ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к новым фосфонатным соединениям, содержащим их композициям, способам их получения и к их применению для лечения ряда медицинских расстройств, например остеопороза и других расстройств метаболизма костей, рака, вирусных инфекций и тому подобного.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Давно известно, что фосфонатные соединения полезны во многих терапевтических аспектах. Особым классом терапевтически полезных фосфонатных соединений являются бифосфонаты, то есть аналоги пирофосфатов, в которых центральный атом кислорода пирофосфатной связи заменен на атом углерода. К этому центральному атому углерода могут быть присоединены различные группы-заместители, образуя производные соединений фосфонатов, которые обладают различной степенью фармакологической эффективности. Эти производные имеют общую структуру
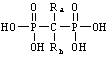
где Ra и Rb могут быть независимо выбраны из гидроксила, амино, сульфгидрила, галогена или множества алкильных или арильных групп либо комбинации таких групп, которые могут быть дополнительно замещены. Примеры включают этидронат, где Ra представляет собой СН3 и Rb представляет собой ОН; клодронат, дихлорметилендифосфоновую кислоту (Cl2MDP), где Ra и Rb представляют собой Cl; памидронат, 3-амино-1-гидроксипропилидендифосфоновую кислоту, где Ra представляет собой этиламино и Rb представляет собой гидроксил; алендронат, 4-амино-1-гидроксибутилидендифосфоновую кислоту, где Ra представляет собой пропиламино и Rb представляет собой гидроксил; ольпадронат, 3-диметиламино-1-гидроксипропилидендифосфоновую кислоту, где Ra представляет собой диметиламиноэтил и Rb представляет собой гидроксил; и амино-ольпадронат (IG-9402), 3-(N,N-диметиламино)-1-аминопропилиденбифосфонат, где Ra представляет собой диметиламиноэтил и Rb представляет собой NH2.
Бифосфонатам и их замещенным производным присуще свойство ингибировать резорбцию кости in vivo. Бифосфонаты также ингибируют апоптоз (запрограмированная смерть клеток) формирующих кость клеток. Следовательно, показания для их применения включают лечение и предупреждение остеопороза, лечение болезни Педжета, рака с костными метестазами, гиперпаратиреоза, ревматоидного артрита, алгодистрофии, грудинно-реберно-кпючичного гиперостоза, болезни Гоше, болезни Энгельманна и некоторых нескелетных расстройств (Papapoulos, S.Е., в Osteoporosis. R. Marcus, D. Feldman and J. Kelsay, eds., Academic Press, San Diego, 1996, p. 1210, Table 1).
Хотя бифосфонаты обладают терапевтически полезными свойствами, как перорально вводимые агенты они имеют фармакологические минусы. Одним из недостатков является низкая доступность при пероральном введении: из желудочно-кишечного тракта всасывается от 0,7 до 5% перорально вводимой дозы. Кроме того, всасывание при пероральном приеме уменьшается при принятии с пищей. Затем, известно, что некоторые имеющиеся в настоящее время в распоряжении бифосфонаты, например FOSAMAXTM (Merck; аледронат натрия), SKELIDTM (Sanofi, тилудронат) и ACTONETM (Procter and Gamble, ризедронат) имеют местную токсичность, вызывая раздражение и изъязвление пищевода. У других бифосфонатов, таких как амино-ольпадронат, отсутствуют антирезорбтивные эффекты (Van Beek, E. et al., J. Bone Miner Res 11(10): 1492-1497 (1996)), однако они ингибируют апоптоз остеоцитов и способны стимулировать образование губчатой структуры кости (Plotkin, L. et al., J Clin Invest 104(10): 1363-1374 (1999) и патент США №5885973). Следовательно, было бы полезно разрабатывать химически модифицированные производные бифосфонатов, которые бы сохраняли или усиливали фармакологическую активность родоначальных соединений, в то же время устраняя или уменьшая их нежелательные побочные эффекты.
Помимо бифосфонатов, известно что монофосфонаты также дают терапевтическую пользу. Одним классом терапевтически полезных монофосфонатов являются противовирусные нуклеотидфосфонаты, такие как, например, цидофовир, циклоцидофовир, адефовир, тенофовир и тому подобные, а также 5'-фосфонаты и метиленфосфонаты азидотимидина, ганцикловира, ацикловира и тому подобных. В соединениях этого типа 5'-гидроксил сахарной группировки или его эквивалент в ациклических нуклеозидах, которые не содержат полной сахарной группировки (ганцикловир, пенцикловир, ацикповир), замещен связью фосфор-углерод. В случае метиленфосфонатов метиленовая группа замещает 5'-гидроксил или его эквивалент, и ее атом углерода, в свою очередь, ковалентно связан с фосфонатом. Ниже представлены различные структуры AZT (азидотимидин), включая соединения, которые предполагается использовать при практическом осуществлении настоящего изобретения. Собственно AZT показан слева. Соединение А представляет собой AZT-монофосфат, который имеет обычную фофсодиэфирную связь между сахаром и фосфатом. В отличие от соединения А в соединениях В (AZT-5'-фосфонат) и С (AZT-5'-метиленфосфонат) отсутствует 5'-гидроксил 3'-азидо-2',3'-дидезоксирибозы, он заменен или связью фосфор-углерод (AZT-фосфонат), или метиленом, присоединенным связью фосфор-углерод (AZT-метиленфосфонат). Соединения В и С являются примерами соединений, полезных при практическом осуществлении настоящего изобретения.
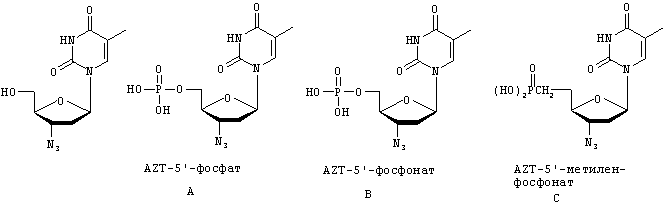
Соединения этого типа могут быть активны как антипролиферативные или противовирусные нуклеотиды. При клеточном метаболизме происходят два дополнительных фосфорилирования с образованием нуклеотид-фосфонат-дифосфата, который представляет собой эквивалент нуклеозид-трифосфатов. Противовирусные нуклеотид-фосфонат-дифосфаты являются селективными ингибиторами вирусных РНК- или ДНК-полимераз или обратных транскриптаз. Следует отметить, что их ингибирующее действие на вирусные полимеразы гораздо больше, чем степень ингибирования ими ДНК-полимераз α, β и γ клеток млекопитающих или РНК-полимераз млекопитающих. Напротив, антипролиферативные нуклеотид-фосфонат-дифосфаты ингибируют ДНК- и РНК-полимеразы раковых клеток и могут показывать гораздо более низкую селективность в сравнении с нормальными клеточными ДНК- и РНК-полимеразами. Так как нуклеотид-фосфонаты плохо всасываются из желудочно-кишечного тракта, они зачастую требуют парентерального введения (например, цидофовир). К тому же отрицательно заряженная фосфонатная группировка может препятствовать проникновению в клетку, давая в результате пониженную противовирусную или антипролиферативную активность. Неожиданно оказалось, что соединения по изобретению могут преодолевать недостатки этого класса агентов.
Известны фармакологически активные агенты, представляющие собой противовирусные фосфонаты; следующие патенты США описывают другие решения для нуклеотид-фосфонатных аналогов: 5672697 (нуклеозид-5'-метиленфосфонаты), 5922695 (противовирусные фосфонометокси-нуклеотидные аналоги), 5977089 (противовирусные фосфонометокси-нуклеотидные аналоги), 6043230 (противовирусные фосфонометокси-нуклеотидные аналоги), 6069249. Ранее было раскрыто получение и применение алкилглицеринфосфатов, ковалентно связанных c не содержащими фосфонат лекарствами, которые имеют амино, карбоксильные, гидроксильные или сульфгидрильные функциональные группы. Эти пролекарства возможно содержат связывающую группу или одну или две дополнительных фосфатных сложноэфирных группировки между лекарственным средством и алкилглицеринфосфатом (патент США №5411947 и патент США на заявку с порядковым номером 08/487081). Известны неполные эфиры хлорметандифосфоновой кислоты (патент США №5376649) и сообщалось о диангидридах клодроната (Ahlmark, et al, J Med Chem 42: 1473-1476 (1999)). Однако обнаружено, что неполные эфиры неспособны высвобождать активный бифосфонат при химическом или биохимическом превращении (Niemi, R. et al., J Chrom В 701:97-102 (1997)). Также описаны пролекарства, содержащие алкилглицеринфосфатные остатки, связанные с противовирусными нуклеозидами (патент США №5223263) или фосфоно-карбоксилатами (патент США №5463092).
Следовательно, имеется постоянная необходимость в менее токсичных более эффективных фармацевтических агентах для лечения множества расстройств, таких как расстройства, вызванные вирусной инфекцией и несоответствующей пролиферацией клеток, например рака. Таким образом, задачей настоящего изобретения является разработка химически модифицированных фосфонатных производных фармакологически активных агентов, например противовирусных и противоопухолевых фармацевтических агентов. Эти модифицированные производные усиливают эффективность родоначального соединения, уменьшая вредные побочные эффекты при введении субъекту, нуждающемуся в этом.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Согласно изобретению предложены аналоги фосфонатных соединений. Соединения фосфонатов, рассматриваемые для применения в соответствии с изобретением, включают те фосфонатные соединения, которые уменьшают резорбцию кости или ингибируют апоптоз остеобластов или остеоцитов, а также те соединения, которые улучшают биологическую активность, селективность или биодоступность нуклеотид-фосфонатных аналогов, которые полезны для лечения рака, различных вирусных инфекций и тому подобного. Соединения по изобретению содержат фосфонаты, ковалентно связанные (непосредственно или непрямо, т.е. через связывающую молекулу) с замещенным или незамещенным алкилглицерином, алкилпропандиолом, алкилэтандиолом или родственной группировкой. В соответствии с другим аспектом настоящего изобретения предложены фармацевтические препараты, содержащие аналоги описанных здесь фосфонатных соединений.
В соответствии с еще одним аспектом настоящего изобретения предложен ряд способов лечения, например способы лечения или предупреждения резорбции кости у млекопитающего, способы усиления остеогенеза путем предупреждения апоптоза остеобластов или остеоцитов, способы увеличения массы и прочности костей, способы лечения вирусных инфекций, способы лечения расстройств, вызванных несоответствующей пролиферацией клеток, например рака, и тому подобное.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
Фиг.1 резюмирует действие соединения по изобретению 1-O-гексадецилоксипропан-алендроната на вызванный дексаметазоном апоптоз клеток остеоцитов MLO-Y4. Столбцы представляют собой среднее значение ± среднеквадратичное отклонение трех независимых измерений. Светлые столбцы обозначают отсутствие дексаметазона, а темные столбцы обозначают присутствие 10-4 М дексаметазона.
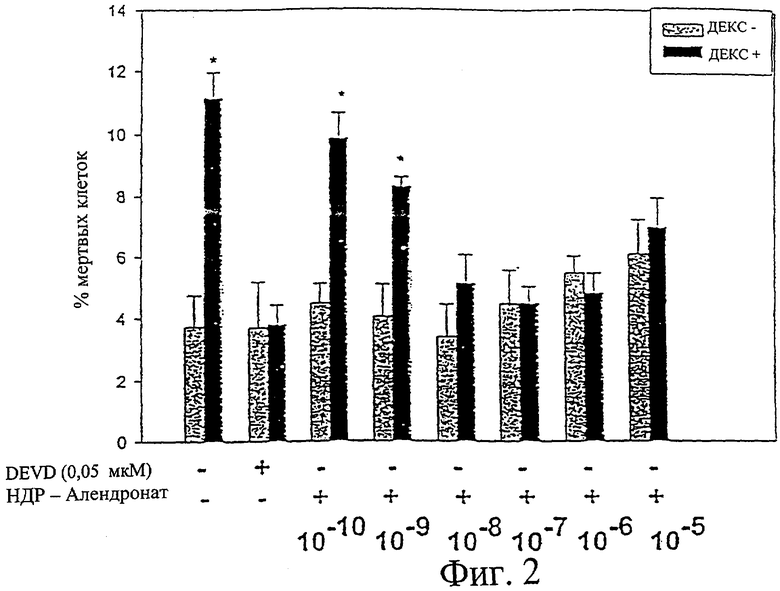
Фиг.2 резюмирует действие соединения по изобретению 1-O-гексадецилоксипропан-алендроната на вызванный дексаметазоном апоптоз клеток свода черепа. Столбцы представляют собой среднее значение ± среднеквадратичное отклонение трех независимых измерений. Серые столбцы обозначают отсутствие дексаметазона, а черные столбцы обозначают присутствие 10-4 М дексаметазона.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
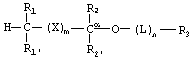
Фосфонатные соединения по изобретению имеют следующую структуру:
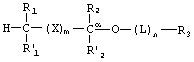
где R1 и R1' независимо представляют собой -Н, возможно замещенный -O(С1-С24)алкил, -O(С1-С24)алкенил, -O(С1-С24)ацил, -S(С1-С24)алкил, -S(C1-С24)алкенил или -S(С1-С24)ацил, причем по меньшей мере один из R1 и R1' не представляет собой -Н и причем указанные алкенильные или ацильные группировки возможно имеют от 1 до 6 двойных связей;
R2 и R2' независимо представляют собой -Н, возможно замещенный -O(С1-С7)алкил, -O(С1-С7)алкенил, -S(С1-С7)алкил, -S(С1-С7)алкенил, -O(C1-С7)ацил, -S(С1-С7)ацил, -N(С1-С7)ацил, -NH(С1-С7)алкил, -N((С1-С7)алкил)2, оксо, галоген, -NH2, -ОН или -SH;
R3 представляет собой фармацевтически активный фосфонат, бифосфонат или фосфонатное производное фармакологически активного соединения, которые связаны с функциональной группой возможного связующего звена L или с имеющимся атомом кислорода при Сα;
X, если присутствует, представляет собой
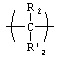
L представляет собой валентную связь или бифункциональную связующую молекулу формулы -J-(CR2)t-G-, где t равно целому числу от 1 до 24, J и G независимо представляют собой -О-, -S-, -С(O)O- или -NH-, a R представляет собой -Н, замещенный или незамещенный алкил или алкенил;
m равно целому числу от 0 до 6 и
n равно 0 или 1.
В предпочтительных воплощениях m равно 0, 1 или 2. В этих предпочтительных воплощениях R2 и R2' предпочтительно представляют собой -Н, а пролекарства представляют собой тогда этандиольные, пропандиольные или бутандиольные производные лекарственного фосфоната. Предпочтительный класс этандиолфосфонатов имеет следующую структуру:
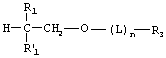
где R1, R1', R3, L и n такие, как определено выше.
Предпочтительный пропандиольный класс имеет следующую структуру:
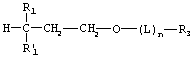
где m=1, a R1, R1', R3, L и n такие, как определено выше в общей формуле.
Предпочтительный глицериновый класс имеет следующую структуру:
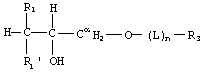
где m=1, R2=H, R2'=OH и R2 и R2' на Сα оба представляют собой -Н. Глицерин представляет собой оптически активную молекулу. При использовании условных обозначений стереоспецифической нумерации для глицерина положение sn-3 является тем положением, которое фосфорилируется глицеролкиназой. В соединениях по изобретению, имеющих глицериновый остаток, группировка -(L)n-R3 может быть присоединена или к sn-3, или к sn-1 положению глицерина.
Во всех классах фармакологически активных агентов по изобретению R1 предпочтительно представляет собой алкоксигруппу, имеющую формулу -O-(СН2)t-СН3, где t равно 0-24. Более предпочтительно t равно 11-19. Наиболее предпочтительно t равно 15 или 17.
Предпочтительные группы R3 включают бифосфонаты, которые известны как клинически полезные, например, следующие соединения:
Этидронат: 1-гидроксиэтилиден-дифосфоновая кислота (EDHP);
Клодронат: дихлорметилен-дифосфоновая кислота (Cl2MDP);
Тилудронат: хлор-4-фенилтиометилен-дифосфоновая кислота;
Памидронат: 3-амино-1-гидроксипропилиден-дифосфоновая кислота (ADP);
Алендронат: 4-амино-1-гидроксибутилиден-дифосфоновая кислота;
Ольпадронат: 3-диметиламино-1-гидроксипропилиден-дифосфоновая кислота (диметил-APD);
Ибандронат: 3-метилпентиламино-1-гидроксипропилиден-дифосфоновая кислота (ВМ 21.0955);
ЕВ-1053: 3-(1-пирролидинил)-1-гидроксипропилиден-дифосфоновая кислота;
Ризедронат: 2-(3-пиридинил)-1-гидроксиэтилиден-дифосфоновая кислота;
Амино-ольпадронат: 3-(N,N-диметиламино-1-аминопропилиден)бифосфонат (IG9402)
и тому подобные.
R3 также может быть выбран из ряда фосфонатсодержащих нуклеотидов (или нуклеозидов, которые можно превратить в их соответствующие производные фосфонаты), которые здесь также предполагается применять для целей настоящего изобретения. Предпочтительные нуклеозиды включают те, которые полезны для лечения расстройств, вызванных несоответствующей пролиферацией клеток, такие как 2-хлор-дезоксиаденозин, 1-β-D-арабинофуранозил-цитидин (цитарабин, ара-Ц), фторуридин, фтордезоксиуридин (флоксуридин), гемцитабин, кладрибин, флударабин, пентостатин (2'-дезоксикоформицин), 6-меркаптопурин, 6-тиогуанин и замещенный или незамещенный 1-β-D-арабинофуранозил-гуанин (ара-Г), 1-β-D-арабинофуранозил-аденозин (ара-А), 1-β-D-арабинофуранозил-уридин (ара-У) и тому подобное.
Нуклеозиды, полезные для лечения вирусных инфекций, также можно превратить в их соответствующие 5'-фосфонаты для применения в качестве группы R3. Такие фосфонатные аналоги типично содержат или фосфонатную (-РО3Н2), или метиленфосфонатную (-СН2-РО3Н2) группу, заместившую 5'-гидроксил в противовирусном нуклеозиде. Некоторыми примерами противовирусных фосфонатов являются производные, полученные путем замены 5'-гидроксила на -РО3Н2:
3'-азидо-3',5'-дидезокситимидин-5'-фосфоновая кислота (AZT-фосфонат)
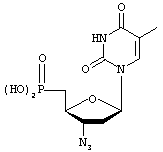
Hakimelahi, G.H.; Moosavi-Movahedi, A.A.; Sadeghi, M.M.; Tsay, S-C.; Hwu, J.R. J. Med. Chem. 1995, 38:4648-4659,
3',5'-дидезокситимидин-2'-ен-5'-фосфоновая кислота (d4T фосфонат)
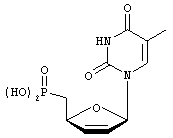
там же,
2',3',5'-тридезоксицитидин-5'-фосфоновая кислота (ddC фосфонат)
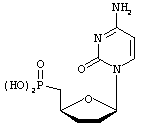
Kofoed, Т., Ismail, A.E.A.A.; Pedersen, E.B.; Nielsen, С. Bull.Soc.Chim. Fr. 1997, 134:59-65,
9-[3-(фосфоно-метокси)пропил]аденин (Адефовир)
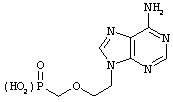
Kim, C.U.; Luh, B.Y.; Misco, P. F.; Bronson, J.J.; Hithcock, M. J.M.; Ghazzouli, I.; Martin, J.C. J. Med. Chem. 1990, 33:1207-1213.
Некоторыми примерами противовирусных фосфонатов являются производные, полученные путем замены 5'-гидроксила на -СН2-РО3Н2:
Ганцикловир-фосфонат
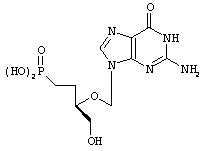
Huffman, J.H.; Sidwell, R.W.; Momson, A.G.; Coombs, J., Reist, E.J. Nucleoside Nucleotides, 1994, 13:607-613.
Ацикловир-фосфонат
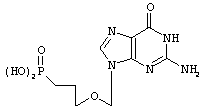
там же,
Ганцикловир-циклофосфонат
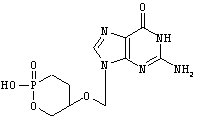
Smee, D.F.; Reist, E.J. Antimicrob. Agents Chemother. 1996, 40:1964-1966,
3'-тиа-2',3'-дидезоксицитидин-5'-фосфоновая кислота
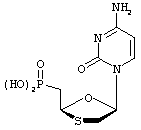
Kraus, J.L; Nucleoside Nucleotides, 1993, 12:157-162.
Другие предпочтительные противовирусные нукпеотидфосфонаты, которые предполагается применять для осуществления на практике настоящего изобретения, получают аналогично из противовирусных нуклеозидов, включающих ddA, ddl, ddG, L-FMAU, DXG, DAPD, L-dA, L-dl, L-(d)T, L-dC, L-dG, FTC, пенцикловир и тому подобное.
Дополнительно противовирусные фосфонаты, такие как цидофовир, цикло-цидофовир, адефовир, тенофовир и тому подобные, можно применять в качестве группы R3 по настоящему изобретению.
Определенные соединения по изобретению обладают одним или более чем одним хиральным центром, например, в сахарных группировках и, таким образом, могут существовать в оптически активных формах. Также, если соединения содержат алкенильную группу или ненасыщенную алкильную или ацильную группировки, то существует возможность наличия цис- и транс-изомерных форм таких соединений. Дополнительные асимметрические атомы углерода могут присутствовать в группе-заместителе, такой как алкильная группа. R- и S-изомеры и их смеси, включая рацемические смеси, а также смеси цис- и транс- изомеров, предусматриваются настоящим изобретением. Подразумевается, что все такие изомеры, а также их смеси включены в это изобретение. Если желателен индивидуальный стереоизомер, то его можно получить хорошо известными в данной области методами, применяя стереоспецифические реакции с исходными материалами, которые содержат асимметрические центры и уже разделены, или же методами, которые дают в результате смеси стереоизомеров и разделением их известными способами.
Существует много фосфонатных соединений, которые можно превратить в производные по изобретению, чтобы улучшить их фармакологическую активность или увеличить их всасывание при пероральном введении, как, например, соединения, описанные в следующих патентах, каждый из которых включен сюда полностью посредством ссылки: патенты США №№3468935 (Этидронат), 4327039 (Памидронат), 4705651 (Алендронат), 4870063 (производные дифосфоновой кислоты), 4927814 (бифосфонаты), 5043437 (фосфонаты азидодидезоксинуклеозидов), 5047533 (ациклические пурин-фосфонатные нуклеотидные аналоги), 5142051 (N-фосфонилметоксиалкильные производные пиримидинового и пуринового оснований), 5183815 (агенты, действующие на кость), 5196409 (бифосфонаты), 5247085 (противовирусные пуриновые соединения), 5300671 (гем-дифосфоновые кислоты), 5300687 (трифторметилбензилфосфонаты), 5312954 (бис- и тетракисфосфонаты), 5395826 (производные гуанидиналкил-1,1-дифосфоновой кислоты), 5428181 (бифосфонатные производные), 5442101 (производные метилендифосфоновой кислоты), 5532226 (трифторметилбензилфосфонаты), 5656745 (аналоги нуклеотидов), 5672697 (нуклеозид-5'-метилен-фосфонаты), 5717095 (аналоги нуклеотидов), 5760013 (аналоги тимидилатов), 5798340 (аналоги нуклеотидов), 5840716 (фосфонатнуклеотидные соединения), 5856314 (тиозамещенные азотсодержащие гетероциклические фосфонатные соединения), 5885973 (ольпадронат), 5886179 (аналоги нуклеотидов), 5877166 (энантиомерно чистые 2-аминопурин-фосфонат-нукпеотидные аналоги), 5922695 (противовирусные фосфонометокси-нукпеотидные аналоги), 5922696 (этиленовые и алленовые фосфонатные производные пуринов), 5977089 (противовирусные фосфонометокси-нуклеотидные аналоги), 6043230 (противовирусные фосфонометокси-нуклеотидные аналоги), 6069249 (противовирусные фосфонометокси-нуклеотидные аналоги), бельгийский патент №672205 (клодронат), европейский патент №753523 (аминозамещенные дифосфоновые кислоты); заявка на европейский патент 186405 (геминальные бифосфонаты) и тому подобное.
Определенные бифосфонатные соединения обладают способностью ингибировать скваленсинтазу и уровни холестерина в сыворотке крови у млекопитающих, включая человека. Примеры таких бифосфонатов описаны, например, в патентах США №№5441946 и 5563128 (Pauls et al. Phosphonate derivatives of lipophilic amines), каждый из которых полностью включен сюда путем ссылки. Аналоги этих ингибирующих скваленсинтазу соединений по изобретению и их применение в лечении нарушений липидного обмена у людей входят в объем настоящего изобретения. Бифосфонаты по изобретению можно применять перорально или местно для предупреждения или лечения заболеваний периодонта, как описано в патенте США №5270365, включенном сюда полностью путем ссылки.
Термин "алкил", как он используется здесь, относится к одновалентному радикалу с прямой или разветвленной цепью или циклическому радикалу, содержащему от 1 до 24 атомов углерода, включая метил, этил, н-пропил, изопропил, н-бутил, изобутил, mpem-бутил, н-гексил и тому подобное.
Как он используется, "замещенный алкил" обозначает алкильные группы, дополнительно несущие один или более чем один заместитель, выбранный из гидрокси, алкокси (низшей алкильной группы), меркапто (низшей алкильной группы), циклоалкила, замещенного циклоалкила, гетероцикла, замещенного гетероцикла, арила, замещенного арила, гетероарила, замещенного гетероарила, арилокси, замещенного арилокси, галогена, трифторметила, циано, нитро, нитрона, амино, амидо, -С(О)Н, ацила, оксиацила, карбоксила, карбамата, сульфонила, сульфурила и тому подобного.
Как он используется здесь, "алкенил" относится к углеводородным группам с прямой или разветвленной цепью, имеющим одну или более чем одну двойную углерод-углеродную связь и содержащим в пределах от примерно 2 до 24 атомов углерода, а "замещенный алкенил" относится к алкенильным группам, дополнительно несущим один или более чем один заместитель, как указано выше.
Как он используется здесь, "арил" относится к ароматическим группам, содержащим в пределах от 6 до 14 атомов углерода, а "замещенный арил" относится к арильным группам, дополнительно несущим один или более чем один заместитель, как указано выше.
Как он используется здесь, "гетероарил" относится к ароматическим группам, содержащим один или более чем один гетероатом (например, N, О, S или тому подобное) как часть структуры кольца и содержащий в пределах от 3 до 14 атомов углерода, а "замещенный гетероарил" относится к гетероарильным группам, дополнительно несущим один или более чем один заместитель, как указано выше.
Термин "связь", или "валентная связь", относится к связи между атомами, состоящей из электронной пары.
Как он используется здесь, термин "фармацевтически приемлемые соли" относится как к солям присоединения кислоты, так и к солям присоединения основания.
Как он используется здесь, термин "пролекарство" относится к производным фармацевтически активных соединений, которые имеют группы, отщепляемые химически или в ходе обмена веществ, и становятся фармацевтически активным соединением в результате сольволиза или в физиологических условиях in vivo.
Фосфонатные аналоги, содержащие терапевтически эффективные фосфонаты (или фосфонатные производные терапевтически эффективных соединений), ковалентно связанные через гидроксильную группу с 1-O-алкилглицерином, 3-O-алкилглицерином, 1-S-алкилтиоглицерином или алкокси-алканолом, могут более эффективно всасываться в желудочно-кишечном тракте, чем родоначальные соединения. Перорально вводимая доза аналога поглощается целиком из желудочно-кишечного тракта млекопитающего, и активное лекарство высвобождается in vivo под воздействием эндогенных ферментов. Фосфонатные аналоги по изобретению также могут иметь более высокую степень биологической активности по сравнению с соответствующими соединениями до получения производных.
Соединения по настоящему изобретению являются усовершенствованием по сравнению с алкилглицеринфосфатными пролекарствами, описанными в предшествующем уровне техники, так как содержащая фосфонат группировка связана непосредственно с алкилглицериновой или алкоксиалканольной группировкой, и потому что наличие фосфонатной связи предотвращает ферментативное превращение в свободное лекарство. В усовершенствованных аналогах могут присутствовать другие связующие звенья между этими группами. Например, бифункциональные связующие звенья, имеющие формулу -O-(СН2)n-С(O)O-, где n равно 1-24, могут связывать фосфонат с гидроксильной группой алкоксиалканольной или алкилглицериновой группировки.
Вышеупомянутое позволяет фосфонату по изобретению достичь большей степени всасывания при пероральном введении. К тому же клеточные ферменты, но не ферменты плазмы или пищеварительного тракта, будут превращать такой конъюгат в свободный фосфонат. Дополнительное преимущество алкокси-алканол-фосфонатов состоит в том, что существенно снижается или исключается тенденция совместно вводимой пищи уменьшать или устранять всасывание фосфонатов, в результате чего достигаются более высокие уровни в плазме и лучшее соблюдение больным схемы лечения.
Соединения по изобретению можно вводить перорально в форме таблеток, капсул, растворов, эмульсий или суспензий, в форме жидкости или твердых частиц при ингаляции, в форме микроинкапсулированных частиц, в виде спрея, через кожу с помощью такого приспособления как трансдермальный пластырь или ректально, например, в форме суппозиториев. Липофильные пролекарственные производные по изобретению особенно хорошо подходят для введения с трансдермальным впитыванием и систем доставки и могут быть использованы в зубной пасте. Введение также можно осуществлять парентерально в форме инъекционных растворов.
Композиции можно приготовить в обычных формах, например капсулах, таблетках, аэрозолях, растворах, суспензиях или вместе с носителями для местного нанесения. Фармацевтические препараты, содержащие соединения по данному изобретению, можно приготовить с помощью традиционных методик, например, как описано в Remington's Pharmaceutical Sciences. 1985.
Применяемый фармацевтический носитель или разбавитель может быть традиционным твердым или жидким носителем. Примерами твердых носителей являются лактоза, сахароза, тальк, желатин, агар, пектин, аравийская камедь, стеарат магния, стеариновая кислота или низшие алкиловые эфиры целлюлозы. Примерами жидких носителей являются сироп, арахисовое масло, оливковое масло, фосфолипиды, жирные кислоты, амины жирных кислот, полиоксиэтилен или вода. Носитель или разбавитель может содержать в себе какой-нибудь известный материал для пролонгированного высвобождения, такой как глицерилмоностеарат или дистеарат, один или в смеси с воском.
Если применяют твердый носитель для перорального введения, то препарат можно таблетировать или поместить в форме порошка или гранул в твердую желатиновую капсулу. Количество твердого носителя варьируется в широких пределах, но обычно составляет от примерно 25 мг до примерно 1 г. Если применяют жидкий носитель, то препарат может быть в форме сиропа, эмульсии, мягкой желатиновой капсулы или стерильной инъекционной жидкости, такой как водная или неводная жидкая суспензия или раствор.
Таблетки готовят путем смешивания активного ингредиента (то есть одного или более чем одного соединения по изобретению) с фармацевтически инертным органическим или неорганическим носителем, разбавителями и/или эксципиентами. Примерами таких эксципиентов, которые можно применять для таблеток, являются лактоза, кукурузный крахмал или его производные, тальк, стеариновая кислота или ее соли. Примерами подходящих эксципиентов для желатиновых капсул являются растительные масла, воски, жиры, полутвердые и жидкие полиолы. Бифосфонатные пролекарства также можно производить в микроинкапсулированной форме.
Для интраназального введения препарат может содержать соединение по изобретению, растворенное или суспендированное в жидком носителе, в частности в водном носителе, для применения в виде аэрозоля. Носитель может содержать солюбилизирующие агенты, такие как пропиленгликоль, поверхностно-активные вещества, усилители всасывания, такие как лецитин или циклодекстрин, или консерванты.
Фармацевтические композиции для парентеральных инъекций по данному изобретению содержат фармацевтически приемлемые стерильные водные или неводные жидкости, дисперсии, суспензии или эмульсии, а также стерильные порошки для растворения с получением стерильных инъекционных растворов или дисперсий непосредственно перед применением.
Подходящими эксципиентами для приготовления растворов и сиропов являются вода, полиолы, сахароза, инвертный сахар, глюкоза и тому подобное. Подходящими эксципиентами для приготовления инъекционных растворов являются вода, полиолы, спирты, глицерин, растительные масла и тому подобное.
Фармацевтические продукты дополнительно могут содержать что-либо из множества таких добавляемых компонентов, как, например, консерванты, солюбилизаторы, стабилизаторы, увлажняющие агенты, эмульгаторы, подсластители, красители, корригенты, буферы, покрывающие агенты, антиоксиданты, разбавители и тому подобное.
Факультативно фармацевтические композиции по изобретению могут содержать соединение, соответствующее общей формуле, в комбинации с одним или более чем одним соединением, проявляющим иную активность, например антибиотиком или другим фармакологически активным материалом. Такие комбинации входят в объем настоящего изобретения.
Согласно данному изобретению предложены способы лечения расстройств млекопитающих, связанных с костным обменом веществ, вирусными инфекциями, несоответствующей пролиферацией клеток и тому подобным. В частности, в этих способах человеку или другому млекопитающему, нуждающемуся в этом, вводят терапевтически эффективное количество пролекарств по данному изобретению. Показания к такому лечению включают лечение возрастного, постклимактерического или вызванного стероидами остеопороза, болезни Педжета, рака с костными метастазами, гиперпаратиреоза, ревматоидного артрита, алгодистрофии, грудинно-реберно-ключичного гиперостоза, болезни Гоше, болезни Энгельманна, некоторых нескелетных расстройств и заболеваний периодонта, вируса иммуннодефицита человека (ВИЧ), гриппа, вируса, простого герпеса (HSV), вируса герпеса человека 6, цитомегаловируса (CMV), вируса гепатита В, вируса Эпштейна-Барра (EBV), вируса ветряной оспы, лимфом, гематологических расстройств, таких как лейкемия, и тому подобное.
В соответствии с одним аспектом по изобретению предложены способы предупреждения или лечения потери кости у млекопитающих, особенно у людей, при которых человеку или млекопитающему вводят терапевтически эффективное количество соединений по данному изобретению. Бифосфонатные пролекарства по изобретению, ингибирующие резорбцию кости, терапевтически полезны для противодействия опосредованной остеокластами резорбции кости или потери кости в условиях, когда бифосфонаты, из которых получено пролекарство, считаются эффективными. Показания к такому лечению включают остеопороз, в частности, у женщин после менопаузы, остеопороз, который сопутствует длительной глюкокортикостероидной терапии, костная болезнь Педжета. Обнаружено, что бифосфонатное соединение клодронат (Ostac, Boehringer-Mannheim, Mannheim, Germany) также снижает костные, а также висцеральные метастазы у пациентов с раком молочной железы при высоком риске отдаленных метастазов (Diel, I.J. et al. (1998) New Engl. J. Med. 339 (60, 357-363). Эффективность бифосфонатных пролекарств по изобретению можно оценить теми же самыми способами, что и эффективность родоначального соединения. Они включают сравнительное измерение минеральной костной плотности поясничного отдела позвоночника, шейки бедра, вертела, предплечья и всего тела наряду с измерениями вертебральных разломов, спинальных деформаций и роста при остеопорозе, сканированием кости или рентгенографической идентификацией повреждений кости при метастазирующей опухоли и тому подобное.
Согласно еще одному аспекту данного изобретения предложены способы увеличения массы и прочности кости у млекопитающих, особенно у людей, при котором вводят стимулирующие анаболизм кости соединения по изобретению, которые ингибируют апоптоз остеобластов и остеоцитов, приводя к более значительным скоростям собственно остогенеза, при этом не изменяя существенно функций остеокластов (Piotkin et al., J Clin Invest 104:1363-1374 (1999), и Van Beek et. al., J Bobe Min Res 11:1492 (1996)).
Согласно еще одному аспекту изобретения предложены способы лечения расстройств, вызванных вирусными инфекциями. Показания к такому лечению включают чувствительные вирусы, такие как вирус иммуннодефицита человека (ВИЧ), грипп, вирус простого герпеса (HSV), вирус герпеса человека 6, цитомегаловирус (CMV), вирус гепатита В и С, вирус Эпштейна-Барра (EBV), вирус ветряной оспы, и заболевания, вызванные ортопокс-вирусами (например, вирусами большой и малой натуральной оспы, коровьей оспы, оспы человека, осповакцины, верблюжьей оспы, обезьяньей оспы и тому подобного), возбудителем геморрагической лихорадки Эбола, вирусом папилломы и тому подобным.
Согласно еще одному аспекту изобретения предложены способы лечения расстройств, вызванных несоответствующей пролиферацией клеток, например рака, такого как меланома, рак легких, рак поджелудочной железы, рак желудка, толстой кишки и прямой кишки, рак предстательной железы и молочной железы, лейкемия, лимфомы и тому подобное. Противопухолевые соединения, которые можно превратить в их нуклеотидфосфонаты для применения в качестве соединений по данному изобретению, включают, но не ограничиваются ими, цитарабин (ара-С), фторуридин, фтордезоксиуридин (флоксуридин), гемцитибин, кладрибин, флударабин, пентостатин (2'-дезоксикоформицин), 6-меркаптопурин, 6-тиогуанин, а также замещенный или незамещенный ара-аденозин (ара-А), ара-гуанозин (ара-Г) и ара-уридин (ара-У). Противоопухолевые соединения по изобретению можно применять одни или в комбинации с другими антиметаболитами или другими классами противоопухолевых лекарств, такими как алкалоиды, ингибиторы топоизомеразы, алкилирующие агенты, противоопухолевые антибиотики и тому подобное.
Пролекарства по изобретению можно вводить перорально, парентерально, местно, ректально и другими путями в соответствующих единицах дозировки, как требуется.
Термин "парентеральный", как он использован здесь, относится к подкожной, внутривенной, внутриартериальной, внутримышечной инъекциям и инъекциям внутрь стекловидного тела глаза или инфузионным методикам.
Термин "местно" включает введение ректально или посредством ингаляционного аэрозоля, а также более общие пути через кожу, слизистые оболочки рта и носа и в зубной пасте.
Термин "эффективное количество" применительно к фосфонатным пролекарствам по изобретению означает количество, которое будет предупреждать или реверсировать указанные выше заболевания. В частности, что касается расстройств, связанных с костным обменом веществ, эффективное количество представляет собой количество, которое будет предупреждать, ослаблять или реверсировать аномальную или чрезмерную резорбцию кости или резорбцию кости, которая встречается у стареющих, особенно в менопаузе, женщин либо предупреждать или противодействовать костным метастазам и висцеральным метастазам при раке молочной железы.
Что касается расстройств, связанных с вирусными инфекциями или несоответствующей пролиферацией клеток, например раком, "эффективное количество" определяют в соответствии с рекомендуемыми дозировками родоначального противовирусного или противоракового соединения. Выбранная доза будет варьироваться в зависимости от активности выбранного соединения, пути введения, тяжести состояния, которое лечат, и состояния и предшествующей истории болезни лечащегося пациента. Знания в данной области позволяют начинать принимать дозы соединения (соединений) на уровнях, более низких, чем необходимо для достижения желаемого терапевтического эффекта, и постепенно увеличивать дозировку, пока желаемый эффект не будет достигнут. Если желательно, эффективную суточную дозу можно разделить на множественные дозы с целью введения, например, от двух до четырех доз в сутки. Следует понимать, однако, что конкретный уровень доз для отдельного пациента будет зависеть от ряда факторов, включающих вес тела, общее состояние здоровья, диету, время, путь введения и комбинирование с другими лекарствами, а также тяжесть заболевания, которое лечат.
В целом, соединения по настоящему изобретению обеспечивают в виде стандартной лекарственной формы, содержащей от 1 до 100% активного ингредиента. Интервал терапевтической дозировки составляет от примерно 0,01 до примерно 1000 мг/кг/сутки, предпочтительно от примерно 0,10 до 100 мг/кг/сутки, при введении пациентам, например людям, в качестве лекарства. Фактические уровни дозировок активных ингредиентов в фармацевтических композициях по данному изобретению можно варьировать, с тем чтобы ввести количество активного соединения(ий), которое эффективно для достижения желаемого терапевтического ответа у конкретного пациента.
Многочисленные эксперименты на животных показали эффективность бифосфонатов в предупреждении потери кости в экспериментальных условиях, имитирующих релевантные клинические расстройства. На основании этих исследований имеются несколько небольших систем модельных животных для оценки эффективности бифосфонатов. Эти тесты также полезны для измерения сравнительной эффективности бифосфонатных пролекарств по изобретению. Оценка бифосфонатной терапии обычно требует определения массы бедренной золы и костной массы, измеренной, например, как трабекулярный объем кости у групп обработанных и необработанных животных. Thomson, D. et al. ((1990) J. Bone and Mineral Res. 5(3):279-286) описывает применение таких способов оценки ингибирования потери кости у иммобилизованных крыс, которых обрабатывали аминогидроксибутан-бифосфонатом. Yamamoto, M. et at. (1993) Calcif Tissue Int 53:278-282, вызывали гипертиреоз у крыс с получением изменений кости, аналогичных таковым у людей с гипертиреозом, и сравнивали группы, которых лечили бифосфонатами и не лечили, биохимически на основании измерений остеокальцина, а также с помощью гистоморфометрического анализа, включая различия в объеме губчатого вещества кости, и гистологического сравнения остеоидных, остеокластных и остеобластных поверхностей в костных срезах. Seedor, J.G. et al. ((1991) J. Bone and Mineral Res. 6(4):339-346) описывает исследования действия алендроната по антагонизации потери кости у овариэктомированых крыс по массе бедренной золы и путем гистоморфометрического анализа объема большеберцовой трабекулярной кости. Анализ Шенка, включающий гистологическое исследование эпифиза растущих крыс, также можно применять в качестве скрининг-анализа. Иллюстративный скрининг-тест для оценки антагонизирующего действия соединений по изобретению на резорбцию кости у лабораторных крыс, у которых с помощью разных приемов нарушили остеогенез, описан в примере 14.
Соединения по изобретению можно получить различными путями, как в общем виде представлено на схемах I-VI. Общие способы этерификации фосфонатов, описанные ниже, приведены только с целью иллюстрации и не должны быть истолкованы как каким-либо образом ограничивающие данное изобретение. Действительно, разработано несколько способов непосредственной конденсации фосфоновых кислот со спиртами (см., например, R.C. Larock, Comprehensive Organic Transformation, VCH, New York, 1989, p. 966, и приводимые там источники информации). Выделение и очистку соединений и промежуточных продуктов, описанных в примерах, можно выполнить, если требуется, с применением любой подходящей методики разделения и очистки, как, например, фильтрации, экстракции, кристаллизации, колоночной флэш-хроматографии, тонкослойной хроматографии, дистилляции или комбинации этих методик. Конкретные иллюстрации подходящих методик разделения и выделения приведены ниже в примерах. Другие эквивалентные методики разделения и выделения, конечно, также могут быть применены.
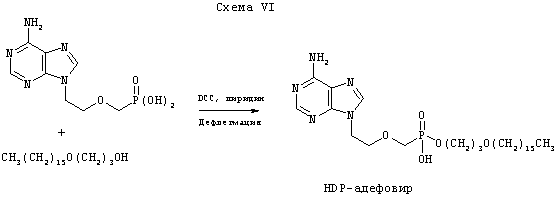
Схема 1 показывает в общих чертах синтез бифосфонатных пролекарств, которые содержат первичную аминогруппу, таких как памидронат или алендронат. Пример 1 дает условия синтеза 1-O-гексадецилоксипропил-алендроната (HDP-алендронат) или 1-O-гексадецилоксипропил-памидроната (HDP-памидронат). В этом способе смесь диметил-4-фталимидобутаноил-фосфоната (1b, получен, как описано в патенте США 5039819) и гексадецилоксипропил-метилфосфита (2) в пиридиновом растворе обрабатывают триэтиламином, получая бифосфонатный тетраэфир 3b, который очищают с помощью хроматографии на силикагеле. Промежуточное соединение 2 получают трансэтерификацией дифенилфосфита, как описано в (Kers, A., Kers, I., Stawinski, J., Sobkowski, M., Kraszewsku, A., Synthesis, April 1995, 427-430). Так, дифенилфосфит в пиридиновом растворе сперва обрабатывают гексадецилоксипропан-1-олом, затем метанолом, в результате чего получают соединение 2.
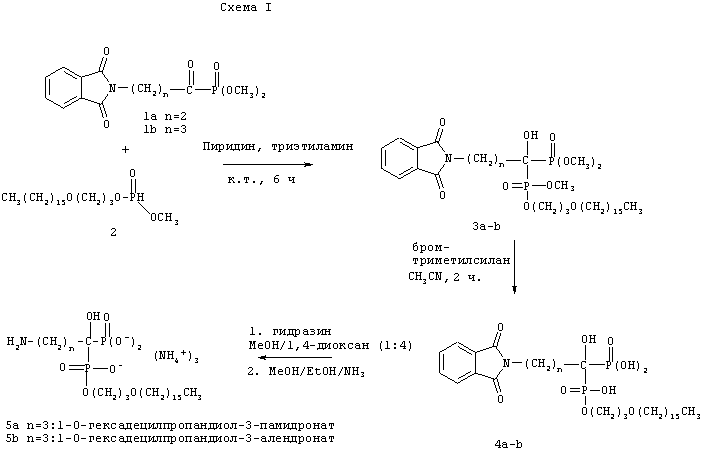
Существенным аспектом данного способа является то, что можно применять другие длинноцепочечные спирты вместо гексадецилоксипропан-1-ола, получая различные соединения по данному изобретению. Обработкой промежуточного соединения 3b бромтриметилсиланом в ацетонитриле селективно расщепляют метиловые эфиры, получая моноэфир 4b. Обработка 4b гидразином в смешанной системе растворителей (20% метанол / 80% 1,4-диоксан) приводит к удалению защитной фталимидной группы, как показано на схеме. Желаемое пролекарство алендроната собирают путем фильтрации и превращают в трехаммонийную соль путем обработки метанольным раствором аммиака.
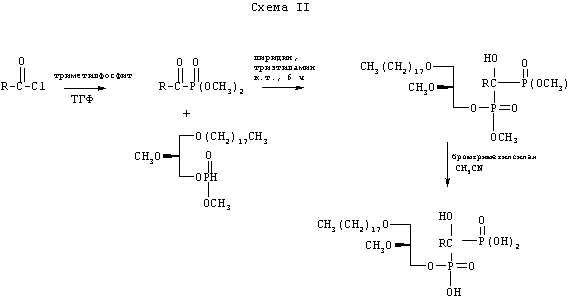
Схема II иллюстрирует синтез аналогов бифосфонатов, у которых отсутствует первичная аминогруппа. В этом случае стадии способа аналогичны таковым на схеме I, за исключением того, что защита фталимидной группой и последующее снятие защиты гидразинолизом не нужны.
Бифосфонаты, имеющие 1-аминогруппу, такие как амино-ольпадронат, можно превратить в аналоги, соответствующие пролекарствам по изобретению, применяя слегка модифицированный способ, показанный на схеме III.
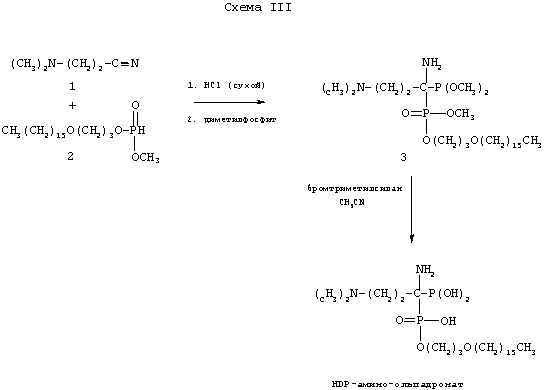
Обработка смеси соединения 2 и 3-(диметиламино)пропионитрила сухим HCl с последующим добавлением диметилфосфита дает тетраэфир 3, который после деметилирования бромтриметилсиланом дает гексадецилоксипропил-1-амино-ольпадронат.
Схема IV иллюстрирует синтез не эфира фосфаната, а бифосфонатного аналога, у которого к первичной аминогруппе присоединена липидная группа родоначального соединения.
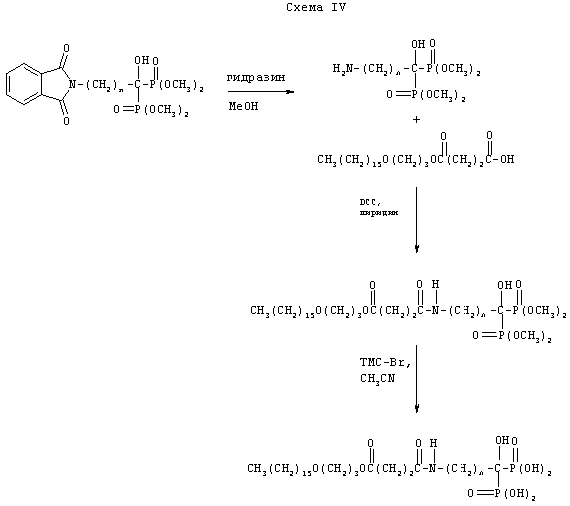
Схема V иллюстрирует общий синтез алкилглицериновых или алкилпропандиольных аналогов цидофовира, цикло-цидофовира и других фосфонатов. Обработкой 2,3-изопропилиденглицерина 1 NaOH в диметилформамиде с последующим взаимодействием с алкилметансульфонатом получают алкиловый эфир 2. Удаление изопропилиденовой группы обработкой уксусной кислотой с последующим взаимодействием с тритилхлоридом в пиридине дает промежуточное соединение 3. Алкилирование промежуточного соединения 3 алкилгалогенидом приводит к соединению 4. Удаление тритильной группы 80%-ной водной уксусной кислотой приводит к O,O-диалкилглицерину 5. В результате бромирования соединения 5 с последующим взаимодействием с натриевой солью цикло-цидофовира или другого фосфонатсодержащего нуклеотида получают желаемый фосфонатный продукт присоединения 7. Размыкание цикла цикло-аддукта осуществляется путем взаимодействия с водным гидроксидом натрия. Предпочтительный пропандиольный класс можно синтезировать, заменяя соединение 5 на 1-O-алкилпропан-3-ол в схеме V. Аналоги тенофовира и адефовира можно синтезировать путем замены цикло-цидофовира (cCDV) на эти нуклеотид-фосфонаты в реакции (f) схемы V. Аналогично этим же способом можно получить и другие нуклеотид-фосфонаты по изобретению.
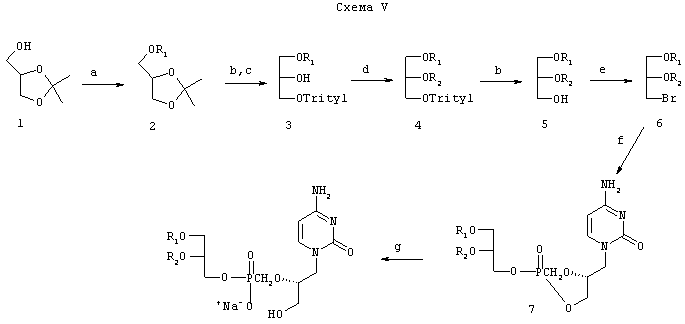
Реагенты: a) NaH, R1OSO2Me, диметилформамид (ДМФ); b) 80% вод. уксусная кислота; с) тритилхлорид, пиридин; d) NaH, R2-Br, ДМФ; е) CBr4, трифенилфосфин; тетрагидрофуран (ТГФ); f) цикло-цидофовир (соль DCMC), ДМФ; д) 0,5 н. NaOH
Схема VI иллюстрирует общий способ синтеза нуклеотид-фосфонатов по изобретению с применением 1-O-гексадецилоксипропил-адефовира в качестве примера. Нуклеотид-фосфонат (5 ммоль) суспендируют в безводном пиридине и добавляют алкоксиалканольное или алкилглицериновое производное и 1,3-дициклогексилкарбодиимид (DCC, 10 ммоль). Смесь нагревают с обратным холодильником и интенсивно перемешивают до тех пор, пока реакция конденсации не завершится (следят с помощью тонкослойной хроматографии). Затем смесь охлаждают и фильтруют. Фильтрат концентрируют при пониженном давлении, остаток абсорбируют на силикагеле и очищают путем колоночной флэш-хроматографии (элюирование смесью приблизительно 9:1 дихлорметан/метанол), получая соответствующий моноэфир фосфоната.
Теперь изобретение будет описано более подробно путем адресации к следующими неограничивающим примерам.
ПРИМЕР 1
Синтез 1-О-гексадецилпропандиол-3-алендроната
А. Гексадецилоксипропилметилфосфит(b)
Гексадецилоксипропилметилфосфит был получен способом, описанным в Kers, A., Kers, I., Stawinski, J., Sobkowski, M., Kraszewsku, A., Synthesis, April 1995, 427-430. К раствору дифенилфосфита (14 г, 60 ммоль) в пиридине (50 мл), поддерживаемому при 0°С, медленно добавили раствор гексадецилоксипропан-1-ола (6,0 г, 20 ммоль) в пиридине (25 мл). Смесь перемешивали в течение 1 часа, перед тем как добавили безводный метанол (10 мл). После перемешивания в течение еще одного часа растворитель выпарили и остаток адсорбировали на силикагеле и хроматографировали, применяя градиентное элюирование (начиная с гексанов и до смеси 20% этилацетат / 80% гексаны), в результате чего получили чистое соединение 2 в виде воскообразного легкоплавкого твердого вещества (4,5 г, выход 60%).
1H-ЯМР (CDCl3, δ): 6.79 (д, 1Н, J = 696 Гц), 4.19 (кв, 2H), 3.78 (д, 3Н), 3.51 (т, 3Н), 3.40 (т, 2H), 1.95 (пент, 2H), 1.25 (шир. с, 28Н), 0.88 (т, 3Н).
В. Гексадецилоксипропил-триметил-4-фталимидобутаноил-фосфонат(3b)
К смеси диметил-4-фталимидобутаноил-фосфоната (1b, 3,0 г. 7,9 ммоль, получен, как описано в патенте США 5039819) и гексадецилоксипропилметилфосфита (2, 2,9 г, 9 ммоль) в пиридине (50 мл) добавили триэтиламин (0,2 г, 2 ммоль). Смесь перемешивали в течение 5 часов при комнатной температуре, затем растворитель удалили в вакууме. Остаток абсорбировали на силикагеле и хроматографировали (этилацетат) с получением 3b (3,5 г, 63%) в виде вязкого масла.
1H-ЯМР (CDCl3): 7.84 (д, 2Н), 7.72 (д, 2Н), 4.45 (м, 1Н), 4.27 (м. 4Н), 4.15 (кв, 2Н), 3.68 (с, 3Н), 3.84 (с, 3Н), 3.71 (т, 2Н), 3.51 (м, 2Н), 3.38 (т, 2Н), 2.04 (м, 2Н), 1.94 (пент, 2Н), 1.54 (м, 2Н), 1.25 (шир. с, 28Н), 0.88 (т, 3Н).
31Р-ЯМР (22.54 (дублет), 21.22 (квартет)).
С. Гексадецилоксипропил-4-фталимидобутаноил-фосфонат(4b)
Соединение 3b, полученное на предыдущей стадии, (3,0 г, 4,3 ммоль), растворили в безводном ацетонитриле (50 мл) и охладили до 0°С. Медленно добавили раствор бромтриметилсилана (3,9 г, 25,5 ммоль) в ацетонитриле (25 мл) и затем этот раствор дополнительно перемешивали в течение 2 часов. Затем смесь медленно вылили в толченый лед. Образовавшийся осадок собирали путем вакуумной фильтрации и сушили в вакууме с получением 1,2 г 4b (выход 42%).
1H-ЯМР (ДМСО-d6): 7.86 (м, 4Н), 3.99 (кв, 2Н), 3.66-3.55 (м, 1Н), 3.54 (м, 2Н), 3.35 (т, 2Н), 3.27 (т, 2Н), 1.89-1.80 (м), 1.72 (пент, 2Н), 1.53-1.40 (м, 2Н), 1.22 (шир.с, 28Н), 0.85 (т,3Н).
31Р-ЯМР: 21.51 (дублет), 19.50 (дублет).
D. 1-О-Гексадецилпропандиол-3-алендронат (5b)
Соединение 4b (300 мг, 0,45 ммоль) растворили в смеси 1,4-диоксана (20 мл) и метанола (5 мл). Затем добавили безводный гидразин и смесь перемешивали при комнатной температуре в течение 4 часов. Выпавший осадок собрали вакуумной фильтрацией и промыли 1,4-диоксаном. Твердое вещество затем суспендировали в этаноле и добавили метанольный раствор аммиака (3 мл). После перемешивания в течение 10 минут полученное в результате твердое вещество собирали путем фильтрации, промыли этанолом и сушили в вакууме. В результате получили 220 мг HDP-алендроната (5b) в виде трехаммонийной соли. Анализ с помощью FT-IR показал отсутствие фталимидной защитной группы. Масс-спекторметрия (МС) с электрораспылением: m/е 532 (МН+), 530 (МН-).
ПРИМЕР 2
Синтез 1-O-гексадецилпропандиол-3-памидроната (5а)
1-O-Гексадецилпропандиол-3-памидронат получают аналогичным способом (по схеме 1), за исключением того что 3-фталамидопропановая кислота применяется для получения диметил-3-фталимидопропаноил-фосфоната (1а). Соединение 1а конденсируют с 2, получая триметилбифосфонат 3а. Снимая защиту так же, как на вышеописанных стадиях С и D, получают HDP-памидронат, как показано.
ПРИМЕР 3
Синтез 1-O-октадецил-2-O-метил-sn-глицеро-3-алендроната
Пролекарства с липофильными группами, отличными от гексадецилоксипропильной, получают путем замены гексадецилоксипропан-1-ола на стадии А примера 1 на различные длинноцепочечные спирты. Например, взаимодействие 1-O-октадецил-2-O-метил-sn-глицерина с дифенилфосфитом в пиридине с последующей обработкой метанолом дает 1-O-октадецил-2-O-метил-sn-глицерилметилфосфит. Конденсация этого диалкилфосфита с фосфонатом 1b с последующим снятием защиты на стадиях С и D дает 1-O-октадецил-2-O-метил-sn-глицеро-3-алендронат. Схема 2 иллюстрирует синтез других бифосфонатных конъюгатов, которые не имеют первичной аминогруппы в боковой цепи. В этом случае не нужны защита фталимидной группой и снятие защиты гидразинолизом.
ПРИМЕР 4
Синтез HDP-амино-ольпадроната
Схема 3 иллюстрирует синтез 1-амино-бифосфонатных конъюгатов. Применяя соединение 2 из примера 1, 3-(диметиламино)пропионитрил и способы, описанные в Orlovskii, V.V.; Vovsi, B.A. J. Gen Chem. USSR (Engl. Transl.) 1976, 46:294-296), получают триметиловый эфир бифосфоната 3. В результате деметилирования бромтриметилсиланом, как описано на стадии С примера 1, получают HDP-амино-ольпадронат.
ПРИМЕР 5
Синтез 1-O-гексадецилпропандиол-3-сукцинил-алендроната
Схема 4 иллюстрирует синтез бифосфонатного конъюгата, у которого липидная группа присоединена к первичной аминогруппе родоначального соединения. Тетраметил-(4-фталимидо-1-гидробутилиден)бифосфонат (2,0 г, 4,4 ммоль) растворили в 0,2 М метанольном растворе гидразина (100 мл) и этот раствор перемешивали при комнатной температуре в течение 3 дней. Смесь сконцентрировали до половины ее объема, когда началось выделение твердого вещества. Это твердое вещество отфильтровали и фильтрат сконцентрировали до сухости. Протонный ЯМР показал, что это соединение представляет собой тетраметил-(4-амино-1-гидробутилиден)бифосфонат. Его сушили над пентаоксидом фосфора при 50°С в течение ночи. К суспензии 1,2 г этого соединения в смеси пиридина (25 мл) и N,N-диметилформамида (25 мл) добавили 3-сукцинил-1-гексадецилоксипропан (1,76 г, 4,4 ммоль). Добавили дициклогексилкарбодиимид (2,52 г, 12,21 ммоль) и эту смесь перемешивали при комнатной температуре в течение 2 дней. Смесь фильтровали, фильтрат абсорбировали на силикагеле и подвергли флэш-хроматографии с возрастающим градиентом метанола в дихлорметане (0%-20%), в результате чего получили сукцинилированное соединение. С него сняли защиту триметилсилилбромидом в ацетонитриле с получением указанного в заголовке соединение, которое очистили кристаллизацией из метанола.
ПРИМЕР 6
Синтез гексадецилоксипропилового и 1-O-октадецил-sn-глицеринового эфиров адефовира
К смеси адефовира (1,36 г, 5 ммоль) и 3-гексадецилокси-1-пропанола (1,8 г, 6 ммоль) в безводном пиридине добавили DCC (2,06 г, 10 ммоль). Смесь нагревали с обратным холодильником и перемешивали в течение 18 часов, затем охладили и отфильтровали. Фильтрат концентрировали при пониженном давлении и остаток нанесли на короткую колонку с силикагелем. В результате элюирования с колонки смесью 9:1 дихлорметан/метанол получили гексадецилоксипропил-адефовир (HDP-ADV) в виде бесцветного порошка.
К смеси адефовира (1,36 г, 5 ммоль) и 1-O-октадецил-sn-глицерина (2,08 г, 6 ммоль) в безводном пиридине (30 мл) добавили DCC (2,06 г, 10 ммоль). Смесь нагревали с обратным холодильником и перемешивали в течение ночи, затем охладили и отфильтровали. Фильтрат концентрировали при пониженном давлении и остаток нанесли на колонку с силикагелем. В результате элюирования с колонки смесью дихлорметан/метанол 9:1 получили 1-O-октадецил-sn-глицерил-3-адефовир.
ПРИМЕР 7
Синтез гексадецилоксипропилового эфира AZT-фосфоната
Фосфонатный аналог AZT (3'-азидо-3',5'-дидезокситимидин-5'-фосфоновая кислота) синтезировали, применяя опубликованную методику (Hakimelahi, G.H.; Moosavi-Movahedi, A.A.; Sadeghi, M.M.; Tsay, S-C.; Hwu, J.R. Journal of Medicinal Chemistry, 1995, 38, 4648-4659).
AZT-фосфонат (1,65 г, 5 ммоль) суспендировали в безводном пиридине (30 мл), затем добавили 3-гексадецилокси-1-пропанол (1,8 г, 6 ммоль) и DCC (2,06 г, 10 ммоль) и эту смесь нагревали с обратным холодильником и перемешивали в течение 6 часов, затем охлаждали и фильтровали. Фильтрат концентрировали при пониженном давлении и остаток нанесли на колонку с силикагелем. В результате элюирования с колонки смесью дихлорметан/метанол 9:1 получили гексадецилоксипропиловый эфир 3'-азидо-3',5'-дидезокситимидин-5'-фосфоновой кислоты.
ПРИМЕР 8
Синтез гексадецилоксипропилового, октадецилоксипропилового, октадецилоксиэтилового и гексадецилового эфиров цикло-цидофовира
К перемешиваемой суспензии цидофовира (1,0 г, 3,17 ммоль) в N,N-диметилформамиде (N,N-ДМФ, 25 мл) добавили N,N-дициклогексил-4-морфолинкарбоксамидин (DCMC, 1,0 г, 3,5 ммоль). Смесь перемешивали в течение ночи до растворения цидофовира. Этот прозрачный раствор затем поместили в капельную воронку и медленно добавили (30 минут) к перемешиваемому горячему пиридиновому раствору (25 мл, 60°С) 1,3-дициклогексилкарбодиимида (1,64 г,7,9 ммоль). Реакционную смесь перемешивали при 100°С в течение 16 часов, затем охладили до комнатной температуры и растворитель удалили при пониженном давлении. Остаток абсорбировали на силикагеле и очищали колоночной флэш-хроматографией, применяя градиентное элюирование (CH2Cl2 + МеОН). Активный в УФ продукт в заключение элюировали смесью CH2Cl2/MeOH/H2O 5:5:1. В результате выпаривания растворителя получили 860 мг белого твердого вещества. 1H- и 31Р-ЯМР спектры показали, что оно представляет собой DCMC-соль цикло-цидофовира (выход 44%).
К раствору цикло-цидофовира (DCMC-соль) (0,5 г, 0,8 ммоль) в безводном ДМФ (35 мл) добавили 1-бром-3-гексадецилоксипропан (1,45 г, 4 ммоль) и смесь перемешивали и грели при 80°С в течение 6 часов. Раствор затем концентрировали в вакууме, остаток абсорбировали силикагелем и очищали колоночной флэш-хроматографией, применяя градиентное элюирование (CH2Cl2+EtOH). Алкилированный продукт элюировали смесью СН2Cl2/EtOH 90:10. Фракции, содержащие чистый продукт, упаривали с получением 260 мг HDP-цикло-цидофовира (выход 55%).
К раствору цикло-цидофовира (DCMC-соль) (1,0 г, 3,7 ммоль) в безводном ДМФ (35 мл) добавили 1-бром-3-октадецилоксипропан (2,82 г, 7,2 ммоль) и смесь перемешивали и нагревали при 85°С в течение 5 часов. Раствор затем концентрировали в вакууме, а остаток абсорбировали силикагелем и очищали колоночной флэш-хроматографией, применяя градиентное элюирование (CH2Cl2+МеОН). Алкилированный продукт элюировали смесью CH2Cl2/MeOH 9:1. Фракции, содержащие чистый продукт, упаривали с получением 450 мг ODP-цикло-цидофовира.
К раствору цикло-цидофовира (cCDV) (DCMC-соль) (1,0 г, 3,7 ммоль) в безводном ДМФ (35 мл) добавили 1-бром-3-октадецилоксиэтан (3,0 г, 7,9 ммоль) и эту смесь перемешивали и нагревали при 80°С в течение 4 часов. Раствор затем сконцентрировали в вакууме, остаток абсорбировали силикагелем и очищали колоночной флэш-хроматографией, применяя градиентное элюирование (CH2Cl2+МеОН). Алкилированный продукт элюировали смесью CH2Cl2/MeOH 9:1. Фракции, содержащие чистый продукт, упаривали с получением 320 мг октадецилоксиэтил-cCDV.
К раствору цикло-цидофовира (DCMC-соль) (0,5 г, 0,8 ммоль) в безводном ДМФ (35 мл) добавили 1-бром-3-гексадекан (1,2 г, 4,0 ммоль) и смесь перемешивали и нагревали при 80°С в течение 6 часов. Раствор затем концентрировали в вакууме и остаток абсорбировали силикагелем и очищали колоночной флэш-хроматографией, применяя градиентное элюирование (CH2Cl2+МеОН). Алкилированный продукт элюировали смесью CH2Cl2/MeOH 9:1. Фракции, содержащие чистый продукт, упаривали с получением 160 мг гексадецил-cCDV.
ПРИМЕР 9
Синтез гексадецилоксипропилового, октадецилоксипропилового, октадецилоксиэтилового и гексадецилового эфиров цидофовира
Гексадецилоксипропил-цикло CDV с предыдущей стадии растворили в 0,5 М NaOH и перемешивали при комнатной температуре в течение 1,5 часа. Затем добавили по каплям 50%-ную водную уксусную кислоту для доведения рН примерно до 9. Выпавший в осадок HDP-CDV выделили фильтрацией, промыли водой, сушили и затем перекристаллизовали (3:1 пара-диоксан/вода) с получением HDP-CDV.
Аналогично, октадецилоксипропиловый, октадецилоксиэтиловый и гексадециловый эфиры cCDV гидролизовали, применяя 0,5 М NaOH, и очищали с получением соответствующих диэфиров цидофовира.
ПРИМЕР 10
Синтез гексадецилоксипропилового эфира цикло-ганцикловир- фосфоната
Циклический фосфонатный аналог ганцикловира получили, применяя опубликованную методику (Reist, E.J.; Sturm, P.A.; Pong, R.Y.; Tanga, M.J. и Sidwell, R.W. Synthesis of acyclonucleoside phosphonates for evaluation as antiviral agents, p. 17-34. In J, C. Martin (ed), Nucleotide Analogues as Antiviral Agents, American Chemical Society, Washingnon, D.C.). После превращения в DCMC-соль в ДМФ цикло-ганцикловир (cGCV)-фосфонат обработали 1-бром-3-гексадецилоксипропаном и смесь нагревали при 80°С в течение 6 часов. В результате выделения алкилированного продукта с помощью флэш-хроматографии получили HDP-цикло-GCV-фосфонат.
ПРИМЕР 11
Синтез гексадецилоксипропилового эфира ганцикловир-фосфоната
Полученный на предыдущей стадии HDP-цикло-GCV-фосфонат растворили в 0,5 М NaOH и перемешивали при комнатной температуре, чтобы превратить в ациклический диэфир. Раствор нейтрализовали 50%-ной водной уксусной кислотой, чтобы осадить продукт, который перекристаллизовывали из смеси 3:1 пара-диоксан/вода.
ПРИМЕР 12
1-O-Гексадецилоксипропан-алендронат ингибирует вызванный дексаметазоном апоптоз клеток остеоцитов MLO-Y4
Клетки остеоциты MLO-Y4 предварительно обрабатывали 1-О-гексадецилоксипропан-алендронатом (HDP-алендронат) в указанной концентрации в течение 1 часа, затем клетки инкубировали в течение 6 часов в присутствии и в отсутствие дексаметазона (конечная концентрация 10-4 М). Процент мертвых клеток определяли с помощью модернизированного трипанового голубого (Plotkin et al., J Clin Invest 104:1363-1374, 1999). Результаты представлены на фиг.1. Столбцы представляют собой среднее значение ± среднеквадратичное отклонение трех независимых измерений. Данные анализировали с помощью одностороннего дисперсионного анализа (ANOVA) (критерий Стьюдент-Кеульс-Ньюмана (Student-Keuls-Newman)). *р<0,05. HDP-алендронат ингибирует вызванный дексаметазоном апоптоз в концентрациях от 10-8 до 10-5 М.
ПРИМЕР 13
1-O-Гексадецилоксипропан-алендронат ингибирует вызванный дексаметазоном апоптоз в клетках свода черепа
Клетки свода черепа получили из новорожденных мышей C57BL/6J и пассировали в культуре ткани. Клетки предварительно обработали HDP-алендронатом в указанной концентрации в течение 1 часа, затем клетки инкубировали в течение 6 часов в присутствии и в отсутствие 10-4 М дексаметазона. Процент мертвых клеток определяли с помощью модернизированного трипанового голубого (Plotkin et al., J Clin Invest 104:1363-1374, 1999). Результаты представлены на фиг.2. Столбцы представляют собой среднее значение ± среднеквадратичное отклонение трех независимых измерений. Данные анализировали путем одностороннего дисперсионного анализа (критерий Стьюдент-Кеульс-Ньюмана). *р<0,05. Предварительная обработка клеток HDP-алендронатом в концентрации 10-8 или больше устраняла вызванное дексаметазоном увеличение процента мертвых клеток (р=<0,05). Клетки, подвергнутые воздействию DEVD (белковый ингибитор апоптоза) в концентрации 0,05 мкМ, а затем дексаметазона, не проявляли увеличения процента мертвых клеток, демонстрируя, что DEVD блокирует вызванный дексаметазоном апоптоз.
ПРИМЕР 14
Ингибирование резорбции кости 1-O-гексадецилпропан-алендронатом у овариэктомированных крыс
Членов групп самок крыс Sprague-Dawley (массой 250-280 г), которых подвергли двусторонней овариоэктомии, лечат либо динатриевой солью 4-амино-1-гидроксибутилиден-1,1-дифосфоновой кислоты, либо 1-O-гексадецилпропандиол-3-алендронатом, которые вводят подкожно в дозах, переходящих от 0 до 8 мг/кг/сутки, в течение периода от 4 до 12 недель. На момент времени двенадцать недель крыс, включая членов контрольной группы, убивают и бедренные кости каждого животного сжигают. Способ введения, альтернативно, может быть пероральным. Определяют массу золы бедренных костей каждой особи и значения сравнивают для каждой группы как показатель массы кости, чтобы определить относительное ингибирование потери кости среди протоколов лечения. Животные, которых лечили 1-O-гексадецилпропан-алендронатом, показывают меньшую потерю массы кости, чем овариэктомированные контроли.
ПРИМЕР 15
Ингибирование резорбции кости 1-O-октадецилоксипропил-алендронатом у людей с остеопорозом
Две группы женщин, находящихся в постклимактерическом периоде, лечат плацебо или 1-O-октадецилоксипропил-алендронатом в дозе от 0,1 до 100 мг/кг/сутки перорально в течение периода от 2 до 3 лет. У членов лечащихся групп постоянно в течение всего курса лечения контролируют минеральную костную плотность, частоту вертебральных разломов, прогрессию вертебральных деформаций, с помощью рентгенографического обследования, и уменьшение роста. Производят сравнения измерений у разных групп, которые лечат, чтобы определить эффективность форм лечения алендронатом в группе, которую лечат. Группа, которую лечили 1-O-октадецилоксипропил-алендронатом, имеет меньшее количество разломов и меньшую скорость уменьшения костной плотности, чем у плацебо-группы.
ПРИМЕР 16
Стимулирование остеогенеза 1-O-октадецилоксипропил-амино-ольпадронатом у людей с вызванным стероидами остеопорозом
Группы пациентов с вызванным стероидами остеопорозом лечат 1-O-октадецилоксипропил-амино-ольпадронатом или плацебо в дозах от 0,1 до 100 мг/кг/сутки перорально в течение периода от 1 месяца до 1 года. У членов лечащихся групп постоянно контролируют в течение всего курса лечения минеральную костную плотность, частоту вертебральных разломов, прогрессию вертебральных деформаций, с помощью рентгенографического обследования, и уменьшение роста. Производят сравнения измерений у разных групп, которые лечат, чтобы определить эффективность лечения 1-O-октадецилоксипропил-амино-ольпадронатом в группе, которую лечат. У пациентов, которых лечили 1-O-октадецилоксипропил-амино-ольпадронатом, костная плотность повышена, а разломы меньше по сравнению с лечением плацебо.
ПРИМЕР 17
Противовирусная активность и селективность фосфонат-нуклеотидных аналогов в отношении цитомегаловируса человека (HCMV)
Противовирусный анализ на HCMV: противовирусные анализы на ДНК HCMV проводили с помощью ДНК-гибридизации, как описывается в Dankner, W.M., Scholl, D., Stanat, S.C., Martin, M., Souke, R.L. and Spector, S.A., J. Virol. Methods 21:293-298, 1990. Кратко, субконфлюэнтные клетки MRC-5 в 24-луночный культуральный планшете предварительно обрабатывали в течение 24 часов различными концентрациями лекарства в минимальной эссенциальной среде Игла (Е-МЕМ), содержащей 2% сыворотку плода коровы (FBS) и антибиотики. Среду удалили и добавили штаммы HCMV в разведении, которое даст результат через 3-4 + цитопатическое действие (СРЕ) в лунках без лекарства через 5 дней. Вирус абсорбировали в течение 1 часа при 37°С, отсасывали и заменяли растворами лекарства. После 5 дней инкубации ДНК HCMV количественно определяли в трех параллелях с помощью гибридизации нуклеиновой кислоты, применяя Набор для определения противовирусной чувствительности в отношении CMV от Diagnostic Hybrids, Inc (Athens, ОН). Среду удалили и осуществляли лизис клеток по инструкциям производителя. После абсорбции лизата фильтры HybriwixTM гибридизовали в течение ночи при 60°С. Фильтры HybriwixTM промывали в течение 30 минут при 73°С и производили подсчет в счетчике гамма-излучения. Результаты выражали как EC50 (50%-ная ингибирующая концентрация).
Предварительные эксперименты были выполнены с 1-O-гексадецилпропандиольными (HDP) производными цидофовира и адефовира, как показано в таблице 1.
Как показывают результаты таблицы 1, 1-O-гексадецилпропандиол-цикло-CDV (HDP-cCDV) оказался более чем в 900 раз активнее CDV или цикпо-CDV. При большей цитотоксичности индекс селективности в отношении HCMV в быстро делящихся клетках составлял более 59000 по сравнению с индексами селективности от 1900 до >2100 для непроизводных CDV. Основываясь на этих многообещающих предварительных результатах, провели дальнейшие эксперименты с использованием дополнительных соединений по изобретению. Эти дальнейшие эксперименты описаны ниже.
Цитотоксичность тестируемых соединений in vitro: субконфлюэнтные клетки фибробласты легкого человека (MRC-5, Американская коллекция типовых культур, Rockvill, MD) в 24-луночных планшетах обрабатывали лекарствами, разведенными в среде Е-МЕМ (Gibco BRL, Grand Island, NY), содержащей 2% сыворотку плода коровы и антибиотики. Через 5 дней инкубирования при 37°С монослой клеток визуально изучали под увеличением и рассчитывали концентрацию лекарства, которая вызывает 50%-ное уменьшение числа клеток.
Данные, полученные в этих экспериментах, показаны в таблице 2.
Таблица 2
Ингибирование репликации цитомегаловируса (CMV) человека в фибробластах легких человека MRC-5 в анализе по уменьшению ДНК
EC50 - 50%-ная эффективная концентрация; CC50 - 50%-ная цитотоксичная концентрация; индекс селективности CC50/EC50. Результаты EC50 представляют собой средние значения 3-6 определений, за исключением того, что для ADV проведена одна репликация в двух параллелях.
Как показывают результаты, приведенные в таблице 2, соединения по изобретению одинаково показывают большую активность и селективность, в сравнении с непроизводными цидофовиром, цикло-цидофовиром и адефовиром.
ПРИМЕР 18
Действие HDP-cCDV на репликацию покс-вирусов in vitro
Активность цидофовира (CDV), цикло-цидофовира (cCDV) и 1-O-гексадецилпропандиол-3-cCDV (HDP-cCDV) тестировали на противовирусную активность в фибробластах крайней плоти человека, инфицированных вирусом коровьей оспы или вирусом осповакцины, путем измерения зависящего от дозы уменьшения цитопатического эффекта (СРЕ). Предварительные значения EC50 для коровьей оспы и осповакцины определяли путем анализа уменьшения СРЕ в клетках фибробластах крайней плоти человека (HFF). Полученные таким образом данные приведены в таблице 3.
Как показано в таблице 3, HDP-cCDV оказался высокоактивным против вируса коровьей оспы со значением EC50=0,11 мкМ по сравнению с 0,97 и 1,8 мкМ для cCDV и CDV, соответственно. В клетках, инфицированных осповакциной, HDP-cCDV оказался чрезвычайно эффективным со значением EC50 меньше 0,03 мкМ по сравнению с 0,72 и 2,1 для cCDV и CDV, соответственно. Основываясь на этих многообещающих предварительных данных, были исследованы действия по изобретению аналогов цидофовира на репликацию других ортопокс-вирусов.
Анализ на противодействие цитопатическому эффекту (СРЕ) покс-вирусов: при каждой концентрации лекарства три лунки, содержащие клетки Vero, инфицировали ортопокс-вирусом в количестве 1000 бляшкообразующих единиц (БОЕ) на лунку, а три других лунки оставили неинфицированными для определения токсичности. Планшеты исследовали и окрашивали после инфицирования вирусом, необработанные клетки показали 4+ СРЕ. К среде добавили нейтральный красный и оценивали размер СРЕ по поглощению нейтрального красного при 540 нм. 50%-ную ингибирующую (EC50) и 50%-ную цитотоксичную (CC50) концентрации определяли из графика доза - ответ. Результаты показаны в таблице 4.
оспа
вакцина
ная оспа,
тяжелая
форма,
Bangladesh
ная оспа,
тяжелая
форма,
Yamada
оспа, легкая
форма,
Garcia
мкМ
EC50 - 50%-ная эффективная концентрация; CC50 - 50%-ная цитотоксичная концентрация для клеток Vero; индекс селективности CC50/EC50; сокращения такие же, как в таблице 2. Результаты представляют собой средние значения трех измерений.
Как показывает таблица 4, соединения по изобретению значительно более активны, чем непроизводные CDV или cCDV, против коровьей оспы, осповакцины и различных штаммов натуральной оспы.
ПРИМЕР 19
Действие 1-O-гексадецилпропандиол-3-адефовира (HDP-ADV) на репликацию ВИЧ-1 in vivo
Предварительные эксперименты по ингибированию репликации ВИЧ-1 соединениями по изобретению выполняли, как описано ниже. Тесты лекарств проводили, как ранее описано в Larder et. al., Antimicrobial Agents & Chemotherapy, 34:436-441, 1990. Клетки НТ4-6С, инфицированные ВИЧ-1LAI, подвергали действию лекарств, как указано, и инкубировали в течение 3 дней при 37°С. Клетки фиксировали кристаллическим фиолетовым для визуализации окрашенных бляшек. Противовирусную активность оценивали как процент контрольных бляшек (без лекарства), определенный в образцах, обработанных лекарством. EC50 представляет собой микромолярную концентрацию, которая уменьшает количество бляшек на 50%. Активность адефовира сравнивали с активностью AZT (зидовудин) и 1-O-гексадецилпропандиол-3-адефовира (HDP-ADV) в клетках НТ4-6С, инфицированных ВИЧ-1. Результаты показаны в таблице 5.
Адефовир проявил умеренную активность со значением EC50=16 мкМ. AZT оказался очень активным, как и ожидалось (EC50=0,007 мкМ), но HDP-ADV оказался наиболее активным из этих трех соединений со значением EC50=0,0001 мкМ, т.е. более чем в пять log активнее, чем адефовир как таковой. Основываясь на этих многообещающих предварительных данных, проводили дальнейшие эксперименты, как описано ниже.
Противовирусный анализ в отношении ВИЧ-1: действие противовирусных соединений на репликацию ВИЧ в клетках HeLa HT4-6C, экспрессирующих CD-4, определяли путем анализа уменьшения бляшек (Larder, B.A., Chesebro, В. и Richman, D.D. Antimicrob. Agents Chemother., 34:436-441, 1990). Кратко, монослои клеток HT4-6C инфицировали 100-300 бляшкообразующими единицами (БОЕ) вируса на лунку в 24-луночных планшетах для микроразведения. Добавляли различные концентрации лекарства к культурной среде, среде Игла, модифицированной по способу Дульбекко, которая содержала 5% сыворотку плода коровы и антибиотики, как указано выше. После инкубирования в течение 3 дней при 37°С монослои фиксировали 10%-ным раствором формальдегида в фосфатном буферном растворе (PBS) и окрашивали 0,25%-ным кристаллическим фиолетовым для визуализации вирусных бляшек. Противовирусную активность оценивали как процент контрольных бляшек, определенный в образцах, обработанных лекарством. Цитотоксичность оценивали по методу, описанному в Hostetler et al., Antiviral Research, 31:59-67, 1996. Результаты показаны в таблице 6.
Таблица 6
Ингибирование репликации ВИЧ в клетках НТ4-6С по уменьшению бляшек
EC50 - 50%-ная эффективная концентрация; CC50 - 50%-ная цитотоксичная концентрация; индекс селективности CC50/EC50. Величины EC50 представляют собой средние значения четырех экспериментов.
Как легко видно по результатам таблицы 6, соединение по изобретению 1-O-гексадецилпропандиол-3-ADV является более активным и селективным, чем адефовир.
ПРИМЕР 20
Действие аналогов цидофовира на репликацию вируса герпеса
Противовирусный анализ в отношении вируса герпеса-1 (HSV-1): субконфлюэнтные клетки MRC-5 в 24-луночных культуральных планшетах инокулировали путем удаления среды и добавления вируса HSV-1 в разведении, которое будет давать 3-4 + СРЕ в лунках без лекарства в течение 20-24 часов. Вирус абсорбировали в течение 1 часа при 37°С, отсасывали и заменяли различными концентрациями лекарств в Е-МЕМ, содержащей 2% сыворотку плода коровы и антибиотики. Примерно через 24 часа инкубирования ДНК HSV количественно определяли в трех параллелях с помощью гибридизации нуклеиновой кислоты с использованием Набора для определения противовирусной чувствительности в отношении HSV от Diagnostic Hybrids, Inc (Athens, ОН). Среду удаляли и осуществляли лизис клеток по инструкциям производителя. После абсорбции лизата фильтры HybriwixTM гибридизовали в течение ночи при 60°С. Фильтры HybriwixTM промывали в течение 30 минут при 73°С и производили подсчет в счетчике гамма-излучения. Цитотоксичность оценивали, как описано в примере 17. Полученные таким образом значения EC50 и CC50 показаны в таблице 7.
Таблица 7
Ингибирование репликации HSV человека в фибробластах легких человека MRC-5 (анализ по уменьшению ДНК)
Сокращения такие же, как в таблице 2. EC50 - 50%-ная эффективная концентрация; CC50 - 50%-ная цитотоксичная концентрация; индекс селективности CC50/EC50. Величины EC50 представляют собой средние значение двух экспериментов, за исключением значения HDP-CDV, которое представляет собой одно измерение, сделанное в двух параллелях.
Как показано в таблице 7, все соединения по изобретению являются более активными против HSV-1, чем нуклеотид-фосфонаты - непроизводные, то есть цидофовир или цикло-цидофовир.
Несмотря на то, что вышеизложенное изобретение описывается достаточно подробно путем иллюстрации и примеров для ясности и понимания, рядовым специалистам в данной области будет очевидно в свете учения данного изобретения, что могут быть сделаны определенные изменения и модификации без отступления от сущности изобретения и объема формулы изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЛЕЧЕНИЕ ИНФЕКЦИИ ВИРУСА ИММУНОДЕФИЦИТА ЧЕЛОВЕКА С ЛЕКАРСТВЕННОЙ УСТОЙЧИВОСТЬЮ | 2000 |
|
RU2265439C2 |
| Производные 1-гидрокси- и 1-метокси-2-(4-нитрофенил)имидазола, обладающие противовирусной активностью в отношении ортопоксвирусов | 2022 |
|
RU2794763C1 |
| ПРОИЗВОДНЫЕ ЭФИРОВ ФОСФОНОВЫХ КИСЛОТ И СПОСОБЫ ИХ СИНТЕЗА | 2011 |
|
RU2581045C2 |
| Нуклеозидные производные 1,3-диаза-2-оксофеноксазина в качестве ингибиторов репликации герпесвирусов. | 2019 |
|
RU2731381C1 |
| ПРИМЕНЕНИЕ 2,4,5-ТРИЗАМЕЩЕННЫХ 1,2,4-ТРИАЗОЛОНОВ В ПРОТИВОВИРУСНОЙ ТЕРАПИИ | 2021 |
|
RU2820287C1 |
| Производные аденозина - ингибиторы репродукции вирусов, относящихся к роду Flavivirus | 2023 |
|
RU2828777C1 |
| Производные аденозина - ингибиторы репродукции вирусов, относящихся к роду Flavivirus | 2024 |
|
RU2839716C1 |
| СЕРУСОДЕРЖАЩИЕ ФОСФОНОВЫЕ КИСЛОТЫ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ ИЛИ ЭФИРЫ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1993 |
|
RU2136691C1 |
| СОЕДИНЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ ДЛЯ ЛЕЧЕНИЯ ВИРУСНЫХ ИНФЕКЦИЙ | 2007 |
|
RU2466729C2 |
| СОЕДИНЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ ДЛЯ ЛЕЧЕНИЯ ВИРУСНЫХ ИНФЕКЦИЙ | 2007 |
|
RU2525392C2 |
Изобретение относится к новым биологически активным фосфонатным соединениям. Описывается фосфонатное соединение, имеющее структуру
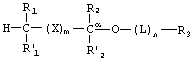
где R1 и R1' независимо представляют собой -Н, возможно замещенный -O(С1-С24)алкил, -O(C1-С24)алкенил, -O(С1-С24)ацил, -S(С1-С24)алкил, -S(С1-С24)алкенил или -S(С1-С24)ацил, где по меньшей мере один из R1 и R1' не представляет собой -Н и где указанные алкенил или ацил возможно имеют от 1 до примерно 6 двойных связей; R2 и R2' независимо представляют собой -Н, возможно замещенный -O(С1-С7)алкил, -O(С1-С7)алкенил, -S(С1-С7)алкил, -S(С1-С7)алкенил, -O(C1-C7)ацил, -S(С1-С7)ацил, -N(С1-С7)ацил, -NH(С1-С7)алкил, -N((С1-С7)алкил)2, оксо, галоген, -NH2, -ОН или -SH; R3 представляет собой фосфонатное производное нуклеозида или бифосфонат; Х представляет собой
L представляет собой валентную связь или бифункциональную связующую молекулу формулы -J-(CR2)t-G-, где t равно целому числу от 1 до 24, J и G независимо представляют собой -О-, -S-, -С(O)O- или -NH-, a R представляет собой -Н, замещенный или незамещенный алкил или алкенил; m равно целому числу от 0 до 6 и n равно 0 или 1. Также описываются фармацевтические композиции, содержащие фосфонатные соединения, способ лечения остеопороза у млекопитающего, способ увеличения минеральной костной плотности, способ предупреждения апоптоза остеобластов и остеоцитов у млекопитающего, способ лечения вирусной инфекции у млекопитающего, способ лечения растущей неоплазмы у млекопитающего и способ модулирования пролиферации клеток. Технический результат - получены новые соединения, обладающие полезными биологическими свойствами. 10 н. и 7 з.п. ф-лы, 2 ил., 7 табл.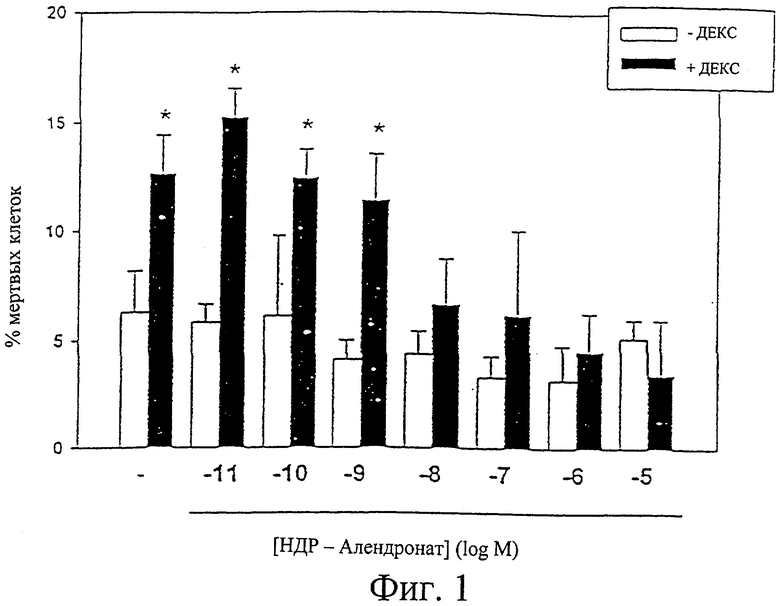
где R1 и R1' независимо представляют собой -Н, возможно замещенный O(С1-С24)алкил, -O(С1-С24)алкенил, -O(С1-С24)ацил, -S(С1-С24)алкил, -S(С1-С24)алкенил или -S(С1-С24)ацил, где по меньшей мере один из R1 и R1' не представляет собой -Н и где указанные алкенил или ацил возможно имеют от 1 до примерно 6 двойных связей;
R2 и R2' независимо представляют собой -Н, возможно замещенный -O(C1-С7)алкил, -O(С1-С7)алкенил, -S(С1-С7)алкил, -S(С1-С7)алкенил, -O(С1-С7)ацил, -S(C1-С7)ацил, -N(C1-C7)ацил, -NH(С1-С7)алкил, -N((C1-C7)алкил)2, оксо, галоген, -NH2, -ОН или -SH;
R3 представляет собой фосфонатное производное нуклеозида или бифосфонат;
Х представляет собой
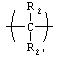
L представляет собой валентную связь или бифункциональную связующую молекулу формулы -J-(CR2)t-G-, где t равно целому числу от 1 до 24, J и G независимо представляют собой -О-, -S-, -С(O)O- или -NH-, a R представляет собой -Н, замещенный или незамещенный алкил или алкенил;
m равно целому числу от 0 до 6; и
n равно 0 или 1.
| Двухполупериодный выпрямитель | 1973 |
|
SU632048A1 |
| Приспособление в пере для письма с целью увеличения на нем запаса чернил и уменьшения скорости их высыхания | 1917 |
|
SU96A1 |
| Прибор, замыкающий сигнальную цепь при повышении температуры | 1918 |
|
SU99A1 |
| US 5627185 А, 06.05.1997 | |||
| Аммонийная соль 9-(4-гидрофосфорил-2-оксабутил)гуанина, обладающая избирательной активностью против вируса простого герпеса | 1988 |
|
SU1594953A1 |

