Данное изобретение относится к новому способу получения хиназолиновых соединений, которые полезны для терапии. Более конкретно, данные соединения полезны для лечения доброкачественной гиперплазии простаты.
В публикации международной патентной заявки WO 98/30560 описан ряд замещенных хинолиновых и хиназолиновых соединений формулы (I), которые нашли применение при лечении доброкачественной гиперплазии простаты
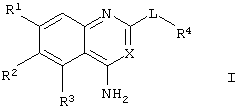
где
R1 представляет С1-4алкокси, необязательно замещенный одним или более атомами фтора;
R2 представляет Н или С1-6алкокси, необязательно замещенный одним или более атомами фтора;
R3 представляет 5- или 6-членное гетероциклическое кольцо, содержащее, по меньшей мере, один гетероатом, выбранный из N, О и S, причем данное кольцо является необязательно замещенным одной или более группами, выбранными из галогена, С1-4алкокси, С1-4алкила и CF3;
R4 представляет 4-, 5-, 6- или 7-членное гетероциклическое кольцо, содержащее, по меньшей мере, один гетероатом, выбранный из N, О и S, причем данное кольцо является необязательно конденсированным с бензольным кольцом или 5- или 6-членным гетероциклическим кольцом, содержащим, по меньшей мере, один гетероатом, выбранный из N, О и S, и причем данная кольцевая система в целом является необязательно замещенной одной или более группами, независимо выбранными из ОН, С1-4алкила, С1-4алкокси, галогена, CONR8R9, SO2NR8R9, (СН2)bNR8R9 и
NHSO2(С1-4алкила), и когда S является членом кольцевой системы, он может быть замещенным одним или двумя атомами кислорода;
R8 и R9 независимо представляют Н, С1-4алкил или вместе с атомом N, к которому они присоединены, они могут представлять 5- или 6-членное гетероциклическое кольцо, содержащее, по меньшей мере, один гетероатом, выбранный из N, О и S;
b представляет 0, 1, 2 или 3;
Х представляет СН или N; и
L отсутствует или представляет циклическую группу формулы Ia,
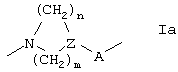
где N связан в положении 2 хинолинового или хиназолинового кольца;
А отсутствует или представляет СО или SO2;
Z представляет СН или N;
m представляет 1 или 2, и, кроме того, когда Z представляет СН, он может означать 0; и
n означает 1, 2 или 3 при условии, что сумма m и n равна 2, 3, 4 или 5; или представляет цепь формулы Ib
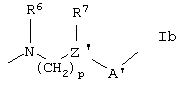
в которой N связан в положении 2 хинолинового или хиназолинового кольца;
А' и Z' имеют те же значения, что и А и Z, выше соответственно;
R6 и R7 независимо представляют Н или С1-4алкил; и
р представляет 1, 2 или 3, и, кроме того, когда Z' представляет СН, он может означать 0.
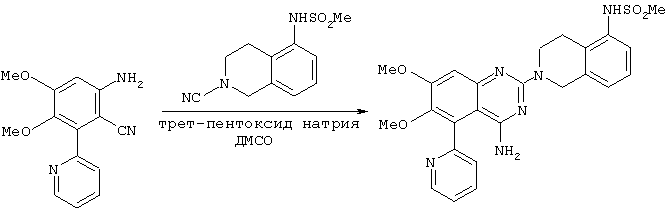
Соединения формулы (I), в которой Х представляет N и L отсутствует, представляют особый интерес. Из этих соединений 4-амино-2-(5-метансульфонамидо-1,2,3,4-тетрагидро-2-изохинолил)-6,7-диметокси-5-(2-пиридил)хиназолин особенно интересен.
В соответствии с WO 98/30560 соединения формулы (I) могут быть получены рядом способов. Однако ни один из этих способов не включает конденсацию двух главных частей молекулы в конвергентном синтезе, при котором образуется хиназолиновое кольцо, и каждый способ имеет недостатки. Например, 4-амино-2-(5-метансульфонамидо-1,2,3,4-тетрагидроизохинол-2-ил)-6,7-диметокси-5-(2-пиридил)хиназолин (соединение примера 19 в WO 98/30560) получают по следующей схеме:
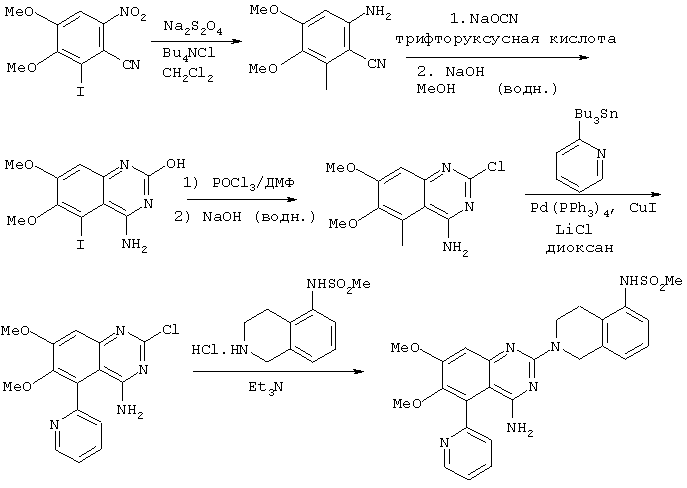
Пути, описанные в WO 98/30560, имеют недостаток, который заключается в том, что они включают использование трибутилоловопиридина в сочетании с йодидом меди и тетракис(трифенилфосфин)палладием. Одной из проблем данного пути синтеза является то, что трибутилоловопиридин очень дорог для приобретения. Данное соединение является токсичным, и существуют проблемы безопасности для работников и связанные с окружающей средой. После использования трудно и дорого избавиться от использованных реагентов из-за вредного действия, которыми обладают оловоорганические соединения на окружающую среду. Еще одной проблемой, связанной с предшествующим способом, является отсутствие конвергенции. Для получения хиназолиновых соединений описанными способами необходим ряд стадий синтеза, причем каждая стадия синтеза приводит как к снижению выхода, так и к увеличению возможности протекания конкурирующих побочных реакций. Таким образом, обычная последовательность реакций требует усилий по очистке продукта и не может дать оптимальный выход.
Еще одной проблемой, связанной с предшествующим способом, описанным в WO 98/30560, является то, что во время реакции в реакторе образуются большие галькоподобные агрегаты. Природа этих агрегатов неясна, но полагают, что они образуются из неорганического материала, образованного различными неорганическими добавками, используемыми во время реакции, такими как хлорид лития или йодид меди. В данном способе существует риск того, что галькоподобные агрегаты могут повредить (вызвать образование трещин) реактор, вызывая протечки реакционной среды и угрозу возгорания или отравления. И, самое меньшее, существует проблема того, что реакция приводит к царапинам на внутренней поверхности реакционного сосуда, являясь, таким образом, причиной преждевременного изнашивания сосуда, плохой теплоотдачи смеси или блокирования.
Недавно сообщалось об использовании раствора метоксида натрия в диоксане для синтеза 2-аминохиназолинов (см. van Muijlwijk-Koezen et al., J. Med. Chem., 2000, vol.43(11), p.2227-2238, в частности получение соединения 4k):
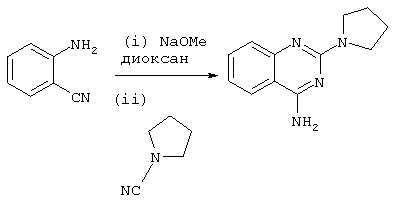
Однако реакция осуществлялась при кипячении с обратным холодильником, и полученный выход составлял только 34%.
Целью данного изобретения является создание способа эффективного синтеза для получения хиназолиновых производных, который исключает проблемы предшествующего способа. Целью также является и обеспечение способа, при котором максимизирована конвергенция (т.е. сведение вместе синтетических фрагментов). Таким образом, цель состоит в создании пути синтеза соединений формулы (I), представляющих наибольший интерес, который дает улучшенный выход относительно существующих путей синтеза. Еще одной целью способа данного изобретения является устранение использования оловоорганических соединений из-за их опасных свойств. Еще одной целью данного изобретения является получение способа, который сводит к минимуму число необходимых стадий синтеза и который устраняет проблему протекания конкурирующих реакций и/или устранения опасных материалов. Желательно также исключить нагревание реакционных смесей, где возможно.
Найден улучшенный путь синтеза хиназолиновых производных формулы (I), представляющих огромный интерес, который отвечает некоторым или всем из вышеуказанных целей.
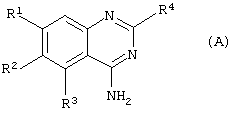
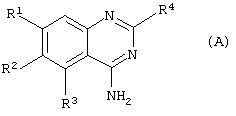
Соответственно данному изобретению представлен способ получения соединения формулы (А) или его фармацевтически приемлемых соли или сольвата
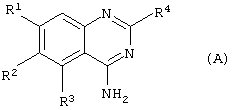
где
R1 представляет С1-4алкокси, необязательно замещенный одним или более атомами фтора;
R2 представляет Н или С1-6алкокси, необязательно замещенный одним или более атомами фтора;
R3 представляет 5- или 6-членное гетероциклическое кольцо, содержащее, по меньшей мере, один гетероатом, выбранный из N, О и S, причем данное кольцо является необязательно замещенным одной или более группами, выбранными из галогена, С1-4алкокси, С1-4алкила и CF3;
R4 представляет 4-, 5-, 6- или 7-членное гетероциклическое кольцо, содержащее, по меньшей мере, один гетероатом, выбранный из N, О и S, причем данное кольцо является необязательно конденсированным с бензольным кольцом или 5- или 6-членным гетероциклическим кольцом, содержащим, по меньшей мере, один гетероатом, выбранный из N, О и S, причем данная кольцевая система в целом является необязательно замещенной одной или более группами, независимо выбранными из ОН, С1-4алкила,
С1-4алкокси, галогена, CONR7R8, SO2NR7R8, (СН2)bNR7R8 и
NHSO2(С1-4алкила), и когда S является членом кольцевой системы, он может быть замещенным одним атомом или двумя атомами кислорода;
R7 и R8 независимо представляют Н, С1-4алкил, или вместе с атомом N, к которому они присоединены, они могут представлять 5- или 6-членное гетероциклическое кольцо, содержащее, по меньшей мере один гетероатом, выбранный из N, О и S;
b представляет 0, 1, 2 или 3;
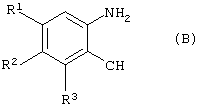
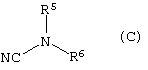
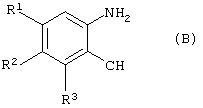
который включает конденсацию соединения формулы (В)
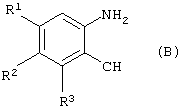
где
R1-R3 имеют значения, указанные выше,
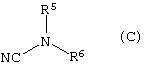
с соединением формулы (С)
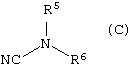
где
R5 и R6, взятые вместе с атомом N, к которому они присоеденены, представляют 4-, 5-, 6- или 7-членное N-содержащее гетероциклическое кольцо, содержащее, по меньшей мере, один гетероатом, выбранный из N, О и S, причем кольцо является необязательно сконденсированным с бензольным кольцом или 5- или 6-членным гетероциклическим кольцом, содержащим, по меньшей мере, один гетероатом, выбранный из N, О и S, причем кольцевая система в целом является необязательно замещенной одной или более группами, независимо выбранными из ОН, С1-4алкила, С1-4алкокси, галогена, CONR7R8, SO2NR7R8, (СН2)bNR7R8 и
NHSO2(С1-4алкила), и когда S является членом кольцевой системы, он может быть замещенным одним или двумя атомами кислорода;
R7, R8 и b имеют значения, указанные выше; и
где необходимо или желательно превращение полученного соединения формулы (А) в фармацевтически приемлемую соль или сольват или превращение полученной соли или сольвата в соединение формулы (А).
Предпочтительно R1 представляет метокси.
Предпочтительно R2 представляет метокси.
Предпочтительно R3 представляет ароматическое кольцо.
Более предпочтительно R3 представляет пиридил, пиримидил, тиенил, фуранил или оксазолил. Более предпочтительно R3 представляет 2-пиридил или 2-пиримидил, причем 2-пиридил наиболее предпочтителен.
Предпочтительно R5 и R6, взятые вместе с атомом N, к которому они присоединены, представляют насыщенное 6-членное N-содержащее кольцо, которое конденсировано с необязательно замещенным бензольным или пиридиновым кольцом. Более предпочтительно R5 и R6 вместе с атомом N, к которому они присоединены, представляют необязательно замещенную тетрагидроизохинолиновую кольцевую систему. Наиболее предпочтительно R5 и R6, взятые вместе с атомом N, с которым они связаны, представляют группу формулы
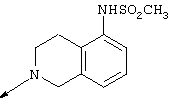
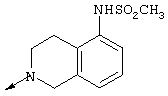
Таким образом, способ предпочтительно используется для получения 4-амино-2-(5-метансульфонамидо-1,2,3,4-тетрагидро-2-изохинолил)-6,7-диметокси-5-(2-пиридил)хиназолина.
Предпочтительно реакция осуществляется в полярном апротонном растворителе например, диметилсульфоксиде.
Предпочтительно реакция осуществляется при температуре в интервале 10-30°С.
Предпочтительно реакция осуществляется в присутствии основания. Более предпочтительно основание является алкоксидом щелочного металла или алкоксидом щелочноземельного металла. Более предпочтительно основание является трет-бутоксидом натрия или трет-пентоксидом натрия, причем последний является наиболее предпочтительным.
В еще одном аспекте данного изобретения представлен способ получения соединения формулы (С), определенной выше, который включает взаимодействие соединения формулы (Е), HNR5R6 (Е) или его кислотно-аддитивной соли, где R5 и R6 имеют значения, указанные выше, с BrCN в присутствии основания амина.
Предпочтительно основанием является три-С1-8алкил, С3-8циклоалкил или гетероциклический амин. Наиболее предпочтительно основанием является N,N-диизопропилэтиламин.
Данное изобретение представляет также альтернативный способ производства соединения формулы (С), определенной выше, который включает взаимодействие соединения формулы (F)
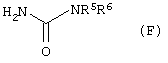
где R5 и R6 имеют значения, указанные в пункте 1, с метансульфонилхлоридом в присутствии пиридина. Соединения формулы (F) могут быть получены из соединений формулы (Е) взаимодействием с цианатом натрия в воде, что проиллюстрировано примером 3А(а). Соединения формулы (Е) или известны, или могут быть получены известными методами.
Предпочтительно два способа получения соединений формулы (С), указанные выше, используются для получения N-(2-циано-1,2,3,4-тетрагидро-5-изохинолил)метансульфонамида.
В другом аспекте данного изобретения представлен способ получения соединения формулы (В)
где
R1-R3 имеют значения, указанные выше;
который включает взаимодействие соединения формулы (D)
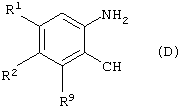
где
R1 и R2 имеют значения, указанные выше; и
R9 является удаляемой группой;
с пиридилборонатом.
Предпочтительно R9 является йодом.
Предпочтительно пиридиновое производное является 2-пиридилборонатом.
Наиболее предпочтительно способ используется для получения 6-амино-3,4-диметокси-2-(2-пиридил)бензонитрила.
Предпочтительно реакция осуществляется в полярном апротонном растворителе. Более предпочтительно полярный апротонный растворитель является тетрагидрофураном или изопропилацетатом.
Предпочтительно реакция присоединения с образованием 6-амино-3,4-диметокси-2-(2-пиридил)бензонитрила осуществляется при температуре выше комнатной. Предпочтительно эта реакция осуществляется в присутствии катализатора. Более предпочтительно катализатор является катализатором палладия(0). Наиболее предпочтительно катализатор производят из ацетата палладия(II) восстановлением in situ. Предпочтительно реакция присоединения осуществляется в присутствии основания. Основанием предпочтительно является карбонат щелочного металла и наиболее предпочтительно им является карбонат калия.
Пиридилборонат может быть получен взаимодействием бромпиридина с триизопропилборатом при температуре ниже комнатной. Предпочтительно данная реакция осуществляется в присутствии основания. Основание предпочтительно является алкиллитиевым реагентом. н-Бутиллитий является предпочтительным алкиллитиевым реагентом.
Соединения формулы (D) могут быть получены из известных соединений или соединений, которые легко получить, используя известные методики, как проиллюстрировано в примере 1.
Данное изобретение, кроме того, представляет соединения формулы (В), определенной выше, и соединения формулы (С), определенной выше.
Данное изобретение иллюстрируется следующими примерами, в которых могут использоваться следующие аббревиатуры:
ДМСО = диметилсульфоксид
ДХМ = дихлорметан
Et = этил
EtOAc = этилацетат
К2ЭДТУ = этилендиаминтетрауксусной кислоты дикалиевая соль
nBuLi = н-бутиллитий
ОАс = ацетат
ТГФ = тетрагидрофуран
Пример 1
Получение 6-амино-2-йодо-3,4-диметоксибензонитрила
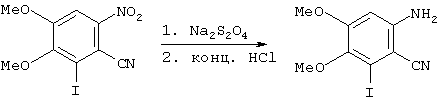
В реакционный сосуд загружали суспензию 6-нитро-2-йодо-3,4-диметоксибензонитрила [см. пример 1(d) WO 98/30560, 10 кг] в этаноле (60 л) при комнатной температуре с раствором дитионита натрия (15,6 кг материала технической марки) в воде (67,5 л) за 45 мин, поддерживая температуру ниже 35°С. Сосуд для добавления промывали водой (10 л). Полученную смесь кипятили с обратным холодильником (примерно 85°С) в течение примерно 90 мин и затем температуру доводили до 65°С. 6М водный раствор соляной кислоты (12,5 л) добавляли за 10 мин и полученную смесь перемешивали при 65°С в течение примерно 5 часов до того, как охлаждали до комнатной температуры. Показатель рН доводили до интервала 7-8, используя 40% раствор гидроксида натрия (2 л), полученную смесь оставляли перемешиваться в течение 3 часов, фильтровали и промывали водой (50 л). Слой осадка взбалтывали до густой суспензии в воде (90 л) в течение ночи при комнатной температуре, фильтровали, промывали водой (100 л) и сушили в вакууме с получением 8,35 кг (92%) соединения, названного в заголовке.
Пример 2
Получение 6-амино-3,4-диметокси-2-(2-пиридил)бензонитрила
(а) Получение 2-пиридилбороната
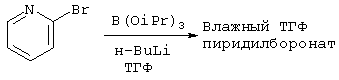
В атмосфере азота перемешиваемый раствор 2-бромпиридина (150 г, 0,95 моль) и триизопропилбората (218 мл, 0,95 моль) в ТГФ охлаждали до -25°С. Добавляли 2,5 М раствор н-BuLi в гексане (378 мл, 0,95 моль) с такой скоростью, чтобы температура не превышала -24°С. После завершения добавления реакционной смеси давали нагреться до комнатной температуры и перемешивали при этой температуре в течение 18 часов. После этого реакционную смесь фильтровали, промывали ТГФ, и процедура, как полагали, завершалась до того, как слой на фильтре полностью высыхал. Часть увлажненного ТГФ бороната использовали в следующей реакции. Анализ увлажненного ТГФ бороната 1Н ЯМР показал соотношение пиридин Н:изопропилметин Н, равное 1:3,75, и что материал содержал 54% мас. растворителя.
(b) Получение 6-амино-3,4-диметокси-2-(2-пиридил)бензонитрила
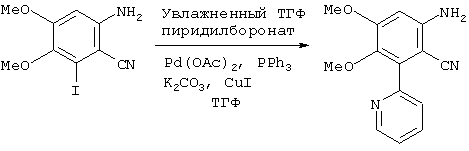
В атмосфере азота к безводному ТГФ (1000 мл) добавляли 6-амино-2-йод-3,4-диметоксибензонитрил (см. пример 1, 50,0 г; 164 ммоль), Pd(ОАс)2 (1,85 г, 8,22 ммоль), PPh3 (трифенилфосфин, 4,31 г, 16,4 ммоль), увлажненный ТГФ боронат [со стадии (а), 286 г, 493 ммоль], CuI (12,5 г, 65 ммоль) и К2СО3 (45,5 г; 328 ммоль). После этого реакционную смесь охлаждали до комнатной температуры и добавляли воду (1000 мл). Затем смесь фильтровали через слой вспомогательного фильтрующего материала Arbocel™ и слой промывали ТГФ (500 мл). Фильтрат затем экстрагировали СН2Cl2 (1000 мл). Водную фазу обратно экстрагировали СН2Cl2 (500 мл) и объединенные экстракты в СН2Cl2 выпаривали в вакууме с получением неочищенного темно-коричневого твердого вещества. Перекристаллизация из EtOAc (250 мл) давала 37,6 г (87%) соединения, названного в заголовке, в виде бежевого твердого вещества.
Пример 2А
Альтернативный способ получения 6-амино-3,4-диметокси-2-(2-пиридил)бензонитрила
(а) Получение N-фенилдиэтаноламинпиридилбороната
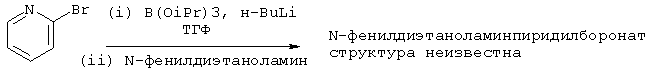 В атмосфере азота перемешиваемый раствор 2-бромпиридина (843 г, 5,33 моль) и триизопропилбората (1,20 кг, 6,40 моль) в ТГФ (6,74 л) охлаждали до -75°С. Добавляли 1,6 М раствор н-BuLi в гексане (4,00 л, 6,40 моль) с такой скоростью, чтобы температура не превышала -67°С. После завершения добавления реакционной смеси давали нагреться до комнатной температуры и перемешивали при этой температуре в течение 16 часов. После этого добавляли раствор N-фенилдиэтаноламина (966 г, 5,33 моль) в ТГФ (966 мл) и полученную смесь кипятили с обратным холодильником в течение 4 часов. Растворитель отгоняли и заменяли изопропанолом до тех пор, пока верхняя температура составляла 76°С (отгонка 11,3 л и добавление 8,4 л изопропанола во время процесса). Смесь охлаждали до комнатной температуры и перемешивали при этой температуре в течение 12 часов. Смесь фильтровали, твердое вещество промывали изопропанолом (1,7 л) и сушили в вакууме в течение ночи при 40°С с получением 1605 г соединения, названного в подзаголовке. Анализ 1Н ЯМР показал соотношение пиридин:N-фенилдиэтаноламин (или литийалкоксида):изопропил, равное 1:1,25:1,55.
В атмосфере азота перемешиваемый раствор 2-бромпиридина (843 г, 5,33 моль) и триизопропилбората (1,20 кг, 6,40 моль) в ТГФ (6,74 л) охлаждали до -75°С. Добавляли 1,6 М раствор н-BuLi в гексане (4,00 л, 6,40 моль) с такой скоростью, чтобы температура не превышала -67°С. После завершения добавления реакционной смеси давали нагреться до комнатной температуры и перемешивали при этой температуре в течение 16 часов. После этого добавляли раствор N-фенилдиэтаноламина (966 г, 5,33 моль) в ТГФ (966 мл) и полученную смесь кипятили с обратным холодильником в течение 4 часов. Растворитель отгоняли и заменяли изопропанолом до тех пор, пока верхняя температура составляла 76°С (отгонка 11,3 л и добавление 8,4 л изопропанола во время процесса). Смесь охлаждали до комнатной температуры и перемешивали при этой температуре в течение 12 часов. Смесь фильтровали, твердое вещество промывали изопропанолом (1,7 л) и сушили в вакууме в течение ночи при 40°С с получением 1605 г соединения, названного в подзаголовке. Анализ 1Н ЯМР показал соотношение пиридин:N-фенилдиэтаноламин (или литийалкоксида):изопропил, равное 1:1,25:1,55.
(b) Получение 6-амино-3,4-диметокси-2-(2-пиридил)бензонитрила
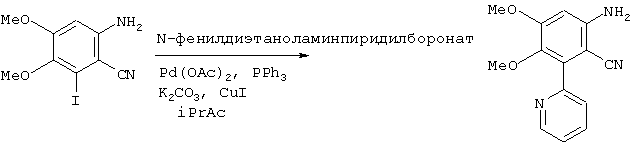 В атмосфере азота 6-амино-2-йод-3,4-диметоксибензонитрил (см. пример 1, 100 г, 0,33 моль) суспендировали в изопропилацетате (1,4 л) при 20°С. В суспензию загружали ацетат палладия (3,69 г, 16 ммоль) с последующей загрузкой трифенилфосфина (17,25 г, 66 ммоль), N-фенилдиэтаноламинпиридилбороната (263,4 г из партии, полученной, как описано выше), йодида меди (25,05 г, 0,13 моль), затем карбоната калия (90,9 г, 0,66 моль) и суспензию кипятили с обратным холодильником в течение 8 часов. Суспензию охлаждали до 70°С и выдерживали при этой температуре в течение ночи. Суспензию охлаждали до 45°С, добавляли тетрагидрофуран (1 л) и суспензию перемешивали при 45°С в течение 1 часа. После этого добавляли вспомогательный фильтрующий материал Arbocel™ и смесь фильтровали через вспомогательный фильтрующий материал Arbocel™. Слой промывали тетрагидрофураном (2×200 мл) и изопропилацетатом (200 мл). Полученный раствор дважды промывали смесью 1:1 5% водного раствора К2ЭДТУ и насыщенным раствором соли (800 мл), затем промывали смесью 1:1 воды и насыщенного раствора соли (800 мл). Органическую фазу отгоняли и заменяли изопропилацетатом так, чтобы оставить конечный объем изопропилацетата, равный 500 мл, который оставляли охлаждаться до комнатной температуры в течение ночи. Полученную суспензию фильтровали с получением светло-коричневого твердого вещества, которое сушили в вакууме в течение ночи при 45°С с выходом 59,6 г (71%) соединения, названного в заголовке.
В атмосфере азота 6-амино-2-йод-3,4-диметоксибензонитрил (см. пример 1, 100 г, 0,33 моль) суспендировали в изопропилацетате (1,4 л) при 20°С. В суспензию загружали ацетат палладия (3,69 г, 16 ммоль) с последующей загрузкой трифенилфосфина (17,25 г, 66 ммоль), N-фенилдиэтаноламинпиридилбороната (263,4 г из партии, полученной, как описано выше), йодида меди (25,05 г, 0,13 моль), затем карбоната калия (90,9 г, 0,66 моль) и суспензию кипятили с обратным холодильником в течение 8 часов. Суспензию охлаждали до 70°С и выдерживали при этой температуре в течение ночи. Суспензию охлаждали до 45°С, добавляли тетрагидрофуран (1 л) и суспензию перемешивали при 45°С в течение 1 часа. После этого добавляли вспомогательный фильтрующий материал Arbocel™ и смесь фильтровали через вспомогательный фильтрующий материал Arbocel™. Слой промывали тетрагидрофураном (2×200 мл) и изопропилацетатом (200 мл). Полученный раствор дважды промывали смесью 1:1 5% водного раствора К2ЭДТУ и насыщенным раствором соли (800 мл), затем промывали смесью 1:1 воды и насыщенного раствора соли (800 мл). Органическую фазу отгоняли и заменяли изопропилацетатом так, чтобы оставить конечный объем изопропилацетата, равный 500 мл, который оставляли охлаждаться до комнатной температуры в течение ночи. Полученную суспензию фильтровали с получением светло-коричневого твердого вещества, которое сушили в вакууме в течение ночи при 45°С с выходом 59,6 г (71%) соединения, названного в заголовке.
Пример 3
Получение N-(2-циано-1,2,3,4-тетрагидро-5-изохинолил)метансульфонамида
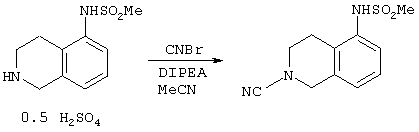
К перемешиваемой густой суспензии N-(1,2,3,4-тетрагидро-5-изохинолил)метансульфонамида гемисульфата (полученного аналогично соединению примера 19(b) в WO 98/30560, но с использованием серной кислоты на последней стадии вместо соляной кислоты; (240 г, 0,88 моль) в ацетонитриле (2400 мл) при 0°С добавляли N,N-диизопропилэтиламин (326 мл, 1,88 ммоль). Добавляли цианогенбромид (99,2 г, 0,94 моль) в течение 20 минут, поддерживая температуру ниже 10°С. Полученной густой суспензии давали нагреться до 20°С и перемешивали при данной температуре в течение 18 часов. После этого периода добавляли воду (2400 мл) и полученную смесь экстрагировали СН2Cl2 (2×2500 мл). Объединенные органические фазы промывали водой (2000 мл) и выпаривали до сухости. Из полученного твердого вещества снова готовили густую суспензию в СН2Cl2 (360 мл) и твердое вещество собирали фильтрованием с получением 158 г (72%) соединения, названного в заголовке.
Пример 3А
Альтернативный способ получения N-(2-циано-1,2,3,4-тетрагидро-5-изохинолил)метансульфонамида
(а) Получение N-(2-карбоксамид-1,2,3,4-тетрагидро-5-изохинолил)метансульфонамида
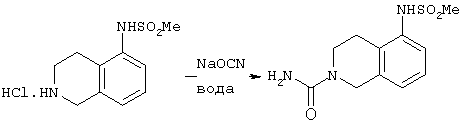
В атмосфере азота N-(1,2,3,4-тетрагидро-5-изохинолил)метансульфонамида гидрохлорид (см. пример 19(b), WO 98/30560, 50 г, 190 моль) суспендировали в воде (250 мл) при 20°С. Медленно в течение 5 минут добавляли раствор цианата натрия (16,1 г, 247 ммоль) в воде (250 мл) и затем смесь перемешивали при комнатной температуре в течение 18 часов. Полученную суспензию фильтровали с получением белого твердого вещества, которое сушили в вакууме в течение ночи при 45°С с выходом 45,0 г (88%) соединения, названного в подзаголовке.
(b) Получение N-(2-циано-1,2,3,4-тетрагидро-5-изохинолил)метансульфонамида
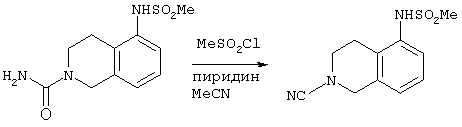
В атмосфере азота N-(2-карбоксамид-1,2,3,4-тетрагидро-5-изохинолил)метансульфонамид [со стадии (а), 10,0 г, 37,1 моль] суспендировали в ацетонитриле (100 мл) при 20°С. К суспензии добавляли метансульфонилхлорид (6,38 г, 55,6 ммоль) и пиридин (7,34 г, 92,8 ммоль). Реакционную смесь перемешивали до образования раствора, затем нагревали до 50°С и выдерживали при этой температуре в течение 4 часов. Раствор охлаждали до комнатной температуры и перемешивали в течение ночи. Растворитель удаляли в вакууме и заменяли водой (60 мл). Полученную суспензию перемешивали в течение 3 часов, затем фильтровали с получением белого твердого вещества. Твердое вещество суспендировали в ацетонитриле (50 мл) и перемешивали при 20°С в течение 3 часов. Суспензию фильтровали с получением твердого белого вещества, которое сушили в вакууме в течение ночи при 45°С с выходом 7,4 г (79%) соединения, названного в заголовке.
Пример 4
4-амино-2-(5-метансульфонамидо-1,2,3,4-тетрагидро-2-изохинолил)-6,7-диметокси-5-(2-пиридил)хиназолин
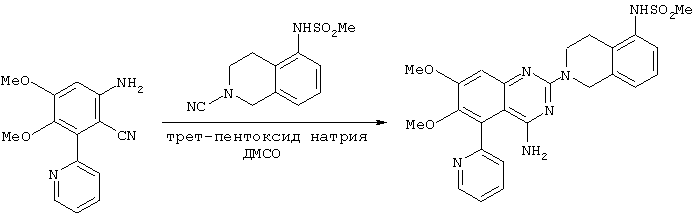 К перемешиваемому раствору 6-амино-3,4-диметокси-2-(2-пиридил)бензонитрила (см. пример 2 или 2А, 7 г, 27 ммоль) и N-(2-циано-1,2,3,4-тетрагидро-5-изохинолил)метансульфонамида (см. пример 3 или 3А, 9 г, 36 ммоль) в ДМСО (21 мл) при комнатной температуре порциями добавляли трет-пентоксид натрия (9,5 г, 92 ммоль) за двадцать минут, поддерживая температуру ниже 30°С. Полученную густую суспензию затем перемешивали в течение 2 часов. После этого добавляли ледяную воду (35 мл) за 1 минуту с последующим добавлением этилацетата (35 мл) перед снижением рН двухфазной смеси до 8,0 с помощью 2М водного раствора HCl (20 мл). Водную фазу экстрагировали этилацетатом (50 мл). Объединенные органические фазы промывали насыщенным раствором NaCl (2×30 мл), доводили объем и перемешивали в течение 3 часов. После этого полученную густую суспензию фильтровали с выходом 10,2 г (74%) соединения, названного в заголовке.
К перемешиваемому раствору 6-амино-3,4-диметокси-2-(2-пиридил)бензонитрила (см. пример 2 или 2А, 7 г, 27 ммоль) и N-(2-циано-1,2,3,4-тетрагидро-5-изохинолил)метансульфонамида (см. пример 3 или 3А, 9 г, 36 ммоль) в ДМСО (21 мл) при комнатной температуре порциями добавляли трет-пентоксид натрия (9,5 г, 92 ммоль) за двадцать минут, поддерживая температуру ниже 30°С. Полученную густую суспензию затем перемешивали в течение 2 часов. После этого добавляли ледяную воду (35 мл) за 1 минуту с последующим добавлением этилацетата (35 мл) перед снижением рН двухфазной смеси до 8,0 с помощью 2М водного раствора HCl (20 мл). Водную фазу экстрагировали этилацетатом (50 мл). Объединенные органические фазы промывали насыщенным раствором NaCl (2×30 мл), доводили объем и перемешивали в течение 3 часов. После этого полученную густую суспензию фильтровали с выходом 10,2 г (74%) соединения, названного в заголовке.
Пример 4А
Альтернативное получение 4-амино-2-(5-метансульфонамидо-1,2,3,4-тетрагидро-2-изохинолил)-6,7-диметокси-5-(2-пиридил)-хиназолина
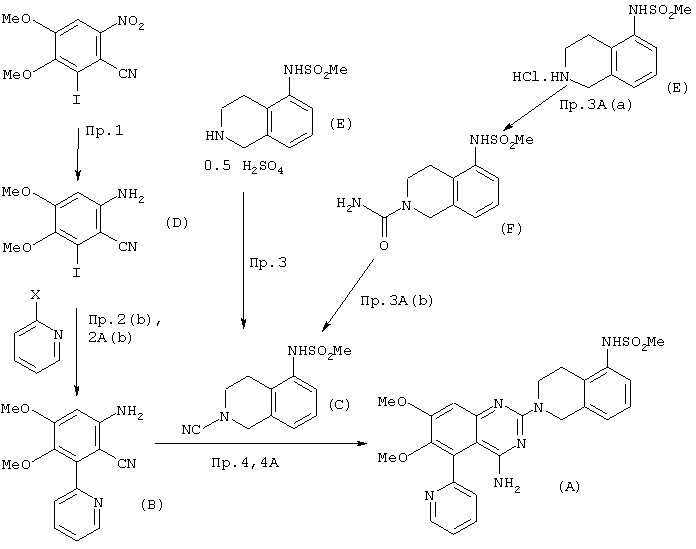
 К перемешиваемому раствору 6-амино-3,4-диметокси-2-(2-пиридил)бензонитрила (см. пример 2 или 2А, 50 г, 196 ммоль) и N-(2-циано-1,2,3,4-тетрагидро-5-изохинолил)метансульфонамида (см. пример 3 или 3А, 63 г, 251 ммоль) в ДМСО (300 мл) при комнатной температуре порциями в течение 120 мин добавляли трет-пентоксид натрия (64,2 г, 582 ммоль), поддерживая температуру ниже 30°С. Полученную густую суспензию затем перемешивали в течение 2 часов. После этого добавляли воду (500 мл) за 5 минут с последующим добавлением изопропилацетата (150 мл). Водную фазу собирали и разделяли с помощью этилацетата (500 мл) перед снижением рН бифазной смеси до интервала 7,0-8,0 с помощью 12М водного раствора HCl (22 мл). Водную фазу экстрагировали этилацетатом (250 мл). Снижали объем объединенных органических экстрактов и заменяли ацетонитрилом с получением конечного объема, равного 500 мл, и перемешивали в течение 13 часов. После этого периода полученную густую суспензию фильтровали с выходом 105 г неочищенного продукта. Данный продукт суспендировали в ацетонитриле (525 мл), кипятили с обратным холодильником в течение одного часа, охлаждали до комнатной температуры и перемешивали в течение 13 часов. Полученную густую суспензию фильтровали с получением 87 г (87%) соединения, названного в заголовке.
К перемешиваемому раствору 6-амино-3,4-диметокси-2-(2-пиридил)бензонитрила (см. пример 2 или 2А, 50 г, 196 ммоль) и N-(2-циано-1,2,3,4-тетрагидро-5-изохинолил)метансульфонамида (см. пример 3 или 3А, 63 г, 251 ммоль) в ДМСО (300 мл) при комнатной температуре порциями в течение 120 мин добавляли трет-пентоксид натрия (64,2 г, 582 ммоль), поддерживая температуру ниже 30°С. Полученную густую суспензию затем перемешивали в течение 2 часов. После этого добавляли воду (500 мл) за 5 минут с последующим добавлением изопропилацетата (150 мл). Водную фазу собирали и разделяли с помощью этилацетата (500 мл) перед снижением рН бифазной смеси до интервала 7,0-8,0 с помощью 12М водного раствора HCl (22 мл). Водную фазу экстрагировали этилацетатом (250 мл). Снижали объем объединенных органических экстрактов и заменяли ацетонитрилом с получением конечного объема, равного 500 мл, и перемешивали в течение 13 часов. После этого периода полученную густую суспензию фильтровали с выходом 105 г неочищенного продукта. Данный продукт суспендировали в ацетонитриле (525 мл), кипятили с обратным холодильником в течение одного часа, охлаждали до комнатной температуры и перемешивали в течение 13 часов. Полученную густую суспензию фильтровали с получением 87 г (87%) соединения, названного в заголовке.
Получение 4-амино-2-(5-метансульфонамидо-1,2,3,4-тетрагидро-2-изохинолил)-6,7-диметокси-5-(2-пиридил)хиназолина в соответствии с вышеприведенными примерами проиллюстрирована на следующей схеме, в которой также представлен номер примера каждой стадии и общая формула, которая охватывает соответствующее соединение
| название | год | авторы | номер документа |
|---|---|---|---|
| 1,4,5-ТРИЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ ИМИДАЗОЛА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1996 |
|
RU2196139C2 |
| ИНГИБИТОР ГЕКСОН-ГЛЮКОКИНАЗЫ И ЕГО ПРИМЕНЕНИЕ | 2020 |
|
RU2797185C2 |
| ПРОИЗВОДНЫЕ ТИЕНО[2,3-d]ПИРИМИДИН-2,4(1Н,3Н)-ДИОНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ЕЕ ПОЛУЧЕНИЯ, СПОСОБ ИММУНОСУПРЕССИИ И СПОСОБ ЛЕЧЕНИЯ ИЛИ СНИЖЕНИЯ РИСКА ОБСТРУКЦИИ ДЫХАТЕЛЬНЫХ ПУТЕЙ | 1998 |
|
RU2225410C2 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛА, ИСПОЛЬЗУЕМЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕИНКИНАЗЫ | 2001 |
|
RU2340611C2 |
| N-СОДЕРЖАЩИЕ ГЕТЕРОАРИЛЬНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ JAK3 КИНАЗЫ | 2010 |
|
RU2553681C2 |
| ИНГИБИТОРЫ ГЛУТАМИНФРУКТОЗО-6-ФОСФАТАМИДОТРАНСФЕРАЗЫ | 2004 |
|
RU2355686C2 |
| ПРОИЗВОДНЫЕ ПИРАЗИНА, ПОЛЕЗНЫЕ В КАЧЕСТВЕ АНТАГОНИСТОВ АДЕНОЗИНОВОГО РЕЦЕПТОРА | 2006 |
|
RU2417994C2 |
| АНАЛОГИ НОНИЦЕПТИНА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2002 |
|
RU2268883C2 |
| (ГЕТЕРО)АРИЛЦИКЛОПРОПИЛАМИНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ LSD1 | 2012 |
|
RU2681211C2 |
| БИЦИКЛИЧЕСКИЕ ПИРИДИНЫ И АНАЛОГИ В КАЧЕСТВЕ МОДУЛЯТОРОВ СИРТУИНА | 2010 |
|
RU2550821C2 |
Данное изобретение относится к новому способу получения хиназолинового соединения формулы (А) или его фармацевтически приемлемой соли или сольвата
где R1 представляет С1-4алкокси; R2 представляет C1-6алкокси; R3 представляет 6-членное гетероциклическое кольцо, содержащее, по меньшей мере, один гетероатом, выбранный из N; R4 представляет 6-членное гетероциклическое кольцо, содержащее, по меньшей мере, один гетероатом, выбранный из N, причем данное кольцо является необязательно конденсированным с бензольным, причем данная кольцевая система в целом является необязательно замещенной группой NHSO2(C1-4алкил). Указанные соединения полезны для лечения доброкачественной гиперплазии простаты. Предложенный способ включает конденсацию соединения формулы (В)
где R1-R3 имеют значения, указанные выше, с соединением формулы (С)
где R5 и R6, взятые вместе с атомом N, к которому они присоединены, представляют 6-членное N-содержащее гетероциклическое кольцо, содержащее, по меньшей мере, один гетероатом, выбранный из N, причем кольцо является необязательно конденсированным с бензольным кольцом, причем кольцевая система в целом является необязательно замещенной группой NHSO2(С1-4алкил), в присутствии основания, выбранного из трет-бутоксида натрия и трет-пентоксида натрия; и где необходимо или желательно, превращение полученного соединения формулы (А) в фармацевтически приемлемую соль или сольват или превращение полученной соли или сольвата в соединение формулы (А). Кроме того, объектами изобретения являются способы получения соединения формулы (С) и соединения формулы (В), а также соединения формул (В) и (С). Технический результат - сведение к минимуму числа необходимых стадий синтеза, устранение протекания конкурирующих реакций и использования опасных материалов. 6 н. и 17 з.п. ф-лы.
где R1 представляет С1-4алкокси;
R2 представляет С1-6алкокси;
R3 представляет 6-членное гетероциклическое кольцо, содержащее, по меньшей мере, один гетероатом, выбранный из N;
R4 представляет 6-членное гетероциклическое кольцо, содержащее, по меньшей мере, один гетероатом, выбранный из N, причем данное кольцо является необязательно конденсированным с бензольным, причем данная кольцевая система в целом является необязательно замещенной группой NHSO2(С1-4алкил),
причем данный способ включает конденсацию соединения формулы (В)
где
R1-R3 имеют значения, указанные выше,
с соединением формулы (С)
где
R5 и R6, взятые вместе с атомом N, к которому они присоединены, представляют 6-членное N-содержащее гетероциклическое кольцо, содержащее, по меньшей мере, один гетероатом, выбранный из N, причем кольцо является необязательно конденсированным с бензольным кольцом и кольцевая система в целом является необязательно замещенной группой NHSO2(С1-4алкил),
в присутствии основания, выбранного из третбутоксида натрия и третпентоксида натрия;
и где необходимо или желательно превращение полученного соединения формулы (А) в фармацевтически приемлемую соль или сольват или превращение полученной соли или сольвата в соединение формулы (А).
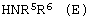
или его кислотно-аддитивной соли, где
R5 и R6 имеют значения, указанные в п.1, с BrCN в присутствии основания амина.
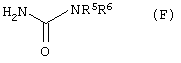
где R5 и R6 имеют значения, указанные в п.1, с метансульфонилхлоридом в присутствии пиридина.
где
R1 и R2 имеют значения, указанные в п.1,
который включает взаимодействие соединения формулы (D)
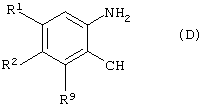
где
R1-R3 имеют значения, указанные в п.1;
R9 является уходящей группой;
с пиридилборонатом.
| Дорожная спиртовая кухня | 1918 |
|
SU98A1 |
| VAN MUIJLWIJK-KOEZEN et al | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Journal of Medical Chemistry | |||
| Зубчатое колесо со сменным зубчатым ободом | 1922 |
|
SU43A1 |
| SEIJAS J.A | |||
| et al | |||
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| Tetrahedron Letters | |||
| Механический грохот | 1922 |
|
SU41A1 |

