Настоящее изобретение относится к соединениям 5-арил-1Н-1,2,4-триазола, к способу их получения и к содержащим их фармацевтическим композициям.
Нестероидные противовоспалительные лекарства (NSAIDs) чаще всего проявляют свое действие за счет ингибирования простагландин-Н-синтазы (PGHS), которая опосредует превращение арахидоновой кислоты в простагландины. Первой стадией этого процесса является окислительная циклизация арахидоновой кислоты до PGE2, после чего следует пероксидное восстановление до PGH2 по второму отличительному сайту связывания. PGHS, обычно известные как циклооксигеназы или СОХ, существуют в двух изоформах, причем каждая играет свою отличную физиологическую роль (Hia, T et al, Proc. Natl. Acad. Sci. USA. 1992, 89, 7384; Holtzman H.J. et al, J. Biol. Chem. 1992, 267, 21438; Herschman H.R., Cancer Metastasis Rev. 1994, 13, 241). Одна из изоформ, СОХ-1, конститутивно продуцируется в различных тканях и, по-видимому, важна для поддержания нормальных физиологических функций, включая поток крови через почки и цитозащиту желудка. Вторая изоформа, СОХ-2, индуцируется различными воспалительными стимулами и, по-видимому, в значительной степени ответственна за высокий уровень продуцирования простагландинов, что приводит к воспалительным процессам (Masferrer J.L. et al., Proc. Natl. Acad. Sci. USA. 1994, 91, 3228; Vane J. et al. Proc. Natl. Acad. Sci. USA. 1994, 91, 2046).
В заявках WO 95/15318, WO 95/15316, патентах США US 5434178, US 5466823, US 5504215, US 5508426 и US 5510496 раскрыты 1,5-диарилпиразолы, обладающие активностью in vitro и in vivo.
Некоторые 1,5-дифенил-1Н-1,2,4-триазолы, такие как соединение (а), обладающие умеренной Сох-2 ингибирующей активностью и противовоспалительным потенциалом, которые не превосходят свойства известных противовоспалительных агентов, были раскрыты в Monatshefte fur Chemie 119, 349-353 (1998).
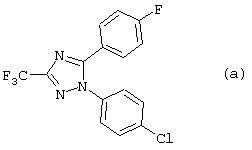
3-циано-1,5-дифенил-1Н-1,2,4-триазолы, такие как соединение (b), о которых сообщалось в Chem. Pharm. Bull. 45(6), 987-995 (1997), являются слабыми и неселективными ингибиторами циклооксигеназы-1 и циклооксигеназы-2.
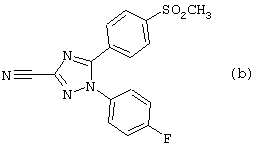
Неожиданно было обнаружено, что некоторые соединения 5-арил-1Н-1,2,4-триазола являются особенно селективными и сильными ингибиторами циклооксигеназы-2.
Соответственно, одной из целей настоящего изобретения является получение соединений 5-арил-1Н-1,2,4-триазола, которые обладали бы эффективной и селективной способностью ингибировать СОХ-2.
Соединения 5-фенил-1Н-1,2,4-триазола настоящего изобретения представлены следующей общей формулой:
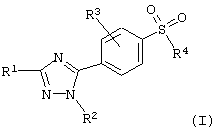
где R1 представляет водород; (C1-C6)алкил; гало (C1-C6) алкил; или фенил, необязательно замещенный одним или несколькими заместителями, выбранными из группы, состоящей из (C1-C4)алкила, галогена, гало(C1-C4)алкила, гидрокси, (C1-C4)алкокси, амино, моно- или ди-(C1-C4)алкиламино, (C1-C4)алкилкарбониламино, (C1-C4)алкилтиокарбониламино, (C1-C4)алкоксикарбониламино, (C1-C4)алкокситиокарбониламино, (C1-C4)алкилсульфонила, (C1-C4)алкилсульфониламино, метилендиокси, нитро и циано;
R2 представляет (C1-C4)алкил; (С3-С8)циклоалкил; фенил или фенил (C1-C4)алкил, где фенил необязательно замещен одним или несколькими заместителями, выбранными из группы, состоящей из (C1-C4)алкила, галогена, гало(C1-C4)алкила, гидрокси, (C1-C4)алкокси, амино, моно- или ди-(C1-C4)алкиламино, (C1-C4)алкилкарбониламино, (C1-C4)алкилтиокарбониламино, (C1-C4)алкоксикарбониламино, (C1-C4)алкокситиокарбониламино, (C1-C4)алкилсульфонила, (C1-C4)алкилсульфониламино, метилендиокси, нитро и циано; или гетероароматический радикал;
R3 представляет водород; галоген; гидрокси; (C1-C6)алкокси; амино; моно- или ди-(C1-С6)алкиламино; (C1-C6)алкилкарбониламино; (C1-C6)алкилтиокарбониламино; (C1-C6)алкоксикарбониламино; (C1-C6)алкокситиокарбониламино; нитро или циано;
R4 представляет (C1-C6)алкил; амино; моно- или ди-(C1-C6)-алкиламино; (C1-C6)алкилкарбониламино; (C1-C6)алкилтиокарбониламино; (C1-C6)алкоксикарбониламино; или (C1-C6)алкокситиокарбониламино; и его фармацевтически приемлемые соли.
Термин "(C1-C4)алкил" или "(C1-C6)алкил" означает линейную или разветвленную углеводородную цепочку, содержащую от 1 до 4 (соответственно 6) атомов углерода, такую как, например, метильный, этильный, пропильный, изопропильный, бутильный, изобутильный, трет-бутильный, пентильный, изопентильный или гексильный радикал.
Термин "(C1-C4)алкокси" или "(C1-C6)алкокси" означает группу OR, в которой R представляет (C1-C4)алкил или (C1-C6)алкил, как указано выше.
Термин "гало (C1-C4) или (C1-C6)алкил" означает (C1-C4) или (C1-C6)алкильный радикал, в котором от 1 до 7 атомов водорода замещены 1-7 атомами галогена, такой как, например, трифторметильный, 2,2,2-трифторэтильный, пентафторэтильный, хлорметильный или бромметильный радикал.
Термин "галоген" означает атом хлора, брома, иода или фтора.
Термин "(C3-C8)циклоалкил" означает насыщенный моноциклический углеводород, содержащий от 3 до 8 атомов углерода, такой как, например, циклопропильный, циклобутильный, циклопентильный, циклогексильный, циклогептильный или циклооктильный радикал.
Термин "гетероароматический радикал" означает 5 или 6-членный моноциклический или 9 или 10-членный бициклический ароматический гетероцикл, содержащий один или два гетероатома, выбранные из N, S и О, такой как, например, пиридинильный, пиридазинильный, пиразинильный, пиримидинильный, пиразолильный, хинолинильный, изохинолинильный, бензимидазолильный, бензоксазолильный, индолильный или индазолильный радикал.
Предпочтительными соединениями формулы (I) являются соединения, в которых:
- R1 представляет водород, (C1-C6)алкил, гало(C1-C6)алкил или фенил;
- R2 представляет (C3-C8)циклоалкил; фенил, необязательно замещенный одним или несколькими заместителями, выбранными из группы, состоящей из галогена, (C1-C4)алкила, (C1-C4)алкокси, гидрокси, нитро, ди-(C1-C4)алкиламино, (C1-C4)алкилсульфониламино, (C1-C4)алкилсульфонила и метилендиокси; фенил(C1-C4)-алкил, где фенил замещен одним или несколькими заместителями, выбранными из группы, состоящей из гидрокси, (C1-C4)алкила и (C1-C4)алкокси; или 5- или 6-членный моноциклический ароматический гетероцикл, содержащий один или два атома азота, серы и/или кислорода;
- R3 представляет водород или галоген;
- R4 представляет (C1-C6)алкил, (C1-C4)алкилкарбониламино или амино. Особенно предпочтительны соединения формулы (I), в которых R1 представляет (C1-C4)алкил или гало (C1-C4)алкил, такой как трифторметил.
Кроме того, особенно предпочтительны соединения формулы (I), в которых R2 представляет фенил, необязательно замещенный одним или несколькими заместителями, выбранными из группы, состоящей из галогена, (C1-C4)алкила, (C1-C4)алкокси, гидрокси, нитро, ди(C1-C4)алкиламино, (C1-C4)алкилсульфониламино, (C1-C4)алкилсульфонила и метилендиокси.
Особенно предпочтительны также соединения формулы (I), в которых R3 представляет водород, и те, в которых R4 представляет (C1-C6)алкил или амино.
Особую ценность представляют следующие соединения:
-1-(4-метоксифенил)-3-метил-5-(4-метилсульфонилфенил)-1Н-1,2,4-триазол;
-1-(4-метоксифенил)-5-(4-метилсульфонилфенил)-3-трифторметил-1H-1,2,4-триазол;
-1-(4-бромфенил)-5-(4-метилсульфонилфенил)-3-трифторметил-1Н-1,2,4-триазол;
-1-(4-метилсульфониламинофенил)-5-(4-метилсульфонилфенил)-3-трифторметил-1H-1,2,4-триазол;
-1-(4-метоксифенил)-5-(4-аминосульфонилфенил)-3-трифторметил-1H-1,2,4-триазол.
Фармацевтически приемлемыми солями соединений формулы (I) являются нетоксичные соли, включая (i) соли, образуемые соединениями формулы (I), содержащими кислотные группы, например, соли щелочных металлов или соли щелочноземельных металлов, такие как соли натрия, соли калия, соли магния и соли кальция, а также соли фармацевтически приемлемых ионов четвертичного аммония или органических аминов, таких как триэтиламин или трис(2-гидроксиэтил)амин и т.п., и (ii) соли, образуемые соединениями формулы (I), содержащими основные группы, например, соли неорганических кислот, таких как хлористоводородная кислота, бромистоводородная кислота, фосфорная кислота, серная кислота, азотная кислота, фосфорная кислота и т.п., или соли органических карбоновых кислот, таких как уксусная кислота, пропионовая кислота, гликолевая кислота, пировиноградная кислота, щавелевая кислота, яблочная кислота, фумаровая кислота, винная кислота, лимонная кислота, бензойная кислота, коричная кислота, миндальная кислота, метансульфоновая кислота и т.п.
Соединениея формулы (I) можно использовать для облегчения боли, для лечения простуды и воспалений при различных состояниях, включая ревматическую лихорадку, симптомы, связанные с гриппом или другими вирусными инфекциями, простуду, боль в области спины и шеи, дисменоррею, головную боль, зубную боль, растяжения и перенапряжения, миозиты, невралгию, синовиты, артриты, включая ревматоидные артриты, дегенеративные заболевания суставов (остеоартриты), подагру и анкилозные спондилиты, тендениты, бурситы, ожоги, повреждения и раны, особенно после хирургических и зубоврачебных процедур. Кроме того, такие соединения могут ингибировать клеточные неопластические превращения и рост опухолевых метастазов, и, следовательно, их можно использовать для лечения наследственного полипоза и рака (рака толстой кишки, легких, пищевода и желудка). Соединения формулы (I) можно также использовать для лечения деменции, включая предстарческую и старческую деменцию, и особенно деменцию, связанную с болезнью Альцгеймера (то есть деменцию Альцгеймера). Соединения формулы (I) также ингибируют сокращения гладкой мускулатуры, вызванные простаноидами, за счет предотвращения синтеза контрактильных простаноидов, и, следовательно, их можно использовать для лечения дисменорреи, при преждевременных родах и при астме.
Благодаря высокой ингибирующей активности в отношении циклооксигеназы-2 (СОХ-2) и/или высокой селективности ингибирования циклооксигеназы-2 по сравнению с ингибированием циклооксигеназы-1, соединения формулы (I) оказываются полезными в качестве альтернативы обычным нестероидным противовоспалительным лекарствам (NSAIDs), особенно в тех случаях, когда такие нестероидные противовоспалительные лекарства могут быть противопоказаны, как например, в случаях, когда у пациентов присутствует пептическая язва, гастрит, местный энтерит, язвенный колит, дивертикулит, или у пациентов с рецидивами поражений желудочно-кишечного тракта; кровотечениями желудочно-кишечного тракта; нарушениями коагуляции, включая анемию, такую как гипопротромбинэмия, гемофилия, или другие проблемы с кровотечениями (включая те, которые связаны ослабленной или нарушенной функцией тромбоцитов); заболеваниями почек (например, нарушенной почечной функцией); с теми пациентами, которым предстоит операция, или теми, кто принимает антикоагулянты; и теми, кто предрасположен к астме, вызываемой NSAID.
Соответственно, следующая цель настоящего изобретения связана с использованием соединений формулы (I) или их фармацевтически приемлемых солей для получения лекарств, которые можно использовать для лечения заболеваний, опосредованных циклооксигеназой, особенно таких заболеваний, которые поддаются лечению с помощью NSAIDs, и которые удобно лечить агентом, который селективно ингибирует СОХ-2 по сравнению с СОХ-1.
Настоящее изобретение относится также к способу лечения вышеуказанных заболеваний, опосредованных циклооксигеназой, включающему введение нуждающемуся в таком лечении субъекту терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли.
Для лечения любого из заболеваний, опосредованных циклооксигеназой, соединения формулы (I) можно вводить, например, перорально, локально, парентерально, путем ингаляций спреями, или ректально в стандартных формах дозировки, содержащих нетоксичные фармацевтически приемлемые носители, адъюванты и средства доставки. Такие лекарственные формы приведены только в качестве примеров, но специалисты-фармацевты могут разработать другие лекарственные формы для введения соединений формулы (I). Термин “парентерально” в том смысле, как он здесь использован, включает подкожные инъекции, внутривенные, внутримышечные инъекции, инъекции в ствол мозга или вливания. Помимо лечения людей соединения формулы (I) можно использовать для лечения теплокровных животных, таких как мыши, крысы, лошади, овцы, собаки, кошки и т.п.
Поэтому следующая цель настоящего изобретения относится к фармацевтическим композициям, включающим терапевтически эффективное количество соединения формулы (I) или его фармацевтически приемлемой соли в качестве активного ингредиента.
Фармацевтические композиции, включающие активный ингредиент, могут быть в форме, пригодной для перорального введения, например, в виде таблеток, лепешек, пастилок, водных или масляных суспензий, диспергируемых порошков или гранул, эмульсий, твердых или мягких желатиновых капсул, сиропов или эликсиров. Композиции, предназначенные для перорального использования, можно получить любым из известных специалистам способов получения фармацевтических композиций, и такие композиции могут включать один или несколько агентов, выбранных из группы, состоящей из подслащивающих агентов, корригентов, красителей и консервантов для придания препаратам элегантного, с фармацевтической точки зрения, вида и приятного вкуса. Таблетки включают активный ингредиент в смеси с нетоксичными фармацевтически приемлемыми эксципиентами, которые пригодны для изготовления таблеток. Эти эксципиенты могут быть, например, инертными разбавителями, такими как карбонат кальция, карбонат натрия, лактоза, фосфат кальция или фосфат натрия; гранулирующими и разрыхляющими агентами, например, кукурузным крахмалом или альгиновой кислотой; связующими агентами, например крахмалом, желатином или аравийской камедью, и смазывающими агентами, например, стеаратом магния, стеариновой кислотой или тальком. Таблетки могут быть без оболочки, или на них известными способами может быть нанесена оболочка для того, чтобы задержать разрушения и абсорбцию в желудочно-кишечном тракте и тем самым обеспечить пролонгированное действие в течение длительного промежутка времени. Например, можно использовать такой продлевающий время действия материал, как моностеарат глицерина или дистеарат глицерина. Покрытие можно также нанести способом, раскрытым в патентах США 4256108, 4166452 и 4265874 для получения осмотических терапевтических таблеток с контролируемым выделением.
Композиции для перорального приема могут быть также в форме твердых желатиновых капсул, где активный ингредиент смешан с инертным твердым разбавителем, например, карбонатом кальция, фосфатом кальция или каолином, или в форме мягких желатиновых капсул, где активный ингредиент смешан с водой или с масляной средой, например арахисовым маслом, жидким парафином или оливковым маслом.
Водные суспензии включают активный ингредиент в смеси с эксципиентами, пригодными для приготовления водных суспензий. Такие эксципиенты являются суспендирующими агентами, например, натрийкарбоксиметилцеллюлозой, метилцеллюлозой, гидроксипропилметилцеллюлозой, альгинатом натрия, поливинилпирролидоном, смолой трагаканта и аравийской камедью; диспергирующими или смачивающими агентами, такими как природные фосфатиды, например лецитин, или продукты конденсации алкиленоксида с жирными кислотами, например полиоксиэтиленстеарат, или продукты конденсации этиленоксида с длинноцепочечными алифатическими спиртами, например гептадекаэтиленоксицетанол, или продукты конденсации этиленоксида с частичными сложными эфирами, полученными из жирных кислот и гекситола, такими как полиэтиленоксисорбитолмоноолеат, или продукты конденсации этиленоксида с частичными сложными эфирами, полученными из жирных кислот и ангидридов гекситола, например полиэтиленоксисорбитанмоноолеат. Водные суспензии могут также включать один или несколько консервантов, например этил- или н-пропил-пара-гидроксибензоат, один или несколько красителей, один или несколько корригентов и один или несколько подслащивающих агентов, таких как сахароза, сахарин или аспартам. Масляные суспензии можно приготовить, суспендируя активный ингредиент в растительном масле, например арахисовом масле, оливковом масле, кунжутном масле или кокосовом масле, или в минеральном масле, таком как жидкий парафин. Масляные суспензии могут включать загуститель, например пчелиный воск, жидкий парафин или цетиловый спирт. Можно добавлять подслащивающие агенты, такие как указанные выше, и корригенты для придания приятного вкуса препаратам для перорального введения. Эти композиции можно законсервировать, добавляя антиоксиданты, такие как аскорбиновая кислота.
Диспергируемые порошки и гранулы, пригодные для приготовления водных суспензий путем добавления воды, содержат активный ингредиент в смеси с диспергирующим или смачивающим агентом, суспендирующим агентом и одним или несколькими консервантами. Примерами подходящих суспендирующих или смачивающих агентов служат уже указанные выше агенты. Могут также присутствовать дополнительные эксципиенты, например подслащивающие агенты, корригенты и красители.
Фармацевтические композиции настоящего изобретения могут также быть в форме эмульсий типа масло-в-воде. Масляной фазой может служить растительное масло, например, оливковое масло, или арахисовое масло, или минеральное масло, например, жидкий парафин, или их смеси. Подходящими эмульгаторами могут быть природные фосфатиды, например, соевые бобы, лецитин и сложные эфиры или частичные эфиры, полученные из жирных кислот и ангидридов гекситола, например сорбитанмоноолеат, и продукты конденсации указанных частичных сложных эфиров и этиленоксида, например полиоксиэтиленсорбитанмоноолеат. Эмульсии могут также включать подсластители и корригенты.
Сиропы и эликсиры могут содержать подслащивающие агенты, например глицерин, пропиленгликоль, сорбит или сахарозу. Такие композиции могут также содержать болеутоляющие средства, консерванты и корригенты и красители.
Фармацевтические композиции могут быть также в форме стерильных водных или масляных суспензий для инъекций. Такие суспензии можно приготовить известными способами, используя такие подходящие диспергирующие или смачивающие агенты и суспендирующие агенты, которые были указаны выше. Стерильные препараты для инъекций могут также быть в форме стерильных растворов или суспензий для инъекций в нетоксичных парентерально приемлемых разбавителях или растворителях, например, в виде раствора в 1,3-бутандиоле. Среди приемлемых носителей и растворителей, которые можно использовать, можно указать воду, раствор Рингера и изотонический раствор хлорида натрия. Кроме того, в качестве растворителя или суспендирующей среды обычно используют стерильные, нелетучие масла. Для этой цели можно использовать любую смесь мягких нелетучих масел, включая синтетические моно- или диглицериды. Кроме того, в препаратах для инъекций находят применение жирные кислоты, такие как олеиновая кислота.
Соединения формулы (I) можно также вводить в форме суппозиториев для ректального введения активного ингредиента. Такие композиции можно приготовить, смешивая активный ингредиент с подходящим не раздражающим эксципиентом, который является твердым при обычных температурах, но становится жидким при ректальных температурах и поэтому плавится в ректуме, выделяя активный ингредиент. Такими материалами являются, например, масло какао и полиэтиленгликоли.
Для местного применения используют кремы, мази, желе, растворы или суспензии и т.п., включающие соединение формулы (I) (для целей рассматриваемой заявки такие формы для местного применения должны включать полоскания для рта и эликсиры).
Для лечения вышеуказанных состояний применимы уровни доз порядка от около 0,01 мг до около 140 мг/кг веса тела в день или в другом варианте от около 0,5 мг до около 1 г для пациента в день. Например, воспаление можно эффективно лечить, вводя от около 0,01 до 50 мг соединения на килограмм веса тела в день или в другом варианте от около 0,5 мг до около 3,5 г для пациента в день, предпочтительно от около 2,5 мг до 1 г для пациента в день.
Количество активного ингредиента, которое можно комбинировать с материалом носителя для получения разовой лекарственной формы, будет меняться в зависимости от подлежащего лечению субъекта и конкретного способа введения. Например, форма, предназначенная для перорального введения человеку, может включать от 0,5 мг до 5 г активного ингредиента, объединенного с подходящим и удобным количеством материала носителя, которое может меняться от около 5 до около 95 вес.% от всей композиции. Стандартная лекарственная форма обычно содержит от около 1 мг до около 1000 мг активного ингредиента, обычно 25, 50, 100, 200, 300, 400, 500, 600, 800 или 1000 мг.
Однако следует учитывать, что конкретный уровень доз для любого конкретного пациента будет зависеть от различных факторов, включая возраст, вес тела, общее состояние здоровья, пол, режим питания, время введения, способ введения, скорость выведения, комбинацию лекарств и степень тяжести конкретного, подлежащего лечению заболевания.
Настоящее изобретение относится далее к способам получения соединений формулы (I). Эти соединения можно получить в соответствии с последовательностями, представленными далее на схемах реакций I, II и III.
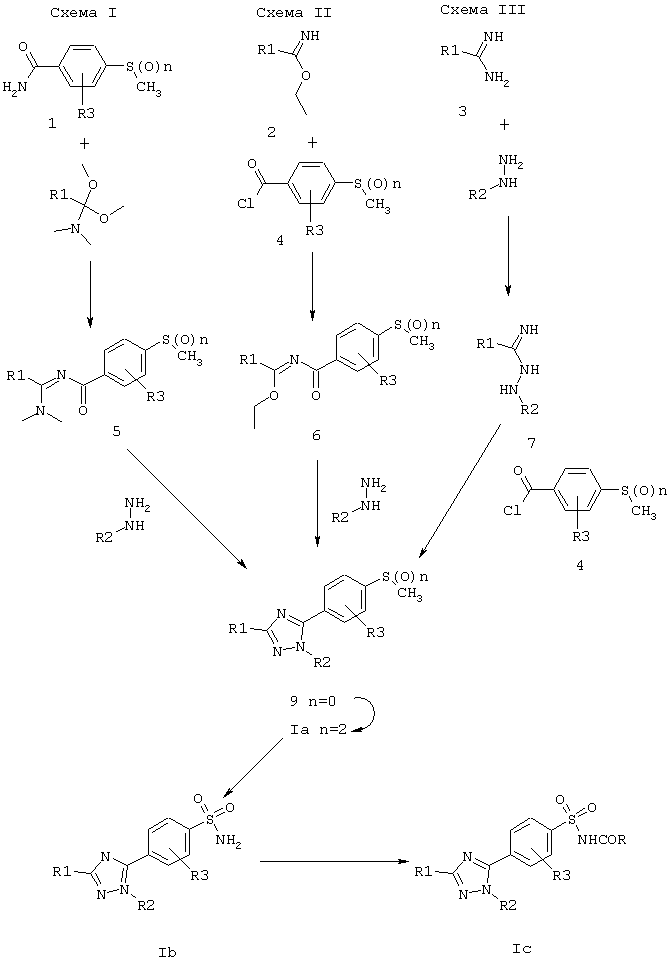
В соответствии со схемой I исходными материалами могут быть производные амидов формулы (1). Их можно получить из соответствующих карбоновых кислот описанными в литературе способами (см. например Org. Synth. col 1,153). Их конденсация с диметилацеталем N,N-диметиламидов, как описано в Synthesis, 119 (1980), приводит к получению N2-ацил-N1,N1-диметиламидинов 5. Конденсация производных 5 с гидразинами в полярном растворителе (например, метаноле, этаноле или т.п.) приводит к получению 1Н-1,2,4-триазольного соединения 9. Если используют гидрохлоридную соль гидразина, добавляют один эквивалент органического основания. Гидразины в основном доступны из коммерческих источников, или их получают из соответствующих аминов известными специалистам способами (Advanced organic chemistry. Jerry March, Wiley, 1985). Затем окисление двумя эквивалентами мета-хлорпербензойной кислоты (МСРВА) в инертном растворителе (например, хлороформе) дает 1Н-1,2,4-триазольные соединения Iа.
В соответствии со схемой II исходными материалами могут быть алкилимидаты 2 или их соли. Реакция алкилимидата с бензоилхлоридом 4 в присутствии органического основания, подобного триэтиламину, приводит к получению N-ацилимидата 6. Такой способ раскрыт в Synthesis, 483 (1983). Реакцию ведут при комнатной температуре в неполярном растворителе, подобном метиленхлориду, хлороформу или толуолу. Циклизация N-ацилимидата 6 с гидразинами для получения IH-1,2,4-триазольного соединения 9 происходит при комнатной температуре без катализатора в неполярном растворителе, подобном метиленхлориду. Если используют гидрохлоридную соль гидразина, добавляют один эквивалент органического основания (подобного триэтиламину). Стадия окисления, подобная представленной на схеме I, приводит к получению 1Н-1,2,4-триазольного соединения 1а.
В соответствии со схемой III исходными материалами могут служить производные амидина 3 или их соли. Они коммерчески доступны, или их можно получить описанными в литературе способами (G.V.BOYD. The chemistry of amidines and imidates, Wiley, vol. 2, chapter 7, 339, 1991). Реакцию гидразинов с производными амидина 3 ведут при комнатной температуре в полярном растворителе (например, в метаноле или этаноле), получая амидразоны 7. Конденсация амидразонов 7 с бензоилхлоридом 4 в присутствии органического основания, подобного пиридину, приводит к получению 1Н-1,2,4-триазольного соединения 9. Эта реакция протекает предпочтительно при температуре кипения с обратным холодильником неполярного растворителя, подобного диоксану. Стадия окисления, подобная представленной не схеме I, приводит к получению 1Н-1,2,4-триазольного соединения Iа.
Обработка арилметилсульфонов Iа основанием и триэтилбораном дает соответствующие перегруппированные сульфоновые кислоты, которые превращают в арилсульфонамиды Ib в процессе окислительного аминирования. Такой способ описан H.Chuang, E.J.Reinhard and D.B.Reitz в Tetrahedron letters, 35 (39), 7201-7204, (1994). Арилметилсульфоны Iа депротонируют небольшим избытком основания, подобного этилмагнийхлориду, при низкой температуре (например, при 0°С) в инертном растворителе, подобном ТГФ, а затем обрабатывают триэтилбораном при температуре кипения с обратным холодильником в течение нескольких часов. В результате обработки гидроксиамин-O-сульфоновой кислотой при комнатной температуре получают арилсульфонамиды Ib.
Сульфонамиды Ib обрабатывают ацетилхлоридом в уксусной кислоте, получая сульфонамиды Iс.
Далее настоящее изобретение будет проиллюстрировано следующими примерами и тестами.
ПРИМЕР 1
1-((3-хлор-4-метил)фенил)-3-метил-5-(4-метилсульфонилфенил)-1Н-1,2,4-триазол
a) Этил N-(4-метилтиобензоил)ацетамидат
К охлаждаемой льдом перемешиваемой суспензии гидрохлорида этилацетамидата (80 г, 0,65 моль) и триэтиламина (175 мл, 1,24 моль) в CH2Cl2 (1000 мл) добавляют по каплям раствор 4-метил-тиобензоилхлорида (110 г, 0,591 моль) (полученный in situ из 4-метилтиобензойной кислоты) в CH2Cl2. Затем реакционную смесь перемешивают в течение ночи при комнатной температуре. Органический слой промывают водой, сушат над сульфатом натрия и упаривают в вакууме. Полученный остаток обрабатывают хроматографически на силикагеле, используя смесь 8/2 гептан/этилацетат в качестве элюента, с получением аморфного твердого вещества (92 г, 65%). Соединение используют на следующей стадии без дополнительной очистки.
1H-ЯМР(ДМСО-d6): 1.30 (т, J=7.2 Гц, 3Н), 1.98 (с, 3Н), 2.5 (с, 3Н), 4.25 (кв, J=7.2 Гц, 2H), 7.35 (дд, J=8.2 Гц, 2Н), 7.85 (д, J=8.2 Гц, 2Н)
b) 1-((3-хлор-4-метил)фенил)-3-метил-5-(4-метилтиофенил)-1Н-1,2,4-триазол
Раствор этил N-(4-метилтиобензоил)ацетамидата (5 г, 21,09 ммоль), (3-хлор-4-метил)фенилгидразина гидрохлорида (4,5 г, 23,20 ммоль) и триэтиламина (3,5 мл, 25,31 ммоль) в CH2Cl2 (25 мл) перемешивают в течение 1,5 час при комнатной температуре. Органический слой промывают водой, сушат над сульфатом натрия и упаривают в вакууме. Полученный остаток обрабатывают хроматографически на силикагеле, используя смесь 8/2 толуол/этилацетат в качестве элюента, и получают масло коричневого цвета (6,4 г), которое кристаллизуют из диизопропилового эфира, получая желто-оранжевый порошок (3,6 г, 52%).
Т. плавления 100°С.
1H-ЯМР (СДСl3): 2.4 (с, 3Н), 2.48 (с, 3Н), 2.50 (с, 3Н), 7.0-7.5 (м, 7Н).
с) 1-((3-хлор-4-метил)фенил)-3-метил-5-(4-метилсульфонилфенил)-1Н-1,2,4-триазол
К раствору 1-((3-хлор-4-метил)фенил)-3-метил-5-(4-метилтиофенил)-1Н-1,2,4-триазола (3,6 г, 10,9 ммоль) в СНСl3 (40 мл) добавляют 2 эквивалента МСРВА (6,3 г, 21,85 ммоль). Реакционную смесь перемешивают в течение 0,5 часа при комнатной температуре, затем добавляют гидросульфит натрия и полученную смесь нейтрализуют NaOH. Органическую фазу выделяют, промывают насыщенным раствором бикарбоната натрия и сушат над сульфатом натрия. В результате упаривания при пониженном давлении получают масло светло-желтого цвета (3,5 г). В результате кристаллизации из этанола получают твердое вещество белого цвета (2,2 г, 56%). Т. плавления 156°С.
1H-ЯМР (СДСl3): 2.45 (с, 3Н), 2.55 (с, 3Н), 3.1 (с, 3Н), 7.0 (дд, 1Н), 7.3 (дд, 1Н), 7.45 (д, 1Н), 7.7 и 7.9 (АВ, 4Н).
Следующие соединения получают, используя способ примера 1, но заменяя (3-хлор-4-метил)фенилгидразингидрохлорид на:
- 4-фторфенилгидразин
- 4-хлорфенилгидразин
- 4-метилфенилгидразин
- фенилгидразин
- 2-хлорфенилгидразин
- 3-хлорфенилгидразин
- 4-трет-бутилфенилгидразин
- 4-бромфенилгидразин
- 4-метоксифенилгидразин
- 2,4-дифторфенилгидразин
- 4-нитрофенилгидразин
- 3,4-дифторфенилгидразин
- 3,4-диметоксифенилгидразин и
- 4-диметиламинофенилгидразин соответственно.
ПРИМЕР 2
1-(4-фторфенил)-3-метил-5-(4-метилсульфонилфенил)-1H-1,2,4-триазол
Т. плавления 180°С
1H-ЯМР(СДСl3): 2.50 (с, 3Н), 3.1 (с, 3Н), 7.05-7.4 (м, 4Н), 7.7 и 7.95 (АВ, 4Н)
МН+=332.
ПРИМЕР 3
1-(4-хлорфенил)-3-метил-5-(4-метилсульфонилфенил)-1H-1,2,4-триазол
Т. плавления 186°С
1H-ЯМР (СДСl3): 2.50 (с, 3Н), 3.1 (с, 3Н), 7.30 и 7.45 (АВ, 4Н), 7.7 и 7.95 (АВ, 4Н).
МН+=348.
ПРИМЕР 4
3-метил-1-(4-метилфенил)-5-(4-метилсульфонилфенил)-1Н-1,2,4-триаэол
Т. плавления 176°С.
1H-ЯМР (СДCl3): 2.4 (с, 3Н), 2.48 (с, 3Н), 3.05 (с, 3Н), 7.2 (м, 4Н), 7.7 и 7.9 (АВ, 4Н).
ПРИМЕР 5
3-метил-5-(4-метилсульфонилфенил)-1-фенил-1Н-1,2,4-триаэол
Т. плавления 146°С.
1H-ЯMP (CДCl3): 2.5 (с, 3Н), 3.05 (с, 3Н), 7.25-7.5 (м, 5Н), 7.7 и 7.9 (АВ, 4Н).
ПРИМЕР 6
1-(2-хлорфенил)-3-метил-5-(4-метилсульфонилфенил)-1H-1,2,4-триазол
Т. плавления 170°С
1H-ЯMP (CДCl3): 2.5 (с, 3Н), 3.05 (с, 3Н), 7.4-7.6 (м, 4Н), 7.7 и 7.9 (АВ, 4Н).
ПРИМЕР 7
1-(3-хлорфенил)-3-метил-5-(4-метилсульфонилфенил)-1Н-1,2,4-триазол
Т. плавления 130°С
1H-ЯМР (СДСl3): 2.5 (с, 3Н), 3.05 (с, 3Н), 7.15 (д, 1Н), 7.25-7.50 (м, 2Н), 7.7 и 7.95 (АВ, 2Н).
ПРИМЕР 8
1-(4-трет-бутилфенил)-3-метил-5-(4-метилсульфонилфенил)-1Н-1,2,4-триазол
Т. плавления 142°С
1H-ЯМР(СДСL3): 1.35 (с, 9Н), 2.55 (с, 3Н), 3.1 (с, 3Н), 7.25 и 7.45 (АВ, 4Н), 7.75 и 7.95 (АВ, 4Н).
ПРИМЕР 9
1-(4-бромфенил)-3-метил-5-(4-метилсульфонилфенил)-1Н-1,2,4-триазол
Т. плавления 188°С
1H-ЯМР(СДСl3): 2.5 (с, 3Н), 3.05 (с, 3Н), 7.20 и 7.60 (АВ, 4Н), 7.70 и 7.95 (АВ, 4Н).
ПРИМЕР 10
1-(4-метоксифенил)-3-метил-5-(4-метилсульфонилфенил)-1Н-1,2,4-триазол
Т. плавления 128°С
1H-ЯМР(СДCl3): 2.5 (с, 3Н), 3.05 (с, 3Н), 3.85 (с, 3Н), 6.95 и 7.25 (АВ, 4Н),7.7 и 7.9 (АВ, 4Н).
ПРИМЕР 11
1-(2,4-дифторфенил)-3-метил-5-(4-метилсульфонилфенил)-1Н-1,2,4-триазол
Т. плавления 160°С
1H-ЯMP(CДCl3): 2.5 (с, 3Н), 3.05 (с, 3Н), 6.9-7.15 (м, 2Н), 7.45-7.60 (м, 1Н), 7.7 и 7.9 (АВ, 4Н).
ПРИМЕР 12
3-метил-5-(4-метилсульфонилфенил)-1-(4-нитрофенил)-1Н-1,2,4-триазол
Т. плавления 180°С
1H-ЯМР (СДСl3): 2.55 (с, 3Н), 3.1 (с, 3Н), 7.55 (д, 2Н), 7.7 (д, 2Н), 7.95 (д, 2Н), 8.30 (д, 2Н).
ПРИМЕР 13
1-(3,4-дифторфенил)-3-метил-5-(4-метилсульфонилфенил)-1Н-1,2,4-триазол
Т. плавления 194°С
1H-ЯМР(СДСl3): 2.5 (с, 3Н), 3.1 (с, 3Н), 7.0-7.35 (м, 3Н), 7.7 и 7.95 (АВ, 4Н).
ПРИМЕР 14
1-(3,4-диметоксифенил)-3-метил-5-(4-метилсульфонилфенил)-1Н-1,2,4-триазол
Т. плавления 186°С
1H-ЯМР (ДМСО d6): 2.40 (с, 3Н), 3.25 (с, 3Н), 3.7 (с, 3Н), 3.8 (с, 3Н), 6.85 (дд, 1Н), 7 (д, 1Н), 7.1 (д, 1Н), 7.7 и 7.95 (АВ, 4Н).
ПРИМЕР 15
1-(4-диметиламинофенил)-3-метил-5-(4-метилсульфонилфенил)-1Н-1,2,4-триазол
Т. плавления 200°С
1H-ЯMP(CДCl3): 2.45 (с, 3Н), 2.50 (с, 3Н), 3.0 (с, 6Н), 6.65 (д, 2Н), 7.15 (2д, 4Н), 7.45 (д, 2Н).
ПРИМЕР 16
1-(4-хлорфенил)-5-(4-метилсульфонилфенил)-3-фенил-1H-1,2,4-триазол
а) Метил N-(4-метилтиобензоил)бензамидат
К охлаждаемой льдом перемешиваемой суспензии метилбензамидата гидрохлорида (5,8 г, 33,8 ммоль) и триэтиламина (9 мл, 62,4 ммоль) в CH2Cl2 (60 мл) по каплям добавляют раствор 4-метилтиобензоилхлорида (5,8 г, 30,7 ммоль) (полученный in situ из 4-метилтиобензойной кислоты) в CH2Cl2 (5,8 мл). Затем реакционную смесь перемешивают при комнатной температуре. Органический слой промывают водой, сушат над сульфатом натрия и упаривают в вакууме. Полученный остаток обрабатывают хроматографически на силикагеле, используя толуол в качестве элюента, и получают аморфное твердое вещество (500 мг, 5%). Соединение используют на следующей стадии без дополнительной очистки.
1H-ЯМР(ДМСО d6): 2.5 (с, 3Н), 4 (с, 3Н), 7.3-7.6 (м, 7Н), 7.9 (д, 2Н).
b) 1-(4-хлорфенил)-5-(4-метилтиофенил)-3-фенил-1Н-1,2,4-триазол
Раствор метил N-(4-метилтиобензоил)бензамидата (500 мг, 1,75 ммоль), гидрохлорида (4-хлор)фенилгидразина (345 мг, 1,92 ммоль) и триэтиламина (0,3 мл, 2,1 ммоль) в CH2Cl2 (2,5 мл) перемешивают в течение 1,5 часов при комнатной температуре. Органический слой разбавляют дихлорметаном, промывают водой, сушат над сульфатом натрия и упаривают в вакууме. Полученное твердое вещество желтого цвета тщательно растирают с толуолом, получая твердое вещество белого цвета (130 мг, 20%).
1Н-ЯМР(СДCl3): 2.5 (с, 3Н), 7.15-7.65 (м, 11Н), 8.25 (дд, 2Н).
c) 1-(4-хлорфенил)-5-(4-метилсульфонилфенил)-3-фенил-1Н-1,2,4-триазол
К раствору 1-(4-хлорфенил)-5-(4-метилтиофенил)-3-фенил-1Н-1,2,4-триазола (130 мг, 0,34 ммоль) в СНСl3 (5 мл) добавляют 2 эквивалента МСРВА (200 мг, 6,88 ммоль). Реакционную смесь перемешивают в течение двух дней при комнатной температуре, затем добавляют гидросульфит натрия и полученную смесь нейтрализуют концентрированной NaOH. После экстрагирования хлороформом органическую фазу промывают водой и сушат над сульфатом натрия. В результате упаривания при пониженном давлении и кристаллизации из этанола получают твердое вещество белого цвета (50 мг, 36%).
Т. плавления 170°С
1Н-ЯМР(СДCl3): 3.1 (с, 3Н), 7.3-7.55 (м, 7Н), 7.8 и 8.0 (АВ, 4Н), 8.15-8.30 (м, 2Н).
ПРИМЕР 17
1-(4-метоксифенил)-5-(4-метилсульфонилфенил)-3-трифторметил-1Н-1,2,4-триазол
a) N-(4-метоксифенил)-трифторацетамидразон
Смесь гидрохлорида 4-метоксифенилгидразина (27,84 г, 159,4 ммоль), трифторацетамидина (25 г, 223,2 ммоль), триэтиламина (22,12 мл, 159,4 ммоль) и метанола (100 мл) перемешивают в атмосфере азота при комнатной температуре в течение 6 часов. Реакционную смесь разбавляют водой (100 мл), экстрагируют этилацетатом (3×100 мл) и объединенные органические слои промывают водой, насыщенным рассолом, и сушат над Na2SO4. В результате флеш-хроматографии на силикагеле (CH2Cl2 в качестве элюента) получают масло коричневого цвета (35 г, 94%), которое используют на следующей стадии без дополнительной очистки.
1H-ЯМР(СДCl3): 3.75 (с, 3Н), 4.35 (шс, 2Н), 6.1 (шс, 1Н), 6.7 и 7.0 (АВ, 4Н).
b) 1-(4-метоксифенил)-5-(4-метилтиофенил)-3-трифторметил-1H-1,2,4-триазол
К раствору N-(4-метоксифенил)трифторацетамидразона (35 г, 0,15 моль) и пиридина (11,6 мл) в диоксане (360 мл) добавляют раствор 4-метилсульфонилбензоилхлорида (26,6 г, 0,142 моль) (полученный in situ из 4-метилтиобензойной кислоты) в диоксане (120 мл). Затем реакционную смесь нагревают при кипении с обратным холодильником в течение ночи. После выпаривания диоксана остаток помещают в дихлорметан, органический слой промывают водой, 0,1 н. НСl, насыщенным рассолом, сушат над сульфатом натрия и упаривают в вакууме. Полученный остаток обрабатывают хроматографически на силикагеле (СН2Сl2 в качестве элюента), получая бесцветное масло (30,9 г, 65%).
1H-ЯМР(ДМСО d6): 2.5 (с, 3Н), 3.35 (с, 3Н), 7.1 и 7.5 (АВ, 4Н), 7.3 и 7.4 (АВ, 4Н).
с) 1-(4-метоксифенил)-5-(4-метилсульфонилфенил)-3-трифторметил-1H-1,2,4-триазол
К раствору 1-(4-метоксифенил)-5-(4-метилтиофенил)-3-трифторметил-1H-1,2,4-триазола (30 г, 0,08 моль) в СН2Сl2 (320 мл) порциями добавляют МСРВА (47,2 г, 0,16 моль). Реакционную смесь перемешивают при комнатной температуре в течение 1,5 часа, затем охлаждают до 0-5°С и осторожно добавляют раствор гидросульфита натрия (500 мл) при поддержании температуры ниже 18-20°С. рН доводят до 8, добавляя 30% NaOH. Эту смесь экстрагируют дихлорметаном, органическую фазу промывают насыщенным рассолом, сушат над сульфатом натрия и упаривают. В результате обработки с помощью флеш-хроматографии на силикагеле (толуол/этилацетат: 8/2 в качестве элюента) и перекристаллизации из этанола получают твердое вещество белого цвета (29,42 г, 90%).
Т. плавления 156°С.
1H-ЯМР(ДМСО d6): 3.29 (с, 3Н), 3.83 (с, 3Н), 7.1 и 7.5 (АВ, 4Н), 7.75 и 8.0 (АВ, 4Н).
МН+=398.
ПРИМЕР 18
1-(4-бромфенил)-5-(4-метилсульфонилфенил)-3-трифторметил-1H-1,2,4-триазол
a) N-(4-бромфенил)трифторацетамидразон
Смесь 4-бромфенилгидразингидрохлорида (7,1 г, 31,8 ммоль), трифторацетамидина (5 г, 44,6 ммоль), триэтиламина (4,5 мл, 31,8 ммоль) и метанола (20 мл) перемешивают в течение ночи при комнатной температуре. Реакционную смесь разбавляют водой, экстрагируют этилацетатом, органический слой промывают водой, насыщенным рассолом, сушат над Na2SO4. В результате флеш-хроматографии на силикагеле (CH2Cl2 в качестве элюента) получают масло оранжевого цвета (6,7 г, 53%), которое используют на следующей стадии без дополнительной очистки.
1H-ЯМР(СДСl3): 4.45 (шс, 2Н), 6.25 (шс, 1Н), 6.9 и 7.35-7.55 (АВ, 4Н).
b) 1-(4-бромфенил)-5-(4-метилсульфонилфенил)-3-трифторметил-1Н-1,2,4-триазол
К раствору N-(4-бромфенил)трифторацетамидразона (6,7 г, 23,7 ммоль) и пиридина (2,1 мл, 26,1 ммоль) в диоксане (40 мл) добавляют раствор 4-метилсульфонилбензоилхлорида (5,96 г, 27,3 ммоль) (полученный in situ из 4-метилсульфонилбензойной кислоты) в диоксане (40 мл). Затем реакционную смесь нагревают при кипении с обратным холодильником в течение 5 часов. После выпаривания диоксана остаток помещают в дихлорметан, органический слой промывают водой, 0,1 н. НСl, насыщенным рассолом, сушат над сульфатом натрия и упаривают в вакууме. Полученный остаток обрабатывают хроматографически на силикагеле, используя смесь 95/5 толуол/диоксан в качестве элюента, затем перекристаллизовывают из этанола, получая твердое вещество белого цвета (2,8 г, 26%).
Т. плавления 198°С
1H-ЯМР(ДМСО d6): 3.29 (с, 3Н), 7.55 (д, 2Н), 7.76 (д, 2Н), 7.8 (дд, 2Н), 8.02 (дд, 2Н).
МН+=446
Следующие соединения получают, используя способ примера 18, но заменяя 4-бромфенилгидразингидрохлорид на:
- 4-нитрофенилгидразин
- 4-фторфенилгидразин
- 4-хлорфенилгидразин
- циклогексилгидразин (полученный по способу NI. Ghali, J.Org.Chem.,1981, 46, 5413) и
- 4-метоксифенилметилгидразин соответственно.
ПРИМЕР 19
1-(4-нитрофенил)-5-(4-метилсульфонилфенил)-3-трифторметил-1H-1,2,4-триазол
1H-ЯМР(ДМСО d6): 3.30 (с, 3Н), 7.85 (т, 4Н), 8.05 (д, 2Н), 8.45 (д, 2Н).
ПРИМЕР 20
1-(4-фторфенил)-5-(4-метилсульфонилфенил)-3-трифторметил-1H-1,2,4-триазол
Т. плавления 230-232°С
1H-ЯМР(ДМСО d6): 3.27 (с, 3Н), 7.4 (т, 2Н), 7.6-7.8 (м, 2Н), 7.8 и 8.0 (АВ, 4Н).
ПРИМЕР 21
1-(4-хлорфенил)-5-(4-метилсульфонилфенил)-3-трифторметил-1H-1,2,4-триазол
Т. плавления 190-192°С
1H-ЯМР(ДМСО d6): 3.3 (с, 3Н), 7.2 (м, 4Н), 7.8 и 8.05 (АВ, 4Н).
ПРИМЕР 22
1-(циклогексил)-5-(4-метилсульфонилфенил)-3-трифторметил-1H-1,2,4-триазол
Т. плавления 136°С
1H-ЯМР(ДМСО d6): 1.15-2.5 (м, 10Н), 3.3 (с, 3Н), 4.3-4.4 (м, 1Н), 8 и 8.15 (АВ, 4Н).
ПРИМЕР 23
1-(4-метоксифенилметил)-5-(4-метилсульфонилфенил)-3-трифторметил-1H-1,2,4-тиазол
Т. плавления 142°С
1H-ЯМР(ДСМО d6): 3.3 (с, 3Н), 3.7 (с, 3Н), 5.5 (с, 2Н), 6,9 и 7.1 (AВ, 4Н), 8 и 8.1 (ДВ, 4Н).
Нижеследующие соединения получают, используя способ примера 18, но заменяя 4-метилсульфонилбензоилхлорид на 2-хлор-4-метилсульфонилбензоилхлорид и 4-бромфенилгидразин на 4-метоксифенилгидразин.
ПРИМЕР 24
5-(2-хлор-4-метилсульфонилфенил)-1-(4-метоксифенил)-3-трифторметил-1H-1,2,4-триазол
Т. плавления 148°С
1Н-ЯМР(ДМСО d6): 3.35 (с, 3Н), 3.80 (с, 3Н), 7 и 7.4 (АВ, 4Н), 8-8.2 (м, 3Н).
ПРИМЕР 25
1-(4-метилсульфониламинофенил)-5-(4-метилсульфонилфенил)-3-трифторметил-1H-1,2,4-триазол
а) 1-(4-аминофенил)-5-(4-метилсульфонилфенил)-3-трифторметил-1Н-1,2,4-триазол
Смесь 1-(4-нитрофенил)-5-(4-метилсульфонилфенил)-3-трифторметил-1Н-1,2,4-триазола (1,2 г, 2,91 ммоль), порошка железа (0,8 г, 14,27 ммоль), хлорида аммония (0,80 г, 1,45 ммоль), этанола (25 мл) и воды (13 мл) нагревают при кипении с обратным холодильником в течение 1 часа, затем охлаждают и фильтруют. Полученный фильтрат выливают в воду, экстрагируют этилацетатом и раствором дихлорметан/метанол. Органические экстракты промывают насыщенным рассолом, сушат над Na2SO4 и упаривают, получая порошок желтого цвета (1 г, 91%), который используют на следующей стадии без дополнительной очистки.
1H-ЯМР(ДМСО d6): 3.30 (с, 3Н), 5.7 (шс, 2Н), 6,65 и 7.18 (АВ, 4Н), 7.75 и 8 (АВ, 4Н).
b) 1-(4-метилсульфониламинофенил)-5-(4-метилсульфонилфенил)-3-трифторметил-1H-1,2,4-триазол
К охлаждаемой льдом перемешиваемой суспензии 1-(4-аминофенил)-5-(4-метилсульфонилфенил)-3-трифторметил-1H-1,2,4-триазола (1 г, 2,61 ммоль) и триэтиламина (0,4 мл, 2,87 ммоль) в CH2Cl2 (20 мл) по каплям добавляют метансульфонилхлорид (0,2 мл, 2,87 ммоль). Затем реакционную смесь перемешивают при комнатной температуре в течение 2 часов. Результаты, полученные с помощью ТСХ, свидетельствуют о присутствии исходного материала. Затем добавляют 0,4 мл метансульфонилхлорида и 10 мг диметиламинопиридина (DМАР) и реакционную смесь нагревают при кипении с обратным холодильником в течение 2 часов. Снова добавляют 0,4 мл метансульфонилхлорида и 10 мг DMAP и смесь перемешивают в течение ночи при комнатной температуре. Реакционную смесь разбавляют водой, экстрагируют CH2Cl2, объединенные органические слои промывают водой, сушат над Na2SO4 и упаривают, получая порошок светло-желтого цвета (1,1 г). Сырой продукт тщательно растирают со смесью дихлорметан/изопропиловый эфир, получая твердое вещество бежевого цвета (0,65 г). Затем раствор этого твердого вещества в 60 мл МеОН/ТГФ (2/1) и 1 н. NaOH (3,6 мл) перемешивают при комнатной температуре в течение 0,25 часа. После выпаривания растворителей, добавления этилацетата и нейтрализации с помощью 1 н. НСl органический слой промывают водой, сушат над сульфатом натрия и упаривают. В результате кристаллизации из пентана и перекристаллизации из смеси изопропиловый эфир/этанол получают твердое вещество светло-розового цвета (0,3 г, 25%).
Т. плавления 188°С
1H-ЯМР(ДМСО d6): 3.15 (с, 3Н), 3.30 (с, 3Н), 7.35 и 7.55 (АВ, 4Н), 7.75 и 8.05 (АВ, 4Н).
МН+=461
ПРИМЕР 26
1,5-ди-(4-метилсульфонилфенил)-3-трифторметил-1H-1,2,4-триазол
a) N-(4-метилсульфонилфенил)трифторацетамидразон
Смесь 4-(метилсульфонил)фенилгидразингидрохлорида (10,1 г, 44,6 ммоль), триэтиламина (6,2 мл, 44,6 ммоль), трифторацетамидина (2,5 г, 22,3 ммоль), ТГФ (40 мл) и метанола (40 мл) перемешивают в течение ночи при комнатной температуре. Реакционную смесь разбавляют водой, экстрагируют этилацетатом, органический слой промывают водой, насыщенным рассолом и сушат над Na2SO4. В результате флеш-хроматографии на силикагеле (циклогексан/этилацетат: 8/2 в качестве элюента) и тщательного растирания с изопропиловым эфиром получают твердое вещество (2,9 г, 46%).
Т. плавления 164°С.
1H-ЯМР (ДМСО d6): 3.1 (с, 3Н), 6.7 (шс, 2Н), 7.05 и 7.7 (АВ, 4Н), 9.25 (с, 1Н).
b) 1,5-ди-(4-метилсульфонилфенил)-3-трифторметил-1H-1,2,4-триазол
К раствору N-(4-метилсульфонилфенил)трифторацетамидразона (4,3 г, 15,28 ммоль) и пиридина (1,4 мл, 16,8 ммоль) в диоксане (30 мл) добавляют раствор 4-метилсульфонилбензоилхлорида (4,3 г, 19,5 ммоль) (полученный in situ из 4-метилсульфонилбензойной кислоты) в диоксане (10 мл). Затем реакционную смесь нагревают при кипении с обратным холодильником в течение 6 часов, затем перемешивают в течение ночи при комнатной температуре. Реакционную смесь фильтруют, концентрируют досуха, разделяют между метиленхлоридом и водой, остаток экстрагируют метиленхлоридом, органический слой промывают 0,1 н. НСl, насыщенным рассолом, сушат над сульфатом натрия и упаривают в вакууме. Полученный остаток обрабатывают хроматографически на силикагеле, используя смесь 8/2 толуол/диоксан в качестве элюента, затем перекристаллизовывают из этанола, получая твердое вещество белого цвета (1,1 г, 26%).
Т. плавления 214°С.
1H-ЯМР (ДМCO d6): 3.29 (с, 3Н), 3.32 (с, 3Н), 7.8 (д, 2Н), 7.9 (д, 2Н), 8.03 (дд, 2Н), 8.12 (дд, 2Н).
МН+=446.
Следующие соединения получают, используя способ примера 26, но заменяя 4-метилсульфонилфенилгидразин на:
- 3,4-диметоксифенилгидразин и
- 3,4-метилендиоксифенилгидразин соответственно.
ПРИМЕР 27
1-(3,4-диметоксифенил)-5-(4-метилсульфонилфенил)-3-трифтор-метил-1H-1,2,4-триазол
Т. плавления 140°С
1H-ЯMP(ДМCO d6): 3.25 (с, 3Н), 3.7 (с, 3Н), 3.8 (с, 3Н), 7.1 (с, 2Н), 7.28 (с, 1Н), 7.8 и 8 (АВ, 4Н).
ПРИМЕР 28
1-(3,4-метилендиоксифенил)-5-(4-метилсульфонилфенил)-3-трифторметил-1H-1,2,4-триазол
Т. плавления 185°С
1H-ЯМР(ДМСО d6): 3.30 (с, 3Н), 6.2 (с, 2Н), 7.1 (с, 2Н), 7.28 (с, 1Н), 7.8 и 8.05 (АВ, 4Н).
ПРИМЕР 29
1-(4-гидроксифенил)-5-(4-метилсульфонилфенил)-3-трифторметил-1Н-1,2,4-триазол
Смесь 1-(4-метоксифенил)-5-(4-метилсульфонилфенил)-3-трифторметил-1H-1,2,4-триазола (10 г, 25,2 ммоль), 48% водного HBr (70 мл) и уксусной кислоты (70 мл) нагревают при 120°С в течение 5,5 часа. Затем добавляют 48% HBr (20 мл) и АсОН (20 мл) и смесь снова нагревают при 120°С в течение 2 часов. После охлаждения раствор выливают в воду (2 л), осадок отфильтровывают, несколько раз промывают водой и сушат. В результате перекристаллизации из этанола получают твердое вещество белого цвета (7,5 г, 78%).
Т. плавления 246°С
1H-ЯМР(ДМСО d6): 3.25 (с, 3Н), 6.9 и 7.35 (АВ, 4Н), 7.75 и 8 (АВ, 4Н), 10.2 (шс, 1Н).
ПРИМЕР 30
1-(4-этоксифенил)-5-(4-метилсульфонилфенил)-3-трифторметил-1Н-1,2,4-триазол
Смесь 1-(4-гидроксифенил)-5-(4-метилсульфонилфенил)-3-трифторметил-1Н-1,2,4-триазола (4 г, 10,4 ммоль), КОН (1,5 г, 26,8 ммоль) и ДМФА (40 мл) перемешивают при комнатной температуре в течение 1 часа. Затем добавляют диэтилсульфат (1,6 мл, 12,2 ммоль), реакционную смесь перемешивают при комнатной температуре в течение 1,5 часа, добавляют NH4OH (20 мл) и смесь выливают в воду (1 л). Осадок отфильтровывают, несколько раз промывают водой и сушат. В результате перекристаллизации из этанола получают твердое вещество белого цвета (3,7 г, 88%).
Т. плавления 112°С.
1H-ЯМР(ДМСО d6): 1.35 (т, 3Н), 3.3 (с, 3Н), 4.10 (кв, 2Н), 7.1 и 7.5 (АВ, 4Н), 7.75 и 8 (АВ, 4Н).
ПРИМЕР 31
1-(2-пиридинил)-5-(4-метилсульфонилфенил)-3-трифторметил-1H-1,2,4-триазол гидрохлорид
a) N-(2-пиридинил)трифторацетамидразон
Смесь 2-гидразинопиридина (5 г, 45,8 ммоль), трифторацетамидина (3,4 г, 30,5 ммоль) и метанола (50 мл) перемешивают в течение ночи при комнатной температуре. Реакционную смесь упаривают досуха. В результате флеш-хроматографии на силикагеле (толуол/этилацетат: 65/35 в качестве элюента) получают аморфное твердое вещество светло-оранжевого цвета (3,1 г, 50%), которое используют на следующей стадии без дополнительной очистки.
1H-ЯМР(ДМСО d6): 6.65 (шс, 3Н), 7 (д, 1Н), 7.1 (т, 1Н), 8.05 (д,1Н), 9.2 (с, 1Н).
b) 1-(2-пиридинил)-5-(4-метилсульфонилфенил)-3-трифторметил-1Н-1,2,4-триазолгидрохлорид
К раствору N-(2-пиридинил)трифторацетамидразона (3,1 г, 15,1 ммоль) в диоксане (15 мл) добавляют раствор 4-метил-сульфонилбензоилхлорида (3,6 г, 16,7 ммоль) (полученный in situ из 4-метилсульфонилбензойной кислоты) в диоксане (15 мл). Затем реакционную смесь кипятят с обратным холодильником в течение 2 часов. После охлаждения реакционную смесь фильтруют и концентрируют досуха. Остаток обрабатывают хроматографически на силикагеле, используя смесь 85/15 толуол/диоксан в качестве элюента, затем перекристаллизовывают из этанола, получая твердое вещество белого цвета (0,94 г, 15%).
Т. плавления 144°С
1H-ЯМР (ДМСО d6): 3.27 (с, 3Н), 7.6 (т, 1Н), 7.8 и 8 (АВ, 4Н), 7.9 (д, 1Н), 8.16 (т, 1Н), 8.46 (д, 1Н).
Следующие соединения получают, используя способ примера 31, но заменяя 2-гидразинопиридин на:
- 3-гидразинопиридин (полученный по способу WO 97/10243) и
- 3-фтор-4-метоксифенилгидразин соответственно.
ПРИМЕР 32
1-(3-пиридинил)-5-(4-метилсульфонилфенил)-3-трифторметил-1H-1,2,4-триазол гидрохлорид
Т. плавления 180°С
1H-ЯМР (ДМСО d6): 3.27 (с, 3Н), 7.6 (м, 1Н), 7.77 (д, 2Н) 7.99-8.09 (м, 3Н), 8.77 (с, 2Н).
ПРИМЕР 33
1-(3-фтор-4-метоксифенил)-5-(4-метилсульфонилфенил)-3-трифторметил-1H-1,2,4-триазол
Т. плавления 180°С
1H-ЯМР (ДМCO d6): 3.30 (с, 3Н), 3. 95 (с, 3Н), 7.35-7.5 (м, 2Н), 7.65 (дд, 1Н), 7.8 и 8.05 (АВ,4Н).
ПРИМЕР 34
1-(4-метоксифенил)-5-(4-аминосульфонилфенил)-3-трифторметил-1Н-1,2,4-триазол
К охлаждаемому льдом раствору 1-(4-метоксифенил)-5-(4-метилсульфонилфенил)-3-трифторметил-1H-1,2,4-триазола (10 г, 25/19 ммоль) в ТГФ (100 мл) по каплям добавляют 2 М раствор н-бутилмагнийхлорида в ТГФ (21 мл, 42 ммоль). Затем реакционную смесь перемешивают при комнатной температуре в течение 3 часов. Реакционную смесь охлаждают до 0°С, по каплям добавляют 1 М раствор триэтилборана в ТГФ (70 мл, 70 ммоль) и реакционную смесь кипятят с обратным холодильником в течение 18 часов. После охлаждения по каплям добавляют раствор гидроксиламин-O-сульфоновой кислоты (12 г, 106 ммоль) и ацетата натрия (17,38 г, 210 ммоль) в H2O (140 мл), поддерживая при этом температуру ниже 15°С. Затем реакционную смесь перемешивают при комнатной температуре в течение 2 часов, остаток экстрагируют этилацетатом (2×100 мл), органический слой промывают насыщенным рассолом, сушат над сульфатом натрия и выпаривают в вакууме. Полученный остаток обрабатывают хроматографически на силикагеле, используя в качестве элюента смесь метиленхлорид/метанол 99/1, затем 98/2, затем перекристаллизовывают из этанола, получая твердое вещество бежевого цвета (2,5 г, 25%).
Т. плавления 228°С.
1H-ЯМР (ДМСО d6): 3.9 (с, 3Н), 7.15 (д, 2Н), 7.5-7.6 (м, 4Н), 7.75 и 7.95 (АВ, 4Н).
ПРИМЕР 35
1-(4-метоксифенил)-5-[(4-(ацетиламино)сульфонил)фенил]-3-трифторметил-1Н-1,2,4-триазол
К суспензии 1-(4-метоксифенил)-5-(4-(аминосульфонил)фенил)-3-трифторметил-1H-1,2,4-триазола (2 г, 5 ммоль) в уксусной кислоте (10 мл) по каплям добавляют ацетилхлорид (10 мл). Затем реакционную смесь нагревают при 80°С в течение 5 часов, концентрируют досуха и остаток обрабатывают хроматографически на силикагеле, используя смесь 98/2 метиленхлорид/метанол в качестве элюента, затем перекристаллизовывают из смеси пентан/метанол, получая твердое вещество белого цвета (1,6 г, 72%).
Т. плавления 85°С
1H-ЯМР (СДСl3): 2.05 (с, 3Н), 3.9 (с, 3Н),7 и 7.3 (АВ, 4Н), 7.75 и 8.05 (АВ, 4Н).
ПРИМЕР 36
1-(4-метоксифенил)-5-(4-метилсульфонилфенил)-1Н-1,2,4-триазол
a) N-(диметиламинометилен)-4-(метилсульфонил)бензамид
Суспензию 4-метилсульфонилбензамида (8 г, 40,2 ммоль) в диметилформамид-диметилацетале (16 мл, 120 ммоль) перемешивают при 120°С в течение 1,75 часа, причем в это время образующийся метанол собирают с помощью обратного холодильника. После охлаждения твердое вещество оранжевого цвета отфильтровывают и сушат (8,92 г, 87%).
Т. плавления 130°С
1H-ЯМР (ДМСО d6): 3.16 (с, 3Н), 3.22 (с, 3Н), 3.27 (с, 3Н), 8 и 8.35 (АВ, 4Н), 8.67 (с, 1Н).
b) 1-(4-метоксифенил)-5-(4-метилсульфонилфенил)-1H-1,2,4-триазол
Смесь N-(диметиламинометилен)-4-(метилсульфонил)бензамида (4 г, 15,7 ммоль), 4-метоксифенилгидразингидрохлорида (2,75 г, 15,7 ммоль), триэтиламина (2,2 мл, 15,7 ммоль) в этаноле (20 мл) нагревают при кипении с обратным холодильником в течение 2,5 часа. После охлаждения реакционную смесь концентрируют досуха, затем разбавляют этилацетатом. Органическую фазу промывают водой и насыщенным рассолом и сушат над сульфатом натрия. Остаток обрабатывают хроматографически на силикагеле, используя смесь 70/30 толуол/диоксан в качестве элюента, затем перекристаллизовывают из этанола, получая твердое вещество светло-оранжевого цвета (0,4 г, 15%).
Т. плавления 182°С
1H-ЯМР (ДМСО d6): 3.26 (с, 3Н), 3.82 (с, 3Н), 7.05 и 7.35 (АВ, 4Н), 7.7 и 8 (АВ,4Н), 8.3 (с, 1Н).
Результаты биологических тестов
Соединения примеров настоящего изобретения тестируют в отношении их способности ингибировать СОХ-1 и/или СОХ-2 активности in vitro. Очищенные СОХ-1 из семенных пузырьков барана и очищенные СОХ-2 из плаценты овец (и то, и другое фирмы Cayman Chemicals) инкубируют в течение 10 минут при 25°С в присутствии их субстрата - арахидоновой кислоты (5 мкМ), с тестовым соединением или без тестового соединения, или в присутствии либо в отсутствие стандартных ингибиторов. Продукт реакции - простагландин Е2 определяют с помощью ферментного иммуноанализа (R&D Systems). Каждая величина является результатом двух определений. Конечная величина ингибирования является средним±стандартная ошибка из, по крайней мере, 3 независимых экспериментов, проведенных в различные дни.
В данной тестовой системе диклофенак, являющейся стандартным неселективным ингибитором как СОХ-1, так и СОХ-2, воспроизводимо демонстрирует присущее ему зависимое от дозы ингибирование активности как СОХ-1, так и СОХ-2 с ИK50, равным 0/54±0,13 мкМ (17) и 0,97±0,14 мкМ (18) соответственно. Стандартный селективный ингибитор СОХ-2 нимесулид тестируют в качестве сравнительного соединения в интервале концентраций от 0,1 до 10 мкМ. При промежуточной концентрации в 1 мкМ, использованной для сравнения, соединения примеров 3, 10, 15, 17, 18, 25 и 34 не продемонстрировали какого-либо существенного ингибирования СОХ-1, но оказались способны селективно ингибировать СОХ-2 (таблица 1).
Соединения примеров 10 и 25 обладают аналогичной нимесулиду эффективностью в той же самой концентрации, а соединения примеров 17, 18 и 34 оказываются даже более эффективными. Соединение примера 18, одно из наиболее эффективных соединений в серии, демонстрирует эффективность, примерно в 10 раз превышающую эффективность нимесулида, так как оно вызывает такое же ингибирование (-53%), что и нимесулид (-50%), но при концентрации, в 10 раз более низкой: 1 мкМ против 10 мкМ соответственно.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПЕСТИЦИДНЫЕ КОМПОЗИЦИИ И СВЯЗАННЫЕ С НИМИ СПОСОБЫ | 2013 |
|
RU2654327C2 |
| ПРОИЗВОДНЫЕ СЛОЖНОГО АМИНОЭФИРА АЛКАЛОИДА И ИХ ЛЕКАРСТВЕННЫЕ КОМПОЗИЦИИ | 2011 |
|
RU2567548C2 |
| ЗАМЕЩЕННЫЕ ПИРАЗОЛОПИРИМИДИНЫ И ЗАМЕЩЕННЫЕ ПУРИНЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ УБИКВИТИН-СПЕЦИФИЧЕСКОЙ ПРОЦЕССИРУЮЩЕЙ ПРОТЕАЗЫ 1 (USP1) | 2019 |
|
RU2833222C2 |
| 2-ЦИАНОПИРИМИДИН-4-ИЛКАРБАМАТ, ИЛИ ПРОИЗВОДНОЕ МОЧЕВИНЫ, ИЛИ ЕГО СОЛЬ И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2019 |
|
RU2788740C2 |
| 1, 2, 4-ТРИАЗОЛЬНОЕ ПРОИЗВОДНОЕ, СПОСОБЫ ЕГО ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2003 |
|
RU2288223C2 |
| СПОСОБ ПОЛУЧЕНИЯ 2-СУЛЬФОНИЛАМИНО-1,2,4,-ТРИАЗОЛО[1,5-a] ПИРИМИДИНОВ | 2007 |
|
RU2325390C1 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ПЕСТИЦИДОВ | 2013 |
|
RU2641916C2 |
| БИЦИКЛИЧЕСКИЕ ЛАКТАМЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2827714C1 |
| ИНГИБИТОР, ПРЕДСТАВЛЯЮЩИЙ СОБОЙ ПРОИЗВОДНОЕ ПИРИДАЗИНА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2020 |
|
RU2807611C2 |
| ПРОИЗВОДНЫЕ N2-(2-ФЕНИЛ)-ПИРИДО[3,4-d]ПИРИМИДИН-2,8-ДИАМИНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ MPS1 ИНГИБИТОРА | 2015 |
|
RU2693460C2 |
Описываются производные 5-арил-1Н-1,2,4-триазола общей формулы I
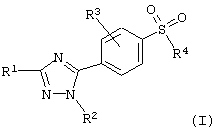
где R1 - водород, (С1-С6)алкил, гало(С1-С6) алкил или фенил;
R2 - (С3-C8)циклоалкил; фенил, где фенил необязательно замещенный одним или несколькими заместителями, выбранными из группы, состоящей из (С1-С4)алкила, галогена, гидрокси, (С1-C4)алкокси, нитро ди-(С1-С4)алкиламино, (C1-C4)алкилсульфонила, (С1-С4) алкилсульфониламино и метилендиокси; фенил(С1-С4)алкил, где фенил замещен (С1-С4)алкокси; или пиридил, или их фармацевтически приемлемые соли и фармацевтическая композиция на их основе. Новые соединения являются эффективными и селективными ингибиторами циклооксигеназы-2(СОХ-2) и могут быть использованы для лечения воспалительных заболеваний. 2 с. и 8 з.п. ф-лы, 1 табл.
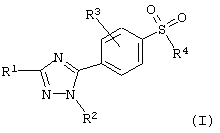
где R1 представляет водород; (C1-C6)алкил; гало(С1-С6) алкил; или фенил;
R2 представляет (С3-C8) циклоалкил; фенил, необязательно замещенный одним или несколькими заместителями, выбранными из группы, состоящей из галогена, (С1-С4)алкила, (С1-С4)алкокси, гидрокси, нитро, ди-(С1-С4)алкиламино, (С1-С4)алкилсульфониламино, (C1-С4)алкилсульфонила и метилендиокси; фенил (С1-С4)-алкил, где фенил замещен (С1-С4)алкокси; или пиридил;
R3 представляет водород или галоген;
R4 представляет (С1-С6)алкил, амино, или (C1-С4)алкилкарбониламино;
или его фармацевтически приемлемая соль.
1-(4-метоксифенил)-3-метил-5-(4-метилсульфонилфенил)-1Н-1,2,4-триазола;
1-(4-метоксифенил)-5-(4-метилсульфонилфенил)-3-трифторметил-1H-1,2,4-триазола;
1-(4-бромфенил)-5-(4-метилсульфонилфенил)-3-трифторметил-1H-1,2,4-триазола;
1-(4-метилсульфониламинофенил)-5-(4-метилсульфонилфенил)-3-трифторметил-1Н-1,2,4-триазола;
1-(4-метоксифенил)-5-(4-аминосульфонилфенил)-3-трифторметил-1Н-1,2,4-триазола.
| WO 9515318 A1, 08.06.1995 | |||
| WO 9500501 A1, 05.01.1995 | |||
| ЕР 0572142 A1, 01.12.1993 | |||
| ПРОИЗВОДНЫЕ ТРИАЗОЛА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ИНСЕКТИЦИДНОЕ И АКАРИЦИДНОЕ СРЕДСТВО | 1994 |
|
RU2131421C1 |

