Область изобретения
Данное изобретение относится к способам лечения инфекции вируса иммунодефицита человека (ВИЧ). В частности, изобретение относится к способам лечения вирусных инфекций, вызываемых вариантами ВИЧ, у которых развилась устойчивость к противовирусным агентам, ингибирующим репликацию указанных вирусов.
Предшествующий уровень техники
Противовирусные агенты успешно используются для лечения множества заболеваний, вызываемых вирусными инфекциями. Эти агенты эффективно излечивают вирусные инфекции посредством ингибирования репликации вируса, например, препятствуя работе вирусных полимеразных ферментов репликации. В частности, инфекции, вызываемые ВИЧ, эффективно излечиваются агентами, которые препятствуют нормальному функционированию вирусной обратной транскриптазы (ОТ), фермента репликации, общего для всех ретровирусов.
Фосфоноформиат (PFA или фоскарнет) является эффективным противовирусным агентом благодаря своей способности ингибировать активность вирусных полимераз, таких как, например, ДНК-и РНК-полимеразы и обратная транскриптаза (Crumpacker, С.S., 1992 The American Journal of Medicine 92(SA): 3S-7S). Продемонстрировано, что фоскарнет действительно эффективно ингибирует развитие вирусов, таких как вирус простого герпеса, гриппа, цитомегаловирус и ретровирусы, например ВИЧ-1 (Bacigalup, et al., 1994 Bone Marrow Transplantation 13: 753-758; Verdonck, et al., 1993 ibid:177-179; Wagstaff, et al., 1994 Drugs 48:199-226).
Фосфоноформиат представляет собой аналог пирофосфата, который является пробочным продуктом нуклеотидной полимеризации и состоит из β и γ фосфатов, отщепляемых от поступающего нуклеотидтрифосфата во время его включения в синтезирующуюся цепь ДНК. Не ограничиваясь какой-либо теорией, полагают, что PFA ингибирует репликацию ретровирусов, таких как ВИЧ-1, посредством связывания с обратной транскриптазой, препятствуя связыванию следующего нуклеозидтрифосфата, блокируя тем самым дальнейший катализ. Использование PFA для лечения вирусных инфекций описано ранее (см., например, Chrisp, et af., Drugs 41:104-129 (1991); Beadle, et al., Antiviral Chem Chemother 9:33-40 (1997); Beadle, et al., Antiviral Chem Chemother 9:33-40 (1988); Hostetler, et al., Antiviral Res. 31:59-67 (1996); Hostetler, et al., Antiviral Chem Chemother:213-220 (2000); Kini, et al., Antiviral Res. 36: 43-53 (1997); Kini, et al., Antiviral Res. 36: 115-124 (1997); Mellors, et al., Mol. Pharmacol. 43:11-16 (1992); Nguyen, et al., Antimicrob. Agents Chemother., 38:2409-2414(1994).
Хотя в лечении СПИДа вследствие использования противоретровирусных агентов был достигнут определенный прогресс, у такого лечения имеет место неблагоприятное последствие, заключающееся в том, что эти противоретровирусные агенты оказывают на ВИЧ-1 селективное давление, которое заставляет их мутировать, приводя к лекарственной нечувствительности и незначительным результатам лечения (Richman, Scientific American, 1988 279:88). Развитие вирусной устойчивости к противоретровирусным лекарственным средствам, используемым для лечения ВИЧ-инфекции, является главной причиной неудач в процессе лечения и ограничивает возможности для применения альтернативных противоретровирусных режимов лечения. Устойчивость возникает ко всем нуклеозидным ингибиторам обратной транскриптазы ВИЧ (NRTI's), включая нуклеозидные аналоги зидовудин (AZT), диданозин (ddl), зальцитабин (ddC), ламивудин (ЗТС), ставудин (d4T) и абакавир, а также ненуклеозидным ингибиторам обратной транскриптазы (NNRTI's), таким как невирапин, делавирдин и ифавиренц (Hirsch, et al., JAMA 1998 279:1984-1991). Кроме того, устойчивость возникала и к ингибиторам протеазы ВИЧ-1, таким как саквинавир, индинавир, ритонавир, агенераза и DMP-450.
Следовательно, имеется потребность в данной области в противовирусных агентах, которые эффективно препятствуют репликации вариантов ВИЧ, у которых развилась устойчивость к ингибированию вирусной репликации в результате применения существующих в настоящее время режимов лечения.
Краткое описание изобретения
Согласно изобретению предложены способы лечения вирусной инфекции у нуждающегося в этом субъекта, при которых указанному субъекту вводят эффективное количество одного или более чем одного липидного аналога фосфоноформиата или тиофосфоноформиата. Липидные аналоги, предусмотренные для практического использования данного изобретения, включают в себя фосфоноформиаты, ковалентно связанные (непосредственно или опосредованно, через линкерную молекулу) с замещенным или незамещенным алкилглицерином, алкилпропандиолом, алкилэтандиолом или сходной группировкой. В частности, согласно изобретению предложены способы лечения вирусных инфекций, вызываемых вирусами, у которых развилась устойчивость к имеющимся в настоящее время противовирусным агентам.
Согласно одному аспекту изобретения предложены способы лечения вирусных инфекций, вызываемых ретровирусами. Согласно частному аспекту изобретения предложены способы лечения вирусных инфекций, вызываемых ретровирусами, у которых развилась устойчивость к противовирусным агентам, таким как, например, ингибиторы обратной транскриптазы и им подобные.
Согласно другому аспекту изобретения предложены способы лечения вирусных инфекций, при которых нуждающемуся в этом субъекту вводят эффективное количество липидного аналога фосфоноформиата в комбинации с азидотимидином (AZT), ингибитором обратной транскриптазы ВИЧ.
Краткое описание графических материалов
На чертеже представлены иллюстративные структуры липидных аналогов фосфоноформиата, предусмотренных для практического использования настоящего изобретения.
Подробное описание изобретения
Согласно данному изобретению предложены способы лечения вирусных инфекций у нуждающегося в этом субъекта. Показания для такого терапевтического вмешательства включают в себя чувствительные вирусы, такие как вирус иммунодефицита человека (ВИЧ), причем обратная транскриптаза указанного вируса приобрела устойчивость к различным ингибиторам обратной транскриптазы, таким как, например, нуклеозидные ингибиторы обратной транскриптазы (NRTI's), ненуклеозидные ингибиторы обратной транскриптазы (NNRTI's) и им подобные.
Способы по данному изобретению включают в себя введение субъекту эффективного количества липидного аналога фосфоноформиата или тиофосфоноформиата, где липидный аналог имеет следующую структуру:
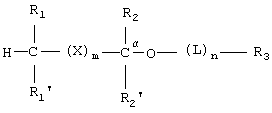
где:
R1 и R1' представляют собой независимо -Н, возможно замещенный -O(С1-С24)алкил, -O(С1-С24)алкенил, -S(С1-С24)алкил, -S(С1-С24)алкенил, -O(С1-С24)ацил, -S(С1-С24)ацил, где по меньшей мере один из R1 и R1' не является -Н, и где указанный алкенил имеет от 1 до примерно 6 двойных связей, и указанный ацил возможно имеет от 1 до примерно 6 двойных связей;
R2 и R2' представляют собой независимо -Н, возможно замещенный -O(С1-С7)алкил, -O(С1-С7)алкенил, -S(С1-С7)алкил, -S(С1-С7)алкенил, -O(C1-С7)ацил, -S(С1-С7)ацил, -N(С1-С7)ацил, -NH(С1-С7)алкил, -N((С1-С7)алкил)2, оксо, галоген, -NH2, -ОН или -SH;
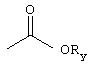
R3 представляет собой фосфоноформиат, который связан либо через свою карбоксильную группу либо через свою фосфонатную группу, с функциональной группой на возможном линкере L или с имеющимся на Сα(кислородом, причем когда R3 связан через свою фосфонатную группу, то тогда карбоксилатная группа указанного фосфоноформиата имеет следующую структуру:
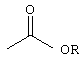
где:
Ry представляет собой -Н или алкил, или
Na+, K+, NH4 + или любой другой физиологически приемлемый катион,
X, когда он присутствует, представляет собой:
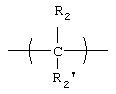
L, когда он присутствует, представляет собой бифункциональную линкерную молекулу, имеющую формулу -J-(CR2)t-G-, где t равно целому числу от 1 до 24, J и G независимо представляют собой -О-, -S-, -С(O)O-или -NH-, и R представляет собой -Н, алкил или алкенил;
m равно целому числу от 0 до 6; и
n равно 0 или 1.
Используемый здесь термин "алкил" обозначает одновалентную прямую или разветвленную цепь или циклический радикал, содержащие от одного до двадцати четырех атомов углерода, включая метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, н-гексил и тому подобное.
Используемый здесь "замещенный алкил" включает в себя алкильные группы, дополнительно несущие один или более чем один заместитель, выбранный из гидрокси, алкокси (из группы низших алкилов), меркапто (из группы низших алкилов), циклоалкила, замещенного циклоалкила, гетероцикла, замещенного гетероцикла, арила, замещенного арила, гетероарила, замещенного гетероарила, арилокси, замещенного арилокси, галогена, трифторметила, циано, нитро, нитрона, амино, амидо, -С(O)Н, ацила, оксиацила, карбоксила, карбамата, сульфонила, сульфонамида, сульфурила и тому подобного.
Используемый здесь "алкенил" обозначает углеводородные группы с прямой или разветвленной цепью, имеющие одну или более чем одну углерод углеродную двойную связь и имеющие длину в пределах примерно от 2 до 24 атомов углерода, а "замещенный алкенил" обозначает алкенильные группы, дополнительно несущие один или более чем один заместитель, как указано выше.
Фосфоноформиаты, предусмотренные для практического использования данного изобретения, включают в себя, например, аддукт бутилового спирта, 1-О-октадецилглицеро-3-PFA (B-PFA), 1-O-октадецил-2-O-метилглицеро-3-PFA (MB-PFA), 1-O-октадецил-2-O-этил-глицеро-3-РРА (EB-PFA) и соединения, описанные в патентах США №5194654, 5411947, 5463092, 5696277, 5744461 и 6002029; а также в заявках США с серийными №№08/487081 и 08/986881, которые включены сюда ссылкой во всей их полноте.
Предпочтительные липиды содержат замещенные глицериновые, пропандиоловые, бутандиоловые и этандиоловые группы. Особенно предпочтительные липиды содержат глицериновые группировки.
Предпочтительные липидные аналоги по изобретению имеют следующую структуру:
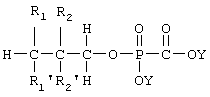
где:
R1 представляет собой O(С18алкил), каждый из R1' и R2' представляет собой -Н, и R2 представляет собой -ОН, Ометил или Оэтил, где получающиеся в результате аналоги обозначены как B-PFA, MB-PFA или EB-PFA, соответственно, и Y представляет собой физиологически приемлемый катион, такой как, например, Na+, K+, NH4 + и тому подобные.
Способы по данному изобретению особенно эффективны для лечения инфекций, вызываемых ретровирусами, такими как, например, ВИЧ-1 и тому подобные.
В предпочтительных воплощениях способы согласно данному изобретению используются для лечения инфекций, вызываемых вирусом иммунодефицита млекопитающих, таким как, например, ВИЧ-1. Способы согласно данному изобретению особенно эффективны для лечения инфекций, вызываемых вариантами ВИЧ-1, которые приобрели устойчивость к ингибитору(ам) обратных транскриптаз (ОТ). Ингибиторы ОТ включают в себя, например, нуклеозидные аналоги зидовудин (AZT), диданозин (ddl), зальцитабин (ddC), ламивудин (ЗТС), ставудин (d4T), эмиривин (FTC), абакавир и тому подобные, а также ненуклеозидные аналоги, такие как, например, невирапин, делавирдин, ифавиренц и тому подобные.
Способы согласно данному изобретению пригодны также для лечения инфекций, вызываемых вариантами ВИЧ-1, у которых развилась устойчивость к ингибиторам протеаз, таким как, например, саквинавир, индинавир, ритонавир, агенераза, DMP-450 и тому подобные.
Согласно данному изобретению также предложены способы предупреждения или лечения патологического состояния, вызванного вирусной инфекцией, у нуждающегося в этом субъекта, при которых указанному субъекту вводят эффективное количество липидного аналога фосфоноформиата.
Кроме того, согласно данному изобретению предложены способы лечения вирусной инфекции у млекопитающего, при котором нуждающемуся в этом субъекту вводят эффективное количество липидного аналога фосфоноформиата в комбинации с ингибитором обратной транскриптазы ВИЧ, таким как, например, зидовудин (AZT, азидодезокситимидин), который селекционирует мутации, которые сенсибилизируют вариант ВИЧ к соединениям PFA по изобретению. Соединения по изобретению, напротив, селекционируют мутации ОТ ВИЧ, которые обращают или уменьшают устойчивость к AZT.
Было показано, что описанные здесь липидные аналоги обладают пероральной биодоступностью (Beadle, et al., Antiviral Chem. Chemo., 1998 9:33-40). Больных СПИДом, инфицированных штаммами ВИЧ с лекарственной устойчивостью, можно лечить предпочтительно посредством перорального введения описанных здесь липидных аналогов. Таким образом, соединения по изобретению могут быть введены множеством путей, например, в форме таблеток, капсул, растворов, эмульсий или суспензий, вдыхаемых жидких или твердых частиц, микроинкапсулированных частиц в виде спрея, через кожу посредством использования, например, трансдермального пластыря, ректально, например, в форме суппозиториев, и тому подобного. Липофильные производные по изобретению особенно хорошо подходят для введения и систем доставки путем трансдермальной абсорбции, а также могут быть использованы в зубной пасте. Введение также может осуществляться парентерально в форме инъецируемых растворов для внутривенного, подкожного, интраперитонеального, цистернального введения и тому подобного.
Фармацевтический носитель или разбавитель, используемый при реализации данного изобретения, может являться традиционным твердым или жидким носителем. Примерами твердых носителей являются лактоза, сахароза, тальк, желатин, агар, пектин, аравийская камедь, стеарат магния, стеариновая кислота или низшие алкиловые эфиры целлюлозы. Примерами жидких носителей являются сироп, арахисовое масло, оливковое масло, фосфолипиды, жирные кислоты, амины жирных кислот, полиоксиэтилен или вода. Носитель или разбавитель может включать в себя любое пролонгирующее высвобождение вещество, известное в данной области, такое как моностеарат или дистеарат глицерина, само по себе или в смеси с воском.
Если для перорального введения используется твердый носитель, то препарат может быть таблетирован или заключен в твердую желатиновую капсулу в форме порошка или шариков. Количество твердого носителя будет широко варьировать, но обычно оно будет составлять от примерно 25 мг до примерно 1 г. Если используется жидкий носитель, то препарат может быть в форме сиропа, эмульсии, мягкой желатиновой капсулы, стерильной инъецируемой жидкости, такой как водная или неводная жидкая суспензия или раствор, и тому подобное.
Таблетки готовят смешиванием активного ингредиента (то есть одного или более чем одного соединения по изобретению) с фармацевтически инертным неорганическим или органическим носителем, разбавителями и/или эксципиентами. Примеры таких эксципиентов, которые могут быть использованы для приготовления таблеток, включают в себя лактозу, кукурузный крахмал или его производные, тальк, стеариновую кислоту или ее соли. Примерами подходящих эксципиентов для желатиновых капсул являются растительные масла, воска, жиры, полутвердые и жидкие полиолы. Липидные аналоги также могут быть изготовлены в микроинкапсулированной форме.
Препарат для интраназального введения может содержать соединение по изобретению, растворенное или суспендированное в жидком носителе, в частности в водном носителе, для аэрозольного применения. Носитель может содержать солюбилизирующие агенты, такие как пропиленгликоль, поверхностно-активные вещества, усилители абсорбции, такие как лецитин или циклодекстрин, или консерванты.
Данное изобретение включает в себя использование фармацевтических композиций, содержащих фармацевтически приемлемые водные или неводные жидкости, дисперсии, суспензии или эмульсии, а также стерильные порошки для разведения в стерильные инъецируемые растворы или дисперсии непосредственно перед использованием парентеральной инъекции. Фармацевтические препараты, содержащие соединения согласно этому изобретению, могут быть приготовлены традиционными способами, например, как описано в Remington's Pharmaceutical Sciences, 1985.
Подходящими эксципиентами для приготовления растворов и сиропов являются вода, полиолы, сахароза, инвертный сахар, глюкоза, липосомы и тому подобное. Подходящими эксципиентами для приготовления инъецируемых растворов являются вода, спирты, полиолы, глицерин, растительные масла и тому подобное.
Фармацевтические продукты могут дополнительно содержать любой из множества дополнительных компонентов, таких как, например, консерванты, солюбилизаторы, стабилизаторы, увлажняющие агенты, эмульгаторы, подсластители, красители, ароматизаторы, буферы, покрывающие агенты, антиоксиданты, разбавители и тому подобное.
Данное изобретение возможно также включает в себя фармацевтические композиции, содержащие жидкий аналог, как он здесь описан, в комбинации с одним или более чем одним соединением, проявляющим другую активность, например антибиотиком или другим фармакологически активным веществом. Такие комбинации и их использование находятся в рамках объема данного изобретения.
Термин "эффективное количество" в применении к липидным аналогам по данному изобретению обозначает количество, которое будет предупреждать или излечивать заболевания, ассоциированные с вирусными инфекциями, указанными выше. "Эффективное количество" определяется по отношению к рекомендованным дозировкам исходного противовирусного соединения. Выбранная дозировка будет изменяться в зависимости от активности выбранного соединения, пути введения, тяжести подлежащего лечению состояния и предпосылок и предшествующей истории болезни пациента, подвергающегося лечению. Однако специалистам в данной области известно, что начинать лечение следует дозами соединения(ий) в концентрациях более низких, чем требуется для достижения желаемого терапевтического эффекта, и постепенно повышать дозировку до тех пор, пока желаемый эффект не будет достигнут. Эффективная суточная доза, если желательно, может быть разделена на кратное число доз для целей введения, например от двух до четырех доз в день. Однако, следует понимать, что индивидуальная дозовый уровень для любого конкретного пациента будет зависеть от множества факторов, включая массу тела, общее состояние здоровья, диету, время и путь введения, комбинирование с другими лекарственными средствами и тяжесть заболевания, подлежащего лечению.
Соединения по данному изобретению обычно отпускают в стандартной лекарственной форме, содержащей от 1% до 100% активного ингредиента. Диапазон терапевтической дозы составляет от примерно 0,01 до примерно 1000 мг/кг/день, причем более предпочтительной является доза от примерно 0,10 мг/кг/день до 100 мг/кг/день, при введении пациентам, например человеку, в качестве лекарственного средства. Фактические дозовые уровни активных ингредиентов в фармацевтических композициях согласно этому изобретению могут быть изменены для того, чтобы вводить такое количество активного компонента(ов), которое эффективно для достижения желаемого терапевтического эффекта у конкретного пациента.
Изобретение описано более подробно посредством следующих неограничивающих примеров.
Примеры
Химические соединения
Следующие соединения были приготовлены в виде 5 мМ липосомальных препаратов, как описано ранее (смотрите Hostetler, et al., Antiviral Chemistry and Chemotherapy 11: 213-220, and Kini, et al., Antiviral Research 36: 43-53): 1-O-октадецил-sn-глицеро-3-РРА (B-PFA), 1-O-октадецил-пропандиол-3-PFA (EB-PFA), 1-O-октадецил-2-O-метил-sn-глицеро-3-PFA (EB-PFA) и 1-О-октадецил-2-O-этил-sn-глицеро-3-РРА (EB-PFA). Соединения хранили при 4°С, а непосредственно перед использованием нагревали до 37°С. 3'-азидо-3'-дезокситимидин (AZT) и фосфоноформиат (PFA) были закуплены в Sigma Chemical Company, St. Louis, Mo. AZT и PFA готовили в виде 10 или 30 мМ исходных растворов в ДМСО (диметилсульфоксид) и стерильной воде, соответственно, и хранили при -20°С. Непосредственно перед использованием соединения нагревали до 37°С и разбавляли до желаемых концентраций средой RPMI 1640.
Клетки
Клетки МТ-2 (AIDS Research and reference Reagent Program, National Institute of Allergy and Infectious Diseases, National Institutes of Health) поддерживали в среде RPMI 1640, обогащенной 10% фетальной коровьей сывороткой (FBS; JRH Biosciences, Lenexa, KS), 10 мМ буфера HEPES, 50 МЕ/мл пенициллина и 50 мкг/мл стрептомицина.
Вирусы
Исходный вирус был получен посредством электропорации (BIO-RAD Gene Pulser®, Hercules, СА) клеток МТ-2 (1,3×107 клеток) 5-10 мкг плазмидной ДНК, кодирующей провирусный клон HIV-1LAI (см. Nguyen, et al., Antimicrob. Agents Chemother. 38:2409-2414, и Peden, et al., Virology 185:661-672). Во время пикового цитопатического эффекта (обычно через 5-7 дней после трансфекции) культуральные надосадочные жидкости собирали и хранили при -80°С. Титры вирусной инфективности определяли в анализах тройных конечных разведении, выполнявшихся на клетках МТ-2 (шесть лунок на разведение). 50%-ную дозу, инфицирующую культуру ткани (TCID50), вычисляли с помощью уравнения Рида-Мьюнча (Reed-Muench, см. Reed, et al., Am. J. Hyg. 27:493-496).
Анализ вирусной чувствительности
Противовирусную активность каждого соединения определяли путем инокуляции клеток МТ-2 клоном HIV-1LAI при множественности заражения (MOI), равной 0,01 TCID50/клетку, с последующей инкубацией в присутствии трехкратных серийных разведении лекарственного средства (3 лунки на разведение) (см. Mellors, et al., Antimicrob. Agents Chemother. 39:1087-1092). Через пять или семь дней после заражения культуральные надосадочные жидкости собирали, лизировали 0,5%-ным тритоном-Х 100 и анализировали на концентрацию антигена р24, используя коммерческий ELISA-анализ (DuPont, NEN Products, Wilmington, Del.). Противовирусную активность соединений выражают как IC50, которая представляет собой концентрацию, необходимую для ингибирования 50% продукции антигена р24. Кратность устойчивости исследуемого вируса вычисляют путем деления IC50 исследуемого вируса на IC50 контрольного вируса HIV-1LAI.
Селекция устойчивых вирусов
Селекцию инициировали инокулированием 1,0×106 клеток МТ-2 при MOI, равной 0,1, HIV-1LAI плазмидного происхождения, которого пассировали в виде бесклеточного вируса 10 раз в клетках МТ-2 в отсутствие соединения (см. Bazmi, et. al., Antimicrobial Agents and Chemotherapy 44:1783-1788). Перед инокуляцией вирусом клетки предварительно обрабатывали лекарственным средством в течение двух часов. Для каждой селекции начальная концентрация составляла IC50 соединения, и селективное давление (то есть концентрацию лекарственного средства) удваивали через каждые три пассажа. Вирусное цитопатическое действие (СРЕ) контролировали ежедневно. При 2+СРЕ (≥2 синцитий на 100 х поле) бесклеточную вирусную надосадочную жидкость собирали и использовали для инициации нового цикла заражения в свежих клетках МТ-2. Пассивируемый вирус регулярно проверяли на снижение чувствительности к соединениям путем определения EC50 по отношению к непассивируемому hiv-1LAI (Mellors, et al., Antimicrob. Agents Chemother. 39:1087-1092).
Генетические анализы
Вирионы собирали из культуральной надосадочной жидкости центрифугированием при 25000×g в течение 1 часа. Тотальную РНК экстрагировали из вирусного осадка, используя TRIZOL® Reagent (Gibco BRL, Grand Island, NY), и ресуспендировали в обработанной диэтилпирокарбонатом стерильной воде. После синтеза кДНК полноразмерный кодирующий участок ОТ амплифицировали, используя ПЦР (см. Bazmi et al., Antimicrobial Agents and Chemotherapy 44: 1783-1788). Весь объем ПЦР-продуктов затем очищали, используя коммерческий набор (Wizard® PCR Purification system, Promega, Madison, Wl) и секвенировали, используя автоматический секвенатор (РЕ Bioscienses, San Francisco, CA).
Получение мутантного рекомбинантного ВИЧ-1
ВИЧ-1, содержащий желаемые мутации, получали олигонуклеотиднаправленным мутагенезом (Altered Sites II, Promega), как описано ранее (Mellors, et al., Molecular Pharm. 41:446-451). После мутагенеза мутантную ОТ субклонировали в клонирующий вектор pxxHIV-1LAI, используя молчащие Xmal-и Xbal-сайты рестрикции на 5' и 3' концах ОТ. ДНК клонов секвенировали, чтобы проверить на присутствие желаемой мутации(ий), и вводили электропорацией в клетки МТ-2, как описано выше, для получения инфекционного мутантного рекомбинантного ВИЧ-1.
Получение рекомбинантного ВИЧ-1 проводили, как описано в указанных выше ссылках Mellors, et al. и Nguyen, et al., которые включены посредством ссылки.
Мутантную ОТ получали олигонуклеотиднаправленным мутагенезом и лигировали в клонирующий вектор pXXHIV-1LAI. Клонирование облегчалось присутствием двух молчащих сайтов рестрикции на 5' и 3' концах pXXHIV-ILAI ОТ. Инфекционный мутантный рекомбинант ВИЧ-1 получали электропорацией клеток МТ-2 мутантными клонами pXXHIV-ILAI. Присутствие желаемых мутаций проверяли секвенированием ДНК.
Штаммы ВИЧ-1, 11163р1 и 11588р1, с устойчивостью к множеству лекарственных средств представляют собой клинические изоляты человека, имеющие указанные мутации в ОТ. Штаммы ВИЧ-1, обработанные липидными аналогами по данному изобретению, включают в себя штаммы дикого типа, например xxLAI и тому подобные, несущие одиночную мутацию, например xxLAIM184V (устойчивый к ЗТС), xxLAIL74V (устойчивый к ddl и ddC), xxLAIK65R (устойчивый к ddl, ddC, DAPD, DXG, тенофовиру и адефовиру), xxLAIT215Y (устойчивый к AZT) и тому подобные, несущие двойные мутации, например xxLAI M41L/T215Y (устойчивый к AZT), xxLAI M184 V/T215Y (устойчивый к ЗТС и AZT), и тому подобные, и несущие множественные мутации, например xxLAI4XAZT ((D67N, K70R, T215Y, K219Q) устойчивый к AZT), xxLAI4XAZT/M184V ((D67N, K70R, M184V, T215Y, K219Q) устойчивый к AZT и ЗТС), xxLAIM5456-12 (устойчивые к AZT+NNRTI: D67N, K70R, K103N, T215Y), xxLAIG2-3g (соустойчивые к А2Т/ЗТС: M41L, D67N, M184V, H208Y, L210W, R211K, L214F, T215Y, 1293V, E297A), 11163 (мультинуклеозидустойчивый вирус: M41L, A62V, V75I, F77L, K103N, F116Y, Q151M, Y181C, M184V), 11588 (мультинуклеозидустойчивый вирус: A62V, K65R, K70R, V75I, F77L, F116Y, Q151M, M184V, K219Q) и тому подобные.
Индексы селективности in vitro для PFA и липидных аналогов PFA были определены на клетках МТ-2 и имеют следующие значения:
Следует отметить, что индекс селективности ((50%-ная цитотоксичекая доза/50%-ная эффективная доза)×100) намного больше для липидных аналогов PFA, чем для самого PFA. Поскольку липидные аналоги характеризуются намного большей пероральной абсорбцией, чем PFA, их можно вводить перорально для контроля репликации устойчивых к лекарственным средствам ВИЧ либо отдельно, либо в комбинации с одним или более чем одним антивирусным агентом, AZT особенно предпочтителен в комбинации с липидными аналогами PFA.
Пример 1
Культуры клеток МТ-2, инфицированные штаммами ВИЧ-1 либо дикого типа (xxLAI), либо перечисленными выше штаммами с лекарственной устойчивостью, обрабатывали липидными аналогами PFA (B-PFA, MB-PFA или EB-PFA). Как видно из Таблицы 1, была определена концентрация в мкМ, необходимая для обеспечения 50%-ного ингибирования репликации штамма ВИЧ с лекарственной устойчивостью (EC50). Эффекты лекарственных средств измеряли путем анализа р24 (вирусный белок) в культуральной среде.
Противовирусная активность B-PFA, MB-PFA и EB-PFA в отношении набора штаммов ВИЧ-1 с лекарственной устойчивостью
EC50, мкМ
Когда соединения по изобретению, B-PFA, MB-PFA или EB-PFA, исследовали в отношении набора вирусов ВИЧ-1 с лекарственной устойчивостью, все зафиксированные значения противовирусных EC50 оказались существенно ниже, чем значение противовирусной EC50 фоскарнета (PFA). За исключением K65R, ODG-PFA имел значения EC50, лежащие в пределах от 0,84 до 4,25 мкМ, против значений в пределах от 7,6 до 37,4 мкМ, в штаммах ВИЧ, имеющих одиночные, двойные и множественные мутации. MB-PFA и EB-PFA были высоко активны в отношении ВИЧ с лекарственной устойчивостью, причем значения EC50 обычно лежали в пределах менее 1 мкМ. У K65R, редкого ddl-устойчивого мутанта, был отмечен низкий уровень устойчивости по сравнению с диким типом (от 3,5 до 14,6 раз). Следует отметить, что большинство штаммов ВИЧ с лекарственной устойчивостью (Таблица 1) были исследованы при множественности заражения (MOI), равной 0,01.
Пример 2
Штаммы с устойчивостью к множеству лекарств исследовали при пятикратно увеличенной MOI, равной 0,05, результаты показаны в Таблице 2.
Противовирусная активность B-PFA, MB-PFA и EB-PFA в отношении набора штаммов ВИЧ-1 с устойчивостью к множеству лекарств
ЕС50, мкМ
Как показано выше, MB-PFA и EB-PFA были высоко активны в отношении штаммов ВИЧ-1 с устойчивостью ко множеству лекарств, клинических изолятов, 11163р1 и 11588pl, даже при пятикратно увеличенной множественности заражения, причем значения ЕС50 обычно лежали в пределах от 0,73 до 1,95 мкМ.
Также была оценена активность трех соединений по изобретению в отношении набора NRTI-устойчивых вариантов ВИЧ-1 (Примеры 3-5). NRTI-устойчивый набор состоял из рекомбинантных вирусов, происходивших от ВИЧ-1LAI, содержащих мутации, дающие устойчивость к нескольким NRTIs, и включал в себя несколько вариантов, устойчивых к различным NRTI (Таблицы 3-5).
Пример 3
Чувствительность NRTI-устойчивых ВИЧ-1 к соединениям PFA по изобретению
EC50 a,б (мкМ) (кратность устойчивости)B
б Среднее ± стандартное отклонение по меньшей мере из трех независимых экспериментов
в Кратность устойчивости относительно вируса дикого типа
г HIV-1LAI кодирующий указанные мутации устойчивости
Из исследованных вирусов только вирусы, содержащие K65R (ddl/DXG-устойчивые), продемонстрировали значительную устойчивость к соединениям по изобретению, а также к немодифицированному (свободному) PFA, причем значения кратности устойчивости лежали в пределах от 3,3 до 8,2 (значения EC50 составили 1,66-14,68 мкМ). ЗТС- и ddl/ddC-устойчивые вирусы (содержащие мутации устойчивости M184V и L74V, соответственно) были чувствительны как к соединениям PFA по изобретению, так и к немодифицированному PFA (кратность устойчивости менее 3,0) (Таблица 3).
Пример 4
Соединения по изобретению также были оценены в отношении набора трех мультинуклеозид-устойчивых (MNR, multinucleoside resistant) вирусов, как показано в Таблице 4.
Чувствительность мультинуклеозид-устойчивых HIV-1 к соединениям PFA по изобретению
ЕС50 a,б (мкМ) (кратность устойчивости)В
б Среднее ± стандартное отклонение из двух или трех независимых экспериментов
в Кратность устойчивости относительно вируса дикого типа
г Вирус дикого типа
д Мультинуклеозид-устойчивый клинический изолят, несущий мутации A62V, V75I, F77L, K103N, F116Y, Q151M, Y181C, M184V
е Мультинуклеозид-устойчивый рекомбинантный вирус, несущий мутации W5I, F77L, F116Y, Q151M
ж Мультинуклеозид-устойчивый рекомбинантный вирус, несущий мутации D67E, S68T, T69S[SA инсерция], T215Y
MNR-набор состоял из вируса, содержащего мутации V75I, F77L, F116Y и Q151M, вируса, содержащего T69S [SA инсерция] и клинического изолята, несущего классический генотип MNR (62V/75I/77L/116Y/151M). Каждое из соединений PFA по изобретению сохраняло эффективность в отношении этих вирусов со значениями кратности устойчивости менее 2. Единственным исключением из этого был вирус, содержащий 75I/77L/116Y/151M, который показал 4,5-кратную устойчивость к EB-PFA.
Пример 5
AZT-устойчивые вирусы включали в себя рекомбинанты, происходящие от HIV-1LAI, с двойными (М4L/TЯ215У) и четверными (D67N, K70R,T215Y,K219Q) мутациями (Таблица 5). Эти AZT-устойчивые вирусы были чувствительны к соединениям по изобретению и к немодифицированному PFA.
Чувствительность AZT-устойчивого HIV-1 к соединениям PFA по изобретению
EC50 а,б (мкМ) (кратность устойчивости)в
б Среднее ± стандартное отклонение из по меньшей мере трех независимых экспериментов
в Кратность устойчивости относительно вируса дикого типа
г HIV-1LAI, кодирующий указанные мутации устойчивости
д 4XAZT=D67N, K70R, T215Y, K219Q
е Молекулярно клонированный изолят, соустойчивый к AZT и 3ТС; M41L, D67N, M184V, H208Y, L210W, R211K, L214F, T215Y, I293V, Е297А
Вирус, содержащий мутации M41L и T215Y, последовательно показал повышенную по сравнению с вирусом дикого типа чувствительность к каждому из соединений по изобретению: изменения кратности значений EC50 лежали в пределах от 0,4 до 0,53 (значения ЕС50 составили от 0,18 до 0,94 мкМ). Вирус, содержащий как четверные мутации устойчивости к AZT, так и мутацию ЗТС-устойчивости M184V (HIV4ХAZT/M184V) или мутацию устойчивости к ненуклеозидному ингибитору обратной транскриптазы (NNRTI) K103N (HIV4ХАZT/K103N), также показал чувствительность к соединениям по изобретению (кратность устойчивости менее 3,0). К тому же молекулярно клонированный клинический изолят, соустойчивый к AZT и ЗТС (G2-3g), был чувствителен к каждому из соединений со значениями кратности устойчивости менее 1,0 (значения ЕС50 составили от 0,30 до 1,67 мкМ).
Пример 6
Вирус, устойчивый к соединениям по изобретению, был отселектирован in vitro серийными пассажами HIV-1LAI в клетках МТ-2 в присутствии возрастающих концентраций соединений по изобретению, как показано в Таблице 6.
Мутации и измененная чувствительность отселектированного in vitro HIV-1, устойчивого к соединениям по изобретению
18
8,97
10,8
ТСА→АСА
СТТ-ТТТ
S117T
L214F
15
2,16
30,9
GTA→ТТА
ATG-→АТА
СТТ→ТТТ
V75L
М1641
L214F
15
3,70
41,1
TGG→GGG
СТТ→ТТТ
W88G
L214F
17
15
122,3
122,2
23,2
23,2
TGG→GGG
ТСА→АСА
W88G
S117T
б Кратность устойчивости относительно исходного вируса (HIV-1LAI пассаж 0)
в Изменение относительно исходного вируса (HIV-1LAI пассаж 0)
Вирус, проявивший 27-кратную устойчивость к MB-PFA, был выделен после 15 циклов бесклеточного пассирования вируса. Секвенирование ДНК гена ОТ (аминокислота (АК) 1-350) из MB-PFA-устойчивого вируса позволило идентифицировать три мутации: V75L, М164I и L241F. Рекомбинантные вирусы, кодирующие мутации V75L, М164I и L241F, были сконструированы и исследованы на чувствительность к MB-PFA. Вирус, содержащий как М1641, так и L241F, проявил 9,3-кратную устойчивость (EC50=8,1 мкМ). Мутация V75L одна или в комбинации с L214F или М164I не давала существенной устойчивости к MB-PFA (менее чем 3-кратная). Селекция вируса, устойчивого к MB-PFA, была повторена; после 15 пассажей отселектированный вирус проявил 31-кратную устойчивость к MB-PFA и кодировал мутации М164I и L214F, но не мутацию V75L.
Вирус, проявивший 10,8-кратную устойчивость к DB-PFA, был выделен после 18 циклов бесклеточного пассирования. Секвенирование ДНК позволило идентифицировать две мутации в ОТ: S117T и L214F. Рекомбинантный вирус, кодирующий S117T, продемонстрировал 10,0-кратную устойчивость к DB-PFA (ЕС50=9,0 мкМ). Добавление мутации L214F к вирусу, кодирующему S117T, не повышало уровня устойчивости к DB-PFA (9,7-кратная).
Вирус, проявивший 41-кратную устойчивость к EB-PFA и имевший мутации W88G и L214F, был выделен после 15 циклов бесклеточного пассирования. Рекомбинантный вирус, кодирующий мутацию W88G, продемонстрировал 9,4-кратную устойчивость, которая возросла до 15,5-кратной устойчивости (EC50=9,1 мкМ) при добавлении мутации L214F. Вирус, устойчивый к EB-PFA, был отселектирован второй раз после 15 пассажей. Этот вирус также содержал мутации W88G и L214F.
HIV-1LAI, служивший в качестве контролей во время селекции с соединениями по изобретению, пассировали в присутствии и в отсутствие немодифицированного PFA. Вирус, продемонстрировавший 23-кратную устойчивость к PFA, был отселектирован в результате двух независимых селекции после 15 и 17 циклов бесклеточного пассирования. Анализ последовательности ДНК ОТ из этих устойчивых к PFA вирусов позволил идентифицировать одиночные мутации в ОТ: W88G (первая селекция) и S117T (вторая селекция). Рекомбинантные вирусы, имевшие мутации W88G и S117T, продемонстрировали 6,2-кратную и 4,7-кратную устойчивость к PFA, соответственно. Ни одна из мутаций, отселектированных соединениями по изобретению или свободным PFA, не была обнаружена у контрольных вирусов, подвергавшихся пассированию в отсутствие лекарственного средства.
Пример 7
Соединения но изобретению также были оценены в отношении набора рекомбинантов, происходящих от HIV-1 LAI, устойчивых к PFA и соединениям PFA по изобретению, как показано в Таблице 7.
Чувствительность к соединениям по изобретения PFA-и PFA/AZT-устойчивых HIV-1
EC50 а,б (мкМ) (кратность устойчивости)в
б Среднее ± стандартное отклонение по меньшей мере из трех независимых экспериментов
в Кратность устойчивости относительно вируса дикого типа
г HIV-1LAI, кодирующий указанные мутации устойчивости
д 4XAZT=D67N, K70R, T215Y, K219Q
Анализ данных Таблицы 7 свидетельствует о том, что устойчивые к PFA вирусы показали сходные уровни устойчивости к соединениям PFA по изобретению. Вирус, содержащий мутацию E89G, был в наименьшей степени чувствителен к соединениям, причем значения кратности устойчивости лежали в пределах от более чем 17,2-кратного до более чем 39,0-кратного. Соединения также были исследованы в отношении набора рекомбинантных вирусов, происходящих от HIV-1LAI, содержащих мутации устойчивости как к PFA, так и к AZT. Как и ранее, вирусы демонстрировали сходные уровни перекрестной устойчивости как к немодифицированному PFA, так и к соединениям PFA по изобретению. В целом, присутствие мутаций устойчивости к AZT снижало уровень перекрестной устойчивости к соединениям PFA по изобретению. На генетическом фоне устойчивости к AZT мутации в кодоне 89 (G или К) в ОТ давали наибольшую степень устойчивости как к немодифицированному PFA, так и к соединениям PFA по изобретению, причем значения кратности устойчивости лежали в пределах от 5,6 до 11,1.
Пример 8
Рекомбинантные вирусы, содержащие мутации, отселектированные соединениями по изобретению, также были оценены на их чувствительность к AZT, как показано в Таблице 8.
Чувствительность мутантного рекомбинантного HIV-1LAI к AZT
б Значения EC50 определены измерением ингибирования продукции р24 антигена в клетках МТ-2
в Среднее ± стандартное отклонение по меньшей мере из трех независимых экспериментов
г Кратность устойчивости относительно вируса дикого типа
д 4XAZT=D67N, K70R, T215Y, K219Q
Ни одна из мутаций, отселектированных соединениями по изобретению, включая L214F, не снижала чувствительности к AZT. Мутация М164I была ассоциирована с повышением чувствительности к AZT (0,6-кратная устойчивость по сравнению с диким типом). Мутации устойчивости, отселектированные соединениями PFA по изобретению, также были введены в устойчивый к AZT исходный вариант (D67N/K70R/T215Y/K219Q), чтобы оценить их влияние на устойчивость к AZT. Мутации S117T, М164I и W88G снижали устойчивость к AZT от 7,3-кратной до 1,4-, 1,1- и 1,5-кратной, соответственно.
Хотя данное изобретение подробно описано посредством иллюстрирования и приведения примеров для целей ясности и понятности, специалистам в данной области в свете этого изобретения будет очевидно, что определенные изменения и модификации могут быть внесены, не выходя за рамки сущности и объема формулы изобретения.
Изобретение относится к области медицины и касается способа лечения вируса иммунодефицита человека с лекарственной устойчивостью путем введения соединения общей формулы I. Способ обеспечивает повышение эффективности лечения. 3 н. и 14 з.п. ф-лы, 1 ил., 8 табл.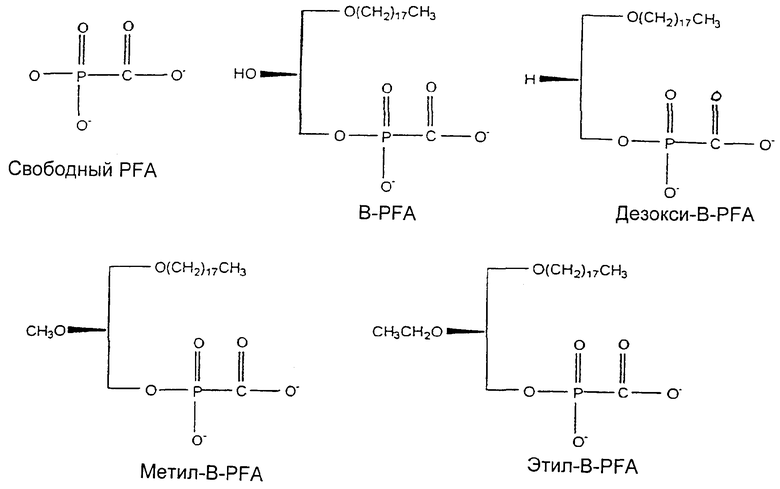
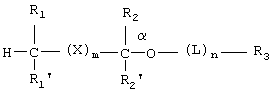
где каждый из R1 и R1' независимо выбран из группы, состоящей из Н, возможно замещенного O(С1-С24)алкила, O(С1-С24)алкенила, S(С1-С24)алкила, S(С1-С24)алкенила, O(С1-С24)ацила, S(С1-С24)ацила, где по меньшей мере один из R1 и R1' не является Н, и где указанный алкенил имеет от 1 до примерно 6 двойных связей и указанный ацил возможно имеет от 1 до примерно 6 двойных связей;
каждый из R2 и R2' независимо выбран из группы, состоящей из Н, возможно замещенного O(С1-С7)алкила, O(С1-С7)алкенила, S(С1-С7)алкила, S(С1-С7)алкенила, O(C1-C7)ацила, S(С1-С7)ацила, N(С1-С7)ацила, NH(C1-С7)алкила, N((С1-С7)алкил)2, оксо, галогена, NH2, ОН и SH;
R3 представляет собой фосфоноформиатную группировку, содержащую карбоксильную группу и фосфонатную группу, где когда R3 связан через фосфонатную группу, карбоксильная группа имеет структуру:
где Ry представляет собой -Н или алкил, или Na+, К+, NH4 +, или любой другой физиологически приемлемый катион,
Х представляет собой группировку, имеющую структуру:
L представляет собой группировку, имеющую структуру -J-(CR2)t-G-, где t равно целому числу от 1 до 24, каждый J и G независимо выбран из группы, состоящей из О, S, С(O)O и NH, и R представляет собой Н, алкил или алкенил;
m равно целому числу от 0 до 6,
n равно 0 или 1,
где вирус иммунодефицита стал устойчивым к ингибиторам обратной транскриптазы до этого лечения.
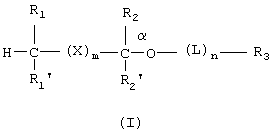
где каждый из R1 и R1' независимо выбран из группы, состоящей из Н, возможно замещенного O(С1-С24)алкила, O(С1-С24)алкенила, S(С1-С24)алкила, S(С1-С24)алкенила, O(С1-С24)ацила, S(С1-С24)ацила, где по меньшей мере один из R1 и R1' не является Н, и где указанный алкенил имеет от 1 до примерно 6 двойных связей и указанный ацил возможно имеет от 1 до примерно 6 двойных связей;
каждый из R2 и R2' независимо выбран из группы, состоящей из Н, возможно замещенного O(С1-С7)алкила, O(С1-С7)алкенила, S(С1-С7)алкила, S(С1-С7)алкенила, O(C1-C7)ацила, S(С1-С7)ацила, N(C1-C7)ацила, NH(C1-С7)алкила, N((С1-С7)алкил)2, оксо, галогена, NH2, ОН и SH;
R3 представляет собой фосфоноформиатную группировку, содержащую карбоксильную группу и фосфонатную группу, где когда R3 связан через фосфонатную группу, карбоксильная группа имеет структуру:
где Ry представляет собой Н или алкил, или Na+, K+, NH47 +, или любой другой физиологически приемлемый катион,
Х представляет собой группировку, имеющую структуру:
L представляет собой группировку, имеющую структуру -J-(CR2)t-G-, где t равно целому числу от 1 до 24, каждый J и G независимо выбран из группы, состоящей из О, S, С(O)O и NH, и R представляет собой Н, алкил или алкенил;
m равно целому числу от 0 до 6,
n равно 0 или 1,
где AZT-устойчивый штамм ВИЧ стал устойчивым до этого лечения.
где каждый из R1 и R1' независимо выбран из группы, состоящей из Н, возможно замещенного O(С1-С24)алкила, O(С1-С24)алкенила, S(С1-С24)алкила, S(С1-С24)алкенила, O(С1-С24)ацила, S(С1-С24)ацила, где по меньшей мере один из R1 и R1' не является Н, и где указанный алкенил имеет от 1 до примерно 6 двойных связей, и указанный ацил возможно имеет от 1 до примерно 6 двойных связей;
каждый из R2 и R2' независимо выбран из группы, состоящей из Н, возможно замещенного O(С1-С7)алкила, O(С1-С7)алкенила, S(С1-С7)алкила, S(С1-С7)алкенила, O(С1-С7)ацила, S(C1-C7)ацила, N(С1-С7)ацила, NH(C1-С7)алкила, N((С1-С7)алкил)2, оксо, галогена, NH2, ОН и SH;
R3 представляет собой фосфоноформиатную группировку, содержащую карбоксильную группу и фосфонатную группу, где когда R3 связан через фосфонатную группу, карбоксильная группа имеет структуру:
где Ry представляет собой -Н или алкил, или Na+, K+, NH4 +, или любой другой физиологически приемлемый катион,
Х представляет собой группировку, имеющую структуру:
L представляет собой группировку, имеющую структуру -J-(CR2)t-G-, где t равно целому числу от 1 до 24, каждый J и G независимо выбран из группы, состоящей из О, S, С(O)O и NH, и R представляет собой Н, алкил или алкенил;
m равно целому числу от 0 до 6,
n равно 0 или 1,
где вирус, вызывающий вирусную инфекцию, стал AZT-устойчивым до этого лечения.
| ФОСФОНОЛИПИДЫ КАРБОНОВЫХ КИСЛОТ И ИХ СОЛИ И АНТИВИРУСНОЕ СРЕДСТВО НА ИХ ОСНОВЕ | 1996 |
|
RU2166508C2 |
| US 5744461 А, 28.04.1998 | |||
| US 5194654 А, 16.03.1993 | |||
| WO 9639831 А, 19.12.1996 | |||
| US 6002029 А, 14.12.1999 | |||
| US 5696277 А, 09.12.1997 | |||
| Автореферат АБД Medline | |||
| Kini | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Antiviral res | |||
| Электрическое сопротивление для нагревательных приборов и нагревательный элемент для этих приборов | 1922 |
|
SU1997A1 |

