В настоящей заявке испрашивается приоритет предварительной заявки на патент Соединенных Штатов №60/747625, поданной 18 мая 2006 года, раскрытие сущности которого включено в данное описание во всей ее полноте.
Область изобретения
Согласно настоящему изобретению предложены новые соединения и фармацевтические композиции, которые действуют как агонисты аденозиновых А2А рецепторов, предоставляя новые способы лечения и диагностики различных медицинских состояний.
Предшествующий уровень техники
Воспалительная реакция предназначена для элиминации вредных агентов из организма. Воспалительную реакцию может инициировать широкий спектр патогенных повреждений, включая инфекцию, аллергены, аутоиммунные раздражители, иммунную реакцию на трансплантированную ткань, ядовитые химические вещества и токсины, ишемию/реперфузию, гипоксию, механическую или термическую травму. Обычно воспаление представляет собой очень локализованное действие, которое способствует удалению, ослаблению путем разведения и изоляции повреждающего агента и поврежденной ткани. Реакция организма становится болезнетворным агентом, когда она приводит к неприемлемому повреждению тканей хозяина в процессе элиминации целевого агента или развитию реакции на травматическое повреждение.
Высвобождение воспалительных цитокинов, таких как фактор некроза опухоли альфа (ФНОα), лейкоцитами является средством, с помощью которого иммунная система борется с инвазиями патогенов, включая инфекции. ФНОα стимулирует экспрессию и активацию факторов адгезии на лейкоцитах и эндотелиальных клетках, примирует нейтрофилы для усиленной воспалительной реакции на вторичные раздражители и усиливает окислительную активность адгезивных нейтрофилов. Кроме того, макрофаги/дендритные клетки действуют как А-клетки, процессирующие антиген для презентации лимфоцитам. Лимфоциты, в свою очередь, стимулируются для действия в качестве провоспалительных цитотоксических клеток.
Обычно цитокины стимулируют нейтрофилы для усиления окислительной (например, супероксид и вторичные продукты) и неокислительной (например, миелопероксидаза и другие ферменты) воспалительной активности. Неприемлемое и чрезмерное высвобождение цитокинов может вызвать контрпродуктивные чрезмерно увеличенные патогенные эффекты посредством высвобождения повреждающих ткани продуктов окислительных и неокислительных процессов.
Несмотря на то, что моноциты медленно собираются в очагах воспаления, при благоприятных условиях моноциты развиваются в долгоживущие тканевые А-клетки и макрофаги. При стимуляции триггерами воспаления моноциты/макрофаги также продуцируют и секретируют ряд цитокинов (включая ФНОα, комплемент, липиды, активные формы кислорода, протеазы и факторы роста, которые модифицируют ткань и регулируют функции окружающих тканей.
Например, показано, что воспалительные цитокины являются патогенными при артрите (С.А.Dinarello, Semin. Immunol., 4, 133 (1992)); ишемии (A.Seekamp et al., Agents-Actions-Supp., 41, 137 (1993)); септическом шоке (D.N.Mannel et al., Rev. Infect. Dis., 9 (suppl. 5), S602-S606 (1987)); астме (N.M.Cembrzynska et al., Am. Rev. Respir. Dis., 147, 291 (1993)); отторжении трансплантированных органов (D.K.Imagawa et al., Transplantation, 51, 57 (1991); рассеянном склерозе (Н.Р.Hartung, Ann. Neurol., 33, 591 (1993)); СПИДе (Т.Matsuyama et al., AIDS, 5, 1405 (1991)); и при щелочном ожоге глаз (F. Miyamoto et al., Opthalmic Res., 30, 168 (1997)). Кроме того, образование супероксида в лейкоцитах связано с усилением репликации вируса иммунодефицита человека (ВИЧ) (S. Legrand-Poels et al., AIDS Res. Hum. Retroviruses, 6, 1389 (1990)).
Серповидно-клеточную анемию исторически рассматривали как заболевание, связанное с дефектами эритроцитов. Недавно было высказано предположение, что широкий спектр клинических проявлений этого заболевания является отчасти следствием хронического воспаления. Эта концепция подкрепляется данными о том, что пациенты со СКА (серповидно-клеточной анемией) демонстрируют множество клинических симптомов хронического воспаления, таких как повышенные уровни цитокинов, присутствие циркулирующих эндотелиальных клеток, повышенный уровень лейкоцитов в крови и увеличение числа клеточных маркеров лейкоцитарной и эндотелиальной активации.
Хорошо известно, что аденозин и некоторые аналоги аденозина, которые неселективно активируют подтипы аденозиновых рецепторов, уменьшают выработку нейтрофилами продуктов окисления при воспалении (B.N.Cronstein et al., Ann. N.Y.Acad. ScL, 451, 291 (1985); P.A.Roberts et al., Biochem. J., 227, 669 (1985); D.J.Schrier et al., J.Immunol, 137, 3284 (1986); B.N.Cronstein et al., Clinical fmmunol, and Immunopath., 42, 76 (1987); M.A.lannone et al., in Topics and Perspective in Adenosine Research, E. Gerlach et al., eds., Springer-Verlag, Berlin, p.286 (1987); S.T.McGarrity et al., J.Leukocyte Biol., 44, 411421 (1988); J. De La Harpe et at., J.Immunol., 143, 596 (1989); S.T.McGarrity et al., J.Immunol., 142, 1986 (1989); и С.Р.Nielson et al., Br. J.Pharmacol., 97, 882 (1989)).
Например, аденозин, как показано, ингибирует высвобождение супероксида из нейтрофилов, стимулированных хемоаттрактантами, такими как синтетический имитатор бактериальных пептидов, f-met-leu-phe (fMLP) (формил-метионил-лейцил-фенилаланин) и С5а-компонент комплемента (B.N.Cronstein et al., J. Immunol., 135, 1366 (1985)). Аденозин может уменьшать значительно усиленный окислительный взрыв ПМН (полиморфноядерных нейтрофилов), первоначально примированный фактором ФНОα и затем стимулированный вторичным раздражителем, таким как f-met-leu-phe (G.W.Sullivan et al., Clin. Res., 41, 172A (1993)). Кроме того, сообщалось, что аденозин может уменьшать скорость репликации ВИЧ в Т-клеточной линии (S.Sipka et al., Acta. Biochim. Biopys. Hung., 23, 75 (1988)). Однако нет никаких данных о том, что аденозин обладает противовоспалительной активностью in vivo (G.S. Firestein et al., Clin. Res., 41, 170A (1993); и B.N.Cronstein et al., Clin. Res., 41, 244A(1993)).
Было высказано предположение, что на нейтрофилах существует больше одного подтипа аденозиновых рецепторов, которые могут обладать противоположными эффектами в отношении высвобождения супероксида (B.N.Cronstein et al., J. Clin. Invest., 85, 1150 (1990)). Существование А2А рецептора на нейтрофилах впервые продемонстрировано Van Calker et al. (D.Van Calker et al., Eur. J.Pharmacology, 206, 285 (1991)).
Проводится прогрессивная разработка соединений, которые являются все более и более эффективными и/или селективными в качестве агонистов аденозиновых А2А рецепторов (AR), на основании анализов радиолигандного связывания и физиологических реакций. Первоначально были разработаны соединения с незначительной селективностью к А2А рецепторам или без нее, такие как сам аденозин или 5'-карбоксамиды аденозина, такие как 5'-N-этилкарбоксамидоаденозин (NECA) (B.N.Cronstein et al., J. Irnmunol., 135, 1366 (1985)). Позднее было показано, что добавление заместителей 2-алкиламино повышает эффективность и селективность, например CV1808 и CGS21680 (M.F. Jarvis et al., J. Pharmacol. Exp.Ther., 251, 888 (1989)). Производные 2-алкоксизамещенного аденозина, такие как WRC-0090 являются даже более эффективными и селективными в качестве агонистов А2А рецептора в коронарных артериях (М. Ueeda et al., J. Med. Chem., 34, 1334 (1991)). 2-Алкилгидразинопроизводные аденозина, например SHA 211 (также называемый WRC-0474), также были определены как агонисты A2A рецептора в коронарных артериях (К. Niiya et al., J. Med. Chem., 35, 4557 (1992)).
Имеется одно сообщение о комбинации относительно неспецифических аналогов аденозина, R-фенилизопропиладенозина (R-PIA) и 2-хлораденозина (Cl-Ado), с ингибитором фосфодиэстеразы (ФДЭ), приводящее к снижению окислительной активности нейтрофилов (М.А.Iannone et al., Topics and Perspectives in Adenosine Research, E. Garlach et al., eds., Springer-Verlag, Berlin, pp.286-298 (1987)). Однако R-PIA и Cl-Ado аналоги на самом деле являются более эффективными активаторами аденозиновых A1 рецепторов, чем аденозиновых A2A рецепторов, и поэтому, вероятно, вызывают побочные эффекты из-за активации A1 рецепторов в сердечной мышце и других тканях, вызывая эффекты, такие как «блокада сердца».
В R.A.Olsson et al. (патент США №5278150) раскрыты селективные агонисты аденозиновых А2 рецепторов формулы, где Rib представляет собой рибозил, R1 может представлять собой Н и R2 может представлять собой циклоалкил. Раскрыто, что соединения являются полезными для лечения гипертензии, атеросклероза и в качестве вазодилататоров.
В Olsson et al. (патент США №5140015) раскрыты некоторые агонисты аденозиновых А2 рецепторов формулы, где C(X)BR2 может представлять собой СН2ОН и R1 может представлять собой алкил- или алкоксиалкил. Раскрыто, что соединения являются полезными в качестве вазодилататоров или в качестве гипотензивных средств.
Linden et al. (патент США №5877180) основан на открытии того, что некоторые воспалительные заболевания, такие как артрит и астма, можно эффективно лечить путем введения соединений, которые являются селективными агонистами аденозиновых A2A рецепторов, предпочтительно в комбинации с ингибитором фосфодиэстеразы IV типа. Согласно воплощению изобретения Linden et al. предложен способ лечения воспалительных заболеваний путем введения эффективного количества аденозинового A2A рецептора следующей формулы: где R и Х являются такими, как описано в патенте.
В одном воплощении изобретение Linden et al. включает введение ингибитора фосфодиэстеразы (ФДЭ) IV типа в комбинации с агонистом аденозинового A2A рецептора. Ингибитор фосфодиэстеразы (ФДЭ) IV типа включает рацемические и оптически активные 4-(полиалкоксифенил)-2-пирролидоны следующей формулы, где R', R18, R19 и Х являются такими, как раскрыто и описано в патенте США №4193926. Ролипрам является примером подходящего ингибитора ФДЭ IV типа, охватываемым вышеуказанной формулой.
В G.Cristalli (патент США №5593975) раскрыты 2-арилэтинильные, 2-циклоалкилэтинильные или 2-гидроксиалкилэтинильные производные, где рибозидный остаток замещен карбоксиамино или замещенным карбоксиамино (R3HNC(О)--). 2-Алкинилпуриновые производные раскрыты в Miyasaka et al. (патент США №4956345), где 2-алкинильная группа замещена (С3-С16)алкилом. Раскрыто, что '975 соединения являются вазодилататорами и ингибируют агрегацию тромбоцитов, и поэтому являются полезными в качестве противоишемических, антиатеросклеротических и гипотензивных агентов.
Однако существует постоянная необходимость в селективных агонистах аденозиновых A2 рецепторов, полезных для терапевтического применения, которые имеют пониженные побочные эффекты. Кроме того, существует постоянная необходимость в селективных агонистах аденозиновых А2 рецепторов, полезных для применения в качестве фармакологических стресс-факторов для визуализации при нагрузке или в других методиках визуализации функции желудочков, которые предпочтительно имеют пониженные побочные эффекты, поскольку они являются химически стабильными и короткодействующими.
Также существует необходимость в новых способах терапии для лечения расстройств, вызванных серповидно-клеточной анемией. Современные терапии являются минимально эффективными и имеют нежелательные побочные эффекты. Соответственно, существует необходимость в соединениях и способах лечения и предупреждения серповидно-клеточной анемии.
Описание изобретения
Согласно настоящему изобретению предложены новые соединения и фармацевтические композиции, которые действуют как агонисты аденозиновых A2A рецепторов.
Согласно настоящему изобретению также предложены новые фармацевтические композиции, содержащие новое соединение и фармацевтически приемлемый носитель.
Согласно настоящему изобретению предложены новые способы лечения и диагностики, использующие эти новые соединения и композиции.
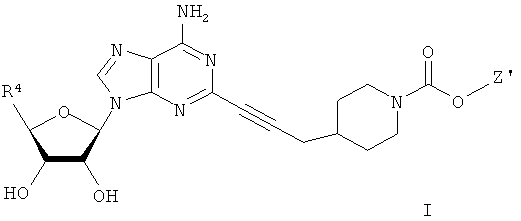
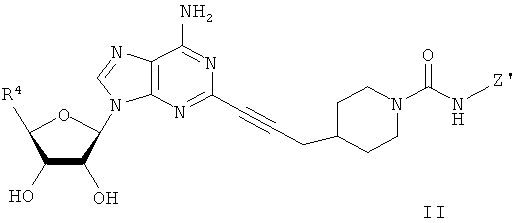
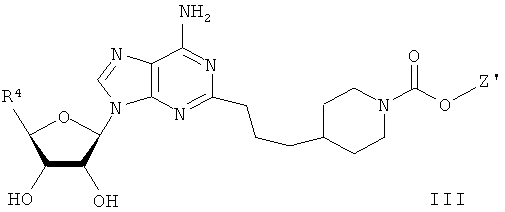
Для решения этих задач в соответствии с одним аспектом настоящего изобретения предложено соединение формулы (I)
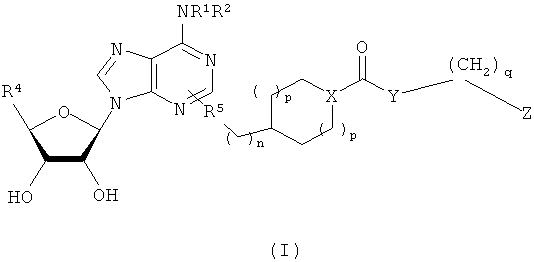
где переменные являются такими, как определено ниже.
Наилучшие варианты осуществления изобретения
Согласно настоящему изобретению предложены новые соединения, действующие как агонисты аденозинового A2A рецептора, и способы применения соединений в способах лечения заболеваний и состояний, в которые вовлечен А2А рецептор и для которых агонизм рецептора дает терапевтическую пользу. Соединения могут быть использованы, например, для лечения воспалительной активности в тканях млекопитающих или для лечения серповидно-клеточной анемии. Воспалительная активность в тканях может иметь место из-за патологических агентов или может иметь место из-за физической, химической или термической травмы или травмы вследствие медицинских процедур, таких как трансплантация органов, тканей или клеток, ангиопластика (РСТА), воспаление вследствие ишемии/реперфузии или имплантация. Соединения по изобретению также могут быть использованы в сочетании с другими способами противовоспалительного лечения или в сочетании с антипатогенными агентами.
В одном воплощении согласно настоящему изобретению предложены новые соединения формулы I или их стереоизомер или фармацевтически приемлемая соль
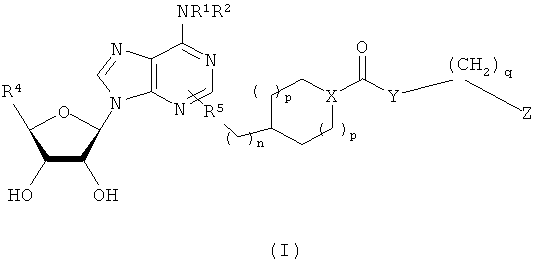
где R1 и R2 независимо выбраны из группы, состоящей из Н, (С1-С8)алкила, (С3-С8)циклоалкила, (С3-С8)циклоалкил(С1-С8)алкилена, арила, арил(С1-С8)алкилена, гетероарила, группы гетероарил(С1-С8)алкилен-, диарил(С1-С8)алкилена и дигетероарил(С1-С8)алкилена, где арильные и гетероарильные кольца возможно замещены 1-4 группами, независимо выбранными из фторо, хлоро, иодо, бромо, метила, трифторметила и метокси;
каждый R независимо выбран из группы, состоящей из Н, С1-С4алкила, циклопропила, циклобутила и (СН2)ациклопропила;
Х представляет собой СН или N при условии, что когда Х представляет собой СН, тогда Z не может быть замещен галогеном С1-C6алкилом, гидроксилом, амино или моно- или ди-(С1-С6алкил)амино;
Y выбран из группы, состоящей из О, NR1, -(OCH2CH2O)mCH2- и
-(NR1CH2CH2O)mCH2-, при условии, что когда Y представляет собой О или NR1, тогда имеется по меньшей мере один заместитель по Z;
Z выбран из группы, состоящей из 5-членного гетероарила, 6-членного арила, 6-членного гетероарила, карбоциклического биарила и гетероциклического биарила, где точка присоединения Y к Z представляет собой атом углерода в Z, где Z замещен 0-4 группами, независимо выбранными из группы, состоящей из F, Cl, Br, I, (С1-С4)алкила, -(CH2)aOR3, -(CH2)aNR3R3, -NHOH, -NR3NR3R3, нитро, -(CH2)aCN, -(CH2)aCO2R3, -(CH2)aCONR3R3, трифторметила и трифторметокси;
альтернативно Y и Z вместе образуют группировку индолил, индолинил, изоиндолинил, тетрагидроизохинолинил или тетрагидрохинолинил, где точкой присоединения является кольцевой атом азота и где указанная группировка: индолил, индолинил, изоиндолинил, тетрагидроизохинолинил или тетрагидрохинолинил, замещена 0-4 группами, независимо выбранными из группы, состоящей из F, Cl, Br, I, C1-C4алкила, -(CH2)aOR3, -(CH2)aNR3R3, -NHOH, -NR3NR3R3, NO2, -(CH2)aCN,
-(CH2)aCO2R3, -(CH2)aCONR3R3, CF3 и OCF3;
R3 независимо выбран из группы, состоящей из Н, (С1-С6)алкила, циклоалкила, арила и гетероарила;
R4 выбран из группы, состоящей из CH2OR, C(O)NRR и CO2R;
R5 выбран из группы, состоящей из СН2СН2, СН=СН и C=C;
а выбран из 0, 1 и 2;
m выбран из 1, 2 и 3;
n выбран из 0, 1 и 2;
каждый р независимо выбран из 0, 1 и 2 и
q выбран из 0, 1 и 2.
В другом воплощении согласно настоящему изобретению предложены новые соединения формулы (Ia) или их фармацевтически приемлемая соль:
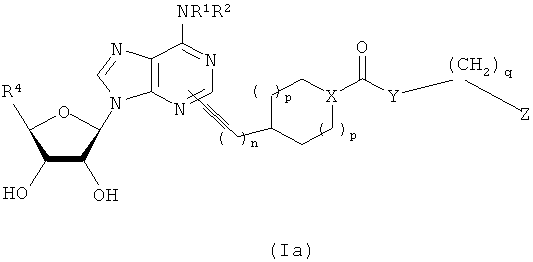
В другом воплощении согласно настоящему изобретению предложены новые соединения формулы (Ib) или их фармацевтически приемлемая соль:
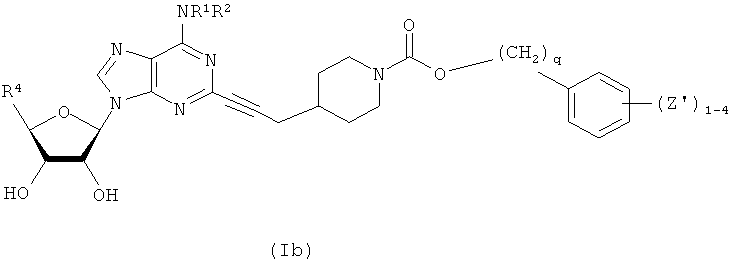
где каждый Z' независимо выбран из группы, состоящей из F, Cl, Br, I, C1-С4алкила,
-(CH2)aOR3, -(CH2)aNR3R3, -NHOH, -NR3NR3R3, NO2, -(CH2)aCN, -(CH2)aCO2R3,
-(CH2)aCONR3R3, CF3 и OCF3.
В другом воплощении согласно настоящему изобретению предложены новые соединения, где R выбран из Н, метила, этила или циклопропила.
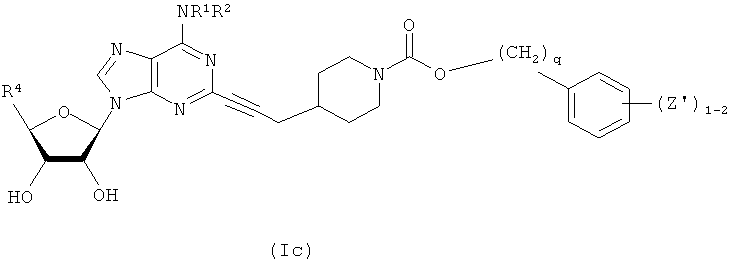
В другом воплощении согласно настоящему изобретению предложены новые соединения формулы (Ic) или их фармацевтически приемлемая соль:
В другом воплощении согласно настоящему изобретению предложены новые соединения, где Z' выбран из группы, состоящей из F, Cl, метила, OR3, NO2, CN, NR3R3 и CO2R3.
В другом воплощении согласно настоящему изобретению предложены новые соединения, где R3 представляет собой метил или водород.
В другом воплощении согласно настоящему изобретению предложены новые соединения, где соединение выбрано из группы, состоящей из соединений под номерами 3, 5-31 и 33-57, показанных в таблице (ниже).
Согласно изобретению предложено соединение формулы I для применения в медицинском лечении, предпочтительно для применения в лечении воспаления или защите тканей млекопитающих от воспаления, такого как воспалительная реакция, например, в результате аллергии, травмы или ишемического/реперфузионного повреждения, а также применение соединения формулы I для производства лекарственного средства для лечения воспалительной реакции вследствие патологического состояния или симптома у млекопитающего, ассоциированного с воспалением.
Млекопитающее или субъект включает человека, лошадь, свинью, собаку и кошку.
Изобретение также включает применение комбинации этих соединений с по меньшей мере одним противовоспалительным соединением. Примером такого соединения является ингибитор фосфодиэстеразы IV типа, и эта комбинация может быть использована для того, чтобы вызвать синергические уменьшения воспалительной реакции, опосредованной лейкоцитами.
Согласно изобретению также предложена фармацевтическая композиция, содержащая эффективное количество соединения формулы (I) или его фармацевтически приемлемой соли в комбинации с фармацевтически приемлемым разбавителем или носителем и, возможно, в комбинации с противовоспалительным соединением. Предпочтительно композиция представлена в виде стандартной лекарственной формы. Носитель может представлять собой жидкий носитель. Композиция может быть адаптирована для перорального, внутривенного, глазного, парентерального, аэрозольного или трансдермального введения.
Композиции по настоящему изобретению могут дополнительно включать ингибитор фосфодиэстеразы IV типа или другое противовоспалительное соединение (например, отличное от ингибитора ФДЭ). Ингибитор фосфодиэстеразы IV типа может представлять собой, например, ролипрам, циломиласт или рофлумиласт.
Дополнительно согласно изобретению предложен терапевтический способ лечения патологического состояния или симптома у млекопитающего, в которые вовлечена активность аденозиновых A2A рецепторов, и агонизм указанных рецепторов является желательным, включающий введение млекопитающему, нуждающемуся в такой терапии, эффективного количества соединения формулы I или его фармацевтически приемлемой соли. Полагают, что активация аденозиновых А2А рецепторов ингибирует воспаление посредством воздействия на нейтрофилы, тучные клетки, моноциты/макрофаги, тромбоциты, Т-клетки и/или эозинофилы. Ингибирование этих клеток воспаления приводит к защите ткани после повреждений ткани.
Кроме того, согласно настоящему изобретению предложен терапевтический способ лечения биологических заболеваний, включающий введение эффективного количества подходящего антибиотического агента, противогрибкового агента или противовирусного агента в сочетании с агонистом аденозиновых A2A рецепторов. Если антипатогенный агент неизвестен, агонист А2А может быть использован отдельно для уменьшения воспаления, которое может возникать во время инфекции, вызванной резистентными к антибиотику бактериями или некоторыми вирусами, такими как вирусы, вызывающие SARS (атипичную пневмонию) или лихорадку Эбола. Возможно способ включает введение ингибитора ФДЭ IV типа. Агонист аденозиновых А2А рецепторов может обеспечить дополнительную терапию для лечения состояний, таких как воспаление, вызванное сепсисом, например гемолитико-уремический синдром человека, при введении с антибиотиками в лечении последствий применения оружия биотерроризма, такого как сибирская язва, туляремия, Escherichia coli, чума и тому подобных. Согласно настоящему изобретению также предложена дополнительная терапия для лечения смертельных бактериальных, грибковых и вирусных инфекций, таких как сибирская язва, туляремия, эшерихиоз и чума, включающая введение антибактериального агента, противогрибкового агента или противовирусного агента в сочетании с селективными агонистами аденозиновых A2A рецепторов.
Согласно настоящему изобретению предложен терапевтический способ лечения биологических заболеваний, которые провоцируют воспаление, либо в отдельности, либо в комбинации с лекарственным препаратом, подавляющим заболевание. Они включают бактерии в комбинации с антибиотиками, включая бактерии, которые вызывают сибирскую язву, туляремию, чуму, болезнь Лайма и сибирскую язву, но не ограничиваясь ими. Также включены вирусы, включая те, которые вызывают RSV (респираторно-синцитиальный вирус), тяжелый острый респираторный синдром (SARS), грипп и лихорадку Эбола, но не ограничиваясь ими, с противовирусной терапией или без нее. Также включены дрожжевые и грибковые инфекции с противодрожжевыми или противогрибковыми агентами или без них.
Антибактериальный агент, противогрибковый агент или противовирусный агент может быть введен совместно (например, одновременно) с агонистом аденозиновых А2А рецепторов, или они могут быть введены либо одновременно, либо в виде смеси, или они могут быть введены последовательно. Последовательное введение агонистов аденозиновых A2A рецепторов можно осуществлять перед введением агента, в пределах нескольких минут или вплоть до примерно 48 часов после введения агента. Предпочтительно введение агонистов аденозиновых A2A рецепторов осуществляют в пределах примерно 24 часов и более предпочтительно в пределах примерно 12 часов.
Способ по изобретению будет также полезен при лечении пациентов с сепсисом, тяжелым сепсисом и, потенциально, синдромом системной воспалительной реакции в дополнение к септическому шоку. Агонисты аденозиновых A2A рецепторов оказывают множественные противовоспалительные эффекты на раннем этапе воспалительного каскада, и, таким образом, короткий курс таких агонистов может принести чрезвычайную пользу при тяжелых, опасных для жизни инфекциях и воспалительных расстройствах у людей, включая ингаляционную форму сибирской язвы, туляремию, эшерихиоз и чуму.
Противовоспалительный эффект агонистов А2А рецепторов подтвержден in vivo в экспериментальных моделях менингита, перитонита и артрита. Потенциально смертельный синдром бактериального сепсиса является все более и более распространенной проблемой в отделениях неотложной помощи. Частота сепсиса и септического шока, в настоящее время на одиннадцатом месте в списке основных причин смерти в Соединенных Штатах, увеличивается. Текущие оценки показывают, что около 900000 новых случаев сепсиса (приблизительно 60% грамотрицательного) возникают в Соединенных Штатах ежегодно с подсчитанным общим коэффициентом смертности 35%. Кроме того, коэффициент смертности, как определено в недавних клинических исследованиях, составляет приблизительно 25%, тогда как 10% пациентов умирает от первопричинного заболевания. Шок развивается в приблизительно 200000 случаях ежегодно с присущим ему коэффициентом смертности 46% (92000 смертей). Сепсис является причиной ежегодно планируемых 5-10 миллиардов долларов на расходы здравоохранения. В настоящее время широко признано, что среди госпитализированных пациентов в отделениях интенсивной терапии некоронарного профиля сепсис является самой распространенной причиной смерти. Септический синдром является проблемой здравоохранения первостепенной важности. Ожидают, что агонисты A2AAR будут использовать в качестве новой и уникальной дополнительной лечебной тактики для снижения заболеваемости и смертности. Полагают, что это лечение улучшит исход при генерализованной форме сибирской язвы, туляремии, эшерихиозе и чуме.
Агонисты аденозиновых A2A рецепторов по изобретению могут ингибировать активацию нейтрофилов, макрофагов и Т-клеток и таким образом уменьшать воспаление, вызванное бактериальными и вирусными инфекциями. Соединения, в сочетании с антибиотиками или противовирусными агентами, могут предотвращать или снижать смертность, вызванную сепсисом или гемолитико-уремическим синдромом или другими воспалительными состояниями. Эффекты агонистов аденозиновых A2A рецепторов усиливаются ингибиторами фосфодиэстеразы IV типа, такими как ролипрам.
Согласно изобретению также предложено соединение формулы I для применения в лекарственной терапии (например, для применения в качестве вспомогательного средства в лечении потенциально смертельных бактериальных инфекций, таких как сибирская язва, туляремия, эшерихиоз, чума, или других бактериальных или вирусных инфекций и лечении общей интоксикации организма, вызванной бактериальными и/или вирусными инфекциями), а также применения соединения формулы I для производства лекарственного средства для уменьшения воспаления, вызванного бактериями или вирусом, или его лечения у млекопитающего, такого как человек. Соединения по изобретению также полезны для лечения уже подвергаемой лечению общей интоксикации организма, когда бактериальные или вирусные агенты вызывают воспаление либо непосредственно, либо в результате лечения, например, антибиотиком или противовирусным агентом.
Сепсис является тяжелым заболеванием, вызванным генерализованной инфекцией в кровяном русле токсин-продуцирующими бактериями или вирусами. Инфекция, которая может проявляться как воспаление, может быть вызвана непосредственно патогенами бактериального или вирусного происхождения или в результате лечения от них, то есть гибели патогенов в результате лечения антибактериальными или противовирусными агентами. Сепсис также можно рассматривать как реакцию организма на инфекцию. Инфекция может быть вызвана микроорганизмами или «микробами» (обычно бактериями), проникающими в организм, может быть ограничена отдельным участком организма (например, абсцесс зуба) или может широко распространяться по кровяному руслу (часто называемая «септицемией» или «заражением крови»).
Общая интоксикация организма или воспалительный шок часто называют септическим шоком, бактериемическим шоком, эндотоксическим шоком, септицемическим шоком или тепловым шоком.
Септический шок является тяжелым аномальным состоянием, которое возникает, когда генерализованная инфекция приводит к низкому кровяному давлению и замедленному кровотоку. Жизненно важные органы, такие как мозг, сердце, почки и печень, могут функционировать неправильно или могут отказать. Септический шок возникает наиболее часто у очень пожилых и совсем молодых людей. Он также встречается у людей с первопричинными заболеваниями. Любой бактериальный организм может вызвать септический шок. Грибы и вирусы также могут вызывать это состояние. Токсины, выделяемые бактериями, грибами или вирусами, могут вызывать прямое повреждение ткани и могут приводить к низкому кровяному давлению и/или плохому функционированию органов. Эти токсины могут также вызывать сильную воспалительную реакцию организма, которая способствует септическому шоку.
В другом аспекте согласно настоящему изобретению также предложен способ лечения тяжелого острого респираторного синдрома (SARS), включающий введение млекопитающему, нуждающемуся в указанной терапии, эффективного противовоспалительного количества агонистов аденозинового A2A рецептора, возможно с ингибитором ФДЭ-IV, таким как ролипрам.
Согласно изобретению также предложены способы лечения серповидно-клеточной анемии путем введения субъекту, страдающему серповидно-клеточной анемией, описанных здесь агонистов A2A.
Согласно настоящему изобретению предложены соединения и способы их применения для выявления наличия и оценки тяжести стенозов коронарных артерий у млекопитающего, такого как человек или домашнее животное. Предпочтительно соединения по изобретению используют в качестве фармакологических стресс-индуцирующих агентов или стресс-факторов, которые полезны при фармакологической стресс-визуализации для выявления и оценки заболевания коронарных артерий. Специфические соединения по изобретению, полезные в качестве стресс-индуцирующих агентов, являются эффективными и селективными в отношении аденозиновых А2А рецепторов, но являются также быстродействующими, так что они быстро выводятся организмом после процесса визуализации.
Таким образом, согласно настоящему изобретению предложен способ выявления наличия и тяжести стенозов коронарных артерий у млекопитающего, такого как человек, включающий (1) введение количества одного или более чем одного соединения общей формулы (I) и (2) осуществление методики на указанном млекопитающем для обнаружения и/или оценки тяжести указанных стенозов коронарных артерий.
Согласно изобретению предложено соединение формулы (I) для применения в медицинских диагностических процедурах, предпочтительно для применения в выявлении наличия и оценки тяжести стенозов коронарных артерий у человека. Согласно настоящему изобретению предложено применение соединения формулы (I) для производства фармакологического вазодилатирующего агента, который может быть использован в клинических перфузионных методиках визуализации для диагностики и оценки степени заболевания коронарных артерий. Предпочтительными перфузионными методиками визуализации являются плоскостная или однофотонная эмиссионная компьютерная томография (SPECT), гамма-сцинтиграфия, позитронная эмиссионная томография (PET), ядерно-магнитная резонансная (ЯМР) томография, магнитно-резонансная интроскопия (MRI), перфузионная контрастная эхокардиография, цифровая субтракционная ангиография (DSA) и сверхскоростная компьютерная томография (CINE СТ).
Согласно изобретению также предложена фармацевтическая композиция, содержащая эффективное количество соединения формулы (I) или его фармацевтически приемлемой соли в комбинации с фармацевтически приемлемым разбавителем или носителем. Предпочтительно композиция представлена в виде стандартной лекарственной формы и может быть адаптирована для парентеральной, например внутривенной инфузии.
Если не оговорено особо, использованы следующие обозначения.
Галогено представляет собой фторо, хлоро, бромо или иодо.
Алкил, алкокси, аралкил, алкиларил и так далее означают как неразветвленные, так и разветвление алкильные группы; но ссылка на отдельный радикал, такой как «пропил», включает только радикал с неразветвленной цепью, а на изомер с разветвленной цепью, такой как «изопропил», делают конкретную ссылку.
Арил означает фенильный радикал или орто-конденсированный бициклический карбоциклический радикал, имеющий примерно от девяти до десяти кольцевых атомов, в котором по меньшей мере одно кольцо является ароматическим. Гетероарил означает радикал моноциклического ароматического кольца, содержащего пять или шесть кольцевых атомов, состоящих из атомов углерода и 1, 2, 3 или 4 гетероатомов, каждый из которых выбран из группы, состоящей из непероксидного кислорода, серы и N(Y), где Y отсутствует или представляет собой Н, О, (С1-С8)алкил, фенил или бензил, а также радикал орто-конденсированного бициклического гетероцикла из примерно восьми-десяти кольцевых атомов, производных от них, в частности бензопроизводное или производное, полученное посредством конденсации с ним дирадикала пропилена, триметилена или тетраметилена.
Гетероарил охватывает моноциклическое ароматическое кольцо, имеющее пять или шесть кольцевых атомов, состоящих из атомов углерода и 1-4 гетероатомов, каждый из которых выбран из группы, состоящей из непероксидного кислорода, серы и N(X), где Х отсутствует или представляет собой Н, О, (С1-С4)алкил, фенил или бензил, или представляет собой заместитель, описанный в другом месте. Гетероарил также охватывает радикал орто-конденсированного бициклического гетероцикла из 8-10 кольцевых атомов, в частности бензопроизводное или производное, полученное посредством конденсации с ним дирадикала пропилена, триметилена или тетраметилена. Необходимо, чтобы хотя бы одно кольцо бициклического гетероарила являлось ароматическим.
Термин «гетероцикл» обычно представляет собой неароматическую гетероциклическую группу, имеющую от 3 до примерно 10 кольцевых атомов, которая может быть насыщенной или частично ненасыщенной, содержащей по меньшей мере один гетероатом (например, 1, 2 или 3), выбранный из группы, состоящей из кислорода, азота и серы. Специфические группы «гетероцикла» включают моноциклические, бициклические или трициклические группы, содержащие один или более чем один гетероатом, выбранный из группы, состоящей из кислорода, азота и серы. Группа «гетероцикл» также может включать одну или более чем одну оксогруппу (=O), присоединенную к кольцевому атому. Неограничивающие примеры гетероциклических групп включают 1,3-диоксолан, 1,4-диоксан, 1,4-дитиан, 2H-пиран, 2-пиразолин, 4Н-пиран, хроманил, имидазолидинил, имидазолинил, индолинил, изохроманил, изоиндолинил, морфолин, пиперазинил, пиперидин, пиперидил, пиразолидин, пиразолидинил, пиразолинил, пирролидин, пирролин, хинуклидин, тиоморфолин и тому подобное.
Термин «карбоциклический биарил» относится к орто-конденсированным бициклическим группировкам, типично содержащим 10 атомов углерода. Примером является нафталин. Термин «гетероциклический биарил», как он использован здесь, относится к орто-конденсированным бициклическим группировкам, содержащим 1-4 гетероатома. Примеры включают индолы, изоиндолы, хинолины, изохинолины, бензофураны, изобензофураны, бензотиофены, бензо[с]тиофены, бензимидазолы, пурины, индазолы, бензоксазол, бензизоксазол, бензотиазол, хиноксалины, хиназолины, циннолины и тому подобное.
Точкой присоединения либо карбоциклического, либо гетероциклического биарила может служить любой кольцевой атом, если позволяет валентность этого атома.
Специфические или предпочтительные значения, перечисленные ниже для радикалов, заместителей и диапазонов, даны только для иллюстрации; они не исключают других определенных значений или других значений в пределах определенных диапазонов для радикалов и заместителей.
Углеродные цепи и их возможно замещенные аналоги могут находиться в форме любой разветвленной цепи, если позволяют валентности и стерические условия атомов. Более конкретно, (С1-С8)алкил может представлять собой метил, этил, пропил, изопропил, бутил, изобутил, втор-бутил, трет-бутил, пентил, 3-пентил, неопентил, гексил, гептил, октил и тому подобное в форме любой разветвленной цепи.
Термин «циклоалкил», как он использован здесь, охватывает бициклоалкил (норборнил, 2.2.2-бициклооктил и так далее) и трициклоалкил (адамантил и так далее), возможно содержащий 1-2 N, О или S. Термин «циклоалкил» также охватывает (циклоалкил)алкил. Таким образом, (С3-С6)циклоалкил может представлять собой циклопропил, циклобутил, циклопентил, циклогексил и тому подобное. (С1-С8)алкокси может представлять собой метокси, этокси, пропокси, изопропокси, бутокси, изобутокси, втор-бутокси, пентокси, 3-пентокси или гексилокси в форме любой разветвленной цепи.
(С2-С6)алкенил может представлять собой винил, аллил, 1-пропенил, 2-пропенил, 1-бутенил, 2-бутенил, 3-бутенил, 1-пентенил, 2-пентенил, 3-пентенил, 4-пентенил, 1-гексенил, 2-гексенил, 3-гексенил, 4-гексенил или 5-гексенил; (С2-С6)алкинил может представлять собой этинил, 1-пропинил, 2-пропинил, 1-бутинил, 2-бутинил, 3-бутинил, 1-пентинил, 2-пентинил, 3-пентинил, 4-пентинил, 1-гексинил, 2-гексинил, 3-гексинил, 4-гексинил или 5-гексинил.
(С1-С6)алканоил может представлять собой ацетил, пропаноил или бутаноил; галогено(С1-С6)алкил может представлять собой иодметил, бромметил, хлорметил, фторметил, трифторметил, 2-хлорэтил, 2-фторэтил, 2,2,2-трифторэтил или пентафторэтил; гидрокси(С1-С6)алкил может представлять собой гидроксиметил, 1-гидроксиэтил, 2-гидроксиэтил, 1-гидроксипропил, 2-гидроксипропил, 3-гидроксипропил, 1-гидроксибутил, 4-гидроксибутил, 1-гидроксипентил, 5-гидроксипентил, 1-гидроксигексил или 6-гидроксигексил.
(С1-С6)алкоксикарбонил (CO2R2) может представлять собой метоксикарбонил, этоксикарбонил, пропоксикарбонил, изопропоксикарбонил, бутоксикарбонил, пентоксикарбонил или гексилоксикарбонил.
(С1-С6)алкилтио может представлять собой метилтио, этилтио, пропилтио, изопропилтио, бутилтио, изобутилтио, пентилтио или гексилтио.
(С2-С6)алканоилокси может представлять собой ацетокси, пропаноилокси, бутаноилокси, изобутаноилокси, пентаноилокси или гексаноилокси; арил может представлять собой фенил, инденил или нафтил и гетероарил может представлять собой фурил, имидазолил, триазолил, триазинил, оксазолил, изоксазолил, тиазолил, изотиазоил, пираксолил, пирролил, пиразинил, тетразолил, пуридил (или его N-оксид), тиенил, пиримидинил (или его N-оксид), индолил, изохинолил (или его N-оксид) или хинолил (или его N-оксид).
Термин «алкилен» относится к двухвалентной неразветвленной или разветвленной углеводородной цепи (например, этилена -СН2СН2-).
Термин «арил(С1-С8)алкилен», например, включает бензил, фенетил, 3-фенилпропил, нафтилметил и тому подобное.
«Проведение лечения» или «лечение» предусматривает лечение болезненного состояния у млекопитающего и включает: (а) предупреждение возникновения болезненного состояния у млекопитающего, в частности, когда такое млекопитающее предрасположено к болезненному состоянию, но диагноз еще не установлен; (б) подавление болезненного состояния, например прекращение его развития; и/или (в) облегчение болезненного состояния, например индуцирование регрессии болезненного состояния до тех пор, пока не будет достигнут желаемый результат. Проведение лечения также включает уменьшение интенсивности симптома заболевания (например, облегчение боли или дискомфорта), при котором подобное уменьшение может или не может непосредственно влиять на заболевание (например причину, передачу, проявление и так далее).
Использованный здесь термин «в сочетании с» относится к совместному введению агента против отторжения с агонистом аденозиновых A2A рецепторов. Совместное введение агента и агонистов аденозиновых A2A рецепторов включает введение агента и агониста либо одновременно, в виде смеси, либо последовательно. Последовательное введение агонистов аденозиновых A2A рецепторов можно осуществлять перед введением агента, в пределах нескольких минут или вплоть до примерно 48 часов перед введением агента. Агонисты аденозиновых A2A рецепторов можно также вводить после введения агента. Предпочтительно введение агонистов аденозиновых А2А рецепторов осуществляют в пределах примерно 24 часов и более предпочтительно в пределах примерно 12 часов.
Количество атомов углерода различных углеводородсодержащих группировок показано посредством префикса, обозначающего минимальное и максимальное число атомов углерода в группировке, то есть префикс Ci-Cj означает группировку, содержащую от числа «i» до числа «j» атомов углерода включительно. Таким образом, например, (С1-С8)алкил относится к алкилу, содержащему от одного до восьми атомов углерода включительно.
Соединения по настоящему изобретению в общем названы согласно номенклатурной системе IUPAC (Международный союз теоретической и прикладной химии) или CAS (Химическая реферативная служба). Могут быть использованы сокращения, которые хорошо известны специалисту в данной области техники (например «Ph» для фенила, «Me» для метила, «Et» для этила, «ч» для часа или часов и «кт» для комнатной температуры).
Специалистам в данной области техники будет понятно, что соединения формулы (I) имеют более одного хирального центра и могут быть выделены в оптически активных и рацемических формах. Предпочтительно рибозидная группировка формулы (I) является производной от D-рибозы. Некоторые соединения могут проявлять полиморфизм. Следует понимать, что настоящее изобретение охватывает любую рацемическую, оптически активную, полиморфную или стереоизомерную форму или смеси этих форм соединения по изобретению, которые обладают полезными свойствами, описанными здесь, и в данной области техники хорошо известно как получить оптически активные формы (например, путем разделения рацемической формы методиками перекристаллизации или ферментативными методиками, путем синтеза из оптически активных исходных веществ, путем хирального синтеза или путем хроматографического разделения при использовании хиральной стационарной фазы) и как определить активность агонистов аденозина при использовании тестов, описанных здесь, или при использовании других подобных тестов, которые хорошо известны в данной области техники.
В числе воспалительных реакций, которые можно лечить (включая профилактическое лечение) соединениями формулы I, возможно с ингибитором ФДЭ IV типа, находятся воспаления, обусловленные: (а) аутоиммунной стимуляцией (аутоиммунными заболеваниями), такими как красная волчанка, рассеянный склероз, бесплодие вследствие эндометриоза, сахарный диабет I типа, включая деструкцию панкреатических островков, приводящую к диабету, и воспалительные последствия диабета, включая язвы ног, болезнь Крона, неспецифический язвенный колит, воспалительное заболевание кишечника, остеопороз и ревматоидный артрит; (б) аллергическими заболеваниями, такими как астма, сенная лихорадка, ринит, аллергический контактный дерматит, вызванный ядоносным сумахом, весенний конъюнктивит и другие опосредованные эозинофилами состояния; (в) кожными заболеваниями, такими как псориаз, контактный дерматит, экзема, инфекционные язвы кожи, заживление открытых ран, целлюлит; (г) инфекционными заболеваниями, включая сепсис, септический шок, энцефалит, инфекционный артрит, эндотоксический шок, грамотрицательный шок, реакция Яриша-Герксгеймера, сибирская язва, чума, туляремия, лихорадка Эбола, опоясывающий лишай, токсический шок, церебральная малярия, бактериальный менингит, острый респираторный дистресс-синдром (ARDS), хроническая обструктивная болезнь легких (ХОБЛ), болезнь Лайма, ВИЧ-инфекция (ФНОα-стимулированная репликация ВИЧ, подавление активности ингибитора обратной транскриптазы фактором некроза опухоли (ФНОα)); (д) истощающими болезнями: кахексией, вызванной раком и ВИЧ; (е) трансплантацией органов, тканей или клеток (например, костного мозга, роговицы, почки, легкого, печени, сердца, кожи, панкреатических островков), включая отторжение трансплантата и болезнь «трансплантат против хозяина»; (ж) неблагоприятными эффектами вследствие лекарственной терапии, включая неблагоприятные эффекты вследствие лечения амфотерицином В, неблагоприятные эффекты вследствие иммуносупрессивной терапии, например лечения интерлейкином-2, неблагоприятные эффекты вследствие лечения моноклональными антителами ОКТЗ, применения контрастных веществ, антибиотиков, неблагоприятные эффекты вследствие лечения ГМ-КСФ (гранулоцитарно-макрофагальным колониестимулирующим фактором), неблагоприятные эффекты лечения циклоспоринами и неблагоприятные эффекты лечения аминогликозидами, стоматит и мукозит вследствие иммуносупрессии; (з) сердечно-сосудистыми состояниями, включая расстройства кровообращения, вызываемые или обостряемые воспалительной реакцией, такие как ишемия, атеросклероз, заболевание периферических сосудов, рестеноз после ангиопластики, воспалительная аневризма аорты, васкулит, инсульт, повреждение спинного мозга, застойная сердечная недостаточность, геморрагический шок, ишемическое/реперфузионное повреждение, спазм сосудов после субарахноидального кровоизлияния, спазм сосудов после инсульта, плеврит, перикардит и сердечно-сосудистые осложнения диабета; (и) диализом, включая перикардит вследствие перитонеального диализа; (к) подагрой; и (л) химической или термической травмой из-за ожогов, кислоты, щелочи и тому подобного.
Дополнительные заболевания включают болезни лошадей, такие как ламинит и воспаление суставов ног у лошадей.
Особый интерес и эффективность имеет применение настоящих соединений для ограничения воспалительных реакций в тех случаях, когда ишемическое/реперфузионное повреждение происходит в результате ангиопластики или тромболизиса. Также особый интерес и эффективность имеет применение настоящих соединений для ограничения воспалительных реакций вследствие трансплантации органов, тканей или клеток, то есть трансплантации аллогенной или ксеногенной ткани млекопитающему-реципиенту, аутоиммунных заболеваний и воспалительных состояний вследствие патологий кровообращения и их лечения, включая ангиопластику, установку стента, установку шунта или пересадку. Неожиданно обнаружили, что введение одного или более чем одного соединения формулы (I) является эффективным после начала воспалительной реакции, например после поражения субъекта патологией или травмой, которая инициирует воспалительную реакцию.
Ткань или клетки, содержащие рецепторные участки связывания лиганда, могут быть использованы для измерения селективности тестируемых соединений по отношению к подтипам специфических рецепторов, количества биологически активного соединения в крови или других физиологических жидкостях или могут быть использованы в качестве инструмента для идентификации потенциальных терапевтических агентов для лечения заболеваний или состояний, ассоциированных с активацией рецепторных участков, путем приведения указанных агентов в контакт с указанными лиганд-рецепторными комплексами и измерения степени замещения лиганда и/или связывания агента или клеточной реакции на указанный агент (например, накопление цАМФ (циклического аденозинмонофосфата)).
В данном описании использованы следующие сокращения:
Специфические ингибиторы фосфодиэстеразы (ФДЭ) IV типа, полезные при практическом осуществлении настоящего изобретения, включают рацемические и оптически активные 4-(полиалкоксифенил)-2-пирролидоны формулы 1, где R1, R18, R19 и Х являются такими, как раскрыто и описано в патенте США №4193926. Ролипрам представляет собой пример подходящего ингибитора ФДЭ IV типа, охватываемого вышеуказанной формулой.
Дополнительные неограничивающие примеры ингибиторов ФДЭ IV, полезных при практическом осуществлении настоящего изобретения, включают соединения, имеющие следующие формулы, и их варианты, но не ограничены ими.
Дополнительно согласно настоящему изобретению предложены фармацевтические композиции, которые включают соединение формулы (I) в комбинации с одним или более чем одним представителем, выбранным из группы, состоящей из следующего: (а) ингибиторы биосинтеза лейкотриенов, ингибиторы 5-липооксигеназы (5-LO) и антагонисты белка, активирующего 5-липоксигеназу (FLAP), выбранные из группы, состоящей из зилейтона; АВТ-761; фенлейтона; тепоксалина; Abbott-79175; Abbott-85761; N-(5-замещенных)тиофен-2-алкилсульфонамидов формулы (5.2.8); 2,6-ди-трет-бутилфенолгидразонов формулы (5.2.10); Zeneca ZD-2138 формулы (5.2.11); SB-210661 формулы (5.2.12); пиридинилзамещенного 2-цианонафталинового соединения L-739,010; 2-цианохинолинового соединения L-746,530; индоловых и хинолиновых соединений МК-591, МК-886 и BAY x 1005; (б) антагонисты рецепторов к лейкотриенам LTB4, LTC4, LTD4 и LTE4, выбранные из группы, состоящей из соединения фенотиазин-3-она L-651,392; амидиносоединения CGS-25019C; бензоксазоламинового соединения онтазоласта; бензолкарбоксимидамидного соединения BIIL 284/260; соединений: зафирлукаста, аблукаста, монтелукаста, пранлукаста, верлукаста (МК-679), RG-12525, Ro-245913, иралукаста (CGP 45715А) и BAY × 7195; (г) ингибиторы 5-липоксигеназы (5-LO); или антагонисты белка, активирующего 5-липоксигеназу (FLAP); (д) двойные ингибиторы 5-липоксигеназы (5-LO) и антагонисты фактора активации тромбоцитов (ФАТ); (е) теофиллин и аминофиллин; (ж) ингибиторы циклооксигеназы-1 (ЦОГ-1) (НПВП (нестероидные противовоспалительные препараты)); и НПВП с оксидом азота; (з) селективный ингибитор ЦОГ-2 рофекоксиб; (и) ингаляционные глюкокортикоиды со сниженными системными побочными эффектами, выбранные из группы, состоящей из преднизона, преднизолона, флунизолида, триамцинолона ацетонида, беклометазона дипропионата, будесонида, флутиказона пропионата и мометазона фуроата; (к) антагонисты фактора активации тромбоцитов (ФАТ); (л) моноклональные антитела, активные против эндогенных воспалительных агентов; (м) агенты против фактора некроза опухоли (анти-ФНОα), выбранные из группы, состоящей из этанерцепта, инфликсимаба и D2E7; (н) ингибиторы молекул адгезии, включая антагонисты VLA-4; (о) иммуносупрессивные агенты, выбранные из группы, состоящей из циклоспорина, азатиоприна и метотрексата; или (п) противоподагрические агенты, выбранные из группы, состоящей из колхицинов.
Примерами фармацевтически приемлемых солей являются соли присоединения органических кислот, образованные кислотами, которые образуют физиологически приемлемый анион, например тозилат, метансульфонат, малат, ацетат, цитрат, малонат, тартрат, сукцинат, бензоат, аскорбат, α-кетоглутарат и α-глицерофосфат. Могут также быть образованы подходящие неорганические соли, включая гидрохлорид, сульфат, нитрат, бикарбонат и карбонат.
Фармацевтически приемлемые соли могут быть получены при использовании стандартных методик, хорошо известных в данной области техники, например путем взаимодействия достаточно основного соединения, такого как амин, с подходящей кислотой, дающей физиологически приемлемый анион. Могут также быть получены соли щелочных металлов (например, натрия, калия или лития) или щелочноземельных металлов (например, кальция) карбоновых кислот.
Соединения формулы I могут быть приготовлены в виде фармацевтической композиции и введены млекопитающему-хозяину, такому как пациент-человек, в разнообразных формах, предназначенных для выбранного пути введения, то есть перорального или парентерального, посредством внутривенного, внутримышечного, местного или подкожного пути введения.
Таким образом, настоящие соединения могут быть введены системно, например перорально, в комбинации с фармацевтически приемлемым носителем, таким как инертный разбавитель или ассимилируемый, пригодный для употребления носитель. Они могут быть заключены в желатиновые капсулы с твердой или мягкой оболочкой, могут быть спрессованы в таблетки или могут быть введены непосредственно в пищевые продукты диеты пациента. Для перорального терапевтического введения активное соединение может быть комбинировано с одним или более чем одним эксципиентом и использовано в форме таблеток для приема внутрь, трансбуккальных таблеток, пастилок, капсул, эликсиров, суспензий, сиропов, облаток и тому подобного. Такие композиции и препараты должны содержать по меньшей мере 0,1% активного соединения. Процентное содержание композиций и препаратов может, конечно, изменяться и может удобным образом составлять от примерно 2 до примерно 60% от массы данной стандартной лекарственной формы. Количество активного соединения в таких терапевтически полезных композициях является таким, чтобы был достигнут эффективный уровень дозы.
Таблетки, пастилки, пилюли, капсулы и тому подобное могут также содержать связующие вещества, такие как трагакантовая камедь, аравийская камедь, кукурузный крахмал или желатин; эксципиенты, такие как дикальцийфосфат; разрыхлитель, такой как кукурузный крахмал, картофельный крахмал, альгиновая кислота и тому подобное; смазывающее вещество, такое как стеарат магния; и подсластитель, такой как сахароза, фруктоза, лактоза или аспартам, или ароматизатор, такой как перечная мята, масло гаультерии или вишневый ароматизатор. Когда стандартная лекарственная форма представляет собой капсулу, она может содержать, кроме веществ вышеприведенного типа, жидкий носитель, такой как растительное масло или полиэтиленгликоль. Другие различные вещества могут присутствовать в качестве оболочек или для иной модификации физической формы твердой стандартной лекарственной формы. Например, таблетки, пилюли или капсулы могут быть покрыты желатином, воском, шеллаком или сахаром и тому подобным. Сироп или эликсир может содержать активное соединение, сахарозу или фруктозу в качестве подсластителя, метил- или пропилпарабены в качестве консервантов, краситель и ароматизатор, такой как вишневая или апельсиновая отдушка. Конечно, любое вещество, применяемое при приготовлении любой стандартной лекарственной формы, должно быть фармацевтически приемлемым и по существу нетоксичным в используемых количествах. Кроме того, активное соединение может быть введено в препараты и устройства длительного высвобождения.
Активное соединение может быть также введено внутривенно или внутрибрюшинно посредством инфузии или инъекции. Растворы активного соединения или его солей могут быть приготовлены в воде, возможно смешанной с нетоксичным поверхностно-активным веществом. Дисперсии могут быть также приготовлены в глицерине, жидких полиэтиленгликолях, триацетине и их смесях и в маслах. При нормальных условиях хранения и применения эти препараты содержат консервант для предотвращения роста микроорганизмов.
Фармацевтические лекарственные формы, подходящие для инъекции или инфузии, могут включать стерильные водные растворы или дисперсии или стерильные порошки, содержащие активный ингредиент, которые предназначены для экстемпорального приготовления стерильных инъекционных или инфузионных растворов или дисперсий, возможно инкапсулированных в липосомы. Во всех случаях конечная лекарственная форма должна быть стерильной, текучей и стабильной в условиях производства и хранения. Жидкая основа или носитель может являться растворителем или жидкой дисперсионной средой, содержащей, например, воду этанол, полиол (например, глицерин, пропиленгликоль, жидкие полиэтиленгликоли и тому подобное), растительные масла, нетоксичные сложные эфиры глицерина и их подходящие смеси. Приемлемую текучесть можно поддерживать, например, посредством образования липосом, посредством поддержания требуемого размера частиц в случае дисперсий или посредством применения поверхностно-активных веществ. Предупреждения деятельности микроорганизмов можно достичь с помощью различных антибактериальных и противогрибковых агентов, например парабенов, хлорбутанола, фенола, сорбиновой кислоты, тимеросала и тому подобных. Во многих случаях будет предпочтительно включать агенты, придающие изотоничность, например сахара, буферные растворы или хлорид натрия. Пролонгированной абсорбции инъекционных композиций можно достичь посредством использования в композициях агентов, замедляющих абсорбцию, например моностеарата алюминия и желатина.
Стерильные инъекционные растворы готовят посредством введения активного соединения в требуемом количестве в соответствующий растворитель с другими различными ингредиентами, которые перечислены выше, в случае необходимости, с последующей стерилизацией путем фильтрации. В случае стерильных порошков для приготовления стерильных инъекционных растворов предпочтительными способами приготовления являются технологии вакуумной сушки и лиофилизации, которые дают порошок активного ингредиента плюс любой дополнительный требуемый ингредиент, присутствующий в предварительно стерилизованных посредством фильтрации растворах.
Для местного введения настоящие соединения могут быть использованы в чистой форме, то есть в тех случаях, когда они представляют собой жидкости. Однако в большинстве случаев будет желательно наносить их на кожу в виде композиций или препаратов в комбинации с дерматологически приемлемым носителем, который может представлять собой твердое вещество, жидкость, или в кожном пластыре.
Полезные твердые носители включают тонкоизмельченные твердые вещества, такие как тальк, глина, микрокристаллическая целлюлоза, диоксид кремния, оксид алюминия и тому подобное. Полезные жидкие носители включают воду, спирты или гликоли или водно-спиртовые/гликолевые смеси, в которых настоящие соединения могут быть растворены или диспергированы в эффективных уровнях дозировки, возможно с помощью нетоксичных поверхностно-активных веществ. Адъюванты, такие как ароматизирующие вещества и дополнительные антимикробные агенты, могут быть добавлены для оптимизации свойств для данного применения. Полученные жидкие композиции могут быть нанесены с гигроскопических прокладок, используемых для импрегнированных бинтов и других повязок, или нанесены распылением на поврежденный участок при использовании помповых или аэрозольных распылителей.
Загустители, такие как синтетические полимеры, жирные кислоты, соли и сложные эфиры жирных кислот, жирные спирты, модифицированные целлюлозы или модифицированные минеральные вещества, также могут быть использованы с жидкими носителями для образования легко распределяющихся паст, гелей, мазей, мыл и тому подобного для применения непосредственно на кожу потребителя.
Примеры полезных дерматологических композиций, которые могут быть использованы для доставки соединений формулы I в кожу, раскрыты Jacquet et al. (патент США №4608392), Geria (патент США №4992478), Smith et al. (патент США №4559157) и Wortzman (патент США №4820508).
Полезные дозировки соединений формулы I могут быть определены посредством сравнения их активности in vitro и активности in vivo в экспериментальных моделях на животных. Способы экстраполяции эффективных дозировок с мышей и других животных на людей известны в данной области техники, например, смотри патент США №4938949. Полезные дозировки ингибиторов ФДЭ IV типа известны в данной области техники. Например, смотри патент США №5877180, столбец 12.
Обычно концентрация соединения(й) формулы I в жидкой композиции, такой как лосьон, будет составлять от примерно 0,1-25 мас.%, предпочтительно от примерно 0,5-10 мас.%. Концентрация в полутвердой или твердой композиции, такой как гель или порошок, будет составлять примерно 0,1-5 мас.%, предпочтительно примерно 0,5-2,5 мас.%.
Количество соединения, или его активной соли, или производного, требуемое для применения в лечении, будет изменяться не только в соответствии с выбранной конкретной солью, но также в соответствии с путем введения, природой состояния, которое подвергают лечению, и возрастом и состоянием пациента и будет оставаться в конечном итоге на усмотрение лечащего врача или клинициста.
Однако в целом подходящая доза будет находиться в пределах от примерно 0,5 до примерно 100 мкг/кг, например от примерно 10 до примерно 75 мкг/кг массы тела в сутки, например от 3 до примерно 50 мкг на килограмм массы тела реципиента в сутки, предпочтительно в пределах от 6 до 90 мкг/кг в сутки, наиболее предпочтительно в пределах от 15 до 60 мкг/кг в сутки.
Соединение удобно вводить в стандартной лекарственной форме, например, содержащей от 5 до 1000 мкг, удобнее от 10 до 750 мкг, наиболее удобно от 50 до 500 мкг активного ингредиента на стандартную лекарственную форму.
В идеале активный ингредиент следует вводить так, чтобы достичь максимальных концентраций активного соединения в плазме от примерно 0,1 до примерно 10 нМ, предпочтительно от примерно 0,2 до 10 нМ, наиболее предпочтительно от примерно 0,5 до примерно 5 нМ. Этого можно достичь, например, путем внутривенной инъекции раствора активного ингредиента в концентрации от 0,05 до 5%, возможно в физиологическом растворе, или перорального введения в виде болюса, содержащего примерно 1-100 мкг активного ингредиента. Желательные уровни в крови можно поддерживать путем длительной инфузии, обеспечивая примерно 0,01-5,0 мкг/кг/ч, или путем прерывистых инфузии, содержащих примерно 0,4-15 мкг/кг активного(ых) ингредиента(ов).
Необходимая доза может быть удобно представлена в виде однократной дозы или в виде разделенных доз, вводимых через соответствующие интервалы, например в виде двух, трех, четырех или более суб-доз в сутки. Сами суб-дозы могут быть дополнительно разделены, например, на ряд прерывистых введений со свободными промежутками, таких как многократные ингаляции из инсуффлятора, или посредством внесения некоторого количества капель в глаз. Например, желательно вводить настоящие композиции внутривенно в течение длительного периода времени после кровоизлияния, которое вызывает воспаление.
Способность данного соединения по изобретению действовать в качестве агониста аденозиновых A2A рецепторов может быть определена с использованием фармакологических моделей, которые хорошо известны в данной области техники, или с использованием тестов, описанных ниже.
Настоящие соединения или композиции, содержащие их, вводят в качестве фармакологических стресс-факторов и используют в сочетании с одной из нескольких неинвазивных диагностических процедур для оценки проявлений перфузии миокарда. Например, аденозин внутривенно может быть использован в сочетании с визуализацией перфузии миокарда с таллием-201 для определения тяжести ишемии миокарда. В этом случае таллий-201 может быть заменен на любое из нескольких различных радиофармацевтических средств (например, радиофармацевтические средства, меченные технецием-99 m (то есть Тс-99m-сестамиби, Тс-99m-тебороксим), радиофармацевтические средства, меченные иодом-123, такие как I-123-IPPA (I123-иодфенилпентадекановая кислота) или BMIPP (бета-метил-иодфенилпентадекановая кислота), рубидий-82, азот-13 и так далее). Аналогично одно из настоящих соединений можно вводить в качестве фармакологического стресс-фактора в сочетании с радионуклидной вентрикулографией для оценки тяжести нарушения сократимости миокарда. В этом случае радионуклидные вентрикулографические исследования могут представлять собой исследование первого прохождения индикатора или синхронизированное с ЭКГ, равновесное исследование правого и/или левого желудочка. Аналогично соединение формулы (I) можно вводить в качестве фармакологического стресс-фактора в сочетании с эхокардиографией для оценки нарушений кинетики стенок миокарда. Аналогично активное соединение можно вводить в качестве фармакологического стресс-фактора в сочетании с инвазивными измерениями коронарного кровотока, например посредством внутрисердечного катетера, для определения функциональной значимости стенозированных коронарных сосудов.
Также предложен способ для диагностики нарушений перфузии миокарда у млекопитающего, включающий: (а) парентеральное введение указанному млекопитающему количества соединения или композиции, как описано выше, и (б) осуществление методики на млекопитающем для выявления наличия стенозов коронарных артерий, оценки тяжести стенозов коронарных артерий или и того, и другого. Миокардиальная дисфункция может представлять собой, например, заболевание коронарных артерий, вентрикулярную дисфункцию и различий в кровотоке через здоровые коронарные сосуды и/или стенозированные коронарные сосуды. Методика для выявления наличия и оценки тяжести заболевания коронарных артерий может представлять собой, например, радиофармацевтическую визуализацию перфузии миокарда, визуализацию функции желудочков или методики измерения скорости коронарного кровотока. Радиофармацевтическая визуализация перфузии миокарда может представлять собой, например, плоскостную сцинтиграфию, однофотонную эмиссионную компьютерную томографию (SPECT), позитронную эмиссионную томографию (PET), ядерно-магнитную резонансную (ЯМР) томографию, перфузионную контрастную эхокардиографию, цифровую субтракционную ангиографию (DSA) и сверхскоростную компьютерную томографию (CINE CT). В сочетании с радиофармацевтической визуализацией перфузии миокарда может быть использован радиофармацевтический агент, и этот радиофармацевтический агент может включать, например, радионуклид, выбранный из группы, состоящей из таллия-201, технеция-99m, азота-13, рубидия-82, иода-123 и кислорода-15. Когда радиофармацевтическая визуализация перфузии миокарда представляет собой сцинтиграфию, радиофармацевтический агент может представлять собой таллий-201. Методика визуализации функции желудочков может представлять собой, например, эхокардиографию, контрастную вентрикулографию или радионуклидную вентрикулографию. Способ измерения скорости коронарного кровотока может представлять собой, например, введение катетера с доплеровским датчиком кровотока, цифровую субтракционную ангиографию и методики радиофармацевтической визуализации. Эти способы диагностики могут также включать стадии: (а) введения человеку путем внутривенной инфузии или путем болюсной инъекции количества соединения или композиции, как описано выше, для обеспечения расширения коронарных артерий; (б) введения человеку радиофармацевтического агента, содержащего таллий-201 или технеций-99m, и (в) проведения у указанного человека сцинтиграфии с целью выявления наличия и оценки тяжести заболевания коронарных артерий. Радиофармацевтический агент может представлять собой, например, Тс-99m-сестамиби.
Способ обычно включает введение одного или более чем одного соединения формулы (I) путем внутривенной инфузии в дозах, которые являются эффективными для обеспечения расширения коронарных артерий (приблизительно 0,25-500, предпочтительно 1-250 мкг/кг/мин). Однако его применение в инвазивных условиях может включать интракоронарное введение лекарственного средства в болюсных дозах 0,5-50 мкг.
Предпочтительные способы включают применение соединения формулы (I) в качестве замены физической нагрузки в сочетании с визуализацией перфузии миокарда для выявления наличия и/или оценки тяжести заболевания коронарных артерий у людей, где визуализацию перфузии миокарда осуществляют посредством любой из нескольких методик, включая радиофармацевтическую визуализацию перфузии миокарда с использованием плоскостной сцинтиграфии или однофотонной эмиссионной компьютерной томографии (SPECT), позитронной эмиссионной томографии (PET), ядерно-магнитной резонансной (ЯМР) томографии, перфузионной контрастной эхокардиографии, цифровой субтракционной ангиографии (DSA) или сверхскоростной компьютерной томографии (CINE СТ).
Также предложен способ, включающий применение соединения формулы (I) в качестве замены физической нагрузки в сочетании с визуализацией для выявления наличия и/или оценки тяжести ишемической вентрикулярной дисфункции у людей, где ишемическую вентрикулярную дисфункцию измеряют посредством любой из нескольких методик визуализации, включая эхокардиографию, контрастную вентрикулографию или радионуклидную вентрикулографию. Миокардиальная дисфункция может представлять собой заболевание коронарных артерий, вентрикулярную дисфункцию, различия в кровотоке через здоровые коронарные сосуды и стенозированные коронарные сосуды и тому подобное.
Также предложен способ, включающий применение соединения формулы (I) в качестве агента, усиливающего коронарный кровоток, в сочетании со способом измерения скорости коронарного кровотока, для оценки способности коронарных артерий к расширению (резервная вместимость) у людей, где скорость коронарного кровотока измеряют посредством любой из нескольких методик, включая введение катетера с доплеровским датчиком кровотока и цифровую субтракционную ангиографию.
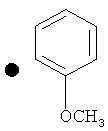
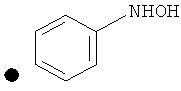
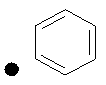
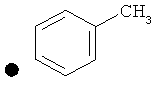
Примеры соединений по изобретению показаны в таблице, приведенной ниже, где соединения под номерами 1, 2, 4 и 32 представлены только в целях сравнения. Соединения являются соединениями формулы (I), если не оговорено особо.
R4=A: CH2OH; В: С(O)Nэтил; С: С(O)Nциклопропил.
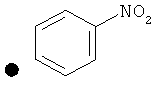
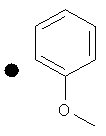
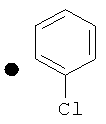
++++<1,0 нМ; +++<10 нМ; ++<100 нМ; +>100 нМ;
 указывает точку присоединения Z группы к Y.
указывает точку присоединения Z группы к Y.
Изобретение будет дополнительно описано посредством ссылки на следующие подробные примеры, которые даны для иллюстрации изобретения и не предназначены для его ограничения.
Примеры
Спектры ядерного магнитного резонанса протонов (1Н ЯМР) записывали на спектрофотометре Gemini-2000 (300 МГц) фирмы Varian (или аналогичном приборе). Величины химических сдвигов выражены в млн-1 (миллионные доли) относительно тетраметилсилана. Для регистрации данных; s=синглет, d=дублет, t=триплет, q=квартет и m=мультиплет. Масс-спектры измеряли на масс-спектрометре серии LCQ Advantage фирмы Finnigan. Аналитическую ВЭЖХ проводили на хроматографе LC10 или LC20 Systemtimes.150 мм) фирмы Shimazdu, как описано ниже. Препаративную ВЭЖХ проводили на хроматографе Discovery HPLC фирмы Shimazdu с колонкой Shim-pack VP-ODS С18 (20×100 мм), работающей при комнатной температуре. Соединения элюировали со скоростью 30 мл/мин градиентом от 20-80% воды (содержащей 0,1% ТФУ) до метанола в течение 15 минут с УФ-детектированием при 254 нм при использовании детектора с перестраиваемой длиной волны SPD10A VP. Все конечные соединения, представленные здесь, как было установлено, достигали более чем 98%-ной чистоты посредством ВЭЖХ. Флэш-хроматографию проводили на силикагеле Silicyle 60A (230-400 меш) или при применении хроматографических колонок многоразового использования и системы от RT Scientific, Manchester N.H. Все реакции проводили в атмосфере азота в высушенной над пламенем стеклянной посуде, если не оговорено особо.
Общий способ 1: типичный способ образования пиперидилалкинов
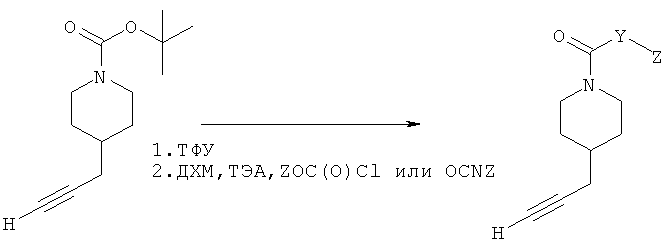
Неразбавленную ТФУ (трисрторуксусную кислоту) (4 мл) добавляли к трет-бутил-4-(проп-2-инил)пиперидин-1-карбоксилату (1,5 г; 18,56 ммоль) при 0°С, раствор оставляли до достижения комнатной температуры и перемешивали в течение дополнительных 3-12 часов. ТФУ удаляли при пониженном давлении до получения густого масла. Это масло охлаждали до 0°С и затем добавляли ДХМ (дихлорметан) (20 мл) и ТЭА (5 мл) с последующим добавлением по каплям соответствующего хлорформиата или изоцианата (~1,5 зкв.). Смесь перемешивали при комнатной температуре в атмосфере N2 в течение 24 ч, протекание реакции контролировали посредством ТСХ и ЖХ/МС. Реакционную смесь обрабатывали посредством фильтрации осадка и промывки 2-3 раза ErOAc с последующим концентрированием при пониженном давлении до получения густого желтого масла. Неочищенную реакционную смесь очищали посредством хроматографии на силикагеле с градиентом Нех(гексан)/ЕtOАс (0-20%). Продукт собирали и концентрировали при пониженном давлении с получением каждый раз желтоватого масла, часто требующего дальнейшей очистки при использовании таких же условий очистки, как указано ранее. Соединения выделяли в виде пахучих желтых масел с выходами 50-90% и характеризовали посредством ЖХ/МС и/или 1H ЯМР-спектроскопии. ВЭЖХ проводили на хроматографе Shimadzu HPLC с использованием колонки Water's Atlantis C18 размером 4,6×150 мм с размером частиц 5 мкм, элюируемой в течение 15 минут линейным градиентом 45-95% МеОН/Н2О (содержащей 0,1% муравьиной кислоты). ТСХ проводили при использовании нейтральных пластин EMD Chemicals Aluminumoxd 60 F254, обрабатываемых 20% EtOAc/Hex и визуализируемых в УФ-свете при длине волны 254 нм или посредством проявления ванилином.
Общий способ 2: типичный способ сочетания Соногаширы алкинов с аналогами 2-иодаденозина
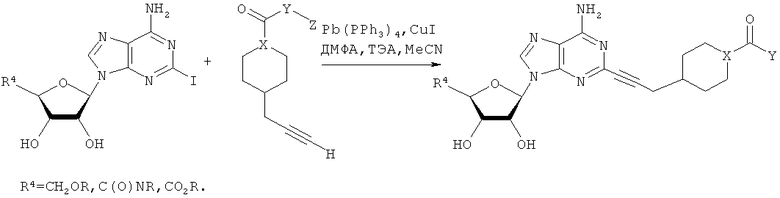
Соответствующий алкин (0,0800 мл; 0,1121 ммоль) добавляли к раствору N-циклопропил-2-иодаденозин-5'-уронамида, N-этил-2-иодаденозин-5'-уронамида или 2-иодаденозина (0,0510 г; 0,1121 ммоль) в ДМФА (5 мл). Затем добавляли ацетонитрил (3 мл) и ТЭА (1 мл), при этом все растворители непосредственно перед использованием дегазировали азотом в течение минимум 3 ч. Смесь перемешивали при комнатной температуре в атмосфере N2/аргон, пока добавляли Pd(PPh3)4 (каталитическое количество, ~0,05 мол.%) и CuI (каталитическое количество, ~0,05 мол.%). Раствор оставляли для перемешивания при комнатной температуре в течение 24-72 ч и протекание реакции контролировали посредством ЖХ/МС. Дополнительные количества алкина, ТЭА, Pd(PPh3)4 и CuI возможно добавляли через 24 ч, если оказывалось, что реакция протекает медленно, чтобы довести реакцию до завершения и израсходовать все нуклеотидное исходное вещество. Затем смесь фильтровали сразу или через 12-24 ч после добавления кремнийсвязанных очистителей, Si-тиола и Si-TAAcOH от Silicycle, которые добавляли для извлечения Cu и палладия. Остаток промывали 2-3 раза смесью МеОН/EtOH и концентрировали при пониженном давлении с получением густого коричневого масла. Неочищенную реакционную смесь очищали посредством хроматографии на силикагеле с градиентом ДХМ/МеОН (0-15%). Продукт собирали и концентрировали при пониженном давлении с получением каждый раз желтоватого маслянистого твердого вещества, часто требующего дальнейшей очистки с использованием колонки С18 (либо препаративной ВЭЖХ, либо стандартной колонки для флэш-хроматографии) с градиентом МеОН/H2O. Соединения выделяли в виде белых твердых веществ с выходами 10-90% и характеризовали посредством ЖХ/МС и/или 1Н/13С ЯМР-спектроскопии. ВЭЖХ проводили на хроматографе Shimadzu HPLC с использованием колонки Water's Atlantis C18 размером 4,6×150 мм с размером частиц 5 мкм, элюируемой в течение 15 минут линейным градиентом 45-95% МеОН/Н2О (содержащей 0,1% муравьиной кислоты). ТСХ проводили при использовании нейтральных пластин EMD Chemicals Aluminumoxd 60 F254, обрабатываемых 10% МеОН/ДХМ и визуализируемых в УФ-свете при длине волны 254 нм или посредством проявления ванилином.
Промежуточное соединение 1
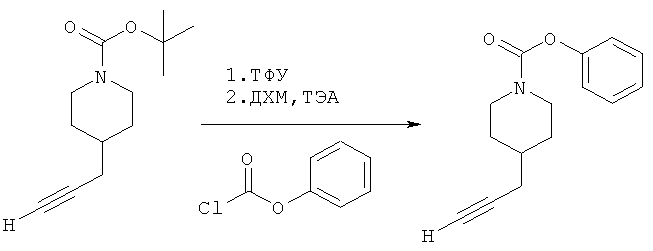
Фенил-4-(проп-2-инил)пиперидин-1-карбоксилат. Фенилхлорформиат (6,2 г; 40,2 ммоль) добавляли к раствору трет-бутил-4-(проп-2-инил)пиперидин-1-карбоксилата (1,65 г; 13,4 ммоль) согласно общему способу 1. Выход = 0,600 г, 34%. m/z MH+=244,08. Время удерживания при ВЭЖХ=10,3 мин.
Промежуточное соединение 2
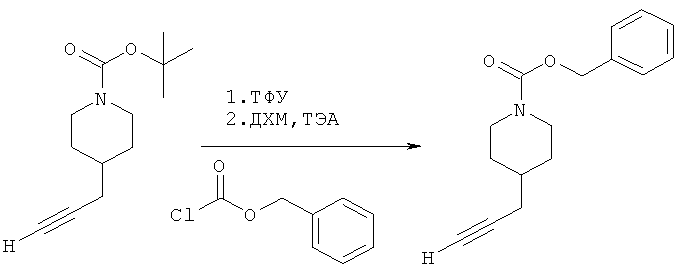
Бензил-4-(проп-2-инил)пиперидин-1-карбоксилат.
Бензилхлорформиат (6,9 г; 40,4 ммоль) добавляли к раствору трет-бутил-4-(проп-2-инил)пиперидин-1-карбоксилата (1,65 г; 13,4 ммоль) согласно общему способу 1. Выход = 0,700 г; 37%. m/z МН+=258,02. Время удерживания при ВЭЖХ=10,2 мин.
Промежуточное соединение 3
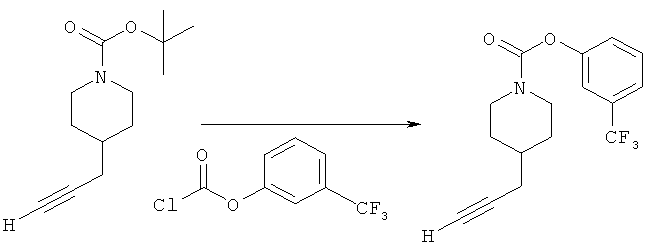
3-(Трифторметил)фенил-4-(проп-2-инил)пиперидин-1-карбоксилат. 3-(Трифторметил)фенилхлорформиат (5,0 г; 29,3 ммоль) добавляли к раствору трет-бутил-4-(проп-2-инил)пиперидин-1-карбоксилата (1,50 г; 6,72 ммоль) согласно общему способу 1. Выход = 1,1031 г; 64%. m/z MH+=302,02. Время удерживания при ВЭЖХ=10,8 мин.
Промежуточное соединение 4
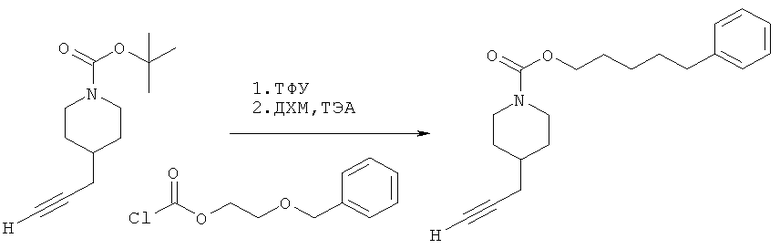
3-(Бензилокси)пропил-4-(проп-2-инил)пиперидин-1-карбоксилат. 3-(Трифторметил)фенилхлорформиат (5,0 г; 23,3 ммоль) добавляли к раствору трет-бутил-4-(проп-2-инил)пиперидин-1-карбоксилата (1,20 г; 5,37 ммоль) согласно общему способу 1. Выход = 1,191 г; 70%. m/z MH+=312,04. Время удерживания при ВЭЖХ=13,9 мин.
Промежуточное соединение 5
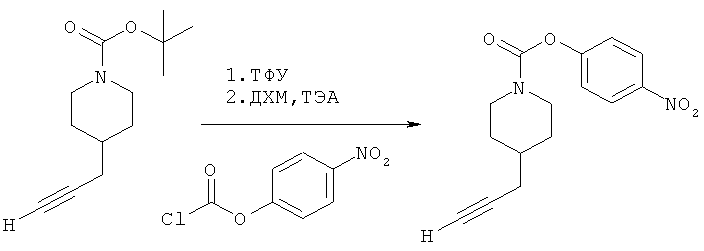
4-(Нитро)фенил-4-(проп-2-инил)пиперидин-1-карбоксилат. 4-(Нитро)фенилхлорформиат (5,0 г; 24,8 ммоль) добавляли к раствору трет-бутил-4-(проп-2-инил)пиперидин-1-карбоксилата (4,25 г; 19,0 ммоль) согласно общему способу 1. Выход = 5,72 г; 80%. m/z MH+=289,04. Время удерживания при ВЭЖХ=10,4 мин.
Промежуточное соединение 6
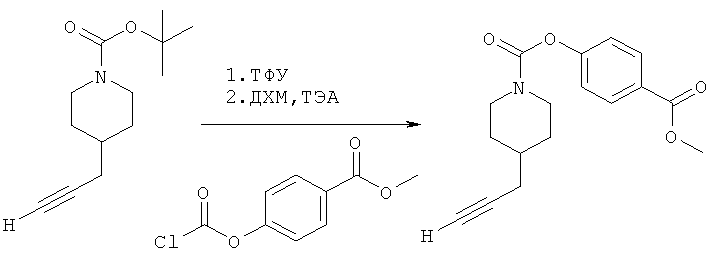
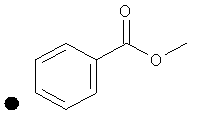
4-(Метоксикарбонил)фенил-4-(проп-2-инил)пиперидин-1-карбоксилат. 4-(Метоксикарбонил)фенилхлорформиат (5,00 г; 23,3 ммоль) добавляли к раствору трет-бутил-4-(проп-2-инил)пиперидин-1-карбоксилата (4,00 г; 17,9 ммоль) согласно общему способу 1. Выход = 5,54 г; 99%. m/z МН+=302,02. Время удерживания при ВЭЖХ=10,8 мин.
Промежуточное соединение 7
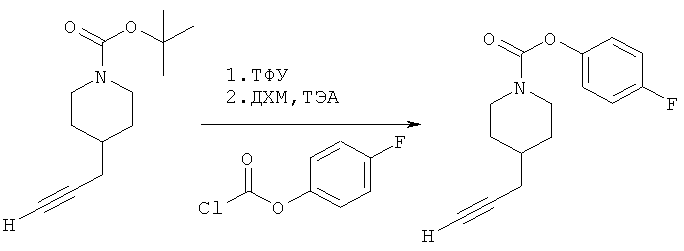
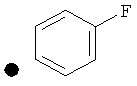
4-(Фтор)фенил-4-(проп-2-инил)пиперидин-1-карбоксилат. 4-(Фтор)фенилхлорформиат (5,00 г; 28,6 ммоль) добавляли к раствору трет-бутил-4-(проп-2-инил)пиперидин-1-карбоксилата (4,90 г; 21,9 ммоль) согласно общему способу 1. Выход = 5,54 г; 99%. m/z MH+=262,03. Время удерживания при ВЭЖХ=10,6 мин.
Промежуточное соединение 8
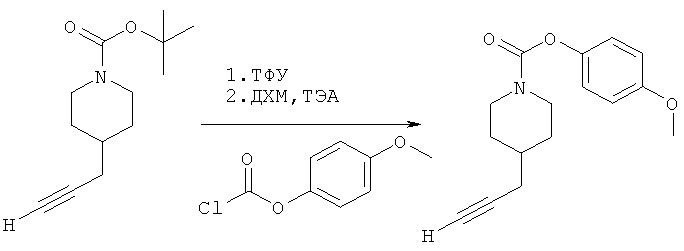
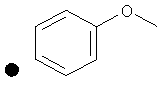
4-(Метокси)фенил-4-(проп-2-инил)пиперидин-1-карбоксилат. 4-(Метокси)фенилхлорформиат (5,00 г; 26,8 ммоль) добавляли к раствору трет-бутил-4-(проп-2-инил)пиперидин-1-карбоксилата (4,60 г; 20,9 ммоль) согласно общему способу 1. Выход=5,02 г; 90%. m/z MH+=274,06. Время удерживания при ВЭЖХ=12,2 мин.
Промежуточное соединение 9
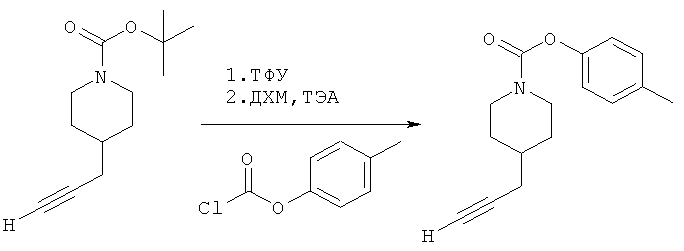
4-(Метил)фенил-4-(проп-2-инил)пиперидин-1-карбоксилат. 4-(Метил)фенилхлорформиат (5,00 г; 29,3 ммоль) добавляли к раствору трет-бутил-4-(проп-2-инил)пиперидин-1-карбоксилата (5,03 г; 22,6 ммоль) согласно общему способу 1. Выход = 5,20 г; 90%. m/z MH+=258,11. Время удерживания при ВЭЖХ=13,0 мин.
Промежуточное соединение 10
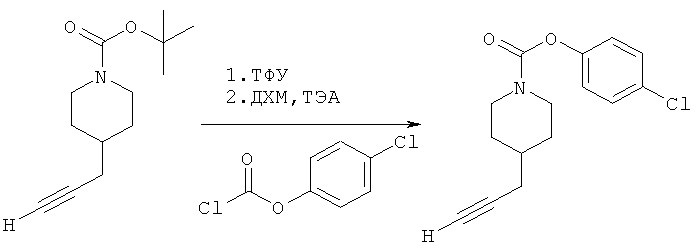
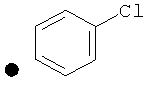
4-(Хлор)фенил-4-(проп-2-инил)пиперидин-1-карбоксилат. 4-(Хлор)фенилхлорформиат (5,00 г; 26,2 ммоль) добавляли к раствору трет-бутил-4-(проп-2-инил)пиперидин-1-карбоксилата (5,50 г; 20,1 ммоль) согласно общему способу 1. Выход=4,83 г; 86%. m/z MH+=278,13. Время удерживания при ВЭЖХ=13,0 мин.
Промежуточное соединение 11
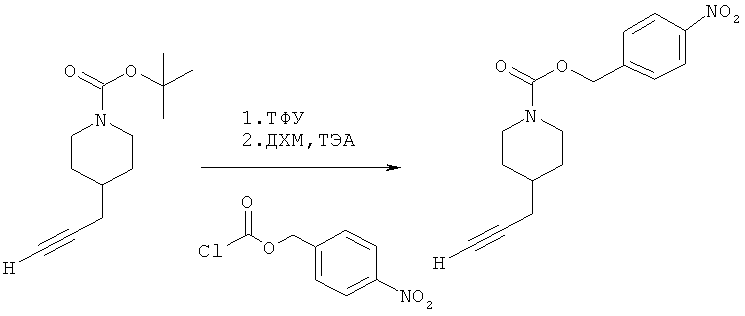
4-(Нитро)бензил-4-(проп-2-инил)пиперидин-1-карбоксилат. 4-(Нитро)бензилхлорформиат (5,00 г; 23,2 ммоль) добавляли к раствору трет-бутил-4-(проп-2-инил)пиперидин-1-карбоксилата (3,98 г; 17,4 ммоль) согласно общему способу 1. Выход = 4,66 г; 86%. m/z MH+=302,98. Время удерживания при ВЭЖХ=12,3 мин.
Промежуточное соединение 12
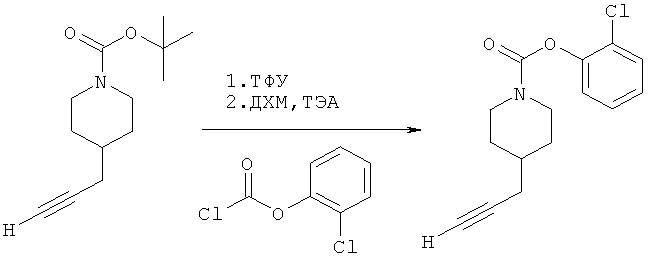
2-(Хлор)фенил-4-(проп-2-инил)пиперидин-1-карбоксилат. 2-(Хлор)фенилхлорформиат (5,00 г; 26,2 ммоль) добавляли к раствору трет-бутил-4-(проп-2-инил)пиперидин-1-карбоксилата (4,50 г; 20,2 ммоль) согласно общему способу 1. Выход = 4,56 г; 82%. m/z MH+=278,13. Время удерживания при ВЭЖХ=9,3 мин.
Промежуточное соединение 13
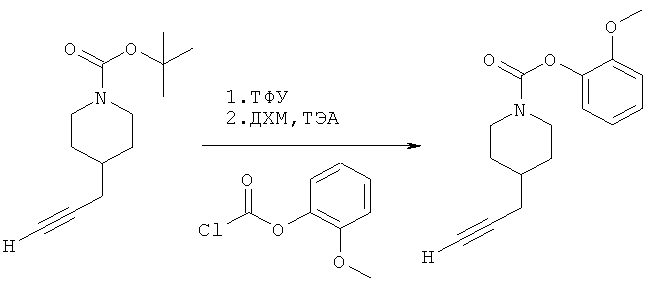
2-(Метокси)фенил-4-(проп-2-инил)пиперидин-1-карбоксилат. 2-(Метокси)фенилхлорформиат (5,00 г; 26,8 ммоль) добавляли к раствору трет-бутил-4-(проп-2-инил)пиперидин-1-карбоксилата (4,60 г; 20,6 ммоль) согласно общему способу 1. Выход = 5,03 г; 89%. m/z MH+=274,04. Время удерживания при ВЭЖХ=8,7 мин.
Общий способ 3: типичный способ образования пиперидилкарбаматов из хлорформиатов, не имеющихся в продаже
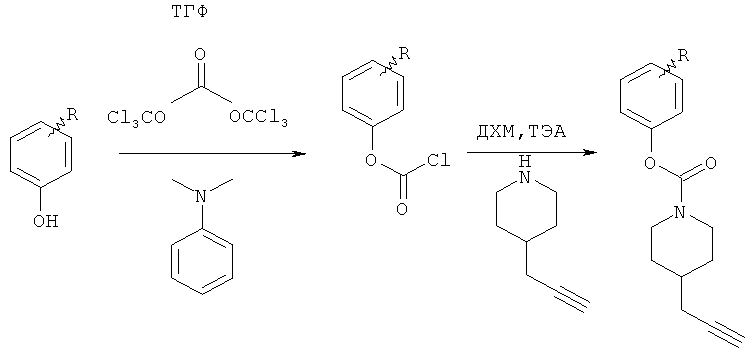
Трифосген (2,849 г; 9,6 ммоль) растворяли в безводном ТГФ (60 мл) и охлаждали до 5°С в атмосфере инертного газа. К этому раствору медленно добавляли дизамещенный фенол (2,96 ммоль) в виде раствора в смеси ТГФ и диметиланилина, 1:1 (общий объем 7,5 мл). Реакционную смесь перемешивали при 5°С в течение 10 минут, а затем перемешивали в течение дополнительных 3 ч при комнатной температуре. Полученную суспензию хлорформиата использовали, как изложено в общем способе 1, с получением соответствующих пиперидилалкинов.
Промежуточное соединение 14
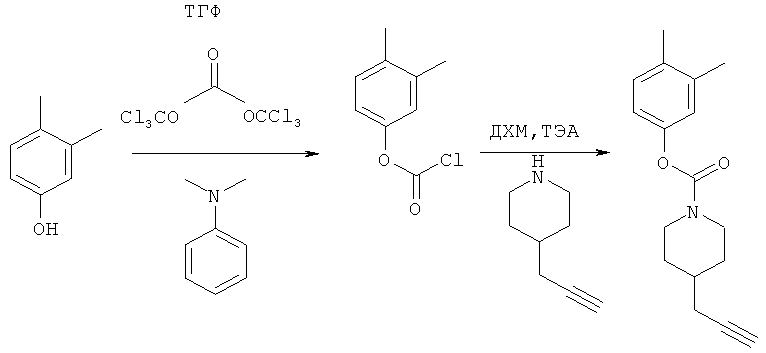
3,4-Диметилфенил-4-(проп-2-инил)пиперидин-1-карбоксилат. 3,4-Диметил-фенол (0,362 г; 2,96 ммоль) добавляли в виде раствора в ТГФ и диметиланилине к смеси трифосгена (0,285 г; 0,96 ммоль) в ТГФ согласно общему способу 3. Выход = 0,350 г; 43%. m/z MH+=272,26. Время удерживания при ВЭЖХ=12,27 мин.
Промежуточное соединение 15
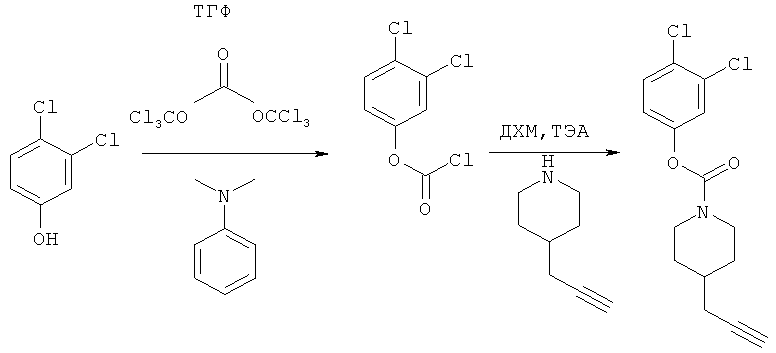
3,4-Дихлорфенил-4-(проп-2-инил)пиперидин-1-карбоксилат. 3,4-Дихлорфенол (4,83 г; 29,6 ммоль) добавляли в виде раствора в ТГФ и диметиланилине к смеси трифосгена (2,849 г; 9,6 ммоль) в ТГФ согласно общему способу 3. Выход = 5,3 г; 57%. m/z MH+=312,14. Время удерживания при ВЭЖХ=12,38 мин.
Промежуточное соединение 16
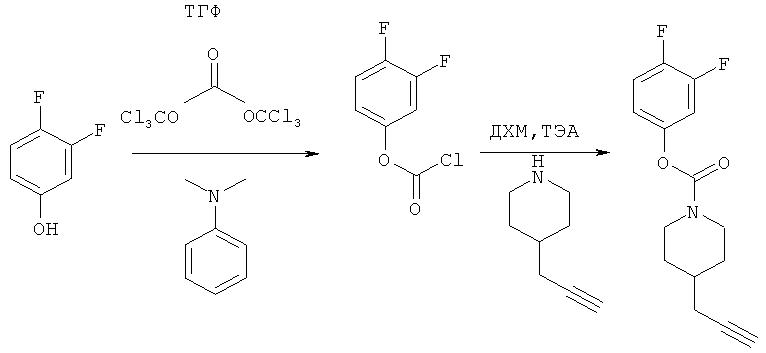
3,4-Дифторфенил-4-(проп-2-инил)пиперидин-1-карбоксилат. 3,4-Дифторфенол (3,85 г; 29,6 ммоль) добавляли в виде раствора в ТГФ и диметиланилине к смеси трифосгена (2,849 г; 9,6 ммоль) в ТГФ согласно общему способу 3. Выход = 4,6 г; 56%. m/z MH+=280,15. Время удерживания при ВЭЖХ=11,88 мин.
Промежуточное соединение 17
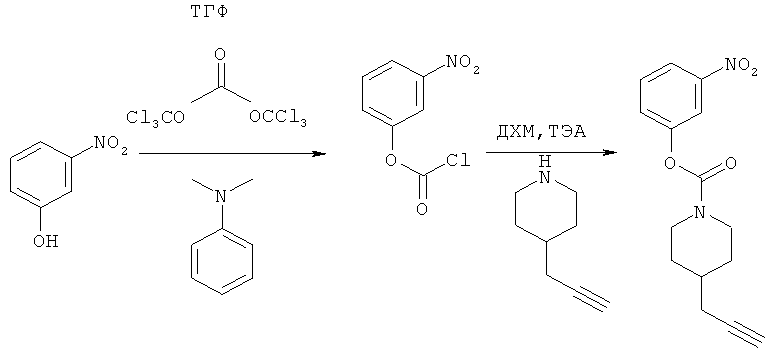
3-Нитрофенил-4-(проп-2-инил)пиперидин-1-карбоксилат. 3-Нитрофенол (4,12 г; 29,6 ммоль) добавляли в виде раствора в ТГФ и диметиланилине к смеси трифосгена (2,849 г; 9,6 ммоль) в ТГФ согласно общему способу 3. Выход = 5,53 г; 86%. m/z MH+=289,18. Время удерживания при ВЭЖХ=11,78 мин.
Промежуточное соединение 18
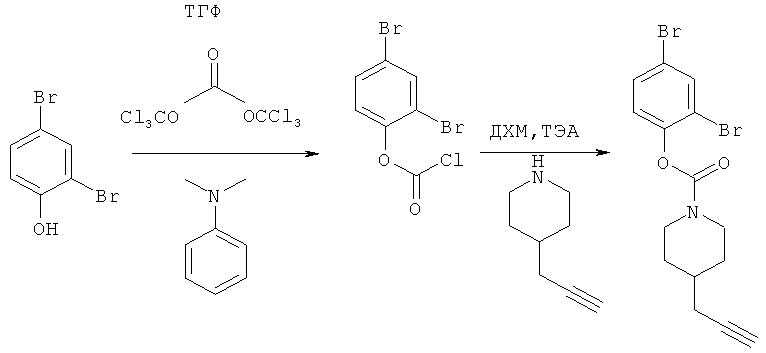
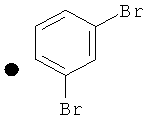
2,4-Дибромфенил-4-(проп-2-инил)пиперидин-1-карбоксилат. 2,4-Дибромфенол (7,45 г; 29,6 ммоль) добавляли в виде раствора в ТГФ и диметиланилине к смеси трифосгена (2,849 г; 9,6 ммоль) в ТГФ согласно общему способу 3. Выход = 6,09 г; 68%. m/z MH+=402,22. Время удерживания при ВЭЖХ=12,49 мин.
Соединение 1
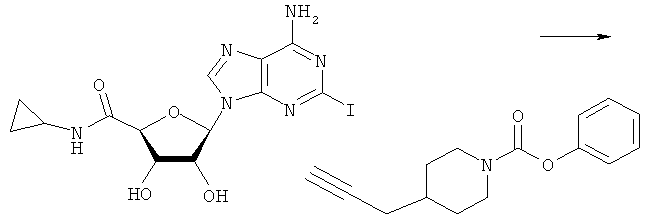
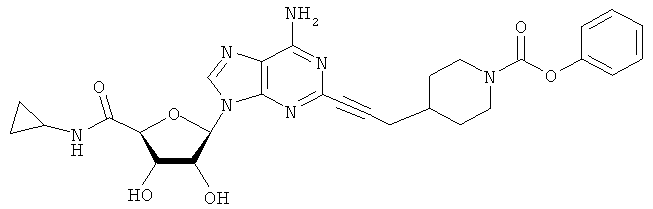
N-Циклопропил-2-{3-(1-(фенилоксикарбонил)-4-пиперидинил]-1-пропин-1-ил}аденозин-5'-уронамид. Партия: JR28-23. Фенил-4-(проп-2-инил)пиперидин-1-карбоксилат, партия JR28-11 (0,0800 мл; 0,1121 ммоль) добавляли к раствору N-циклопропил-2-иодаденозин-5'-уронамида (0,0510 г; 0,1121 ммоль) согласно общему способу 2. Выход = 19,0 мг; 31%. 1H ЯМР (CD3OD) δ 8.40 (s, 1H), 8.20 (s, 1H), 7.39-7.07 (dt, 5H), 6.02 (d, 1H), 4.39 (m, 1H), 3.31-2.90 (2×t, s, 4H), 2.71 (m, 4H), 2.49 (m, 1H), 1.98 (bd, 4H), 1.43-1.18 (2×m, 5H), 0.95-0.75 (2×m, 4H). m/z MH+=562,19. Время удерживания при ВЭЖХ=9,3 мин.
Соединение 2
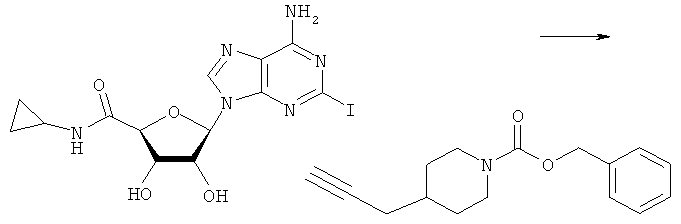
N-Циклопропил-2-{3-[1-(бензилоксикарбонил)-4-пиперидинил]-1-пропин-1-ил}аденозин-5'-уронамид. Партия: JR28-25. Бензил-4-(проп-2-инил)пиперидин-1-карбоксилат, партия JR28-13 (0,0800 мл; 0,1121 ммоль) добавляли к раствору N-циклопропил-2-иодаденозин-5'-уронамида (0,0510 г; 0,1121 ммоль) согласно общему способу 2. Выход = 26,2 мг; 41%. 1H ЯМР (CD3OD) δ 8.39 (s, 1Н), 8.33 (s, 1H), 7.34-7.32 (dt, 5H), 6.01 (d, 1H), 5.10 (m, 2H), 4.81-4.79 (m, 1H), 4.4 (s, 2H), 4.20-4.15 (d, 2H), 2.50 (2×m, 4H), 2.42 (d, 1H), 1.84-1.85 (b d, 4H), 1.30-1.27 (b m, 5H) 0.76-53 (2×m, 4H). 13С (CD3OD) δ 173.72, 157.17, 156.97, 150.81, 147.86, 142.99, 138.23, 129.53, 129.06, 128.83, 120.30, 90.33, 86.41, 85.75, 74.74, 73.89, 68.23, 45.13, 36.60, 32.56, 26.63, 23.38, 6.96. m/z MH+=576,19. Время удерживания при ВЭЖХ=9,8 мин.
Соединение 3
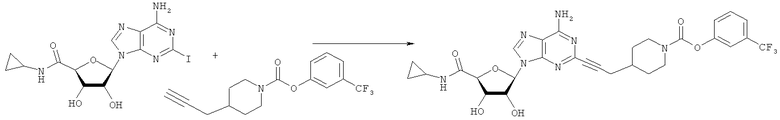
N-Циклопропил-2-{3-[1-(3-(трифторметил)феноксикарбонил)-4-пиперидинил]-1-пропин-1-ил}аденозин-5'-уронамид. Партия: JR28-71. 4-Трифторметил-4-(проп-2-инил)пиперидин-1-карбоксилат, партия JR28-57 (0,080 мл; 0,1121 ммоль) добавляли к раствору N-циклопропил-2-иодаденозин-5'-уронамида (0,0500 г; 0,1121 ммоль) согласно общему способу 2. Выход = 32,0 мг; 46%. 1H ЯМР (CD3OD) δ 8.41 (s, 1H), 7.60-7.34 (m, 4H), 6.02 (d, 1H), 4.84 (m, 1H), 4.31 (m, 2H), 3.00 (2×m, 2H), 2.99-2.94 (2×m, 4H), 2.71 (m, 1H), 2.46 (d, 4H), 1.98-1.30 (2×m, 5H), 0.77-0.58 (2×m, 4H). m/z MH+=630,18. Время удерживания при ВЭЖХ=10,5 мин.
Соединение 4
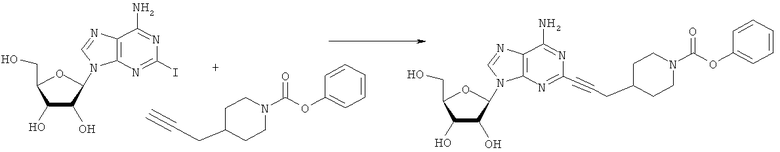
2-{3-[1-(Феноксикарбонил)пиперидин-4-ил]пропин-1-ил}аденозин. Партия: АВ-9-97. Фенил-4-(проп-2-инил)пиперидин-1-карбоксилат, партия JR28-11 (0,329 г; 1,352 ммоль) добавляли к раствору 2-иодаденозина (0,513 г; 1,305 ммоль) согласно общему способу 2. Выход: 54,5 мг; 82%. 1Н ЯМР (CD3OD) δ 8.30 (s, 1H), 7.38-7.06 (3×m, 5H), 5.94 (d, 1H), 4.71 (m, 1H), 4.32-4.17 (2×m, 7Н), 3.92-3.72 (2×d, 2H), 3.10-2.90 (2×bt, 4H), 2.48 (d, 5H), 1.93 (2×m, 5H). m/z МН+=509,18. Время удерживания при ВЭЖХ=5,3 мин.
Соединение 5
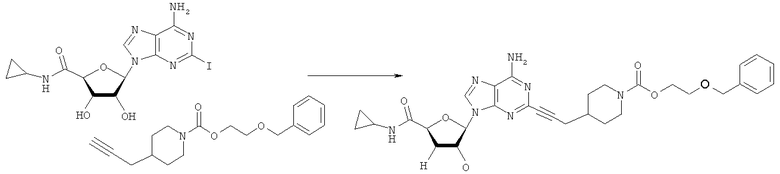
N-Циклопропил-2-{3-[1-(2-(бензилокси)этоксикарбонил)-4-пиперидинил]-1-пропин-1-ил}аденозин-5'-уронамид. Партия: JR28-69. 2-(Бензилокси)этил-4-(проп-2-инил)пиперидин-1-карбоксилат, партия JR28-55 (0,080 мл; 0,1121 ммоль) добавляли к раствору N-циклопропил-2-иодаденозин-5'-уронамида (0,0500 г; 0,1121 ммоль) согласно общему способу 2. Выход = 20,0 мг; 29%. 1H ЯМР (CD3OD) δ 8.20 (s, 1H), 7.55 (s, 1H), 7.33-7.30 (m, 5H), 6.02 (d, 1H), 4.86 (m, 1H), 4.53 (m, 2H), 4.41-4.13 (4×m, 8H), 3.74-3.66 (m, 4H), 2.70 (m, 1H), 2.41-2.36 (d, 4H), 1.87-1.26 (2×m, 5H), 0.76-0.54 (2×m, 4H). m/z MH+=620,22. Время удерживания при ВЭЖХ=9,6 мин.
Соединение 6
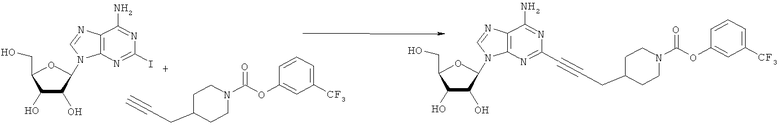
2-{3-[1-((3-Трифторметил)феноксикарбонил)пиперидин-4-ил]пропин-1-ил}аденозин. Партия: АВ-9-129. 4-Трифторметил-4-(проп-2-инил)пиперидин-1-карбоксилат, партия JR28-57 (0,844 г; 2,71 ммоль) добавляли к раствору 2-иодаденозина (0,978 г; 2,488 ммоль) согласно общему способу 2. Выход: 1,205 г; 84%. 1H ЯМР (CD3OD) δ 8.40 (s, 1H), 7.59-7.37 (3×m, 4H), 5.95 (d, 1H), 4.71 (m, 2H), 4.31-4.16 (2×m, 5H), 3.91-3.75 (2×d, 2H), 3.09-2.94 (2×bt, 4H), 2.48 (d, 5H), 1.96-1.44 (2×m, 5H). m/z MH+=577,13. Время удерживания при ВЭЖХ=11,2 мин.
Соединение 7
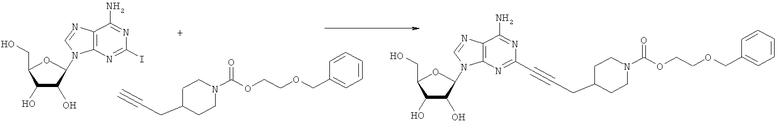
2-{3-[1-((2-Бензилокси)этоксикарбонил)пиперидин-4-ил]пропин-1-ил}аденозин. Партия: АВ-9-131. 2-(Бензилокси)этил-4-(проп-2-инил)пиперидин-1-карбоксилат, партия JR28-55 (0,780 г; 2,59 ммоль) добавляли к раствору 2-иодаденозина (0,936 г; 2,381 ммоль) согласно общему способу 2. Выход: 0,575 г; 43%. 1H ЯМР (CD3OD) δ 8.29 (s, 1Н), 7.31 (m, 5H), 5.93 (d, 1H), 4.54 (s, 2H), 4.70-4.14 (3×m, 5H), 3.87-3.66 (2×m, 6H), 2.80 (bs, 4H), 2.42 (d, 5H), 1.89-1.50 (2×m, 5H). m/z MH+=567,17. Время удерживания при ВЭЖХ=8,0 мин.
Соединение 8
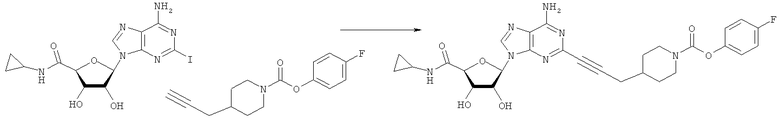
N-Циклопропил-2-{3-[1-(4-фторфеноксикарбонил)-4-пиперидинил]-1-пропин-1-ил}аденозин-5'-уронамид. Партия: JR28-97. 4-Фторфенокси-4-(проп-2-инил)пиперидин-1-карбоксилат, партия JR28-87 (0,150 мл; 0,1681 ммоль) добавляли к раствору N-циклопропил-2-иодаденозин-5'-уронамида (0,075 г; 0,1681 ммоль) согласно общему способу 2. Выход = 43,0 мг; 44%. 1H ЯМР (CD3OD) δ 8.41 (s, 1H), 7.65-7.61 (m, 4H), 7.01 (d, 1H), 6.02 (d, 1H), 4.83 (m, 1H), 4.41-4.38 (m, 2H), 3.33-2.95 (3×m, 6H), 2.71 (m, 1H), 2.48-2.46 (d, 4H), 1.97-1.92 (2×m, 5H), 0.78-0.55 (2×m, 4H). m/z MH+=580,24. Время удерживания при ВЭЖХ=9,3 мин.
Соединение 9
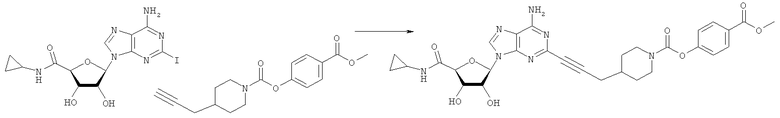
N-Циклопропил-2-{3-[1-(4-метоксикарбонилфеноксикарбонил)-4-пиперидинил]-1-пропин-1-ил}аденозин-5'-уронамид. Партия: JR28-95. 4-(Метоксикарбонил)фенил-4-(проп-2-инил)пиперидин-1-карбоксилат, партия JR28-85 (0,150 мл; 0,2241 ммоль) добавляли к раствору N-циклопропил-2-иодаденозин-5'-уронамида (0,100 г; 0,2241 ммоль) согласно общему способу 2. Выход = 68,9 мг; 50%. 1H ЯМР (CD3OD) δ 8.40 (s, 1H), 8.04-8.02 (m, 3H), 7.23-7.21 (d, 2H), 6.02 (d, 1H), 4.81 (m, 1H), 4.32 (m, 2H), 3.89 (s, 4H), 3.09-2.71 (2×bt, 4H), 2.72 (m, 1H), 2.49 (d, 4H), 1.98-1.43 (2×m, 5H), 0.78-0.54 (2×m, 4H). m/z МН+=620,25. Время удерживания при ВЭЖХ=9,5 мин.
Соединение 10
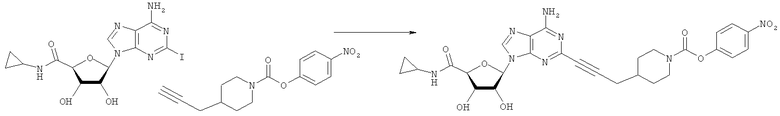
N-Циклопропил-2-{3-[1-(4-нитрофенилоксикарбонил)-4-пиперидинил]-1-пропин-1-ил}аденозин-5'-уронамид. Партия: JR28-93. 4-Нитрофенил-4-(проп-2-инил)пиперидин-1-карбоксилат, партия JR28-83 (0,150 мл; 0,2241 ммоль) добавляли к раствору N-циклопропил-2-иодаденозин-5'-уронамида (0,100 г; 0,2241 ммоль) согласно общему способу 2. Выход = 47,0 мг; 35%. 1H ЯМР (CD3OD) δ 8.41 (s, 1H), 8.29-8.24 (m, 3Н), 7.38-7.34 (d, 2H), 6.02 (d, 1H), 4.80 (m, 1H), 4.42-4.19 (m, 4H), 3.12-2.95 (2×bt, 4H), 2.71 (m, 1H), 2.49 (d, 4H), 1.98-1.47 (2×m, 5H), 0.78-0.54 (2×m, 4H). m/z MH+=607,26. Время удерживания при ВЭЖХ=9,1 мин.
Соединение 11
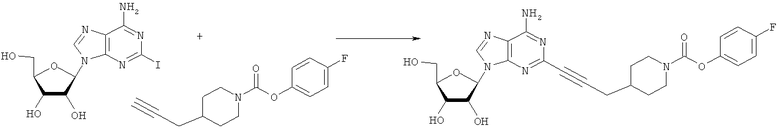
2-{3-[1-((4-Фторфенокси)карбонил)пиперидин-4-ил]пропин-1-ил}аденозин. Партия: АВ-9-147. 4-Фторфенил-4-(проп-2-инил)пиперидин-1-карбоксилат, партия JR28-97 (0,700 г; 2,68 ммоль) добавляли к раствору 2-иодаденозина (0,812 г; 2,065 ммоль) согласно общему способу 2. Выход: 0,902 г; 83%. 1H ЯМР (CD3OD) δ 8.34 (s, 1H), 7.11 (m, 4H), 5.98 (d, 1H), 4.76 (m, 2H), 4.37-4.19 (2×m, 3Н), 3.95-3.75 (2×d, 2H), 3.07-2.93 (2×bt, 4H), 2.47 (d, 5H), 1.98-1.44 (2×m, 5H). m/z MH+=527,19. Время удерживания при ВЭЖХ=7,6 мин.
Соединение 12
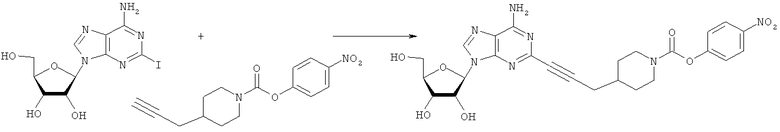
2-{3-[1-((4-Нитрофенокси)карбонил)пиперидин-4-ил]пропин-1-ил}аденозин. Партия: АВ-9-149. 4-Нитрофенил-4-(проп-2-инил)пиперидин-1-карбоксилат, партия JR28-97 (0,630 г; 2,185 ммоль) добавляли к раствору 2-иодаденозина (0,727 г; 1,849 ммоль) согласно общему способу 2. Выход: 1,192 г; 65%. 1H ЯМР (CD3OD) δ 8.26 (m, 1H), 8.25-7.34 (2×m, 4H), 5.94 (d, 1H), 4.72 (m, 2H), 4.32-4.14 (2×m, 3Н), 3.91-3.71 (2×d, 2H), 3.17-2.90 (2×bt, 4H), 2.48 (d, 5H), 1.45 (m, 5H). m/z MH+=554,15. Время удерживания при ВЭЖХ=7,3 мин.
Соединение 13
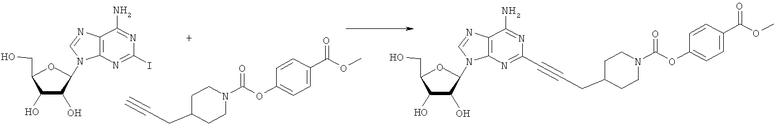
2-{3-[1-((4-Метоксикарбонилфенокси)карбонил)пиперидин-4-ил]пропин-1-ил}аденозин. Партия: АВ-9-151. 4-(Метоксикарбонил)фенил-4-(проп-2-инил)пиперидин-1-карбоксилат, партия JR28-85 (0,670 г; 2,223 ммоль) добавляли к раствору 2-иодаденозина (0,675 г; 1,717 ммоль) согласно общему способу 2. Выход: 0,648 г; 67%. 1H ЯМР (CD3OD) δ 8.30 (s, 1H), 8.04-7.20 (2×m, 4H), 5.94 (d, 1H), 4.69 (m, 2H), 4.32-3.71 (4×m, 8H), 3.91-2.71 (2×d, 2H), 3.16-2.90 (2×bt, 4H), 2.48 (d, 5H), 1.98-1.45 (2×m, 5H). m/z MH+=567,14. Время удерживания при ВЭЖХ=7,9 мин.
Соединение 14
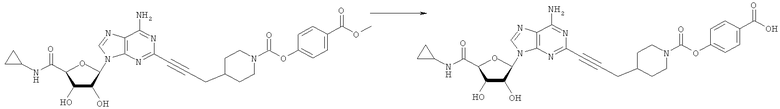
N-Циклопропил-2-{3-[1-(4-карбоксифеноксикарбонил)-4-пиперидинил]-1-пропин-1-ил}аденозин-5'-уронамид. Партия: JR28-115. N-Циклопропил-2-{3-[1-(4-метоксикарбонилфеноксикарбонил)-2-пиперидинил]-1-пропин-1-ил}аденозин-5'-уронамид, партия JR28-95, ATL360 (0,0060 г; 0,0097 ммоль) добавляли к раствору ТГФ (0,5 мл) и H2O (1 мл). Добавляли 1 н. LiOH (1 мл) и смесь перемешивали при комнатной температуре в течение 3 часов. Смесь упаривали с получением чистого белого твердого вещества. Выход = 5,0 мг; 85%. 1H ЯМР (CD3OD) δ 8.38 (m, 2H), 8.04-8.01 (m, 3Н), 7.20-7.17 (d, 2H), 6.02 (d, 1H), 4.81 (m, 1H), 4.41-4.20 (m, 4H), 3.14-2.95 (2×bt, 4H), 2.74 (m, 1H), 2.49 (d, 4Н), 1.98-1.44 (2×m, 5H), 0.87-0.56 (2×m, 4H). m/z MH+=606,32. Время удерживания при ВЭЖХ=10,4 мин.
Соединение 15
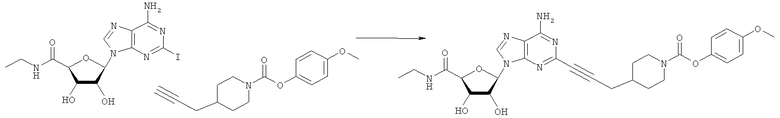
N-Этил-2-{3-[1 -(4-метоксифеноксикарбонил)-4-пиперидинил]-1-пропин-1-ил}аденозин-5'-уронамид. Партия: АР26-28. 4-(Метоксикарбонил)фенил-4-(проп-2-инил)пиперидин-1-карбоксилат, партия JR28-103 (0,250 мл; 0,4606 ммоль) добавляли к раствору N-этил-2-иодаденозин-5'-уронамида (0,200 г; 0,4606 ммоль) согласно общему способу 2. Выход = 98,3 мг; 37%. m/z MH+=580,25. Время удерживания при ВЭЖХ=8,0 мин.
Соединение 16
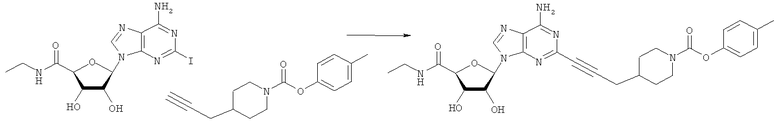
N-Этил-2-{3-[1-(4-метилфеноксикарбонил)-4-пиперидинил]-1-пропин-1-ил}аденозин-5'-уронамид. Партия: АР26-29. 4-(Метил)фенил-4-(проп-2-инил)пиперидин-1-карбоксилат, партия JR28-105 (0,150 мл; 0,2303 ммоль) добавляли к раствору N-этил-2-иодаденозин-5'-уронамида (0,100 г; 0,2303 ммоль) согласно общему способу 2. Выход = 38,0 мг; 29%. 1H ЯМР (CD3OD) δ 8.30 (s, 1Н), 8.20 (s, 1Н), 7.17-6.93 (2×d, 4H), 6.00 (d, 1H), 4.71 (m, 1H), 4.45-4.29 (m, 4H), 3.53-2.85 (2×2 m, 2×bt, 6H), 2.31 (d, 3H), 1.96-1.87 (m, 4H), 1.40 (3×m, 8H). m/z MH+=564,26. Время удерживания при ВЭЖХ=8,5 мин.
Соединение 17
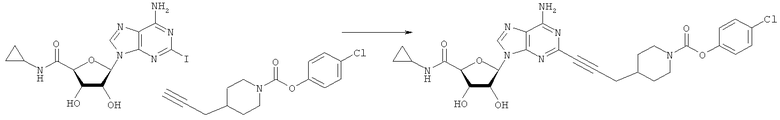
N-Циклопропил-2-{3-[1-(4-хлорфеноксикарбонил)-4-пиперидинил]-1-пропин-1-ил}аденозин-5'-уронамид. Партия: АР26-30. 4-(Хлор)фенил-4-(проп-2-инил)пиперидин-1-карбоксилат, партия JR28-107 (0,150 мл; 0,2303 ммоль) добавляли к раствору N-циклопропил-2-иодаденозин-5'-уронамида (0,100 г; 0,2303 ммоль) согласно общему способу 2. Выход = 70,4 мг; 53%. 1H ЯМР (CD3OD) δ 8.41 (s, 2H), 7.37-7.08 (2×d, 4H), 6.02 (d, 1H), 4.82 (m, 1H), 4.42-4.12 (2×m, 4H), 3.11-2.92 (2×2 bt, 4H), 2.71 (m, 1H), 2.48-1.88 (d, 4H), 2.01-1.42 (2×m, 5H), 0.78-0.55 (2×m, 4H). m/z MH+=596,27. Время удерживания при ВЭЖХ=8,4 мин.
Соединение 18
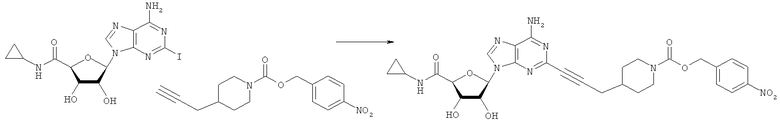
N-Циклопропил-2-{3-[1-(4-нитробензилоксикарбонил)-4-пиперидинил]-1-пропин-1-ил}аденозин-5'-уронамид. Партия: АР26-31. 4-(Нитро)бензил-4-(проп-2-инил)пиперидин-1-карбоксилат, партия JR28-109 (0,150 мл; 0,2303 ммоль) добавляли к раствору N-циклопропил-2-иодаденозин-5'-уронамида (0,100 г; 0,2303 ммоль) согласно общему способу 2. Выход = 42,8 мг; 31%. 1H ЯМР (CD3OD) δ 8.40 (s, 1H), 8.22-7.56 (2×d, 4H), 8.16 (s, 1H), 6.02 (d, 1H), 5.23 (s, 2H), 4.81 (m, 1H), 4.40 (m, 2H), 3.29-2.85 (3×m, 6H), 2.70 (m, 1H), 1.91-1.19 (2×m, 5H), 0.77-0.53 (2×m, 4H). 13C (CD3OD) δ 172.56, 156.04, 155.34, 147.84, 146.72, 144.81, 141.85, 128.05, 123.49, 89.20, 84.61, 73.30, 72.81, 65.71, 44.11, 35.38, 31.37, 25.47, 22.25, 5.80. m/z MH+=621,26. Время удерживания при ВЭЖХ=7,9 мин.
Соединение 19
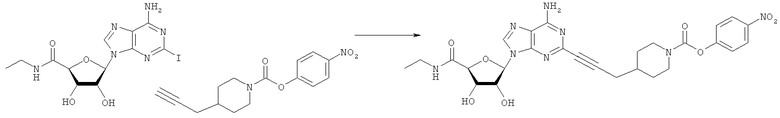
N-Этил-2-{3-[1-(4-нитрофенилоксикарбонил)-4-пиперидинил]-1-пропин-1-ил}аденозин-5'-уронамид. Партия: АР26-27. 4-(Нитро)фенил-4-(проп-2-инил)пиперидин-1-карбоксилат, партия JR28-83 (0,250 мл; 0,4306 ммоль) добавляли к раствору N-этил-2-иодаденозин-5'-уронамида (0,200 г; 0,4606 ммоль) согласно общему способу 2. Выход = 247,5 мг; 90%. 1H ЯМР (CD3OD) δ 8.30-7.34 (4×m, 6H), 6.00 (d, 1H), 4.83 (s, 1H), 4.31-4.20 (3×m, 4H), 3.12-2.96 (2×bt, 6H), 4.29 (d, 4H), 1.98-1.44 (2×m, 5H), 1.19 (s, 3H). m/z MH+=595,25. Время удерживания при ВЭЖХ=7,9 мин.
Соединение 20
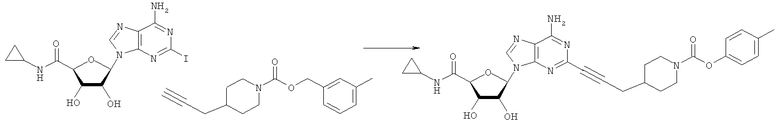
N-Циклопропил-2-{3-[1-(4-метилфенилоксикарбонил)-4-пиперидинил]-1-пропин-1-ил}аденозин-5'-уронамид. Партия: АР26-43. 4-(Метил)фенил-4-(проп-2-инил)пиперидин-1-карбоксилат, партия JR28-105 (0,259 г; 1,006 ммоль) добавляли к раствору N-циклопропил-2-иодаденозин-5'-уронамида (0,109 г; 0,2443 ммоль) согласно общему способу 2. Выход = 125,7 мг; 89%. 1H ЯМР (CD3OD) δ 8.40 (s, 2H), 7.17-6.93 (2×d, 4H), 6.02 (d, 1H), 4.80 (m, 1H), 4.38-4.21 (2×m, 4H), 3.05-2.91 (2×bt, 4H), 2.71 (m, 1H), 2.67 (s, 3H) 2.31 (s, 4H), 1.96-1.41 (2×m, 5H), 0.77-0.57 (2×m, 4H). 13C (CD3OD) δ 173.66, 157.18, 155.71, 150.61, 147.88, 143.01, 136.20, 130.73, 122.54, 90.34, 86.36, 85.75, 74.73, 73.89, 26.60, 23.38, 22.11, 20.78, 6.950, 4.575. m/z MH+=576,35. Время удерживания при ВЭЖХ=10,0 мин.
Соединение 21
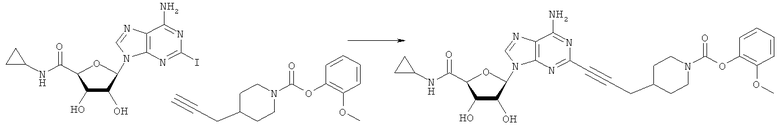
N-Циклопропил-2-{3-[1-(2-метоксифенилоксикарбонил)-4-пиперидинил]-1-пропин-1-ил}аденозин-5'-уронамид. Партия: АР26-44. 2-(Метокси)фенил-4-(проп-2-инил)пиперидин-1-карбоксилат, партия АР26-39 (0,209 г; 0,7647 ммоль) добавляли к раствору N-циклопропил-2-иодаденозин-5'-уронамида (0,112 г; 0,2510 ммоль) согласно общему способу 2. Выход=94,5 мг; 64%. 1H ЯМР (CD3OD) δ 8.41 (s, 1H), 8.00 (s, 1H), 7.20-6.89 (2×t, d, 4H), 6.03 (d, 1H), 4.80 (m, 1H), 3.79 (s, 3H), 3.34-2.85 (4×m, 8H), 2.70 (m, 1H), 2.49 (d, 4H), 1.96-1.30 (2×m, 5H), 0.78-0.55 (2×m, 4H). 13С (CD3OD) δ 173.71, 157.17, 155.53, 155.45, 153.13, 153.08, 143.05, 141.91, 127.63, 127.60, 124.06, 121.68, 113.68, 92.68, 113.69, 90.30, 86.41, 85.76, 74.76, 56.38, 36.57, 34.79, 32.51, 26.68, 23.39, 6.95. m/z MH+=592,29. Время удерживания при ВЭЖХ=9,2 мин.
Соединение 22
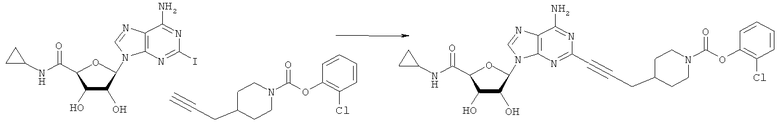
N-Циклопропил-2-{3-[1-(2-хлорфенилоксикарбонил)-4-пиперидинил]-1-пропин-1-ил}аденозин-5'-уронамид. Партия: АР26-45. 2-(Хлор)фенил-4-(проп-2-инил)пиперидин-1-карбоксилат, партия АР26-38 (0,303 г; 1,091 ммоль) добавляли к раствору N-циклопропил-2-иодаденозин-5'-уронамида (0,108 г; 0,2420 ммоль) согласно общему способу 2. Выход = 45,8 мг; 32%. 1Н ЯМР (CD3OD) δ 8.43 (s, 2H), 7.46-7.19 (2×m, d, 4H), 6.03 (d, 1H), 4.83 (m, 1H), 4.39-4.20 (2×m, 4H), 3.29-2.98 (2×bt, 4H), 2.64 (m, 1H), 2.46 (d, 4H), 1.42-1.19 (2×m, 5H), 0.78-0.56 (2×m, 4H). m/z MH+=596,29. Время удерживания при ВЭЖХ=9,7 мин.
Соединение 23
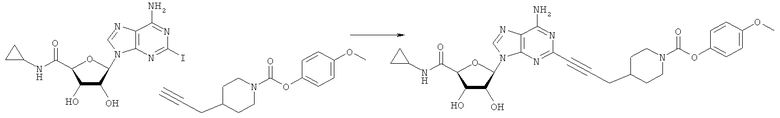
N-Циклопропил-2-{3-[1-(4-метилфенилоксикарбонил)-4-пиперидинил]-1-пропин-1-ил}аденозин-5'-уронамид. Партия: АР26-42. 4-(Метокси)фенил-4-(проп-2-инил)пиперидин-1-карбоксилат, партия JR28-103 (0,243 г; 0,8890 ммоль) добавляли к раствору N-циклопропил-2-иодаденозин-5'-уронамида (0,110 г; 0,2465 ммоль) согласно общему способу 2. Выход = 27,8 мг; 19%. 1H ЯМР (CD3OD) δ 7.97 (s, 2H), 7.00-6.85 (2×m, 4H), 6.03 (d, 1H), 4.83 (s, 1H), 4.41-4.21 (2×m, 4H), 3.76 (s, 3H), 3.12-2.85 (2×m, 4H), 2.70 (m, 1H), 2.48 (d, 4H), 1.99-1.24 (2×m, 5H), 0.77-57 (2×m, 4H). m/z MH+=592,33. Время удерживания при ВЭЖХ=9,5 мин.
Соединение 24
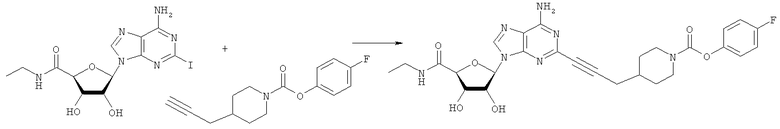
N-Этил-2-{3-[1-(4-фторфенилоксикарбонил)-4-пиперидинил]-1-пропин-1-ил}аденозин-5'-уронамид. Партия: АР26-47. 4-(Фтор)фенил-4-(проп-2-инил)пиперидин-1-карбоксилат, партия JR28-87 (0,291 г; 1,114 ммоль) добавляли к раствору N-этил-2-иодаденозин-5'-уронамида (0,119 г; 0,2741 ммоль) согласно общему способу 2. Выход = 98,3 мг; 63%. 1H ЯМР (CD3OD) δ 8.30 (s, 1Н), 7.97 (s, 1H), 7.11-7.08 (2×m, 4H), 6.00 (d, 1H), 4.75 (s, 1H), 4.45-4.29 (2×m, 4H), 3.47 (m, 6H), 3.12-2.85 (3×m, 4H), 2.48 (d, 4H), 1.96-1.39 (2×m, 5H), 1.28 (t, 3H). m/z МН+=568,15. Время удерживания при ВЭЖХ=9,6 мин.
Соединение 25
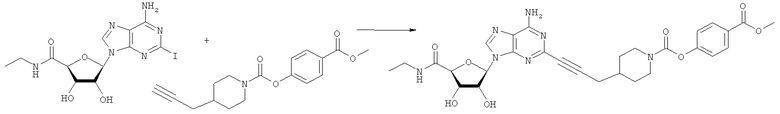
N-Этил-2-{3-[1-(4-метоксикарбоксифенилоксикарбонил)-4-пиперидинил]-1-пропин-1-ил}аденозин-5'-уронамид. Партия: АР26-48. 4-(Метоксикарбокси)фенил-4-(проп-2-инил)пиперидин-1-карбоксилат, партия JR28-85 (0,244 г; 0,8097 ммоль) добавляли к раствору N-этил-2-иодаденозин-5'-уронамида (0,109 г; 0,2510 ммоль) согласно общему способу 2. Выход = 77,8 мг; 51%. 1H ЯМР (CD3OD) δ 8.30 (s, 1H), 8.04-8.015 (d, 3H), 7.24-7.21 (d, 2H), 6.00 (d, 1H), 4.80 (m, 1H), 4.46-4.31 (2×m, 3H), 3.89 (s, 4H), 3.47 (m, 6H), 2.49 (d, 4H), 1.94-1.40 (2×m, 5H), 1.20 (t, 3H). 13С (CD3OD) δ 178.79, 178.29, 172.11, 172.09, 156.74, 154.58, 143.41, 143.36, 131.94, 128.40, 125.07, 122.93, 90.55, 88.76, 74.94, 74.62, 73.28, 68.27, 52.64, 45.81, 36.44, 35.18, 34.69, 26.49, 25.29, 15.30. m/z MH+=608,15. Время удерживания при ВЭЖХ=9,7 мин.
Соединение 26
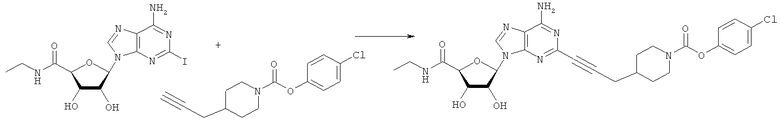
N-Этил-2-{3-[1-(4-хлорфенилоксикарбонил)-4-пиперидинил]-1-пропин-1-ил}аденозин-5'-уронамид. Партия: АР26-49. 4-(Хлор)фенил-4-(проп-2-инил)пиперидин-1-карбоксилат, партия JR28-107 (0,281 г; 1,012 ммоль) добавляли к раствору N-этил-2-иодаденозин-5'-уронамида (0,116 г; 0,2672 ммоль) согласно общему способу 2. Выход = 139,5 мг; 89%. 1H ЯМР (CD3OD) δ 8.29 (s, 1Н), 7.37-7.07 (2×m, 4H), 5.99 (d, 1H), 4.73 (m, 1H), 4.45 (s, 2H), 4.34-4.17 (2×m, 2H), 3.06-2.92 (2×bt, 6H), 2.47 (d, 4H), 1.96-1.37 (2×m, 5H), 1.19 (t, 3H). 13C (CD3OD) δ 172.31, 157.31, 155.04, 151.528, 147.64, 143.39, 131.72, 130.30, 130.30, 124.51, 124.51, 120.73, 90.55, 86.41, 86.14, 82.93, 74.95, 73.28, 45.75, 45.38, 36.45, 35.19, 32.63, 32.63, 26.51, 15.33. m/z MH+=584,17. Время удерживания при ВЭЖХ=10,4 мин.
Соединение 27
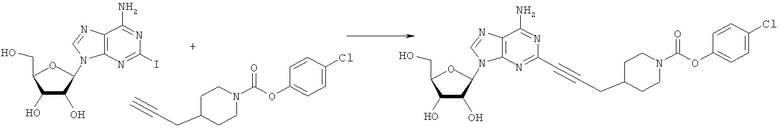
2-{3-[1-((4-Хлор)феноксикарбонил)пиперидин-4-ил]пропин-1-ил}аденозин. Партия: АВ-10-015. 4-Хлорфенил-4-(проп-2-инил)пиперидин-1-карбоксилат, партия JR28-107 (0,624 г; 2,247 ммоль) добавляли к раствору 2-иодаденозина (0,656 г; 1,669 ммоль) согласно общему способу 2. Выход: 0,445 г; 49%. 1H ЯМР (CD3OD) δ 8.30 (s, 1H), 7.37-7.07 (2×m, 4H), 5.93 (d, 1H), 4.71 (m, 2H), 4.31-4.17 (2×m, 3H), 3.91-3.75 (2×d, 2H), 3.07-2.93 (2×bt, 4H), 2.48 (d, 5H), 1.97-1.40 (2×m, 5H). m/z MH+=543,09. Время удерживания при ВЭЖХ=7,3 мин.
Соединение 28
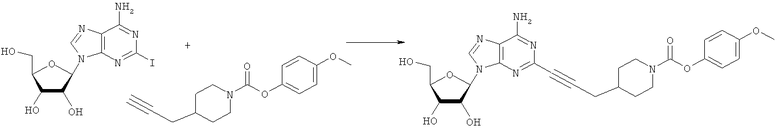
2-{3-[1-((4-Метокси)феноксикарбонил)пиперидин-4-ил]пропин-1-ил}аденозин. Партия: АВ-10-017. 4-Метоксифенил-4-(проп-2-инил)пиперидин-1-карбоксилат, партия JR28-103 (597 г; 2,184 ммоль) добавляли к раствору 2-иодаденозина (0,649 г; 1,651 ммоль) согласно общему способу 2. Выход: 0,294 г; 33%. 1H ЯМР (CD3OD) δ 8.30 (s, 1H), 7.00-6.87 (2×m, 4H), 5.94 (d, 1H), 4.73 (m, 2H), 4.33-4.14 (2×m, 3Н), 3.92-3.76 (2×d, 5H), 3.10-2.90 (2×bt, 4H), 2.46 (d, 5H), 1.91-1.40 (2×m, 5H). m/z MH+=539,14. Время удерживания при ВЭЖХ=5,5 мин.
Соединение 29
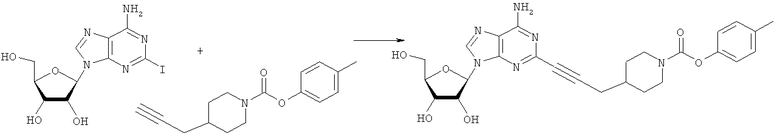
2-{3-[1-((4-Метил)феноксикарбонил)пиперидин-4-ил]пропин-1-ил}аденозин. Партия: АВ-10-019. 4-Метилфенил-4-(проп-2-инил)пиперидин-1-карбоксилат, партия JR28-105 (0,548 г; 2,130 ммоль) добавляли к раствору 2-иодаденозина (0,629 г; 1,600 ммоль) согласно общему способу 2. Выход: 0,236 г; 28%. 1H ЯМР (CD3OD) δ 8.29 (s, 1H), 7.14-6.92 (2×m, 4H), 5.95 (d, 1H), 4.75 (m, 2H), 4.34-4.16 (2×m, 3Н), 3.91-3.71 (2×d, 2H), 3.10-2.90 (2×bt, 4H), 2.28 (s, 3Н), 2.43 (d, 5H), 1.92-1.41 (2×m, 5H). m/z MH+=523,16. Время удерживания при ВЭЖХ=6,8 мин.
Соединение 30
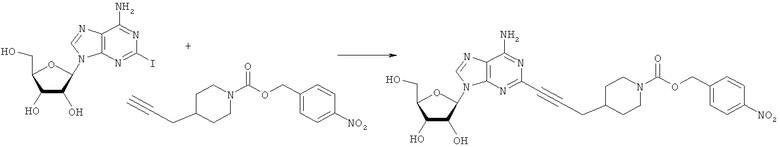
2-{3-[1 -((4-Нитро)бензилоксикарбонил)пиперидин-4-ил]пропин-1-ил}аденозин. Партия: АВ-10-021. 4-Нитробензил-4-(проп-2-инил)пиперидин-1-карбоксилат, партия JR28-109 (0,743 г; 2,458 ммоль) добавляли к раствору 2-иодаденозина (0,658 г; 1,674 ммоль) согласно общему способу 2. Выход: 0,694 г; 42%. 1H ЯМР (CD3OD) δ 8.29 (s, 1H), 8.18-7.56 (2×d, 4H), 5.94 (d, 1H), 5.23 (s, 2H), 4.69 (m, 2H), 4.31-4.14 (2×m, 3Н), 3.84-3.74 (2×d, 2H), 3.10-2.90 (bs, 4H), 2.44 (d, 5H), 1.91-1.40 (2×m, 5H). m/z MH+=568,10. Время удерживания при ВЭЖХ=5,8 мин.
Соединение 31
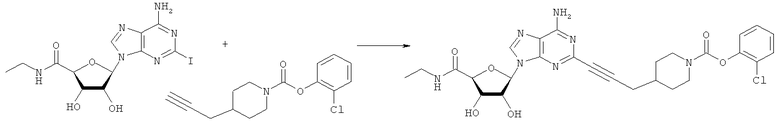
N-Этил-2-{3-[1-(2-хлорфенилоксикарбонил)-4-пиперидинил]-1-пропин-1-ил}аденозин-5'-уронамид. Партия: АР26-57. 2-(Хлор)фенил-4-(проп-2-инил)пиперидин-1-карбоксилат, партия АР26-38 (0,296 г; 1,066 ммоль) добавляли к раствору N-этил-2-иодаденозин-5'-уронамида (0,127 г; 0,2925 ммоль) согласно общему способу 2. 1H ЯМР (CD3OD) δ 9.10 (s, 1H), 8.30 (s, 1H), 7.46-7.20 (3×m, 5H), 6.00 (d, 1H), 4.74 (m, 1H), 4.47-4.18 (3×m, 4H), 3.48 (m, 1H), 3.99-2.97 (2×bt, 5H), 2.94 (d, 4H), 1.98-1.41 (2×m, 5H), 1.23 (t, 3H). m/z MH+=584,17. Время удерживания при ВЭЖХ=10,0 мин.
Соединение 32
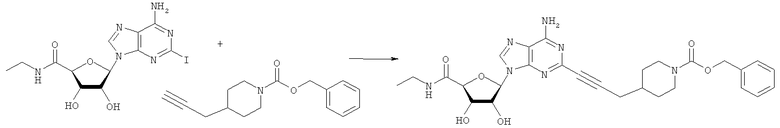
N-Этил-2-{3-[1-(бензилоксикарбонил)-4-пиперидинил]-1-пропин-1-ил}аденозин-5'-уронамид. Партия: АР26-58. Бензил-4-(проп-2-инил)пиперидин-1-карбоксилат, партия JR28-13 (0,236 г; 0,6643 ммоль) добавляли к раствору N-этил-2-иодаденозин-5'-уронамида (0,124 г; 0,2856 ммоль) согласно общему способу 2. m/z МН+=564,18. Время удерживания при ВЭЖХ=10,1 мин.
Соединение 33
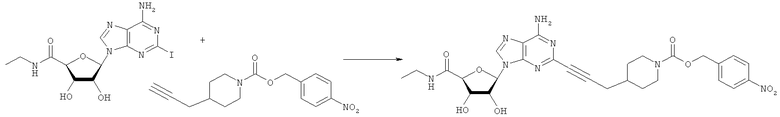
N-Этил-2-{3-[1-(4-нитробензилоксикарбонил)-4-пиперидинил]-1-пропин-1-ил}аденозин-5'-уронамид. Партия: АР26-59. 4-(Нитро)бензил-4-(проп-2-инил)пиперидин-1-карбоксилат, партия JR28-107 (0,281 г; 1,012 ммоль) добавляли к раствору N-этил-2-иодаденозин-5'-уронамида (0,116 г; 0,2672 ммоль) согласно общему способу 2. 1H ЯМР (CD3OD) δ 8.30 (s, 1H), 8.22 (d, 3H), 7.59 (d, 2H), 5.99 (d, 1H), 5.23 (s, 2H), 4.71 (m, 1H), 4.45 (m, 2H), 4.30 (2×m, 2H), 3.47 (m, 2H), 3.31-2.89 (2×m, 4H), 2.44 (d, 4H), 1.92-1.23 (2×d, 5H), 1.17 (t, 3H). m/z MH+=609,20. Время удерживания при ВЭЖХ=9,8 мин.
Соединение 34
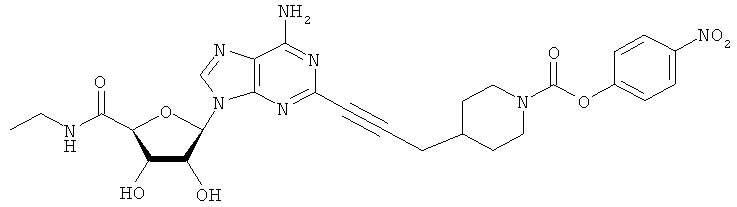
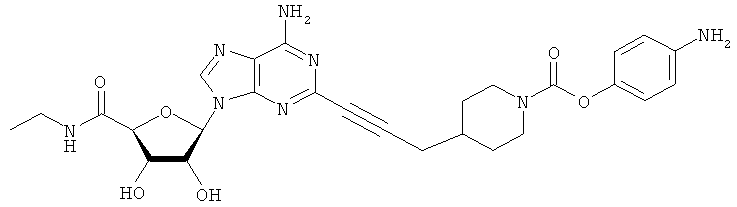
N-Этил-2-{3-[1-(4-аминофеноксикарбонил)-4-пиперидинил]-1-пропин-1-ил}аденозин-5'-уронамид. Партия: АР26-35. N-Этил-2-{3-[1-(4-нитрофенилоксикарбонил)-4-пиперидинил]-1-пропин-1-ил}аденозин-5'-уронамид, партия АР26-27 (0,00960 г; 0,0161 ммоль) добавляли к раствору цинковой пыли (0,0250 г; 0,3823 ммоль) в 2 мл формиата аммония. Затем смесь перемешивали при 50°С в течение 3 ч, упаривали и очищали согласно общему способу 2. m/z MH+=565,12. Время удерживания при ВЭЖХ=5,8 мин.
Соединение 35
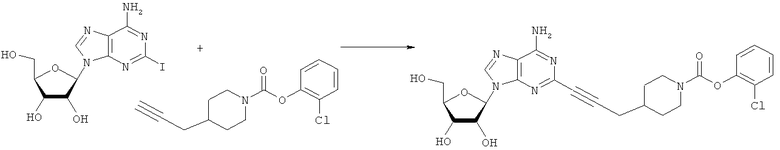
2-{3-[1-((2-Хлор)феноксикарбонил)пиперидин-4-ил]пропин-1-ил}аденозин. 2-Хлорфенил-4-(проп-2-инил)пиперидин-1-карбоксилат (0,588 г; 2,117 ммоль) добавляли к раствору 2-иодаденозина (0,602 г; 1,531 ммоль) согласно общему способу 2. Выход 733 мг; 88%. 1H ЯМР (CD3OD) δ 8.30 (s, 1H), 7.45-7.40, 7.33-7.26, 7.22-7.16 (3×m, 4H), 5.95 (d, 1H, J=6,3 Гц), 4.74 (t, 1H), 4.41-4.30 (m, 2H), 4.20-4.11 (m, 2H), 3.94-3.86, 3.78-3.71, (2×m, 2H), 3.08, 2.92 (2×br t, 2H), 2.43 (d, 2H, J=6,2 Гц), 2.01-1.78, 1.54-1.28 (2×m, 5H). 13С ЯМР (CD3OD) δ 157.2, 154.3, 150.1, 148.9, 147.4, 142.6, 131.1, 129.0, 128.3, 127.9, 125.4, 120.4, 91.3, 88.2, 86.7, 82.2, 75.5, 72.6, 64.7, 63.5, 45.8, 36.4, 32.4, 26.5, 25.2. LRMS ESI (M+H+) 543,1. Время удерживания при ВЭЖХ=6,2 мин.
Соединение 36
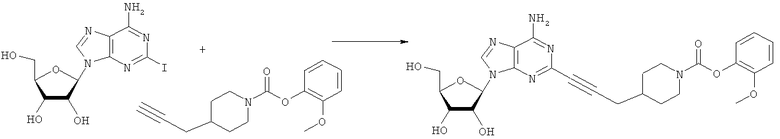
2-{3-[1 -((2-Метокси)феноксикарбонил)пиперидин-4-ил]пропин-1-ил}аденозин. 2-Метоксифенил-4-(проп-2-инил)пиперидин-1-карбоксилат (0,565 г; 2,067 ммоль) добавляли к раствору 2-иодаденозина (0,650 г; 1,653 ммоль) согласно общему способу 2: выход 775 мг; 87%. 1H ЯМР (CD3OD) δ 8.30 (s, 1H), 7.24-7.17, 7.09-7.04, 6.97-6.91 (3×m, 4H), 5.96 (d, 1H, J=6,5 Гц), 4.74 (dd, 1H, J=5,1, J=6,4 Гц), 4.43-4.32 (m, 2H), 4.25-4.16 (m, 2H), 3.96-3.89, 3.82 (s, 3H), 3.80-3.74, (2×m, 2H), 3.07, 2.92 (2×br t, 2H), 2.51 (d, 2H, J=6,0 Гц), 2.03-1.85, 1.60-1.35 (2×m, 5H). 13С ЯМР (CD3OD) δ 157.3, 155.5, 153.1, 150.2, 147.5, 142.6, 141.9, 127.6, 124.1, 121.7, 120.4, 113.7, 91.3, 88.3, 86.7, 82.2, 75.5, 72.6, 64.7, 63.5, 56.4, 45.8, 36.6, 32.5, 26.5, 25.2. LRMS ESl (M+H+) 539,1. Время удерживания при ВЭЖХ=5,1 мин.
Соединение 37
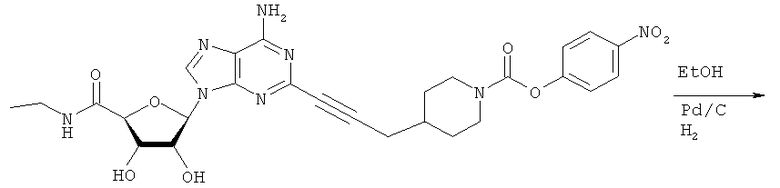
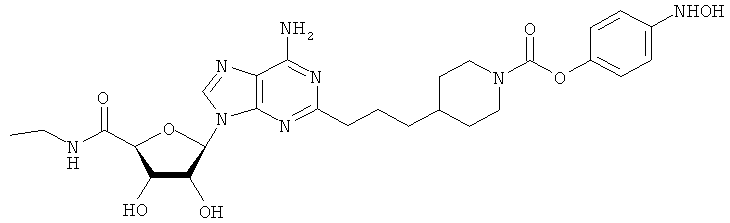
N-Этил-2-[3-[1-(4-гидроксиамино)фенил-4-пропилпиперидин]}-1-карбоксилат]аденозин-5'-уронамид. Партия: АР26-53. N-Этил-2-{3-[1-(4-нитрофеноксикарбонил)-4-пиперидинил]-1-пропин-1-ил}аденозин-5'-уронамид, партия АР26-27 (0,010 г; 0,0168 ммоль) добавляли к раствору Pd/C (0,0250 г; 0,3823 ммоль) в 3 мл этанола. Смесь затем перемешивали в течение 12 ч в атмосфере H2, фильтровали и очищали согласно общему способу 2. m/z МН+=584,16. Время удерживания при ВЭЖХ=10,4 мин.
Соединение 38
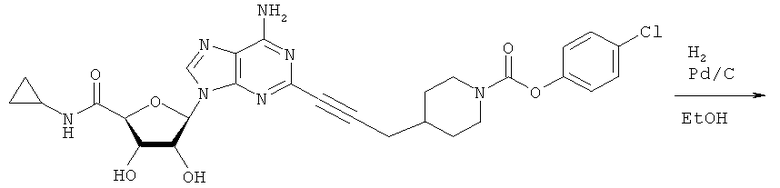
N-Циклопропил-2-[3-[1-(феноксикарбонил)-4-пропилпиперидин]}-1-карбоксилат]аденозин-5'-уронамид. Партия: АР26-67. N-Циклопропил-2-{3-[1-(4-хлорфеноксикарбонил)-4-пиперидинил]-1-пропин-1-ил}аденозин-5'-уронамид, ATL370, партия АР26-30 (0,0056 г; 0,00940 ммоль) добавляли к раствору Pd/C (каталитическое количество) в 3 мл этанола. Смесь затем перемешивали в течение 12 ч в атмосфере Н2, фильтровали и очищали согласно общему способу 2. m/z MH+=566,25. Время удерживания при ВЭЖХ=8,4 мин.
Соединение 39
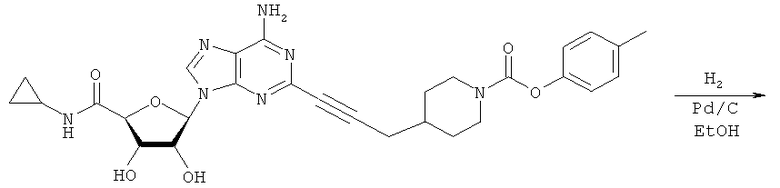
N-Циклопропил-2-[3-[1-(4-метилфеноксикарбонил)-4-пропилпиперидин]}-1-карбоксилат]аденозин-5'-уронамид. Партия: АР26-66. N-Циклопропил-2-{3-[1-(4-метилфеноксикарбонил)-4-пиперидинил]-1-пропин-1-ил}аденозин-5'-уронамид, ATL376, партия АР26-43 (0,0072 г; 0,0125 ммоль) добавляли к раствору Pd/C (каталитическое количество) в 3 мл этанола. Смесь затем перемешивали в течение 12 ч в атмосфере Н2, фильтровали и очищали согласно общему способу 2. m/z MH+=580,25. Время удерживания при ВЭЖХ=9,3 мин.
Соединение 40
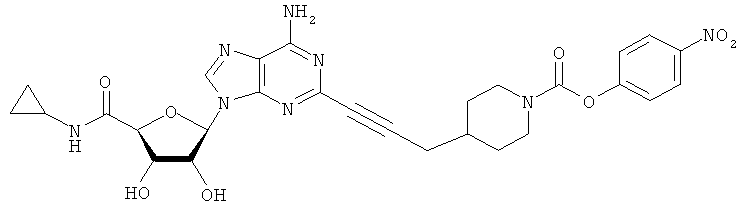
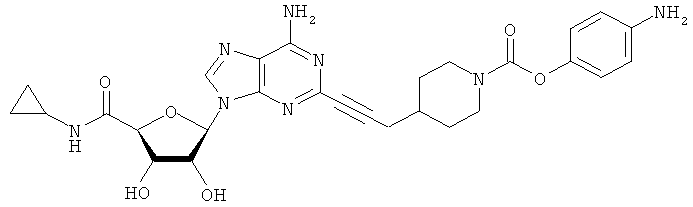
N-Циклопропил-2-[3-[1-(4-нитрофеноксикарбонил)-4-пропилпиперидин]}-1-карбоксилат]аденозин-5'-уронамид. Партия: АР26-71.
N-Циклопропил-2-{3-[1-(4-аминофеноксикарбонил)-4-пиперидинил]-1-пропин-1-ил}аденозин-5'-уронамид, ATL361, партия JR28-93 (0,0052 г; 0,00857 ммоль) добавляли к раствору Pd/C (каталитическое количество) в 3 мл этанола. Смесь затем перемешивали в течение 12 ч в атмосфере Н2, фильтровали и очищали согласно общему способу 2. m/z MH+=577,19. Время удерживания при ВЭЖХ=5,6 мин.
Соединение 41
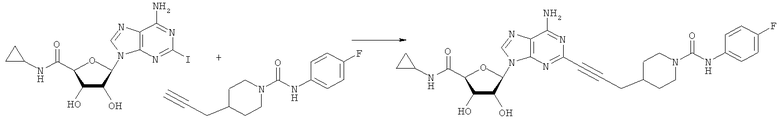
N-Циклопропил-2-{3-[1-(4-фторфенилоксикарбоксимидил)-4-пиперидинил]-1-пропин-1-ил}аденозин-5'-уронамид. Партия: АР26-72. 4-Фторфенил-4-(проп-2-инил)пиперидин-1-мочевину, партия АР26-64 (0,287 г; 1,103 ммоль) добавляли к раствору N-циклопропил-2-иодаденозин-5'-уронамида (0,076 г; 0,1703 ммоль) согласно общему способу 2. Выход = 58,6 мг; 60%. 1H ЯМР (CD3OD) δ 8.40 (s, 1H), 8.02 (s, 1H), 7.88-6.98 (2×m, 4H), 6.02 (d, 2H), 4.41 (m, 1H), 4.22-2.69 (4×m, 9H), 2.46 (2, 1H), 2.18 (m, 4H), 1.95-1.25 (2×m, 5Н), 0.77-0.57 (2×m, 4H). m/z MH+=579,19. Время удерживания при ВЭЖХ=8,4 мин.
Соединение 42
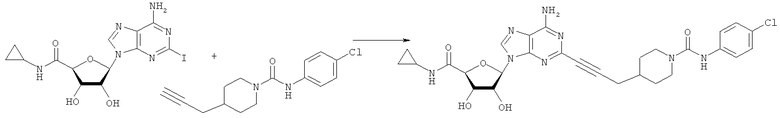
N-Циклопропил-2-{3-[1-(4-хлорфенилоксикарбоксимидил)-4-пиперидинил]-1-пропин-1-ил}аденозин-5'-уронамид. Партия: АР26-73. 4-Хлорфенил-4-(проп-2-инил)пиперидин-1-мочевину, партия АР26-63 (0,304 г; 1,098 ммоль) добавляли к раствору N-циклопропил-2-иодаденозин-5'-уронамида (0,074 г; 0,1658 ммоль) согласно общему способу 2. Выход = 25,2 мг; 26%. 1H ЯМР (CD3OD) δ 8.70 (s, 1H), 8.40 (s, 1H), 7.35-7.24 (2×d, 4H), 6.01 (d, 2H), 4.47 (m, 1H), 4.18-2.71 (4×m, 8H), 2.47 (d, 1H), 1.96-1.28 (2×m, 5H), 0.77-0.55 (2×m, 4H). m/z MH+=595,16. Время удерживания при ВЭЖХ=9,4 мин.
Соединение 43
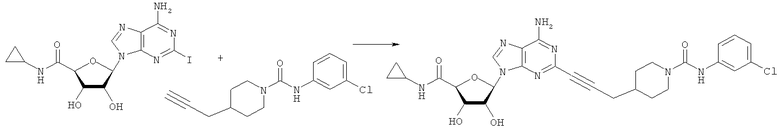
N-Циклопропил-2-{3-[1-(3-хлорфенилоксикарбоксимидил)-4-пиперидинил]-1-пропин-1-ил)аденозин-5'-уронамид. Партия: АР26-74. 3-Хлорфенил-4-(проп-2-инил)пиперидин-1-карбоксилат, партия АР26-62 (0,305 г; 1,102 ммоль) добавляли к раствору N-циклопропил-2-иодаденозин-5'-уронамида (0,085 г; 0,1905 ммоль) согласно общему способу 2. Выход = 25,5 мг; 23%. 1H ЯМР (CD3OD) δ 8.40 (s, 1H), 8.0 (s, 1H), 7.65-7.21 (2×m, 4H), 6.04 (d, 2Н), 4.23 (s, 1H), 3.74-2.96 (4×m, 8H), 2.71 (m, 1H), 2.46 (d, 4H), 1.96-1.29 (2×m, 5H), 0.78-0.56 (2×m, 4H). m/z MH+=595,15. Время удерживания при ВЭЖХ=9,4 мин.
Соединение 44
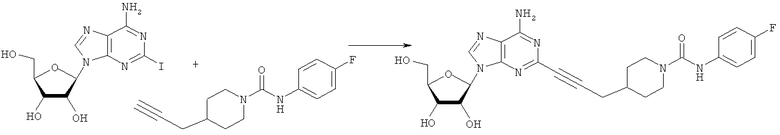
2-{3-[1-(4-Фторфеноксикарбамидил)пиперидин-4-ил]пропин-1-ил}аденозин. N-(4-Фторфенил)-4-(проп-2-инил)пиперидин-1-карбоксамид (1,122 г; 4,31 ммоль) добавляли к раствору 2-иодаденозина (0,571 г; 1,452 ммоль) согласно общему способу 2. Выход 14 мг; 2%. 1H ЯМР (CD3OD) δ 8.35 (s, 1H), 7.35-7.27 (m, 2Н), 7.03-6.93 (m, 2Н), 5.93 (d, 1H, J=6,3 Гц), 4.72 (br t, 1H), 4.34-4.29 (m, 1H), 4.23-4.13 (m, 3Н), 3.89 (dd, 1H, J=2,1 Гц, J=12,6 Гц), 3.73 (dd, 1H, J=2,3, J=12,6 Гц), 2.88 (t, 2Н), 2.42 (d, 2Н, J=6,2 Гц), 1.97-1.74, 1.44-1.25 (2×m, 5H). LRMS ESI (M+H+) 526,1. Время удерживания при ВЭЖХ=6,8 мин.
Соединение 45
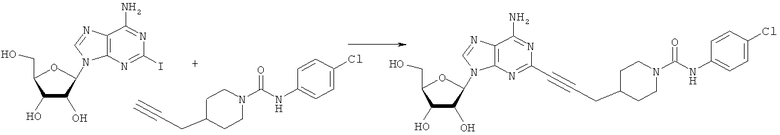
2-{3-[1-(4-Хлорфеноксикарбамидил)пиперидин-4-ил]пропин-1-ил}аденозин. N-(4-Хлорфенил)-4-(проп-2-инил)пиперидин-1-карбоксамид (1,458 г; 5,27 ммоль) добавляли к раствору 2-иодаденозина (0,569 г; 1,447 ммоль) согласно общему способу 2. Выход 16 мг; 2%. 1H ЯМР (CD3OD) δ 8.35 (s, 1H), 7.34 (d, 2Н, J=9,1 Гц), 7.22 (d, 2Н, J=9,1 Гц), 5.93 (d, 1H, J=6,5 Гц), 4.71 (t, 1H), 4.33-4.28 (m, 1H), 4.24-4.13 (m, 3Н), 3.89 (dd, 1H, J=2,3 Гц, J=12,6 Гц), 3.73 (dd, 1H, J=2,5, J=12,7 Гц), 2.90 (t, 2Н), 2.44 (d, 2Н, J=6,2 Гц), 1.99-1.78, 1.46-1.26 (2×m, 5H). Время удерживания при ВЭЖХ=9,4 мин.
Соединение 46
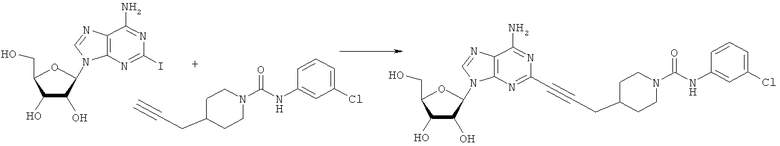
2-{3-[1-(3-Хлорфеноксикарбамидил)пиперидин-4-ил]пропин-1-ил}аденозин. N-(3-Хлорфенил)-4-(проп-2-инил)пиперидин-1-карбоксамид (1,603 г; 5,79 ммоль) добавляли к раствору 2-иодаденозина (0,640 г; 1,628 ммоль) согласно общему способу 2. Выход 344 мг; 39%. 1H ЯМР (CD3OD) δ 8.30 (s, 1Н), 7.49 (t, 1H, J=1,9 Гц), 7.28-7.15 (m, 2H), 6.98-6.93 (m, 1H), 5.93 (d, 1H, J=63 Гц), 4.72 (t, 1H), 4.33-4.29 (m, 1H), 4.24-4.14 (m, 3Н), 3.89 (dd, 1H, J=2,2 Гц, J=12,6 Гц), 3.73 (dd, 1H, J=2,5, J=12,7 Гц), 2.88 (t, 2H, J=12,5 Гц), 2.42 (d, 2H, J=6,2 Гц), 1.97-1.76, 1.44-1.26 (2×m, 5H). 13С ЯМР (CD3OD) δ 157.3, 157.2, 150.1, 147.5, 142.8, 142.6, 135.1, 130.7, 123.5, 121.5, 120.5, 119.7, 91.3, 88.2, 86.9, 82.1, 75.5, 72.6, 63.5, 45.4, 36.7, 32.6, 26.5. LRMS ESI (М+Н+) 542,1. Время удерживания при ВЭЖХ=9,7 мин.
Соединение 47
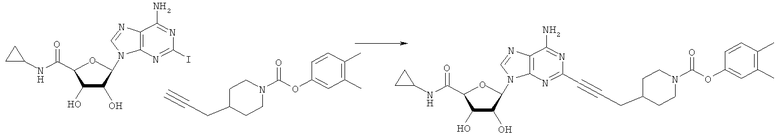
N-Циклопропил-2-{3-[1-(3,4-диметилфеноксикарбонил)-4-пиперидинил]-1-пропин-1-ил}аденозин-5'-уронамид. Диметилфенил-4-(2-пропин-1-ил)пиперидин-1-карбоксилат, хх (230 мг; 0,8325 ммоль) добавляли к раствору N-циклопропил-2-иодаденозин-5'-уронамида (0,100 г; 0,231 ммоль) согласно общему способу 2. Выход = 23,0 мг; 17%. m/z MH+=590,25. Время удерживания при ВЭЖХ=11,82 мин.
Соединение 48
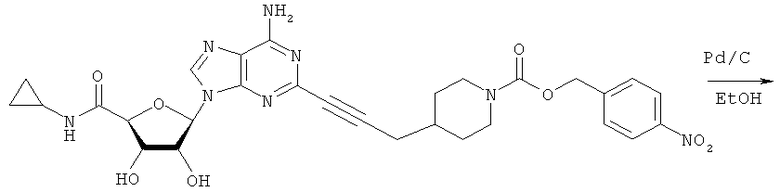
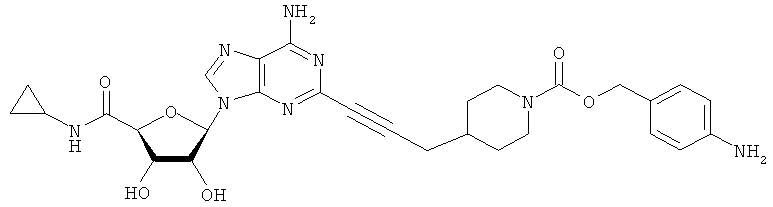
N-Циклопропил-2-{3-(1-((4-амино)бензилоксикарбонил)-4-пиперидинил]пропин-1-ил}аденозин-5'-уронамид. Партия: АР32-030. N-циклопропил-2-{3-[1-(4-нитробензилкарбонил)-4-пиперидинил]-1-пропин-1-ил}аденозин-5'-уронамид, партия АР26-30 (0,0015 г; 0,0156 ммоль) добавляли к раствору Pd/C (каталитическое количество) в EtOH (3 мл). Раствор оставляли перемешиваться в течение 12 часов в атмосфере Н2 при комнатной температуре и затем концентрировали и очищали посредством препаративной ВЭЖХ с получением белого твердого вещества, m/z MH+=592,01, время удерживания при ВЭЖХ=11 мин.
Соединение 49
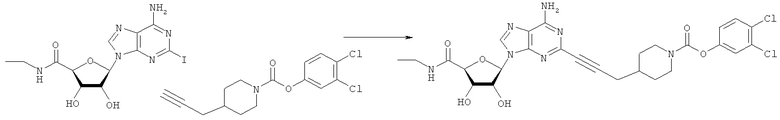
N-Этил-2-{3-[1-(3,4-дихлорфеноксикарбонил)-4-пиперидинил]-1-пропин-1-ил}аденозин-5'-уронамид. Дихлорфенил-4-(2-пропин-1-ил)пиперидин-1-карбоксилат, ×× (0,287 мг; 0,918 ммоль) добавляли к раствору N-этил-2-иодаденозин-5'-уронамида (0,110 г; 0,255 ммоль) согласно общему способу 2. Выход = 15,0 мг; 10%. m/z MH+=618,18. Время удерживания при ВЭЖХ=12,39 мин.
Соединение 50
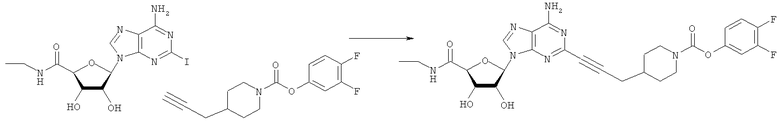
N-Этил-2-{3-(1-(3,4-дифторфеноксикарбонил)-4-пиперидинил]-1-пропин-1-ил}аденозин-5'-уронамид. Дифторфенил-4-(2-пропин-1-ил)пиперидин-1-карбоксилат, ×× (0,257 мг; 0,918 ммоль) добавляли к раствору N-этил-2-иодаденозин-5'-уронамида (0,110 г; 0,255 ммоль) согласно общему способу 2. Выход = 23,0 мг; 16%. m/z MH+=586,28. Время удерживания при ВЭЖХ=10,88 мин.
Соединение 51
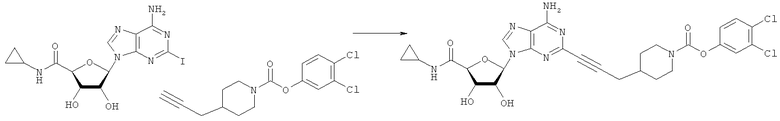
N-Циклопропил-2-{3-[1-(3,4-хлорфеноксикарбонил)-4-пиперидинил]-1-пропин-1-ил}аденозин-5'-уронамид. Дихлорфенил-4-(2-пропин-1-ил)пиперидин-1-карбоксилат, ×× (0,260 мг; 0,8325 ммоль) добавляли к раствору N-циклопропил-2-иодаденозин-5'-уронамида (0,100 г; 0,231 ммоль) согласно общему способу 2. Выход = 38,0 мг; 26,4%. m/z MH+=630,19. Время удерживания при ВЭЖХ=12,18 мин.
Соединение 52
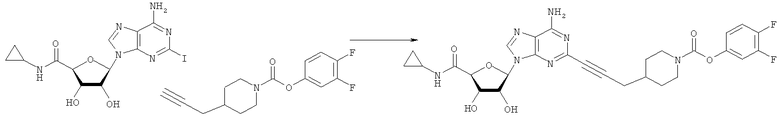
N-Циклопропил-2-{3-[1-(3,4-фторфеноксикарбонил)-4-пиперидинил]-1-пропин-1-ил}аденозин-5'-уронамид. Дифторфенил-4-(2-пропин-1-ил)пиперидин-1-карбоксилат, ×× (0,233 мг; 0,8325 ммоль) добавляли к раствору N-циклопропил-2-иодаденозин-5'-уронамида (0,100 г; 0,231 ммоль) согласно общему способу 2. Выход = 32,0 мг; 24,0%. m/z MH+=598,20. Время удерживания при ВЭЖХ=10,65 мин.
Соединение 53
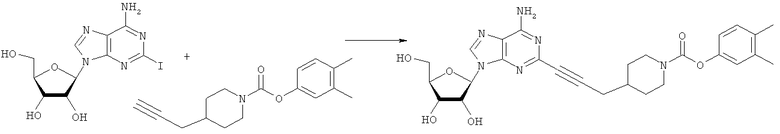
2-{3-[1-((3,4-Диметил)феноксикарбонил)пиперидин-4-ил]пропин-1-ил}аденозин. 3,4-Диметилфенил-4-(проп-2-инил)пиперидин-1-карбоксилат (1,430 г; 5,27 ммоль) добавляли к раствору 2-иодаденозина (0,620 г; 1,577 ммоль) согласно общему способу 2. Выход 252 мг; 30%. 1H ЯМР (CD3OD) δ 8.30 (s, 1Н), 7.08 (d, 1H, J=8,2 Гц), 6.84 (d, 1H, J=2,3 Гц), 6.78 (dd, 1H, J=2,4, J=8,1 Гц), 5.93 (d, 1H, J=6,5 Гц), 4.71 (dd, 1H, J=5,1 Гц, J=6,5 Гц), 4.38-4.27 (m, 2H), 4.25-4.14 (m, 2H), 3.89 (dd, 1H, J=2,3 Гц, J=12,6 Гц), 3.74 (dd, 1H, J=2,6, J=12,6 Гц), 3.04, 2.90 (2×br t, 2H), 2.46 (d, 2H, J=6,2 Гц), 2.23, 2.22 (2×s, 6H), 2.02-1.79, 1.52-1.26 (2×m, 5H). 13С ЯМР (CD3OD) δ 157.3, 155.8, 150.8, 150.2, 147.5, 142.6, 138.8, 134.7, 131.1, 123.7, 120.5, 119.9, 91.4, 88.3, 86.7, 82.3, 75.5, 72.6, 63.5, 45.5, 36.5, 32.5, 26.5, 19.8, 19.1. LRMS ESI (M+H+) 537,2. Время удерживания при ВЭЖХ=10,9 мин.
Соединение 54
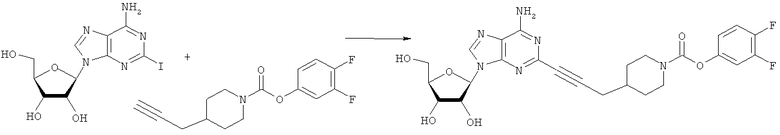
2-{3-[1-((3,4-Дифтор)феноксикарбонил)пиперидин-4-ил]пропин-1-ил}аденозин. 3,4-Дифторфенил-4-(проп-2-инил)пиперидин-1-карбоксилат (1,830 г; 6,55 ммоль) добавляли к раствору 2-иодаденозина (0,611 г; 1,554 ммоль) согласно общему способу 2: выход 201 мг; 24%. 1Н ЯМР (CD3OD) δ 8.30 (s, 1H), 7.25 (m, 1H), 7.12 (m, 1H), 6.96-6.89 (m, 1H), 5.93 (d, 1H, J=6,5 Гц), 4.71 (dd, 1H, J=5,1 Гц, J=6,4 Гц), 4.35-4.24 (m, 2H), 4.23-4.13 (m, 2H), 3.89 (dd, 1H, J=2,3 Гц, J=12,6 Гц), 3.74 (dd, 1H, J=2,5, J=12,6 Гц), 3.05, 2.91 (2×br t, 2H), 2.46 (d, 2H, J=6,2 Гц), 2.01-1.79, 1.52-1.27 (2×m, 5H). LRMS ESI (М+Н+) 545,2. Время удерживания при ВЭЖХ=9,1 мин.
Соединение 55
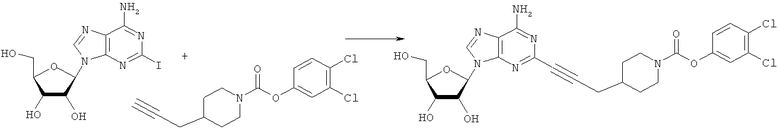
2-{3-[1-((3,4-Хлор)феноксикарбонил)пиперидин-4-ил]пропин-1-ил}аденозин. 3,4-Дихлорфенил-4-(проп-2-инил)пиперидин-1-карбоксилат (1,730 г; 5,54 ммоль) добавляли к раствору 2-иодаденозина (0,657 г; 1,671 ммоль) согласно общему способу 2. Выход 427 мг; 44%. 1H ЯМР (CD3OD) δ 8.30 (s, 1H), 7.50 (d, 1H, J=8,8 Гц), 7.36 (d, 1H, J=2,6 Гц), 7.08 (dd, 1H, J=2,7, J=8,8 Гц), 5.93 (d, 1H, J=6,5 Гц), 4.71 (dd, 1H, J=5,1 Гц, J=6,3 Гц), 4.35-4.14 (m, 4H), 3.89 (dd, 1H, J=2,3 Гц, J=12,6 Гц), 3.74 (dd, 1H, J=2,5, J=12,6 Гц), 3.06, 2.92 (2 x br t, 2H), 2.46 (d, 2H, J=6,3 Гц), 2.03-1.80, 1.54-1.28 (2×m, 5H). 13C ЯМР (CD3OD) δ 157.3, 154.5, 152.0, 150.3, 147.6, 142.6, 133.5, 131.8, 130.0, 125.2, 123.0, 120.5, 91.4, 88.3, 86.6, 82.3, 75.5, 72.6, 63.5, 45.8, 36.4, 32.5, 26.4. LRMS ESI (M+H+) 577,1. Время удерживания при ВЭЖХ=7,6 мин.
Соединение 56
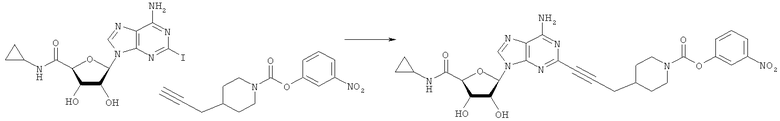
N-Циклопропил-2-{3-[1-(3-нитрофеноксикарбонил)-4-пиперидинил]-1-пропин-1-ил}аденозин-5'-уронамид. Нитрофенил-4-(2-пропин-1-ил)пиперидин-1-карбоксилат, ×× (0,240 мг; 0,8325 ммоль) добавляли к раствору N-циклопропил-2-иодаденозин-5'-уронамида (0,100 г; 0,231 ммоль) согласно общему способу 2. Выход = 15,0 мг; 11,0%. m/z МН+=607,26. Время удерживания при ВЭЖХ=9,17 мин.
Соединение 57
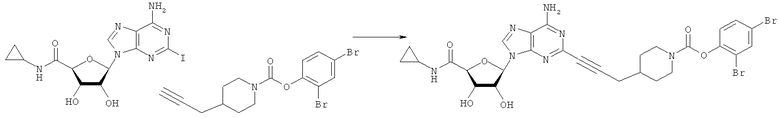
N-Циклопропил-2-{3-[1-(2,4-бромфеноксикарбонил)-4-пиперидинил]-1-пропин-1-ил}аденозин-5'-уронамид. Дибромфенил-4-(2-пропин-1-ил)пиперидин-1-карбоксилат, ×× (0,333 мг; 0,8325 ммоль) добавляли к раствору N-циклопропил-2-иодаденозин-5'-уронамида (0,100 г; 0,231 ммоль) согласно общему способу 2. Выход = 2,0 мг; 1,3%. m/z MH+=708,08. Время удерживания при ВЭЖХ=11,04 мин.
Приготовление клеточных культур и мембран. Клетки Sf9 (клетки бабочки-совки Spodoptera frugiperda) культивировали в среде Грейса, обогащенной 10% фетальной бычьей сывороткой, 2,5 мкг/мл амфотерицина В и 50 мкг/мл гентамицина, в атмосфере 50% N2/50% O2. Инфицирование вирусом проводили при плотности 2,5×106 клеток/мл с множественностью инфекции два для каждого использованного вируса. Инфицированные клетки получали через 3 суток после инфицирования и дважды промывали в забуференном фосфатом физиологическом растворе (PBS) для насекомых (PBS рН 6,3). Затем клетки ресуспендировали в лизирующем буфере (20 мМ HEPES (N-2-гидроксиэтилпиперазин-N-2-этансульфоновая кислота) рН 7,5, 150 мМ NaCl, 3 мМ MgCl2, 1 мМ β-меркаптоэтанола (ВМЕ), 5 мкг/мл лейпептина, 5 мкг/мл пепстатина А, 1 мкг/мл апротинина и 0,1 мМ PMSF (фенилметилсульфонилфторид)) и мгновенно замораживали для хранения при -80°С. Клетки размораживали на льду, доводили до общего объема 30 мл в лизирующем буфере и разрушали посредством кавитации N2 (600 ф/кв дюйм (4,14 МПа) в течение 20 минут). Проводили низкоскоростное центрифугирование для удаления всех нелизированных клеток (1000×g в течение 10 минут) с последующим высокоскоростным центрифугированием (17000×g в течение 30 минут). Осадок после окончательного центрифугирования гомогенизировали в буфере, содержащем 20 мМ HEPES рН 8, 100 мМ NaCl, 1% глицерина, 2 мкг/мл лейпептина, 2 мкг/мл пепстатина А, мкг/мл апротинина, 0,1 мМ PMSF и 10 мкМ ГДФ (гуанозиндифосфат), при использовании небольшого стеклянного гомогенизатора с последующим пропусканием через иглу 26 калибра. Мембраны делили на аликвоты, мгновенно замораживали в жидком N2 и хранили при -80°С. Мембраны из клеток, стабильно экспрессирующих человеческий А1 AR (клетки СНО K1 (клетки яичников китайских хомячков)) или А3 AR (клетки НЕК 293 (клетки почек человека)), получали, как описано Robeva et al. (1996).
Анализ связывания радиолигандов. Связывание радиолигандов с рекомбинантными человеческими А2А рецепторами в мембранах клеток Sf9 проводили при использовании либо меченого радиоактивным изотопом агониста 125I-APE (Luthin et al., 1995), либо меченого радиоактивным изотопом антагониста 125I-ZM241385 (125I-ZM). Для обнаружения высокого сродства, ГТФγS-чувствительного состояния A1 и А3 AR, авторы использовали агонист 125I-АВА (Linden et al., 1985; Linden et al, 1993). Эксперименты по связыванию проводили в трех повторностях с 5 мкг (A2A) или 25 мкг (А1 и А3) мембранного белка в общем объеме 0,1 мл НЕ буфера (20 мМ HEPES и 1 мМ ЭДТА (этилендиаминтетраацетат)) с 1 Ед/мл аденозиндезаминазы и 5 мМ MgCl2 с 50 мкМ ГТФγS или без него. Мембраны инкубировали с радиолигандами при комнатной температуре в течение трех часов (для агонистов) или двух часов (для антагонистов) в 96-луночных GF/C фильтровальных планшетах Millipore Multiscreen®, и анализы прекращали путем быстрой фильтрации в харвестер клеток (Brandel, Gaithersburg, MD) с последующими промывками 4×150 мкл в течение 30 секунд ледяным 10 мМ Трис-HCl, рН 7,4, 10 мМ MgCl2. Неспецифическое связывание измеряли в присутствии 50 мкМ NECA. Анализы конкурентного связывания проводили, как описано Robeva et al (1996), при использовании 0,5-1 нМ 125I-APE, 125I-ZM241385 или 125I-ABA. Авторы обнаружили, что иногда является важным заменять наконечники для пипеток после каждого серийного разведения для предотвращения перенесения на наконечники сильных гидрофобных соединений. Значения К, для связывания конкурирующих соединений в одном сайте получали из значений IC50 с поправкой на исчерпание радиолигандов и конкурирующих соединений, как описано ранее (Linden, 1982).
Linden J (1982). Calculating the Dissociation Constant of an Unlabeled Compound From the Concentration Required to Displace Radiolabel Binding by 50%. J Cycl Nucl Res 8: 163-172.
Linden J, Patel A and Sadek S (1985). [125I]Aminobenzyladenosine, a New Radioligand With Improved Specific Binding to Adenosine Receptors in Heart. Circ Res 56: 279-284.
Linden J, Taylor HE, Robeva AS, Tucker AL, Stehle JH, Rivkees SA, Fink JS and Reppert SM (1993). Molecular Cloning and Functional Expression of a Sheep Аз Adenosine Receptor With Widespread Tissue Distribution. Mol Pharmacol 44: 524-532.
Luthin DR, Olsson RA, Thompson RD, Sawmiller DR and Linden J (1995). Characterization of Two Affinity States of Adenosine A2A Receptors With a New Radioligand, 2-[2-(4-Amino-3-[125I]lodophenyl)Ethylamino]Adenosine. Mol Pharmacol 47:307-313.
Robeva AS, Woodard R, Luthin DR, Taylor HE and Linden J (1996). Double Tagging Recombinant A1- and A2A-Adenosine Receptors With Hexahistidine and the FLAG Epitope. Development of an Efficient Generic Protein Purification Procedure. Biochem Pharmacol 51: 545-555.
Хемилюминесцентные методы
Усиленная люминолом хемилюминесценция, мера окислительной активности нейтрофилов, зависит как от продуцирования супероксида, так и от активации гранулярного фермента миелопероксидазы. Свет излучается нестабильными высокоэнергетическими видами кислорода, такими как хлорноватистая кислота и синглетный кислород, генерируемый активированными нейтрофилами.
Очищенные человеческие нейтрофилы (2×106 клеток/мл), суспендированные в сбалансированном солевом растворе Хенкса, содержащем 0,1% человеческий сывороточный альбумин (HSA), аденозиндезаминазу (1 Ед/мл) и ролипрам (100 нМ), инкубировали (37°С) на водяной бане в течение 15 минут с rhФHO-α (человеческий рекомбинантный фактор некроза опухоли-α) (10 Ед/мл). После инкубирования аликвоты по 100 мкл ПМН (полиморфноядерных нейтрофилов) переносили в лунки (96-луночные планшеты с белыми стенками и прозрачным дном для культур тканей Costar #3670; 2 лунки/условие), содержащие 50 мкп HSA и люминол (конечная концентрация 100 М) с агонистом аденозина или без него (конечные концентрации агониста 0,01-1000 нМ). Планшет инкубировали в течение 5 мин (37°С) и затем добавляли во все лунки fMLP (50 мкл в HSA; конечная концентрация 1 М).
Максимальную хемилюминесценцию определяли с помощью счетчика-анализатора Victor 1420 Multilabel Counter в режиме хемилюминесценции при использовании компьютерной программы Wallac Workstation. Данные представлены в виде максимальной хемилюминесценции как процент активности в отсутствие агониста аденозина. ЕС50 определяли с использованием компьютерной программы PRISM. Все соединения тестировали с ПМН от трех разных доноров. Эти данные представлены в таблице.
Действие агонистов A2A на окислительную активность нейтрофилов. f-met-leu-phe (fMLP), люминол, супероксиддисмутазу, цитохром С, фибриноген, аденозиндезаминазу и трипановый синий получали от Sigma Chemical. Фиколл-гипак закупали у ICN (Aurora, ОН) и Cardinal Scientific (Santa Fe, NM) и Accurate Chemicals и Scientific (Westerbury, NY). Эндотоксин (липополисахарид; Е coli К235) получен от List Biologicals (Campbell, CA). Сбалансированный солевой раствор Хенкса (HBSS) и набор ЛАЛ-реактивов (лизат амебоцитов Limulus) получены от BioWittaker (Walkersville, MD).
Человеческий сывороточный альбумин (HSA) получен от Cutter Biological (Elkhart, IN). Человеческий рекомбинантный фактор некроза опухоли-α поставлялся Dianippon Pharmaceutical Co. Ltd. (Osaka, Japan). ZM241385 (4-(2-[7-амино-2-(2-фурил)-[1,2,4]триазоло[2,3-а][1,3,5]триазин-5-иламино]этил)фенол) был любезно предоставлен Simon Poucher, Zeneca Pharmaceuticals, Cheshire, UK. Исходные растворы (1 мМ и 10 мМ в ДМСО) готовили и хранили при -20°С.
Получение человеческих нейтрофилов. Очищенные нейтрофилы (98% нейтрофилов и >95% жизнеспособных согласно отсутствию окрашивания трипановым синим), содержащие <1 тромбоцита на 5 нейтрофилов и <50 пг/мл эндотоксина (тест с лизатом амебоцитов Limulus), получали из нормальной гепаринизированной (10 Ед/мл) венозной крови посредством одностадийного метода разделения с помощью фиколл-гипака (A.Ferrante et al., J. Immunol. Meih 36, 109(1980)).
Хемилюминесценция при высвобождении воспалительных активных видов кислорода из примированных и стимулированных человеческих нейтрофилов
Усиленная люминолом хемилюминесценция, мера окислительной активности нейтрофилов, зависит как от продуцирования супероксида, так и от активации гранулярного фермента миелопероксидазы. Свет излучается нестабильными высокоэнергетическими видами кислорода, генерируемыми активированными нейтрофилами. Очищенные нейтрофилы (5-10×105 клеток/мл) инкубировали в сбалансированном солевом растворе Хенкса, содержащем 0,1% человеческий сывороточный альбумин (1 мл) с тестируемым агонистом А2А с ролипрамом или без него и с фактором некроза опухоли-α или без него (1 Ед/мл), в течение 30 минут при 37°С на встряхиваемой водяной бане. Затем считывали усиленную люминолом (1×10-4 М) стимулированную f-met-leu-phe (1 мкМ) хемилюминесценцию с помощью фотометра Chronolog® (Crono-log Corp., Havertown, PA) при 37°С в течение 2-4 минут. Хемилюминесценция представлена в виде относительного максимума излучаемого света (высота кривой) в сравнении с образцами с фактором некроза опухоли-α и без агониста или ролипрама.
Все публикации, патенты и патентные документы включены в данное описание посредством ссылки, как если бы индивидуально включенные ссылкой. Изобретение описано со ссылкой на различные конкретные и предпочтительные воплощения и методики. Однако следует понимать, что многие изменения и модификации могут быть сделаны, оставаясь в пределах сущности и объема изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| СЕЛЕКТИВНЫЕ АНТАГОНИСТЫ АДЕНОЗИНОВЫХ A РЕЦЕПТОРОВ | 2007 |
|
RU2467009C2 |
| ПРОИЗВОДНЫЕ ЦИКЛИЧЕСКИХ АМИНОВ В КАЧЕСТВЕ АНТАГОНИСТОВ РЕЦЕПТОРА ЕР4 | 2011 |
|
RU2565596C2 |
| КОМПОЗИЦИИ ТЕТРАГИДРОХИНОЛИНОВ В КАЧЕСТВЕ ИНГИБИТОРОВ БРОМОДОМЕНА ВЕТ | 2014 |
|
RU2727169C2 |
| БИЦИКЛИЧЕСКИЕ ЛАКТАМЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2716136C2 |
| ПИРАЗОЛОПИРИМИДИНОВЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРА КИНАЗЫ | 2017 |
|
RU2714206C1 |
| ИНГИБИТОРЫ ВЗАИМОДЕЙСТВИЯ МЕНИН-MLL | 2017 |
|
RU2829484C2 |
| ПРОИЗВОДНЫЕ ХИНОЛИНА И СОДЕРЖАЩИЕ ИХ ИНГИБИТОРЫ MELK | 2011 |
|
RU2582610C2 |
| ПОЛИЦИКЛИЧЕСКИЕ АНТАГОНИСТЫ TLR7/8 И ИХ ПРИМЕНЕНИЕ В ЛЕЧЕНИИ ИММУННЫХ РАССТРОЙСТВ | 2016 |
|
RU2783748C2 |
| СОЕДИНЕНИЕ, СПОСОБНОЕ К СВЯЗЫВАНИЮ С РЕЦЕПТОРОМ S1P, И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2004 |
|
RU2390519C2 |
| ПРОИЗВОДНЫЕ ТИЕНО[2,3-с]ХИНОЛИНА-4-ОНА, ПОЛЕЗНЫЕ ПРИ ЛЕЧЕНИИ РВК-ЗАВИСИМОГО ЗАБОЛЕВАНИЯ | 2011 |
|
RU2574398C2 |
Изобретение относится к агонистам А2A рецепторов, представленных соединением формулы (I) или его фармацевтически приемлемой солью
 ,
,
в которой R1 и R2 независимо представляют собой Н; каждый R независимо выбран из группы, состоящей из Н, С1-С4алкила, циклопропила, циклобутила и (СН2)ациклопропила; Х представляет собой N; Y выбран из группы, состоящей из О, NR1,
-(ОСН2СН2О)mСН2- и -(NR1CH2CH2O)mCH2-, при условии, что когда Y представляет собой О или NR1, тогда имеется по меньшей мере один заместитель по Z; Z представляет собой 6-членный арил, где Z замещен 0-4 группами, независимо выбранными из группы, состоящей из F, Cl, Вr, I, (С1-С4)алкила, -(CH2)аОR3,
-(CH2)aNR3R3, -NHOH, нитро, -(CH2)aCN,
-(CH2)aCO2R3, -(CH2)aCONR3R3, трифторметила и трифторметокси; R3 независимо выбран из группы, состоящей из Н, (С1-С6)алкила и циклоалкила; R4 выбран из группы, состоящей из CH2OR, C(O)NRR и CO2R; R5 выбран из группы, состоящей из СН2СН2, СН=СН и С≡С; а выбран из 0, 1 и 2; m выбран из 1, 2 и 3; n означает 1; каждый р независимо означает 1 и q выбран из 0 и 1. Изобретение также относится к фармацевтической композиции, обладающей активностью агониста аденозиновых А2A рецепторов, содержащей указанное выше соединение, а также изобретение относится к применению указанного выше соединения в производстве лекарственного средства для лечения заболевания, связанного с А2A рецепторами. 3 н. и 9 з.п. ф-лы, 1 табл.
1. Соединение формулы (I) или его фармацевтически приемлемая соль:
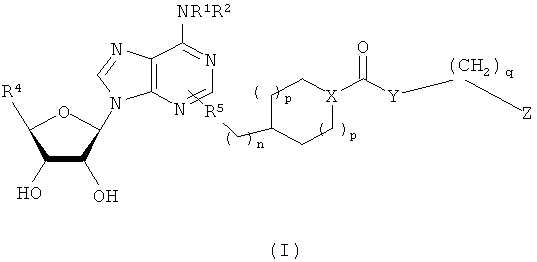
где R1 и R2 независимо представляют собой Н;
каждый R независимо выбран из группы, состоящей из Н, С1-С4алкила, циклопропила, циклобутила и (СН2)ациклопропила;
Х представляет собой N;
Y выбран из группы, состоящей из О, NR1, -(ОСН2СН2O)mСН2- и
-(NR1CH2CH2O)mCH2-, при условии, что когда Y представляет собой О или NR1, тогда имеется по меньшей мере один заместитель по Z;
Z представляет собой 6-членный арил, где Z замещен 0-4 группами, независимо выбранными из группы, состоящей из F, Cl, Вr, I, (С1-С4)алкила, -(CH2)aOR3, -(CH2)aNR3R3, -NHOH, нитро, -(CH2)aCN, -(CH2)aCO2R3, -(CH2)aCONR3R3, трифторметила и трифторметокси;
R3 независимо выбран из группы, состоящей из Н, (С1-С6)алкила и циклоалкила;
R4 выбран из группы, состоящей из CH2OR, C(O)NRR и CO2R;
R5 выбран из группы, состоящей из СН2СН2, СН=СН и С≡С;
а выбран из 0, 1 и 2;
m выбран из 1, 2 и 3;
n означает 1;
каждый р независимо означает 1; и
q выбран из 0 и 1.
2. Соединение по п.1, представляющее собой соединение формулы (Iа) или его фармацевтически приемлемую соль:
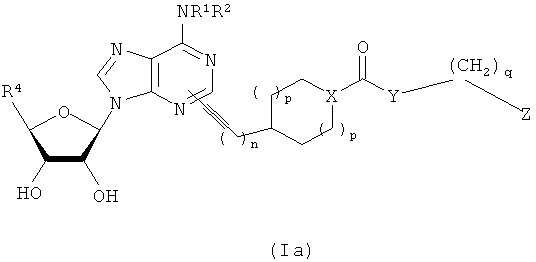
3. Соединение по п.2, представляющее собой соединение формулы (Ib) или его фармацевтически приемлемую соль:
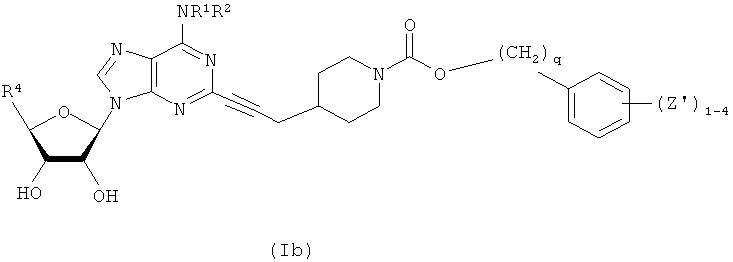
где каждый Z' независимо выбран из группы, состоящей из F, Cl, Br, I, C1-С4алкила,
-(СН2)аОR3, -(CH2)aNR3R3, -NHOH, NO2, -(CH2)aCN, -(CH2)aCO2R3, -(CH2)aCONR3R3, СF3 и ОСF3.
4. Соединение по п.3, где R выбран из Н, метила, этила или циклопропила.
5. Соединение по п.3, представляющее собой соединение формулы (Iс) или его фармацевтически приемлемую соль:
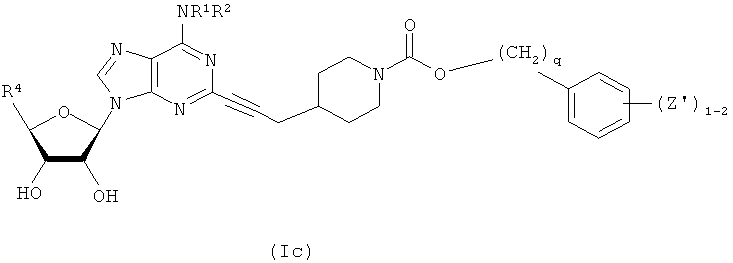
6. Соединение по п.5, где Z' выбран из группы, состоящей из F, Cl, метила, OR3, NO2, CN, NR3R3 и CO2R3.
7. Соединение по п.5, где R3 представляет собой метил или водород.
8. Соединение по п.1, выбранное из группы, состоящей из соединений под номерами 3, 5-31 и 33-57, представленных в таблице 1.
9. Соединение по п.1, выбранное из
или его фармацевтически приемлемой соли.
10. Соединение по любому из пп.1-8 или 9 для применения в качестве агониста аденозиновых А2A рецепторов.
11. Фармацевтическая композиция, обладающая активностью агониста аденозиновых А2A рецепторов, содержащая соединение по любому из пп.1-8 или 9 и фармацевтически приемлемый носитель.
12. Применение соединения по любому из пп.1-8 или 9 в производстве лекарственного средства для лечения заболевания, связанного с А2А - рецепторами.
| Пломбировальные щипцы | 1923 |
|
SU2006A1 |
| Способ и приспособление для нагревания хлебопекарных камер | 1923 |
|
SU2003A1 |
| RU 2004106635 A, 27.07.2005. | |||

