Изобретение относится к области антивирусной терапии и, в частности, к ненуклеозидным соединениям, которые ингибируют ВИЧ обратную транскриптазу и применяются для лечения болезней, опосредуемых вирусом иммунодефицита человека (ВИЧ). В изобретении предлагаются новые соединения пиразола согласно формуле I для лечения или профилактики болезней, опосредуемых ВИЧ, СПИД или СПИД-ассоциированным комплексом, и применение названных соединений в монотерапии или комбинационной терапии.
Вирус иммунодефицита человека ВИЧ является агентом, инициирующим синдром иммунодефицита (СПИД), болезнь, характеризующуюся разрушением иммунной системы, в особенности CD4 +Т-клеток, и сопровождающуюся восприимчивостью к посторонним инфекциям. ВИЧ инфекция ассоциируется с предшествующим СПИД-ассоциированным комплексом (ARC), синдромом, характеризующимся такими симптомами, как устойчивая генерализованная лимфаденопатия, лихорадка и потеря веса.
Подобно другим ретровирусам, ВИЧ геном кодирует предшественников белка, известных как gag и gag-pol, которые подвергаются воздействию вирусной протеазы с получением протеазы, обратной транскриптазы (ОТ), эндонуклеазы/интегразы и зрелых структурных белков вирусного ядра. Прерывание этого процесса препятствует нормальному продуцированию инфекционного вируса. Поэтому значительные усилия направляются на контроль ВИЧ посредством ингибирования кодирующих вирусы ферментов.
В настоящее время имеющаяся химиотерапия направлена на два критических вирусных фермента: ВИЧ протеазу и ВИЧ обратную транскриптазу. (J.S.G.Montaner et al. Antiretroviral терапии: 'the state of the art', Biomed & Pharmacother. 1999, 53: 63-72; R.W.Shafer and D.A.Vuitton, Highly active retroviral терапии (HAART) for the лечения of infection with human immunodeficiency virus type, Biomed. & Pharmacother. 1999, 53: 73-86; E. De Clercq, New Developments in Anti-ВИЧ Chemotherap.Curr. Med. Chem. 2001, 8:1543-1572). Два основных класса ингибиторов обратной транскриптазы (ОТИ) подразделяются на: нуклеозидные ингибиторы обратной транскриптазы (NRTI) (НИОТ) и ненуклеозидные ингибиторы обратной транскриптазы (ННИОТ). В настоящее время CCR5 ко-рецептор выбран в качестве потенциальной мишени для анти-ВИЧ химиотерапии (D. Chantry, Expert Opin. Emerg. Drugs 2004, 9(1): 1-7; С.G.Barber Curr. Opin. Invest. Drugs, 2004, 5(8): 851-861; D. Schols Curr. Topics Med. Chem., 2004, 4(9): 883-893; N.A.Meanwell and J.F.Kadow Curr. Opin. Drug Discov. Dev., 2003, 6(4): 451-461).
Типичным представителем НИОТ является 2',3'-дидеоксинуклеозид (ddN), аналоги которого могут быть фосфорилированы перед взаимодействием с вирусной ОТ. Соответствующие трифосфаты функционируют как конкурентные ингибиторы или альтернативные субстраты для вирусной ОТ. После внедрения в нуклеиновые кислоты нуклеозидные аналоги прерывают процесс элонгации цепи. ВИЧ обратная транскриптаза обладает способностью конструирования ДНК, которая дает возможность резистентным штаммам преодолеть блокаду посредством разрыва нуклеозидного аналога и продолжения элонгации. В настоящее время клинически используются НИОТ, включающие: зидовудин (AZT), диданозин (ddI), зальцитабин (ddC), ставудин (d4T), ламивудин (3ТС) и тенофовир (РМРА).
ННИОТ впервые были открыты в 1989 г. ННИОТ представляют собой аллостерические ингибиторы, которые обратимо связываются с несубстратно-связывающим участком ВИЧ обратной транскриптазы посредством измененной части активного участка или блокирования полимеразной активности (R.W.Buckheit, Jr., Non-nucleoside reverse transcriptase inhibitors: perspectives for novel therapeutic compounds and strategies for treatment of HIV infection, Expert Opin. Investig. Drugs, 2001, 10(8)1423-1442; E. De Clercq, The role of non-nucleoside reverse transcriptase inhibitors (NNRTIs) in the therapy of HIV infection, Antiviral Res., 1998, 38:153-179; E. De Clercq, New Developments in Anti-HIV Chemotherapy, Current medicinal Chem., 2001, 8(13):1543-1572; G.Moyle, The Emerging Roles of Non-Nucleoside Reverse Transcriptase Inhibitors in Antiviral Therapy, Drags, 2001, 61 (1): 19-26). Несмотря на то, что в лабораторных условиях были получены свыше тридцати структурных классов ННИОТ, только три соединения были утверждены для применения в ВИЧ терапии: эфавиренз, невирапин и делавирдин.
Первоначально, при рассматрении ННИОТ в качестве перспективного класса соединений в условиях in vitro и in vivo быстро было установлено, что ННИОТ обладают низким барьером к преодолению лекарственной резистентности ВИЧ штаммов и специфической для этого класса токсичности. Лекарственная резистентность часто развивается только исходя из единственной точечной мутации в обратной транскриптазе. Хотя комбинационная терапия с НИОТ, ИР (ингибиторы протеазы) и ННИОТ имеет во многих случаях низшие вирусные нагрузки и оказывает прогрессирующее действие при лечении болезни, значительные терапевтические проблемы остаются нерешенными (R.М.Gulick, Eur. Soc. Clin. Microbiol, and Inf. Dis., 2003, 9(3): 186-193). Смешанная (коктельная) терапия является эффективной не для всех пациентов, часто возникают потенциально серьезные неблагоприятные реакции, и быстро репродуцирующий вирус ВИЧ оказывается искусным при создании мутантных лекарственно-резистентных вариантов дикого типа протеазы и обратной транскриптазы. Поэтому остается потребность в безопасных лекарствах с активностью против дикого типа и обычных распространенных резистентных штаммов ВИЧ.

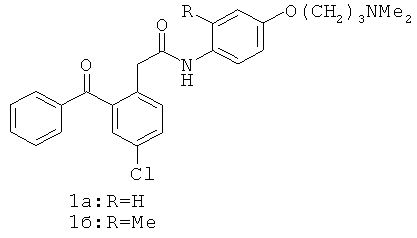
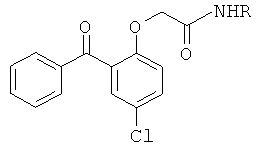
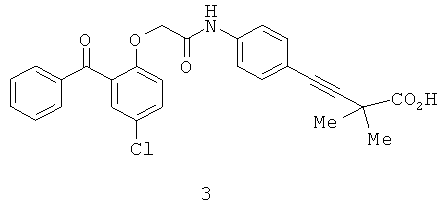
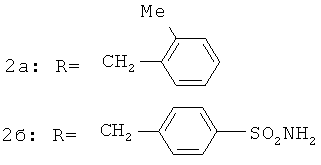
Было найдено, что 2-бензоилфенил-N-[фенил]ацетамидные соединения (1а) и (1б) ингибируют ВИЧ-1 обратную транскриптазу (P.G.Wyatt et al., J. Med. Chem., 1995, 38(10):1657-1665). Дальнейшее массовое обследование идентифицировало родственные соединения, например, 2-бензоилфенокси-N-[фенил]ацетамид (2а) и сульфонамидное производное (2б), которые ингибируют обратную транскриптазу (J. Н. Chan et al., J. Med Chem., 2004, 47(5):1175-1182; K.R.Romines et al., J. Med. Chem., 2006, 49(2): 727-739; C.L.Webster et al., WO 01/17982). P.Bonneau et al. в патенте США 2006/0069261, опубликованном 30 марта 2006 г., раскрывают производные 4-{4-[2-(2-бензоилфенокси)ацетиламино]фенил}-2,2-диметилбут-3-иновой кислоты (3), которые ингибируют ВИЧ обратную транскриптазу
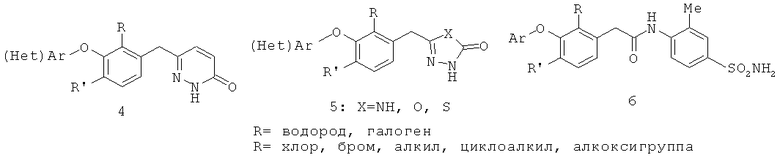
Пиридазинон в качестве ненуклеозидного ингибитора обратной транскриптазы (4) описан: J.P.Dunn et al. в патенте США 2004/0198736, опубликованном 23 марта 2004 г., и J.P.Dunn et al. в патенте США 2005/021554, опубликованном 22 марта 2005 г.
5-Аралкил-2,4-дигидро-[1,2,4]триазол-3-он, 5-аралкил-3Н-[1,3,4]оксадиазол-2-он и 5-аралкил-3Н-[1,3,4]тиадиазол-2-он в качестве ненуклеозидных ингибиторов обратной транскриптазы (5) описаны J.P.Dunn et al. в патенте США 2004/0192704, опубликованном 23 марта 2004 г., и J.P. Dunn et al. в патенте США 2006/0025462, опубликованном 27 июня 2005 г. Родственные соединения описаны Y.D.Saito et al. в публикации США 60/722 335. Фенилацетамид в качестве ненуклеозидного ингибитора обратной транскриптазы (6) раскрыт J.P.Dunn et al. в патенте США 2005/0239881, опубликованной 27 октября 2005 г., а методы лечения ретровирусной инфекции фенилацетамидными соединениями описаны J.P.Dunn et al. в патенте США 2005/0239880, опубликованной 27 октября 2005 г.; Т.Mirzadegan и Т.Silva в публикации США 60/728 443, опубликованной 19 октября 2005 г.; и Z.К.Sweeney и Т.Silva в публикации США 60/728 609 от 19 октября 2005 г. Эти публикации входят в объем настоящего изобретения в виде ссылок.
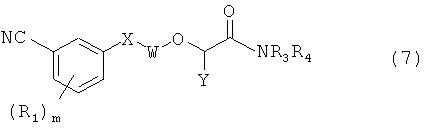
В публикации WO 2006/067587 от 26 июня 2006 г. L.Н.Jones et al. раскрывают производные простых биарильных эфиров (7) и включающие их композиции, которые связаны с ферментом обратная транскриптаза и являются ее модуляторами, в частности ингибиторами.
Пиразольные соединения, которые ингибируют ВИЧ обратную транскриптазу, были раскрыты L.Н.Jones et al. в WO 2002/085860, опубликованном 31 октября 2002 г. под заголовком "Preparation of aryloxy derivatives as reverse transcriptase inhibitors for treating HIV", H.L.Jones et al. in WO 2004/031178, опубликованном 15 апреля 2008 г. под заголовком " Preparation of pyrazole amides for treating HIV infections", D.A.Price et al. in WO 2004/031178, опубликованном 15 апреля 2004 г. под заголовком "Preparation of pyrazole derivatives as HIV reverse transcriptase inhibitors", P.J.Edwards et al in WO 2004/031178, опубликованном 15 апреля 2004 г. под заголовком "Preparation of pyrazole derivatives as therapeutic agents for HIV mediated diseases", O. Barba and L.H.Jones in WO 2004/029042, опубликованном 8 апреля 2004 г. WO 2004/0408 под заголовком "Preparation of pyrazole derivatives as reverse transcriptase inhibitors" and R.G.Corbau et al. in WO 2002/004424, опубликованном 17 января 2002 г. под заголовком "Pyrazole derivatives useful as reverse transcriptase inhibitor, for the лечения of HIV infection, and their use, formulations, and preparation". Все раскрытые пиразольные соединения ингибируют ВИЧ обратную транскриптазу.
В.W.Dymock et al. in WO 2002/100853, опубликованном 12 декабря 2002 г. под заголовком "Preparation of pyrazole derivatives as HIV reverse transcriptase inhibitors", и J.Dunn et al. в WO 2004/074257, опубликованном 2 сентября 2002 г. под заголовком "Preparation of pyrazole derivatives as non-nucleoside reverse transcriptase inhibitors for the treatment of HIV disorders and compositions thereof", также раскрывают пиразольные соединения, которые ингибируют ВИЧ ОТ.
М.J.Genin et al. (J. Med. Chem., 2000 43(5): 1034-1040) раскрывают пиразольные соединения с активностью против P236L мутанта, резистентного в отношении препарата делавирадина.
Настоящее изобретение включает соединения, которые проявляют потенциальную активность против дикого типа и мутантной ВИЧ обратной транскриптазы. Изобретение включает соединение согласно формуле I
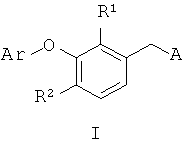
где:
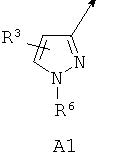
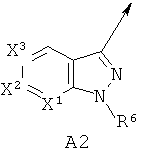
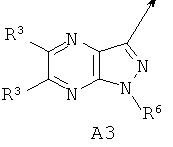
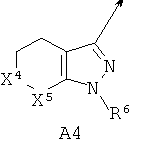
А выбирают из (i) А1, (ii) А2, где (а) X1, X2 и X3 независимо обозначают CR3, (б) один из X1 и X2 обозначает N или N-O, а другой из X1 и X2 вместе с X3 обозначают независимо CR3, (в) X1 и один из X2 или X3 обозначают N, а другой из X2 или X3 обозначает CR3, (г) один из X1 и X2 обозначает NR5, а другой из X1 и X2 обозначает С(=O), X3 обозначает CR3 и связь между X1 и X2 является простой связью; (iii) A3, или (iv) А4, где один из X4 и X5 обозначает NR5, другой из X4 и X5 обозначает С(=O);
R1 обозначает фтор или водород;
R2 обозначает водород, галоген, C1-6алкил, C3-7циклоалкил, C1-6галоалкил, C1-6алкоксигруппу или C1-6алкилсульфонил;
Ar обозначает фенил, замещенный от одной до трех групп, независимо выбранных в каждом случае из группы, включающей водород, галоген, цианогруппу, C1-6алкил, C1-6алкоксигруппу, C1-6галоалкил и C3-7циклоалкил;
R3 независимо выбран в каждом случае из группы, включающей: (i) водород, (ii) гидроксигруппу, (iii) C1-6алкоксигруппу, (iv) галоген, (v) NR4aR4b, (vi)
C1-6ациламиногруппу, (vii) C1-6алкилсульфониламиногруппу, (viii) цианогруппу, (ix) нитрогруппу, (x) NHNH2; и (xi) фенил, необязательно замещенный от одного до трех заместителями, независимо выбранными из группы, включающей галоген, C1-6алкил, C1-6алкоксигруппу, цианогруппу, нитрогруппу;
R4a и R4b независимо обозначают водород или C1-6алкил;
R5 обозначает водород или C1-3алкил;
R6 обозначает водород или C1-6алкил; и,
их фармацевтически приемлемую соль.
Соединения формулы I используются в качестве ингибиторов ВИЧ обратной транскриптазы и предоставляют метод для профилактики и лечения ВИЧ инфекций и лечения СПИД и/или СПИД-ассоциированного комплекса (ARC). ВИЧ подвергается легким мутациям своего генетического кода, приводящим к штаммам с пониженной чувствительностью к терапии современными терапевтическими средствами. Соединения формулы I применяются для лечения пациентов, инфицированных штаммом ВИЧ с, по крайней мере, одной мутацией, сравнимой с диким типом вируса. Настоящее изобретение относится также к композициям, содержащим соединения формулы I, используемым для профилактики и лечения ВИЧ инфекций и лечения СПИД и/или СПИД-ассоциированного комплекса (ARC). Настоящее изобретение, кроме того, относится к соединениям формулы I, которые применяются в монотерапии или комбинационной терапии с другими антивирусными агентами.
В одном варианте по настоящему изобретению предлагается соединение согласно формуле I, где R1, R2, R3, R4a, R4b, R5, R6, Ar, Al, A2, A3, A4, X1, X2, X4, X4, X5 имеют значения, описанные в данном тексте выше. Фраза "как определено в описании выше" относится к наиболее широкому определению для каждой группы, представленной в кратком изложении сущности изобретения. В других вариантах, приведенных ниже, в каждом варианте присутствуют заместители, которые неполностью подпадают под определение, данное в кратком изложении сущности изобретения.
В одном варианте по настоящему изобретению предлагается соединение согласно формуле I, где R2 обозначает водород, галоген, C1-6алкил, C3-7циклоалкил или
C1-6алкоксигруппу; и R1, R3, R4a, R4b, R5, R6, Ar, A1, A2, A3, A4, X1, X2, X4, X4, Х5 имеют значения, представленные выше.
В другом варианте по настоящему изобретению предлагается соединение согласно формуле I, где А обозначает А1 или А2.
В другом варианте по настоящему изобретению предлагается соединение согласно формуле I, где А обозначает А1.
В другом варианте по настоящему изобретению предлагается соединение согласно формуле I, где А обозначает А2 и X1, X2 и X3 независимо обозначают CR3.
В другом варианте по настоящему изобретению предлагается соединение согласно формуле I, где А обозначает А2, X1, X2 и X3 независимо обозначает CR3, R1 обозначает фтор; R2 обозначает C1-6алкил, C1-6алкоксигруппу или галоген; R6 обозначает водород и Ar обозначает 3-цианофенил, необязательно замещенный одной или двумя группами, независимо выбранными из галогена, цианогруппы и C1-6 галоалкила.
В другом варианте по настоящему изобретению предлагается соединение согласно формуле I, где А обозначает А2, X1, X2 и X3 обозначают независимо CR3, R1 обозначает фтор; R2 обозначает бром или хлор; R6 обозначает водород и Ar обозначает 3-цианофенил, необязательно замещенный одной или двумя группами, независимо выбранными из галогена, цианогруппы и C1-6 галоалкила.
В другом варианте по настоящему изобретению предлагается соединение согласно формуле I, где А обозначает А2, один из X1 или X2 обозначает N или N-О, другой из вместе с X3 независимо обозначает CR3.
В другом варианте по настоящему изобретению предлагается соединение согласно формуле I, где А обозначает А2, один из X1 или X2 обозначает N или N-О, другой из X1 и X2 наряду с X3 независимо обозначает CR3, R1 обозначает фтор; R2 обозначает C1-6алкил, C1-6алкоксигруппу или галоген; R5 и R6 обозначают водород и Ar обозначает 3-цианофенил, необязательно замещенный одной или двумя группами, независимо выбранными из галогена, цианогруппы и C1-6галоалкила.
В другом варианте по настоящему изобретению предлагается соединение согласно формуле I, где А обозначает А2, один из X1 или X2 обозначает N или N-О, другой из X1 и X2 наряду с X3 обозначают независимо CR3, R1 обозначает фтор; R2 обозначает C1-6алкил, C1-6алкоксигруппу или галоген; R5 и R6 обозначают водород и Ar обозначает 3-цианофенил, необязательно замещенный галогеном, цианогруппой или C1-6галоалкилом, находящимся в м-положении по отношению к цианозаместителю.
В другом варианте по настоящему изобретению предлагается соединение согласно формуле I, где А обозначает А2, один из X1 или X2 обозначает N или N-О, другой из X1 и X2 наряду с X3 обозначают независимо CR3, R1 обозначает фтор; R2 обозначает хлор или бром; R5 и R6 обозначают водород и Ar обозначает 3-цианофенил, необязательно замещенный галогеном, цианогруппой или C1-6галоалкилом, находящимся в м-положении по отношению к цианозаместителю.
В другом варианте по настоящему изобретению предлагается соединение согласно формуле I, где А обозначает А2, один из X1 или X2 обозначает NR5, другой из X1 или X2 обозначает С(=O), X3 обозначает CR3 и связь между X1 и X2 представляет собой простую связь.
В другом варианте по настоящему изобретению предлагается соединение согласно формуле I, где А обозначает А2, X1 и X2 обозначают N и X3 обозначает CR3.
В другом варианте по настоящему изобретению предлагается соединение согласно формуле I, где А обозначает А2, один из X1 и X2 обозначает N и X3 обозначает CR3, R1 обозначает фтор; R2 обозначает C1-6алкил, C1-6алкоксигруппу или галоген; R5 и R6 обозначают водород и Ar обозначает 3-цианофенил, необязательно замещенный галогеном, цианогруппой или C1-6 галоалкилом, находящимся в м-положении по отношению к цианозаместителю.
В другом варианте по настоящему изобретению предлагается соединение согласно формуле I, где А обозначает А2, один из X1 и X2 обозначают N и X3 обозначает CR3, R1 обозначает фтор; R2 обозначает C1-6алкил, C1-6алкоксигруппу или галоген; R5 и R6 обозначают водород и Ar обозначает 3-цианофенил, необязательно замещенный галогеном, цианогруппой или C1-6галоалкилом, находящимся в м-положении по отношению к цианозаместителю.
В другом варианте по настоящему изобретению предлагается соединение согласно формуле I, где А обозначает А2, один из X1 и X2 обозначает N и X3 обозначает CR3, R1 обозначает фтор; R2 обозначает бром или хлор; R5 и R6 обозначают водород и Ar обозначает 3-цианофенил, необязательно замещенный галогеном, цианогруппой или C1-6алоалкилом, находящимся в м-положении по отношению к цианозаместителю.
В другом варианте по настоящему изобретению предлагается соединение согласно формуле I, где А обозначает А2, X1 и X2 обозначают N и X3 обозначает CR3.
В другом варианте по настоящему изобретению предлагается соединение согласно формуле I, где А обозначает A3, R1 обозначает фтор; R2 обозначает C1-6алкил, C1-6алкоксигруппу или галоген; и Ar обозначает 3-цианофенил, необязательно замещенный галогеном, цианогруппой или C1-6 галоалкилом, находящимся в м-положении по отношению к цианозаместителю.
В другом варианте по настоящему изобретению предлагается соединение согласно формуле I, где А обозначает A3, R1 обозначает фтор; R2 обозначает C1-6алкил, C1-6алкоксигруппу или галоген; R6 обозначает водород и Ar обозначает 3-цианофенил, необязательно замещенный галогеном, цианогруппой или C1-6 галоалкилом, находящимся в м-положении по отношению к цианозаместителю.
В другом варианте по настоящему изобретению предлагается соединение согласно формуле I, где А обозначает А4 и один из X4 или X5 обозначает NR5, другой из X4 или X5 обозначает С(=O).
В еще одном варианте по настоящему изобретению предлагается соединение, выбранное из группы соединений от I-1 до I-63, представленных в таблице 1.
В одном другом варианте по настоящему изобретению предлагается способ лечения ВИЧ инфекции, или профилактики ВИЧ инфекции, или лечение СПИД или СПИД-ассоциированного комплекса (ARC), заключающийся во введении пациенту, нуждающемуся в этом, терапевтически эффективного количества соединения согласно формуле I, где R1, R2, R3, R4a, R4b, R5, R6, Ar, Al, A2, A3, A4, X1, X2, X4, X4, X5 имеют значения, обозначенные в описании выше.
В еще одном другом варианте по настоящему изобретению предлагается способ лечения ВИЧ инфекции, или профилактики ВИЧ инфекции, или лечения СПИД или СПИД-ассоциированного комплекса (ARC), заключающийся во введении пациенту, нуждающемуся в этом, терапевтически эффективного количества, по крайней мере, одного соединения, выбранного из группы, включающей ингибиторы ВИЧ протеазы, нуклеозидные ингибиторы обратной транскриптазы, ненуклеозидные ингибиторы обратной транскриптазы, CCR5 антагонисты и ингибиторы вирусной гибридизации вместе с соединением согласно формуле I, где R1, R2, R3, R4a, R4b, R5, R6, Ar, A1, A2, A3, A4, X1, X2, X4, X4, X5 имеют значения, обозначенные в описании выше.
В еще одном варианте по настоящему изобретению предлагается способ лечения ВИЧ инфекции, или профилактики ВИЧ инфекции, или лечения СПИД или СПИД-ассоциированного комплекса (ARC), заключающийся в совместном введении пациенту, нуждающемуся в этом, терапевтически эффективного количества, по крайней мере, одного соединения, выбранного из группы, включающей зидовудин, ламивудин, диданозин, зальцитабин, ставудин, рескриптор, сустива, вирамун, эфавиренц, невирапин, делавердин, сакьюнавир, ритонавир, нелфинавир, индинавир, ампренавир и лопинавир и эфувиртид вместе с соединением согласно формуле I, где R1, R2, R3, R4a, R4b, R5, R6, Ar, A1, A2, A3, A4, X1, X2, X4, X4, X5 имеют значения, обозначенные в описании выше.
В другом варианте по настоящему изобретению предлагается способ ингибирования ВИЧ обратной транскриптазы у пациента, инфицированного ВИЧ, заключающийся во введении соединения согласно формуле I, где R1, R2, R3, R4a, R4b, R5, R6, Ar, A1, A2, A3, A4, X1, X2, X4, X4, X5 имеют значения, обозначенные в описании выше.
В еще одном другом варианте по настоящему изобретению предлагается способ ингибирования ВИЧ обратной транскриптазы у пациента, инфицированного штаммом ВИЧ, экспрессирующим обратную транскриптазу, по крайней мере, с одной мутацией, сравнимой с диким типом ВИЧ, заключающийся во введении соединения согласно формуле I, где R1, R2, R3, R4a, R4b, R5, R6, Ar, A1, A2, A3, A4, X1, X2, X4, X4, X5 имеют значения, обозначенные в описании выше.
В еще одном другом варианте по настоящему изобретению предлагается способ ингибирования ВИЧ обратной транскриптазы у пациента, инфицированного штаммом ВИЧ, проявляющим пониженную восприимчивость к эфавиренцу, невирапину или делавердину, заключающийся во введении соединения согласно формуле I, где R1, R2, R3, R4a, R4b, R5, R6, Ar, A1, A2, A3, A4, X1, X2, X4, X4, X5 имеют значения, обозначенные в описании выше.
В другом варианте по настоящему изобретению предлагается фармацевтическая композиция, включающая соединение согласно формуле I, где R1, R2, R3, R4a, R4b, R5, R6, Ar, A1, A2, A3, A4, X1, X2, X4, X4, X5 имеют значения, обозначенные в описании выше, и, по крайней мере, один носитель, наполнитель или разбавитель.
Обозначения "а" или "an" относятся в данном описании к одному или более единицам; например (a compound) соединение относится к одному или более соединениям или, по крайней мере, к одному соединению. Как таковые, термины "а" (или "an"), "один или более", и "по крайней мере, один" могут быть взаимозаменяемыми в данном описании.
Предполагается, что описанные в данном тексте определения могут добавляться друг к другу, образовывая химически соответствующие комбинации, такие как «гетероалкиларил», «галоалкилгетероарил», «арилалкилгетероциклил», «алкилкарбонил», «алкоксиалкил» и подобные им. Когда термин «алкил» используется в качестве суффикса, следующего за другим термином, как в термине «фенилалкил», или «гидроксиалкил», его следует относить к алкильной группе по определению выше, при этом замещенным от одного до двух заместителями, выбранными из других групп, имеющих специфические наименования. Так, например, термин «фенилалкил» относится к алкильной группе, содержащей от одного до двух фенильных заместителя, и таким образом включающей бензил, фенилэтил и бифенил. Термин "гетероциклилалкил" относится к алкильной группе, содержащей от одного до двух гетероциклических заместителя. Термин «алкиламиноалкил» обозначает алкильную группу, содержащую от одного до двух алкиламинозаместителей. Термин «гидроксиалкил» включает 2-гидроксиэтил, 2-гидроксипропил, 1-(гидроксиметил)-2-метилпропил, 2-гидроксибутил, 2,3-дигидроксибутил, 2-(гидроксиметил), 3-гидроксипропил и так далее. Соответственно, по определению, данному в описании, термин «гидроксиалкил» используется для определения подгруппы гетероалкильных групп, описанных ниже. Термин «(ар)алкил» относится либо к незамещенной алкильной группе, либо к аралкильной группе. Термин «(гетеро)арил» относится либо к арильной, либо к гетероарильной группе.
Термины «необязательный» или «необязательно» обозначают, что описанные случай или обстоятельство впоследствии могут иметь место, но не происходят, и что описание включает как примеры, где случай или обстоятельство встречаются, так и примеры, в которых они не встречаются. Например, «необязательная связь» означает, что связь может присутствовать, а может и не присутствовать, и что описание включает, простую, двойную или тройную связи.
Термин "необязательная связь" означает, что связь может или не может присутствовать, и что описание включает простую, двойную или тройную связи. Если заместитель обозначает "связь" или "отсутствие", это значит, что атомы, соединенные с заместителями, связаны напрямую.
Технические и научные термины, используемые в данном описании, имеют смысл, в общем понятный любому специалисту в области техники, к которой относится настоящее изобретение, если не указано иначе. Ссылки даются на различные методологии и материалы, известные специалисту в данной области техники. Стандартные работы, на которые даются ссылки, включают общие принципы фармакологии, например, Goodman and Gilman's The Pharmacological Basis of Therapeutics, 10th Ed., McGraw Hill Companies Inc., New York (2001). Любые подходящие материалы и/или методы, известные специалистам в данной области техники, могут быть использованы при осуществлении настоящего изобретения. Однако описаны предпочтительные материалы и методы. Материалы, реагенты и подобные им, на которые даются ссылки в настоящем описании, и примеры могут быть получены из коммерческих источников, если не указано иначе.
Как принято в настоящем описании, или в переходной фазе, или в пунктах формулы изобретения, термины "включает(ют)" и "включая" должны интерпретироваться, как имеющие начальное-конечное значение. То-есть, термины могут интерпретироваться синонимически фразами "имеющие по крайней мере" или "включающие по крайней мере". Когда в контексте рассматривается процесс, термин "включая" подразумевает, что процесс включает, по крайней мере, перечисленные стадии, но может также включать дополнительные стадии. Когда в контексте рассматривается соединение или композиция, термин "включая" подразумевает, что соединение или композиция включают, по крайней мере, перечисленные свойства или компоненты, но могут также включать дополнительные свойства или компоненты.
Когда любой переменный заместитель (например, R1, R4a, Ar, X1 или Het) встречается более одного раза в любом фрагменте или формуле изображенных и описанных соединений, применяемых или испрашиваемых в настоящем изобретении, их определение в каждом случае является независимым от их определения в каждом другом случае. Также и комбинации заместителей и/или переменных допустимы только, если такие соединения приводят к стабильным соединениям.
"Стабильным" соединением является соединение, которое может быть получено и выделено, и структура и свойства которого остаются или могут быть сохранены по существу неизменными на период времени, достаточный для использования соединения в целях, описанных в данном тексте (например, терапевтическое или профилактическое введение субъекту).
Если точно не установлено противоположное, все диапазоны, упомянутые в данном описании, включаются. Например, гетероциклическое кольцо, описанное как содержащее "от 1 до 4 гетероатомов", представляет собой кольцо, которое может содержать 1, 2, 3 или 4 гетероатома. Также должно быть понятно, что любой диапазон, упомянутый в данном описании, включает в себя все субдиапазоны внутри этого диапазона. Так, например, предполагается, что арил или гетероарил, описанные, как необязательно замещенные "от 1 до 5 заместителями", включают любой арил, необязательно замещенный от 1 до 4 заместителями, от 1 до 3 заместителями, от 1 до 2 заместителями, от 2 до 5 заместителями, от 2 до 4 заместителями, от 2 до 3 заместителями, от 3 до 5 заместителями, от 3 до 4 заместителями, от 4 до 5 заместителями, 1 заместителем, 2 заместителями, 3 заместителями, 4 заместителями и 5 заместителями.
Термин «ацил», используемый в данном описании, относится к группе формулы
-C(=O)R, где R обозначает водород или низший алкил, как определено в описании.
«C1-3ацил» относится к ацильной группе, как определено в описании, где R обозначает C1-3алкил.
Термин "ациламиногруппа", используемый в данном описании, относится к группе формулы -NHC(=O)R, где R обозначает водород или низший алкил, как определено в описании.
Термин "алкил", используемый в данном описании, относится к неразветвленному или разветвленному, насыщенному, моновалентному углеводородному остатку, содержащему от 1 до 10 атомов углерода. Термин "низший алкил" относится к линейному или разветвленному углеводородному остатку, содержащему от 1 до 6 атомов углерода. Термин "С1-10алкил", используемый в данном описании, относится к алкилу, содержащему от 1 до 10 атомов углерода.
Термины "алкилсульфониламиногруппа" и "арилсульфониламиногруппа", используемые в данном описании, относятся к группе формулы -NR'S(=O)2R, где R обозначает алкил или арил, соответственно, R' обозначает водород или C1-3алкил, и при этом алкил и арил имеют значения по определению выше.
Термины «аминогруппа», «алкиламиногруппа» и «диалкиламиногруппа», используемые в данном описании, относятся к -NH2, -NHR и -NR2, соответственно, и R обозначает алкил по определению выше. Две алкильные группы, присоединенные к атому азота в диалкильном фрагменте, могут быть одинаковыми или различными. Термины «аминоалкил», «алкиламиноалкил» и «диалкиламиноалкил», используемые в данном описании, относятся к NH2(CH2)n-, RHN(CH2)n- и R2N(CH2)n-, соответственно, где n обозначает от 1 до 10, и R обозначает алкил по определению в описании выше. Термин «C1-6алкиламиногруппа», используемый в данном описании, относится к аминоалкилу, где алкил обозначает C1-6. Термин «фениламиногруппа», используемый в данном описании, относится к -NHPh, где Ph представляет собой необязательно замещенную фенильную группу.
Термин «циклоалкил», используемый в данном описании, относится к насыщенному углеводородномуу кольцу, содержащему от 3 до 8 атомов углерода, например, циклопропилу, циклобутилу, циклопентилу, циклогексилу, циклогептилу или циклооктилу. «C3-5циклоалкил», используемый в данном описании, относится к циклоалкилу, содержащему от 3 до 5 атомов углерода в карбоциклическом кольце.
Термин «алкоксигруппа», используемый в данном описании, относится к -О-алкильной группе, где алкил обозначен по определению выше, такой как метоксигруппа, этоксигруппа, н-пропилоксигруппа, изо-пропилоксигруппа, н-бутилоксигруппа, изо-бутилоксигруппа, трет-бутилоксигруппа, пентилоксигруппа, гексилоксигруппа, включая их изомеры. Термин «низшая алкоксигруппа», используемый в данном описании, относится к алкоксигруппе с «низшей алкильной» группой, как она определена ранее. «C1-10алкоксигруппа» относится к -O-алкилу, где алкил обозначает C1-10.
Термин «цианогруппа», используемый в данном описании, относится к углероду, соединенному с атомом азота тройной связью, например, -C≡N. Термин «нитрогруппа», используемый в данном описании, относится к группе -NO2.
Термин «галоалкил», используемый в данном описании, относится к неразветвленной или разветвленной алкильной группе по определению выше, где 1, 2, 3 или более атомов водорода замещены галогеном. «C1-3 галоалкил», используемый в данном описании, относится к галоалкилу, содержащему от 1 до 3 атомов углерода и от 1 до 8 галоидных заместителей. В качестве примеров могут служить 1-фторметил, 1-хлорметил, 1-бромметил, 1-йодометил, трифторметил, трихлорметил, трибромметил, трийодметил, 1-фторэтил, 1-хлорэтил, 1-бромэтил, 1-йодэтил, 2-фторэтил, 2-хлорэтил, 2-бромэтил, 2-йодэтил, 2,2-дихлорэтил, 3-бромпропил или 2,2,2-трифторэтил.
Термин "галоалкоксигруппа", используемый в данном описании, относится к -O-галоалкильной группе, где галоалкил имеет значение по определению выше.
Термин "галоген" или "гало", используемый в данном описании, относится к фтору, хлору, брому или йоду.
Термины «гидроксиалкил» и «алкоксиалкил», используемые в данном описании, относятся к алкильным радикалам по определению выше, где от 1 до 3 атомов водорода у различных атомов углерода замещены одной или несколькими гидроксильными группами или алкоксигруппами, соответственно. «С1-6гидроксиалкил» относится к C1-6алкильной группе по определению выше, где от 1 до 3 атомов водорода у различных атомов углерода замещены одной или несколькими гидроксильными группами.
Термин «C1-6карбоксиалкил», используемый в данном описании, относится к C1-6алкильной группе по определению выше, где один или два атома водорода у различных атомов углерода замещены карбоксильными группами. Группа NRaRb по определению в пункте 1 формулы изобретения является группой, где Ra обозначает карбоксиалкильную группу, которая включает, не лимитируя, натуральные аминокислоты глицин, аланин, валин, лейцин и изолейцин.
Термины « азетидин», «пирролидин», «пиперидин» и «азепин» относятся к 4-, 5-, 6- или 7-членным циклоалканам, соответственно, где один атом углерода заменен атомом азота.
Термин "арил", используемые в данном описании, относится к фенильному кольцу, которое может быть необязательно замещено одним или более, предпочтительно, одним или тремя заместителями, независимо выбранными из гидроксигруппы, тиогруппы, цианогруппы, алкила, алкоксигруппы, низшей галоалкоксигруппы, алкилтиогруппы, галогена, галоалкила, гидроксиалкила, нитрогруппы, алкоксикарбонила, аминогруппы, алкиламиногруппы, диалкиламиногруппы, аминоалкила, алкиламиноалкила и диалкиламиноалкила, алкилсульфонила, арилсульфинила, алкиламиносульфонила, ариламиносульфонила, алкилсульфониламиногруппы, арилсульфониламиногруппы, карбамоила, алкилкарбамоила и диалкилкарбамоила, арилкарбамоила, алкилкарбониламиногруппы, арилкарбониламиногруппы, если не указано иначе. Альтернативно, два соседних атома арильного кольца могут быть замещенны метилендиоксигруппой или этилендиоксигруппой. Термин "арилоксигруппа", используемый в данном описании, относится к необязательно замещенному фенолу.
Термин "азаиндазолы" относится в общем к конденсированному пиразолу с одним атомом азота в любом положении в кольце, сконденсированном с пиразолом, (например, 1Н-пиразоло[3,4-с]пиридин-3-ил или 1Н-пиразоло[3,4-b]пиридин-3-ил) и термин "диазадиазолы" относится в общем к конденсированному пиразолу с двумя атомами азота в любом положении в кольце, сконденсированном с пиразолом, (например, 1Н-пиразоло[3,4-d]пиримидин-3-ил).
Термин "инертный органический растворитель" или "инертный растворитель" подразумевает растворитель, который является инертным в условиях проводимой реакции. В случае реакции бензальдоксима с основанием инертный растворитель является таким, который не имеет кислого протона и не реагирует с трифторнитробензолом. Примеры инертных растворителей включают эфирные растворители и углеводороды. Термин "основание" относится к органическому или неорганическому основанию, достаточно сильному, чтобы депротонизировать фенол II. Примеры таких оснований многочисленны и хорошо известны из области техники.
Алкилбромацетат (синтетический эквивалент уксусной кислоты) является производным уксусной кислоты с уходящей группой при α-углеродном атоме, которая способна к замене фенолятной солью. Несмотря на применении в описании в данной реакции этилбромацетата, аналогично могут быть использованы другие сложные эфиры. Сложный эфир может быть также заменен амидом, включая производные анилида, описанные в данном тексте.
Термин «дикий тип» штамма, используемый в данном описании, относится к ВИЧ вирусным штаммам, которые обладают доминантным генотипом, находящимся в натуральных условиях в нормальной популяции, которая не может подвергаться воздействию ингибиторов обратной транскриптазы. Термин «дикого типа обратная транскриптаза», используемый в данном описании, относится к обратной транскриптазе, экспрессируемой штаммом дикого типа, который был секвенирован и включен в SwissProt базу данных под номером Р03366.
Термин "пониженная чувствительность", используемый в данном описании, относится приблизительно к 10-кратному или более, изменению в чувствительности данного вирусного изолята по сравнению с чувствительностью, проявляемой вирусом дикого типа в одинаковых экспериментальных системах.
Термин «нуклеозидные и нуклеотидные ингибиторы обратной транскриптазы» (НИОТ), используемый в данном описании, относится к нуклеозидам и нуклеотидам и их аналогам, которые ингибируют активность ВИЧ-1 обратной транскриптазы, фермента, который катализирует конверсию вирусной геномной ВИЧ-1 РНК в провирусную ВИЧ-1 ДНК. Достигнутый прогресс в развитии ОТИ и ПИ (PI) ингибиторов представлен в: F.М. Uckun and О.J.D'Cruz, Exp.Opin. Ther. Pat., 2006, 16:265-293; L. Menendez-Arias, Eur. Pharmacother., 2006, 94-96 and S. Rusconi and O. Vigano, Future Drugs, 2006, 3(1): 79-88.
Типичные применяемые НИОТ включают: зидовудин (AZT; RETROVIR®); диданозин (ddl; VIDEX®); зальцитабин (ddC; HIVID®); ставудин (d4T; ZERIT®); ламивудин (3TC; EPIVIR®); абакавир (1592U89; ZIAGEN®); адефовир дипивоксил (бис(РОМ)-РМЕА; PREVON®); лобукавир (BMS-180194), ингибитор нуклеозидной обратной транскриптазы, описанный в публикациях ЕР-0358154 и ЕР-0736533; ВСН-10652, ингибитор обратной транскриптазы (в форме рацемической смеси ВСН-10618 и ВСН-10619), разработанный фирмой Biochem Pharma; эмитрицитабин [(-)-FTC], разработанный фирмой Triangle Pharmaceuticals; β-L-FD4 (также называемый β-L-D4C и имеющий наименование β-L-2',3'-диклеокси-5-фторцитидин), лицензированный фирмой Vion Pharmaceuticals; DAPD, пуриновый нуклеозид, (-)-β-D-2,6,-диаминопуриндиоксолан, описанный в публикации ЕР-0656778 и лицензированный фирмой Triangle Pharmaceuticals; и лоденозин (FddA), 9-(2,3-дидеокси-2-фтор-β-D-трео-пентофуранозил)аденин, кислотностабильный, основанный на пурине, ингибитор обратной транскриптазы, разработанный фирмой US Bioscience Inc.
Типичные применяемые ННИОТ включают: невирапин (BI-RG-587; VIRAMUNE®); делавирадин (ВНАР, U-90152; RESCRIPTOR®); эфавиренц (DMP-266, SUSTIVA®); PNU-142721, фуропиридинтиопиримидин, разработанный фирмой Pfizer; AG-1549 (разработанный Shionogy #S-1153); 5-(3,5-дихлорфенил)тио-4-изопропил-1-(4-пиридил)метил-1Н-имидазол-2-илметилкарбонат), раскрытый в публикации WO 96/10019; МКС-442, (1-(этоксиметил)-5-(1-метилэтил)-6-(фенилметил)-(2,4(1Н,3Н)-пиримидиндион); и (+)-каланолид A (NSC-675451) и В, производные кумарина, описанные в публикации US 5 489 697; этравирин (ТМС-125; 4-[6-амино-5-бром-2-(4-цианофениламино)пиримидин-4-илокси]-3,5-диметилбензонитрил) и DAPY (ТМС278; 4-{4-[4-((Е)-2-циановинил)-2,6-диметилфениламино]пиримидин-2-иламино}бензонитрил) от фирмы Tibotec-Virco и Johnson (Johnson & BILR-355 BS (12-этил-8-[2-(1-гидроксихинолин-4-илокси)этил]-5-метил-11,12-дигидро-5Н-1,5,10,12-тетраазадибензо[a,e]циклооктен-6-он от фирмы Boehringer-Ingleheim; PHI-236 (7-бром-3-[2-(2,5-диметоксифенил)этил]-3,4-дигидро-1Н-пиридо[1,2-а][1,3,5]триазин-2-тион) и PHI-443 (1-(5-бромпиридин-2-ил)-3-(2-тиофен-2-илэтил)тиомочевина) от фирмы Paradigm Pharmaceuticals.
Термин «ингибитор протеазы» (ИП), используемый в данном описании, подразумевает ингибитор ВИЧ-1 протеазы, фермента, требуемого для протеолитического расщепления вирусных полипротеиновых предшественников (например, вирусных GAG и GAG Pol полипротеинов) на отдельные функциональные белки, найденные в инфекционном ВИЧ-1. Ингибиторы ВИЧ протеазы включают соединения, имеющие пептидомиметическую структуру с большой молекулярной массой (7600 дальтон) и в основном пептидным характером, например, CRIXIVAN®, а также непептидные ингибиторы протеазы, например, VIRACEPT®.
Особенно подходящие ингибиторы протеазы включают препарат сакьюнавир в твердых гелевых капсулах, выпускаемый под маркой INVIRASE®, и препарат в мягких гелевых капсулах, выпускаемый под маркой FOKTOVASE(от фирмы Roche; ритонавир (АВТ-538), выпускаемый под маркой NORVIR от фирмы Abbott Laboratories; лопинавир (АВТ-378) также от фирмы Abbott; KALETRA®, являющийся композицией, включающей лоринавир и субтерапевтическую дозу ритонавира от фирмы Abbott Laboratories; индинавир (МК-639), выпускаемый под маркой CRIXIVAN® от фирмы Merck &Co.; нелфнавир (AG-1343), выпускаемый под маркой VIRACEPT® от фирмы Agouron Pharmaceuticals, Inc.; ампренавир (141W94), выпускаемый под маркой AGENERASE®от фирмы Vertex Pharmaceuticals, Inc. и GSK; типранавир (PNU-140690), выпускаемый под маркой APTIVUS® от фирмы BI; лазинавир (BMS-234475/CGP-61755) от фирмы BMS; BMS-2322623, азапептид, разработанный фирмой BMS в виде вторичной генерации ВИЧ-1 ИП; GW-640385X (VX-385), совместно разработанный фирмами GSK и Vertex; AG-001859 в доклинической разработке фирмы Agouron/Pfizer; SM-309515, разработанный фирмой Sumitomo Pharmaceuticals.
Дополнительно ингибиторы протеазы в доклинической разработке включают N-циклоалкилглицины от фирмы BMS, α-гидроксиарилбутанамиды от фирмы Enanta Pharmaceuticals; производные α-гидрокси-γ-[[(карбоциклически- или гетероциклически-замещенного)амино)карбонил]алканамида; γ-гидрокси-2-(фторалкиламинокарбонил)-1-пиперазинпентаамиды от фирмы Merck; производные дигидропирона и гидроксиэтиламиносульфонамиды α- и β-аминокислот от фирмы Pfizer; и производные L-лизина, замещенного N-аминокислотой, от фирмы Procyon. Вход ВИЧ в клетки-мишени требуют наличия CD-4 рецептора поверхности клетки и CCR5 (М-тропические штаммы) и CXCR4 (Т-тропические штаммы) хемокиновых со-рецепторов. Хемокиновый антагонизм, который блокирует вирусное присоединение к хемокинам, использует ингибиторы вирусной инфекции. Такеда идентифицировал ТАК-779 в качестве потенциального CCR5 антагониста. (М.Shiraishi et al., J.Med. Chem., 2000, 43 (10): 2049-2063; M.Babba et al. Proc. Nat. Acad. Sci. USA, 1999 96: 5698-5703) и TAK-220 (C. Tremblay et al. Antimicrob. Agents Chemother., 2005, 49 (8): 3483-3485). В публикациях WO 00/39125 (D.R.Armour et al.) и WO 01/90106 (M.Perros et al.) раскрыты гетероциклические соединения, являющиеся потенциальными и селективными антагонистами CCR5. Миравикор (UK 427 857; MVC), предоставленный фирмой Pfizer в III фазе клинических испытаний, показал активность против ВИЧ-1 изолятов и лабораторных штаммов (P.Dorr et al., Antimicrob. Agents Chemother., 2005, 49 (11): 4721-4732; A. Wood and D. Armour, Prog. Med. Chem., 2005, 43: 239-271; C.Watson et al., Mol. Pharm., 2005, 67 (4): 1268-1282; M. J. Macaktney et al., 43rd Intersci. Conf. Antimicrob. Agents Chemother., September 14-17, 2003, Abstract H-875). Фирмой Шеринг предложен препарат Sch-351125 (SCH-C) в I/II фазах клинических исследований и сообщается о получении более перспективного следующего соединения, викровирока (Sch-417690, SCH-D) в I фазе исследований. (S.W.McCrombie et al., WO 00066559; В.M.Baroudy et al. WO 00066558; A. Palani et al, J. Med. Chem., 2001, 44 (21): 3339-3342; J.R.Tagat et al, J. Med. Chem., 2001, 44 (21): 3343-3346; J.A.Esté, Cur. Opin. Invest. Drugs, 2002, 3 (3): 379-383; J.M.Struzki et al. Proc. Nat. Acad Sci. USA, 2001, 98: 12718-12723). Фирма Merck раскрыла получение S-оксида (2S)-2-(3-хлорфенил)-1-N-(метил)-N-(фенилсульфонил)амино]-4-[спиро(2,3-дигидробензотиофен-3,4-пиперидин-1'-ил)бутана (1) и родственных производных с хорошим сродством к CCR5 рецептору и потенциальной ВИЧ активностью. (P.Е.Finke et al., Bioorg. Med. Chem. Lett., 2001, 11: 265-270; P.E.Finke et al., Bioorg. Med. Chem. Lett., 2001, 11: 2469-2475; P.E.Finke et al., Bioorg. Med. Chem. Lett., 2001, 11: 2475-2479; J.J.Hale et al., Bioorg. Med. Chem. Lett., 2001, 11: 2741-22745; D.Kim et al., Bioorg. Med. Chem. Lett., 2001, 11: 3099-3102) C.L.Lynch et al. Org Lett. 2003, 5: 2473-2475; R.S.Veazey et al. J. Exp.Med. 2003, 198: 1551-1562. Препарат GSK-873140 (ONO-4128, E-913, AK-602) был предъявлен в программе, проводимой при Kumamoto University (K.Maeda et al. J.Biol. Chem., 2001, 276: 35194-35200; H. Nakata et al. J.Virol., 2005, 79(4): 2087-2096) и был предложен для клинических испытаний. В публикациях WO 00/166525; WO 00/187839; WO 02/076948; WO 02/076948; WO 02/079156, WO 2002/070749, WO 2003/080574, WO 2003/042178, WO 2004/056773, WO 2004/018425 фирма Astra Zeneca раскрывает 4-аминопиперидиновые соединения, которые являются CCR5 антагонистами. В патенте США 2005/0176703, опубликованном 11 августа 2005 г., S.D.Gabriel и D.М.Rotstein раскрыли гетероциклический CCR5 антагонист, способный предотвращать вход ВИЧ в клетку. В патенте США 2006/0014767, опубликованном 19 января 2006 г., Е.K.Lee et al. раскрыли гетероциклический CCR5 антагонист, также способный предотвращать вход ВИЧ в клетку.
Прикрепление ингибиторов эффективно блокирует взаимодействие между белками вирусной оболочки и химокиновыми рецепторами или CD40 белком. TNX-355 является гуманизированным IgG4 моноклональным антителом, которое связывается с конформационным эпитопом на домене 2 CD4. (L.С.Burkly et al., J.Immunol. 1992, 149: 1779-87). TNX-355 может ингибировать вирусное прикрепление CCR5-, CXCR4- и двойственных/смешанных тропических ВИЧ-1 штаммов. (Е. Godofsky et al., In Vitro Activity of the Humanized Anti-CD4 Monoclonal Antibody, TNX-355, against CCR5, CXCR4, and Dual-Tropic Isolates and Synergy с Enfuvirtide, 45th Annual Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC), December 16-19, 2005, Washington DC.
Abstract # 3844; D. Norris et al. TNX-355 in Combination with Optimized Background Regime (OBR) Exhibits Greater Antiviral Activity than OBR Alone in HIV-Treatment Experienced Patients, 45th Annual Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC). December 16-19, 2005, Washington DC. Abstract # 4020).
Макромолекулярные виды терапии, включающие антитела, растворимые рецепторы и их биологически активные фрагменты, стали потенциально важным дополнением к стандартным низкомолекулярным лекарствам (О.Н.Brekke and I. Sandlie Nature Review Drug Discov., 2003, 2: 52-62; A.M.Reichert, Nature Biotech., 2001, 19: 819-821). Антитела с высокой специфичностью и сродством могут быть избирательно направлены на экстрацеллюлярные белки, незаменимые для вирусной клеточной гибридизации. CD4, CCR5 и CXCR4 могут служить мишенью для антител, которые ингибируют вирусную гибридизацию.
V. Roschke et al. (Characterization of a Panel of Novel Human Monoclonal Antibodies that Specifically Antagonize CCR5 and Block ВИЧ-1 Entry, 44th Annual Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC). October 29, 2004, Washington DC. Abstract # (2871) описывают моноклональные антитела, которые связаны с CCR5 рецептором, и ингибируют HIV вход в клетки, экспрессирующие CCR5 рецептор. L. Wu и С.R МасКау раскрывают в патенте США 09/870 932, опубликованном 30 мая 2001 г., моноклональные антитела 5С7 и 2D7, которые связаны с CCR5 рецептором таким образом, что способны ингибировать ВИЧ инфекцию клетки. W.С.Olsen et al. (J. Virol., 1999, 73 (5): 4145-4155) описывают способность ингибирования: (i) ВИЧ-1 вход в клетку, (ii) слияние с мембраной, опосредованное ВИЧ-1 оболочкой, (iii) gp120 связывание с CCR5 и (iv) СС-хемокиновую активность. Синергизм между анти-CCR5 антителом Pro140 и низкомолекулярными CCR5 антагонистами описан Murga et al. (3rd IAS Conference on HIV Pathogenesis and Treatment, Abstract TuOa.02.06. July 24-27, 2005, Rio de Janeiro, Brazil). Были выделены анти-CCR5 антитела, которые ингибируют вход ВИЧ-1 в клетку, и описаны М. Brandt et al. в патенте США 11/394 439, опубликованном 31 марта, 2006 г.
FUZEON®(Т-20, DP-178, пентафьюзид) раскрыт в патенте США 5464933. Т-20 и аналог, Т-1249, являются аналогами ВИЧ gp41 фрагмента, которые эффективно ингибируют конформационное превращение, требуемое для ВИЧ слияния. Т-20 утвержден и поставляется фирмами Roche и Trimeris. FUZEON вводится в виде непрерывной подкожной инфузии или инъекции в комбинационной терапии с другими классами анти-ВИЧ лекарственных препаратов.
Другие антивирусные агенты, которые могут быть использованы в ВИЧ терапии, включают гидроксимочевину, рибавирин, IL-2, IL-12, пентафузид. Гидромочевина (Droxia), ингибитор рибонуклеозид трифосфатредуктазы, фермент, включенный в активацию Т-клеток, раскрыт фирмой NCl Pharmaceuticals и разработан фирмой Bristol-Myers Squibb; как было найдено при доклиническом исследовании, он оказывает синергетический эффект на активность диданозина и был изучен совместно со ставудином. IL-2 раскрыт в публикациях Ajinomoto ЕР-0142268, Takeda ЕР-0176299 и Chiron US Pat. Nos. RE 33653, 4530787, 4569790, 4604377, 4748234, 4752585 и 4949314, и производится под названием PROLEUKIN®(альдеслейкин) фирмой Chiron Corp. в виде лиофилизованного порошка для IV инфузии или подкожного введения. IL-12 раскрыт в WO 96/25171 и производится фирмами Roche и Wyeth Pharmaceuticals. Рибавирин, 1-β-D-рибофуранозил-1Н-1,2,4-триазол-3-карбоксамид описан в патенте США 4 211 771 и производится фирмой ICN Pharmaceuticals.
В общем, аббревиатуры, используемые в этой заявке, включают: ацетил (Ас), азо-бис-изобутирилнитрил (АИБН), 9-борбицикло[3.3.1]нонан (9-ББН или ББН), трет-бутоксикарбонил (Бок), ди-трет-бутилпирокарбонат или бок-ангидрид (БОК2О), бензил (Bn), бутил (Bu), бензилоксикарбонил (CBS или Z), диимидазолкарбонил (КДИ), 1,4-диазабицикло[2.2.2]октан (ДАБЦО), диэтиламиносульфуртрифторид (ДАСТ), дибензилиденацетон (dba), 1,5-диазацикло[4.3.0]нон-5-ен (ДБН), 1,8-диазабицикло[5.4.0]ундец-7-ен (ДБУ), N,N'-дициклогексилкарбодиимид (ДЦК), 1,2-дихлорэтан (ДХЭ), дихлорметан (ДХМ), диэтилазодикарбоксилат (ДЭАД), ди-изо-пропилазодикарбоксилат (ДИАД), ди-изо-бутилалюминийгидрид (ДИБАL или ДИБАL-Н), ди-изо-пропилэтиламин (ДИПЭА), N,N-диметилацетамид (ДМА), 4-N,N-диметиламинопиридин (ДМАП), N,N-диметилформамид (ДМФ), диметилсульфоксид (ДМСО), 1,1'-бис-(дифенилфосфино)этан (dppe), 1,1'-бис-(дифенилфосфино)ферроцен (dppf), гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (ЭДСI), этил (Et), этилацетат (EtOAc), этанол (EtOH), этиловый эфир 2-этокси-2Н-хинолин-1-карбоновой кислоты (EEDQ), эквивалент (экв.), диэтиловый эфир (Et2O), уксусная кислота (HOAc), 1-N-гидроксибензотриазол (ГОБТ), атмосфера (атм), высокоэффективная жидкостная хроматография (ВЭЖХ), литийгексаметилдисилазан (LiГМДС), метанол (МеОН), температура плавления (т.пл.), MeSO2- (мезил или Ms), метил (Me), ацетонитрил (MeCN), м-хлорпербензойная кислота (МХПБК), масс-спектр (МС), метил-трет-бутиловый эфир (МТБЭ), N-бромсукцинимид (N-БС), N-карбоксиангидрид (N-KA), N-хлорсукцинимид (N-XC), N-метилморфолин (N-ММ), N-метилпирролидон (N-МП), хлорхромат пиридиния (ПХХ), дихромат пиридиния (ПДХ), 1,2-дихлорэтан (DCE), пропил (Pr), фенил (Ph), изо-пропил (i-Pr), фунт на дюйм (psi), пиридин (pyr), комнатная температура (кт или КТ), трет-бутилдиметилсилил или t-BuMe2Si, (ТБДМС), триэтиламин (Et3N или ТЭА), трифлат или CF3SO2- (Tf), трифторуксусная кислота (ТФК), 2,2,6,6-тетраметилгептан-2,6-дион (ТМГД), тетрафторборат О-бензотриазол-1-ил-N,N,N',N'-тетраметилурония (ТБТУ), тонкослойная хроматография (ТСХ), тетрагидрофуран (ТГФ), триметилсилил или Me3Si (ТМС), моногидрат п-толуолсульфокислоты (TsOH или п-TsOH), 4-Me-C6H4SO2- или тозил (Ts), N-уретан-N-карбоксиангидрид (UNCA). Стандартная номенклатура, включающая префиксы нормальный (н-), изо (i-), вторичный (втор-), третичный (трет-) и нео-, имеет в виду их стандартные значения при использовании с алкильным фрагментом (J. Rigaudy and D.P.Klesney, Nomenclature in Органический Chemistry, IUPAC 1979 Pergamon Press, Oxford.).
Соединения по настоящему изобретению могут быть получены различными методами, представленными на иллюстративных синтетических схемах, приведенных и описанных ниже. Исходные вещества и реагенты, используемые при получении этих соединений, в основном либо доступны через коммерческие фирмы такие, как Aldrich Chemical Co., или могут быть получены методами, известными специалистам в области техники, приведенными в следующих публикациях: Fieser and Fieser's Reagents for Organic Synthesis; Wiley & Sons: New York, Volumes 1-21; R. C. LaRock, Comprehensive Organic Transformations, 2nd edition Wiley-VCH, New York 1999; Comprehensive Organic Synthesis, B.Trost and I. Fleming (Eds.) vol. 1-9 Pergamon, Oxford, 1991; Comprehensive Heterocyclic Chemistry, A.R.Katritzky and C.W.Rees (Eds) Pergamon, Oxford 1984, vol. 1-9; Comprehensive Heterocyclic Chemistry II, A.R.Katritzky and C. W. Rees (Eds) Pergamon, Oxford 1996, vol. 1-11; and Organic Reactions, Wiley (Sons: New York, 1991, Volumes 1-40. Представленные далее синтетические реакционные схемы только иллюстрируют некоторые методы, которыми могут быть синтезированы соединения по настоящему изобретению, при этом возможно осуществление различных модификаций этих синтетических реакционных схем специалистом в области техники, имеющей отношение к тематике данной заявки.
Исходные и промежуточные вещества могут быть при необходимости выделены и очищены с использованием традиционных методик, включая, не лимитируя, фильтрование, дистилляцию, кристаллизацию, хроматографию и подобные им методы. Такие вещества могут быть охарактеризованы с использованием стандартных средств, включая физические константы и спектральные данные.
Если не указано иначе, реакции, описанные в данном тексте, проводятся в атмосфере инертного газа при атмосферном давлении в температурном интервале приблизительно от -78°С до 150°С, более предпочтительно приблизительно от 0°С до 125°С, и наиболее предпочтительно и стандартно при комнатной температуре или температуре окружающей среды, например, около 20°С. Специалист в области техники способен найти оптимальные реакционные условия для каждой реакции без специально поставленного эксперимента.
Некоторые соединения в следующих схемах изображены с обобщенными заместителями, однако, специалисту в области техники легко понять, что природа R групп может варьироваться с выходом на различные соединения, рассматриваемые в настоящем изобретении. Кроме того, реакционные условия служат в качестве примера, при этом альтернативные условия также хорошо известны. Реакционные последовательности в следующих примерах не подразумевают ограничения объема изобретения, изложенного в формуле изобретения.
Примеры показательных соединений, включенных в настоящее изобретение и входящих в объем изобретения, приведены в следующих таблицах. Эти примеры и методы получения, которые затем приведены, дают возможность специалистам в области техники более ясно понять и практически ознакомиться с настоящим изобретением. Они не подразумевают ограничения объема изобретения, но выполняют только иллюстративную и представительную функции.
Соединения формулы I проявляют таутомеризм. Таутомерные соединения могут существовать в виде двух или более превращающихся друг в друга разновидностей соединения. Прототропные таутомеры образуются в результате миграции ковалентно связанного атома водорода между двумя атомами. Таутомеры в общем находятся в равновесии и попытка разделить их на индивидупльные таутомеры обычно дает смесь, чьи химические и физические свойства соответствуют смеси соединений. Положение равновесия зависит от химических особенностей молекулы. Например, во многих алифатических альдегидах и кетонах, таких как ацетальдегид, кетонная форма доминирует, в то время как в фенолах преобладает энольная форма. В общем прототропические таутомеры включают кето/энольные (-С(=O)-СН-/-С(-ОН)=СН-), амид/имидокислотные (-C(=O)-NH-/-C(-OH)=N-) и амидиновые (-C(=NR)-NH-/-C(-NHR)=N-) татомеры. Последние две формы особенно часто встречаются в гетероарильных и гетероциклических кольцах, и настоящее изобретение включает все таутомерные формы соединений.
В общем, используемая в этой заявке номенклатура базируется на AUTONOM™(v.4.0, компьютаризированной системе института Бельштейна для формирования IUРАС систематической номенклатуры. Если есть расхождение между изображенной структурой и данным этой структуре наименованием, изображенной структуре следует отдавать предпочтение.
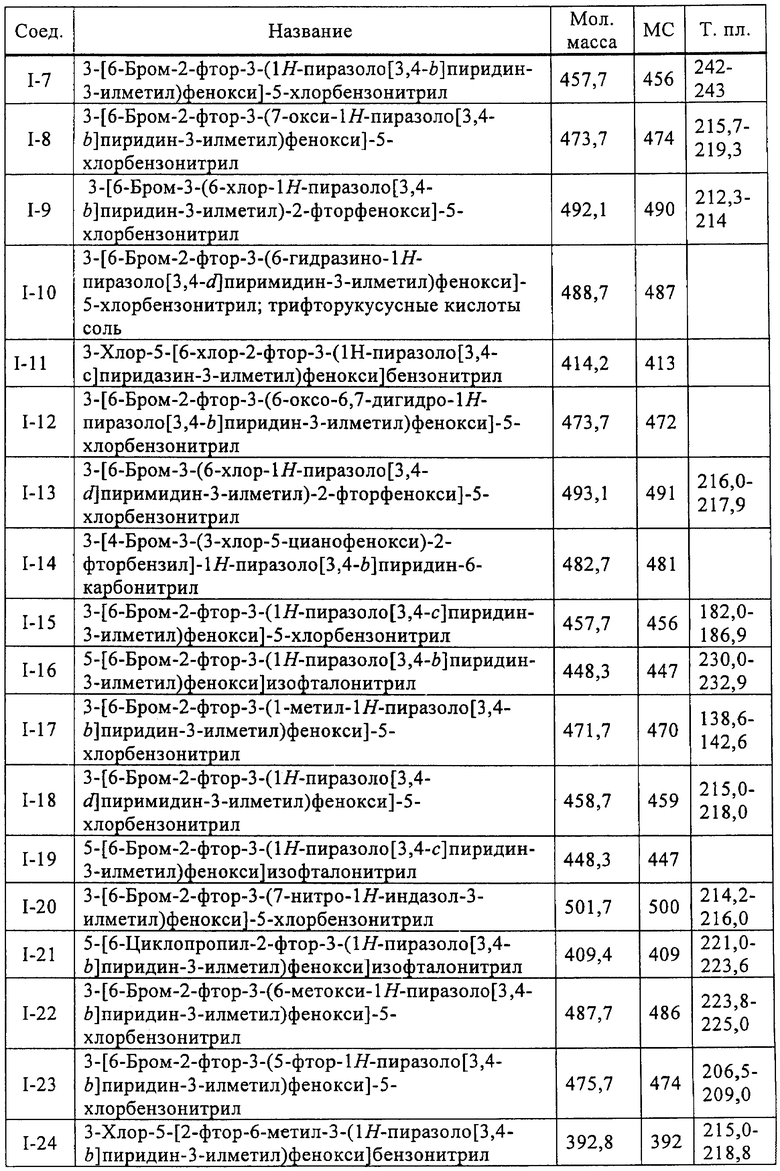

Исходными соединениями для пиразолов, индазолов, азаиндазолов и ди-азаиндазолов по настоящему изобретению являются сложные эфиры 2,4-дизамещенных-3-арилоксифенилуксусных кислот, например, (10а), которые могут быть получены методом, описанным в примере А. Некоторые варианты по настоящему изобретению являются неконденсированными пиразолами, которые легко получаются путем циклизации β-кетоэфиров и гидразина (см., пример 1) или биалкилирования металлированного пиразола (пример 2). Конденсированные пиразолы, раскрытые в данном описании, могут быть стандартно получены путем интрамолекулярной циклизации гидразина и α-оксоароматического кольца, имеющего замещаемый остаток в о-положении по отношению к оксо-заместителю. Замещаемым остатком обычно является галоген; однако, другие уходящие группы (такие, как мезилат или замещенное арилокси-кольцо) также используются в таких реакциях. Требуемый α-оксо-2-гало-(гетеро)арильный фрагмент (14б) получают посредством конденсации Кляйзена соединения (10а) и необязательно замещенного сложного эфира 2-галобензойной кислоты (например, 12). Стандартная схема включает реакцию 2-галоарилкарбоновой кислоты с диимидазолкарбонилом, приводящую к активированному производному кислоты in situ, которое, в свою очередь, эффективно конденсируется с (10а) в присутствии основания, давая β-кетоэфир (14а), который омыляют и декарбоксилируют с образованием сединения (14б). Альтернативно, сложный эфир (10а) может быть превращен в соответствующий аьдегид (10б) посредством прямого восстановления соединения (10б) или восстановления соответствующего первичного спирта и повторным окислением спирта в соединение (10б). Короче говоря, производные карбоновой кислоты могут быть иногда непосредственно восстановлены в карбоксальдегиды с помощью гидридов металлов таких, как ДИБАL-Н или LiAlH(O-трет-Bu)3 в инертном растворителе таком, как углеводородный растворитель, обычно при пониженных температурах. Более жесткие условия обычно приводят к соответствующему первичному спирту, который легко окисляется до альдегида с помощью целого ряда окислительных агентов, хорошо известных из области техники (см., например, J.March, Advanced Organic Chemistry. John Wiley & Sons, New York, 1992, p.1167-1172). Присоединение металл(гетеро)ароматического соединения к карбонилу и последующее окисление образующегося продукта также приводит к соединению (14б). Циклизация о-замещенных ацетофенонов с гидразином позволяет получить индазолы (X. Li et al., J. Med Chem., 2003, 46(26): 5663-5673; B.R.Henke et al., J. Med. Chem., 1997, 40(17): 2706-2725; S.Caron and E.Vazquez, Org. Proc. Res. Develop., 2001, 5(6):587-592). Реакция может быть проведена в любом инертном растворителе, который имеет температуру кипения, достаточно высокую, чтобы в результате приводить к имину и замещению уходящей группы.
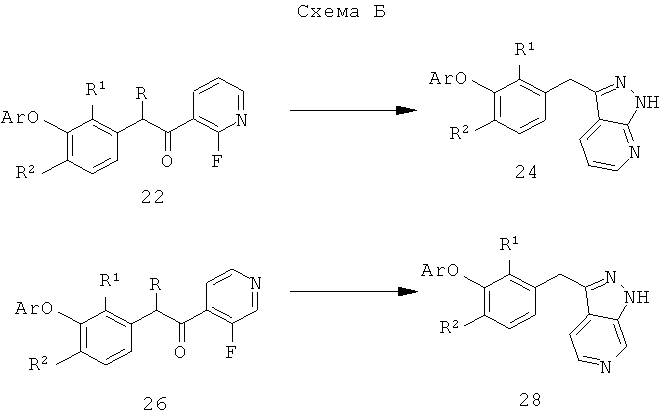
Настоящее изобретение также включает конденсированные аза- и ди аза-индазольные соединения. 1H-пиразоло[3,4-б]пиридин-3-илметильные соединения могут быть получены из промежуточного продукта 3-ацил-2-галопиридина (22) посредством циклизации с гидразином (В.М.Lynch et al., Can. J. Chem., 1988, 66(3):420-428). Требуемые соединения доступны с помощью конденсации Кляйзена (приводя к соединению (22), где R обозначает алкоксикарбонил) и последующего декарбоксилирования (приводя к соединению (22), где R обозначает Н), исходя из 2-галоникотиновой кислоты (например, пример 6). Таким образом обработка 3-{6-бром-2-фтор-3-[2-(2-фторпиридин-3-ил)-2-оксоэтил]фенокси}-5-хлорбензонитрила (22), (R=Н, R1=F, R2=Br, Ar=3-хлор-5-цианофенил) гидразином приводит к образованию имина и замещению лабильного фтора на пиридиновое кольцо с получением соединения (I-7). 2-Хлорзамещенная никотиновая кислота также может быть использована для введения пиразольного кольца. Альтернативно, пиридиновые производные, которые могут быть селективно металлизированы в 3-положении, могут быть конденсированы с альдегидом и вновь окислены до кетона (см. пример 7). Оптимальное осуществление процесса зависит от доступности соответствующего реагента. 1Н-пиразоло[3,4-с]пиридин-3-илметильные производные получают аналогично из производных 3-галоизоникотиновой кислоты (см. пример 16) или из 3-гало-4-металлпиридинов (см. пример 12). Аналоги 1Н-пиразоло[3,4-d]пиримидин-3-ила получают из 4-хлорпиримидин-5-карбоновой кислоты посредством конденсации Кляйзена с последующей интрамолекулярной конденсацией с гидразином, которые приводят к соединению (1-18). 6-Хлор-1Н-пиразоло[3,4-d]пиримидинильные соединения получают подобным образом, исходя из 2,4-дихлорпиримидин-5-карбоновой кислоты. Замена 2-хлорзаместителя гидразином во время циклизации приводит к выделению соли трифторуксусной кислоты 3-[6-бром-2-фтор-3-(6-гидразино-1Н-пиразоло[3,4-а]пиримидин-3-илметил)фенокси]-5-хлорбензонитрила (1-10) в качестве побочного продукта реакции. Соединения формулы I, где А обозначает
1Н-пиразоло[3,4-с]пиридазин-3-илметил (А=А2, X1=Х2=N, X3=СН) получают аналогично, за исключением того, что конденсацию Кляйзена проводят с 3-(2,4-дифторфенокси)пиридазин-4-карбоновой кислотой (см. пример 21), вместо 2-фторникотиновой кислоты.
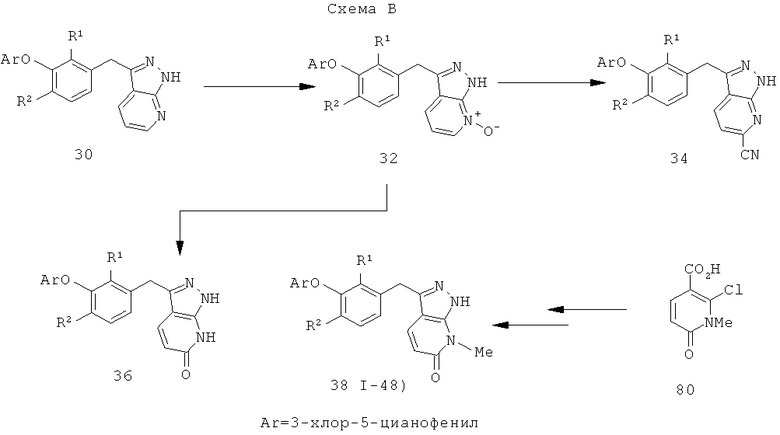
Дополнительное замещение в промежуточной 2-галоникотиновой кислоте приводит к ряду пиразоло[3,4-b]пиридин-3-ильных производных, например, соединений (I-22) и (I-23). Альтернативно, пиразоло[3,4-b]пиридиновое кольцо может быть далее замещено после обработки конденсированного кольца. Окисление азота пиридина в соответствующий N-оксид проводится с помощью м-хлорпербензойной кислоты. Известны другие подходящие для применения в этой реакции окислители, например, перекись водорода и перкислоты. N-оксид превращают далее в соответствующий нитрил (1-14), используя цианид натрия и ТМС-C1 (Н. Vorbrügen and К. Krolikiewicz, Synthesis, 1983 316-18), или в соответствующее 6-оксо-6,7-дигидро-1Н-пиразоло[3,4-b]пиридин-3-ильное производное (I-12) с применением ангидрида трифторуксусной кислоты (ТФАА) (K.Konno et al., Heterocycles, 1986, 24(8):2169-2172). 6-Оксо-6,7-дигидро-1Н-пиразоло[3,4-b]пиридин-3-ильные производные с N-алкильным заместителем на азоте пиридона получают аналогично, посредством двухстадийного процесса, включающего конденсацию Кляйзена и последующую интрамолекулярную конденсацию с гидразином, исходя из N-алкилированного пиридона (80). Активирование соединения (40) с помощью КДИ приводит в результате к замещению хлорида на свободный имидазол, который далее замещается гидразином во время циклизации (см. пример 9).
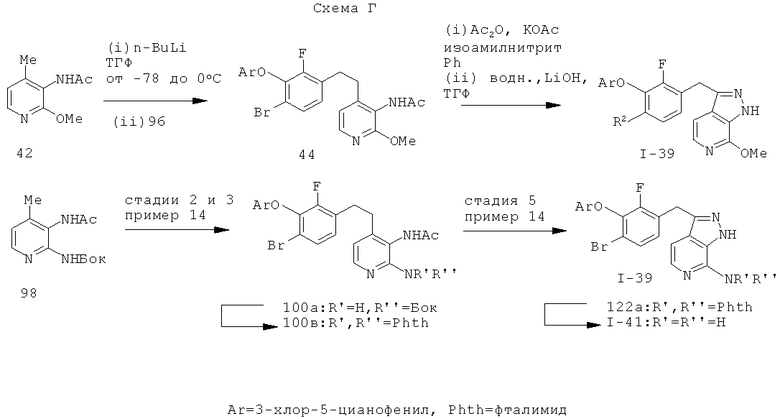
Некоторые замещенные пиразоло[3,4-b]пиридин-3-илметильные соединения были получены посредством синтеза Якобсона-Хубера (P. Marakos et al., SynLett, 1997, 561; С.Ruechardt and V.Hassmann, Annalen, 1980, 908-927; C.Ruechardt and V.Hassmann, Synthesis, 1972, 375; P. Jacobson and L. Huber, Chem. Ber., 1908, 41:667). Применяя эти условия ариламин превращают в соль диазония, которая подвергается затем интрамолекулярной циклизации с получением аза-индазола. Требуемые пиколиновые производные (42) и (98) могут быть получены согласно литературным источникам. Депротонизация гетероарилметильной группы проводится с помощью н-BuLi, после чего полученное в результате гетероарилметиллитиевое промежуточное соединение алкилируется соединением (96), давая соединение (44), которое вводится в реакцию Якобсона с получением соединения (I-39).
Соединения по настоящему изобретению могут быть приготовлены в виде разнообразных дозированных форм для орального введения. Оральное введение может осуществляться в форме таблеток, таблеток в оболочке, драже, твердых и мягких желатиновых капсул, растворов, эмульсий, сиропов или суспензий. Соединения по настоящему изобретению эффективны при введении другими методами, включая непрерывное капельное внутривенное введение, топическое, парентеральное, внутримышечное, внутривенное, подкожное, трансдермальное (которое может включать агент, активирующий проницаемость), трансбуккальное, назальное, ингаляционное и суппозитарное введение, наряду с другими методами введения. Предпочтительным методом введения является обычное оральное введение с использованием стандартного регламента суточного дозирования, которое может регулироваться в соответствии с заболеванием и ответом пациента на активный ингредиент.
Соединение или соединения по настоящему изобретению, а также их фармацевтически приемлемые соли, вместе с одним или более стандартных наполнителей, носителей или разбавителей, могут быть превращены в форму фармацевтических композиций и единичных доз. Фармацевтические композиции и формы единичных доз могут включать стандартные ингредиенты в стандартных пропорциях, с или без дополнительных активных соединениий или элементов, при этом единичная доза может содержать любое подходящее эффективное количество активного ингредиента, пропорциональное рекомендованной к применению суточной дозе. Фармацевтические композиции могут применяться в твердом виде, таком как таблетки или наполненные капсулы, полутвердом виде, порошках, в поддерживающих высвобождение формах, или в виде жидкостей, таких как растворы, суспензии, эмульсии, эликсиры, или наполненные капсулы для орального применения; или в форме суппозиториев для ректального или вагинального введения; или в форме стерильных иньекционных растворов для парентерального введения. Типичный препарат должен содержать приблизительно от 5 до 95 мас.% активного соединения или соединений. Термин «препарат» или «дозируемая форма» предполагает включение как твердой, так и жидкой формы активного соединения, и каждый специалист в области техники должен понимать, что активный ингредиент может существовать в виде различных форм препаратов, в зависимости от органа или ткани, являющихся мишенями, и от необходимой дозы и фармакокинетических параметров.
Термин «вспомогательное вещество», используемый в данном описании, относится к соединению, которое применяется при изготовлении фармацевтической композиции, в основном безопасной, нетоксичной и не являющейся нежелательной ни с биологической, ни с любой другой точки зрения, и включающей вспомогательные вещества, которые приемлемы для ветеринарного использования, а также в медицинской фармакологии. Термин «вспомогательное вещество», используемый в данном описании, включает как одно, так и более одного вспомогательного вещества.
Фраза «фармацевтически приемлемая соль» соединения подразумевает соль, являющуюся фармацевтически приемлемой и обладающей необходимой фармакологической активностью исходного соединения. Такие соли включают: (1) кислотно-аддитивные соли, образованные с неорганическими кислотами, такими как хлористоводородная кислота, бромистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и им подобные; или образованные с органическими кислотами, такими как уксусная кислота, пропионовая кислота, гексановая кислота, циклопентанпропионовая кислота, гликолевая кислота, пировиноградная кислота, молочная кислота, малоновая кислота, янтарная кислота, яблочная кислота, малеиновая кислота, фумаровая кислота, винная кислота, лимонная кислота, бензойная кислота, 3-(4-гидроксибензоил)бензойная кислота, коричная кислота, миндальная кислота, метансульфоновая кислота, этансульфоновая кислота, 1,2-этандисульфоновая кислота, 2-гидроксиэтансульфоновая кислота, бензолсульфоновая кислота, 4-хлорбензолсульфоновая кислота, 2-нафталинсульфоновая кислота, 4-толуолсульфоновая кислота, камфорсульфоновая кислота, 4-метилбицикло[2.2.2]-окт-2-ен-1- кислота, глюкогептоновая кислота, 3-фенилпропионовая кислота, триметилуксусная кислота, трет-бутилуксусная кислота, лаурилсерная кислота, глюконовая кислота, глутаминовая кислота, гидроксинафтойная кислота, салициловая кислота, стеариновая кислота, муконовая кислота и им подобные; или (2) соли, образующиеся, когда кислотный протон, присутствующий в исходном соединении, либо замещается ионом металла, например, ионом щелочного металла, ионом щелочноземельного металла или ионом алюминия; либо координируется с органическим основанием, таким как этаноламин, диэтаноламин, триэтаноламин, N-метилглюкамин, и им подобным. N-Ацилсульфонамиды имеют кислотный протон, который в принципе может образовать соль с органическим или неорганическим катионом.
Предпочтительными фармацевтически приемлемыми солями являются соли, образующиеся из уксусной кислоты, хлористоводородной кислоты, серной кислоты, метансульфоновой кислоты, малеиновой кислоты, фосфорной кислоты, винной кислоты, лимонной кислоты, с ионами натрия, калия, кальция, цинка и магния. Следует понимать, что все ссылки на фармацевтически приемлемые соли включают формы с присоединенным растворителем (сольваты) или кристаллические формы (полиморфотропы), как определо в описании, этих кислотно-аддитивных солей.
Твердые формы препаратов включают порошки, таблетки, пиллеты, капсулы, облатки, суппозитории и дисперсные гранулы. Твердый носитель может представлять собой одну или более субстанций, которые могут действовать как разбавители, вкусовые агенты, растворители, смазки, суспендирующие агенты, связующие агенты, консерванты, дезинтегрирующие агенты или инкапсулирующий материал. В порошках носителем обычно является тонко размельченное твердое вещество, которое смешивается с тонко размельченным активным компонентом. В таблетках активный компонент обычно смешивается с носителем, обладающим необходимой связывающей способностью, в соответствующих пропорциях, и запрессовывается в таблетки с необходимой формой и размером. Подходящие носители включают, не лимитируя, карбонат магния, стеарат магния, тальк, сахар, лактозу, пектин, декстрин, крахмал, желатин, трагакант, метилцеллюлозу, натрий карбоксиметилцеллюлозу, низкоплавкий воск, масло какао и им подобные. Препараты в твердой форме могут содержать в дополнение к активному компоненту красители, отдушки, стабилизаторы, буферы, искусственные и натуральные подсластители, дисперсанты, загустители, солюбилизирующие агенты и им подобные.
Жидкие препараты пригодны также для орального введения, включая жидкие препараты, содержащие эмульсии, сиропы, эликсиры, водные растворы и водные суспензии. Они включают твердые формы препаратов, предназначенные для превращения в жидкую форму незадолго до употребления. Эмульсии могут быть получены в растворах, например, в водных растворах пропиленгликоля, или могут содержать эмульгирующие агенты, такие как лецитин, сорбитанмоноолеат или аравийскую камедь. Водные растворы могут быть получены растворением активного компонента в воде и добавлением соответствующих красителей, улучшителей вкуса, стабилизаторов и загущающих агентов. Водные суспензии могут быть получены диспергированием тонкоизмельченного активного компонента в воде с вязким материалом, таким как натуральные или синтетические смолы, полимеры, метилцеллюлоза, натрийкарбоксиметилцеллюлоза, и другие, хорошо известные суспендирующие агенты.
Соединения по настоящему изобретению могут быть приготовлены для парентерального введения (например, путем иньекции, например, болюсной инъекции или непрерывной инфузии) и могут содержаться в виде единичной дозы в ампулах, предварительно наполненных шприцах, в контейнерах небольшого объема или контейнерах, содержащих многократную дозу, с добавленным консервантом. Композиции могут быть приготовлены в виде таких форм, как суспензии, растворы или эмульсии, в масляных или водных растворах, например, в виде растворов в водном полиэтиленгликоле. Примеры масляных или неводных носителей, разбавителей и растворителей включают пропиленгликоль, полиэтиленгликоль, растительные масла (например, оливковое масло), и инъекционные органические сложные эфиры (например, этилолеат), и могут содержать такие агенты, как консерванты, смачивающие агенты, эмульсифицирующие или суспендирующие, стабилизирующие и/или диспергирующие агенты. Альтернативно, активный агент может находиться в порошкообразной форме, полученной посредством асептического выделения стерильного твердого вещества или лиофилизации из раствора, для получения из него перед использованием соответствующего раствора в стерильной воде.
Соединения по настоящему изобретению могут быть приготовлены для местного эпидермического введения в виде мазей, кремов или лосьонов или трансдермальных пластырей. Мази и кремы могут быть, например, приготовлены на водной или масляной основе с добавлением соответствующих загущающих и/или желирующих агентов. Лосьоны могут быть приготовлены на водной или масляной основе, и, в общем, также содержать один или более эмульсирующих, стабилизирующих, диспергирующих, суспендирующих, загущающих или красящих агентов. Препараты, подходящие для топического введения через рот, включают леденцы, содержащие активные агенты с вкусовыми добавками, обычно на основе сахарозы и аравийской камеди, или тракаганта; пастилки, содержащие активный ингредиент на инертной основе, такой как желатин и глицерин, или сахароза и аравийская камедь; и ополаскиватели рта, включающие активный ингредиент в соответствующем жидком носителе.
Соединения по настоящему изобретению могут быть приготовлены для введения в виде суппозиториев. При этом низкоплавкий воск такой, как смесь глицеридов жирных кислот или масла какао, сначала расплавляют, а затем гомогенно диспергируют в этом растворе активный компонент, например, посредством перемешивания. Затем расплавленную гомогенную смесь переносят в формы соответствующего размера и оставляют для самопроизвольного охлаждения и затвердевания.
Соединения по настоящему изобретению могут быть приготовлены для вагинального введения в виде пессариев, тампонов, кремов, гелей, паст, пен или спреев, содержащих дополнительно к активному ингредиенту носители, хорошо известные из области техники.
Соединения по настоящему изобретению могут быть приготовлены для назального введения. Растворы или суспензии вводят непосредственно в носовую полость посредством стандартных способов с помощью капельницы, пипетки или спрея. Препараты могут быть изготовлены в форме единичной или мультиплетной дозы. В последнем случае при применении капельницы или пипетки пациенту вводится соответствующий, определенный объем раствора или суспензии. В случае использования спрея введение осуществляется, например, дозированным разбрызгиванием жидкости с помощью пульверизатора.
Соединения по настоящему изобретению могут быть приготовлены для аэрозольного введение, в частности, в респираторный тракт, включая интраназальное введение. Соединения при этом должны иметь небольшие размеры частиц, например, порядка пяти (5) микрон или менее. Частицы такого размера могут быть получены известными из области техники методами, например, с помощью микронизации. Активный ингредиент помещается в баллон под давлением с соответствующим пропеллентом таким, как хлорфторуглерод (ХФУ), например, дихлордифторметан, трихлорфторметан или дихлортетрафторэтан, двуокись углерода или другой подходящий газ. Аэрозоль обычно может содержит также поверхностно-активное вещество такое, как лецитин. Доза лекарства может регулироваться с помощью измерительного клапана. Альтернативно, активные ингредиентыы могут быть представлены в виде сухого порошка, например, порошкообразной смеси на основе лактозы, крахмала, производных крахмала, гидроксипропилметилцеллюлозы и поливинилпирролидина (ПВП). Для введения в назальную полость порошковый носитель должен быть в форме геля. Порошковые композиции могут находиться в форме единичной дозы, например, в капсулах или картриджах, например, из желатина, или упакованы в блистеры, из которых порошок может быть введен посредством ингаляции.
При необходимости препараты могут быть получены с кишечно-растворимой адаптацией для поддержания или контроля высвобождения активного ингредиент при введении. Например, соединения по настоящему изобретению могут быть сформированы для трансдермальной или подкожной доставки лекарства. Эта система доставки является удобной, когда необходимо поддержание высвыбождения соединения и когда соблюдение пациентом режима и схемы лечения являются решающими. Соединения при трансдермальных методах введения часто соединяют с приклеивающейся к коже твердой подложкой. Соединение может быть также скомбинировано с усилителем его проникающей способности, например, азоном (1-додецилазациклогептан-2-он). Механизмы систем поддержания высвыбождения закрепляются подкожно в субдермальный слой хирургически или иньекционно. Субдермальные имплантаты инкапсулируют соединение в жидкую растворимую мембрану, например, силиконовый полимер или биодеградируемый полимер, например, полиакриловую кислоту.
Подходящие препараты наряду с фармацевтическими носителями, разбавителями и наполнителями описаны в Remington: The Science and Practice of Pharmacy, 1995, edited by E.W.Martin, Mack Publishing Company, 19th edition, Easton, Pennsylvania. Специалист в данной области техники может модифицировать технику приготовления препаратов для получения многочисленных рецептур, предназначенных для каждого отдельного способа введения, чтобы избежать превращения композиций по настоящему изобретению в нестабильные препараты и ухудшить их терапевтическую активность.
Модификация настоящих соединений, повышающих их растворимость в воде или другом растворителе может сопровождаться незначительными модификациями, например, образованием соли, сложно-эфирной этерификацией и т.д., которые хорошо известны любому специалисту в области техники. Также специалисту в области техники хорошо известны способы введения и режим дозирования отдельного соединения, чтобы управлять фармакокинетикой настоящих соединений с максимально благоприятным эффектом для пациента.
Термин «терапевтически эффективное количество», используемый в настоящем описании, подразумевает количество, требуемое, чтобы снизить симптомы болезни у пациента. Статус ВИЧ инфекции может быть проконтролирован измерением вирусной нагрузки (РНК) или мониторингом уровней Т-клеток. Доза может быть отрегулирована в зависимости от индивидуальных требований в каждом конкретном случае. Такая доза может варьироваться в широких пределах в зависимости от многочисленных факторов таких, как тяжесть болезни, подлежащей лечению, возраст и общее состояние здоровья пациента, лечение пациента другими лекарственными средствами, пути и формы введения лекарственного средства, включающие предпочтения и опыт лечащего врача. Для орального введения дневная доза составляет приблизительно от 0,01 до 100 мг/кг массы тела в день и является соответствующей для монотерапии и/или комбинационной терапии. Предпочтительные суточные дозы составляют приблизительно от 0,1 до 500 мг/кг массы тела, более предпочтительные приблизительно от 0,1 до 100 мг/кг массы тела и наиболее предпочтительно приблизительно от 1,0 до 10 мг/кг массы тела в день. Таким образом, для пациента с массой тела 70 кг доза введения составляет приблизительно от 7 мг до 0,7 г в день. Суточная доза может быть введена в виде разовой дозы или единичных доз, обычно в количестве от 1 до 5 доз в день. Обычно лечение начинается с доз, меньших, чем оптимальная доза соединения. Кроме того, дозу увеличивают небольшими порциями, пока не будет достигнут оптимальный эффект для индивидуального пациента. Любой специалист в лечении болезней, описанных в данном изобретении, может установить, без дополнительного эксперимента, на основании профессиональных знаний, опыта и приведенных в настоящей заявке данных, терапевтически эффективное количество соединений по настоящему изобретению для данной болезни и пациента.
В вариантах осуществления изобретения активное соединение или соль могут быть введены в сочетании с другим антивирусным агентом таким, как нуклеозидный ингибитор обратной транскриптазы, другой ненуклеозидный ингибитор обратной транскриптазы или ингибитор ВИЧ протеазы. Когда активное соединение или его производное или соль вводится в комбинации с другим антивирусным агентом, активность может возрастать по сравнению с активностью исходного соединения. Когда лечение представляет собой комбинационную терапию, такое введение может быть совместным или последовательным в отношении нуклеозидных производных. Термин «совместное введение», используемый в данном описании, включает введение агентов в одно и то же время или в разное время. Введение двух или более агентов в одно и то же время достигается посредством введения разовой дозы препарата, содержащего от двух или более активных ингредиентов, или посредством последовательного одновременного введения двух или более дозированных форм с одним активным агентом.
Следует понимать, что ссылки в данном тексте на лечение распространяются на профилактику, а также лечение существующих болезненных состояний, и что лечение животных включает лечение человека, а также других животных. Кроме того, лечение ВИЧ инфекции, как определено в данном описании, включает также лечение или профилактику болезни или болезненного состояния, связанного или опосредованного ВИЧ инфекцией или его клиническими симптомами.
Фармацевтические препараты представляют собой предпочтительно единичные дозированные формы. В такой форме препарат подразделен на единичные дозы, содержащие соответствующие количества активного компонента. Единичная доза может быть упакована, при этом пакет может содержать отдельные количества препарата в виде пакетированных таблеток, капсул и порошков в запаянных ампулах. Также единичная дозированная форма может быть представлена в виде капсул, таблеток, облаток или леденцов, или может представлять собой соответствующее число любого из них в пакетированной форме.
Данные примеры и методики получения, которые описаны далее, представлены, чтобы дать возможность специалисту в области техники более отчетливо понять и практически уяснить данное изобретение. Они не лимитируют объема изобретения, а скорее иллюстрируют и представляют его.
Примеры
Показательный пример А: 3-Арилоксифенилукусусные кислоты [4-Хлор-3-(3-хлор-5-цианофенокси)-2-фторфенил]укусусная кислота (R-1) и хлорангидрид [4-хлор-3-(3-хлор-5-цианофенокси)-2-фторфенил]укусусной кислоты (R-2)
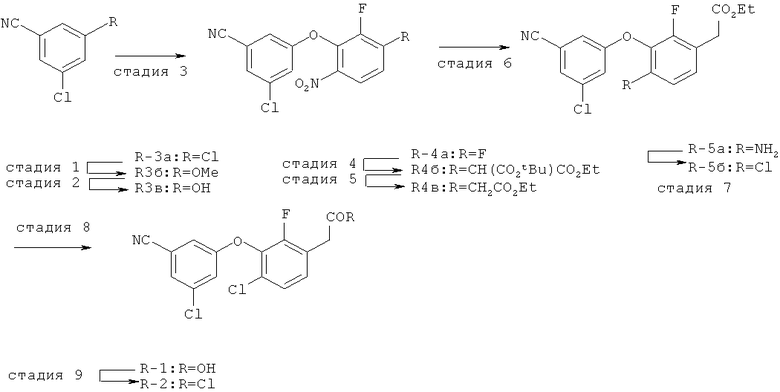
стадия 1: в круглодонную колбу объемом 100 мл помещают в токе азота 3,5-дихлорбензонитрил (R-3a) (7,0 г, 40,69 ммолей) и безводный ДМФ (75 мл), затем добавляют метоксид натрия (2,26 г, 44,76 ммолей) и образовавшийся раствор перемешивают далее при КТ в течение 24 ч. Когда реакция завершится, в реакционную емкость добавляют по каплям водный раствор 10%-ной HCl. Сырую смесь экстрагируют ЕtOАс и последовательно промывают водным раствором кислоты, водой и рассолом. EtOAc экстракты высушивают (Na2SO4), фильтруют и растворитель удаляют в вакууме, получая сырое твердого вещества, которое перекристаллизовывают из смеси гексан/ацетон, получая 5,9 г (выход 86%) соединения (R-3б).
стадия 2: в круглодонную колбу объемом 250 мл помещают (R-3б) (7,0 г, 41,766 ммолей) и 2,4,6-коллидин (100 мл). Смесь нагревают до температуры 170°С, добавляют Lil (16,76 г, 125,298 ммолей) и реакционную смесь нагревают в течение 4 ч. Когда соединение (R-3б) полностью прореагирует, реакцию охлаждают до КТ и останавливают с помощью 10%-ного водного раствора HCl. Образовавшуюся смесь экстрагируют EtOAc и промывают водой и рассолом. EtOAc экстракт высушивают над сульфатом натрия и фильтруют. Растворитель удаляют в вакууме, получая желтое масло, которое очищают с помощью хроматографии на силикагеле, элюируя смесью EtOAc/гексан (в соотношении 10:90) и получая 6,0 г (выход 94%) соединения (R-3в).
стадия 3: в круглодонную колбу объемом 250 мл помещают соединение (R-3в) (6,0 г, 39,070 ммолей) и безводный ТГФ (100 мл) и раствор охлаждают до температуры 0°С. К охлажденному раствору прибавляют трет-бутоксид натрия (46,89 г, 4,51 ммолей) и образовавшийся раствор перемешивают в течение 1 ч. Затем по каплям добавляют 2,3,4-трифторнитробензол (6,92 г, 39,070 ммолей), поддерживая при этом температуру реакции при 0°С, пока фенол полностью не прореагирует. После этого смесь гасят, добавляя 10%-ный водный раствор HCl, и образовавшуюся смесь перемешивают дополнительно в течение 1 ч. Затем смесь экстрагируют ЕtO Ас и промывают водой и рассолом. Этилацетатный слой высушивают (Na2SO4.) и фильтруют. Растворитель удаляют в вакууме, получая желтое масло, которое очищают с помощью колоночной хроматографии на силикагеле, элюируя смесью гексан/EtOAc (в соотношении 92:8) и получая 10 г (выход 82%) соединения (R-4a).
стадия 4: к раствору трет-бутилэтилмалоната (10,31 г, 54,80 ммолей) и безводного N-МП (200 мл), охлажденного до температуры 0°С, при перемешивании в атмосфере азота добавляют NaH (40%-ный в минеральном масле) (1,84 г, 76,70 ммолей). Смесь оставляют перемешиваться при температуре 0°С дополнительно в течение 1 ч. бис-Ариловый эфир (R-4a) (15,00 г, 49,80 ммолей) добавляют затем в реакционную емкость и перемешивают в атмосфере азота при КТ до завершения реакции, после чего смесь гасят добавлением 10%-ного водного раствора HCl при КТ. Затем смесь экстрагируют EtOAc и промывают водой и рассолом. EtOAc экстракт высушивают (Na2SO4) и фильтруют, растворитель удаляют в вакууме, получая соединение (R-4б) в виде светло-желтого масла, которое используется на следующей стадии без дальнейшей очистки.
стадия 5: Сложный диэфир (R-4б) (24,0 г, 50,117 ммолей) растворяют в дихлорэтане (300 мл) и ТФК (6,29 г, 55,13 ммолей) и нагревают при температуре 75°С в течение 24 ч. Затем смесь охлаждают до КТ и растворитель вместе с избытком ТФК удаляют в вакууме. Сырое масло растворяют в ДХМ, охлаждают до температуры 0°С и добавляют водный раствор NaHCO3. Затем смесь экстрагируют ДХМ и промывают водой и рассолом. ДХМ экстракт высушивают (Na2SO4), фильтруют и растворитель удаляют в вакууме, получая желтое масло. Сырое масло очищают с помощью хроматографии на силикагеле, элюируя смесью гексан/EtOAc (в соотношении 90:10) и получая 15,0 г (выход 80%) соединения (R-4в).
стадия 6: в круглодонную колбу объемом 250 мл помещают соединение (R-4в) (8,0 г, 21,12 ммолей) и абсолютный EtOH. В реакционную емкость добавляют NH4Cl (2,26 г, 42,244 ммолей), воду (30 мл) и железо (1,17 г, 21,12 ммолей). Реакцию перемешивают и нагревают при температуре 80°С в течение 4 ч. Когда соединение (R-4в) прореагирует, гетерогенную смесь фильтруют через слой CELITE® и остаток на фильтре промывают EtOAc. Водный фильтрат экстрагируют EtOAc и промывают водой и рассолом. Объединенные EtOAc экстракты высушивают над Na2SO4 и фильтруют. Затем растворитель удаляют в вакууме, получая светлое масло, которое очищают с помощью хроматографии на силикагеле, элюируя смесью гексан/ EtOAc (в соотношении 85: 15) и получая 6,0 г (выход 87%) соединения (R-5a).
стадия 7: в круглодонную колбу объемом 100 мл помещают безводный MeCN (15 мл) в постоянном токе азота. К этой смеси добавляют Cu(II)Cl2 (0,083 г, 0,624 ммолей) и трет-бутилнитрил (0,064 г, 0,624 ммолей). Смесь нагревают при температуре 70°С в течение 30 мин. К этой смеси добавляют соединение (R-5а) (0,100 г, 0,624 ммолей) одной порцией и продолжают перемешивание дополнительно в течение 2 ч. После того как исходные вещества прореагируют, реакционную смесь охлаждают до КТ и гасят 10%-ным водным раствором HCl. Затем смесь экстрагируют EtOAc и объединенные экстракты промывают водой и рассолом. EtOAc экстракт высушивают (Na2SO4) и фильтруют. Растворитель удаляют в вакууме, получая слегка коричневое масло, которое очищают с помощью хроматографии на силикагеле, элюируя смесью гексан/EtOAc (в соотношении 96:4) и получая 0,080 г (выход 76%) соединения (R-5б).
стадия 8: в высушенную в печи круглодонную колбу объемом 100 мл, продутую азотом, помещают соединение (R-5б) (2,0 г, 5,43 ммолей) и ТГФ (20 мл) и перемешивают смесь в токе азота. В реакционную емкость добавляют затем LiOH (0,46 г, 10,86 ммолей) с последующим добавлением 5 мл деионизированной воды. Реакцию перемешивают в течение 1 ч в непрерывном токе азота. Гомогенную смесь охлаждают до температуры 0°С и гасят 10%-ным водным раствором HCl, после чего реакционную смесь перемешивают дополнительно в течение 15 мин. Затем сырую смесь экстрагируют EtOAc и промывают водой и рассолом. Органические экстракты высушивают (Na2SO4) и фильтруют. Растворитель удаляют в вакууме и сырую кислоту (R-1) используют без дальнейшей очистки.
стадия 9: в круглодонную колбу объемом 100 мл помещают соединение (R-1) (0,200 г, 0,520 ммолей) и ДХМ (5 мл) и раствор перемешивают в токе азота при КТ. Затем к раствору добавляют по каплям тионилхлорид (0,061 г, 0,520 ммолей), а потом одну каплю ДМФ. Реакцию перемешивают в течение 1 ч при КТ. Избыток растворителя и тионилхлорида удаляют в вакууме, получая карбоновую кислоту (R-2) в виде сырого желтого масла, которое используют в следующей реакции без дальнейшей очистки.
Общий метод получения трет-бутилфенилацетатов
К охлажденному льдом раствору этилового или метилового сложного эфира замещенной фенилукусусной кислоты в ТГФ добавляют водный раствор LiOH·H2O (1,5 экв.). Реакционную смесь перемешивают при КТ и прохождение гидролиза отслеживают с помощью ТСХ или ВЭЖХ. Когда реакция завершается, добавляют 1-молярный раствор HCl и EtOAc, органическую фазу промывают рассолом, высушивают, фильтруют и выпаривают, получая соответствующую карбоновую кислоту.
К раствору карбоновой кислоты в трет-бутаноле в инертной атмосфере добавляют ДМАП (0,3 экв. и ди-трет-бутилдикарбонат (Бок ангидрид, 2 экв.). Реакцию перемешивают при КТ до прекращения выделения газа и завершения реакции. Растворитель удаляют в вакууме и продукт очищают с помощью хроматографии на силикагеле.
4-Хлор-3-(3,5-дицианофенокси)-2-фторфенил [уксусная кислота (R-7) и хлорангидрид 4-хлор-3-(3,5-дицианофенокси)-2-фторфенилуксусной кислоты (R-8)
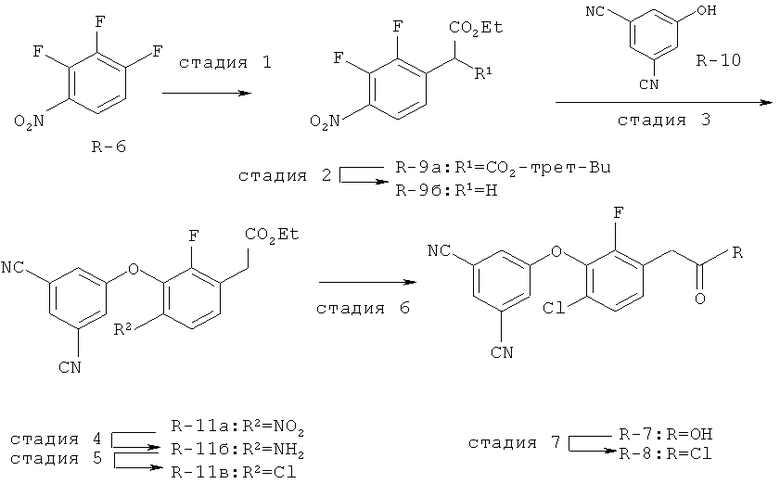
стадии 1 и 2: этил-2,3-дифтор-4-нитрофенилацетат (R-9б)
К охлажденному льдом раствору трет-бутилэтилмалоната (фирма Alfa Aesar) (31,2 г, 166 ммолей) в N-МП (300 мл), охлажденному до температуры 0°С в атмосфере азота, добавляют NaH (60%-ная масляная дисперсия, 13,1 г, 218 ммолей), поддерживая при этом температуру ниже 20°С. После добавления раствор выдерживают в течение 20 мин. К этому раствору добавляют по каплям 2,3,4-трифторнитробензол (R-6) (фирма Oakwood Products Inc.) (26,6 г, 163 ммолей) в N-МП (50 мл), поддерживая при этом температуру ниже 20°С (реация экзотермична). После завершения добавления реакцию выдерживают в течение 2 ч при КТ и реакционную смесь добавляют к водному раствору NH4Cl (1,5 л), экстрагируют EtOAc (трижды по 200 мл), промывают пять раз водой (400 мл), высушивают (MgSO4) и выпаривают. Сырой замещенный малоновый эфир (R-9a) используют без дальнейшей очистки.
Сложный эфир (R-9a) растворяют в ДХМ (400 мл) и добавляют ТФК (100 мл), полученный раствор нагревают при температуре 40°С в течение 16 ч. Затем реакционную смесь охлаждают до КТ и растворители выпаривают. Сырой продукт растворяют в EtOAc (400 мл), промывают последовательно водным раствором NaHCO3, водой и рассолом, высушивают (MgSO4) и выпаривают. Образовавшееся масло очищают с помощью хроматографии на силикагеле, элюируя смесью 5% EtOAc/гексаны и получая соединение (R-9б) в виде золотистого масла (выход 11,9 г, 30%), которое кристаллизуется при стоянии.
стадия 3: раствор безводного ТГФ (100 мл) и соединения (R-10) (10,00 г, 69,38 ммолей), охлажденного до температуры 0°С, обрабатывают трет-бутоксидом натрия (7,34 г, 76,32 ммолей). Затем смесь перемешивают в течение 30 мин при температуре 0°С, после чего добавляют соединение (R-9б) (17,01 г, 69,38 ммолей) и перемешивают в течение 3 ч. Реакцию останавливают с помощью 10%-ного водного раствора НС1. Сырую смесь затем экстрагируют EtOAc и объединенные экстракты промывают водой и рассолом. Органическую фазу высушивают (Na2SO4) и фильтруют. Растворитель удаляют в вакууме, получая сырое масло, которые очищают с помощью хроматографии на силикагеле, элюируя смесью гексаны/EtOAc (в соотношении 90:10) и получая 20 г (выход 78%) соединения (R-11а).
Введение хлорзаместителя (стадии 4 и 5) проводят, как описано на стадиях 6 и 7 при получении соединения (R-1). Гидролиз эфира и получение хлорангидрида кислоты (стадии 7 и 8), проводимых с помощью методов, описанных на стадиях 8 и 9 при получении соединения (R-2), приводят к соединениям (R-7) и (R-8).
Этиловый эфир [4-хлор-3-(3-циано-5-дифторметоксифенокси)-2-фторфенил]уксусной кислоты (R-12)
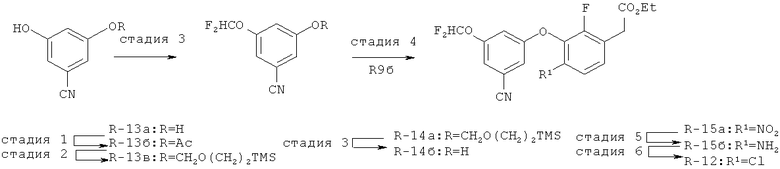
стадия 1: Уксусный ангидрид (30 мл, 4 экв.) добавляют к раствору соединения (R-13a) (10,36 г, 77 ммолей) в безводном пиридине (60 мл), охлажденному до температуры 0°С в атмосфере азота. Затем реакцию нагревают до КТ и перемешивают в течение 16 ч. Летучие компоненты удаляют в вакууме, а оставшееся масло растворяют в EtOAc, промывают водой, 5%-ным раствором HCl, рассолом и высушивают (MgSO4). Летучие компоненты удаляют, получая 14,5 г (выход 86%) диацетата. Затем диацетат (14 г, 64 ммолей) растворяют в смеси EtOH (100 мл) и бензола (100 мл) и охлаждают до 0°С. После этого добавляют по каплям раствор КОН (3,6 г, 1 экв.) в EtOH. Через 1 ч раствор переносят в охлажденный льдом насыщенный раствор NH4Cl, экстрагируют диэтиловым эфиром и промывают рассолом. Эфирные экстракты концентрируют и очищают с помощью хроматографии на силикагеле, элюируя смесью гексан/EtOAc с градиентом от 0% до 25% EtOAc, получая 10 г соединения (R-13б) (выход 88%).
стадия 2: (2-Триметилсилилэтокси)метилхлорид (2,2 мл, 1,1 экв.) добавляют к раствору соединения (R-13б) (2,0 г, 11,3 ммолей) и ДИПЭА (2,4 мл, 1,2 экв.) в ДХМ (50 мл), охлажденному до температуры 0°С. Затем раствор нагревают до КТ, перемешивают в течение 16 ч и переносят в насыщенный раствор NaHCO3. Водный раствор экстрагируют ДХМ и объединенные органические экстракты промывают водой и рассолом и высушивают (MgSO4). Растворители удаляют в вакууме и ацетилированный продукт растворяют в смеси воды (8 мл) и ТГФ (32 мл), после чего добавляют LiOH·H2O (0,71 г, 1,5 экв.). Смесь перемешивают в течение 2 ч, подкисляют до рН 5 и экстрагируют этиловым эфиром. Органический слой высушивают (MgSO4) и выпаривают, получая 2,5 г (выход 80%) соединения (R-13b).
стадия 3: F2ClCCO2Na (2,84 г, 2,3 экв.) добавляют к раствору Cs2CO3 (3,69 г, 1,4 экв.), соединения (R-13в) (2,26 г, 8,09 ммолей), ДМФ (32 мл) и воды (2 мл).
Полученную смесь нагревают при температуре 100°С в течение 2 ч, охлаждают до КТ и переносят в насыщенный раствор NH4Cl. Затем раствор экстрагируют смесью EtOAc и гексанов и органический слой промывают рассолом и высушивают (MgSO4). Сырой продукт очищают с помощью хроматографии на силикагеле, элюируя смесью EtOAc/гексан с градиентом от 0 до 10% EtOAc, получая 1,83 г (выход 70%) соединения (R-14a). Затем дифторметиловый эфир (R-14a) растворяют в МеОН (30 мл) и добавляют 5,6 мл 1-молярного раствора НС1. Раствор нагревают при температуре 50°С в течение 5 ч и перемешивают при КТ в течение 16 ч. Летучие компоненты выпаривают, а водный остаток распределяют между ДХМ и водой. Водный слой экстрагируют ДХМ и объединенные экстракты промывают водой и рассолом. Летучие компоненты удаляют в вакууме, получая 780 мг (выход 73%) соединения (R-14б).
Конденсацию соединения (R-14б) с соединением (R-9б) проводят методом, описанном на стадии 3 при получении соединения (R-7). Восстановление нитрогруппы (стадия 5), диазотирование амина и замещение хлоридом (стадия 6), гидролиз эфира и превращение кислоты в хлорангидрид кислоты осуществляют методом, описанном на стадиях 6-9 при получении соединения (R-2).
Этиловый эфир [4-хлор-3-(3-циано-5-метоксифенокси)-2-фторфенил]уксусной кислоты получают подобным же образом за исключением использования на стадии 4 вместо соединения (R-14б) 3-циано-5-метоксифенола (CAS Reg. No. 124993-53-9).
Этиловый эфир [4-хлор-3-(3-циано-5-дифторметилфенокси)-2-фторфенил]укусусной кислоты (R-16a)
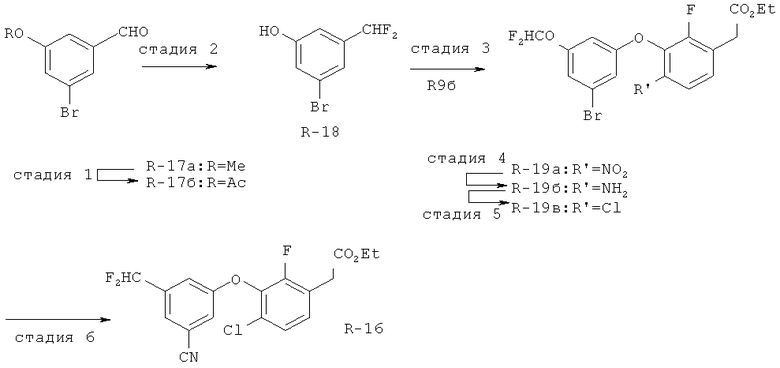
стадия 1: Раствор BBr3 (29,1 мл 1-молярного раствора в ДХМ, 29,1 ммолей) медленно добавляют к раствору соединения (R-17a) (2,5 г, 11,62 ммолей, СAS Reg. No. 262450-65-7) в безводном ДХМ (25 мл), в атмосфере азота при температуре -78°С. Оранжевый раствор нагревают до КТ, перемешивают в течение 2 ч и переносят на лед. Затем смесь экстрагируют ДХМ (100 мл) и органический слой промывают H2O (50 мл) и рассолом (50 мл). Растворители выпаривают, а оставшееся масло очищают с помощью хроматографии на силикагеле, элюируя смесью EtOAc/гексаны с градиентом от 0% до 20% EtOAc, получая нужный фенол. К раствору этого фенола в пиридине (10 мл) в атмосфере аргона медленно добавляют уксусный ангидрид (0,6 мл, 6,33 ммолей). Через 2 ч летучие компоненты удаляют, получая 3-бром-5-формилфенилацетат (R-17б), с выходом 1,02 г (40%).
стадия 2: ДАСТ (диэтиламиносульфуртрифторид) (1,02 мл, 7,69 ммолей) добавляют к раствору 3-бром-5-формилфенилацетата (R-17б) (1,1 г, 4,52 ммолей) в ДХМ (5 мл) в атмосфере азота в NALGENE® колбе. Затем добавляют EtOH (0,013 мл, 0,23 ммолей) и смесь перемешивают в течение 16 ч. После этого реакционную смесь медленно добавляют к водному насыщенному раствору NaHCO3. После окончания добавления вносят ДХМ (50 мл) и слои разделяют. Органический слой промывают рассолом (30 мл) и высушивают (MgSO4). Растворитель удаляют, получая желтое масло, которое растворяют в ТГФ (15 мл) и Н2О (4 мл). Затем добавляют LiOH·H2O (474 мг, 11,3 ммолей) и реакционную смесь перемешивают при КТ в течение 2 ч. Раствор затем по каплям переносят в 5%-ный водный раствор НС1 (50 мл) и смесь экстрагируют EtOAc (трижды по 30 мл). Объединенные органические фракции промывают рассолом (30 мл) и высушивают (MgSO4). Выпаривание летучих компонентов приводит к маслу, которое очищают с помощью хроматографии на силикагеле, элюируя смесью EtOAc/гексаны с градиентом от 0% до 25% EtOAc, получая 800 мг (выход 79%) соединения (R-18).
Конденсацию фенола (R-18) с соединением (R-9б) (стадия 3) проводят методом, описанном на стадии 3 при получении соединения (R-7). Восстановление нитрогруппы (стадия 4), диазотирование амина и замещение хлоридом (стадия 5), приводящее к соединению (R-19б), осуществляют методом, описанном на стадиях 6 и 7 при получении соединения (R-2).
стадия 6: раствор соединения (R-19b) (757 мг, 1,73 ммолей), Pd[P(Ph)3]4(0) (300 мг, 0,26 ммолей) и цианида цинка (122 мг, 1,04 ммолей) в ДМФ (8 мл) в атмосфере азота нагревают при температуры 80°С в течение 4 ч. Затем реакционную смесь охлаждают до КТ и прибавляют к 2-молярному водному раствору NH4OH. Далее раствор экстрагируют смесью EtOAc/гексаны (в соотношении 1:1) (трижды по 30 мл), объединенные органические фракции промывают H2O (трижды по 20 мл) и высушивают (MgSO4). Растворитель выпаривают, а оставшееся масло очищают с помощью хроматографии на силикагеле, элюируя смесью EtOAc/гексаны с градиентом от 0% до 25% EtOAc, получая 580 мг (выход 87%) соединения (R-16).
Гидролиз этилового эфира и превращение в хлорангидрид кислоты могут быть проведены, как описано на стадиях 8-9 при получении соединения (R-2).
Этиловый эфир [3-(3-бром-5-цианофенокси)-4-хлор-2-фторфенил]уксусной кислоты (R-20в)
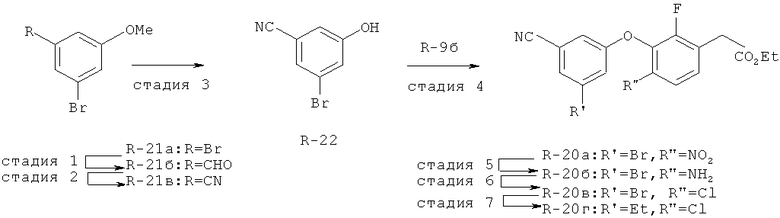
стадия 1: н-BuLi (2,6 мл 1,6-молярного раствора, 1,1 экв.) медленно добавляют к раствору соединения (R-21a) (1,0 г, 3,8 ммолей, CAS Reg. No. 74137-36-3) в Et2O (20 мл), охлажденному до температуры -78°С, в атмосфере азота. Раствор перемешивают в течение 45 мин, после чего добавляют ДМФ с помощью шприца. Затем раствор медленно нагревают до КТ, переносят в насыщенный раствор NH4Cl и экстрагируют этиловым эфиром. Фазу, окрашенную в оранжевый цвет, промывают рассолом, высушивают (MgSO4), фильтруют и выпаривают, получая 0,80 г (выход 98%) соединения (R-21б).
стадия 2: раствор альдегида (R-21б) (12,0 г, 56 ммолей), NH2OH·HCl (19,4 г, 5 экв.), EtOH (100 мл) и пиридина (10 мл) нагревают при температуре 65°С в течение 16 ч. Затем смесь охлаждают до КТ и распределяют между смесью 50% EtOAc/гексаны и водой. Органический слой промывают рассолом и высушивают (MgSO4). Летучие компоненты затем выпаривают, получая 12,4 г (выход 97%) оксима. Это вещество растворяют в безводном диоксане (100 мл) и пиридине (26 мл, 6 экв.). Раствор охлаждают до температуры 0°С, добавляют ТФКА (15 мл, 2 экв.) и смесь оставляют самопроизвольно нагреваться до КТ. Затем раствор перемешивают в течение 2 дней и нагревают при температуре 60°С в течение 1 ч, после чего смесь охлаждают до КТ и осторожно переносят в ледяную воду. Смесь экстрагируют дихлорметаном и объединенные органические слои промывают водой, 1-молярным раствором HCl и рассолом. Затем органический слой высушивают (MgSO4) и выпаривают, получая 10,4 г (выход 90%) соединения (R-21в).
стадия 3: безводный коллидин (100 мл) вносят в сухую колбу, содержащую соединение (R-21в) (10,4 г, 49 ммолей) и LiI (19,6 г, 3 экв.). Раствор нагревают в атмосфере азота при температуре 150°С в течение ночи, охлаждают до КТ и переносят в охлажденный льдом 1-молярный раствор HCl. Затем смесь экстрагируют раствором EtOAc/гексаны (в соотношении 1:1), промывают водой и высушивают (MgSO4). Концентрирование в вакууме дает 8,7 г (выход 89%) соединения (R-22).
Конденсацию фенола (R-22) с соединением (R-9б) (стадия 4) проводят методом, описанном на стадии 3 при получении соединения (R-7). Восстановление нитрогруппы (стадия 5), диазотирование амина и замещение хлоридом (стадия 6), приводящее к соединению (R-20в), осуществляют методом, описанном на стадиях 6 и 7 при получении соединения (R-2).
Этиловый эфир [4-хлор-3-(3-циано-5-этилфенокси)-2-фторфенил]уксусной кислоты (R-20 г) получают обработкой ТГФ раствора соединения (R-20 г) с помощью Pd(dppf)Cl2, ДИБАL-Н (1-молярный раствор в толуоле), методика, использующая диэтилцинк, описана при получении соединения (R-31).
Этиловый эфир [4-бром-3-(3-хлор-5-цианофенокси)-2-фторфенил]уксусной кислоты (R-23a) и [4-бром-3-(3-хлор-5-цианофенокси)-2-фторфенил]уксусная кислота(R-23б)
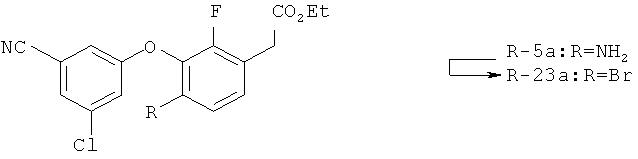
Круглодонную колбу объемом 150 мл загружают MeCN (50 мл), CuBr2 (2,8 г, 12,61 ммолей) и трет-бутилнитрилом (1,4 г, 13,76 ммолей), дегазируют, наполняют аргоном и нагревают смесь до температуры 70°С. К этой смеси добавляют по каплям раствор соединения (R-5a) (4,0 г, 11,47 ммолей), растворенного в MeCN (20 мл). Затем реакционную смесь перемешивают при температуре 70°С в течение 4 ч, а потом охлаждают до температуры 0°С. Реакцию останавливают с помощью 10%-ного раствора HCl (30 мл) и экстрагируют EtOAc. Объединенные экстракты последовательно промывают 10%-ным раствором HCl и рассолом. Оранжевый экстракт высушивают (Na2SO4), фильтруют, и летучие растворители удаляют в вакууме, получая черное масло, которое очищают с помощью хроматографии на силикагеле, элюируя смесью гексаны/EtOAc (в соотношении 95:5) и получая 2,5 г (выход 52,8%) соединения (R-23a). Гидролиз этилового эфира проводят по методике, описанной на стадии 8 примера 1, получая карбоновую кислоту (R-23б).
Этиловый эфир [4-бром-3-(3,5-дицианофенокси)-2-фторфенил]уксусной кислоты (R-24) получают из соединения (R-11б) путем восстановления нитрогруппы, как описано на стадии 6 при получении соединения (R-5a), и диазотирования амина и замещения бромом, как описано в случае соединения (R-23a).
Этиловый эфир [4-бром-3-(3-пиано-5-дифторметилфенокси)-2-фторфенил]уксусной кислоты (R-25)
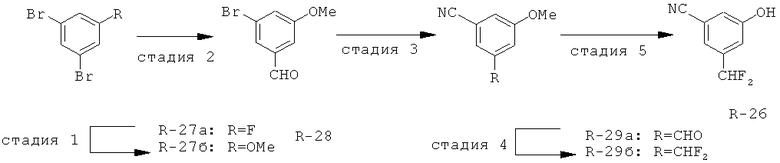
стадия 1: раствор соединения (R-27a) (CAS Reg. No. 1435-51-4), MeONa (1 экв.) и ДМФ перемешивают в течение ночи в атмосфере азота при КТ. Летучие растворители удаляют в вакууме, а остаток распределяют между Et2O и водой. Органическую фазу промывают 5%-ным раствором NaOH, водой и рассолом, высушивают (MgSO4), фильтруют и выпаривают, получая соединение (R-276).
стадия 2: К раствору соединения (R-276) (60 г, 0,2256 моля) и безводного Et2O (1 л), охлажденного до температуры -78°С, в атмосфере аргона добавляют по каплям в течение 30 мин н-BuLi (100 мл, 0,2482 моля, 2,5-молярный раствор в гексане). Желтый раствор перемешивают при температуре -78°С в течение 20 мин. Затем в реакционную смесь добавляют по каплям сухой ДМФ (19 мл, 248,2 ммолей) в течение 15 мин, после чего реакцию перемешивают при температуре -78°С в течение 10 мин перед удалением охлаждающей бани и оставляют самопроизвольно нагреваться до температуры -30°С в течение 30 мин. Затем реакционную колбу помещают на водяную баню и нагревают до температуры -10°С. После этого реакционную смесь медленно переносят в охлажденный насыщенный водный раствор NH4Cl (400 мл). Органический слой отделяют, а водную фазу трижды экстрагируют Et2O. Объединенные экстракты промывают водой, высушивают (MgSO4), фильтруют и выпаривают, получая масло, которое твердеет при стоянии. Сырой продукт очищают с помощью хроматографии на силикагеле, элюируя смесью гексан/EtOAc с градиентом от 3 до 5% EtOAc, получая соединение (R-28).
стадия 3: Цианирование соединения (R-28) для получения соединение (R-29а) проводят с помощью Zn(CN)2, Pd(PPri3)4(0) и ДМФ, как описано на стадии 6 при получении соединения (R-16).
стадия 4: ДАСТ (диэтиламиносульфуртрифторид) (21,04 мл, 519 ммолей) добавляют к раствору соединения (R-29a) (15,1 г, 94 ммолей) и ДХМ в NALGENE® колбе в атмосфере азота. Затем добавляют EtOH (0,013 мл, 0,23 ммолей) и смесь перемешивают в течение 16 ч. После этого реакционную смесь медленно вносят в водный насыщенный раствор NaHCO3. После окончания добавляют ДХМ (50 мл) и слои разделяют. Органический слой промывают рассолом (30 мл) и высушивают (MgSO4). Растворитель удаляют, а сырой продукт дважды очищают с помощью ускоренной хроматографии на силикагеле, элюируя смесью EtOAc/гексаны с градиентом от 0% до 10% EtOAc и получая соединение (R-29б) в виде белого твердого вещества.
стадия 5: метиловый эфир (R-29б) деметилируют в растворе 48%-ного водного раствора HBr и ледяной HOAc, нагревая при температуре 120°С до полного завершения деметилирования. Затем удаляют летучие компоненты, а остаток распределяют между водой и ДХМ, получая соединение (R-26).
Конденсацию соединения (R-26) с соединением (R-9б) проводят методом, описанном на стадии 3 при получениисоединения (R-7). Восстановление нитрогруппы осуществляют методом, описанном на стадии 6 при получении соединения (R-2). Диазотирование и замещение диазогруппы бромом осуществляют методом, описанном для соединения (R-23a), получая соединение (R-25).
Этиловый эфир [3-(3-хлор-5-цианофенокси)-2-фтор-4-метилфенил]уксусной кислоты (R-31)
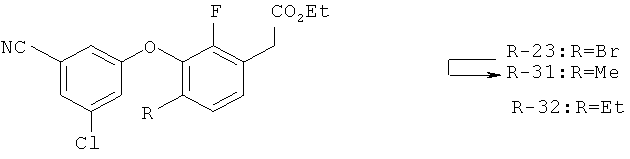
К дегазированному, охлажденному льдом раствору ТГФ (15 мл) и Pd(dppf)Cl2 (0,09 г, 0,121 ммолей) добавляют ДИБАL-Н (0,012 ммолей, 1-молярный раствор в толуоле). Затем реакционную смесь оставляют самопроизвольно нагреваться до КТ. После этого добавляют раствор соединения (R-23a) (1,0 г, 2,42 ммолей), а затем диметилцинк (1-молярный раствор в ТГФ, 4,240 ммолей). Реакцию нагревают до температуры 65°С в течение 4 ч, охлаждают до КТ и останавливают с помощью водного раствора NH4Cl. Образовавшуюся смесь экстрагируют EtOAc и промывают последовательно раствором NH4Cl и рассолом. EtOAc экстракт высушивают (Na2SO4), фильтруют и легколетучий растворитель удаляют в вакууме, получая темно-коричневое масло, которое очищают с помощью хроматографии на силикагеле, элюируя смесью гексан/EtOAc (в соотношении 95:5) и получая 0,50 г (выход 59%) соединения (R-31).
Этиловый эфир [3-(3-циано-5-дифторметилфенокси)-2-фтор-4-метилфенил]уксусной кислоты (R-33) получают из соединения (R-25) с применением методики, описанной выше для соединения (R-31).
Этиловый эфир [3-(3,5-дицианофенокси)-2-фтор-4-метилфенил]уксусной кислоты (R-34) получают из соединения (R-24) с применением методики, описанной выше для соединения (R-31).
Этиловый эфир [3-(3-хлор-5-цианофенокси)-4-этил-2-фторфенил]уксусной кислоты (R-32) получают из соединения (R-23) с применением методики, описанной выше для соединения (R-31), за исключением использования диэтилцинка вместо диметилцинка.
Этиловый эфир [3-(3,5-дицианофенокси)-4-этил-2-фторфенил]уксусной кислоты (R-36) получают из соединения (R-24) с применением методики, описанной выше для соединения (R-31), за исключением использования диэтилцинка вместо диметилцинка.
Этиловый эфир [3-(3-циано-5-дифторметилфенокси)-4-этил-2-фторфенил]уксусной кислоты (R-37) получают из соединения (R-25) с применением методики, описанной выше для соединения (R-31), за исключением использования диэтилцинка вместо диметилцинка.
Этиловый эфир [3-(3-хлор-5-цианофенокси)-4-циклопропил-2-фторфенил]уксусной кислоты (R-38)
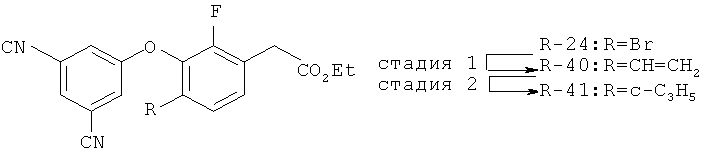
стадия 1: К раствору соединения (R-24) (0,80 г, 1,99 ммолей) и Pd(PPh3)4 (0,23 г, 0,10 экв.) в толуоле (10 мл) с помощью шприца добавляют трибутилвинилолово (0,635 мл, 1,1 экв.) и раствор нагревают с обратным холодильником в течение 5 ч. Затем реакцию охлаждают до КТ и переносят реакционную смесь в насыщенный водный раствор NH4Cl, после чего экстрагируют EtOAc. Органический слой промывают Н2О и рассолом, высушивают (MgSO4) и выпаривают. Образовавшееся серовато-коричневое твердое вещество очищают с помощью хроматографии на силикагеле, элюируя смесью EtOAc/гексан с градиентом от 0 до 25% EtOAc и получая при этом 0,60 г (выход 85%) соединения (R-40).
стадия 2: Диэтиловый эфир (18 мл), Н2О (10 мл) и твердый КОН (3 г) объединяют в колбе Эрленмейера и охлаждают до температуры 0°С. Затем порциями добавляют нитрозомочевину (1,17 г, 10 экв.) и перемешивают смесь в течение 1 ч. Эфирную фазу декантируют со слоя КОН и выдерживают при температуре 0°С. В разделительной колбе эфир (R-40) (0,4 г, 1,14 ммолей) и Pd(OAc)2 (0,01 г, 0,05 экв.) растворяют в Et2O (10 мл) и ДХМ (5 мл) и охлаждают до температуры 0°С. Затем декантированный эфирный раствор диазометана вносят в эту смесь и перемешивают в течение 3 ч. Раствор фильтруют через CELITE® и SiO2 и концентрируют, получая 0,40 г (выход 95%) соединения (R-41).
Этиловый эфир [3-(3-хлор-5-цианофенокси)-4-циклопропил-2-фторфенил]уксусной кислоты (R-41a) получают аналогично за исключением использования этилового эфира [4-бром-3-(3-хлор-5-цианофенокси)-2-фторфенил]уксусной кислоты (R-23a) вместо соединения (R-24).
[3-(3-Хлор-5-цианофенокси)-2-фтор-4-метокси-фенил]уксусная кислота (R-42б)
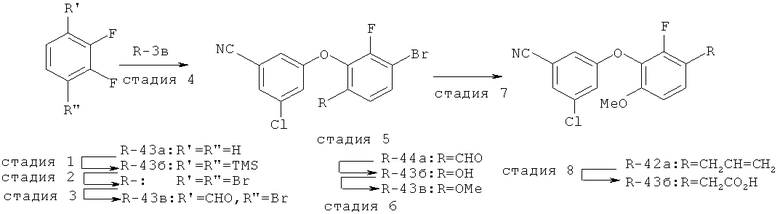
стадия 1: раствор ди-изопропиламина (150 мл, 108,3 г,1,07 молей) в ТГФ (500 мл) охлаждают до температуры -78°С и в атмосфере азота добавляют в течение 15 мин н-BuLi (100 мл, 1,00 моль, 10-молярный раствор в гексане). Затем образовавшуюся смесь перемешивают в течение 30 мин при температуре -78°С, после чего добавляют смесь соединения (R-43a) (45 мл, 52,110 г, 0,457 моля) и хлортриметилсилана (130,0 мл, 111,28 г, 1,024 молей) со скоростью, позволяющей поддерживать внутреннюю температуру реакции ниже -50°С. Раствор перемешивают при температуре -78°С в течение 1 ч и затем реакцию останавливают при этой же температуре путем добавления 1-молярного раствора H2SO4, разбавляют МТБЭ и реакционную смесь насыщают твердым NaCl. Затем фазы разделяют и водную фазу экстрагируют МТБЭ (300 мл). Объединенные органические экстракты высушивают (MgSO4), фильтруют и растворители выпаривают, получая 118 г (выход 100%) соединения (R-43б) в виде белого твердого вещества.
стадия 2: К неразбавленному брому (76,9 мл, 1,50 молей), охлажденному до температуры 0°С на ледяной бане, добавляют порциями в твердом виде соединение (R-43б) (126,23 г, 0,500 молей), поддерживая внутреннюю температуру реакции в интервале от 20 до 45°С (внимание: реакция экзотермична!). Затем реакционную смесь перемешивают при температуре 58°С течение 2 ч. Через 1 ч после этого дополнительно добавляют бром (45,48 г) и через капельную воронку добавляют циклогексан (10 мл). Реакционную смесь затем охлаждают до температуры 0°С и медленно переносят в охлажденный льдом насыщенный водный раствор NaHSO3. После этого образовавшуюся смесь насыщают твердым NaCl, экстрагируют МТБЭ (дважды: по 500 мл и 200 мл), высушивают (MgSO4) и концентрируют в вакууме, получая 191 г соединения (R-43в). Затем реакционную смесь перегоняют приблизительно при 60 мбар, получая 161,53 г бесцветной жидкости, кипящей при температуре 110°С и содержащей около 11% монобромпроизводного. После этого продукт снова перегоняют через шаровую барботажную колонку приблизительно при 50 мбар, что позволяет получить 141,3 (выход 78,5%) соединения (R-43в) с температурой кипения 93-94°С и чистотой >99,6.
стадия 3: получение изо-PrMgCl·LiCl. Образец LiCl (4,56 г, 107,6 ммолей) высушивают в высоком вакууме с помощью струйной воздушной сушилки в течение 10 мин. К сухому твердому веществу в атмосфере азота при температуре 23°С добавляют изо-PrMgCl (53,8 мл, 107,6 ммолей, 2-молярный раствор в ТГФ) и образовавшуюся смесь перемешивают при температуре 23°С в течение 3 дней.
К раствору соединения (R-43в) (1,29 мл, 10 ммолей) в ТГФ (5 мл) при температуре -40°С добавляют раствор изо-PrMgCl·LiCl (5,5 мл, 11 ммолей, 2-молярный раствор в ТГФ) со скоростью, позволяющей поддерживать внутреннюю температуру реакции ниже -30°С. Перемешивание продолжают в интервале температур от -35 до -30°С в течение 1 ч, а затем нагревают при температуре -7°С дополнительно в течение 1 ч. Затем реакционную смесь охлаждают до температуры -30°С, сразу одной порцией добавляют ДМФ (1,00 мл, 13 ммолей) (температура при этом поднимается до -23°С) и продолжают перемешивание в течение 3,5 ч в интервале температур от -25 до +15°С. Реакционную смесь затем переносят в 1-молярный раствор H2SO4 со льдом, образовавшуюся смесь насыщают твердым NaCl и дважды экстрагируют МТБЭ. Объединенные экстракты высушивают (MgSO4), фильтруют и концентрируют в вакууме, получая 2,17 г (выход 98%) соединения (R-43 г) в виде белого твердого вещества.
стадия 4: К раствору соединения (R-3в) (3,84 г), порошкообразного K2CO3 (4,2 г) и н-бутилнитрила добавляют соединение (R-43 г) (5,57 г). Затем реакционную смесь нагревают с обратным холодильником в течение 4,5 ч до завершения реакции по данным ГХ/МС. Реакционную смесь после этого охлаждают, переносят в воду и затем добавляют EtOAc. Образовавшуюся смесь оставляют до самопроизвольного разделения на два слоя. При этом на границе раздела фаз и вдоль стенок верхнего слоя присутствует некоторое количество кристаллов, которые отфильтровывают и промывают водой и гексаном. Затем фильтрат выпаривают в вакууме, остаток переносят в изопропиламин (IPА) и снова выпаривают. Твердое вещество растирают с гексаном и фильтруют. Маточный раствор выпаривают, а остаток очищают с помощью хроматографии на силикагеле, элюируя смесью гексан/EtOAc (в соотношении 80:20). Продукт растирают с изопропиламином, фильтруют и промывают гексаном. Фракции продукта объединяют, получая 1,45 г (выход 83%) соединения (R-44a).
стадия 5: ТФКА (8,88, 4,231 ммолей) вносят в круглодонную колбу объемом 100 мл и перемешивают при температуре 0°С. Затем добавляют через капельную воронку перекись водорода (0,290, 8,46 ммолей, 30%-ная) и перемешивают в течение 2 ч при температуре 0°С, получая трифторперукусусную кислоту (ТФПК).
К раствору соединения (R-44a) (2,0, 5,64 ммолей) в ДХМ (20 мл), перемешиваемому при температуре 0°С, добавляют KH2PO4 (15,35 г, 112,82 ммолей). Затем к этой суспензии добавляют по каплям при температуре 0°С ТФПК и реакцию снова перемешивают в течение 48 ч. После того как исходные вещества прореагируют, реакционную смесь охлаждают до температуры 0°С, разбавляют рассолом и гасят водным 10%-ным раствором бисульфита натрия. Образовавшуюся смесь затем экстрагируют ДХМ, промывают рассолом, высушивают (Na2SO4), фильтруют и растворитель удаляют в вакууме, получая желтое твердое вещество, которое очищают с помощью хроматографии на силикагеле, элюируя смесью гексан/EtOAc (в соотношении 92:8) и получая 1,8 г (выход 94%) соединения (R-44б).
стадия 6: К раствору соединения (R-44б) (1,8 г, 5,26 ммолей) в ДМФ (15 мл) добавляют Cs2CO3 (3,43 г, 10,52 ммолей) и MeI (0,74 г, 5,26 ммолей). Затем реакционную смесь перемешивают при температуре 85°С в течение 12 ч. Когда соединение (R-44б) полностью прореагирует, реакционную смесь охлаждают до КТ, экстрагируют EtOAc и объединенные экстракты промывают водой и рассолом. EtOAc экстракт высушивают (Na2SO4), фильтруют и концентрируют в вакууме, получая соединение (R-44в) в виде желтого масла, которое используют на следующей стадии без дальнейшей очистки.
стадия 7: В сухую, продутую азотом круглодонную колбу объемом 100 мл, загружают соединение (R-44в) (1,6 г, 4,50 ммолей) и безводный ТГФ (20 мл). Затем смесь охлаждают до температуры -20°С и добавляют по каплям раствор изо-PrMgCl·LiCl (5,40 мл, 5,40 молей, 2-молярный в ТГФ, см. стадию 3). Реакцию перемешивают в течение 2 ч при температуре -20°С, добавляют раствор CuCN LiCl (0,100 мл, 0,100 молей, 1-молярный в ТГФ) и перемешивание продолжают при температуре -20°С. К этой смеси добавляют аллилбромид (1,08 г, 9,0 ммолей) и смесь перемешивают дополнительно в течение 2 ч. Затем реакцию останавливают добавлением водного раствора NH4Cl, после чего смесь экстрагируют EtOAc и промывают водой и рассолом. Экстракты высушивают (Na2SC4), фильтруют, и растворитель удаляют в вакууме, получая желтое масло. Сырой продукт очищают с помощью хроматографии на силикагеле, элюируя смесью гексан/EtOAc (в соотношении 95:5) и получая 1 г (выход 70%) соединения (R-42a).
стадия 8: К раствору соединения (R-42a) (0,100 г, 0,315 ммолей), EtOAc (2 мл), MeCN (2 мл) и воды (3 мл) добавляют NaIO4 (0,437 г, 2,050 ммолей) и RuCl3 (0,001 г, 0,006 ммоля). Когда соединение (R-42a) полностью прореагирует, сырую смесь фильтруют через слой CELITE®, промывают EtOAc и объединенные EtOAc экстракты промывают рассолом, высушивают (Na2SO4), фильтруют и выпаривают в вакууме, получая 0,090 г (выход 85%) соединения (R-42б) в виде желтого твердого вещества, которое переносят в EtOAc и промывают рассолом. EtOAc раствор высушивают (Na2SO4) и фильтруют, растворитель удаляют в вакууме, получая соединение (R-42б) в виде желтого твердого вещества (выход 0,090 г, 85%).
[3-(3,5-Дицианофенокси)-2-фтор-4-метоксифенил]уксусная кислота (R-45) и [3-(3-циано-5-дифторметилфенокси)-2-фтор-4-метоксифенил]уксусная кислота (R-46) могут быть получены аналогично за исключением использования соединений (R-10) и (R-26), соответственно, вместо 3-хлор-5-гидроксибензонитрила.
Пример 1: 3-Хлор-5-[6-хлор-2-фтор-3-(5-фенил-1H-пиразол-3-илметил)фенокси]бензонитрил (1-4)
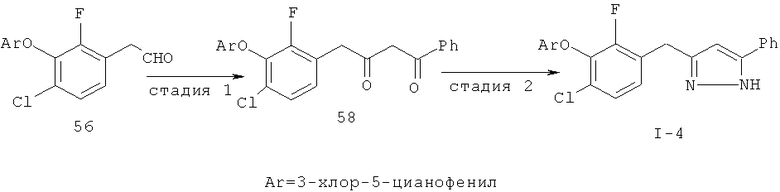
3-Хлор-5-[6-хлор-2-фтор-3-(2-оксоэтил)фенокси]бензонитрил (56) может быть получен восстановлением соединения (R-5б) дибораном, после чего образовавшийся спирт снова окисляют до спирта с прмощью СrО3-пиридина.
стадия 1: к раствору соединения (56)(0,40 г, 1,2 ммоля) в ДХМ (2 мл) добавляют раствор, полученный присоединением SnCl2 (0,035 г, 0,15 экв.) к раствору фенилдиазоацетата (0,16 г, 0,9 экв.) в ДХМ (3 мл). Суспензию перемешивают при КТ в течение ночи и добавляют еще одну порцию SnCl2 (35 мг, 0,15 экв.) к реакционной смеси. Через 1 ч раствор переносят в воду, экстрагируют ЕtOАс и объединенные органические экстракты промывают, высушивают (Na2SO4), фильтруют и концентрируют. Продукт очищают с помощью хроматографии на силикагеле, элюируя смесью EtOAc/гексан с градиентом от 5% до 20% ЕTO и получая 0,27 г (выход 50%) соединения (58).
стадия 2: моногидрат гидразина (0,15 мл, 5 экв.) добавляют к раствору соединения (58) (0,27 г, 0,6 ммолей) в EtOH (6 мл). Затем раствор нагревают с обратным холодильником в течение 1 ч, после чего реакционную смесь концентрируют. Сырой продукт очищают с помощью хроматографии на силикагеле, элюируя смесью EtOAc/гексан с градиентом от 10% до 40% ЕtOАс, получая 0,24 г (выход 90%) соединения (1-4).
3-[6-Бром-2-фтор-3-(5-фенил-1Н-пиразол-3-илметил)фенокси]-5-дифторметилбензонитрил (1-5) может быть получен аналогично из 3-хлор-5-[6-бром-2-фтор-3-(2-оксоэтил)фенокси]бензонитрила.
Пример 2: 3-[4-Хлор-3-(3-хлорфенокси)бензил]-1H-пиразол (1-2)
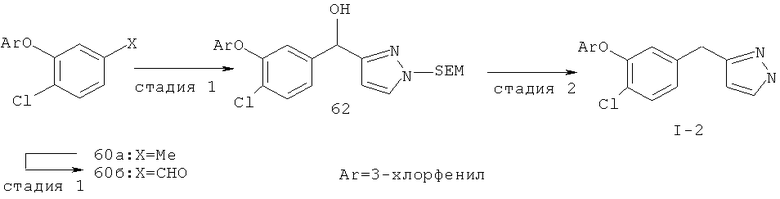
стадия 1. смесь соединения (60а) (1,37 г, 4,13 ммолей), бромсукцинимида (1,16 г, 6,5 ммолей) и борнитрила алюминия (39 мг) в ССl4 (20 мл) нагревают с обратным холодильником в течение 12 ч в атмосфере азота. Затем смесь охлаждают, фильтруют и летучие компоненты выпаривают. Остаток очищают с помощью колоночной хроматографии на силикагеле, элюируя смесью гексанов и получая 1,26 г (выход 74%) нужного дибромида. Дибромид растворяют в EtOH (40 мл) и добавляют раствор AgNO3 (2,5 г) в H2O (10 мл), при этом немедленно образуется белый осадок. Смесь нагревают при температуре 100°С в течение 45 мин. Раствор охлаждают до КТ, фильтруют через CELITE® и концентрируют. Остаток распределяют между EtOAc и водой, и органический слой высушивают (Na2SO4), фильтруют и концентрируют, получая 0,91 г (выход 100%) соединения (60б).
стадия 2: н-BuLi (2 мл 1,6-молярный раствор в ТГФ) добавляют по каплям к раствору 1-(2-триметилсиланилэтоксиметил)-1Н-имидазола (0,40 г, 2 ммоля) в ТГФ (2 мл) при температуре -78°С. Раствор перемешивают в течение 5 мин, а затем добавляют по каплям раствор соединения (60б) (0,43 г, 1,6 ммолей) в ТГФ (2 мл). Раствор потом нагревают до температуры 0°С, переносят в охлажденный льдом водный раствор NH4Cl и экстрагируют EtOAc (50 мл). Органический слой промывают рассолом, высушивают (Na2SO4), фильтруют и концентрируют. Сырой продукт очищают с помощью хроматографии на силикагеле, элюируя смесью EtOAc/гексан с градиентом от 20% до 30% EtOAc и получая 0,37 г (выход 51%) соединения (62).
стадия 3: раствор конц. НС1 (4 мл) в Н2О (4 мл) добавляют к раствору соединения (62) (0,37 г, 0,82 ммоля) в МеОН (5 мл). Раствор нагревают при температуре 65°С в течение 1,5 ч, охлаждают и переносят на лед. Затем смесь нейтрализуют NaHCO3, экстрагируют EtOAc, высушивают и концентрируют. Остаток очищают с помощью колоночной хроматографии, элюируя смесью 5% МеОН/ДХМ и получая 0,21 г (выход 77%) освобожденного от защитной группы пиразола. Этот продукт (0,17 г, 0,5 ммолей) растворяют в ДХМ (2 мл) и Et3SiH (2 мл) и добавляют ТФК (1 мл), после чего смесь перемешивают при температуре 60°С в течение 3 ч, а затем при КТ в течение ночи. К смеси дополнительно добавляют 2 мл EtSiH и 1 мл ТФК и смесь нагревают с обратным холодильником в течение 5 ч. Затем раствор охлаждают до КТ, переносят в суспензию размельченного льда с NaHCO3 и экстрагируют EtOAc. Органический слой промывают водой, высушивают (Na2SO4) и концентрируют. Остаток очищают с помощью колоночной хроматографии на силикагеле, элюируя смесью 50% EtOAc/гексаны и получая 0,14 г (выход 85%) соединения (1-2).
Пример 3: 3-Хлор-5-[6-хлор-2-фтор-3-(1H-индазол-3-илметил)фенокси]бензонитрил (I-3)
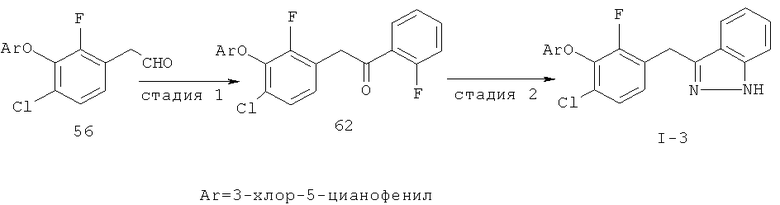
стадия 1: изо-PrMgCl (1,7 мл 2-молярный раствор, 1,1 экв.) добавляют к раствору 2-фторбромбензола (0,33 мл, 1 экв.) в ТГФ (2 мл) и охлаждают до температуры 0°С. Раствор перемешивают при 0°С в течение 1,25 ч, затем охлаждают до температуры -78°С и добавляют по каплям раствор соединения (56) (0,99 г, 3 ммоля) в ТГФ (2 мл). Затем реакционную смесь медленно нагревают до температуры 0°С и переносят в холодный водный раствор NH4Cl. Полученную смесь экстрагируют эфиром и объединенные органические экстракты промывают, высушивают, фильтруют и концентрируют в вакууме. Сырой остаток очищают с помощью хроматографии на силикагеле, элюируя смесью EtOAc/гексан с градиентом от 0% до 25% EtOAc и получая 0,57 г (выход 44%) о-фторфенильного аддукта. Часть этого аддукта (0,26 г, 0,62 ммолей) растворяют в ДХМ (3 мл) и одной порцией добавляют к ней Десс-Мартин перйодинан (0,32 г, 1,2 экв.). Через 4 ч реакционную смесь переносят в насыщенный водный раствор Na2S2O4. Затем смесь экстрагируют ДХМ, промывают, высушивают и концентрируют. Сырой продукт очищают с помощью хроматографии на силикагеле, элюируя смесью EtOAc/гексан с градиентом от 0% до 20% EtOAc и получая 0,23 г (выход 87%) соединения (62).
стадия 2: гидразин (0,24 мл, 10 экв.) добавляют к раствору соединения (62) (0,32 г, 0,77 ммолей) в смеси диоксана (3,6 мл) и EtOH (0,4 мл). Через 2 ч летучие компоненты удаляют.Очистка с помощью ВЭЖХ дает 0,04 г (выход 13%) соединения (I-3).
3-[6-Бром-2-фтор-3-(1H-индазол-3-илметил)фенокси]-5-дифторметилбензонитрил (1-6) получают аналогично из соединения (R-25) и 2-фторбензойной кислоты, последовательно используя конденсацию Кляйзена и гидразиновую циклизацию.
Пример 4: 3-[6-бром-2-фтор-3-(7-нитро-1H-индазол-3-илметил)фенокси]-5-хлорбензонитрил (I-20), 3-[3-(7-амино-1H-индазол-3-илметил)-6-бром-2-фторфенокси]-5-хлорбензонитрил (I-35), N-{3-[4-бром-3-(3-хлор-5-цианофенокси)-2-фторбензил]-1H-индазол-7-ил}ацетамид (I-36), N-{3-[4-бром-3-(3-хлор-5-цианофенокси)-2-фторбензил1-1H-индазол-7-ил)метансульфонамид (I-38)
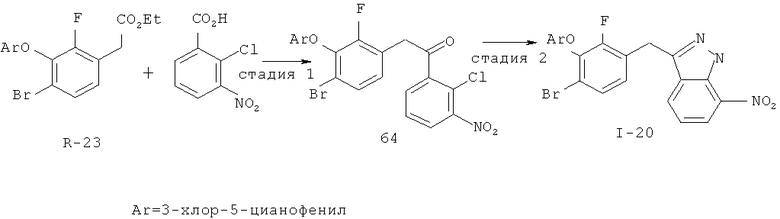
стадия 1: КДИ (0,16 г, 1 экв.) добавляют к раствору 2-хлор-3-нитробензойной кислоты (CAS Reg No. 3970-35-2) (0,19 г, 0,92 ммоля) в ДМФ (3 мл). Раствор нагревают при температуре 50°С в течение 45 мин, охлаждают до температуры -10°С и добавляют раствор соединения (R-23a) (0,40 г, 1 экв.) в ДМФ (2 мл), а затем NaH (0,13 г, 55%-ная суспензия в минеральном масле, 3,2 экв.), после чего реакцию нагревают до КТ. Затем реакционную смесь переносят в насыщенный водный раствор NH4Cl и экстрагируют EtOAc. Органические экстракты высушивают (MgSO4), фильтруют и выпаривают. Остаток растворяют в смеси ДМСО (5 мл) и рассола (0,3 мл) и нагревают при температуре 150°С в течение 20 мин. Затем раствор переносят в насыщенный раствор LiCl, водный раствор экстрагируют EtOAc, высушивают (MgSO4), фильтруют и выпаривают. Сырой продукт очищают с помощью хроматографии на силикагеле, элюируя смесью EtOAc/гексан с градиентом от 5% до 50% EtOAc и получая 0,42 г (выход 92%) соединения (64).
стадия 2: гидразин (0,040 мл, 3 экв.) добавляют к раствору соединения (64) (0,22 г, 0,42 ммоля) в диоксане (3 мл). Через 2 ч реакционную смесь распределяют между водой и EtOAc. Выпаривание органического слоя дает масло, которое очищают с помощью хроматографии на силикагеле, элюируя смесью EtOAc/гексан с градиентом от 50% до 100% EtOAc и получая 0,120 г (выход 58%) соединения (I-20).
К раствору соединения (I-20) (0,70 г, 139 ммолей) в ЕtOH (400 мкл) и Н2O (100 мкл) добавляют NH4Cl (0,031 г, 4,2 экв.) и Fe порошок (0,032 г, 4,2 экв.). После нагревания в течение 30 мин при температуре 90°С реакционную смесь охлаждают до КТ, фильтруют через слой CELITE® и концентрируют в вакууме. Сырой продукт очищают с помощью хроматографии на силикагеле, элюируя смесью EtOAc/гексан с градиентом от 33% до 70% EtOAc и получая 0,068 г (выход 91%) соединения (I-35).
К раствору соединения (I-35) (0,017 г, 0,036 ммоля) в НОАс (180 мкл) медленно добавляют Ас2О (0,0042 г, 1,15 экв.) в НОАс (180 мкл). Затем образовавшийся раствор нагревают при температуре 80°С в течение 30 мин, затем охлаждают до КТ и концентрируют в вакууме. Сырой продукт очищают с помощью препаративной тонкослойной хроматографии, используя смесь 5% МеОН/ДХМ и получая 0,015 г (выход 81%) соединения (I-36). N-{3-[4-Бром-3-(3-хлор-5-цианофенокси)-2-фторбензил]-1H-индазол-7-ил}метансульфонамид (I-38) получают аналогично, используя смесь метансульфонилхлорида с триэтиламином вместо Ас2О/НОАс.
Пример 5: 3-[4-Бром-3-(3-хлор-5-цианофенокси)-2-фторбензил]-1H-индазол-7-карбонитрил (I-30)
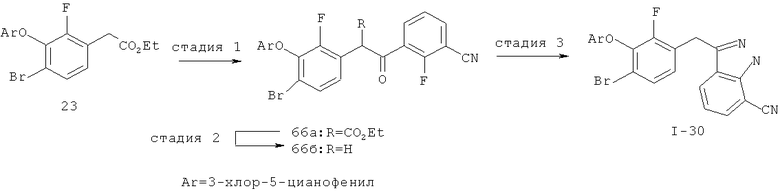
стадия 1: смесь КДИ (0,556 г, 1,1 экв.), 3-циано-2-фторфенилуксусной кислоты (0,567 г, 1,1 экв.), соединения (23) (1,24 г, 3,12 ммолей) и NaH (0,240 г, 3,20 экв.) в ДМФ (31 мл) вводят в реакцию, как описано в примере 1. Сырой продукт очищают с помощью хроматографии на силикагеле, элюируя смесью 25% EtOAc/гексан и получая 0,700 г (выход 41%) соединения (66а).
стадия 2: смесь соединения (66а) (0,70 г, 1,28 ммолей), ДМСО (7,8 мл) и Н2О (0,4 мл) вводят в реакцию, как описано на стадии 2 примера 1, получая 0,626 г (выход 100%) соединения (66б).
стадия 3: смесь соединения (66б) (0,20 г, 0,41 ммолей), гидразина (0,039 мл, 3 экв.), EtOH (0,052 мл) и диоксана (3,5 мл) вводят в реакцию, как описано на стадии 3 примера 1, получая 0,119 г (выход 60%) соединения (I-30).
Пример 6: 3-[6-бром-2-фтор-3-(1H-пиразоло[3,4-6]пиридин-3-илметил)фенокси]-5-хлорбензонитрил (I-7)
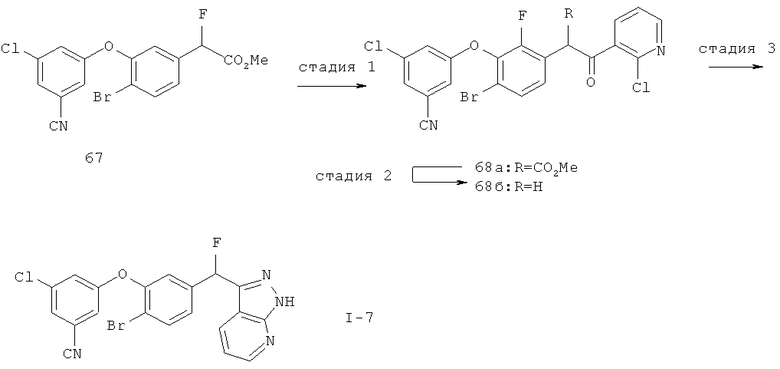
стадия 1: К раствору 2-хлорникотиновой кислоты (1,96 г, 12,5 ммолей) в ДМФ (63 мл) добавляют КДИ (2,02 г, 12,5 ммолей) и раствор нагревают при температуре 50°С в течение 2 ч, после чего реакционную смесь охлаждают до температуры -10°С, добавляют последовательно раствор соединения (67) (4,51 г, 11,3 ммолей) в ДМФ (46 мл) и твердый NaH (1,45 г, 36,2 ммолей) (метиловый эфир соединения (67) получают методом, описанным для получения соединения (R-23a) за исключением использования метил-трет-бутилмалоната вместо этил-трет-бутилмалоната). Затем реакционную смесь перемешивают при температуре -10°С в течение 15 мин, нагревают до КТ и перемешивают в течение 14 ч. После этого реакционную смесь распределяют между NH4Cl и EtOAc, водный слой экстрагируют EtOAc и объединенные органические экстракты промывают 1-нормальным раствором НС1, рассолом, высушивают (MgSO4), фильтруют и концентрируют в вакууме. Сырой продукт очищают с помощью колоночной хроматографии на силикагеле, элюируя смесью EtOAc/гексан с градиентом от 25 до 30% EtOAc и получая 3,25 г (выход 53%) соединения (68а).
стадия 2: раствор соединения (68а) (3,25 г, 6,04 ммолей) в ДМСО (35 мл) и Н2О (1,7 мл) перемешивают на предварительно нагретой до температуры 150°С масляной бане в течение 30 мин. Затем реакционную смесь распределяют между EtOAc и насыщенным водным раствором NaHCO3. Водную фазу экстрагируют EtOAc (трижды по 50 мл) и объединенные органические экстракты высушивают (MgSO4), фильтруют и концентрируют в вакууме, получая 2,45 г (выход 85%) соединения (686) в виде желтого масла.
стадия 3: К раствору соединения (686) (2,3 г, 4,8 ммолей) в диоксане (41 мл) и EtOH (6 мл) добавляют гидразин (1,50 мл, 10 экв.) и реакционную смесь нагревают при температуре 100°С в течение 2 ч, после чего реакционную смесь охлаждают до КТ и растворитель удаляют. Остаток распределяют между смесью 10% МеОН/ДХМ и насыщенным водным раствором NaHCO3. Затем водный слой снова экстрагируют смесью 10% МеОН/ДХМ и объединенные органические экстракты высушивают (MgSO4), фильтруют и концентрируют в вакууме, получая желтое твердое вещество, которое растирают со смесью 30% EtOAc/гексаны, получая 1,91 г (выход 87%) соединения (1-7) в виде белого твердого вещества.
3-Хлор-5-[6-хлор-2-фтор-3-(1H-пиразоло[3,4-b]пиридин-3-илметил)фенокси]бензонитрил (I-46) получают аналогично из этилового эфира [4-хлор-3-(3-хлор-5-цианофенокси)-2-фторфенокси]уксусной кислоты (R-5б).
5-[6-Бром-2-фтор-3-(1H-пиразоло[3,4-6]пиридин-3-илметил)фенокси]изофталонитрил (I-16) получают аналогично из этилового эфира [4-бром-3-(3,5-дицианофенокси)-2-фторфенил]уксусной кислоты (R-39).
3-Хлор-5-[2-фтор-6-метил-3-(1H-пиразоло[3,4-6]пиридин-3-илметил)фенокси]бензонитрил (I-24) получают аналогично из этилового эфира [3-(3-хлор-5-цианофенокси)-2-фтор-4-метил-фенил]уксусной кислоты (R-31).
5-[6-Циклопропил-2-фтор-3-(1H-пиразоло[3,4-b]пиридин-3-илметил)фенокси]изофталонитрил (I-21) получают аналогично из этилового эфира [4-циклопропил-3-(3,5-дицианофенокси)-2-фторфенил]уксусной кислоты (R-41).
3-Хлор-5-[6-этил-2-фтор-3-(1H-пиразоло[3,4-b]пиридин-3-илметил)фенокси]бензонитрил (I-25) получают аналогично из этилового эфира [3-(3,5-дицианофенокси)-4-этил-2-фторфенил]уксусной кислоты (R-32).
3-Хлор-5-[6-циклопропил-2-фтор-3-(1H-пиразоло[3,4-b]пиридин-3-илметил)фенокси]бензонитрил (I-33) получают аналогично из этилового эфира 3-(3-хлор-5-цианофенокси)-4-циклопропил-2-фторфенил]уксусной кислоты (R-41).
3-[6-Хлор-2-фтор-3-(1H-пиразоло[3,4-b]пиридин-3-илметил)фенокси]-5-дифторметилбензонитрил (I-28) получают аналогично из этилового эфира [4-хлор-3-(3-циано-5-дифторметилфенокси)-2-фторфенил]уксусной кислоты (R-16).
3-[6-Бром-2-фтор-3-(1H-пиразоло[3,4-b]пиридин-3-илметил)фенокси]-5-дифторметилбензонитрил (I-29) получают аналогично из этилового эфира [4-бром-3-(3-циано-5-дифторметилфенокси)-2-фторфенил]уксусной кислоты (R-25).
3-Дифторметил-5-[2-фтор-6-метил-3-(1H-пиразоло[3,4-b]пиридин-3-илметил)фенокси]бензонитрил (I-40) получают аналогично из этилового эфира [3-(3-циано-5-дифторметилфенокси)-2-фтор-4-метил-фенил]уксусной кислоты (R-33).
5-[6-Этил-2-фтор-3-(1H-пиразоло[3,4-b]пиридин-3-илметил)фенокси]изофталонитрил (I-45) получают аналогично из этилового эфира [3-(3,5-дицианофенокси)-4-этил-2-фторфенил]уксусной кислоты (R-36).
3-[6-Бром-2-фтор-3-(6-метокси-1H-пиразоло[3,4-b]пиридин-3-илметил)фенокси]-5-хлорбензонитрил (I-22) получают аналогично за исключением замены на стадии (1) 2-хлорникотиновой кислоты 2-хлор-6-метоксиникотиновой кислотой.
3-[6-Бром-2-фтор-3-(5-фтор-1H-пиразоло[3,4-b]пиридин-3-илметил)фенокси]-5-хлорбензонитрил (I-23) получают аналогично за исключением замены на стадии (1) 2-хлорникотиновой кислоты 2-хлор-5-фторникотиновой кислотой.
3-Дифторметил-5-[2-фтор-6-метокси-3-(1H-пиразоло [3,4-b]пиридин-3-илметил)фенокси]бензонитрил (I-27) получают аналогично из метилового эфира [3-(3-3-хлор-5-цианофенокси)-2-фтор-4-метоксифенил]уксусной кислоты (R-426).
Пример 7: 3-[6-Бром-2-фтор-3-(1H-пиразоло[3,4-b]пиридин-3-илметил)фенокси]-5-хлорбензонитрил (I-7) (через реакцию Гриньяра)
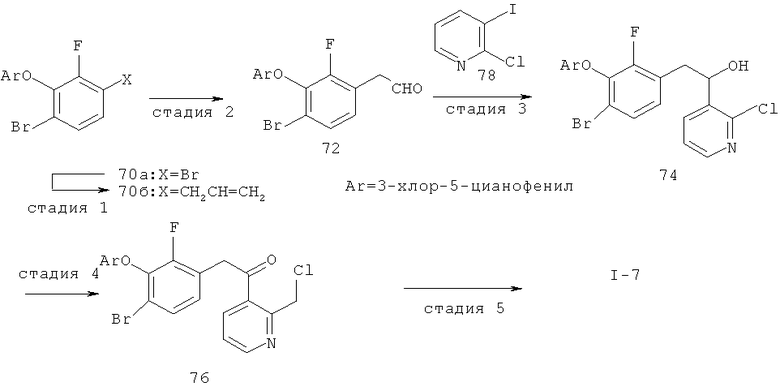
стадия 1: к раствору соединения (70а) (8,0 г, 19,7 ммолей) в толуоле при температуре -78°С медленно добавляют изо-PrMgCl (1,23 мл 2-молярного раствора в ТГФ). Затем смесь выдерживают в течение 4,5 ч, добавляют раствор CuCN·2LiCl (4 мл 1-молярного раствора в ТГФ) и реакционную смесь нагревают при температуре -30°С в течение 15 мин. Раствор затем охлаждают до температуры -50°С и сразу же добавляют аллилбромид (3,41 мл, 2 экв.). После этого смесь нагревают до КТ, переносят в раствор NH4Cl и экстрагируют Et2O. Органические экстракты промывают рассолом, высушивают (MgSO4) и выпаривают. Сырой продукт очищают с помощью колоночной хроматографии на силикагеле, элюируя смесью 5% EtOAc/гексан и получая 5,5 г (выход 76%) соединения (70б).
стадия 2: Озон медленно барбатируют через раствор соединения (70б) (5,8 г, 15,8 ммолей), ДХМ (105 мл) и МеОН (55 мл), охлажденного до температуры -78°С. Через 40 мин, когда раствор приобретает голубой цвет, введение озона прекращают и через реакционную смесь пропускаю азот в течение 15 мин. Затем с помощью шприца в реакционную смесь вводят Me2S (11,6 мл, 10 экв.), раствор нагревают до 0°С и выдерживают в течение 2 ч. Раствор выпаривают и сразу же переносят в колонку с силикагелем, а затем элюируют смесью EtOAc/гексан с градиентом от 10% до 35% EtOAc, получая 4,2 г (выход 72%) соединения (72).
стадия 3: раствор изо-PrMgCl в ТГФ (1 экв., 2-молярный раствор) добавляют по каплям к раствору соединения (78) (0,23 г, 0,96 ммоля) в ТГФ (3 мл), охлажденного до температуры -40°С, в атмосфере N2. Раствор перемешивают в течение 30 мин и добавляют по каплям раствор соединения (72) (0,35 г, 1 экв.) в ТГФ (3 мл). Затем реакционную смесь нагревают до 0°С, выдерживают в течение 1 ч и переносят по каплям в буферный водный раствор (рН 7). Водную смесь экстрагируют EtOAc, объединенные экстракты промывают рассолом, высушивают (MgSO4) и концентрируют в вакууме. Сырой продукт очищают с помощью хроматографии на силикагеле, элюируя смесью EtOAc/гексан с градиентом от 0% до 35% EtOAc и получая 0,21 г (выход 45%) соединения (74).
стадия 4: К раствору соединения (74) (0,77 г, 0,19 ммоля) в ДХМ (9 мл), охлажденному до 0°С, добавляют реактив Десса-Мартина перйодинан (0,81 г, 1,2 экв.). Затем смесь перемешивают в течение 4 ч, гасят с помощью NaHCO3 (1 г), после чего органическую фазу отделяют и выпаривают. Оставшееся твердое вещество очищают посредством хроматографии на силикагеле, элюируя смесью EtOAc/гексан с градиентом от 0% до 35% EtOAc и получая 0,48 г (выход 62%) соединения (I-7), идентичного соединению, описанному в примере 6.
Пример 8: 3-[6-Бром-2-фтор-3-(6-оксо-6,7-дигидро-1H-пиразоло[3,4-b]пиридин-3-илметил)фенокси]-5-хлорбензонитрил (I-12)
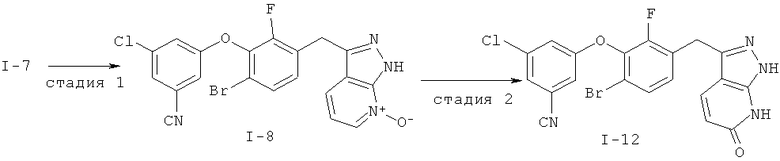
стадия 1: пиразолопиридин (I-7) (0,10 г, 0,22 ммолей) суспендируют в ДХМ (4 мл) и добавляют МХПБК (м-перхлорбензойную кислоту) (0,055 г, 1,1 экв.), после чего суспензию перемешивают в течение ночи. Затем дополнительно добавляют к реакционной смеси 30 мл МХПБК в ДХМ (4 мл). Через 2 ч растворитель удаляют, а оставшееся твердое вещество растирают с Et2O, отделяют фильтрованием и промывают 10 мл Et2O, получая 0,90 г соединения (I-8).
стадия 2: к раствору соединения (I-8) (0,100 г, 0,212 ммоля) в ДХМ (1 мл), охлажденному до 0°С, добавляют ТФКА (0,562 мл). Через 1 ч растворитель удаляют, получая пенообразное коричневое твердое вещество, которое очищают с помощью обращенно-фазовой хроматографии на силикагеле, получая 0,010 г (выход 10%) соединения (I-12) в виде белого твердого вещества.
3-[6-Бром-2-фтор-3-(5-фтор-7-окси-1H-пиразоло[3,4-b]пиридин-3-илметил)фенокси]-5-хлорбензонитрил (I-26) и 3-[6-бром-2-фтор-3-(5-фтор-6-оксо-6,7-дигидро-1H-пиразоло[3,4-b]пиридин-3-илметил)фенокси]-5-хлорбензонитрил (I-34) получают аналогично из соединения (I-23).
Пример 9: 3-[6-Бром-2-фтор-3-(7-метил-6-оксо-6,7-дигидро-1H-пиразоло[3,4-b]пиридин-3-илметил)фенокси]-5-хлорбензонитрил (I-48)
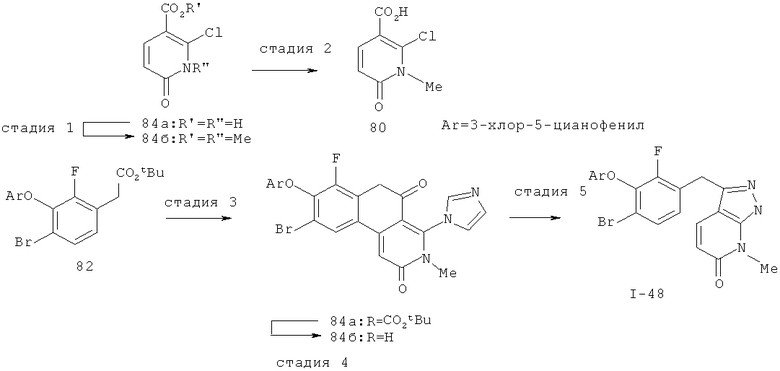
2-Хлор-6-оксо-1,6-дигидропиридин-3-карбоновую кислоту получают, как описано A.G.Beaman и О.N.Miller в патенте США 3682932.
стадия 1: Триметилсилилдиазометан (27,6 мл, 2-молярный раствор в гексане, 6 экв.) осторожно добавляют в течение 10 мин к охлажденному льдом раствору соединения (78а) (1,60 г, 9,20 ммолей) в ДХМ (45 мл) и МеОН (45 мл). Затем реакционную смесь гасят НОАс и концентрируют в вакууме. После этого продукт очищают с помощью хроматографии на силикагеле, элюируя смесью EtOAc/ДХМ с градиентом от 5 до 10% EtOAc и получая 1,25 г (выход 68%) соединения (78б).
стадия 2: раствор LiOH·H2O (54 мг, 1,2 экв.) в H2O (3,3 мл) медленно добавляют к раствору соединения (78б) (0,200 г, 1,1 ммолей) в ТГФ (10 мл) при КТ. После перемешивания в течение 15 ч реакционную смесь подкисляют до рН 1 с помощью 2-молярного раствора НС1, разбавляют водой и экстрагируют EtOAc. Объединенные экстракты промывают рассолом, высушивают (MgSO4) и концентрируют в вакууме, получая 0,180 г слегка загрязненного соединения (80).
стадии 3 и 4: к раствору соединения (80) (0,120 г, 0,691 ммолей) в ДМФ (1,2 мл) добавляют КДИ (0,123 г, 1,1 экв.) одной порцией. После нагревания в течение 30 мин при температуре 50°С раствор охлаждают до 0°С и вводят раствор соединения (82) (0,335 г, 1,1 экв.) в ДМФ (2.3 мл). Затем медленно добавляют NaH (0,094 г, 60%-ный раствор в минеральном масле, 3,4 экв.) одной порцией и реакционную смесь оставляют самопроизвольно нагреваться до КТ в течение 2 ч, после чего смесь снова охлаждают до 0°С, гасят насыщенным водным раствором NH4Cl, разбавляют водой и экстрагируют EtOAc. Объединенные экстракты промывают рассолом, высушивают (MgSO4) и концентрируют в вакууме. Остаток затем снова растворяют в ДХМ (6,9 мл) и ТФК (3,5 мл), перемешивают в течение 15 ч и концентрируют в вакууме. Сырой продукт очищают с помощью хроматографии на силикагеле, элюируя смесью МеОН/ДХМ с градиентом от 1% до 5% МеОН и получая 0,225 г (выход 60%) соединения (84б).
стадия 5: Гидразин (40 мкл, 3 экв.) медленно добавляют к раствору соединения (84б) (0,225 г, 0,420 ммоля) в 1,4-диоксане (4,2 мл) при КТ. Через 1 ч реакционную смесь концентрируют и очищают с помощью хроматографии на силикагеле, элюируя смесью МеОН/ДХМ с градиентом от 1 до 5% МеОН и получая 0,087 г (выход 43%) соединения (I-48).
Пример 10: 3-[4-Бром-3-(3-хлор-5-цианофенокси)-2-фторбензил]-1H-пиразоло[3,4-b]пиридин-6-карбонитрил (I-14)
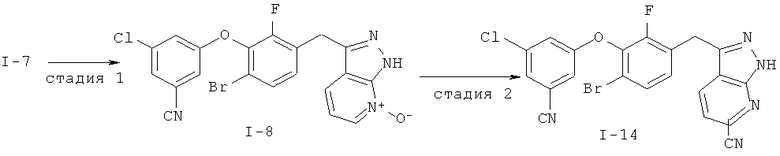
стадия 1: соединение (I-7) превращают в соединение (I-8) с 85%-ным выходом, как описано в стадии 1 примера 8.
стадия 2: к раствору соединения (I-8) (0,120 г, 0,253 ммоля) в ДМФ (6 мл) добавляют последовательно NaCN (0,050 г, 1,01 ммолей), ТЭА (0,175 мл, 5 экв.) и триметилсилилхлорид (0,128 мл, 4 экв.). Образовавшуюся коричневую реакционную смесь нагревают при температуре 110°С в течение 5 ч. Затем реакционную смесь распределяют между Н2O и EtOAc и органический слой высушивают (MgSO4), фильтруют и концентрируют в вакууме. Сырой продукт очищают с помощью колоночной хроматографии на силикагеле, элюируя смесью МеОН/ДХМ (оба содержат 1% концентрированного NH4OH) и получая 0,018 г (выход 15%) соединения (I-14).
Пример 11: 3-[6-бром-2-фтор-3-(1-метил-1H-пиразоло[3,4-b]пиридин-3-илметил)фенокси]-5-хлорбензонитрил (I-17)
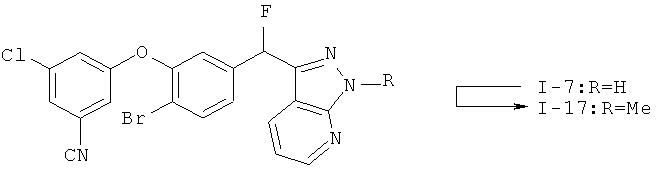
К раствору соединения (I-7) (0,100 г, 0,218 ммолей) в ДМФ (2 мл), охлажденному до температуры 0°С, добавляют последовательно NaH (0,010 г, 1,2 экв.) и МеI (0,016 мл, 1,2 экв., по каплям). Через 1 ч реакционную смесь постепенно нагревают до КТ и перемешивают в течение 16 ч. Затем реакционную смесь распределяют между водным раствором NH4Cl и EtOAc. Органический слой промывают 1-нормальным раствором НСl, водный слой снова экстрагируют EtOAc, объединенные органические экстракты высушивают (MgSO4), фильтруют и выпаривают. Сырой продукт очищают с помощью хроматографии на силикагеле, элюируя смесью EtOAc/гексаны (20% EtOAc) и получая 0,055 г (выход 53%) соединения (I-17).
Пример 12: 3-[6-Бром-2-фтор-3-(1H-пиразоло[3,4-с]пиридин-3-илметил)фенокси]-5-хлорбензонитрил (I-15)
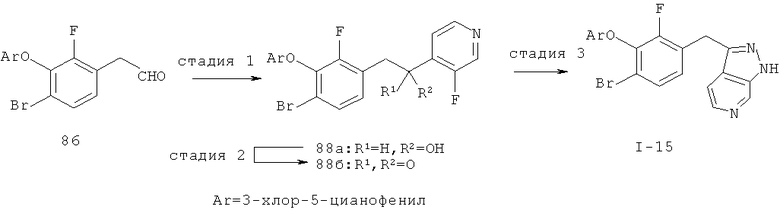
стадии 1 и 2: проводят аналогично стадиям 3 и 4 примера 7 за исключением использования на стадии (3) 3-фтор-4-йодпиридина вместо 2-хлор-3-йодпиридина.
стадия 3: гидразин (0,288 мл, 5 экв.) добавляют к раствору соединения (88б) (0,85 г, 1,8 ммолей) в диоксане (9 мл) и EtOH (0,5 мл), после чего раствор нагревают при температуре 80°С в течение 3 ч. Затем раствор распределяют между EtOAc и водой, отделяют органический слой и выпаривают, получая остаток в виде масла, которое очищают с помощью хроматографии на силикагеле, элюируя смесью МеОН/ДХМ с градиентом от 0% до 5% МеОН и получая 0,24 г (выход 29%) соединения (I-15).
5-[6-Бром-2-фтор-3-(1H-пиразоло[3,4-с]пиридин-3-илметил)фенокси|изофталонитрил (I-19) получают аналогично, исходя из этилового эфира [4-бром-3-(3,5-дицианофенокси)-2-фторфенил]уксусной кислоты (R-39).
3-[6-Бром-2-фтор-3-(1H-пиразоло[3,4-с]пиридин-3-илметил)фенокси]-5-дифторметилбензонитрил (I-31) получают аналогично, исходя из этилового эфира [4-бром-3-(3-циано-5-дифторметилфенокси)-2-фторфенил]уксусной кислоты (R-25).
3-[6-Хлор-2-фтор-3-(1H-пиразоло[3,4-с]пиридин-3-илметил)фенокси]-5-дифторметилбензонитрил (I-32) получают аналогично исходя из этилового эфира [4-хлор-3-(3-циано-5-дифторметилфенокси)-2-фторфенил]уксусной кислоты (R-16).
Пример 13: 3-[6-Бром-2-фтор-3-(1H-пиразоло[3,4-d]пиримидин-3-илметил)фенокси]-5-хлорбензонитрил (I-18)
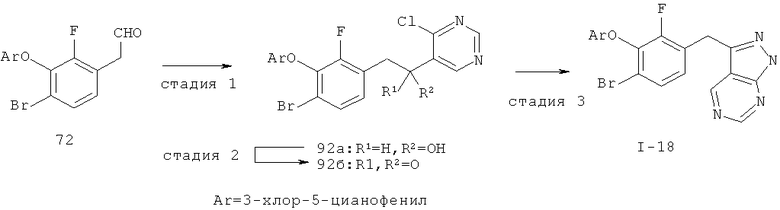
стадия 1: 5-бром-4-хлорпиримидин (0,890 г, 1,0 экв.) растворяют в толуоле (40 мл) и охлаждают до температуры -40°С, после этого добавляют по каплям изопропилмагнийхлорид (2,5 мл, 1,1 экв.) и раствор перемешивают при температуре -20°С в течение 1 ч. Затем добавляют к реакционной смеси раствор соединения (72) (1,7 г, 4,6 ммолей) в толуоле (8 мл), перемешивают при 0°С в течение 3 ч, переносят в NH4Cl и экстрагируют EtOAc. Органический слой промывают рассолом, высушивают (Na2SO4) и концентрируют в вакууме. Продукт очищают с помощью хроматографии на силикагеле, элюируя смесью EtOAc/гексан с градиентом от 0 до 40% и получая 0,35 г (выход 16%) соединения (92а) в виде белого твердого вещества.
стадия 2: к раствору соединения (92а) (0,35 г, 0,73 ммоля) в ДХМ (5 мл), охлажденному до температуры 0°С, добавляют перйодинан (реактив Десса-Мартина) (0,374 г, 1,2 экв.). Затем образовавшийся коричневый раствор перемешивают при 0°С в течение 4 ч и выпаривают. Продукт очищают с помощью хроматографии на силикагеле, элюируя смесью EtOAc/гексан с градиентом от 0 до 30% EtOAc и получая 0,220 г (выход 63%) соединения (92б) в виде желтого масла.
стадия 3: К раствору соединения (92б) (0,22 г, 0,456 ммоля), диоксана (5 мл) и EtOH (0,7 мл) добавляют с помощью шприца гидразин (0,075 мл, 5 экв.). Затем реакцию перемешивают в течение 3 ч, переносят в NH4Cl и экстрагируют смесью 10% МеОН/ДХМ. Органический слой промывают рассолом, высушивают (Na2SO4) и выпаривают, получая грязно-белое твердое вещество, которое растирают со смесью 70% EtOAc/гексаны, получая 0,035 г (выход 15%) соединения (I-18) в виде белого порошка.
3-[6-Бром-3-(6-хлор-1H-пиразоло [3,4-d]пиримидин-3-илметил)-2-фторфенокси]-5-хлорбензонитрил (1-13) получают аналогично за исключением использования на стадии (1) 5-бром-2,4-дихлорпиримидина вместо 5-бром-4-хлорпиримидина. Конечную циклизацию с гидразином проводят следующим образом:
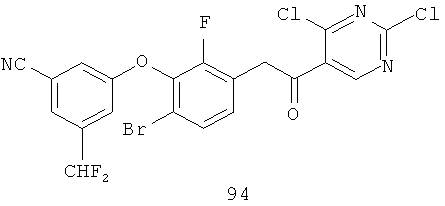
стадия 3: ДИПЭА (15 мкл) добавляют к раствору соединения (94) (0,25 г, 0,5 ммоля) в диоксане (3 мл). Гидразин (15 мкл, 1 экв.) медленно добавляют к этому раствору, а затем дополнительно вносят ДИПЭА (200 мкл). Через 2 ч раствор переносят в воду и водную смесь экстрагируют EtOAc. Органические экстракты высушивают (MgSO4), фильтруют и выпаривают, получая остаток, который очищают с помощью хроматографии на силикагеле, элюируя смесью EtOAc/гексан с градиентом от 20% до 70%) и получая 0,18 г (выход 76%) соединения (I-13).
Пример 14: 3-[3-(7-Амино-1H-пиразоло[3,4-с]пиридин-3-илметил)-6-бром-2-фторфенокси]-5-хлорбензонитрил (I-41), (схема Г)
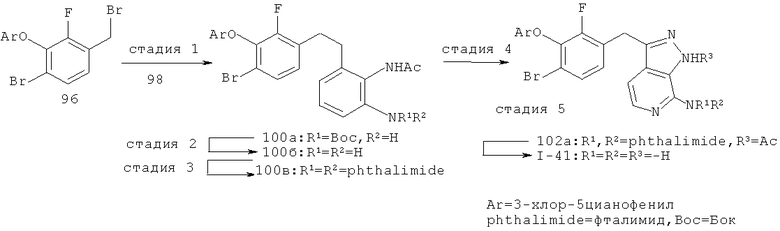
трет-Бутиловый эфир (3-ацетиламино-4-метилпиридин-2-ил)карбаминовой кислоты (98) получают в три стадии из 2-амино-4-метил-3-нитропиридина, используя методику: Townsend, Heterocycles, 2002, 57:2335-2343.
3-(6-Бром-3-бромметил-2-фторфенокси)-5-хлорбензонитрил (96) получают в четыре стадии, как описано ниже:
Гидрид натрия (42 мг, 60%-ная суспензия, 1,05 экв.) добавляют к раствору соединения (R-3в) (153 мг, 1 ммоль) в диметилацетамиде (1 мл). Раствор выдерживают при температуре 50°С в течение 30 мин и добавляют соединения (R-43в) (2,7 г, 10 экв.). Затем раствор нагревают при температуре 125°С в течение 2 ч, а потом охлаждают до КТ. Реакционную смесь разбавляют EtOAc и промывают 10%-ным раствором H2SO4. Концентрация органического слоя и очистка с помощью хроматографии на силикагеле, элюируя смесью 10% EtOAc/гексан, дает 0,331 г (выход 82%) 3-хлор-5-(3,6-дибром-2-фторфенокси)бензонитрила (103).
Раствор изо-PrMgCl (17,3 мл, 2-молярный раствор в ТГФ, 1,75 экв.) медленно добавляют к раствору соединения (103) (8,0 г, 19,7 ммолей) в толуоле (160 мл) при температуре -78°С. Раствор выдерживают в течение 1,5 ч, а затем с помощью шприца переносят в колбу, содержащую ДМФ (2,3 мл, 1,5 экв.) в толуоле (30 мл). Затем раствор гасят водным раствором NH4Cl и разбавляют EtOAc. Органический слой отделяют, промывают рассолом, высушивают и выпаривают. Сырой продукт очищают с помощью хроматографии на силикагеле, элюируя смесью 20% EtOAc/гексаны и получая 6,5 г (выход 98%) 3-(6-бром-2-фтор-3-формилфенокси)-5-хлорбензонитрила (105) в виде желтого твердого вещества.
NaBH4; (0,64 г, 1,5 экв.) добавляют порциями к перемешиваемому раствору соединения (105) (4,0 г, 11,3 ммолей) в смеси ТГФ (20 мл) и МеОН (10 мл) при КТ. Затем реакционную смесь перемешивают в течение 12 ч и гасят добавлением насыщенного водного раствора NH4Cl. Водную часть смеси экстрагируют EtOAc и органический раствор промывают водой и выпаривают. Сырой продукт очищают с помощью хроматографии на силикагеле, элюируя смесью EtOAc/гексан с градиентом от 20% до 50% EtOAc и получая 1,9 г (выход 47%) 3-(6-бром-2-фтор-3-гидроксиметилфенокси)-5-хлорбензонитрила (107) в виде желтого масла.
Раствор PBr3 (0,74 мл 1-молярного раствора в ДХМ, 1,1 экв.) добавляют к раствору соединения (107) (0,24 г, 0,67 ммолей) в ДХМ (5 мл). Смесь перемешивают при КТ в течение 1 ч и затем концентрируют в вакууме. Остаток растворяют в EtOAc, промывают насыщенным водным раствором NaHСО3 и высушивают (MgSO4). Выпаривание летучих компонентов приводит к 0,17 г (выход 60%) соединения (96).
стадии 1 и 2: BuLi (6,35 мл, 2,5-молярный раствор в гексане, 4 экв.) медленно добавляют к раствору соединения (98) (1,05 г, 3,97 ммолей) в ТГФ (100 мл), охлажденному до температуры -78°С. Через 15 мин раствор нагревают до 0°С в течение 15 мин, а затем снова охлаждают до температуры -78°С. Бромид (96) (2,5 г, 1,5 экв.) в ТГФ (15 мл) медленно вносят в реакционную смесь и перемешивают при КТ в течение 2 ч. Смесь затем снова охлаждают до 0°С, гасят насыщенным раствором NH4Cl, разбавляют водой и экстрагируют EtOAc. Органические экстракты промывают рассолом, высушивают (MgSO4) и концентрируют в вакууме. Сырой продукт очищают с помощью хроматографии на силикагеле, элюируя смесью EtOAc/гексан с градиентом от 20% до 75% EtOAc и получая 0,700 г частично очищенного соединения (100а). Полученный остаток растворяют в ДХМ (21,5 мл) и обрабатывают ТФК (830 мкл) в течение 15 ч. Затем реакционную смесь концентрируют в вакууме и очищают с помощью хроматографии на силикагеле, элюируя смесью МеОН/ДХМ с градиентом от 3% до 9% МеОН и получая 0,220 г частично очищенного соединения (100б).
стадия 3: К раствору соединения (100б) (0,200 г, около 0,397 ммоля) из стадий (1 и 2) и ДХМ (4 мл), охлажденному до температуры 0°С, добавляют ТЭА (330 мкл, 6 экв.), а затем фталоилдихлорид (100 мкл, 1,7 экв.), после чего реакционную смесь нагревают до КТ. Через 1 ч добавляют насыщенный раствор NH4Cl, смесь разбавляют Н2О и затем экстрагируют EtOAc. Органические слои промывают рассолом, высушивают (MgSO4) и концентрируют в вакууме. Продукт очищают с помощью хроматографии на силикагеле, элюируя смесью EtOAc/гексан и получая 0,110 г (выход 44%, 4% через 3 стадии) соединения (100в).
стадия 4: ацетат калия (26 мг, 1,5 экв.) и Ас2О (50 мкл, 3 экв.) добавляют к раствору соединения (100в) (0,110 г, 1,74 ммолей) в бензоле (2,3 мл). Затем раствор нагревают до температуры 80°С и медленно добавляют изо-амилнитрит (32 мкл, 1,4 экв.). Температуру повышают до 95°С и через 4 ч добавляют еще одну порцию КОАс, Ас2О и изо-амилнитрита. Через 14 ч раствор охлаждают до КТ, фильтруют через слой CELITE® и концентрируют в вакууме. Затем продукт очищают с помощью хроматографии на силикагеле, элюируя смесью EtOAc/гексан с градиентом от 33% до 66% EtOAc и получая 0,100 г (выход 89%) соединения (102а) в виде смеси ацетамидных региоизомеров в соотношении 2:1.
стадия 5: гидразин (7,9 мкл, 2,2 экв.) медленно добавляют к раствору соединения (102а) (0,074 г, 0,114 ммоля) в EtOH (2,3 мл). Через 1 ч реакционную смесь разбавляют EtOH (500 мкл). Через 2 ч раствор разбавляют EtOAc, фильтруют через CELITE® и концентрируют в вакууме. Сырой продукт очищают с помощью препаративной ВЭЖХ, получая 0,019 г (выход 19%) соединения (I-41).
Пример 15: 3-[6-Бром-2-фтор-3-(7-метокси-1H-пиразоло[3,4-с1пиридин-3-илметил)фенокси]-5-хлорбензонитрил (I-39), схема Г)
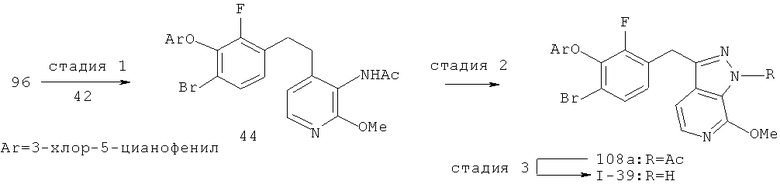
3-Ацетамидо-2-метокси-4-метилпиридин (42) получают в три стадии из 2-хлор-4-метил-3-нитропиридина по методике, описанной Townsend et al. (SynLett., 2002, 9:1479-1482).
стадия 1: н-бутиллитий (3,1 мл, 2,5-молярный раствор в гексане, 2,1 экв.) медленно прибавляют к раствору соединения (42) (0,665 г, 3,69 ммолей) в ТГФ (37 мл) при температуре -78°С. Через 15 мин раствор нагревают при температуре -50°С в течение 2 ч, затем медленно добавляют раствор соединения (96) (2,32 г, 1,5 экв.) в ТГФ (17 мл) и реакционную смесь перемешивают при 0°С в течение 2 ч. После этого смесь гасят насыщенным водным раствором NH4Cl, разбавляют водой и затем экстрагируют EtOAc. Объединенные органические слои промывают рассолом, высушивают (MgSO4) и концентрируют в вакууме. Полученный продукт очищают с помощью хроматографии на силикагеле, элюируя смесью МеОН/ДХМ с градиентом от 1% до 5% МеОН и получая 0,077 г (выход 4%) соединения (44).
стадия 2: Ацетат калия (10,2 мг, 1,2 экв.) и Ac2O (27,4 мг, 3,1 экв.) прибавляют к раствору соединения (44) (4,5 мг, 0,087 ммолей) в бензоле (1,7 мл). Раствор нагревают до температуры 80°С и затем медленно добавляют изо-амилнитрит (32 мкл, 1,6 экв.). После этого реакционную смесь нагревают при температуре 95°С в течение 6 ч, охлаждают до КТ, фильтруют через слой CELITE® и концентрируют в вакууме. Сырой продукт очищают с помощью препаративной ТСХ на силикагеле и выделяют с помощью смеси 50% EtOAc/гексан, получая 0,021 г (выход 50%) соединения (108а) в виде смеси ацетамидных региоизомеров в соотношении 2:1.
стадия 3: раствор LiOH·H2O (1,3 мг, 1,1 экв.) в Н2O (300 мкл) медленно добавляют к раствору соединения (108) (0,015 г, 0,028 ммолей) в ТГФ (300 мкл) при 0°С. Через 30 мин добавляют насыщенный водный раствор NH4Cl, реакционную смесь разбавляют водой и экстрагируют EtOAc. Объединенные экстракты промывают рассолом, высушивают (MgSO4) и концентрируют в вакууме. Сырой продукт очищают с помощью препаративной ТСХ на силикагеле и выделяют, используя смесь 33% EtOAc/гексаны и получая 0,002 г (выход 13%) соединения (I-39).
Пример 16: 3-Хлор-5-[6-хлор-3-(7-хлор-1H-пиразоло[3,4-c]пиридин-3-илметил)-2-фторфенокси]бензонитрил (I-42) и 3-[4-хлор-3-(3-хлор-5-цианофенокси)-2-фторбензил]-1H-пиразоло[3,4-с]пиридин-7-карбонитрил (I-43)
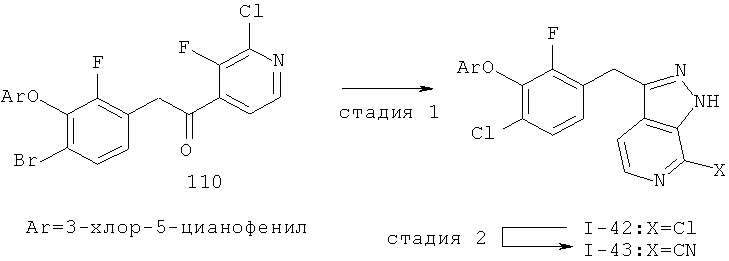
Кетон (110) получают из 2-хлор-3-фтор-изоникотиновой кислоты и соединения (R-5б) методом, описанным на стадии 1 примера 4.
стадия 1: Кетон (110) (0,50 г, 1,10 ммолей) растворяют в диоксане (10 мл) и EtOH (1,4 мл), а затем добавляют с помощью шприца гидразин (0,042 г, 1,1 экв.). Реакцию перемешивают при температуре 70°С в течение 4 ч, после чего раствор охлаждают до КТ, переносят в водный раствор NаНСО3 и экстрагируют смесью 10% МеОН/ДХМ. Органический слой промывают рассолом, высушивают (MgSO4) и выпаривают, получая 0,340 г (выход 70%) соединения (I-42).
стадия 2: Реакционную колбу, содержащую соединение (I-42) (0,033 г, 0,074 ммолей), Zn (CN)2 (0,0052 г, 0,6 экв.), металлический Zn (0,0029 г, 0,6 экв.), Pd (dba)3 (0,007 г, 0,1 экв.), dppf (0,0082 г, 0,2 экв.) и ДМА нагревают при температуре 105°С в течение 3 ч, после чего реакционную смесь фильтруют и выпаривают, а сырой продукт очищают с помощью препаративной ТСХ на силикагеле (0-15% МеОН/ДХМ), получая 0,005 г (выход 15%) соединения (I-43).
5-[6-Хлор-3-(7-циано-1H-пиразоло[3,4-с]пиридин-3-илметил)-2-фторфенокси]изофталонитрил (I-44) получают аналогично, исходя из этилового эфира [4-хлор-3-(3,5-дицианофенокси)-2-фторфенил]уксусной кислоты (R-11в).
Пример 17: 3-[6-Бром-3-(6-хлор-1H-пиразоло[3,4-b]пиридин-3-илметил)-2-фторфенокси]-5-хлорбензонитрил (I-9)
К раствору 3-[6-бром-2-фтор-3-(2-оксоэтил)фенокси]-5-хлорбензонитрила (72) (0,35 г, 0,96 ммолей) в ТГФ добавляют раствор реактива Гриньяра, полученного из изо-PrMgCl (0,52 мл 2-молярного раствора, 1,1 экв.) и 3-йод-2,6-дихлорпиридина (0,28 г, 1,05 экв.), как описано в примере 2. Очистка полученного продукта с помощью хроматографии дает 0,28 г (выход 57%) спирта. Окисление образовавшегося спирта с помощью перйодинана (реактив Десса-Мартина) (выход 47%) проводят, как описано на стадии 4 примера 7, а циклизацию гидразином (выход 39%) проводят, как описано на стадии 3 примера 6, при этом получают соединение (I-9).
Пример 18: 3-[6-Бром-2-фтор-3-(6-метокси-1H-пиразоло[3.4-b]пиридин-3-илметил)фенокси]-5-хлорбензонитрил (I-22)
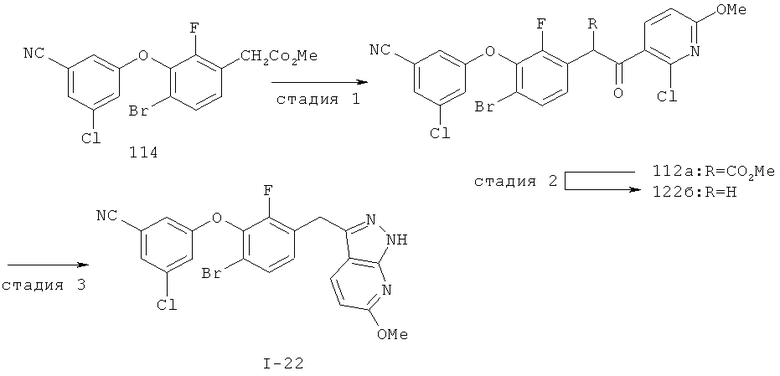
стадия 1: 2-Хлор-6-метоксиникотиновую кислоту (0,23 г, 1,1 экв.) смешивают с КДИ (0,20 г, 1,1 экв.) в ДМФ (5 мл) и нагревают при температуре 50°С в течение 1 ч. Затем реакцию охлаждают до температуры -10°С и добавляют сначала раствор соединения (114) (0,45 г, 1,13 ммолей) в ДМФ (5 мл), а потом NaH (0,14 г, 3,2 экв.). После добавления реакцию перемешивают при КТ в течение 3 ч. Затем реакционную смесь переносят в насыщенный водный раствор NH4Cl, экстрагируют смесью EtOAc/гексаны (в соотношении 1:1, 50 мл), промывают Н2O и рассолом, высушивают (MgSO4) и концентрируют в вакууме. Сырой продукт очищают с помощью хроматографии на силикагеле, элюируя смесью EtOAc/гексан с градиентом от 0 до 15% EtOAc и получая 0,35 г (выход 54%) соединения (112а) в виде белого твердого вещества.
стадия 2: Раствор соединения (112а), ДМСО (3 мл) и Н2O (0,15 мл) нагревают при температуре 150°С в течение 2 ч. Затем реакционную смесь охлаждают до КТ, переносят в насыщенный LiCl раствор и экстрагируют EtOAc. Объединенные экстракты промывают водой и рассолом, высушивают (Na2SO4) и концентрируют в вакууме. Сырой продукт очищают с помощью хроматографии на силикагеле, элюируя смесью EtOAc/гексан с градиентом от 0 до 12% EtOAc) и получая 0,200 г (выход 35%, общий на стадиях 1 и 2) соединения (112б) в виде белого твердого вещества.
стадия 3: к раствору соединения (112б) (0,2 г, 0,392 ммоля) в диоксане (5 мл) и EtOH (0,7 мл) добавляют с помощью шприца гидразин (0,086 мл, 7,0 экв.). Затем реакцию перемешивают при температуре 80°С в течение 2,5 ч, раствор охлаждают до КТ, переносят в водный раствор NH4Cl и экстрагируют смесью 10%МеОН/ДХМ. Объединенные экстракты промывают рассолом, высушивают (Na2SO4) и выпаривают, получая 0,087 г (выход 46%) соединения (I-22).
Пример 19: 3-[6-Бром-2-фтор-3-(5-фтор-7-окси-1H-пиразоло[3,4-b]пиридин-3-илметил)фенокси]-5-хлорбензонитрил (I-34)
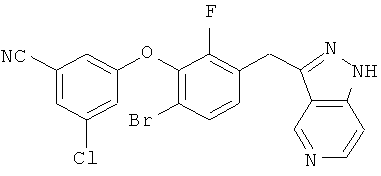
Названное в заголовке соединение получают, используя методики стадий 1-3 примера 6, за исключением замены на стадии (1) 2-хлорникотиновой кислоты 4-хлорникотиновой кислотой (CAS Reg. No. 10177-29-4).
Пример 20: 3-[6-Бром-2-фтор-3-(1Н-пиразоло[3,4-b]пиразин-3-илметил)фенокси]-5-хлорбензонитрил (I-53)
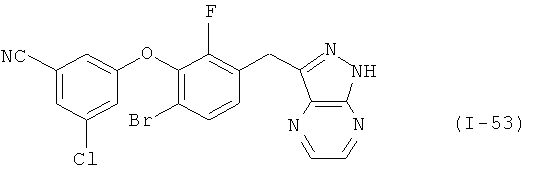
3-[6-Бром-2-фтор-3-(1Н-пиразоло[3,4-b]пиразин-3-илметил)фенокси]-5-хлорбензонитрил (I-53) получают, используя методики стадий 1-3 примера 6, за исключением замены на стадии (1) 2-хлорникотиновой кислоты 3-хлор-пиразинкарбоновой кислотой (CAS Reg No. 27398-39-6).
Пример 21: 3-[6-Бром-2-фтор-3-(1Н-пиразоло[3,4-с]пиридазин-3-илметил)фенокси1-5-хлорбензонитрил (I-49)
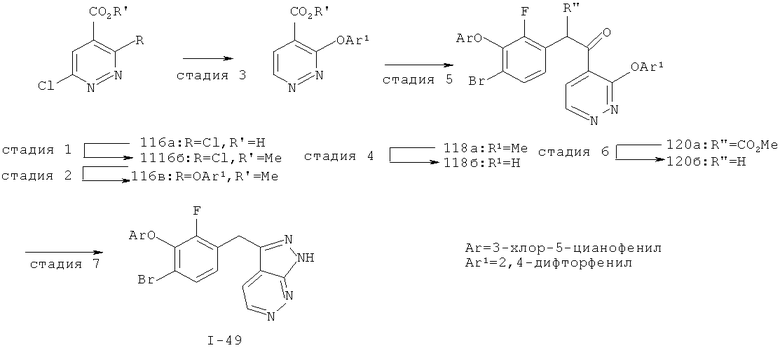
стадия 1: к раствору 3,6-дихлор-4-карбоксипиридазина (7,5 г, 38,9 ммолей, фирма Aldrich) в ДХМ (30 мл) и МеОН (10 мл), охлажденному до температуры 0°С, медленно, через пипетку добавляют раствор (триметилсилил)диазометана (2-молярный раствор в гексане) до тех пор, пока не возникнет устойчивое желтое окрашивание реакционной смеси. После добавления растворители удаляют в вакууме. Сырой продукт очищают с помощью хроматографии на силикагеле, элюируя смесью EtOAc/гексан с градиентом от 10 до 25% EtOAc и получая 3,89 г (выход 86%) соединения (116б) в виде коричневого масла, превращающегося в твердое вещество при стоянии.
стадия 2: Гидрид натрия (1,53 г, 38,27 ммолей) суспендируют в сухом ТГФ (70 мл) в атмосфере азота, охлажденном до температуры 0°С, и добавляют по каплям с помощью шприца 2,4-дифторфенол (3,31 мл, 34,94 ммолей). После добавления смесь перемешивают в течение 15 мин, затем охлаждающую баню удаляют, выдерживают реакционную смесь в течение 30 мин, в конце раствор опять охлаждают до температуры 0°С и добавляют с помощью шприца раствор соединения (116б) (6,89 г, 33,28 ммолей) в сухом ТГФ (20 мл). Затем образовавшуюся смесь перемешивают при КТ в течение ночи, нагревают при температуре 50°С в течение 3 ч, охлаждают до КТ и добавляют насыщенный водный раствор NH4Cl (40 мл), а вслед за ним воду (60 мл). Смесь затем трижды экстрагируют EtOAc, высушивают (MgSO4), фильтруют и выпаривают. Сырой продукт очищают с помощью хроматографии на силикагеле, элюируя смесью EtOAc/гексан с градиентом от 10 до 20% EtOAc и получая 8,15 г (выход 82%) соединения (116в) в виде слегка желтого масла.
стадия 3: к раствору соединения (116в) (8,15 г, 127,11 ммолей) в МеОН (40 мл) добавляют формиат аммония (8,55 г, 1,1 экв.), а затем 10%-ный Pd/C (500 мг). Смесь нагревают при температуре 50°С в течение 20 мин, а затем при 60°С в течение 35 мин. После этого смесь охлаждают до КТ и фильтруют через 2 см слой CELITE®, который затем тщательно промывают МеОН. Легколетучие растворители выпаривают, а остаток распределяют между ДХМ (80 мл) и H2O. ДХМ слой отделяют, а водный слой дважды экстрагируют ДХМ и водой (80 мл). Объединенные экстракты высушивают (MgSO4), фильтруют и выпаривают. Сырой продукт очищают с помощью хроматографии на силикагеле, элюируя смесью EtOAc/гексан с градиентом от 10 до 50% EtOAc и получая 5,5 г (выход 76%) соединения (118а) в виде полувязкого желтого масла.
стадия 4: к раствору соединения (118а) (5 г, 18,78 ммолей) в ТГФ (40 мл) и МеОН (10 мл) добавляют водный раствор LiOH (21,6 мл, 1-молярный раствор). Смесь перемешивают в течение 15 мин, после чего реакция завершается по данным ТСХ. Затем смесь концентрируют, остаток разбавляют Н2O (25 мл) и ТГФ (20 мл) и подкисляют смесь до рН 2-3, используя 10%-ную НС1. Образовавшееся твердое вещество отделяют фильтрованием, промывают водой (50 мл) и EtOAc (30 мл), получая 4,08 г (выход 86%) соединения (118б) в виде белого порошка.
стадия 5: к раствору соединения (118б) (605 мг, 2,4 ммолей) в ДМФ (10 мл) добавляют КДИ (410 мг, 2,5 ммоля). Затем смесь нагревают при температуре 50°С в атмосфере аргона в течение 1,5 ч, охлаждают до температуры -10°С и добавляют с помощью шприца раствор соединения (67) (1 г, 2,5 ммоля) в ДМФ (5 мл). Потом во время энергичного перемешивания добавляют NaH (336 мг, 8,4 ммолей) тремя порциями с промежутками в 20 мин. Оранжевый раствор перемешивают еще в течение 10 мин, затем охлаждающую баню удаляют и смесь перемешивают в течение 1 ч при КТ. Затем реакционную смесь разбавляют насыщенным водным раствором NH4Cl (20 мл), водой (30 мл) и EtOAc (50 мл) и взбалтывают. EtOAc фазу промывают рассолом (50 мл) и рассольный раствор экстрагируют EtOAc (дважды по 30 мл). Объединенные экстракты высушивают (MgSO4), фильтруют и выпаривают. Сырой продукт очищают с помощью хроматографии на силикагеле, элюируя смесью EtOAc/гексан с градиентом от 40 до 100% EtOAc и получая 685 мг (выход 45%) соединения (120а) в виде оранжевой пены.
стадия 6: к раствору соединения (120а) (670 мг, 1,06 ммолей) в ДМСО (8 мл) добавляют воду (0,4 мл) и рассол (10 капель). Смесь нагревают при температуре 145°С (температура масляной бани) в атмосфере аргона в течение 10 мин. Затем раствор охлаждают до КТ и добавляют воду (60 мл), EtOAc (30 мл) и Et2O (30 мл). В эту смесь при перемешивании вводят NaCl (2 г). Смесь опять перемешивают, после чего отделяют органическую фазу, промывают рассолом (50%) и рассольный раствор экстрагируют смесью EtOAc/Et2O (в соотношении 1:1, дважды по 50 мл). Объединенные органические фазы высушивают (MgSO4), фильтруют и выпаривают. Сырой продукт очищают с помощью препаративной ТСХ, выделяя его смесью 40% EtOAc/гексаны и получая 380 мг (выход 62%) соединения (120б) в виде слегка желтой пены.
стадия 7: к раствору соединения (120б) (100 мг, 0,17 ммолей) в МеОН (2 мл) добавляют трет-бутилкарбазат (45 мг, 2 экв.), а затем ледяную НОАс (0,03 мл). Полученную смесь нагревают при температуре 60°С в течение 5 ч и затем перемешивают при КТ в течение ночи. После этого смесь распределяют между ДХМ (20 мл) и 5%-ным водным раствором NaHCO3 (20 мл). Водную фазу снова экстрагируют ДХМ (дважды по 20 мл) и объединенные органические экстракты высушивают (MgSO4), фильтруют и выпаривают. Остаток растворяют в ТГФ (4 мл) в емкости для микроволнового нагревания, добавляют ДБУ (0,04 мл, 1,5 экв.) к образовавшемуся раствору и нагревают его в течение 10-12 мин при 150°С в микроволновой печи. Затем смесь распределяют между EtOAc (40 мл), водой (30 мл) и насыщенным водным раствором NH4Cl (5 мл). Органическую фазу отделяют, а водную фазу экстрагируют EtOAc (дважды по 30 мл). Объединенные экстракты высушивают (MgSO4), фильтруют и выпаривают. Сырой продукт очищают с помощью препаративной ТСХ, выделяя продукт смесью 6% МеОН/ДХМ и получая 45 мг (выход 58%) соединения (1-49) в виде грязно-белого порошка.
Трифторацетат 5-[6-бром-2-фтор-3-(1H-пиразоло[3,4-с]пиридазин-3-илметил)фенокси]изофталонитрила (I-52) получают аналогично за исключением замены на стадии 5 соединения (67) соединением (R-24).
5-[6-Этил-2-фтор-3-(1H-пиразоло[3,4-с]пиридазин-3-илметил)фенокси]изофталонитрил (I-54) получают аналогично за исключением замены на стадии 5 соединения (67) соединением (R-36).
3-Хлор-5-[6-этил-2-фтор-3-(1H-пиразоло[3,4-с]пиридазин-3-илметил)фенокси]бензонитрил (I-55) получают аналогично за исключением замены на стадии 5 соединения (67) соединением (R-32).
3-Хлор-5-[6-циклопропил-2-фтор-3-(1H-пиразоло[3,4-с]пиридазин-3-илметил)фенокси]бензонитрил (I-56) получают аналогично за исключением замены на стадии 5 соединения (67) соединением (R-42a).
5-[6-Циклопропил-2-фтор-3-(1H-пиразоло[3,4-с]пиридазин-3-илметил)фенокси]изофталонитрил (I-57) получают аналогично за исключением замены на стадии 5 соединения (67) соединением (R-41).
Пример 22: 3-[6-Бром-2-фтор-3-(6-метил-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-илметил)фенокси]-5-хлорбензонитрил (I-51)
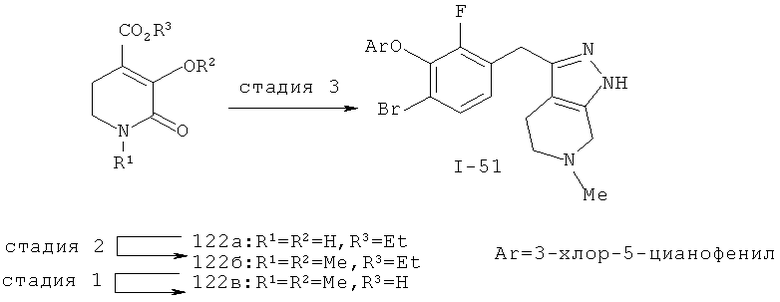
Исходное соединение (122а) получают методом, описанным by S.D.Barrett et al., Org. Prep.Proc. Int., 1997, 29(3):330-334.
стадии 1 и 2: Йодметан (0,75 мл, 2 экв.) медленно добавляют к раствору соединения (122а) (1,10 г, 5,94 ммолей) и KOtBu (1,34 г, 2 экв.) в толуоле (30 мл). После перемешивания в течение ночи (18 ч) загустевшую смесь гасят 6-молярным раствором НCl (30 мл), разбавляют водой и экстрагируют EtOAc. Органические слои промывают рассолом, высушивают (MgSO4) и концентрируют. Полученный остаток растворяют в ДХМ (13 мл) и МеОН (13 мл) НС1 при 0°С. К этому раствору осторожно добавляют триметилсилилдиазометан (5 мл, 2-молярный раствор в смеси гексанов, 4 экв.) в течение 10 мин. Затем реакционную смесь гасят добавлением НОАс и концентрируют в вакууме. Энольный эфир (122б) очищают с помощью хроматографии на силикагеле, элюируя смесью EtOAc/ДХМ с градиентом от 25 до 50% EtOAc и получая 0,250 г (выход 20%) соединения (122б). Часть этого вещества (200 мг, 0,94 ммолей) растворяют в ТГФ (9 мл) и добавляют раствор LiOH·Н2О (47 мг, 1,2 экв.) в H2O (3 мл). После перемешивания в течение ночи раствор подкисляют до рН 1 с помощью 10%-ного раствора HCl и экстрагируют EtOAc. Объединенные органические слои промывают рассолом, высушивают (MgSO4) и концентрируют, получая 120 мг (выход 69%) соединения (122в).
5-Метокси-1-метил-6-оксо-1,2,3,6-тетрагидропиридин-4-карбоновую кислоту (122в) превращают в соединение (I-51), используя методику, описанную на стадиях 3-5 примера 9. В этом случае замещения метокси-заместителя имидазолом не наблюдалось.
Пример 23: 3-[6-Бром-2-фтор-3-(6-метил-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-илметил)фенокси]-5-хлорбензонитрил (1-50)
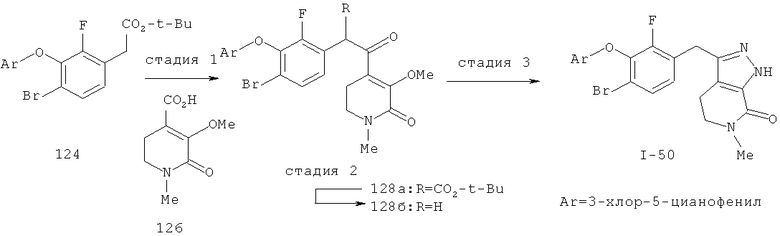
стадии 1 и 2: КДИ (0,116 г, 1,1 экв.) добавляют одной порцией к раствору кислоты (126) (0,120 г, 0,65 ммолей) в ДМФ (1,1 мл). После нагревания в течение 30 мин при температуре 50°С раствор охлаждают до температуры 0°С и добавляют раствор соединения (124) (0,315 г, 1,1 экв.) в ДМФ (2,3 мл). Затем гидрид натрия (0,088 г, 60%-ный в минеральном масле, 3,4 экв.) медленно приливают в реакционную смесь и оставляют ее самопроизвольно нагреваться до КТ в течение 2 ч. Смесь затем снова охлаждают до 0°С, гасят насыщенным раствором NH4Cl, разбавляют водой и экстрагируют EtOAc. Объединенные органические экстракты промывают рассолом, высушивают (MgSO4) и концентрируют. Полученный остаток снова растворяют в ДХМ (6,5 мл) и обрабатывают ТФК (3,25 мл) в течение 3 ч. После этого реакционную смесь концентрируют и остаток очищают с помощью хроматографии на силикагеле, элюируя смесью МеОН/ДХМ с градиентом от 1 до 5% МеОН и получая 0,180 г (выход 37%) соединения (128б).
стадия 3: Гидразин (53 мкл, 5 экв.) медленно добавляют к раствору соединения (128б) (0,170 г, 0,335 ммолей) в 1,4-диоксане (3,4 мл) при КТ. Затем реакционную смесь нагревают до температуры 50°С в течение 3 ч, охлаждают до КТ и концентрируют. Сырой продукт очищают с помощью хроматографии на силикагеле, элюируя смесью EtOAc/гексан с градиентом от 40 до 100% EtOAc и получая 0,060 г (выход 37%) соединения (I-50).
Пример 24: 3-Хлор-5-[6-дифторметил-2-фтор-3-(1Н-пиразоло[3,4-с]пиридазин-3-илметил)фенокси]бензонитрил (I-60)
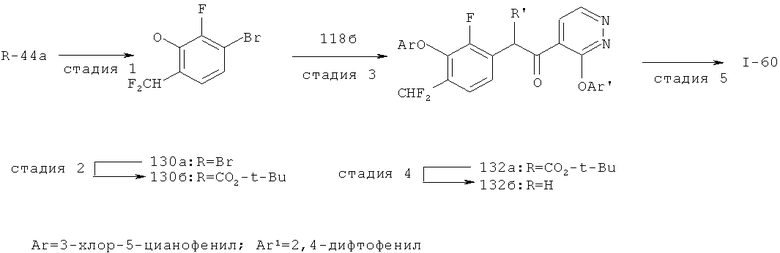
стадия 1: к раствору соединения (R-44a) (3,2 г, 9,04 ммолей) в ДХМ (12 мл) добавляют последовательно диэтиламиносульфуртрифторид (3,2 г, 2,2 экв.) и EtOH (0,02 г, 0,05 экв.) и реакционную смесь перемешивают в течение 16 ч. Затем реакционную смесь распределяют между водным раствором NaHCO3 и ДХМ. Органический слой промывают последовательно водой и рассолом, высушивают (Na2SO4), фильтруют и выпаривают, получая 1,9 г (выход 56%) соединения (130а).
стадия 2: к раствору соединения (130а) (1,9 г, 5,045 ммолей) и Pd(0)[P(трет-Bu)3]2 (0,39 г, 0,15 экв.) в диоксане (30 мл) при КТ добавляют хлорид 2-трет-бутокси-2-оксоэтилцинка (25 мл; 0,5-молярный раствор в эфире) и образовавшийся раствор перемешивают при КТ в течение 6 ч. Реакционную смесь распределяют между водным раствором НCl и EtOAc. Органический слой промывают последовательно водой и рассолом, высушивают (Na2SO4), фильтруют и выпаривают. Сырой продукт очищают с помощью хроматографии на силикагеле, элюируя смесью EtOAc/гексан с градиентом от 2 до 12% EtOAc и получая 0,65 г (выход 30%) соединения (130б).
стадия 3: К раствору соединения (118б) (0,088 г, 1,1 экв.) и ДМФ (1 мл) добавляют КДИ (0,06 г, 1,15 экв.) и раствор нагревают при температуре 50°С в течение 1 ч. В реакционную смесь, охлажденную до температуры -25°С, вносят раствор соединения (130б) (0,13 г, 0,316 ммолей) в ДМФ (1 мл) и NaH (0,04 г, 3,2 экв.). Затем реакционную смесь медленно нагревают до КТ и перемешивают в течение 6 ч, после чего ее распределяют между насыщенным водным раствором NaHCO3 и смесью EtOAc/гексаны (в соотношении 1:1). Органический слой промывают последовательно H2O и рассолом, высушивают (Na2SO4), фильтруют и выпаривают, получая 0,200 г (выход 98%) соединения (132а).
стадия 4: раствор соединения (132а) (0,2 г, 0,31 ммолей) и п-TsOH (0,015 г, 0,25 экв.) в толуоле (2,5 мл) нагревают при температуре 130°С в течение 2 ч. Затем реакционную смесь охлаждают, переносят в раствор бикарбоната натрия и экстрагируют EtOAc. Органический слой промывают последовательно водой и рассолом, высушивают (Na2SO4), фильтруют и выпаривают. Сырой продукт очищают с помощью хроматографии на силикагеле, элюируя смесью EtOAc/гексан с градиентом от 15 до 50% EtOAc и получая 0,15 г (выход 89%) соединения (132б).
стадия 5: раствор соединения (132б) (0,15 г, 0,274 ммолей), п-TsOH (0,10 г, 2 экв.) и гидразина (0,03 мл, 2 экв.) в ИПА (2 мл) нагревают при температуре 80°С в течение 16 ч. В реакционную смесь, охлажденную до температуры 0°С, добавляют Н2О (2,6 мл). Величину рН образовавшегося раствора доводят приблизительно до 9 с помощью 20%-ного раствора Na2CO3, затем разбавляют H2O (5 мл) и нагревают при КТ в течение 1 ч. Мутную смесь переносят в EtOAc и органический слой промывают последовательно водой и рассолом, высушивают (Na2SO4), фильтруют и выпаривают. Сырой продукт очищают с помощью хроматографии на силикагеле, элюируя смесью МеОН/ДХМ с градиентом от 2,5 до 10% МеОН. Извлеченный продукт растирают со смесью EtOAc/гексаны, получая 0,040 г (выход 34%) соединения (I-60).
Пример 25: 3-Хлор-5-[2-фтор-6-метансульфонил-3-(1Н-пиразоло[3,4-с]пиридазин-3-илметил)фенокси]бензонитрил (1-63)
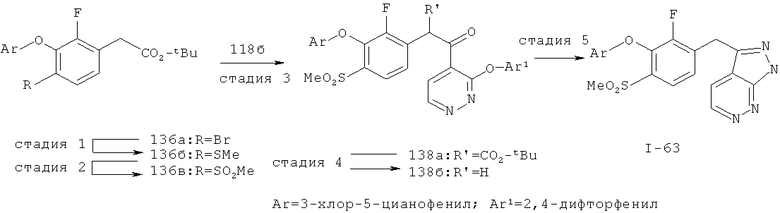
стадия 1: к раствору соединения (136а) (4,03 г, 9,15 ммолей) в м-ксилоле (60 мл) добавляют K2CO3 (846 мг, 6,12 ммолей), Pd2(dba)3 (840 мг, 0, 92 ммолей), ксантфоса (Xantphos) (600 мг, 1,04 ммолей, CASRN 161265-03-8) и NaSMe (810 мг, 11,56 ммолей). Смесь дегазируют, а затем нагревают при температуре 135°С в атмосфере аргона в течение 20 ч. После этого реакцию охлаждают до КТ и добавляют рассол (80 мл). Затем смесь экстрагируют EtOAc (80 мл), водную фазу снова экстрагируют EtOAc (дважды по 70 мл), высушивают (MgSO4), фильтруют и концентрируют в вакууме. Сырой продукт очищают с помощью хроматографии на силикагеле и получая 2,3 г соединения (136б) в виде желтого масла.
стадия 2: к раствору соединения (136б) (2,4 г, 5,88 ммолей) в МеОН (60 мл) и ТГФ (8 мл), охлажденному до температуры 0°С (ледяная баня), добавляют по каплям OKCONE® (7,35 г, 11,96 ммолей), растворенный в воде (22 мл). После добавления смесь перемешивают в течение 15 мин и затем удаляют охлаждающую баню. Образовавшуюся смесь перемешивают в течение ночи, а затем нагревают при температуре 50°С в течение 4 ч. Реакционную смесь охлаждают далее до КТ и водный насыщенный раствор NaHCO3 добавляют по каплям до прекращения пенообразования. Затем добавляют воду (20 мл) и смесь экстрагируют EtOAc (40 мл). Экстракты промывают рассолом (40 мл) и промывной рассол снова экстрагируют EtOAc (дважды по 30 мл). Объединенные EtOAc экстракты высушивают (MgSO4), фильтруют и концентрируют в вакууме, получая 2,5 г соединения (136в) в виде бело-желтого твердого вещества.
стадия 3: к раствору соединения (118б) (274 мг, 1,1 ммолей) в сухом ДМФ (8 мл) добавляют КДИ (188 мг, 1,2 ммолей). Смесь нагревают при температуре 50°С в течение 2 ч, а затем охлаждают до температуры -10°С. С помощью шприца к смеси добавляют раствор соединения (136в) (500 мг, 1,14 ммолей) в ДМФ (5 мл). В охлажденную смесь вносят затем NaH (152 мг, 3,81 ммолей, 60%-ный раствор в минеральном масле) в течение 20 мин тремя эквивалентными порциями. Закончив добавление, смесь перемешивают в течение 15 мин, после чего удаляют охлаждающую баню и перемешивание продолжают в течение 1 ч. Затем к раствору осторожно прибавляют насыщенный водный раствор NH4Cl (5 мл), потом воду (30 мл) и EtOAc (40 мл). Реакционную смесь перемешивают и EtOAc фазу отделяют. Водную фазу вновь экстрагируют EtOAc (дважды по 30 мл). Объединенные экстракты высушивают (MgSO4), фильтруют и выпаривают. Сырой продукт очищают с помощью хроматографии на силикагеле, элюируя смесью EtOAc/гексан с градиентом от 50 до 100% EtOAc и получая 0,256 г соединения (138а) в виде желтого пенообразного твердого вещества.
стадия 4: к раствору соединения (138а) (256 мг, 0,38 ммолей) в анизоле (5 мл) добавляют порошкообразный борный ангидрид (133 мг). Смесь нагревают при температуре 140°С в течение 1 ч, затем охлаждают до КТ и концентрируют в вакууме. Остаток охлаждают (ледяная баня) и распределяют между водой (25 мл) и EtOAc (25 мл). Смесь перемешивают при КТ в течение 1 ч, а затем взбалтывают. EtOAc фазу промывают рассолом (25 мл), а водную фазу вновь экстрагируют EtOAc (дважды по 20 мл). Объединенные органические экстракты высушивают (MgSO4), фильтруют и концентрируют, получая 0,215 г соединения (138б) в виде бледного оранжево-желтого твердого вещества.
стадия 5: к раствору соединения (138б) (215 мг, 0,38 ммолей) в ИРА (2 мл) добавляют п-TsOH (144 мг) и гидразингидрат (0,04 мл, 85%-ный). Затем смесь нагревают при температуре 80°С в атмосфере азота в течение 18 ч. После этого реакционную смесь охлаждают до КТ и последовательно добавляют воду (3,5 мл), 20%-ный водный раствор Na2CO3 (0,5 мл), затем дополнительно воду (1,5 мл). Смесь перемешивают в течение 5 мин и затем оставляют на 1,5 ч. Образовавшийся осадок (65 мг) отделяют фильтрованием. Вторую порцию осадка (130 мг) извлекают из фильтрата. Эти полутвердые осадки объединяют, адсорбируют на SiO2 пластине и с помощью препаративной ТСХ выделяют, используя EtOAc и получая 0,055 г соединения (I-63) в виде бледного бело-оранжевого твердого вещества.
Пример 26: 3-[6-Хлор-2-фтор-3-(1Н-пиразоло]3,4-с]пиридазин-3-илметил)фенокси]-5-дифторметилбензонитрил
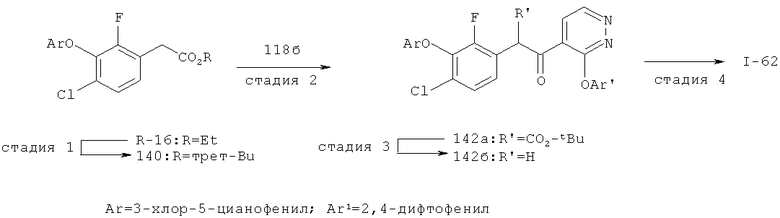
стадия 1: соединение (R-16) гидролизуют до соответствующей карбоновой кислоты с помощью LiOH в водном растворе ТГФ, перемешивая при КТ в течение 3 ч, стандартная методика, превращающая кислоту в ее трет-бутиловый эфир, включает перемешивание раствора трет-BuOH, раствора кислоты, Бок-ангидрида и ДМАР в течение 2 ч. Сырой продукт очищают с помощью хроматографии на силикагеле, элюируя смесью 5% EtOAc/гексан и получая соединение (140).
стадия 2: к раствору соединения (118б) (0,485 г, 1,92 ммолей) и ДМФ (9 мл), внесенного в высушенную в пламени колбу, добавляют КДИ (0,326 г, 2,01 ммолей), и раствор нагревают при температуре 50°С в течение 65 мин, а затем охлаждают до температуры 0°С и добавляют раствор соединения (140) (0,720 г, 1,75 ммолей) в небольшом количестве ДМФ, а затем NaH (0,189 г, 4,72 ммолей, 50%-ная дисперсия в минеральном масле). Реакционную смесь перемешивают в течение 1 ч, после чего переносят в насыщенный водный раствор NH4Cl. Выпавшее в осадок твердое вещество отделяют, промывают водой и высушивают в вакууме, получая 0,978 г соединения (142а) в виде коричневого твердого вещества.
стадия 3: К раствору соединения (142а) (0,978 г, 1,51 ммолей) в анизоле (7,5 мл) добавляют борный ангидрид (0,527 г, 7,57 ммолей) и образовавшийся раствор нагревают при температуре 140°С в течение 2 ч. Затем реакционную смесь охлаждают на ледяной бане и раствор распределяют между EtOAc и Н2О. Органическую фазу отделяют, промывают рассолом, высушивают, фильтруют и концентрируют в вакууме. Сырой продукт очищают с помощью хроматографии на силикагеле, элюируя смесью 1% МеОН/ДХМ и получая 0,580 г соединения (142б).
стадия 4: суспензию соединения (142б) (0,580 г, 1,06 ммолей) и п-толуолсульфокислоты (0,404 г, 2,13 ммолей) в изопропиламине (5 мл) перемешивают при КТ в течение 20 мин. Затем раствор перемешивают при температуре 80°С до завершения реакции. Реакционную смесь охлаждают на ледяной бане, а затем последовательно добавляют к ней H2O (10,6 мл), 20%-ный водный раствор Na2CO3 (2 мл) и H2O (5,3 мл). Образовавшуюся смесь перемешивают при КТ в течение 1 ч. Выпавший осадок отделяют, промывают H2O и высушивают в вакууме, получая 89 мг соединения (I-62).
3-[6-Хлор-2-фтор-3-(1Н-пиразоло[3,4-с]пиридазин-3-илметил)фенокси]-5-метилбензонитрил (I-61) получают методом, описанным на стадиях (2-4) настоящего примера из трет-бутил [4-хлор-3-(3-циано-5-метилфенокси)-2-фторфенил] ацетата (144). Исходное соединение (144) для синтеза получают аналогично методике, описанной для соединения (R-1) в показательном примере, за исключением замены на стадии (3) соединения (R-3в) 3-гидрокси-5-метилбензонитрилом (CASRN 95658-81-4).
Пример 27: Анализ гетерополимерной ВИЧ обратной транскриптазы: определение IC50 ингибитора
Анализ ВИЧ-1 ОТ проводят в 96-ячеистых Millipore MultiScreen MADVNOB50 планшетах с использованием очищенного рекомбинантного фермента и поли(rA)/олиго(dT)16 матричного праймера с общим объемом 50 мкл. Составными компонентами анализа являются: 50 мМ Tris/HCl, 50 мМ NaCl, 1 мМ ЭДТК, 6 мМ MgCl2, 5 мкМ dТТФ, 0,15 мк Ci [3Н] dTTФ, 5 мкг/мл поли(rА), прегибридизированной до 2,5 мкг/мл олиго (dT)16, и в диапазоне концентраций ингибитора с конечной концентрацией 10% ДМСО. Реакции инициируют добавлением 4 нМ ВИЧ-1 ОТ и после инкубирования при температуре 37°С в течение 30 мин, их останавливают добавлением 50 мкл охлажденной льдом 20%-ной трихлоруксусной кислоты (ТХК), после чего оставляют самопроизвольно осаждаться при температуре 4°С в течение 30 мин. Осадок отделяют вакуумированием в планшете и последовательно промывают трижды по 200 мкл 10%-ной ТСА и дважды по 200 мкл 70%-ным этанолом. Окончательно планшеты высушивают и радиоактивность считывают в Packard TopCounter после добавления 25 мкл сцинтилляционной жидкости на ячейку. Величины IC50 были подсчитаны с помощью графика: % ингибирования в зависимости от log10 концентраций ингибитора.
Показательные величины IC50 представлены ниже Значения IC50 всех соединений за исключением I-1 и I-2 составляли менее 100 наномолей
Пример 28: Анализ гомополимерной ВИЧ обратной транскриптазы: определение IC50 ингибитора
Анализ ВИЧ-1 ОТ проводят в 96-ячеистых Millipore filtermet NOBSO планшетах с использованием очищенного рекомбинантного фермента и поли(rА)/олиго(dТ)16 матричного праймера с общим объемом 50 мкл. Составными компонентами анализа являются: 50 мМ Tris/HCl, 50 мМ NaCl, 1 мМ ЭДТК, 6 мМ MgCl2, 5 мкМ dЛТФ, 0,1 мк Ci [3Н] dTTФ, 5 мкг/мл поли(rA), прегибридизированной до 2,5 мкг/мл олиго (dT)16, и в диапазоне концентраций ингибитора с конечной концентрацией 10% ДМСО. Реакции инициируют добавлением 5 нМ ВИЧ-1 ОТ и после инкубирования при температуре 37°С в течение 30 мин, их останавливают добавлением 50 мкл охлажденной льдом 20%-ной трихлоруксусной кислоты (ТХК), после чего оставляют самопроизвольно осаждаться при температуре 4°С в течение 30 мин. Осадок отделяют вакуумированием в планшете и последовательно промывают дважды по 200 мкл 10%-ной ТХК и дважды по 200 мкл 70%-ным этанолом. Окончательно планшеты высушивают и радиоактивность считывают в Wallac Microbeta 1450 после добавления 15 мкл сцинтилляционной жидкости на ячейку. Величины IC50 (таблица 3) были подсчитаны с помощью графика: % ингибирования в зависимости от log10 концентраций ингибитора.
Пример 29: Фармацевтические композиции
Фармацевтические композиции соединения для введения различными способами получены, как описано в этом примере.
Композиция для орального введения (А)
Ингредиенты смешивают и вносят в капсулы, содержащие приблизительно 100 мг каждая; одна капсула соответствует общей суточной дозе.
Композиция для орального ведение (Б)
Ингредиенты смешивают и гранулируют, используя в качестве растворителя метанол. Композиции затем высушивают и превращают в таблетки (содержащие приблизительно 20 мг активного соединения) в соответствующей таблетирующей машине.
Композиция для орального введение (В)
Ингредиенты смешивают с образованием суспензии для орального введения.
Парентеральные композиции (Г)
Активный ингредиент растворяют в порции воды для иньекции. Затем при перемешивании добавляют достаточное для получения изотонического раствора количество хлорида натрия. Раствор доводят до нужной массы, используя оставшуюся воду для иньекции, фильтруют через мембранный фильтр (0,2 микрона) и расфасовывают в стерильных условиях. Суппозиторные композиции (R)
Инградиенты расплавляют вместе и перемешивают на паровой бане, и прессуют в пресс-формах, содержащих 2,5 г общей массы.
Все ингредиенты, за исключением воды, объединяются и нагреваются приблизительно до температуры 60°С при перемешивании. Затем добавляется достаточное количество воды приблизительно при этой же температуре при энергичном перемешивании с целью получения эмульсии градиентов, а затем снова добавляют воду до 100 г.
Особенности, раскрытые в вышеприведенном описании или последующей формуле изобретения, выраженные в специфических формах или терминах, выполняют функцию раскрытия, или способ или процесс для достижения раскрытых результатов, как соответствующих, и могут быть в отдельности или в любой комбинации таких функций утилизированы для реализации изобретения в его разнообразных формах.
Вышеприведенное изобретение описано в некоторых деталях путем иллюстрации и примера для целей ясности и понимания. Каждому специалисту в данной области техники должно быть очевидно, что изменения и модификации можно осуществить, не выходя за рамки данного изобретения, объем которого определяется формулой изобретения. Поэтому должно быть ясным, что вышеприведенное описание предназначено для иллюстрации, а не для ограничения. Объем изобретения поэтому определяется не ссылкой на вышеприведенное описание, а должен вместо этого определяться ссылкой на пункты формулы изобретения наряду с полным объемом эквивалентов, которые встречаются в этих пунктах.
Все патенты, патентные заявки и публикации, цитируемые в этой заявке, полностью включены посредством ссылок в ее объем в той же самой степени, как если бы они были представлены в виде индивидуальных патента, патентной заявки или публикации.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИТОРЫ СЕРИН/ТРЕОНИН КИНАЗЫ ДЛЯ ЛЕЧЕНИЯ ГИПЕРПРОЛИФЕРАТИВНЫХ ЗАБОЛЕВАНИЙ | 2013 |
|
RU2644947C2 |
| ИМИДАЗО[1,5-а] ПИРИМИДО[5,4-d][1] БЕНЗАЗЕПИНОВЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ МОДУЛЯТОРОВ РЕЦЕПТОРА GABA A | 2002 |
|
RU2287531C2 |
| ИНГИБИТОРЫ ТИРОЗИНКИНАЗЫ БРУТОНА | 2014 |
|
RU2653500C2 |
| СОЕДИНЕНИЯ ГЕТЕРОАРИЛА В КАЧЕСТВЕ ИНГИБИТОРОВ ТКБ И ИХ ПРИМЕНЕНИЕ | 2015 |
|
RU2742122C2 |
| ИНГИБИТОРЫ ТИРОЗИНКИНАЗЫ БРУТОНА | 2010 |
|
RU2542585C2 |
| ПРОИЗВОДНЫЕ 2-АЗА-БИЦИКЛО[3.3.0]ОКТАНА | 2008 |
|
RU2478099C2 |
| СОЕДИНЕНИЯ ПИРИДИНОНА В КАЧЕСТВЕ СЕЛЕКТИВНЫХ ЦИТОТОКСИЧЕСКИХ АГЕНТОВ ПРОТИВ КЛЕТОК, ИНФИЦИРОВАННЫХ ВИЧ | 2020 |
|
RU2816090C2 |
| ФЕНОКСИМЕТИЛЬНЫЕ ПРОИЗВОДНЫЕ | 2016 |
|
RU2746481C1 |
| НОВЫЕ ТИЕНОПИРИМИДИНОВЫЕ ПРОИЗВОДНЫЕ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2014 |
|
RU2605403C2 |
| ИНГИБИТОРЫ КАНАЛОВ С ТРАНЗИТОРНЫМ ПОТЕНЦИАЛОМ НА ОСНОВЕ ОКСАДИАЗОЛОВ | 2019 |
|
RU2818244C2 |
Настоящее изобретение относится к соединениям, которые могут быть применены для лечения ВИЧ инфекции, или профилактики ВИЧ инфекции, или лечения СПИД или СПИД-ассоциированного комплекса. Соединения по изобретению представляют собой соединения формулы I, где А обозначает A1, А2, A3 или А4 и R1, R2, R3, R4a, R4b, R5, R6, Ar, X1, X2, X4, X4 и X5 имеют значения, приведенные в формуле изобретения. Эти соединения обладают ингибирующей активностью в отношении обратной транскриптазы ВИЧ. Настоящее изобретение также относится к фармацевтической композиции, содержащий названные соединения. 2 н. и 20 з.п. ф-лы, 3 табл., 29 пр.
1. Соединение формулы I
где А выбран из группы, состоящей из:
(i)A1,
(ii) А2, где
(а)Х1,X2 и X3 независимо обозначают CR3,
(б) один из X1 и X2 обозначает N или N→O, а другой из X1 и X2 наряду с X3 независимо обозначают CR3;
(в) X1 и один из X2 или X3 обозначают N, а другой из X2 или X3 обозначает CR3;
(г) один из X1 и X2 обозначает NR5, другой из X1 и X2 обозначает С(=O), X3 обозначает CR3, и связь между X1 и X2 представляет собой простую связь; или
(iii) A3, и
(iv) А4, где один из X4 и X5 обозначает NR5, другой из X4 и X5 обозначает С(=O);
R1 обозначает фтор или водород;
R2 обозначает водород, галоген, С1-6алкил, С3-7циклоалкил, C1-6галогеналкил, С1-6алкоксигруппу или С1-6алкилсульфонил;
Аr обозначает фенил, замещенный от одной до трех групп, независимо выбранных в каждом случае из группы, включающей водород, галоген, цианогруппу, С1-6алкил, С1-6алкоксигруппу, С1-6галогеналкил и С3-7циклоалкил;
R3 независимо выбран в каждом случае из группы, включающей:
(i) водород,
(ii) гидроксигруппу,
(iii) С1-6алкоксигруппу,
(iv) галоген,
(v) NR4aR4b,
(vi) C1-6ациламиногруппу,
(vii) С1-6алкилсульфониламиногруппу,
(viii) цианогруппу,
(ix) нитрогруппу,
(x) NHNH2; и
(xi) фенил, необязательно замещенный от одного до трех заместителями, независимо выбранными из группы, включающей галоген, C1-6алкил, С1-6алкоксигруппу, цианогруппу, нитрогруппу;
R4a и R4b независимо обозначают водород или С1-6алкил;
R5 обозначает водород или С1-3алкил;
R6 обозначает водород или С1-6алкил; а также
3-[6-бром-2-фтор-3(1Н-пиразол[4,3-с]пиридин-3-илметил)фенокси]-5-хлорбензонитрил
и их фармацевтически приемлемые соли.
2. Соединение по п.1, где А обозначает А1 или А2.
3. Соединение по п.2, где А обозначает А1.
4. Соединение по п.1, где А обозначает А2, и X1, X2 и X3 независимо обозначают CR3.
5. Соединение по п.4, где R1 обозначает фтор; R2 обозначает С1-6алкил, C1-6алкоксигруппу или галоген, R6 обозначает водород, и Ar обозначает 3-цианофенил, необязательно замещенный одной или двумя группами, независимо выбранными из галогена, цианогруппы и С1-6галогеналкила.
6. Соединение по п.5, где R2 обозначает бром или хлор.
7. Соединение по п.1, где А обозначает А2, один из X1 или X2 обозначает N или N-O, другой из X1 и X2 наряду с X3 независимо обозначают CR3.
8. Соединение по п.7, где R1 обозначает фтор; R2 обозначает С1-6алкил, C1-6алкоксигруппу или галоген; Ar обозначает 3-цианофенил, необязательно замещенный одной или двумя группами, независимо выбранными из галогена, цианогруппы и С1-6галогеналкила; R5 и R6 обозначают водород.
9. Соединение по п.8, где Ar обозначает 3-цианофенил, необязательно замещенный галогеном, цианогруппой или С1-6галогеналкилом, расположенным в метаположении по отношению к заместителю циано.
10. Соединение по п.9, где R2 обозначает бром или хлор.
11. Соединение по п.1, где А обозначает А2, один из X1 или X2 обозначает NR5, другой из X1 или X2 обозначает С(=O), X3 обозначает CR3, и связь между X1 и X2 представляет собой простую связь.
12. Соединение по п.1, где А обозначает А2, X1 и X2 обозначают N, и X3 обозначает CR3.
13. Соединение по п.12, где R1 обозначает фтор; R2 обозначает С1-6алкил, С1-6алкоксигруппу или галоген; Ar обозначает 3-цианофенил, необязательно замещенный одной или двумя группами, независимо выбранными из галогена, цианогруппы и С1-6галогеналкила; и R6 обозначает водород.
14. Соединение по п.13, где Ar обозначает 3-цианофенил, необязательно замещенный галогеном, цианогруппой или С1-6галогеналкилом, расположенным в метаположении по отношению к заместителю циано.
15. Соединение по п.14, где R2 обозначает бром или хлор.
16. Соединение по п.1, где А обозначает А2, X1 и X2 обозначают N, и X3 обозначает CR3.
17. Соединение по п.1, где А обозначает A3, R1 обозначает фтор; R2 обозначает С1-6алкил, С1-6алкоксигруппу или галоген; и Ar обозначает 3-цианофенил, необязательно замещенный одной или двумя группами, независимо выбранными из галогена, цианогруппы и С1-6галогеналкила.
18. Соединение по п.17, где R6 обозначает водород.
19. Соединение по п.1, где А обозначает А4, и один из X4 или X5 обозначает NR5, другой из X4 или X5 обозначает С(=O).
20. Соединение по п.1, которое является соединением, представляющим собой:
5-(4-Метил-3-феноксибензил)-2Н-пиразол-3-ол,
3-[4-Хлор-3-(3-хлорфенокси)бензил]-1Н-пиразол,
3-Хлор-5-[6-хлор-2-фтор-3-(1Н-индазол-3-илметил)фенокси]бензонитрил,
3-Хлор-5-[6-хлор-2-фтор-3-(5-фенил-1Н-пиразол-3-илметил)фенокси]-бензонитрил,
3-[6-Бром-2-фтор-3-(5-фенил-1Н-пиразол-3-илметил)фенокси]-5-дифторметилбензонитрил,
3-[6-Бром-2-фтор-3-(1Н-индазол-3-илметил)фенокси]-5-дифторметилбензонитрил,
3-[6-Бром-2-фтор-3-(1Н-пиразоло[3,4-b]пиридин-3-илметил)фенокси]-5-хлорбензонитрил,
3-[6-Бром-2-фтор-3-(7-окси-1Н-пиразоло[3,4-b]пиридин-3-илметил)-фенокси]-5-хлорбензонитрил,
3-[6-Бром-3-(6-хлор-1Н-пиразоло [3,4-b] пиридин-3-илметил)-2-фторфенокси]-5-хлорбензонитрил,
Трифторацетат 3-[6-бром-2-фтор-3-(6-гидразино-1Н-пиразоло [3,4-d]пиримидин-3-илметил)фенокси]-5-хлорбензонитрила,
3-Хлор-5-[6-хлор-2-фтор-3-(1Н-пиразоло[3,4-с]пиридазин-3-илметил)-фенокси]бензонитрил,
3-[6-Бром-2-фтор-3-(6-оксо-6,7-дигидро-1Н-пиразоло[3,4-b]пиридин-3-илметил)фенокси]-5-хлорбензонитрил,
3-[6-Бром-3-(6-хлор-1Н-пиразоло[3,4-е]пиримидин-3-илметил)-2-фторфенокси]-5-хлорбензонитрил,
3-[4-Бром-3-(3-хлор-5-цианофенокси)-2-фторбензил]-1Н-пиразоло[3,4-b]пиридин-6-карбонитрил,
3-[6-Бром-2-фтор-3-(1Н-пиразоло[3,4-с]пиридин-3-илметил)фенокси]-5-хлорбензонитрил,
5-[6-Бром-2-фтор-3-(1Н-пиразоло[3,4-b]пиридин-3-илметил)фенокси]-изофталонитрил,
3-[6-Бром-2-фтор-3-(1-метил-1Н-пиразоло[3,4-b]пиридин-3-илметил)фенокси]-5-хлорбензонитрил,
3-[6-Бром-2-фтор-3-(1Н-пиразоло[3,4-d]пиримидин-3-илметил)фенокси]-5-хлорбензонитрил,
5-[6-Бром-2-фтор-3-(1Н-пиразоло[3,4-с]пиридин-3-илметил)фенокси]-изофталонитрил,
3-[6-Бром-2-фтор-3-(7-нитро-1Н-индазол-3-илметил)фенокси]-5-хлорбензонитрил,
5-[6-Циклопропил-2-фтор-3-(1Н-пиразоло[3,4-b]пиридин-3-илметил)-фенокси]изофталонитрил,
3-[6-Бром-2-фтор-3-(6-метокси-1Н-пиразоло[3,4-b]пиридин-3-илметил)-фенокси]-5-хлорбензонитрил,
3-[6-Бром-2-фтор-3-(5-фтор-1Н-пиразоло[3,4-b]пиридин-3-илметил)-фенокси]-5-хлорбензонитрил,
3-Хлор-5-[2-фтор-6-метил-3-(1Н-пиразоло[3,4-b]пиридин-3-илметил)-фенокси]бензонитрил,
3-Хлор-5-[6-этил-2-фтор-3-(1Н-пиразоло[3,4-b]пиридин-3-илметил)-фенокси] бензонитрил,
3-[6-Бром-2-фтор-3-(5-фтор-7-окси-1Н-пиразоло[3,4-b]пиридин-3-илметил)-фенокси]-5-хлорбензонитрил,
3-Дифторметил-5-[2-фтор-6-метокси-3-(1Н-пиразоло[3,4-b]пиридин-3-илметил)фенокси]бензонитрил,
3-[6-Хлор-2-фтор-3-(1Н-пиразоло[3,4-b]пиридин-3-илметил)фенокси]-5-дифторметилбензонитрил,
3-[6-Бром-2-фтор-3-(1Н-пиразоло[3,4-b]пиридин-3-илметил)фенокси]-5-дифторметилбензонитрил,
3-[4-Бром-3-(3-хлор-5-цианофенокси)-2-фторбензил]-1Н-индазол-7-карбонитрил,
3-[6-Бром-2-фтор-3-(1Н-пиразоло[3,4-с]пиридин-3-илметил)фенокси]-5-дифторметилбензонитрил,
3-[6-Хлор-2-фтор-3-(1Н-пиразоло[3,4-с]пиридин-3-илметил)фенокси]-дифторметилбензонитрил,
3-Хлор-5-[6-циклопропил-2-фтор-3-(1Н-пиразоло[3,4-b]пиридин-3-илметил)фенокси]бензонитрил,
3-[6-Бром-2-фтор-3-(5-фтор-6-оксо-6,7-дигидро-1Н-пиразоло[3,4-b]пиридин-3-илметил)фенокси]-5-хлорбензонитрил,
3-[3-(7-Амино-1Н-индазол-3-илметил)-6-бром-2-фторфенокси]-5-хлорбензонитрил,
N-{3-[4-Бром-3-(3-хлор-5-цианофенокси)-2-фторбензил]-1Н-индазол-7-ил}ацетамид,
3-[6-Бром-2-фтор-3-(1Н-пиразоло[4,3-с]пиридин-3-илметил)фенокси]-хлорбензонитрил,
N-{3-[4-Бром-3-(3-хлор-5-цианофенокси)-2-фторбензил]-1Н-индазол-7-ил}метансульфонамид,
3-[6-Бром-2-фтор-3-(7-метокси-1Н-пиразоло[3,4-с]пиридин-3-илметил)-фенокси]-5-хлорбензонитрил,
3-Дифторметил-5-[2-фтор-6-метил-3-(1Н-пиразоло[3,4-b]пиридин-3-илметил)фенокси]бензонитрил,
3-[3-(7-Амино-1Н-пиразоло[3,4-с]пиридин-3-илметил)-6-бром-2-фторфенокси]-5-хлорбензонитрил,
3-Хлор-5-[6-хлор-3-(7-хлор-1Н-пиразоло[3,4-с]пиридин-3-илметил)-2-фторфенокси]бензонитрил,
3-[4-Хлор-3-(3-хлор-5-цианофенокси)-2-фторбензил]-1Н-пиразоло [3,4 с]пиридин-7-карбонитрил,
5-[6-Хлор-3-(7-циано-1Н-пиразоло[3,4-с]пиридин-3-илметил)-2-фторфенокси]изофталонитрил,
5-[6-Этил-2-фтор-3-(1Н-пиразоло[3,4-b]пиридин-3-илметил)фенокси]-изофталонитрил,
3-Хлор-5-[6-хлор-2-фтор-3-(1H-пиразоло[3,4-b]пиридин-3-илметил)-фенокси]бензонитрил,
3-[6-Бром-2-фтор-3-(7-метил-6-оксо-6,7-дигидро-1Н-пиразоло[3,4-b]пиридин-3-илметил)фенокси]-5-хлорбензонитрил,
Трифторацетат 3-[6-бром-2-фтор-3-(7-метил-6-оксо-6,7-дигидро-1Н-пиразоло[3,4-b]пиридин-3-илметил)фенокси]-5-хлорбензонитрила, 3-[6-Бром-2-фтор-3-(1Н-пиразоло[3,4-с]пиридазин-3-илметил)фенокси]-5-хлорбензонитрил,
3-[6-Бром-2-фтор-3-(6-метил-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-илметил)фенокси]-5-хлорбензонитрил,
Трифторацетат 3-[6-бром-2-фтор-3-(6-метил-7-оксо-4,5,6,7-тетрагидро-1Н-пиразоло[3,4-с]пиридин-3-илметил)фенокси]-5-хлорбензонитрила,
Трифторацетат 5-[6-бром-2-фтор-3-(1Н-пиразоло[3,4-с]пиридазин-3-илметил)фенокси]изофталонитрила,
3-[6-Бром-2-фтор-3-(1Н-пиразоло[3,4-b]пиразин-3-илметил)фенокси]-5-хлорбензонитрил,
5-[6-Этил-2-фтор-3-(1Н-пиразоло[3,4-с]пиридазин-3-илметил)фенокси]-изофталонитрил,
3-Хлор-5-[6-этил-2-фтор-3-(1Н-пиразоло[3,4-с]пиридазин-3-илметил)-фенокси]бензонитрил,
3-Хлор-5-[6-циклопропил-2-фтор-3-(1Н-пиразоло[3,4-с]пиридазин-3-илметил)фенокси]бензонитрил,
5-[6-Циклопропил-2-фтор-3-(1Н-пиразоло[3,4-с]пиридазин-3-илметил)-фенокси]изофталонитрил,
3-Хлор-5-[2-фтор-6-метокси-3-(1Н-пиразоло[3,4-с]пиридазин-3-илметил)-фенокси]бензонитрил,
5-[6-Хлор-2-фтор-3-(1Н-пиразоло[3,4-с]пиридазин-3-илметил)фенокси]-изофталонитрил,
3-Хлор-5-[6-дифторметил-2-фтор-3-(1Н-пиразоло[3,4-с]пиридазин-3-илметил)фенокси]бензонитрил,
3-[6-Хлор-2-фтор-3-(1Н-пиразоло[3,4-с]пиридазин-3-илметил)фенокси]-5-метилбензонитрил,
3-[6-Хлор-2-фтор-3-(1Н-пиразоло[3,4-с]пиридазин-3-илметил)фенокси]-5-дифторметилбензонитрил, или
3-Хлор-5-[2-фтор-6-метансульфонил-3-(1Н-пиразоло[3,4-с]пиридазин-3-илметил)фенокси]бензонитрил.
21. Соединение по одному из пп.1-20 для применения в качестве лекарственного средства, обладающего ингибирующей активностью в отношении обратной транскриптазы ВИЧ.
22. Фармацевтическая композиция, обладающая ингибирующей активностью в отношении обратной транскриптазы ВИЧ, включающаяся соединение по одному из пп.1-20 в терапевтически эффективном количестве и по крайней мере один носитель, наполнитель или разбавитель.
| RU 2005128832, 27.04.2006 | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛА ДЛЯ ЛЕЧЕНИЯ ВИРУСНЫХ ЗАБОЛЕВАНИЙ | 2001 |
|
RU2270832C2 |

