Глюкокиназа (GK) является одной из четырех гексокиназ, найденных у млекопитающих [Colowick, S.P., в сборнике "The Enzymes", Vol.9 (P.Boyer, ред.). Academic Press, Нью-Йорк, NY, pages 1-48, 1973]. Гексокиназы катализируют первую стадию метаболизма глюкозы, то-есть превращение глюкозы в глюкозо-6-фосфат. Глюкокиназа имеет ограниченное распределение в клетке, находясь, в основном, в панкреатических бета-клетках и клетках паренхимы печени. Кроме того, GK является ферментом, контролирующим скорость метаболизма глюкозы в этих двух типах клеток, которые, как известно, играют решающую роль в гомеостазе глюкозы всего организма [Chipkin, S.R., Kelly, K.L. и Ruderman, N.B. в Joslin′s Diabetes (C.R.Khan and G.C.Wier, ред.), Lea и Febiger, Филадельфия, PA, стр. 97-115, 1994]. Концентрация глюкозы, при которой GK проявляет половину максимальной активности, составляет примерно 8 мМ. Три другие гексокиназы насыщаются глюкозой при намного меньших концентрациях (<1 мМ). Поэтому поступление глюкозы по пути метаболизма GK растет, в то время как концентрация глюкозы в крови возрастает от уровня концентрации при голоде (5 мМ) до уровня концентрации, возникающей после приема пищи (≈10-15 мМ), именно после содержащей углеводы пищи [Printz, R.G., Magnuson, М.А. и Granner, D.К. в Ann. Rev. Nutrition, Vol. 13 (R.E. Olson, D.M. Bier и D.B. McCormick, ред.). Annual Review, Inc., Palo Alto, CA, стр. 463-496, 1993]. Эти данные более десяти лет назад внесли вклад в гипотезу, что GK функционирует в качестве глюкозного сенсора в бета-клетках и гепатоцитах (Meglasson, M.D. и Matschinsky, F.M., Amer. J.Physiol., 246, Е1-Е13, 1984). Недавно исследования с применением трансгенных животных подтвердили, что GK действительно играет решающую роль в гомеостазе глюкозы всего организма. Животные, которые не экспрессируют GK, умирают в течение дней появления на свет от сильного диабета, в то время как животные, сверхэкспрессирующие GK, имеют улучшенную толерантность к глюкозе (Grupe, A., Hultgren, В., Ryan, А. и др.. Cell, 83, 69-78, 1995; Ferrie, Т., Riu, E., Bosch, F. и др., FASEB J., 10, 1213-1218, 1996). Увеличение выделения глюкозы связывают через GK в бета-клетках с увеличенной секрецией инсулина и в гепатоцитах с увеличенным отложением гликогена и, возможно, с уменьшенным продуцированием глюкозы.
Полученные данные, что диабет типа II достигших зрелости молодых людей (MODY-2) вызывается потерей функциональных мутаций в гене GK, предполагают, что GK также функционирует у людей в качестве глюкозного сенсора (Liang, Y., Kesavan, P., Wang, L. и др., Biochem. J., 309, 167-173, 1995). Дополнительное доказательство, поддерживающее важную роль GK в регуляции метаболизма глюкозы у людей, было представлено путем идентификации больных, экспрессирующих мутантную форму GK с повышенной ферментативной активностью. У этих больных обнаруживается устойчивая гипогликемия, связанная с несоответственно повышенным уровнем инсулина в плазме (Glaser, В., Kesavan, P., Heyman, М. и др. New England J. Med., 338, 226-230, 1998). В то время как мутации гена GK не обнаружены у большинства больных диабетом типа II, соединения, которые активируют GK и, тем самым, повышают чувствительность сенсорной системы GK, будут, кроме того, полезны при лечении гипергликемических признаков всех видов диабета типа II. Активаторы глюкокиназы будут усиливать течение процесса метаболизма глюкозы в бета-клетках и гепатоцитах, что будет сочетаться с повышенной секрецией инсулина. Такие средства будут полезны для лечения диабета типа II.
Настоящее изобретение предоставляет соединение, включающее амид формулы
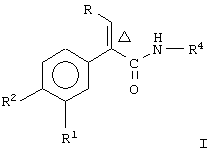
где R1 и R2 означают независимо друг от друга водород, галоген, аминогруппу, нитрогруппу, перфтор(низш.)алкил, (низш.)алкилтиогруппу, перфтор(низш.)алкилтиогруппу, (низш.)алкилсульфонил, перфтор(низш.)алкилсульфонил, (низш.)алкилсульфонилметил или (низш.)алкилсульфинил;
R означает -(CH2)m-R3 или низший алкил, содержащий от 2 до 4 атомов углерода;
R3 означает циклоалкил, содержащий от 3 до 8 атомов углерода;
R4 означает 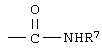
или незамещенное или однозамещенное пяти- или шестичленное гетероароматическое кольцо, связанное с помощью углеродного атома кольца с указанной аминогруппой, причем это пяти- или шестичленное гетероароматическое кольцо содержит от 1 до 2 гетероатомов, выбранных из группы, состоящей из серы или азота, где один гетероатом, являясь азотом, расположен рядом со связующим углеродным атомом кольца, и азанное однозамещенное гетероароматическое кольцо замещено при другом углеродном атоме кольца, несмежном с упомянутым связующим атомом углерода кольца, заместителем, который выбирается из группы, состоящей из галогена или
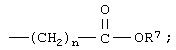
m означает 0 или 1;
n означает 0, 1, 2, 3 или 4;
R7 означает водород или низший алкил и
Δ означает транс-конфигурацию относительно двойной связи,
или его фармацевтически приемлемую соль.
Соединения формулы I являются активаторами глюкокиназы и полезны для повышения секреции инсулина при лечении диабета типа II.
Данное изобретение предоставляет соединение, включающее амид формулы
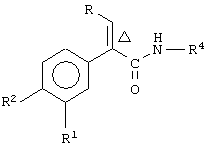
где R1 и R2 означают независимо друг от друга водород, галоген, аминогруппу, нитрогруппу, перфтор(низш.)алкил, (низш.)алкилтиогруппу, перфтор(низш.)алкилтиогруппу, (низш.)алкилсульфинил, (низш.)алкилсульфонил, (низш.)алкилсульфонилметил или перфтор(низш.)алкилсульфонил;
R означает -(CH2)m-R3 или низший алкил, содержащий от 2 до 4 атомов углерода;
R3 означает циклоалкил, содержащий от 3 до 8 атомов углерода;
R4 означает 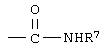
или незамещенное или однозамещенное пяти- или шестичленное гетероароматическое кольцо, связанное с помощью углеродного атома кольца с указанной аминогруппой, причем это пяти- или шестичленное гетероароматическое кольцо содержит от 1 до 2 гетероатомов, выбранных из группы, состоящей из серы или азота, где один гетероатом, являющийся азотом, расположен рядом со связующим углеродным атомом кольца, и указанное однозамещенное гетероароматическое кольцо замещено при другом углеродном атоме кольца, несмежном с упомянутым связующим углеродным атомом кольца, заместителем, который выбирается из группы, состоящей из галогена или
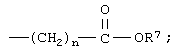
m означает 0 или 1;
n означает 0, 1, 2, 3 или 4;
R7 означает водород или низший алкил и
Δ означает транс-конфигурацию относительно двойной связи,
или его фармацевтически приемлемую соль, которые пригодны в качестве активаторов для повышения секреции инсулина при лечении диабета типа II. Согласно этому изобретению было найдено, что соединения формулы I, имеющие транс-конфигурацию относительно двойной связи, обладают такой глюкокиназной активностью. С другой стороны, соединения формулы I с цис-конфигурацией относительно двойной связи не обладают глюкокиназной активностью.
Когда применяется термин "цис" в данной заявке, это означает, что два наибольших заместителя, присоединенных по двойной связи, находятся по одну сторону относительно двойной связи. Термин "транс", используемый в этой заявке, означает, что наибольшие заместители, присоединенные по двойной связи, располагаются по разные стороны относительно двойной связи и имеют E-конфигурацию.
Настоящее изобретение также относится к фармацевтической композиции, состоящей из соединения формулы I и фармацевтически приемлемого носителя и/или адъюванта. Кроме того, настоящее изобретение относится к применению таких соединений для получения лекарств для лечения диабета типа II. Настоящее изобретение также относится к способам получения соединений формулы I. Кроме этого, данное изобретение относится к методу терапевтического лечения диабета типа II, методу, включающему введение соединения формулы I человеку или животному.
Используемый в данной заявке термин "низший алкил" включает алкильные группы и с прямой, и с разветвленной цепью, содержащие от 1 до 7 атомов углерода, как, например, метил, этил, пропил, изопропил, предпочтительно метил и этил, наиболее предпочтительно метил.
Используемый здесь термин "галоген или галоид", если не указано иначе, означает все четыре галогена, то-есть фтор, хлор, бром и иод.
Как здесь упоминается, "перфтор(низш.)алкил" означает любую низшую алкильную группу, где все атомы водорода низшей алкильной группы замещены или заменены фтором. Среди предпочтительных перфтор(низш.)алкильных групп находятся трифторметил, пентафторэтил, гептафторпропил и т.д., наиболее предпочтителен трифторметил.
Используемый здесь термин "арил" означает одноядерные ароматические углеводородные группы, как, например, фенил, толил и т.д., которые могут быть незамещенными или замещенными по одному или нескольким положениям галогеном, нитрогруппой, низшим алкилом или (низш.)алкоксильными заместителями, и многоядерные арильные группы, как, например, нафтил, антрил и фенантрил, которые могут быть незамещенными или могут быть замещены одной или несколькими из вышеупомянутых групп. Предпочтительными арильными группами являются замещенные или незамещенные одноядерные арильные группы, в частности фенил. Используемый здесь термин "(низш.)алкоксигруппа" включает алкоксигруппы и с прямой, и с разветвленной цепью, содержащие от 1 до 7 атомов углерода, как, например, метоксигруппа, этоксигруппа, пропоксигруппа, изопропоксигруппа, предпочтительно метокси- и этоксигруппа.
Термин "аралкил" означает алкильную группу, предпочтительно низший алкил, в котором один из атомов водорода может быть замещен арильной группой. Примерами аралкильных групп являются бензил, 2-фенилэтил, 3-фенилпропил, 4-хлорбензил, 4-метоксибензил и им подобные группы.
Используемый здесь термин "низшая алкановая кислота" означает низшие алкановые кислоты, содержащие от 2 до 7 атомов углерода, как, например, пропионовая кислота, уксусная кислота и им подобные. Термин "низший алканоил" означает одновалентные алканоильные группы, содержащие от 2 до 7 углеродных атомов, как, например, пропионоил, ацетил и им подобные.
Термин "ароевые кислоты" означает арилалкановые кислоты, где арил является таким, как указано выше, и алкановая кислота содержит от 1 до 6 атомов углерода. Термин "ароил" означает остатки ароевой кислоты, где арил является таким, как указано здесь ранее, с удаленной от карбоксильной части гидроксильной группой. Среди предпочтительных ароильных групп называют бензоил.
Во время протекания реакции различные функциональные группы, как, например, свободные карбоксильные или гидроксильные группы, защищают с помощью общепринятых, способных подвергаться гидролизу сложноэфирных или простых эфирных защитных групп. Используемый здесь термин "способные подвергаться гидролизу сложноэфирные или простые эфирные защитные группы" означает любой сложный эфир или простой эфир, применяемый обычно для защиты карбоновых кислот или спиртов, которые могут быть подвергнуты гидролизу с образованием соответственно гидроксильной или карбоксильной группы. Примерами сложноэфирных групп, используемых для указанных целей, являются такие, в которых ацильные остатки являются производными низшей алкановой кислоты, арил(низш.)алкановой кислоты или (низш.)алкандикарбоновой кислоты. Среди активированных кислот, которые могут использоваться для образования таких групп, находятся ангидриды кислот, галоидангидриды кислот, предпочтительно хлорангидриды кислот или бромангидриды кислот, производные от арильных или низших алкановых кислот. Примерами ангидридов являются ангидриды, производные монокарбоновой кислоты, как, например, уксусный ангидрид, ангидрид бензойной кислоты и ангидриды низших алкандикарбоновых кислот, например янтарный ангидрид, а также эфиры хлормуравьиной кислоты, например, предпочтительны трихлорформиат, этилхлорформиат. Подходящими простыми эфирными защитными группировками для спиртов являются, например, простые тетрагидропираниловые эфиры, как, например, простые 4-метокси-5,6-дигидрокси-2Н-пираниловые эфиры. Другими являются простые ароилметиловые эфиры, как, например, бензиловый, бензгидрильный или трифенилметиловый простые эфиры, или простые α-(низш.)алкокси(низш.)алкиловые эфиры, например метоксиметиловый или аллиловый простые эфиры, или простые алкилсилиловые эфиры, как, например, простой триметилсилиловый эфир.
Термин "защитная группировка для аминогруппы" означает любую общепринятую защитную группировку для аминогруппы, которая может быть расщеплена с образованием свободной аминогруппы. Предпочтительными защитными группами являются общепринятые защитные группировки для аминогруппы, используемые в пептидном синтезе. Особо предпочтительны те защитные группы для аминогруппы, которые расщепляются в слабокислых условиях при рН от 2,0 до 3. Особенно предпочтительны такие защитные группы для аминогруппы, как, например, трет-бутоксикарбонилкарбамат, бензилоксикарбонилкарбамат, 9-флуоренилметилкарбамат.
Гетероароматическое кольцо, обозначенное R4, может быть незамещенным или однозамещенным пяти- или шестичленным гетероароматическим кольцом, содержащим от 1 до 2 гетероатомов, выбранных из группы, состоящей из азота или серы, и связанным с помощью углеродного атома кольца с указанным аминным остатком амидной группы. Гетероароматическое кольцо содержит первый азотный гетероатом рядом со связующим углеродным атомом кольца, и, если имеются другие гетероатомы, то они могут быть серой или азотом. Предпочтительными гетероароматическими кольцами являются пиридинил, пиримидинил и тиазолил, наиболее предпочтительны пиридинил и тиазолил. Эти гетероароматические кольца, которые представляют R4, связаны через углеродный атом кольца с амидной группой и образуют амиды формулы I. Кольцевой углеродный атом гетероароматического кольца, который соединяется амидной связью с образованием соединения формулы I, не может содержать какого-либо заместителя. Когда R4 является незамещенным или однозамещенным пяти- или шестичленным гетероароматическим кольцом, предпочтительными кольцами являются те, которые содержат азотный гетероатом рядом со связующим атомом углерода кольца и второй гетероатом рядом со связующим атомом углерода кольца или по соседству с упомянутым первым гетероатомом.
Термин "фармацевтически приемлемые соли", используемый здесь, включает любую соль как с неорганическими, так и с органическими фармацевтически приемлемыми кислотами, как, например, соляная кислота, бромистоводородная кислота, азотная кислота, серная кислота, фосфорная кислота, лимонная кислота, муравьиная кислота, малеиновая кислота, уксусная кислота, янтарная кислота, винная кислота, метансульфокислота, п-толуолсульфокислота и им подобные кислоты. Термин "фармацевтически приемлемые соли" также включает любую фармацевтически приемлемую соль с основанием, как, например, соли с аминами, соли с триалкиламинами и им подобные. Соли могут быть совсем легко получены специалистами в данной области при использовании стандартных методик.
Соединение формулы I по данному изобретению представляет два предпочтительных вида, то-есть соединение формулы
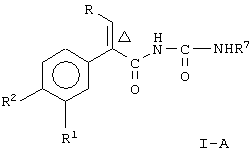
где Δ, R, R1, R2 и R7 являются такими, как указано выше;
и соединение формулы
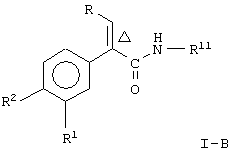
где R, R1, R2 и Δ являются такими, как указано выше;
R11 является незамещенным или однозамещенным пяти- или шестичленным гетероароматическим кольцом, связанным с помощью углеродного атома кольца с указанной аминогруппой, это пяти- или шестичленное гетероароматическое кольцо содержит от 1 до 2 гетероатомов, выбранных из группы, состоящей из серы или азота, причем один гетероатом является азотом, расположенным рядом со связующим углеродным атомом кольца; упомянутое однозамещенное гетероароматическое кольцо имеет заместитель при атоме углерода кольца, другом, а несмежном с упомянутым связующим атомом углерода, заместитель, выбранный из группы, состоящей из галогена или
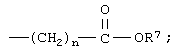
n означает 0, 1, 2, 3 или 4 и
R7 означает водород или низший алкил.
В соответствии с одним предпочтительным примером воплощения соединения формулы I R может быть низшим алкилом, содержащим от 2 до 4 углеродных атомов. В другом предпочтительном примере осуществления R может быть -(CH2)m-R3, где R3 и m являются такими, как указано выше. Предпочтительными гетероциклическими остатками R3 являются циклопентил, циклогексил, циклогептил и циклооктил, более предпочтительны циклопентил, циклогексил и циклогептил. В одном предпочтительном варианте осуществления изобретения R3 означает циклопентил, в другом предпочтительном варианте осуществления R3 означает циклогексил. В предпочтительном варианте осуществления изобретения m является 1, в другом предпочтительном варианте осуществления m означает 0. Предпочтительные гетероароматические кольца R11 согласно представленному изобретению являются незамещенными или однозамещенными пиридинилом или тиазолилом. В одном предпочтительном варианте осуществления изобретения гетероароматическое кольцо R11 является незамещенным или однозамещенным пиридинилом, в другом предпочтительном варианте осуществления изобретения гетероароматическое кольцо R11 означает незамещенный или однозамещенный тиазолил. В соответствии с дальнейшими предпочтительными вариантами осуществления изобретения гетероароматическое кольцо R11 является либо незамещенным, либо имеет один заместитель в виде галогена или -(CH2)n-C(O)-OR7, где n и R7 являются такими, как указано выше. Предпочтительные заместители R1 и R2 выбираются независимо из группы, включающей водород, галоид, нитрогруппу, перфтор(низш.)алкил, (низш.)алкилсульфонил и (низш.)алкилсульфонилметил. В предпочтительном варианте осуществления изобретения один из R1 и R2 означает галоид, (низш.)алкилсульфонил или (низш.)алкилсульфонилметил и другой означает водород, галоид, нитрогруппу или перфтор(низш.)алкил. В более предпочтительном варианте осуществления изобретения один из R1 и R2 означает (низш.)алкилсульфонил и другой означает водород, галоид, нитрогруппу или перфтор(низш.)алкил. Предпочтительным остатком R7 является низший алкил.
В соответствии с одним примером воплощения соединения формулы I-A R может быть циклоалкильной группой, которая содержит от 3 до 8 атомов углерода, предпочтительно циклогексилом (соединение I-A1). В различные примеры воплощения циклогексиламидов соединения I-A1 включены те соединения, где один из R1 и R2 означает водород, галоген, (низш.)алкилсульфонил или перфтор(низш.)алкил и другой из упомянутых R1 и R2 означает галоген, (низш.)алкилсульфонил или перфтор(низш.)алкил, и особенно те соединения, где один из R1 и R2 означает водород или (низш.)алкилсульфонил или перфтор(низш.)алкилсульфонил и другой означает (низш.)алкилсульфонил или перфтор(низш.)алкил. Другим примером воплощения соединения формулы I-A являются те соединения, где R является низшей алкильной группой, содержащей от 2 до 4 атомов углерода (соединения формулы I-A2). Среди примеров воплощения соединений формулы I-A2 находятся те соединения, где один из R1 и R2 означает водород, галоген, (низш.)алкилсульфонил или перфтор(низш.)алкил и другой из упомянутых R1 и R2 означает галоген, (низш.)алкилсульфонил или перфтор(низш.)алкил.
Примером воплощения соединения формулы I-B являются такие соединения, где R11 является незамещенным или однозамещенным тиазольным кольцом. Когда R11 означает незамещенное тиазольное кольцо, R может быть низшей алкильной группой, содержащей от 2 до 4 атомов углерода (соединение I-B1). Среди примеров воплощения соединений формулы I-B1 имеются такие соединения, где один из R1 или R2 означает водород, (низш.)алкилсульфонил, (низш.)алкилсульфонилметил, перфтор(низш.)алкил, галоген, нитрогруппу и другой из упомянутых R1 или R2 означает (низш.)алкилсульфонил, (низш.)алкилсульфонилметил, перфтор(низш.)алкил, галоген или нитрогруппу, и предпочтительно те соединения формулы IB-1, где один из R1 и R2 означает водород, (низш.)алкилсульфонил и другой из упомянутых R1 и R2 означает (низш.)алкилсульфонил.
Примером воплощения соединения формулы I-B являются те соединения, где R означает циклоалкил, содержащий от 3 до 8 атомов углерода (соединение IB-2).
Среди примеров воплощения соединений формулы I-B2 имеются такие соединения, где циклоалкильной группой является циклопентил (IВ-2(а)). Примером воплощения соединений I-В2(а) являются такие соединения формулы IВ-2(а), где R11 является незамещенным тиазольным кольцом (соединения IB-2а(1)). Среди примеров воплощения соединения IВ-2а(1) имеются такие соединения, где один из упомянутых R1 и R2 означает водород, (низш.)алкилсульфонил, (низш.)алкилсульфонилметил, перфтор(низш.)алкил, галоген или нитрогруппу и другой из упомянутых R1 и R2 означает (низш.)алкилсульфонил, (низш.)алкилсульфонилметил, перфтор(низш.)алкил, галоген или нитрогруппу, и особенно предпочтительными примерами воплощения соединений IВ-2(а)(1) являются соединения, где
а) один из R1 или R2 означает (низш.)алкилсульфонил и другой означает водород, нитрогруппу, (низш.)алкилсульфонил, галоген или перфтор(низш.)алкил;
б) один из R1 и R2 означает галоген, водород или перфтор(низш.)алкил и другой означает перфтор(низш.)алкил или галоген и
в) один из R1 и R2 означает (низш.)алкилсульфонилметил и другой означает водород, (низш.)алкилсульфонилметил или галоген.
Среди примеров воплощения соединения формулы IВ-2а имеются такие соединения, где R11 является однозамещенным тиазолильным кольцом, что включает соединения, где R11 является галоидзамещенным тиазольным кольцом (соединения формулы IВ-2(а)(2)). Среди примеров воплощения соединений формулы IВ-2(а)(2) имеются такие соединения, где один из R1 и R2 является (низш.)алкилсульфонилом, водородом или галогеном и другой является (низш.)алкилсульфонилом или галогеном.
Другим примером воплощения соединений IB-2 являются те соединения, где R означает циклогексил (соединения IВ-2(b)). Среди примеров воплощения соединений IВ-2(b) имеются такие соединения, где R11 является незамещенным тиазолильным кольцом (соединение 1В-2(b)(1)). Среди предпочтительных соединений IB-2(b) имеются такие соединения, где один из R1 или R2 означает водород, (низш.)алкилсульфонил, (низш.)алкилсульфонилметил, перфтор(низш.)алкил, галоген, нитрогруппу и другой означает (низш.)алкилсульфонил, (низш.)алкилсульфонилметил, перфтор(низш.)алкил, галоген или нитрогруппу, в частности
(а) где один из R1 или R2 означает (низш.)алкилсульфонил и другой означает водород, нитрогруппу, (низш.)алкилсульфонил, галоген или перфтор(низш.)алкил;
(б) где один из R1 и R2 означает галоген, водород или перфтор(низш.)алкил и другой означает перфтор(низш.)алкил или галоген и
(в) где один из R1 и R2 означает (низш.)алкилсульфонилметил и другой означает водород, (низш.)алкилсульфонилметил или галоген.
Другим примером воплощения соединения IВ-2(b) являются такие соединения, где R11 является однозамещенным тиазолильным циклом и, в частности, галоидзамещенным циклом (соединение IВ-2(b)(2)). Среди примеров воплощения соединений IВ-2(b)(2) имеются такие соединения, где один из R1 и R2 означает (низш.)алкилсульфонил и другой означает галоген, перфтор(низш.)алкил или водород.
Другим примером воплощения соединения IB-2 являются те соединения, где R означает циклогептил (соединение IB-2(d)) или циклооктил (соединение IВ-2(е)). Примером воплощения соединений (соединение IB-2(d) и соединение IВ-2(е)) являются те соединения, где R11 означает незамещенный тиазолил (соединения IB-2(d)(1) и IВ-2(е)(1)) соответственно. В этом случае соединениями IB-2(d)(1) и IВ-2(е)(1), которые предпочтительны, являются такие соединения, где один из R1 и R2 означает (низш.)алкилсульфонил, водород, галоген или перфтор(низш.)алкил и другой означает (низш.)алкилсульфонил, галоген или перфтор(низш.)алкил.
Другим примером воплощения соединения IB-2(d) и соединения IВ-2(е) являются такие соединения, где R11 означает однозамещенное тиазолильное кольцо и заместителем является галоид. В этих случаях один из R1 и R2 может быть водородом, (низш.)алкилсульфонилом, перфтор(низш.)алкилом или галогеном и второй может быть галогеном, (низш.)алкилсульфонилом или перфтор(низш.)алкилом. В соединениях IB-2(d) и IB-2(e) R11 является монозамещенным тиазолилом, заместителем может быть
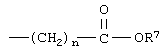
где n и R7 являются такими, как указано выше.
В случае таких соединений один из R1 и R2 в этих соединениях может быть (низш.)алкилсульфонилом и второй из упомянутых R1 и R2 является (низш.)алкилсульфонилом или водородом.
Другим классом соединений формулы IB являются такие соединения, где R означает -CH2-R3 и R3 является таким, как указано выше. Среди соединений, включенных в рамки данного варианта осуществления изобретения, имеются соединения, где R означает группу -СН2-циклогексил (соединение IB-3). Соединения IB-3 включают такие, где R11 является замещенным или незамещенным тиазолильным кольцом и, в особенности, те соединения, где R11 является незамещенным тиазолильным кольцом и где заместителем в тиазолильном кольце является
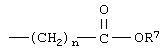
где n и R7 являются такими, как указано выше.
В этом случае предпочтительны соединения, где один из R1 и R2 означает (низш.)алкилсульфонил и другой означает (низш.)алкилсульфонил или водород. В соответствии с примером воплощения соединения формулы IB R может быть циклопентилом. Пример воплощения этого класса включает соединения, где R11 является незамещенным или однозамещенным пиридинильным кольцом. Предпочтительным примером воплощением этого класса являются те соединения, где один из R1 и R2 означает водород, (низш.)алкилсульфонил или галоген и другой из упомянутых R1 и R2 означает (низш.)алкилсульфонил или галоген.
В соответствии с настоящим изобретением соединения формулы IA и IB могут быть получены из следующих соединений формул:
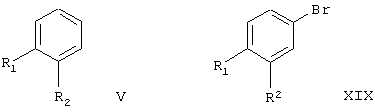
где R1 и R2 являются такими, как указано выше.
Согласно данному изобретению соединения формулы IA и IB получают из соединений формулы V по следующей реакционной схеме:
Схема 1
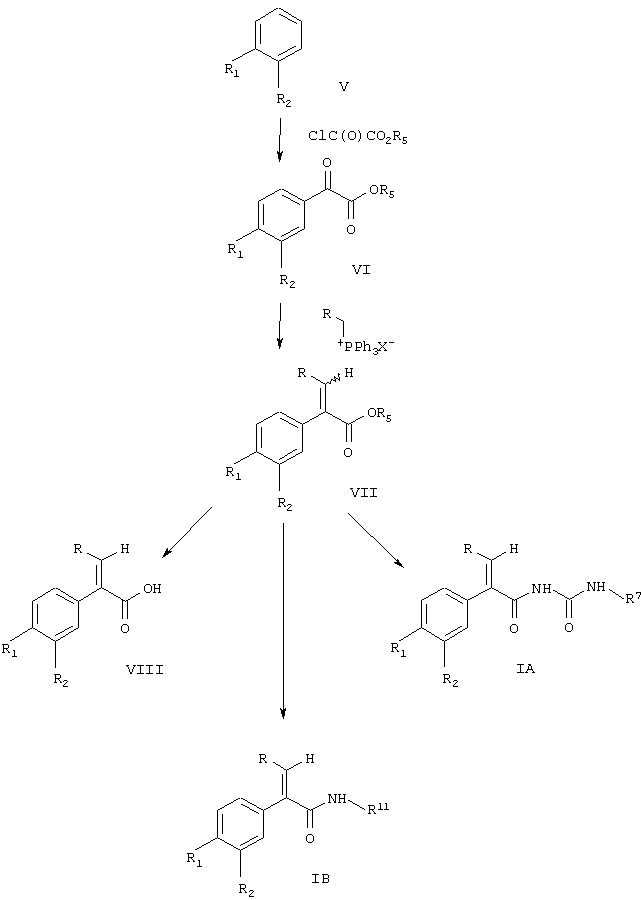
где R, R1, R2, R7 и R11 являются такими, как указано выше;
R5 вместе с связанным с ним атомом кислорода образует способную подвергаться гидролизу защитную группу для кислоты и
Х означает галоген.
Соединение формулы V или XIX, где один из R1 и R2 означает нитрогруппу, тиогруппу, аминогруппу, галоген и другой означает водород, являются известными веществами. Замещенные амином соединения формулы V или XIX могут быть превращены в соединения с другими заместителями либо до, либо после превращения в соединения формулы IA или IB. Для этого аминогруппы могут быть продиазотированы с образованием соответствующего диазониевого соединения, которое in situ может быть подвергнуто реакции с желаемым (низш.)алкилтиолом, перфтор(низш.)алкилтиолом (см., например, Baleja, J.D., Synth. Соmm. 1984, 14, 215; Giam, C.S., Kikukawa, K.,J. Chem.Soc, Chem. Comm. 1980, 756; Kau, D., Krushniski, J.H., Robertson, D.W., J.Labelled Compd Rad. 1985, 22, 1045; Oade, S., Shinhama, K., Kim, Y.H., Bull Chem Soc Jpn. 1980, 53, 2023; Baker, B.R., и др., J.Org. Chem. 1952, 17, 164) для получения соответствующих соединений формул V и XIX, где один из заместителей является (низш.)алкилтиогруппой, перфтор(низш.)алкилтиогруппой и другой является водородом. Если желательно, (низш.)алкилтиопроизводные или перфтор(низш.)алкилтиопроизводные могут быть превращены путем окисления в соответствующие (низш.)алкилсульфонил- или перфтор(низш.)алкилсульфонилзамещенные соединения формулы V или XIX. Для осуществления такого превращения может быть использован любой принятый способ окисления алкилтиозаместителей в сульфоны. Если желательно получить соединения с перфтор(низш.)алкильными группами формулы V или XIX, в качестве исходных веществ могут быть применены соответствующие галоидзамещенные соединения формулы V или XIX. Любой принятый способ превращения галоидсодержащей ароматической группы в соответствующую перфтор(низш.)алкильную группу может быть применен для осуществления такого превращения (см.,например, Katayama, Т., Umeno, M., Chem. Lett. 1991, 2073; Reddy, G.S., Tam., Organometallics, 1984, 3, 630; Novak, J., Salemink, C.A., Synthesis, 1983, 7, 597; Eapen, K.C.. Dua, S.S., Tamboroski, C., J. Org. Chem. 1984, 49, 478; Chen, Q.-Y., Duan, J.-X., J. Chem.Soc. Chem. Comm. 1993, 1389; Clark, J.H., McClinton, M.A., Jone, C.W., Landon, P., Bishop, D., Blade, R.J., Tetrahedron Lett.1989, 2133; Powell, R.L., Heaton, С.А., патент US №5113013).
Соединения формулы V или XIX, где оба заместителя R1 и R2 являются аминогруппами, могут быть получены из соответствующего динитросоединения формулы V или XIX. Для проведения такого превращения может быть использован любой принятый способ восстановления нитрогруппы в аминогруппу. Соединение формулы V или XIX, где оба R1 и R2 означают аминогруппы, могут быть применены для получения соответствующего соединения формулы V или XIX, где оба R1 и R2 означают иод или бром, используя реакцию диазотирования. Для осуществления такого превращения может быть применен любой принятый способ превращения аминогруппы в группу в виде иода или брома (см., например, Lucas, H.J., Kennedy, E.R., Org. Synth. Coll., Vol, II, 1943, 351). Если желательно получить соединения формулы V или XIX, где оба R1 и R2 означают (низш.)алкилтиогруппу или перфтор(низш.)алкилтиогруппу, соединение формулы V или XIX, где R1 и R2 являются аминогруппами, могут быть использованы в качестве исходного вещества. Любой общепринятый способ превращения ариламиногруппы в арилтиоалкильную группу может быть применен для осуществления такого превращения. Если желательно получить соединение формулы V или XIX, где R1 и R2 означают (низш.)алкилсульфонил или перфтор(низш.)алкилсульфонил, соответствующие соединения формулы V или XIX, где R1 и R2 означают (низш.)алкилтиогруппу или перфтор(низш.)алкилтиогруппу, могут быть использованы в качестве исходных веществ. Для осуществления такого превращения может быть использован любой традиционный способ окисления алкилтиозаместителей в сульфоны. Если желательно получить соединения формулы V или XIX, где оба R1 и R2 замещены перфтор(низш.)алкильными группами, соответствующие галоидзамещенные соединения формулы V или XIX могут быть использованы в качестве исходных веществ. Для осуществления такого превращения может быть использован любой общепринятый способ превращения ароматической галоидсодержащей группы в соответствующую перфтор(низш.)алкильную группу.
Соединения формулы V или XIX, где один из R1 и R2 означает нитрогруппу, а другой означает галоид, известны из литературы (см. для 4-хлор-3-нитрофенилуксусной кислоты, Tadayuki, S., Hiroki, M., Shinji, U., Mitsuhiro, S., патент JP 71-99504, Chemical Abstracts 80:59716; см. для 4-нитро-3-хлорфенилуксусной кислоты, Zhu, J.; Beugelmans, R.; Bourdet, S.; Chastanet, J.; Rousssi, G., J. Org. Chem. 1995, 60, 6389; Beugelmans, R.; Bourdet, S.; Zhu, J., Tetrahedron Lett. 1995, 36, 1279). Так, если желательно получить соединение формулы V или XIX, где один из R1 и R2 означает нитрогруппу и другой означает (низш.)алкилтиогруппу или перфтор(низш.)алкилтиогруппу, в качестве исходного соединения может быть использовано соответствующее соединение, где один из R1 и R2 является нитрогруппой, а другой хлором. В этой реакции может быть использован любой общепринятый способ нуклеофильного замещения ароматической хлорсодержащей группы (низш.)алкилтиолом (см., например, Singh, P.; Batra, M.S.; Singh, H., J. Chem. Res.-S 1985 (6), S 204; Ono, M.; Nakamura, Y.; Sata, S.; Itoh, I., Chem. Lett, 1988, 1393; Wohrle, D.; Eskes, M.; Shigehara, K.; Yamada, A, Synthesis, 1993, 194; Sutter, M.; Kunz, W., патент US 5169951). Когда доступны соединения формулы V или XIX, где один из R1 и R2 означает нитрогруппу и другой означает (низш.)алкилтиогруппу или перфтор(низш.)алкилтиогруппу, они могут быть превращены в соответствующие соединения формулы V или XIX, где один из R1 и R2 означает нитрогруппу и другой означает (низш.)алкилсульфонил или перфтор(низш.)алкилсульфонил, используя стандартные методики окисления. Если желательно получить соединения формулы V или XIX, где один из R1 и R2 означает аминогруппу и другой означает (низш.)алкилтиогруппу или перфтор(низш.)алкилтиогруппу, в качестве исходного вещества может быть применено соответствующее соединение, где один из R1 и R2 означает нитрогруппу и другой означает (низш.)алкилтиогруппу или перфтор(низш.)алкилтиогруппу. Для осуществления такого превращения может быть использован любой традиционный способ восстановления связанной с ароматическим ядром нитрогруппы в аминогруппу. Если желательно получить соединения формулы V или XIX, где один из R1 и R2 означает (низш.)алкилтиогруппу и другой означает перфтор(низш.)алкилтиогруппу, в качестве исходного вещества может быть использовано соответствующее соединение, где один из R1 и R2 означает аминогруппу и другой означает (низш.)алкилтиогруппу или перфтор(низш.)алкилтиогруппу. Для осуществления такого превращения может быть применен любой общепринятый способ, предусматривающий диазотирование аминогруппы при ароматическом кольце и взаимодействие полученного при этом соединения in situ с желаемым (низш.)алкилтиолом. Если желательно получить соединения формулы V или XIX, где один из R1 и R2 означает (низш.)алкилсульфонил и другой означает перфтор(низш.)алкилсульфонил, в качестве исходных веществ могут быть использованы соответствующие соединения, где один из R1 и R2 означает (низш.)алкилтиогруппу и другой означает перфтор(низш.)алкилтиогруппу. Для осуществления такого превращения может быть применен любой стандартный способ окисления простой ароматической тиоэфирной группы в соответствующий сульфон. Если желательно получить соединения формулы V или XIX, где один из R1 и R2 означает галоген и другой означает (низш.)алкилтиогруппу или перфтор(низш.)алкилтиогруппу, в качестве исходных веществ могут быть использованы соответствующие соединения, где один из R1 и R2 означает аминогруппу и другой означает (низш.)алкилтиогруппу или перфтор(низш.)алкилтиогруппу. Для осуществления такого превращения может быть использован любой общепринятый способ диазотирования ароматического амина и превращения in situ в ароматическое галоидное соединение. Если желательно получить соединения формулы V или XIX, где один из R1 и R2 означает галоид и другой означает (низш.)алкилсульфонил или перфтор(низш.)алкилсульфонил, в качестве исходных веществ могут быть использованы соответствующие соединения, где один из R1 и R2 означает галоид и другой означает (низш.)алкилтиогруппу или перфтор(низш.)алкилтиогруппу. Для осуществления такого превращения может быть использован любой стандартный метод окисления простого ароматического тиоэфира в соответствующий сульфон. Если желательно получить соединение формулы V или XIX, где один из R1 и R2 означает нитрогруппу и другой означает аминогруппу, в качестве исходного вещества может быть использовано соединение формулы V или XIX, где один из R1 и R2 означает нитрогруппу и другой означает хлор. Хлорный заместитель при фенильном кольце может быть превращен в иодный заместитель (см., например, Bunnett, J.F.; Conner, R.M.; Org. Synth. Coll Vol V, 1973, 478; Clark, J.H.; Jones, C.W., J.Chem. Soc. Chem. Commun. 1987, 1409), который в свою очередь может реагировать с переносящим азид агентом с образованием соответствующего азида (см., например, Suzuki, Н.; Miyoshi, K.; Shinoda, M., Bull. Chem. Soc. Jpn, 1980, 53, 1765). Этот азид может быть восстановлен обычным способом в амин при восстановлении обычно применяемым восстанавливающим средством для превращения азидов в амины (см., например, Soai, К.; Yokoyama, S.; Ookawa, A., Synthesis, 1987, 48).
Для получения соединения, где R1 и/или R2 означают (низш.)алкилсульфонилметил в соединении формулы I, можно исходить из известного соединения формулы V, где один или оба R1 и R2 означают метил. Метильные группы в этих соединениях могут быть бромированы с помощью любых стандартных средств для бромирования метильных групп при фенильном кольце. Это бромированное соединение затем обрабатывают натриевой солью (низш.)алкилтаола (как, например, тиометилат натрия) для получения (низш.)алкилтиометильного производного. Чтобы получить (низш.)алкилсульфонилметильный заместитель можно применять для осуществления данного превращения любой принятый способ окисления заместителей в виде (низш.)алкилтиогрупп в сульфоны, как, например, описанный выше.
Заместители, которые представляют собой R1 и R2, могут быть присоединены к кольцу после образования соединений формул IA и IB. Следовательно, все из описанных реакций для получения различных заместителей R1 и R2 в соединении формулы I могут быть осуществлены на соединениях формул IA и IB после их образования.
Соединения формулы IA и IB получают из соединения формулы V или XIX, как представлено в схемах 1 или 2. На первой стадии этой реакции в схеме 1 соединение формулы V подвергается реакции с оксалилхлоридом, где свободная, способная гидролизоваться органическая кислотная группа в оксалилхлориде защищается с помощью любых общепринятых защитных групп для кислоты. К предпочтительным защитным группам для кислот относятся способные подвергаться гидролизу сложные эфиры оксалилхлорида. Защитная группа образуется с помощью R5. Взаимодействие защищенного оксалилхлорида с соединением формулы V для получения соединения формулы VI проводится по реакции Фриделя-Крафтса. При проведении этой реакции может использоваться любое из условий, принятое для осуществления реакции Фриделя-Крафтса. В этой реакции R1 и R2 не могут быть нитрогруппой. С другой стороны, R1 и R2 могут быть аминогруппой. Однако до осуществления реакции эта аминогруппа должна быть защищена с помощью обычной, способной подвергаться гидролизу защитной группировки для аминогруппы. На некой последующей стадии реакции эти защитные группировки для аминогруппы могут быть удалены и аминогруппы превращены в нитрогруппы, как описано здесь ранее.
Соединение формулы VI может реагировать с галоидной солью трифенилфосфония формулы IX по реакции Виттига с образованием соединения формулы VII. При проведении этой реакции любое из условий, обычных для проведения реакции Виттига, может быть использовано для успешного осуществления такого синтеза из соединения формулы VI и соединения формулы IX с целью получения соединения формулы VII. Соединение формулы VII получается в виде образовавшейся по реакции Виттига смеси цис- и транс-изомеров по отношению к двойной связи. Смесь цис- и транс-изомеров соединения формулы VII непосредственно гидролизуется в соединение формулы VIII. При этой реакции гидролиза соединение формулы VIII получается преобладающе в виде транс-изомера в этой смеси. Кроме того, образующийся при этой реакции гидролиза транс-изомер получается в виде твердого вещества, в то время как цис-изомер получается в виде маслянистого вещества. Принимая это во внимание, очень легко отделить транс-изомер, используя обычные способы кристаллизации из этой смеси для получения соединения формулы VIII в виде чистого транс-изомера, по существу свободного от соответствующего цис-изомера. Такая кристаллизация может проводиться на данной ступени или на следующих ступенях реакции при образовании соединений формулы IA или IB. Поэтому с помощью такой методики соединение формулы IA и IB может быть получено в чистой транс-форме, в значительной степени свободной от соответствующего цис-изомера.
При выделении транс-изомера очистку лучше осуществлять путем гидролиза защитной группы -OR5 до соответствующего соединения формулы VIII в виде свободной кислоты и выделения этой свободной кислоты с помощью кристаллизации в виде транс-изомера, свободного от соответствующего цис-изомера. При получении соединения формулы IB в виде его транс-формы предпочтительно осуществлять процедуру кристаллизации с соединением формулы VIII. С другой стороны, очистка кристаллизацией может быть проведена, используя соединения формулы IB и IA. Поскольку транс-изомеры этих соединений являются твердыми веществами и цис-изомеры являются маслянистыми веществами, любой общепринятый способ кристаллизации может быть применен для успешного проведения такой очистки.
На следующей стадии этого способа соединение формулы VIII присоединяют к соединению формулы

где R11 является таким, как указано выше,
для получения соединения формулы IB. Эта реакция сочетания может быть проведена с использованием любого из обычных способов путем взаимодействия кислоты с первичным амином для получения амида. С другой стороны, соединение формулы VII может быть непосредственно соединено с соединением формулы XIV для получения соединения формулы IB без каких-либо промежуточных стадий гидролиза.
При получении соединения формулы IA соединение формулы VII подвергают реакции с

Эта реакция может быть осуществлена путем превращения соединения формулы VII в соответствующую свободную кислоту путем удаления защитной группы R5 с образованием карбоновой кислоты. Карбоновая кислота формулы VIII может быть превращена в соответствующий амид при превращении кислоты в хлорангидрид и затем при реакции этого хлорангидрида кислоты с аммиаком. При этой процедуре могут быть использованы условия, общепринятые для превращения кислоты в ее хлорангидрид. Этот хлорангидрид кислоты затем подвергается реакции с алкилизоцианатом формулы XV для получения аддукта с мочевиной формулы IA. Любой обычный способ взаимодействия алкилизоцианата с амидом для образования связи с мочевиной использует соединение формулы IA.
Соединение формулы IA может быть образовано в виде смеси цис- и транс-изомеров, если соединение формулы VII не было очищено. Если желательно, очистка может быть осуществлена и в случае соединения формулы IA для получения соединения формулы IA в виде полностью транс-изомера, освобожденного от цис-изомера. Таким же способом, каким могут быть очищены соединение формулы IB или соединение формулы VIII, может быть очищено соединение формулы IA для получения полностью транс-изомера.
В соответствии с другим вариантом осуществления данного изобретения соединение формулы VII также может быть получено по следующей реакционной схеме 2. Эта реакционная схема применима для получения соединений формулы IA и IB, где один или оба R1 и R2 означают нитрогруппу. Реакция сочетания может быть легко проведена с любой из указанных групп R1 и R2, в частности, когда R1 и R2 являются нитрогруппами.
Схема 2
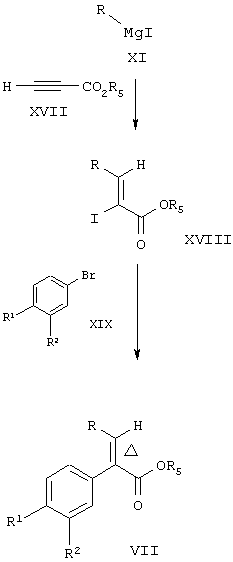
где R5 вместе со связанным с ним атомом кислорода образует способную подвергаться гидролизу защитную группу карбоновой кислоты, R, R1, R2 и Δ являются такими, как указано выше.
По схеме 2 соединение формулы XI может быть генерировано in situ или из соответствующего магнийорганического реагента, или из цинкорганического реагента и растворимого медного реагента (CuCN и 2LiCl) (см., например, Knochel, P.; Singer, R.D., Chem. Rev. 1993, 93, 2117). Затем соединение формулы XI добавляют к соединению формулы XVII с целью получения путем 1,4-конъюгированного присоединения высоко регио- и стереоселективным образом винилмедного промежуточного соединения, которое при иодолизе с помощью иода образовывало соединение формулы XVIII, в котором R и иод находятся в син-отношении друг к другу. Соединение формулы XVIII затем реагирует с активированным металлическим цинком (см., например, Knochel, P.; Janakiram Rao. С., Tetrahedron, 1993, 49, 29) для получения винилцинкового промежуточного соединения, которое затем подвергают взаимодействию с бромистым или йодистым соединением формулы XIX в присутствии источника Pd(0) для получения соединения формулы VII. Когда проводят эту реакцию, ароматический заместитель присоединяется таким образом, чтобы в соединении формулы VII имела место транс-конфигурация относительно двойной связи.
Все соединения формулы I, которые включают приведенные далее в примерах соединения, активировали глюкокиназу in vitro при использовании методики из примера А. Таким образом, они усиливают течение процесса метаболизма глюкозы, что вызывает повышенную секрецию инсулина. Поэтому соединения формулы I являются активаторами глюкокиназы, полезными для увеличения секреции инсулина.
Были исследованы следующие приведенные в виде примеров соединения и было показано, что они являются отличными активаторами глюкокиназы при исследовании активности in vivo при введении в соответствии с анализом, описанным в примере Б:
(Е)-3-циклопентил-2-(4-метансульфонилфенил)-N-тиазол-2-илакриламид;
(Е)-3-циклогексил-2-(4-метансульфонилфенил)-N-тиазол-2-илакриламид;
(Е)-3-циклогептил-2-(4-метансульфонилфенил)-N-тиазол-2-илакриламид;
(Е)-2-(3-хлор-4-метансульфонилфенил)-3-циклопентил-N-тиазол-2-илакриламид;
(Е)-3-циклогексил-2-(4-метансульфонил-3-трифторметилфенил)-N-тиазол-2-илакриламид;
(Е)-3-циклогексил-2-(4-метансульфонил-3-нитрофенил)-N-тиазол-2-илакриламид;
(Е)-N-(5-бромтиазол-2-ил)-3-циклогептил-2-(4-метансульфонилфенил)акриламид;
(Е)-2-(3-хлор-4-метансульфонилфенил)-3-циклопентил-N-пиридин-2-илакриламид;
(Е)-N-(5-бромпиридин-2-ил)-3-циклогексил-2-(4-метансульфонил-3-трифторметилфенил)акриламид;
тиазол-2-иламид (Е)-4-циклопентил-2-(4-метансульфонилфенил)бут-2-еновой кислоты;
метиловый эфир (Е)-2-[4-циклопентил-2-(4-метансульфонилфенил)бут-2-еноиламино]тиазол-4-карбоновой кислоты и
тиазол-2-иламид (Е)-4-циклопентил-2-(4-метансульфонил-3 -трифторметилфенил)бут-2-еновой кислоты.
На основании их способности активировать глюкокиназу соединения вышеприведенной формулы I могут быть использованы в качестве лекарственных средств для лечения диабета типа II. Поэтому, как упомянуто ранее, лекарственные средства, содержащие соединение формулы I, также являются объектом настоящего изобретения, как и способ приготовления таких лекарственных средств, способ, который предусматривает включение одного или нескольких соединений формулы I и, если желательно, одного или нескольких других терапевтически ценных веществ в галеновую форму для введения.
Фармацевтические композиции могут быть введены перорально, например, в виде таблеток, таблеток с покрытием, драже, твердых и мягких желатиновых капсул, растворов, эмульсий или суспензий. Введение может также быть проведено через прямую кишку, например, используя суппозитории, местно или чрескожно, например, используя мази, кремы, гели или растворы, или парентерально, например внутривенно, внутримышечно, подкожно, внутриоболочечно или чрескожно, используя, например, инъекционные растворы. Кроме того, введение может быть проведено подъязычно или в виде аэрозоля, например в виде распыляемого раствора. Для приготовления таблеток, таблеток с покрытием, драже или твердых желатиновых капсул соединения по настоящему изобретению могут быть смешаны с фармацевтически инертными неорганическими или органическими наполнителями. Примеры подходящих наполнителей для таблеток, драже или твердых желатиновых капсул включают лактозу, кукурузный крахмал или их производные, тальк или стеариновую кислоту или ее соли. Подходящие наполнители для использования с мягкими желатиновыми капсулами включают, например, растительные масла, воски, жиры, полутвердые или жидкие полиолы и т.д.; в соответствии с природой активных ингредиентов может, однако, возникнуть такое обстоятельство, что для мягких желатиновых капсул совсем не нужен наполнитель. Для приготовления растворов и сиропов наполнители, которые могут быть использованы, включают, например, воду, полиолы, сахарозу, инвертный сахар и глюкозу. Для инъецируемых растворов наполнители, которые могут быть использованы, включают, например, воду, спирты, полиолы, глицерин и растительные масла. Для суппозиториев и для местного или чрескожного применения наполнители, которые могут быть использованы, включают, например, природные или отверждающие масла, воски, жиры и полутвердые или жидкие полиолы. Фармацевтические композиции могут также содержать консерванты, солюбилизирующие средства, стабилизирующие средства, смачивающие средства, эмульгаторы, подсластители, красители, отдушки, соли для изменения осмотического давления, буферы, средства для покрытия или антиоксиданты. Как упоминалось ранее, они могут также содержать другие терапевтически ценные средства. Необходимым условием является то, чтобы все используемые в приготовлении препаратов адъюванты были нетоксичны.
Предпочтительными формами применения являются внутривенное, внутримышечное или пероральное введение, наиболее предпочтительно пероральное введение. Дозировки, в которых вводятся соединения формулы (I) в эффективных количествах, зависят от природы специфически активных ингредиентов, возраста и потребностей больного и способа введения. Обычно рассматриваются дозы, составляющие примерно 1-100 мг/кг массы тела в день.
Изобретение будет лучше понято из следующих примеров, которые приведены с целью иллюстрации и не ограничивают изобретение, определяемое пунктами формулы изобретения, приведенной далее.
ПРИМЕРЫ
Примеры биологической активности
Пример A: in vitro глюкокиназная активность
Анализ глюкокиназы: глюкокиназа (GK) подвергалась испытанию путем связывания продуцирования глюкозо-6-фосфата с генерированием восстановленного никотинамидадениндинуклеотида (NADH) с помощью глюкозо-6-фосфат-дегидрогеназы (G6PDH, 0,75-1 к-единиц/мг; фирма Boehringer Mannheim, Индианаполис, IN) из Leuconostoc mesenteroides в качестве связывающего фермента (схема 3).
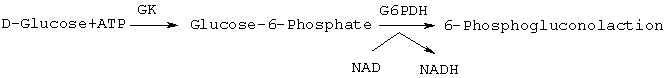
Схема 3
Рекомбинант GK1 из печени человека была экспрессирована в Е.Coli в виде составного белка глутатион-S-трансферазы (GST-GK) [Liang и др., 1995] и очищена путем аффинной хроматографии на колонке с глутатион-сефарозой 4В, используя методику, предоставленную производителем (Amersham Pharmacia Biotech, Piscataway, NJ). Проведенные ранее исследования продемонстрировали, что ферментативные свойства нативной GK и GST-GK являются по существу идентичными (Liang и др., 1995); Neet и др., 1990).
Испытание проводили при 25°С в плоскодонном 96-ячеечном планшете для культуры клеток ткани от Costar (Кембридж, МА) при конечном объеме при инкубировании в 120 мкл. Инкубационная смесь содержала 25 мМ буфер с N-2-гидроксиэтилпиперазин-N′-2-этансульфоновой кислотой (Hepes) (pH 7,1), 25 мМ КСl, 5 мМ D-глюкозу, 1 мМ аденозинтрифосфат (АТР), 1,8 мМ никотинамидаденин-динуклеотид (NAD), 2 мМ хлорид магния, 1 мкМ сорбит-6-фосфат, 1 мМ дитиотреитол, исследуемое соединение или 10% диметилсульфоксид (ДМСО), 1,8 единицы/мл G6-PDH и GK (см. ниже). Все органические реагенты имели чистоту >98% и были предоставлены фирмой Boehringer Mannheim, за исключением D-глюкозы и Hepes, которые были получены от фирмы Sigma Chemical Co, St Louis, МО. Исследуемые соединения растворяли в ДМСО и прибавляли в инкубационную смесь без GST-GK в объеме 12 мкл, чтобы получить конечную концентрацию ДМСО в 10%. Эту смесь предварительно инкубировали в камере с регулируемой температурой спектрофотометра с микропланшетом SPECTRAmax 250 (Molecular Devices Corporation, Sunnyvale, CA) в течение 10 минут до установления температурного равновесия и затем начинали реакцию при прибавлении 20 мкл GST-GK.
После прибавления фермента повышение оптической плотности (ОП) при 340 нм регистрировали в течение 10-минутного инкубационного периода в качестве критерия активности GK. Прибавляли GST-GK в количестве, достаточном для получения увеличения ОП340 от 0,08 до 0,1 единицы за 10-минутный период инкубирования в ячейках, содержащих 10% ДМСО, но без исследуемого соединения. Предварительные эксперименты установили, что скорость реакции GK была линейной за этот период времени даже в присутствии активаторов, которые приводили к 5-кратному возрастанию активности GK. Активность GK в контрольных ячейках сравнивали с активностью в ячейках, содержащих исследуемые активаторы GK, и рассчитывали концентрацию активатора, приводящую к 50% возрастанию активности GK, то-есть SC1,5. Все соединения формулы I, описанные в примерах синтеза, имели SC1,5, меньшую или равную 30 мкМ.
Пример Б: in vivo активность
Протокол in vivo скрининга активатора глюкокиназы
Мышам линии C57BL/6J перорально принудительно вводят активатор глюкокиназы (GK) в дозе 50 мг/кг массы тела после двухчасового периода голодания. Определение глюкозы в крови проводят пять раз в течение шестичасового периода исследования после введения.
Мышей (n=6) взвешивают и подвергают голоданию в течение двухчасового периода перед пероральной обработкой. Активаторы GK готовятся в концентрации 6,76 мг/мл в носителе Gelucire (этанол : Gelucire 44/14 : полиэтиленгликоль (ПЭГ) 400 достаточное количество, 4 : 66 : 30 об./мас./об.). Мышам перорально вводят по 7,5 мкл лекарственной формы на грамм массы тела до дозы, равной 50 мг/кг. Непосредственно перед дозированием для отсчета показаний глюкозы в крови отбирается предварительная проба (время ноль) путем отрезания небольшого кусочка хвоста животного (~1 мм) и отбора 15 мкл крови в гепаринизированную капиллярную трубку для анализа. После введения активатора GK из той же раны хвоста берутся через 1, 2, 4 и 6 часов после дозирования дополнительные пробы крови для определения глюкозы в крови. Результаты интерпретируются при сравнении средних значений глюкозы в крови у шести обработанных наполнителем мышей со значениями у шести обработанных активатором GK мышей в течение шестичасового периода исследования. Соединения считаются активными, если они показывают статистически значительное (p≤0,05) уменьшение глюкозы в крови по сравнению с наполнителем для двух последовательных временных точек анализа.
Пример 1
Тиазол-2-иламид (Е)-2-(4-метансульфонилфенил)пент-2-еновой кислоты
Смесь хлористого лития (1,7 г, 40 ммолей, предварительно высушенного при 130°С в высоком вакууме в течение 2 ч) и цианида меди (1,78 г, 20 ммолей) в безводном тетрагидрофуране (20 мл) перемешивали при 25°С в атмосфере аргона в течение 10 минут до получения прозрачного раствора. Реакционную смесь охлаждали до -70°С и затем медленно обрабатывали 1 М раствором этилмагнийбромида в тетрагидрофуране (20 мл, 20 ммолей). После прибавления реакционной смеси давали нагреться до -30°С и перемешивали при этой температуре 5 минут. Полученную в результате реакционную смесь опять охлаждали до -70°С и затем медленно обрабатывали метиловым эфиром пропиоловой кислоты (1,52 г, 18 ммолей). Реакционную смесь перемешивали 4 часа при температуре от -40 до -30°С и затем охлаждали до температуры -70 - -60°С, в это время реакционную смесь медленно обрабатывали раствором иода (6,86 г, 27 ммолей) в безводном тетрагидрофуране (20 мл). После прибавления раствора иода баню для охлаждения убирали и реакционной смеси давали нагреться до 25оС, при этой температуре ее перемешивали 1 ч. Реакционную смесь затем выливали в раствор, состоящий из насыщенного водного раствора хлористого аммония (90 мл) и гидроокиси аммония (10 мл), и органическое соединение экстрагировали диэтиловым эфиром (3×50 мл). Объединенные органические экстракты последовательно промывали насыщенным водным раствором тиосульфата натрия (1×100 мл) и насыщенным водным раствором хлористого натрия (1×100 мл). Органический слой сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме. Хроматография с помощью системы Biotage (ФЛЭШ 40М, двуокись кремния, 19/1 гексаны/диэтиловый эфир) приводила к метиловому эфиру (Е)-2-иодпентеновой кислоты (2,9 г, 67%) в виде бесцветного масла: масс-спектрометрия высокого разрешения с ионизацией электронным ударом (ЭИ-МСВР) m/е вычислено для С6Н9IO2 (М+) 239,9647, найдено 239,9646.
Смесь цинковой пыли (2,36 г, 36 ммолей, фирма Aldrich, -325 меш) и безводного тетрагидрофурана (3 мл) в атмосфере аргона обрабатывали 1,2-дибромэтаном (0,28 г, 1,5 ммоля). Цинковую суспензию затем нагревали с помощью струйной воздушной сушилки до бурного кипения, давали охладиться и опять нагревали. Эту операцию повторяли трижды, чтобы убедиться, что цинковая пыль проактивирована. Суспензию активированной цинковой пыли затем обрабатывали триметилсилилхлоридом (163 мг, 1,5 ммоля) и суспензию перемешивали 15 мин при 25оС. Реакционную смесь затем обрабатывали по каплям раствором метилового эфира (Е)-2-иодпентеновой кислоты (2,9 г, 12 ммолей) в безводном тетрагидрофуране (3 мл) в течение 3 мин. Реакционную смесь затем перемешивали при 40-45°С в течение 1 часа и затем перемешивали в течение ночи при 25°C. Реакционную смесь затем разбавляли безводным тетрагидрофураном (10 мл) и перемешивание прекращали, чтобы дать возможность осаждаться избытку цинковой пыли (~2 ч). В отдельном реакционном сосуде перемешивали бис(дибензилиденацетон)палладий(0) (135 мг, 0,25 ммоля) и трифенилфосфин (260 мг, 1 ммоль) в безводном тетрагдрофуране (16 мл) при 25°С под аргоном в течение 10 мин и затем обрабатывали 4-бромфенилметилсульфоном (2,11 г, 9 ммолей) и свежеприготовленным цинковым производным в тетрагидрофуране. Полученный в результате раствор цвета красного кирпича нагревали при 50°С в течение 24 ч. Реакционную смесь затем охлаждали до 25°С и затем выливали в насыщенный водный раствор хлористого аммония (100 мл), и органическое соединение экстрагировали этилацетатом (3×50 мл). Объединенные органические экстракты промывали насыщенным водным раствором хлористого натрия (1×100 мл), сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме. Хроматография с помощью системы Biotage (ФЛЭШ 40М, двуокись кремния, 3/2 гексаны/этилацетат) приводила к метиловому эфиру (Е)-2-(4-метансульфонилфенил)пентеновой кислоты (1,88 г, 78%) в виде вязкого масла желтого цвета: ЭИ-МСВР m/е вычислено для C13H16O4S (М+) 268,0769, найдено 268,0772.
Раствор метилового эфира (Е)-2-(4-метансульфонилфенил)пентеновой кислоты (1,83 г, 6,82 ммоля) в этаноле (30 мл) обрабатывали 1 н. раствором гидроокиси натрия (15 мл). Раствор нагревали при 45-50°С в течение 15 ч, к этому времени анализ реакционной смеси с применением тонкослойной хроматографии указывал на отсутствие исходного вещества. Реакционную смесь концентрировали в вакууме для удаления этанола. Остаток разбавляли водой (50 мл) и экстрагировали диэтиловым эфиром (1×50 мл) для удаления любых нейтральных примесей. Водный слой затем подкисляли 1 н. водным раствором соляной кислоты и полученную кислоту экстрагировали этилацетатом (2×70 мл). Объединенные органические слои промывали насыщенным водным раствором хлористого натрия (1×100 мл), сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме, получали (Е)-2-(4-метансульфонилфенил)пентеновую кислоту (1,43 г, 82%) в виде твердого вещества черного цвета: ЭИ-МСВР m/е вычислено для C12H14O4S (M+H)+ 254,0621, найдено 254,0623.
Раствор трифенилфосфина (1,23 г, 4,7 ммоля) в хлористом метилене (15 мл) охлаждали до 0°С и затем обрабатывали N-бромсукцинимидом (836 мг, 4,7 ммоля). Реакционную смесь перемешивали при 0°С в течение 30 мин и затем обрабатывали раствором (Е)-2-(4-метансульфонилфенил)пентеновой кислоты (703 мг, 2,76 ммоля) в хлористом метилене (5 мл). Прозрачный раствор перемешивали 10 мин при 0°С и затем оставляли нагреваться до 25°С, при этой температуре его перемешивали 1,5 часа. Реакционную смесь затем обрабатывали 2-аминотиазолом (829 мг, 8,28 ммоля) и полученную в результате суспензию перемешивали 15 ч при 25°С. Реакционную смесь затем концентрировали в вакууме для удаления хлористого метилена и остаток разбавляли этилацетатом (100 мл) и 1 н. водным раствором соляной кислоты (100 мл). Два слоя разделяли и водный слой экстрагировали этилацетатом (1×50 мл). Объединенные органические экстракты последовательно промывали насыщенным водным раствором бикарбоната натрия (2×50 мл) и насыщенным водным раствором хлористого натрия (1×100 мл), сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме. Хроматография с помощью системы Biotage (ФЛЭШ 40М, двуокись кремния, 4/1-1/1 гексаны/этилацетат) приводила к тиазол-2-иламиду (Е)-2-(4-метансульфонилфенил)пент-2-еновой кислоты (150 мг, 16%) в виде твердого кристаллического вещества: tпл 155-158°C; ЭИ-МСВР m/е вычислено для C15H16N2O3S2 (М+) 336,0602, найдено 336, 0601.
Пример 2
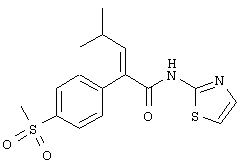
Тиазол-2-иламид (Е)-2-(4-метансульфонилфенил)-4-метилпент-2-еновой кислоты
Смесь хлористого лития (1,69 г, 40 ммолей, предварительно высушенного при 130°С в высоком вакууме 2 ч) и цианида меди (1,79 г, 20 ммолей) в безводном тетрагидрофуране (20 мл) перемешивали при 25°С под аргоном в течение 10 мин для получения прозрачного раствора. Реакционную смесь охлаждали до -70°С и затем медленно обрабатывали 2 М раствором изопропилмагнийхлорида в тетрагидрофуране (10 мл, 20 ммолей). После прибавления реакционной смеси давали нагреться до -30°C и при этой температуре ее перемешивали 5 мин. Полученную в результате реакционную смесь опять охлаждали до -70°С и затем медленно обрабатывали метиловым эфиром пропиоловой кислоты (1,52 г, 18 ммолей). Реакционную смесь перемешивали 4 ч при температуре от -40 до -30°С и затем охлаждали до -70 - -60°С, в это время реакционную смесь медленно обрабатывали раствором иода (6,86 г, 27 ммолей) в безводном тетрагидрофуране (20 мл). После прибавления раствора иода охлаждающую баню удаляли и реакционной смеси давали нагреться до 25°С, при этой температуре ее перемешивали 1 ч. Затем реакционную смесь выливали в раствор, содержащий насыщенный водный раствор хлористого аммония (90 мл) и гидроокись аммония (10 мл), и органическое соединение экстрагировали диэтиловым эфиром (3×50 мл). Объединенные органические экстракты последовательно промывали насыщенным водным раствором тиосульфата натрия (1×100 мл) и насыщенным водным раствором хлористого натрия (1×100 мл). Органический слой сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме. Хроматография с помощью системы Biotage (ФЛЭШ 40М, двуокись кремния, 20/1 гексаны/диэтиловый эфир) приводила к метиловому эфиру (Е)-2-иод-4-метилпентеновой кислоты (2,23 г, 49%) в виде бесцветного масла: ЭИ-МСВР m/е вычислено для C7H11IO2 (M+) 253,9804, найдено 253,9805.
Смесь цинковой пыли (1,71 г, 26 ммолей, фирма Aldrich, - 325 меш) и безводного тетрагидрофурана (2 мл) обрабатывали под аргоном 1,2-дибромэтаном (0,28 г, 1,5 ммоля). Цинковую суспензию затем нагревали до бурного кипения с помощью струйной воздушной сушилки, давали охладиться и опять нагревали. Эту процедуру повторяли три раза, чтобы убедиться, что цинковая пыль проактивирована. Суспензию активированной цинковой пыли затем обрабатывали триметилсилилхлоридом (163 мг, 1,5 ммоля) и суспензию перемешивали 15 мин при 25°С. Реакционную смесь затем обрабатывали по каплям раствором метилового эфира (Е)-2-иод-4-метилпентеновой кислоты (2,22 г, 8,7 ммоля) в безводном тетрагидрофуране (3 мл) в течение 2 минут. Реакционную смесь затем перемешивали при 40-45°С в течение 1 ч и затем перемешивали в течение ночи при 25°С. Реакционную смесь затем разбавляли безводным тетрагидрофураном (8 мл) и перемешивание прекращали, чтобы дать возможность осаждаться избытку цинковой пыли (~2 ч). В отдельной реакционной колбе перемешивали бис(дибензилиденацетон)палладий(0) (81 мг, 0,15 ммоля) и трифенилфосфин (156 мг, 0,6 ммоля) в безводном тетрагидрофуране (15 мл) при 25°С под аргоном в течение 10 мин и затем обрабатывали 4-бромфенилметилсульфоном (1,64 г, 7 ммолей) и свежеприготовленным цинковым производным в тетрагидрофуране. Полученный в результате раствор цвета красного кирпича нагревали при 50°С в течение 24 ч. Реакционную смесь затем охлаждали до 25°С и затем выливали в насыщенный водный раствор хлористого аммония (100 мл), и органическое соединение экстрагировали этилацетатом (3×50 мл). Объединенные органические экстракты промывали насыщенным водным раствором хлористого натрия (1×100 мл), сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме. Хроматография с помощью системы Biotage (ФЛЭШ 40М, двуокись кремния, 3/2 гексаны/этилацетат) приводила к метиловому эфиру (Е)-2-(4-метансульфонилфенил)-4-метилпентеновой кислоты (1,876 г, 95%) в виде вязкого масла желтого цвета: ЭИ-МСВР m/е вычислено для C14H18O4S (M+) 282,0926, найдено 282,0933.
Раствор метилового эфира (Е)-2-(4-метансульфонилфенил)-4-метилпентеновой кислоты (1,83 г, 6,48 ммоля) в этаноле (35 мл) обрабатывали 1 н. водным раствором гидроокиси натрия (15 мл). Раствор нагревали при 45-50°С в течение 15 ч, к этому времени анализ реакционной смеси с помощью тонкослойной хроматографии указывал на отсутствие исходного вещества. Реакционную смесь концентрировали в вакууме для удаления этанола. Остаток разбавляли водой (50 мл) и экстрагировали диэтиловым эфиром (1×50 мл) для удаления любых нейтральных примесей. Водный слой затем подкисляли 1 н. водным раствором соляной кислоты и полученную в результате кислоту экстрагировали этилацетатом (2×70 мл). Объединенные органические слои промывали насыщенным водным раствором хлористого натрия (1×100 мл), сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме, получали (Е)-2-(4-метансульфонилфенил)-4-метилпентеновую кислоту (1,6 г, 92%) в виде твердого вещества белого цвета: tпл 179-182°С; ЭИ-МСВР m/е вычислено для C13H16O4S (M+H)+ 269,0847, найдено 269,0858.
Раствор трифенилфосфина (1,11 г, 4,24 ммоля) в хлористом метилене (15 мл) охлаждали до 0°С и затем обрабатывали N-бромсукцинимидом (755 мг, 4,24 ммоля). Реакционную смесь перемешивали при 0°С в течение 30 минут и затем обрабатывали раствором (Е)-2-(4-метансульфонилфенил)-4-метилпентеновой кислоты (655 мг, 2,12 ммоля) в хлористом метилене (4 мл). Прозрачный раствор перемешивали 10 мин при 0°С и затем ему давали нагреться до 25°С, при этой температуре его перемешивали 1,5 ч. Реакционную смесь затем обрабатывали 2-аминотиазолом (636 мг, 6,36 ммоля) и полученную в результате суспензию перемешивали 15 ч при 25°С. Реакционную смесь затем концентрировали в вакууме для удаления хлористого метилена и остаток разбавляли этилацетатом (100 мл) и 1 н. водным раствором соляной кислоты (100 мл). Два слоя разделяли и водный слой экстрагировали этилацетатом (1×50 мл). Объединенные органические экстракты последовательно промывали насыщенным водным раствором бикарбоната натрия (2×50 мл) и насыщенным водным раствором хлористого натрия (1×100 мл), сушили над безводным сульфатом магния,фильтровали и упаривали в вакууме. Хроматография с помощью системы Biotage (ФЛЭШ 40М, двуокись кремния, 4/1-1/1 гексаны/этилацетат) приводила к неочищенной смеси соединений (365 мг). Эту смесь растворяли в этилацетате (5 мл) и диэтиловом эфире (5 мл) и затем обрабатывали гексанами (10 мл). Твердый осадок отфильтровывали и промывали гексанами, получали тиазол-2-иламид (Е)-2-(4-метансульфонилфенил)-4-метилпент-2-еновой кислоты (219 мг, 29%) в виде аморфного твердого вещества: ЭИ-МСВР m/е вычислено для C16H18N2O3S2 (M+) 350,0759, найдено 350,0754.
Пример 3
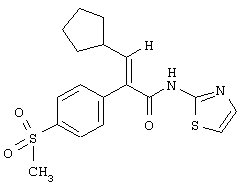
(Е)-3-Циклопентил-2-(4-метансульфонилфенил)-N-тиазол-2-илакриламид
Смесь хлористого алюминия (412,65 г, 3,09 моля) в хлористом метилене (1,11 л) охлаждали до 0°С и перемешивали до растворения твердого вещества. Реакционную смесь затем медленно обрабатывали этилоксалилхлоридом (300 мл, 2,69 моля) и получающаяся в результате реакционная смесь меняла цвет от желтого до оранжевого. Реакционную смесь затем медленно обрабатывали раствором тиоанизола (300 мл, 2,56 моля) в хлористом метилене (244 мл) малыми порциями в течение 1 ч. При прибавлении тиоанизола температуру реакционной смеси поддерживали ниже 10°С. Полученной реакционной смеси давали нагреться до 25°С, при этой температуре перемешивали 1 ч. Реакционную смесь затем опять охлаждали до 0°С и затем медленно обрабатывали льдом/водой (800 мл) в течение 1 ч. Реакционную смесь затем переносили в делительную воронку порциями в 1 л. Однолитровые порции непрерывно экстрагировали хлористым метиленом до отсутствия в водном слое продукта по данным тонкослойной хроматографии. Объединенные органические слои сушили над сульфатом магния, фильтровали и концентрировали в вакууме, получали этиловый эфир (4-метилсульфанилфенил)оксоуксусной кислоты (481,67 г, 84%)в виде жидкости желтого цвета, который использовали без дальнейшей очистки: ЭИ-МСВР m/е вычислено для С11H12О3S (М+) 224,0507, найдено 224,0500.
Раствор иодметилциклопентана (129,38 г, 0,616 моля) и трифенилфосфина (161,54 г, 0,616 моля) в ацетонитриле (308 мл) кипятили с обратным холодильником в течение 9 дней. Реакционную смесь оставляли охлаждаться до 25°С и затем концентрировали в вакууме для получения твердого вещества. Твердое вещество растирали с диэтиловым эфиром и затем фильтровали. Твердый остаток хорошо промывали диэтиловым эфиром до отсутствия в промывной жидкости иодметилциклопентана и трифенилфосфина по данным тонкослойной хроматографии. Полученное в результате твердое вещество сушили на воздухе и получали иодистый циклопентилметилтрифенилфосфоний (266,92 г, 92%) в виде твердого вещества светло-желтого цвета: tпл 195-198°C; МСВР с бомбардировкой быстрыми атомами (FAB) m/e вычислено для С24Н26Р (М+Н)+ 345,1772, найдено 345,1784.
Суспензию иодистого циклопентилметилтрифенилфосфония (151,73 г, 0,321 моля) в безводном тетрагидрофуране (494 мл) охлаждали до 0°С и затем медленно обрабатывали 1,0 М раствором бис(триметилсилил)амида лития (309 мл, 0,309 моля). Реакционную смесь ярко-оранжевого цвета перемешивали 1 ч при 0°С. Реакционную смесь затем обрабатывали небольшими порциями раствора этилового эфира (4-метилсульфанилфенил)оксоуксусной кислоты (55,42 г, 0,247 моля) в безводном тетрагидрофуране (100 мл). Полученную в результате реакционную смесь перемешивали при 0°С в течение 30 мин и затем давали ей нагреться до 25°С, при этой температуре перемешивали 6 часов. Затем реакционную смесь разбавляли водой (500 мл), в это время реакционная смесь имела рН=11. В реакционной смеси устанавливали рН=6 с помощью 10% водного раствора соляной кислоты и затем реакционную смесь оставляли на ночь при 25°С. Реакционную смесь концентрировали в вакууме для удаления тетрагидрофурана и затем разбавляли диэтиловым эфиром (1 л). Начинал осаждаться осадок и реакционную смесь оставляли на 1 ч при 25°С. Твердый осадок отфильтровывали и хорошо промывали диэтиловым эфиром. Полученный в результате фильтрат из двух слоев переносили в делительную воронку и слои разделяли. Водный слой далее экстрагировали диэтиловым эфиром (1×500 мл).
Объединенные органические слои промывали насыщенным водным раствором хлористого натрия (1×500 мл), сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Очистка с использованием слоя силикагеля (силикагель 60 фирмы Merck, 230-400 меш, 9/1 гексаны/этилацетат) приводила к этиловому эфиру 3-циклопентил-2-(4-метилсульфанилфенил)акриловой кислоты (58,93 г, 82%) в виде масла желтого цвета, состоящего из смеси изомеров (E):(Z)=1,44:1. Вещество использовали без дальнейшего разделения и идентификации.
Раствор смеси изомеров этилового эфира 3-циклопентил-2-(4-метилсульфанилфенил)акриловой кислоты [58,93 г, 0,203 моля, (E):(Z)=1,44:1] в муравьиной кислоте (203 мл) охлаждали до 0°С и затем медленно обрабатывали 30%-ным водным раствором перекиси водорода (62,2 мл, 0,609 моля). Реакционную смесь перемешивали при 0°С в течение 30 мин, затем смеси давали нагреться до 25°С, при этой температуре перемешивали 2 ч. Реакционную смесь опять охлаждали до 0°С и затем медленно обрабатывали насыщенным водным раствором бисульфита натрия (1 л). Реакционную смесь затем экстрагировали этилацетатом (2×700 мл). Объединенные органические слои промывали насыщенным водным раствором хлористого натрия (1×700 мл), сушили над сульфатом магния, фильтровали и концентрировали в вакууме, получали этиловый эфир 3-циклопентил-2-(4-метансульфонилфенил)акриловой кислоты (65,02 г, 99%) в виде масла желтого цвета, состоящего из смеси изомеров (E):(Z)=1,63:1. Продукт использовали без дальнейшей очистки и идентификации.
Раствор смеси изомеров этилового эфира 3-циклопентил-2-(4-метансульфонилфенил)акриловой кислоты [65,02 г, 0,202 моля, (E):(Z)=1,63:1] в метаноле (504 мл) обрабатывали 1 н. водным раствором гидроокиси натрия (423 мл, 0,423 моля). Реакционную смесь перемешивали 20 ч при 25°С, к этому времени тонкослойная хроматография указывала на присутствие исходного вещества. Реакционную смесь затем концентрировали в вакууме для удаления некоторого количества метанола (300 мл). Полученную в результате реакционную смесь кипятили с обратным холодильником 1 ч, к этому времени тонкослойная хроматография указывала на отсутствие исходного вещества. Затем реакционную смесь концентрировали в вакууме для удаления метанола. Оставшийся водный слой подкисляли до рН=1 с помощью концентрированной соляной кислоты и затем экстрагировали этилацетатом (2×1 л). Объединенные органические слои сушили над сульфатом магния, фильтровали и концентрировали в вакууме, получали 3-циклопентил-2-(4-метансульфонилфенил)акриловую кислоту (62,58 г) в виде твердого вещества кремового цвета, состоящего из смеси изомеров (E):(Z)=16,2:1. Твердое вещество кремового цвета обрабатывали этилацетатом (200 мл) и полученную в результате суспензию нагревали до кипения. Полученное в результате твердое вещество белого цвета в жидкости светло-желтого цвета, представляющей этилацетат, оставляли охлаждаться до 25°С. Твердое вещество отфильтровывали и получали чистую (Е)-3-циклопентил-2-(4-метансульфонилфенил)акриловую кислоту (41,18 г, 69%) в виде твердого вещества белого цвета, tпл 200-202°С; ЭИ-МСВР m/е вычислено для C15H18O4S (M+) 294,0926, найдено 294,0921.
Раствор N,N-диметилформамида (17,5 мл, 226,61 ммоля) в безводном тетрагидрофуране (420 мл) охлаждали до -25°С в атмосфере азота и затем обрабатывали оксалилхлоридом (18,8 мл, 215,42 ммоля). Раствор становился мутным вскоре после прибавления оксалилхлорида. Реакционной смеси давали нагреться до 25°С. При нагревании до 25°С выделение газа начиналось около -20°С и осаждались твердые частицы белого цвета по мере повышения температуры нагревания. Реакционную смесь перемешивали при 25°С в течение 15 мин, в результате получали густую суспензию из твердых частиц белого цвета. Реакционную смесь затем охлаждали опять до -25°С и обрабатывали раствором (Е)-3-циклопентил-2-(4-метансульфонилфенил)акриловой кислоты (41,18 г, 139,88 ммоля) в безводном тетрагидрофуране (300 мл) в течение 10 мин. После завершения прибавления раствора (Е)-3-циклопентил-2-(4-метансульфонилфенил)акриловой кислоты реакционной смеси давали нагреться до 0°С, при этой температуре ее перемешивали 1 ч. За время нахождения при 0°С плотные твердые частицы частично растворялись и образовывалась мелкозернистая суспензия из твердых частиц белого цвета. Через 1 ч выдержки при 25°С реакционную смесь охлаждали до -45°С. Реакционную смесь затем обрабатывали путем введения через трубку в течение 10 мин предварительно охлажденного (-45°С) раствора 2-аминотиазола (44,97 г, 449,02 ммоля) и триэтиламина (62,6 мл, 449,02 ммоля) в тетрагидрофуране (280 мл). Реакционнаясмесь меняла цвет от суспензии белого цвета до светло-коричневого цвета после завершения прибавления раствора 2-аминотиазола/триэтиламина. Реакционной смеси давали затем нагреться до 0°С в течение 15 мин, используя баню со льдом/водой. Затем реакционной смеси давали нагреться до 25°С в течение 30 мин и затем перемешивали 1 ч при 25°С. После этого реакционную смесь охлаждали до -25°С и затем обрабатывали 1 М водным раствором лимонной кислоты (250 мл), и полученной реакционной смеси давали нагреться до 25°С. Реакционную смесь фильтровали через слой целита для удаления осевших твердых частиц. Целит промывали этилацетатом до отсутствия продукта в промывочной жидкости по данным тонкослойной хроматографии. Фильтрат в виде двух слоев переносили в делительную воронку и слой разделяли. Водный слой экстрагировали этилацетатом (1×500 мл). Органический слой концентрировали в вакууме для удаления тетрагидрофурана и полученный в результате остаток разбавляли этилацетатом (700 мл). Объединенные органические слои последовательно промывали 2 М водным раствором бисульфата натрия (3×200 мл), насыщенным водным раствором хлористого натрия (1×200 мл), 10%-ным водным раствором карбоната калия (4×200 мл) и насыщенным водным раствором хлористого натрия (1×300 мл). Органический слой затем сушили над сульфатом магния, фильтровали и концентрировали в вакууме. Флэш-хроматография (силикагель 60 фирмы Merck, 70-230 меш, 3/2 гексаны/этилацетат) приводила к (Е)-3-циклопентил-2-(4-метансульфонилфенил)-N-тиазол-2-илакриламиду (27,93 г, 53%) в виде твердого вещества белого цвета: tпл 172-173°C; FAB-MCBP m/e вычислено для C18H20N2O3S2 (M+H)+ 377,0993, найдено 377,0986.
Пример 4
(Е)-3-Циклогексил-2-(4-метансульфонилфенил)-N-тиазол-2-илакриламид
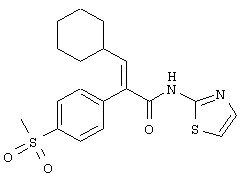
Смесь цинковой пыли (16,34 г, 250 ммолей, фирма Aldrich, -325 меш) и безводного тетрагидрофурана (6 мл) обрабатывали в атмосфере аргона 1,2-дибромэтаном (0,94 г, 5 ммолей). Цинковую суспензию затем нагревали до бурного кипения с помощью струйной воздушной сушилки, давали охладиться и опять нагревали. Эту процедуру повторяли три раза, чтобы убедиться, что цинковая пыль проактивирована. Суспензию активированной цинковой пыли затем обрабатывали триметилсилилхлоридом (0,54 г, 5 ммолей) и суспензию перемешивали 15 мин при 25°С. Реакционную смесь затем обрабатывали по каплям раствором иодистого циклогексила (21 г, 100 ммолей) в безводном тетрагидрофуране (30 мл) в течение 15 минут. Во время прибавления температура поднималась до 60°С. Реакционную смесь затем перемешивали 3 ч при 40-45°С. Реакционную смесь затем охлаждали до 25°С и разбавляли безводным тетрагидрофураном (60 мл). Перемешивание прекращали, чтобы дать возможность осаждаться избытку цинковой пыли (~3 ч). В отдельной реакционной колбе перемешивали смесь хлористого лития (8,48 г, 200 ммолей, предварительно высушенного при 130°С в высоком вакууме в течение 3 ч) и цианида меди (8,95 г, 100 ммолей) в безводном тетрагидрофуране (110 мл) 10 мин при 25°С до получения прозрачного раствора. Реакционную смесь охлаждали до -70°С и затем медленно обрабатывали свежеприготовленным цинковым раствором, используя шприц. После прибавления реакционной смеси давали нагреться до 0°С, при этой температуре перемешивали 5 мин. Реакционную смесь опять охлаждали до -70°С и затем медленно обрабатывали метиловым эфиром пропиоловой кислоты (7,56 г, 90 ммолей). Полученную в результате реакционную смесь перемешивали 15 ч при температуре от -70 до -50°С и затем медленно обрабатывали раствором иода (34,26 г, 135 ммолей) в безводном тетрагидрофуране (30 мл), поддерживая температуру от 70 до -60°С. После прибавления раствора иода охлаждающую баню удаляли и реакционной смеси давали нагреться до 25°С, при этой температуре перемешивали 2 ч. Реакционную смесь затем выливали в раствор, состоящий из насыщенного раствора хлористого натрия (400 мл) и гидроокиси аммония (100 мл), и органическое соединение экстрагировали этилацетатом (3×250 мл). Объединенные органические экстракты последовательно промывали насыщенным водным раствором тиосульфата натрия (1×500 мл) и насыщенным водным раствором хлористого натрия (1×500 мл), сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме. Флэш-хроматография (силикагель 60 фирмы Merck, 230-400 меш, 9/1 гексаны/этилацетат) приводила к метиловому эфиру (Е)-3-циклогексил-2-иодакриловой кислоты (26,3 г, 99%) в виде масла светло-розового цвета: ЭИ-МСВР m/е вычислено для С10Н15IO2 (М+) 294,0117, найдено 294,0114.
Смесь цинковой пыли (2,6 г, 40 ммолей, фирма Aldrich, -325 меш) и безводного тетрагидрофурана (3 мл) обрабатывали в атмосфере аргона 1,2-дибромэтаном (0,37 г, 2 ммоля). Цинковую суспензию затем нагревали до бурного кипения с помощью струйной воздушной сушилки, давали охладиться и опять нагревали. Эту процедуру повторяли три раза, чтобы убедиться, что цинковая пыль проактивирована. Суспензию активированной цинковой пыли затем обрабатывали триметилсилилхлоридом (217 мг, 2 ммоля) и суспензию перемешивали 15 мин при 25°С. Реакционную смесь затем обрабатывали по каплям раствором метилового эфира (Е)-3-циклогексил-2-иодакриловой кислоты (5,88 г, 20 ммолей) в безводном тетрагидрофуране (5 мл) в течение 5 минут. Во время прибавления температура поднималась до 50°С. Реакционную смесь затем перемешивали при 40-45°С в течение 1 ч и затем перемешивали в течение ночи при 25°С. Реакционную смесь затем разбавляли безводным тетрагидрофураном (10 мл) и перемешивание прекращали, чтобы дать возможность осаждаться избытку цинковой пыли (~2 ч). В отдельной реакционной колбе перемешивали под аргоном 10 мин при 25°С бис(дибензилиденацетон)палладий(0) (270 мг, 0,5 ммоля) и трифенилфосфин (520 мг, 2 ммоля) в безводном тетрагидрофуране (25 мл) и затем обрабатывали 4-бромфенилметилсульфоном (4,23 г, 18 ммолей) и свежеприготовленным цинковым производным в тетрагидрофуране. Полученный в результате раствор цвета красного кирпича нагревали при 50°С в течение 24 ч, к этому времени при анализе с помощью тонкослойной хроматографии в реакционной смеси не обнаруживали исходного вещества. Реакционную смесь охлаждали до 25°C и затем выливали в насыщенный водный раствор хлористого аммония (150 мл), и органическое соединение экстрагировали этилацетатом (3х100 мл). Объединенные органические экстракты промывали насыщенным водным раствором хлористого натрия (1×200 мл), сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме. Флэш-хроматография (силикагель 60 фирмы Merck, 230-400 меш, 3/2 гексаны/этилацетат) приводила к метиловому эфиру (Е)-3-циклогексил-2-(4-метансульфонилфенил)акриловой кислоты (5,79 г, 99%) в виде низкоплавкого твердого вещества белого цвета: ЭИ-МСВР m/е вычислено для C17H22O4S (M+) 322,1238, найдено 322,1236.
Раствор метилового эфира (Е)-3-циклогексил-2-(4-метансульфонилфенил)акриловой кислоты (5,7 г, 17,95 ммоля) в этаноле (65 мл) обрабатывали 1 н. водным раствором едкого натра (54 мл). Раствор нагревали при 45-50°С в течение 15 ч, к этому времени анализ с помощью тонкослойной хроматографии указывал на отсутствие исходного вещества. Реакционную смесь затем концентрировали в вакууме для удаления этанола, остаток разбавляли водой (100 мл) и экстрагировали диэтиловым эфиром (1×150 мл) для удаления любых нейтральных примесей. Водный слой подкисляли 1 н. водным раствором соляной кислоты. Полученную в результате кислоту экстрагировали этилацетатом (2×150 мл). Объединенные органические слои промывали насыщенным водным раствором хлористого натрия (1×250 мл), сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме, получали (Е)-3-циклогексил-2-(4-метансульфонилфенил)акриловую кислоту (5,18 г, 94%) в виде твердого вещества белого цвета: tпл 195-197°С; ЭИ-МСВР m/е вычислено для C16H20O4S (М+Н)+ 309,1160, найдено 309,1165.
Раствор трифенилфосфина (8,79 г, 33,52 ммоля) в хлористом метилене (100 мл) охлаждали до 0°С и затем обрабатывали N-бромсукцинимидом (5,97 г, 33,52 ммоля). Реакционную смесь перемешивали при 0°С в течение 30 мин и затем обрабатывали раствором (Е)-3-циклогексил-2-(4-метансульфонилфенил)акриловой кислоты (5,17 г, 16,76 ммоля) в хлористом метилене (20 мл). Прозрачный раствор перемешивали 15 мин при 0°С и затем давали ему нагреться до 25°С, при этой температуре перемешивали 1,5 ч. Реакционную смесь затем обрабатывали 2-аминотиазолом (5,04 г, 50,3 ммоля) и полученную в результате суспензию перемешивали 2 дня при 25°С. Реакционную смесь концентрировали в вакууме для удаления хлористого метилена и остаток разбавляли этилацетатом (250 мл) и 1 н. водным раствором соляной кислоты (150 мл). Два слоя разделяли и водный слой экстрагировали этилацетатом (1×100 мл). Объединенные органические экстракты последовательно промывали насыщенным водным раствором бикарбоната натрия (1×150 мл) и насыщенным водным раствором хлористого натрия (1×250 мл), сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме. Флэш-хроматография (силикагель 60 фирмы Merck, 230-400 меш, 8,5/1,5-3/2 гексаны/этилацетат) приводила к (Е)-3-циклогексил-2-(4-метансульфонилфенил)-N-тиазол-2-илакриламиду (2,8 г, 42%) в виде аморфного твердого вещества: tпл 167-169°С; ЭИ-МСВР m/е вычислено для C19H22N2O3S2 (M+) 390,1072, найдено 390,1073.
Пример 5
(Е)-3-Циклогептил-2-(4-мегансульфонилфенил)-N-тиазол-2-илакриламид
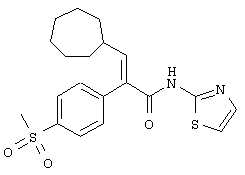
Смесь металлического магния (4,81 г, 200 ммолей) и безводного тетрагидрофурана (10 мл) обрабатывали в атмосфере аргона раствором 1,2-дибромэтана (0,94 г, 5 ммолей) в безводном тетрагидрофуране (5 мл). Полученную реакционную смесь перемешивали 10 мин для активирования металлического магния. Реакционную смесь затем обрабатывали по каплям раствором циклогептилбромида (17,7 г, 100 ммолей) в безводном тетрагидрофуране (30 мл), одной пятой частью в течение 5 мин. Полученную в результате реакционную смесь перемешивали 5-10 мин для инициирования экзотермической реакции. Оставшуюся часть циклогептилбромида прибавляли затем по каплям, поддерживая температуру внутри ниже 50°С. После завершения прибавления раствор перемешивали 1 ч и затем разбавляли безводным тетрагидрофураном (80 мл). В отдельной реакционной колбе перемешивали при 25°С в атмосфере аргона в течение 10 мин смесь хлористого лития (8,48 г, 200 ммолей, предварительно высушенного при 130°С в высоком вакууме в течение 3 ч) и цианида меди (8,96 г, 100 ммолей) в безводном тетрагидрофуране (110 мл) до получения прозрачного раствора. Реакционную смесь охлаждали до -70°С и затем медленно обрабатывали свежеприготовленным циклогептилмагнийбромидом. После прибавления реакционной смеси давали нагреться до -10°С, при этой температуре перемешивали 5 мин. Полученную в результате реакционную смесь опять охлаждали до -70°С и затем обрабатывали метиловым эфиром пропиоловой кислоты (7,57 г, 90 ммолей). Реакционную смесь перемешивали 15 ч при температуре от -70 до -50°С и затем медленно обрабатывали раствором иода (34,3 г, 135 ммолей) в безводном тетрагидрофуране (30 мл), поддерживая температуру от -70 до -60°С. После прибавления раствора иода охлаждающую баню удаляли и реакционной смеси давали нагреться до 25°С, при этой температуре перемешивали 2 ч. Реакционную смесь затем выливали в раствор, состоящий из насыщенного водного раствора хлористого аммония (400 мл) и гидроокиси аммония (100 мл), и органическое соединение экстрагировали этилацетатом (3×200 мл). Объединенные экстракты последовательно промывали насыщенным водным раствором тиосульфата натрия (1×400 мл) и насыщенным водным раствором хлористого натрия (1×400 мл). Органический слой затем сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме. Флэш-хроматография (силикагель 60 фирмы Merck, 230-400 меш, 20/1 -10/1 гексаны/этилацетат) приводила к метиловому эфиру (Е)-3-циклогептил-2-иодакриловой кислоты (17,86 г, 64%) в виде бесцветного масла: ЭИ-МСВР m/е вычислено для С11Н17IO2 (М+) 308,0273, найдено 308,0273.
Смесь цинковой пыли (2,6 г, 40 ммолей, фирма Aldrich, -325 меш) и безводного тетрагидрофурана (3 мл) обрабатывали в атмосфере аргона 1,2-дибромэтаном (0,38 г, 2 ммоля). Цинковую суспензию затем нагревали до бурного кипения с помощью струйной воздушной сушилки, давали охладиться и опять нагревали. Эту процедуру повторяли три раза, чтобы убедиться, что цинковая пыль проактивирована. Суспензию активированной цинковой пыли затем обрабатывали триметилсилилхлоридом (220 мг, 2 ммоля) и суспензию перемешивали 15 мин при 25°С. Реакционную смесь затем обрабатывали по каплям раствором метилового эфира (Е)-3-циклогептил-2-иодакриловой кислоты (6,16 г, 20 ммолей) в безводном тетрагидрофуране (5 мл) в течение 10 минут. Реакционную смесь затем перемешивали при 40-45°С в течение 1 ч и затем перемешивали в течение ночи при 25°С. Реакционную смесь затем разбавляли безводным тетрагидрофураном (10 мл) и перемешивание прекращали, чтобы дать возможность осаждаться избытку цинковой пыли (~2 ч). В отдельной реакционной колбе перемешивали бис(дибензилиденацетон)палладий(0) (270 мг, 0,5 ммоля) и трифенилфосфин (520 мг, 2 ммоля) в безводном тетрагидрофуране (25 мл) при 25°С в атмосфере аргона в течение 10 мин и затем обрабатывали 4-бромфенилметилсульфоном (4,23 г, 18 ммолей) и свежеприготовленным цинковым производным в тетрагидрофуране. Полученный в результате раствор цвета красного кирпича нагревали при 50°С в течение 24 ч. Реакционную смесь охлаждали до 25°С и затем выливали в насыщенный водный раствор хлористого аммония (150 мл), и органическое соединение экстрагировали этилацетатом (3×150 мл). Объединенные органические экстракты промывали насыщенным водным раствором хлористого натрия (1×300 мл), сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме. Флэш-хроматография (силикагель 60 фирмы Merck, 230-400 меш, 4/1-1/1 гексаны/этилацетат) приводила к метиловому эфиру (Е)-3-циклогептил-2-(4-метансульфонилфенил)акриловой кислоты (6,01 г, 99%) в виде вязкого масла желого цвета: ЭИ-МСВР m/е вычислено для C18H24O4S (M+) 336,1395, найдено 336,1395.
Раствор метилового эфира (Е)-3-циклогептил-2-(4-метансульфонилфенил)акриловой кислоты (6,01 г, 17,8 ммоля) в этаноле (65 мл) обрабатывали 1 н. водным раствором едкого натра (55 мл). Раствор нагревали при 45-50°С в течение 15 ч, к этому времени анализ с помощью тонкослойной хроматографии указывал на отсутствие исходного вещества. Реакционную смесь затем концентрировали в вакууме для удаления этанола. Остаток разбавляли водой (100 мл) и экстрагировали диэтиловым эфиром (1×150 мл) для удаления любых нейтральных примесей. Водный слой подкисляли 1 н. водным раствором соляной кислоты и полученную в результате кислоту экстрагировали этилацетатом (2×150 мл). Объединенные органические слои промывали насыщенным водным раствором хлористого натрия (1×150 мл), сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме, получали (Е)-3-циклогептил-2-(4-метансульфонилфенил)акриловую кислоту (4,99 г, 86%) в виде твердого вещества белого цвета: tпл 164-166°С; ЭИ-МСВР m/е вычислено для C17H22O4S (М+Н)+ 322,1239, найдено 322,1237.
Раствор трифенилфосфина (8,08 г, 30,8 ммоля) в хлористом метилене (100 мл) охлаждали до 0°С и затем обрабатывали N-бромсукцинимидом (5,48 г, 30,8 ммоля). Реакционную смесь перемешивали при 0°С в течение 30 мин и затем обрабатывали раствором (Е)-3-циклогептил-2-(4-метансульфонилфенил)акриловой кислоты (4,97 г, 15,41 ммоля) в хлористом метилене (20 мл). Прозрачный раствор перемешивали 15 мин при 0°С и затем давали ему нагреться до 25°С, при этой температуре перемешивали 1,5 ч. Реакционную смесь затем обрабатывали 2-аминотиазолом (4,63 г, 46,23 ммоля) и полученную в результате суспензию перемешивали 2 дня при 25°С. Реакционную смесь концентрировали в вакууме для удаления хлористого метилена и остаток разбавляли этилацетатом (250 мл) и 1 н. водным раствором соляной кислоты (150 мл). Два слоя разделяли и водный слой экстрагировали этилацетатом (1×150 мл). Объединенные органические экстракты последовательно промывали насыщенным водным раствором бикарбоната натрия (1×250 мл) и насыщенным водным раствором хлористого натрия (1×200 мл), сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме. Флэш-хроматография (силикагель 60 фирмы Merck, 230-400 меш, 5/1-3/2 гексаны/этилацетат) приводила к (Е)-3-циклогептил-2-(4-метансульфонилфенил)-N-тиазол-2-илакриламиду (2,7 г, 43%) в виде аморфного твердого вещества. Это соединение растворяли в ацетонитриле (~55 мл) и хранили в течение ночи при 25°C. Твердое вещество отфильтровывали и промывали ацетонитрилом (5 мл), получали (Е)-3-циклогептил-2-(4-метансульфонилфенил)-N-тиазол-2-илакриламид (2,1 г, 33%) в виде кристаллического твердого вещества: tпл 163-165°С; ЭИ-МСВР m/е вычислено для C20H24N2O3S2 (M+) 404,1253, найдено 404,1251.
Пример 6
(Е)-3-Циклооктил-2-(4-метансульфонилфенил)-N-тиазол-2-илакриламид
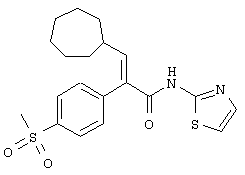
Смесь металлического магния (1,94 г, 80 ммолей) и безводного тетрагидрофурана (3 мл) обрабатывали под аргоном раствором 1,2-дибромэтана (0,56 г, 3 ммоля) в безводном тетрагидрофуране (2 мл). Полученную реакционную смесь перемешивали 10 минут для активирования металлического магния. Реакционную смесь затем обрабатывали по каплям раствором циклооктилбромида (7,64 г, 40 ммолей) в безводном тетрагидрофуране (15 мл), одной пятой частью в течение 5 мин. Полученную в результате реакционную смесь перемешивали 5-10 мин для инициирования экзотермической реакции. Оставшуюся часть раствора циклооктилбромида прибавляли затем по каплям, поддерживая температуру внутри ниже 50°С. После завершения прибавления раствор перемешивали 1 ч и затем разбавляли безводным тетрагидрофураном (30 мл). В отдельной реакционной колбе смесь хлористого лития (3,39 г, 80 ммолей, предварительно высушенного при 130°С в высоком вакууме 3 ч) и цианида меди (3,58 г, 40 ммолей) в безводном тетрагидрофуране (40 мл) перемешивали при 25°С в атмосфере аргона в течение 10 мин до получения прозрачного раствора. Реакционную смесь охлаждали до -70°С и затем медленно обрабатывали свежеприготовленным циклооктилмагнийбромидом. После прибавления реакционной смеси давали нагреться до -10°С, при этой температуре перемешивали 5 мин. Полученную в результате реакционную смесь опять охлаждали до -70°С и затем обрабатывали метиловым эфиром пропиоловой кислоты (3,02 г, 36 ммолей). Реакционную смесь перемешивали 15 ч при температуре от -70 до -50°С и затем медленно обрабатывали раствором иода (15,22 г, 60 ммолей) в безводном тетрагидрофуране (15 мл), поддерживая температуру от -70 до -60°С. После прибавления раствора иода охлаждающую баню удаляли и реакционной смеси давали нагреться до 25°С, при этой температуре перемешивали 2 ч. Реакционную смесь затем выливали в раствор, состоящий из насыщенного водного раствора хлористого аммония (200 мл) и гидроокиси аммония (50 мл), и органическое соединение экстрагировали этилацетатом (3×100 мл). Объединенные органические экстракты последовательно промывали насыщенным водным раствором тиосульфата натрия (1×200 мл) и насыщенным водным раствором хлористого натрия (1×200 мл). Органический слой затем сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме. Хроматография с помощью системы Biotage (ФЛЭШ 40М, двуокись кремния, 20/1-10/1 гексаны/диэтиловый эфир) приводила к метиловому эфиру (Е)-3-циклооктил-2-иодакриловой кислоты (5,04 г, 43%) в виде бесцветного масла: ЭИ-МСВР m/е вычислено для С12Н19IO2 (М+) 322,0430, найдено 322,0432.
Смесь цинковой пыли (1,3 г, 20 ммолей, фирма Aldrich, -325 меш) и безводного тетрагидрофурана (3 мл) обрабатывали в атмосфере аргона 1,2-дибромэтаном (0,38 г, 2 ммоля). Цинковую суспензию затем нагревали до бурного кипения с помощью струйной воздушной сушилки, давали охладиться и опять нагревали. Эту процедуру повторяли три раза, чтобы убедиться, что цинковая пыль проактивирована. Суспензию активированной цинковой пыли затем обрабатывали триметилсилилхлоридом (220 мг, 2 ммоля) и суспензию перемешивали 15 мин при 25°С. Реакционную смесь затем обрабатывали по каплям раствором метилового эфира (Е)-3-циклооктил-2-иодакриловой кислоты (3,22 г, 10 ммолей) в безводном тетрагидрофуране (4 мл) в течение 10 минут. Реакционную смесь затем перемешивали при 40-45°С в течение 1 ч и затем перемешивали в течение ночи при 25°С. Реакционную смесь затем разбавляли безводным тетрагидрофураном (8 мл) и перемешивание прекращали, чтобы дать возможность осаждаться избытку цинковой пыли (~2 ч). В отдельной реакционной колбе перемешивали под аргоном 10 мин при 25°С бис(дибензилиденацетон)палладий(0) (135 мг, 0,25 ммоля) и трифенилфосфин (260 мг, 1 ммоль) в безводном тетрагидрофуране (10 мл) и затем обрабатывали 4-бромфенилметилсульфоном (2,12 г, 9 ммолей) и свежеприготовленным цинковым производным в тетрагидрофуране. Полученный в результате раствор цвета красного кирпича нагревали при 50°С в течение 24 ч. Реакционную смесь охлаждали до 25°С и затем выливали в насыщенный водный раствор хлористого аммония (100 мл) и органическое соединение экстрагировали этилацетатом (3×75 мл). Объединенные органические экстракты промывали насыщенным водным раствором хлористого натрия (1×200 мл), сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме. Флэш-хроматография (силикагель 60 фирмы Merck, 230-400 меш, 4/1 -1/1 гексаны/этилацетат) приводила к метиловому эфиру (Е)-3-циклооктил-2-(4-метансульфонилфенил)акриловой кислоты (2,85 г, 90%) в виде полутвердого вещества светло-желтого цвета: ЭИ-МСВР m/е вычислено для C19H26O4S (M+) 350,1552, найдено 350,1554.
Раствор метилового эфира (Е)-3-циклооктил-2-(4-метансульфонилфенил)акриловой кислоты (2,82 г, 8,05 ммоля) в этаноле (30 мл) обрабатывали 1 н. водным раствором едкого натра (20 мл). Раствор нагревали при 45-50°С в течение 15 ч, к этому времени анализ с помощью тонкослойной хроматографии указывал на отсутствие исходного вещества. Реакционную смесь концентрировали в вакууме для удаления этанола. Остаток разбавляли водой (100 мл) и экстрагировали диэтиловым эфиром (1×75 мл) для удаления любых нейтральных примесей. Водный слой подкисляли 1 н. водным раствором соляной кислоты и полученную в результате кислоту экстрагировали этилацетатом (2×100 мл). Объединенные органические слои промывали насыщенным водным раствором хлористого натрия (1×150 мл), сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме, получали (Е)-3-циклооктил-2-(4-метансульфонилфенил)акриловую кислоту (2,64 г, 97%) в виде твердого вещества светло-желтого цвета: ЭИ-МСВР m/е вычислено для C18H24O4S (M+) 336,1395, найдено 336,1390.
Раствор трифенилфосфина (2,09 г, 8 ммолей) в хлористом метилене (25 мл) охлаждали до 0°С и затем обрабатывали N-бромсукцинимидом (1,42 г, 8 ммолей). Реакционную смесь перемешивали при 0°С в течение 30 мин и затем обрабатывали раствором (Е)-3-циклооктил-2-(4-метансульфонилфенил)акриловой кислоты (1,345 г, 4 ммоля) в хлористом метилене (10 мл). Прозрачный раствор перемешивали 15 мин при 0°С и затем давали ему нагреться до 25°С, при этой температуре его перемешивали 1,5 ч. Реакционную смесь затем обрабатывали 2-аминотиазолом (1,2 г, 12 ммолей) и полученную в результате суспензию перемешивали 2 дня при 25°С.
Реакционную смесь затем концентрировали в вакууме для удаления хлористого метилена и остаток разбавляли этилацетатом (100 мл) и 1 н. водным раствором соляной кислоты (100 мл). Два слоя разделяли и водный слой экстрагировали этилацетатом (1×50 мл). Объединенные органические экстракты последовательно промывали насыщенным водным раствором бикарбоната натрия (1×150 мл) и насыщенным водным раствором хлористого натрия (1×100 мл), сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме. Флэш-хроматография (силикагель 60 фирмы Merck, 230-400 меш, 5/1-3/2 гексаны/этилацетат) приводила к (Е)-3-циклооктил-2-(4-метансульфонилфенил)-N-тиазол-2-илакриламиду (1,22 г, 73%) в виде аморфного твердого вещества: ЭИ-МСВР m/е вычислено для C21H26N2O3S2 (M+) 418,1385, найдено 418,1385.
Пример 7
(Е)-N-(5-Бромтиазол-2-ил)-3-циклопентил-2-(4-метансульфонилфенил)акриламид
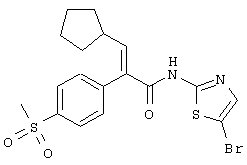
Суспензию (Е)-3-циклопентил-2-(4-метансульфонилфенил)-N-тиазол-2-илакриламида (получен в примере 3, 3,044 г, 1,17 ммоля) и N-бромсукцинимида (0,20 г, 1,17 ммоля) в четыреххлористом углероде (4 мл) при 25°С обрабатывали перекисью бензоила (14,17 мг, 0,058 ммоля). Полученную в результате смесь нагревали до 90°С, при этой температуре перемешивали в течение ночи. Реакционной смеси давали охладиться до 25°С и затем концентрировали в вакууме. Остаток растворяли в этилацетате (50 мл). Органическую фазу затем промывали водой (1×50 мл) и насыщенным водным раствором хлористого натрия (1×50 мл), сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме. Хроматография с помощью системы Biotage (ФЛЭШ 40М, двуокись кремния, 4/1-1/1 гексаны/этилацетат) приводила к (E)-N-(5-бромтиазол-2-ил)-3-циклопентил-2-(4-метансульфонилфенил)акриламиду (115 мг, 22%) в виде твердого вещества белого цвета: tпл 202-205°C.
Пример 8
(Е)-3-Циклопентил-2-(3,4-дихлорфенил)-N-тиазол-2-илакриламид
Смесь хлористого алюминия (16,81 г, 126,05 ммоля) в хлористом метилене (105 мл) охлаждали до 5°С и перемешивали до растворения твердого вещества. Реакционную смесь затем медленно обрабатывали метилоксалилхлоридом (8,1 мл, 88,24 ммоля) и полученную реакционную смесь перемешивали при 5°С в течение 30 минут. Реакционную смесь затем медленно обрабатывали 1,2-дихлорбензолом (12,35 г, 84,04 ммоля). Полученной в результате реакционной смеси давали нагреться до 25°С, при этой температуре ее перемешивали 6 ч. Реакционную смесь затем хранили при 0°С в течение 15 ч. Реакционную смесь медленно выливали в лед/воду (400 мл). Слои встряхивали и разделяли. Водный слой далее экстрагировали хлористым метиленом (1×200 мл). Объединенные органические слои промывали насыщенным водным раствором бикарбоната натрия (1×200 мл) и водой (1×100 мл), сушили над сульфатом магния, фильтровали и концентрировали в вакууме. Флэш-хроматография (силикагель 60 фирмы Merck, 230-400 меш, 9/1 гексаны/этилацетат) приводила к метиловому эфиру (3,4-дихлорфенил)оксоуксусной кислоты (0,78 г, 4%) в виде твердого вещества желтого цвета: tпл 58,2-63°С; ЭИ-МСВР m/е вычислено для С9Н6Сl2O3 (М+) 231,9694, найдено 231,9699.
Суспензию йодистого циклопентилметилтрифенилфосфония (полученного в примере 3, 3,95 г, 8,37 ммоля) в безводном тетрагидрофуране (10 мл) охлаждали до 0°С и затем обрабатывали по каплям 1,0 М раствором бис(триметилсилил)амида натрия (8,4 мл, 8,37 ммоля). Реакционную смесь ярко-оранжевого цвета перемешивали при 0°С в течение 45 мин. Реакционную смесь затем обрабатывали раствором метилового эфира (3,4-дихлорфенил)оксоуксусной кислоты (1,30 г, 5,58 ммоля) в тетрагидрофуране (4 мл). Полученной в результате реакционной смеси давали нагреться до 25°С, при этой температуре перемешивали 64 часа. Реакционную смесь затем концентрировали в вакууме для удаления тетрагидрофурана. Остаток разбавляли водой (150 мл) и затем экстрагировали диэтиловым эфиром (1×200 мл). Органический слой сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Флэш-хроматография (силикагель 60 фирмы Merck, 70-230 меш, 19/1 гексаны/этилацетат) приводила к метиловому эфиру 3-циклопентил-2-(3,4-дихлорфенил)акриловой кислоты (821,1 мг, 49%) в виде масла желтого цвета, состоящего из смеси (E):(Z)-изомеров в отношении 4,5:1. Смесь изомеров использовали без дальнейшего разделения и идентификации.
Раствор изомерной смеси метилового эфира 3-циклопентил-2-(3,4-дихлорфенил)акриловой кислоты [821,1 мг, 2,74 ммоля, (E):(Z)=4,5:1] в тетрагидрофуране (3,4 мл) обрабатывали 0,8 М водным раствором гидроокиси лития (3,4 мл, 2,74 ммоля). Реакционную смесь перемешивали при 25°С в течение 17 ч и затем кипятили с обратным холодильником 4 ч. Реакционной смеси давали охладиться до 25°С и затем ее концентрировали в вакууме для удаления тетрагидрофурана. Оставшийся водный слой подкисляли до рН=2 с помощью 10%-ного водного раствора соляной кислоты и затем экстрагировали этилацетатом (2×150 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Флэш-хроматография (силикагель 60 фирмы Merck, 70-230 меш, 1/1 гексаны/этилацетат) приводила к чистой (Е)-3-циклопентил-2-(3,4-дихлорфенил)акриловой кислоте (205,4 мг, 26%) в виде твердого вещества белого цвета: tпл 119-120°С; ЭИ-МСВР m/е вычислено для С14Н14Сl2О2 (М+) 284,0371, найдено 284,0370.
Раствор (Е)-3-циклопентил-2-(3,4-дихлорфенил)акриловой кислоты (73,9 мг, 0,26 ммоля), гексафторфосфата O-бензотриазол-1-ил-N,N,N′,N′-тетраметилурония (108,1 мг, 0,29 ммоля) и N,N-диизопропилэтиламина (136 мкл, 0,78 ммоля) в безводном N,N-диметилформамиде (1,3 мл) перемешивали при 25°С в течение 15 мин и затем обрабатывали 2-аминотиазолом (51,9 мг, 0,52 ммоля). Полученную в результате реакционную смесь перемешивали при 25°С в течение 21 ч. Реакционную смесь затем концентрировали в вакууме для удаления N,N-диметилформамида. Остаток затем разбавляли этилацетатом (100 мл). Органический слой промывали 10%-ным водным раствором соляной кислоты (1×100 мл), насыщенным водным раствором бикарбоната натрия (1×100 мл) и насыщенным водным раствором хлористого натрия (1×100 мл). Органический слой сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Флэш-хроматография (силикагель 60 фирмы Merck, 70-230 меш, 4/1 гексаны/этилацетат) приводила к двум изомерным продуктам. Продукт с более высоким значением Rf соответствовал желаемому (Е)-3-циклопентил-2-(3,4-дихлорфенил)-N-тиазол-2-илакриламиду (15,3 мг, 16%), выделенному в виде твердого воскообразного вещества белого цвета: tпл 57-59°С; ЭИ-МСВР m/е вычислено для С17Н16Сl2N2ОS (М+) 366,0360, найдено 366,0360.
Пример 9
(Е)-2-(3-Хлор-4-метансульфонилфенил)-3-циклопентил-N-тиазол-2-илакриламид
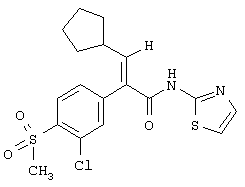
Раствор хлористого алюминия (34,8 г, 261,4 ммоля) в хлороформе (120 мл) в атмосфере аргона охлаждали до 0°С и затем прибавляли по каплям раствор этилоксалилхлорида (18,7 мл, 167,5 ммолей) в хлороформе (120 мл). Реакционную смесь перемешивали при 0°С в течение 30 мин и затем обрабатывали по каплям раствором 2-хлортиоанизола (25,0 г, 156,5 ммоля) в хлороформе (120 мл). Получающаяся в результате реакционная смесь приобретала красный цвет. Реакционной смеси давали нагреться до 25°С, при этой температуре перемешивали еще 3,5 ч. Реакционную смесь затем медленно смешивали с водой (500 мл) и после прибавления воды реакционная смесь приобретала желтый цвет. Полученный в результате раствор затем экстрагировали хлороформом (3×50 мл). Органическую фазу сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Флэш-хроматография (силикагель 60 фирмы Merck, 230-400 меш, 80/20 гексаны/этилацетат) приводила к этиловому эфиру (3-хлор-4-метилсульфанилфенил)оксоуксусной кислоты (31,37 г, 77%) в виде масла желтого цвета.
Суспензию йодистого циклопентилметилтрифенилфосфония (полученного в примере 3, 725 мг, 1,53 ммоля) в тетрагидрофуране (10 мл) охлаждали до 0°С и затем обрабатывали 1,0 М раствором бис(триметилсилил)амида натрия в тетрагидрофуране (2,14 мл, 2,14 ммоля). Полученную в результате реакционную смесь красного цвета перемешивали 45 мин при 0°С и затем медленно обрабатывали раствором этилового эфира (3-хлор-4-метилсульфанилфенил)оксоуксусной кислоты (355 мг, 1,37 ммоля) в тетрагидрофуране (5 мл). Реакционную смесь нагревали до 25°С, при этой температуре перемешивали 20 ч. Реакционную смесь затем разбавляли водой (50 мл) и экстрагировали диэтиловым эфиром (3×25 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Хроматография с помощью системы Biotage (Флэш 12М, двуокись кремния, 80/20 гексаны/этилацетат) приводила к этиловому эфиру 2-(3-хлор-4-метилсульфанилфенил)-3-циклопентилакриловой кислоты (267 мг, 60%) в виде масла желтого цвета, состоящего из смеси 2:1 (Е):(Z)-изомеров. Изомерную смесь использовали без дальнейшего разделения и идентификации.
Раствор смеси изомеров этилового эфира 2-(3-хлор-4-метилсульфанилфенил)-3-циклопентилакриловой кислоты [100 мг, 0,31 ммоля, (E):(Z)=2:1] в хлористом метилене (5 мл) охлаждали до 0°С и затем обрабатывали 3-хлорнадбензойной кислотой (содержание 80%, 157 мг, 0,729 ммоля). Реакционную смесь перемешивали при 0°С в течение 3,5 ч и затем разбавляли хлористым метиленом (25 мл). Органическую фазу промывали насыщенным водным раствором карбоната натрия (2×10 мл) и насыщенным водным раствором хлористого натрия (2×10 мл). Органический слой сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Хроматография с помощью системы Biotage (Флэш 12М, двуокись кремния, 80/20 гексаны/этилацетат) приводила к этиловому эфиру 2-(3-хлор-4-метансульфонилфенил)-3-циклопентилакриловой кислоты (95 мг, 86%) в виде бесцветного масла, состоящего из смеси 2:1 (Е):(Z)-изомеров. Смесь изомеров использовали без дальнейшего разделения и идентификации.
Раствор смеси изомеров этилового эфира 2-(3-хлор-4-метансульфонилфенил)-3-циклопентилакриловой кислоты [500 мг, 1,40 ммоля, (E):(Z)=2:1] в этаноле (16 мл) обрабатывали раствором гидроокиси калия (393,6 мг, 7,00 ммолей) в воде (3,7 мл). Раствор желтого цвета перемешивали 3 часа при 25°С и затем концентрировали в вакууме для удаления этанола. Оставшийся водный слой подкисляли 1 н. водным раствором соляной кислоты до рН=2 и затем экстрагировали хлористым метиленом (3×15 мл). Объединенные органические слои затем сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Флэш-хроматография (силикагель 60 фирмы Merck, 230-400 меш, 75/25 гексаны/этилацетат плюс 1% уксусная кислота) приводила к (Е)-2-(3-хлор-4-метансульфонилфенил)-3-циклопентилакриловой кислоте (458 мг, 99%, 95% составляет Е-изомер) в виде пенообразного вещества белого цвета: FAB-MCBP m/e вычислено для C15H17ClO4S (М+Н)+ 329,0614, найдено 329,0628.
Раствор трифенилфосфина (120 мг, 0,46 ммоля) в хлористом метилене (5 мл) охлаждали до 0°С и затем медленно обрабатывали N-бромсукцинимидом (92 мг, 0,52 ммоля). Реакционную смесь перемешивали при 0°С до тех пор, как реакционная смесь становилась гомогенной. Полученную в результате реакционную смесь светло-пурпурного цвета затем обрабатывали (Е)-2-(3-хлор-4-метансульфонилфенил)-3-циклопентилакриловой кислотой (100 мг, 0,30 ммоля) и реакционную смесь перемешивали при 0°С в течение 20 мин. Реакционной смеси затем давали нагреться до 25°С, при этой температуре перемешивали 30 мин. После этого реакционную смесь обрабатывали 2-аминотиазолом (46 мг, 0,46 ммоля) и пиридином (0,044 мл, 0,55 ммоля) и полученную в результате реакционную смесь перемешивали при 25°С в течение 16 ч. Реакционную смесь затем разбавляли водой (10 мл) и экстрагировали хлористым метиленом (3×15 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Флэш-хроматография (силикагель 60 фирмы Merck, 230-400 меш, 70/30 гексаны/этилацетат) приводила к (Е)-2-(3-хлор-4-метансульфонилфенил)-3-циклопентил-N-тиазол-2-илакриламиду (63 мг, 50%) в виде масла желтого цвета: ЭИ-МСВР m/e вычислено для C18H19ClN2O3S2(M+) 410,0526, найдено 410,0529.
Пример 10
(Е)-2-(3-Бром-4-метансульфонилфенил)-3-циклопентил-N-тиазол-2-илакриламид
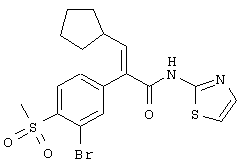
Раствор изоамилнитрита (8,06 мл, 60 ммолей) в диметилдисульфиде (36,02 мл, 400 ммолей) при 25°С медленно обрабатывали 2,4-диброманилином (4,8 г, 20 ммолей). Реакция протекала экзотермически с выделением газа. Полученную в результате реакционную смесь коричневого цвета нагревали до 80-90°С в течение 2 ч, к этому времени анализ реакционной смеси с использованием тонкослойной хроматографии указывал на отсутствие исходного вещества. Реакционную смесь охлаждали до 25°С и затем концентрировали в вакууме. Полученный в результате остаток растворяли в этилацетате (200 мл). Органический слой промывали последовательно 1 н. водным раствором соляной кислоты (1×200 мл) и насыщенным водным раствором хлористого натрия (1×200 мл), сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме. Очистка с применением слоя силикагеля (силикагель 60 фирмы Merck, 230-400 меш, 4/1 гексаны/этилацетат) приводила к 2,4-дибромтиоанизолу (11,04 г, 99%) в виде коричневого масла: ЭИ-МСВР m/е вычислено для С7Н6Вr2S (М+) 279,8623, найдено 279,8619.
Раствор 2,4-дибромтиоанизола (11,04 г, 39,15 ммоля) в хлористом метилене (280 мл) охлаждали до -10°С и затем обрабатывали 3-хлорнадбензойной кислотой (содержание 86%, 20,26 г, 117,4 ммоля). Реакционную смесь перемешивали при -10°С в течение 10 мин и затем давали ей нагреться до 25°С, при этой температуре перемешивали в течение ночи. К этому времени анализ реакционной смеси с применением тонкослойной хроматографии указывал на отсутствие исходного вещества. Реакционную смесь затем фильтровали и твердый остаток промывали хлористым метиленом (1×100 мл). Фильтрат затем разбавляли 1 н. раствором гидроокиси натрия (100 мл) и два слоя разделяли. Органический слой концентрировали в вакууме, получали твердое вещество коричневого цвета. Твердое вещество коричневого цвета растворяли в этилацетате (200 мл). Органический слой промывали последовательно насыщенным водным раствором бикарбоната натрия (2×100 мл) и насыщенным водным раствором хлористого натрия (1×100 мл), сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме до получения сиропа. Этот сироп обрабатывали диэтиловым эфиром и гексанами до получения твердого вещества белого цвета. Полученное в результате твердое вещество собирали при фильтрации, получали 2,4-дибромфенилметилсульфон (10,3 г, 84%) в виде твердого вещества белого цвета: tпл 124-126°С; ЭИ-МСВР m/е вычислено для С7Н6Вr2O2S (М+) 311,8455, найдено 311,8455.
Смесь хлористого лития (8,48 г, 200 ммолей, предварительно высушенного при 130°С в высоком вакууме 2 ч) и цианида меди (8,96 г, 100 ммолей) в безводном тетрагидрофуране (100 мл) перемешивали при 25°С под аргоном в течение 10 мин до получения прозрачного раствора. Реакционную смесь охлаждали до -70°С и затем медленно обрабатывали 2 М раствором циклопентилмагнийхлорида в диэтиловом эфире (55 мл, 110 ммолей). После прибавления реакционной смеси давали нагреться до -30°С и при этой температуре перемешивали 5 мин. Полученную в результате реакционную смесь опять охлаждали до -70°С и затем медленно обрабатывали метиловым эфиром пропиоловой кислоты (7,99 г, 95 ммолей). Реакционную смесь перемешивали при температуре от -60 до -50°С в течение ночи и затем охлаждали до -70 - -60°С, в это время реакционную смесь медленно обрабатывали раствором иода (34,3 г, 135 ммолей) в безводном тетрагидрофуране (30 мл). После прибавления раствора иода охлаждающую баню удаляли и реакционной смеси давали нагреться до 25°С, при этой температуре перемешивали 2 ч. Затем реакционную смесь выливали в раствор, содержащий насыщенный водный раствор хлористого аммония (200 мл) и гидроокись аммония (50 мл), и органическое соединение экстрагировали диэтиловым эфиром (3×100 мл). Объединенные органические экстракты последовательно промывали насыщенным водным раствором тиосульфата натрия (1×300 мл) и насыщенным водным раствором хлористого натрия (1×300 мл). Органический слой сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме. Флэш-хроматография (силикагель 60 фирмы Merck, 230-400 меш, 20/1 гексаны/диэтиловый эфир) приводила к метиловому эфиру (Е)-3-циклопентил-2-иодакриловой кислоты (25,8 г, 97%) в виде масла желтого цвета: ЭИ-МСВР m/е вычислено для С9Н13IO2 (М+) 279,9960, найдено 279,9961.
Смесь цинковой пыли (650 мг, 10 ммолей, фирма Aldrich, -325 меш) и безводного тетрагидрофурана (1 мл) обрабатывали в атмосфере аргона 1,2-дибромэтаном (187 мг, 1,5 ммоля). Цинковую суспензию затем нагревали до бурного кипения с помощью струйной воздушной сушилки, давали охладиться и опять нагревали. Эту процедуру повторяли три раза, чтобы убедиться, что цинковая пыль проактивирована. Суспензию активированной цинковой пыли затем обрабатывали триметилсилилхлоридом (108 мг, 1 ммоль) и суспензию перемешивали 15 мин при 25°С. Реакционную смесь затем обрабатывали по каплям раствором метилового эфира (Е)-3-циклопентил-2-иодакриловой кислоты (660 мг, 2,25 ммоля) в безводном тетрагидрофуране (2 мл) в течение 3 минут. Реакционную смесь затем перемешивали при 40-45°С в течение 1 ч и затем перемешивали в течение ночи при 25°С. Реакционную смесь затем разбавляли безводным тетрагидрофураном (4 мл) и перемешивание прекращали, чтобы дать возможность осаждаться избытку цинковой пыли (~2 ч). В отдельной реакционной колбе перемешивали бис(дибензилиденацетон)палладий (0) (37 мг, 0,07 ммоля) и трифенилфосфин (72 мг, 0,3 ммоля) в безводном тетрагидрофуране (6 мл) при 25°С в атмосфере аргона в течение 10 мин и затем обрабатывали 2,4-дибромфенилметилсульфоном (1,05 г, 3,5 ммоля) и свежеприготовленным цинковым производным в тетрагидрофуране. Полученный в результате раствор цвета красного кирпича нагревали при 40-45°С в течение выходных дней в конце недели. Реакционную смесь охлаждали до 25°С и затем выливали в насыщенный водный раствор хлористого аммония (50 мл), и органическое соединение экстрагировали этилацетатом (3×35 мл). Объединенные органические экстракты промывали насыщенным водным раствором хлористого натрия (1×100 мл), сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме. Хроматография с помощью системы Biotage (ФЛЭШ 40М, двуокись кремния, 5/1 гексаны/этилацетат) приводила к метиловому эфиру (Е)-3-циклопентил-2-(3-бром-4-метансульфонилфенил)акриловой кислоты (1,03 г, 77,6%) в виде масла светло-желтого цвета.
Раствор метилового эфира (Е)-3-циклопентил-2-(3-бром-4-метансульфонилфенил)акриловой кислоты (357 мг, 0,92 ммоля) в этаноле (6 мл) обрабатывали 1 н. водным раствором гидроокиси натрия (2 мл). Раствор нагревали при 45-50°С в течение 15 часов, к этому времени анализ реакционной смеси с помощью тонкослойной хроматографии указывал на отсутствие исходного вещества. Реакционную смесь концентрировали в вакууме для удаления этанола. Остаток разбавляли водой (10 мл) и экстрагировали диэтиловым эфиром (1×30 мл) до удаления любых нейтральных примесей. Водный слой затем подкисляли 1 н. водным раствором соляной кислоты и полученную в результате кислоту экстрагировали этилацетатом (2×20 мл). Объединенные органические слои промывали насыщенным водным раствором хлористого натрия (1×50 мл), сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме, получали (Е)-3-циклопентил-2-(3-бром-4-метансульфонилфенил)акриловую кислоту (339 г, 98%) в виде аморфного твердого вещества: ЭИ-МСВР m/е вычислено для C15H17BrO4S (M+) 372,0031, найдено 372,0028.
Раствор трифенилфосфина (467 мг, 1,78 ммоля) в хлористом метилене (8 мл) охлаждали до 0°С и затем обрабатывали N-бромсукцинимидом (317 мг, 1,78 ммоля). Реакционную смесь перемешивали при 0°С в течение 30 мин и затем обрабатывали раствором (Е)-3-циклопентил-2-(3-бром-4-метансульфонилфенил)акриловой кислоты (334 мг, 0,89 ммоля) в хлористом метилене (4 мл). Реакционную смесь перемешивали 15 мин при 0°С и затем давали нагреться до 25°С, при этой температуре перемешивали 1,5 ч. Реакционную смесь затем обрабатывали 2-аминотиазолом (713 мг, 7,12 ммолей) и полученную в результате суспензию перемешивали 2 дня при 25°С. Реакционную смесь затем концентрировали в вакууме для удаления хлористого метилена и остаток разбавляли этилацетатом (40 мл) и 1 н. водным раствором соляной кислоты (50 мл). Два слоя разделяли и водный слой экстрагировали этилацетатом (1×25 мл). Объединенные органические экстракты последовательно промывали 1 н. водным раствором соляной кислоты (1×50 мл), насыщенным водным раствором бикарбоната натрия (1×50 мл) и насыщенным водным раствором хлористого натрия (1×50 мл). Органический слой сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме. Хроматография с помощью системы Biotage (ФЛЭШ 40S, двуокись кремния, 3,1 гексаны/этилацетат) приводила к чистому (Е)-2-(3-бром-4-метансульфонилфенил)-3-циклопентил-N-тиазол-2-илакриламиду (71 мг; 17,5%) в виде аморфного твердого вещества белого цвета: ЭИ-МСВР m/е вычислено для C18H19BrN2O3S2 (M+) 454,0020, найдено 454,0025.
Пример 11
(Е)-3-Циклогексил-2-(3,4-дифторфенил)-N-тиазол-2-илакриламид
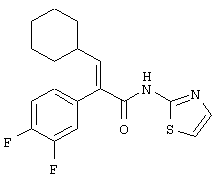
Смесь цинковой пыли (980 мг, 15 ммолей, фирма Aldrich, -325 меш) и безводного тетрагидрофурана (3 мл) обрабатывали в атмосфере аргона 1,2-дибромэтаном (0,37 г, 2 ммоля). Цинковую суспензию затем нагревали до бурного кипения с помощью струйной воздушной сушилки, давали охладиться и опять нагревали. Эту процедуру повторяли три раза, чтобы убедиться, что цинковая пыль проактивирована. Суспензию активированной цинковой пыли затем обрабатывали триметилсилилхлоридом (82 мг, 0,75 ммоля) и суспензию перемешивали 15 мин при 25°С. Реакционную смесь затем обрабатывали по каплям раствором метилового эфира (Е)-3-циклогексил-2-иодакриловой кислоты (получен в примере 4, 1,47 г, 5 ммолей) в безводном тетрагидрофуране (1,5 мл) в течение 3 минут. Во время прибавления температура поднималась до 45°С. Реакционную смесь затем перемешивали при 40-45°С в течение 1 ч и затем перемешивали в течение ночи при 25°С. Реакционную смесь затем разбавляли безводным тетрагидрофураном (5 мл) и перемешивание прекращали, чтобы дать возможность осаждаться избытку цинковой пыли (~2 ч). В отдельной реакционной колбе перемешивали бис(дибензилиденацетонпалладий(0) (54 мг, 0,1 ммоля) и трифенилфосфин (104 мг, 0,4 ммоля) в безводном тетрагидрофуране (10 мл) при 25°С под аргоном в течение 10 мин и затем обрабатывали 3,4-дифториодбензолом (960 мг, 4 ммоля) и свежеприготовленным цинковым производным в тетрагидрофуране. Полученный в результате раствор цвета красного кирпича нагревали 15 ч при 25°С, к этому времени анализ реакционной смеси с помощью тонкослойной хроматографии указывал на отсутствие исходного вещества. Реакционную смесь затем выливали в насыщенный водный раствор хлористого аммония (50 мл) и органическое соединение экстрагировали диэтиловым эфиром (2×50 мл). Объединенные органические экстракты промывали насыщенным водным раствором хлористого натрия (1×50 мл), сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме. Хроматография с помощью системы Biotage (ФЛЭШ 40М, двуокись кремния, 5/1 гексаны/диэтиловый эфир) приводила к метиловому эфиру (Е)-3-циклогексил-2-(3,4-дифторфенил)акриловой кислоты (1,06 г, 95%) в виде масла: ЭИ-МСВР m/е вычислено для C16H18F2O2(M+) 280,1275, найдено 280,1275.
Раствор метилового эфира (Е)-3-циклогексил-2-(3,4-дифторфенил)акриловой кислоты (0,55 г, 1,97 ммоля) в этаноле (10 мл) обрабатывали 1 н. водным раствором гидроокиси натрия (4 мл). Раствор нагревали при 40°С в течение 15 часов, к этому времени анализ реакционной смеси с помощью тонкослойной хроматографии указывал на отсутствие исходного вещества. Реакционную смесь затем концентрировали в вакууме для удаления этанола, остаток разбавляли водой (30 мл) и затем подкисляли 1 н. водным раствором соляной кислоты. Полученную в результате кислоту экстрагировали этилацетатом (2×30 мл). Объединенные органические слои промывали насыщенным водным раствором хлористого натрия (1×50 мл), сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме, получали (Е)-3-циклогексил-2-(3,4-дифторфенил)акриловую кислоту (0,51 г, 97%) в виде твердого вещества белого цвета: tпл 119-121°C; ЭИ-МСВР m/е вычислено для C15H16F2O2 (M+H)+ 267,1196, найдено 267,1200.
Раствор трифенилфосфина (847 мг, 3,2 ммоля) в хлористом метилене (10 мл) охлаждали до 0°С и затем обрабатывали N-бромсукцинимидом (575 мг, 3,2 ммоля). Реакционную смесь перемешивали при 0°С в течение 30 мин и затем обрабатывали раствором (Е)-3-циклогексил-2-(3,4-дифторфенил)акриловой кислоты (507 мг, 1,9 ммоля) в хлористом метилене (4 мл). Прозрачный раствор перемешивали 10 мин при 0°С и затем давали нагреться до 25°С, при этой температуре его перемешивали 1 ч. Реакционную смесь затем обрабатывали 2-аминотиазолом (476 мг, 4,75 ммоля) и полученную в результате суспензию перемешивали 15 ч при 25°С. Реакционную смесь затем концентрировали в вакууме для удаления хлористого метилена и остаток разбавляли этилацетатом (75 мл). Органический слой последовательно промывали 1 н. водным раствором соляной кислоты (2×30 мл), насыщенным водным раствором бикарбоната натрия (2×30 мл) и насыщенным водным раствором хлористого натрия (1×50 мл). Органический слой затем сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме. Хроматография с помощью системы Biotage (ФЛЭШ 40М, двуокись кремния, 8/1-4/1 гексаны/этилацетат) приводила к (Е)-3-циклогексил-2-(3,4-дифторфенил)-N-тиазол-2-илакриламиду (520 мг; 78%) в виде твердого аморфного вещества: ЭИ-МСВР m/е вычислено для C18H18F2N2OS (M+) 348,1108, найдено 348,1104.
Пример 12
(Е)-3-Циклогексил-2-(4-метансульфонил-3-трифторметилфенил)-N-тиазол-2-илакриламид
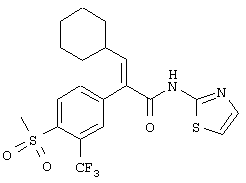
Раствор изоамилнитрита (4,02 мл, 30 ммолей) в диметилдисульфиде (19,8 мл, 220 ммолей) при 25°С медленно обрабатывали 4-бром-2-(трифторметил)анилином (4,8 г, 20 ммолей). Реакция протекала экзотермически с выделением газа. Полученную в результате реакционную смесь коричневого цвета нагревали до 80-90°С в течение 2 ч, к этому времени анализ реакционной смеси с помощью тонкослойной хроматографии указывал на отсутствие исходного вещества. Реакционную смесь охлаждали до 25°С и затем концентрировали в вакууме. Полученный в результате остаток растворяли в этилацетате (200 мл). Органический слой промывали последовательно 1 н. водным раствором соляной кислоты (1×200 мл) и насыщенным водным раствором хлористого натрия (1×200 мл), сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме. Хроматография с помощью системы Biotage (ФЛЭШ 40, двуокись кремния, 8/1 гексаны/этилацетат) приводила к 4-бром-1-метилсульфанил-2-трифторметилбензолу (4,73 г, 87%) в виде масла коричневого цвета: ЭИ-МСВР m/е вычислено для С8Н6ВrF3S (М+) 269,9326, найдено 269,9327.
Раствор 4-бром-1-метилсульфанил-2-трифторметилбензола (4,71 г, 17,4 ммоля) в хлористом метилене (100 мл) охлаждали до -10°С и затем обрабатывали 3-хлорнадбензойной кислотой (содержание 86%, 9,0 г, 52,2 ммоля). Реакционную смесь перемешивали при -10°С в течение 10 мин и затем давали нагреться до 25°С, при этой температуре перемешивали в течение ночи. К этому времени анализ реакционной смеси с помощью тонкослойной хроматографии указывал на отсутствие исходного вещества. Реакционную смесь затем фильтровали и твердый остаток промывали хлористым метиленом (1×50 мл). Фильтрат концентрировали в вакууме. Полученный в результате остаток растворяли в этилацетате (100 мл). Органический слой промывали последовательно насыщенным водным раствором бикарбоната натрия (2×100 мл) и насыщенным водным раствором хлористого натрия (1×100 мл), сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме, получали твердое вещество желтого цвета. Перекристаллизация из хлористого метилена (20 мл), диэтилового эфира (10 мл) и гексанов приводила к 4-бром-1-метансульфонил-2-трифторметилбензолу (3,46 г, 57%) в виде твердого вещества белого цвета: tпл 110-112°С; ЭИ-МСВР т/е вычислено С8Н6ВrF3О2S (М+) 301,9224, найдено 301,9223.
Смесь цинковой пыли (1,3 г, 20 ммолей, фирма Aldrich, -325 меш) и безводного тетрагидрофурана (2 мл) обрабатывали в атмосфере аргона 1,2-дибромэтаном (187 мг, 1 ммоль). Цинковую суспензию затем нагревали до бурного кипения с помощью струйной воздушной сушилки, давали охладиться и опять нагревали. Эту процедуру повторяли три раза, чтобы убедиться, что цинковая пыль проактивирована. Суспензию активированной цинковой пыли затем обрабатывали триметилсилилхлоридом (110 мг, 1 ммоль) и суспензию перемешивали 15 мин при 25°С. Реакционную смесь затем обрабатывали по каплям раствором метилового эфира (Е)-3-циклогексил-2-иодакриловой кислоты (получен в примере 4, 2,5 г, 8,5 ммолей) в безводном тетрагидрофуране (3 мл) в течение 5 минут. После прибавления реакционную смесь перемешивали при 40-45°С в течение 1 ч и затем перемешивали в течение ночи при 25°С.
Реакционную смесь затем разбавляли безводным тетрагидрофураном (4 мл) и перемешивание прекращали, чтобы дать возможность осаждаться избытку цинковой пыли (~2 ч). В отдельной реакционной колбе перемешивали под аргоном 10 мин при 25°С бис(дибензилиденацетон)палладий(0) (108 мг, 0,2 ммоля) и трифенилфосфин (209 мг, 0,8 ммоля) в безводном тетрагидрофуране (10 мл) и затем обрабатывали 4-бром-1-метансульфонил-2-трифторметилбензолом (2,12 г, 7 ммолей) и свежеприготовленным цинковым производным в тетрагидрофуране. Полученный в результате раствор цвета красного кирпича нагревали 2 дня при 40-45°С. Реакционную смесь охлаждали до 25°С, затем выливали в насыщенный водный раствор хлористого аммония (100 мл) и органическое соединение экстрагировали этилацетатом (3×75 мл). Объединенные органические экстракты промывали насыщенным водным раствором хлористого натрия (1×100 мл), сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме. Хроматография с помощью системы Biotage (ФЛЭШ 40М, двуокись кремния, 9/1-3/1 гексаны/этилацетат) приводила к метиловому эфиру (Е)-3-циклогексил-2-(4-метансульфонил-3-трифторметилфенил)акриловой кислоты (2,7 г, 99%) в виде вязкого масла: ЭИ-МСВР m/е вычислено для C18H21F3O4S (M+) 391,1191, найдено 391,1200.
Раствор метилового эфира (Е)-3-циклогексил-2-(4-метансульфонил-3-трифторметилфенил)акриловой кислоты (1,8 г, 4,6 ммоля) в этаноле (20 мл) обрабатывали 1 н. водным раствором гидроокиси натрия (15 мл). Раствор нагревали при 45-50°С в течение 15 часов, к этому времени анализ реакционной смеси с помощью тонкослойной хроматографии указывал на отсутствие исходного вещества. Реакционную смесь затем концентрировали в вакууме для удаления этанола, остаток разбавляли водой (40 мл) и затем экстрагировали диэтиловым эфиром (1×50 мл) для удаления любых нейтральных примесей. Водный слой подкисляли 1 н. водным раствором соляной кислоты. Полученную в результате кислоту экстрагировали этилацетатом (2×75 мл). Объединенные органические слои промывали насыщенным водным раствором хлористого натрия (1×100 мл), сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме, получали (Е)-3-циклогексил-2-(4-метансульфонил-3-трифторметилфенил)акриловую кислоту (1,74 г, 99%) в виде твердого вещества белого цвета: tпл 62-64°С; ЭИ-МСВР m/е вычислено для C17H19F3O4S (М+Н)+ 377,1034, найдено 377,1041.
Раствор трифенилфосфина (1,39 г, 5,3 ммоля) в хлористом метилене (50 мл) охлаждали до 0°С и затем обрабатывали N-бромсукцинимидом (0,94 г, 5,3 ммоля). Реакционную смесь перемешивали при 0°С в течение 30 мин и затем обрабатывали раствором (Е)-3-циклогексил-2-(4-метансульфонил-3-трифторметилфенил)акриловой кислоты (1,00 г, 2,66 ммоля) в хлористом метилене (10 мл). Прозрачный раствор перемешивали 15 мин при 0°С и затем давали ему нагреться до 25°С, при этой температуре перемешивали 1,5 ч. Реакционную смесь затем обрабатывали 2-аминотиазолом (800 мг, 7,98 ммоля) и полученную в результате суспензию перемешивали 2 дня при 25°С. Реакционную смесь концентрировали в вакууме для удаления хлористого метилена и остаток разбавляли этилацетатом (100 мл) и 1 н. водным раствором соляной кислоты (100 мл). Два слоя разделяли и водный слой экстрагировали этилацетатом (1×50 мл). Объединенные органические экстракты последовательно промывали 1 н. водным раствором соляной кислоты (1×100 мл), насыщенным водным раствором бикарбоната натрия (1×100 мл) и насыщенным водным раствором хлористого натрия (1×100 мл). Органический слой затем сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме. Хроматография с помощью системы Biotage (ФЛЭШ 40М, двуокись кремния, 5/1-3/2 гексаны/этилацетат) приводила к (Е)-3-циклогексил-2-(4-метансульфонил-3-трифторметилфенил)-N-тиазол-2-илакриламиду (367 мг, 30%) в виде аморфного твердого вещества: ЭИ-МСВР m/е вычислено для С20Н21F3N2О3S2 (М+) 458,0946, найдено 458,0947.
Пример 13
(Е)-N-(5-Бромтиазол-2-ил)-3-циклогексил-2-(4-метансульфонил-3-трифторметилфенил)акриламид
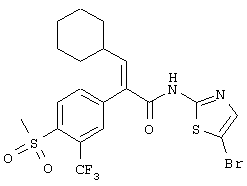
Суспензию (Е)-3-циклогексил-2-(4-метансульфонил-3-трифторметилфенил)-N-тиазол-2-илакриламида (получен в примере 12, 150 мг, 3,27 ммоля) и N-бромсукцинимида (69 мг, 0384 ммоля) в четыреххлористом углероде (2 мл) при 25°С обрабатывали перекисью бензоила (4,65 мг, 0,02 ммоля). Полученную в результате реакционную смесь нагревали до 90°С, при этой температуре перемешивали в течение ночи. Реакционной смеси давали охладиться до 25°С и затем концентрировали в вакууме. Остаток растворяли в этилацетате (25 мл). Органическую фазу затем промывали водой (1×30 мл) и насыщенным водным раствором хлористого натрия (1×30 мл), сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме. Хроматография с помощью системы Biotage (ФЛЭШ 40S, двуокись кремния, 4/1 гексаны/этилацетат) приводила к (Е)-N-(5-бромтиазол-2-ил)-3-циклогексил-2-(4-метансульфонил-3-трифторметилфенил)акриламиду (59 мг, 33%) в виде аморфного твердого вещества белого цвета.
Пример 14
(Е)-3-Циклогексил-2-(4-метансульфонил-3-нитрофенил)-N-тиазол-2-илакриламид
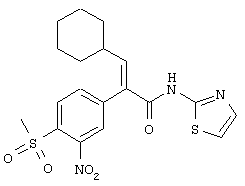
Раствор изоамилнитрита (2,01 мл, 15 ммолей) в диметилдисульфиде (9,9 мл, 110 ммолей) при 25°С медленно обрабатывали 4-бром-2-нитроанилином (2,17 г, 10 ммолей). Реакция протекала экзотермически с выделением газа. Полученную в результате реакционную смесь коричневого цвета нагревали до 80-90°С в течение 2 ч, к этому времени анализ реакционной смеси с помощью тонкослойной хроматографии указывал на отсутствие исходного вещества. Реакционную смесь охлаждали до 25°С и затем концентрировали в вакууме. Полученный в результате остаток растворяли в этилацетате (100 мл). Органический слой промывали последовательно 1 н. водным раствором соляной кислоты (1×100 мл) и насыщенным водным раствором хлористого натрия (1×100 мл), сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме. Хроматография с помощью системы Biotage (ФЛЭШ 40М, двуокись кремния, 6/1-5/1 гексаны/этилацетат) приводила к 5-бром-2-тиометоксинитробензолу (1,9 г, 76%) в виде твердого вещества коричневого цвета: ЭИ-МСВР m/е вычислено для C7H6BrNO2S (M+) 246,9372, найдено 246, 9368.
Раствор 5-бром-2-тиометоксинитробензола (1,37 г, 5,5 ммоля) в хлористом метилене (40 мл) охлаждали до -10°С и затем обрабатывали 3-хлорнадбензойной кислотой (содержание 86%, 2,80 г, 16,56 ммоля). Реакционную смесь перемешивали при -10°С в течение 10 мин и затем давали нагреться до 25°С, при этой температуре перемешивали 2 ч. К этому времени анализ реакционной смеси с помощью тонкослойной хроматографии указывал на отсутствие исходного вещества. Реакционную смесь затем концентрировали в вакууме. Полученный в результате остаток растворяли в этилацетате (100 мл). Органический слой промывали последовательно насыщенным водным раствором бикарбоната натрия (2×100 мл) и насыщенным водным раствором хлористого натрия (1×100 мл), сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме. Хроматография с помощью системы Biotage (ФЛЭШ 40 М, двуокись кремния, 3/1 гексаны/этилацетат) приводила к неочищенному 4-бром-2-нитрофенилметилсульфону (1,5 г) в виде твердого вещества. Это твердое вещество растворяли в хлористом метилене, обрабатывали гексанами и затем фильтровали для получения чистого 4-бром-2-нитрофенилметилсульфона (0,98 г, 63%) в виде твердого вещества белого цвета: tпл 175-177°С; ЭИ-МСВР m/е вычислено для С7Н6ВrNO4S (М+) 278,9201, найдено 278,9210.
Смесь цинковой пыли (650 мг, 10 ммолей, фирма Aldrich, -325 меш) и безводного тетрагидрофурана (1 мл) обрабатывали в атмосфере аргона 1,2-дибромэтаном (187 мг, 1 ммоль). Цинковую суспензию затем нагревали до бурного кипения с помощью струйной воздушной сушилки, давали охладиться и опять нагревали. Эту процедуру повторяли три раза, чтобы убедиться, что цинковая пыль проактивирована. Суспензию активированной цинковой пыли затем обрабатывали триметилсилилхлоридом (110 мг, 1 ммоль) и суспензию перемешивали 15 мин при 25°С. Реакционную смесь затем обрабатывали по каплям раствором метилового эфира (Е)-3-циклогексил-2-иодакриловой кислоты (получен в примере 4, 1,2 г, 4,2 ммоля) в безводном тетрагидрофуране (2 мл) в течение 5 минут. Реакционную смесь затем перемешивали при 40-45°С в течение 1 ч и затем перемешивали в течение ночи при 25°С. Реакционную смесь затем разбавляли безводным тетрагидрофураном (4 мл) и перемешивание прекращали, чтобы дать возможность осаждаться избытку цинковой пыли (~2 ч). В отдельной реакционной колбе перемешивали под аргоном 10 мин при 25°С бис(дибензилиденацетон)палладий(0) (54 мг, 0,1 ммоля) и трифенилфосфин (104 мг, 0,4 ммоля) в безводном тетрагидрофуране (4 мл) и затем обрабатывали 4-бром-2-нитрофенилметилсульфоном (0,94 г, 3,35 ммоля) и свежеприготовленным цинковым производным в тетрагидрофуране. Полученный в результате раствор цвета красного кирпича нагревали 15 ч при 50°С. Реакционную смесь затем охлаждали до 25°С, затем выливали в насыщенный водный раствор хлористого аммония (70 мл) и органическое соединение экстрагировали этилацетатом (3×50 мл). Объединенные органические экстракты промывали насыщенным водным раствором хлористого натрия (1×100 мл), сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме. Хроматография с помощью системы Biotage (ФЛЭШ 40М, двуокись кремния, 9/1-3/1 гексаны/этилацетат) приводила к метиловому эфиру (Е)-3-циклогексил-2-(4-метансульфонил-3-нитрофенил)акриловой кислоты (1 г, 82%) в виде аморфного твердого вещества белого цвета: ЭИ-МСВР m/е вычислено для С17Н21NО6S (М+) 367,1090, найдено 367,1091.
Раствор метилового эфира (Е)-3-циклогексил-2-(4-метансульфонил-3-нитро фенил)акриловой кислоты (597 мг, 1,62 ммоля) в этаноле (10 мл) обрабатывали 1 н. водным раствором гидроокиси натрия (8 мл). Раствор нагревали при 45-50°С в течение 15 часов, к этому времени анализ реакционной смеси с помощью тонкослойной хроматографии указывал на отсутствие исходного вещества. Реакционную смесь затем концентрировали в вакууме для удаления этанола. Остаток разбавляли водой (20 мл) и затем экстрагировали диэтиловым эфиром (1×50 мл) для удаления любых нейтральных примесей. Водный слой подкисляли 1 н. водным раствором соляной кислоты и полученную в результате кислоту экстрагировали этилацетатом (2×50 мл). Объединенные органические слои промывали насыщенным водным раствором хлористого натрия (1×100 мл), сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме, получали (Е)-3-циклогексил-2-(4-метансульфонил-3-нитрофенил)акриловую кислоту (0,514 г, 90%) в виде твердого вещества белого цвета: tпл 244-247°С; ЭИ-МСВР m/е вычислено для С16Н19NО6S (М+) 353,0933, найдено 353,0929.
Раствор трифенилфосфина (720 мг, 2,75 ммоля) в хлористом метилене (25 мл) охлаждали до 0°С и затем обрабатывали N-бромсукцинимидом (490 мг, 2,75 ммоля). Реакционную смесь перемешивали при 0°С в течение 30 мин и затем обрабатывали раствором (Е)-3-циклогексил-2-(4-метансульфонил-3-нитрофенил)акриловой кислоты (485 мг, 1,37 ммоля) в хлористом метилене (5 мл). Реакционную смесь перемешивали 15 мин при 0°С и затем давали ей нагреться до 25°С, при этой температуре перемешивали 1,5 ч. Реакционную смесь затем обрабатывали 2-аминотиазолом (412 мг, 4,12 ммоля) и полученную в результате суспензию перемешивали 2 дня при 25°С. Реакционную смесь затем концентрировали в вакууме для удаления хлористого метилена и остаток разбавляли этилацетатом (70 мл) и 1 н. водным раствором соляной кислоты (50 мл). Два слоя разделяли и водный слой экстрагировали этилацетатом (1×50 мл). Объединенные органические экстракты последовательно промывали 1 н. водным раствором соляной кислоты (1×100 мл), насыщенным водным раствором бикарбоната натрия (1×100 мл) и насыщенным водным раствором хлористого натрия (1×100 мл). Органический слой сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме. Хроматография с помощью системы Biotage (ФЛЭШ 40S, двуокись кремния, 5/1-3/2 гексаны/этилацетат) приводила к (Е)-3-циклогексил-2-(4-метансульфонил-3-нитрофенил)-N-тиазол-2-илакриламиду (122 мг, 20%) в виде аморфного твердого вещества: ЭИ-МСВР m/е вычислено для C19H21N3O5S2 (M+) 435,0923, найдено 435,0923.
Пример 15
(Е)-2-(3-Хлор-4-метансульфонилметилфенил)-3-циклогексил-N-тиазол-2-илакриламид
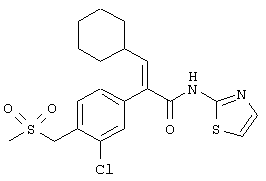
Суспензию 2-хлор-4-иодтолуола (7,57 г, 30 ммолей) и N-бромсукцинимида (5,34 г, 30 ммолей) в четыреххлористом углероде (40 мл) обрабатывали перекисью бензоила (0,3 г, 1,2 ммоля). Реакционную смесь затем нагревали при 90°С в течение 15 ч, к этому времени анализ реакционной смеси с помощью тонкослойной хроматографии указывал на отсутствие исходного материала. Реакционную смесь затем охлаждали до 25°С и концентрировали в вакууме. Полученный в результате остаток розового цвета растворяли в этилацетате (200 мл). Органический слой промывали последовательно водой (2×100 мл) и насыщенным водным раствором хлористого натрия (1×100 мл), сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме. Хроматография с помощью системы Biotage (ФЛЭШ 40М, двуокись кремния, гексаны) приводила к бромистому 2-хлор-4-иодбензилу (4,83 г, 48%) в виде твердого вещества белого цвета: tпл 44-45,5°С; ЭИ-МСВР m/е вычислено для С7Н5ВrСlI (М+) 329,8308, найдено 329,8319.
Раствор бромистого 2-хлор-4-иодбензила (4,82 г, 14,54 ммоля) в N,N-диметилформамиде (30 мл) обрабатывали тиометилатом натрия (2,04 г, 29,08 ммоля). После прибавления раствор становился мутным и приобретал желтую окраску. Полученную в результате реакционную смесь перемешивали 3 ч при 25°С. Реакционную смесь затем разбавляли этилацетатом (100 мл). Органический слой последовательно промывали водой (2×100 мл) и насыщенным водным раствором хлористого натрия (1×100 мл), сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме, получали 2-хлор-4-иодбензилметилсульфид (4,24 г, 97%) в виде бесцветного масла, которое использовали без дальнейшей очистки: ЭИ-МСВР m/е вычислено для C8H8ClIS (M+) 297,9080, найдено 297,9078.
Раствор 2-хлор-4-иодбензилметилсульфида (4,24 г, 14,2 ммоля) в хлористом метилене (100 мл) охлаждали до -5°С и затем обрабатывали 3-хлорнадбензойной кислотой (содержание 86%, 7,35 г, 42,6 ммоля). Реакционную смесь перемешивали при -5°С в течение 15 мин и затем давали нагреться до 25°С, при этой температуре смесь перемешивали 3 ч. В это время анализ реакционной смеси с помощью тонкослойной хроматографии указывал на отсутствие исходного вещества. Твердый осадок отфильтровывали и затем промывали хлористым метиленом (1×50 мл). Фильтрат затем концентрировали в вакууме и полученный в результате остаток растворяли в смеси этилацетата (20 мл) и диэтилового эфира (100 мл). Органический слой промывали последовательно насыщенным водным раствором бикарбоната натрия (2×100 мл), насыщенным водным раствором бисульфита натрия (1×100 мл) и насыщенным водным раствором хлористого натрия (1×100 мл). Органический слой затем сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме. Хроматография с помощью системы Biotage (ФЛЭШ 40М, двуокись кремния, 10/1/10 гексаны/этилацетат/хлористый метилен) приводила к 2-хлор-4-иодбензилметилсульфону (3,67 г, 78%) в виде твердого вещества белого цвета: tпл 125-127°С; ЭИ-МСВР m/е вычислено для C8H8ClIO2S (M+) 329,8979, найдено 329,8969.
Смесь цинковой пыли (650 мг, 10 ммолей, фирма Aldrich, -325 меш) и безводного тетрагидрофурана (2 мл) обрабатывали в атмосфере аргона 1,2-дибромэтаном (187 мг, 1 ммоль). Цинковую суспензию затем нагревали до бурного кипения с помощью струйной воздушной сушилки, давали охладиться и опять нагревали. Эту процедуру повторяли три раза, чтобы убедиться, что цинковая пыль проактивирована. Суспензию активированной цинковой пыли затем обрабатывали триметилсилилхлоридом (110 мг, 1 ммоль) и суспензию перемешивали 15 мин при 25°С. Реакционную смесь затем обрабатывали по каплям раствором метилового эфира (Е)-3-циклогексил-2-иодакриловой кислоты (получен в примере 4, 1,17 г, 4 ммоля) в безводном тетрагидрофуране (2 мл) в течение 5 минут. Реакционную смесь затем перемешивали при 40-45°С в течение 1 ч и затем перемешивали в течение ночи при 25°С. Реакционную смесь затем разбавляли безводным тетрагидрофураном (4 мл) и перемешивание прекращали, чтобы дать возможность осаждаться избытку цинковой пыли (~2 ч). В отдельной реакционной колбе перемешивали под аргоном 10 мин при 25°С бис(дибензилиденацетон)палладий(0) (54 мг, 0,1 ммоля) и трифенилфосфин (104 мг, 0,4 ммоля) в безводном тетрагидрофуране (4 мл) и затем обрабатывали 2-хлор-4-иодбензилметилсульфоном (0,85 г, 2,57 ммоля) и свежеприготовленным цинковым производным в тетрагидрофуране. Полученный в результате раствор цвета красного кирпича нагревали 2 дня при 50°С. Реакционную смесь охлаждали до 25°С и затем выливали в насыщенный водный раствор хлористого аммония (50 мл), и органическое соединение экстрагировали этилацетатом (3×30 мл). Объединенные органические экстракты промывали насыщенным водным раствором хлористого натрия (1×100 мл), сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме. Хроматография с помощью системы Biotage (ФЛЭШ 40М, двуокись кремния, 9/1-3/1 гексаны/этилацетат) приводила к метиловому эфиру (Е)-3-циклогексил-2-(3-хлор-4-(метилен)метилсульфонилфенил)акриловой кислоты (0,94 г, 98%) в виде аморфного твердого вещества белого цвета: ЭИ-МСВР m/е вычислено для C18H23ClO4S (M+) 370,1005, найдено 370,1001.
Раствор метилового эфира (Е)-3-циклогексил-2-(3-хлор-4-(метилен)метилсульфонилфенил)акриловой кислоты (887 мг, 2,4 ммоля) в этаноле (10 мл) обрабатывали 1 н. водным раствором гидроокиси натрия (8 мл). Раствор нагревали при 45-50°С в течение 15 часов, к этому времени анализ реакционной смеси с помощью тонкослойной хроматографии указывал на отсутствие исходного вещества. Реакционную смесь затем концентрировали в вакууме для удаления этанола. Остаток разбавляли водой (20 мл) и экстрагировали диэтиловым эфиром (1×50 мл) для удаления любых нейтральных примесей. Водный слой подкисляли 1 н. водным раствором соляной кислоты и полученную в результате кислоту экстрагировали этилацетатом (2×50 мл). Объединенные органические слои промывали насыщенным водным раствором хлористого натрия (1×100 мл), сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме, получали (Е)-3-циклогексил-2-(3-хлор-4-(метилен)метилсульфонилфенил)акриловую кислоту (0,847 г, 99%) в виде твердого вещества белого цвета: tпл 105-108°С; ЭИ-МСВР m/е вычислено для C17H21ClO4S (M+) 356,0849, найдено 356,0844.
Раствор трифенилфосфина (1,23 г, 4,69 ммоля) в хлористом метилене (15 мл) охлаждали до 0°С и затем обрабатывали N-бромсукцинимидом (830 мг, 4,69 ммоля). Реакционную смесь перемешивали при 0°С в течение 30 мин и затем обрабатывали раствором (Е)-3-циклогексил-2-(3-хлор-4-(метилен)метилсульфонилфенил)акриловой кислоты (837 мг, 2,34 ммоля) в хлористом метилене (6 мл). Реакционную смесь перемешивали 15 мин при 0°С и затем давали ей нагреться до 25°С, при этой температуре перемешивали 1,5 ч. Реакционную смесь затем обрабатывали 2-аминотиазолом (702 мг, 7,02 ммоля) и полученную в результате суспензию перемешивали 2 дня при 25°С. Реакционную смесь затем концентрировали в вакууме для удаления хлористого метилена и остаток разбавляли этилацетатом (70 мл) и 1 н. водным раствором соляной кислоты (50 мл). Два слоя разделяли и водный слой экстрагировали этилацетатом (1×50 мл). Объединенные органические экстракты последовательно промывали 1 н. водным раствором соляной кислоты (1×100 мл), насыщенным водным раствором бикарбоната натрия (1×100 мл) и насыщенным водным раствором хлористого натрия (1×100 мл). Органический слой сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме. Хроматография с помощью системы Biotage (ФЛЭШ 40М, двуокись кремния, 5/1 - 3/2 гексаны/этилацетат) приводила к чистому (Е)-2-(3-хлор-4-метансульфонилметилфенил)-3-циклогексил-N-тиазол-2-илакриламиду (596 мг, 58%) в виде твердого вещества белого цвета: tпл 218-221 С; ЭИ-МСВР m/е вычислено для C20H23ClN2O3S2 (M+) 438,0839, найдено 438,0834.
Пример 16
(Е)-N-(5-Бромтиазол-2-ил)-3-циклогептил-2-(4-метансульфонилфенил)акриламид
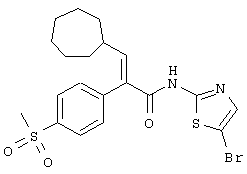
Суспензию (Е)-3-циклогептил-2-(4-метансульфонилфенил)-N-тиазол-2-илакриламида (получен в примере 5, 202 мг, 0,5 ммоля) и N-бромсукцинимида (89 мг, 0,5 ммоля) в четыреххлористом углероде (2 мл) при 25°С обрабатывали перекисью бензоила (6 мг, 0,025 ммоля). Полученную в результате реакционную смесь нагревали до 90°С и перемешивали при этой температуре в течение ночи.
Реакционной смеси давали охладиться до 25°С и затем концентрировали в вакууме. Остаток растворяли в этилацетате (25 мл). Органическую фазу затем промывали водой (1×30 мл) и насыщенным водным раствором хлористого натрия (1×30 мл), сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме. Хроматография с помощью системы Biotage (ФЛЭШ 40М, двуокись кремния, 4/1 гексаны/этилацетат) приводила к (E)-N-(5-бромтиазол-2-ил)-3-циклогептил-2-(4-метансульфонилфенил)акриламиду (86 мг, 36%) в виде твердого вещества белого цвета: tпл 159-163°С.
Пример 17
(Е)-3-Циклогептил-2-(4-метансульфонил-3-трифторметилфенил)-N-тиазол-2-илакриламид
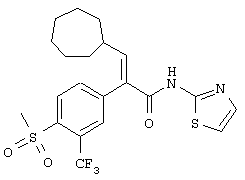
Смесь цинковой пыли (390 мг, 6 ммолей, фирма Aldrich, -325 меш) и безводного тетрагидрофурана (1 мл) обрабатывали в атмосфере аргона 1,2-дибромэтаном (94 мг, 0,5 ммоля). Цинковую суспензию затем нагревали до бурного кипения с помощью струйной воздушной сушилки, давали охладиться и опять нагревали. Эту процедуру повторяли три раза, чтобы убедиться, что цинковая пыль проактивирована. Суспензию активированной цинковой пыли затем обрабатывали триметилсилилхлоридом (55 мг, 0,5 ммоля) и суспензию перемешивали 15 мин при 25°С. Реакционную смесь затем обрабатывали по каплям раствором метилового эфира (Е)-3-циклогептил-2-иодакриловой кислоты (получен в примере 5, 616 мг, 2 ммоля) в безводном тетрагидрофуране (2 мл). После прибавления реакционную смесь перемешивали при 40-45°С в течение 1 ч и затем перемешивали в течение ночи при 25°С. Реакционную смесь затем разбавляли безводным тетрагидрофураном (2 мл) и перемешивание прекращали, чтобы дать возможность осаждаться избытку цинковой пыли (~2 ч). В отдельной реакционной колбе перемешивали бис(дибензилиденацетон)палладий(0) (27 мг, 0,05 ммоля) и трифенилфосфин (52 мг, 0,2 ммоля) в безводном тетрагидрофуране (4 мл) при 25°С в атмосфере аргона в течение 10 мин и затем обрабатывали 4-бром-1-метансульфонил-2-трифторметилбензолом (получен в примере 12, 303 мг, 1 ммоль) и свежеприготовленным цинковым производным в тетрагидрофуране. Полученный в результате раствор цвета красного кирпича нагревали 24 ч при 40-45°С. Реакционную смесь затем охлаждали до 25°С и затем выливали в насыщенный водный раствор хлористого аммония (30 мл), и органическое соединение экстрагировали этилацетатом (3×25 мл). Объединенные органические экстракты промывали насыщенным водным раствором хлористого натрия (1×100 мл), сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме. Хроматография с помощью системы Biotage (ФЛЭШ 40М, двуокись кремния, 4/1 гексаны/этилацетат) приводила к метиловому эфиру (Е)-3-циклогептил-2-(4-метансульфонил-3-трифторметилфенил)акриловой кислоты (387 мг, 95%) в виде вязкого масла.
Раствор метилового эфира (Е)-3-циклогептил-2-(4-метансульфонил-3-трифторметилфенил)акриловой кислоты (387 мг, 0,96 ммоля) в этаноле (6 мл) обрабатывали 1 н. водным раствором гидроокиси натрия (2 мл). Раствор нагревали при 45-50°С в течение 15 часов, к этому времени анализ реакционной смеси с помощью тонкослойной хроматографии указывал на отсутствие исходного вещества. Реакционную смесь затем концентрировали в вакууме для удаления этанола и остаток разбавляли водой (20 мл) и экстрагировали диэтиловым эфиром (1×30 мл) для удаления любых нейтральных примесей. Водный слой подкисляли 1 н. водным раствором соляной кислоты. Полученную в результате кислоту экстрагировали этилацетатом (2×35 мл). Объединенные органические слои промывали насыщенным водным раствором хлористого натрия (1х100 мл), сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме, получали (Е)-3-циклогептил-2-(4-метансульфонил-3-трифторметилфенил)акриловую кислоту (268 мг, 72%) в виде твердого вещества коричневого цвета: tпл 151-156°C.
Раствор трифенилфосфина (341 мг, 1,3 ммоля) в хлористом метилене (7 мл) охлаждали до 0°С и затем обрабатывали N-бромсукцинимидом (231 мг, 1,3 ммоля). Реакционную смесь перемешивали при 0°С в течение 30 мин и затем обрабатывали раствором (Е)-3-циклогептил-2-(4-метансульфонил-3-трифторметилфенил)акриловой кислоты (255 мг, 0,65 ммоля). После перемешивания 15 мин при 0°С реакционная смесь становилась прозрачной. Прозрачному раствору давали нагреться до 25°С, при этой температуре его перемешивали 1,5 ч. Реакционную смесь затем обрабатывали 2-аминотиазолом (193 мг, 1,95 ммоля) и полученную в результате суспензию перемешивали 2 дня при 25°С. Реакционную смесь концентрировали в вакууме для удаления хлористого метилена и остаток разбавляли этилацетатом (50 мл) и 1 н. водным раствором соляной кислоты (50 мл). Два слоя разделяли и водный слой экстрагировали этилацетатом (1×30 мл). Объединенные органические экстракты последовательно промывали 1 н. водным раствором соляной кислоты (1×50 мл), насыщенным водным раствором бикарбоната натрия (1×50 мл) и насыщенным водным раствором хлористого натрия (1×50 мл). Органический слой затем сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме. Хроматография с помощью системы Biotage (ФЛЭШ 40S, двуокись кремния, 4/1-2/1 гексаны/этилацетат) приводила к чистому (Е)-3-циклогептил-2-(4-метансульфонил-3-трифторметилфенил)-N-тиазол-2-илакриламиду (133 мг, 43%) в виде твердого аморфного вещества.
Пример 18
(Е)-N-(5-Бромтиазол-2-ил)-3-циклогептил-2-(4-метансульфонил-3-трифторметилфенил)акриламид
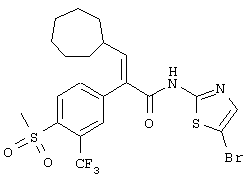
Суспензию (Е)-3-циклогептил-2-(4-метансульфонил-3-трифторметилфенил)-N-тиазол-2-илакриламида (получен в примере 17, 63 мг, 0,133 ммоля) и N-бромсукцинимида (26 мг, 0,146 ммоля) в четыреххлористом углероде (2 мл) при 25°С обрабатывали перекисью бензоила (2 мг, 0,006 ммоля). Полученную в результате реакционную смесь нагревали до 90°С и перемешивали при этой температуре в течение ночи. Реакционной смеси давали охладиться до 25°С и затем концентрировали в вакууме. Остаток растворяли в этилацетате (25 мл). Органическую фазу затем промывали водой (1×30 мл) и насыщенным водным раствором хлористого натрия (1×30 мл), сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме. Хроматография с помощью системы Biotage (ФЛЭШ 40S, двуокись кремния, 5/1 гексаны/этилацетат) приводила к (Е)-N-(5-бромтиазол-2-ил)-3-циклогептил-2-(4-метансульфонил-3-трифторметилфенил)акриламиду (35,5 мг, 48%) в виде твердого аморфного вещества белого цвета.
Пример 19
(Е)-2-(3-Хлор-4-метансульфонилфенил)-3-циклопентил-N-пиридин-2-илакриламид
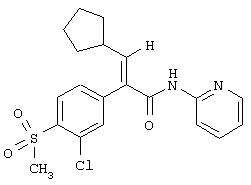
Раствор трифенилфосфина (266 мг, 1,01 ммоля) в хлористом метилене (11 мл) охлаждали до 0°С и затем обрабатывали N-бромсукцинимидом (204 мг, 1,15 ммоля). Реакционную смесь перемешивали при 0°С до тех пор, когда реакционная смесь становилась гомогенной. Полученную в результате реакционную смесь светло-розового цвета затем обрабатывали раствором (Е)-2-(3-хлор-4-метансульфонилфенил)-3-циклопентилакриловой кислоты (получена в примере 9, 222 мг, 0,68 ммоля) и реакционную смесь перемешивали 20 мин при 0°С. Реакционной смеси затем давали нагреться до 25°С, при этой температуре перемешивали 30 мин. По истечении этого времени реакционную смесь обрабатывали 2-аминопиридином (95 мг, 1,01 ммоля) и пиридином (0,098 мл, 1,22 ммоля) и полученную в результате реакционную смесь перемешивали 16 ч при 25°С. Реакционную смесь затем разбавляли водой (10 мл) и экстрагировали хлористым метиленом (3×15 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Хроматография с помощью системы Biotage (ФЛЭШ, 40S, двуокись кремния, 75/25 гексаны/этилацетат) приводила к (Е)-2-(3-хлор-4-метансульфонилфенил)-3-циклопентил-N-пиридин-2-илакриламиду (70 мг, 25%) в виде твердого стекловидного вещества желтого цвета: ЭИ-МСВР m/е вычислено для C20H21ClN2O3S (M+) 404,0961, найдено 404,0962.
Пример 20
(Е)-N-(5-Бромпиридин-2-ил)-3-циклогексил-2-(4-метансульфонил-3-трифторметилфенил)акриламид
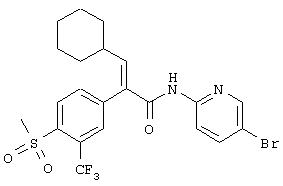
Раствор трифенилфосфина (525 мг, 2 ммоля) в хлористом метилене (12 мл) охлаждали до 0°С и затем обрабатывали N-бромсукцинимидом (356 мг, 2 ммоля). Реакционную смесь перемешивали при 0°С в течение 30 мин и затем обрабатывали (Е)-3-циклогексил-2-(4-метансульфонил-3-трифторметилфенил)акриловой кислотой (получена в примере 12, 376 мг, 1 ммоль). Реакционную смесь перемешивали 15 мин при 0°С и затем давали ей нагреться до 25°С, при этой температуре перемешивали 1,5 ч. Реакционную смесь затем обрабатывали 2-амино-5-бромпиридином (519 мг, 3 ммоля) и полученную в результате суспензию перемешивали 3 дня при 25°С. Реакционную смесь концентрировали в вакууме для удаления хлористого метилена и остаток разбавляли этилацетатом (50 мл) и 1 н. водным раствором соляной кислоты (50 мл). Два слоя разделяли и водный слой экстрагировали этилацетатом (1×30 мл). Объединенные органические экстракты последовательно промывали насыщенным водным раствором бикарбоната натрия (1×50 мл) и насыщенным водным раствором хлористого натрия (1×50 мл). Органический слой затем сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме. Хроматография с помощью системы Biotage (ФЛЭШ 40S, двуокись кремния, 4/1-2/1 гексаны/этилацетат) приводила к (Е)-N-(5-бромпиридин-2-ил)-3-циклогексил-2-(4-метансульфонил-3-трифторметилфенил)акриламиду (44 мг, 8,3%) в виде твердого аморфного вещества: ЭИ-МСВР m/е вычислено для С22Н22ВrF3N2O3S (М+) 530,0487, найдено 530,0484.
Пример 21
Тиазол-2-иламид (Е)-4-циклопентил-2-(4-метансульфонилфенил)бут-2-еновой кислоты
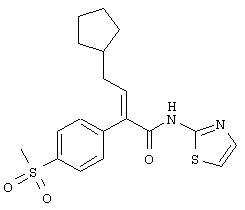
Смесь цинковой пыли (3,92 г, 60 ммолей, фирма Aldrich, -325 меш) и безводного тетрагидрофурана (4 мл) обрабатывали в атмосфере аргона 1,2-дибромэтаном (0,56 г, 3 ммоля). Цинковую суспензию затем нагревали до бурного кипения с помощью струйной воздушной сушилки, давали охладиться и опять нагревали. Эту процедуру повторяли три раза, чтобы убедиться, что цинковая пыль проактивирована. Суспензию активированной цинковой пыли затем обрабатывали триметилсилилхлоридом (0,32 г, 3 ммоля) и суспензию перемешивали 15 мин при 25°С. Реакционную смесь затем обрабатывали по каплям раствором иодистого циклопентилметила (4,2 г, 20 ммолей) в безводном тетрагидрофуране (7 мл) в течение 5 мин. Во время прибавления температура поднималась до 50°С, реакционную смесь перемешивали при 40-45°С в течение ночи. Реакционную смесь затем охлаждали до 25°С и разбавляли безводным тетрагидрофураном (5 мл). Перемешивание прекращали, чтобы дать возможность осаждаться избытку цинковой пыли (~2 ч). В отдельной реакционной колбе смесь хлористого лития (1,7 г, 40 ммолей, предварительно высушенного при 130°С в высоком вакууме 2 ч) и цианида меди (1,79 г, 20 ммолей) в безводном тетрагидрофуране (20 мл) перемешивали 10 мин при 25°С для получения прозрачного раствора. Реакционную смесь охлаждали до -70°С и затем медленно обрабатывали свежеприготовленным раствором цинка с помощью шприца. После прибавления реакционной смеси давали нагреться до -30°С, при этой температуре перемешивали 5 мин. Реакционную смесь опять охлаждали до -70°С и затем медленно обрабатывали метиловым эфиром пропиоловой кислоты (1,52 г, 18 ммолей). Реакционную смесь перемешивали 4 ч при температуре от -40 до -30°С и затем медленно обрабатывали раствором иода (6,85 г, 27 ммолей) в безводном тетрагидрофуране (10 мл), поддерживая температуру от -70 до -60°С. После прибавления раствора иода охлаждающую баню удаляли и реакционной смеси давали нагреться до 25°С, при этой температуре перемешивали 1 ч. Реакционную смесь затем выливали в раствор, состоящий из насыщенного водного раствора хлористого аммония (90 мл) и гидроокиси аммония (10 мл), и органическое соединение экстрагировали диэтиловым эфиром (3×50 мл). Объединенные органические экстракты последовательно промывали насыщенным водным раствором тиосульфата натрия (1×100 мл) и насыщенным водным раствором хлористого натрия (1×100 мл), сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме. Хроматография с помощью системы Biotage (ФЛЭШ 40М, двуокись кремния, 9/1 гексаны/диэтиловый эфир) приводила к метиловому эфиру (Е)-4-циклопентил-2-иодбут-2-еновой кислоты (4,56 г, 86%) в виде бесцветного масла: ЭИ-МСВР m/е вычислено для С10Н15IO2 (М+) 294,0116, найдено 294,0114.
Смесь цинковой пыли (0,98 г, 15 ммолей, фирма Aldrich, -325 меш) и безводного тетрагидрофурана (3 мл) обрабатывали в атмосфере аргона 1,2-дибромэтаном (0,14 г, 0,75 ммоля). Цинковую суспензию затем нагревали до бурного кипения с помощью струйной воздушной сушилки, давали охладиться и опять нагревали. Эту процедуру повторяли три раза, чтобы убедиться, что цинковая пыль проактивирована. Суспензию активированной цинковой пыли затем обрабатывали триметилсилилхлоридом (82 мг, 0,75 ммоля) и суспензию перемешивали 15 мин при 25°С. Реакционную смесь затем обрабатывали по каплям раствором метилового эфира (Е)-4-циклопентил-2-иодбут-2-еновой кислоты (1,47 г, 5 ммолей) в безводном тетрагидрофуране (1,5 мл) в течение 3 мин. После прибавления реакционную смесь перемешивали при 40-45°С в течение 1 ч и затем перемешивали в течение ночи при 25°С. Реакционную смесь затем разбавляли безводным тетрагидрофураном (5 мл) и перемешивание прекращали, чтобы дать возможность осаждаться избытку цинковой пыли (~2 ч). В отдельной реакционной колбе перемешивали бис(дибензилиденацетон)палладий(0) (54 мг, 0,1 ммоля) и трифенилфосфин (104 мг, 0,4 ммоля) в безводном тетрагидрофуране (10 мл) 10 мин при 25°С в атмосфере аргона и затем обрабатывали 4-бромфенилметилсульфоном (0,94 г, 4 ммоля) и свежеприготовленным цинковым производным в тетрагидрофуране. Полученный в результате раствор цвета красного кирпича нагревали 24 ч при 50°С, к этому времени анализ реакционной смеси с помощью тонкослойной хроматографии указывал на отсутствие исходного вещества. Реакционную смесь затем охлаждали до 25°С и затем выливали в насыщенный водный раствор хлористого аммония (75 мл), и органическое соединение экстрагировали диэтиловым эфиром (3×50 мл). Объединенные эфирные экстракты промывали насыщенным водным раствором хлористого натрия (1×100 мл), сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме. Хроматография с помощью системы Biotage (ФЛЭШ 40М, двуокись кремния, 3/7 гексаны/диэтиловый эфир) приводила к метиловому эфиру (Е)-4-циклопентил-2-(4-метансульфонилфенил)бут-2-еновой кислоты (1,10 г, 86%) в виде бесцветного масла: ЭИ-МСВР m/е вычислено для C17H22O4S (М+) 322,1235, найдено 322,1239.
Раствор метилового эфира (Е)-4-циклопентил-2-(4-метансульфонилфенил)бут-2-еновой кислоты (1,00 г, 3,1 ммоля) в этаноле (17 мл) обрабатывали 1 н. водным раствором гидроокиси натрия (7 мл). Раствор нагревали при 45-50°С в течение 15 часов, к этому времени анализ реакционной смеси с помощью тонкослойной хроматографии указывал на отсутствие исходного вещества. Реакционную смесь затем концентрировали в вакууме для удаления этанола, остаток разбавляли водой (30 мл) и экстрагировали диэтиловым эфиром (1×50 мл) для удаления любых нейтральных примесей. Водный слой подкисляли 1 н. водным раствором соляной кислоты. Полученную в результате кислоту экстрагировали этилацетатом (2×30 мл). Объединенные органические слои промывали насыщенным водным раствором хлористого натрия (1×50 мл), сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме, получали (Е)-4-циклопентил-2-(4-метансульфонилфенил)бут-2-еновую кислоту (0,95 г, 99%) в виде твердого вещества белого цвета: tпл 162-165°С; ЭИ-МСВР m/е вычислено для C16H20O4S (M+H)+ 309,1160, найдено 308,1158.
Раствор трифенилфосфина (672 мг, 2,56 ммоля) в хлористом метилене (7,5 мл) охлаждали до 0°С и затем обрабатывали N-бромсукцинимидом (456 мг, 2,56 ммоля). Реакционную смесь перемешивали при 0°С в течение 30 мин и затем обрабатывали раствором (Е)-4-циклопентил-2-(4-метансульфонилфенил)бут-2-еновой кислоты (545,5 мг, 1,47 ммоля) в хлористом метилене (4 мл). Прозрачный раствор перемешивали 10 мин при 0°С и затем давали ему нагреться до 25°С, при этой температуре перемешивали 1 ч. Реакционную смесь затем обрабатывали 2-аминотиазолом (378 мг, 3,76 ммоля) и полученную в результате суспензию перемешивали при 25°С в течение выходных дней в конце недели. Реакционную смесь концентрировали в вакууме для удаления хлористого метилена и остаток разбавляли этилацетатом (75 мл) и 1 н. водным раствором соляной кислоты (100 мл). Два слоя разделяли и водный слой экстрагировали этилацетатом (1×50 мл). Объединенные органические экстракты последовательно промывали насыщенным водным раствором бикарбоната натрия (2×50 мл) и насыщенным водным раствором хлористого натрия (1×100 мл), сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме. Хроматография с помощью системы Biotage (ФЛЭШ 40М, двуокись кремния, 4/1-1/1 гексаны/этилацетат) приводила к тиазол-2-иламиду (Е)-4-циклопентил-2-(4-метансульфонилфенил)бут-2-еновой кислоты (200 мг, 35%) в виде твердого вещества белого цвета: tпл 173-176°C; ЭИ-МСВР m/е вычислено для C19H22N2O3S2 (M+) 390,1071, найдено 390,1072.
Пример 22
Метиловый эфир (Е)-2-[4-циклопентил-2-(4-метансульфонилфенил)бут-2-еноиламино]тиазол-4-карбоновой кислоты
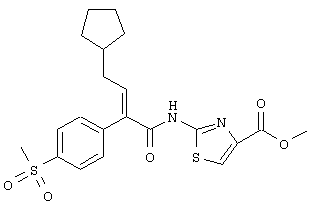
Раствор трифенилфосфина (525 мг, 2 ммоля) в хлористом метилене (25 мл) охлаждали до 0°С и затем обрабатывали N-бромсукцинимидом (355 мг, 2 ммоля). Реакционную смесь перемешивали при 0°С в течение 30 мин и затем обрабатывали (Е)-4-циклопентил-2-(4-метансульфонилфенил)бут-2-еновой кислотой (получена в примере 21, 308 мг, 1 ммоль). Прозрачный раствор перемешивали 10 мин при 0°С и затем ему давали нагреться до 25°С, при этой температуре перемешивали 1 ч. Реакционную смесь затем обрабатывали метиловым эфиром 2-аминотиазол-4-карбоновой кислоты (400 мг, 2,52 ммоля) и полученную в результате суспензию перемешивали при 25°С в течение выходных дней в конце недели. Реакционную смесь концентрировали в вакууме для удаления хлористого метилена и остаток разбавляли этилацетатом (50 мл) и 1 н. водным раствором соляной кислоты (50 мл). Два слоя разделяли и водный слой экстрагировали этилацетатом (1×25 мл). Объединенные органические экстракты последовательно промывали насыщенным водным раствором бикарбоната натрия (2×50 мл) и насыщенным водным раствором хлористого натрия (1×100 мл), сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме. Хроматография с помощью системы Biotage (ФЛЭШ 40М, двуокись кремния, 3/1-1/1 гексаны/этилацетат) приводила к метиловому эфиру (Е)-2-[4-циклопентил-2-(4-метансульфонилфенил)бут-2-еноиламино]тиазол-4-карбоновой кислоты (250 мг, 56%) в виде твердого вещества белого цвета: tпл 85-90°С; ЭИ-МСВР m/е вычислено для C21H24N2O5S2 (М+) 448,1127, найдено 448, 1117.
Пример 23
Этиловый эфир (Е)-2-[4-циклопентил-2-(4-метансульфонилфенил)бут-2-еноиламино]тиазол-5-карбоновой кислоты
Раствор трифенилфосфина (787 мг, 3 ммоля) в хлористом метилене (40 мл) охлаждали до 0°С и затем обрабатывали N-бромсукцинимидом (534 мг, 3 ммоля). Реакционную смесь перемешивали при 0°С в течение 30 мин и затем обрабатывали (Е)-4-циклопентил-2-(4-метансульфонилфенил)бут-2-еновой кислотой (получена в примере 21, 462 мг,1,5 ммоля). Прозрачный раствор перемешивали 10 мин при 0°С и затем ему давали нагреться до 25°С, при этой температуре перемешивали 1 ч. Реакционную смесь затем обрабатывали этиловым эфиром 2-аминотиазол-5-карбоновой кислоты (774 мг, 4,5 ммоля) и полученную в результате суспензию перемешивали при 25°С в течение выходных дней в конце недели. Реакционную смесь концентрировали в вакууме для удаления хлористого метилена и остаток разбавляли этилацетатом (70 мл) и 1 н. водным раствором соляной кислоты (70 мл). Два слоя разделяли и водный слой экстрагировали этилацетатом (1×50 мл). Объединенные органические экстракты последовательно промывали насыщенным водным раствором бикарбоната натрия (1×100 мл) и насыщенным водным раствором хлористого натрия (1×100 мл), сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме. Хроматография с помощью системы Biotage (ФЛЭШ 40М, двуокись кремния, 3/1-1/1 гексаны/этилацетат) приводила к этиловому эфиру (Е)-2-[4-циклопентил-2-(4-метансульфонилфенил)бут-2-еноиламино]тиазол-5-карбоновой кислоты (250 мг, 36%) в виде аморфного твердого вещества белого цвета: ЭИ-МСВР m/е вычислено для C22H26N2O5S2 (М+) 462,1283, найдено 462.1282.
Пример 24
Тиазол-2-иламид (Е)-4-циклопентил-2-(3,4-дифторфенил)бут-2-еновой кислоты
Смесь цинковой пыли (0,98 г, 15 ммолей, фирма Aldrich, -325 меш) и безводного тетрагидрофурана (3 мл) обрабатывали в атмосфере аргона 1,2-дибромэтаном (0,14 г, 0,75 ммоля). Цинковую суспензию затем нагревали до бурного кипения с помощью струйной воздушной сушилки, давали охладиться и опять нагревали. Эту процедуру повторяли три раза, чтобы убедиться, что цинковая пыль проактивирована. Суспензию активированной цинковой пыли затем обрабатывали триметилсилилхлоридом (82 мг, 0,75 ммоля) и суспензию перемешивали 15 мин при 25°С. Реакционную смесь затем обрабатывали по каплям раствором метилового эфира (Е)-4-циклопентил-2-иодбут-2-еновой кислоты (получен в примере 21, 1,47 г, 5 ммолей) в безводном тетрагидрофуране (1,5 мл) в течение 3 мин. После прибавления реакционную смесь перемешивали при 40-45°С в течение 1 ч и затем перемешивали в течение ночи при 25°С. Реакционную смесь затем разбавляли безводным тетрагидрофураном (5 мл) и перемешивание прекращали, чтобы дать возможность осаждаться избытку цинковой пыли (~2 ч). В отдельной реакционной колбе перемешивали при 25°С под аргоном в течение 10 мин бис(дибензилиденацетон)палладий(0) (54 мг, 0,1 ммоля) и трифенилфосфин (104 мг, 0,4 ммоля) в безводном тетрагидрофуране (10 мл) и затем обрабатывали 3,4-дифториодбензолом (0,96 г, 4 ммоля) и свежеприготовленным цинковым производным в тетрагидрофуране. Полученный в результате раствор цвета красного кирпича нагревали 15 ч при 25°С, к этому времени анализ реакционной смеси с помощью тонкослойной хроматографии указывал на отсутствие исходного вещества. Реакционную смесь охлаждали до 25°С и затем выливали в насыщенный водный раствор хлористого аммония (50 мл), и органическое соединение экстрагировали диэтиловым эфиром (2×50 мл). Объединенные эфирные экстракты промывали насыщенным водным раствором хлористого натрия (1×50 мл), сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме. Хроматография с помощью системы Biotage (ФЛЭШ 40М, двуокись кремния, 4/1 гексаны/диэтиловый эфир) приводила к метиловому эфиру (Е)-4-циклопентил-2-(3,4-дифторфенил)бут-2-еновой кислоты (0,82 г, 73%) в виде вязкого масла: ЭИ-МСВР m/е вычислено для C16H18F2O2 (M+) 280,1275, найдено 280,1275.
Раствор метилового эфира (Е)-4-циклопентил-2-(3,4-дифторфенил)бут-2-еновой кислоты (0,80 г, 2,85 ммоля) в этаноле (14 мл) обрабатывали 1 н. водным раствором гидроокиси натрия (6 мл). Раствор нагревали при 40°С в течение 15 часов, к этому времени анализ реакционной смеси с помощью тонкослойной хроматографии указывал на отсутствие исходного вещества. Реакционную смесь затем концентрировали в вакууме для удаления этанола и остаток разбавляли водой (30 мл) и затем экстрагировали диэтиловым эфиром (1×50 мл) для удаления любых нейтральных примесей. Водный слой подкисляли 1 н. водным раствором соляной кислоты. Полученную в результате кислоту экстрагировали этилацетатом (2×50 мл). Объединенные органические слои промывали насыщенным водным раствором хлористого натрия (1×80 мл), сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме, получали (Е)-4-циклопентил-2-(3,4-дифторфенил)бут-2-еновую кислоту (0,65 г, 86%) в виде бесцветного масла: ЭИ-МСВР m/е вычислено для C15H16F2O2 (М+Н)+ 267,1196, найдено 267,1195.
Раствор трифенилфосфина (1,05 г, 4 ммоля) в хлористом метилене (15 мл) охлаждали до 0°С и затем обрабатывали N-бромсукцинимидом (712 мг, 4 ммоля). Реакционную смесь перемешивали при 0°С в течение 30 мин и затем обрабатывали раствором (Е)-4-циклопентил-2-(3,4-дифторфенил)бут-2-еновой кислоты (0,63 г, 2,36 ммоля) в хлористом метилене (4 мл). Прозрачный раствор перемешивали 15 мин при 0°С и затем ему давали нагреться до 25°С, при этой температуре перемешивали 1,5 ч. Реакционную смесь затем обрабатывали 2-аминотиазолом (0,59 г, 5,9 ммоля) и полученную в результате суспензию перемешивали при 25°С в течение выходных дней в конце недели. Реакционную смесь концентрировали в вакууме для удаления хлористого метилена и остаток разбавляли этилацетатом (100 мл) и 1 н. водным раствором соляной кислоты (100 мл). Два слоя разделяли и водный слой экстрагировали этилацетатом (1×50 мл). Объединенные органические экстракты последовательно промывали насыщенным водным раствором бикарбоната натрия (2×50 мл) и насыщенным водным раствором хлористого натрия (1×100 мл), сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме. Хроматография с помощью системы Biotage (ФЛЭШ 40М, двуокись кремния, 8/1 гексаны/этилацетат) приводила к тиазол-2-иламиду (Е)-4-циклопентил-2-(3,4-дифторфенил)бут-2-еновой кислоты (435 мг, 53%) в виде аморфного твердого вещества: ЭИ-МСВР m/е вычислено для C18H18F2N2OS (M+) 348,1108, найдено 348,1103.
Пример 25
Тиазол-2-иламид (Е)-4-циклопентил-2-(4-метансульфонил-3-трифторметилфенил)бут-2-еновой кислоты
Смесь цинковой пыли (0,65 г, 10 ммолей, фирма Aldrich, -325 меш) и безводного тетрагидрофурана (2 мл) обрабатывали в атмосфере аргона 1,2-дибромэтаном (140 мг, 0,75 ммоля). Цинковую суспензию затем нагревали до бурного кипения с помощью струйной воздушной сушилки, давали охладиться и опять нагревали. Эту процедуру повторяли три раза, чтобы убедиться, что цинковая пыль проактивирована. Суспензию активированной цинковой пыли затем обрабатывали триметилсилилхлоридом (82 мг, 0,75 ммоля) и суспензию перемешивали 15 мин при 25°С. Реакционную смесь затем обрабатывали по каплям раствором метилового эфира (Е)-4-циклопентил-2-иодбут-2-еновой кислоты (получен в примере 21, 1,03 г, 3,5 ммоля) в безводном тетрагидрофуране (1,5 мл) в течение 3 мин. После прибавления реакционную смесь перемешивали при 40-45°С в течение 1 ч и затем перемешивали в течение ночи при 25°С. Реакционную смесь затем разбавляли безводным тетрагидрофураном (3 мл) и перемешивание прекращали, чтобы дать возможность осаждаться избытку цинковой пыли (~2 ч). В отдельной реакционной колбе перемешивали при 25°С в атмосфере аргона в течение 10 мин бис(дибензилиденацетон)палладий(0) (54 мг, 0,1 ммоля) и трифенилфосфин (104 мг, 0,4 ммоля) в безводном тетрагидрофуране (10 мл) и затем обрабатывали 4-бром-1-метансульфонил-2-трифторметилбензолом (получен в примере 12, 0,76 г, 2,5 ммоля) и свежеприготовленным цинковым производным в тетрагидрофуране. Полученный в результате раствор цвета красного кирпича нагревали 15 ч при 25°С. Реакционную смесь затем выливали в насыщенный водный раствор хлористого аммония (50 мл) и органическое соединение экстрагировали этилацетатом (2×50 мл). Объединенные органические экстракты промывали насыщенным водным раствором хлористого натрия (1×50 мл), сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме. Хроматография с помощью системы Biotage (ФЛЭШ 40М, двуокись кремния, 2/1 гексаны/этилацетат) приводила к метиловому эфиру (Е)-4-циклопентил-2-(4-метансульфонил-3-трифторметилфенил)бут-2-еновой кислоты (0,85 г, 87%) в виде вязкого масла: ЭИ-МСВР m/е вычислено для C18H21F3O4S (M+) 390,1113, найдено 390,1113.
Раствор метилового эфира (Е)-4-циклопентил-2-(4-метансульфонил-3-трифторметилфенил)бут-2-еновой кислоты (0,82 г, 2,1 ммоля) в этаноле (10 мл) обрабатывали 1 н. водным раствором гидроокиси натрия (5 мл). Раствор нагревали при 40°С в течение 15 часов, к этому времени анализ реакционной смеси с помощью тонкослойной хроматографии указывал на отсутствие исходного вещества. Реакционную смесь затем концентрировали в вакууме для удаления этанола и остаток разбавляли водой (30 мл) и экстрагировали диэтиловым эфиром (1×50 мл) для удаления любых нейтральных примесей. Водный слой подкисляли 1 н. водным раствором соляной кислоты. Полученную в результате кислоту экстрагировали этилацетатом (2×50 мл). Объединенные органические слои промывали насыщенным водным раствором хлористого натрия (1×80 мл), сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме, получали (Е)-4-циклопентил-2-(4-метансульфонил-3-трифторметилфенил)бут-2-еновую кислоту (0,73 г, 92%) в виде смолистого твердого вещества: ЭИ-МСВР m/е вычислено для С17Н19F3O4S (M)+ 376,0243, найдено 376,0261.
Раствор трифенилфосфина (550 мг, 2,1 ммоля) в хлористом метилене (25 мл) охлаждали до 0°С и затем обрабатывали N-бромсукцинимидом (374 мг, 2,1 ммоля). Реакционную смесь перемешивали при 0°С в течение 30 мин и затем обрабатывали раствором (Е)-4-циклопентил-2-(4-метансульфонил-3-трифторметилфенил)бут-2-еновой кислотой (395 мг, 1,05 ммоля) в хлористом метилене (5 мл). Прозрачный раствор перемешивали 15 мин при 0°С и затем ему давали нагреться до 25°С, при этой температуре перемешивали 1,5 ч. Реакционную смесь затем обрабатывали 2-аминотиазолом (320 мг, 3,2 ммоля) и полученную в результате суспензию перемешивали при 25°С в течение выходных дней в конце недели. Реакционную смесь концентрировали в вакууме для удаления хлористого метилена и остаток разбавляли этилацетатом (50 мл) и 1 н. водным раствором соляной кислоты (50 мл). Два слоя разделяли и водный слой экстрагировали этилацетатом (1×30 мл). Объединенные органические экстракты последовательно промывали насыщенным водным раствором бикарбоната натрия (2×50 мл) и насыщенным водным раствором хлористого натрия (1×100 мл). Органический слой затем сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме. Хроматография с помощью системы Biotage (ФЛЭШ 40М, двуокись кремния, 1/1 гексаны/этилацетат) приводила к тиазол-2-иламиду (Е)-4-циклопентил-2-(4-метансульфонил-3-трифторметилфенил)бут-2-еновой кислоты (77 мг, 16%) в виде аморфного твердого вещества: ЭИ-МСВР m/е вычислено для С20Н21F3N2О3S2 (М+) 458,0946, найдено 458,0946.
Пример 26
(Е)-1-[2-(3,4-Дихлорфенил)-4-метилпент-2-еноил]-3-метилмочевина
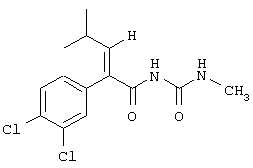
Смесь хлористого алюминия (16,81 г, 126,05 ммоля) в хлористом метилене (105 мл) охлаждали до 5°С и перемешивали до растворения твердого вещества. Реакционную смесь затем медленно обрабатывали метилоксалилхлоридом (8,1 мл, 88,24 ммоля) и полученную реакционную смесь перемешивали при 5°С в течение 30 минут. Реакционную смесь затем медленно обрабатывали 1,2-дихлорбензолом (12,35 г, 84,04 ммоля). Полученной в результате реакционной смеси давали нагреться до 25°С, при этой температуре ее перемешивали 6 ч. Реакционную смесь затем хранили при 0°С в течение 15 ч. Реакционную смесь медленно выливали в лед/воду (400 мл). Встряхивали и разделяли слои. Водный слой далее экстрагировали хлористым метиленом (1×200 мл). Объединенные органические слои промывали насыщенным водным раствором бикарбоната натрия (1×200 мл) и водой (1×100 мл), сушили над сульфатом магния, фильтровали и концентрировали в вакууме. Флэш-хроматография (силикагель 60 фирмы Merck, 230-400 меш, 9/1 гексаны/этилацетат) приводила к метиловому эфиру (3,4-дихлорфенил)оксоуксусной кислоты (0,78 г, 4%) в виде твердого вещества желтого цвета: tпл 58,2-63°С; ЭИ-МСВР m/е вычислено для С9Н6Сl2O3 (М+) 231,9694, найдено 231,9699.
Суспензию бромистого изобутилтрифенилфосфония (2,02 г, 4,96 ммоля) в безводном тетрагидрофуране (5,4 мл) охлаждали до 0°С и затем обрабатывали по каплям 1,0 М раствором бис(триметилсилил)амида натрия (5 мл, 4,96 ммоля). Реакционную смесь ярко-оранжевого цвета перемешивали при 0°С в течение 1 ч. Реакционную смесь затем обрабатывали раствором метилового эфира (3,4-дихлорфенил)оксоуксусной кислоты (0,77 г, 3,30 ммоля) в тетрагидрофуране (3 мл). Полученной в результате реакционной смеси давали нагреться до 25°С, при этой температуре ее перемешивали 15 ч. Реакционную смесь смешивали с водой (10 мл) и затем концентрировали в вакууме для удаления тетрагидрофурана. Остаток далее разбавляли водой (50 мл) и затем экстрагировали этилацетатом (2×75 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Флэш-хроматография (силикагель 60 фирмы Merck, 230-400 меш, 97/3 гексаны/этилацетат) приводила к метиловому эфиру 2-(3,4-дихлорфенил)-4-метилпент-2-еновой кислоты (749 мг, 83%) в виде вязкого масла желтого цвета, содержащего смесь (E):(Z)-изомеров в отношении 3,5:1. Смесь изомеров использовали без дальнейшего разделения и идентификации.
Смесь изомеров метилового эфира 2-(3,4-дихлорфенил)-4-метилпент-2-еновой кислоты [749,0 мг, 2,74 ммоля, (E):(Z)=3,5:1] и метилмочевины (812,6 мг, 10,97 ммоля) обрабатывали раствором метилата магния в метаноле (7,4 мас.%, 16 мл, 10,97 ммоля). Полученную в результате реакционную смесь кипятили с обратным холодильником 15 часов. Реакционной смеси давали охладиться до 25°С и затем фильтровали через целит. Целит тщательно промывали этилацетатом. Фильтрат концентрировали в вакууме. Флэш-хроматография (силикагель 60 фирмы Merck, 230-400 меш, 9/1 гексаны/этилацетат) приводила к неочищенной 1-[2-(3,4-дихлорфенил)-4-метилпент-2-еноил]-3-метилмочевине (280,2 мг) в виде твердого вещества белого цвета. Повторная флэш-хроматография (силикагель 60 фирмы Merck, 230-400 меш, 3/2 гексаны/диэтиловый эфир) снова приводила к неочищенной 1-[2-(3,4-дихлорфенил)-4-метилпент-2-еноил]-3-метилмочевине (114,6 мг) в виде твердого вещества белого цвета. Перекристаллизация из гексанов/этилацетата приводила к чистой (Е)-1-[2-(3,4-дихлорфенил)-4-метилпент-2-еноил]-3-метилмочевине (24,7 мг, 3%) в виде твердого вещества белого цвета: tпл 177-178°C; FAB-MCBP m/е вычислено для C14H16Cl2N2O2 (M+H)+ 315,0667, найдено 315,0652.
Пример 27
(Е)-1-[3-Циклогексил-2-(4-метансульфонил-3-трифторметилфенил)акрилоил]-3-метилмочевина
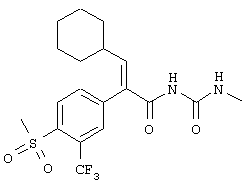
Раствор изоамилнитрита (4,02 мл, 30 ммолей) в диметилдисульфиде (19,8 мл, 220 ммолей) при 25°С медленно обрабатывали 4-бром-2-(трифторметил)анилином (4,8 г, 20 ммолей). Реакция протекала экзотермически с выделением газа. Полученную в результате реакционную смесь коричневого цвета нагревали до 80-90°С в течение 2 ч, к этому времени анализ реакционной смеси с помощью тонкослойной хроматографии указывал на отсутствие исходного вещества. Реакционную смесь охлаждали до 25°С и затем концентрировали в вакууме. Полученный в результате остаток растворяли в этилацетате (200 мл). Органический слой последовательно промывали 1 н. водным раствором соляной кислоты (1×200 мл) и насыщенным водным раствором хлористого натрия (1×200 мл), сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме. Хроматография с помощью системы Biotage (ФЛЭШ 40М, двуокись кремния, 8/1 гексаны/этилацетат) приводила к 4-бром-1-метилсульфанил-2-трифторметилбензолу (4,73 г, 87%) в виде масла коричневого цвета: ЭИ-МСВР m/е вычислено для С8Н6ВrF3S (М+) 269,9326, найдено 269,9327.
Раствор 4-бром-1-метилсульфанил-2-трифторметилбензола (4,71 г, 17,4 ммоля) в хлористом метилене (100 мл) охлаждали до -10°С и затем обрабатывали 3-хлорнадбензойной кислотой (содержание 86%, 9,0 г, 52,2 ммоля). Реакционную смесь перемешивали при -10°С в течение 10 мин и затем давали ей нагреться до 25°С, при этой температуре перемешивали в течение ночи. К этому времени анализ реакционной смеси с помощью тонкослойной хроматографии указывал на отсутствие исходного вещества. Реакционную смесь затем фильтровали и твердый осадок промывали хлористым метиленом (1×50 мл). Фильтрат концентрировали в вакууме. Полученный в результате остаток растворяли в этилацетате (100 мл). Органический слой промывали последовательно насыщенным водным раствором бикарбоната натрия (2×100 мл) и насыщенным водным раствором хлористого натрия (1×100 мл), сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме, получали твердое вещество желтого цвета. Перекристаллизация из хлористого метилена (20 мл), диэтилового эфира (10 мл) и гексанов приводила к 4-бром-1-метансульфонил-2-трифторметилбензолу (3,46 г, 57%) в виде твердого вещества белого цвета: tпл 110-112°С; ЭИ-МСВР m/е вычислено для С8Н6ВrF3O2S (М+) 301,9224, найдено 301,9223.
Смесь цинковой пыли (16,34 г, 250 ммолей, фирма Aldrich, -325 меш) и безводного тетрагидрофурана (6 мл) обрабатывали в атмосфере аргона 1,2-дибромэтаном (0,94 г, 5 ммолей). Цинковую суспензию затем нагревали до бурного кипения с помощью струйной воздушной сушилки, давали охладиться и опять нагревали. Эту процедуру повторяли три раза, чтобы убедиться, что цинковая пыль проактивирована. Суспензию активированной цинковой пыли затем обрабатывали триметилсилилхлоридом (0,54 г, 5 ммолей) и суспензию перемешивали 15 мин при 25°С. Реакционную смесь затем обрабатывали по каплям раствором циклогексилиодида (21 г, 100 ммолей) в безводном тетрагидрофуране (30 мл) в течение 15 мин. Во время прибавления температура повышалась до 60°С. Реакционную смесь затем перемешивали 3 ч при 40-45°С. Реакционную смесь затем охлаждали до 25°С и разбавляли безводным тетрагидрофураном (60 мл). Перемешивание прекращали, чтобы дать возможность осаждаться избытку цинковой пыли (~3 ч). В отдельной реакционной колбе смесь хлористого лития (8,48 г, 200 ммолей, предварительно высушенного при 130°С в высоком вакууме 3 ч) и цианида меди (8,95 г, 100 ммолей) в безводном тетрагидрофуране (110 мл) перемешивали 10 мин при 25°С до получения прозрачного раствора. Реакционную смесь охлаждали до -70°С и затем медленно обрабатывали свежеприготовленным цинковым раствором, используя шприц. После прибавления реакционной смеси давали нагреться до 0°С, при этой температуре перемешивали 5 минут. Реакционную смесь опять охлаждали до -70°С и затем медленно обрабатывали метиловым эфиром пропиоловой кислоты (7,56 г, 90 ммолей). Полученную в результате реакционную смесь перемешивали 15 ч при температуре от -70 до -50°С и затем медленно обрабатывали раствором иода (34,26 г, 135 ммолей) в безводном тетрагидрофуране (30 мл), поддерживая температуру от -70 до -60°С. После прибавления раствора иода удаляли охлаждающую баню и реакционной смеси давали нагреться до 25°С, при этой температуре перемешивали 2 ч. Реакционную смесь затем выливали в раствор, состоящий из насыщенного водного раствора хлористого аммония (400 мл) и гидроокиси аммония (100 мл), и органическое соединение экстрагировали этилацетатом (3×250 мл). Объединенные органические экстракты последовательно промывали насыщенным водным раствором тиосульфата натрия (1×500 мл) и насыщенным водным раствором хлористого натрия (1×500 мл), сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме. Флэш-хроматография (силикагель 60 фирмы Merck, 230-400 меш, 9/1 гексаны/диэтиловый эфир) приводила к метиловому эфиру (Е)-3-циклогексил-2-иодакриловой кислоты (26,3 г, 99%) в виде масла светло-розового цвета: ЭИ-МСВР m/е вычислено для С10Н15IO2 (М+) 294,0117, найдено 294,0114.
Смесь цинковой пыли (1,3 г, 20 ммолей, фирма Aldrich, -325 меш) и безводного тетрагидрофурана (2 мл) обрабатывали в атмосфере аргона 1,2-дибромэтаном (187 мг, 1 ммоль). Цинковую суспензию затем нагревали до бурного кипения с помощью струйной воздушной сушилки, давали охладиться и опять нагревали. Эту процедуру повторяли три раза, чтобы убедиться, что цинковая пыль проактивирована. Суспензию активированной цинковой пыли затем обрабатывали триметилсилилхлоридом (110 мг, 1 ммоль) и суспензию перемешивали 15 мин при 25°С. Реакционную смесь затем обрабатывали по каплям раствором метилового эфира (Е)-3-циклогексил-2-иодакриловой кислоты (2,5 г, 8,5 ммоля) в безводном тетрагидрофуране (3 мл) в течение 5 минут. После прибавления реакционную смесь перемешивали 1 ч при 40-45°С и затем перемешивали в течение ночи при 25°С. Реакционную смесь затем разбавляли безводным тетрагидрофураном (4 мл) и перемешивание прекращали, чтобы дать возможность осаждаться избытку цинковой пыли (~2 ч). В отдельной реакционной колбе перемешивали бис(дибензилиденацетон)палладий(0) (108 мг, 0,2 ммоля) и трифенилфосфин (209 мг, 0,8 ммоля) в безводном тетрагидрофуране (10 мл) 10 мин при 25°С в атмосфере аргона и затем обрабатывали 4-бром-1-метансульфонил-2-трифторметилбензолом (2,12 г, 7 ммолей) и свежеприготовленным цинковым производным в тетрагидрофуране. Полученный в результате раствор цвета красного кирпича нагревали 2 дня при 40-45°С. Реакционную смесь охлаждали до 25°С и затем выливали в насыщенный водный раствор хлористого аммония (100 мл), и органическое соединение экстрагировали этилацетатом (3×75 мл). Объединенные органические экстракты промывали насыщенным водным раствором хлористого натрия (1×100 мл), сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме. Хроматография с помощью системы Biotage (ФЛЭШ 40М, двуокись кремния, 9/1-3/1 гексаны/этилацетат) приводила к метиловому эфиру (Е)-3-циклогексил-2-(4-метансульфонил-3-трифторметилфенил)акриловой кислоты (2,7 г, 99%) в виде вязкого масла: ЭИ-МСВР m/е вычислено для C18H21F3O4S (М+) 391,1191, найдено 391,1200.
Раствор метилового эфира (Е)-3-циклогексил-2-(4-метансульфонил-3-трифторметилфенил)акриловой кислоты (1,8 г, 4,6 ммоля) в этаноле (20 мл) обрабатывали 1 н. водным раствором гидроокиси натрия (15 мл). Раствор нагревали при 45-50°С в течение 15 ч, к этому времени анализ реакционной смеси с помощью тонкослойной хроматографии указывал на отсутствие исходного вещества. Реакционную смесь затем концентрировали в вакууме для удаления этанола, остаток разбавляли водой (40 мл) и экстрагировали диэтиловым эфиром (1×50 мл) для удаления любых нейтральных примесей. Водный слой подкисляли 1 н. водным раствором соляной кислоты. Полученную в результате кислоту экстрагировали этилацетатом (2×75 мл). Объединенные органические слои промывали насыщенным водным раствором хлористого натрия (1×100 мл), сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме, получали (Е)-3-циклогексил-2-(4-метансульфонил-3-трифторметилфенил)акриловую кислоту (1,74 г, 99%) в виде твердого вещества белого цвета: tпл 62-64°С; ЭИ-МСВР m/е вычислено для C17H19F3O4S (М+Н)+ 377,1034, найдено 377,1041.
Раствор (Е)-3-циклогексил-2-(4-метансульфонил-3-трифторметилфенил)акриловой кислоты (282 мг, 0,75 ммоля) во фторбензоле (1 мл) и N,N-диметилформамиде (3 мкл) при 25°С обрабатывали по каплям оксалилхлоридом (81 мкл, 0,9 ммоля) в течение 2-3 мин. Прозрачный раствор перемешивали 1 ч при 25°С и затем обрабатывали метилмочевиной (167 мг, 2,25 ммоля). Полученную в результате суспензию нагревали при 70°С (температура в бане) в течение 10 минут и затем обрабатывали пиридином (121 мкл, 1,5 ммоля). Реакционную смесь затем перемешивали 20 ч при 70°С. Реакционную смесь затем охлаждали до 25°С и разбавляли этилацетатом (50 мл) и 3 н. водным раствором соляной кислоты (40 мл). Два слоя разделяли и водный слой экстрагировали этилацетатом (1×20 мл). Объединенные органические экстракты последовательно промывали насыщенным водным раствором бикарбоната натрия (1×50 мл) и насыщенным водным раствором хлористого натрия (1×50 мл), сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме. Хроматография с помощью системы Biotage (ФЛЭШ 40М, двуокись кремния, 4/1 гексаны/этилацетат) приводила к (Е)-1-[3-циклогексил-2-(4-метансульфонил-3-трифторметилфенил)акрилоил]-3-метилмочевине (104 мг, 32%) в виде твердого вещества белого цвета: tпл 199-202°C, ЭИ-МСВР m/е вычислено для С19Н23F3Н2O4S (М+) 432,1331, найдено 432,1332.
Пример А
Таблетки, содержащие следующие ингредиенты, могут быть получены общепринятым способом.
Ингредиенты, мг на таблетку:
Пример Б
Капсулы, содержащие следующие ингредиенты, могут быть получены общепринятым способом.
Ингредиенты, мг в капсуле:
| название | год | авторы | номер документа |
|---|---|---|---|
| АКТИВАТОРЫ ГЛЮКОКИНАЗЫ | 2000 |
|
RU2242469C2 |
| ПРОИЗВОДНЫЕ ТИОАМИДА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2000 |
|
RU2245874C2 |
| АЗАИНДОЛЫ В КАЧЕСТВЕ АКТИВАТОРОВ ГЛЮКОКИНАЗЫ | 2010 |
|
RU2593369C2 |
| ЗАМЕЩЕННЫЕ 3-ЦИАНОТИОФЕНАЦЕТАМИДЫ В КАЧЕСТВЕ АНТАГОНИСТОВ РЕЦЕПТОРА ГЛЮКАГОНА | 2004 |
|
RU2336274C2 |
| ЗАМЕЩЕННЫЕ ПИРРОЛИДИН-2-КАРБОКСАМИДЫ | 2011 |
|
RU2564022C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ 2,6-ДИАМИНОПИРИДИН-3-ОНА | 2005 |
|
RU2385866C2 |
| ПРОИЗВОДНЫЕ ПИРРОЛИДИНА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1990 |
|
RU2024506C1 |
| СОЕДИНЕНИЯ, СОДЕРЖАЩИЕ КОНДЕНСИРОВАННОЕ БИЦИКЛИЧЕСКОЕ ЯДРО, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ИНГИБИРОВАНИЯ ПРЕВРАЩЕНИЯ ФЕРМЕНТА АНГИОТЕНЗИНА И НЕЙТРАЛЬНОЙ ЭНДОПЕПТИДАЗЫ | 1994 |
|
RU2125056C1 |
| Способ получения спирозамещенных производных глутарамида или их фармацевтически допустимых солей | 1987 |
|
SU1612996A3 |
| 2-САХАРИНИЛМЕТИЛАРИЛКАРБОКСИЛАТЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ) И КОМПОЗИЦИЯ, ИНГИБИРУЮЩАЯ АКТИВНОСТЬ ПРОТЕОЛИТИЧЕСКОГО ФЕРМЕНТА | 1991 |
|
RU2114843C1 |
Изобретение относится к транс-олефиновому активатору глюкокиназы, представляющему собой соединение, выбранное из группы, состоящей из олефинового амида формулы
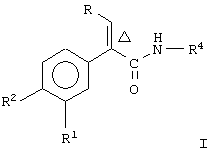
где R1 и R2 означают независимо друг от друга водород, галоген, нитрогруппу, перфтор(низш.)алкил, (низш.)алкилсульфонил или (низш.)алкилсульфонилметил; R означает -(CH2)m-R3 или низший алкил, содержащий от 2 до 4 атомов углерода; R3 означает циклоалкил, содержащий от 3 до 8 атомов углерода; R4 означает
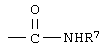
или незамещенное или однозамещенное пяти- или шестичленное гетероароматическое кольцо, связанное с помощью углеродного атома кольца с указанной аминогруппой, причем это пяти- или шестичленное гетероароматическое кольцо содержит от 1 до 2 гетероатомов, выбранных из группы, состоящей из серы или азота, где один гетероатом, являясь азотом, расположен рядом со связующим кольцевым углеродным атомом, и указанное однозамещенное гетероароматическое кольцо замещено при углеродном атоме кольца, несмежном с упомянутым связующим атомом углерода, заместителем, выбранным из группы, состоящей из галогена и
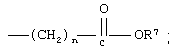
m означает 0 или 1; n означает 0, 1, 2, 3 или 4; R7 означает водород или низший алкил и Δ означает транс-конфигурацию относительно двойной связи, или его фармацевтически приемлемую соль. Также изобретение относится к фармацевтической композиции, способу профилактического или терапевтического лечения диабета типа II и к способам получения соединений формулы I. Изобретение позволяет получить активаторы глюкокиназы, которые повышают секрецию инсулина при лечении диабета типа II. 5 н. и 20 з.п. ф-лы.
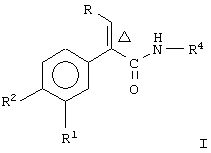
где R1 и R2 означают независимо друг от друга водород, галоген, нитрогруппу, перфтор(низш.)алкил, (низш.)алкилсульфонил или (низш.)алкилсульфонилметил;
R означает -(CH2)m-R3 или низший алкил, содержащий от 2 до 4 атомов углерода;
R3 означает циклоалкил, содержащий от 3 до 8 атомов углерода;
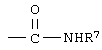
R4 означает
или незамещенное или однозамещенное пяти- или шестичленное гетероароматическое кольцо, связанное с помощью углеродного атома кольца с указанной аминогруппой, причем это пяти- или шестичленное гетероароматическое кольцо содержит от 1 до 2 гетероатомов, выбранных из группы, состоящей из серы или азота, где один гетероатом, являясь азотом, расположен рядом со связующим кольцевым углеродным атомом; и указанное однозамещенное гетероароматическое кольцо замещено при углеродном атоме кольца, не смежном с упомянутым связующим атомом углерода, заместителем, выбранным из группы, состоящей из галогена и
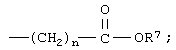
m означает 0 или 1;
n означает 0, 1, 2, 3 или 4;
R7 означает водород или низший алкил; и
Δ означает транс-конфигурацию относительно двойной связи;
или его фармацевтически приемлемая соль.
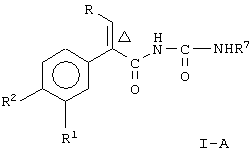
где Δ, R, R1 и R2 и R7 являются такими, как указано в п.1.
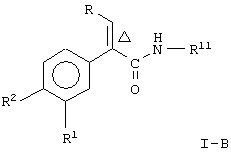
где R, R2, R1 и Δ являются такими, как указано в п.1, и
R11 является незамещенным или однозамещенным пяти- или шестичленным гетероароматическим кольцом, связанным с помощью углеродного атома кольца с указанной аминогруппой, причем это пяти- или шестичленное гетероароматическое кольцо содержит 1-2 гетероатома, выбранных из группы, состоящей из серы или азота, где один гетероатом, являющийся азотом, расположен рядом со связующим углеродным атомом кольца; и упомянутое однозамещенное гетероароматическое кольцо замещено при углеродном атоме кольца, не смежном с упомянутым связующим атомом углерода, заместителем, выбранным из группы, состоящей из галогена или
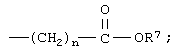
n означает 0, 1, 2, 3 или 4; и
R7 означает водород или низший алкил.
(Е)-1-[3-циклогексил-2-(4-метансульфонил-3-трифторметилфенил)акрилоил)-3-метил-мочевины;
(Е)-1-[2-(3,4-дихлорфенил)-4-метилпент-2-еноил]-3-метилмочевины;
тиазол-2-иламида (Е)-2-(4-метансульфонилфенил)пент-2-еновой кислоты;
тиазол-2-иламида (Е)-2-(4-метансульфонилфенил)-4-метилпент-2-еновой кислоты;
(Е)-3-циклопентил-2-(4-метансульфонилфенил)-N-тиазол-2-илакриламида;
(Е)-2-(3-хлор-4-метансульфонилфенил)-3-циклопентил-N-тиазол-2-илакриламида;
(Е)-2-(3-бром-4-метансульфонилфенил)-3-циклопентил-N-тиазол-2-илакриламида;
(Е)-3-циклопентил-2-(3,4-дихлорфенил)-N-тиазол-2-илакриламида;
(Е)-N-(5-бромтиазол-2-ил)-3-циклопентил-2-(4-метансульфонилфенил)акриламида;
(Е)-3-циклогексил-2-(4-метансульфонилфенил)-N-тиазол-2-илакриламида;
(Е)-3-циклогексил-2-(4-метансульфонил-3-нитрофенил)-N-тиазол-2-илакриламида;
(Е)-3-циклогексил-2-(4-метансульфонил-3-трифторметилфенил)-N-тиазол-2-ил-акриламида;
(Е)-3-циклогексил-2-(3,4-дифторфенил)-N-тиазол-2-илакриламида;
(Е)-2-(3-хлор-4-метансульфонилметилфенил)-3-циклогексил-N-тиазол-2-илакриламида;
(Е)-N-(5-бромтиазол-2-ил)-3-циклогексил-2-(4-метансульфонил-3-трифторметил-фенил)акриламида;
(Е)-3-циклогептил-2-(4-метансульфонилфенил)-N-тиазол-2-илакриламида;
(Е)-3-циклооктил-2-(4-метансульфонилфенил)-N-тиазол-2-илакриламида;
(Е)-N-(5-бромтиазол-2-ил)-3-циклогептил-2-(4-метансульфонилфенил)акриламида;
(Е)-3-циклогептил-2-(4-метансульфонил-3-трифторметилфенил)-N-тиазол-2-ил-акриламида;
(Е)-N-(5-бромтиазол-2-ил)-3-циклогептил-2-(4-метансульфонил-3-трифторметил-фенил)акриламида;
тиазол-2-иламида(Е)-4-циклопентил-2-(4-метансульфонилфенил)бут-2-еновой кислоты;
метилового эфира (Е)-2-[4-циклопентил-2-(4-метансульфонилфенил)бут-2-еноил-амино]тиазол-4-карбоновой кислоты;
этилового эфира (Е)-2-[4-циклопентил-2-(4-метансульфонилфенил)бут-2-еноил-амино]тиазол-5-карбоновой кислоты;
(Е)-2-(3-хлор-4-метансульфонилфенил)-3-циклопентил-N-тиазол-2-илакриламид;
(Е)-2-(3-хлор-4-метансульфонилфенил)-3-циклопентил-N-пиридин-2-илакриламид;
(Е)-N-(5-бромпиридин-2-ил)-3-циклогексил-2-(4-метансульфонил-3-трифторметил-фенил)акриламида;
тиазол-2-иламида(Е)-4-циклопентил-2-(3,4-дифторфенил)бут-2-еновой кислоты
и
тиазол-2-иламида (Е)-4-циклопентил-2-(4-метансульфонил-3-трифторметилфенил)бут-2-еновой кислоты.
(Е)-3-циклопентил-2-(4-метансульфонилфенил)-N-тиазол-2-илакриламида;
(Е)-3-циклогексил-2-(4-метансульфонилфенил)-N-тиазол-2-илакриламида;
(Е)-3-циклогептил-2-(4-метансульфонилфенил)-N-тиазол-2-илакриламида;
(Е)-2-(3-хлор-4-метансульфонилфенил)-3-циклопентил-N-тиазол-2-илакриламида;
(Е)-3-циклогексил-2-(4-метансульфонил-3-трифторметилфенил)-N-тиазол-2-ил-акриламида;
(Е)-3-циклогексил-2-(4-метансульфонил-3-нитрофенил)-N-тиазол-2-илакриламида;
(Е)-N-(5-бромтиазол-2-ил)-3-циклогептил-2-(4-метансульфонилфенил)акриламида;
(Е)-2-(3-хлор-4-метансульфонилфенил)-3-циклопентил-N-пиридин-2-илакриламида;
(Е)-N-(5-бромпиридин-2-ил)-3-циклогексил-2-(4-метансульфонил-3-трифторметил-фенил)акриламида;
тиазол-2-иламида(Е)-4-циклопентил-2-(4-метансульфонилфенил)бут-2-еновой кислоты;
метилового эфира(Е)-2-[4-циклопентил-2-(4-метансульфонилфенил)бут-2-еноил-амино]тиазол-4-карбоновой кислоты и тиазол-2-иламида(Е)-4-циклопентил-2-(4-метансульфонил-3-трифторметилфенил)бут-2-еновой кислоты.
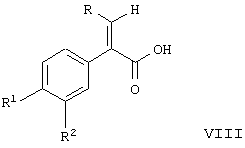
где R, R1 и R2 являются такими, как в п.1, или соединения формулы VII
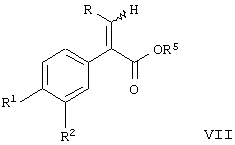
где R, R1 и R2 являются такими, как указано в п.1, и R5 вместе со связанным с ним атомом кислорода образует способную гидролизоваться защитную группу для кислоты, с соединением формулы XIV

где R11 является таким, как указано в п.1, для получения соединения формулы I-B
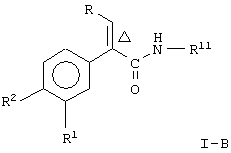
где R, R1, R2, R11 и Δ являются такими, как указано в п.3,
и последующее, в случае необходимости, превращение остатка R1 и/или R2 в другой остаток R1 и/или R2, как указано в п.1.
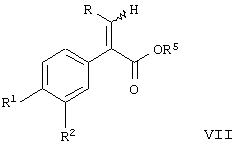
где R, R1 и R2 являются такими, как указано в п.1, и R5 вместе со связанным с ним атомом кислорода образует способную гидролизоваться защитную группу для кислоты, с соединением формулы XV
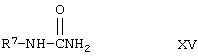
где R7 является таким, как указано в п.1,
для получения соединения формулы I-A
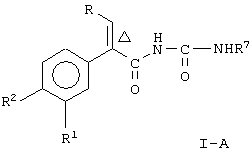
где R, R1, R2 и Δ являются такими, как указано в п.3,
и последующее, в случае необходимости, превращение остатка R1 и/или R2 в другой остаток R1 и/или R2, как указано в п.1.
| ГАЛОГЕНЗАМЕЩЕННЫЕ N-БЕНЗГИДРИЛ- N -(ТРИФТОРАЦЕТИЛ) МОЧЕВИНЫ, ПРОЯВЛЯЮЩИЕ ПРОТИВОСУДОРОЖНУЮ АКТИВНОСТЬ | 1991 |
|
SU1829341A1 |
| Способ получения производных 3-хинолинкарбоновой кислоты | 1975 |
|
SU566522A3 |
| US 5510478 A, 23.04.1996. | |||

