуре в течение 2 часов. Реакцию останавливали смесью воды и льда (10 см3) и проводили экстракцию дихлорметаном (3× 4 см3). Объединенную органическую фазу промывали насыщенным водным раствором кислого углекислого натрия (3× 3 см3) и соляным раствором (2× 3 см3) и высушивали над Na2SO4. Растворитель удаляли при пониженном давлении и остаток подвергали хроматографической очистке на силикагелевой колонке с использованием дихлорметана/метанола (98:2, о/о) в качестве элюента с получением нуклеозида 45 в виде твердого вещества белого цвета (0,12 г, 69%). δ H (СDСl3) 9,25 (1Н, шс, NH), 7,93 (1Н, д, J 7,2, 6-Н), 7,50-7,15 (9Н, м, DMT), 6,88-6,78 (4Н, м, DMT), 5,63 (1Н, с, 1’-Н), 5,59 (1Н, д, J 8,0, 5-Н), 4,48 (1Н, с, 2’-Н), 4,26 (1Н, с, 3’-Н), 3,88 (1Н, д, J 8,1, 1’’-Нa), 3,85-3,55 (7Н, м, 1’’-Hb, ОСН3), 3,58-3,40 (2Н, м, 5’-На, 5’-Hb). δ C (CDCl3) 164,10 (С-4), 158,60 (DMT), 150,45 (С-2), 147,53 (DMT), 144,51 (С-6), 139,72, 135,49, 135,37, 130,20, 129,28, 128,09, 127,85, 127,07 (DMT), 113,39, 113,17 (DMT), 101,79 (С-5), 88,20, 87,10, 86,87 (С-1’, С-4’, DMT), 79,25, 71,79, 69,70, 58,13 (С-2’, С-3’, С-5’, С-1’’), 55,33 (ОСН3). FAB-MS m/z 559 [М+Н]+.
Пример 48
(1R,3R,4R,7S)-4-(2-цианоэтокси(диизопропиламино)фосфинокси)-1-(4,4’-диметокситритилоксиметил)-3-(урацил-1-ил)-2,5-диоксабицикло[2.2.1]гептан (46)
К раствору соединения 45 (0,07 г, 0,125 ммоль) в безводном дихлорметане (2 см3) при комнатной температуре добавляли N,N-диизопропилэтиламин (0,1 см3) и 2-цианоэтил-N,N-диизопропилфосфорамидохлоридит (0,07 см3, 0,32 ммоль). После перемешивания в течение 1 часа реакцию останавливали добавлением МеОН (2 см3) и полученную смесь разбавляли этилацетатом (5 см3) и промывали последовательно насыщенным водным раствором кислого углекислого натрия (3× 2 см3) и соляным раствором (3× 2 см3) и высушивали над Na2SO4. Растворитель выпаривали при пониженном давлении и остаток подвергали хроматографической очистке на силикагелевой колонке с использованием дихлорметана/метанола (99:1, о/о) в качестве элюента с получением пены белого цвета. Эту пену растворяли в дихлорметане (2 см3) и полученный продукт осаждали из петролейного эфира (10 см3, охлажден до -30° С) при мощном перемешивании. Преципитат отбирали фильтрованием и высушивали с получением соединения 46 в виде твердого вещества белого цвета (0,055 г, 58%). δ P (CDCl3) 149,18, 149,02.
Пример 49
9-(2-O-ацетил-4-С-ацетоксиметил-3,5-ди-O-бензил-β -D-рибофуранозил)-2-N-изобутирилгуанин (47)
К перемешиваемой суспензии аномерной смеси 33 (1,28 г, 5,6 ммоль) и 2-N-изобутирилгуанина (1,8 г, 3,7 ммоль) в безводном дихлорэтане (600 см3) добавляли N,N-бис(триметилсилил)ацетамид (4 см3, 16,2 ммоль). Реакционную смесь перемешивали с нагреванием с обратным холодильником в течение 1 часа. По каплям добавляли триметилсилилтрифлат (1,5 мл, 8,28 ммоль) и полученный раствор перемешивая нагревали с обратным холодильником в течение 2 часов. Реакционную смесь оставляли остывать до комнатной температуры в течение 1,5 часов. После разбавления дихлорметаном до 250 см3 смесь промывали насыщенным водным раствором кислого углекислого натрия (200 см3) и водой (250 см3). Растворитель удаляли при пониженном давлении и остаток подвергали хроматографической очистке на силикагелевой колонке с использованием в качестве элюентов 1,25% (200 см3) и 1,5% (750 см3) метанола в дихлорметане (о/о) с получением 2,10 г (87%) твердого вещества белого цвета, которое, исходя из данных 1H-ЯМР, состоит из трех изомеров (их соотношение 12,5:2,5:1). Основной продукт, образовавшийся в описанных условиях, очевидно, является соединением 47 (Р.Garner, S.Ramakanth, 1988, J. Org. Chem., 53, 1294; H.Vorbruggen, K.Krolikiewicz, B.Bennua, 1981, Chem. Ber., 114, 1234). Отдельные изомеры не выделяли, а полученную смесь использовали на следующих этапах. Для соединения 47: δ H (СDСl3) 12,25 (шс, NHCO), 9,25 (шс, NH), 7,91 (с, 8-Н), 7,39-7,26 (м, Bn), 6,07 (д, J 4,6, 1’-Н), 5,80 (дд, J 5,8, 4,7, 2’-Н), 4,72 (д, J 5,9, 3’-Н), 4,59-4,43 (м, Вn, 1’’-На), 4,16 (д, J 12,1, 1’’-Hb), 3,70 (д, J 10,1, 5’-Ha, 3,58 (д, J 10,1, 5’-Hb), 2,65 (м, СНСО), 2,05 (с, СОСН3), 2,01 (с, СОСН3), 1,22 (д, J 6,7, СН3СН), 1,20 (д, J 7,0, СН3СН). δ C (СDСl3) 178,3 (СОСН), 170,6, 179,8 (СОСН3), 155,8, 148,2, 147,6 (гуанин), 137,6, 137,2 (гуанин, Bn), 128,5, 128,4, 128,2, 128,1, 128,0, 127,8, 127,7 (Bn), 121,2 (гуанин), 86,2, 86,0 (С-1’, С-4’), 77,8 (С-3’), 74,9, 74,5, 73,7, 70,4 (Bn, C-2’, C-5’), 63,5 (С-1’), 36,3 (СОСН), 20,8, 20,6 (СОСН3), 19,0 (СН3СН). Для смеси изомеров: FAB-MS m/z 648 [М+Н]+, 670 [М+Na]+. Вычислено: С - 60,8; Н - 6,0; N - 10,4. Для C33H36N5O9 рассчитано: С - 61,3; Н - 5,6; N - 10,8%.
Пример 50
9-(3,5-ди-O-бензил-4-С-гидроксиметил-β -D-рибофуранозил)-2-N-изобутирилгуанин (48)
Раствор смеси, полученной в примере 49, содержащий соединение 47 (2,10 г, 3,25 ммоль), в THF/пиридине/метаноле (2:3:4, о/о) (40 см3) охлаждали до -10° С и к перемешиваемому раствору добавляли метоксид натрия (320 мг, 5,93 ммоль). Полученную реакционную смесь перемешивали при 10° С в течение 30 минут и нейтрализовывали добавлением 2 см3 уксусной кислоты. Растворитель выпаривали при пониженном давлении и остаток дважды экстрагировали в системе дихлорметан/вода (2× 100 см3). Органические фракции объединяли и выпаривали при пониженном давлении. После выпаривания вместе с толуолом остаток подвергали хроматографической очистке на силикагелевой колонке в градиенте (2-7%) метанола в дихлорметане (о/о) с получением твердого вещества белого цвета (1,62 г). В соответствии с данными 1H-ЯМР, оно содержит три изомера (в соотношении 13,5:1,5:1). Для основного продукта 48: δ H (СD3ОD) 8,07 (с, 8-Н) 7,36-7,20 (м, Bn), 6,05 (д, J 3,9, 1’-Н), 4,81 (д, J 11,5, Bn), 4,75 (м, 2’-H), 4,56 (д, J 11,5, Bn), 4,51-4,43 (м, Bn, 3’-H), 3,83 (д, J 11,7, 1’’-На), 3,65 (д, J 11,7, 1’’-Hb), 3,64 (д, J 10,6, 5’-На), 3,57 (д, J 10,3, 5’-Hb), 2,69 (м, СНСО), 1,20 (6Н, д, J 6,8, СН3СН). δ c (СD3ОD) 181,6 (СОСН), 157,3, 150,2, 149,5 (гуанин), 139,4, 139,3, 139,0 (гуанин, Bn), 129,5, 129,4, 129,3, 129,2, 129,1, 129,0, 128,9, 128,8 (Bn), 121,2 (гуанин), 90,7, 89,6 (С-1’, C-4’), 79,2 (С-3’), 75,8, 74,5, 74,3, 72,2 (Bn, C-2’, С-5’), 63,1 (C-1’’), 36,9 (СОСН), 19,4 (СН3СН), 19,3 (CH3СН). Для смеси изомеров: FAB-MS m/z 564 [М+Н]+.
Пример 51
Промежуточное соединение 49
Раствор смеси, полученной в примере 50, содержащий соединение 48 (1,6 г), в безводном пиридине (6 см3), перемешивали при -20° С и добавляли р-толуолсульфонилхлорид (0,81 г, 4,27 ммоль). Раствор перемешивали при -20° С в течение 1 часа и при -25° С в течение 2 часов. Затем смесь разбавляли дихлорметаном до 100 см3 и немедленно промывали водой (2× 100 см3). Органическую фазу отделяли и выпаривали при пониженном давлении. Остаток подвергали хроматографической очистке с использованием дихлорметана/метанола в качестве элюента (1-2%, о/о) с получением промежуточного соединения 49 (980 мг). После элюции соединения 49 из колонки исходную смесь, содержащую соединение 48 (510 мг), элюировали 8% метанолом в дихлорметане (о/о). Полученный материал концентрировали, высушивали при пониженном давлении и обрабатывали таким же образом, как было описано выше, с получением дополнительно 252 мг того же промежуточного соединения. Промежуточное соединение (1,23 г) очищали методом ВЭЖХ в силикагеле (картридж PrepPak, загруженный порасилом, 15-20 мкм, 125А, скорость потока 60 см3/мин, элюент 0-4% метанол в дихлорметане, о/о, продолжительность элюции 120 минут). Фракции, содержащие интермедиат 49, объединяли и концентрировали с получением твердого вещества белого цвета (1,04 г). В соответствии с данными 1Н-ЯМР оно содержит два основных продукта, а именно 1’’-О- и 2’’-O-моносилилированные производные в соотношении примерно 2:1. FAB-MS m/z 718 [М+Н]+. Вычислено: С - 60,4; Р - 5,8; N - 9,3. Для С36Н39N5О9S рассчитано: С - 60,2; Н - 5,5; N - 9,8%.
Пример 52
(1S,3R,4R,7S)-7-бензилокси-1-бензилоксиметил-3-(2-N-изобутирилгуанин-9-ил)-2,5-диоксабицикло[2.2.1]гептан (50)
К раствору промежуточного соединения 49 (940 мг) в безводном THF (20 см3) добавляли 60%-ную суспензию гидрида натрия (в/о, 130 мг, 3,25 ммоль) и полученную смесь перемешивали при комнатной температуре в течение 1 часа. Добавляли уксусную кислоту (0,25 мл) и смесь концентрировали при пониженном давлении. Остаток растворяли в дихлорметане (100 см3) и промывали водой (2× 100 см3). Отделяли органическую фазу и выпаривали ее при пониженном давлении. Остаток очищали в силикагеле методом колончатой хроматографии с использованием метанола/дихлорметана (1-1,5%, о/о) в качестве элюента с получением нуклеозида 50 в виде твердого вещества белого цвета (451 мг, 57%). δ H (СDСl3) 12,25 (1Н, шс, NHCO), 10,12 (1Н, шс, NH), 7,84 (1Н, с, 8-Н), 7,31-7,15 (10Н, м, Bn), 5,72 (1Н, с, 1’-Н), 4,60-4,46 (5Н, м, Вn, 2’-Н), 4,14 (1Н, с, 3’-Н), 4,02 (1Н, д, J 7,9, 1’-На), 3,85 (1H, д, J 7,9, 1’-Hb), 3,78 (2Н, с, 5’-Н), 2,81 (1Н, м, СНСО), 1,24 (3H, д, J 6,8, СН3СН), 1,22 (3H, д, J 6,4, СН3СН). δ C (CDCl3) 179,5 (СОСН), 155,6, 148,1, 147,3 (гуанин), 137,3, 136,9, 136,0 (гуанин, Bn), 128,4, 128,3, 127,9, 127,8, 127,5, 127,4 (Bn), 121,2 (гуанин), 87,1, 86,2 (С-1’, С-4’), 77,0 (С-3’), 73,6, 72,5, 72,1 (Вn, С-2’, С-5’), 64,9 (C-1’’), 36,1 (СОСН), 19,0 (СН3СН), 18,9 (СН3СН). FAB-MS m/z 546 [М+Н]+. Получено: С - 63,3; Н - 5,9; N - 12,5. Для С29Н30N5О6 рассчитано: С - 64,0; Н - 5,6; N - 12,9%.
Альтернативный способ получения соединения 50. G1AQ.
К суспензии соединения 78 (1,5 г, 2,51 ммоль) и N-2-изобутирилгуанина (0,93 г, 4,06 ммоль) в сухом DCM (50 мл) добавляли BSA (N,O-бистриметилсилилацетамид: 3,33 мл, 13,5 ммоль) и полученную смесь нагревали с обратным холодильником в течение 2 часов. К этой смеси добавляли триметилсилилтрифлат (1,25 мл, 6,9 ммоль) и продолжали нагревание полученной смеси с обратным холодильником еще в течение 2 часов. Смесь оставляли остывать до комнатной температуры, разбавляли DCM до 200 мл и промывали насыщенным водным раствором кислого углекислого натрия NаНСО3 и водой. После хроматографической очистке на силикагелевой колонке (1-2,5% метанола в дихлорметане) получили 1,05 г (55%) желаемого изомера G1AQ, а еще 380 мг изомеров, характеризующихся более высокой подвижностью, конвертировали в G1AQ путем повторения описанной выше процедуры. К раствору G1AQ (1,05 г в 12 мл метанола) добавляли гидроокись аммония (12 мл 25%-ного водного раствора) и полученную смесь перемешивали при комнатной температуре в течение 1 часа. После концентрирования продукт очищали хроматографически на силикагелевой колонке (1-3% метанола в дихлорметане) с получением 700 мг соединения G3 в виде твердого вещества белого цвета. 70 мг G3 в безводном THF (15 мл) обрабатывали гидридом натрия (225 мг 60% суспензии в минеральном масле). Через полчаса реакцию останавливали добавлением 1,25 мл уксусной кислоты и полученную смесь концентрировали при пониженном давлении. Остаток растворяли в дихлорметане, промывали в NaHCO3 и воде и подвергали хроматографической очистке в силикагеле в градиенте 0,5-3% метанола в дихлорметане. Выход соединения 50 составил 400 мг (75%).
Пример 53
(1S,3R,4R,7S)-7-гидрокси-1-гидроксиметил-3-(2-N-изобутирилгуанин-9-ил)-2,5-диоксабицикло[2.2.1]гептан (51)
Смесь нуклеозида 50 (717 мг, 1,31 моль) и 10% палладия на углероде (500 мг) суспендировали в метаноле (8 см3) при комнатной температуре. Полученную смесь несколько раз дегазировали при пониженном давлении и помещали в атмосферу водорода. После перемешивания в течение 24 часов смесь подвергали хроматографической очистке с использованием метанола/дихлорметана (8-20%, о/о) в качестве элюента с получением нуклеозида 51 в виде стекловидного вещества (440 мг, 92%). δ H (CD3OD) 8,12 (1Н, шс, 8-Н), 5,86 (1Н, с, 1’-Н), 4,50 (1Н, с, 2’-Н), 4,30 (1Н, с, 3’-Н), 4,05 (1Н, д, J 8,0, 1’’-На), 3,95 (2Н, с, 5’-Н), 3,87 (1Н, д, J 7,9, 1’’-Hb), 2,74 (1Н, м, СНСО), 1,23 (6Н, д, J 6,9, СН3СН). δ C (CD3OD, сигналы углеводородной части спектра) 90,2, 87,6 (C-1’, С-4’), 81,1 (С-3’), 72,9, 71,3 (С-2’, С-5’), 58,2 (С-1’’), 37,1 (СОСН), 19,5 (СН3СН). FAB-MS m/z 366 [M+H]+.
Пример 54
(1R,3R,4R,7S)-1-(4,4’-диметокситритилоксиметил)-7-гидрокси-3-(2-N-изобутирилгуанин-9-ил)-2,5-диоксабицикло[2.2.1]гептан (52)
Смесь соединения 51 (440 мг, 1,21 ммоль) и 4,4’-диметокситритилхлорида (573 мг, 1,69 ммоль) растворяли в безводном пиридине (7 см3) и перемешивали при комнатной температуре в течение 4 часов. Смесь выпаривали при пониженном давлении с получением масла. Экстракцию проводили в системе дихлорметан/вода (1:1, о/о, 40 см3). Отделяли органическую фазу и концентрировали ее с получением раствора в минимальном количестве дихлорметана, содержащего 0,5% пиридина (о/о), который загружали на силикагелевую хроматографическую колонку, уравновешенную тем же растворителем. Продукт элюировали в градиенте метанола (0,6-2%, о/о) в дихлорметане, содержащем 0,5% пиридина (о/о) с получением соединения 52 в виде твердого вещества белого цвета (695 мг, 86%). δ H (СDСl3) 12,17 (1Н, шс, NHCO), 10,09 (1Н, шс, NH), 7,87 (1Н, с, 8-Н), 7,42-6,72 (13Н, м, DMT), 5,69 (1Н, с, 1’-Н), 4,59 (1Н, с, 2’-Н), 4,50 (1Н, с, 3’-Н), 3,98 (1Н, д, J 8,1, 1’’-Нa), 3,69-3,39 (9Н, м, DMT, 5’-H, 1’’-Hb), 2,72 (1Н, м, СНСО), 1,17 (6Н, д, J 6,8, СН3СН). δ C (СDСl3) 179,8 (СОСН), 158,8, 144,5, 135,6, 135,5, 130,1, 128,1, 127,7, 126,9, 113,2 (DMT), 155,8, 147,9, 147,5, 137,0, 120,8 (гуанин), 87,6, 86,4, 86,1 (С-1’, C-4’, DMT), 79,7 (С-3’), 72,6, 71,4 (С-2’, С-5’), 59,8 (C-1’’), 55,2 (DMT), 36,1 (СОСН), 19,1, 18,8 (СН3СН). FAB-MS m/z 668 [М+Н]+.
Пример 55
(1R,3R,4R,7S)-7-(2-цианоэтокси(диизопропиламино)фосфинокси)-1-(4,4’-диметокситритилоксиметил)-3-(2-N-изопропионилгуанин-9-ил)-2,5-диоксабицикло[2.2.1]гептан (53)
Соединение 52 (670 мг, 1,0 моль) растворяли при комнатной температуре в безводном дихлорметане (5 см3), содержащем N,N-диизопропилэтиламин (0,38 см3, 4 ммоль). По каплям при перемешивании добавляли 2-цианоэтил-N,N-диизопропилфосфорамидохлоридит (0,36 см3, 2,0 ммоль). Через 5 часов добавляли метанол (2 см3) и полученную смесь разбавляли дихлорметаном до 100 см3 и промывали насыщенным водным раствором кислого углекислого натрия (50 см3). Отделяли органическую фазу и растворитель выпаривали при пониженном давлении. Остаток растворяли в минимальном количестве смеси дихлорметана и петролейного эфира (1:1, о/о), содержащего 0,5% пиридина (о/о), и загружали силикагелевую колонку, уравновешенную тем же растворителем. Колонку промывали дихлорметаном/петролейным эфиром/пиридином (75:25:0,5, о/о, 250 см3) и продукт элюировали с использованием градиента метанола (0-1%, о/о) в дихлорметане, содержащем 0,5% пиридина (о/о). Фракции, содержащие основной продукт, выпаривали и затем выпаривали вместе с толуолом. Остаток растворяли в безводном дихлорметане (5 см3) и осаждали петролейным эфиром (10 см3) с получением соединения 53 в виде твердого вещества белого цвета (558 мг, 64%) после фильтрации и высушивания. δ P (CDCl3) 148,17, 146,07. FAB-MS m/z 868 [M+1]+.
Пример 56
1-(2-O-ацетил-4-С-ацетоксиметил-3,5-ди-O-бензил-β -D-рибофуранозил)-4-N-бензоилцитозин (54)
К перемешиваемому раствору аномерной смеси 33 (4,0 г, 8,22 ммоль) и 4-N-бензоилцитозина (2,79 г, 13,0 ммоль) добавляли N,O-бис(триметилсилил)ацетамид (8,16 см3, 33,0 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа и охлаждали до 0° С. По каплям добавляли триметилсилилтрифлат (3,0 см3, 16,2 ммоль) и полученную смесь перемешивали при 60° С в течение 2 часов. Последовательно добавляли насыщенный водный раствор кислого углекислого натрия (3× 20 см3) и соляной раствор (2× 20 см3) и отделенную органическую фазу высушивали над Na2SO4. Растворитель удаляли при пониженном давлении и остаток подвергали хроматографической очистке на силикагелевой колонке с использованием дихлорметана/метанола (99:1, о/о) в качестве элюента с получением соединения 54 в виде белого твердого вещества (3,9 г, 74%). δ H (СDСl3) 8,28 (1Н, д, J 7,5, 6-Н), 7,94-7,90 (2Н, м, Bz), 7,65-7,25 (13Н, м, Bn, Bz), 7,16 (1Н, д, J 7,1, 5-Н), 6,22 (1H, д, J 2,8, 1’-Н), 5,51 (1H, дд, J 2,8, 5,8, 2’-Н), 4,62 (1H, д, J 11,6, Bn), 4,51 (1H, д, J 12,3, 1’’-Ha), 4,49-4,34 (4Н, м, 3’ -Н, Bn), 4,21 (1H, д, J 12,3, 1’’-Hb), 3,85 (1H, д, J 10,3, 5’-На), 3,47 (1H, д, J 10,3, 5’-Hb), 2,13 (3H, с, СОСН3), 2,06 (3H, с, СОСН3). δ C (CDCl3) 170,52, 169,61 (С=O), 166,83, 162,27 (С-4, С=O), 154,26 (С-2), 145,26 (С-6), 137,25, 136,93, 133,18, 129,0, 128,75, 128,51, 128,45, 128,18, 128,10, 127,89, 127,71 (Bn, Bz), 96,58 (С-5), 89,42, 86,52 (С-1’, С-4’), 76,21, 75,10, 74,17, 73,70, 69,70, 63,97 (С-2’, С-3’, Bn, С-5’, C-1’’), 20,82 (СОСН3). FAB-MS m/z 664 [M+Na]+, 642 [М+Н]+. Получено: С - 65,0; Н - 5,7; N - 6,5. Для С35Н35N3O9 рассчитано: С - 65,5; Н - 5,5; N - 6,5%.
Пример 57
1-(3-,5-ди-O-бензил-4-С-гидроксиметил-β -D-рибофуранозил)-4-N-бензоилцитозин (55)
К перемешиваемому раствору нуклеозида 54 (3,4 г, 5,3 ммоль) в метаноле (20 см3) добавляли метоксид натрия (0,663 г, 11,66 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 10 минут и затем нейтрализовывали 20%-ной водной соляной кислотой. Растворитель частично выпаривали и остаток экстрагировали дихлорметаном (3× 50 см3).
Объединенную органическую фазу промывали насыщенным водным раствором кислого углекислого натрия (3× 20 см3) и высушивали над Nа2SO4. Растворитель удаляли при пониженном давлении и остаток подвергали хроматографической очистке на силикагелевой колонке с использованием дихлорметана/метанола (98,5:1,5, о/о) в качестве элюента с получением соединения 55 в виде твердого вещества белого цвета (1,6 г, 54%). δ H (CDCl3) 9,95 (1Н, шс, NH), 8,33 (1Н, д, J 7,4, 6-Н), 7,98 (2Н, м, Bz), 7,60-7,12 (14Н, м, Bn, Bz, 5-H), 6,17 (1H, д, J 1,6, 1’-Н), 4,78 (1H, д, J 11,8, Bn), 4,48-4,27 (5Н, м, Bn, 2’-Н, 3’-Н), 3,85 (1H, д, J 11,8, 5’-На), 3,66-3,61 (2Н, м, 5’-Нb, 1’’-На), 3,47 (1Н, д, J 10,4, 1’’-Hb). δ C (CDCl3) 167,5, 162,31 (С-4, С=O), 155,36 (С-2), 145,34 (С-6), 137,49, 137,08, 133,09, 133,01, 128,94, 128,67, 128,48, 128,30, 128,01, 127,90, 127,80 (Bn, Bz), 96,53 (С-5), 93,97, 89,35 (С-1’, C-4’), 76,06, 75,28, 73,70, 72,76, 70,26, 62,44 (С-2’, C-3’, Bn, С-5’, С-1’’). FAB-MS m/z 558 [М+Н]+.
Пример 58
Промежуточное соединение 56
Раствор нуклеозида 55 (2,2 г, 3,94 ммоль) в безводном тетрагидрофуране (60 см3) перемешивали при -20° С и суспензию 60%-ного гидрида натрия в минеральном масле (в/о, 0,252 г, 6,30 ммоль) добавляли семью порциями в течение 45 минут. Раствор перемешивали при температуре -20° С в течение 15 минут и после этого маленькими порциями добавляли р-толуолсульфонилхлорид (0,901 г, 4,73 ммоль). Полученный раствор перемешивали в течение 4 часов при -20° С. Дополнительно добавляли гидрид натрия (0,252 г, 6,30 ммоль) и р-толуолсульфонилхлорид (0,751 г, 3,93 ммоль). Реакционную смесь выдерживали при -20° С в течение 48 часов. Реакцию останавливали добавлением смеси льда и воды (50 см3), после чего проводили экстрагирование дихлорметаном (3× 60 см3). Объединенную органическую фазу промывали насыщенным водным раствором кислого углекислого натрия (3× 20 см3) и высушивали над Na2SO4. Растворитель выпаривали при пониженном давлении и остаток подвергали хроматографической очистке на силикагелевой колонке с использованием дихлорметана/метанола (99:1, о/о) в качестве элюента с получением промежуточного соединения 56 (1,80 г).
Пример 59
(1S,3R,4R,7S)-7-бензилокси-1-бензилоксиметил-3-(4-N-бензоилцитозин-1-ил)-2,5-диоксабицикло[2.2.1]гептан (57)
Промежуточное соединение 56 (1,80 г) растворяли в безводном DMF (30,0 см3) и 60%-ную суспензию гидрида натрия в минеральном масле (в/о, 0,16 мг, 3,9 ммоль) добавляли пятью порциями в течение 30 минут при 0° С. Реакционную смесь перемешивали при комнатной температуре в течение 36 минут. Реакцию останавливали добавлением смеси льда и воды (70 см3) и полученную в результате смесь экстрагировали дихлорметаном (3× 50 см3). Объединенную органическую фазу промывали насыщенным водным раствором кислого углекислого натрия (3× 30 см3) и высушивали над Nа2SO4. Растворитель удаляли при пониженном давлении и остаток подвергали хроматографической очистке на силикагелевой колонке с использованием дихлорметана/метанола (99,5:0,5, о/о) в качестве элюента с получением соединения 57 в виде твердого вещества белого цвета (1,08 г, 79%). δ н (CDCl3) 8,95 (1Н, шс, NH), 8,20 (1Н, д, J 7,5, 6-Н), 7,95-7,92 (2Н, м, Bz), 7,66-7,22 (14Н, м, Bn, Bz, 5-Н), 5,78 (1Н, с, 1’-Н), 4,70-4,65 (3H, м, 2’-Н, Bn), 4,60 (1Н, д, J 11,6, Bn), 4,47 (1H, д, J 11,6, Bn), 4,05-3,78 (5Н, м, 3’-Н, 5’-На, 1’’-На, 5’-Нb, 1’’-Hb). δ C (СDCl3) 167,0, 162,36 (С-4, С=O), 154,5 (С-2), 144,58 (С-6), 137,46, 136,93, 133,35, 132,93, 129,11, 128,67, 128,50, 128,16, 128,11, 127,68, 127,60 (Bn), 96,35 (С-5), 88,38, 87,67 (C-1’, C-4’), 76,14, 75,70, 73,79, 72,27, 72,09, 64,34 (Bn, C-5’, С-1’’, С-2’, С-3’). FAB-MS m/z 540 [М+Н]+. Получено: С - 68,0; Н - 5,5, N - 7,5. Для C31H29N3O6 рассчитано: С - 69,0; Н - 5,4; N - 7,8%.
Пример 60
(1S,3R,4R,7S)-7-гидрокси-1-гидроксиметил-3-(цитозин-1-ил)-2,5-2,5-диоксабицикло[2.2,1]гептан (57А)
К раствору нуклеозида 57 (0,3 г, 0,55 ммоль) в безводном метаноле (22 см3) добавляли 1,4-циклогексадиен (5,0 см3) и 10% палладий на углероде (0,314 г). Полученную смесь, перемешивая, нагревали с обратным холодильником в течение 18 часов. Добавляли дополнительно 10% палладий на углероде (0,380 г) и 1,4-циклогексадиен (5,5 см3) и смесь нагревали с обратным холодильником в течение 54 часов. Реакционную смесь отфильтровывали через подушечку силикагеля, которую после этого промывали метанолом (1500 см3). Объединенный фильтрат выпаривали в условиях пониженного давления и остаток подвергали хроматографической очистке на силикагелевой колонке с использованием дихлорметана/метанола (92,5:7,5, о/о) в качестве элюента с получением соединения 57А в виде твердого вещества белого цвета (0,051 г, 36%). δ H ((СD3)2SO) 7,73 (1Н, д, J 7,7, 6-Н), 7,12-7,20 (2Н, шс, NH2), 5,74 (1Н, д, J 7,7, 5-Н), 5,61 (1Н, шс, 3’-ОН), 5,39 (1Н, с, 1’-Н), 5,12 (1Н, м, 5’-ОН), 4,08 (1Н, с, 2’-Н), 3,80 (1Н, д, J 7,7, 1’’-Ha), 3,81 (1Н, с, 3’-Н), 3,74 (2Н, м, 5’-На, 5’-Hb), 3,63 (1Н, д, J 7,7, 1’’-Hb). δ C ((СO3)2SO) 165,66 (С-4), 154,58 (С-2), 139,68 (С-6), 93,19 (С-5), 88,42, 86,73 (С-1’, С-4’), 78,87, 70,85, 68,32, 56,04 (С-2’, С-1’’, С-3’, С-5’). FAB-MS m/z 256 [М+Н]+.
Пример 61
Промежуточное соединение 57В
К нуклеозиду 57А (0,030 г, 0,11 ммоль), суспендированному в безводном пиридине (2,0 см3), добавляли триметилсилилхлорид (0,14 см3, 1,17 ммоль) и перемешивали при комнатной температуре в течение 1 часа. Бензоилхлорид (0,07 см3, 0,58 ммоль) добавляли при 0° С и полученную смесь перемешивали при комнатной температуре в течение 2 часов. После охлаждения реакционной смеси до 0° С добавляли воду (3,0 см3). После перемешивания в течение 5 минут добавляли водный раствор аммиака (1,5 см3, 32%, в/о) и продолжали перемешивание при комнатной температуре в течение 30 минут. Полученную смесь выпаривали при пониженном давлении и остаток подвергали хроматографической очистке на силикагелевой колонке с использованием дихлорметана/метанола (97,5:2,5, о/о) в качестве элюента с получением промежуточного соединения 57В в виде твердого вещества белого цвета (0,062 г).
Пример 62
(1R,3R,4R,73)-1-(4,4’-диметокситритилоксиметил)-7-гидрокси-3-(4-N-бензоилцитозин-1-ил)-2,5-диоксабицикло[2.2.1]-гептан (57С)
К раствору промежуточного соединения 57В (0,042 г, 0,11 ммоль) в безводном пиридине (1,5 см3) добавляли 4,4’-диметокситритилхлорид (0,06 г, 0,17 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 3,5 часов, охлаждали до 0° С и добавляли насыщенный водный раствор кислого углекислого натрия (20 см3). Экстракцию проводили дихлорметаном (3× 10 см3). Объединенную органическую фазу высушивали над Na2SO4 и упаривали досуха при пониженном давлении. Остаток подвергали хроматографической очистке на силикагелевой колонке с использованием дихлорметана/метанола/пиридина (98,0:1,5:0,5, о/о) в качестве элюента с получением нуклеозида 57С в виде твердого вещества белого цвета (0,031 г, ≈ 63% от 57А). δ H (C5D5N) 12,32 (1Н, шс, NHCO), 8,75-7,06 (20Н, м, DMT, Bz, H-5, H-6), 6,24 (1H, c, 1’-H), 5,11 (1-H, c, 2’-H), 4,90 (1H, с, 3’-Н), 4,38 (1H, д, J 7,6, 1’’-На), 4,10 (1H, д, J 7,6, 1’’-Hb), 4,02 (1H, д, J 10,6, 5’-На), 3,87 (1H, д, J 10,6, 5’-Hb), 3,77, 3,76 (2 × 3Н, 2 × с; 2 × ОСН3). δ C (C5D5N) 169,00 (NHCO), 164,24 (С-2), 159,39 (DMT), 150,5, 145,62 (DMT), 144,31, 132,89, 130,82, 130,72, 129,09, 128,89, 128,60, 113,96 (DMT), 96,96, 89,01, 87,18, 79,91, 72,56, 70,25 (С-5, C-1’, C-4’, С-2’, С-1’’, С-3’), 59,51 (С-5’), 55,33 (ОСН3). FAB-MS m/z 662 [М+Н]+.
Пример 63
(1R,3R,4R,7S)-7-(2-цианоэтокси(диизопропиламино)фосфинокси)-1-(4,4’-диметокситритилоксиметил)-3-(4-N-бензоилцитозин-1-ил)-2,5-диоксабицикло[2.2.1]гептан (57D)
К раствору нуклеозида 57С (0,025 г, 0,03 ммоль) в безводном дихлорметане (1,5 см3) добавляли N,N-диизопропилэтиламин (0,03 см3, 0,17 ммоль), а после этого по каплям добавляли 2-цианоэтил-N,N-диизопропилфосфорамидохлоридит (0,02 см3, 0,09 ммоль). После перемешивания при комнатной температуре в течение 5 часов реакционную смесь охлаждали до 0° С, добавляли дихлорметан/пиридин (10,0 см3, 99,5:0,5, о/о) и проводили промывание с использованием насыщенного водного раствора кислого углекислого натрия (3× 8 см3). Органическую фазу отделяли, высушивали над Nа2SO4 и упаривали досуха при пониженном давлении. Остаток подвергали кроматографической очистке на силикагелевой колонке с использованием дихлорметана/метанола/пиридина (99,0:0,5:0,5, о/о) в качестве элюента с получением амидита 57D в виде светло-желтого маслянистого вещества (0,038 г). δ P (CDCl3) 147,93.
Пример 64
9-(2-O-ацетил-4-С-ацетилоксиметил-3,5-ди-O-бензил-β -D-рибофуранозил)-6-N-бензоиладенин (58)
К перемешиваемой суспензии аномерной смеси 33 (5,0 г, 10,3 ммоль) и 6-N-бензоиладенина (3,76 г, 15,7 ммоль) в безводном дихлорметане (200 см3) добавляли N,O-бис(триметилсилил)ацетамид (15,54 см3, 61,8 ммоль). Реакционную смесь перемешивали с нагреванием с обратным холодильником в течение 1 часа и затем охлаждали до комнатной температуры. По каплям добавляли триметилсилилтрифлат (7,0 см3, 38,7 ммоль) и полученную смесь нагревали с обратным холодильником в течение 20 часов. Реакционную смесь затем оставляли остывать до комнатной температуры и объем смеси снижали на 75% при пониженном давлении. Добавляли дихлорметан (250 см3) и полученный раствор промывали насыщенным водным раствором кислого углекислого натрия (3× 50 см3) и водой (50 см3). Органическую фазу высушивали над Na2SO4 и выпаривали при пониженном давлении. Остаток подвергали хроматографической очистке на силикагелевой колонке с использованием дихлорметана/метанола (99,5:0,5, о/о) в качестве элюента с получением нуклеозида 58 в виде твердого вещества белого цвета (3,65 г, 52%). δ H (CDCl3) 9,25 (1Н, шс, NH), 8,71 (1Н, с, 8-Н), 8,24 (1H, с, 2-Н), 8,0 (2Н, д, J 7,5, Bz), 7,60-7,23 (13Н, м, Bn, Bz), 6,35 (1H, д, J 4,6, 1’-Н), 5,99 (1H, дд, J 4,9, 5,3, 2’-Н), 4,78 (1H, д, J 5,6, 3’-Н), 4,64-4,42 (5Н, м, Bn, 1’’-На), 4,25 (1H, д, J 12,1, 1’’-Hb), 3,72 (1H, д, J 10,1, 5’-На), 3,56 (1Н, д, J 10,1, 5’-Hb), 2,07 (3H, с, СОСН3), 2,02 (3H, с, СОСН3). δ C (CDCl3) 170,42, 169,72 (СОСН3), 164,60 (NHCO), 152,51 (С-6), 151,45 (С-2), 149,46 (С-4), 141,88 (С-8), 137,04, 137,00, 133,50, 132,60, 128,86, 128,66, 128,53, 128,41, 128,38, 128,18, 128,06, 127,91, 127,88, 127,79, 127,63, 123,26 (Bz, Bn, C-5), 86,38 (С-1’), 86,25 (С-4’), 77,74, 74,74, 74,44, 73,48 (С-2’, С-3’, 2 × Bn), 70,11 (С-1’’), 63,42 (C-5’), 20,70, 20,54 (СОСН3). FAB-MS m/z 666 [М+Н]+.
Пример 65
9-(3,5-ди-O-бензил-4-С-гидроксиметил-β -D-рибофуранозил)-6-N-бензоиладенин (59)
К перемешиваемому раствору нуклеозида 58 (4,18 г, 6,28 ммоль) в метаноле (50 см3) добавляли метоксид натрия (0,75 г, 13,8 ммоль) при 0° С. Реакционную смесь перемешивали в течение 2 часов и добавляли лед. Смесь нейтрализовывали с использованием 20%-ного водного раствора соляной кислоты. Проводили экстракцию дихлорметаном (3× 75 см3), отделяли органическую фазу, высушивали ее над Na2SO4 и выпаривали при пониженном давлении. Остаток подвергали хроматографической очистке на силикагелевой колонке с использованием дихлорметана/метанола (98,5:1,5 о/о) в качестве элюента с получением нуклеозида 59 в виде твердого вещества белого цвета (2,68 г, 73%). δ H (СDCl3) 9,42 (1Н, шс, NH), 8,58 (1Н, с, Н-8), 8,16 (1Н, с, 2-Н), 7,96 (2Н, д, J 7,2, Bz), 7,52-7,08 (13Н, м, Bn, Bz), 6,18 (1Н, д, J 2,5, 1’-Н), 4,85-4,38 (4Н, м, Вn, 2’-Н, 3’-Н), 4,33 (2Н, с, Bn) 3,90 (1Н, д, J 11,9, 1’’-Ha), 3,71 (1Н, д, J 1,8, 1’’-Hb), 3,50-3,39 (2Н, м, 5-Н). δ C (CDCl3) 164,98 (NHCO), 152,19 (С-6), 151,00 (С-2), 149,34 (С-4), 142,28 (С-8), 137,32, 137,25, 133,46, 132,70, 128,69, 128,49, 128,40, 128,11, 128,03, 127,94, 127,83, 127,62, (Bz, Bn), 122,92 (С-5), 90,94, 88,75 (С-1’, С-4’), 77,65, 74,08, 73,44, 73,20, 71,12, 62,39 (C-1’’, С-5’, С-2’, С-3’, 2 × Bn). FAB-MS m/z 582 [M+H]+. Получено: С - 65,6; Н - 5,5; N - 11,1, Для C32H31N5O6 рассчитано: С - 66,1; Н - 5,4; N - 12,0%.
Пример 66
Промежуточное соединение 60
Раствор нуклеозида 59 (2,43 г, 4,18 ммоль) в безводном тетрагидрофуране (25 см3) перемешивали при -20° С и 60%-ную суспензию гидрида натрия в минеральном масле (в/о, 0,28 г, 7,0 ммоль) добавляли четырьмя порциями в течение 30 минут. После перемешивания в течение 1 часа небольшими порциями добавляли р-толуолсульфонилхлорид (1,34 г, 7,0 моль). Полученную смесь перемешивали при -10° С в течение 15 часов. Добавляли охлажденную льдом воду (50 см3) и проводили экстракцию дихлорметаном (3× 50 см3). Объединенную органическую фазу промывали насыщенным водным раствором кислого углекислого натрия (2× 25 см3), высушивали над Na2SO4 и выпаривали при пониженном давлении. Остаток подвергали хроматографической очистке на силикагелевой колонке с использованием дихлорметана/метанола (99:1, о/о) в качестве элюента с получением промежуточного соединения 60 (1,95 г).
Пример 67
(1S,3R,4R,7S)-7-бензилокси-1-бензилоксиметил-3-(6-N-бензоиладенин-9-ил)-2,5-диоксабицикло[2.2.1]гептан (61)
Промежуточное соединение 60 (1,90 г) растворяли в безводном DMF (20 см3) и 60%-ную суспензию гидрида натрия в минеральном масле (в/в, 0,16 г, 3,87 ммоль) добавляли небольшими порциями при 0° С. Полученную смесь перемешивали при комнатной температуре в течение 10 часов и затем концентрировали при пониженном давлении. Остаток растворяли в дихлорметане (75 см3), промывали насыщенным водным раствором кислого углекислого натрия (2× 25 см3), высушивали над Na2SO4 и выпаривали при пониженном давлении. Остаток подвергали хроматографической очистке на силикагелевой колонке с использованием дихлорметана/метанола (99:1, о/о) в качестве элюента с получением нуклеозида 61 в виде твердого вещества белого цвета (1,0 г, ≈ 44% от 59). δ H (СDСl3) 8,71 (Н, с, 8-Н), 8,23 (1Н, с, 2-Н), 8,02 (2Н, м, J 7,0, Bz), 7,99-7,19 (13Н, м, Bn, Bz), 6,08 (1H, с, 1’-Н), 4,78 (1H, с, 2’-Н), 4,61-4,50 (4Н, м, 2 × Bn), 4,24 (1H, с, 3’-Н), 4,12 (1H, д, J 7,8, 1’’-Ha), 4,00 (1H, д, J 7,9, 1’’-Hb), 3,85-3,78 (2Н, м, 5’-На, 1’’-Hb). δ C (СDСl3) 164,61 (NHCO), 152,32 (С-6), 150,61 (С-2), 149,35 (С-4), 140,67 (С-8), 137,24, 136,76, 133,33, 132,66, 128,68, 128,39, 128,29, 127,94, 127,77, 127,51 (Bn, Bz), 123,43 (С-5), 87,14, 86,52 (C-1’, C-4’), 77,21, 76,77, 73,56, 72,57, 72,27, 64,65 (С-2’, С-3’, C-1’’, 2 × Bn, C-5’). FAB-MS m/z 564 [M+H]+. Получено: С - 66,2; Н - 5,5; N - 11,4. Для С32Н29N5O5 рассчитано: С - 66,2; Н - 5,2; N - 12,4%.
Пример 68
(1S,3R,4R,7S)-7-гидрокси-1-гидроксиметил-3-(аденин-9-ил)-2,5-диоксабицикло[2.2.1]гептан (61А)
К перемешиваемому раствору нуклеозида 61 (0,80 г, 1,42 ммоль) в безводном дихлорметане (30 см3) при -78° С по каплям добавляли ВСl3 (1 М раствор в гексане, 11,36 см3, 11,36 ммоль) в течение 30 минут. Полученную смесь перемешивали при -78° С в течение 4 часов, вновь по каплям добавляли ВСl3 (1 М раствор в гексане, 16,0 см3,16,0 ммоль) и смесь перемешивали при -78° С в течение 3 часов. Затем температуру реакционной смеси медленно увеличивали до комнатной температуры и перемешивание продолжали в течение 30 минут. Добавляли метанол (25,0 см3) при -78° С и смесь перемешивали при комнатной температуре в течение 12 часов. Полученную смесь выпаривали при пониженном давлении и остаток подвергали хроматографической очистке на силикагелевой колонке с использованием дихлорметана/метанола (92:8, о/о) в качестве элюента с получением нуклеозида 61А в виде твердого вещества белого цвета (0,332 г, 84%). δ H ((CD3)2SO) 8,22 (1H, с, 8-Н), 8,15 (1H, с, 2-Н), 7,33 (2Н, с, NH2), 5,89 (1H, с, 1’-Н), 5,83 (1H, д, J 4,2, 3’-ОН), 5,14 (1H, т, J 5,9, 5’-ОН), 4,14 (1H, с, 2’-H), 4,-25 (1H, Д, J 4,2, 3’-Н), 3,92 (1H, д, J 7,8, 1’’-На), 3,81-3,41 (3H, м, 5’-Ha, 5’-Нb, 1’’-Нb). δ C ((СO3)2SO) 155,90 (С-6), 152,64 (С-2), 148,35 (С-4), 137,72 (С-8), 118,94 (С-5), 88,48, 85,17 (С-1’, С-4’), 79,09, 71,34, 69,83, 56,51 (С-2’, С-3’, С-1’’, С-5’). FAB-MS m/z 280 [М+Н]+.
Пример 69
(1S,3R,4R,7S)-7-гидрокси-1-гидроксиметил-3-(6-N-бензоиладенин-9-ил)-2,5-диоксабицикло[2.2.1]гептан (61В)
К перемешиваемому раствору нуклеозида 61А (0,32 г, 1,15 ммоль) в безводном пиридине (1 см3) добавляли триметилсилилхлорид (0,73 см3, 5,73 ммоль) и полученную смесь перемешивали при комнатной температуре в течение 20 минут. Бензоилхлорид (0,67 см3, 5,73 ммоль) добавляли при 0° С и реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь охлаждали до 0° С и добавляли охлажденную льдом воду (15,0 см3). После перемешивания в течение 5 минут добавляли 32% (в/о) водный раствор аммиака (1,5 см3) и смесь перемешивали в течение 30 минут. Полученную смесь упаривали досуха и остаток растворяли в воде (25 см3). После выпаривания смеси при пониженном давлении и остаток подвергали хроматографической очистке на силикагелевой колонке с использованием дихлорметана/метанола (97:3, о/о) в качестве элюента с получением нуклеозида 61В в виде твердого вещества белого цвета (0,55 г). FAB-MS m/z 384 [M+H]+.
Пример 70
(1R,3R,4R,7S)-7-гидрокси-1-(4,4’-диметокситритилоксиметил)-3-(6-N-бензоиладенин-9-ил)-2,5-диоксабицикло[2.2.1]-гептан (61С)
К перемешиваемому раствору соединения 61В (0,50 г) в безводном пиридине (20 см3) добавляли 4,4’-диметокситритилхлорид (0,71 г, 2,09 ммоль) и 4-N,N-димeтилaминoпиpидин (DMAP) (0,1 г). После перемешивания при комнатной температуре в течение 2 часов и в течение 1 часов при 50° С реакционную смесь охлаждали до 0° С и добавляли насыщенный водный раствор кислого углекислого натрия (100 см3). После экстракции дихлорметаном (3× 50 см3) объединенную органическую фазу высушивали над N2SO4 и выпаривали при пониженном давлении. Остаток подвергали хроматографической очистке на силикагелевой колонке с использованием дихлорметана/метанола/пиридина (98,0:1,5:0,5) в качестве элюента с получением нуклеозида 61С в виде твердого вещества белого цвета (0,36 г, ≈ 50% от 61А). δ H (C5D5N) 12,52 (NHCO), 9,10 (2Н, д, J 7,7, Bz), 8,88 (1Н, с, 8-Н), 8,50-7,11 (17Н, м, DMT, Bz, 2-Н), 6,65 (1H, с, H-1’), 5,25 (2Н, с, Н-2’, Н-3’), 4,71 (1Н, д, J 7,8, 1’’-На), 4,56 (1H, д, J 7,8, 1’’-Нb), 4,20 (1H, д, J 10,8, 5’-На), 4,07 (1H, д, J 10,8, 5’-Нb), 3,82, 3,81 (2 × 3Н, 2 × с, 2 × ОСН3). δ C (C5D5N) 167,56 (NHCO), 159,24 (С-6), 152,50, 152,08, 151,81, 145,84, 141,45, 136,52, 136,28, 132,55, 130,76, 130,70, 129,32, 128,85, 128,76, 128,46, 127,38, 126,33 (DMT, Bz, C-2, C-4, C-8), 113,84 (С-5), 88,64, 87,20, 86,85, 80,52, 73,13, 72,16, 60,86 (С-1’, С-4’, DMT,С-2’, С-3’; С-1’’, С-5’), 55,24 (ОСН3). FAB-MS m/z 686 [М+Н]+. Получено: C - 68,3; Н - 5,0; N - 9,7. Для С39Н35N5О7 расчитано: С - 68,3; Н - 5,1; N - 10,2%.
Пример 71
(1R,3R,4R,7S)-7-(2-цианоэтокси(диизопропиламино)фосфинокси-1-(4,4’-диметокситритилоксиметил)-3-(6-N-бензоиладенин-9-ил)-2,5-диоксабицикло[2,2.1]гептан (61D)
К раствору соединения 61С (190 мг, 0,277 ммоль) в безводном диклорметане (1,5 см3) добавляли N,N-диизопропилэтиламин (0,16 см3, 0,94 ммоль) и 2-цианоэтил-N,N-диизoпpoпилфосфорамидохлоридит (0,1 см3, 0,42 ммоль) при 0° С. Смесь оставляли нагреваться до комнатной температуры и перемешивали в течение 5 часов. Раствор разбавляли дихлорметаном (50 см3), промывали насыщенным водным раствором кислого углекислого натрия (2× 30 см3) и выпаривали при пониженном давлении. Продукты выделяли методом ТЭЖХ в силикагеле: картридж PrepPak, 25× 100 мм, загружена Prep Nova-Pak® HR Silica, 6 мкм, градиент раствора B в растворе A (от 0% до 15% в течение 25 минут и от 15% до 100% в течение еще 25 минут; раствор А - петролейный эфир/дихлорметан/пиридин – 60:39,6:0,4, о/о/о; раствор В этилацетат). Фракции, содержащие два основных продукта (время удерживания 30-40 минут) объединяли, концентрировали при пониженном давлении, выпаривали вместе с безводным толуолом (2× 40 см3) и высушивали в вакууме в течение ночи. Остаток растворяли в безводном дихлорметане (4 см3) и осаждали добавлением этого раствора в безводный петролейный эфир (80 см3) при интенсивном перемешивании. Преципитат собирали фильтрованием, промывали петролейным эфиром (2× 20 см3) и высушивали при пониженном давлении с получением соединения 61D (178 мг, 73%) в виде твердого вещества белого цвета. δ р (CD3CN) 148,42, 147,93.
Пример 72
1-(2,3-О-изопропилиден-4-С-(4-толуолсульфонилоксиметил)-β -D-рибофуранозил)уридин (62)
К перемешиваемому раствору 1-(2,3-О-изопропилиден-4’-С-гидроксиметил-β -О-рибофуранозил)уридина (2,0 г, 6,4 ммоль) (R.Youssefyeh, D.Tegg, J.P.H.Verheyden, G.H.Jones & J.G.Moffat, 1977, Tetrahedron Lett., 5, 435; G.H.Jones, M.Taniguchi, D.Tegg & J.G.Moffat, 1979, J. Org. Chem., 44, 1309) в безводном пиридине (28 см3) добавляли р-толуолсульфонилхлорид (1,46 г, 7,3 ммоль), растворенный в безводном пиридине (10 см3) при -30° С. Через 30 минут реакционную смесь оставляли нагреваться до комнатной температуры и продолжали перемешивание при комнатной температуре в течение 12 часов. Реакцию останавливали метанолом (4 см3), добавляли силикагель (2 г) и при пониженном давлении удаляли растворитель. Остаток подвергали хроматографической очистке на силикагелевой колонке с использованием градиента 0-3% метанола в дихлорметане (о/о) в качестве элюента с получением нуклеозида 62 в виде твердого вещества палево-красного цвета (1,8 г, 60%). δ H (СDСl3) 9,80 (1Н, шс, NH), 7,80 (2Н, д, J 8,3, Ts), 7,46 (1Н, д, J 8,1, 6-Н), 7,35 (2Н, д, J 8,01, Ts), 5,72 (1Н, д, J 8,0, 5-Н), 5,54 (1Н, д, J 3,5, 1’-H), 5,08 (1Н, дд, J 3,5, 6,4, 2’-Н), 4,94 (1Н, д, J 6,4, 3’-Н), 4,18 (2Н, с, 1”-H), 3,82-3,70 (2Н, м, 5’-Н), 2,45 (3H, с, Ts), 1,46, 1,29 (6Н, с, СН3). δ C (CDCl3) 163,6 (С-4), 150,4 (С-2), 145,2 (С-6), 142,9, 132,5, 129,9, 128,0 (Ts), 114,7 (ОСО), 102,6 (С-5), 94,9, 87,6, 83,9, 81,5 (С-4’, С-1’, С-3’, С-2’), 68,7, 63,5 (C-1’’, C-5’), 26,4, 24,7 (СН3), 21,7 (Ts). FAB-MS m/z 469 [М+Н]+.
Пример 73
1-(4-С-(р-толуолсульфонилоксиметил-β -D-рибофуранозил)-уридин (63)
Нуклеозид 62 (1,33 г, 2,83 ммоль) растворяли в 80%-ной уксусной кислоте (25 см3) и перемешивали при 80° С в течение 3 часов, после чего растворитель удаляли при пониженном давлении. Остаток выпаривали вместе с этанолом (10 см3) и очищали методом хроматографии на силикагелевой колонке с использованием градиента 0-2% метанола в дихлорметане (о/о) в качестве элюента с получением нуклеозида 63 в виде твердого вещества белого цвета (391 мг, 33%). δ H (CD3ОD) 7,81 (1Н, д, J 8,1, 6-Н), 7,77 (1Н, д, J 8,4, Ts), 7,40 (2Н, д, J 8,6, Ts), 5,74 (1Н, д, J 6,6, 1’-Н), 5,69 (1Н, д, J 8,1, 5-Н), 4,17-4,33 (2Н, м, 2’-Н, 3’-Н), 3,67-3,62 (2Н, м, 1’’-Н), 3,26-3,20 (2Н, м, 5’-Н), 2,43 (3H, с, Ts). δ C (CD3OD) 166,0 (С-4), 153,0 (С-2), 146,5 (С-6), 142,5, 130,9, 129,15 (Ts), 103,1 (С-5), 89,0, 87,2 (С-1’, С-4’), 75,1, 72,7, 71,3, 63,8 (C-1’’, C-3’, C-2’, C-5’), 21,6 (Ts).
Пример 74
(1S,3R,4R,7S)-7-гидрокси-1-гидроксиметил-3-(урацил-1-ил)-2,5-диоксабицикло[2.2.1]гептан (44)
К перемешиваемому раствору нуклеозида 63 (64 мг, 0,14 ммоль) в безводном DMF (2 см3) медленно добавляли гибрид натрия (8,4 мг, 21 моль, 60%-ная суспензия в минеральном масле, в/о) в безводном DMF (2 см3) при 0° С. Реакционную смесь затем нагревали до 120° С в течение 6 часов. После остановки реакции водой (2 см3) растворители удаляли при пониженном давлении и остаток подвергали хроматографической очистке на силикагелевой колонке с использованием в качестве элюента градиента 5-7% метанола в дихлорметане (о/о) с получением нуклеозида 44 в виде твердого вещества белого цвета (9 мг, 29%). Данные ядерно-магнитного резонанса согласуются с теми, которые были ранее приведены для соединения 44.
Пример 75
(1S,3R,4R,7S)-7-ацетокси-1-ацетоксиметил-3-(тимин-1-ил)-2,5-диоксабицикло[2.2.1]гептан (64)
К перемешиваемому раствору нуклеозида 37 (209,8 мг, 0,78 ммоль) в безводном пиридине (2,5 см3) добавляли уксусный ангидрид (0,3 см3, 3,23 ммоль) и каталитическое количество DMAP (5 мг). После перемешивания в течение 2 часов добавляли этанол (4 см3) и полученную смесь выпаривали при пониженном давлении. Остаток вновь растворяли в дихлорметане и промывали насыщенным водным раствором кислого углекислого натрия (7 см3). Органическую фазу высушивали над Na2SO4 и выпаривали при пониженном давлении. Остаток подвергали хроматографической очистке на силикагелевой колонке с использованием дихлорметана/метанола (93:3, о/о) в качестве элюента с получением соединения 64 в виде твердого вещества белого цвета (249 мг, 90%). δ C (СDСl3) 169,59, 163,20, 149,50, 133,55, 110,64, 87,05, 85,38, 77,84, 71,70, 71,02, 58,60, 20,62, 20,53, 12,78. FAB-MS m/z 355 [M+H]+.
Пример 76
(1S,3R,4R,7S)-1-гидроксиметил-3-(5-метил-4-N-бензоилцитозин-1-ил)-7-гидрокси-2,5-диоксабицикло[2.2.1]гептан (65)
К раствору нуклеозида 64 (232,7 мг, 0,66 ммоль) в безводном ацетонитриле (3 см3) добавляли раствор 1,2,4-триазола (420 мг, 6,1 ммоль,) и РОСl3 (0,12 см3, 1,3 ммоль) в безводном ацетонитриле (5 см3). Реакционную смесь охлаждали до 0° С и добавляли безводный триэтиламин (0,83 см3), после чего смесь выдерживали при комнатной температуре в течение 90 минут. Добавляли триэтиламин (0,54 см3) и воду (0,14 см3) и полученную реакционную смесь перемешивали в течение 10 минут и выпаривали при пониженном давлении. Остаток растворяли в EtOAc и промывали насыщенным водным раствором кислого углекислого натрия (2× 9 см3) и водой (9 см3). Водную фазу экстрагировали дихлорметаном (3× 20 см3). Объединенную органическую фазу выпаривали при пониженном давлении и полученный остаток вновь растворяли в диоксане (4 см3), после чего добавляли 32%-ный водный раствор аммиака (0,7 см3). После перемешивания в течение 3 часов реакционную смесь выпаривали при пониженном давлении и выпаривали вместе с безводным пиридином. Остаток растворяли в безводном пиридине (3,6 см3) и добавляли бензоилхлорид (0,42 см3, 3,6 ммоль). После перемешивания в течение 2 часов реакцию останавливали добавлением воды (1 см3) и реакционную смесь выпаривали при пониженном давлении. Остаток затем вновь растворяли в EtOAc и промывали водой (3× 7 см3). Органическую фазу упаривали досуха при пониженном давлении и остаток растворяли в пиридине/метаноле/воде (13:6:1, о/о/о 14 см3) при 0° С и добавляли 2 М раствор едкого натра в пиридине/метаноле/воде (13:6:1, о/о/о 7 см3). После 20-минутного перемешивания реакционную смесь выпаривали при пониженном давлении. Остаток подвергали хроматографической очистке на силикагелевой колонке с использованием в качестве элюента дихлорметана/метанола (95:5, о/о) с получением нуклеозида 65 в виде пены желтого цвета (94,6 мг, 38%). δ H (CD3OD) 8,21 (1Н, шс), 8,02 (1H, м), 7,84-7,9 (1Н, м), 7,41-7,58 (4Н, м), 5,61 (1H, с), 4,36 (1H, с), 4,10 (1H, с), 3,98 (1H, д, J 8,0), 3,94 (2Н, с), 3,78 (1H, д, J 7,9), 2,11 (3H, д, J 1,0). δ C (CD3OD, выборочные сигналы) 133,66, 132,90, 130,63, 129,50, 129,28, 128,65, 90,71, 88,86, 80,57, 72,47, 70,22, 57,53, 14,01. FAB-MS m/z 374 [М+Н]+.
Пример 77
(1R,3R,4R,7S)-1-(4,4’-диметокситритилоксиметил)-3-(5-метил-4-N-бензоилцитозин-1-ил)-7-O-(2-цианоэтокси(диизопропиламино)фосфинокси)2,5-диоксабицикло[2.2.1]гептан (66)
К перемешиваемому раствору нуклеозида 65 (82 мг, 0,22 ммоль) в безводном пиридине (1,5 см3) добавляли 4,4’-диметокситритилхлорид (80 мг, 0,24 ммоль) и продолжали перемешивание при комнатной температуре в течение 12 часов. Дополнительно добавляли 4,4’-диметокситритилхлорид (80 мг, 0,24 ммоль) и продолжали перемешивание в течение еще 12 часов. Добавляли метанол (0,5 см3) и реакционную смесь выпаривали при пониженном давлении. Остаток подвергали хроматографической очистке на силикагелевой колонке с использованием дихлорметана/метанола/пиридина (98,5:1,0:0,5, о/о/о. Полученное промежуточное соединение (FAB-MS m/z 676) (50,2 мг) выпаривали вместе с безводным ацетонитрилом и растворяли в безводном дихлорметане (0,62 см3). Добавляли N,N-диизопропилэтиламин (0,1 см3) с последующим добавлением 2-цианоэтил-N,N-диизопропилфосфорамидохлоридита (0,3 см3, 0,11 ммоль). После перемешивания при комнатной температуре в течение 3 часов добавляли воду (1 см3) и полученную в результате смесь разбавляли этилацетатом (10 см3), промывали насыщенным водным раствором кислого углекислого натрия (3× 6 см3) и соляным раствором (3× 6 см3). Органическую фазу высушивали над Na2SO4 и выпаривали при пониженном давлении. Остаток очищали методом ВЭЖХ в силикагеле. В результате преципитации, описанной выше при получении соединения 53, получали соединение 66 в виде твердого вещества белого цвета (29,5 мг, 0,03 ммоль, 14%). δ P (СН3СN) 148,46, 147,49.
Пример 78
9-(4-гидроксиметил)-2,3-О-изопропилиден-β -D-рибофуранозил)-6-N-бензоиладенин (67)
Смесь оксалилхлорида (0,93 мл, 10,75 ммоль) и дихлорметана (25 мл) охлаждали до -70° С. По каплям при интенсивном перемешивании добавляли диметилсульфоксид (ДМСО, 1,7 мл, 22 ммоль). Полученную смесь перемешивали в течение 10 минут при -70° С и в течение 5 минут добавляли раствор 9-(2,3-O-изопропилиден-β -D-рибофуранозил)-6-N-бензоиладенина (3,4 г, 8,27 ммоль) в диметилсульфоксиде/дихлорметане (1:9, о/о, 20 мл). Смесь перемешивали при -60° С в течение 30 минут. Добавляли триэтиламин (7 мл, 50,3 ммоль) и полученную смесь оставляли нагреваться до комнатной температуры. Раствор разбавляли дихлорметаном (50 мл) и промывали водой (3× 100 мл). Водную фракцию дополнительно промывали дихлорметаном (100 мл). Органическую фазу концентрировали до уровня маслянистой массы, выпаривали вместе с диоксаном (50 мл) и вновь растворяли в диоксане (30 мл). Добавляли 37%-ный водный раствор формальдегида (2,95 мл, 33,4 ммоль) и 2 М водный раствор едкого натра (8,26 мл), затем смесь перемешивали при комнатной температуре в течение 10 минут и охлаждали до 0° С. Добавляли борогидрид натрия (640 мг, 16,9 ммоль) и реакционную смесь оставляли нагреваться до комнатной температуры на 15 минут. Реакцию останавливали добавлением уксусной кислоты (5 мл) и к смеси добавляли дихлорметан и насыщенный водный раствор кислого углекислого натрия (по 100 мл каждого). Органическую фазу промывали водой (100 мл), концентрировали в вакууме и полученный продукт выделяли методом хроматографии на силикагелевой колонке (2,5× 25 см), используя в качестве элюента градиент 2-3,2% метанола в дихлорметане (о/о). Выход соединения 67 в виде твердого вещества белого цвета составил 1,85 г (50,7%). δ H (CDCl3) 8,72 (1Н, с), 8,14 (1H, с), 8,03-8,00 (2Н, м), 7,60-7,57 (1H, м), 7,56-7,46 (2Н, м), 6,00 (1H, д, J 4,9), 5,35 (1H, дд, J’5,8, J’’5,0), 5,13 (1H, д, J 5,8), 3,87-3,78 (4Н, м), 1,65 (3H, с), 1,38 (3H, с). δ C (CDCl3) 164,8, 152,2, 150,4, 150,2, 142,6, 133,3, 132,9, 128,8, 128,0 124,1, 114,7, 93,3, 90,2, 83,8, 82,5, 65,3, 62,9, 27,3, 25,1. FAB-MS m/z 442 [М+Н]+, 464 [M+Na]+.
Альтернативный синтез соединения 67
К раствору 2’,3’-O-изопропилиденаденозина (15 г) в безводном пиридине (250 мл) добавляли триметилсилилхлорид (15,5 мл). Реакционную смесь перемешивали при комнатной температуре в течение 20 минут и охлаждали до 0° С. По каплям добавляли бензоилхлорид (17 мл) и полученную смесь выдерживали при комнатной температуре в течение 2-3 часов. Добавляли воду (50 мл) и 25%-ный водный раствор гидроксида аммония (100 мл) и продолжали перемешивание в течение 3 часов. Затем смесь концентрировали при пониженном давлении, выпаривали вместе с толуолом (2× 200 мл) и дихлорметаном и добавляли насыщенный водный раствор кислого углекислого натрия. Органическую фазу упаривали досуха с получением твердого вещества желтого цвета. Рекристаллизация из этанола привела к получению промежуточного соединения в виде твердого вещества белого цвета (12,5 г, ≈ 80%). Оксалилхлорид (4,68 мл) в сухом дихлорметане (120 мл) охлаждали до -70° С. ДМСО (8,5 мл) добавляли при интенсивном перемешивании. Позднее (через 10 минут) по каплям в течение 20 минут добавляли раствор промежуточного соединения (17 г) в 10% ДМСО/дихлорметане (100 мл), синтез которого описан выше. Смесь оставляли до повышения температуры до -50° С на период 30 минут после того, как реакция была остановлена добавлением триэтиламина (35 мл). К реакционной смеси добавляли дихлорметан (200 мл), который промывали водой (3× 200 мл). Промежуточное соединение концентрировали в вакууме, выпаривали вместе с диоксаном и вновь растворяли в диоксане (120 мл). Добавляли формальдегид (37%) и 2 М водный раствор едкого натра (40 мл) и реакционную смесь перемешивали в течение 1 часа. Смесь нейтрализовывали уксусной кислотой (6 мл) и добавляли дихлорметан (400 мл) и насыщенный водный раствор кислого углекислого натрия (400 мл). Концентрировали органическую фазу. Соединение 67 очищали хроматографически в силикагеле в градиенте 1,5-5,0% метанола в дихлорметане. Выход соединения 67 составил 8,5 (46%). Данные соответствовали тому, что было выше определено в данном примере.
Пример 79
9-(2,3-О-изопропилиден-4-(р-толуолсульфонилоксиметил)-β -D-рибофуранозил)-6-N-бензоиладенин (68) и 9-(4-гидроксиметил-2,3-О-изопропилиден-5-O-(р-толуолсульфонил)-β -D-рибофуранозил)-6-N-бензоиладенин
Смесь соединения 67 (1,95 г, 4,42 ммоль) и р-толуолсульфонилхлорида (1,26 г, 6,63 ммоль) растворяли в 10 мл безводного пиридина при 0° С. Реакционную смесь перемешивали в течение 4 часов и затем разбавляли дихлорметаном (100 мл), промывали водой (2× 100 мл) и концентрировали при пониженном давлении. Очистку смеси проводили на хроматографической силикагелевой колонке с использованием градиента (1-4%) метанола в дихлорметане, что позволило выделить исходный материал 67 (360 мг, 18,5%) и два структурных изомера, а именно 68 (менее полярный изомер; 971 мг, 36,7%) и 9-(4-гидроксиметил-2,3-О-изопропилиден-5-O-(4’-толуолсульфонил)-β -D-рибофуранозил-N6-бензоиладенин (более полярный изомер; 352 мг, 13,1) в виде твердых веществ белого цвета. Для 68: δ H (СDСl3) 8,69 (1Н, с), 8,11 (1Н, с), 8,00 (2Н, м), 7,79 (2Н, м), 7,58-7,55 (1Н, м), 7,50-7,46 (2Н, м), 7,34-7,32 (2Н, м), 5,88 (1Н, д, J 4,9), 5,35 (1Н, дд, J’5,8, J’’5,0), 5,13 (1H, д, J 5,8), 3,87-3,78 (4Н, м), 1,65 (3H, с), 1,38 (3H, с). δ C (СDСl3) 164,7, 152,0, 150,2, 150,1, 144,9, 142,5, 133,2, 132,7, 132,3, 129,6, 128,6, 127,9, 127,8, 123,9, 114,6, 93,1, 87,9, 83,4, 81,6, 68,3, 64,4, 27,1, 25,0, 21,5. FAB-MS: m/z 596 [М+Н]+.
Пример 80
9-(4-(р-толуолсульфонилоксиметил)-β -D-рибофуранозил)-6-N-бензоиладенин (69)
Раствор соединения 68 (940 мг, 1,58 ммоль) в 10 мл 90%-ного водной трифторуксусной кислоты выдерживали в течение 30 минут при комнатной температуре и концентрировали в вакууме в маслянистую массу. После выпаривания вместе с метанолом (2× 20 мл) и толуолом (20 мл) смесь очищали на хроматографической силикагелевой колонке (2× 25 см) в градиенте метанола (2-5%) в дихлорметане с получением соединения. 69 (825 мг, 94%) в виде твердого вещества белого цвета. δ H (метанол-d4) 8,67 (1Н, с), 8,53 (1Н, с), 8,05 (2Н, д, J 7,7), 7,75 (2Н, д, J 8,2), 7,63 (1Н, м), 7,53 (2Н, м), 7,32 (2Н, д, J 8,0), 5,94 (1Н, д, J 7,1), 4,92 (1Н, дд, J’7,1, J’’5,3), 4,41 (1Н, д, J 5,1), 4,38 (1Н, д, J 10,2), 4,28 (1Н, д, J 10,2), 3,80 (1Н, д, J 12,0), 3,68 (1Н, д, J 12,0), 2,35 (3H, с). δ C (метанол-d4) 168,2, 152,9, 150,8, 151,2, 146,4, 144,9, 134,7, 134,1, 134,0, 130,8, 129,7, 129,4, 129,1, 125,1, 90,0, 88,4, 75,3, 73,1, 71,1, 64,2, 21,6. FAB-MS: m/z 556 [М+Н]+.
Пример 81
9-(4-р-толуолсульфонилоксиметил)-3,5-O-(тетраизопропилдисилоксан-1,3-диил)-β -D-рибофуранозил)-6-N-бензоиладенин (70)
К раствору соединения 69 (780 мг, 1,40 ммоль) в безводном пиридине (7 мл) добавляли 1,3-дихлор-1,1,3,3-тетраизопропилдисилоксан (500 мл, 1,57 ммоль) при 0° С. После перемешивания в течение 2 часов при 0° С добавляли дополнительно 1,3-дихлор-1,1,3,3-тетраизопропилдисилоксан (50 мл, 0,16 ммоль). Реакционную смесь оставляли нагреваться до комнатной температуры, разбавляли дихлорметаном (100 мл) и промывали водой (2× 100 мл). Органическую фазу концентрировали и остаток очищали методом препаративной ВЭЖХ (картридж PrepPak, Porasil, 15-20 мкм, 125 А; элюент - 0-3% метанола в дихлорметане (о/о) в течение 120 минут; скорость потока 60 мл/мин). После концентрирования в вакууме получали 870 мг (78%) соединения 70 в виде твердого вещества белого цвета. δ H (CDCl3) 8,65 (1Н, с), 8,03 (2Н, м), 8,00 (1Н, с), 7,83 (2Н, д, J 8,4), 7,58 (1Н, м), 7,49 (2Н, м), 7,34 (2Н, д, J 8,4), 5,87 (1Н, с), 5,70 (1H, д, J 6,2), 4,68 (1Н, д, J 6,2), 4,59 (1H, д, J 10,8), 4,31 (1H, д, J 11,0), 3,91 (2Н, с), 2,45 (3H, с), 1,03-0,84 (28Н, м). δ C (CDCl3) 164,9, 152,2, 150,5, 150,0, 144,7, 142,9, 133,5, 132,9, 132,8, 129,7, 128,8, 128,1, 128,0, 123,6, 90,3, 85,3, 76,0, 75,0, 68,7, 65,2, 21,6, 17,5, 17,4, 17,2, 17,1, 17,0, 16,9, 13,1, 13,0, 12,5, 12,4. FAB-MS: m/z 798 [М+Н]+.
Пример 82
9-(2-O,4-С-метилен-3,5-O-(тетраизопропилдисилокса-1,3-диил)-β -D-рибофуранозил)-6-N-бензоиладенин (71)
Раствор соединения 70 (540 мг, 0,68 ммоль) в безводном THF (20 мл) охлаждали до 0° С и при перемешивании добавляли гидрид натрия (130 мг 60%-ной суспензии в минеральном масле, 3,25 ммоль). Реакционную смесь перемешивали в течение 30 минут и затем нейтрализовывали добавлением 750 мкл уксусной кислоты. Добавляли дихлорметан (50 мл), реакционную смесь промывали насыщенным водным раствором кислого углекислого натрия (2× 50 мл) и концентрировали при пониженном давлении. Остаток загружали на силикагелевую колонку (2,5× 25 см) и продукт элюировали с использованием градиента концентраций метанола (0,5-1,2%) в дихлорметане в качестве элюента с получением соединения 71 (356 мг, 84%) в виде пены белого цвета. δ H (СDСl3) 8,77 (1Н, с), 8,28 (1Н, с), 8,03 (2Н, м), 7,59 (1Н, м), 7,50 (2Н, м), 6,08 (1Н, с), 4,86 (1Н, с), 4,56 (1Н, с), 4,14 (1Н, д, J 13,2), 4,06 (1Н, д, J 7,7), 3,97 (1Н, д, J 13,2), 3,89 (1Н, д, J 7,7), 1,12-0,95 (28Н, м). δ C (CDCl3) 164,8, 152,6, 150,5, 149,6, 140,7, 133,6, 132,7, 128,7, 127,9, 123,1, 89,4, 86,5, 78,9, 71,7, 71,2, 56,7, 17,3, 17,1, 17,0, 16,9, 16,8, 13,3, 13,1, 12,5, 12,2. FAB-MS: m/z 626 [М+Н]+.
Пример 83
7-гидрокси-1-гидроксиметил-3-(6-N-бензоиладенин-9-ил)-2,5-диоксабицикло[2.2.1]гептан (61В)
Триэтиламинтрисгидрофторид (300 мл, 1,84 ммоль) добавляли к раствору соединения 71 (420 мг, 0,067 ммоль) в безводном THF (7 мл). Реакционную смесь выдерживали при комнатной температуре в течение 1 часа и концентрировали в маслянистое вещество, которое очищали на хроматографической силикагелевой колонке (2× 25 см) с использованием для элюции градиента 4-10% метанола в дихлорметане (о/о). Выход соединения 61В в виде твердого вещества белого цвета составил 232 мг (92%). Данные ядерно-магнитного резонанса были идентичны тем, которые были получены ранее для 61В.
Пример 84
1-(3,5-ди-O-бензил-4-С-(р-толуолсульфонилоксиметил)-2-O-р-толуолсульфонил-β -D-рибофуранозил)тимин (72)
Раствор 1-(3,5-ди-O-бензил-4-С-(гидроксиметил)-β -D-рибофуранозил)-тимин 35 (1,48 г, 3,16 ммоль), DMAP (1,344 г, 0,011 моль) и р-толуолсульфонилхлорида (1,45 г, 7,6 ммоль) в дихлорметане (20 мл) перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь разбавляли дихлорметаном (30 мл) и промывали насыщенным водным раствором кислого углекислого натрия (3× 20 мл) и хлорида натрия (2× 25 мл). Органическую фазу высушивали над Na2SO4, отфильтровывали и выпаривали при пониженном давлении. Остаток подвергали хроматографической очистке на силикагелевой колонке с использованием метанола/дихлорметана (1:99, о/о) в качестве элюента с получением нуклеозида 72 (1,95 г, 80%) в виде твердого вещества белого цвета. FAB-MS m/e 776. δ C (СDСl3) 162,9, 149,8 (С-2, С-4), 145,8, 145,2 (2 × Ts), 136,9, 136,8 (2 × Bn), 134,3 (С-6), 132,1, 132,0, 130,0, 129,9, 129,0 128,9, 128,4, 128,3, 128,2, 128,0, 127,7 (2 × Ts, 2 × Bn), 111,2 (С-5), 85,3, 84,0 (С-1’, С-4’), 78,9, 78,3, 75,2, 74,3, 72,7 69,1 (С-2’, C-3’, С-5’, С-1’’, 2 × Bn), 21,7 (2 × СН3), 11,9 (СН3); Аналитический расчет для C39H40N2S2O11: С - 60,30; Н - 5,19; N - 3,61. Получено: С - 59,95; Н - 5,11, N - 3,81.
Пример 85
1-(2-бензиламино-2-дезокси-3,5-ди-O-бензил-2-N,4-С-метилен-β -D-рибофуранозил)тимин (73)
Раствор соединения 72 (8,6 г, 11,1 моль) в бензиламине (10 мл) перемешивали при 130° С в течение 36 часов. Реакционную смесь напрямую подвергали хроматографической очистке в силикагеле с использованием метанола/дихлорметана (1:99, о/о) в качестве элюента с получением нуклеозида 73 (1,79 г, 30%) в виде твердого вещества белого цвета. FAB-MS m/e 540. δ C (СDСl3) 163,9, 149,8 (С-2, С-4), 139,2, 137,6, 137,3 (3 × Bn), 135,6 (С-6), 128,5, 128,4, 128,3, 128,2, 128,0, 127,7, 127,0 (3 × Bn), 109,6 (С-5), 88,2, 86,3 (С-1’, С-4’), 76,7, 73,8, 72,0, 66,0, 63,8, 57,9, 57,8 (C-2’, С-3’, С-5’, С-1’’, 3 × Bn), 12,2 (СН3). Аналитический расчет для С32Н33N3O5·0,5H2O: С - 70,06; Н - 6,25; N - 7,66. Получено: С - 70,00; Н – 6,06; N - 7,50;
Пример 86
1-(2-амино-2-дезокси-2-N,4-С-метилен-β -D-рибофуранозил)-тимин (74)
К раствору нуклеозида 73 (1,62 г, 0,003 моль) в этаноле (1.50 мл) добавляли 20% гидроокиси палладия на углероде (3 г) и полученную суспензию перемешивали в течение 5 дней в атмосфере водорода. Катализатор отфильтровывали через силикагель и промывали метанолом (20 мл). Объединенный фильтрат концентрировали при пониженном давлении с получением твердого вещества белого цвета, которое отфильтровывали и промывали метанолом/дихлорметаном (1:4, о/о) с получением монобензилированного промежуточного соединения (0,82 г, 76%). FAB-MS m/e 360 [М+Н]+. 13С-ЯМР (ДМСО-d6, 250 МГц): 163,7, 149,8 (С-2, С-4), 138,2 (Bn), 134,9 (С-6), 128,2, 127,5, 127,4 (Bn), 107,8 (С-5), 87,8, 87,6 (С-1’, 0-4’), 72,7, 68,9, 65,9, 61,7, 49,4 (С-2’, С-3’, С-5’, С-1’’, Bn), 11,9 (СН3). Аналитический расчет для C18H21N3O6: С - 60,16; Н - 5,89; N - 11,69. Получено: С - 59,86; Н - 5,61; N - 11,56. Смесь этих промежуточных соединений (0,1 г, 0,29 ммоль), формата аммония (0,085 г, 1,35 ммоль), 10% палладия на углероде (130 мг) в безводном метаноле (7 мл) нагревали с обратным холодильником в течение 2 часов. Катализатор отфильтровывали через силикагель и промывали метанолом (15 мл); объединенный фильтрат концентрировали досуха при пониженном давлении. Остаток подвергали хроматографческой очистке с использованием для элюции метанола/дихлорметана (1:9, о/о) с получением титульного соединения 74 (0,053 г, 71%) в виде твердого вещества белого цвета. FAB-MS m/e 270.
δ H (ДМСО-d6) 11,29 (шс, 1H, NH), 7,73 (д, 1H, J 1,1, 6-Н), 5,31 (с, 1H, 1’-Н), 5,29 (шс, 1Н, 3’-ОН), 5,13 (м, 1Н, 5’-ОН), 3,81 (с, 1H, 3’-Н), 3,69 (м, 2Н, 5’-Н), 3,23 (с, 1Н, 2’-Н), 2,88 (д, 1H, J 9,8, 1’’-Ha), 2,55 (d, 1H, J 9,8, 1’’-Нb), 1,77 (д, 3Н, J 0,8, СН3). δ C (ДМСО-d6) 164,0, 150,1 (С-2, С-4), 135,6 (С-6), 107,8 (С-5), 89,5, 87,9 (С-1’, С-4’), 68,7, 61,9, 57,1, 49,4, (С-2’, C-3’, С-5’, С-1’’). Аналитический расчет для C11H15N3O5·0,5H2O: С - 47,48; Н - 5,80; N - 15,10. Получено: С - 47,54; Н - 5,30; N - 14,79.
Альтернативный метод конверсии 73 в 74
К раствору соединения 73 (0,045 г, 0,0834 ммоль) в метаноле (6 мл) добавляли 10% палладий на углероде (0,118 г) и также добавляли тремя порциями в течение 3 часов формат аммония (0,145 г, 0,0023 моль). Суспензию нагревали с обратным холодильником в течение 4,5 часов. Катализатор отфильтровывали через силикагель и промывали метанолом (4× 3 мл). Объединенный фильтрат концентрировали и остаток подвергали хроматографической очистке на силикагелевой колонке с использованием для элюции метанола/дихлорметана (1:9, о/о) с получением нуклеозида 74 (0,015 г, 67%). Данные спектрального анализа согласовывались с данными, ранее приведенными в примере, описывающем соединение 74.
Пример 87
1-(2-амино-2-дезокси-2-N,4-С-метилен-2-N-трифторацетил-β -D-рибофуранозил)тимин (74-СОСF3)
К суспензии нуклеозида 74 (0,050 г, 0,186 ммоль) в метаноле (2 мл) добавляли DMAP (0,013 мг, 0,106 ммоль) и этилтрифторацетат (0,029 мл, 0,242 ммоль) и полученную смесь перемешивали при комнатной температуре в течение 2,5 часов. Растворитель удаляли при пониженном давлении и остаток подвергали хроматографической очистке на силикагелевой колонке с использованием для элюции метанола/дихлорметана (2,5:97/5, о/о) с получением титульного нуклеозида 74-СОСF3 в виде твердого вещества белого цвета после выпаривания растворителей при пониженном давлении (0,055 г, 81%). FAB-MS m/z 366 [М+Н]+. 13С-ЯМР (CD3OD, 62,9 МГц) δ 166,5, 157,7 (q, 2JC,F 37,5 Гц, COCF3), 157,6 (q, 1JC,F 37,2 Гц, СОСF3), 151,8, 136,8, 136,8, 117,6 (d, 1JC,F 287,5 Гц, СF3), 117,5 (d, 2JC,F 286,5 Гц, СF3), 110,8, 110,8, 90,7, 89,3, 87,7, 87,3, 70,1, 68,6, 66,2, 66,2, 64,5, 57,9, 53,3, 12,7. Аналитический расчет для С13Н14N3О6F3: С – 42,8; Н - 3,9; N - 11,5. Получено: С - 42,5; Н - 4,0; N - 11,2.
Пример 88
1-(2-амино-2-дезокси-5-O-4,4’-диметокситритил-2-N,4-С-метилен-2-N-трифторацетил-β -D-рибофуранозил)тимин (74-DMT)
К раствору нуклеозида 74-СОСF3 (0,030 г, 0,082 ммоль) в безводном пиридине (0,6 мл) при 0° С по каплям (в течение 20 минут) добавляли 4,4’-диметоксиметилхлорид (0,054 г, 0,159 ммоль), растворенный в безводном пиридине/дихлорметане (0,6 мл, 1:1, о/о) и полученную смесь перемешивали в течение 10 часов при комнатной температуре. Добавляли смесь льда и воды (5 мл) и полученную в результате смесь экстрагировали дихлорметаном (3× 5 мл). Объединенную органическую фазу промывали насыщенным водным раствором кислого углекислого натрия (3× 2 мл), высушивали над Na2SO4 и отфильтровывали. Фильтрат упаривали досуха при пониженном давлении и остаток подвергали хроматографической очистке на силикагеле с использованием в качестве элюента метанола/дихлорметана/пиридина (1,5:98,0:0,5, о/о) с получением нуклеозида 74-DMT в виде твердого вещества белого цвета после выпаривания растворителей при пониженном давлении (0,051 г, 93%). FAB-MS m/z 667 [М]+, 668 [М+Н]+. FAB-HRMS, расчет для С34H32N3О8F
Пример 89
1-(2-амино-3-О-(2-цианоэтокси(диизопропиламино)фосфино-2-дезокси)-5-O-4,4’-диметокситритил-2-N,4-С-метилен-2-N-трифторацетил-β -D-рибофуранозил)тимин (74А)
К раствору нуклеозида 74-DMT (0,121 г, 0,181 ммоль) в безводном дихлорметане (2 мл) добавляли N,N-диизопропилэтиламин (0,093 мл, 0,54 ммоль) и 2-цианоэтил-N,N-диизопропилфосфорамидохлоридит (0,057 мл, 0,26 ммоль) при 0° С и полученную смесь перемешивали при комнатной температуре в течение 10 часов. Смесь разбавляли дихлорметаном (20 мл), экстрагировали насыщенным водным раствором кислого углекислого натрия (3× 10 мл), высушивали над N2SO4 и отфильтровывали. Фильтрат упаривали досуха при пониженном давлении и остаток подвергали хроматографической очистке на силикагеле с использованием в качестве элюента метанола/дихлорметана/пиридина (1,5:98,0:0,5, o/о/о) с получением сырого продукта (0,107 г) после выпаривания растворителей при пониженном давлении. Остаток растворяли в безводном дихлорметане (1 мл) и по каплям добавляли при интенсивном перемешивании в петролейный эфир (60-80° С, 30 мл) при -30° С, осаждали нуклеотид 74А с получением после фильтрации твердого вещества белого цвета (0,090 г, 57%). FAB-MS m/z 868 [М+Н]+, 890 [M+Na]+. 31Р-ЯМР (СD3СN, 121,5 МГц) δ 150,4, 150,2, 148,8, 149,1.
Пример 90
1-(2-амино-2-N,4-С-метилен-3,5-O-(тетраизопропилдисилоксан-1,3-диил)-β -D-рибофуранозил)тимин (74В)
К раствору нуклеозида 74 (0,20 г, 0,74 ммоль) в безводном пиридине (3 мл) при -15° С добавляли по каплям (в течение 3 часов) 1,3-дихлор-1,1,3,3-тетраизопропилдисилоксан (0,305 мл, 0,0011 моль) и полученную смесь перемешивали при комнатной температуре в течение 10 часов. Добавляли МеОН (3 мл) и смесь упаривали досуха при пониженном давлении. Остаток подвергали хроматографической очистке на силикагеле с использованием в качестве элюента метанола/дихлорметана (1:99, о/о) с получением нуклеозида 74В в виде твердого вещества белого цвета после выпаривания растворителей при пониженном давлении (0,370 мг, 97%). FAB-MS m/z 512 [М+Н]+. 13H-ЯМР ((СD3)2SO, 400 МГц) δ 11,37 (шс, 1Н), 7,48 (с, 1Н), 5,32 (с, 1Н), 4,06 (д, 1Н, J 13,5 Гц), 4,00 (с, 1Н), 3,84 (д, 1Н, J 13,5 Гц), 3,41 (с, 1Н), 2,92 (д, 1Н, J 10,2 Гц), 2,64 (д, 1Н, J 10,2 Гц), 1,74 (с, 3Н), 1,10-0,92 (м, 28Н). 13С-ЯМР ((СD)3SO2, 62,9 МГц) 6 163,8, 149,8, 134,1, 107,9, 89,5, 87,9, 70,1, 61,1, 57,9, 49,3, 17,2, 17,2, 17,0, 16,9, 16,8, 16,7, 12,6, 12,2, 11,7. Аналитический расчет для С23Н41N3О6Si2: С - 54,0; Н - 8,1; N - 8,2. Получено: С - 54,0; Н - 8,3; N - 7,8.
Пример 91
1-(2-дезокси-2-метиламино-2-N,4-С-метилен-3,5-O-(тетра-изопропилдисилоксан-1,3-диил)-β -D-рибофуранозил)тимин (74С)
К раствору нуклеозида 74В (0,33 г, 0,64 ммоль) в безводном THF/дихлорметане (4:1, о/о) при -10° С по каплям (в течение 30 минут) добавляли 1,8-диазабицикло[5.4.0]ундецен-7 (DBU, 0,125 мл, 0,836 ммоль) и метилиодид (0,05 мл, 0,79 ммоль) и полученную смесь перемешивали в течение 48 часов при 10° С. Также по каплям (в течение 15 минут) в реакционную смесь дополнительно добавляли DBU (0,05 мл, 0,33 ммоль) и метилиодид (0,02 мл, 0,32 ммоль) и перемешивали в течение 24 часов при 10° С. Смесь упаривали досуха при пониженном давлении и остаток подвергали хроматографической очистке в силикагеле с использованием в качестве элюента метанола/дихлорметана (1:99, о/о) с получением нуклеозида 74С в виде твердого вещества белого цвета после выпаривания растворителей (0,25 г, 74%). FAB-MS m/z 526 [М+Н]+. 1H-ЯМР (СDСl3, 400 МГц) δ 8,19 (шс, 1H), 7,65 (д, 1Н, J 1,3 Гц), 5,59 (с, 1H), 4,11 (с, 1Н), 4,05 (д, 1Н, J 13,2 Гц), 3,87 (д, 1H, J 13,2 Гц), 3,44 (с, 1H), 2,98 (д, 1H, J 9,5 Гц), 2,71 (д, 1H, J 9,5 Гц), 2,72 (с, 3Н), 1,91 (д, 1H, J 1,1 Гц), 1,12-0,96 (м, 28Н), 13С-ЯМР (CDCl3, 62,9 МГц) δ 163,7, 149,6, 135,2, 109,7, 90,9, 85,7, 71,4, 67,3, 58,6, 58,2, 41,2, 17,5, 17,4, 17,3, 17,2, 17,1, 16,9, 13,3, 13,1, 13,0, 12,6, 12,1. Аналитический расчет для C24H44N3O6Si2·0,25H2O: С - 54,4; Н - 8,3; N - 7,9. Получено: С – 54,4; Н - 8,1; N – 7,7.
Пример 92
1-(2-дезокси-2-метиламино-2-N,4-С-метилен-β -D-рибофуранозил)тимин (74D)
К раствору нуклеозида 74С (0,40 г, 0,76 ммоль) в THF при комнатной температуре добавляли раствор фторида тетрабутиламмония в THF (1,0 М, 2,2 мл, 2,2 ммоль) и полученную реакционную смесь перемешивали в течение 20 минут, после чего добавляли смесь пиридина, воды и метанола (6 мл, 3:1:1, о/о/о). Реакционную смесь добавляли к смоле Dowex 50× 200 (2,2 г, Н+ (пиридиниум) форма, 100-200 меш), суспендированной в пиридине/воде/метаноле (6 мл, 3:1:1, о/о) и полученную в результате смесь перемешивали в течение 20 минут. После фильтрации остаток промывали в пиридине/воде/метаноле (3× 3 мл, 3:1:1, о/о) и объединенный фильтрат упаривали досуха при пониженном давлении с получением маслянистого остатка после совместного выпаривания с метанолом (2× 5 мл). В результате хроматографической очистки на силикагелевой колонке с использованием в качестве элюента метанола/дихлорметана (1:49, о/о) получили нуклеозид 74D в виде твердого вещества белого цвета после выпаривания растворителей при пониженном давлении (0,17 г, 79%). FAB-MS m/z 284 [M+H]+. FAB-HRMS, расчет для С12Н18N3О
Пример 93
1-(2-дезокси-5-O-4,4’-диметокситритил-2-метиламино-2-N,4-С-метилен-β -D-рибофуранозил)тимин (74Е)
К раствору нуклеозида 74D (0,135 г, 0,477 ммоль) в безводном пиридине (1,5 мл) при 0° С по каплям (в течение 20 минут) добавляли раствор 4,4’-диметокситритилхлорида (0,238 г, 0,702 ммоль) в безводном пиридине/дихлорметане (1,0 мл, 1:1, о/о) и полученную в результате реакционную смесь перемешивали в течение 10 часов при комнатной температуре. Добавляли смесь воды и льда (5 мл) и полученную смесь экстрагировали дихлорметаном (3× 10 мл). Объединенную органическую фазу промывали насыщенным водным раствором бикарбоната натрия (3× 5 мл), высушивали над N2SO4 и отфильтровывали. Фильтрат упаривали досуха и остаток подвергали хроматографической очистке на силикагелевой колонке с использованием в качестве элюента метанола/дихлорметана/пиридина (1:98:1, о/о/о) с получением нуклеозида 74Е в виде твердого вещества белого цвета после выпаривания растворителей при пониженном давлении (0,20 г, 72%). FAB-MS m/z 586 [М+Н]+. 1H-ЯМР (C5D5N, 400 МГц) δ 13,2 (шс, 1Н), 7,98 (д, 1H, J 1,3 Гц), 7,98-7,00 (м, 13Н), 6,12 (с, 1H), 4,78 (д, 1Н, J 3,7 Гц), 3,88-3,79 (м, 4Н), 3,71 (с, 3Н), 3,71 (с, 3Н), 3,29 (д, 1H, J 9,3 Гц), 2,84 (д, 1H, J 9,3 Гц), 2,81 (с, 3Н), 1,85 (д, 3Н, J 0,9 Гц). 13С-ЯМР (C5D5N, 62,9 МГц) δ 165,1, 159,2, 151,4, 145,9, 136,5, 136,4, 130,8, 130,7, 128,7, 128,4, 127,4, 113,8, 109,6, 89,8, 86,8, 85,1, 72,0, 68,7, 60,9, 59,4, 55,2, 40,1, 13,1. Аналитический расчет для C33H35N3O7·0,25H2O: С - 67,2; Н - 6,1; N - 7,1. Получено: С - 67,2; Н - 6,2; N - 6,9.
Пример 94
1-(3-O-(2-цианоэтокси(диизопропиламино)фосфино)-5-O-4,4’-диметокситритил-2-метиламино-2-N,4-С-метилен-2-дезокси-β -D-рибофуранозил)тимин (74F)
К раствору нуклеозида 74Е (0,130 г, 0,222 ммоль) в безводном дихлорметане (2 мл) при 0° С добавляли N,N-диизопропилэтиламин (0,088 мл, 0,514 ммоль) и 2-цианоэтил-N,N-диизопропилфосфорамидохлоридит (0,065 мл, 0,291 ммоль) и полученную смесь перемешивали в течение 10 часов при комнатной температуре. Добавляли дихлорметан (30 мл) и реакционную смесь экстрагировали насыщенным водным раствором кислого углекислого натрия (3× 10 мл), высушивали над Na2SO4 и отфильтровывали. Фильтрат упаривали досуха и остаток подвергали хроматографической очистке на силикагелевой колонке с использованием в качестве элюента метанола/дихлорметана/пиридина (0,5:98,5:1,0, o/о/о) с получением сырого продукта (0,120 г) после выпаривания растворителей при пониженном давлении. Остаток растворяли в безводном дихлорметане (1 мл) и, при мощном перемешивании по каплям добавляя петролейный эфир (60-80° С, 30 мл) при -30° С, преципитировали нуклеотид 74F с получением после фильтрации твердого вещества белого цвета (0,090 г, 52%). 31Р-ЯМР (CD3CN, 121,5 МГц) δ 147,7.
Пример 95
1-(3,5-ди-O-бензил-4-С-(р-толуолсульфонилоксиметил)-2-O-р-толуолсульфонил-β -D-рибофуранозил)урацил (75)
К перемешиваемому раствору 1-(3,5-ди-O-бензил-4-С-гидроксиметил-β -D-рибофуранозил)урацила 41 (3,55 г, 7,81 ммоль) в дихлорметане (50 см3) добавляли DMAP (3,82 г) и р-толуолсульфонилхлорид (4,47 г, 23,5 ммоль) при комнатной температуре. Смесь перемешивали в течение 2 часов и добавляли дихлорметан (100 см3). Реакционную смесь промывали в насыщенном водном растворе кислого углекислого натрия (2× 75 см3) и высушивали над Na2SO4. Органическую фазу выпаривали при пониженном давлении и подвергали хроматографической очистке на силикагелевой колонке с использованием в качестве элюента дихлорметана/метанола (99,5:0,5, о/о) с получением нуклеозида 75 (4,65 г, 78%) в виде твердого вещества белого цвета. δ H (CDCl3) 8,49 (1Н, шс, NH), 7,67 (1Н, д, J 8,3, 6-Н), 7,51-7,03 (18Н, м, Bn, Ts), 6,0 (1Н, д, J 7,6, 1’-Н), 5,05 (1Н, м, 2’-Н), 4,91 (2Н, м, 5-Н, Bn), 4,56 (2Н, м, Bn), 4,42 (1Н, д, J 10,4, Bn), 4,31 (1Н, д, J 4,9, 3’-Н), 4,05 (2Н, м, 1’’-Н), 3,75-3,64 (2Н, м, 5’’-Н), 2,41 (3H, с, СН3), 2,34 (3H, с, СН3). δ С (CDCl3) 162,2 (С-4), 149,5 (С-2), 146,0, 145,3 (Ts), 139,0 (С-6), 136,7, 131,9, 130,0, 129,9, 128,9, 128,7, 128,5, 128,4, 128,3, 128,2, 128,0, 127,6 (Bn, Ts) 102,7 (С-5), 85,5 (1’-C), 84,4 (4’-C), 79,2, 78,3, 75,1, 74,3, 72,4, 69,1 (Bn, 3’-C, 2’-C, 5’-C, 1’’-C), 21,7, 21,6 (Ts). FAB-MS m/z 763. Получено: С - 61,2; Н - 4,4; N - 3,3. Для С38Н38N2O11S2 рассчитано: С - 59,8; Н - 5,0; N - 3,6.
Пример 96
1-(2-дезокси-3,5-ди-O-бензил-2S,4-С-метилен-2-меркапто-β -D-рибофуранозил)тимин (76)
К перемешиваемому раствору нуклеозида 75 (3,70 г, 4,86 ммоль) в DMF (40 см3) добавляли тиоацетат калия (0,83 г, 7,28 ммоль). Полученную смесь перемешивали и нагревали до 110° С в течение 80 часов. После выпаривания при пониженном давлении добавляли воду (100 см3). Проводили экстракцию дихлорметаном (4× 50 см3) и объединенную органическую фазу высушивали над Nа2SO4, отфильтровывали и выпаривали при пониженном давлении. Остаток подвергали хроматографической очистке в силикагеле с использованием в качестве элюента дихлорметана/метанола (99,6:0,4, о/о) с получением нуклеозида 76 (1,65 г, 75%) в виде твердого вещества белого цвета. δ H (СDСl3) 9,08 (1Н, шс, NH), 7,98 (1Н, d, J 8,1, 6-Н), 7,39-7,20 (10Н, м, Вn), 5,85 (1Н, с, 1’-Н), 5,26 (1Н, д, J 8,1, 5-Н), 4,61 (1Н, д, J 11,4, 5’-H), 4,56 (2Н, с, Вn), 4,45 (1Н, д, J 11,4, Bn), 4,14 (1Н, д, J 1,7, 3’-Н), 3,82 (2Н, м, Bn), 3,72 (1Н, д, J 1,9, 2’-H), 3,02 (1Н, д, J 9,9, 1’’-Ha), 2,78 (1Н, д, J 9,9, 1’’-Нb). δ C (CDCl3) 163,4 (С-4), 150,0 (С-2), 139,9 (С-6), 137,2, 136,8, 128,6, 128,5, 128,2, 127,9, 127,7 (Bn), 100,8 (С-5), 90,8, 88,8 (C-1’, С-4’), 76,5, 73,8, 72,0, 70,0 (2 × Bn, С-3’, С-5’), 49,52 (C-2’), 35,63 (C-1’’). FAB-MS m/z 453. Получено: С - 63,4; Н - 5,1; N - 5,9. Для C24H24N2O2S рассчитано: С - 63,7; Н – 5,3; N - 6,1.
Пример 97
1-(2-O-р-толуолсульфонил-4-С-(р-толуолсульфонилоксиметил)-β -D-рибофуранозил)урацил (76А)
К раствору соединения 75 (0,80 г, 1,0 ммоль) в абсолютном этаноле (2 см3) добавляли 20% гидроксид палладия на углероде (0,80 г) и полученную смесь несколько раз дегазировали водородом и перемешивание продолжали в атмосфере водорода в течение 48 часов. Катализатор отфильтровывали и полученный фильтрат выпаривали при пониженном давлении. Остаток подвергали хроматографической очистке на силикагелевой колонке с использованием в качестве элюента дихлорметана/метанола (99:1, о/о) с получением нуклеозида 76А (0,30 г, 49%) в виде твердого вещества белого цвета. δ H (CD3OD) 7,67 (4Н, м), 7,45 (1Н, д, J 8,2 Гц), 7,34 (4Н, м), 5,86 (1Н, д, J 8,0 Гц), 5,40 (1Н, д, J 8,1 Гц), 4,95 (1Н, м), 4,35 (1Н, д, J 5,0 Гц), 4,17 (2Н, м), 3,61 (2Н, с), 2,40 (6Н, с). δ C (CD3OD) 165,4, 151,6, 147,5, 146,6, 141,3, 134,0, 133,8, 131,4, 130,9, 129,2, 128,9, 103,7, 88,0, 85,4, 80,7, 72,4, 71,0, 64,3, 21,7, 21,6. FAB-MS m/z 583 [М+Н]+.
Пример 98
1-(3,5-O-(тетраизопропилдисилокса-1,3-диил)-2-O-р-толуолсульфонил-4-С-(р-толуолсульфонилоксиметил)-β -D-рибофуранозил)урацил (76В)
К перемешиваемому раствору нуклеозида 76А (0,27 г, 0,46 ммоль) в безводном пиридине (4 см3) добавляли 1,3-дихлор-1,1,3,3-тетраизопропилдисилоксан (0,22 см3, 0,70 ммоль). После 48-часового перемешивания реакционную смесь охлаждали до 0° С и добавляли насыщенный водный раствор кислого углекислого натрия (15 см3). Смесь экстрагировали дихлорметаном (3× 10 см3) и объединенную органическую фазу высушивали над N2SO4 и отфильтровывали. Растворители выпаривали при пониженном давлении и остаток подвергали хроматографической очистке в силикагеле с использованием в качестве элюента дихлорметана/метанола (99,5:0,5, о/о) с получением нуклеозида 76В (0,37 г, 97%) в виде твердого вещества белого цвета. δ H (СDСl3) 8,70 (1Н, шс), 7,80 (4Н, м), 7,36 (4Н, м), 6,98 (1Н, д, J 8,1 Гц), 5,64 (1Н, д, J 8,0 Гц), 5,18 (2Н, м), 4,98 (1Н, д, J 7,0 Гц), 4,39-4,32 (2Н, м), 3,92-3,76 (2Н, с), 2,45 (6Н, с), 1,27-0,66 (28Н, м). δ C (СDСl3) 162,9, 149,3, 145,6, 144,8, 143,9, 132,9, 130,1, 129,9, 128,2, 128,1, 102,2, 94,6, 84,7, 80,4, 72,8, 67,8, 64,6, 21,7, 17,3, 17,2, 17,1, 16,9, 16,8, 13,1, 12,8, 12,3. FAB-MS m/z 825 [М+Н]+.
Пример 99
1-(2-дезокси-2-меркапто-2-S,4-С-метилен-3,5-O-(тетраизопропилдисилокса-1,3-диил)-β -D-рибофуранозил)урацил (76С)
К перемешиваемому раствору нуклеозида 76В (0,26 г, 0,32 ммоль) в DMF (5 см3) добавляли тиоацетат калия (0,054 г, 0,47 ммоль). Полученную реакционную смесь перемешивали при 110° С в течение 20 часов. После выпаривания реакционной смеси при пониженном давлении добавляли воду (20 см3). Проводили экстракцию дихлорметаном (3× 10 см3) и объединенную органическую фазу высушивали над Na2SO4, отфильтровывали и выпаривали при пониженном давлении. Остаток подвергали хроматографической очистке в силикагеле с использованием в качестве элюента дихлорметана/метанола (99,25:0,75, о/о) с получением нуклеозида 76С (0,125 г, 77%) в виде твердого вещества белого цвета. δ H (СDСl3) 8,55 (1Н, шс), 8,02 (1Н, д, J 8,1 Гц), 5,82 (1Н, с, 1’-Н), 5,65 (1Н, д, J 8,1 Гц), 4,37 (1Н, д, J 2,1 Гц), 4,10 (1Н, д, J 13,2 Гц), 3,90 (1H, д, J 13,1 Гц), 3,53 (1Н, д, J 2,1 Гц), 2,92 (1Н, д, J 10,1 Гц), 2,74 (1Н, д, J 10,0 Гц), 1,30-0,80 (28Н, м). δ C (CDCl3) 163,2, 149,8, 139,6, 100,9, 91,4, 90,7, 71,5, 59,8, 51,5, 34,4, 17,5, 17,3, 17,1, 16,9, 15,5, 13,6, 13,3, 13,1, 12,9, 12,3. FAB-MS m/z 515 [М+Н]+.
Пример 100
1-(2-дезокси-2-меркапто-2-S,4-С-метилен-β -D-рибофуранозил)урацил (76D)
К перемешиваемому раствору нуклеозида 76С (25 мг, 0,049 ммоль) в THF (1,0 см3) добавляли раствор фторида тетрабутиламмония (0,20 см3 в 1 М растворе THF, 0,20 ммоль) при 0° С. После перемешивания реакционной смеси при 0° С в течение 1 часа добавляли воду (5 см3) и смесь выпаривали. Остаток подвергали хроматографической очистке в силикагеле с использованием в качестве элюента дихлорметана/метанола (97:3, о/о) с получением нуклеозида 76D (9,0 мг, 69%) в виде твердого вещества белого цвета. δ H (CD3OD) 8,19 (1Н, д, J 8,1 Гц, 6-Н), 5,77 (1H, с, 1’-Н), 5,65 (1H, д, J 8,1 Гц, 5-Н), 4,31 (1H, д, J 2,1 Гц, 3’-Н), 3,86 (2Н, с, 5’-Н), 3,53 (1H, д, J 2,2 Гц, 2’-Н), 2,93 (1H, д, J 10,3 Гц, 1’’-На), 2,73 (1H, д, J 10,3 Гц, 1’’-Нb). δ C (CD3OD) 166,5, 152,0, 141,7, 101,2, 92,1, 92,0, 71,4, 59,9, 53,6, 35,4. FAB-MS m/z 273 [М+Н]+.
Пример 101
1-(2-дезокси-5-O-(4,4’-диметокситритил)-2-меркапто-2-S,4-С-метилен-β -D-рибофуранозил)урацил (76Е)
К раствору соединения 76D (0,2 г, 0,37 ммоль) в безводном пиридине (5 см3) добавляли 4,4’-диметокситритилхлорид (0,186 г, 0,55 ммоль) при комнатной температуре. Полученный раствор перемешивали в течение 5 часов, после чего реакционную смесь охлаждали до 0° С. Добавляли насыщенный водный раствор кислого углекислого натрия (30 см3) и полученную смесь экстрагировали дихлорметаном (3× 50 см3). Объединенную органическую фазу отделяли и высушивали над Nа2SO4. Растворители удаляли при пониженном давлении и остаток подвергали хроматографической очистке на силикагелевой колонке с использованием в качестве элюента дихлорметана/метанола/пиридина (98,5:1,0:0,5, o/o/o) с получением нуклеозида 76Е в виде твердого вещества буровато-белого цвета (0,175 г, 83%). δ C (СDСl3) 164,5, 159,4, 151,6, 145,7, 139,9, 136,4, 136,0, 135,6, 130,9, 130,8, 128,8, 128,5, 128,4, 127,5, 127,4, 122,7, 113,9, 101,5, 91,7, 90,2, 87,6, 71,8, 61,9, 55,3, 53,7, 36,2, 30,6. FAB-MS m/z 574 [М]+, 575 [М+Н]+. Получено: С - 65,2; Н - 5,4; N - 5,0. Для С31Н30N2О7S рассчитано: С - 64,8; Н - 5,3; N - 4,9%.
Пример 102
1-(3-O-(2-цианоэтокси(диизопропиламино)фосфино)-(2-дезокси-5-O-(4,4’-диметокситритил)-2-меркапто-2-S,4-С-метилен-β -D-рибофуранозил)урацил (76F)
К раствору соединения 76Е (0,160 г, 0,28 ммоль) в безводном дихлорметане (2 см3) при 0° С добавляли N,N-диизопропилэтиламин (0,27 см3) и 2-цианоэтил-N,N-диизопропилфосфорамидохлоридит (97 мг, 0,42 ммоль). Перемешивание продолжали при комнатной температуре в течение 5 часов. Реакционную смесь охлаждали до 0° С и добавляли насыщенный водный раствор кислого углекислого натрия (30 см3). Проводили экстракцию дихлорметаном (3× 20 см3) и объединенную органическую фазу высушивали над Na2SO4 и упаривали досуха при пониженном давлении. Остаток подвергали хроматографической очистке на силикагелевой колонке с использованием в качестве элюента дихлорметана/метанола/пиридина (99:0,5:0,5, о/о) с получением пены белого цвета. Этот остаток растворяли в дихлорметане (2 см3) и полученный продукт осаждали из легких фракций перегонки нефти (100 см3, охлажденные до -40° С) при мощном перемешивании. Преципитат собирали фильтрованием и окончательно высушивали с получением нуклеозида 76F в виде твердого вещества белого цвета (95 мг, 44%). δ P (СDСl3) 148,9, 149,0.
Пример 103
3,5-ди-О-бензил-1,2-O-изопропилиден-4-С-(р-толуолсульфонилоксиметил)-β -D-рибофураноза (77)
Раствор 3,5-ди-O-бензил-4-С-гидроксиметил-1,2-O-изопропилиден-α -D-рибофуранозы 31 (15,38 г, 38,4 ммоль), безводного пиридина (20 см3) и безводного дихлорметана (80 мл) перемешивали при -5° С. р-Толуолсульфонилхлорид (8,75 г, 46,0 ммоль), растворенный в безводном дихлорметане (8 см3), добавляли в течение 15 минут. Полученный раствор перемешивали при комнатной температуре в течение 17 часов. Реакцию останавливали охлажденной льдом водой (200 см3). Проводили экстракцию дихлорметаном (5× 150 см3) и объединенную органическую фазу промывали насыщенным водным раствором кислого углекислого натрия (3× 100 см3) и соляным раствором (3× 100 см3), высушивали над Nа2SO4, отфильтровывали и выпаривали при пониженном давлении. Остаток подвергали хроматографической очистке на силикагелевой колонке с использованием в качестве элюента дихлорметана/метанола (98,5:1,5, о/о) с получением соединения 77 в виде прозрачного маслянистого вещества (17,4 г, 82%). δ H (CDCl3) 7,79-7,19 (14Н, м, Bn), 5,66 (1Н, д, J 3,6, 1-Н), 4,69-4,20 (8Н, м, Bn, 5-На, 5-Нb, 3-Н, 2-Н), 3,53 (1Н, д, J 10,3, 1’-На), 3,46 (1Н, д, J 10,3, 1’-Нb), 2,40 (3H, с, СН3), 1,29 (3H, с, СН3), 1,26 (3H, с, СН3). δ C (CDCl3) 144,6, 137,9, 137,3, 133,0, 129,8, 128,4, 128,3, 128,1, 128,0, 127,9, 127,7, 127,6 (ароматический), 113,6 (С(СН3)2), 104,2, (С-1), 84,7 (С-4), 79,0, 78,7, 73,7, 72,7, 70,7, 70,2 (Bn, С-2, C-3, C-5, C-1’), 26,3, 26,0 (С(СН3)2), 21,6 (СН3). FAB-MS m/z 555 [М+Н]+. Получено: С - 64,8; Н - 6,2. Для С30Н34O8S рассчитано: С - 64,9; Н - 6,1%.
Пример 104
1,2-ди-О-ацетил-3,5-ди-O-бензил-4-С-(р-толуолсульфонилоксиметил)-α ,β -D-рибофураноза (78)
Раствор фуранозы 77 (17,4 г, 31,4 ммоль) в 80%-ной уксусной кислоте (250 см3) перемешивали при 60° С в течение 20 часов. Растворитель удаляли в вакууме и остаток выпаривали вместе с толуолом (3× 20 см3). Остаток вновь растворяли в безводном пиридине (100 см3). Добавляли уксусный ангидрид и полученный раствор перемешивали при комнатной температуре в течение 15 часов. Реакцию останавливали добавлением смеси льда и воды (200 см3) и реакционную смесь экстрагировали дихлорметаном (4× 150 см3). Объединенную органическую фазу промывали насыщенным водным раствором кислого углекислого натрия (2× 125 см3) и соляным раствором (3× 150 см3), высушивали над Na2SO4, отфильтровывали и выпаривали при пониженном давлении. Остаток подвергали хроматографической очистке на силикагелевой колонке с использованием в качестве элюента дихлорметана/метанола (98,5:1,5, о/о) с получением соединения 78 (соотношение α :β ≈ 1:1 в виде прозрачного маслянистого вещества (13,5 г, 72%). δ C (СDСl3) 169,8, 169,6, 69,4, 168,8 (С=O), 144,7, 137,7, 137,5, 132,8, 129,7, 129,6, 128,5, 128,4, 128,3, 128,2, 128,0, 127,8, 127,7, 127,6 (Bn), 97,4, 94,2 (С-1), 86,4, 84,2 (С-4), 78,9, 77,5, 74,5, 74,1, 73,7, 73,5, 71,8, 70,6, 70,5, 69,6, 69,5 (Bn, C-2, C-3, C-1’), 21,6, 21,0, 20,8, 20,6, 20,4 (СОСН3, С(СН3)2). FAB-MS m/z 599 [М+Н]+.
Альтернативная процедура получения соединения 78
3-О-бензил-1,2:5,6-ди-О-изопропилиден-α -О-глюкофураноза (30В)
К раствору 1,2:5,6-ди-О-изопропилиден-α -D-аллофуранозы (30А) (полученной от Pfanstiehl Lab. Inc.) (40 г) в диметилформамиде при 0° С добавляли маленькими порциями гидрид натрия. Реакционную смесь перемешивали в течение 1 часа и по каплям добавляли бензилбромид в течение 1 часа. Полученную реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Для остановки реакции добавляли метанол и диметилформамид удаляли под давлением. Полученный сироп экстрагировали этилацетатом и промывали соляным раствором. После выпаривания этилацетата получали полутвердое вещество (93%). Гомогенизировали с помощью ТСХ.
3-О-бензил-1,2-O-изопропилиден-α -D-глюкофураноза (30С)
Частичный гидролиз соединения 30В (50 г) проводили в 75%-ной уксусной кислоте в течение 20 часов. В результате концентрирования до меньшего объема и экстрагирования этилацетатом получили соединение 30С (40 г, 90%). Гомогенизировали с помощью ТСХ.
3-О-бензил-1,2-O-изопропилиден-α -D-рибопентодиальдофураноза (30D)
Раствор соединения 30С (40 г) в воде/метаноле (1:1) медленно добавляли при перемешивании к раствору периодата натрия в воде при 0° С. Реакционную смесь перемешивали в течение 2 часов, добавляли этиленгликоль и смесь экстрагировали этилацетатом. Высушенный экстракт выпаривали с получением соединения 30D (32 г, 89%). Гомогенизировали с помощью ТСХ. На этом этапе важным является добавление метанола, что обеспечивает завершение реакции.
3-О-бензил-4-(гидроксиметил)-1,2-O-изопропилиден-α -D-эритропентофураноза (30Е)
Водный 37%-ный раствор формальдегида и 1N раствор едкого натра добавляли при 0° С к перемешиваемому раствору соединения 30D (32 г) в воде и тетрагидрофуране (1:1), реакцию продолжали в течение 16 часов, экстрагировали этилацетатом и промывали соляным раствором. После выпаривания органическая фаза образовывала сироп, который из смеси эфира и петролейного эфира кристаллизовался в виде твердого вещества белого цвета (23 г), в то время как фильтрат представлял собой маслянистое вещество, кристаллизовавшееся в низкоплавкое твердое вещество (10 г), при общем выходе соединения 30Е 92%: 23 г (твердое вещество белого цвета было до 99% очищено с помощью тонкослойной хроматографии), 10 г низкоплавкого твердого вещества (включает более подвижные в ТСХ примеси, приблизительная чистота 75%). На этом этапе очень важным с точки зрения завершения реакции является добавление тетрагидрофурана.
3,5-ди-O-бензил-4-С-гидроксиметил-1,2-O-изопропилиден-α -D-рибофураноза (31)
В результате бензилирования соединения 30Е (20 г) с использованием гидрида натрия (60%) и бензилбромида при -10° С получали смесь двух изомеров. Применяя колончатую хроматографию получали основной изомер соединения 31 (14 г, 54%). Гомогенизировали с помощью ТСХ.
3,5-ди-О-бензил-1,2-O-изопропилиден-4-С-тозил-α -D-рибофураноза (77)
Раствор соединения 31 (12,5 г) в пиридине при 0° С обрабатывали р-толуолсульфонилхлоридом и реакционную смесь выдерживали при комнатной температуре в течение 14-16 часов. В результате удаления пиридина, экстракции метиленхлоридом и насыщенным раствором бикарбоната натрия получили 77 (14 г, 80%). Гомогенизировали с помощью ТСХ.
1,2-ди-О-ацетил-3,5-O-бензил-4-С-тозил-D-рибофураноза (78)
Проводили гидролиз соединения 77 (14 г) 75%-ной уксусной кислотой при 65° С в течение 18 часов. Растворитель удаляли под давлением и остаток обрабатывали этанолом (3× 100 мл), толуолом (3× 50 мл) и безводным пиридином (2× 50 мл). (Это соединение 78 кристаллизовали из петролейного эфира в виде твердого вещества белого цвета). Остаток отбирали в сухой пиридин и обрабатывали уксусным ангидридом при комнатной температуре в течение 8 часов. В результате экстракции этилацетатом и насыщенным бикарбонатом натрия с последующей промывкой в соляном растворе получили соединение 78 в виде меси аномеров α и β (12 г, 83%). Прямое сравнение репрезентативных образцов соединения 78 (данные ТСХ, ВЭЖХ, ЯМР) подтвердили его идентичность и чистоту.
Пример 105
1-(2-O-ацетил-35-ди-O-бензил-4-С-(р-толуолсульфонилоксиметил)-β -D-рибофуранозил)тимин (79)
К перемешиваемому раствору аномерной смеси 78 (12,8 г, 21,4 ммоль) и тимина (5,38 г, 42,7 ммоль) в безводном ацетонитриле (182 см3) добавляли N,O-бис(триметилсилил)ацетамид (31,68 мл, 128,23 ммоль). Полученную реакционную смесь перемешивали в течение 1 часа при комнатной температуре и затем перемешивание продолжали при 60° С в течение 1,5 часов. После охлаждения до 0° С по каплям добавляли триметилсилилтрифлат (6,57 мл, 30,33 ммоль) и реакционную смесь перемешивали при 60° С в течение 10 часов. Реакционную смесь нейтрализовывали с использованием охлажденного на льду насыщенного водного раствора кислого углекислого натрия (90 мл). Реакционную смесь отфильтровывали и полученный фильтрат концентрировали при пониженном давлении до половины исходного объема. Проводили экстракцию дихлорметаном (4× 200 см3). Объединенную органическую фазу промывали насыщенным водным раствором кислого углекислого натрия (3× 150 см3) и соляным раствором (3× 150 мл), высушивали над Na2SO4, отфильтровывали и выпаривали при пониженном давлении. Остаток подвергали хроматографической очистке на силикагелевой колонке с использованием в качестве элюента дихлорметана/метанола (от 99:1 до 98:2, о/о) с получением нуклеозида 79 в виде твердого вещества белого цвета (13,1 г, 92%). δ H (СDСl3) 9,04 (с, 1Н, NH), 7,73-7,19 (15Н, м, 6-Н, ароматическое), 5,94 (1Н, д, J 5,5, 1’-Н), 5,37 (1Н, д, J 5,6, 2’-Н), 4,57-4,40 (5Н, м, 3’-Н, 5’-На, 5’-Нb, Bn), 4,14 (2Н, с, Bn), 3,75 (1Н, д, J 10,2, 1’’-На), 3,57 (1Н, д, J 10,2, 1’’-Нb), 2,41 (3H, с, СН3С6Н5), 2,02 (3H, с, СОСН3), 1,54 (3H, с, СН3). δ C (CDCl3) 169,8 (С=O), 163,5 (С-4), 150,2 (С-2), 145,0, 136,8, 135,6, 132,1, 129,7, 128,5, 128,0, 127,9, 127,8, 127,5 (ароматическое), 113,5 (С-5), 86,8, 85,3, 77,6, 74,6, 74,3, 73,6, 70,8, 68,8 (Bn, С-1’, С-3’, С-2’, С4’), 21,3 (СН3), 20,5 (СОСН3), 11,8 (СН3). FAB-MS m/z 665 [M+Н]+. Получено: С - 61,2; H - 5,3; N - 4,1; S - 4,7. Для С34Н36О10NS рассчитано: С - 61,4; Н - 5,4; N - 4,2; S - 4,8.
Пример 106
1-(3,5-ди-O-бензил-4-С-(р-толуолсульфонилоксиметил)-β -D-рибофуранозил)тимин (80)
Нуклеозид 79 (13,1 г, 19,7 ммоль) растворяли в растворе аммиака в метаноле (200 см3, приготовлен путем разбавления насыщенного раствора аммиака в метаноле равным объемом метанола) и перемешивали при комнатной температуре в течение 4 часов. После этого реакционную смесь выпаривали и остаток растворяли в дихлорметане (400 см3). Органическую фазу промывали соляным раствором (3× 150 см3), высушивали над Nа2SO4, отфильтровывали и выпаривали при пониженном давлении. Остаток подвергали хроматографической очистке на силикагелевой колонке с использованием в качестве элюента дихлорметана/метанола (99,5:0,5, о/о) с получением нуклеозида 80 в виде твердого вещества белого цвета (10,7 г, 87%). δ H (CDCl3) 9,66 (с, 1Н, NH), 7,71-7,21 (15Н, м, 6-Н, ароматическое), 5,72 (1Н, д, J 5,1, 1’-Н), 4,75, 4,55 (2Н, каждый д, J 11,5, Bn), 4,51 (2Н, с, Bn), 4,37 (1Н, т, J 5,4, 2’-Н), 4,30-4,12 (3H, м, 3’-Н, Bn), 3,76 (1Н, д, J 10,2, 1’’-На), 3,59 (1Н, д, J 10,2, 1’’-Нb), 2,39 (3H, с, СН3С6Н5), 1,48 (3H, с, СН3). δ C (CDCl3) 163,8 (С-4), 150,9 (С-2), 145,0, 137,0, 136,9, 135,9, 132,3, 129,8, 128,7, 128,6, 128,2, 128,1, 128,0, 127,6 (ароматическое), 111,0 (С-5), 89,6, 85,3, 78,4, 74,5, 73,8, 71,1, 69,7 (Bn, С-1’, С-3’, С-2’, С-4’, С-1’’), 21,6 (СН3), 12,0 (СН3). FAB-MS m/z 623 [М+Н]+. Получено: С - 61,5; Н - 5,2; N - 4,4; S - 5,2. Для С32Н34O9N2S рассчитано: С - 61,7; Н - 5,4; N - 4,5; S - 5,1.
Пример 107
(1S,3R,4R,7S)-7-бензилокси-1-бензоилоксиметил-3-(тимин-1-ил)-2,5-диоксабицикло[2.2.1]гептан (36)
К перемешиваемому раствору нуклеозида 80 (10,65 г, 17,1 ммоль) в безводном DMF (150 см3) 60%-ную суспензию гидрида натрия в минеральном масле (0,9 г, 22,2 ммоль) добавляли маленькими порциями при 0° С. Реакционную смесь перемешивали при 0° С в течение 15 часов, после чего дополнительно добавляли 60% гидрид натрия (0,205 г, 5,12 ммоль) и реакционную смесь перемешивали еще в течение 22 часов при 0° С. Добавляли метанол (20 см3) и реакционную смесь после этого концентрировали при пониженном давлении до половины исходного объема. Добавляли охлажденную льдом воду (300 см3) и проводили экстракцию дихлорметаном (5× 150 см3). Объединенную органическую фазу промывали насыщенным водным раствором кислого углекислого натрия (3× 40 см3) соляным раствором (3× 40 см3), высушивали над Na2SO4, отфильтровывали и выпаривали при пониженном давлении. Остаток подвергали хроматографической очистке на силикагелевой колонке с использованием в качестве элюента дихлорметана/метанола (99,5:0,5, о/о) с получением нуклеозида 36 в виде твердого вещества белого цвета (7,1 г, 92%). Данные спектрального анализа соответствуют данным, ранее полученным для соединения 36. Получено: С - 66,2; Н - 5,8; N - 6,1. Для C25H26N2O6 рассчитано: С - 66,6; Н - 5,8; N - 6,2.
Пример 108
3,5-ди-О-бензил-1,2-O-изопропилиден-4-С-метансульфонилоксиметил-α -D-рибофураноза (200)
К перемешиваемому раствору фуранозы 31 (2,16 г, 5,39 ммоль) в безводном пиридине (3 мл) при 0° С добавляли по каплям метансульфонилхлорид (0,61 мл, 16,0 ммоль). Реакционную смесь перемешивали в течение 20 минут при комнатной температуре, реакцию останавливали смесью льда и воды (300 мл) и экстрагировали дихлорметаном (2× 300 мл). Объединенные экстракты промывали насыщенным водным раствором кислого углекислого натрия (300 мл) и высушивали над МgSO4. Растворитель удаляли путем перегонки при пониженном давлении и остаток подвергали хроматографической очистке в силикагеле с использованием дихлорметана в качестве элюента с получением соединения 200 в виде маслянистого вещества (2,55 г, 99%). 1H-ЯМР (СDСl3): δ 7,37-7,24 (10Н, м, Bn), 5,78 (1Н, д, J 3,8 Гц, Н-1), 4,85 (1Н, д, J 11,7 Гц, Bn), 4,73 (1Н, д, J 11,9 Гц, Bn), 4,64 (1Н, дд, J 4,0, 5,3 Гц, Н-2), 4,54 (1Н, д, J 11,9 Гц, Н-5’), 4,52 (1Н, д, J 11,9 Гц, Bn), 4,46 (1H, д, J 11,9 Гц, Н-5’), 4,41 (1H, д, J 11,8 Гц, Bn), 3,60 (1H, д, J 10,4 Гц, Н-5), 3,50 (1H, д, J 10,5 Гц, Н-5), 3,06 (3H, с, SO2СН3), 1,68 (3H, с, СН3), 1,34 (3H, с, СН3). 13С-ЯМР (CDСl3): δ 137,79, 137,31, 128,54, 128,48, 128,16, 128,01, 127,87, 127,79 (Bn), 113,66 (С(СН3)2), 104,46 (С-1), 84,88 (С-4), 78,48, 78,41 (С-2, С-3), 73,65, 72,63, 70,78, 70,16 (Bn, С-5, С-5’), 37,84 (SO2СН3), 26,20 (СН3), 25,69 (СН3). MS FAB: 501 (M+Na, 100%). Получено: С - 60,37; Н - 6,29; S - 6,53. Для С24Н30О8S рассчитано: С - 60,24; Н - 6,32; S - 6,70%.
Пример 109
Метил-3,5-ди-O-бензил-4-С-метансульфонилоксиметил-α -D-рибофураноза (201)
Раствор фуранозы 200 (1,133 г, 2,37 ммоль) в метанольной соляной кислоте (20%, в/о, 31,7 мл) и воду (4,4 мл) перемешивали при комнатной температуре в течение 2 часов. После нейтрализовывания бикарбонатом натрия (насыщенный раствор) полученную смесь экстрагировали дихлорметаном (2× 150 мл). Объединенные экстракты промывали водой (150 мл) и высушивали над МgSO4. Растворитель удаляли путем перегонки при пониженном давлении и остаток подвергали хроматографической очистке в силикагеле с использованием в качестве элюента дихлорметана/метанола (99:1) с получением соединения 201 (в смеси аномеров α :β ≈ 1:2) в виде прозрачного маслянистого вещества (1,018 г, 95%). 1H-ЯМP (CDCl3): δ 7,39-7,22 (м, Bn), 4,86 (шс, Bn), 4,69-3,99 (м, Bn, Н-5’, Н-1, Н-2, Н-3), 3,68 (д, J 8,9 Гц, Н-5 β ), 3,51 (д, J 9,8 Гц, H-5 α ), 3,46 (с, ОСН3α), 3,34 (д, J 9,1 Гц, Н-5 β ), 3,32 (д, J 9,7 Гц, Н-5 α ), 3,28 (с, ОСН3β), 2,97 (3H, с, SO2СН3 β ), 2,93 (3H, с, SO2СН3 α ). 13С-ЯМР (CDCl3): δ 137,74, 136,98, 128,70, 128,64, 128,58, 128,56, 128,37, 128,21, 128,15, 128,09, 127,98, 127,86, 127,83 (Bn), 107,54 (С-1 β ), 103,39 (C-1 α ), 84,65, 83,18, 81,90, 78,87 (С-4, С-3), 75,04, 74,07, 73,73, 73,70, 73,38, 72,56, 72,11, 70,85, 70,55, 70,20 (C-2, Bn, C-5, C-5’), 55,90 (ОСН3 α ), 54,96 (ОСН3 β ), 37,18 (SО2СН3 β ), 37,07 (SО2СН3 α ). MS FAB: 475 (M+Na, 25%). Получено: С - 58,40; Н – 6,33. Для С24Н30О8S рассчитано: С - 58,39; Н - 6,24%.
Пример 110
(3R)- и (3S)-(1S,4R,7S)-7-бензилокси-1-бензилоксиметил-3-метокси-2,5-диоксабицикло[2.2.1]гептан (202 и 203)
Раствор соединения 201 (3,32 г, 7,34 ммоль) в безводном DMF (25 мл) перемешивали при 0° С и добавляли 60%-ную суспензию гидрида натрия в минеральном масле (700 мг, 16,9 ммоль). Полученную реакционную смесь перемешивали при комнатной температуре в течение 90 минут, реакцию останавливали водой (300 мл) и экстрагировали диэтиловым эфиром (2× 300 мл). Объединенные экстракты промывали водой (200 мл) высушивали над MgSO4. Растворитель удаляли путем перегонки при пониженном давлении и остаток подвергали хроматографической очистке в силикагеле с использованием дихлорметана в качестве элюента с получением соединений 202 и 203 в виде прозрачных маслянистых веществ (1,571 г, 60% и 0,111 г, 30%, соответственно. (1S,3R,4R,7S)-7-бензилокси-1-бензилоксиметил-3-метокси-2,5-диоксабицикло[2.2.1]гептан (202). 1H-ЯМР (CDCl3): δ 7,36-7,26 (10Н, м, Bn), 4,81 (1Н, с, Н-1), 4,65 (1Н, д, J 11,9 Гц, Bn), 4,61 (2Н, с, Bn), 4,56 (1Н, д, J 11,9 Гц, Bn), 4,11 (1H, с, Н-2), 4,09 (1H, с, Н-3), 4,01 (1H, д, J 7,5 Гц, H-5’), 3,80-3,77 (3H, м, Н-5’, Н-5), 3,39 (3H, с, ОСН3). 13С-ЯМР (CDCl3): δ 138,05, 137,36, 128,47, 128,44, 127,88, 127,73, 127,63 (Bn), 104,97 (С-1), 85,13 (С-4), 79,16 (С-3), 77,18 (С-2), 73,64 (Bn), 72,26, 72,10 (Bn, С-5’), 66,50 (С-5), 55,34 (ОСН3). MS FAB: 379 (M+Na, 28%). Получено: С - 70,55; Н - 6,97. Для C21H24O5 рассчитано: С - 70,77; Н - 6,79%. (1S,3S,4R,7S)-7-бензилокси-1-бензилоксиметил-3-метокси-2,5-диоксабицикло[2.2.1]гептан (203). 1H-ЯМР (СDСl3): δ 7,36-7,26 (10Н, м, Bn), 5,00 (1H, с, Н-1), 4,67-4,54 (4Н, м, Bn), 4,18 (1H, с, Н-2), 3,99 (1H, с, Н-3), 3,99-3,90 (2Н, м, Н-5’), 3,75-3,68 (2Н, м, Н-5), 3,49 (3H, с, ОСН3). 13С-ЯМР (CDCl3): δ 137,83, 137,53, 128,51, 128,48, 127,96, 127,82, 127,71, 127,62 (Bn), 104,05 (С-1), 88,44 (С-4), 79,54 (С-3), 77,16 (С-2), 73,68 (Bn), 72,61 (С-5’), 72,24 (Bn), 65,73 (С-5), 56,20 (ОСН3). MS FAB: 379 (M+Na, 100%).
Пример 111
(1R,2S,3S)-2-бензилокси-3-бензилоксиметил-1-(метокси(тимин-1-ил)метил)-3-триметилсилилокситетрагидрофуран (204)
Раствор соединения 202 (216 мг, 0,606 ммоль) и тимина (153 мг, 1,22 ммоль) в безводном ацетонитриле (9,3 мл) добавляли (N,O-бис(триметилсилил)ацетамид (BSA, 0,90 мл, 3,6 ммоль) и перемешивали, нагревая с обратным холодильником в течение 15 минут. Раствор охлаждали до 0° С и по каплям добавляли триметилсилилтрифлат (0,153 мл, 0,777 ммоль). После перемешивания при комнатной температуре в течение 18 часов и при 60° С еще в течение 24 часов, реакцию останавливали насыщенным водным раствором кислого углекислого натрия (20 мл) и проводили экстракцию дихлорметана (2× 50 мл). Объединенные экстракты промывали насыщенным водным раствором кислого углекислого натрия (50 мл) и высушивали над MgSO4. Растворитель удаляли при пониженном давлении и остаток подвергали хроматографической очистке в силикагеле с использованием в качестве элюента дихлорметана/метанола (98:2) с получением соединения 204 (смесь диастереомеров ≈ 1,7:1) в виде твердого вещества (196 мг, 67%). 1H-ЯМР (СDСl3): δ 7,36-7,14 (м, Bn, Н-6), 5,77 (1H, д, J 7,9 Гц, H-1’), 5,57 (1Н, д, J 5,8 Гц, H-1’), 4,68-4,43 (м, Bn, H-2’), 4,12-3,68 (м, Н-5’, Н-5’, Н-3’), 3,32 (с, ОСН3), 3,24 (с, ОСН3), 1,93 (д, J 0,9 Гц, СН3), 1,86 (д, J 1,1 Гц, СН3), 0,14 (с, Si(СН3)3), 0,12 (с, Si(СН3)3). 13С-ЯМР (СDСl3): δ 163,68, 163,55 (С-4), 151,58, 151,07 (С-2), 137,84, 137,74, 137,32 (Bn), 135,93, 135,10 (С-6), 128,57, 128,42, 128,41, 128,10, 127,95, 127,85, 127,77, 127,74 (Bn), 111,38, 111,01 (С-5), 86,89, 85,61, 85,40, 84,72, 83,40, 83,31, 82,10 (C-1’, С-2’, С-3’, С-4’), 75,20, 73,98, 73,62, 73,59, 72,55, 72,13, 71,04, 70,74 (Bn, C-5’, С-5’’), 56,82, 56,54 (ОСН3), 12,47, 12,38 (СН3), 1,72, 1,69 (Si(СН3)3). MS FAB: 555 (M+H, 65%), 577 (M+Na, 70%). Получено: С - 62,76; Н - 6,88; N - 4,94. Для C29H38N2O7Si рассчитано: С - 62,79; Н - 6,90; N - 5,05%.
Пример 112
(1R,2S,3S)-2-бензилокси-3-бензилоксиметил-1-(метокси(6-N-бензоиладенин-9-ил)-метил)-3-триметилсилилокситетрагидрофуран (205)
Раствор соединения 202 (240 мг, 0,673 ммоль) и 6-N-бензоиладенина (301 мг, 1,26 ммоль) в безводном ацетонитриле (8,2 мл) добавляли BSA (0,67 мл, 2,7 ммоль) и перемешивали при комнатной температуре в течение 1 часов. Раствор охлаждали до 0° С и добавляли по каплям триметилсилилтрифлат (0,25 мл, 1,33 ммоль). После перемешивания при 65° С в течение 18 часов реакцию останавливали добавлением насыщенного водного раствора кислого углекислого натрия (50 мл), экстрагировали в дихлорметане (2× 50 мл). Объединенные экстракты высушивали над MgSO4. Растворитель удаляли при пониженном давлении и остаток подвергали хроматографической очистке в силикагеле с использованием в качестве элюента дихлорметана/метанола (98:2) с получением соединения 205 (смесь диастереомеров ≈ 1,8:1) в виде твердого вещества (185 мг, 41%). 1H-ЯМР (СDСl3): δ 8,78 (с, Н-8), 8,21 (с, Н-2), 8,17 (с, Н-2), 8,03-8,00 (м, Bz), 7,61-7,49 (м, Bz), 7,36-7,23 (м, Bn), 7,07-7,04 (м, Bz), 5,85 (1H, д, J 7,9 Гц, H-1’), 5,76 (1Н, д, J 6,0 Гц, H-1’), 4,74-4,40 (м, Bn, Н-2’), 4,22-3,62 (m, Н-5’, Н-5’, Н-3’), 3,33 (с, ОСН3), 3,24 (с, ОСН3), 0,15 (с, Si(СН3)3), 0,14 (с, Si(СН3)3). 13С-ЯМР (СDСl3): δ 164,68 (HNC=O), 153,17, 152,99 (С-6), 149,47 (С-2), 141,82, 141,66 (С-8), 137,74, 137,71, 137,65 (Bn), 133,87, 132,87, 132,78 (Bz), 128,97, 128,93, 128,45, 128,42, 128,38, 128,14, 127,97, 127,88, 127,82, 127,78 (Bn, Bz), 123,66, 122,85 (С-5), 86,41, 86,23, 85,70, 85,24, 84,78, 83,73, 83,58, 82,79 (С-1’, С-2’, С-3’, 0-4’), 75,32, 74,55, 73,61, 72,18, 71,98, 70,85, 70,59 (Bn, C-5’, C-5’’), 57,23, 57,04 (ОСН3), 1,78 (Si(СН3)3). MS FAB: 668 (М+Н, 50%), 690 (M+Na, 100%). Получено: С - 64,07; Н - 6,01; N - 9,94. Для С29Н38N2O7Si· 0,5Н2О рассчитано: С - 63,88; Н - 6,25; N - 10,34%.
Пример 113
(1R,2R,3R)-2-бензилокси-3-бензилоксиметил-3-гидрокситетрагидрофурфураль (206)
Раствор соединения 202/203 (252 мг, 0,707 ммоль) в 80%-ной уксусной кислоте (3,8 мл) перемешивали при 90° С в течение 2 часов, после чего растворитель удаляли путем перегонки при пониженном давлении. Остаток выпаривали вместе с толуолом (3× 10 мл) с получением соединения 206 в виде маслянистого вещества (242 мг, 100%). 1H-ЯМР (СDСl3): δ 9,66 (1Н, д, J 0,8 Гц, Н-1), 7,36-7,25 (10Н, м, Bn), 4,68 (1Н, д, J 11,9 Гц, Bn), 4,60-4,39 (5Н, м, Bn, Н-2, Н-3), 3,98-3,92 (2Н, м, Н-5), 3,85 (1Н, д, J 9,3 Гц, Н-5’), 3,52 (1Н, д, J 9,2 Гц, Н-5’). 13С-ЯМР (CDCl3): δ 203,64 (С-1), 137,39, 137,19, 128,61, 128,54, 128,29, 128,12, 127,87, 127,83 (Bn), 87,17, 87,05 (С-4, С-2), 80,98 (С-3), 75,00, 73,70, 71,86 (Bn, С-5’), 67,84 (С-5). MS FAB: 707 (2× M+Na, 100%).
Пример 114
(2S,3S,4R,7S)-3-ацетокси-7-бензилокси-1-бензилоксиметил-2,5-диоксабицикло[2.2.1]гептан (207)
К перемешиваемому раствору соединения 206 (230 мг, 0,672 ммоль) в безводном пиридине (2,0 мл) добавляли уксусный ангидрид (0,18 мл, 1,91 ммоль). Полученную реакционную смесь перемешивали при комнатной температуре в течение 23 часов, добавляли воду (0,13 мл) и растворитель удаляли путем перегонки при пониженном давлении. Остаток выпаривали с толуолом (3× 10 мл) и подвергали хроматографической очистке в силикагеле с использованием в качестве элюента дихлорметана/метанола (99:1) с получением соединения 207 в виде прозрачного маслянистого вещества (56,7 мг, 23%). 1Н-ЯМР (СDСl3): δ 7,38-7,26 (10Н, м, Bn), 6,00 (1Н, с, Н-1), 4,68 (1Н, д, J 12,0 Гц, Bn), 4,62 (1Н, д, J 12,2 Гц, Bn), 4,60 (1Н, д, J 12,4 Гц, Bn), 4,56 (1Н, д, J 12,2 Гц, Bn), 4,17 (1Н, с, Н-2), 4,14 (1Н, с, Н-3), 4,01 (1Н, д, J 7,7 Гц, Н-5’), 3,81-3,78 (3H, м, Н-5’, Н-5), 20,06 (3H, с, СОСН3). 13С-ЯМР (CDCl3) δ 169,18 (С=O), 137,92, 137,48, 128,52, 128,45, 128,03, 127,77, 127,73, 127,68 (Bn), 95,95 (С-1), 86,49 (С-4), 78,27, 76,58 (С-3, С-2), 73,65 (Bn), 72,26, 71,96 (Bn, С-5’), 65,49 (С-5), 20,98 (СОСН3). MS FAB: 407 (M+Na, 55%). Получено: С - 68,80; Н - 6,11. Для С22H24О6, рассчитано: С - 68,74; Н - 6,29%.
Пример 115
(1S,3S,4R,7S)-3-(6-N-бензоиладенин-9-ил)-7-бензилокси-1-бензилоксиметил-2,5-диоксабицикло[2.2.1]гептан (208)
К раствору фуранозы 207 (167 мг, 0,434 ммоль) и 6-N-бензоиладенина (194 мг, 0,813 ммоль) в безводном ацетонитриле (5,3 мл) добавляли BSA (0,43 мл, 1,76 ммоль) и перемешивали при комнатной температуре в течение 1 часа. Полученный раствор охлаждали до 0° С и добавляли по каплям триметилсилилтрифлат (0,16 мл, 0,86 ммоль). После перемешивания при 65° С в течение 2 часов реакцию останавливали насыщенным водным раствором кислого углекислого натрия (40 мл) и реакционную смесь экстрагировали дихлорметаном (2× 50 мл). Объединенные экстракты высушивали над MgSO4. Растворитель удаляли при пониженном давлении и остаток подвергали хроматографической очистке в силикагеле с использованием в качестве элюента дихлорметана/метанола (98:2) с получением соединения 208 в виде твердого вещества (111 мг, 45%). 1Н-ЯМР (CDCl3): δ 8,82 (1Н, с, Н-8), 8,14 (1Н, с, Н-2), 7,59-7,26 (15Н, м, Bz, Bn), 6,74 (1Н, с, H-1’), 4,92 (1Н, с, Н-2’), 4,74-4,39 (4Н, м, Bn), 4,42 (1Н, с, Н-3’), 4,19-4,10 (2Н, м, Н-5’), 3,92 (1Н, д, J 11,8 Гц, Н-5’), 3,88 (1Н, д, J 11,5 Гц, Н-5’). MS FAB: 564 (М+Н, 100%).
Пример 116
Метил-2-O-ацетил-3,5-ди-O-бензил-4-С-метансульфонилоксиметил-D-рибофуранозид (209)
К перемешиваемому раствору соединения 201 (687 мг, 1,52 ммоль) в безводном пиридине (4 мл) при 0° С по каплям добавляли уксусный ангидрид (0,43 мл, 4,56 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 дней, реакцию останавливали насыщенным водным раствором кислого углекислого натрия (75 мл) и экстрагировали дихлорметаном (150+75 мл). Объединенные экстракты высушивали над MgSO4, растворитель удаляли путем перегонки при пониженном давлении и остаток подвергали хроматографической очистке в силикагеле с использованием дихлорметана в качестве элюента с получением соединения 209 в виде прозрачного маслянистого вещества (β :α ≈ 3:1, 750 мг, 100%). MS FAB: 463 (М-ОСН3, 100%), 517 (M+Na, 28%). Получено: С - 58,53; Н - 6,16. Для C24H30O9S рассчитано С - 58,29; Н - 6,11%. Метил-2-О-ацетил-3,5-ди-O-бензил-4-С-метансульфонилоксиметил-β -D-рибофуранозид (209β ). 1H-ЯМР (CDCl3): δ 7,36-7,18 (10Н, м, Bn), 5,27 (1Н, д, J 4,9 Гц, Н-2), 4,88 (1H, с, Н-1), 4,55-4,44 (6Н, м, Н-5’, Bn), 4,35 (1H, д, J 5,0 Гц, Н-3), 3,73 (1H, д, J 9,2 Гц, Н-5), 3,38 (1H, д, J 9,3 Гц, Н-5), 3,30 (3H, с, ОСН3), 2,95 (3H, с, SО2СН3), 2,11 (3H, с, ОССН3). 13С-ЯМР (CDCl3) δ 169,91 (С=O), 137,83, 137,28, 128,49, 128,44, 127,99, 127,87, 127,77 (Bn), 105,40 (С-1), 82,65, 81,05, 74,55, 73,62, 73,56, 71,86, 70,22 (С-2, С-3, С-4, С-5, С-5’, Bn), 55,03 (ОСН3), 37,14 (SО2СН3), 20,73 (ОССН3). Метил-2-О-ацетил-3,5-ди-O-бензил-4-С-метансульфонилоксиметил-α -D-рибофуранозид (209α ). 1H-ЯMP (СDСl3): δ 7,36-7,18 (10Н, м, Bn), 5,09 (1Н, д, J 4,5 Гц, Н-1), 4,95 (1Н, дд, J 4,5, 6,8 Гц, Н-2), 4,65-4,44 (6Н, м, Н-5’, Bn), 4,27 (1Н, д, J 6,6 Гц, Н-3), 3,49 (1Н, д, J 9,9 Гц, Н-5), 3,46 (3H, с, ОСН3), 3,36 (1Н, д, J 9,9 Гц, Н-5), 2,92 (3H, с, SO2СН3), 2,14 (3H, с, ОССН3). 13С-ЯМР (СDСl3) δ 170,41 (С=O), 137,59, 137,28, 128,56, 128,51, 128,49, 128,44, 127,98, 127,88 (Bn), 102,35 (С-1), 84,25, 77,53, 74,66, 73,67, 72,12, 70,39, 70,28 (С-2, С-3, С-4, С-5, C-5’, Bn), 56,07 (ОСН3), 36,94 (SО2СН3), 20,63 (ОССН3).
Пример 117
Фенил-2-О-ацетил-3,5-ди-O-бензил-4-С-метансульфонилоксиметил-1-тио-β -D-рибофуранозид (210)
Метод а. К перемешиваемому раствору соединения 209 (738 мг, 1,49 ммоль) в безводном дихлорметане (6,4 мл) добавляли фенилтиотриметилсилан (2,42 мл, 12,8 ммоль) и охлаждали до 0° С. По каплям добавляли триметилсилилтрифлат (0,67 мл, 3,67 ммоль) и полученный раствор перемешивали при комнатной температуре в течение 4 часов. Реакцию останавливали добавлением насыщенного водного раствора кислого углекислого натрия (100 мл) и экстрагировали дихлорметаном (2× 200 мл). Объединенные экстракты высушивали над MgSO4 и растворитель удаляли путем перегонки при пониженном давлении. Остаток подвергали хроматографической очистке в силикагеле с использованием дихлорметана в качестве элюента с получением соединения 210 в виде прозрачного маслянистого вещества (564 мг, 86%) и непрореагировавшего исходного материала (191 мг, 26%). Метод b. К перемешиваемому раствору соединения 211 (86 мг, 0,165 ммоль) в безводном дихлорметане (0,49 мл) добавляли фенилтиотриметилсилан (0,16 мл, 0,825 ммоль) и охлаждали до 0° С. Добавляли триметилсилилтрифлат (0,037 мл, 0,206 моль) и полученный раствор перемешивали при комнатной температуре в течение 2 часов. Реакцию останавливали добавлением насыщенного водного раствора кислого углекислого натрия (15 мл) и полученную смесь экстрагировали дихлорметаном (2× 25 мл). Объединенные экстракты высушивали над МgSO4 и растворитель удаляли путем перегонки при пониженном давлении. Остаток подвергали хроматографической очистке в силикагеле с использованием дихлорметана в качестве элюента с получением соединения 210 в виде прозрачного маслянистого вещества (75 мг, 79%). 1H-ЯМР (СDСl3): δ 7,47-7,19 (15Н, м, Bn, SPh), 5,48 (1Н, д, J 3,6 Гц, Н-2), 5,34 (1H, дд, J 3,7, 5,2 Гц, Н-1), 4,54-4,36 (7Н, м, Н-3, Н-5’, Bn), 3,66 (1H, д, J 9,7 Гц, Н-5), 3,48 (1H, д, J 9,5 Гц, Н-5), 2,89 (3H, с, SО2СН3), 2,09 (3H, с, ОССН3). 13С-ЯМР (СDСl3): δ 169,93 (С=O), 137,69, 137,08, 132,65, 132,45, 129,15, 128,53, 128,52, 128,18, 128,14, 128,08, 127,91, 127,85 (Bn, SPh), 87,99, 84,35, 80,34, 75,33, 74,20, 73,67, 70,83, 69,34 (С-1, С-2, С-3, С-4, С-5, С-5’, Bn), 37,27 (SO2СН3), 20,68 (ОССН3). MS FAB: 463 (M-SPh, 100%), 595 (M+Na, 24%). Получено: С - 61,17; Н - 5,55. Для С29Н32О8S2 рассчитано: С - 60,82; Н - 5,63%.
Пример 118
1,2-ди-О-ацетил-3,5-ди-O-бензил-4-С-метансульфонилоксиметил-D-рибофураноза (211)
Раствор соединения 201 (150 мг, 0,313 ммоль) в 80%-ной уксусной кислоте (1,5 мл) перемешивали при 90° С в течение 3 часов. Растворитель удаляли путем перегонки при пониженном давлении и остаток выпаривали вместе с этанолом (3× 5 мл), толуолом (3× 5 мл) и пиридином (3× 5 мл). Остаток вновь растворяли в безводном пиридине (0,62 мл), добавляли уксусный ангидрид (0,47 мл) и полученный раствор перемешивали при комнатной температуре в течение 16 часов. Реакцию останавливали водой (50 мл) и полученную реакционную смесь экстрагировали дихлорметаном (2× 50 мл). Объединенные экстракты промывали насыщенным водным раствором кислого углекислого натрия (50 мл) и высушивали над MgSO4. Растворитель выпаривали и остаток подвергали хроматографической очистке на силикагелевой колонке с использованием дихлорметана в качестве элюента с получением соединения 211 в виде маслянистого вещества (99 мг, 60%). 1H-ЯМР (СDСl3): δ 7,39-7,21 (м, Bn), 6,38 (д, J 4,6 Гц, Н-1 β ), 6,15 (с, Н-1 α ), 5,35 (д, J 4,9 Гц, Н-2 α ), 5,17 (дд, J 6,3, 4,9 Гц, Н-2 β ), 4,69-4,23 (м, H-3, Bn), 3,64 (д, J 9,7 Гц, Н-5 α ), 3,52 (д, J 10,1 Гц, Н-2 β ), 3,45 (д, J 9,7 Гц, Н-5 α ), 3,39 (д, J 9,9 Гц, Н-2 β ), 2,99 (с, SO2CH3 α ), 2,96 (с, SO2CH3 β ), 2,14, 2,13, 2,06, 1,90 (4× с, СОСН3). 13С-ЯМР (CDCl3): δ 169,68, 169,00 (С=O), 137,68, 137,05, 128,60, 128,55, 128,50, 128,21, 128,12, 128,04, 127,94, 127,82, 127,79 (Bn), 99,35 (С-1 α ), 94,24 (С-1 β ), 86,36 (С-4 β ), 84,28 (С-4 α ), 79,15, 77,47, 74,58, 74,06, 73,73, 73,56, 71,67, 70,57, 70,19, 69,84 (Bn, C-2, С-3, С-5, С-5’), 37,61 (SO2СН3 β ), 37,48 (SO2CH3 α ), 21,07, 20,74, 20,63, 20,39 (СОСН3). MS FAB: 545 (M+Na, 13%). Получено: С - 57,70; Н - 5,56. Для С25Н30О10S рассчитано: С - 57,46; Н - 5,79%.
Пример 119
(3R)- и (3S)-(1S,4R,7S)-7-бензилокси-1-бензилоксиметил-3-фенилтио-2,5-диоксабицикло[2.2.1]гептан (212)
Раствор соединения 210 (553 мг, 0,966 ммоль) в метаноле, насыщенном аммиаком (35 мл), перемешивали при комнатной температуре в течение 2 часов, после чего растворитель удаляли путем перегонки при пониженном давлении. Остаток вновь растворяли в безводном DMF (3,5 мл) и раствор перемешивали при 0° С. Добавляли 60%-ную суспензию гидрида натрия (118 мг, 2,88 ммоль) и полученную реакционную смесь перемешивали при комнатной температуре в течение 12 часов. Реакцию останавливали добавлением насыщенного водного раствора кислого углекислого натрия (100 мл) и полученную в результате смесь экстрагировали дихлорметаном (2× 100 мл). Объединенные экстракты высушивали над MgSO4 и растворитель удаляли путем перегонки при пониженном давлении. Остаток подвергали хроматографической очистке в силикагеле с использованием дихлорметана в качестве элюента с получением соединения 212 в виде прозрачного маслянистого вещества (404 мг, 96%). MS FAB: 435 (М+Н, 35%), 457 (M+Na, 16%). Получено: С - 71,76; Н - 6,18. Для С26Н26O4S рассчитано: C - 71,86; Н - 6,03%. (1S,3R,4R,7S)-7-бензилокси-1-бензилоксиметил-3-фенилтио-2,5-диоксабицикло[2.2.1]гептан (212β ). 1H-ЯМР (СDСl3): δ 7,46-7,26 (15Н, м, Bn, SPh), 5,35 (1Н, с, Н-1), 4,68-4,56 (4Н, м, Bn), 4,31 (1Н, с, Н-2), 4,10 (1Н, с, Н-3), 4,09 (1Н, д, J 7,3 Гц, Н-5’), 3,93 (1H, д, J 7,8 Гц, Н-5’), 3,79 (2Н, м, Н-5). 13С-ЯМР (СDСl3): δ 138,03, 137,45, 133,42, 132,36, 129,19, 128,55, 128,46, 128,05, 127,84, 127,83, 127,76 (Bn, SPh), 89,96 (С-1), 87,18 (С-4), 79,71 (С-2), 79,40 (С-3), 73,64 (Bn), 73,23 (C-5’), 72,30 (Bn), 66,31 (С-5). (1S,3S,4R,7S)-7-бензилокси-1-бензилоксиметил-3-фенилтио-2,5-диоксабицикло[2.2.1]гептан (212α ). 1H-ЯМР (CDCl3): δ 7,52-7,19 (15Н, м, Bn, SPh), 5,52 (1H, с, Н-1), 4,70-4,50 (4Н, м, Bn), 4,41 (1Н, с, Н-2), 4,18 (1Н, д, J 7,8 Гц, Н-5’), 4,08 (1H, д, J 8,4 Гц, Н-5’), 4,07 (1H, с, Н-3), 3,78 (1H, д, J 11,3 Гц, Н-5), 3,72 (1H, д, J 11,5 Гц, Н-5). 13С-ЯМР (CDCl3): δ 137,89, 137,46, 135,29, 130,93, 129,13, 128,99, 128,57, 128,48, 127,81, 127,76, 127,58, 126,95 (Bn, SPh), 91,87 (С-1), 88,59 (С-4), 80,07, 79,14 (С-2, С-3), 73,65, 73,40, 72,04 (Bn, C-5’), 65,62 (С-5).
Пример 120
(3R)- и (3S)-(1S,4R,7S)-7-бензилокси-1-бензилоксиметил-3-(тимин-1-ил)2,5-диоксабицикло[2.2.1]гептан (36+213)
Тимин (175 мг, 1,38 ммоль) перемешивали в гексаметилдисилазане (6,8 мл) с нагреванием с обратным холодильником и добавляли сульфат аммония (5 мг). После 16-часового перемешивания прозрачный раствор охлаждали до 40° С и растворитель удаляли путем перегонки при пониженном давлении. К остатку добавляли раствор соединения 212 (201 мг, 0,463 ммоль) в безводном дихлорметане (4,6 мл) и молекулярное сито с диаметром пор 4А (180 мг). После перемешивания при комнатной температуре в течение 10 минут добавляли NBS (107 мг, 0,602 ммоль) и полученную реакционную смесь перемешивали в течение еще 30 минут. Реакцию останавливали насыщенным водным раствором тиосульфата натрия (25 мл) и экстракт высушивали над МgSO4 и выпаривали, а остаток подвергали хроматографической очистке в силикагеле с использованием в качестве элюента дихлорметана/метанола (97:3) с получением продукта 36+213 в виде смеси аномеров (β :α ≈ 1:2) (127 мг, 61%). 1H-ЯМР (СDСl3): δ 7,49 (д, J 0,9 Гц, Н-6 β ), 7,46 (д, J 1,0 Гц, Н-6 α ), 7,39-7,25 (м, Bn), 5,94 (с, H-1’ α ), 5,64 (с, H-1’ β ), 4,71-4,50 (м, Bn, Н-2’), 4,23 (с, Н-3’ α ), 4,16 (д, J 8,6 Гц, Н-5’’ α ), 4,09-3,78 (м, Н-5’, Н-5’’, Н-3’ β ), 1,94 (д, J 0,9 Гц, СН3 α ), 1,62 (д, J 1,2 Гц, СН3 β ). MS FAB: 551 (М+Н, 96%).
Пример 121
(3R)- и (3S)-(1S,4R,7S)-7-гидрокси-1-гидроксиметил-3-(тимин-1-ил)2,5-диоксабицикло[2.2.1]гептан (37+214)
Раствор соединений 36+213 (175 мг, 0,39 ммоль) в этаноле (2,7 мл) перемешивали при комнатной температуре и добавляли 20% гидроксид палладия на углероде (50 мг). Полученную смесь несколько раз дегазировали аргоном и помещали в атмосферу водорода. После перемешивания в течение 18 часов реакционную смесь подвергали хроматографической очистке на силикагелевой колонне с использованием в качестве элюента дихлорметана/метанола (95:5) с получением смеси соединений 37 и 214 (1:1,2) (26 мг, 25%). 1H-ЯМР (CD3OD): δ 7,78 (д, J 1,3 Гц, Н-6 α ), 7,73 (д, J 1,2 Гц, Н-6 β ), 5,88 (с, H-1’ α ), 5,53 (с, H-1’ β ), 4,38 (с, Н-2’ α ), 4,34 (с, Н-3’ α ), 4,26 (с, R-2’ β ), 4,08-3,69 (м, Н-5’, Н-5’’, Н-3’ β ), 1,92 (д, J 1,2 Гц, СН3 α ), 1,88 (d, J 1,1 Гц, СН3 β ). 13С-ЯМР (CD3OD): δ 138,00 (С-6 α ), 136,96 (С-6 β ), 110,80 (С-5 β ), 110,08 (С-5 α ), 92,49, 89,01 (C-4’, С-1’ α ), 90,46, 88,37 (С-4’, С-1’β ), 80,89, 74,27, 73,34 (С-2’, С-3’, С-5’ α ), 80,59, 72,47, 70,39 (С-2’, С-3’, С-5’ β ), 59,29 (С-5’’ α ), 57,61 (C-5’’ β ), 12,52 (СН3 α ), 12,39 (СН3 β ). MS EI: 270 (М+, 100%).
Получение LNA-фосфорамидитов
Пример 122
4-N-бензоил-LNА-С [(1R,3R,4R,7S)-3-(4-N-бензоилцитозин-1-ил)-(гидроксиметил)-7-гидрокси-2,5-диоксабицикло{2.2.1}-гептан]
LNA-C (формула Z) помещали в абсолютный этанол и нагревали с обратным холодильником. К нагреваемому раствору добавляли бензойный ангидрид (2 эквивалента) и реакционную смесь подвергали ВЭЖХ (элюент: 20% ацетонитрил в 0,1 М ТЕАА, рН 7,0, скорость потока 1 мл/мин, аналитическая колонка Novapak C-18). Дополнительно добавляли ангидрид с интервалами 0,5-2 часа до тех пор, пока при проведении ВЭЖХ не прекращалось увеличение количества получаемого продукта. Реакционную смесь концентрировали на роторном испарителе. Остаток повторно промывали эфиром, отфильтровывали и высушивали с получением твердого вещества белого цвета. Выход составил 45%.
Основной метод диметокситритилирования защищенных по основанию LNA-нуклеозидов (LNА-СBz, LNA-T, LNA-GiBu, LNA-ABz).
Защищенные по основанию LNA-нуклеозиды выпаривали вместе с пиридином (2× ) и перемешивали с диметокситритилхлоридом (1,5 эквивалента) в пиридине (≈ 10 мл/г нуклеозида). Реакцию продолжали в ВЭЖХ (50% ацетонитрил в 0,1 М ТЕАА, рН 7,0 в течение 5 минут, 50-100% ацетонитрил в течение 10 минут и 100% ацетонитрил в течение 5 минут; скорость потока 1 мл/мин; колонка Novapak C-18). Когда прореагировавшим оказывалось более 95% исходного материала, реакционную смесь помещали на лед. Реакцию останавливали добавлением холодного насыщенного NаНСО3 (≈ 15 мл × объем пиридина). Смесь экстрагировали дихлорметаном (3 х половину объема бикарбоната натрия). Органические экстракты объединяли, высушивали над Na2SO4, отфильтровывали и концентрировали на роторном испарителе.
Остаток высушивали в вакууме и подвергали хроматографической очистке в силикагеле с использованием 0,5% пиридина и 0-2% метанола в дихлорметане в качестве элюентов. Фракции, содержащие очищенные продукты, объединяли и концентрировали на роторном испарителе. Остаток выпаривали вместе с безводным ацетонитрилом (3х) и высушивали в вакууме.
Основной метод фосфитилирования защищенных LNA-нуклеозидов
Защищенные по своему основанию диметокситритил-LNA-нуклеозиды выпаривали вместе с безводным дихлорметаном (2х) и помещали в безводный дихлорметан (10 мл на 1 г нуклеозидов в вариантах A, G и Т и ≈ 30 мл/г для варианта С). К этой смеси добавляли сначала бис(диизопропиламино) (2-цианоэтил)фосфит (1,05-1,10 эквивалента), а затем тетразол (0,95 эквивалента). Смесь перемешивали при комнатной температуре и реакцию продолжали в ВЭЖХ (70% ацетонитрил в 0,1 М ТЕАА, рН 7,0 в течение 2 минут, 70-100% ацетонитрил в течение 8 минут и 100% ацетонитрил в течение 5 минут; скорость потока 1 мл/мин; колонка Novapak С-18). Когда в реакции образовывалось более 90% амидита и его количество переставало увеличиваться, реакционную смесь помещали на лед. Ее разводили дихлорметаном (примерно 15-20-кратный объем по отношению к исходному объему) и промывали холодным насыщенным раствором бикарбоната натрия (2х) и затем холодным соляным раствором (1х). Органическую фазу высушивали над Na2SO4 отфильтровывали и концентрировали на роторном испарителе. Остаток выпаривали с безводным ацетонитрилом (3х) и высушивали в вакууме в течение ночи. ВЭЖХ-чистота варьировалась в пределах 93-98%.
Получение LNА-нуклеозид-5’-трифосфатов
Пример 123
Синтез LNA-нуклеозид-5’-трифосфатов (Tetrahedron Lett., 1988, 29, 4525)
В полипропиленовой пробирке размером 13× 100 мм нуклеозиды 37, 44, 51, 4-N-бензоилированный 57А или 61В (93,8 мкмоль) суспендировали в 1 мл пиридина (высушенного с помощью СаН2). Раствор упаривали досуха в вакуумной центрифуге в высоком вакууме. Остаток дважды ресуспендировали в ацетонитриле (высушенном с помощью СаН2) и упаривали досуха. Нуклеозид суспендировали в 313 мкл триметилфосфата (высушенного с использованием 4А-молекулярного сита), к которому добавляли 30,1 мг Proton Sponge™ (1,5 эквивалента). Смесь уплотняли, центрифугировали и охлаждали до 0° С добавляли РОСl3 (9,8 мкл, 1,1 эквивалент) при центрифугировании. Реакцию осуществляли при 0° градусов в течение 2,5 часов. В это время 469 мкмоль пирофосфата натрия (5 эквивалентов) растворяли в 5 мл воды и пропускали через 5 мл ионобменной смолы Dow 50 H+. Когда вытекающий продукт становился кислым, его отбирали в 220 мкл трибутиламина и выпаривали с образованием сиропа. ТВА-пирофосфат трижды выпаривали вместе с сухим ацетонитрилом. Наконец, сухой пирофосфат растворяли в 1,3 мл DMF (4А-сита). Через 2,5 часа, за которые проходила реакция, ТВА-пирофосфат и 130 мкл трибутиламина добавляли к раствору нуклеозидов при интенсивном вращении. Через 1 минуту реакцию останавливали добавлением 3 мл 0,1 М триэтилацетата аммония (рН 7,5). По данным хроматографии Mono-Q было показано наличие 49% нуклеозид-5’-трифосфатов. Реакционную смесь разбавляли до 100 мл водой и адсорбировали на Q-сефарозную ионобменную колонку, промывали водой и элюировали с использованием линейного градиента 0-700 мМ NaCl в 5 мМ фосфата натрия (рН 7,5). Фракции, содержащие трифосфаты, тестировали методом ионобменной хроматографии с колонками Mono-Q. Фракции, содержащие трифосфаты, объединяли и концентрировали до состояния насыщения NaCl. Продукт обессоливали с помощью картриджа C18. Количественный анализ трифосфатов осуществляли с помощью УФ-спектроскопии и формировали в виде конечного раствора с концентрацией 10 мМ. Выходы составили 17-44%. В описанном методе были получены LNA-нуклеозиды оснований U, Т, A, G и С.
Получение LNA-модифицированных олигонуклеотидов
Пример 124
Синтез олигонуклеотидов, включающих LNA формул V, X, Y и ZT, ZU, ZG, ZC, ZA, ZMeC
Бициклические нуклеозид-3’-O-фосфорамидитные аналоги 8, 19, 30, 39, 46, 53, 57D, 61D и 66, равно как и доступные на коммерческой основе 3’-O-фосфорамидиты были использованы для синтеза являющихся примерами вариантов LNA-олигонуклеотидов по настоящему изобретению (в количественном масштабе 0,2-5 мкмоль), в составе которых имелись бы один или несколько LNA вариантов V, X, Y и ZT, ZU, ZG, ZC, ZA, ZMeC. Чистоту и состав синтезируемых LNA-олигонуклеотидов проверяли с применением метода капиллярного гель-электрофореза и(или) ВЭЖХ, и(или) MALDI-MS. В целом для всех мономеров были подтверждены удовлетворительные параметры реакций сочетания. Наиболее высокий уровень эффективности сочетания (примерно 95-100%) были отмечены для нуклеозидов 39, 46, 53, 57D, 61D и 66 (соответствующих LNA-мономерам формулы Z), для которых наиболее удовлетворительные результаты были получены тогда, когда осуществляли синтез полностью LNA-модифицированных олигонуклеотидов или когда LNA инкорпорировали в ДНК- или РНК-олигонуклеотиды, не модифицированные какими-либо иными способами, или LNA вносили в олигонуклеотид, полностью состоящий из фосфотиоатных мономеров. LNA-олигонуклеотиды растворяли в чистой воде и концентрацию определяли в единицах OD260. Растворимость во всех случаях была идеальной. Для синтеза стандартных ДНК/РНК-олигонуклеотидов и частичного модифицированных LNA-олигомеров использовали стандартные CPG- или полистиреновые подложки. Для синтеза полностью модифицированных LNA-олигомеров (например, такого как 5’-d(GTGATATGC)-3’) использовали специальную твердую подложку BioGenex Universal CPG Support (BioGenex, США) или же использовали LNA-производные подложки.
Пример 125
Синтез фосфотиоатных-LNА-олигонуклеотидов
Полностью фосфотиоатные-LNA-олигонуклеотиды (табл. 7) синтезировали с помощью автоматического ДНК-синтезатора с использованием примерно тех же условий, которые были описаны ранее (пример 124). В качестве агента для сульфирования использовали реагент Бьюкажа. Выходы реакции поэтапного сочетания мономеров превысил 98%. После завершения синтеза освобождение от защитных групп и отделение от твердой подложки эффективно осуществляли с использованием концентрированного раствора аммиака (55° С, 14 часов).
Пример 126
Синтез 2’-тио-LNA-олигонуклеотидов
1’-тио-LNA-олигонуклеотиды (включающие мономер US “тиоварианта” формулы Z, показанного на фиг.2; фиг.37, табл. 8) синтезировали с использованием автоматического ДНК-синтезатора на основе стандартных протоколов (пример 124). Выход поэтапного сочетания для амидита 76F составил примерно 85% (продолжительность реакции сочетания 12 минут; как предполагается, повышение чистоты амидита 76F должно обусловить повышение выхода реакции сочетания). По завершении синтеза освобождение от защитных групп и отделение от твердой подложки эффективно осуществляли с использованием концентрированного раствора аммиака (55° С, 8 часов).
Пример 127
Синтез 2’-амино-LNA-олигонуклеотидов
С применением процедур, сходных с теми, которые были описаны в примере 126, 2’-амино-LNA-олигонуклеотиды (включающие мономер ТNH и мономер ТNMe’ “аминовариантов” формулы Z, показанных на фиг.2; фиг.35 и 36) были эффективно синтезированы на автоматическом ДНК-синтезаторе с использованием амидитов 74А и 74F (выходы поэтапного сочетания ≥ 98%).
Пример 128
Мечение LNA-олигомеров флуоресцеином
LNA-олигомеры (формулы Z, показанной на фиг.2) AL16 (5’-d(TGTG-TGAAATTGTTAT)-3’; полужирное выделение обозначает LNA-модифицированные олигонуклеотиды) и AL17 (5’-d(ATAAAGTGTAAAG)-3’; полужирное выделение обозначает LNA-модифицированные олигонуклеотиды) были успешно помечены флуоресцеином с использованием набора реактивов системы FluoroAmp T4 Kinase Green Oligonucleotide Labeling System, используемых в соответствии с рекомендациями производителя (Promega). Вкратце, 16 нмоль LNA-олигомера (либо AL16, либо AL17) помечали 5’-тиофосфатом в 50 мкл реакционного буфера, содержащего киназу T4 и γ -S-АТФ. Реакцию инкубировали при 37° С в течение 2 часов. Тиофосфорилированные LNA-олигомеры осаждали добавлением 5 мкл преципитата для олигонуклеотидов (Promega) и 165 мкл охлажденного на льду (до -20° С) 95%-ного этанола. После центрифугирования центрифугаты промывали 500 мкл охлажденного на льду (до -20° С) 70%-ного этанола и растворяли в 25 мкл буфера PBSE. Свежеприготовленный раствор 5-малеимидфлуоресцеина (50 мкг в 5 мкл ДМСО) добавляли к тиофосфорилированным LNA-олигомерам и реакционную смесь инкубировали при 68° С в течение 30 минут. К каждому LNA-олигомеру дополнительно добавляли 5-малеимидфлуоресцеина (50 мкг в 5 мкл ДМСО) и реакционную смесь инкубировали еще в течение 60 минут. После инкубации к каждой реакционной смеси добавляли по 10 мкл преципитата для олигонуклеотидов, а затем по 180 мкл охлажденного на льду (до -20° С) 70% этанола и 100 мкл N,N-диметилформамида. Помеченные флуоресцеином LNA-олигомеры очищали с помощью ВЭЖХ с обращенной фазой со следующими параметрами: колонка Delta-Pak С-18, поры 300 А, размеры 0,4× 30 см; элюент 0-50% ацетонитрил в 0,04 М триэтиламмонийном буфере (рН 7,0); скорость потока 1,5 мл/мин. Фракции, содержащие LNA-олигомеры, объединяли и выпаривали при пониженном давлении (используя вакуумную центрифугу, оборудованную масляным насосом) в течение 12 часов.
Данные гибридизации
Пример 129
Термостабильность олигонуклеотидов, включающих мономеры формул V, X, Y и ZT, ZU, ZG, ZC, ZA, ZMeC
Термостабильность LNA-модифицированных олигонуклеотидов определяли спектрофотометрически с использованием спектрофотометра, оборудованного терморегулируемым элементом Пелтье. Гибридизационные смеси по 1 мл получали с использованием одного из трех буферов (10 мМ Na2HPO4, pH 7,0, 100 мМ NaCl, 0,1 мM EDTA/10 мМ Na2HPO4, pH 7,0, 0,1 мМ EDTA/3 M хлорид тетраметиламмония, 10 мМ Na2HPO4, pH 7,0, 0,1 мМ EDTA) и эквимолярные (1 мкМ или 1,5 мкМ) количества различных LNA-модифицированных олигонуклеотидов и комплементарных им или включающих несоответствия ДНКили РНК-олигонуклеотидов. Идентичные гибридизационные смеси, в которых используются немодифицированные олигонуклеотиды, приготавливали в качестве контрольных. Величины Тm определяли как первую производную кривой диссоциации. В табл. 1-4 обобщены полученные результаты (LNA-варианты показаны полужирным шрифтом). На фиг.2 показано использование мономерных LNA. Номенклатура V, X, Y и ZT, ZU, ZG, ZC, ZA, ZMeC соответствует структурам V, X, Y и Z, показанным на фиг.2. В таблицах обозначены основания, входящие в состав LNA-мономеров. Кроме того, для тио- и аминовариантов LNA-структур формула Z в последних двух таблицах в соответствии с используемой номенклатурой обозначает ZTS и ZTNH, соответственно.
LNA-включающие Z-структуры были протестированы практически полностью (см. табл. 1). Когда три ZT-остатка вносили в состав олигонуклеотида смешанной последовательности, то величина Тm, определяемая в буфере, содержащем NaCl, и в отношении комплементарных ДНК-олигонуклеотидов (10), и в отношении комплементарных РНК-олигонуклеотидов (16) была существенно выше (грубые оценки для РНК и ДНК - 7° С и 5° С, соответственно, в расчете на 1 модификацию) по сравнению с величиной Тm соответствующих дуплексов, образуемых с немодифицированными олигонуклеотидами (1 и 8). Сходные результаты были получены при анализе LNA-включающих ZT-остатков, a также одного из остатков ZG (21 и 24В) или ZU (25), ZC (69), ZMeC (65) и ZA (58). Когда в состав РНК- или ДНК-олигонуклеотидных мишеней вносили ошибки, то величина Тm для LNA-модифицированных олигонуклеотидов во всех случаях существенно снижалась (11-15А и 17; 18-20 и 22-24А; 26-31; 57 и 59-60; 63-64 и 66, и 67): это со всей определенностью указывает на то, что LNA-модифицированные олигонуклеотиды гибридизуют со своими последовательностями-мишенями, подчиняясь правилам модели Уотсона-Крика, основывающейся на образовании водородных связей. Во всех случаях снижение Тm для LNA-модифицированных олигонуклеотидов в ответ на внесение ошибок в мишень было таким же или даже более выраженным по сравнению с тем, что определяется для соответствующих немодифицированных олигонуклеотидов (2-7 и 9; 33-38): это подтверждает то, что LNA-модифицированные олигонуклеотиды являются по меньшей мере столь же специфичными, как и их немодифицированные аналоги. Снижение ионной силы гибридизационного раствора (с 10 мМ Na2HPO4, pH 7,0, 100 мМ NaCl, 0,1 мМ EDTA до 10 мМ Na2HPO4, pH 7,0, 0,1 мМ EDTA) обусловливает снижение величины Тm для LNA-модифицированных олигонуклеотидов в отношении комплементарных им ДНК-олигонуклеотидов (40, 41) или РНК-олигонуклеотидов (49А, 41А). Сходный эффект наблюдается при анализе немодифицированных олигонуклеотидов и комплементарных им ДНК-олигонуклеотидов (39) или РНК-олигонуклеотидов (39А).
Добавление 3 М хлорида тетраметиламмония (ТМАС) в состав гибридизационного буфера приводит к существенному повышению Тm LNA-модифицированных олигонуклеотидов по отношению к комплементарным им ДНК-олигонуклеотидам (10, 21, 25). Более того, присутствие ТМАС приводит к сглаживанию различий в величинах Тm, определяемых для различных олигонуклеотидов, которые выявлялись при использовании NaCl-содержащего буфера (диапазон наименьшей и наибольшей Тm 44° С и 49° С, соответственно, для NaCl-буфера против, соответственно, 56° С и 57° С в ТМАС-буфере). Внесение ошибок приводит к существенному снижению Тm LNA-модифицированных олигонуклеотидов в отношении их ДНК-мишеней (11-13, 18-20 и 26-28). Сходная картина наблюдается при анализе немодифицированных контрольных олигонуклеотидов (1-4 и 32-35).
Данные, полученные в варианте с низкосолевыми буферами, показывают, что LNA-модифицированные олигонуклеотиды проявляют чувствительность к ионной силе гибридизационного буфера, сходную с таковой, характерной и для нормальных олигонуклеотидов. Исходя из данных, полученных с ТМАС-буфером, показано, что ТМАС проявляет эффект выравнивания показателей Тm для LNA-модифицированных олигонуклеотидов, сходный с таким же эффектом, выявляющимся в анализе немодифицированных LNA-олигонуклеотидов. LNA-модифицированные олигонуклеотиды в обоих этих гибридизационных буферах сохраняют параметры своей высокоточной специфичности.
Полностью LNA-модифицированные олигонуклеотиды, включающие все четыре мономера (71 и 75), почти полностью модифицированные LNA-олигонуклеотиды (модифицированные по всем положениям, за исключением 3’-концевого ДНК-нуклеозида), включающие и ZG, и ZT (41 и 41А), и частично модифицированные олигонуклеотиды, включающие центральный блок ZT и ZG (40 и 40А), также проявляют существенно повышенный уровень аффинности по сравнению с немодифицированным контрольным олигонуклеотидом (39 и 39А; 1 и 8). Это указывает на то, что LNA формулы Z перспективны с точки зрения конструирования и полностью, и частично модифицированных олигонуклеотидов. Заявители отмечают, что почти полностью модифицированный олигомер (41 и 41А) проявляет беспрецендентно высокий уровень аффинности по отношению и к РНК- (>93° С), и к ДНК-олигонуклеотидам (83° С). Сходная исключительно высокая аффинность (в отношении обоих типов олигонуклеотидов) была выявлена в анализе почти полностью модифицированного LNA-олигомера, включающего исключительно вариант ZT (табл. 1: 52 и 53), и полностью модифицированного LNA-олигомера (71 и 75). Аффинность частично модифицированного поли-Т-олигонуклеотида зависела от положений, занятых внесенными ZT, и от их числа (44-51). Хотя величины Тm в отношении РНК-мишеней (45, 47, 49 и 51) во всех случаях были выше по сравнению с соответствующими немодифицированными олигонуклеотидами (43), те же варианты проявляют меньшие Тm, в отношении ДНК-мишени (46). Поскольку олигонуклеотид со смешанной последовательностью, включающий три ZT-остатка, проявлял в существенной степени повышенный уровень аффинности в отношении к своим ДНК- (10) и РНК-мишеням (16) по сравнению с немодифицированными контрольными олигонуклеотидами (1 и 8), это подтверждает, что другие, в отличие от характерных для модели Уотсона-Крика, мотивы (такие как, например, мотив связывания Хогстена) “открыты” для поли-Т-олигонуклеотидов, а также что такие мотивы связывания по крайней мере в некоторой степени восприимчивы к конкретной структуре модифицированного олигонуклеотида. Во всех случаях внесение отдельных ошибочных (несоответствующих) оснований в состав комплекса, образованного полностью модифицированным остатками ZT поли-Т-олигонуклеотидом и ДНК-мишенью (54-56), обусловливал существенное падение Тm.
Олигонуклеотиды, включающие LNA формул либо V (табл. 2), либо Х (табл. 3), либо Y (табл. 4), были проанализированы в контексте анализа полностью и частично модифицированных поли-Т-последовательностей. Полностью модифицированные олигонуклеотиды формулы V и Y проявляют увеличение Тm (хотя выраженное в меньшей степени по сравнению с ZT-модифицированными олигонуклеотидами) по отношению как к РНК-мишеням (табл. 2, 14, и табл. 4, 14), так и к ДНК-мишеням (табл. 2, 13, и табл. 4, 13) по сравнению с немодифицированными олигонуклеотидами (табл. 1, 42 и 43). Частично модифицированные олигонуклеотиды, включающие мономеры формул V и Y проявляли сходные свойства по отношению к частично модифицированным олигонуклеотидам, включающим ZT: по-видимому, это объясняется гомополимерной природой последовательности, которая была определена выше. Олигонуклеотиды, включающие XT, во всех случаях проявляют в значительной степени сниженную Тm по сравнению с контрольными ДНК-олигонуклеотидами.
Пример 130
Образование стабильных гибридов полностью модифицированным LNA-олигонуклеотидом с комплементарным ДНК и в параллельной, и в антипараллельной ориентациях
Полностью модифицированный LNA-олигонуклеотид гибридизовали с комплементарной ему ДНК в обоих вариантах ориентации - параллельной и антипараллельной. В состав гибридизационных растворов (1 мл) входили 10 мМ Na2HPO4 (рН 7,0), 100 мМ NaCl и 0,1 мМ EDTA и 1 мкМ каждого из двух олигонуклеотидов. Как показано в табл. 1, и антипараллельная (71), и параллельная ориентация связывания (77) приводили к образованию стабильных дуплексов. Из этих двух вариантов вариант с антипараллельной ориентацией приводил к наибольшей стабильности. Однако даже дуплекс с параллельной ориентацией существенно более стабилен по сравнению с соответствующим антипараллельным дуплексом с участием немодифицированных ДНК-олигонуклеотидов (табл. 1, 1).
Пример 131
Возможность использования LNA-мономеров для увеличения аффинности РНК-олигомеров в отношении комплементарных им нуклеиновых кислот
Термостабильность комплексов, образованных 9-мерным РНК-олигонуклеотидом, включающим три LNA-T-мономера (Z7), и комплементарными ДНК- или РНК-олигонуклеотидами, определяли спектрофотометрически. В состав гибридизационных растворов (1 мл) входили 10 мМ Na2HPO4 (pH 7,0), 100 мМ NaCl и 0,1 мМ EDTA и 1 мкМ каждого из двух олигонуклеотидов. В качестве контроля были сформированы идентичные гибридизационные смеси, использующие немодифицированные РНК-олигонуклеотиды. Как видно из табл. 5, LNA-модифицированный РНК-олигонуклеотид гибридизует и с комплементарным ДНК-олигонуклеотидом (1), и с комплементарным РНК-олигонуклеотидом (3). Как ранее было установлено для LNA-модифицированных ДНК-олигонуклеотидов, аффинность по связыванию LNA-модифицированных РНК-олигонуклеотидов наиболее выражена в отношении РНК-комплемента (3). В обоих случаях аффинность LNA-модифицированного РНК-олигонуклеотида оказывается существенно более высокой по сравнению с немодифицированным контролем (2 и 4). В табл. 5 также показано, что в случае с LNA-модифицированными РНК-олигонуклеотидами специфичность в отношении и ДНК-, и РНК-мишеней сохраняется.
Пример 132
Спаривание оснований LNA-LNA
РНК- или ДНК-олигонуклеотиды, включающие по три мономера ZT-LNA, или олигонуклеотиды, полностью модифицированные мономерами LNA-Z, гибридизовали с комплементарными немодифицированными ДНК-олигонуклеотидами или ДНК-олигонуклеотидами, включающими по три мономера LNA-ZA: величину Тm получаемых гибридов определяли спектрофотометрически. В состав гибридизационных растворов (1 мл) входили 10 мМ Nа2НРO4 (рН 7,0), 100 мМ NaCl и 0,1 мМ EDTA и 1 мкМ каждого из двух олигонуклеотидов. Как видно из табл. 6, все LNA-модифицированные олигонуклеотиды гибридизовали с комплементарными им немодифицированными ДНК-олигонуклеотидами (2 и 3), равно как и с комплементарными LNA-модифицированными олигонуклеотидами (4, 5 и 6). Как было установлено ранее, присутствие LNA-мономеров в одной из цепей гибрида (2 и 3) обусловливает существенное повышение Тm по сравнению с немодифицированным контрольным молекулярным гибридом (1). Присутствие пар оснований LNA-LNA в гибридном дуплексе обусловливает даже большее повышение величины Тm (4 и 5). Более того, характеризующийся высоким уровнем стабильности гибрид может быть образован между полностью модифицированным LNA-олигонуклеотидом и олигонуклеотидом, частично модифицированным остатками LNA-ZA (6). Это является первым примером спаривания в парах LNA-LNA в гибридной молекуле.
Пример 133
Проявление полностью LNA-фосфомонотиоатным олигонуклеотидом относительно меньшего снижения термостабильности по отношению к комплементарными ДНК и РНК по сравнению с полностью ДНК-фосфотиоатным ДНК-олигонуклеотидом
Термостабильность полностью ДНК-фосфомонотиоатного олигонуклеотида, включающего три мономера LNA-ZT (LNA-олигонуклеотид), и соответствующий полностью ДНК-фосфомонотиоатный контрольный олигонуклеотид по отношению к комплементарным ДНК и РНК оценивали при соблюдении тех же условий, которые были описаны в примере 132, однако EDTA была исключена (табл. 7). Было установлено, что полностью LNA-фосфомоно-тиоатный олигонуклеотид, включающий три мономера LNA-ZT, проявляет лишь ненамного сниженную термостабильность (табл. 7, 3 и 4) при сравнении с соответствующим контрольным LNA-олигонуклеотидом (табл. 1, 10 и 16). Соответствующий полностью ДНК-фосфомонотиоатный олигонуклеотид (табл. 7, 1 и 2) проявлял существенное снижение термостабильности при сравнении с соответствующим контрольным ДНК-олигонуклеотидом (табл. 1, 1 и 8). Это может иметь перспективное применение полностью или частично LNA-фосфомонотиоатных олигонуклеотидов в “антисмысловой терапии” и в других терапевтических подходах. Таким образом, была подтверждена совместимость LNA-мономеров и немодифицированных мономеров в составе фосфомонотиоатного олигонуклеотида. Можно ожидать, что такие конструкции будут проявлять и активность РНКазы-Н, и резистентность к нуклеазам в дополнение к улучшению параметров гибридизации за счет присутствия LNA.
Пример 134
Проявление 2’-тиo-LNA свойств распознавания нуклеиновых кислот, сравнимых с такими свойствами самих LNA (Z-мономер)
Условия гибридизации соответствовали тем, которые описаны в примере 132, за исключением EDTA. Результаты, полученные для 2’-тио-LNA (табл. 8), отчетливо показывают наличие положительного эффекта в отношении термостабильности дуплексов по отношению и к ДНК, и к РНК, в результате внесения 2’-тио-LNA-мономера US (мономеры, соответствующие формуле Z, показанной на фиг.2, при том, что “мостик” метиленоксигруппы замещен “мостиком” метилентиогруппы). Этот эффект (Δ Тm ≈ +5° С в расчете на одну модификацию, в отношении ДНК-мишени; Δ Тm ≈ +8° С в расчете на одну модификацию, в отношении РНК-мишени) сравним с тем, который был выявлен для исходной LNA. Эта картина в некоторой степени запутывается при одновременном внесении двух модификаций (2’-тиогруппа и урацил вместо тимина). Однако с учетом того, что ранее заявители выявили идентичность температур диссоциации LNA-мономеров тимина и урацила, и с учетом того, что присутствие в контрольных молекулах 2’-дезоксиуридина вместо тимидина должно обусловливать проявление меньших величин Тm, то такое сравнение допустимо.
Пример 135
Проявление 2’-амино-LNА (мономера 2TNH) и 2’-метиламино-LNA (мономера ZTNMe) свойств по распознаванию нуклеиновых кислот, сравнимых с теми, которые характерны для исходных LNA (мономер Z)
Условия гибридизации соответствовали тем, которые описаны в примере 132, за исключением EDTA. Данные по параметрам диссоциации дуплексов для вариантов с 2’-aмино-LNA (табл. 9) отчетливо показывают наличие позитивного эффекта в отношении термостабильности дуплексов с участием и ДНК, и РНК, обусловливаемого внесением 2’-амино-LNA-мономеров ТNH или ТNMe (мономеры, соответствующие формуле Z, показанной на фиг.2, при том, что “мостик” метиленоксигруппы замещен “мостиком” метиленаминогруппы или “мостиком” метилен(N-метил)аминогруппы, соответственно). Этот эффект (Δ Тm ≈ +3° С в расчете на одну модификацию, в отношении ДНК-мишени; Δ Тm ≈ от +6° С до 8° С в расчете на одну модификацию, в отношении РНК-мишени) сравним с тем, который был выявлен для исходной LNA. Заслуживает внимание и то, что повышенная термостабильность также наблюдается в варианте с олигомером, включающим смесь 2’-алкиламино-LNA-мономеров и неалкилированных 2’-aмино-LNA-мономеров.
LNA и LNA-модифицированные олигонуклеотиды, как субстраты для ферментов.
Пример 136
3’-экзонуклеолитическая стабильность олигомеров 5’-V
Раствор олигонуклеотидов (0,2 OD) в 2 мл буфера (0,1 М Трис-HCl, рН 8,6, 0,1 М NaCl, 14 мМ MgCl2) обрабатывали при 25° С 1,2 ед. SVPDE (фосфодиэстераза змеиного яда). В процессе обработки было отмечено увеличение поглощения при 260 нм. В то время как немодифицированный по-литимидин-14 полностью разрушался за 10 минут действия фермента, варианты 5’-V
Пример 137
LNA-модифицированные олигомеры, как субстраты для полинуклеотидкиназы Т4.
По 20 пикомолей каждой затравки (FP2: 5’-GGTGGTTTGTTTG-3’: ДНК-зонд), (AL2: 5’-GGTGGTTTGTTTG-3’: LNA-нуклеозиды выделены полужирным шрифтом) и (AL3: 5’-GGTGGTTTGTTTG-3’: LNA-нуклеозиды выделены полужирным шрифтом) смешивали с полинуклеотидкиназой Т4 (5 ед.: New England Biolabs) и 6 мкл γ -32P-АТФ (300 Ки/ммоль, Amershaiti) в буфере, содержащем 70 мМ Трис-НСl (рН 7,6), 10 мМ MgCl2, 5 мМ дитиотреутола (конечный объем 20 мкл). Образцы инкубировали при 37° С в течение 40 минут и после этого нагревали до 65° С на 5 минут. К каждой реакционной смеси добавляли по 2 мкл тРНК (1 мкг/мкл), 29 мкл 3 М ацетата аммония и 100 мкл этанола. Реакционные смеси инкубировали при -20° С в течение 30 минут и помеченные олигонуклеотиды преципитировали центрифугированием при 15000g в течение 30 минут. Осадок ресуспендировали в 20 мкл воды. Образцы (1 мкл) смешивали с загрузочным буфером (формамид [рН 8,0], 0,1% ксиленцианола FF, 0,1% бромфенолового синего и 10 мМ EDTA) и подвергали электрофорезу в денатурирующем полиакриламидном геле (16% раствор акриламида/бисакриламида, 7 М мочевины, 1xTBE и 0,1 мМ EDTA) в ТВЕ-буфере (90 мМ Трис-НСl [рН 8,3], 90 мМ борной кислоты и 2,5 мМ двухнатриевой EDTA). Гель высушивали в гелевой сушке (BioRad, модель 583) и подвергали радиоавтографическому анализу с использованием рентгеновской пленки (пленка CL-XPosure, Pierce 34075) в течение 20 минут. Полученные результаты показаны на фиг.6 (FP2 - дорожки 1 и 2; AL2 - дорожки 3 и 4; AL3 - дорожки 5 и 6). На основе данных экспериментов можно сделать три вывода. Во-первых, можно заключить, что частично и полностью модифицированные LNA-олигомеры являются эффективными имитаторами нативных нуклеиновых кислот с точки зрения их способности служить субстратом для ферментов, специфичных в отношении нуклеиновых кислот, таких как полинуклеотидкиназа. Во-вторых, можно сделать вывод о том, что LNA-модифицированные олигомеры могут эффективно преципитироваться в методиках, обычно применяемых для осаждения обычных нуклеиновых кислот. Действительно, относительные уровни интенсивности сигналов для немодифицированных (дорожки 1, 2), частично модифицированных (дорожки 3, 4) и полностью модифицированных олигомеров (дорожки 5, 6) на рентгенограммах подтверждают, что, чем большее LNA-нуклеозидов входит в состав стандартного ДНК-олигонуклеотида, тем более эффективно он может быть преципитирован в солевой-спиртовой процедуре. В-третьих, сходные положения сигналов на рентгенограммах, соответствующих немодифицированным, частично и полностью модифицированным олигонуклеотидам, показывает, что инкорпорация LNA-нуклеозидов в состав ДНК-олигомеров не приводит к изменению его электрофоретической подвижности в полиакриламидном геле.
Пример 138
3’-концевое мечение LNA-включающих олигонуклеотидов с использованием терминальной дезоксинуклеотидилтрансферазы
Олигонуклеотиды, в состав которых входят LNA-мономеры, были подвергнуты 3’-концевому мечению с использованием фермента терминальной дезоксинуклеотидилтрансферазы. Последовательности и степень модификаций с использованием LNA были следующими (мономеры LNA обозначены полужирным шрифтом):

Олигонуклеотид (50 пкмоль) инкубировали с участием 250 мкКи α -32Р-ддАТФ (3000 Ки/ммоль) и 100 ед. терминальной дезоксинуклеотидилтрансферазы в 250 мкл 100 мМ какодилатного буфера, рН 7,2, 2 мМ CoCl2 и 0,2 мМ 2-меркаптоэтанола при 37° С в течение 2 часов. Реакцию останавливали добавлением формамид-содержащего загрузочного буфера и нагреванием до 100° С на 5 минут перед помещением на лед. Образцы (0,2 пкмоль) пропускали по 19%-ному акриламидному гелю, содержащему 7 М мочевины: уровень включения радиоактивности в бэнды олигонуклеотидов количественно определяли с помощью фосфорочувствительного датчика (Molecular Dynamics). Полученные результаты показали наличие включения радиоактивности во всех тестированных случаях, включая олигонуклеотид, характеризующийся наивысшим количеством LNA: контроль - 94,9%, (1) - 39,7%, (2) - 83,7%, (3) - 31,7%. Делается заключение о том, что LNA-модифицированные олигонуклеотиды являются субстратом для фермента терминальной дезоксинуклеотидилтрансферазы.
Пример 139
Способность терминальной дезоксинуклеотидилтрансферазы (TdT) обеспечивать концевые присоединения в модифицированных олигонуклеотидах в зависимости от конструкции олигомера.
Следующие 15-нуклеотидные затравки и смесь 8-32-мерных олигонуклеотидных маркеров были помечены по 5’-концу [γ -33P]-АТФ и полинуклеотидкиназы Т4 (где LNA-мономеры выделены полужирным шрифтом):
Реакционные смеси кипятили на протяжении 5 минут после проведения мечения с целью исключения остаточной полинуклеотидкиназой активности. По 4 пкмоль каждой из помеченных затравок, 25 ед. терминальной дезоксинуклеотидилтрансферазы и 16 мкМ дАТФ инкубировали в 25 мкл 100 мМ какодилатного буфера (рН 7,2), 2 мМ CoCl2 и 0,2 мМ 2-меркаптоэтанола в течение 90 минут при 37° С. Реакции останавливали добавлением формамид-содержащего стоп-раствора и продукты реакций пропускали по 19%-ному полиакриламидному гелю, содержащему 7 М мочевины и помеченные маркеры. Проводили авторадиографический анализ высушенного геля с использованием пленки Biomax. Как показано на фиг.22, для вариантов Р1 (дорожка 2), Р2 (дорожка 4) и PZ1 (дорожка 3) во всех случаях выявляется удлинение “хвоста”, оцениваемое более чем в 70 нуклеотидов, с учетом параметров 8-32-нуклеотидных маркерных фрагментов (дорожки 1 и 6). При тех же условиях реакции концевой достройки варианта PZ2 не происходит. Делается заключение о том, что фермент TdT будет толерантен к присутствию LNA-мономеров в пределах олигонуклеотида, но не самом его 3’-конце.
Пример 140
LNА-тимидин-5’-трифосфат (LNA-TTP), как субстрат для терминальной дезоксинуклеотидилтрансферазы (TdT)
С целью проанализировать способность трифосфата в составе LNA-TTP (пример 123) распознаваться терминальной дезоксинуклеотидилтрансферазой в качестве субстрата, была осуществлена реакция “концевой достройки” олигонуклеотида. 15-нуклеотидную затравку (ее последовательность: 5’-TGC ATG TGC TGG AGA-3’) и смесь 8-32-мерных олигонуклеотидов помечали по 5’-концам с использованием [γ -33Р]-АТФ и полинуклеотидкиназы Т4. Реакционные смеси кипятили на протяжении 5 минут после проведения мечения с целью исключения остаточной полинуклеотидкиназной активности. 4 пкмоль помеченной затравки, 25 ед. терминальной дезоксинуклеотидилтрансферазы и 32, 64 или 128 мкМ дТТФ или LNA-TTP инкубировали в 25 мкл 100 мМ какодилатного буфера (рН 7,2), 2 мМ CoCl2 и 0,2 мМ 2-меркаптоэтанола в течение 90 минут при 37° С. Реакции останавливали добавлением формамид-содержащего стоп-раствора и продукты реакций пропускали по 19%-ному полиакриламидному гелю, содержащему 7 М мочевины и помеченные маркеры. Проводили авторадиографический анализ высушенного геля с использованием пленки Biomax. Как показано на фиг.10, во всех случаях в реакциях с 32 мкМ дТТФ (дорожка В), 64 мкМ дТТФ (дорожка С) или 128 мкМ дТТФ (дорожка D) происходила “хвостовая достройка” олигонуклеотида, на что указывает сравнение с 8-32-мерными олигонуклеотидными маркерами (крайние левая и правая дорожки), размер которой оценивается более чем в 100 нуклеотидов. Реакции с LNA-TTP (32 мкМ дТТФ (дорожка Е), 64 мкМ дТТФ (дорожка F) или 128 мкМ дТТФ (дорожка G)) во всех случаях обусловливали удлинение затравок на одно основание, а примерно в половине случаев удлинение имело еще один нуклеотид. Эти результаты в значительной степени сходны с теми, которые были получены для рибонуклеотидов и TdT. Делается заключение о том, что LNA-производные трифосфаты могут распознаваться и инкорпорироваться в ДНК-олигонуклеотид с участием фермента TdT. Это наблюдение, согласно которому LNA-TTP может связываться с полимеразой, подтверждает перспективность успешного использования производных LNA-мономеров в качестве нуклеозид-содержащих лекарственных средств.
Пример 141
Возможность включения LNA-аденозин-, -цитозин-, -гуанозини- и -уридин-5’-трифосфатов (LNA-АТФ, LNA-ЦТФ, LNA-ГТФ, LNA-УТФ) в цепь ДНК под контролем не имеющего экзонуклеазной активности фрагмента Кленова ДНК-полимеразы I.
Тест на достройку затравки был применен для оценки LNA-NTP (см. пример 123) и рибонуклеотидов, как потенциальных субстратов для свободного фрагмента Кленова ДНК-полимеразы I. В данном тесте было применено 5’-концевое 33Р-мечение 15-нуклеотидной затравки, гибридизуемой с одной из четырех различных 24-нуклеотидных матриц. Последовательности затравки и матриц таковы (LNA-мономеры выделены полужирным шрифтом):

Один пкмоль 33Р-помеченной затравки гибридизовали с 2 пкмоль матрицы в × 2 буфере Кленова. К этой смеси добавляли либо 4 мкМ дНТФα S, либо 500 мкМ LNA-HTP, либо смесь 4 мкМ дНТФα S и 500 мкМ LNA-HTP. К каждой реакции добавляли по 2 ед. фрагмента Кленова ДНК-полимеразы. К каждой из реакционных смесей добавляли по 2 миллиед. неорганической пирофосфатазы. Также объединяли затравку, матрицу и фермент в контрольном варианте. Все реакции проводили при общем объеме 20 мкл. Реакционные смеси инкубировали при 37° С в течение 3 минут. Затем реакции останавливали добавлением 10 мкл стоп-раствора формамид-EDTA. Продукты реакций разделяли в 19%-ном полиакриламидном геле с добавлением 7 М мочевины и полученные фрагменты диагностировали по размеру, сравнивая с “лестницей” 33Р-помеченных 8-32-олигонуклеотидных эталонов и используя для съемки рентгенографическую пленку Kodak Biomax.
На фиг.20 показаны данные опыта с LNA-УТФ для матрицы 1. Показанные треки (1-12) соответствуют следующим реакциям: внесение LNA-УТФ с участием фрагмента Кленова. Дорожка 1 - затравка, матрица и фермент. Дорожка 2 - то же плюс дТТФα S. Дорожка 3 - плюс LNA-УТФ. Дорожка 4 - плюс дТТФα S и дГТФα S. Дорожка 5 - плюс LNA-УТФ и дГТФα S. Дорожка 6 - плюс дАТФα S, дГТФα S и дТТФα S. Дорожка 7 - плюс LNA-УТФ, дЦТФα S, дГТФα S и дТТФα S. Дорожка 8 - плюс дГТФα S. Дорожка 9 - плюс дЦТФα S, дГТФα S и дТТФα S. Дорожка 10 - плюс LNA-УТФ, дАТФα S, дЦТФα S и дГТФα S. Дорожка 11 - плюс дАТФα S, дЦТФα S и дГТФα S. Дорожка 12 - все 4 дНТФα S. Крайние дорожки соответствуют 8-32-мерным олигонуклеотидным маркерам, использованным для определения длины тестированных продуктов.
Как видно из фиг.20, LNA-УТФ специфическим образом инкорпорируется как нуклеотид “Т”. Дальнейшая достройка по 3’-концу, на котором находится LNA-УТФ, в значительной степени замедлена.
На фигуре 21 показаны результаты опытов с LNA-АТФ, LNA-ЦТФ и LNA-ГТФ с использованием матриц 2-4. Треки (1-21) соответствуют следующим реакциям. Дорожки 1, 7, 13 и 17 - затравка, матрица и фермент. Дорожка 2 - то же плюс дГТФα S. Дорожка 3 - плюс дАТФα S и дГТФα S. Дорожка 4 - плюс LNA-ГТФ. Дорожка 5 - плюс дГТФα S и LNA-АТФ. Дорожка 6 - плюс LNA-АТФ и LNA-ГТФ. Дорожка 8 - плюс дАТФα S. Дорожка 9 - плюс дАТФα S и дЦТФα S. Дорожка 10 - плюс LNA-АТФ. Дорожка 11 - плюс дЦТФα S и LNA-АТФ. Дорожка 12 - плюс дАТФα S и LNA-ЦТФ. Дорожка 14 - плюс дТТФα S. Дорожка 15 - плюс дГТФα S и дТТФα S. Дорожка 16 - плюс дТТФα S и LNA-ГТФ. Дорожка 18 - плюс дЦТФα S. Дорожка 19 - плюс дЦТФα S и дТТФα S. Дорожка 20 - плюс LNA-ЦТФ. Дорожка 21 - дТТФα S и LNA-ЦТФ. Крайние дорожки соответствуют 8-32-мерным олигонуклеотидным маркерам, использованным для определения длины тестированных продуктов.
Эксперименты с использованием матрицы 2 (треки 1-6) показывают, что LNA-ГТФ способны образовывать продукт “+1” при эффективном удлинении затравки (4-й трек). Добавление дГТФα S и LNA-АТФ в большинстве случаев приводит к образованию продукта “-1-2” (5-й трек). Это обусловливается включением дГТФα S с образованием продукта “+1” и последующей достройкой за счет LNA-АТФ. Имеется указание на присутствие небольшого количества продукта “+3”, что обусловливается последующим включением LNA-АТФ. Эксперименты с матрицей 3 (треки 7-12) показывают, что LNA-АТФ эффективно инкорпорируется с образованием продукта “+1” (10-й трек). Достройка этого продукта с участием дЦТФα S замедлена (11-й трек). Добавление дАТФα S и LNA-ЦТФ приводит к образованию продуктов “+2” и “+3” (12-й трек). Отсутствие сколько-нибудь существенного количества продукта “+1” указывает на то, что добавление первого мономера LNA-ЦТФ эффективно, в то время как добавление второго остатка LNA-ЦТФ замедлено. Результаты экспериментов с матрицами 1 (треки 13-16) и 4 (треки 17-21) указывают на наличие тенденций, сходных с таковыми у других матриц. LNA-ЦТФ эффективно инкорпорируется с образованием продукта “+1” при использовании матрицы (20-й трек). Достройка этого продукта с участием дТТФα S опять-таки оказывается замедленной (21-й трек). Добавление LNA-ГТФ и дТТФα S в реакции с матрицей 1 обусловливает образование продукта “+2” (16-й трек). Опять-таки эти данные указывают на то, что добавление отдельного LNA-трифосфата является весьма эффективным, однако добавление последующего LNA-трифосфата замедлено.
Пример 142
Возможность использования LNA-мономеров для повышения резистентности олигонуклеотида к расщеплению экзонуклеазой III.
С целью протестировать параметры устойчивости LNA-включающих олигонуклеотидов к расщеплению экзонуклеазой III, была проведена следующая реакция. Следующие 15-нуклеотидные затравки и 8-32-мерные олигонуклеотидные эталоны были подвергнуты 5’-концевому мечению с использованием [γ -33P]-ATФ и полинуклеотидкиназы Т4 (где LNA-мономеры выделены полужирным шрифтом):

Реакционные смеси кипятили на протяжении 5 минут после проведения мечения с целью исключения остаточной полинуклеотидкиназной активности. По 8 пкмоль каждой затравки гибридизовали с 25 пкмоль матрицы (ее последовательность: 3’-ACG ТАС ACG АСС ТСТ АСС TTG СТА-5’) в 2х буфере Кленова. К каждой реакции добавляли по 10 ед. экзонуклеазы III. Также ставили контрольные варианты, в которые вместо экзонуклеазы добавляли 1 мкл воды. Реакционные смеси инкубировали при 37° С в течение 5 минут. Реакции останавливали добавлением 10 мкл стоп-раствора формамид-EDTA. Реакции нагревали до 95° С на 3 минуты с последующей загрузкой в 19%-ный полиакриламидный гель, содержащий 7 М мочевины. Гель фиксировали смесью 10% уксусной кислотой и 10% метанолом с последующим переносом на бумагу ЗММ и высушиванием. Высушенный гель анализировали на фосфорном датчике в течение 3 часов. Этот сканер работал на базе прибора Molecular Dynamics Storm 860, оборудованном программой ImageQuant. Проведенное сканирование гелей показало, что в отсутствие экзонуклеазы интенсивность сигналов для полноразмерных затравок Р2 и PZ2 составила, соответственно, 99% и 96%. В присутствие фермента после 5-минутной инкубации сохранялось лишь 20% сигнала полноразмерной затравки Р2. Однако для полноразмерной затравки PZ2 при том же режиме обработки сохранялось 62% интенсивности сигнала. Это указывает на то, что единственный LNA-мономер, присоединенный с 3’-конца олигонуклеотида, придает ему повышенную устойчивость к действию экзонуклеазы III.
Применение в ПЦР.
Пример 143
LNA-мономеры могут быть использованы для существенного увеличения эффективности применения биотинилированных ДНК-олигонуклеотидов при специфичном по последовательности отборе (“отлове”) ПЦР-амплификонов в МТР-формате. Два помеченных дигоксигенином ампликона, основанные на последовательности плазмиды pUC19, были получены с помощью ПЦР следующим образом.
Реакционная смесь ПЦР для 1-го ампликона:
1 мкл pUC19 (1нг/мкл),
1 мкл обратной затравки (5’-AACAGCTATGACCATG-3’) (20 мкМ),
1 мкл прямой затравки (5’-GTAAAACGACGGCCAGT-37) (20 мкМ),
10 мкл дНТФ-mix (2 мМ дАТФ, 2 мМ дЦТФ, 2 мМ дГТФ и 6 мМ ДУТФ),
1,5 мкл дигоксигенин-11-дУТФ (1 мМ),
10 мкл 10х буфера Taq (Boehringer Mannheim, включает MgCl2),
1 мкл Taq-полимеразы (Boehringer Mannheim) (5 ед./мкл),
дистиллированная вода 100 мкл.
Реакционная смесь ПЦР для 2-го ампликона:
1 мкл pUC19 (1нг/мкл),
0,4 мкл затравки 3 (5’-GATAGGTGCCTCACTGAT-3’) (50 мкМ),
0,4 мкл затравки 4 (5’-GTCGTTCGCTCCAAGCTG-3’) (50 мкМ),
10 мкл дНТФ-mix (2 мМ дАТФ, 2 мМ дЦТФ, 2 мМ дГТФ и 6 мМ дУТФ),
1,5 мкл дигоксигенин-11-дУТФ (1 мМ),
10 мкл 10х буфера Taq (Boehringer Mannheim, включает MgCl2),
1 мкл Taq-полимеразы (Boehringer Mannheim) (5 ед./мкл),
дистиллированная вода 100 мкл.
Параметры ПЦР (используется амплификатор модели Perkin-Elmer 9600): 94° С - 5 минут; добавление полимеразы; 94° С - 1 минута, 45° С - 1 минута, 70° С - 2 минуты (29 циклов); 72° С - 10 минут.
По 10 мкл каждой ПЦР анализировали в стандартном агарозном геле, в котором были обнаружены ожидаемые фрагменты длиной 100 и 500 нуклеотидов.
10 мкл помеченного дигоксигенином ампликона 1 или ампликона 2 смешивали с 5 пкмоль 5’-биотинилированного отборочного зонда в 1xSSC (0,15 М NaCl, 15 мМ цитрата натрия, рН 7,0) при общем объеме 450 мкл. Были использованы следующие отборочные зонды: B-DNA1 (биотин-ATGC-CTGCAGGT-CGAC-3’; ДНК-зонд, специфичный в отношении 1-го амплификона), B-DNA2 (биотин-GGTGGTTTGTTTG-3’; ДНК-зонд, специфичный в отношении 2-го ампликона), B-LNA2 (биотин-GGTGGT-TTGTTTG-3’; LNA-нуклеозиды выделены полужирным шрифтом; LHA-зонд, специфичный в отношении 2-го ампликона). Реакции нагревали до 95° С на 5 минут с целью денатурации ампликонов и оставляли остывать при 25° С на 15 минут, обеспечивая гибридизацию между зондом и последовательностью-мишенью ампликона. После гибридизации 190 мкл каждой реакционной смеси переносили на покрытый стрептавидином планшет (Pierce, № по каталогу 15124) и инкубировали в течение 1 часа при 37° С. После промывки планшета фосфатно-солевым буфером (ФСБТ: 0,15 М Na+, pH 7,2, 0,05% Твин-20, 3× 300 мкл) добавляли 200 мкл антидигоксигениновых антител, помеченных пероксидазой (Boehringer Mannheim, разведение 1:1000 в фосфатно-солевом буфере). Планшеты инкубировали в течение 30 минут при 37° С и промывали (ФСБТ, 3× 300 мкл). Лунки тестировали на пероксидазную активность путем добавления 100 мкл раствора субстрата (0,1 М цитратфосфатного буфера, рН 5,0, 0,66 мг/мл орто-фенилендиаминдигидрохлорида, 0,012% перекиси водорода). Реакцию останавливали через 8 минут добавлением 10 мкл серной кислоты (0,5 М) и считывали параметры поглощения при 492 нм с использованием микропланшетного датчика. Как показано на фиг.3, немодифицированные биотин-ДНК-отборочные зонды (B-DNA1 и B-DNA2) в обоих случаях проявляют себя в соответствии с ожидаемым, т.е. каждый из них отбирает только “свой” ПЦР-ампликон. По сравнению с зондом B-DNA1 зонд B-DNA2 характеризуется большей неэффективностью в отборе “своего” амплификона. Однако отборочная способность зонда B-DNA2 может быть резко увеличена путем замещения 12 из 13 дезоксирибонуклеозидов на соответствующие LNA-нуклеозиды. Как показано на фиг.3, использование зонда В-LNA2 вместо зонда B-DNA2 обусловливает более чем 10-кратное повышение чувствительности в проведенном тесте. В то же время зонд B-LNA2 сохраняет способность, свойственную немодифицированному зонду B-DNA2, эффективно различать родственные и неродственные ампликоны, обеспечивая тем самым специфичность LNA-олигомеров. Делается заключение, что (1) биотин, ковалентно присоединенный к LNA-модифицированному олигомеру, сохраняет способность связываться со стрептавидином, (2) LNA-модифицированные олигомеры эффективны в тесте на МТР-опосредованном отборе ампликонов, и (3) LNA предоставляет возможность резкого увеличения эффективности применения стандартных ДНК-олигонуклеотидов для аффинного отбора ПЦР-ампликонов.
Пример 144
Способность LNA-замещенных олигомеров отбирать соответствующие им ПЦР-ампликоны по механизму “инвазии цепи”.
Два идентичных состава по 10 мкл реакций с ампликоном 1 или 2 (приготовленные так же, как описано в примере 143) смешивали с 1, 5 или 25 пкмоль отборочного зонда B-LNA2 (биотин-GGTGGTTTGTTTG-3’; LNA-нуклеозиды выделены полужирным шрифтом; LHA-зонд, специфичный в отношении 2-го ампликона) в 1xSSC (0,15 M NaCl, 15 мМ цитрата натрия, рН 7,0) при общем объеме 450 мкл. Один набор реакций нагревали до 95° С на 5 минут с целью денатурации ампликонов и оставляли остывать при 25° С на 15 минут, обеспечивая, гибридизацию между зондом и последовательностью-мишенью ампликона. Другой набор реакций оставляли без этапа денатурации. По 190 мкл из каждой реакции переносили на покрытый стрептавидином планшет (Pierce, № по каталогу 15124) и инкубировали в течение 1 часа при 37° С. После промывки планшета фосфатно-солевым буфером (ФСБТ: 0,15 M Na+, рН 7,2, 0,05% Твин-20, 3× 300 мкл) добавляли 200 мкл антидигоксигениновых антител, помеченных пероксидазой (Boehringer Manneim разведение 1:1000 в фосфатно-солевом буфере). Планшеты инкубировали в течение 30 минут при 37° С и промывали (ФСБТ, 3× 300 мкл). Лунки тестировали на пероксидазную активность путем добавления 100 мкл раствора субстрата (0,1 М цитратфосфатного буфера, рН 5,0, 0,66 мг/мл орто-фенилендиаминдигидрохлорида, 0,012% перекиси водорода). Реакцию останавливали через 10 минут добавлением 10 мкл серной кислоты (0,5 М) и считывали параметры поглощения при 492 нм с использованием микропланшетного датчика. Когда ампликоны перед гибридизацией с отборочным зондом денатурировали (фиг.4А), были определены такие же эффективность и специфичность по последовательности, как в эксперименте, описанном в примере 143. Повышение концентрации зонда B-LNA2 с 1 до 5 пкмоль приводит к увеличению эффективности отбора. Дальнейшее увеличение до 25 пкмоль зонда приводит к снижению интенсивности сигнала. Это наблюдение соответствует насыщению потенциальных сайтов связывания биотина со стрептавидином-МТР. В варианте, когда ампликоны накануне гибридизации с отборочным зондом не денатурировали (фиг.4В), также был отмечен эффективный и специфичный по последовательности отбор ампликона. Действительно, полученные данные показывают, что отбор ампликонов без предварительной денатурации столь же эффективен и специфичен, как и отбор после денатурации. Это отчетливо подтверждает, что зонд Bio-LNA2 способен связываться со своей последовательностью-мишенью по механизму “инвазии цепи”. Как представляется, приведен первый пример специфичного по последовательности распознавания двухцепочечной ДНК в физиологичных по солевой концентрации условиях с использованием смешанного пурин-пиримидинового зонда. Помимо потенциального существенного упрощения ряда базовых исследовательских и диагностических процедур, данное неожиданное свойство LNA-модифицированных олигонуклеотидов может быть перспективным с точки зрения принципиального улучшения способов создания новых высокоэффективных лекарственных средств, используемых в технологии “антисмысловой терапии”, в частности, в т.н. “антигенном подходе”.
Пример 145
Эффективный специфичный по последовательности отбор ПЦР-ампликона LNA-замещенным олигомером, иммобилизованным на твердой подложке.
Лунки покрытого стрептавидином микротитровального планшета (Boehringer Mannheim) инкубировали в течение 1 часа либо с 5 пкмоль зонда B-DNA2 (биотин-GGTGGTTTGTTTG-3’; ДНК-зонд, специфичный в отношении 2-го ампликона), либо зонда В-LNA2 (биотин-GGTGGTTTGTTTG-3’; LNA-нуклеозиды выделены полужирным шрифтом; LHA-зонд, специфичный в отношении 2-го ампликона) при общем объеме 100 мкл в 1xSSC (0,15 М NaCl, 15 мМ цитрата натрия, рН 7,0). В целом четыре лунки инкубировали с зондом B-DNA2, четыре лунки - с зондом B-LNA2 и четыре лунки инкубировали с “чистым” буфером. После инкубации лунки трижды промывали в 1xSSC. Помеченные дигоксигенином ампликон-1 (60 мкл) или амплификон-2 (60 мкл) смешивали с 540 мкл 1xSSC, денатурировали нагреванием до 95° С на 5 минут и переносили (по 100 мкл) в лунки микротитровального планшета. По две лунки, содержащие либо B-DNA2, либо B-LNA2, либо не содержащие отборочного зонда, “получали” ампликон-1 и по две лунки, содержащие либо B-DNA2, либо B-LNA2, либо не содержащие отборочного зонда, “получали” ампликон-2. После 1 часа выдерживания при 37° С планшет трижды промывали фосфатно-солевым буфером (ФСБТ: 0,15 М Na+, рН 7,2, 0,05% Твин-20, 3× 300 мкл) и добавляли 200 мкл антидигоксигениновых антител, помеченных пероксидазой (Boehringer Mannheim, разведение 1:1000 в фосфатно-солевом буфере). Планшеты инкубировали в течение 30 минут при 37° С и промывали (ФСБТ, 3× 300 мкл). Лунки тестировали на пероксидазную активность путем добавления 100 мкл раствора субстрата (0,1 М цитратфосфатного буфера, рН 5,0, 0,66 мг/мл орто-фенилендиаминдигидрохлорида, 0,012% перекиси водорода). Реакцию останавливали через 6 минут добавлением 10 мкл серной кислоты (0,5 М) и считывали параметры поглощения при 492 нм с использованием микропланшетного датчика. Как показано на фиг.5, LNA-модифицированный отборочный зонд (В-LNA2) отбирает специфичный для него ампликон (ампликон-2) с очень высокой степенью эффективности и существенно лучше (с чувствительностью, большей примерно в 5 раз), чем соответствующий немодифицированный ДНК-отборочный зонд (B-DNA2). Не было выявлено сигнала в варианте, когда зонд B-LNA2 инкубировали с “чужим” ампликоном (ампликоном-1), что подтверждает очень точную специфичность зонда B-LNA2. Делается заключение о том, что LNA-амплифицированные олигонуклеотиды эффективны в специфичном по последовательности отборе ПЦР-ампликонов в варианте, когда они иммобилизованы на твердую поверхность. Также заключается, что использование LNA-амплифицированных олигонуклеотидов вместо стандартных ДНК-олигонуклеотидов позволит повысить показатели получаемых сигналов над уровнем фонового шума. Таким образом, LNA предоставляют способ существенного улучшения применимости современных тестов, основанных на использовании ДНК, в которых используются иммобилизованные отборочные зонды, например, в формате “иммобилизованных наборов”, в котором множественные зонды используются для одновременной детекции наличия нескольких различающихся нуклеотидных последовательностей в образце.
Пример 146
Возможность использования полностью перемешанных LNA-мономеров для существенного увеличения эффективности использования иммобилизованных биотинилированных ДНК-олигонуклеотидов для специфичного по последовательности отбора ПЦР-ампликонов в МТР-формате.
Три помеченных дигоксигенином ампликона из последовательности онкогена N-ras (см.: Nucl. Acids Res., 1985, 13 [14], 52-55) были сформированы с применением ПЦР-амплификации.
ПЦР-затравки:
Прямая затравка 5’-CCAGCTCTCAGTAGTTTAGTACA-3’, нуклеотиды 701-723 в соответствии с опубликованной последовательностью.
Обратная затравка 910-bр: 5’-GTAGAGCTTTCTGGTATGACACA-3’, нуклеотиды 1612-1590 (обратная последовательность в соответствии с опубликованной последовательностью).
Обратная затравка 600-bр: 5’-TAAGTCACAGACGTATCTCAGAC-3’ нуклеотиды 1331-1308 (обратная последовательность в соответствии с опубликованной последовательностью).
Обратная затравка 200-bр: 5’-CTCTGTTTCAGACATGAACTGCT-3’ нуклеотиды 909-886 (обратная последовательность в соответствии с опубликованной последовательностью).
Реакционная смесь ПЦР для ампликонов N-ras. 2,3 мкл геномной ДНК плаценты человека (440 нг/мкл), 50 мкл 10× ПЦР-буфера (без MgCl2, Perkin-Elmer), 30 мкл 25 мМ MgCl2, 50 мкл смеси нуклеотидов дНТФ-mix (по 2 мМ дАТФ, дГТФ, дЦТФ и 1,8 мМ дТТФ), 10 мкл 1 мМ дигоксигенин-11-дУТФ, 10 мкл 25 мкМ прямой затравки, 10 мкл 25 мкМ обратной затравки, 5 мкл 3 ед./мкл реактива AmpliTaq Gold (Perkin Elmer) и вода до 500 мкл. Условия ПЦР. Указанную выше реакционную смесь готовили для каждого из ПЦР-продуктов N-ras. Единственным различием было включение различных обратных затравок 910-bp, 600-bp и 200-bр. ПЦР-смеси разделяли на аликвоты по десяти ПЦР-пробиркам и помещали в амплификатор модели Perkin-Elmer 9600, соблюдая следующие условия амплификации: 95° С - 3 минуты; 95° С - 1 минута, 55° С - 2 минуты, 72° С - 3 минуты (30 циклов); 72° С - 10 минут; 4° С - выдержка. По 10 мкл каждой ПЦР анализировали в стандартном агарозном геле и наблюдали присутствие фрагментов ожидаемого размера - 910, 60 и 200 пар нуклеотидов. Условия тестирования. Лунки покрытого стрептавидином микротитровального планшета (Boehringer Mannheim; связывающая способность - 20 пкмоль биотина на лунку) инкубировали в течение 1 часа в 5xSSCT (0,75 М NaCl, 75 мМ цитрата, рH 7,0, 0,1% Твин-20) при 37° C с 1 пкмоль зонда DNA-Nras-Cap-A (биотин-5’-ТТССАСАGСАСАА-3’), LNA/DNA-Nras-Сар-А (биотин-5’-ТТССАСАGСАСАА-3’), LNA-Nras-Cap-A (биотин-5’-TTCCACAGCACAA-3’), DNA-Nras-Cap-B (биотин-5’-AGAGCGАТААСА-3’), LNA/DNA-Nras-Cap-B (биотин-5’-AGAGCCGATAACA-3’) или LNA-Nras-Cap-B (биотин-5’AGAGCCGATAACA-3’); полужирным шрифтом выделены LNA-нуклеозиды. Отборочные зонды Nras-Cap-A обеспечивают отбор ампликонов Nras-910, Nras-600 и Nras-200. Отборочные зонды Nras-Cap-B обеспечивают отбор ампликонов Nras-910 и Nras-600. После инкубации различных отборочных зондов лунки промывали буфером 5xSSCT и по 5 мкл нативных или денатурированных (95° С в течение 5 минут и 10 минут на льду) помеченных дигоксигенином ампликонов (Nras-910, Nras-600 или Nras-200) в 95 мкл 1xSSCT (0,15 М NaCl, 15 мМ цитрата, рН 7,0, 0,1% Твин-20) добавляли в расчете на каждую лунку и инкубировали в течение 1 часа при 37° С. Лунки трижды.промывали фосфатно-солевым буфером (1хФСБТ: 0,15 М Na+, рН 7,2, 0,05% Твин-20) и инкубировали в течение получаса при 37° С с 200 мкл антидигоксигениновых антител, помеченных пероксидазой (Boehringer Mannheim, разведение) 1:1000 в 1хФСБТ). Наконец, лунки трижды промывали в 1хФСБТ и тестировали на пероксидазную активность путем добавления 100 мкл раствора субстрата (0,1 М цитрат-фосфатного буфера, рН 5,0, 0,66 мг/мл орто-фенилендиаминдигидрохлорида, 0,012% перекиси водорода). Реакцию останавливали через 9 минут добавлением 100 мкл 0,5 М серной кислоты и четырежды разводили в H2SO4 с последующим считыванием параметров поглощения при 492 нм с использованием микропланшетного датчика. Как показано на фиг.23А, отборочные зонды, имеющие в своем составе по 12 LNA-нуклеозидов (LNA-Nras-Cap-A и LNA-Nras-Сар-В), высокоэффективно отбирают специфичные для них амплификоны без предварительной денатурации (нативные ампликоны). Отборочные зонды, в составе которых имеются по 4 LNA-нуклеозида (LNA/-DNA-Nras-Cap-A и LNA/DNA-Nras-Cap-B), отбирают те же самые амплификоны с меньшей эффективностью, а “чистые” отборочные ДНК-зонды (DNA-Nras-Cap-A и DNA-Nras-Сар-В) вообще не отбирают специфичные для них ампликоны. Контрольный ампликон (Nras-200) не отбирается зондами LNA-Nras-Cap-B и LNA/DNA-Nras-Cap-B: это подтверждает высокоточную специфичность LNA-включающих отборочных зондов. На фиг.23В показаны данные такого же эксперимента, проведенного на материале денатурированных ампликонов. По сути та же картина отличается от предыдущей лишь тем, что эффективность отбора ампликонов в целом возрастает. Делается заключение о том, что LNA-модифицированные олигонуклеотиды предоставляют способ конструирования отборочных зондов, которые будут эффективно отбирать ампликоны без их предварительной денатурации, т.е. осуществлять отбор по механизму вытеснения цепи. Такая их способность обеспечивает высокий уровень специфичности в современных форматах детекции ампликонов, основанных на последовательностях ДНК.
Пример 147
LNA-модифицированные олигонуклеотиды, как затравки для полимераз нуклеиновых кислот
Способность LNA-модифицированного олигонуклеотида (5’-GGTGGTTTGTTTG-3’: LNA-нуклеозиды выделены полужирным шрифтом) служить затравкой для зависимого от матрицы каталитического удлинения (достройки) исследовали с использованием трех различных типов полимераз. Это - обратная транскриптаза вируса M-MuLV (Boehringer Mannheim), которая может быть использована и с РНК-матрицей, и с ДНК-матрицей, фрагмент Кленова (ДНК-полимеразы кишечной палочки), который представляет тип стандартных ДНК-полимераз, и термостабильная полимераза BM-Taq (Boehringer Mannheim). В качестве контроля были проведены реакции достройки идентичной немодифицированной ДНК-затравки (5’-GGTGGTTTGTTTG-3’). LNA- и ДНК-затравки были помечены с использованием γ -32P-АТФ в соответствии с описанным выше в примере 137. В качестве матрицы использовали 50-мерный ДНК-олигонуклеотид - 5’-AAAAATCGACGCTCAAGTC-AGAAAAGCATCTCACAAACAAACAAACCACC-3’. В реакции с полимеразой M-MuLV (Boehringer Mannheim) включали 2 мкл помеченной LNA-затравки или ДНК-затравки (10 мкМ), 2 мкл ДНК-матрицы (10 мкМ), 2 мкл 2 мМ дНТФ, 2 мкл 10х буфера (500 мМ Трис-HCl, 300 мМ KCl, 60 мМ MgCl2, 100 мМ дитиотреитола, рН 8,3 (37° С)), 1 мкл фермента (20 ед./мкл) и воду до 20 мкл. Реакционные смеси имкубировали при 37° С в течение 60 минут. Реакция с полимеразой Кленова включала 2 мкл помеченной LNA-затравки или ДНК-затравки (10 мкМ), 2 мкл ДНК-матрицы (10 мкМ), 2 мкл 2 мМ дНТФ, 2 мкл 10х буфера (100 мМ Трис-HCl, 50 мМ MgCl2, 75 мМ дитиотреитола, pH 7,5), 1 мкл фермента (10 ед./мкл) и воду до 20 мкл. Реакционные смеси инкубировали при 37° С в течение 60 минут. Реакции с полимеразой BM-Taq (Boehringer Mannheim) включали 2 мкл помеченной LNA-затравки или ДНК-затравки (10 мкМ), 2 мкл ДНК-матрицы (10 мкМ), 2 мкл 2 мМ дНТФ, 2 мкл 10х буфера (100 мМ Трис-HCl, 50 мМ КС1, 15 мМ MgCl2, pH 8,3), 1 мкл фермента (5 ед./мкл) и воду до 20 мкл. Реакционные смеси инкубировали при исходной температуре 37° С, которую повышали со скоростью 1° С в минуту до 60° С, при которой смеси выдерживали 30 минут. По окончании инкубации реакции останавливали добавлением 10 мкл загрузочного буфера (0,25% бромфенолового синего - в/о, 0,25% ксиленцианола - в/о, 80% формамида - о/о). Образцы нагревали до 95° С на 1 минуту, помещали на лед и по 2 мкл загружали в 8%-ный полиакриламидный гель, который подвергали электрофорезу на оборудовании Life Technologies Inc. BRL, модель 52. После электрофореза гель высушивали на пластинке и подвергали авторадиографическому анализу (рентгенографическая пленка Kodak X-Omat AR). Как показано на фиг.7, отчетливые сходные продукты выявляются при обоих типах затравок (LNA и ДНК) при использовании фрагмента Кленова (дорожки 3) или полимеразы BM-Taq (дорожки 5). При использовании обратной транскриптазы M-MuLV (дорожки 2) продукт достройки может быть выявлен только в варианте с использованием LNA-затравки. Помеченные LNA- и ДНК-затравки, которые не подвергались каталитической достройке, показаны на дорожках 1, 4 и 6. Делается заключением о том, что инкорпорация LNA-нуклеозидов в состав стандартных ДНК-олигонуклеотидов не предотвращает распознавания полимеразами нуклеиновых кислот образующегося с их участием дуплексов “олигонуклеотид/матрица”. Также заключается, что LNA-моди-фицированные олигонуклеотиды могут служить столь же эффективными затравками, что и немодифицированные ДНК-олигонуклеотиды.
Пример 148
LNA-модифицированные олигонуклеотиды, как затравки для процессов амплификации матриц
Способность LNA-модифицированных олигонуклеотидов служить в качестве затравок для ПЦР-амплификации анализировали с использованием трех олигомеров, различающихся только по числу внесенных в их состав LNA-нуклеозидов, и они включали: 4 LNA-нуклеозида (затравка AL2 - 5’-GGTGGTTTGTTTG-3’: LNA-нуклеозиды выделены полужирным шрифтом), 1 LNA-нуклеозид (затравка AL10 - 5’-GGTGGTTTGTTTG-3’: LNA-нуклеозиды выделены полужирным шрифтом) и с отсутствием LNA-нуклеозидов (затравка FP2 - 5’-GGTGGTTTGTTTG-3’). В реакциях ПЦР (100 мкл) либо отсутствовала матрица (контроль), либо они включали 0,01 нг, 0,1 нг или 1 нг матрицы (плазмида pUC19), 0,2 мкМ обратной затравки (5’-GTGGTTCG-CTCCAAGCTG-3’), 0,2 мкМ прямой затравки AL2, AL10 или FP2, по 200 мкМ дАТФ, дГТФ, дЦТФ и дТТФ, 10 мМ Трис-НСl (рН 8,3), 1,5 мМ MgCl2, 50 мМ КСl и 2,5 ед. полимеразы BM-Taq. Всего было осуществлено 50 циклов: 94° С - 1 минута, 45° С - 1 минута, 72° С - 1,5 минуты (после первых 30 циклов добавляли 2,5 ед. Taq-полимеразы); использовали амплификатор Techne Genius. По окончании последнего цикла реакции инкубировали при 72° С в течение 3 минут и оставляли при 4° С на ночь. К 30 мкл каждой реакционной смеси добавляли 6 мкл загрузочного буфера (0,25%, в/о бромфенолового синего и 40%, о/о глицерина) и образцы (вместе размерным маркером Amplisize™ ) загружали в 2%-ный агарозный гель и подвергали электрофорезу в течение 45 минут при напряжении 150 В. Наконец, гель окрашивали бромистым этидием и фотографировали. Как показано на фиг.8, в ПЦР, в которой использовались немодифицированная прямая затравка FP2 и немодифицированная обратная затравка, образовывались выявляемые ампликоны правильного размера при всех протестированных количествах матрицы (дорожка 9 - 0,01 нг матрицы; дорожка 10 - 0,1 нг; и дорожка 11 - 1 нг). В контрольной (без матрицы) реакции сигнал вовсе отсутствовал (дорожка 12). В варианте с заменой прямой затравки FP2 на затравку, включающую по середине 1 LNA-нуклеозид (AL10), ампликоны также выявляются при всех тестированных количествах матрицы (дорожка 5 - 0,01 нг матрицы; дорожка 6 - 0,1 нг; и дорожка 7 - 1 нг). Это отчетливо говорит о том, что затравка AL10 поддерживает амплификацию по экспоненциальному типу, т.е. затравка AL10 может как достраиваться, так и использоваться сама со себе в качестве матрицы. Опять-таки, в контрольной реакции без матрицы (дорожка 8) амплификоны не образовывались. В варианте с заменой прямой затравки FP2 на затравку, включающую по середине 4 LNA-нуклеозида (AL2), ампликоны правильного размера не были выявлены ни в одной из реакций (дорожка 1 - 0,01 нг матрицы; дорожка 2 - 0,1 нг; дорожка 3 - 1 нг; дорожка 4 - матрица отсутствует). Однако при наивысшей концентрации матрицы (1 нг) в геле обнаруживается бэнд высокомолекулярного фрагмента (дорожка 3). По-видимому, это является артефактом влияния затравки RP1, на что указывает контрольная реакция, в которой каждая из затравок AL2 (дорожка А), AL10 (дорожка В), FP2 (дорожка С) и RP1 (дорожка D) были протестированы по их способности образовывать ампликон при наивысшем количестве матрицы (1 нг). Поскольку для AL2 была показана способность служить затравкой (пример 147), то отсутствие выявляемых ампликонов отчетливо подтверждает, что она не может работать в качестве матрицы, т.е. наличие блока из 4 соседних LNA-нуклеозидов предотвращает продвижение полимеразы и тем самым “переводит” реакцию в амплификацию линейного типа (продукт которой не может быть выявлен применяемыми экспериментальными приемами). Делается заключение о том, что LNA-модифицированные олигонуклеотиды могут быть использованы в качестве затравок в ПЦР-амплификации. Также заключается, что степень амплификации (градуированная в пределах от полностью экспоненциальной до линейной амплификации) может контролироваться за счет параметров конструирования LNA-модифицированного олигонуклеотида. Заявители отмечают, что возможность блокировки продвижения полимеразы путем внесения LNA-нуклеозидов в состав затравки обеспечит образование амплификонов, несущих одноцепочечные (“выступающие”) концы. Такие концы могут эффективно использованы в гибридизации без денатурации ампликона, и такое свойство сможет найти очень широкое применение.
Пример 149
Возможность ковалентной иммобилизации LNA-модифицированного олигомера, включающего 5’-антрахинон, на твердой подложке с использованием облучения и эффективность иммобилизованного олигомера в отборе комплементарного ДНК-олигонуклеотида 25 пкмоль/мкл или 12,5 пкмоль/мкл антрахинон-ДНК-олигомера (5’-AQ-CAG CAG TCG АСА GAG-3’) или антрахинон-LNA-модифицированного ДНК-олигомера (5’-AQ-CAG CAG TCG АСА GAG-3’: LNA-олигомер подчеркнут) раскапывали (1 мкл/пятно) в 0,2 М LiCl на поликарбонатной пластинке (Nunc). Олигомеры облучали в течение 15 минут мягким ультрафиолетом. После облучения пластинку трижды промывали водой Milli-Q и высушивали на воздухе. 25 мл 0,5 пкмоль/мкл комплементарного биотинилированного олигомера (5’-биотин-СТС TGT CGA CTG CTG-3’) гибридизовали с иммобилизованными олигомерами в 5xSSCT (75 мМ цитрата, 0,75 М NaCl, рН 7,7, 0,1% Твин-20) при 50° С в течение 2 часов. После четырехкратной промывки в 1xSSCT и однократной промывки в фосфатно-солевом буфере (ФСБТ: 0,15 М Na+, рН 7,2, 0,05% Твин-20) на пластинку наносили 25 мл ФСБТ, содержащего 0,06 мкг/мл соединенной со стрептавидином пероксидазы хрена и 1 мкг/мл стрептавидина. Пластинку инкубировали в течение 30 минут и промывали 4 раза в 25 мл ФСБТ. Затем пластинку проявляли хемолюминесцентным субстратом (SuperSignal; Pierce) в соответствии с рекомендациями производителя и использовали рентгенографическую пленку (CL-XPosure, Pierce 34075). Как показано на фиг.9, и AQ-ДНК-олигомер, и AQ-LNA-модифицированный ДНК-олигомер дают отчетливый сигнал. Делается заключение о том, что соединенные с антрахиноном LNA-модифицированные ДНК-олигомеры могут быть эффективно присоединены к твердой поверхности путем облучения, при том, что такие олигомеры, присоединенные таким образом, способны гибридизовать с своими комплементарными ДНК-олигонуклеотидными мишенями.
Пример 150
Гибридизация и наборная детекция с использованием различных LNA-модифицированных Сγ 3-помеченных октомеров.
Приготовление пластинок. Стеклянные пластинки аминосиланировали с использованием 10%-ного раствора аминопропилтриэтоксисилана в ацетоне с последующей промывкой ацетоном. На пластинки были раскапаны следующие олигонуклеотиды:

Для каждого олигонуклеотида было нанесено по 10 повторяющихся пятен объемом приблизительно 1 нл (каждое пятно) из каждой пипетки на каждую из 12 пластинок.

Пластинки и условия гибридизации:
пластинки 1, 2 и 3 гибридизовали с зондом aZ1 @ 300 фкмоль/мкл, 30 фкмоль/мкл, 3 фкмоль/мкл;
пластинки 4, 5 и 6 гибридизовали с зондом aZ2 @ 300 фкмоль/мкл, 30 фкмоль/мкл, 3 фкмоль/мкл;
пластинки 7, 8 и 9 гибридизовали с зондом aZ3 @ 300 фкмоль/мкл, 30 фкмоль/мкл, 3 фкмоль/мкл;
пластинки 10, 11 и 12 гибридизовали с последовательностью 16 @ 300 фкмоль/мкл, 30 фкмоль/мкл, 3 фкмоль/мкл.
Зонд, разбавленный в 30 мкл гибридизационного буфера (5× SSC, 7% лаурилсаркозина натрия) раскапывали по длине каждой пластинки, накрывали покровным стеклом и помещали в пластиковый контейнер на поверхность пластиковой вставки, которая расположена на бумажном полотенце, смоченном водой. Контейнеры закрывали алюминиевой фольгой с целью недопущения попадания света и инкубировали при +4° С в течение ночи.
Промывка пластинок. Покровные стекла удаляли и пластинки вставляли в держатель (6 пластинок на 1 держатель), который помещали в стеклянные чашки, завернутые в фольгу:
После промывки пластинки высушивали потоком воздуха и сканировали. Уровень флуоресценции определяли на слайд-сканере и полученные данные анализировали с использованием программы ImageQuant (Molecular Dynamics). Как показано на фиг.11, отсутствие связывания Сγ 3-зондов отмечается по отношению к олигонуклеотиду 3, включающему ошибку, при использовании и немодифицированного зонда (пластинки 10-12), и однократно LNA-модифицированного зонда aZ1 (пластинки 1-3), и однократно LNA-модифицированного зонда aZ2 (пластинки 4-6), и трехкратно модифицированного зонда aZ3 (пластинки 7-9) (т.е. выявляемый сигнал в отношении ошибочного олигонуклеотида сравним с фоновым сигналом). Интенсивность этих сигналов отчетливо коррелирует с количеством остатков LNA, имеющихся в составе зондов, и с концентрацией этих использованных зондов. Каждый остаток LNA-T увеличивает интенсивность сигнала приблизительно вдвое по сравнению с сигналом нормального ДНК-олигонуклеотидного зонда: т.е. зонды aZ1 и aZ2 дают удвоенный сигнал последовательности 16, а зонд aZ3 - в 8 раз более сильный сигнал последовательности 16. Различение правильной и ошибочной мишеней оказывается хорошим при использовании замещений остатками LNA-T, а увеличение интенсивности сигнала обеспечивает лучшую распознаваемость ошибочных мишеней.
Пример 151
Гибридизация и детекция концевых ошибок в наборном тесте с использованием LNA-модифицированных Сγ 3-помеченных октомеров.
Приготовление пластинок. Стеклянные пластинки аминосиланировали с использованием 10%-ного раствора аминопропилтриэтоксисилана в ацетоне с последующей промывкой ацетоном. На пластинки были раскапаны в количестве 1 пкмоль/мкл следующие олигонуклеотиды:

Для каждого олигонуклеотида было нанесено по 10 повторяющихся пятен объемом приблизительно 1 нл (каждое пятно) из каждой из 6 пипеток на каждую из 12 пластинок.

В обозначения зондов, включающих LNA-мономеры, был включен префикс 35Z, который также входит в идентификационный номер последовательности.
Отдельные LNA-мономеры определены выделением курсивом/полужирным и расположены по 3’и 5’-концам LNA-мономеров.
Пластинки и условия гибридизации. Каждый зонд гибридизовали на отдельной пластинке и концентрации всех зондов составили 1 фкмоль/мкл. Каждый зонд разводили в гибридизационном буфере (5× SSC, 7% лаурилсаркозин натрия), 30 мкл которого раскапывали по длине пластинки, которые затем накрывали покровным стеклом и помещали в пластиковый контейнер на поверхность пластиковой вставки, которая расположена на бумажном полотенце, смоченном водой. Контейнеры закрывали алюминиевой фольгой с целью недопущения попадания света и инкубировали при +4° С в течение ночи.
Промывки пластинок.
Покрывные стекла удаляли и пластинки помещали в держатель (8 пластинок на 1 держатель, которые помещали в стеклянные чашки, обернутые в фольгу. Все пластинки промывали в 5× SSC дважды по 5 минут при температуре +4° С. После промывки пластинки высушивали в потоке воздуха и сканировали. Уровень флуорисценции анализировали в программе Image Quant Molecular Dynamics).
Заключение. Как показано на фиг.12 и 13, зонды, включающие LNA-нуклеозиды по своим концам 5’ и 3’, в большинстве случаев проявляют существенно лучшие параметры в различении правильных и ошибочных последовательностей-мишеней по сравнению с соответствующими им немодифицированными олигонуклеотидами.
Для ДНК-олигонуклеотидов ошибки С=Т наиболее трудны для различения, например, тогда, когда зонд, характеризующийся последовательностью 1, гибридизует с последовательностью-мишенью 132 и когда зонд, характеризующийся последовательностью 5, гибридизует с последовательностью-мишенью 134. Другие ошибки выявляемы, такие как ошибки Т=Т и G=T, хотя сигналы в этих случаях оказываются более слабыми: например, в случаях, когда зонды, характеризующиеся последовательностями 5 и 6, гибридизуют с последовательностью-мишенью 135. LNA-олигомеры приводят к существенному снижению интенсивности этих ошибочных сигналов для С=Т и Т=Т до уровней, сопоставимых с уровнями, характерными для других типов ошибок. Относительная интенсивность сигналов для зондов, имеющих последовательности 1, 2 и 3, оказывается сходной для ДНК- и LNA-олигомеров. Однако для последовательностей зондов 4, 5 и 6 LNA-олигомеры проявляют существенно более высокую интенсивность сигналов в случаях, когда они гибридизуют со своими “правильными” последовательностями-мишенями, соответственно, 9, 133 и 134.
Пример 152
Гибридизация и детекция концевых ошибок в наборном тесте с использованием AT- и полностью LNA-модифицированных Сγ 3-помеченных октомеров
Приготовление пластинок.
Стеклянные пластинки аминосиланировали с использованием 10%-ного раствора аминопропилтриэтоксисилана в ацетоне с последующей промывкой ацетоном. На пластинки были раскапаны в количестве 1 пкмоль/мкл следующие олигонуклеотиды:

Для каждого олигонуклеотида было нанесено по 10 повторяющихся пятен объемом приблизительно 1 нл (каждое пятно) из каждой из 6 пипеток на каждую из 36 пластинок.

В обозначениях зондов, включающих LNA-мономеры, использованы префиксы ATZ- или A11Z-, которые являются частью номера последовательности. Отдельные LNA-мономеры обозначены курсивом в описаниях LNA-олигомеров.
Пластинки и условия гибридизации. Каждый зонд гибридизовали на отдельной пластинке и концентрации всех зондов составили 1 фкмоль/мкл. Каждый зонд разводили в гибридизационном буфере (5× SSC, 7% лаурилсаркозин натрия), 30 мкл которого раскапывали по длине пластинки, которые затем накрывали покровным стеклом и помещали в пластиковый контейнер на поверхность пластиковой вставки, которая расположена на бумажном полотенце, смоченном водой. Контейнеры закрывали алюминиевой фольгой с целью недопущения попадания света и инкубировали при комнатной температуре в течение ночи.
Промывка пластинок. Покровные стекла удаляли и пластинки помещали в держатель (9 пластинок на 1 держатель, которые помещали в стеклянные чашки, обернутые в фольгу. Все пластинки промывали в 5× SSC дважды по 5 минут при комнатной температуре. После промывки пластинки высушивали в потоке воздуха и сканировали. Уровень флуоресценции анализировали с помощью слайд-сканера и полученные данные анализировали в программе ImageQuant (Molecular Dynamics).
Заключение. Как показано на фиг.15А, 15В и 15С, средний уровень интенсивности гибридизации ДНК при комнатной температуре составил примерно 10% от уровня интенсивности, достигаемого при использовании олигонуклеотидов, LNA-модифицированных по AT или всем нуклеозидам. Не обнаружено сигналов на пластинках, которые гибридизовали с ДНК-зондами 5 и 6. Следовательно, такие условия не являются оптимальными для ДНК-зондов. Однако распознавание правильных и ошибочных последовательностей при использовании при заменах LNA-нуклеозидов по основаниям А и Т было высокоэффективным. Уровень жесткости условий гибридизации для полностью LNA-модифицированных олигонуклеотидов может не быть столь высоким для различения правильных и ошибочных последовательностей, который требуется для столь же хорошего различения с использованием AT-LNA-олигонуклеотидов. Олигонуклеотиды, включающие LNA-модификации, работают очень хорошо, а наиболее трудными для идентификации являются такие ошибки:
зонд 1 в отношении последовательности-мишени 135= СТ-ошибка;
зонд 2 в отношении последовательности-мишени 131= GT-ошибка;
зонд 3 в отношении последовательности-мишени 15= АА-ошибка;
зонд 4 в отношении последовательности-мишени 131= СТ-ошибка;
зонд 5 в отношении последовательности-мишени 135= ТТ-ошибка;
зонд 6 в отношении последовательности-мишени 135= GT-ошибка;
зонд 6 в отношении последовательности-мишени 133= GA-ошибка.
При использовании AT-LNA-олигонуклеотидов хорошие результаты по различению были получены тогда, когда интенсивность ошибочных сигналов обычно составляла 50% от интенсивности правильных сигналов. Для таких ошибок полностью LNA-модифицированные олигомеры проявляют интенсивность сигналов на ошибках на уровне 50-70% от уровня интенсивности правильных сигналов. В целом модифицирование с использованием LNA позволяет использовать более высокие температуры для гибридизации и промывки, при том, что концевые ошибки будут определены. Эти результаты по крайней мере столь же хороши, как те, которые были получены с ДНК-зондами, гибридизуемыми при 4° С (см. пример 151).
Пример 153
Использование [α -33P]-dдНТФs и ДНК-полимеразной системы ThermoSequenase™ для секвенирования ДНК-матриц, включающих мономеры LNA-T.
Радиоактивно помеченные реакции концевого секвенирования были сформированы с целью определения способности мономера LNA-T служить в качестве матрицы для ДНК-полимеразы. 15-мерная затравка (последовательность: 5’-TGC ATG TGC TGG AGA-3’) была использована для прайминга следующих коротких олигонуклеотидных последовательностей (мономер LNA обозначены полужирным шрифтом):

Были сформированы следующие реакционные смеси:
Смесь для матрицы 1:
× 16 буфера ThermoSequenase 2 мкл
1 пкмоль/мкл матрицы 1 6 мкл
вода 4 мкл
ДНК-полимераза ThermoSequenase (4 ед./мкл) 2 мкл
Общий объем 20 мкл
Смесь для матрицы TZ1: × 16 буфера ThermoSequenase 2 мкл
затравка, 2 пкмоль/мкл 6 мкл
матрица TZ1, 1 пкмоль/мкл 6 мкл
вода 4 мкл
ДНК-полимераза ThermoSequenase (4 ед./мкл) 2 мкл
Общий объем 20 мкл
В каждую эппендорфскую пробирку добавляли по 2 мкл смеси нуклеотидов (7,5 мкМ каждого из дНТФ). В пробирки 1 и 5 добавляли по 0,5 мкл [α -33P]-ddATP. В пробирки 2 и 6 добавляли по 0,5 мкл [α -33P]-ddCTP. В пробирки 3 и 7 добавляли по 0,5 мкл [α -33P]-ddGTP. В пробирки 4 и 8 добавляли по 0,5 мкл [α -33P]-ddTTP. В каждую из пробирок 1-4 добавляли по 4,5 мкл смеси для матрицы 1. В каждую из пробирок 1-4 добавляли по 4,5 мкл смеси для матрицы TZ1. Все реакции инкубировали при 60° С в течение 3 минут. Реакции останавливали добавлением 4 мкл стоп-раствора формамида/EDTA. Реакционные смеси нагревали до 95° С в течение 3 минут с последующей загрузкой в 19%-ный полиакриламидный гель, содержащий 7 М мочевины. Гели фиксировали в 10%-ной уксусной кислоты и 10% метанола с последующим переносом на бумагу ЗММ и высушивали. Высушенные гели экспонировали с использованием рентгенографической пленки Kodak Biomax.
Полученные результаты представлены на фиг.18 (дорожки 1-4) и фиг.19 (дорожки 5-8). Эти дорожки соответствуют следующим реакциям: (фигура 18): дорожка 1 - ddATP; дорожка 2 - ddCTP; дорожка 3 - ddGTP; дорожка 4 - ddTTP; дорожка 5 - олигонуклеотидные маркеры, состоящие из 8-32 оснований; (фигура 19): дорожка А - олигонуклеотидные маркеры, состоящие из 8-32 оснований; дорожка 5 - ddATP; дорожка 6 - ddCTP; дорожка 7 - ddCTP; дорожка 8 - ddTTP.
Как видно из фиг.18 и 19, полные последовательности обеих матриц могут быть легко прочитаны и отсканированы. Искомой является последовательность 5’-TGG AAC GTA-3’, которая соответствует последовательности матрицы 3’-АСС TTG СТА-5’. Это указывает на то, что единственный мономер LNA-T может служить матрицей для ДНК-полимераз. Мономер LNA-T конкретно определяется как “Т” с учетом включения ddATP.
Применение в терапии
Пример 154
Возможность внесения в клетки LNA-модифицированных олигомеров. Эксперименты с радиоактивно помеченными LNA-олигомерами 10 пкмоль олигодезоксирибонуклеотида (ОДН) (ОДН №10: 5’-ТТА ACG TAG GTG CTG GAC TTG TCG CTG TTG ТАС ТТ-3’: 35-нуклеотид, комплементарный участку последовательности гена катепсина-D человека) и по 10 пкмоль двух LNA-олиго-нуклеотидов - AL16 (5’-d[TGT GTG AAA TTG TTA Т]-3’; LNA-нуклеозиды выделены полужирным шрифтом) и AL17 (5’-d[ATA AAG TGT AAA G]-3’; LNA-нуклеозиды выделены полужирным шрифтом) смешивали в полинуклеотидкиназой Т4 (10 ед.; BRL, № по каталогу 510-8004SA), 5 мкл γ -32Р-АТФ (5000 Ки/ммоль, 10 мкКи/мкл, Amersham) в киназном буфере (50 мМ Трис-HCl, рН 7,6, 10 мМ MgCl2, 5 мМ дитиотреитола, 0,1 мМ EDTA). Образцы инкубировали в течение 45 минут при 37° С и после этого нагревали до 68° С на 10 минут и затем переносили в +0° С. Не включившиеся нуклеотиды удаляли путем пропускания через колонки Chroma Spin TE-10 (Clontech, № по каталогу К1320-1). Выходы составили 5× 105 имп./мин на 1 мкл, 2× 105 имп/мин на 1 мкл и 0,8× 105 имп/мин на 1 мкл для, соответственно, ОДН №1, AL16 и AL17. Клетки MCF-7 опухоли молочной железы человека, исходно полученные из банка культур Human Cell Culture Bank (Mason Res. Inst., Rockville, США), культивировали в культуральной среде DME/F12 (1:1), в которую добавляли 1% инактивированной нагреванием плодной телячьей сыворотки (Gibco BRL), 6 нг/мл бычьего инсулина (Novo) и 2,5 мМ глутамакса (Life Technologies) в 25-см2 колбах для клеточных культур (Nunclon, NUNC) и инкубировали во влажном инкубаторе при 37° С в атмосфере 5% СО2, 20% О2, 75% N2. Клетки MCF-7 в момент проведения эксперимента находились на стадии 40%-ного слияния. Небольшое количество (менее 0,1 пкмоль) обработанных киназой олигонуклеотидов смешивали с 1,5 мкг плазмиды pEGFP-NI (Clontech, № по каталогу 60851) и смешивали с 100 мкл разбавленного трансфекционного агента FuGENE6 (Boehringer Mannheim, № по каталогу 1-814443): разведение - 5 мкл FuGENE6 в 95 мкл культуральной среды DME/F12 без сыворотки. Смесь “FuGENE6/ДНК/олигомер” напрямую добавляли в культуральную среду (5 мл) с растущими клетками MCF-7 и инкубировали с клетками в течение 18 часов, точно соблюдая рекомендации производителя. Были поставлены три группы экспериментов: (1) ОДН №10 + pEGFP-NI; (2) AL16 + pEGFP-NI; (3) AL17 + pEGFP-NI. Поглощение клетками ДНК/LNA-материала анализировали путем удаления содержащей смесь “FuGENE6/AHK/onHroMep” культуральной среды (аликвоту переносили в контейнер для подсчет сцинтилляций). Клетки однократно ополаскивали фосфатно-солевым буфером (ФСБ), добавляли свежую культуральную среду и анализировали клетки с помощью флуоресцентного микроскопа. Приблизительно 30% трансфицированных клеток содержали флуоресцирующий зеленым цветом материал: это указывает на то, что приблизительно 30% этих клеток поглотили плазмиду pEGFP-NI и экспрессируют репортерный зеленый флуоресцирующий белок, кодируемый этой плазмидой. После флуоресцентной микроскопии из культуральных колб удаляли слившиеся клетки MCF-7. Вкратце, удаляли культуральную среду, затем клетки промывали раствором 0,25%-ного трипсина (Gibso BRL), 1 мМ EDTA в ФСБ (без ионов Мg2+ и Са2+), добавляли 1 мл трипсина/EDTA и клетки инкубировали в течение 10 минут при 37° С. В процессе инкубации клетки “размягчались” и могли быть легко ресуспендированы и перенесены в емкость сцинтилляционного счетчика. Затем клетки полностью суспендировали, добавляя 10 мл сцинтилляционной смеси Optifluor (Packard, № по каталогу 6013199), и материал обсчитывали на сцинтилляционном счетчике Wallac-1409. Получены следующие результаты: (1) ОДН №10+ pEGFP-NI: приблизительно 1,4% добавочной радиоактивности ассоциировалось с клеточным материалом; (2) AL16 + pEGFP-NI: приблизительно 0,8% добавочной радиоактивности ассоциировалось с клеточным материалом; (3) AL17 + pEGFP-NI: приблизительно 0,4% добавочной радиоактивности ассоциировалось с клеточным материалом. Делается вывод о том, что 0,4-0,8% добавленных LNA-олигомеров были поглощены клетками.
Пример 155
Эффективная доставка LNA в живые клетки MCF-7 опухоли молочной железы человека.
С целью повышения эффективности поглощения LNA клетками MCF-7 человека были протестированы различные трансфекционные агенты и использовались разные концентрации 5’F1ТС-помеченных LNA и ДНК. Были исследованы олигонуклеотиды, описанные в таблице.

Зонды AL16 и AL17 были каталитически помечены FITC в соответствии с описанным в примере 128. Олигонуклеотиды EQ3009-01 и EQ3008-01 были помечены FITC с применением стандартных твердофазных химических реакций. Были протестированы три трансфекционных агента - FuGENE6 (Boehringer Mannheim, № по каталогу 1-814443), SuperFect (Quiagen, № по каталогу 301305) и Lipofectin (Gibco BRL, № по каталогу 18292-011). Клетки MCF-7 опухоли молочной железы человека культивировали так же, как это описано ранее (пример 154). За три дня до проведения экспериментов клетки высевали при плотности приблизительно 8 тысяч клеток на 1 см2. В зависимости от варианта эксперимента клетки MCF-7 высевали в стандартные колбы Т25 (Nunc, Life Technologies, № по каталогу 163371А), 24-луночные планшеты (Nunc, Life Technologies, № по каталогу 143982А) или флаконы-слайды (Nunc, Life Technologies, № по каталогу 170920А). Эксперименты проводили на этапе 30-40%-ного слияния клеток. Поглощение клетками LNA и ДНК анализировали в бессывороточных условиях, т.е. нормальную, содержащую сыворотку культуральную среду DME/F12 удаляли и заменяли на среду DME/F12 без сыворотки перед тем, как трансфекционную смесь добавляли к клеткам. В этих условиях агент SuperFect проявил токсичность в отношении клеток MCF-7. Трансфекционные смеси, содержащие SuperFect и плазмидная ДНК (pEGFP-N1, Clontech, № по каталогу 6085-1), ДНК-олигомер или LNA-олигомер, были в равной степени токсичными в отношении клеток MCF-7. В противоположность агенту SuperFect, агенты FuGene6 и Lipofectin работали эффективно в отношении плазмидной ДНК (pEGFP-N1). Однако только липофектин был способен обеспечивать эффективную доставку олигонуклеотидов в живые клетки MCF-7. Вкратце, эффективная доставка FITC-помеченных LNA и ДНК в клетки MCF-7 была отмечена при культивировании клеток в культуральной среде DME/F12, содержащей 1% плодной телячьей сыворотки, до примерно 40%-ного слияния. Агент Lipofectin затем разбавляли в 40 раз в культуральной среде DME/F12, лишенной сыворотки и комбинировали с олигомером до концентрации 750 нМ этого олигомера. Олигонуклеотиднолипофектиновый комплекс оставляли образовываться на 15 минут при комнатной температуре и далее разбавляли бессывороточной культуральной средой до конечной концентрации олигомера 250 нМ и 0,8 мкг/мл липофектина. Затем культуральную среду удаляли из клеток и заменяли на культуральную среду, содержащую олигонуклеотиднолипофектиновый комплекс. Клетки инкубировали при 37° С в течение 6 часов, однократно ополаскивали в бессывороточной среде DME/F12 и инкубировали еще в течение 18 часов в среде DME/F12 с сывороткой (1% FCS) при 37° С. Результаты эксперимента оценивали либо напрямую на живых клетках в культуральных колбах или на 24-луночных планшетах, либо на клетках, культивируемых во флаконах-слайдах и зафиксированных в 4%-ном охлажденном на льду PFA. Во всех случаях использовали флуоресцентный микроскоп модели Leica DMRB, оборудованный высокоразрешающей камерой CCD. Результаты, полученные на живых клетках, показаны на фиг.16, а результаты, полученные на фиксированных клетках, культивировавшихся во флаконах-слайдах, показаны на фиг.17. Во всех случаях клетки, показанные на фиг.16 и 17, были трансфицированы FITC-помеченной LNA-молекулой AL16, Путем подсчета общего числа клеток и клеток, флуоресцирующих зеленым цветом, в нескольких зрительных полях было установлено, что FITC-помеченные LNA молекулы AL16 были трансфицированы приблизительно в 35% клеток MCF-7. Важным является то, что была показана предпочтительная локализация LNA в ядрах клеток (фиг.17). Это важно, потому что ядерное поглощение флуоресцирующих олигонуклеотидов коррелирует с их потенциальной антисмысловой активностью (Stein С.A. et al., 1997, ″ Making sense of antisense: A debate", b ″ HMS Beagle: a BioMedNet Publ." - hmsbeagle.соm/06/cutege/overwiev.htm (адрес в Интернете)). Увеличение количества олигомера и липофектина до конечной концентрации 1250 нМ олигомера и 4 мкг/мл липофектина приводит лишь к незначительному увеличению доли флуоресцирующих зеленым цветом клеток. Увеличение этой концентрации далее становится токсичным для клеток. Сходные результаты были получены при анализе других LNA и FITC-помеченных ДНК-олигонуклеотидов (см. вышеприведенную таблицу). Делается заключение о том, что: (1) LNA может быть эффективно доставлен в живые клетки MCF-7 опухоли молочной железы с использованием трансфекции, опосредуемой липофектином; (2) постоянно высокая доля клеток (30% и более) трансфицируется при использовании конечной концентрации 250 нМ LNA и 0,8 мкг липофектина на 1 мл культуральной среды, не содержащей сыворотки. Увеличение концентрации LNA и липофектина до 5-кратного эффективность трансфекции увеличивает незначительно; (3) процедура обеспечивает трансфекцию LNA в ядра клеток, что, в соответствии с известным из литературы, указывает на перспективность трансфекции LNA с целью достижения антисмысловых эффектов в клетках.
Пример 156
Возможность трансфекции LNA-модифицированных олигонуклеотидов в клетки. Эксперимент с LNA-олигомерами, помеченными флуоресцеином
Два LNA-олигомера AL16 (5’-TGT GTG AAA TTG ТТА Т-3’; LNA-нуклеозиды выделены полужирным шрифтом) и AL17 (5’-АТА AAG TGT AAA G-3’: LNA-нуклеозиды выделены полужирным шрифтом) помечали флуоресцеином в соответствии с описанным в примере 128. Клетки MCF-7 опухоли молочной железы человека культивировали так же, как это было описано в примере 154. Были поставлены 3 экспериментальных варианта: (1) приблизительно 1,5 мкг FITC-помеченного AL16; (2) приблизительно 1,5 мкг FITC-помеченного AL17; и (3) приблизительно 0,75 мкг FITC-помеченного AL16 и 0,75 мкг плазмиды pRSV-β Gal (плазмида, экспрессирующая бактериальный ген lacZ, который кодирует фермент β -галактозидазу: Tulchinsky et al., 1992, Proc. Natl. Acad. Sci. USA, 89, 9146-9150). Два LNA-олигомера и смесь LNA-плазмиды смешивали с FuGENE6 и добавляли к клеткам MCF-7 в соответствии с описанным в примере 154. После инкубации в течение 18 часов клеточное поглощение LNA-олигомеров оценивали с помощью флуоресцентного микроскопа в отношении культур клеток. В части обработанных клеток отмечено присутствие зеленого флуоресцентного сигнала (см. фиг.16): это указывает на то, что клетки поглотили помеченные флуоресцеином LNA. В этом отношении помеченный флуоресцеином AL16 обладал предпочтительными параметрами по отношению к помеченному флуоресцеином AL17. После проведения флуоресцентной микроскопии культуральную среду удаляли из клеток, обрабатывавшихся и помеченным флуоресцеином AL16, и плазмидой pRSV-β Gal. Клетки промывали однократно ФСБ, фиксировали 2% (о/о) формальдегидом, 0,2% (о/о) глутарового альдегида при 4° С в течение 5 минут, и содержащие β -галактозидазу клетки окрашивались в синий цвет при воздействии реактивом Х-gal (5-бром-4-хлор-3-индоил-β -D-галактопиранозид), который бесцветный объект окрашивает в синий цвет в присутствие β -галактозидазной активности. Окрашивание реактивом Х-gal показало, что плазмида pRSV-β Gal эффективно трансфицируется внутрь клеток. Делается заключение о том, что помеченные флуоресцеином LNA-олигомеры поглощались клетками.
Пример 157
Относительная стабильность LNA-модифицированных олигонуклеотидов в условиях клеточной культуры.
После флуоресцентной микроскопии, описанной в примере 156, клетки, трансфицировавшиеся только помеченным флуоресцеином AL16-LNA, оставляли инкубироваться в течение 3 дополнительных дней. За этот период времени число флуоресцирующих зеленым цветом клеток не изменялось. Делается вывод о том, что помеченные флуоресцеином LNA-олигомеры характеризуются хорошими параметрами стабильности в условиях, существующих в клеточной культуре.
Пример 158
Блокирование антисмысловыми “замкнутыми нуклеиновыми кислотами” (LNA) индуцируемого [D-Ala2]-дельторфином обезболивания у “находящихся в сознании” крыс в тесте на “тепловодный хвостовой щелчок”.
Самцам крыс линии Sprague-Dawley (вес тела 300 г) внутриоболочечно имплантировали полиэтиленовый катетер с последующим восстановительным периодом в течение 5 дней, после чего проводили инъецирование (включая контроль). Антисмысловые LNA-конструкции (12,5 и 2,5 мкг на одну инъекцию) вводили в объеме 5 мкл дважды в день (в 8 часов утра и в 5 часов вечера) в течение 3 дней. Не было выявлено каких-либо неспецифических проявлений или токсичности, на что указывают наблюдения за локомоторной активностью и взвешивания животных. На следующий день после последней инъекции крысам инъецировали [D-Ala2]-дельторфин (60 мкг, внутриоболочечно) и тестировали на “тепловодный щелчок” (реакцию попадания хвоста животного в воду, подогретую до 52° С), позволяющий оценивать произошедшее обезболивание, опосредуемое δ -опиоидными рецепторами. На фиг.14 приведены данные, являющиеся величинами, усредненными по 6-8 животным в пределах каждой из опытных групп (данные конвертированы в процентный показатель наиболее вероятного ответа, %МРЕ). Статистический анализ был проведен по Крускалль-Уоллесу в программе ANOVA с последующим сопоставлением экспериментальных вариантов с контрольными. Как показано на фиг.14, дельторфин обусловливает мощный обезболивающий эффект у животных, которым вводили солевой раствор. Этот эффект на статистически значимом уровне подавлялся обеими группами антисмысловых LNA-конструкций (12,5 и 2,5 мкг) по сравнению с контролем, которому вводили имитационный солевой раствор.
LNA-твердые подложки
Пример 159
Базовый метод получения DMT-LNA-нуклеозидсукцинатов.
Защищенные по основанию DMT-LNA-нуклеозиды и янтарный ангидрид (1,5 эквивалента) помещали в безводный этилендихлорид (≈ 10 мл/г нуклеозида). К этой смеси добавляли триэтиламин (2 эквивалента) и полученную смесь перемешивали при комнатной температуре. После этого реакционную смесь подвергали ВЭЖХ (при тех же самых условиях, которые применялись в реакции тритилирования). После завершения реакции (>95%) реакционную смесь концентрировали, выпаривали вместе с этилендихлоридом и ацетонитрилом и высушивали в вакууме с целью удаления триэтиламина. Остаток растворяли в этилендихлориде или этилацетате (≈ 100 мл/г исходного нуклеозида), промывали холодной 10%-ной лимонной кислотой (3× 80 мл/г) и холодной водой (3× 80 мл/г). Органический слой высушивали над безводным сульфатом натрия, отфильтровывали и концентрировали в присутствие 1-2 эквивалентов триэтиламина или без него. Оставшееся твердое вещество выпаривали вместе с безводным ацетонитрилом (2-3х) и высушивали в вакууме с получением продукта в виде твердого вещества белого цвета. Базовый метод получения LNA-нуклеозидных подложек. Защищенный по основанию DMT-LNA-нуклеозидсукцинат (в виде свободной кислоты или ее триэтиламмонийной соли, 65 мкмоль на 1 г подложки), аминопроизводную подложку (Primer Support™ 30HL, 160 мкмоль аминогрупп на 1 г подложки), DMAP (3 мг/г) и 1-(3-[диметиламино]пропил)-3-этилкарбодиимидгидрохлорид (80 мг на 1 г подложки) помещали в безводный пиридин (6 мл/г подложки). К этой смеси добавляли триэтиламин (16 мкл/г подложки) и полученную смесь выдерживали в шейкере при 150 об/мин в течение ночи. Подложку отфильтровывали, промывали метанолом (3× 10 мл/г подложки) и дихлорметаном (3× 10 мл/г подложки). После высушивания на воздухе подложку высушивали в вакууме в течение получаса. К этому материалу добавляли 6% DMAP в безводном ацетонитриле (смесь-А, ≈ 3 мл/г подложки) и смесь 20% уксусного ангидрида/30% 2,4,6-коллидина/50% ацетонитрила (смесь-В, ≈ 3 мл/г подложки). Смесь помещали в шейкер на 5 часов. Подложку отфильтровывали, промывали безводным дихлорметаном (2× 10 мл/г подложки) и высушивали так же, как перед этим. Ее ресуспендировали в перемешанных смеси-А и смеси-В (общий объем 6 мл на 1 г подложки) и выдерживали в шейкере на протяжении ночи. Подложку отфильтровывали, промывали метанолом (6× 10 мл/г подложки), дихлорметаном (3× 10 мл/г подложки) и высушивали на воздухе. Далее ее высушивали в вакууме в течение 5-6 часов. Количественные параметры загружаемости определяли в тесте с диметокситритилом: они были определены на уровне приблизительно 40 мкмоль/г.
Пример 160
Синтез первой цепи кДНК с использованием поли-дТ затравок, включающие мономеры LNA-T.
Реакции были сформированы с целью определения способности полидезоксириботимидиновых затравок, включающих LNA-T-мономеры, затравлять синтез первой цепи кДНК. Следующие поли-дТ затравки были протестированы (LNA-мономеры выделены полужирным шрифтом):
В качестве контроля использовали поли-дТ затравку, входящую в состав набора реактивов для мечения кДНК - RPK0140 kit Сγ Dye cDNA label-ling kit (Amersham Pharmacia Biotech).
Реакции были сформированы для каждой из вышеназванных затравок следующим образом:
мРНК резуховидки Arabidopsis, 0,5 мкг/мкл 1 мкл
поли-дТ затравка, 8 пкмоль/мкл 2 мкл
× 5 обратнотранскриптазный буфер AMV 4 мкл
вода 1 мкл
общий объем 8 мкл
Эту смесь затем нагревали до 75° С в течение 3 минут и затем оставляли остывать до комнатной температуры в течение по крайней мере 10 минут.
Затем к каждой реакционной смеси добавляли следующее:
пирофосфат натрия, 80 мМ 1 мкл
ингибитор рибонуклеазы плаценты человека, 20 ед./мкл 1 мкл
раствор дНТФ, 0,5 мМ 7 мкл
[α -33P]-дАТФ, 10 мКи/мл, 3000 Ки/ммоль 2 мкл
обратная транскриптаза AMV, 20 ед./мкл 1 мкл
Общий объем 20 мкл
Реакции инкубировали при 42° С в течение 2 часов. Затем их нагревали до 95° С на 3 минуты с последующей загрузкой в 6%-ный полиакриламидный гель, содержащий 7 М мочевины. Гели фиксировали в 10% уксусной кислоты + 10% метанола с последующим переносом на бумагу 3ММ и высушиванием. Высушенные гели экспонировали на рентгенографическую пленку Kodak Biomax в течение ночи.
Отпечатки отчетливо показали, что LNA-включающие олигонуклеотидные затравки RTZ1-4 были способы эффективно затравлять синтез кДНК. Количество и интенсивность кДНК-продуктов, образовавшихся в поставленных реакциях, были такими же, как и у продуктов, полученных с использованием прикрепленных контрольных дТ-олигонуклеотидов. Небольшое количество кДНК образовывалось и с затравки RTZ 5, однако выход был существенно меньшим по сравнению с выходом с контрольной олигонуклеотидной затравки.
Пример 161
Эффективное функционирование LNA-модифицированных олигонуклеотидов, ковалентно присоединенных к сефарозным шарикам, при специфичном по последовательности отборе молекул РНК.
Три олигонуклеотида были синтезированы химическими методами (Amy Mueller), которые использовали для оценки связывания с поли-р(А):
- NH2-(T8)-T - контроль,
- NH2-(T15)-T - контроль,
- NH2(LNA-T8)-Т - тест
По 200 нмоль каждого олигонуклеотида объединяли с 50 мг препаративной, активированной цианобромидом сефарозы-4В (Pharmacia) с учетом инструкции производителей. Для блокирования непрореагировавших сайтов связывания на сефарозе использовали 100 нМ Трис, рН 8,0.
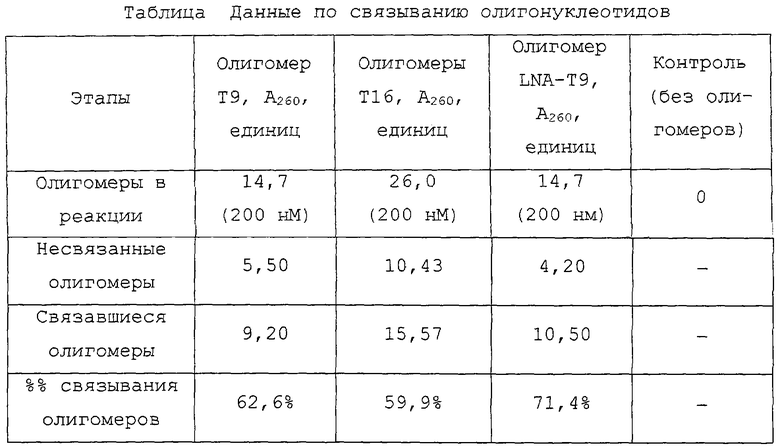
Смолы со связанными олигонуклеотидами разделяли на две порции (≈ 25 мг смолы в каждой) для анализа связывания полирибоаденозина в двух повторностях. Для связывания использовали тест-реактивы Poly-(rA) Pharmacia, № по каталогу 27-4110-01: растворяли при плотности поглощения 28,2 единиц/мл (А260) в буфере связывания. По пять единиц A260 были связаны с дупликатными порциями по 25 мг для каждого варианта смолы со связанными на ней олигонуклеотидами в расчете на SOP QC 5543. Несвязанные “пропущенные” поли-(рибоА) количественно определяли по поглощению при А260 и использовали для определения количества связанного реагента. Характер связанного поли-(рибоА) определяли путем промывки в низкосолевом буфере и нескольких элюций. Как показано в табл. 10, шарики, покрытые и LNA, и ДНК, были эффективны в связи с отбором (связыванием на себе) полирибоаденозиновых молекул-мишеней. Однако шарики, покрытые LNA, более прочно связывали поли-(рибоА)-мишень по сравнению с шариками, покрытыми ДНК: на это указывают различия в параметрах элюции различных вариантов шариков. Делается заключение о том, что (2) LNA-нонатимидиновый олигонуклеотид эффективен в отборе молекул РНК, включающих мотив, состоящий из остатков аденина, и (2) отобранные (связанные) молекулы РНК связыватся более прочно с LNA-Т9-олигомером по сравнению с прочностью связывания с контрольными 9- и 16-тиминовыми ДНК-олигонуклеотидами.

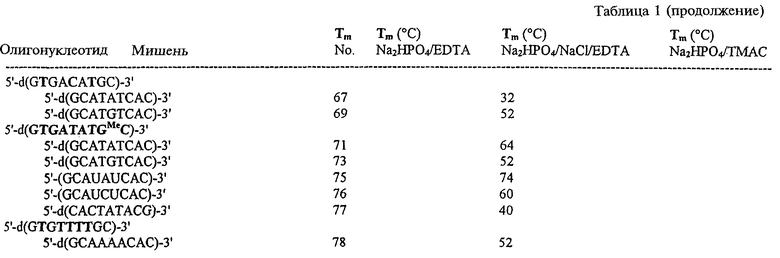

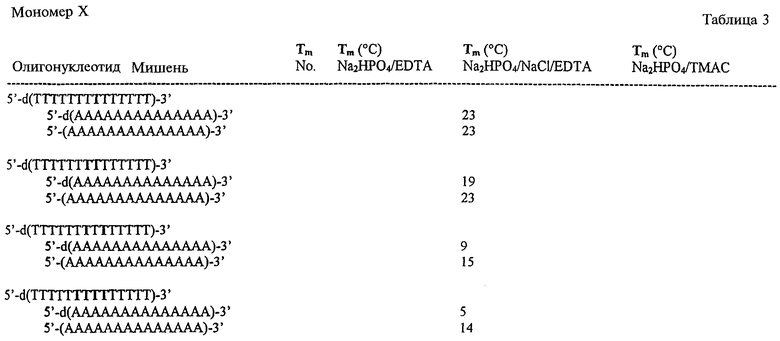
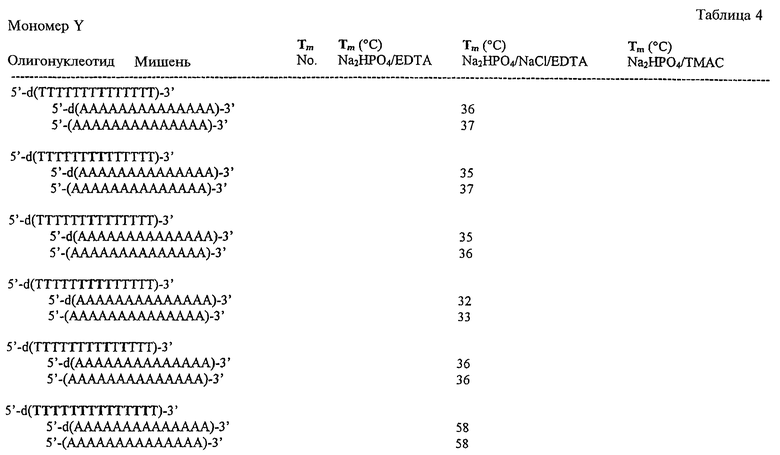
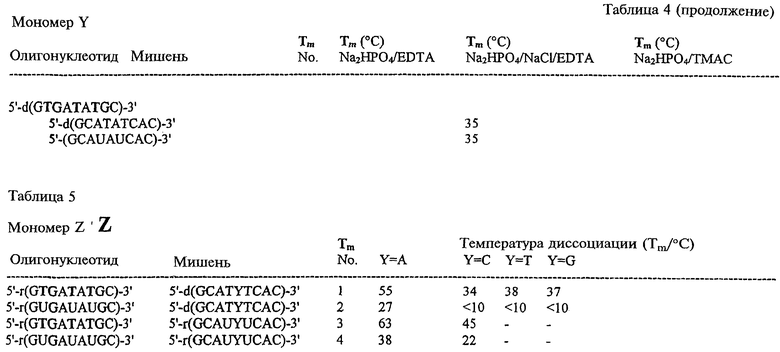




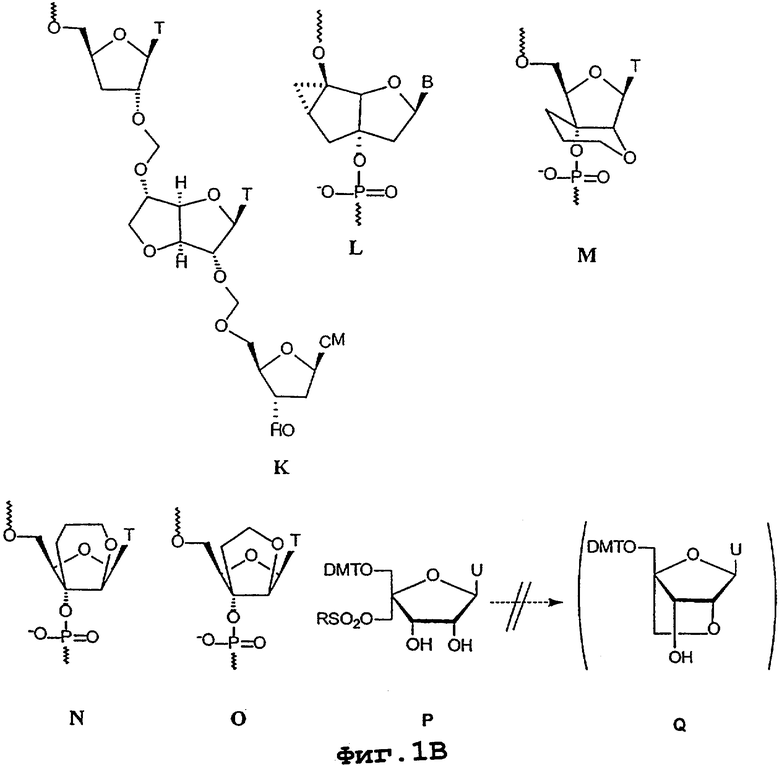
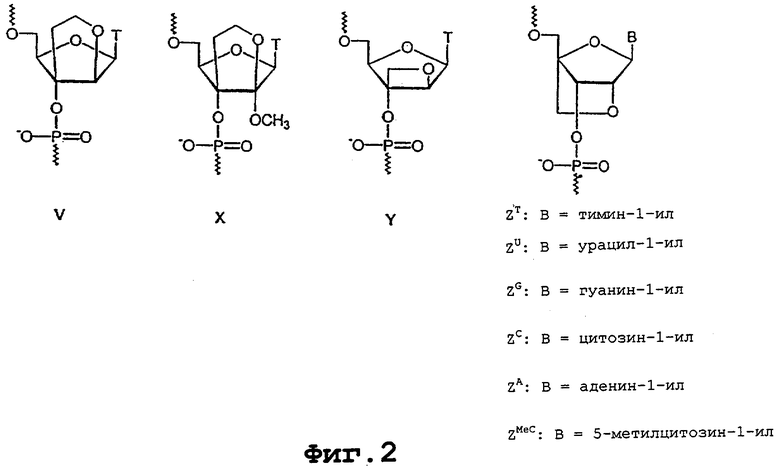
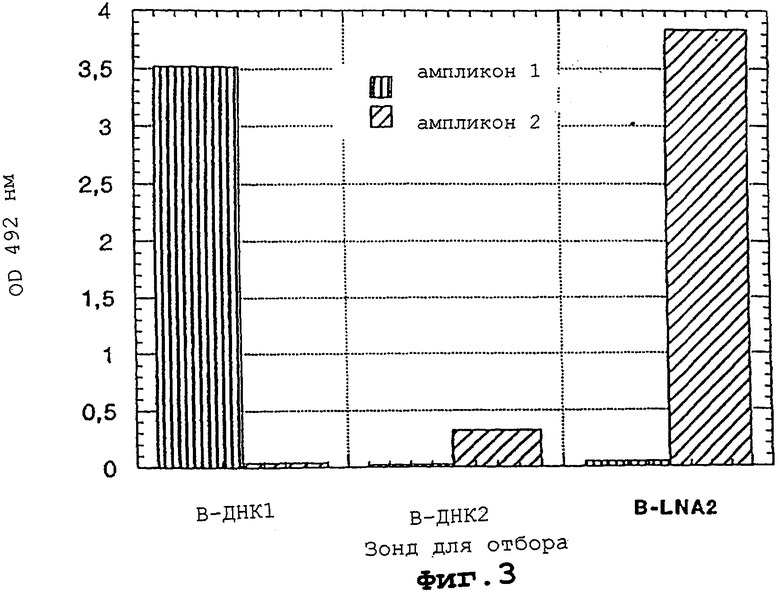
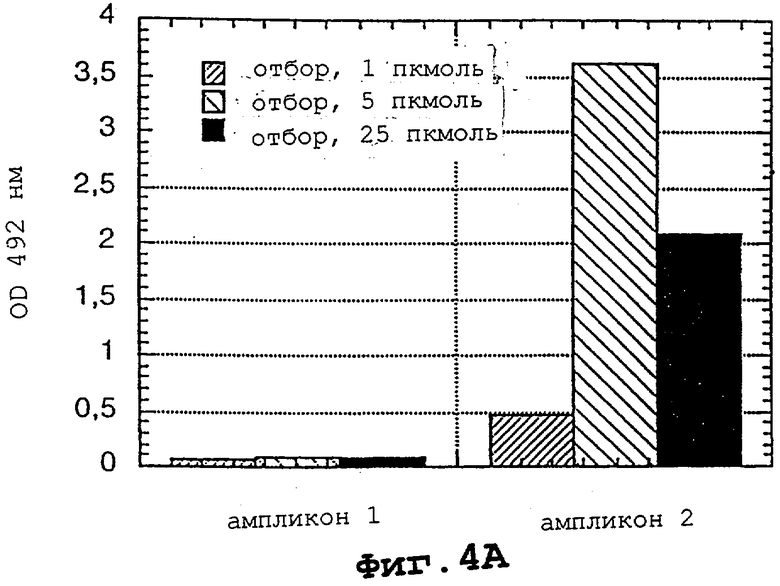
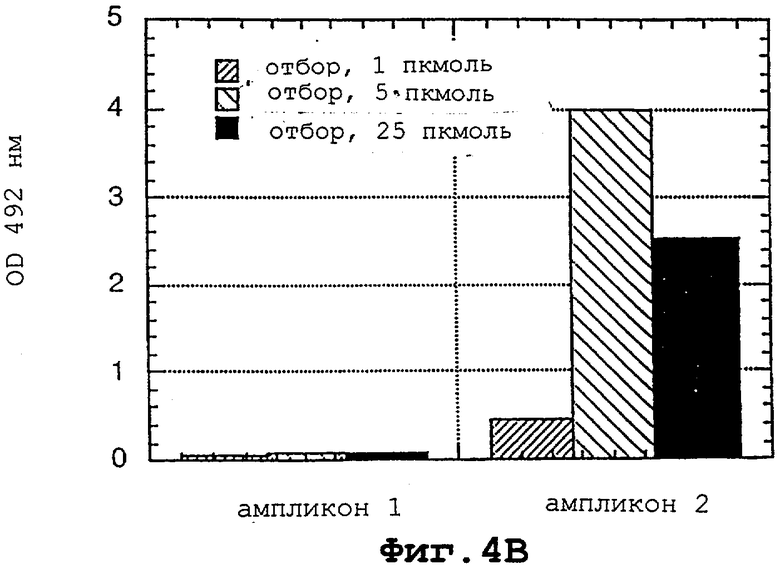
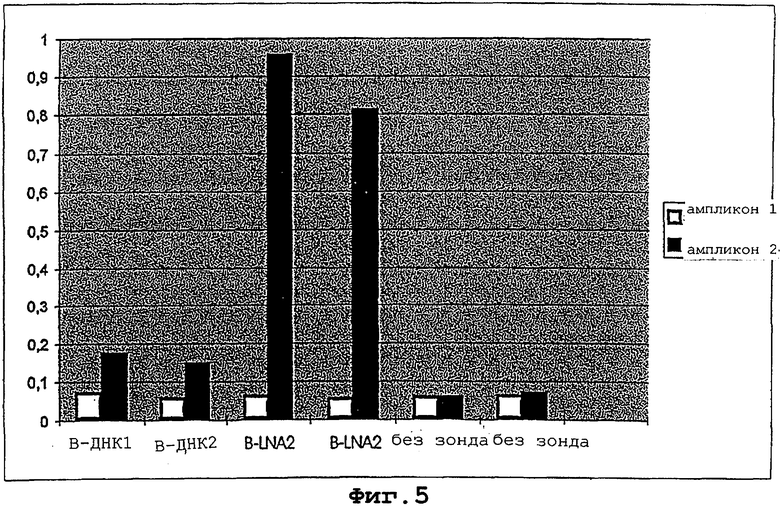



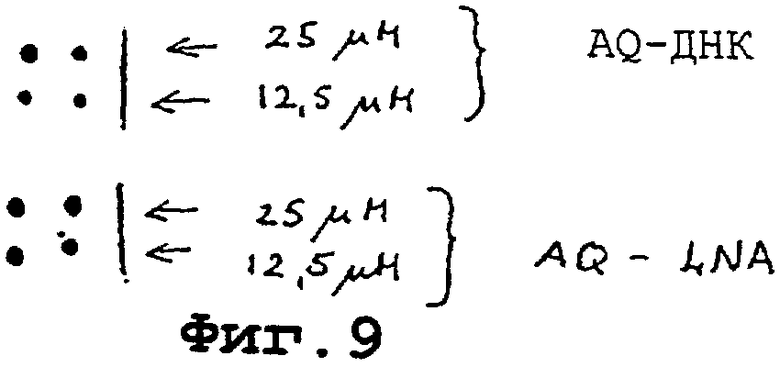

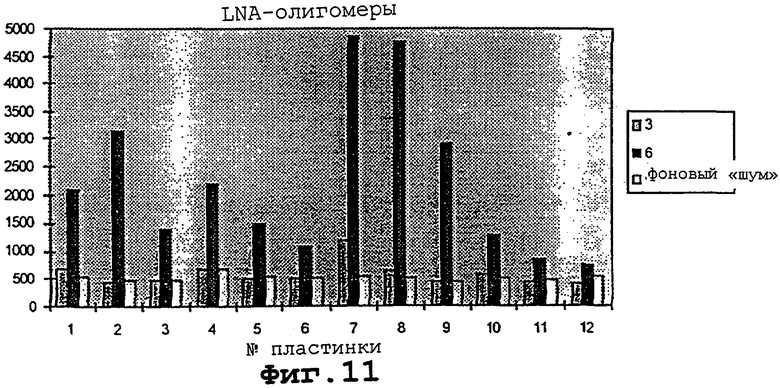
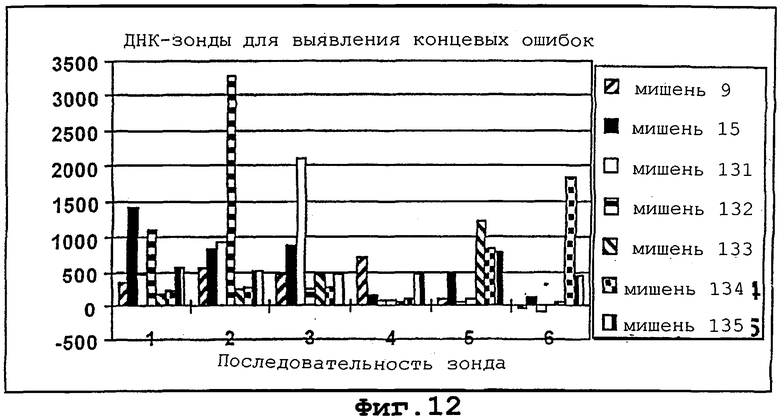
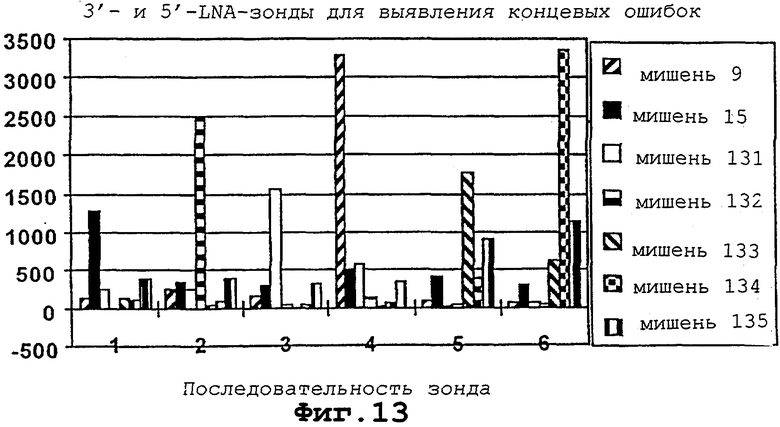
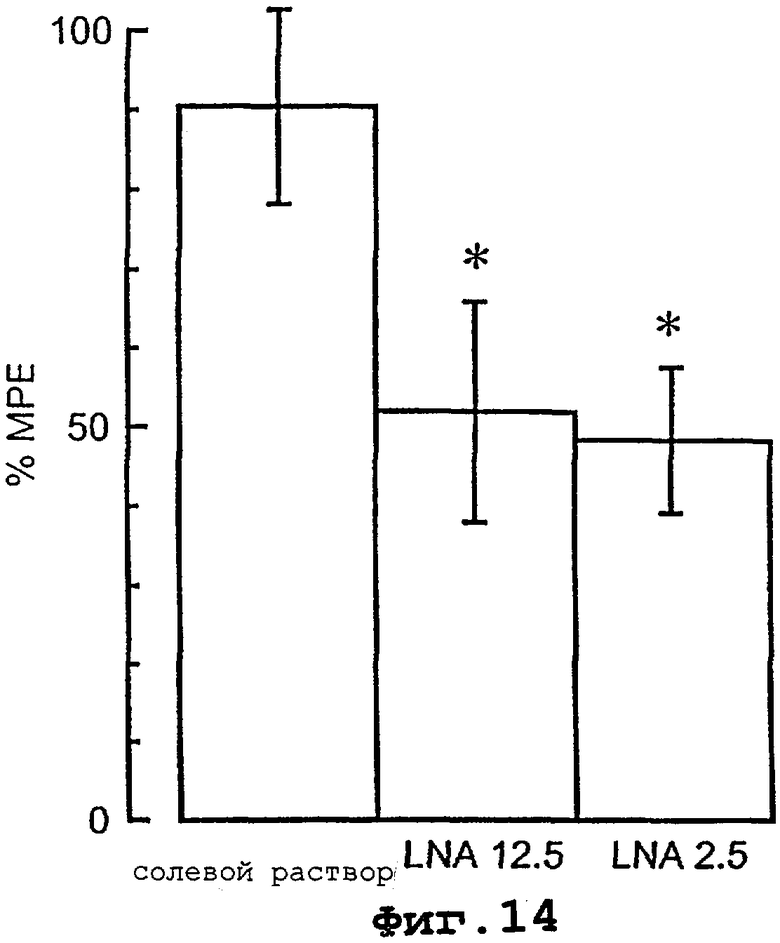
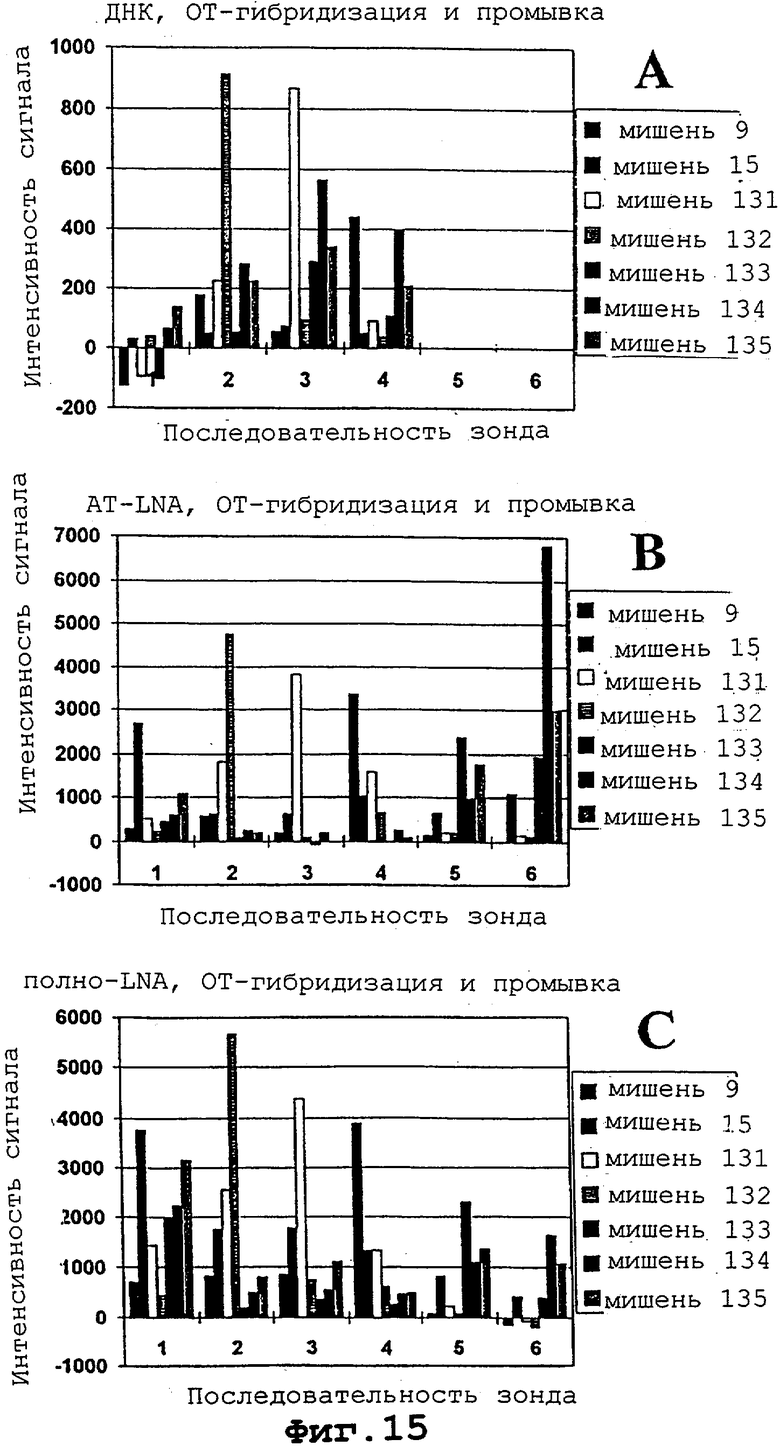
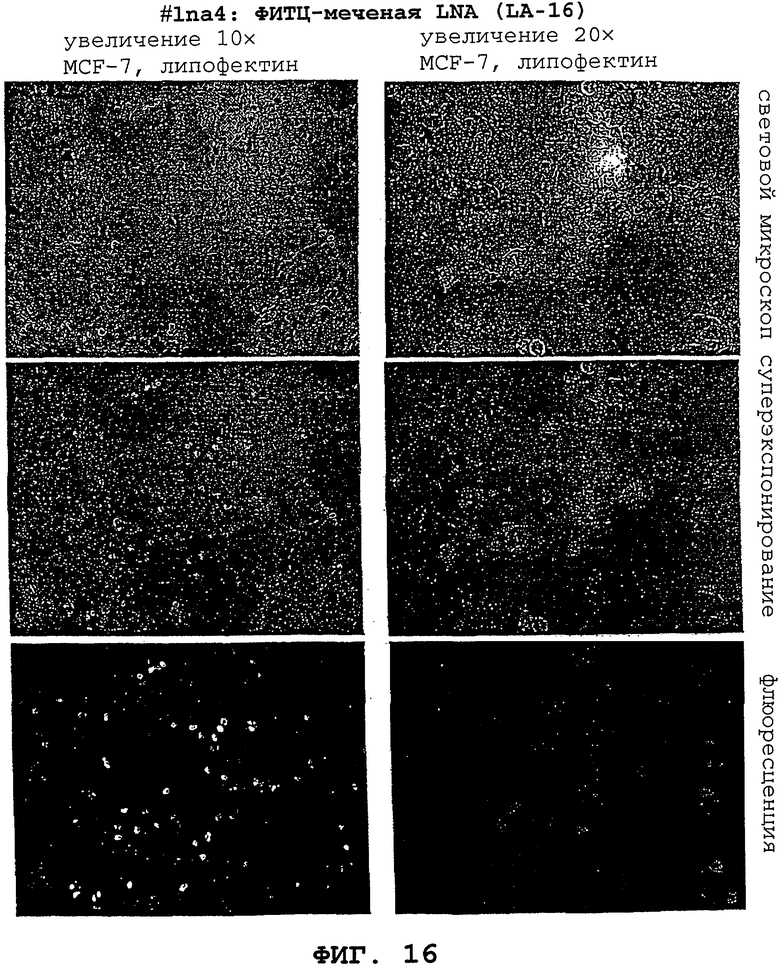
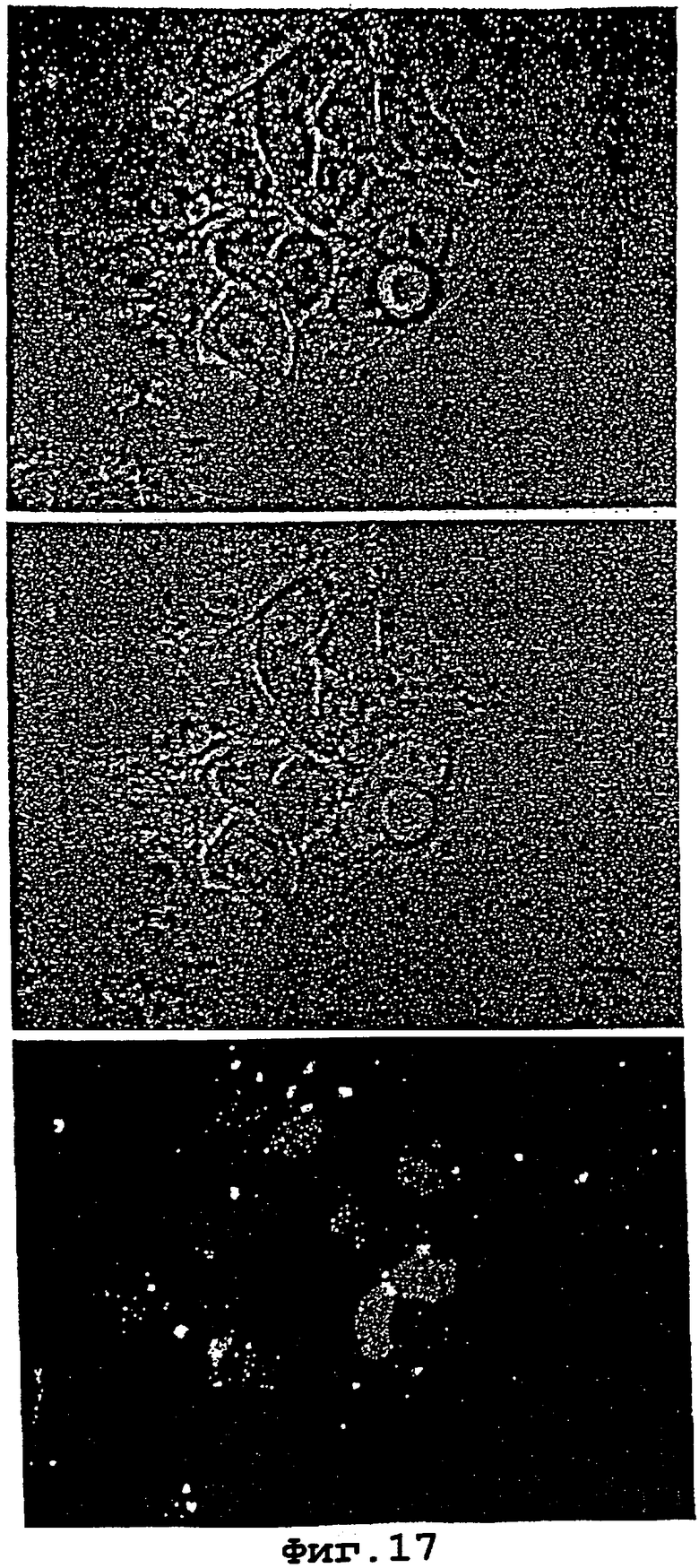


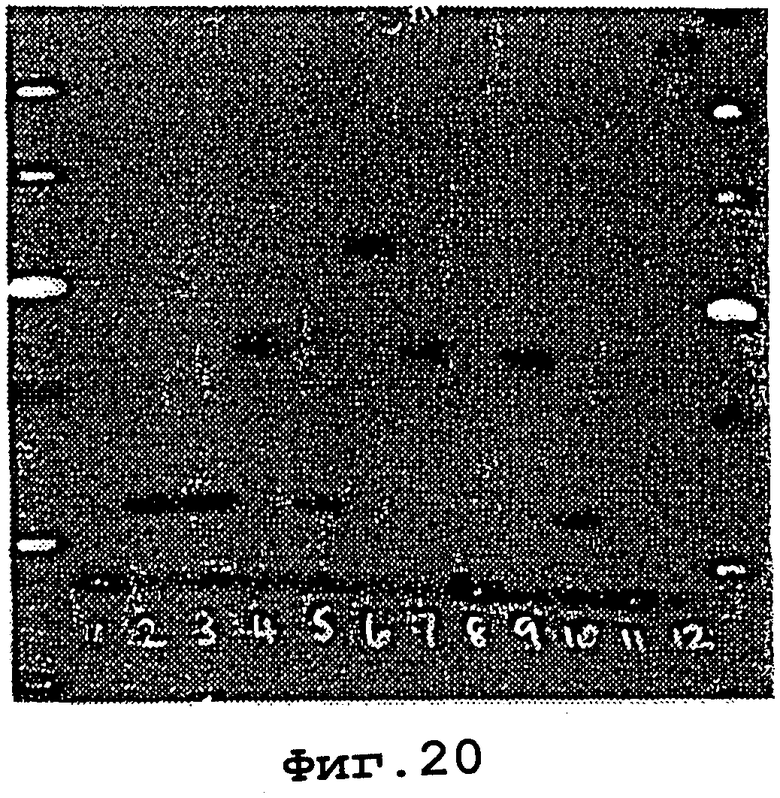


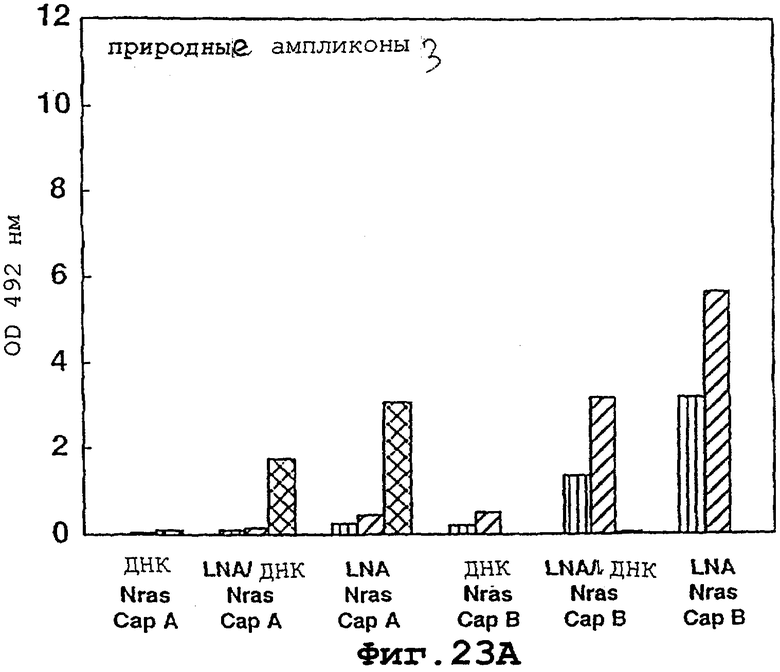
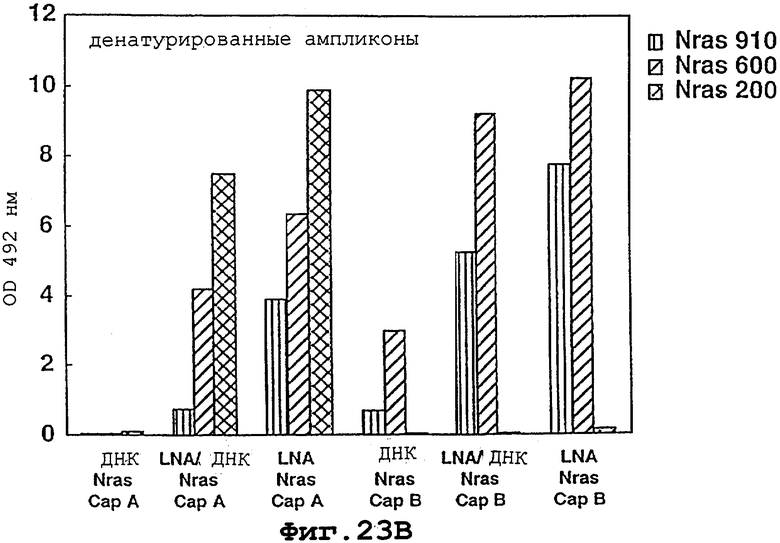
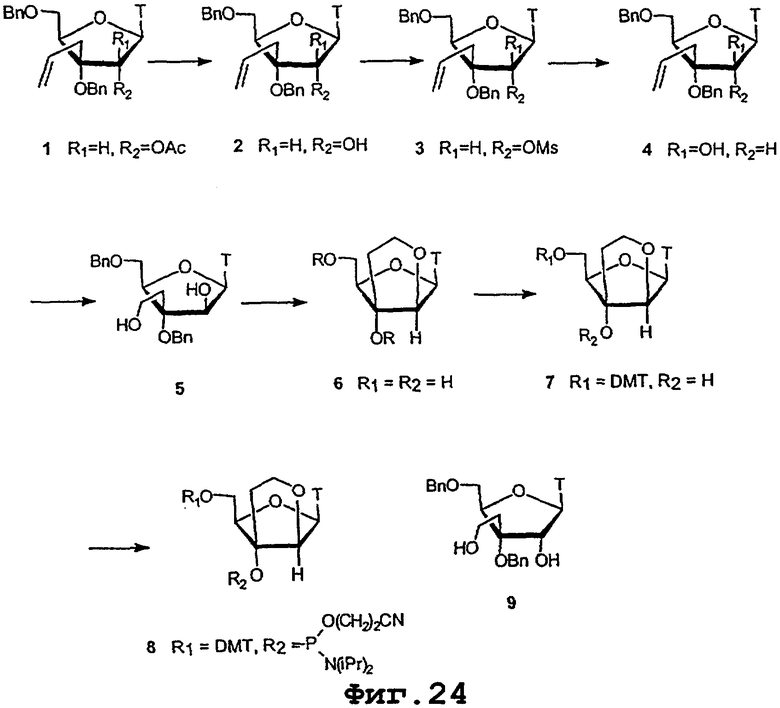
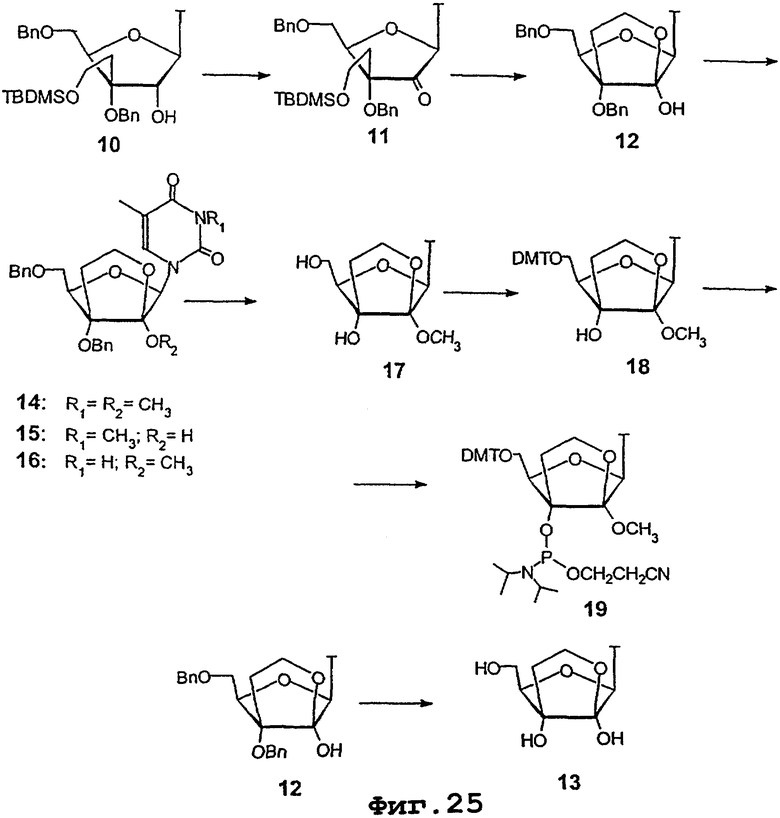
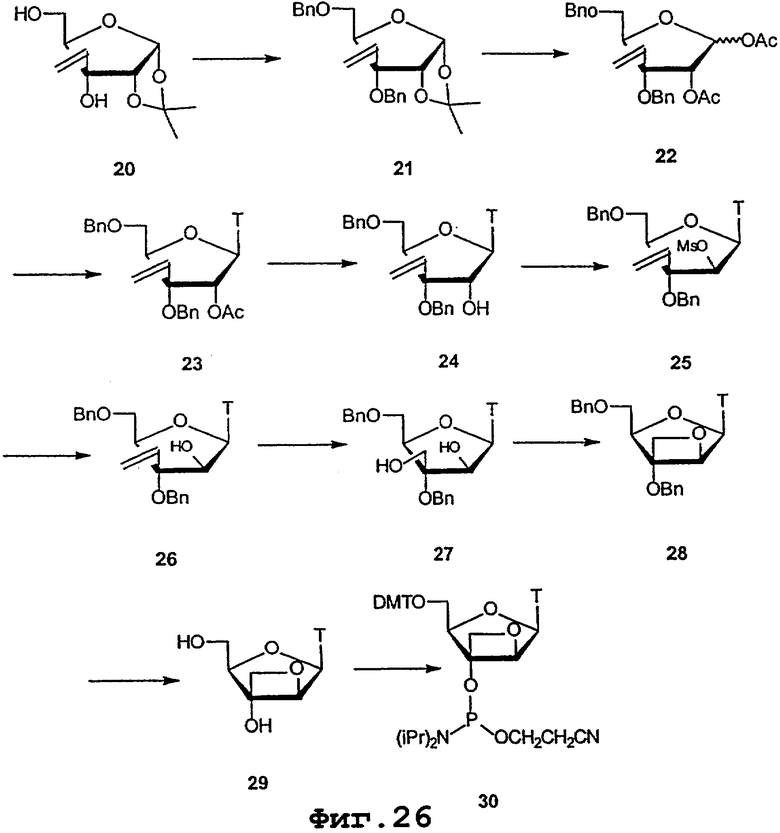
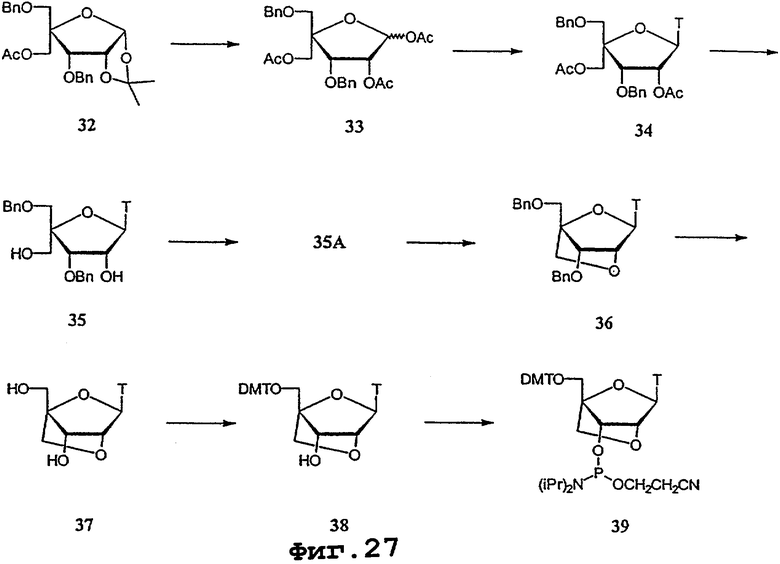
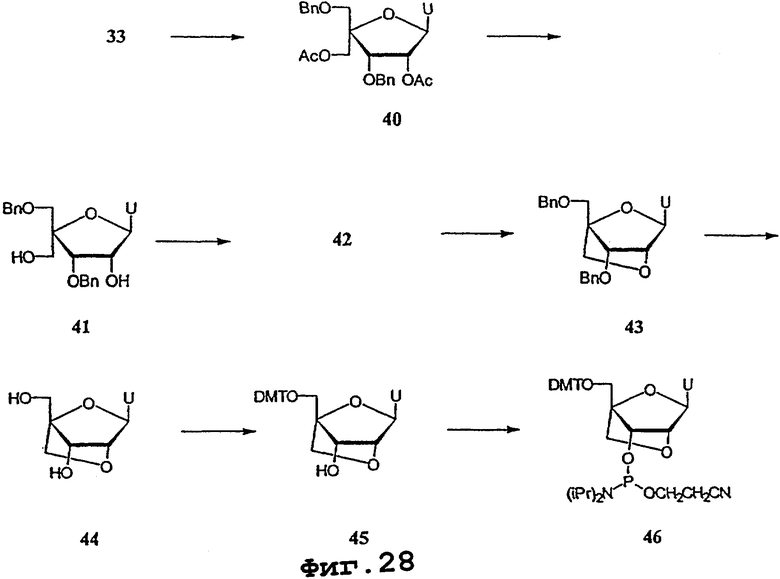
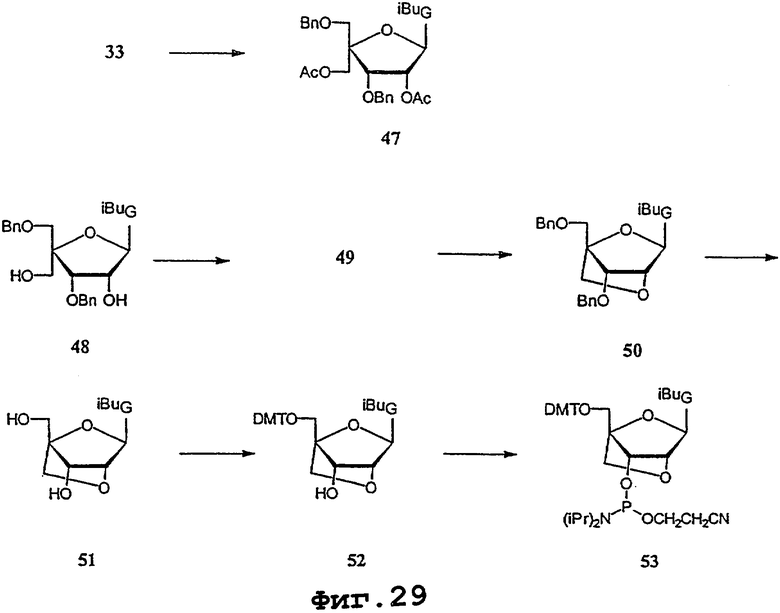
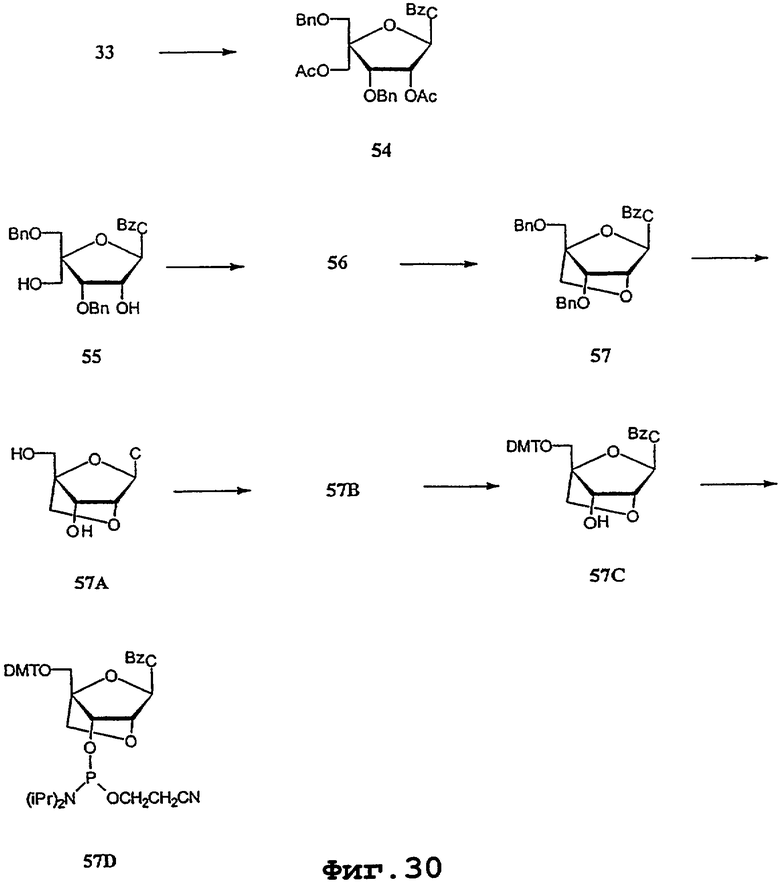
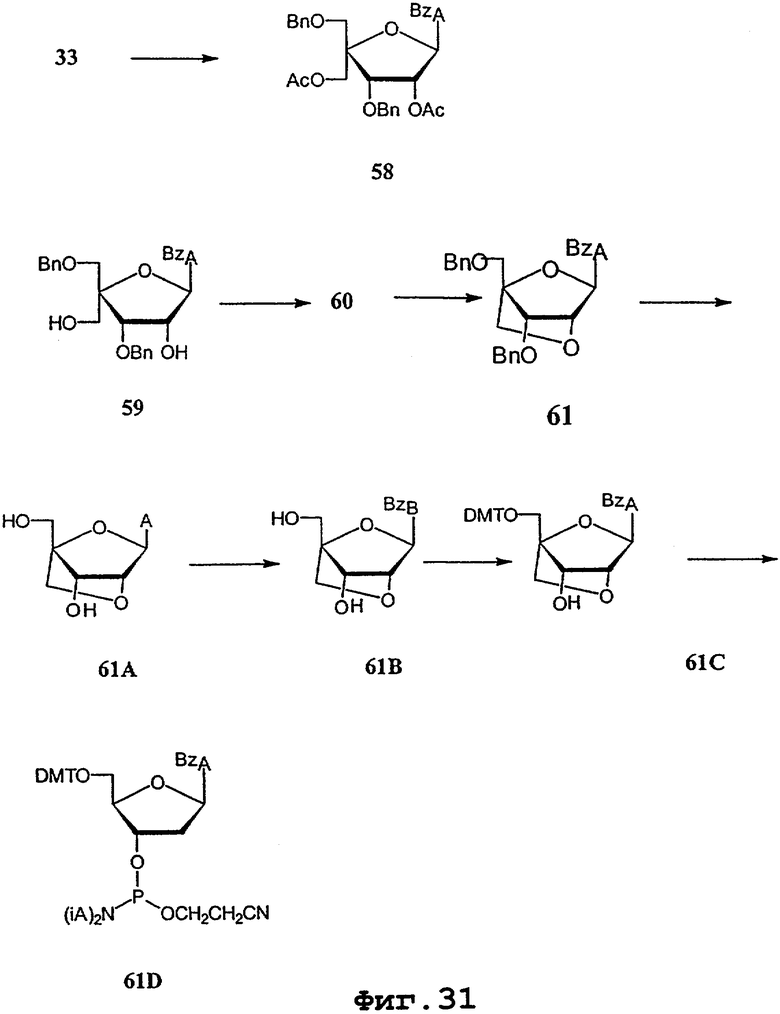
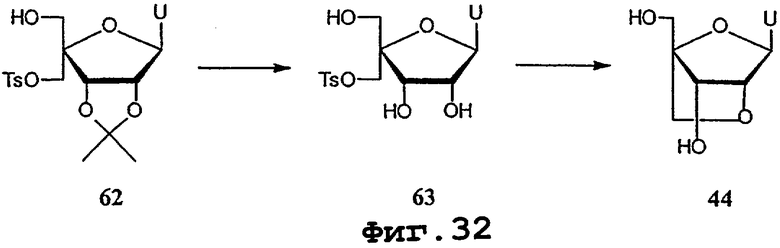
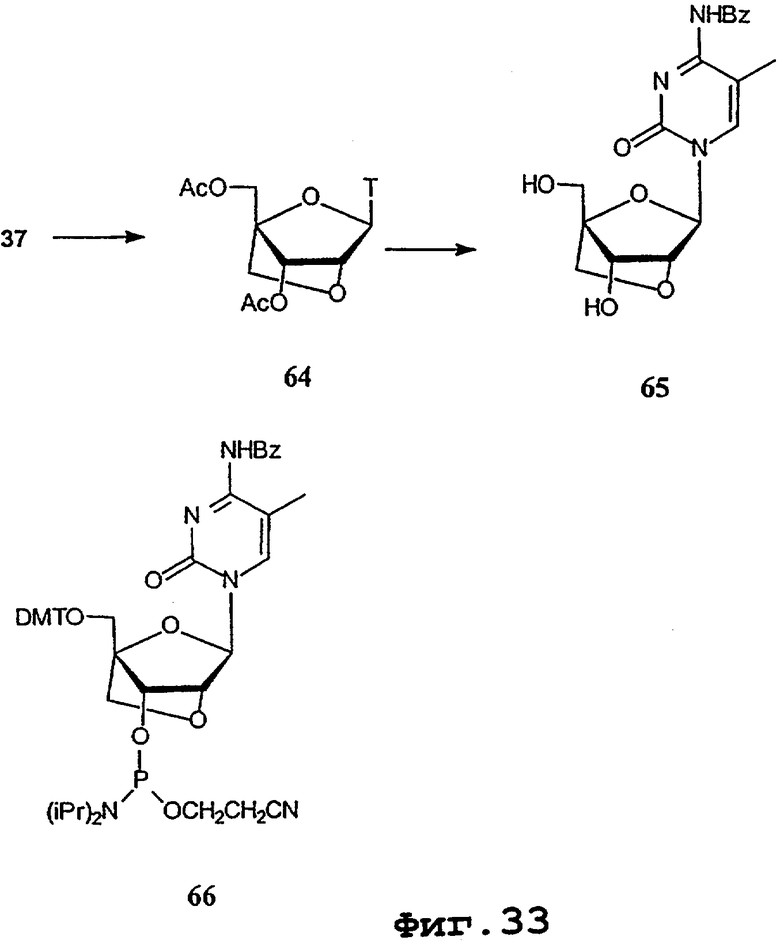
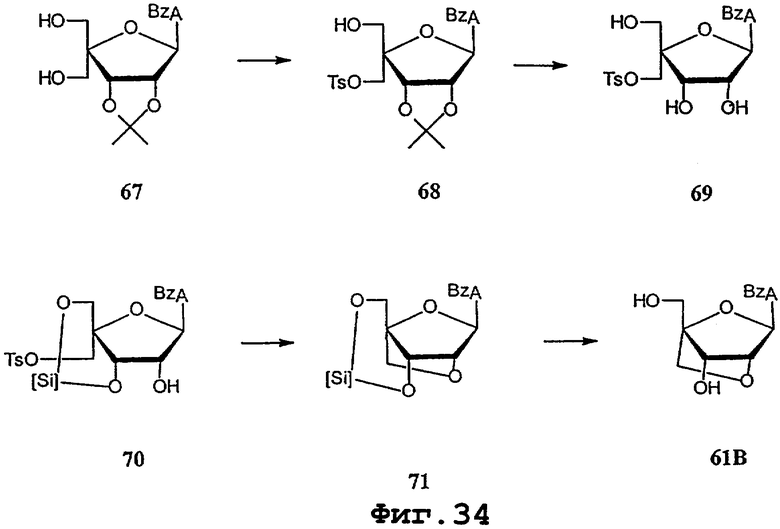
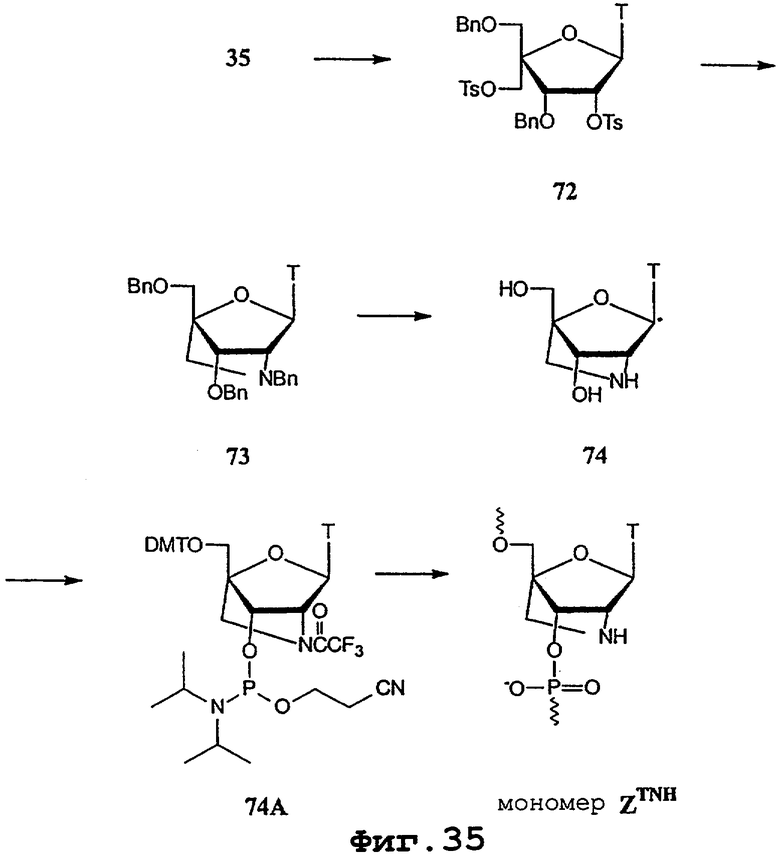
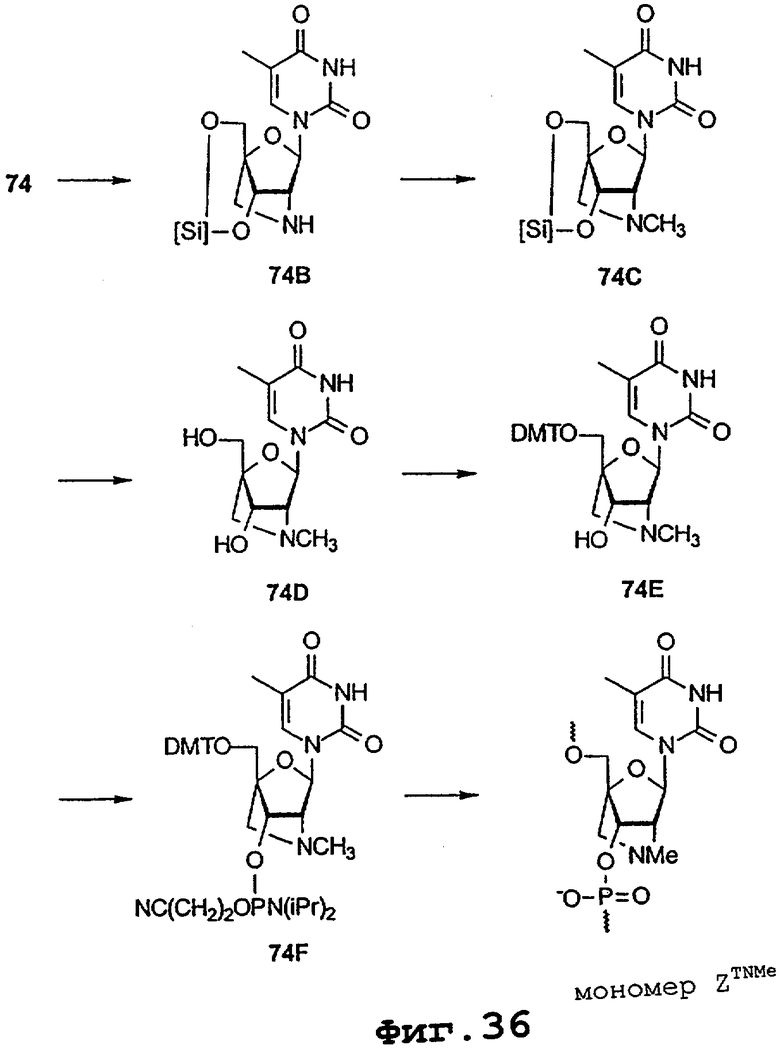
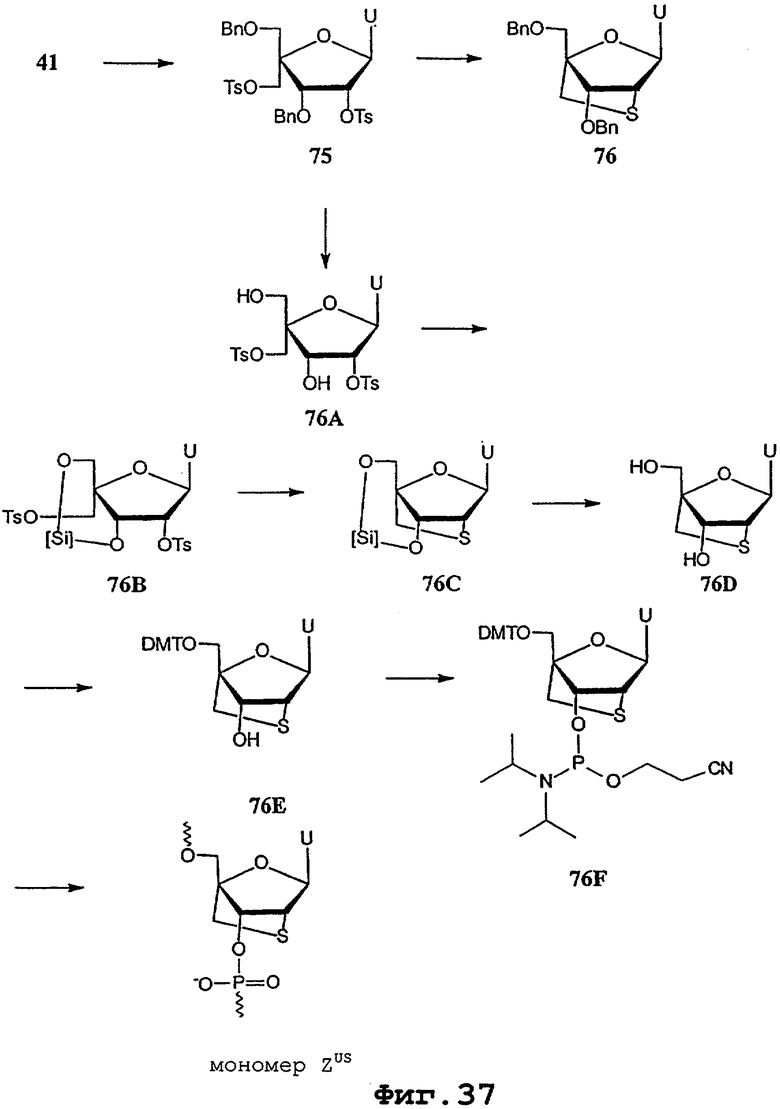
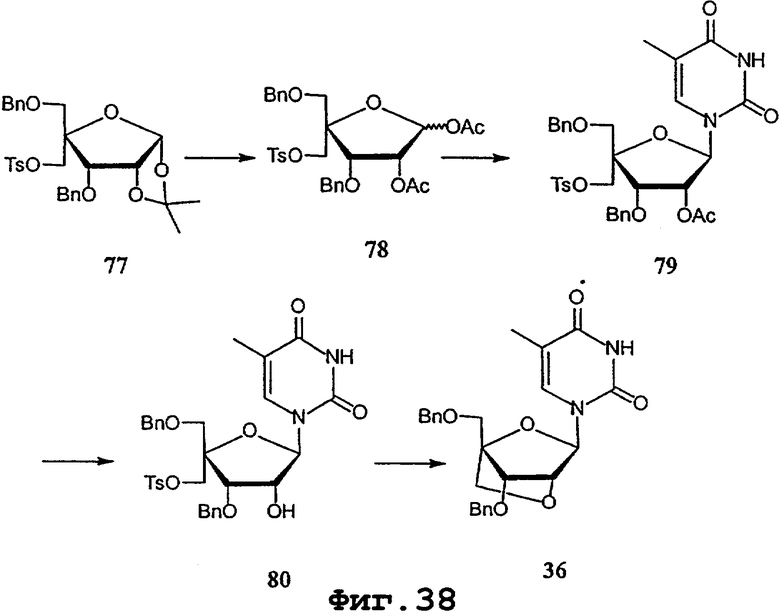
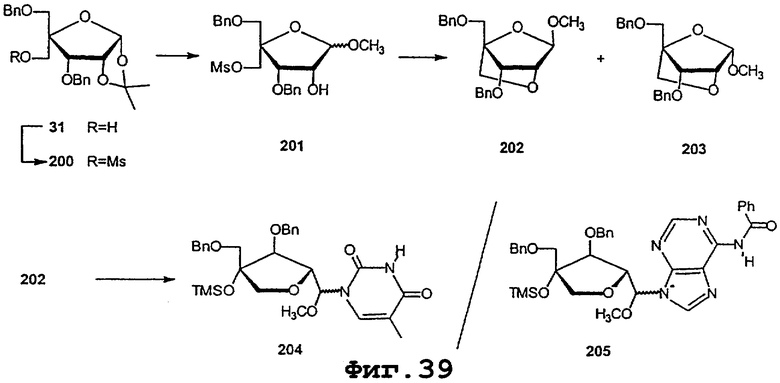
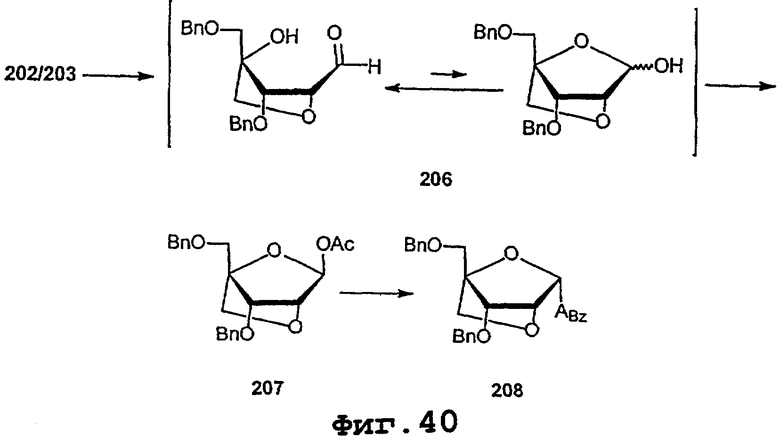
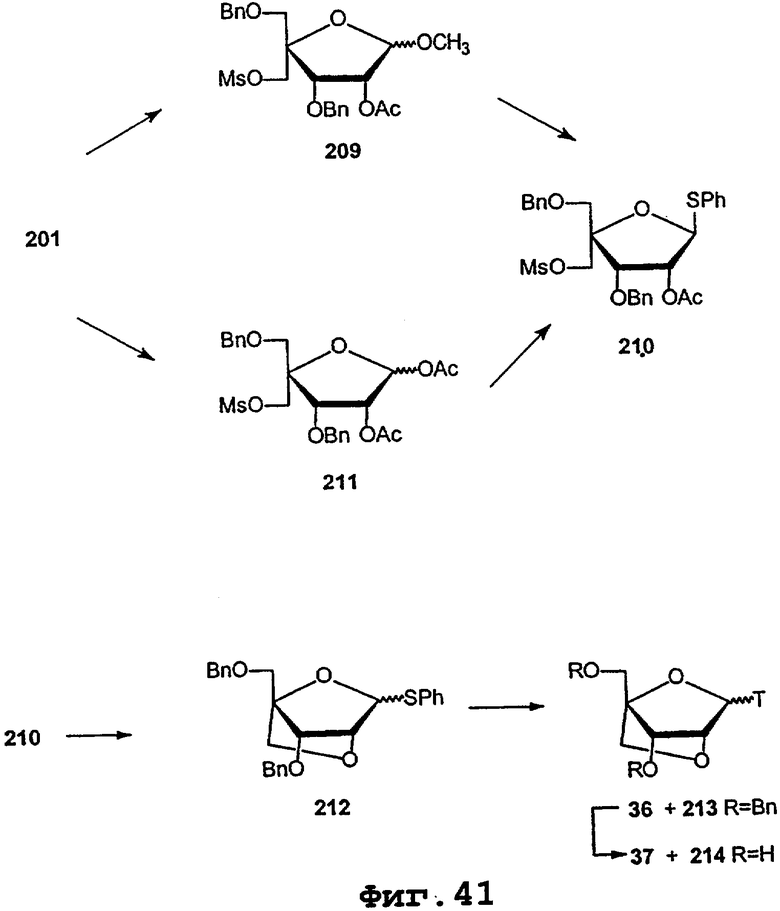
Изобретение относится к LNA-модифицированному олигонуклеотиду, включающему по крайней мере один нуклеозидный аналог (LNA) общей формулы I, где X - -О-; В - нуклеотидное основание; Р - место присоединения межнуклеозидного “мостика” или 5’-концевая группа, которую выбирают из гидроксила, монофосфата, дифосфата и трифосфата; R3 или R3* - межнуклеозидный мостик 3’-концевая группа; и R2* и R4* - бирадикал, выбираемый из -(CR*R*)r-O-(CR*R*)s-, -(CR*R*)r-S-(CR*R*)s-, -(CR*R*)r-N(R*)-(CR*R*)s-, при этом каждый из R1*, R2, R3*, R3, R5* и R5, не участвующих в образовании бирадикала или межнуклеозидного “мостика”, обозначает водород, галоген, гидрокси, меркапто, амино, азидо; или R2 и R3 - бирадикал -(CR*R*)r-O-(CR*R*)S-, при этом R2* выбирают из водорода, гидрокси и необязательно замещенной С1-6алкокси группы, a R1*, R4*, R5 и R5* - водород; где каждый из r и s равен 0 - 4, при условии, что сумма r+s равна 1 - 4, а каждый R* представляет собой водород или C1-6алкил; или его основной соли или кислотно-аддитивной соли. Также предложен соответствующий нуклеозидный аналог (LNA) и материал твердой подложки с иммобиблизованным на нем LNA. LNA-модифицированные олигонуклеотиды обладают повышенным уровнем аффиности и специфичности в отношении комплементарных ДНК и РНК-олигомеров. 3 н. и 59 з.п. ф-лы, 41 ил., 14 табл.
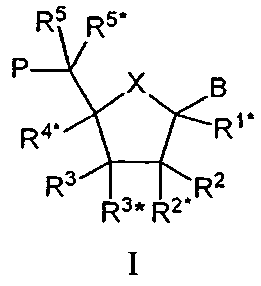
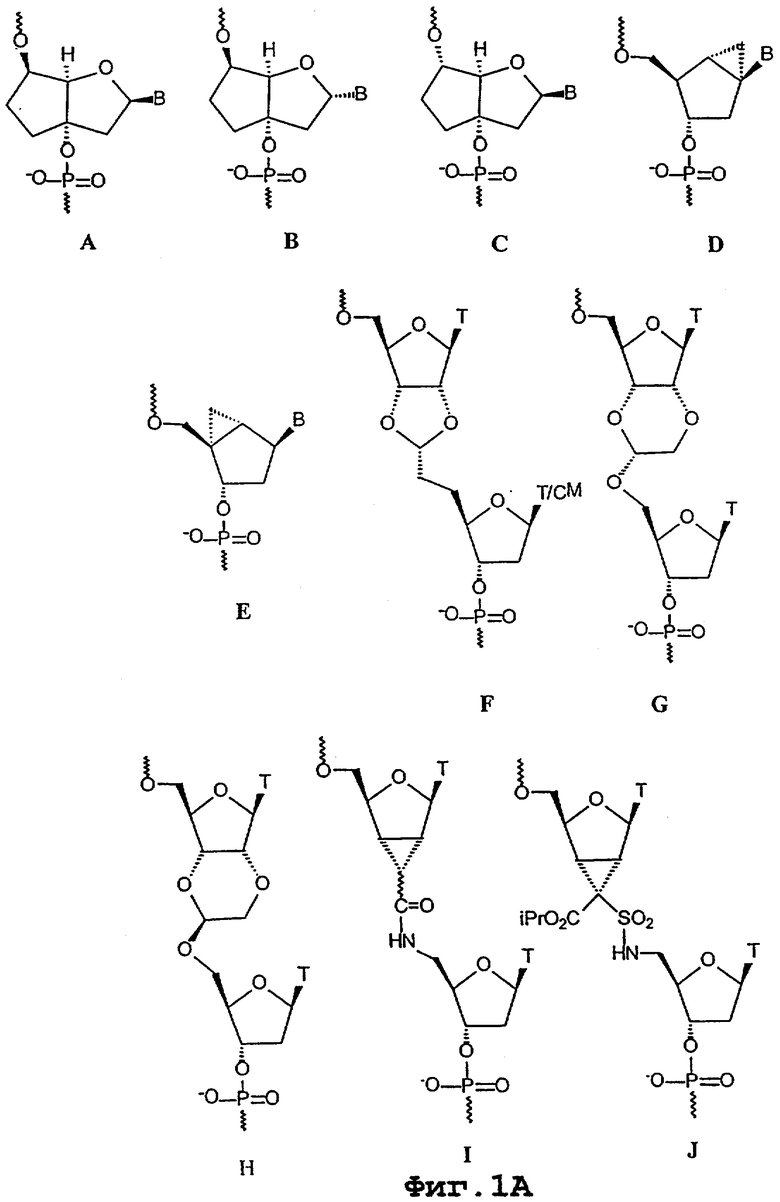

где X представляет -О-;
В представляет нуклеотидное основание;
Р обозначает положение радикала для формирования межнуклеозидного “мостика” со следующим мономером или 5’-концевую группу, где указанную 5’-концевую группу выбирают из гидроксила, монофосфата, дифосфата и трифосфата;
R3 или R3* представляют группу Р*, которая обозначает межнуклеозидный мостик с предыдущим мономером или 3’-концевую группу; и
негеминальные заместители R2* и R4* обозначают бирадикал, выбираемый из -(CR*R*)r-O-(CR*R*)s-, -(CR*R*)r-S-(CR*R*)s-, -(CR*R*)r-N(R*)-(CR*R*)s-, при этом каждый из заместителей R1*, R2, R3*, R3, R5* и R5, не участвующих в образовании бирадикала или межнуклеозидного “мостика”, обозначает водород, галоген, гидрокси, меркапто, амино, азидо, или
негеминальные заместители R2 и R3 обозначают бирадикал -(CR*R*)r-O-(CR*R*)S-, при этом R2* выбирают из водорода, гидрокси и необязательно замещенной С1-6алкоксигруппы, a R1*, R4*, R5 и R5* обозначают водород;
где каждый из r и s равен 0 - 4, при условии, что сумма r+s равна 1 - 4, а каждый R* представляет собой водород или C1-6алкил;
или его основная соль или кислотно-аддитивная соль; причем олигонуклеотид может включать фотохимически активную группу, термохимически активную группу, хелатирующую группу, репортерную группу или лиганд, которые обуславливают его прямую или непрямую детекцию или иммобилизацию на твердой подложке.
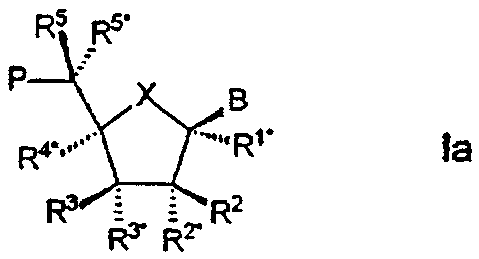
при том, что Р, Р*, В, X, R1*, R2, R2*, R3, R4*, R5 и R5* определены в п.1.
G-[Nu-L]n(0)-{[LNA-L]m(q)-[Nu-L]n(q)}q-G* V
где q равно 1-50;
каждый из n(0),..., n(q) независимо друг от друга равен 0-100;
каждый из m(l),..., m(q) независимо друг от друга равен 1-100;
при условии, что сумма n(0),..., n(q) и m(1),..., m(q) равна 2-150;
G обозначает 5’-концевую группу;
каждый Nu независимо обозначает природный нуклеозид;
каждый LNA независимо является нуклеозидным аналогом;
каждый L независимо обозначает межнуклеозидный “мостик” между двумя группами, выбираемыми из Nu и LNA, или L вместе с G* образует 3’-концевую группу; и
каждый LNA-L независимо обозначает нуклеозидный аналог общей формулы I
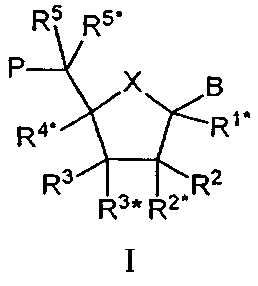
при том, что заместители В, Р, Р*, R1*, R2, R2*, R3, R5 и R5* и X определены в пп. 1-15.
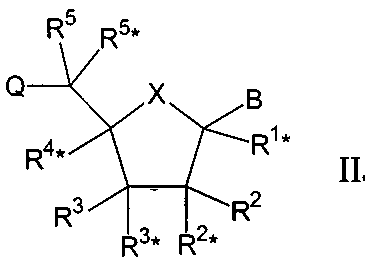
где X представляет собой -О-;
В представляет собой нуклеотидное основание, которое необязательно защищено по своим функциональным составляющим;
R3 или R3* представляет собой группу Q*, которую выбирают из гидрокси, Prot-О-, Act-O;
Q представляет собой гидроксил, монофосфат, дифосфат, трифосфат, Prot-O- или Act-O, где
Рrоt обозначает защитную группу для –ОН; Act обозначает активационную группу для -ОН;
негеминальные заместители R2* и R4* обозначают бирадикал, выбираемый из -(CR*R*)r-O-(CR*R*)s-, -(CR*R*)r-S-(CR*R*)s-, -(CR*R*)r-N(R*)-(CR*R*)s-, при этом каждый из заместителей R1*, R2, R3*, R3, R5* и R5, не участвующих в образовании бирадикала, Q или Q*, обозначает водород, гидрокси, меркапто, амино, азидо; или
негеминальные заместители R2 и R3 обозначают бирадикал -(CR*R*)r-O-(CR*R*)S-, при этом R2* выбирают из водорода, гидроксила и необязательно замещенной C1-6алкоксигруппы, a R1*, R4*, R5 и R5* обозначают водород; где
каждый из r и s равен 0 - 4, при условии, что сумма r+s равна 1 - 4, а каждый R* представляет собой водород или C1-6алкил;
или его основная соль или кислотно-аддитивная соль.
(lR,2R,4R,5S)-l-(2-циaнoэтoкcи(диизoпpoпилaминo) фocфинoкcи)-2-(4,4’-диметокситритилоксиметил)-4-(тимин-1-ил)-3,6-диоксабицикло[3.2.0]гептана,
(lR,3R,4R,7S)-7-(2-циaнoэтoкcи(диизoпpoпилaминo) фocфинoкcи)-l-(4,4’-диметокситритилоксиметил)-3-(тимин-1-ил)-2,5-диоксабицикло[2.2.1]гептана,
(lR,3R,4R,7S)-7-(2-циaнoэтoкcи(диизoпpoпилaминo) фocфинoкcи)-l-(4,4’-диметокситритилоксиметил)-3-(2-N-изопропионилгуанин-9-ил)-2,5-диоксабицикло[2.2.1]гептана,
(1R,3R,4R,7S)-7-(2-цианоэтокси(диизопропиламино) фосфинокси)-1-(4,4’-диметокситритилоксиметил)-3-(4-N-бензоилцитозин-1-ил)-2,5-диоксабицикло[2.2.1]гептана,
(1R,3R,4R,7S)-7-(2-цианоэтокси(диизопропиламино) фосфинокси)-1-(4,4’-диметокситритилоксиметил)-3-(6-N-бензоиладенин-9-ил)-2,5-диоксабицикло[2.2.1 ]гептана,
(1R,3R,4R,7S)-7-(2,2-диметокситритилоксиметил)-3-(5-метил-4-N-бензоилцитозин-1-ил)-2,5-диоксабицикло[2.2.1]гептана,
1-(2-амино-3-О-(2-цианоэтокси(диизопропиламино)фосфино-2-дезокси)-5-О-4,4’-диметокситритил-2-N,4-С-метилен-2-N-трифторацетил-β-D-рибофуранозил)тимина,
1-(3-О-(2-цианоэтокси(диизопропиламино)фосфино)-5-О-4,4’-диметокситритил-2-метиламино-2-N,4-С-метилен-2-дезокси-β-D-рибофуранозил)тимина,
1-(3-О-(2-цианоэтокси(диизопропиламино)фосфино)-2-дезокси-5-О-(4,4’-диметокситритил)-2-меркапто-2-S,4-С-метилен-β-D-рибофуранозил)урацила.
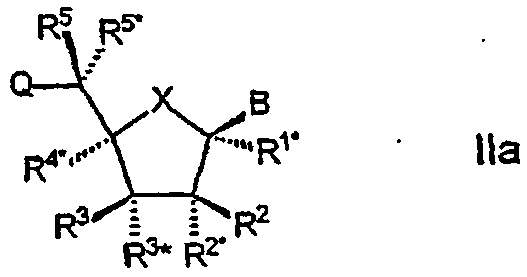
где заместители Q, В, R1*, R2, R2*, R3, R3*, R4*, R5 и R5* определены в п. 38.
(1R,3R,4R,7S)-7-(2-цианоэтокси(диизопропиламино) фосфинокси)-1-(4,4’-диметокситритилоксиметил)-3-(тимин-1-ил)-2,5-диокса-бицикло[2.2.1]гептана,
(lR,3R,4R,7S)-7-гидpoкcи-l-(4,4’-димeтoкcитpитилoкcимeтил)-3-(тимин-l-ил)-2,5-диоксабицикло-[2.2.1]гептан-7-0-(2-хлорфенилфосфатa) и
(lR,3R,4R,7S)-7-гидpoкcи-l-(4,4’-димeтoкcитpитилoкcимeтил)-3-(тимин-l-ил)-2,5-диоксабицикло-[2.2.1]гептан-7-0-(Н-фосфоната)
и соответствующих -3-(цитозин-1-ил)ьных, -3-(урацил-1-ил)ьных, -3-(аденин-1-ил)ьных и -3-(гуанин-3-ил)ьных аналогов.
Приоритет по пунктам и признакам:
| P | |||
| Nielsen et al | |||
| JOURNAL OF THE CHEMICAL SOCIETY, CHEMICAL COMMUNICATIONS, № 9, 7 MAY 1997, PP | |||
| Устройство для регистрации замерзания и оттаивания почвы | 1923 |
|
SU825A1 |
| V.E.MARQUEZ ET AL | |||
| JOURNAL OF MEDICINAL CHEMISTRY, V | |||
| Машина для изготовления проволочных гвоздей | 1922 |
|
SU39A1 |
| Предохранительное устройство для паровых котлов, работающих на нефти | 1922 |
|
SU1996A1 |
| ДВУХЛОПАСТНОЙ ВЕТРЯК | 1925 |
|
SU3739A1 |
| R.KUMAR ET AL | |||
| Bioorg.Med.Chem.Lett, V.8, 1998, PP | |||
| Электрический конденсатор переменной емкости | 1925 |
|
SU2219A1 |
| S.K | |||
| SINGH ET AL | |||
| CHEMICAL COMMUNICATIONS, № 4, 21 FEB | |||
| Способ и аппарат для получения гидразобензола или его гомологов | 1922 |
|
SU1998A1 |
| Снеговая лыжа для самолетов | 1913 |
|
SU455A1 |
| A.A.KOSHKIN ET AL | |||
| TETRAHEDRON, V | |||
| Видоизменение прибора для получения стереоскопических впечатлений от двух изображений различного масштаба | 1919 |
|
SU54A1 |
| ПЕРЕНОСНОЙ ПРИБОР ДЛЯ ФРЕЗЕРОВАНИЯ, ШЛИФОВКИ И Т.П. РАБОТ | 1924 |
|
SU3607A1 |
| S.OBIKA ET AL | |||
| TETRAHEDRON LETTERS, 1997, V | |||
| Способ сужения чугунных изделий | 1922 |
|
SU38A1 |
| КУХОННЫЙ ОЧАГ С ПРИСПОСОБЛЕНИЕМ ДЛЯ ВОДЯНОГО ОТОПЛЕНИЯ | 1927 |
|
SU8735A1 |
| S.OBIKA ET AL | |||
| TETRAHEDRON LETTERS, 1999, V | |||
| Приспособление с иглой для прочистки кухонь типа "Примус" | 1923 |
|
SU40A1 |
| ТЕЛЕФОННОЕ УСТРОЙСТВО | 1926 |
|
SU6465A1 |
| Дорожная спиртовая кухня | 1918 |
|
SU98A1 |
| РЕЗИСТЕНТНЫЕ К НУКЛЕАЗЕ ОЛИГОНУКЛЕОЗИДЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И РЕЗИСТЕНТНЫЙ К НУКЛЕАЗЕ НУКЛЕОЗИДНЫЙ ДИМЕР | 1991 |
|
RU2131436C1 |

