Настоящая заявка претендует на приоритет предварительной заявки на патент США с порядковым номером 61/378743, поданной 31 августа 2010, содержание которой в полном объеме включено в настоящую заявку в качестве ссылки.
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение главным образом относится к способам, подходящим для синтеза замещенных тозилоксиметилфосфонатов. Изобретение применимо, например, в области синтетической органической химии и фармации.
ПРЕДПОСЫЛКИ ИЗОБРЕТНИЯ
Применение пролекарств получило широкое распространение с конца 1950-х для повышения биодоступности лекарственных средств, а также для их направленной доставки после перорального введения. Пролекарство представляет собой соединение, которое претерпевает превращение в организме, перед тем как оказать терапевтическое действие. Стратегия применения пролекарств основана на химической модификации действующего вещества путем присоединения фрагментов, образующих пролекарство, к фармакологически активной форме, причем в идеальном случае пролекарства должны преодолевать биохимические и физические препятствия, мешающие переносу исходного вещества. Ограниченная биодоступность при пероральном приеме обычно является следствием плохой способности проникать через мембраны, низкой растворимости в воде (в жидких средах желудочно-кишечного тракта) или значительного пресистемного метаболизма.
В течение длительного времени полагали, что абсорбция большинства лекарственных средств в кишечнике происходит за счет пассивной диффузии, при которой определяющим фактором является растворимость лекарственных средств в липидах. Однако было показано, что многие водорастворимые соединения хорошо проникают через клеточные оболочки за счет действия механизмов переноса, опосредованных специфическими носителями. Эти мембранные переносчики играют ключевую роль при определении действия на клетки или организмы широкого круга растворенных веществ, включая нутриенты и продукты клеточного обмена, а также молекулы лекарственных средств. Определенные усилия были направлены на то, чтобы улучшить биодоступность лекарственных средств с помощью различных пролекарственных фрагментов, направляющих различные системы активного переноса, присутствующие в тонком кишечнике. Примеры систем переноса включают пептидные переносчики, органические катионные переносчики, органические анионные переносчики, переносчики глюкозы, переносчики витаминов, переносчики желчных кислот, переносчики жирных кислот, переносчики фосфатов, переносчики монокарбоновых кислот, переносчики бикарбонатов, переносчики ABC, переносчики нуклеозидов и переносчики аминокислот, которые описаны H.-C. Shi et al., в обзоре R.Mannhold, H.Kubinyi, G.Folkers, Eds., Methods and Principles in Medicinal Chemistry, Wiley-VCH, Weinheim, 2003; pp.245-278, включенном в настоящую заявку посредством ссылки. Все эти переносчики в основном расположены на мембранах щеточной каймы, имеют различное распределение вдоль желудочно-кишечного тракта и демонстрируют различную специфичность в отношении субстратов.
Цидофовир [(S)-1-(3-гидрокси-2-фосфонилметоксипропил)цитозин, HPMPC] был одобрен для применения в клинической практике с целью лечения связанного со СПИДом цитомегаловирусного ретинита. Цидофовир известен с точки зрения широкого спектра активности против практически всех ДНК вирусов. Было показано, что этот препарат обладает терапевтическим потенциалом не только против цитомегаловируса, но также против других вирусов герпеса, например, вируса простого герпеса (HSV), вируса varcella-zoster (опоясывающего лишая), вируса Эпштейна-Барра (EBV) и вирусов герпеса человека 6, 7 и 8 типов. Кроме того, он обладает противовирусной активностью против аденовирусов, паповавирусов, таких как папилломавирус и полиомавирус, поксвирусов, таких как вирус натуральной оспы (этиологический агент натуральной оспы) и других ортопоксвирусов, таких как вирус оспы обезьян и иридовирус.
Настоящее изобретение, в частности, относится к способам синтеза липидных пролекарств цидофовира. Идеальный способ синтеза производных цидофовира мог бы, например, позволить получить продукт с высокой чистотой и высоким выходом. Предпочтительно, такой способ мог бы позволить избежать или минимизировать использование очистки хроматографическими методами. Настоящее изобретение направлено на обеспечение одной или нескольких из этих желаемых характеристик.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к морфологической форме (форме A) [[(S)-2-(4-амино-2-оксо-1(2H)-пиримидинил)-1-(гидроксиметил)этокси]метил]моно[3-(гексадецилокси)пропилового] эфира фосфоновой кислоты (далее в тексте заявки “CMX001”). Форма A CMX001 характеризуется картиной дифракции рентгеновских лучей, включающей пики при углах 2θ примерно 5,5, 19,3, 20,8 и 21,3 градуса.
В одном из вариантов осуществления форма A характеризуется картиной дифракции рентгеновских лучей, дополнительно включающей пики при углах 2θ примерно 17,8 и 23,3 градуса.
В одном из вариантов осуществления форма A характеризуется картиной дифракции рентгеновских лучей, включающей пики при углах 2θ примерно 5,5, 17,8, 19,3, 20,8, 21,3 и 23,3 градуса.
В одном из вариантов осуществления форма A характеризуется картиной дифракции рентгеновских лучей, включающей пики при углах 2θ примерно 5,5, 13,5, 17,8, 19,0, 19,3, 20,5, 20,8, 21,3, 23,3, 23,9, 24,9 и 25,9 градуса.
В одном из вариантов осуществления форма A характеризуется картиной дифракции рентгеновских лучей, включающей пики при углах 2θ примерно 5,5, 11,0, 13,5, 14,3, 17,8, 18,3, 19,0, 19,3, 20,2, 20,5, 20,8, 21,3, 22,1, 22,7, 23,3, 23,9, 24,3, 24,9, 25,6 и 25,9 градуса.
В другом варианте осуществления форма A характеризуется картиной дифракции рентгеновских лучей, по существу аналогичной картине, приведенной на фиг.4.
В другом варианте осуществления форму A получают способом очистки, включающим перекристаллизацию неочищенного препарата [[(S)-2-(4-амино-2-оксо-1(2H)-пиримидинил)-1-(гидроксиметил)этокси]метил]моно[3-(гексадецилокси)пропилового] эфира фосфоновой кислоты из органического растворителя, такого как спирт (например, метанол, этанол и изопропанол). Предпочтительно, этот органический растворитель представляет собой метанол.
В одном из вариантов осуществления форма A имеет чистоту более 91%, например, более 92,5%, более 95%, более 96%, более 97% или более 97,5%.
В одном из вариантов осуществления форма A имеет чистоту более 98%, например, более 98,5%, более 99%, более 99,2%, более 99,5% или более 99,8%.
В другом варианте осуществления форма A содержит менее 1,5% N4-алкилированного продукта, например, менее 1,0% N4-алкилированного продукта, или менее 0,5% N4-алкилированного продукта.
В другом варианте осуществления форма A не содержит N4-алкилированного продукта.
Кроме того, в настоящем изобретении описаны способы получения эфиров замещенной фосфоновой кислоты. Например, в одном из вариантов осуществления изобретение относится к способам получения CMX001, например, формы A CMX001. Предпочтительно, чтобы такие способы обеспечивали возможность получения CMX001 (например, формы A) с высокой чистотой и в большом количестве без необходимости очистки методом хроматографии.
Далее, в одном из вариантов осуществления в изобретении описан усовершенствованный способ получения [[(S)-2-(4-амино-2-оксо-1(2H)-пиримидинил)-1-(гидроксиметил)этокси]метил]моно[3-(гексадецилокси)пропилового]эфира фосфоновой кислоты (CMX001). Усовершенствованный способ включает обработку (S)-N1-[(2-гидрокси-3-трифенилметокси)пропил]цитозина (в настоящей заявке “CMX212”) ди-трет-бутоксидом магния, затем обработку натриевой солью моно[3-(гексадецилокси)пропилового]эфира P-[[[(4-метилфенил)сульфонил]окси]метил]фосфоновой кислоты (в настоящей заявке “CMX203”) с образованием [[(S)-2-(4-амино-2-оксо-1(2H)-пиримидинил)-1-(гидроксиметил)-2-(трифенилметокси)этил]метил]моно[3-(гексадецилокси)пропилового] эфира фосфоновой кислоты (в настоящей заявке “CMX225”). [[(S)-2-(4-амино-2-оксо-1(2H)-пиримидинил)-1-(гидроксиметил)-2-(трифенилметокси)этил]метил]моно[3-(гексадецилокси)пропиловый] эфир фосфоновой кислоты (CMX225) подвергают взаимодействию с реагентом для удаления защитной группы, получая [[(S)-2-(4-амино-2-оксо-1(2H)-пиримидинил)-1-(гидроксиметил)этокси]метил]моно[3-(гексадецилокси)пропиловый] эфир фосфоновой кислоты (CMX001).
В другом варианте осуществления изобретение относится к способу синтеза очищенного [[(S)-2-(4-амино-2-оксо-1(2H)-пиримидинил)-1-(гидроксиметил)этокси]метил]моно[3-(гексадецилокси)пропилового]эфира фосфоновой кислоты (CMX001), где указанный способ включает:
(a) взаимодействие цитозина с (S)-тритил глицидиловым эфиром в присутствии карбоната металла и первого подходящего органического растворителя с образованием (S)-N1-[(2-гидрокси-3-трифенилметокси)пропил]цитозина (CMX212);
(b) взаимодействие (S)-N1-[(2-гидрокси-3-трифенилметокси)пропил]цитозина (CMX212) с натриевой солью моно[3-(гексадецилокси)пропилового]эфира P-[[[(4-метилфенил)сульфонил]окси]метил]фосфоновой кислоты (CMX203) в присутствии ди-трет-бутоксида магния и второго подходящего органического растворителя с образованием [[(S)-2-(4-амино-2-оксо-1(2H)-пиримидинил)-1-(гидроксиметил)-2-(трифенилметокси)этил]метил]моно[3-(гексадецилокси)пропилового] эфира фосфоновой кислоты (CMX225);
(c) взаимодействие [[(S)-2-(4-амино-2-оксо-1(2H)-пиримидинил)-1-(гидроксиметил)-2-(трифенилметокси)этил]метил]моно[3-(гексадецилокси)пропилового] эфира фосфоновой кислоты (CMX225) с реагентом для удаления защитной группы в присутствии метанола с образованием неочищенного [[(S)-2-(4-амино-2-оксо-1(2H)-пиримидинил)-1-(гидроксиметил)этокси]метил]моно[3-(гексадецилокси)пропилового] эфира фосфоновой кислоты (CMX001); и
(d) перекристаллизацию неочищенного [[(S)-2-(4-амино-2-оксо-1(2H)-пиримидинил)-1-(гидроксиметил)этокси]метил]моно[3-(гексадецилокси)пропилового]эфира фосфоновой кислоты (CMX001) из третьего подходящего органического растворителя.
Кроме того, настоящее изобретение относится к способам получения замещенных тозилоксиметилфосфонатов. Например, в одном из вариантов осуществления в изобретении описаны способы получения натриевой соли моно[3-(гексадецилокси)пропилового]эфира P-[[[(4-метилфенил)сульфонил]окси]метил]фосфоновой кислоты (CMX203). Предпочтительно, чтобы такие способы давали возможность получать CMX203 с высокой чистотой и в большом количестве без необходимости очистки методом хроматографии.
Далее, в другом варианте осуществления настоящее изобретение относится к усовершенствованному способу получения натриевой соли моно[3-(гексадецилокси)пропилового]эфира P-[[[(4-метилфенил)сульфонил]окси]метил]фосфоновой кислоты (CMX203). Усовершенствование включает гашение реакционной смеси, полученной при взаимодействии алкоксиалканола и (дихлорфосфорил)метил 4-метилбензолсульфоната, бикарбонатом натрия с последующим доведением значения pH до 2,0 и выделением желаемого продукта. Желаемый продукт отделяют с помощью дихлорметана и концентрируют. После концентрирования желаемый продукт повторно растворяют в 2-пропаноле и добавляют гидроксид натрия. Завершают осаждение желаемого продукта из 2-пропанола.
В другом варианте осуществления изобретение относится к способу синтеза натриевой соли моно[3-(гексадецилокси)пропилового]эфира P-[[[(4-метилфенил)сульфонил]окси]метил]фосфоновой кислоты (CMX203) с высоким выходом, где указанный способ включает:
(a) взаимодействие (дихлорфосфорил)метил 4-метилбензолсульфоната с 3-(гексадецилокси)пропан-1-олом в присутствии пиридина или триэтиламина в первом подходящем растворителе с образованием соответствующей реакционной смеси;
(b) гашение полученной смеси подходящим реагентом и водой;
(c) доведение значения pH полученной смеси до 2.0 с образованием неочищенного продукта; и
(d) растворение неочищенного продукта во втором подходящем растворителе.
В некоторых вариантах осуществления натриевую соль моно[3-(гексадецилокси)пропилового]эфира P-[[[(4-метилфенил)сульфонил]окси]метил]фосфоновой кислоты (CMX203) синтезируют с выходом, равным или превышающим примерно 73% относительно 3-(гексадецилокси)пропан-1-ола.
В другом варианте осуществления настоящее изобретение относится к способу синтеза гексадецил метансульфоната с высоким выходом, где указанный способ включает взаимодействие 1-гексадеканола с метансульфонилхлоридом в присутствии амина в подходящем растворителе.
В следующем варианте осуществления изобретение относится к способу синтеза 3-(гексадецилокси)пропан-1-ола с высоким выходом, где указанный способ включает взаимодействие 1,3-пропандиола с гексадецил метансульфонатом в присутствии гидрида металла в N-метилпирролидиноне (NMP).
В следующем варианте осуществления в изобретении описан способ синтеза натриевой соли моно[3-(гексадецилокси)пропилового]эфира P-[[[(4-метилфенил)сульфонил]окси]метил]фосфоновой кислоты (CMX203). Указанный способ включает взаимодействие (дихлорфосфорил)метил 4-метилбензолсульфоната с 3-(гексадецилокси)пропан-1-олом в присутствии пиридина в подходящем растворителе с образованием соответствующей реакционной смеси. Полученную смесь гасят соответствующим реагентом и водой. Затем доводят значение pH погашенной реакционной смеси до 2, получая неочищенный продукт. Затем этот неочищенный продукт растворяют в 2-пропаноле и гидроксиде натрия, получая 3-(гексадецилокси)пропил тозилметилфосфонат.
В еще одном варианте осуществления в изобретении описан способ синтеза натриевой соли моно[3-(гексадецилокси)пропилового]эфира P-[[[(4-метилфенил)сульфонил]окси]метил]фосфоновой кислоты (CMX203). Этот способ включает приведение в контакт (дихлорфосфорил)метил 4-метилбензолсульфоната с алкоксиалканолом в присутствии подходящего основания в подходящем растворителе с получением алкоксиалкилзамещенного тозилоксиметилфосфоната. Этот алкоксиалкилзамещенный тозилоксиметилфосфонат гасят подходящим реагентом и водой. Затем доводят значение pH погашенного алкоксиалкилзамещенного тозилоксиметилфосфоната до 2, получая неочищенный продукт. Затем этот неочищенный продукт растворяют в реагенте, подходящем для перекристаллизации, и гидроксиде натрия, получая желаемый алкоксиалкилзамещенный тозилоксиметилфосфонат.
В другом варианте осуществления второй подходящий растворитель на стадии (d) дополнительно обрабатывают гидроксидом натрия.
В другом варианте осуществления алкоксиалканол представляет собой 3-(гексадецилокси)пропан-1-ол, подходящим основанием является пиридин, подходящим растворителем является дихлорметан, реагент для гашения реакционной смеси представляет собой бикарбонат натрия и растворителем для перекристаллизации является 2-пропанол.
В следующем варианте осуществления изобретение относится к способу синтеза (дихлорфосфорил)метил 4-метилбензолсульфоната путем:
(a) взаимодействия диэтил (тозилокси)метилоксифосфоната и ацетонитрила с бромтриметилсиланом и нагревания с получением соответствующей смеси; и
(b) добавления к полученной смеси дихлорметана и оксалилхлорида с получением (дихлорфосфорил)метил 4-метилбензолсульфоната.
В другом варианте осуществления в смесь, полученную на стадии (b), добавляют катализатор (например, N,N-диметилформамид), получая (дихлорфосфорил)метил 4-метилбензолсульфонат.
В следующем варианте осуществления изобретение относится к способу синтеза 3-(гексадецилокси)пропан-1-ола путем:
(a) взаимодействия в N-метилпирролидиноне 1,3-пропандиола с гидридом натрия с образованием соответствующей смеси; и
(b) добавления раствора гексадецил метансульфоната в N-метилпирролидиноне с образованием 3-(гексадецилокси)пропан-1-ола.
В еще одном варианте осуществления изобретение относится к способу синтеза гексадецил метансульфоната путем:
(a) взаимодействия 1-гексадеканола, дихлорметана и диизопропилэтиламина с образованием соответствующей смеси; и
(b) добавления к полученной смеси метансульфонилхлорида с получением гексадецил метансульфоната.
В другом аспекте изобретение относится к способу синтеза CMX001 (например, формы A) путем:
(a) взаимодействия (S)-N1-[(2-гидрокси-3-(PG-O)пропил]цитозина с натриевой солью моно[3-(гексадецилокси)пропилового]эфира P-[[[(4-метилфенил)сульфонил]окси]метил]фосфоновой кислоты в присутствии ди-трет-бутоксида магния и подходящего органического растворителя A с образованием [3-(гексадецилокси)пропил]гидро[[[(S)-1-(4-амино-2-оксопиримидин-1(2H)-ил)-3-(PG-O)пропан-2-ил]окси]метил]фосфоната; и
(b) взаимодействия [3-(гексадецилокси)пропил]гидро[[[(S)-1-(4-амино-2-оксопиримидин-1(2H)-ил)-3-(PG-O)пропан-2-ил]окси]метил]фосфоната с реагентом для удаления защитной группы в присутствии подходящего органического растворителя B с образованием [[(S)-2-(4-амино-2-оксо-1(2H)-пиримидинил)-1-(гидроксиметил)этокси]метил]моно[3-(гексадецилокси)пропилового] эфира фосфоновой кислоты; где PG означает фрагмент, защищающий гидроксильную группу.
В одном из вариантов осуществления фрагмент PG можно удалять в кислой среде.
В одном из вариантов осуществления PG означает трифенилметил, монометокситритил или диметокситритил.
В одном из вариантов осуществления реагентом для удаления защитной группы является хлористый водород.
В одном из вариантов осуществления подходящий органический растворитель A представляет собой N,N-диметилформамид.
В одном из вариантов осуществления ди-трет-бутоксид магния имеет чистоту свыше 98%.
В одном из вариантов осуществления подходящий растворитель B является спиртом, таким как метанол.
В одном из вариантов осуществления способ дополнительно включает перекристаллизацию [[(S)-2-(4-амино-2-оксо-1(2H)-пиримидинил)-1-(гидроксиметил)этокси]метил]моно[3-(гексадецилокси)пропилового]эфира фосфоновой кислоты из подходящего для перекристаллизации органического растворителя.
В одном из вариантов осуществления подходящий для перекристаллизации органический растворитель является неводным.
В одном из вариантов осуществления подходящий для перекристаллизации органический растворитель является нетоксичным.
В одном из вариантов осуществления подходящий для перекристаллизации органический растворитель является фармацевтически приемлемым.
В одном из вариантов осуществления подходящий для перекристаллизации органический растворитель представляет собой метанол.
В одном из вариантов осуществления способ дополнительно включает синтез (S)-N1-[(2-гидрокси-3-(PG-O)пропил]цитозина путем:
взаимодействия цитозина с (S)-2-(PG-O-метил)оксираном в присутствии карбоната металла и подходящего органического растворителя C с образованием (S)-N1-[(2-гидрокси-3-(PG-O)пропил]цитозина.
В одном из вариантов осуществления карбонат металла представляет собой карбонат калия.
В одном из вариантов осуществления подходящий органический растворитель C представляет собой N,N-диметилформамид.
В еще одном аспекте настоящее изобретение относится к соединениям, предназначенным для лечения вирусной инфекции у субъекта, например, иммунодефицитного субъекта, имеющим структуру формулы (I):
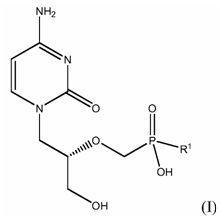
где:
R1 представляет собой незамещенный или замещенный C1-C6 алкоксил, или незамещенный или замещенный C1-C30 алкокси-С1-C6-алкоксил; или их энантиомерам, диастереомерам, рацематам или смесям, причем соединение формулы (I) имеет чистоту более 91% или находится в форме A. В одном из вариантов осуществления чистота соединения формулы (I) составляет >92%, >93%, >94%, >95%, >97,5%, >98%, >99% или >99,5%. В другом варианте осуществления это соединение находится в форме A. В еще одном варианте осуществления соединение находится в форме A и имеет чистоту более 91% (например, >92%, >93%, >94%, >95%, >97,5%, >98%, >99% или >99,5%). В одном из вариантов осуществления соединение формулы (I) получают перекристаллизацией неочищенного соединения из подходящего для перекристаллизации растворителя, описанного в настоящей заявке. В другом варианте осуществления это соединение не является гидратом. В еще одном варианте осуществления это соединение является сольватом, например, сольватом с метанолом, сольватом с этанолом или сольватом с изопропанолом.
В следующем аспекте изобретение относится к соединениям, предназначенным для предупреждения вирусных инфекций у субъекта, например, иммунодефицитного субъекта, имеющим структуру формулы (I):
где:
R1 представляет собой незамещенный или замещенный C1-C6 алкоксил, или незамещенный или замещенный C1-C30 алкокси-С1-C6-алкоксил; или их энантиомерам, диастереомерам, рацематам или смесям, причем соединение формулы (I) имеет чистоту более 91% или находится в форме A. В одном из вариантов осуществления чистота соединения формулы (I) составляет >92%, >93%, >94%, >95%, >97,5%, >98%, >99% или >99,5%. В другом варианте осуществления это соединение находится в форме A. В еще одном варианте осуществления соединение находится в форме A и имеет чистоту более 91% (например, >92%, >93%, >94%, >95%, >97,5%, >98%, >99% или >99,5%). В одном из вариантов осуществления соединение формулы (I) получают перекристаллизацией неочищенного соединения из подходящего для перекристаллизации растворителя, описанного в настоящей заявке. В другом варианте осуществления это соединение не является гидратом. В еще одном варианте осуществления это соединение является сольватом, например, сольватом с метанолом, сольватом с этанолом или сольватом с изопропанолом.
В другом варианте осуществления вирусная инфекция, которую предполагается лечить или предупреждать, невосприимчива к лечению или профилактике другими нуклеозидфосфонатами, например, цидофовиром, циклическим цидофовиром, тенофовиром, адефовиром и т.д. Альтернативно или дополнительно, эти другие нуклеозидфосфонаты (например, цидофовир (CDV)) демонстрируют токсическое побочное действие (например, нефротоксичность) у указанных иммунодефицитных субъектов.
В другом варианте осуществления субъект инфицирован по меньшей мере одним вирусом. Этот вирус может быть выбран из группы, состоящей из: вируса иммунодефицита человека (HIV), вируса гриппа, вируса простого герпеса (HSV), вируса герпеса человека 6 (HHV-6), цитомегаловируса (CMV), вирусов гепатита B и C, вируса Эпштейна-Барра (EBV), вируса varicella zoster, вирусов натуральной и малой оспы, вируса осповакцины, вируса оспы, вируса коровьей оспы, вируса оспы верблюдов, вируса оспы обезьян, вируса Эбола, вируса папилломы, аденовируса или вируса полиомы, включая вирус Джона Каннингема (JVC), вируса BK и вакуолизирующего обезьяньего вируса 40 или обезьяньего вируса 40 (SV40). В другом варианте осуществления субъект инфицирован по меньшей мере одним вирусом с двухцепочечной ДНК.
В другом варианте осуществления субъект инфицирован вирусом или любой комбинацией двух или более вирусов, выбранных из группы, состоящей из человеческого CMV (HCMV), вируса BK, HHV-6, аденовируса и EBV.
В другом варианте осуществления субъект инфицирован двумя или более вирусами, по меньшей мере один из которых представляет собой, например, вирус с двухцепочечной ДНК, и вирусы проявляют синергическое действие. Например, эти вирусы представляют собой вирусы HCMV и BK.
В другом варианте осуществления соединение формулы (I), имеющее чистоту более 91% или находящееся в форме A, используется для лечения вирусной инфекции (например, инфекции, вызванной вирусом с двухцепочечной ДНК) у субъекта, где указанная инфекция невосприимчива к гидрохлориду валганцикловира (или ганцикловиру), или где у указанного субъекта проявляются побочные эффекты гидрохлорида валганцикловира (или ганцикловира). В качестве альтернативы или дополнения, соединение формулы (I), имеющее чистоту более 91% или находящееся в форме A, используется для лечения цитомегаловируса (CMV) после лечения ганцикловиром, например, если CMV инфекция появилась во время лечения. Пациент может быть пациентом с трансплантатом стволовых клеток костного мозга, в частности, если существует опасность (реальная или гипотетическая) токсичности ганцикловира в отношении костного мозга у данного пациента.
В другом варианте осуществления субъектом является млекопитающее. В еще одном варианте осуществления субъектом является человек.
В следующем варианте осуществления соединение формулы (I), имеющее чистоту более 91% или находящееся в форме A, вводят перорально, например, в дозе от примерно 0,01 мг/кг до примерно 10 мг/кг или более, например, до 100 мг/кг. В другом варианте осуществления указанное соединение формулы (I), имеющее чистоту более 91% или находящееся в форме A, вводят указанному субъекту в дозе примерно 0,01, 0,05, 0,1, 0,5, 1, 1,5, 2, 2,5, 3, 3,5, 4, 4,5, 5, 5,5, 6, 6,5, 7, 7,5, 8, 8,5 9, 9,5 или 10 мг/кг или более, или в любом диапазоне, ограниченном приведенными значениями.
В другом варианте осуществления изобретение относится также к применению соединения формулы (I), имеющего чистоту более 91% или находящегося в форме A, для производства лекарственного средства для терапевтического и/или профилактического лечения вирусной инфекции у субъекта, например, иммунодефицитного субъекта.
В другом варианте осуществления изобретение относится к способу терапевтического и/или профилактического лечения вирусной инфекции у субъекта, например, иммунодефицитного субъекта, где указанный способ включает введение субъекту соединения формулы (I), имеющего чистоту более 91% или находящегося в форме A.
В следующем варианте осуществления изобретение также относится к пероральной дозированной форме, содержащей соединение формулы (I), имеющее чистоту более 91% или находящееся в форме A, предназначенной для терапевтического и/или профилактического лечения вирусной инфекции у субъекта, где указанная пероральная дозированная форма, при введении человеку в дозе 2 мг/кг указанного соединения, обеспечивает AUC0-inf указанного соединения от примерно 2000 до примерно 4000 ч*нг/мл, например, от примерно 2500 до примерно 3000 ч*нг/мл.
В другом варианте осуществления изобретение также относится к пероральной дозированной форме, содержащей соединение формулы (I), имеющее чистоту более 91% или находящееся в форме A, предназначенной для терапевтического и/или профилактического лечения вирусной инфекции у субъекта, где указанная пероральная дозированная форма, при введении человеку в дозе 2 мг/кг указанного соединения, обеспечивает Cmax указанного соединения от примерно 100 до примерно 500 нг/мл, например, от примерно 200 до примерно 400 ч*нг/мл.
В другом варианте осуществления изобретение относится также к пероральной дозированной форме, содержащей соединение формулы (I), имеющее чистоту более 91% или находящееся в форме A, для терапевтического и/или профилактического лечения вирусной инфекции у субъекта, где указанная пероральная дозированная форма, при введении человеку в дозе 2 мг/кг указанного соединения формулы (I) и метаболизме указанного соединения формулы (I) до цидофовира, обеспечивает Cmax указанного цидофовира, которая составляет менее чем примерно 30% от Cmax указанного соединения формулы (I), например, менее чем примерно 20% от Cmax указанного соединения формулы (I).
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
На фиг.1 изображена дифрактограмма рентгеновских лучей (XRD) порошка CMX001 в форме A (образец № 1).
На фиг.2 изображена XRD CMX001, форма A (образец №2).
На фиг.3 изображена XRD CMX001, форма A (образец №3).
На фиг.4 изображена XRD CMX001, форма A (образец №4).
На фиг.5 изображена XRD CMX001, форма A (образец №5).
На фиг.6 изображена XRD CMX001, форма B (образец №6).
На фиг.7 изображено наложение рентгеновских дифрактограмм формы A (образец №4) и формы B (образец №6).
На фиг.8(a)-(d) изображены 1H-ЯМР спектры формы A (образец №5).
На фиг.8(e)-(f) изображены 31P-ЯМР спектры формы A (образец №5).
На фиг.9 изображена ВЭЖХ хроматограмма формы A (образец №5).
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В настоящем описании и приложенной формуле изобретения формы единственного числа “a”, “an” и “the” включают упоминание форм множественного числа, если контекст явно не указывает на противоположное. Так, например, ссылка на «реагент» включает не только единственный реагент, но также комбинацию или смесь двух или более различных реагентов, ссылка на «заместитель» включает единственный заместитель, а также два или более заместителей и т.п.
Имеется в виду, что в настоящем описании фразы «например», «к примеру», «такой как» или «включающий» предваряют примеры, которые дополнительно проясняют более общее определение. Эти примеры приведены лишь для содействия пониманию раскрытия изобретения и не служат цели какого бы то ни было ограничения объема изобретения. Кроме того, в настоящем описании термины «может», «необязательный», «необязательно» или «необязательно может» означают, что описанное после этого термина обстоятельство может иметь или не иметь места, т.е. это описание включает примеры, в которых это обстоятельство имеет место, а также примеры, в которых это обстоятельство не имеет места. Например, фраза «необязательно присутствует» означает, что объект может присутствовать или не присутствовать, и, таким образом, это описание включает примеры, в которых объект присутствует, а также примеры, в которых объект не присутствует.
В описании и формуле настоящего изобретения будет использоваться следующая терминология, в соответствии с приведенными ниже определениями.
В настоящем описании фраза «имеющий формулу» или «имеющий структуру» не предназначена служить для ограничения и применяется в таком же смысле, в которым обычно применяется термин «включающий». Термин «независимо выбранный из» используется в настоящей заявке для указания на то, что упомянутые элементы, например, группы R или подобные, могут быть одинаковыми или различными.
Термин «алкил» в настоящей заявке относится к разветвленной или неразветвленной насыщенной углеводородной группе, как правило, хотя и необязательно, содержащей от 1 до примерно 24 атомов углерода, такой как метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, октил, децил и т.п., а также к циклоалкильной группе, такой как циклопентил, циклогексил и т.п. Как правило, хотя и необязательно, алкильные группы в настоящей заявке могут содержать от 1 до примерно 18 атомов углерода, и такие группы могут содержать от 1 до примерно 12 атомов углерода. Термин «низший алкил» означает алкильную группу, включающую от 1 до 6 атомов углерода, например, 1, 2, 3, 4, 5 или 6 атомов углерода. Термин «замещенный алкил» относится к алкилу, замещенному одной или несколькими замещающими группами, и термины «гетероатом-содержащий алкил» и «гетероалкил» относятся к алкильному заместителю, в котором по меньшей мере один атом углерода заменен гетероатомом, как более подробно описано ниже.
Термин «алкенил» используется в настоящей заявке для обозначения линейной, разветвленной или циклической углеводородной группы, содержащей от 2 до примерно 24 атомов углерода, включающей по меньшей мере одну двойную связь, такой как этенил, н-пропенил, изопропенил, н-бутенил, изобутенил, октенил, деценил, тетрадеценил, гексадеценил, эйкозенил, тетракозенил и т.п. Как правило, хотя и необязательно, алкенильная группа в настоящем изобретении может содержать от 2 до примерно 18 атомов углерода и, например, может содержать от 2 до 12 атомов углерода. Термин «низший алкенил» означает алкенильную группу, содержащую от 2 до 6 атомов углерода. Термин «замещенный алкенил» относится к алкенилу, замещенному одной или несколькими замещающими группами, и термины «гетероатом-содержащий алкенил» и «гетероалкенил» относятся к алкенилу, в котором по меньшей мере один атом углерода заменен гетероатомом.
Термин «алкинил» используется в настоящей заявке для обозначения линейной или разветвленной углеводородной группы, содержащей от 2 до примерно 24 атомов углерода, включающей по меньшей мере одну тройную связь, такой как этинил, н-пропинил и т.п. Как правило, хотя и необязательно, алкинильная группа в настоящем изобретении может содержать от 2 до примерно 18 атомов углерода и, например, такая группа может также содержать от 2 до 12 атомов углерода. Термин «низший алкинил» означает алкинильную группу, содержащую от 2 до 6 атомов углерода. Термин «замещенный алкинил» относится к алкинилу, замещенному одной или несколькими замещающими группами, и термины «гетероатом-содержащий алкинил» и «гетероалкинил» относятся к алкинилу, в котором по меньшей мере один атом углерода заменен гетероатомом.
Термин «алкокси» в настоящем описании означает алкильную группу, связанную через один концевой фрагмент простого эфира; т.е. «алкокси» группа может быть представлена в виде -O-алкил, где алкил соответствует данному выше определению. «Низшая алкокси» группа означает алкоксигруппу, содержащую от 1 до 6 атомов углерода и включает, например, метокси, этокси, н-пропокси, изопропокси, трет-бутилокси и т.д. Заместители, обозначенные как «C1-C6 алкокси» или «низшие алкокси» в настоящей заявке могут содержать, например, от 1 до 3 атомов углерода, и, в качестве дополнительного примера, такие заместители могут содержать 1 или 2 атома углерода (т.е. метокси и этокси).
Термин «арил» в настоящем описании, и если не указано иное, относится к ароматическому заместителю, как правило, хотя и необязательно, содержащему от 5 до 30 атомов углерода и включающему одно ароматическое кольцо или несколько ароматических колец, которые конденсированы друг с другом, связаны непосредственно или через промежуточные группы (так что разные ароматические кольца связаны с общей группой, например, метиленовым или этиленовым фрагментом). Арильные группы могут, например, содержать от 5 до 20 атомов углерода и, в качестве еще одного примера, арильные группы могут содержать от 5 до 12 атомов углерода. Например, арильные группы могут содержать одно ароматическое кольцо или два конденсированных или связанных ароматических кольца, например, фенил, нафтил, бифенил, дифениловый эфир, дифениламин, бензофенон и т.п. Термин «замещенный арил» относится к арильному фрагменту, замещенному одной или несколькими замещающими группами, и термины «гетероатом-содержащий арил» и «гетероарил» относятся к арильному заместителю, в котором по меньшей мере один атом углерода заменен гетероатомом, как более подробно будет описано ниже. Если не указано иное, термин «арил» включает незамещенные, замещенные и/или гетероатом-содержащие ароматические заместители.
Термин «аралкил» относится к алкильной группе с арильным заместителем, и термин «алкарил» относится к арильной группе с алкильным заместителем, где «алкил» и «арил» соответствуют данным выше определениям. Как правило, аралкильные и алкарильные группы в настоящем изобретении содержат от 6 до 30 атомов углерода. Аралкильные и алкарильные группы могут, например, содержать от 6 до 20 атомов углерода, и, в качестве еще одного примера, такие группы могут содержать от 6 до 12 атомов углерода.
Термин «амино» в настоящей заявке относится к группе -NZ1Z2, где Z1 и Z2 являются атомами водорода или неводородными заместителями, включающими, например, алкил, арил, алкенил, аралкил и их замещенные и/или гетероатом-содержащие варианты.
Термины «гало» или «галоген» используются в традиционном смысле для обозначения замещающих атомов хлора, брома, фтора или йода.
Термин «гетероатом-содержащий», например, «гетероатом-содержащая алкильная группа» (именуемая также «гетероалкильной» группой) или «гетероатом-содержащая арильная группа» (именуемая также «гетероарильной» группой), относится к молекуле, соединительной группе или заместителю, в которых один или несколько атомов углерода заменены атомами, отличными от углерода, например, азотом, кислородом, серой, фосфором или кремнием, обычно азотом, кислородом или серой. Подобным образом, термин «гетероалкил» относится к алкильному заместителю, который является гетероатом-содержащим, термин «гетероциклический» относится к циклическому заместителю, который является гетероатом-содержащим, термины «гетероарил» или «гетероароматический» соответственно относятся к «арилу» и «ароматическим» заместителям, которые являются гетероатом-содержащими, и т.п. Примеры гетероалкильных групп включают алкоксиарил, алкилсульфанил-замещенный алкил, N-алкилированный аминоалкил и т.п. Примеры гетероарильных заместителей включают пирролил, пирролидинил, пиридинил, хинолинил, индолил, фурил, пиримидинил, имидазолил, 1,2,4-триазолил, тетразолил и т.д., и примерами гетероатом-содержащих алициклических групп являются пирролидино, морфолино, пиперазино, пиперидино, тетрагидрофуранил и т.д.
Термин «гидрокарбил» относится к одновалентному гидрокарбильному радикалу, содержащему от 1 до примерно 30 атомов углерода, в том числе, от 1 до примерно 24 атомов углерода, от 1 до примерно 18 атомов углерода и от 1 до примерно 12 атомов углерода, включая линейные, разветвленные, циклические, насыщенные и ненасыщенные фрагменты, такие как алкильные группы, алкенильные группы, арильные группы и т.п. Термин «замещенный гидрокарбил» относится к гидрокарбилу, замещенному одной или несколькими замещающими группами, и термин «гетероатом-содержащий гидрокарбил» относится к гидрокарбилу, в котором по меньшей мере один атом углерода заменен гетероатомом.
Под термином «замещенный», например, «замещенный гидрокарбил», «замещенный алкил», «замещенный арил» и т.п., который встречается в некоторых из приведенных выше определений, подразумевается, что в гидрокарбиле, алкиле, ариле или другом фрагменте, по меньшей мере один атом водорода, связанный с атомом углерода (или другим атомом), заменен одним или несколькими неводородными заместителями. Примеры таких заместителей включают, не ограничиваясь ими, функциональные группы и гидрокарбильные фрагменты C1-C24 алкил (включая C1-C18 алкил, C1-C12 алкил и C1-C6 алкил), C2-C24 алкенил (включая C2-C18 алкенил, C2-C12 алкенил и C2-C6 алкенил), C2-C24 алкинил (включая C2-C18 алкинил, C2-C12 алкинил и C2-C6 алкинил), C5-C30 арил (включая C5-C20 арил и C5-C12 арил) и C6-C30 аралкил (включая C6-C20 аралкил и C6-C12 аралкил).
Под термином «функциональная группа», который встречается в некоторых из приведенных выше определений, подразумевается группа, не являющаяся водородом, содержащая один или несколько функциональных фрагментов, не являющихся углеводородными группами. Примеры функциональных групп включают, не ограничиваясь перечисленными: галоген, гидроксил, сульфгидрил, C1-C24 алкокси, C2-C24 алкенилокси, C2-C24 алкинилокси, C5-C20 арилокси, ацил (включая C2-C24 алкилкарбонил (-CO-алкил) и C6-C20 арилкарбонил (-CO-арил)), ацилокси (-O-ацил), C2-C24 алкоксикарбонил (-(CO)-O-алкил), C6-C20 арилоксикарбонил (-(CO)-O-арил), галогенкарбонил ((-CO)-X, где X означает галоген), C2-C24 алкилкарбонато (-O-(CO)-O-алкил), C6-C20 арилкарбонато (-O-(CO)-O-арил), карбокси (-COOH), карбоксилато (-COO-), карбамоил (-(CO)-NH2), монозамещенный C1-C24 алкилкарбамоил (-(CO)-NH(C1-C24 алкил)), дизамещенный алкилкарбамоил (-(CO)-N(C1-C24 алкил)2), монозамещенный арилкарбамоил (-(CO)-NH-арил), тиокарбамоил (-(CS)-NH2), карбамидо (-NH-(CO)-NH2), циано (-C≡N), изоциано (-N+≡C-), цианато (-O-C≡N), изоцианато (-O-N+≡C-), изотиоцианато (-S-C≡N), азидо (-N=N+=N-), формил (-(CO)-H), тиоформил (-(CS)-H), амино (-NH2), моно- и ди-(C1-C24 алкил)-замещенный амино, моно- и ди-(C5-C20 арил)-замещенный амино, C2-C24 алкиламидо (-NH-(CO)-алкил), C5-C20 ариламидо (-NH-(CO)-арил), имино (-CR=NH, где R=водород, C1-C24 алкил, C5-C20 арил, C6-C20 алкарил, C6-C20 аралкил и т.д.), алкилимино (-CR=N(алкил), где R=водород, алкил, арил, алкарил и т.д.), арилимино (-CR=N(арил), где R=водород, алкил, арил, алкарил и т.д.), нитро (-NO2), нитрозо (-NO), сульфо (-SO2-OH), сульфонато (-SO2-O-), C1-C24 алкилсульфанил (-S-алкил, также именуемый «алкилтио»), арилсульфанил (-S-арил, также именуемый «арилтио»), C1-C24 алкилсульфинил (-(SO)-алкил), C5-C20 арилсульфинил (-(SO)-арил), C1-C24 алкилсульфонил (-SO2-алкил), C5-C20 арилсульфонил (-SO2-арил), фосфоно (-P(O)(OH)2), фосфонато (-P(O)(O-)2), фосфинато (-P(O)(O-)), фосфо (-PO2) и фосфино (-PH2), моно- и ди(C1-C24 алкил)-замещенный фосфино, моно- и ди(C5-C20 арил)-замещенный фосфино; а также гидрокарбильные фрагменты C1-C24 алкил (включая C1-C18 алкил, C1-C12 алкил и C1-C6 алкил), C2-C24 алкенил (включая C2-C18 алкенил, C2-C12 алкенил и C2-C6 алкенил), C2-C24 алкинил (включая C2-C18 алкинил, C2-C12 алкинил и C2-C6 алкинил), C5-C30 арил (включая C5-C20 арил и C5-C12 арил) и C6-C30 аралкил (включая C6-C20 аралкил и C6-C12 аралкил). Кроме того, упомянутые выше функциональные группы, если конкретная группа допускает такое замещение, могут быть дополнительно замещены одной или несколькими дополнительными функциональными группами, или одним или несколькими гидрокарбильными фрагментами, такими как фрагменты, конкретно перечисленные выше. Аналогично, упомянутые выше гидрокарбильные фрагменты могут быть дополнительно замещены одной или несколькими функциональными группами или дополнительными гидрокарбильными фрагментами, такими как фрагменты, конкретно перечисленные выше.
Следует учитывать, что некоторые из приведенных выше определений могут частично перекрываться, так что некоторые химические фрагменты могут подпадать под более чем одно определение.
Если термин «замещенный» находится перед перечнем возможных замещенных групп, имеется в виду, что этот термин применяется к каждому члену этого перечня. Например, фразу «замещенный алкил и арил» следует интерпретировать как «замещенный алкил и замещенный арил».
Когда говорят, что два фрагмента «соединены», имеется в виду, что эта фраза включает случаи, когда эти два фрагмента непосредственно соединены друг с другом, а также случаи, когда между двумя фрагментами присутствует линкер. Линкер может включать такие группы, как гетероатомы, C1-C24 алкилен (включая C1-C18 алкилен, C1-C12 алкилен и C1-C6 алкилен), C2-C24 алкенилен (включая C2-C18 алкенилен, C2-C12 алкенилен и C2-C6 алкенилен), C2-C24 алкинилен (включая C2-C18 алкинилен, C2-C12 алкинилен и C2-C6 алкинилен), C5-C30 арилен (включая C5-C20 арилен и C5-C12 арилен) и C6-C30 аралкилен (включая C6-C20 аралкилен и C6-C12 аралкилен).
Настоящее изобретение относится к способам синтеза эфиров замещенных фосфоновых кислот. Далее, в некоторых аспектах изобретение относится к способам получения соединений, имеющих структуру формулы (I):
где R1 представляет собой незамещенный или замещенный C1-C6 алкокси-, или незамещенный или замещенный C1-C30 алкокси-C1-C6-алкокси-; или их энантиомеров, диастереомеров, рацематов или смесей.
В другом варианте осуществления R1 означает C10-C30 алкокси-C2-C4-алкокси-.
В другом варианте осуществления R1 означает гексадецилоксипропилокси-.
Кроме того, изобретение относится к способам синтеза замещенных фосфонатов, в частности, замещенных тозилоксиметилфосфонатов. Затем, в некоторых аспектах изобретение относится к способам получения соединений, имеющих структуру формулы (II):
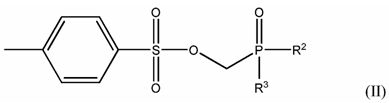
где:
R2 означает незамещенный или замещенный C1-C6 алкокси-, или незамещенный или замещенный C1-C30 алкокси-C1-C6-алкокси-;
R3 означает OR4 или O-A+;
R4 означает H или незамещенный или замещенный C1-C6 алкил; и
A+ означает Li+, Na+ или K+.
В другом варианте осуществления R3 означает O-A+ и R2 представляет собой C10-C30 алкокси-C2-C4-алкокси-. Например, A+ означает Na+ и R2 представляет собой C10-C30 алкокси-пропилокси-.
В другом варианте осуществления R3 означает O-A+ и R2 представляет собой гексадецилоксипропилокси-.
Соединения структурной формулы (I) предпочтительно получают реакцией алкилирования, в которой принимают участие CMX212 и соединение структурной формулы (II).
Соединения структурной формулы (II) предпочтительно выделяют из подходящего растворителя, например дихлорметана, после добавления гасящего реагента и доведения pH до 2,0.
Настоящее изобретение относится к способам синтеза соединений формул (I) и (II). В настоящем изобретении также разработаны детализированные способы синтеза различных соединений, раскрытых в настоящем изобретении, в соответствии с приведенными ниже схемами, которые рассмотрены в примерах.
В тексте настоящей заявки, если указано, что композиции имеют, включают или содержат определенные компоненты, считается, что композиции также состоят в основном из или состоят из перечисленных компонентов. Аналогично, если указано, что способы или процессы имеют, включают или содержат определенные стадии, эти способы также состоят в основном из или состоят из указанных стадий. Далее, следует понимать, что порядок осуществления стадий или порядок выполнения определенных действий не играет существенной роли до тех пор, пока изобретение остается работоспособным. Более того, две или несколько стадий или операций можно осуществлять одновременно.
Способы синтеза по настоящему изобретению подходят для широкого спектра функциональных групп, поэтому могут использоваться исходные вещества с различными заместителями. Способы по настоящему изобретению, как правило, позволяют получать желаемое конечное соединение на завершающей или одной из завершающих стадий общего способа, хотя в некоторых случаях может оказаться желательным дальнейшее преобразование соединения в его фармацевтически приемлемую соль, эфир или пролекарство.
Методика A: Синтез [[(S)-2-(4-амино-2-оксо-1(2H)-пиримидинил)-1-(гидроксиметил)этокси]метил]моно[3-(гексадецилокси)пропилового] эфира фосфоновой кислоты (CMX001)
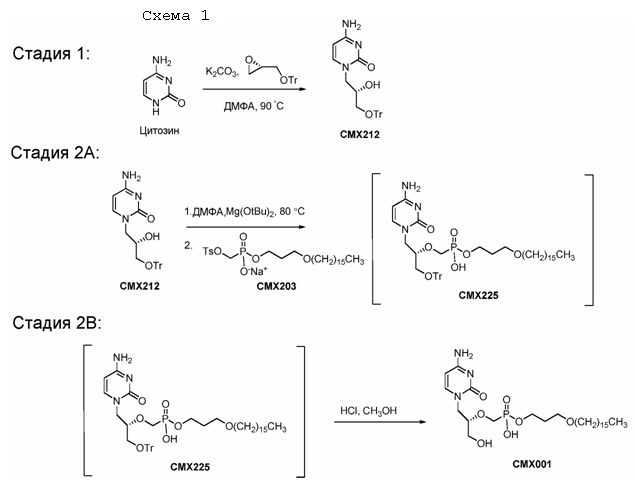
Стадия 1: Синтез (S)-N1-[(2-гидрокси-3-трифенилметокси)пропил]цитозина (CMX212)
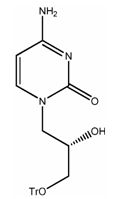
Это соединение получают взаимодействием цитозина с (S)-тритил глицидиловым эфиром в присутствии небольшого количества подходящего основания, такого как карбонат металла (например, карбонат калия), в подходящем органическом растворителе (например, N,N-диметилформамиде, трет-амиловом спирте) при подходящей температуре (например, от 60 до 120°C) до завершения реакции, обычно в течение примерно 4-14 часов, например, приблизительно 8-10 часов.
Стадии 2A и 2B: Синтез [[(S)-2-(4-амино-2-оксо-1(2H)-пиримидинил)-1-(гидроксиметил)этокси]метил]моно[3-(гексадецилокси)пропилового]эфира фосфоновой кислоты (CMX001)
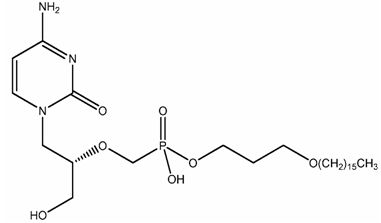
Это соединение получают взаимодействием CMX212 с CMX203 в присутствии подходящего основания, такого как алкоксид металла (например, ди-трет-бутоксид магния, трет-бутоксид натрия, трет-бутоксид лития, трет-амилалкоксид натрия, трет-бутоксид калия, метоксид натрия), гидрид металла (например, гидрид натрия, гидрид калия) или амид металла (например, бис(триметилсилил)амид лития), в подходящем органическом растворителе (например, в N,N-диметилформамиде, N,N-диметилацетамиде, диметилсульфоксиде, 1-метил-2-пирролидиноне) при подходящей температуре (например, от 50 до 110°C) до завершения реакции, обычно в течение примерно 0,25-5 часов, например, приблизительно от двух до четырех часов. Неочищенную реакционную смесь подвергают обработке водой. Неочищенный продукт экстрагируют подходящим органическим растворителем (например, этилацетатом, изопропилацетатом, дихлорметаном и т.д.) и органический растворитель концентрируют, получая неочищенный CMX225. Неочищенный CMX225 подвергают взаимодействию с подходящим реагентом для снятия защитной группы (например, хлористым водородом, ацетилхлоридом) в органическом растворителе (например, метаноле) до завершения реакции, обычно от одного до шести часов, например, от двух до трех часов. Неочищенный CMX001 подвергают перекристаллизации с использованием подходящей системы растворителей (например, метанол/ацетон/вода, этанол, метанол). Ди-трет-бутоксид магния коммерчески доступен от Chemetall (Kings Mountain, NC).
Следует понимать, что хотя для осуществления алкилирования CMX212 подходит широкий диапазон условий, наиболее предпочтительными являются определенные условия проведения реакции, поскольку они позволяют получить наибольшее количество продукта и/или приводят к продукту, имеющему наибольшую чистоту. В частности, предпочтительным алкоксидом металла является ди-трет-бутоксид магния.
Следует иметь в виду, что для завершения преобразования CMX225 в CMX001 необходимо осуществить реакцию снятия защиты. В частности, необходимо удалить O-защитную группу (т.е. тритил) для получения свободного гидроксила, присутствующего в CMX001. Таким образом, в одном из вариантов осуществления, CMX001 получают путем снятия защиты с CMX225 с помощью газообразного хлористого водорода.
Следует учесть, что хотя несколько способов известного уровня техники, которые используются для синтеза CMX001, приводят к получению соли CMX001, например, натриевой соли CMX001, в настоящем изобретении разработаны прямые способы синтеза CMX001 в форме свободной кислоты без промежуточного образования соли.
Один из предпочтительных вариантов осуществления настоящего изобретения описан в методике A. Эта методика представляет собой усовершенствованный способ получения [[(S)-2-(4-амино-2-оксо-1(2H)-пиримидинил)-1-(гидроксиметил)этокси]метил]моно[3-(гексадецилокси)пропилового]эфира фосфоновой кислоты.
Далее по тексту описаны стадии 1, 2A и 2B.
Что касается стадии 1 методики A, то (S)-N1-[(2-гидрокси-3-трифенилметокси)пропил]цитозин (CMX212) получают взаимодействием цитозина с (S)-тритил глицидиловым эфиром в присутствии небольшого количества подходящего основания, такого как карбонат металла (например, карбонат калия), в подходящем растворителе (например, ДМФА, трет-амиловом спирте) при подходящей температуре (например, от 60 до 120°C) до завершения реакции. В предпочтительных методиках очистка CMX212 колоночной хроматографией не является необходимой.
В другом варианте осуществления синтез CMX212 приводит к повышению выхода по сравнению с другими способами, известными в технике. Например, синтез CMX212 проходит с выходом более 55%, 60%, 65%, 70%, 75%, 80%, 85% или 90%.
Далее, что касается стадии 2A методики A, промежуточный продукт, т.е. [[(S)-2-(4-амино-2-оксо-1(2H)-пиримидинил)-1-(гидроксиметил)-2-(трифенилметокси)этил]метил]моно[3-(гексадецилокси)пропиловый] эфир фосфоновой кислоты (CMX225), получают взаимодействием CMX212 с CMX203 в присутствии подходящего основания, такого как алкоксид металла (например, ди-трет-бутоксид магния, трет-бутоксид натрия, трет-бутоксид лития), в подходящем растворителе (например, ДМФА, N,N-диметилацетамиде, диметилсульфоксиде, 1-метил-2-пирролидиноне) при подходящей температуре (например, от 50 до 110°C) до завершения реакции. Полученную смесь подвергают экстракции водой (например, в кислой среде). Затем CMX225 экстрагируют подходящим органическим растворителем (например, этилацетатом, изопропилацетатом, дихлорметаном). В предпочтительных способах очистка колоночной хроматографией не является необходимой. Например, стадию водной экстракции можно использовать для того, чтобы избежать очистки колоночной хроматографией.
В другом варианте осуществления алкилирование CMX212 не приводит к значительному алкилированию 4-аминогруппы.
В другом варианте осуществления стадия 2A методики A приводит к менее чем 5% выходу бис-алкилированного CMX212. В другом варианте осуществления стадия 2A методики A приводит к менее чем 4% выходу бис-алкилированного CMX212. В следующем варианте осуществления стадия 2A методики A приводит к менее чем 3% выходу бис-алкилированного CMX212. В следующем варианте осуществления стадия 2A методики A приводит к менее чем 2% выходу бис-алкилированного CMX212. В следующем варианте осуществления стадия 2A методики A приводит к менее чем 1,5% выходу бис-алкилированного CMX212. В следующем варианте осуществления, стадия 2A методики A приводит к менее, чем 1% выходу бис-алкилированного CMX212. В следующем варианте осуществления стадия 2A методики A приводит к менее чем 0,75% выходу бис-алкилированного CMX212. В еще одном варианте осуществления стадия 2A методики A приводит к менее чем 0,5% выходу бис-алкилированного CMX212.
В другом варианте осуществления CMX212 образуется с чистотой более 90%, например, более 92,5%, более 95%, более 97,5% или более 99%.
В другом варианте осуществления CMX212 образуется с примесью цитозина не более 10%, например, с примесью цитозина не более 7,5%, с примесью цитозина не более 5%, с примесью цитозина не более 2,5%, с примесью цитозина не более 1%.
В другом варианте осуществления CMX203 образуется с чистотой более 80%, например, более 82,5%, более 85%, более 87,5%, более 90,0%, более 92,5%, более 95%, более 97,5% или более 99%.
В другом варианте осуществления используется алкоксид металла с чистотой более 85%, например, с чистотой более 87,5%, с чистотой более 90%, с чистотой более 92,5%, с чистотой более 95%, с чистотой более 97,5% или с чистотой более 99%.
В другом варианте осуществления алкоксид металла представляет собой ди-трет-бутоксид магния.
В следующем варианте осуществления используется ди-трет-бутоксид магния с чистотой более 85%. Например, с чистотой более 87,5%, с чистотой более 90%, с чистотой более 92,5%, с чистотой более 95%, с чистотой более 97,5% или с чистотой более 99%.
В следующем варианте осуществления алкоксид металла представляет собой ди-трет-бутоксид магния и степень преобразования CMX212 и CMX203 в CMX225 превышает 80%, 85%, 90% или 95%.
В следующем варианте осуществления температура, подходящая для проведения стадии 2A методики A, составляет около 80°C и реакция завершается примерно за 4 часа.
В следующем варианте осуществления водный раствор, используемый для водной экстракции, представляет собой водный раствор HCl.
В следующем варианте осуществления подходящий органический растворитель для экстракции CMX225 представляет собой изопропилацетат.
В следующем варианте осуществления после стадии водной экстракции используется перегонка в вакууме.
В еще одном варианте осуществления растворитель (например, изопропилацетат или ДМФА) меняют на метанол.
Далее, что касается стадии 2B методики A, [[(S)-2-(4-амино-2-оксо-1(2H)-пиримидинил)-1-(гидроксиметил)этокси]метил]моно[3-(гексадецилокси)пропиловый]эфир фосфоновой кислоты (CMX001) получают взаимодействием CMX225 с подходящим реагентом для снятия защиты (например, хлористым водородом, ацетилхлоридом) в подходящем растворителе (например, метаноле) до завершения реакции. CMX001 подвергают перекристаллизации с использованием подходящей системы растворителей (например, метанол:ацетон:вода, этанол, метанол).
В другом варианте осуществления снятие защиты соединения CMX225 осуществляют газообразным хлористым водородом.
В следующем варианте осуществления температуру, при которой проводят реакцию снятия защиты, поддерживают в диапазоне от 0 до 20°C, например, от 5 до 15°C.
В другом варианте осуществления реакционную смесь стадии 2B методики A гасят водой и значение pH доводят примерно до 2,3-2,7, например, до приблизительно 2,5.
В другом варианте осуществления перекристаллизация CMX001 из подходящей системы растворителей приводит к получению вещества с чистотой более 91% (например, >92%, >93%, >94%, >95%, >97,5%, >98%, >99% или >99,5%).
В другом варианте осуществления перекристаллизация CMX001 из подходящей системы растворителей позволяет получить форму A. Предпочтительно, форма A имеет чистоту более 91% (например, >92%, >93%, >94%, >95%, >97,5%, >98%, >99% или >99,5%).
В одном из вариантов осуществления форма A не является гидратом.
В другом варианте осуществления форма A является сольватом, например, сольватом с метанолом, сольватом с этанолом или сольватом с изопропанолом.
В следующем варианте осуществления форма A является нестехиометрическим сольватом, например, сольватом с метанолом, сольватом с этанолом или сольватом с изопропанолом.
В следующем варианте осуществления форма A является десольватированным сольватом, например, десольватированным сольватом с метанолом, десольватированным сольватом с этанолом или десольватированным сольватом с изопропанолом.
В другом варианте осуществления перекристаллизация CMX001 из подходящей системы растворителей приводит к получению вещества с чистотой >99% AUC ВЭЖХ (по площади под кривой на хроматограмме ВЭЖХ).
В другом варианте осуществления в синтезе CMX001 не используется колоночная хроматография.
В другом варианте осуществления CMX001 выделяют в форме свободной кислоты.
В другом варианте осуществления CMX001 подвергают перекристаллизации из метанола.
В другом варианте осуществления CMX001 подвергают перекристаллизации и выделяют из метанола при температуре не ниже 20°C.
В другом варианте осуществления выполнение стадий 2A и 2B методики A приводит к получению менее 5% N4-алкилированного CMX001. В следующем варианте осуществления выполнение стадий 2A и 2B методики A приводит к получению менее 4% N4-алкилированного CMX001. В следующем варианте осуществления выполнение стадий 2A и 2B методики A приводит к получению менее 3% N4-алкилированного CMX001. В следующем варианте осуществления выполнение стадий 2A и 2B методики A приводит к получению менее 2% N4-алкилированного CMX001. В следующем варианте осуществления выполнение стадий 2A и 2B методики A приводит к получению менее 1,5% N4-алкилированного CMX001. В следующем варианте осуществления выполнение стадий 2A и 2B методики A приводит к получению менее 1% N4-алкилированного CMX001. В следующем варианте осуществления выполнение стадий 2A и 2B методики A приводит к получению менее 0,75% N4-алкилированного CMX001. В следующем варианте осуществления выполнение стадий 2A и 2B методики A приводит к получению менее 0,5% N4-алкилированного CMX001. В следующем варианте осуществления выполнение стадий 2A и 2B методики A приводит к получению менее 0,4% N4-алкилированного CMX001. В еще одном варианте осуществления выполнение стадий 2A и 2B методики A приводит к получению менее 0,3% N4-алкилированного CMX001.
В другом варианте осуществления на стадии 2A вместо трет-бутоксида магния используют другой алкоксид металла (например, трет-бутоксид калия), и содержание N4-алкилированного CMX001 оказывается существенно выше (например, по меньшей мере в пять раз выше), чем при использовании трет-бутоксида магния.
Методика B: натриевая соль моно[3-(гексадецилокси)пропилового]эфира P-[[[(4-метилфенил)сульфонил]окси]метил]фосфоновой кислоты (CMX203)
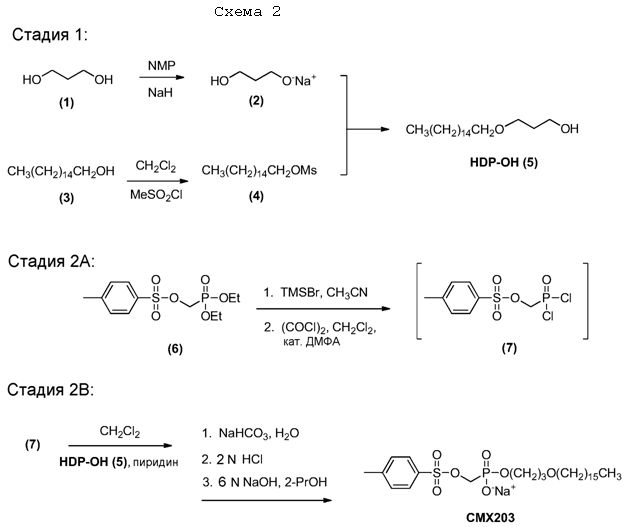
Стадия 1: Синтез 3-(гексадецилокси)пропан-1-ола (5)
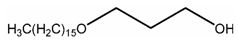
Гексадецил метансульфонат (4) получают взаимодействием 1-гексадеканола (3) с метансульфонилхлоридом в присутствии подходящего основания, такого как амин (например, диизопропилэтиламин), в подходящем растворителе (например, дихлорметане) при подходящей температуре реакции (например, от температуры ниже комнатной до 30°C) до завершения реакции, обычно от 0,5 до четырех часов, например, от одного до двух часов. 3-(Гексадецилокси)пропан-1-ол (5) получают взаимодействием 1,3-пропандиола (1) с соединением (4) в присутствии подходящего основания, такого как гидрид металла (например, гидрид натрия), в подходящем растворителе (например, N-метилпирролидиноне (NMP)) при подходящей температуре реакции (например, при температуре от комнатной до повышенной) до завершения реакции, обычно в течение от 12 до 28 часов.
Стадии 2A и 2B: Синтез натриевой соли моно[3-(гексадецилокси)пропилового]эфира P-[[[(4-метилфенил)сульфонил]окси]метил]фосфоновой кислоты (CMX203)
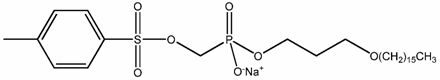
CMX203 получают взаимодействием диэтил (тозилокси)метилфосфоната (6) с бромтриметилсиланом в подходящем растворителе (например, ацетонитриле) при подходящей температуре реакции (например, при температуре от комнатной до повышенной) до завершения реакции, обычно от одного до четырех часов, например, от одного до двух часов. Полученную смесь подвергают взаимодействию с галогенирующим агентом (например, оксалилхлоридом) в подходящем растворителе (например, дихлорметане) в присутствии подходящего катализатора (например, N,N-диметилформамида) при подходящей температуре (например, при комнатной температуре) до завершения реакции, обычно от 8 до 20 часов, например, от 12 до 16 часов. Полученный (дихлорфосфорил)метил 4-метилбензолсульфонат (7) подвергают взаимодействию с (гексадецилокси)пропан-1-олом (5) в подходящем растворителе (например, дихлорметане) до завершения реакции. Диэтил (тозилокси)метилфосфонат является коммерчески доступным от Lacamas Laboratories (Portland, OR).
Другим предпочтительным вариантом осуществления настоящего изобретения является методика B. Эта методика относится к усовершенствованному способу получения натриевой соли моно[3-(гексадецилокси)пропилового]эфира P-[[[(4-метилфенил)сульфонил]окси]метил]фосфоновой кислоты (CMX203). Далее по тексту будут описаны стадии 1, 2A и 2B.
Что касается стадии 1 методики B, то 3-(гексадецилокси)пропан-1-ол (5) получают взаимодействием 1,3-пропандиола (1) с гексадецил метансульфонатом (4) в присутствии подходящего основания, такого как гидрид металла (например, гидрид натрия), в подходящем растворителе (например, NMP) при подходящей температуре реакции (например, при температуре от комнатной до повышенной) до завершения реакции.
В другом варианте осуществления 3-(гексадецилокси)пропан-1-ол (5) подвергают перекристаллизации из ацетонитрила.
В следующем варианте осуществления используют гексадеканол (3) с высокой чистотой. Например, гексадеканол (3) имеет чистоту более 95%, чистоту более 96%, чистоту более 97%, чистоту более 98%, чистоту более 99% или чистоту более 99,5%.
В следующем варианте осуществления используется NMP с высокой чистотой. Конкретно, NMP не включает химическую примесь бутиролактона. Например, NMP имеет чистоту более 95%, чистоту более 96%, чистоту более 97%, чистоту более 98%, чистоту более 99% или чистоту более 99,5%.
После выполнения стадии 1 осуществляют стадию 2A методики B, на которой получают промежуточный продукт (дихлорфосфорил)метил 4-метилбензолсульфонат (7) путем взаимодействия диэтил (тозилокси)метилфосфоната (6) с бромтриметилсиланом в подходящем растворителе (например, ацетонитриле) при подходящей температуре реакции (например, при температуре от комнатной до повышенной) до завершения реакции. Полученную смесь подвергают взаимодействию с галогенирующим агентом (например, оксалилхлоридом) в подходящем растворителе (например, дихлорметане) в присутствии подходящего катализатора (например, N,N-диметилформамида) при подходящей температуре (например, комнатной температуре) до завершения реакции.
После этого осуществляют стадию 2B методики B, на которой CMX203 получают взаимодействием соединения (7) с соединением (5) в подходящем растворителе (например, дихлорметане) с добавлением пиридина при подходящей температуре (например от -5 до 5°C) до завершения реакции. Полученную смесь гасят подходящим растворителем (например, водой). Перед выделением продукта к реакционной смеси добавляют насыщенный раствор бикарбоната натрия и доводят pH до 2,0 кислотой (например, хлористоводородной кислотой), и образуется CMX203 в форме свободной кислоты. Затем органический слой отделяют, концентрируют и после этого растворяют в подходящем растворителе (например, 2-пропаноле) и добавляют гидроксид натрия, чтобы преобразовать свободную кислоту в CMX203. CMX203 собирают в виде осадка. В предпочтительных способах очистка колоночной хроматографией не является необходимой. Например, органический слой, отделенный после регулирования значения pH, не требует очистки колоночной хроматографией.
В другом варианте осуществления стадия 2B методики B включает: гашение реакционной смеси гасящим реагентом (например, бикарбонатом натрия); и доведение значения pH до 2 кислотой (например, хлористоводородной кислотой) с последующим отделением слоя, содержащего CMX203 в форме свободной кислоты.
В другом варианте стадия 2B методики B включает использование в качестве растворителя дихлорметана, а не другого растворителя (например, диэтилового эфира).
При синтезе CMX203 в качестве побочного продукта образуется тозилоксиметилфосфоновая кислота (“CMX247”), и ее удаляют при перекристаллизации из 2-пропанола или одной из систем растворителей, описанных в примере 5.
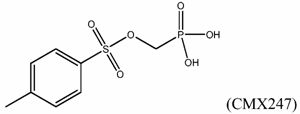
В другом варианте осуществления CMX203 подвергают перекристаллизации из 2-пропанола. В другом варианте осуществления CMX203 подвергают перекристаллизации из системы растворителей, описанной в примере 5.
В другом варианте осуществления перекристаллизация CMX203 из подходящей системы растворителей обеспечивает получение вещества с чистотой ≥99%.
В другом варианте осуществления перекристаллизация CMX203 из подходящей системы растворителей обеспечивает получение продукта с содержанием CMX247≤1%, например, ≤0,5%, ≤0,25%, ≤0,1% или ≤0,01%.
В другом варианте осуществления изобретение относится к композициям (например, пероральным дозированным формам) с желаемыми фармакокинетическими характеристиками. Кроме того, эти композиции обеспечивают возможность того, чтобы метаболизм соединения формулы (I), имеющего чистоту более 91% или находящегося в форме A, протекал так, чтобы содержание метаболита (т.е. цидофовира) в крови оставалось ниже того уровня, при котором возникает нефротоксичность.
Настоящее изобретение позволяет получать соединения с высокой степенью чистоты или в определенной морфологической форме (например, форме A), композиции, описанные в настоящей заявке, и обеспечивает способы лечения или профилактики одной или нескольких вирусных инфекций у субъекта, например, иммунодефицитного субъекта.
Иммунодефицитные субъекты включают реципиентов трансплантированных органов, пациентов, которым осуществляют гемодиализ, пациентов с раковыми заболеваниями, пациентов, принимающих иммуносупрессоры, и ВИЧ-инфицированных пациентов. Настоящее изобретение охватывает терапевтическое и/или профилактическое лечение иммунодефицитных субъектов, а также субъектов, для которых существует опасность стать иммунодефицитными, но у которых не проявляются симптомы иммунодефицита. Примеры субъектов, у которых имеется риск развития иммунодефицита, включают, не ограничиваясь только ими, субъектов, принимающих иммуносупрессоры или химиотерапевтические средства, субъектов, у которых имеются раковые заболевания, а также субъектов, инфицированных ВИЧ.
Фармацевтические композиции
Настоящее изобретение относится также к фармацевтическим композициям, содержащим соединение формулы (I) или (II) в комбинации с по меньшей мере одним фармацевтически приемлемым эксципиентом или носителем.
«Фармацевтическая композиция» представляет собой состав, содержащий соединение по настоящему изобретению в форме, подходящей для введения субъекту. В одном из вариантов осуществления фармацевтическая композиция находится в насыпной форме или в дозированной форме. Дозированная форма является любой из широкого круга форм, таких как капсулы, IV мешки, таблетки, насос для подачи разовой дозы на аэрозольном ингаляторе или флаконе. Количество действующего ингредиента (например, состава раскрытого в изобретении соединения или его соли, гидрата, сольвата или изомера) в дозированной форме композиции является эффективным количеством и меняется в зависимости от конкретного вида лечения, в котором применяется эта дозированная форма. Специалист в данной области поймет, что в некоторых случаях необходимо осуществлять стандартные изменения дозировки в зависимости от возраста и состояния пациента. Дозировка будет также зависеть от пути введения. В изобретении рассматриваются различные пути введения, включая пероральный, легочный, ректальный, парентеральный, трансдермальный, подкожный, внутривенный, внутримышечный, интраперитонеальный, ингаляционный, буккальный, сублингвальный, интраплевральный, интратекальный, интраназальный и т.п. Дозированные формы для местного или трансдермального введения соединения по настоящему изобретению включают порошки, спреи, мази, пасты, кремы, лосьоны, гели, растворы, пластыри и устройства для ингаляции. В одном из вариантов осуществления действующее соединение в стерильных условиях смешивают с фармацевтически приемлемым носителем и с любыми консервантами, буферными веществами или пропеллентами, которые необходимо добавить в состав.
В настоящем описании фраза «фармацевтически приемлемый» относится к тем соединениям, материалам, композициям, носителям и/или дозированным формам, которые с точки зрения обоснованного мнения медицины подходят для применения в контакте с тканями людей и животных, не проявляя избыточной токсичности, раздражающего действия, аллергических реакций или других проблем или осложнений, и соответствуют разумному соотношению польза/риск.
«Фармацевтически приемлемый эксципиент или носитель» означает эксципиент или носитель, который применим для получения фармацевтической композиции, которая является в основном безопасной, нетоксичной и не является нежелательной ни с биологической, ни с какой-либо другой точки зрения, и этот термин включает эксципиенты, которые подходят для ветеринарного применения, а также для фармацевтического применения в лечении людей. Термин «фармацевтически приемлемый эксципиент» в настоящем описании и формуле изобретения включает как один, так и более чем один такой эксципиент.
Фармацевтическая композиция по настоящему изобретению имеет такой состав, который подходит для намеченного пути введения. Примеры путей введения включают парентеральный, например, внутривенный, интрадермальный, подкожный, пероральный (например, ингаляционный), трансдермальный (местный) и трансмукозальный пути введения. Растворы или суспензии, используемые для парентерального, интрадермального или подкожного применения, могут включать следующие компоненты: стерильный разбавитель, такой как вода для инъекций, солевой раствор, нелетучие масла, полиэтиленгликоли, глицерин, пропиленгликоль или другие синтетические растворители; антибактериальные агенты, такие как бензиловый спирт или метилпарабены; антиоксиданты, такие как аскорбиновая кислота или бисульфит натрия; хелатообразующие агенты, такие как этилендиаминтетрауксусная кислота; буферные средства, такие как ацетаты, цитраты или фосфаты, а также средства для регулирования тоничности, такие как хлорид натрия или декстроза. Значение pH можно регулировать добавлением кислот или оснований, например, хлористоводородной кислоты или гидроксида натрия. Парентеральный препарат можно помещать в ампулы, одноразовые шприцы и флаконы, содержащие несколько доз, изготовленные из стекла или пластмассы.
Термин «терапевтически эффективное количество» в настоящем описании относится к количеству фармацевтического агента, достаточному, чтобы вылечить, облегчить или предотвратить выявленное заболевание или состояние, или для того, чтобы проявить заметное терапевтическое или ингибирующее действие. Это действие можно обнаружить любым способом анализа, известным в технике. Точное значение эффективного количества для субъекта будет зависеть от массы тела и размеров субъекта, и его здоровья; природы и тяжести состояния, подвергаемого лечению; а также терапевтического средства или комбинации терапевтических средств, выбранных для введения. Терапевтически эффективное количество для данной ситуации можно определить с помощью стандартных экспериментов, которые находятся в пределах навыков и компетенции клинициста. В предпочтительном аспекте подвергаемое лечению заболевание или состояние представляет собой вирусную инфекцию.
Для любого соединения терапевтически эффективное количество можно первоначально определить либо с помощью исследований на клеточных культурах, например, клетках новообразований, либо на животных моделях, обычно крысах, мышах, кроликах, собаках или свиньях. Кроме того, животные модели могут быть использованы для определения подходящей концентрации и пути введения. Эту информацию затем можно использовать для определения подходящих доз и путей введения людям. Терапевтическую/профилактическую эффективность и токсичность, например, ED50 (дозу, которая проявляет терапевтическую эффективность для 50% популяции) и LD50 (дозу, которая оказывается летальной для 50% популяции), можно определить по стандартным фармацевтическим методикам на клеточных культурах или экспериментальных животных. Соотношение между дозами, проявляющими токсический и терапевтический эффекты, именуют терапевтическим индексом, и этот индекс можно выразить как отношение LD50/ED50. Предпочтительными являются фармацевтические композиции, которые демонстрируют высокие значения терапевтического индекса. Дозировка может меняться в указанном диапазоне в зависимости от применяемой дозированной формы, восприимчивости пациента и пути введения.
Дозировку и путь введения подбирают таким образом, чтобы обеспечить желаемые уровни действующего агента (агентов) или чтобы поддержать желаемый эффект. Факторы, которые можно при этом принимать во внимание, включают тяжесть болезненного состояния, общее состояние здоровья субъекта, его возраст, массу тела и пол, рацион питания, время и частоту введения, комбинацию (комбинации) лекарственных средств, чувствительность реакции и переносимость/восприимчивость к терапии. Фармацевтические композиции длительного действия можно вводить каждые 3-4 дня, каждую неделю или раз в две недели, в зависимости от периода полужизни и скорости выведения конкретного состава.
Фармацевтические композиции, содержащие действующие соединения по настоящему изобретению, можно изготавливать общеизвестными способами, например, путем обычного смешивания, растворения, гранулирования, изготовления драже, отмучивания, эмульгирования, инкапсулирования, включения в носитель или лиофилизации. Фармацевтические композиции можно получать стандартным способом с использованием одного или нескольких фармацевтически приемлемых носителей, в том числе, эксципиентов и/или вспомогательных компонентов, которые облегчают переработку действующих соединений в готовые препараты, и которые могут быть использованы в фармацевтических целях. Подходящий состав безусловно будет зависеть от выбранного пути введения.
Фармацевтические композиции, подходящие для применения в форме инъекций, включают стерильные водные растворы (в случае, если компоненты состава растворимы в воде) или дисперсии, а также стерильные порошки для получения стерильных растворов или дисперсий для немедленного введения. В случае внутривенного введения, подходящие носители включают физиологический солевой раствор, бактериостатическую воду, Cremophor EL™ (BASF, Parsippany, N.J.) или фосфатный буферный солевой раствор (PBS). Во всех случаях композиция должна быть стерильной и должна иметь такую текучесть, чтобы ее можно было легко вводить с помощью шприца. Она должна сохранять устойчивость в условиях производства и хранения, и ее необходимо защитить от загрязняющего действия микроорганизмов, таких как бактерии и грибки. Носитель может представлять собой растворитель или дисперсионную среду, содержащую, например, воду, этанол, полиолы (например, глицерин, пропиленгликоль и жидкий полиэтиленгликоль и т.п.), а также их подходящую смесь. Может поддерживаться необходимая текучесть, например, путем использования покрытия, такого как лецитин, сохранением требуемого размера частиц в случае дисперсии, а также с помощью ПАВ. Предотвратить действие микроорганизмов можно с помощью различных антибактериальных и противогрибковых агентов, например, парабенов, хлорбутанола, фенола, аскорбиновой кислоты, тимеросала и т.п. Во многих случаях будет предпочтительно включение в композицию изотонических агентов, например, сахаров, полиспиртов, таких как манит, сорбит, хлорида натрия. Продолжительной абсорбции композиций для инъекции можно добиться включением в композицию агента, который замедляет абсорбцию, например, моностеарата алюминия и желатина.
Стерильные растворы для инъекций могут быть получены путем включения действующего соединения, взятого в необходимом количестве, в подходящем растворителе с одним из перечисленных выше ингредиентов или их комбинацией, при необходимости, с последующей стерилизацией фильтрованием. Как правило, дисперсии получают введением действующего соединения в стерильный носитель, который содержит основную дисперсионную среду и другие необходимые ингредиенты из числа перечисленных выше. В случае стерильных порошков для получения стерильных растворов для инъекции, способами их получения являются вакуумная сушка и лиофилизация, что позволяет получить порошок действующего ингредиента в комбинации с любыми дополнительными желаемыми ингредиентами, образовавшимися из их раствора, полученного ранее стерильным фильтрованием.
Пероральные композиции обычно включают инертный разбавитель или пригодный в пищу фармацевтически приемлемый носитель. Они могут быть заключены в желатиновые капсулы или спрессованы в таблетки. Для перорального терапевтического введения действующее соединение может быть включено в носитель и использовано в форме таблеток, пастилок или капсул. Пероральные композиции можно также получать с использованием жидкого носителя, с целью применения в качестве полоскания для рта, причем соединение в жидком носителе применяют перорально, полоща рот, и выплевывают или проглатывают. В композицию в качестве ее составной части можно включать фармацевтически приемлемые связующие агенты и/или адъюванты. Таблетки, пилюли, капсулы, пастилки и т.п. могут содержать любой из следующих ингредиентов или соединений сходной природы: связующий компонент, такой как микрокристаллическая целлюлоза, смола трагаканта или желатин; эксципиент, такой как крахмал или лактоза, дезинтегрирующий агент, такой как альгиновая кислота, Primogel или кукурузный крахмал; смазывающее средство, такое как стеарат магния или Sterotes; средство для улучшения скольжения, такое как коллоидный диоксид кремния; подсластитель, такой как сахароза или сахарин; или вкусовое средство, такое как перечная мята, метилсалицилат или апельсиновая добавка.
Для введения с помощью ингаляции соединения доставляют в форме аэрозоля из контейнера, находящегося под давлением, или распылителя, который содержит подходящий пропеллент, например, газ, такой как диоксид углерода, или ингалятора.
Системное введение можно также осуществлять трансмукозальным или трансдермальным путем. С целью трансмукозального или трансдермального введения в составе используются проникающие средства (пенетранты), которые подходят для того барьера, который необходимо преодолеть. Такие пенетранты широко известны в технике и включают, например, в случае трансмукозального введения, детергенты, соли желчных кислот и производные фусидовой кислоты. Трансмукозальное введение можно осуществить с помощью назальных спреев или суппозиториев. Для трансдермального введения действующие соединения включают в состав мазей, бальзамов, гелей или кремов, которые хорошо известны в технике.
Действующие соединения можно включать в состав вместе с фармацевтически приемлемыми носителями, которые будут препятствовать быстрому выведению соединения из организма, как в случае составов с регулируемым высвобождением, в число которых входят имплантаты и микроинкапсулированные системы доставки. Можно применять биоразрушаемые, биосовместимые полимеры, такие как этиленвинилацетат, полиангидриды, полигликолевая кислота, коллаген, полиортоэфиры и полимолочная кислота. Способы получения таких составов должны быть очевидны специалисту в данной области техники. Эти продукты можно также приобрести у Alza Corporation и Nova Pharmaceuticals, Inc. Липосомальные суспензии (включая липосомы, нацеленные на инфицированные клетки с моноклональными антителами против вирусных антигенов) также могут быть использованы в качестве фармацевтически приемлемых носителей. Их можно получать способами, известными специалистам в данной области техники, например, описанными в патенте США 4522811.
Особенно предпочтительно получать пероральные или парентеральные композиции в виде дозированных форм, для легкости введения и однородности дозировки. Термин «дозированная лекарственная форма» в настоящей заявке относится к физически дискретным единицам, предназначенным для введения разовой дозы субъекту, подвергаемому лечению; причем каждая единица содержит заранее определенное количество действующего соединения, рассчитанное на получение желаемого терапевтического эффекта, в сочетании с необходимым фармацевтическим носителем. Конкретные характеристики дозированных лекарственных форм по настоящему изобретению определяются и непосредственно зависят от индивидуальных характеристик действующего соединения и конкретного терапевтического эффекта, которого необходимо добиться.
При терапевтическом применении дозировки фармацевтических композиций по настоящему изобретению меняются в зависимости от лекарственного средства, возраста, массы и клинического состояния принимающего их пациента, причем опыт и мнение клинициста или практикующего врача, назначающего терапию, среди прочих факторов влияет на выбор дозировки. Дозировки могут находиться в пределах от примерно 0,01 мг/кг до примерно 100 мг/кг. В предпочтительных аспектах дозировки могут находиться в диапазоне от примерно 0,1 мг/кг до примерно 10 мг/кг. В одном из аспектов вводимая доза будет находиться в пределах от примерно 1 мг до примерно 1 г; от примерно 10 мг до примерно 500 мг; от примерно 20 мг до примерно 400 мг; от примерно 40 мг до примерно 400 мг; или от примерно 50 мг до примерно 400 мг, в виде одной дозы, нескольких доз или при непрерывном введении (где указанная доза может быть отрегулирована с учетом массы пациента в кг, поверхности тела в м2 и возраста в годах). В некоторых вариантах осуществления количество действующего соединения на единицу дозированной форы может составлять от 0,1 мг до примерно 1000 мг, например, приблизительно 0,1, 0,5, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 15, 20, 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95 или 100 мг или более. В одном из вариантов осуществления это количество может составлять примерно 20 мг. В одном из вариантов осуществления это количество может составлять примерно 50 мг.
В другом варианте осуществления изобретение относится к композициям (например, фармацевтическим композициям) с желаемыми фармакокинетическими характеристиками. Например, композиции по настоящему изобретению могут обеспечивать такое содержание соединения формулы (I) в крови, которое, после метаболизма до терапевтически активной формы (т.е. цидофовира), приводит к содержанию метаболита, которое не вызывает токсичности (например, нефротоксичности).
Эффективное количество фармацевтического агента представляет собой такое количество, которое обеспечивает улучшение, объективно обнаруживаемое клиницистом или другим квалифицированным наблюдателем. В настоящем описании термин «эффективная дозировка» относится к количеству действующего соединения, которое вызывает желаемый биологический эффект у субъекта или в клетке.
В другом варианте осуществления CMX001 или другую композицию по настоящему изобретению можно вводить субъекту в виде единственной дозы. В другом варианте осуществления CMX001 или другую композицию по настоящему изобретению можно вводить субъекту в виде многократных доз. Многократные дозы можно вводить регулярно, например, каждые 12 часов, каждый день, каждые 2 дня, каждые 3 дня, каждые 4 дня, каждые 5 дней, каждые 6 дней, каждые 7 дней, каждые 8 дней, каждые 9 дней, каждые 10 дней, каждые 11 дней, каждые 12 дней, каждые 13 дней, каждые 14 дней или каждые 15 дней. Например, введение можно осуществлять два раза в неделю. Кроме того, при каждом отдельном введении можно применять ту же самую или другую дозу.
Например, субъекту можно вводить первую дозу в 2 мг/кг и затем одну или несколько дополнительных доз по 2 мг/кг. Например, субъекту можно вводить первую дозу 2 мг/кг и затем одну или несколько дополнительных доз по 1 мг/кг. Например, субъекту можно вводить первую дозу 2 мг/кг и затем одну или несколько дополнительных доз по 3 мг/кг. Например, субъекту можно вводить первую дозу 4 мг/кг и затем одну или несколько дополнительных доз по 4 мг/кг.
Введение многократных доз можно осуществлять через различные промежутки времени. Например, первые 2, 3, 4, 5, 6, 7 или 8 или более доз можно вводить с интервалом в 6 дней, а остальные дозы вводить с интервалом в 7 дней. Например, первые 2, 3, 4, 5, 6, 7 или 8 или более доз можно вводить с интервалом в 7 дней, а остальные дозы вводить с интервалом в 3 дня.
В другом варианте осуществления изобретение относится к пероральной дозированной форме, содержащей соединение формулы (I), имеющее чистоту более 91% или находящееся в форме A, и предназначенной для терапевтического и/или профилактического лечения вирусной инфекции у субъекта, где указанная пероральная дозированная форма, при введении человеку в дозе 2 мг/кг указанного соединения, обеспечивает AUC0-inf указанного соединения от примерно 2000 до примерно 4000 ч*нг/мл, например, от приблизительно 2500 до приблизительно 3000 ч*нг/мл. В некоторых вариантах осуществления AUC0-inf указанного соединения составляет примерно 2000, 2100, 2200, 2300, 2400, 2500, 2600, 2700, 2800, 2900, 3000, 3100, 3200, 3300, 3400, 3500, 3600, 3700, 3800, 3900 или 4000 ч*нг/мл или находится в любом диапазоне в указанных пределах. AUC0-inf можно определить любым из способов, хорошо известных в технике, и как описано в примерах настоящей заявки.
В другом варианте осуществления изобретение относится к пероральной дозированной форме, содержащей соединение формулы (I), имеющее чистоту более 91% или находящееся в форме A, предназначенной для терапевтического и/или профилактического лечения вирусной инфекции у субъекта, где указанная пероральная дозированная форма, при введении человеку в дозе 2 мг/кг указанного соединения, обеспечивает Cmax указанного соединения от примерно 100 до примерно 500 нг/мл, например, от приблизительно 200 до приблизительно 400 нг/мл. В некоторых вариантах осуществления Cmax указанного соединения составляет примерно 100, 110, 120, 130, 140, 150, 160, 170, 180, 190, 200, 210, 220, 230, 240, 250, 260, 270, 280, 290, 300, 310, 320, 330, 340, 350, 360, 370, 380, 390, 400, 410, 420, 430, 440, 450, 460, 470, 480, 490 или 500 нг/мл, или находится в любом диапазоне в указанных пределах. Cmax можно определить любым из способов, хорошо известных в технике, и как описано в примерах настоящей заявки.
В другом варианте осуществления изобретение относится к пероральной дозированной форме, содержащей соединение формулы (I), имеющее чистоту более 91% или находящееся в форме A, предназначенной для терапевтического и/или профилактического лечения вирусной инфекции у субъекта, где указанная пероральная дозированная форма, при введении человеку в дозе 2 мг/кг указанного соединения формулы (I) и метаболизме указанного соединения формулы (I) до цидофовира, обеспечивает Cmax указанного цидофовира, которая составляет менее примерно 30% Cmax указанного соединения формулы (I), например, менее примерно 20% Cmax указанного соединения формулы (I). В некоторых вариантах осуществления Cmax метаболита (т.е. цидофовира) составляет менее примерно 50%, 45%, 40%, 35%, 30%, 25%, 20%, 15% или 10% Cmax соединения формулы (I).
В другом варианте осуществления изобретение относится к пероральной дозированной форме, содержащей соединение формулы (I), имеющее чистоту более 91% или находящееся в форме A, где введение человеку дозы 2 мг/кг указанного соединения формулы (I) обеспечивает AUC0-inf цидофовира от примерно 1000 до примерно 5000 ч*нг/мл, например, от приблизительно 1500 до приблизительно 4000 ч*нг/мл. В некоторых вариантах осуществления AUC0-inf цидофовира составляет примерно 1000, 1100, 1200, 1300, 1400, 1500, 1600, 1700, 1800, 1900, 2000, 2100, 2200, 2300, 2400, 2500, 2600, 2700, 2800, 2900, 3000, 3100, 3200, 3300, 3400, 3500, 3600, 3700, 3800, 3900, 4000, 4100, 4200, 4300, 4400, 4500, 4600, 4700, 4800, 4900 или 5000 ч*нг/мл или находится в любом диапазоне в указанных пределах.
В другом варианте осуществления изобретение относится к пероральной дозированной форме, содержащей соединение формулы (I), имеющее чистоту более 91% или находящееся в форме A, где введение человеку дозы 2 мг/кг указанного соединения формулы (I) обеспечивает Cmax цидофовира от примерно 10 до примерно 100 нг/мл, например, от приблизительно 20 до приблизительно 70 нг/мл. В некоторых вариантах осуществления Cmax соединения формулы (I) составляет примерно 10, 20, 30, 40, 50, 60, 70, 80, 90 или 100 нг/мл или находится в любом диапазоне в указанных пределах.
В некоторых вариантах осуществления пероральная дозированная форма обеспечивает более чем одну из указанных выше фармакокинетических характеристик, например, AUC0-inf или Cmax соединения формулы (I) или метаболита (т.е. цидофовира), или отношение Cmax метаболита (т.е. цидофовира) к соединению формулы (I), например, 2, 3, 4 или более фармакокинетических характеристик в любой комбинации.
Фармакокинетическое поведение композиции будет до некоторой степени меняться от одного субъекта к другому в пределах популяции субъектов. Приведенные выше значения для композиций по настоящему изобретению основаны на средних характеристиках в популяции. Имеется в виду, что настоящее изобретение охватывает композиции, которые в среднем попадают в приведенные диапазоны, хотя предполагается, что некоторые субъекты могут выходить за пределы указанных диапазонов.
Фармацевтические композиции по настоящему изобретению могут быть помещены в контейнер, упаковку или дозатор совместно с инструкциями для введения.
Соединения по настоящему изобретению, кроме того, способны образовывать соли. Все эти формы также входят в объем заявленного изобретения.
В настоящем описании термин «фармацевтически приемлемые соли» относится к производным соединений по настоящему изобретению, в которых исходное соединение модифицировано путем получения его соли с кислотой или основанием. Примеры фармацевтически приемлемых солей включают, не ограничиваясь ими, соли, образованные минеральными или органическими кислотами и основными остатками, например аминами, соли, образованные щелочами или органическими основаниями и кислотными остатками, например, карбоновыми кислотами и т.п. Фармацевтически приемлемые соли включают обычные нетоксичные соли или четвертичные аммониевые соли исходных соединений, образованные, например, нетоксичными неорганическими или органическими кислотами. Например, такие обычные нетоксичные соли включают, не ограничиваясь ими, соли, полученные из неорганических или органических кислот, выбранных из 2-ацетоксибензойной, 2-гидроксиэтансульфоновой, уксусной, аскорбиновой, бензолсульфоновой, бензойной, бикарбоновой, угольной, лимонной, этилендиаминтетрауксусной, этандисульфоновой, 1,2-этансульфоновой, фумаровой, глюкогептоновой, глюконовой, глутаминовой, гликолевой, гликолиларсаниловой, гексилрезорциновой, гидрабаминовой, бромистоводородной, хлористоводородной, йодистоводородной, гидроксималеиновой, гидроксинафтойной, изетионовой, молочной, лактобионовой, лаурилсульфоновой, малеиновой, яблочной, миндальной, метансульфоновой, напсиловой, азотной, щавелевой, памовой, пантотеновой, фенилуксусной, фосфорной, полигалактуроновой, пропионовой, салициловой, стеариновой, субуксусной, янтарной, сульфаминовой, сульфаниловой, серной, дубильной, винной, толуолсульфоновой и общераспространенных аминокислот, например, глицина, аланина, фенилаланина, аргинина и т.д.
Другие примеры фармацевтически приемлемых солей включают соли гексановой (капроновй) кислоты, циклопентанпропионовой кислоты, пировиноградной кислоты, малоновой кислоты, 3-(4-гидроксибензоил)бензойной кислоты, коричной кислоты, 4-хлорбензолсульфоновой кислоты, 2-нафталинсульфоновой кислоты, 4-толуолсульфоновой кислоты, камфорсульфоновой кислоты, 4-метилбицикло-[2.2.2]-окт-2-ен-1-карбоновой кислоты, 3-фенилпропионовой кислоты, триметилуксусной кислоты, трет-бутилуксусной кислоты, муконовой кислоты и т.п. Настоящее изобретение охватывает также соли, образованные в случае, если в исходном соединении присутствует кислотный протон, и он либо замещен ионом металла, например, ионом щелочного металла, ионом щелочноземельного металла или ионом алюминия; либо координирован с органическим основанием, таким как этаноламин, диэтаноламин, триэтаноламин, трометамин, N-метилглюкамин, диэтиламин, диэтиламиноэтанол, этилендиамин, имидазол, лизин, аргинин, морфолин, 2-гидроксиэтилморфолин, дибензилэтилендиамин, триметиламин, пиперидин, пирролидин, бензиламин, гидроксид тетраметиламмония и т.п.
Следует понимать, что все ссылки на фармацевтически приемлемые соли включают формы, содержащие растворитель (сольваты), или различные кристаллические формы (полиморфы) указанной соли, определенные в настоящем описании.
Соединения по настоящему изобретению можно также получать в виде сложных эфиров, например, фармацевтически приемлемых сложных эфиров. Например, функциональную группу карбоновой кислоты в соединении можно преобразовать в соответствующий сложный эфир, например, метиловый, этиловый или другой сложный эфир. Кроме того, спиртовую группу соединения можно преобразовать в соответствующий сложный эфир, например, ацетат, пропионат или другой сложный эфир.
Соединения по настоящему изобретению можно также получать в виде пролекарств, например, фармацевтически приемлемых пролекарств. Термин «пролекарство» используется в настоящей заявке для обозначения соединения, которое высвобождает исходное действующее лекарственное соединение in vivo. Известно, что поскольку пролекарства улучшают многочисленные желаемые качества фармацевтических препаратов (например, растворимость, биодоступность, легкость производства и т.д.), соединения по настоящему изобретению можно доставлять в форме пролекарств. Таким образом, имеется в виду, что настоящее изобретение охватывает пролекарства заявленных соединений, способы их доставки и содержащие их композиции. Имеется в виду, что «пролекарства» включают любые ковалентно связанные носители, которые высвобождают исходное действующее лекарственное средство по настоящему изобретению in vivo, если это пролекарство вводят субъекту. Пролекарства в настоящем изобретении получают модификацией функциональных групп, присутствующих в соединении, таким образом, чтобы модифицированные фрагменты расщеплялись либо при стандартной обработке, либо in vivo, образуя исходное соединение. Пролекарства включают соединения по настоящему изобретению, в которых гидрокси, амино, сульфгидрильная, карбокси или карбонильная группа связаны с любой группой, которую можно отщепить in vivo с образованием свободного гидроксила, свободной аминогруппы, свободного сульфгидрила, свободной карбоксигруппы или свободной карбонильной группы, соответственно.
Примеры пролекарств включают, не ограничиваясь перечисленными, сложные эфиры (например, ацетаты, диалкиламиноацетаты, формиаты, фосфаты, сульфаты и бензоаты) и карбаматы (например, N,N-диметиламинокарбонил), образованные функциональными гидроксигруппами, сложные эфиры (например, этиловые сложные эфиры, эфиры морфолиноэтанола), образованные карбоксильными функциональными группами, N-ацильные производные (например, N-ацетил), N-основания Манниха, основания Шиффа и енаминоны, образованные функциональными аминогруппами, оксимы, ацетали, кетали и эфиры енолов, образованные альдегидными и кетонными функциональными группами соединений по настоящему изобретению и т.п., см. Bundegaard, H., Design of Prodrugs, p1-92, Elsevier, New York-Oxford (1985).
Соединения по настоящему изобретению или их фармацевтически приемлемые соли, сложные эфиры или пролекарства вводят перорально, назально, трансдермально, пульмонально, ингаляционно, буккально, сублингвально, интраперитонеально, подкожно, внутримышечно, внутривенно, ректально, интраплеврально, интратекально и парентерально. В одном из вариантов осуществления соединение вводят перорально. Специалист в данной области техники должен понимать преимущества тех или иных путей введения.
Схему введения соединений по настоящему изобретению выбирают в соответствии с рядом факторов, включая тип, вид, возраст, массу, пол и медицинское состояние пациента; тяжесть состояния, подвергаемого лечению; путь введения; функцию печени и почек пациента; и конкретное применяемое соединение или его соль. Рядовой врач или ветеринар способен легко определить и назначить эффективное количество лекарственного средства, необходимого для предотвращения, противодействия или остановки прогрессирования данного состояния.
Методики получения составов и введения раскрытых соединений по настоящему изобретению можно найти в Remington: the Science and Practice of Pharmacy, 19th edition, Mack Publishing Co., Easton, PA (1995). В одном из вариантов осуществления описанные в заявке соединения и их фармацевтически приемлемые соли используются в фармацевтических препаратах в комбинации с фармацевтически приемлемым носителем или разбавителем. Подходящие фармацевтически приемлемые носители включают твердые инертные наполнители или разбавители, а также стерильные водные или органические растворы. Соединения по настоящему изобретению должны присутствовать в таких фармацевтических композициях в количествах, достаточных для того, чтобы обеспечить желаемые дозировки в указанных выше по тексту диапазонах.
Следует понимать, что способы, раскрытые в настоящем описании, подходят для получения желаемых соединений как в небольших, так и в значительных объемах. В предпочтительных вариантах осуществления способов, описанных в настоящей заявке, фосфонаты можно получать в значительном объеме, например, в объеме, характерном для промышленного производства, а не в экспериментальном/лабораторном объеме. Например, периодический вариант осуществления способов по настоящему изобретению позволяет получать порции фосфоната массой по меньшей мере 1 г, или по меньшей мере 5 г, или по меньшей мере 10 г, или по меньшей мере 100 г, или по меньшей мере 1 кг, или по меньшей мере 100 кг. Кроме того, эти способы дают возможность получать фосфонат, имеющий чистоту по меньшей мере 98% или по меньшей мере 98,5 по данным ВЭЖХ. В предпочтительных вариантах осуществления настоящего изобретения эти продукты получают в результате последовательности реакций, которая не включает очистку с помощью любой формы хроматографии (например, газовой хроматографии, ВЭЖХ, препаративной ЖХ, эксклюзионной хроматографии и т.п.).
Все патенты, заявки на патенты и публикации, упомянутые в настоящем описании, включены в заявку во всей полноте посредством ссылки. Однако если патент, патентная заявка или публикация, содержащие прямые определения, включены в настоящую заявку посредством ссылки, следует понимать, что упомянутые прямые определения применимы только к включенному патенту, патентной заявке или публикации, в которую включены эти определения, а не к остальной части текста настоящей заявки, в частности, к формуле изобретения настоящей заявки.
Следует понимать, что хотя изобретение и было описано в связи с конкретными предпочтительными вариантами его осуществления, предшествующее описание, а также последующие примеры предназначены для иллюстрации, а не для ограничения объема настоящего изобретения. Специалист в данной области техники должен понять, что в изобретении могут быть осуществлены различные изменения и произведена замена на эквиваленты, без выхода за пределы объема изобретения, и, кроме того, другие аспекты преимущества и модификации будут ясны специалисту в той области, к которой относится изобретение.
Все процентные доли и отношения, использованные в настоящей заявке, если не указано иное, относятся к массовым долям и отношениям. Другие особенности и преимущества настоящего изобретения очевидны из различных примеров, приведенных ниже по тексту. Приведенные примеры иллюстрируют различные составные части и методики, применимые при практическом осуществлении настоящего изобретения. Эти примеры не ограничивают заявленное изобретение. На основе настоящего описания специалист в данной области способен выявить и использовать другие составные части и методики, которые подходят для практического воплощения настоящего изобретения.
ПРИМЕРЫ
Если не указано иное, аналитические инструменты и параметры, использованные для исследования соединений, описанных в примерах, являются следующими:
Данные ЯМР регистрировали на приборе Jeol 300, модель JNM-ECP300. Использовали следующие образцы. Образцы CMX212 для ЯМР готовили в D6-ДМСО (~10 мг/мл). Образцы CMX001 для ЯМР готовили в виде насыщенных растворов CMX001 в D3-MeOD.
Данные рентгеноструктурного анализа (XRD) регистрировали на приборе Rigaku Ultima IV с источником излучения CuKα (40 КВ, 44 мА) в диапазоне углов 2-тета от 2 до 50 градусов при скорости сканирования 2 градуса/мин и шаге дискретизации 0,020 градуса. Порошкообразные образцы помещали на стандартные держатели для образцов (стеклянные пластины). При исследовании использовали устройство для вращения образца (60 об/мин).
Для ВЭЖХ использовали прибор Agilent 1100 Series. Подробности проведения анализа были следующими:
Реагенты и материалы
Вода (H2O), хроматографического качества.
Метанол (MeOH), хроматографического качества.
Ацетат аммония, степени чистоты по стандарту ACS.
Динатрия этилендиаминтетраацетат (ЭДТА), степени чистоты по стандарту ACS.
Цитозин эталонный стандарт.
ACC-338.1 (CMX212) эталонный стандарт.
Бис-тритил, стандарт загрязнений.
Шприц, снабженный 0,45-мкм фильтром для шприцев.
Колонка: Phenomenex Synergi Polar-RP, 150 мм × 3 мм, размер частиц 4 мкм.
Получение подвижной фазы
50 мМ буферный раствор:
Смешивали 3,85 г ацетата аммония и 18,6 мг динатрий ЭДТА с 1000 мл воды и перемешивали до растворения твердых веществ. Фильтровали через 0,45-мкм фильтр.
Подвижная фаза A: (35/65) 50 мМ буферный раствор/метанол
Смешивали 350 мл 50 мМ буферного раствора и 650 мл метанола. Тщательно перемешивали и обрабатывали ультразвуком в течение 5 мин для дегазации.
Подвижная фаза B: 100% метанол
Параметры работы
Детектирование: УФ при 274 нм или 225 нм;
Объем впрыска: 5 мкл;
Температура колонки: 30°C;
Скорость потока: 0,8 мл/мин;
Градиент:
Время анализа образца: 35 минут.
Приблизительные значения времени удерживания:
Пример 1
Получение (S)-N1-[(2-гидрокси-3-трифенилметокси)пропил] цитозина (CMX212)
В инертной атмосфере, например, атмосфере азота, при комнатной температуре в реактор помещали (S)-тритил глицидиловый эфир (40,0 кг, 126,4 моль), цитозин (12,8 кг, 115,2 моль), карбонат калия (1,7 кг, 12,3 моль) и безводный N,N-диметилформамид (51,2 кг) и нагревали при 85-95°C в течение 9 часов. Реакционную смесь охлаждали до 60-70°C и гасили толуолом (150,4 кг). Полученную суспензию охлаждали до температуры от -5 до 0°C, фильтровали, промывали толуолом (25,6 кг) и затем промывали ацетоном (3×25,7 кг). Остаток на фильтре суспендировали в ацетоне (128,0 кг) и нагревали примерно при 56°C в течение 30 минут, затем охлаждали до температуры ниже 0°C и фильтровали. Осадок на фильтре промывали ацетоном (25,6 кг) и высушивали в вакууме при 45°C до постоянной массы, получая 32,4 кг (65,8%) CMX212 в виде твердого вещества белого или не совсем белого цвета. Обычная чистота по данным ВЭЖХ (AUC - площадь под кривой) составляла >99% на длине волны 274 нм и >98% на длине волны 225 нм. Спектр 1H-ЯМР соответствовал структуре. Температура плавления=215°C (с разложением).
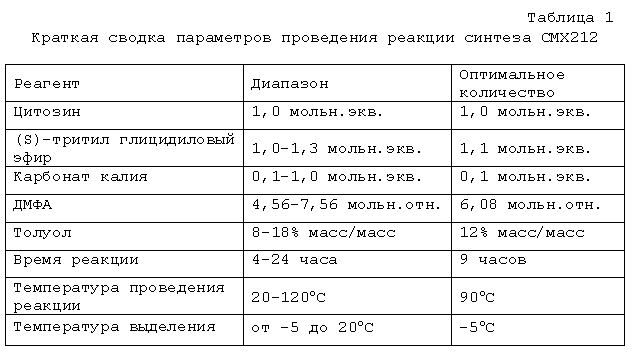
Пример 2
Получение [[(S)-2-(4-амино-2-оксо-1(2H)-пиримидинил)-1-(гидроксиметил)этокси]метил]моно[3-(гексадецилокси)пропилового]эфира фосфоновой кислоты (CMX001)
В атмосфере инертного газа, например азота, при комнатной температуре в реактор помещали CMX212 (14,5 кг, 33,9 моль), CMX203 (21,2 кг, 37,2 моль), ди-трет-бутоксид магния (6,1 кг, 35,7 моль) и N,N-диметилформамид (44,9 кг). Реакционную смесь нагревали при 77-83°C в течение трех-четырех часов. Реакционную смесь концентрировали отгонкой летучих продуктов до тех пор, пока не была удалена приблизительно половина N,N-диметилформамида. Концентрат разбавляли изопропилацетатом (120,1 кг) и объединенную органическую фракцию последовательно промывали 0,5М HCl (примерно 40 галлонов) и насыщенным раствором соли (40 галлонов). Из органической фазы отгоняли изопропилацетат. Концентрат разбавляли метанолом (87,0 кг) и повторно концентрировали для удаления остаточного изопропилацетата. Типичная чистота по данным ВЭЖХ (AUC) неочищенного CMX225 составляла >92% на длине волны 225 нм и >94% на длине волны 274 нм. Концентрат, содержащий неочищенный CMX225, разбавляли метанолом (76,8 кг) и вводили в реактор газообразный хлористый водород (3,8 кг) под поверхность растворителя. (Важно регулировать скорость введения газообразного HCl, поддерживая температуру реакции в пределах от 5 до 15°C). Когда добавление HCl завершалось, температуру реакционной смеси поддерживали ниже 18°C в течение 2 часов и затем фильтровали для удаления нерастворимого вещества. Фильтрат разбавляли водой (30,5 галлона) и доводили pH смеси до 2,3-2,7 1,0 н. гидроксидом натрия. Твердые вещества отделяли фильтрованием и последовательно промывали водой (11,1 галлона) и ацетоном (2×29,0 кг). Осадок на фильтре суспендировали в ацетоне (101,5 кг) при температуре примерно 40°C в течение 1 часа, затем фильтровали и промывали ацетоном (2×29,0 кг). Типичная чистота неочищенного CMX001 по данным ВЭЖХ (AUC) составляла >97% на длине волны 274 нм и >76% на длине волны 225 нм. Неочищенный продукт нагревали при 70°C в метаноле (101,5 кг), получая гомогенный раствор, охлаждали до 15-25°C в течение 2 часов, фильтровали и промывали метанолом (29,2 кг). Типичная чистота по данным ВЭЖХ (AUC) составляла >98% после первой перекристаллизации. Второй раз продукт подвергали перекристаллизации из метанола (75,9 кг), фильтровали, промывали метанолом (29,0 кг) и высушивали в вакууме при 50°C до постоянной массы, получая 14,8 кг (77,7%) CMX001 в виде твердого вещества от не совсем белого до белого цвета. Типичная чистота по данным ВЭЖХ (AUC) составляла >99% (см. фиг.9) Спектр 1H-ЯМР соответствовал структуре (см. фиг.8(a)-(d)).
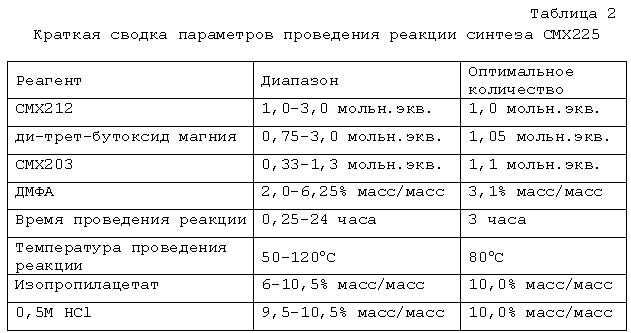
Другие основания, используемые для получения CMX225, включают трет-бутоксид натрия, трет-бутоксид лития, трет-бутоксид калия, гидрид натрия, метоксид натрия и трет-амил алкоксид натрия.
Другие растворители, используемые для получения CMX225, включают: ДМСО, HMPA, DMA и NMP.
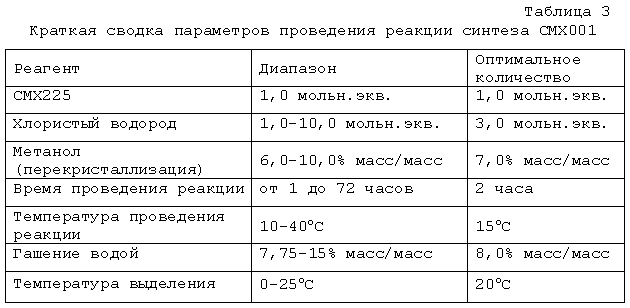
Другие кислоты, используемые для удаления тритильной защитной группы, включают: ацетилхлорид.
Другие растворители, используемые для получения CMX001, включают: дихлорметан.
Другие растворители, используемые для перекристаллизации, включают: метанол:ацетон:вода, этанол, метанол:ацетон, метанол:вода.
Отмечено, что если при сочетании CMX212 и CMX203 использовали трет-бутоксид калия, то содержание образовавшегося N4-алкилированного (или бис-алкилированного) побочного продукта было значительно выше (например, по меньшей мере в пять раз выше), чем при использовании трет-бутоксида магния.
Пример 3
Получение гексадецил метансульфоната (4)
В атмосфере инертного газа, например азота, в реактор помещали 1-гексадеканол 3 (3,78 кг), безводный дихлорметан (40 л) и диизопропилэтиламин (2,21 кг). Реакционную смесь охлаждали до температуры от -5 до +5°C и добавляли метансульфонилхлорид (1,87 кг) с регулируемой скоростью в течение 2 часов, чтобы гарантированно поддерживать температуру реакционной смеси ниже 5°C. По окончании добавления смесь нагревали до 20-30°C и перемешивали в течение одного-двух часов. За ходом реакции следили с помощью ГХ-МС и считали реакцию оконченной, когда степень преобразования достигала ≥95%. Поддерживая температуру реакционной смеси в пределах от 20 до 30°C, разбавляли смесь водой (15 л). Органический слой отделяли, промывали водой (0,50 кг) и концентрировали досуха, получая соединение 4 в виде твердого вещества светло-желтого цвета (4,87 кг, 96%). Типичная чистота по данным ВЭЖХ (AUC) составляла >95%. Спектр 1H-ЯМР соответствовал структуре.
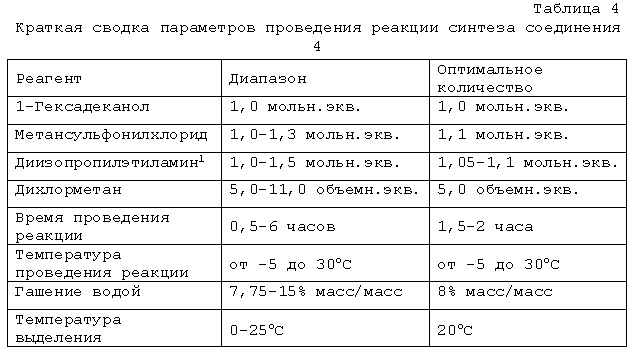

Пример 4
Получение 3-(гексадецилокси)пропан-1-ола (5)
В атмосфере инертного газа, например азота, в реактор помещали 1,3-пропандиол (4,07 кг) и NMP (30 л). Реакционную смесь охлаждали до температуры от -5 до 5°C и выдерживали в атмосфере азота. Осторожно порциями добавляли гидрид натрия (1,07 кг, 60% в минеральном масле). По окончании добавления реакционную смесь перемешивали при комнатной температуре в течение еще 2 часов. При температуре 20-55°C к реакционной смеси медленно добавляли раствор гексадецил метансульфоната 4 (4,39 кг) в NMP (10 л). Полученный раствор перемешивали в течение 12-28 часов при 20-35°C. За ходом реакции наблюдали с помощью ГХ-МС и реакцию считали завершенной, когда степень преобразования достигала ≥95%. Реакционную смесь охлаждали до температуры от -5 до 5°C и медленно разбавляли водой (15 л). Реакционную смесь экстрагировали этилацетатом (2×25 л). Органическую фазу промывали водой (20 л), высушивали над Na2SO4 и концентрировали, получая масло коричневого цвета. Неочищенный продукт растворяли в метаноле (20 л) и оставляли при температуре от 20 до 30°C на 12 часов. Выделившиеся в результате этого твердые примеси отделяли фильтрованием и отбрасывали, и фильтрат концентрировали. К концентрату добавляли ацетонитрил (40 л) и выдерживали смесь при температуре от 5 до 15°C в течение 16 часов. Твердое вещество отделяли фильтрованием и высушивали при 25-30°C, получая соединение 5 в виде твердого вещества белого цвета (3,1 кг, 77%). Типичная чистота по данным ВЭЖХ (AUC) составляла >95%. Спектр 1H-ЯМР соответствовал структуре.
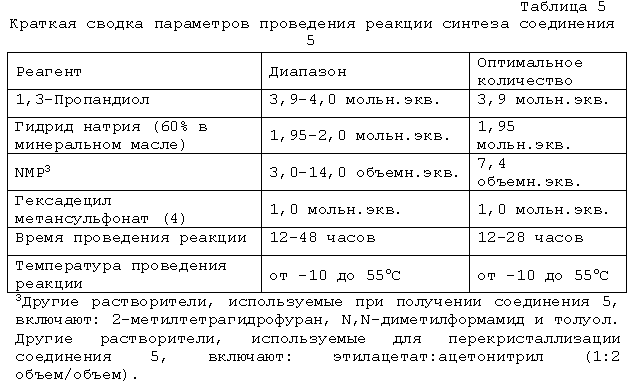
Пример 5:
Получение натриевой соли моно[3-(гексадецилокси)пропилового]эфира P-[[[(4-метилфенил)сульфонил]окси]метил]фосфоновой кислоты (CMX203)
В атмосфере инертного газа, например азота, в реактор помещали диэтил (тозилокси)метилоксифосфонат (6, 4,00 кг) и безводный ацетонитрил (36 л). К реакционной смеси осторожно добавляли бромтриметилсилан (6,65 кг). По окончании добавления реакционную смесь перемешивали при 20-30°C в течение 1 часа, затем нагревали до 55°C и перемешивали в течение еще двух часов. Смесь охлаждали до 20-30°C и концентрировали в вакууме. Концентрат растворяли в безводном дихлорметане (36 л) и медленно, в течение двух часов, добавляли оксалилхлорид (5,52 кг). Наблюдалось выделение избыточного газа из реакционной смеси. Реакционную смесь перемешивали в течение двух часов и добавляли N,N-диметилформамид (ДМФА) (2,0 мл). После добавления ДМФА реакционную смесь перемешивали в течение 12-16 часов. Реакционную смесь концентрировали, получая (дихлорфосфорил)метил 4-метилбензолсульфонат (7) в виде масла коричневого цвета. Соединение (7) растворяли в безводном дихлорметане (36 л) и добавляли 3-(гексадецилокси)пропан-1-ол (5) (3,25 кг). Реакционную смесь охлаждали до температуры от -5 до 5°C и по каплям добавляли пиридин (2,56 кг). После завершения реакции смесь перемешивали при 20-30°C в течение двух часов и затем гасили медленным добавлением воды (5,0 л). Первые 500 мл воды добавляли в течение 30 минут из-за сильного выделения тепла. Остаток воды добавляли в течение 15 минут. Насыщенный раствор бикарбоната натрия добавляли в течение 30 минут и продолжали перемешивание в течение одного часа. Значение pH смеси доводили до 2,0 6 н. хлористоводородной кислотой. Органический слой отделяли и водную фазу экстрагировали дихлорметаном (10 л). Объединенные органические слои промывали водой (2×10 л), высушивали над сульфатом натрия и концентрировали, получая масло коричневого цвета. Неочищенный продукт растворяли в 2-пропаноле (50 л) и добавляли 6 н. гидроксид натрия (4,0 мл). Раствор выдерживали при 20-30°C в течение трех дней. Осадок собирали фильтрованием и промывали 2-пропанолом (12 л). Осадок на фильтре высушивали в вакууме, получая CMX203 в виде твердого вещества белого цвета (4,50 кг, 73% относительно 3-(гексадецилокси)пропан-1-ола (5)).
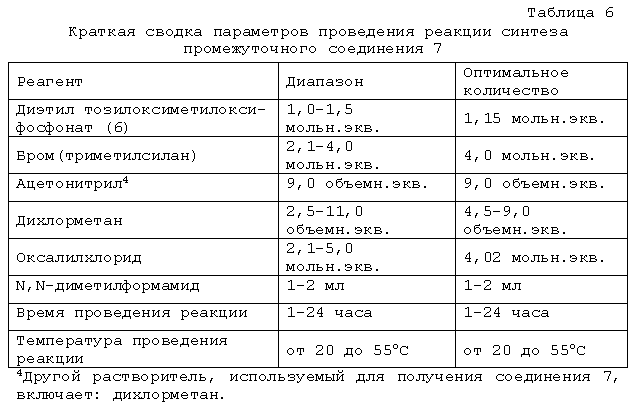


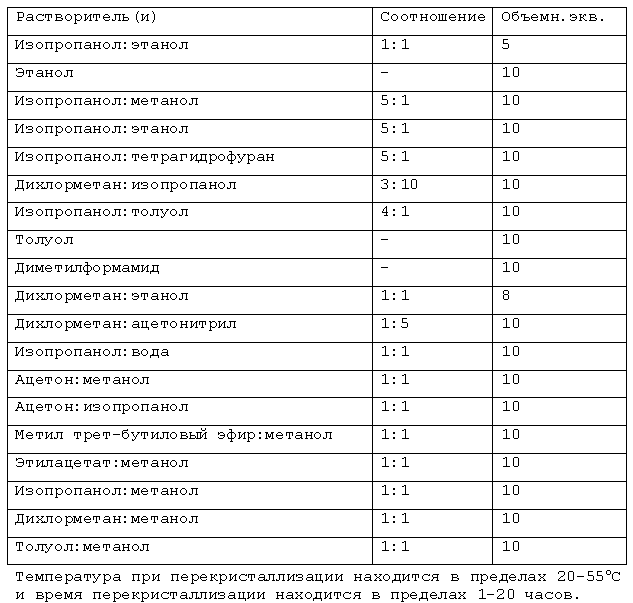
Пример 6
Получение (S)-N1-[(2-гидрокси-3-трифенилметокси)пропил]цитозина (CMX212)
В атмосфере азота при комнатной температуре в реактор помещали цитозин, (S)-тритил глицидиловый эфир, карбонат калия и безводный N,N-диметилформамид. Реакционную смесь нагревали и выдерживали при температуре 85-95°C до завершения реакции, после чего охлаждали до 60-70°C. Затем реакционную смесь гасили водой, перемешивали при комнатной температуре и фильтровали. Влажный твердый продукт высушивали азеотропной отгонкой с толуолом, охлаждали до 25±5°C и фильтровали. Твердый продукт суспендировали в ацетоне при 35±5°C, фильтровали и высушивали в вакууме при 50±5°C до получения продукта с содержанием остаточного растворителя менее чем или равным 0,5%. Выход: примерно от 36,1 кг до 40,9 кг (84,4-95,6 моль) CMX212; 75-85% в расчете на цитозин. Наблюдение за ходом реакции: ВЭЖХ - завершение реакции исходное вещество (цитозин) составляет ≤5% AUC.
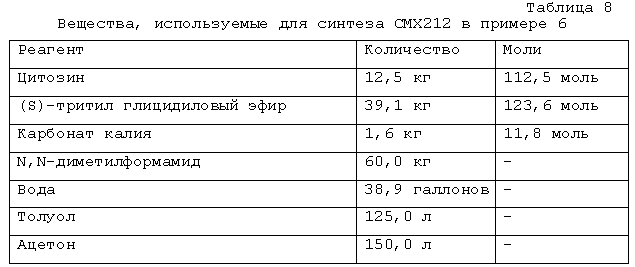
Пример 7
Получение [[(S)-2-(4-амино-2-оксо-1(2H)-пиримидинил)-1-(гидроксиметил)этокси]метил]моно[3-(гексадецилокси)пропилового]эфира фосфоновой кислоты (CMX001)
В атмосфере азота при комнатной температуре в реактор помещали CMX212, CMX203, ди-трет-бутоксид магния и безводный N,N-диметилформамид. Реакционную смесь нагревали и выдерживали при температуре 77-83°C до завершения реакции. Реакционную смесь концентрировали приблизительно до половины исходного объема, затем охлаждали до температуры в пределах 25-30°C. В реактор добавляли изопропилацетат, экстрагировали 0,5М HCl и затем насыщенным раствором соли. Органическую фазу выдерживали в вакууме, отгоняя изопропилацетат. Добавляли метанол и концентрировали реакционную смесь, удаляя весь остаточный изопропилацетат. Полученный промежуточный продукт (CMX225) растворяли в метаноле, охлаждали и вводили в реактор газообразный хлористый водород, пропуская его в объем раствора в метаноле. Реакционную смесь перемешивали до завершения взаимодействия, после чего фильтровали, отделяя все нерастворимые вещества. Смесь гасили водой и доводили pH примерно до 2,5, добавляя гидроксид натрия. Полученное твердое вещество отделяли фильтрованием, промывали водой и затем ацетоном. Твердые продукты суспендировали в ацетоне примерно при 40°C и фильтровали. Неочищенный продукт подвергали перекристаллизации из метанола и высушивали. Продукт второй раз подвергали перекристаллизации из метанола и высушивали в вакууме примерно при 50°C до остаточного содержания растворителя в продукте менее чем или равного 0,5%. Выход: приблизительно 12,4-14,3 кг (22,0-25,4 моль) CMX001; 65-75% в расчете на CMX212. Наблюдение за ходом реакции: ВЭЖХ - стадия 2A: CMX212 составляет ≤5% AUC; стадия 2B: N4-алкилированный побочный продукт (примесь) составляет ≤1% AUC.
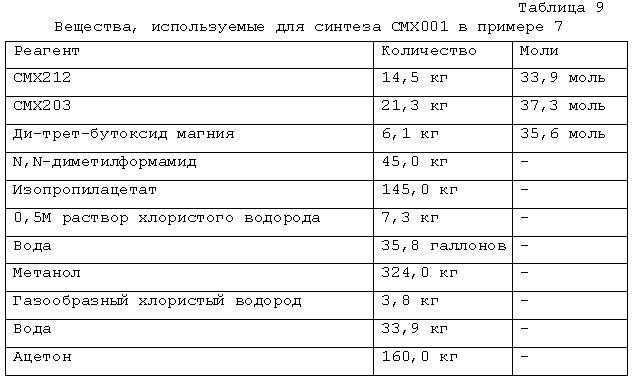
Кроме того, было отмечено, что если для сочетания с CMX203 вместо CMX212 использовали (S)-N1-[(2,3-дигидрокси)пропил]цитозин, то в тех же условиях реакции не происходило образования CMX001.
Пример 8
Способ получения [[(S)-2-(4-амино-2-оксо-1(2H)-пиримидинил)-1-(гидроксиметил)этокси]метил]моно[3-(гексадецилокси)пропилового]эфира фосфоновой кислоты (CMX001), описанный в WO 2005/08788
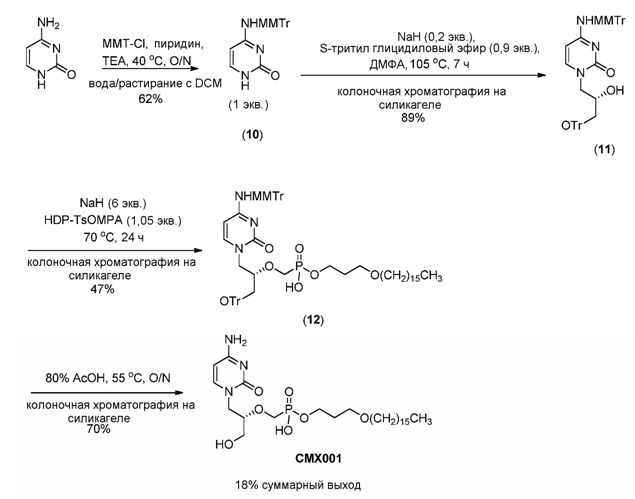
Синтез N4-монометокситритил-O3'-тритил-дигидроксипропилцитозина (11)
N4-монометокситритилцитозин (10) (3,44 г, 8,9 ммоль) и гидрид натрия (0,043 г, 1,78 ммоль) перемешивали в диметилформамиде (ДМФА) (50 мл) при комнатной температуре в течение одного часа. Полученную реакционную смесь обрабатывали (S)-тритил глицидиловым эфиром (2,5 г, 8,0 ммоль) и нагревали до 105°C в течение семи часов. Неочищенный продукт растворяли в хлороформе, промывали водой, концентрировали и очищали колоночной хроматографией, получая желаемый продукт с выходом 89%.
Синтез гексадецилоксипропилового эфира N4-монометокситритил-O3'-тритил-цидофовира (12)
К раствору N4-монометокситритил-O3'-тритил-дигидроксипропилцитозина (0,70 г, 1,0 ммоль) в ДМФА (10 мл) добавляли гидрид натрия (0,14 г, 6,0 ммоль). К полученному раствору добавляли гексадецилоксипропиловый эфир толуолсульфонилоксиметилфосфоната (HDP-TsOMPA) (0,82 г, 1,05 ммоль) и перемешивали смесь при 70°C в течение 24 часов, после чего охлаждали до комнатной температуры. Смесь экстрагировали хлороформом и промывали водой, высушивали, концентрировали и очищали колоночной хроматографией на силикагеле, получая желаемый продукт с выходом 47%.
Синтез гексадецилоксипропил-цидофовира (CMX001)
Гексадецилоксипропиловый эфир N4-монометокситритил-O3'-тритил-цидофовира (0,38 г, 0,35 ммоль) обрабатывали 80% уксусной кислотой (10 мл) и перемешивали при 55°C в течение ночи. Растворитель выпаривали и остаток очищали колоночной хроматографией на силикагеле (20% метанол в дихлорметане), получая желаемый продукт с выходом 70%. Суммарный выход CMX001 составил 18%.
Пример 9
Способ получения гексадецилоксипропилового эфира толуолсульфонилоксиметилфосфоната (HDP-TsOMPA), описанный в WO 2005/08788
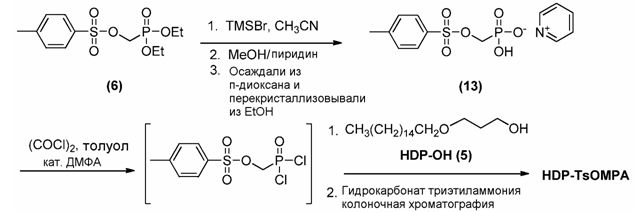
Синтез толуолсульфонилоксиметилфосфоната пиридиния (13)
Диэтил толуолсульфонилоксиметилфосфонат (6) (1,0 г, 3,1 ммоль) растворяли в сухом ацетонитриле (25 мл), охлаждали смесь на водяной бане и перемешивали магнитной мешалкой. Одной порцией добавляли бромтриметилсилан (1,42 г, 9,3 ммоль). Смесь перемешивали в течение 4 часов. Выпаривали растворитель, получая густое масло. Добавляли метанол/пиридин (30 мл) и перемешивали смесь в течение 30 мин. Выпаривали растворитель и остаток объединяли с п-диоксаном и перемешивали. Собирали кристаллы белого цвета и осуществляли перекристаллизацию из этанола (EtOH), получая 750 мг продукта (73%).
Синтез гексадецилоксипропилового эфира толуолсульфонилоксиметилфосфоната (HDP-TsOMPA)
К раствору толуолсульфонилоксиметилфосфоната пиридиния (1,0 г, 3,0 ммоль) в сухом толуоле (20 мл) одной порцией добавляли оксалилхлорид (0,39 мл, 4,5 ммоль) и ДМФА (0,02 мл, 0,3 ммоль). Раствор перемешивали при комнатной температуре в течение 1 часа. Толуол и избыток оксалилхлорида удаляли в вакууме. Остаток повторно растворяли в толуоле (10 мл). Добавляли 3-гексадецилокси-1-пропанол (5) (0,81 мл, 2,7 ммоль). Смесь перемешивали при комнатной температуре в течение ночи. К смеси добавляли гидрокарбонат триэтиламммония в качестве буфера (10 мл) и перемешивали смесь в течение 30 минут. Растворители выпаривали. Остаток растворяли в хлороформе (50 мл), промывали водой (2×10 мл) и выпаривали растворитель, получая 1 грамм неочищенного продукта. Примеси удаляли колоночной флэш-хроматографией (силикагель, 15% EtOH/дихлорметан), выход=0,60 г (40%).
Из описания, приведенного в WO 2005/08788 не ясно, являлась ли HDP-TsOMPA триэтиламмониевой солью, натриевой солью, свободной кислотой или их смесью.
Пример 10
Получение [[(S)-2-(4-амино-2-оксо-1(2H)-пиримидинил)-1-(гидроксиметил)этокси]метил]моно[3-(гексадецилокси)пропилового]эфира фосфоновой кислоты (CMX001) с использованием циклического цидофовира по методике, описанной в US 6716825
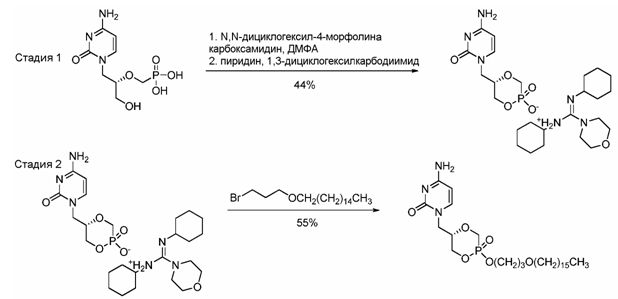
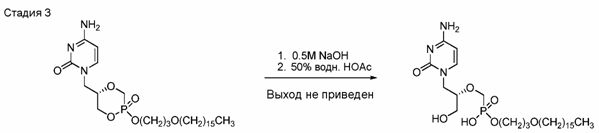
Как описано в патенте США 6716825, на стадии 1 приведенной выше схемы цидофовир суспендировали в N,N-ДМФА и добавляли карбоксамид N,N'-дициклогексил-4-морфолина. Смесь перемешивали в течение ночи для растворения цидофовира. Затем полученный прозрачный раствор помещали в капельную воронку и медленно (30 мин) добавляли при перемешивании к горячему (60°C) раствору 1,3-дициклогексилкарбодиимида в пиридине. Полученную реакционную смесь перемешивали при 100°C в течение 16 ч, охлаждали до комнатной температуры и удаляли растворители при пониженном давлении. Реакционную смесь адсорбировали на силикагель и очищали продукт колоночной флэш-хроматографией, используя градиентное элюирование (CH2Cl2 и MeOH) и затем смесью 5:5:1 CH2Cl2/MeOH/H2O. Фракции, содержащие продукт, объединяли и концентрировали в вакууме. Продукт, т.е. DCMC соль циклического цидофовира, выделяли с выходом 44%. На стадии 2 раствор соли циклический цидофовир DCMC в сухом N,N-ДМФА помещали в 1-бром-3-гексадецилоксипропан и перемешивали смесь при нагревании до 80°C в течение 6 ч. Раствор концентрировали в вакууме, остаток адсорбировали силикагелем и очищали колоночной флэш-хроматографией, используя градиентное элюирование (CH2Cl2 и этанол). Продукт элюировали смесью 90:10 CH2Cl2/EtOH. Фракции, содержащие чистый продукт, концентрировали в вакууме. Гексадецилоксипропил-циклический цидофовир получали с выходом 55%. На стадии 3 гексадецилоксипропил-циклический цидофовир растворяли в 0,5М NaOH и перемешивали при комнатной температуре в течение 1,5 ч. Затем по каплям добавляли 50% водный раствор уксусной кислоты, доводя значение pH до 9. Выпавший в осадок гексадецилоксипропил-цидофовир отделяли фильтрованием, промывали водой и высушивали. Продукт кристаллизовали из смеси 3:1 п-диоксан/вода, получая гексадецилоксипропил-цидофовир (HDP-CDV, т.е. CMX001).
Пример 10A
Повторяли способ, описанный в US 6716825 согласно примеру 10, описанным ниже образом с небольшими изменениями
Синтез гексадецилоксипропил-циклического цидофовира
К гетерогенному раствору циклического цидофовира (39 г, 0,131 моль, 1 экв., приобретен у Gilead Sciences) в N,N-ДМФА (2,0 л) в атмосфере азота добавляли N,N'-дициклогексил-4-морфолинкарбоксамидин (DCMC) (0,131 моль, 38,51 г, 1 экв.). Полученную смесь перемешивали в течение ночи. В результате этого большая часть твердых веществ в реакционной смеси растворялась. К полученному раствору добавляли 1-бром-3-гексадецилоксипропан (238,45 г, 0,656 моль, 5 экв.). Реакционную смесь нагревали до 80°C и перемешивали в течение 6 часов. Неочищенную реакционную смесь концентрировали в вакууме при 80°C. Неочищенную реакционную смесь абсорбировали силикагелем (300 г) и очищали колоночной хроматографией (800 г SiO2). Колонку элюировали дихлорметаном (6 л), удаляя избыток 1-бром-3-гексадецилоксипропана. Затем растворитель заменяли смесью 9:1 CH2Cl2:EtOH (32 л). Продукт выходил вместе с этой системой растворителей. Фракции 30-39 объединяли, получая гексадецилоксипропил-циклический цидофовир (17,8 г) с выходом 25%.
Синтез гексадецилоксипропил-цидофовира (HDP-CDV) с доведением pH до значения 5,5 после гидролиза
К гексадецилоксипропил-циклическому цидофовиру (4,00 г, 1,0 эквивалент) добавляли 0,5 н. гидроксид натрия (118 мл, 8,0 эквивалентов). Раствор перемешивали при комнатной температуре в течение 1,5 часов. В результате гидролиза раствор оставался мутным. Значение pH доводили до 2,78 (желаемое значение pH составляло 2,5) медленным добавлением концентрированной уксусной кислоты (200 мл). При этом значении pH реакционная смесь становилась раствором. Затем значение pH доводили до 5,5, используя 3 н. NaOH, и перемешивали смесь в течение ночи при комнатной температуре. Полученное твердое вещество отделяли фильтрованием и высушивали на воздухе (получали 3,76 г неочищенного продукта). Этот продукт кристаллизовали, растворяя его в 100 мл смеси 3:1 п-диоксан/вода при 65°C. Гетерогенную реакционную смесь фильтровали и оставляли стоять при комнатной температуре в течение ~2 ч, после чего помещали на ночь в холодильник.
Затем продукт фильтровали и высушивали на воздухе в течение 1,5 часов. Фильтровальную воронку помещали в вакуумную печь и высушивали продукт при 46°C в течение 48 часов. В результате реакции было получено 0,93 г аморфного твердого вещества не совсем белого цвета.
Синтез гексадецилоксипропил-цидофовира (HDP-CDV) с доведением pH до значения 4,51 после гидролиза
К гексадецилоксипропил-циклическому цидофовиру (4,00 г, 1,0 эквивалент) добавляли 0,5 н. гидроксид натрия (118 мл, 8,0 эквивалентов). Раствор перемешивали в течение 1,5 часов. В результате гидролиза раствор оставался мутным. Значение pH доводили до 4,51 (целевое значение pH составляло 4,5), добавляя концентрированную уксусную кислоту (9 мл). Полученное твердое вещество отделяли фильтрованием (отделение твердого вещества фильтрованием занимало менее 3 минут) и высушивали в вакууме в течение ночи. Получали 3,5 г твердого вещества белого цвета (мелкий порошок). Это вещество кристаллизовали из смеси п-диоксан/вода (3:1). Первая попытка использования этой смеси в количестве 10 мл/г при 65°C оказалась безуспешной. При этом соотношении растворилось лишь очень незначительное количество вещества. Использовали дополнительные 85 мл смеси диоксан:вода (3:1). Гетерогенный раствор фильтровали в горячем состоянии через воронку из спеченного стекла. Фильтрование заняло примерно 20 секунд. Раствору давали остыть до комнатной температуры и затем помещали в холодильник на ночь. Выделившееся твердое вещество отделяли фильтрованием (фильтрование заняло менее 1 минуты). Полученное твердое вещество (белый порошок) высушивали в вакуумной печи при 46°C в течение 48 часов (643 мг).
Синтез гексадецилоксипропил-цидофовира (HDP-CDV) с доведением pH до значения 3,51 после гидролиза
К гексадецилоксипропил-циклическому цидофовиру (4,00 г, 1,0 эквивалент) добавляли 0,5 н. гидроксид натрия (118 мл, 8,0 эквивалентов). Раствор перемешивали при комнатной температуре в течение 1,5 часов. В результате гидролиза раствор оставался мутным. Значение pH доводили до 3,51, добавляя по каплям концентрированную уксусную кислоту (55 мл), и оставляли реакционную смесь стоять при комнатной температуре в течение 3 часов. Продукт отделяли фильтрованием и высушивали на воздухе в течение ночи. Полученное твердое вещество белого цвета с утра оставалось влажным. Считая, что масса этого твердого вещества белого цвета в полностью сухом состоянии могла бы составить ~3,6 г, в расчете на массу двух других высушенных неочищенных продуктов, описанных выше, кристаллизацию начинали, добавляя 40 мл смеси 3:1 п-диоксан/вода и нагревая до 65°C. Продолжали добавление смеси 3:1 п-диоксан/вода порциями по 10 мл, вновь повышая каждый раз температуру до 65°C. Продукт почти полностью растворился после добавления в общей сложности 110 мл смеси 3:1 диоксан/вода при 65°C. Смесь фильтровали и хранили при комнатной температуре в течение 4 часов, затем помещали в холодильник на ночь.
Продукт фильтровали и высушивали на воздухе в течение 1,5 ч, получая 1,61 г твердого вещества белого цвета. Образец массой 200 мг помещали обратно в воронку из спеченного стекла и высушивали на воздухе в течение 48 ч. Остальные 1,41 г высушивали в вакуумной печи при 46°C в течение 48 ч. Образец, высушенный на воздухе на фильтре из спеченного стекла, весил 0,27 г (116-018B). Образец массой 1,41 г, высушенный в вакуумной печи, весил 1,29 г (116-018A). Оба образца представляли собой твердые кристаллические вещества белого цвета.
Кроме того, обнаружили, что если pH после гидролиза довести до 9, используя концентрированную уксусную кислоту, то не наблюдалось осаждения HDP-CDV, в противоположность тому, что описано в патенте US 6716825. Кроме того, обнаружили, что если значение pH после гидролиза превышало 3,5, наблюдалось образование смеси HDP-CDV в форме свободной кислоты и натриевой соли HDP-CDV. Помимо этого, оказалось, что если довести значение pH до 2,78, то наблюдалось образование раствора (из гетерогенного раствора). Не желая ограничиваться каким-либо объяснением, это, наиболее вероятно, происходит за счет протонирования аминогруппы цитозина образовавшегося HDP-CDV ацетата, с растворением образующегося соединения в реакционном растворителе. Все образцы HDP-CDV, полученные в этом примере, обладали более низкой чистотой (79-91% масс/масс) по сравнению с образцами высокой чистоты (>99% масс/масс), полученными в примерах 2 или 7.
Пример 11
Исследование кристаллических форм CMX001
Исследование дифракции рентгеновских лучей на порошке CMX001 было выполнено для четырех образцов морфологической формы A CMX001, полученных способом, описанным выше в примере 2, т.е. образцов №№1-5 и одного образца формы B CMX001, т.е. образца №6, полученного перекристаллизацией неочищенного CMX001, синтезированного по методике, описанной в US 6716825 (т.е. с использованием смеси 3:1 п-диоксан/вода) согласно примеру 10A. Морфологическая форма B была охарактеризована как нестехиометрический гидрат CMX001.
Данные для каждого из образцов приведены ниже в таблицах 10-15, и дифрактограммы всех образцов приведены на фиг.1-6
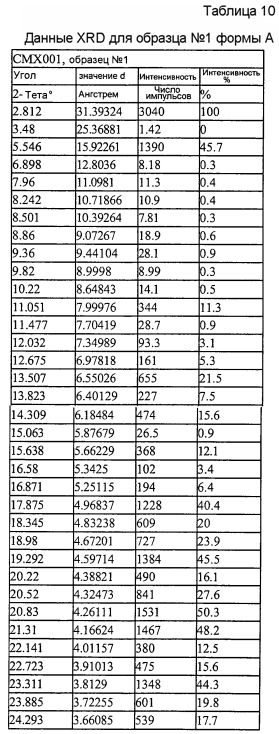
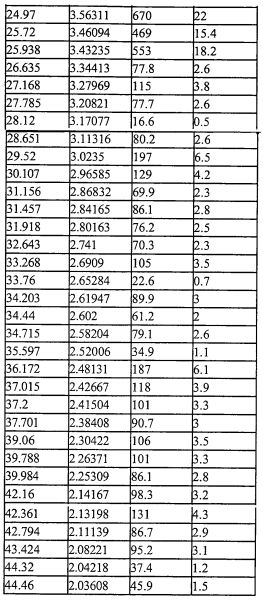
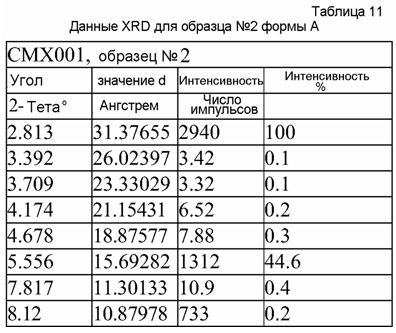
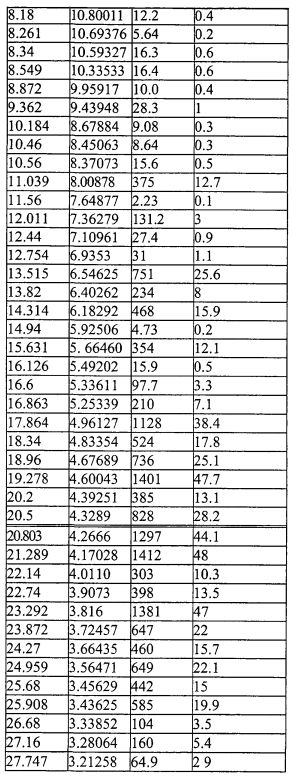
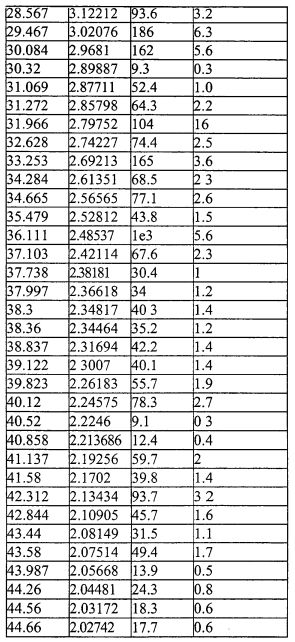
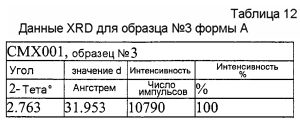
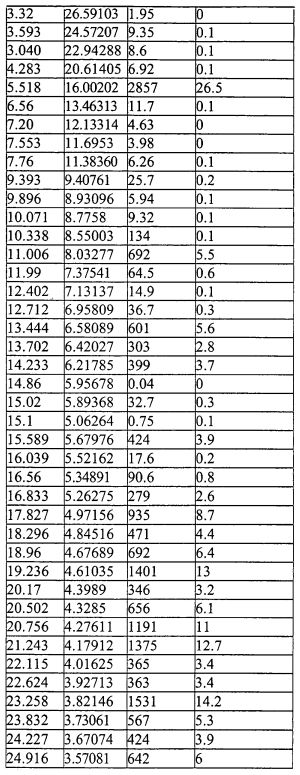
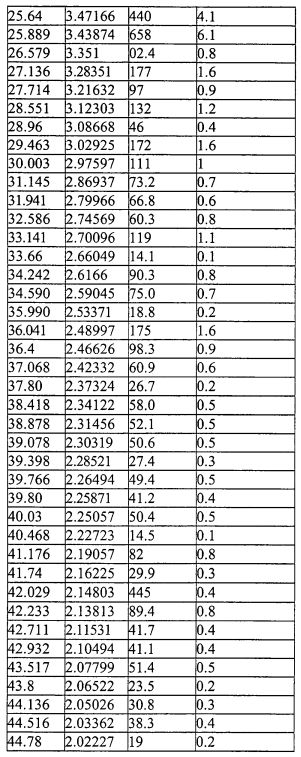
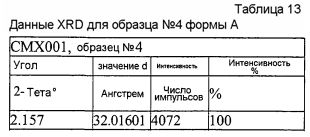
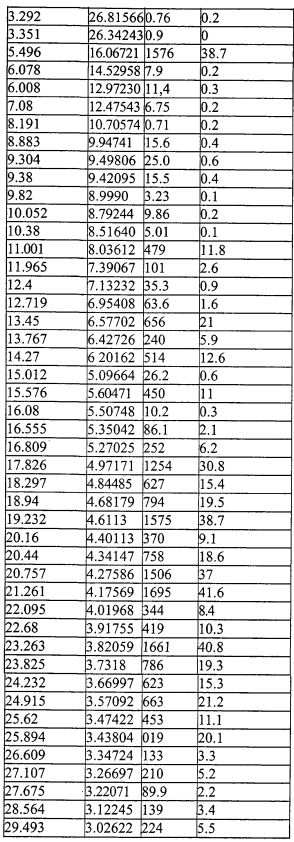
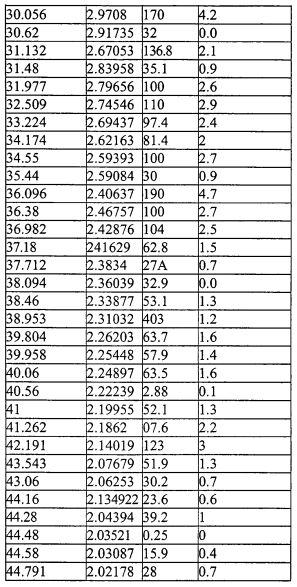
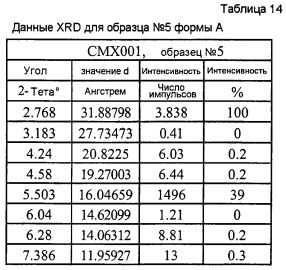
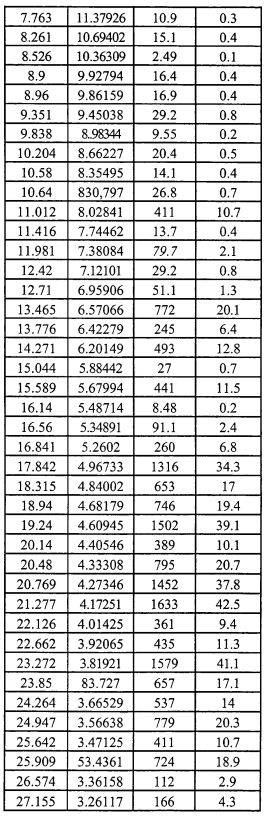
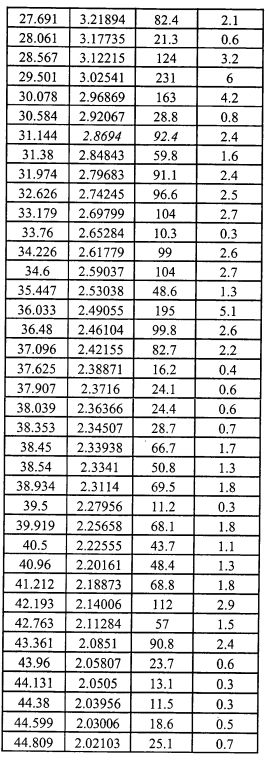
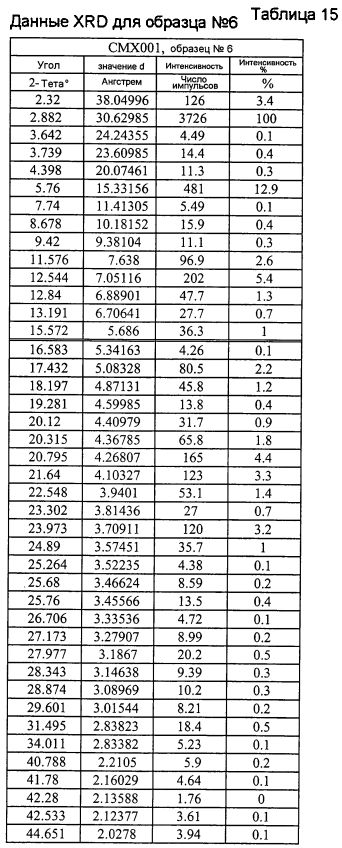
Как видно из данных по дифракции рентгеновских лучей на порошке, образцы №№1-5 формы A демонстрируют в основном одинаковые дифракционные картины. Фактически, все образцы, полученные способом, описанным в примере 2, по данным рентгеноструктурных исследований, представляли собой одну и ту же форму A. Кроме того, как следует из наложения рентгеновских дифрактограмм, приведенного на фиг.7, форма A и форма B демонстрируют явные различия в картинах дифракции рентгеновских лучей на порошке. Сводка отличающихся особенностей этих дифрактограмм показана ниже в таблице 16.
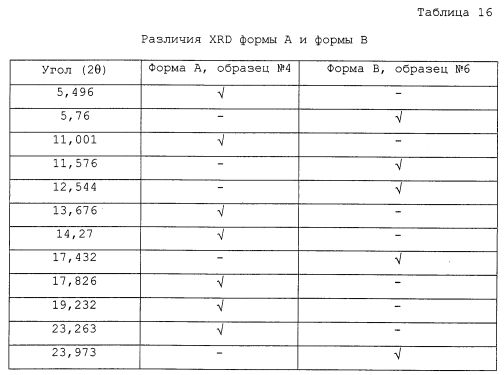
Пример 12
Составы таблеток
Было разработано несколько составов таблеток для CMX001. Таблетки прессовали из стандартной смеси, меняя содержание лекарственного вещества, для получения таблеток с различным содержанием действующего начала. Дозированные формы, содержавшие 20 мг, 50 мг и 100 мг, соответственно, представляли собой круглые, двояковыпуклые таблетки, имевшие размеры 7,3 мм × 3,5 мм, 7,9 мм × 3,8 мм и 10,5 мм × 4,4 мм соответственно. CMX001 в форме свободной кислоты включали в состав прессованных таблеток с немедленным высвобождением, содержащих 20, 50 или 100 мг CMX001 (см. таблицы 17 и 18).
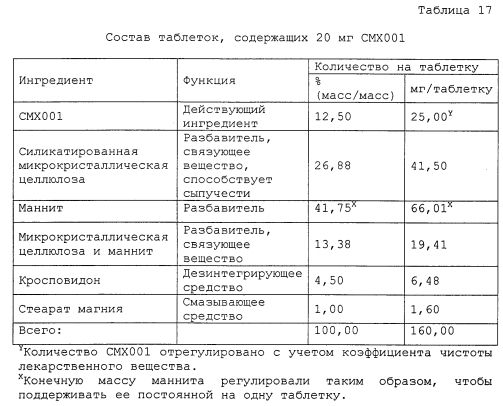
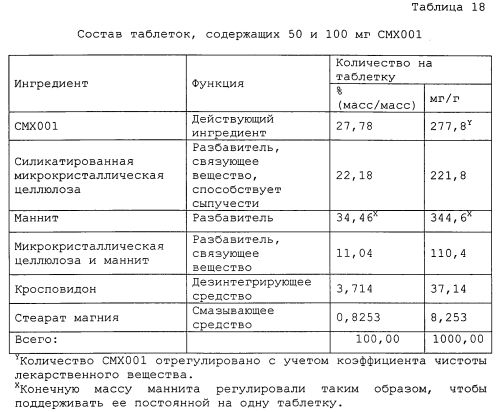
Пример 13
Исследование стабильности
Проводили исследование стабильности 50 мг и 100 мг таблеток. В таблицах 19 и 20 показаны результаты для 50 мг и 100 мг таблеток, соответственно:
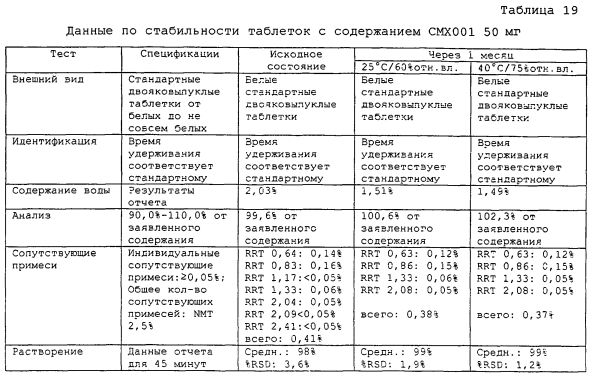
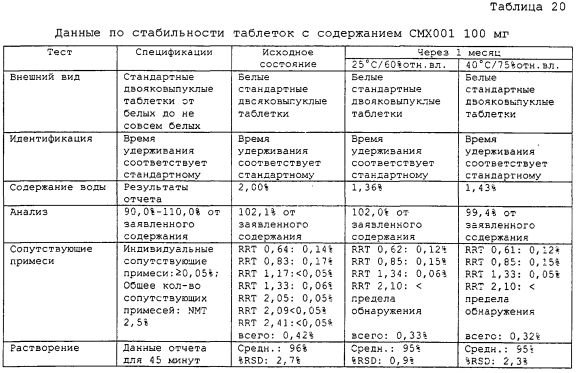
Пример 14
Моноаммониевая соль CMX001
CMX001 в форме свободной кислоты преобразовывали в моноаммониевую соль, используя следующую методику. Снабжали 5-литровую круглодонную колбу механической мешалкой, датчиком температуры и адаптером для введения газа. В колбу помещали CMX001 в форме свободной кислоты (87,3 г, 0,155 моль), 2-пропанол (180 мл) и 28-30% гидроксид аммония (13 мл). Реакционную смесь перемешивали и доводили до кипения с обратным холодильником (62-80°C), добиваясь растворения (10 мин). Примечание: Раствор не следует перемешивать более 15 минут при кипении с обратным холодильником. Раствору давали остыть до температуры менее 25°C в течение 16±8 ч. Смесь охлаждали до температуры 5±5°C в течение минимум 1 ч. Продукт фильтровали и промывали охлажденным 2-пропанолом (5±5°C, 430 мл). Продукт, твердое вещество белого цвета, высушивали при 30-35°C в течение 25 ч 10 мин ±2 ч. Выход: примерно 86,8 г (0,15 моль) моноаммониевой соли CMX001, что составляет 96,7% от теоретического в расчете на CMX001 в форме свободной кислоты. Наблюдение за ходом реакции: pH раствора аммониевой соли CMX001.
ЭКВИВАЛЕНТЫ
Настоящее изобретение можно воплотить в других конкретных формах, не отступая от сути изобретения или его существенных характеристик. Поэтому описанные выше варианты осуществления следует считать во всех смыслах иллюстративными, а не ограничивающими объем изобретения, описанного в настоящей заявке. Таким образом, объем настоящего изобретения определяется приложенной формулой изобретения, а не предшествующим описанием, и считается, что все изменения, которые соответствуют смыслу и попадают в рамки эквивалентности пунктам формулы изобретения, охвачены настоящим изобретением.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБЫ ПОЛУЧЕНИЯ ОПТИЧЕСКИ АКТИВНОГО ЦИС-2-ГИДРОКСИМЕТИЛ-4-(ЦИТОЗИН-1'-ИЛ)-1,3-ОКСАТИОЛАНА ИЛИ ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫХ СОЛЕЙ | 2005 |
|
RU2430919C2 |
| ФОСФОНАТНЫЕ СОЕДИНЕНИЯ | 2000 |
|
RU2258707C2 |
| СПОСОБ ПОЛУЧЕНИЯ КОНДЕНСИРОВАННЫХ ПРОИЗВОДНЫХ ИМИДАЗОЛА | 2016 |
|
RU2734818C2 |
| ПРОИЗВОДНЫЕ 1,6-ДИАЗАБИЦИКЛО[3.2.1]ОКТАН-7-ОНА И ИХ ПРИМЕНЕНИЕ ПРИ ЛЕЧЕНИИ БАКТЕРИАЛЬНЫХ ИНФЕКЦИЙ | 2012 |
|
RU2570423C2 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ И СОЛЬВАТЫ АВТ-263 ДЛЯ ПРИМЕНЕНИЯ В ЛЕЧЕНИИ ЗАБОЛЕВАНИЙ, СВЯЗАННЫХ С БЕЛКОМ BCL-2 | 2010 |
|
RU2551376C2 |
| КОМПОЗИЦИИ ИЛИ ПРОЛЕКАРСТВА ПРОТИВОМИКРОБНЫХ И РОДСТВЕННЫХ ПРОТИВОМИКРОБНЫМ СОЕДИНЕНИЙ, ОБЛАДАЮЩИЕ ВЫСОКОЙ ПРОНИКАЮЩЕЙ СПОСОБНОСТЬЮ | 2010 |
|
RU2586287C2 |
| СПОСОБ СИНТЕЗА АНАЛОГОВ ГОРМОНОВ ЩИТОВИДНОЙ ЖЕЛЕЗЫ И ИХ ПОЛИМОРФОВ | 2013 |
|
RU2668960C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, ВКЛЮЧАЮЩИЕ ИНГИБИТОРЫ НЭП (НЕЙТРАЛЬНОЙ ЭНДОПЕПТИДАЗЫ), ИНГИБИТОРЫ ЭНДОГЕННОЙ ПРОДУЦИРУЮЩЕЙ ЭНДОТЕЛИН СИСТЕМЫ И ИНГИБИТОРЫ ГМГ (ГИДРОКСИМЕТИЛГЛУТАРИЛ)СоА РЕДУКТАЗЫ | 2005 |
|
RU2410118C2 |
| СПОСОБ СИНТЕЗА АНАЛОГОВ ГОРМОНОВ ЩИТОВИДНОЙ ЖЕЛЕЗЫ И ИХ ПОЛИМОРФОВ | 2021 |
|
RU2802282C2 |
| 4'-ЗАМЕЩЕННЫЕ НУКЛЕОЗИДНЫЕ ИНГИБИТОРЫ ОБРАТНОЙ ТРАНСКРИПТАЗЫ И ИХ ПОЛУЧЕНИЕ | 2016 |
|
RU2720811C2 |
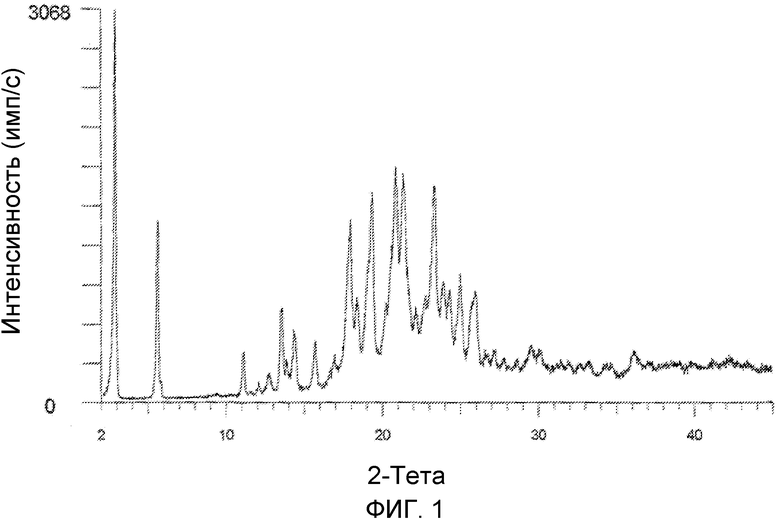
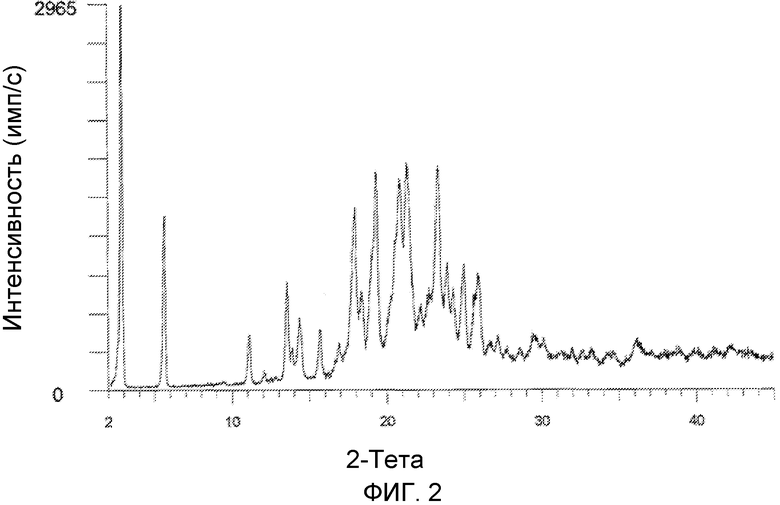
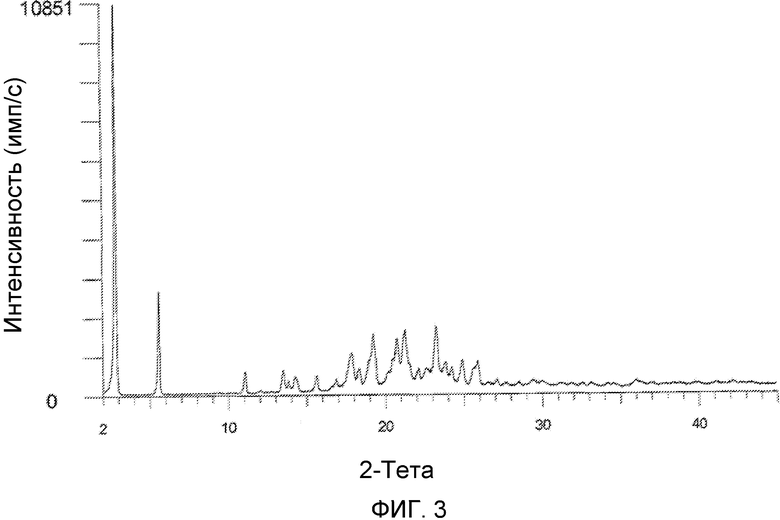
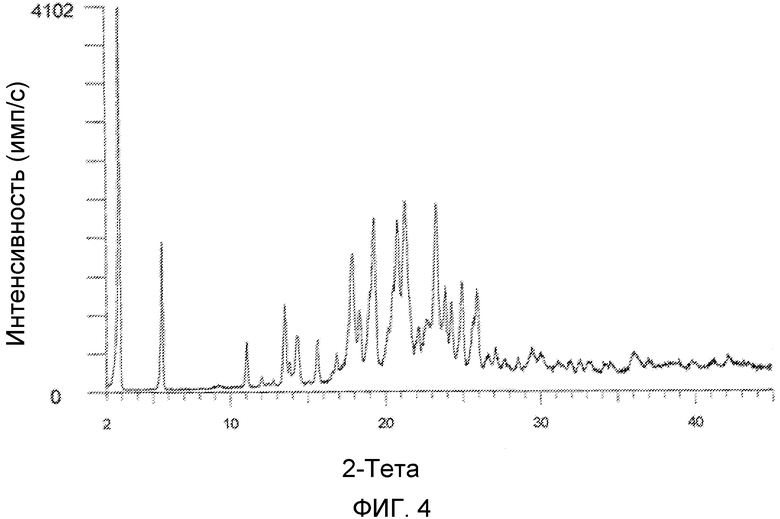
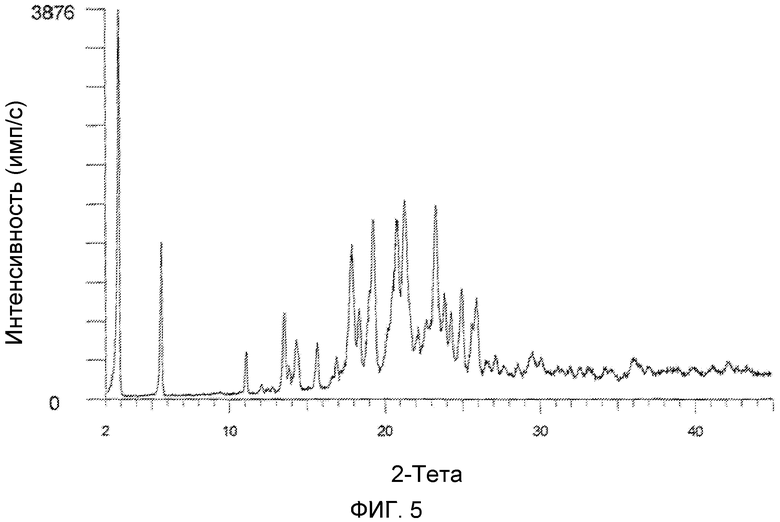
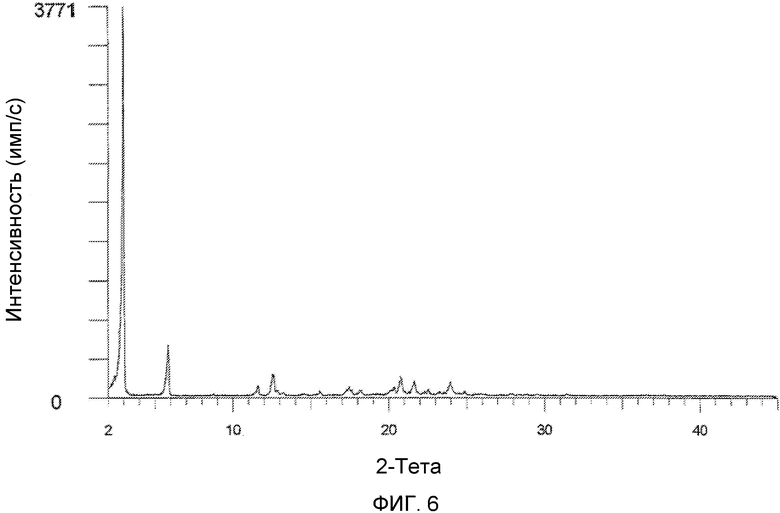
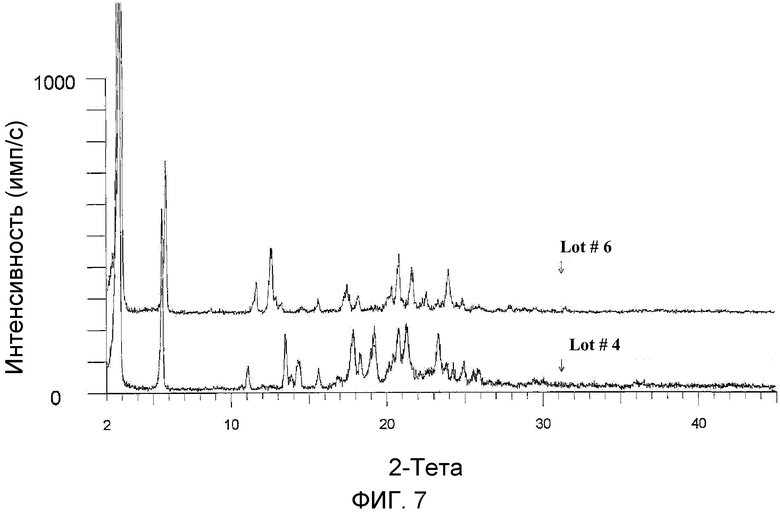
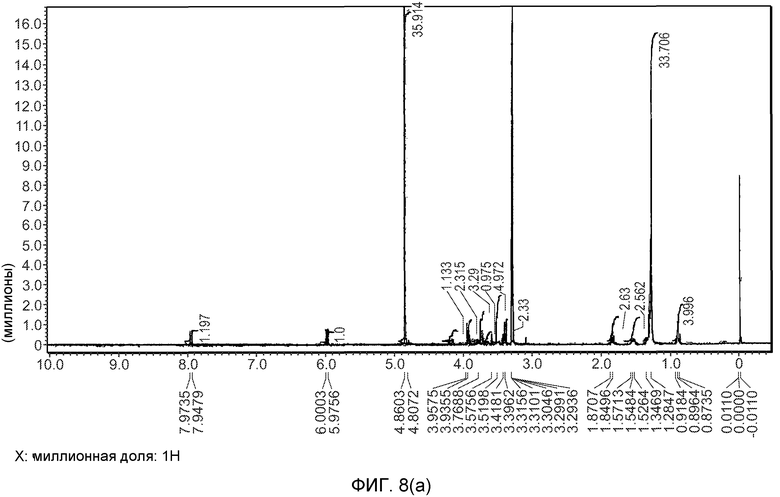
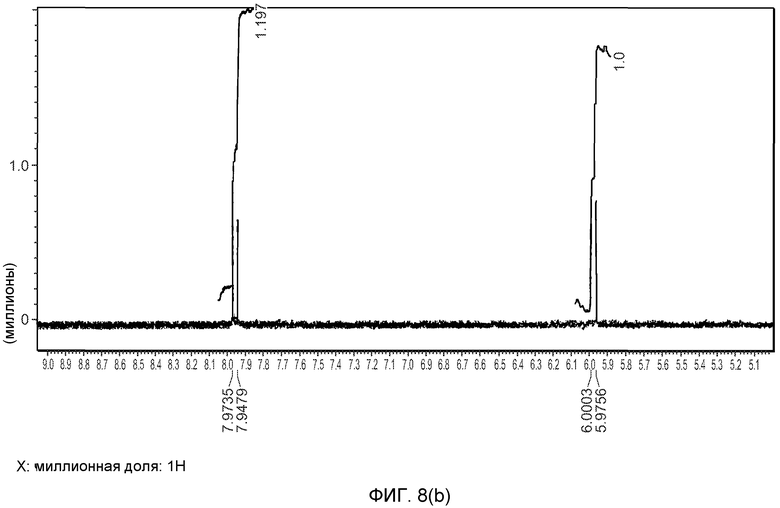
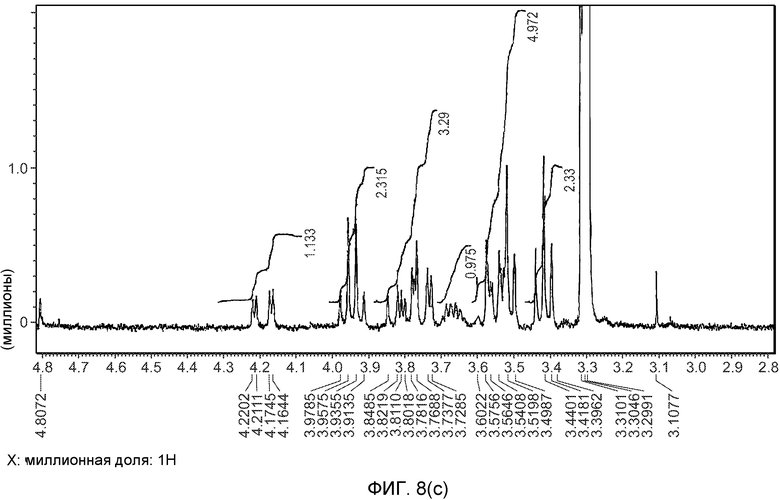
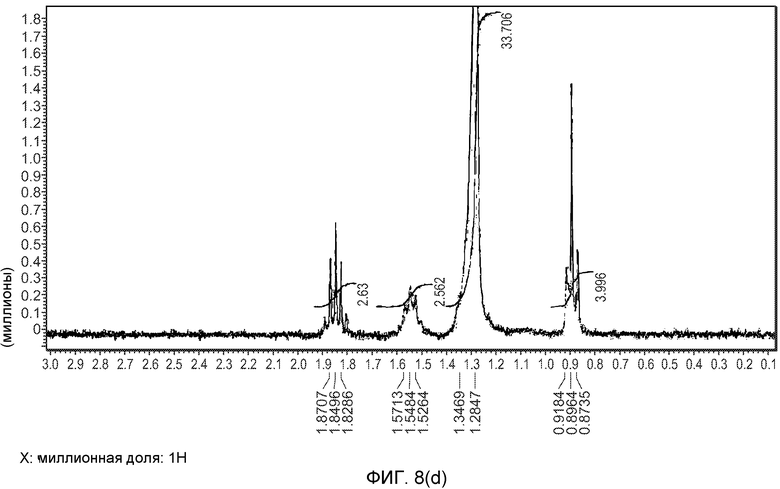
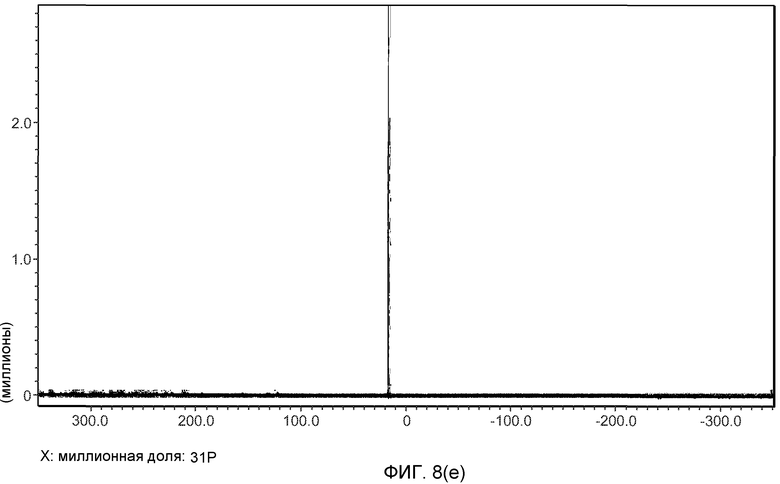
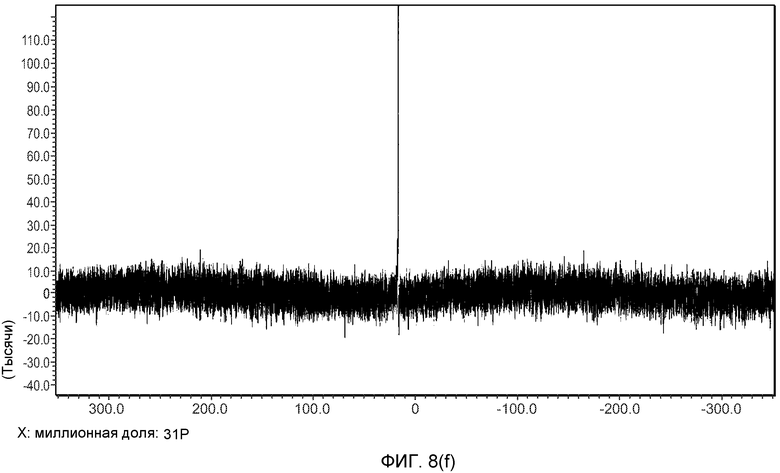
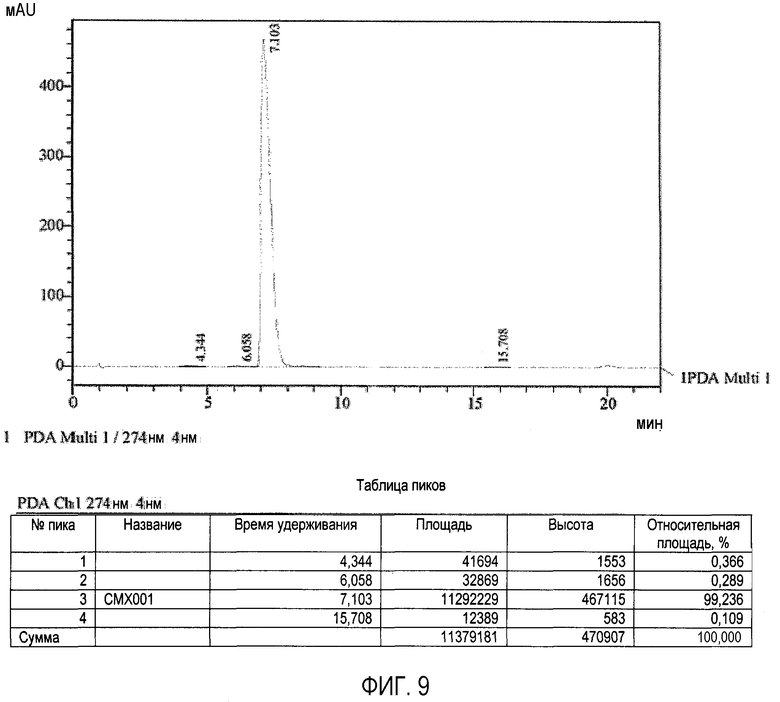
Изобретение относится к новой форме [[(S)-2-(4-амино-2-оксо-1(2Н)-пиримидинил)-1-(гидроксиметил)этокси]метил]моно[3-(гексадецилокси)пропилового]эфира фосфоновой кислоты, характеризующейся картиной дифракции рентгеновских лучей, включающей пики при углах 2θ примерно 5,5, 19,3, 20,8 и 21,3 градуса и чистотой более 91%, которая может быть использована в фармацевтической промышленности, а также к способу ее получения. Предложенный способ включает взаимодействие цитозина с (S)-тритил глицидиловым эфиром в присутствии карбоната металла с образованием (S)-N1-[(2-гидрокси-3-трифенилметокси)пропил]цитозина; взаимодействие (S)-N1-[(2-гидрокси-3-трифенилметокси)пропил]цитозина с натриевой солью моно[3-(гексадецилокси)пропилового]эфира р-[[[(4-метилфенил)сульфонил]окси]метил]фосфоновой кислоты в присутствии ди-трет-бутоксида магния с образованием [[(S)-2-(4-амино-2-оксо-1(2Н)-пиримидинил)-1-(гидроксиметил)-2-(трифенилметокси)этил]метил]моно[3-(гексадецилокси)пропилового]эфира фосфоновой кислоты; удаление защитной группы с получением [[(S)-2-(4-амино-2-оксо-1(2Н)-пиримидинил)-1-(гидроксиметил)этокси]метил]моно[3-(гексадецилокси)пропилового]эфира фосфоновой кислоты и его перекристаллизацию из метанола, этанола, изопропанола, системы метанол:ацетон:вода, метанол:ацетон или метанол:вода. Предложена новая удобная для изготовления лекарственных форм форма биологически активного соединения и новый эффективный способ ее получения. 4 н. и 13 з.п. ф-лы, 9 ил., 14 пр., 22 табл.
1. Морфологическая форма [[(S)-2-(4-амино-2-оксо-1(2Н)-пиримидинил)-1-(гидроксиметил)этокси]метил]моно[3-(гексадецилокси)пропилового]эфира фосфоновой кислоты (форма А), характеризующаяся картиной дифракции рентгеновских лучей, включающей пики при углах 2θ примерно 5,5, 19,3, 20,8 и 21,3 градуса и чистотой более 91%.
2. Морфологическая форма по п. 1, характеризующаяся картиной дифракции рентгеновских лучей, в основном сходной с изображенной на фиг. 4.
3. Морфологическая форма по п. 1, полученная с применением способа очистки, включающего перекристаллизацию неочищенного препарата [[(S)-2-(4-амино-2-оксо-1(2Н)-пиримидинил)-1-(гидроксиметил)этокси]метил]моно[3-(гексадецилокси)пропилового]эфира фосфоновой кислоты из метанола.
4. Способ синтеза морфологической формы по п. 1, включающий:
(a) взаимодействие цитозина с (S)-тритил глицидиловым эфиром в присутствии карбоната металла и N,N-диметилформамида или трет-амилового спирта с образованием (S)-N1-[(2-гидрокси-3-трифенилметокси)пропил]цитозина;
(b) взаимодействие (S)-N1-[(2-гидрокси-3-трифенилметокси)пропил]цитозина с натриевой солью моно[3-(гексадецилокси)пропилового]эфира р-[[[(4-метилфенил)сульфонил]окси]метил]фосфоновой кислоты в присутствии ди-трет-бутоксида магния и N, N-диметилформамида, N,N-диметилацетамида, диметилсульфоксида или 1-метил-2-пирролидинона с образованием [[(S)-2-(4-амино-2-оксо-1(2Н)-пиримидинил)-1-(гидроксиметил)-2-(трифенилметокси)этил]метил]моно[3-(гексадецилокси)пропилового]эфира фосфоновой кислоты;
(c) взаимодействие [[(S)-2-(4-амино-2-оксо-1(2Н)-пиримидинил)-1-(гидроксиметил)-2-(трифенилметокси)этил]метил]моно[3-(гексадецилокси)пропилового]эфира фосфоновой кислоты с реагентом для удаления защитной группы в присутствии метанола с получением неочищенного [[(S)-2-(4-амино-2-оксо-1(2Н)-пиримидинил)-1-(гидроксиметил)этокси]метил]моно[3-(гексадецилокси)пропилового]эфира фосфоновой кислоты; и
(d) перекристаллизацию неочищенного [[(S)-2-(4-амино-2-оксо-1(2Н)-пиримидинил)-1-(гидроксиметил)этокси]метил]моно[3-(гексадецилокси)пропилового]эфира фосфоновой кислоты из метанола, этанола, изопропанола, системы метанол:ацетон:вода, метанол:ацетон или метанол:вода.
5. Способ по п. 4, где дитрет-бутоксид магния имеет чистоту более 98%.
6. Способ по п. 4, где полученный [[(S)-2-(4-амино-2-оксо-1(2Н)-пиримидинил)-1-(гидроксиметил)этокси]метил]моно[3-(гексадецилокси)пропиловый]эфир фосфоновой кислоты имеет чистоту >99%.
7. Способ по п. 4, где полученный продукт включает менее 1,5% N4-алкилированного соединения.
8. Способ синтеза натриевой соли моно[3-(гексадецилокси)пропилового]эфира р-[[[(4-метилфенил)сульфонил]окси]метил]фосфоновой кислоты, включающий:
(a) взаимодействие (дихлорфосфорил)метил 4-метилбензолсульфоната с 3-(гексадецилокси)пропан-1-олом в присутствии основания в дихлорметане с получением соответствующей реакционной смеси;
(b) гашение полученной смеси бикарбонатом натрия и водой c получением неочищенного продукта; и
(c) растворение неочищенного продукта в 2-пропаноле.
9. Способ по п. 8, где 2-пропанол на стадии (с) обрабатывают гидроксидом натрия.
10. Способ по п. 8, где натриевая соль моно[3-(гексадецилокси)пропилового]эфира Р-[[[(4-метилфенил)сульфонил]окси]метил]фосфоновой кислоты не содержит примеси:
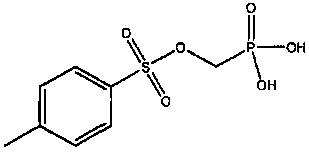
11. Способ по п. 8, дополнительно включающий синтез (дихлорфосфорил)метил 4-метилбензолсульфоната путем:
(a) взаимодействия диэтил(тозилокси)метилоксифосфоната и ацетонитрила с бромтриметилсиланом при нагревании с образованием соответствующей реакционной смеси; и
(b) добавления к полученной смеси дихлорметана и оксалилхлорида с получением (дихлорфосфорил)метил 4-метилбензолсульфоната.
12. Способ по п. 8, дополнительно включающий синтез 3-(гексадецилокси)пропан-1-ола путем:
(a) взаимодействия 1,3-пропандиола с гидридом натрия в N,N-диметилформамиде с образованием соответствующей реакционной смеси; и
(b) добавления к полученной смеси раствора гексадецил метансульфоната в N,N-диметилформамиде с получением 3-(гексадецилокси)пропан-1-ола.
13. Способ по п. 12, дополнительно включающий синтез гексадецил метансульфоната путем:
(а) взаимодействия 1-гексадеканола, дихлорметана и диизопропилэтиламина с образованием соответствующей смеси; и
(b) добавления к полученной смеси метансульфонилхлорида с получением гексадецилметансульфоната.
14. Способ синтеза [[(S)-2-(4-амино-2-оксо-1(2Н)-пиримидинил)-1-(гидроксиметил)этокси]метил]моно[3-(гексадецилокси)пропилового]эфира фосфоновой кислоты, включающий:
взаимодействие (S)-N1-[(2-гидрокси-3-(PG-О)пропил]цитозина с натриевой солью моно[3-(гексадецилокси)пропилового]эфира р-[[[(4-метилфенил)сульфонил]окси]метил]фосфоновой кислоты в присутствии ди-трет-бутоксида магния и N,N-диметилформамида с образованием [3-(гексадецилокси)пропил]гидро[[[(S)-1-(4-амино-2-оксопиримидин-1(2Н)-ил)-3-(PG-О)пропан-2-ил]окси]метил]фосфоната; и
взаимодействие [3-(гексадецилокси)пропил]гидро[[[(S)-1-(4-амино-2-оксопиримидин-1(2Н)-ил)-3-(PG-О)пропан-2-ил]окси]метил]фосфоната с реагентом для удаления защитной группы в присутствии метанола образованием [[(S)-2-(4-амино-2-оксо-1(2Н)-пиримидинил)-1-(гидроксиметил)этокси]метил]моно[3-(гексадецилокси)пропилового]эфира фосфоновой кислоты; где PG означает фрагмент, защищающий гидроксильную группу.
15. Способ по п. 14, где дитрет-бутоксид магния имеет чистоту более 98%.
16. Способ по п. 14, дополнительно включающий перекристаллизацию [[(S)-2-(4-амино-2-оксо-1(2Н)-пиримидинил)-1-(гидроксиметил)этокси]метил]моно[3-(гексадецилокси)пропилового]эфира фосфоновой кислоты из подходящего для перекристаллизации органического растворителя с получением морфологической формы указанной фосфоновой кислоты, характеризующейся картиной дифракции рентгеновских лучей, включающей пики при углах 2θ примерно 5,5, 19,3, 20,8 и 21,3 градуса.
17. Способ по п. 14, дополнительно включающий синтез (S)-N1-[(2-гидрокси-3-(PG-О)пропил]цитозина путем:
взаимодействия цитозина с (S)-2-(PG-O-метил)оксираном в присутствии карбоната металла и N,N-диметилформамида с образованием (S)-N1-[(2-гидрокси-3-(PG-О)пропил]цитозина.
| WO 2008133966 A1, 06.11.2008 | |||
| US 20070003516 A1, 04.01.2007 | |||
| US 6716825 B2, 06.04.2004 | |||
| Устройство для использования энергии волн с целью подъема воды | 1928 |
|
SU11948A1 |

