Область техники, к которой относится изобретение
Настоящее изобретение относится к производному бензимидазола, более конкретно к производному бензимидазола, используемому в качестве ингибитора активности человеческой химазы.
Уровень техники
Химаза представляет собой нейтральную протеазу, присутствующую в гранулах мастоцитов и вовлекается в различные биологические реакции, в которых участвуют мастоциты. Так например, имеются сообщения о том, что химаза выполняет разнообразные функции, включающие промотирование (стимулирование) дегрануляции мастоцитов, активацию интерлейкина-1β (IL-1β), активацию матричной протеазы, разложение фибронектина и коллагена IV, промотирование выделения трансформирующего ростового фактора-β (TGF-β), активацию субстанции Р и вазоактивного кишечного полипептида (VIP), конверсию ангиотензина I (Ang I) в ангиотензин II (Ang II) и конверсию эндотелина.
На основании вышесказанного, ингибиторы указанной химазной активности рассматриваются, как многообещающие профилактические и/или терапевтические агенты для лечения таких респираторных заболеваний, как бронхиальная астма, таких воспалительных и аллергических заболеваний, как аллергический ринит, атопический дерматит и крапивница, таких сердечно-сосудистых заболеваний, как склеротические сосудистые поражения, вазоконстрикция, нарушения периферического кровообращения, почечная и сердечная недостаточность, таких метаболических заболеваний костей и хрящей, как ревматоидный артрит и остеоартрит.
Хотя известные примеры ингибиторов химазной активности включают производные триазина (не прошедшая экспертизы опубликованная заявка на патент Японии №8-208654), производные гидантоина (не прошедшая экспертизы опубликованная заявка на патент Японии №9-31061), производное имидазолидина (Международная патентная публикация № WO 96/04248), производное хиназолина (Международная патентная публикация № WO 97/11941), производное гетероциклического амида ((Международная патентная публикация № WO 96/33974), производное цефама (не прошедшая экспертизы опубликованная заявка на патент Японии №10-087493), производное фенола (не прошедшая экспертизы опубликованная заявка на патент Японии №10-087567), производное гетероциклического амида (Международная патентная публикация № WO 98/18794), производное ацетоамида (Международная патентная публикация №WO 98/09949), производное гетероциклического амида (не прошедшая экспертизы опубликованная заявка на патент Японии №10-007661), производное ангидрида кислоты (не прошедшая экспертизы опубликованная заявка на патент Японии № 11-049739), производное гетероциклического амида (Международная публикация № WO 99/32459) и производное ацетоамида (Международная патентная публикация № WO 99/41277), перечисленные соединения и соединение настоящего изобретения имеют совершенно различные структуры.
Описанные ингибиторы химазы далеки от успешного применения в результате того, что они обладают недостаточной активностью или имеют неустойчивую структуру. Однако соединение настоящего изобретения обладает чрезвычайно высокой активностью и демонстрирует превосходные кинетические свойства в крови, что делает его очень ценным лекарственным средством.
С другой стороны, пример технологии, относящейся к соединению настоящего изобретения, приведен в описании патента США №5124336. В указанном описании раскрывается производное бензимидазола, как соединение, обладающее антагонистической активностью в отношении тромбоксанового рецептора. Однако соединение, раскрытое в цитированном описании, не содержит гетероарильной группы, замещенной в бензимидазольном кольце, и рассматриваемый документ не содержит никаких упоминаний об активности указанного соединения в отношении человеческой химазы. Кроме этого, хотя производное бензимидазола описано, как противоопухолевый агент в не прошедшей экспертизу опубликованной заявке на патент Японии №01-265089, в цитированном документе не содержится упоминаний об ингибирующей активности рассматриваемого соединения в отношение химазной активности.
Раскрытие сущности изобретения
Цель настоящего изобретения состоит в разработке нового соединения, обладающего свойствами ингибитора активности человеческой химазы, который может применяться в клинических условиях.
В результате широких и ранее проведенных исследований, направленных на выполнение поставленной задачи, авторы настоящего изобретения разработали производное бензимидазола или его медицински применимую соль, отвечающие следующей ниже общей формуле (1), имеющие совершенно отличную структуру от известных соединений и, тем самым, создали настоящее изобретение:
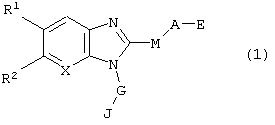
в которой R1 и R2 могут иметь одинаковые или различные значения и, независимо друг от друга, представляют собой атом водорода, атом галогена, тригалометильную группу, циано группу, гидроксильную группу, алкильную группу, содержащую 1-4 углеродных атома, алкокси группу, содержащую 1-4 углеродных атома, или R1 и R2 вместе представляют группы -O-СН2-O-, -O-СН2СН2-O-, или -СН2СН2CH2- (причем указанные группы, могут быть замешены одной или более алкильными группами, содержащими 1-4 углеродных атома);
А представляет собой замещенную или незамещенную, линейную, циклическую или разветвленную алкиленовую или алкениленовую группу, содержащую 1-7 углеродных атомов, которые могут чередоваться с одной или более группами -О-, -S-, -SO2-, и NR3- (где R3 представляет собой атом водорода или линейную/разветвленную алкильную группу, содержащую 1-6 углеродных атомов); заместитель, который может содержаться в указанных группах, выбирают из атома галогена, гидроксильной группы, нитро группы, циано группы, линейной или разветвленной алкильной группы, содержащей 1-6 углеродных атомов, линейной или разветвленной алкокси группы, содержащей 1-6 углеродных атомов (включая случай, когда две соседних группы образуют ацетальную связь, а именно, тот случай, когда алкильные части двух геминальных алкокси групп соединяются друг с другом с образованием кольца), линейной или разветвленной алкилтио группы, содержащей 1-6 углеродных атомов, линейной или разветвленной алкилсульфонильной группы, содержащей 1-6 углеродных атомов, линейной или разветвленной ацильной группы, содержащей 1-6 углеродных атомов, линейной или разветвленной ациламино группы, содержащей 1-6 углеродных атомов, тригалометильной группы, тригалометокси группы, фенильной группы, оксогруппы, и феноки группы, которая может быть замещена одним или более атомами галогена; причем один или более из перечисленных заместителей могут, независимо друг от друга, соединяться по произвольными положениями алкиленовой или алкениленовой групп, при условии, что исключается случай, когда М представляет собой простую связь, а гидроксильная и фенольная группы одновременно связаны, в качестве заместителей, с теми углеродными атомами фрагмента А, которые связаны с М;
Е представляет собой группы -COOR3, -SO3R3, -CONHR3, -SO2NHR3, тетразол-5-ильную группу, 5-оксо-1,2,4-оксадиазол-3-ильную группу или 5-оксо-1,2,4-тиадиазол-3-ильную группу (где R3 имеет указанные выше значения);
G представляет собой замещенную или незамещенную, линейную или разветвленную алкиленовую группу, содержащую 1-6 углеродных атомов, которые могут чередоваться с одним или более -О-, -S-, -SO2- и NR3- (где R3 имеет указанные выше значения. При наличии указанных атомов или групп атомов, они не находятся в непосредственном соединении с бензимидазольным кольцом); причем заместитель на указанной алкиленовой группе выбирают из атома галогена, гидроксильной группы, нитро группы, циано группы, линейной или разветвленной группы, содержащей 1-6 углеродных атомов, линейной или разветвленной алкокоси группы, содержащей 1-6 углеродных атомов (включая случай, когда две соседние группы образуют ацетальную связь), тригалометильной группы, тригалометокси группы, фенильной группы, и оксо группы;
М представляет собой простую связь или группу -S(O)m-, где m представляет собой целое число в интервале 0-2;
J представляет собой замещенную или незамещенную гетероциклическую группу, содержащую 4-10 углеродных атомов и один или более гетероатомов в кольце, выбранных из группы, состоящей из атома кислорода, атома азота или атома серы, при условии исключения имидазольного кольца и незамещенного пиридинового кольца; заместитель в указанной ароматической гетероциклической группе выбирают из атома галогена, гидроксильной группы, нитро группы, циано группы, линейной или разветвленной алкильной группы, содержащей 1-6 углеродных атомов, линейной или разветвленной алкокси группы, содержащей 1-6 углеродных атомов (включая случай, когда две соседних группы образуют ацетальную связь), линейную или разветвленную алкилтио группу, содержащую 1-6 углеродных атомов, линейную или разветвленную алкилсульфонильную группу, содержащую 1-6 углеродных атомов, линейную или разветвленную ацильную группу, содержащую 1-6 углеродных атомов, линейную или разветвленную ациламино группу, содержащую 1-6 углеродных атомов, линейную или разветвленную анилидную группу, тригалометильную группу, тригалометокси группу, фенильную группу, или оксо группу, группу COOR3, и фенокси группу, которая может быть замещена одним или более атомами галогена; причем один или более из указанных заместителей могут быть замещены по произвольным позициям кольца; и
Х представляет собой метиновую группу (-СН=) или атом азота.
Предпочтительные варианты реализации изобретения
Заместители в соединении настоящего изобретения, представленном приведенной выше формулой (1) имеют значения, указанные ниже.
R1 и R2 могут иметь одинаковые или различные значения и, независимо друг от друга, представляют собой атом водорода, атом галогена, тригалометильную группу, циано группу, гидроксильную группу, алкильную группу, содержащую 1-4 углеродных атома, алкокси группу, содержащую 1-4 углеродных атома. С другой стороны, R1 и R2 вместе представляют собой группы -O-СН2-O-, -O-СН2СН2-O-, или -CH2СН2СН2-, и в этом случае, указанные группы могут быть замещены одной или более алкильными группами, содержащими 1-4 углеродных атома.
Конкретными примерами указанных алкильных групп, содержащих 1-4 углеродных атома, в качестве R1 и R2, могут служить метальная группа, этильная группа, н- или изо-пропильная группа, а также н-, изо-, втор- или третбутильная группа. Предпочтительным примером является метильная группа. Конкретными примерами алкокси групп, содержащих 1-4 углеродных атома, могут служить метокси группа, этокси группа, н- или изопропокси группа, а также н-, изо-, втор- или трет-бутокси группа.
Предпочтительные примеры R1 и R2 включают атом водорода, атом галогена, тригалометильную группу, циано группу, гидроксильную группу, алкильную группу, содержащую 1-4 углеродных атома, и алкокси группу, содержащую 1-4 углеродных атома. Более предпочтительные примеры включают атом водорода, атом галогена, тригалометильную группу, циано группу, алкильную группу, содержащую 1-4 углеродных атома и алкокси группу, содержащую 1-4 углеродных атома, еще более предпочтительные примеры включают атом водорода, атом хлора, атом фтора, трифторметильную группу, метильную группу, метокси группу и этокси группу, тогда как особенно предпочтительные примеры включают атом водорода, метильную группу и метокси группу.
А представляет собой замещенную или незамещенную, линейную циклическую или разветвленную алкиленовую или алкениленовую группу, содержащую 1-7 углеродных атома. Примерами незамещенной, линейной, циклической или разветвленной алкиленовой группы, содержащей 1-7 углеродных атомов, могут служить метиленовая группа, этиленовая группа, н-, или изопропиленовая группа, 2,2-диметилпропиленовая группа, н-, изо-, или трет-бутиленовая группа, 1,1-диметалбутиленовая группа, н-пентиленовая группа и циклогексиленовая группа. Более предпочтительные примеры включают этиленовую группу, н-пропиленовую группу, 2,2-диметилпропиленовую группу и н- или трет-бутиленовую группу. Еще более предпочтительные примеры включают н-пропиленовую группу и 2,2-диметилпропиленовую группу. Особенно предпочтительным примером может служить н-пропиленовая группа. Примеры незамещенной линейной или разветвленной алкиленовой группы, содержащей 1-7 углеродных атомов, включают виниленовую группу, пропениленовую группу, бутениленовую группу, и пентениленовую группу.
Хотя указанная алкиленовая группа или алкениленовая группа могут чередоваться с одним или более -O-, -S-, -SO2- и =NR3- (где R3 представляет собой атом водорода либо линейную или разветвленную алкильную группу, содержащую 1-6 углеродных атомов), указанные атомы или группы атомов непосредственно не связаны с М. Специальные примеры включают чередующиеся этиленовые группы, пропиленовые группы или н-, или трет-бутиленовые группы. Более конкретные примеры включают -СН2OCH2-, -СН2OCH2СН2-, -CH2SCH2-, -CH2SCH2CH2-, -CH2SO2CH2-, -CH2SO2CH2CH2-, CH2NR4CH2-, и -CH2NR4CH2CH2-. Предпочтительные примеры включают -CH2OCH2-, -CH2SCH2-, И -CH2SO2CH2-.
Возможный заместитель в указанной алкиленовой группе выбирают из атома галогена, гидроксильной группы, нитро группы, циано группы, линейной или разветвленной алкильной группы, содержащей 1-6 углеродных атомов, линейной или разветвленной алкокси группы, содержащей 1-6 углеродных атомов (включая случай, когда две соседние группы образуют ацетальную связь), линейной или разветвленной алкилтиогруппы, содержащей 1-6 углеродных атомов, линейной или разветвленной алкилсульфонильной группы, содержащей 1-6 углеродных атомов, линейной или разветвленной ацильной группы, содержащей 1-6 углеродных атомов, линейной или разветвленной ациламиногруппы, содержащей 1-6 углеродных атомов, тригалометильной группы, тригалометокси группы, фенильной группы, оксогруппы, и феноксигруппы, которая может быть замещена одним или более атомами галогена. Один или более из указанных заместителей могут, независимо друг от друга, быть связаны по произвольным положениям алкиленовой группы или алкениленовой группы, при условии, что исключен случай, когда М представляет собой простую связь, а гидроксильная группа и фенильная группа одновременно связаны в качестве заместителей с теми атомами углерода в А, которые связаны с М.
Примерами атомов галогена могут служить атом фтора, атом хлора, атом брома и атом иода. Предпочтительными примерами являются атом фтора и атом хлора.
Конкретные примеры линейной или разветвленной алкильной группы, содержащей 1-6 углеродных атомов, включают метильную группу, этильную группу, н- или изопропильную группу, а также н-, изо-, втор-, или трет-бутильную группу, тогда как предпочтительными примерами могут служить метильная группа и этильная группа. Более предпочтительным примером является метильная группа.
Конкретные примеры линейной или разветвленной алкокси группы, содержащей 1-6 углеродных атомов, включают метокси группу, этокси группу, н-, или изопропокси группу, а также н-, изо-, втор- или трет-бутоксигруппу, тогда как предпочтительные примеры включают метокси группу и этокси группу. Еще более предпочтительным примером может служить метокси группа.
Конкретные примеры линейной или разветвленной алкилтиогруппы, содержащей 1-6 углеродных атомов, включают метилтио группу, этилтио группу, н-, или изопропилтио группу, а также н-, изо-, втор- или трет-бутилтиогруппу, а предпочтительные примеры представляют собой метилтио группу и этилтио группу. Более предпочтительным примером может служить метилтио группа.
Конкретными примерами линейной или разветвленной алкилсульфонильной группы, содержащей 1-6 углеродных атомов, могут служить метилсульфонильная группа, этилсульфонильная группа, н- или изопропилсульфонильная группа, а также н-, изо-, втор- или трет-бутилсульфонильная группа, а предпочтительными примерами могут служить метилсульфонильная группа и этилсульфонильная группа. Более предпочтительный пример представляет собой метилсульфонильную группу.
Примерами линейной или разветвленной ацильной группы, содержащей 1-6 углеродных атомов, могут служить ацетильная группа, ацетилкарбонильная группа, н- или изопропилкарбонильная группа и н-, изо-, втор- или трет-бутилкарбонильная группа, а предпочтительными примерами являются ацетильная группа и этилкарбонильная группа.
Более предпочтительным примером может служить ацетильная группа.
Конкретные примеры линейной или разветвленной ациламино группы, содержащей 1-6 углеродных атомов, включают ацетиламино группу, этилкарбониламино группу, н- или изопропилкарбониламино группу, а также н-, изо-, втор- или трет-бутилкарбониламино группу, а предпочтительными примерами могут служить ацетиламино группа и этилкарбониламино группа. Более предпочтительным примером является ацетиламино группа.
Конкретными примерами тригалометильной группы являются трифторметильная группа, трибромметильная группа и трихлорметильная группа. Предпочтительным примером является трифторметильная группа.
В частности А, предпочтительно, представляет собой замещенную или незамещенную, линейную, циклическую или разветвленную алкиленовую группу, содержащую 1-7 углеродных атомов {хотя они могут чередоваться с одним или более из -О-, -S-, -SO2-, и -NR3- (где NR3 имеет указанные выше значения), указанные атомы или группы атомов непосредственно не связаны с М}. Предпочтительные примеры включают -СН2СН2-, -СН2СН2СН2-, -СН2С(=O)СН2-, -CH2OCH2-, -CH2SCH2, -CH2S(=O)CH2-, -CH2CF2CH2-, -CH2SO2CH2-, -СН2СН2СН2СН2-, -СН2С(СН3)2СН2-, -CH2SO2CH2CH2-, -СН2С(=O)СН2СН2, -СН2С(=O)(СН3)2СН2-, и -СН2С(=O)С(=O)СН2-. Более предпочтительными примерами могут служить -СН2СН2-, -СН2СН2СН2-, -СН2С(=O)СН2-, -СН2OCH2-, -CH2SCH2, -CH2S(=O)CH2-, -CH2CF2CH2-, -CH2SO2CH2-, и -СН2С(СН3)2СН2-. Еще более предпочтительными примерами могут служить -СН2СН2-, -СН2СН2СН2-, и -СН2С(СН3)2СН2-. Особенно предпочтительным примером является -СН2СН2СН2-.
Е представляет собой -COOR3, -SO3R3, -CONHR3, -SO2NHR3, тетразол-5-ил, 5-оксо-1,2,4-оксодиазол-3-ил или 5-оксо-1,2,4-тиадиазол-3-ильную группу (где R3 представляет собой атом водорода либо линейную или разветвленную алкильную группу, содержащую 1-6 углеродных атомов).
Примерами R3 могут служить атом водорода, метильная группа, этильная группа, н- или изопропильная группа, а также н-, изо-, втор- или трет-бутильная группа. Предпочтительными примерами являются атом водорода, метильная группа и этильная группа. Особенно предпочтительный пример представляет собой атом водорода.
В частности, предпочтительными примерами Е могут служить OCOOR3, -SO2R3, а также тетразол-5-ильная группа. Более предпочтительным примером является группа -COOR3. Особенно предпочтительным примером может служить группа =СООН.
G представляет собой замещенную или незамещенную, линейную или разветвленную алкиленовую группу, содержащую 1-6 углеродных атомов, которые могут чередоваться с одним или более из -О-, -S-, -SO2-, и -NR3. Где NR3 имеет указанные выше значения. В случае наличия указанных гетероатомов или атомных групп, они непосредственно не связаны с бензимидазольным кольцом. Заместители в алкиленовой группе выбирают из атома галогена, гидроксильной группы, нитро группы, циано группы, линейной или разветвленной алкильной группы, содержащей 1-6 углеродных атомов, линейной или разветвленной алкокси группы, содержащей 1-6 углеродных атомов (включая случай, когда две соседние группы образуют ацетальную связь), тригалометильной группы, тригалометоксигруппы, фенильной группы и оксогруппы. Конкретные примеры G включают -СН2-, -СН2СН2-, -СН2СО-, СН2СН2O-, -CH2CONH-, -СО-, -SO2-, -CH2SO2-, -CH2S-, и -CH2CH2S-, тогда как предпочтительные примеры включают -СН2-, -СН2СН2-, -СН2СО- и -СН2СН2O-. Более предпочтительные примеры представляют собой -СН2- и -СН2СН2-, и особенно предпочтительный пример включает -СН2-. Указанные группы связаны с положением слева от атома 1 (атом N) бензимидазольного кольца, и справа относительно J.
М представляет собой простую связь или группу -S(O)m, где m представляет собой целое число в интервале 0-2. Предпочтительными примерами М являются -S- и -SO2-. Особенно предпочтительный пример представляет собой -S-.
J представляет собой замещенную или незамещенную гетероциклическую группу, содержащую 1-4 углеродных атома и один или более кольцевых гетероатомов, выбранных из группы, состоящей из атома кислорода, атома азота и атома серы. Однако исключаются имидазольное кольцо и незамещенное пиридиновое кольцо. Кроме этого, J ограничено теми группами, которые могут быть синтезированы химическим методом.
Конкретные примеры незамещенных гетероциклических групп, содержащих 4-10 углеродных атомов и один или более кольцевых гетероатомов, выбранных из группы, состоящей из атома кислорода, атома азота и атома серы, включают фурильную группу, тиенильную группу, тиазолильную группу, пиримидинильную группу, оксазолильную группу, изоксазолильную группу, бензофурильную группу, бензимидазолильную группу, хинолильную группу, изохинолильную группу, хиноксалинильную группу, бензоксадиазолильную группу, бензотиазиазолильную группу, индолильную группу, бензотиазолильную группу, бензотиенильную группу и бензоизооксазолильную группу. Предпочтительный пример представляет собой бициклическое гетероциклическое кольцо. Более предпочтительными примерами могут служить бензофурильная группа, бензоимидазолильная группа, хинолильная группа, изохинолильная группа, хиноксалинильная группа, бензоксадиазолильная группа, бензотиазолильная группа, индолильная группа, бензотиазолильная группа, бензотиенильная группа и бензоизоксазолильная группа, тогда как особенно предпочтительный пример представляет собой бензотиенильную группу или индолильную группу.
Заместитель для ароматической гетероциклической группы выбирают из атома галогена, гидроксильной группы, нитро группы, циано группы, линейной или разветвленной алкильной группы, содержащей 1-6 углеродных атомов, линейной или разветвленной алкокси группы, содержащей 1-6 углеродных атомов (включая случай, когда две соседних группы образуют ацетальную связь), линейной или разветвленной алкилтио группы, содержащей 1-6 углеродных атомов, линейной или разветвленной алкилсульфонильной группы, содержащей 1-6 углеродных атомов, линейной или разветвленной ацильной группы, содержащей 1-6 углеродных атомов, линейной или разветвленной ациламино группы, содержащей 1-6 углеродных атомов, замещенной или незамещенной анилидной группы, тригалометильной группы, тригалометоксигруппы, фенильной группы, и фенокси группы, которая может быть замещена одним или более атомами галогена. Одна или более из указанных замещающих групп, независимо друг от друга, может быть связана с произвольными позициями кольца.
Примерами атомов галогена может служить атом фтора, атом хлора, атом брома и атом иода. Предпочтительным примером является атом фтора и атом хлора.
Конкретные примеры линейной или разветвленной алкильной группы, содержащей 1-6 углеродных атомов, включают метильную группу, этильную группу, н- или изопропильную группу, а также н-, изо-, втор- или трет-бутильную группу, а предпочтительные примеры представляют собой метильную группу и этильную группу. Более предпочтительным примером может служить метильная группа.
Конкретными примерами линейных или разветвленных алкокси групп, содержащих 1-6 углеродных атомов, могут служить метокси группа, этокси группа, н- или изопропилоксигруппа, н-, изо-, втор- или трет-бутилокси группа, а предпочтительными примерами могут служить метокси группа и этокси группа. Более предпочтительным примером является метокси группа.
Конкретные примеры линейной или разветвленной алкилтио группы, содержащей 1-6 углеродных атомов, включают метилтио группу, этилтио группу, н- или изопропилтио группу, а также н-, изо-, втор- или трет-бутилтио группу, а предпочтительные примеры включают метилтио группу и этилтио группу. Более предпочтительным примером служит метилтио группа.
Конкретные примеры линейной или разветвленной алкилсульфонильной группы, содержащей 1-6 углеродных атомов, включают метилсульфонильную группу, этилсульфонильную группу, н- или изопропилсульфонильную группу, а также н-, изо-, втор- или трет-бутилсульфонильную группу, а предпочтительные примеры включают метилсульфонильную группу и этилсульфонильную группу. Более предпочтительным примером служит метилсульфонильная группа.
Конкретные примеры линейной или разветвленной ацильной группы, содержащей 1-6 углеродных атомов, включают ацетильную группу, этилкарбонильную группу, н- или изопропилкарбонильную группу, а также н-, изо-, втор- или трет-бутилкарбонильную группу, а предпочтительные примеры включают ацетильную и этилкарбонильную группу. Более предпочтительным примером служит ацетильная группа.
Конкретные примеры линейной или разветвленной ациламино группы, содержащей 1-6 углеродных атомов, включают ацетиламино группу, этилкарбониламиногруппу, н- или изопропилкарбониламино группу, а также н-, изо-, втор- или трет-бутилкарбониламино группу, а предпочтительные примеры включают ацетиламино группу и этилкарбониламино группу. Более предпочтительным примером служит ацетиламино группа.
Конкретные примеры тригалометильной группы включают трифторметильную группу, трибромметильную группу и трихлорметильную группу.
Х представляет собой -СН= группу или атом азота, причем предпочтительным примером является -СН= группа.
Предпочтительные примеры соединений, представленных указанной выше формулой (1), включают различные группы соединений, составленные из комбинаций каждой из групп, предварительно описанных в качестве предпочтительных примеров.
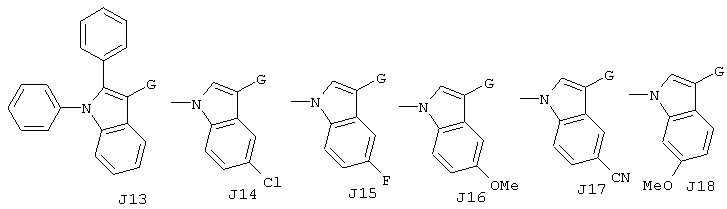
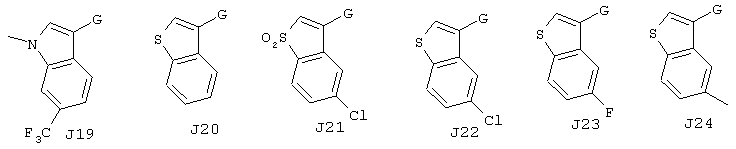
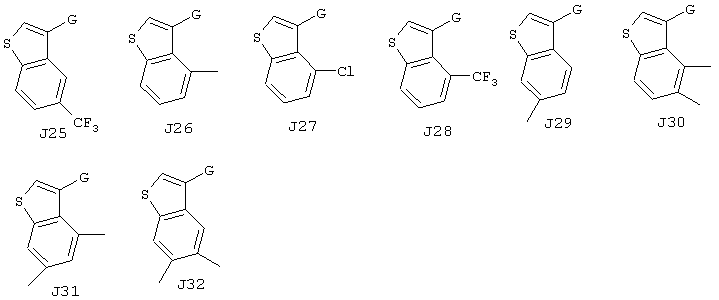
Хотя авторы не имеют намерения ограничивать указанные группы, те из них, что представлены в таблице 1 являются особенно предпочтительными. Особенно предпочтительные примеры таких соединений в таблице 1 включают соединения №№34, 38, 39, 41, 42, 52, 54, 56, 58, 59, 63, 135, 137, 148, 152, 154, 244, 340, 436, 514, 519, 521, 532, 534, 536, 538, 615, 628, 1112 и 1114.
Кроме этого, А1-А3 и J1-J32 в таблице 1 представляют собой группы, отвечающие следующим ниже формулам. Хотя Е, G, M, m, X имеют указанные выше значения, в представленных ниже формулах даны репрезентативные примеры указанных фрагментов, а именно, Е представляет собой СООН, G представляет собой СН2, М представляет собой S (при m, равном 0), или простую связь (обозначенную в таблице, как "-"), а Х представляет собой группу -СН=. Однако настоящее изобретение не ограничивается рассматриваемыми соединениями.
В том случае, когда Е представляет собой COOR3, а М представляет собой S, производное бензимидазола (1) настоящего изобретения может быть получено по способу синтеза (А) или способу синтеза (В), которые описаны ниже:
Способ синтеза (А)

где Z представляет собой галоген или группу аммония, a R1, R2, R3, A, G, J и Х имеют указанные выше значения.
Производное ортофенилендиамина (а2) получают восстановлением нитро группы производного 2-нитроанилина (а1). После реакции с CS2 и получения соединения (а3), это соединение реагирует с галогенидным производным сложного эфира (а4) с получением (а5), после чего проводят реакцию с галогенидным производным или солью аммония (а6) с получением соединения (а7) настоящего изобретения. Если необходимо, то путем гидролиза может быть получено производное безимидазола (а8), в котором R3 представляет собой атом водорода.
Восстановление нитрогруппы можно осуществлять в соответствие с условиями обычного каталитического восстановления, в реакции с газообразным водородом при температуре в интервале от комнатной до 100°С, в присутствии такого катализатора, как Pd-C в кислых, нейтральных или щелочных условиях. Кроме этого, реакцию также можно осуществлять по способу, в котором проводят обработку цинком или оловом в кислых условиях, или по методу, в котором используют цинковый порошок в нейтральных или щелочных условиях.
Реакцию между производным ортофенилендиамина (а2) и CS2 можно проводить согласно способу, например, описанному в The Journal of organic chemistry (J.Org. Chem.), 1954, т.19, стр.631-637 (пиридиновый раствор), или в The Journal of medical chemistry (J.Med.Chem.), 1993, т.36, стр.1175-1187 (этанольный раствор).
Реакцию между производным тиобензимидазола (а3) и галоидным эфиром (а4) можно проводить путем перемешивания при температуре 0-200°С в присутствии такого основания, как NaH, Et3N, NaOH или К2СО3 в соответствие с условиями обычной реакции S-алкилирования.
Реакцию между производным тиобензимидазола (а5) и галоидным производным или солью аммония (а6) можно проводить путем перемешивания при температуре 0-200°С в присутствии такого основания, как NaH, Et3N, NaOH, К2СО3 или Ca2СО3 в соответствие с условиями обычной реакции N-алкилирования или N-ацилирования.
Реакцию удаления карбокси-защитной группы R3 предпочтительно проводить с применением способа гидролиза, в котором используют такую щелочь, как гидроксид лития или такую кислоту, как хлористоводородная или трифторуксусная кислота.
Способ синтеза (В)
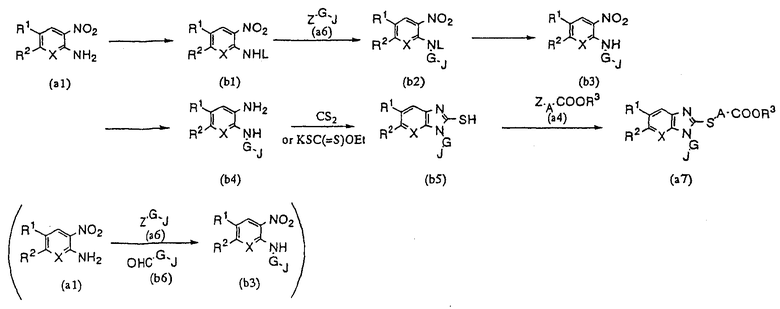
Согласно представленной схеме, (b1) получают путем защиты аминогруппы производного 2-нитроанилина (а1) с помощью подходящей защитной группы L. Затем это вещество реагирует с галоидным производным или солью аммония (а6) с образованием (b2), а (b3) получают удалением защитной группы L. Производное ортофенилендиамина (b4) получают восстановлением нитро группы соединения (b3). После реакции этого соединения с CS2 или KCS(=S)Oet и получения соединения (b5), последнее соединение реагирует с производным галоидного сложного эфира (а4) с целью получения производного бензимидазола (а7) настоящего изобретения. Полученное соединение, по необходимости, может быть подвергнуто гидролизу с образованием производного бензимидазола настоящего изобретения, в котором R3 представляет собой атом водорода.
Соединение (b3) также может быть непосредственно получено в реакции незащищенного галоидного производного, соли аммония (а6) или производного альдегида (b6) с производным 2-нитроанилина (а1). Примеры защитной группы L включают трифторацетильную группу, ацетильную группу, трет-бутоксикарбонильную группу и бензильную группу. Реакцию между производным 2-нитроанилина (а1) и производным альдегида (b6) можно осуществлять, как обычное восстановительное аминирование при температуре 0-200°С в среде такого растворителя, как этанол, метанол или дихлорметан, с использованием таких соединений с большим числом атомов водорода, как LiAlH4, NaBH4, NaBH3CN или NaBH(OAc)3, или такого восстанавливающего агента, как диборан. Реакцию между производным ортофенилендиамина (b4) и CS2 можно проводить тем же методом, что в способе синтеза (А), тогда как реакцию с KCS(=S)Oet можно проводить по методике, описанной, например, в Organic Synthesis (OS), 1963, т.4, стр.569-570. Другие реакции могут осуществляться так же, как в способе синтеза (А).
В том случае, когда Е представляет собой COOR3, М представляет собой S, a G представляет собой амидную связь, производное бензимидазола (1) настоящего изобретения может быть получено согласно следующему способу синтеза (С):
Способ синтеза (С):
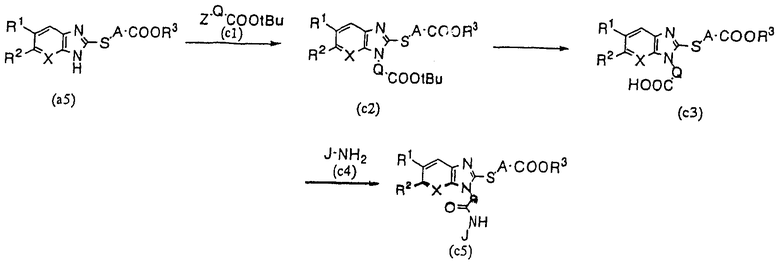
где Q представляет собой метиленовую группу, фениленовую группу, и т.п., Z представляет собой галоген, a R1, R2, R3, A, J и Х имеют указанные выше значения, при условии, что R3 в кислотных условиях представляет собой такую неактивную защитную группу, как метильная группа или этильная группа.
Соединение (с2) получают по реакции производного тиобензимидазола (а5) с галоидным производным трет-бутилового эфира (с1). Затем это соединение подвергают гидролизу в кислых условиях с получением (с3). Последнее соединение подвергают конденсации с производным амина (с4) с целью получения соединения (с5) настоящего изобретения. Это вещество, в случае необходимости, может быть подвергнуто гидролизу с целью получения производного бензимидазола настоящего изобретения, в котором R3 представляет собой атом водорода.
При осуществлении конденсационного аминирования использовали обычный способ с применением конденсирующего агента. Примерами конденсирующих агентов могут служить DCC, DIPC, TDC=WSCl, WSCl·HCl, ВОР и DPPA, причем перечисленные соединения могут использоваться по отдельности, или в комбинации с HONSu, HOBt или HOOBt. Реакцию проводят при температуре 0-200°С в среде такого подходящего растворителя, как ТГФ, хлороформ или третбутанол. Другие реакции могут проводиться так же, как и в способе синтеза (А).
В том случае, когда Е представляет собой COOR3, М представляет собой S, а G содержит простую эфирную связь, производное бензимидазола (1) настоящего изобретения может быть получено согласно следующему способу синтеза (D):
Способ синтеза (D):
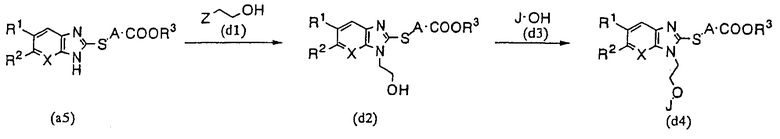
где Z представляет собой галоген, a R1, R2, R3, A, J и Х имеют указанные выше значения.
Соединение (d2) получают в реакции, например, галоген производного спирта (d1) с тиобензимидазольным соединениям (а5). Далее, это соединение реагирует с производным фенола (d3) с целью получения соединения (d4) настоящего изобретения. Если необходимо, то полученное соединение может быть подвергнуто гидролизу с образованием производного бензимидазола, в котором R3 представляет собой атом водорода.
Реакцию этерификации проводят по методике Mitsunobu и аналогичных реакций при температуре 0-200°С, в среде такого подходящего растворителя, как N-метилморфолин или ТГФ, с использованием такого производного фосфина, как трифенилфосфин или трибутилфосфин и такого азосоединения, как DEAD или TMAD. Другие реакции могут быть осуществлены так же, как способе синтеза (А).
В том случае, когда Е представляет собой тетразол-5-ил, а М представляет собой S, производное бензимидазола (1) настоящего изобретения может быть получено согласно следующему способу синтеза (Е):
Способ синтеза (Е):

где R1, R2, R3, A, G, J и X имеют указанные выше значения.
Нитрильную форму (е1) превращают в тетразольную форму (е2) по реакции с различными производными азида. Примерами азидных производных могут служить такие производные азида триалкилолова, как азид триметилолова, азотистоводородная кислота и ее аммониевые соли. При использовании органического производного азида олова, должно использоваться строго 1-4 моля в расчете на соединение (е1). Кроме этого, при использовании азотистоводородной кислоты или ее аммониевых солей, следует использовать 1-5 молей азида натрия и хлористого аммония или такого третичного амина, как триэтиламин, в расчете на соединение (е1). Каждую из указанных реакций проводят при температуре 0-200°С в присутствии такого растворителя, как толуол, бензол или ДМФ.
Производное бензимидазола (1) настоящего изобретения может быть получено согласно следующему способу синтеза (F), в том случае, когда М представляет собой SO или SO2:
Метод синтеза (F):
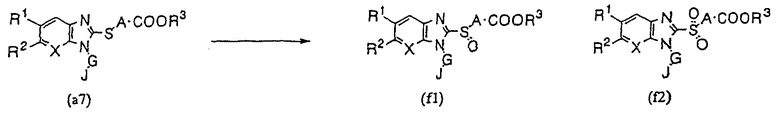
где R1, R2, R3, A, G, J и Х имеют указанные выше значения.
Сульфоксидное производное (а7) и/или сульфоновое производное (f2) получают в реакции производного бензимидазола (а7) с пероксидным производным в среде подходящего растворителя. Примерами используемого пероксидного соединения могут служить надбензойная кислота, м-хлорнадбензойная кислота, надуксусная кислота, а также пероксид водорода, тогда как примеры подходящего растворителя включают хлороформ и дихлорметан. Величина используемого соотношения между соединением (а7) и пероксидным соединением не имеет конкретных ограничений, но хотя такое соотношение может выбираться в широких пределах, обычно, предпочтительно, использовать соотношение 1,2-5 молей. Каждую реакцию обычно проводят при 0-50°С и, предпочтительно, при температуре в интервале от 0° до комнатной температуры, и каждая из рассматриваемых реакций завершается за 4-20 часов.
Производное бензимидазола (1) настоящего изобретения может быть получено в соответствие со следующим способом синтеза (G) в том случае, когда М представляет собой простую связь:
Способ синтеза (G):
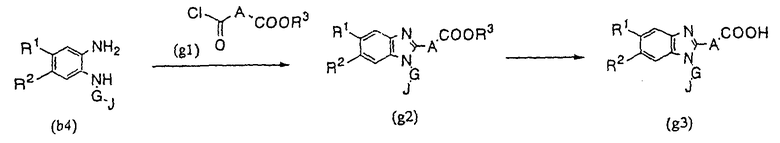
где, X, A, G, J и R3 имеют указанные выше значения.
В рассматриваемом случае, производное бензимидазола (g2) может быть получено по реакции известного производного хлорангидрида (g1) с диаминовым соединением (b4). В случае необходимости, в результате гидролиза группы - COOR3 соединения (g2) может быть получено производное бензимидазола (g3), в котором R3 представляет собой атом водорода.
Кроме этого, реакция циклизации описана в Journal of medical chemistry (J. Med. Chem.) 1993, т.36, стр.1175-1187.
Помимо этого, Z-G-J, описанные в способах синтеза (А)-(F), могут быть синтезированы различными методами, описанными в большом числе публикаций.
Так, например, галогенидное производное бензотиофена может быть синтезировано со ссылкой на следующие литературные и патентные источники.
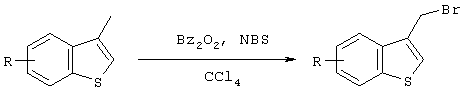
J. Chem. Soc. (1965), 774
J. Chem. Soc. Perkin Trans 1, (1972), 3011
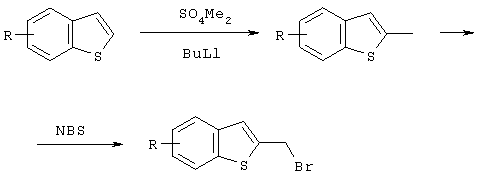
JACS, 74, 664 (1951); Патент US 4282227
Рассматриваемые соединения также могут быть синтезированы по методикам, описанным в следующих литературных источниках и патентных публикациях. Причем рассматриваемые соединения могут быть синтезированы не только в реакциях, описанных в следующей ниже литературе, но также и путем комбинирования таких типичных реакций, как окисление-восстановление или галогенирование ОН группы.
J. Chem. Soc. (1965), 774; Bull Chem. Soc. Jpn (1968), 41, 2215.
Опубликованная не прошедшая экспертизы заявка на патент Японии №10-298180; Sulfur Reports (1999), т.22,1-47; J. Chem. Soc. comm. (1988), 888. J. Heterocyclic. Chem., 19, 859; (1982); Synthetic Communication; (1991), 21, 959. Tetrahedron Letters (1992), т.33, №49, 7499; Synthetic Communication (1993), 23 (6), 743. Опубликованная не прошедшая экспертизы заявка на патент Японии 2000-239270; J. Med. Chem. (1985), 28, 1896; Arch Pharm (1975), 308, 7, 513; Khim Gerotsikl Soedin (1973), 8, 1026. Bull. Chem. Soc. Jpn. (1997), 70, 891; J. Chem. Soc. Perkin 1 (1973), 750; J. Chem. Soc. Chem. Comm. (1974), 849; J. Chem. Soc. Comm. (1972), 83.
В частности гидроксиметильная форма в положении 3 бензотиофена может быть легко синтезирована по методике, описанной в J. Chem. Soc. Chem. Comm. (1974), 849.
Что касается иодидов, то Cl- и Br-содержащие формы могут быть получены в результате галогенного обмена с NaI и т.д.
Кроме этого, четвертичная аммониевая соль бензотиофена может быть синтезирована в реакции такого подходящего амина, как диметиламин с упомянутым выше галоидным производным бензотиофена. Это вещество также может быть синтезировано следующим способом:
Способ синтеза (Н)
 где R представляет собой один или более заместителей, в вышеупомянутом J, причем их число может быть произвольным и указанные заместители могут не зависеть друг от друга.
где R представляет собой один или более заместителей, в вышеупомянутом J, причем их число может быть произвольным и указанные заместители могут не зависеть друг от друга.
Производное циклического бензотиофена (h3) получают путем превращения амино группы производного 2-нитрианилина (h1) в циано группу (h2) и реакции с этил 2-меркаптоацетатом. Карбоновую кислоту (h5) получают в реакции цианирования амино группы в цианоформу (h4) с последующим гидролизом сложного эфира. После этого, карбоновую кислоту декарбоксилируют с образованием соединения (h6). Далее, циано группу подвергают восстановлению с целью получения аминной формы (h7) с последующим N-диметилированием с образованием соединения (h8), после чего проводят N-метилирование с образованием четвертичной соли (h9).
В результате планирования амино группы производного 2-нитроанилина (hi) путем превращения амино группы в диазоний с использованием, например, хлористоводородной кислоты или сульфита натрия и последующей реакции с хлористой медью (I) и цианидом калия получают цианоформу.
Реакцию превращения цианоформы (h2) в производное бензотиофена (h3) можно проводить с целью получения производного циклического бензотиофена (h3) в результате нагревания с 2-меркаптоацетатом, в среде такого подходящего растворителя, как ДМФ, в присутствии подходящего основного реагента следуя при этом методике, описанной, например, в Synthetic Communications, 23(6), 743-748 (1993); или Farmaco, Ed. Sci., 43, 1165 (1988).
Что касается реакции цианирования соединения (h3), то (h3) может быть переведено в цианоформу (h4) в реакции цианида меди и трет-бутилсульфита в среде такого подходящего растворителя, как ДМСО при соответствующих температурных условиях.
Гидролиз сложного эфира может быть осуществлен традиционными способами. Так например, карбоновая кислота (h5) может быть получена путем гидролиза сложного эфира в таком подходящем растворителе, как ТТФ-МеОН в присутствии такого подходящего основного реагента, как гидроксид натрия.
Реакцию декарбоксилирования карбоновой кислоты можно проводить путем нагревания в таком подходящем растворителе, как хинолин в присутствии медного катализатора.
Восстановление циано группы в амино группу можно проводить, например, путем восстановления в таком подходящем растворителе, как Et2O-THF при подходящей температуре с использованием подходящего восстанавливающего агента, например, литийалюминий гидрида.
Метилирование амино группы можно проводить, например, путем нагревания в муравьиной кислоте или водном растворе формалина.
Превращение амино группы в четвертичную соль можно проводить, например, путем реакции с йодистым метилом в среде этанола.
Четвертичная аминная соль индола может быть синтезирована, например, в соответствие со следующим способом:
Способ синтеза (К)
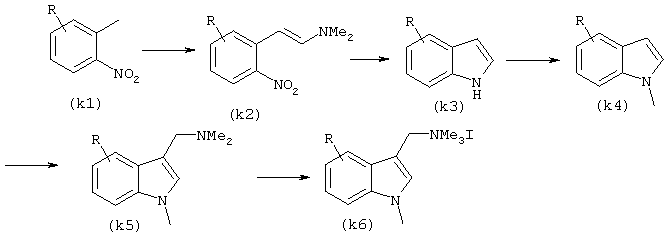
где R представляет собой один или более заместителей в указанном выше J, причем число заместителей может быть произвольным и указанные заместители могут не зависеть друг от друга.
Нитроформу (k1) превращают в енамин (k2) путем енанимирования с последующим превращением в индольную форму (k3) в результате индольной циклизации согласно способу Reissert. Форму (k5) с диметиламинометильным заместителем в положении 3 получают по реакции Манниха с последующим N-диметилированием, после чего проводят N-метилирование с целью получения соли четвертичного аммония (k6).
Реакцию образования енамина можно проводить, например, путем нагревания производного О-нитротолуола (k1) с диметилацеталью N,N-диметилформамида и пирролидином в среде такого подходящего растворителя как ДМФ.
Реакцию циклизации индола можно проводить при комнатной температуре с использованием газообразного водорода в присутствии никеля Рэнея в среде такого подходящего растворителя, как толуол.
N-метилирование можно осуществлять, например, путем нагревания в ДМФ с применением трет-бутоксикалия или диметилоксалата.
Диметиламинометилирование в положение 3 можно осуществлять, например, с использованием реакции Манниха, проводя ее при комнатной температуре в смешанном растворителе диоксануксусная кислота, с использованием водного раствора формалина или водного раствора диметиламина.
Кроме этого, производное индола может быть синтезировано по методике согласно Heterocycles, vol.22, # 1,195 (1984).
Кроме этого, бензотиофен, индол и другие гетероциклические галогениды и четвертичные соли могут быть синтезированы по другим методикам, описанным в литературе, например, согласно Hetericyclic compound chemistry (Kondansha Scientific, H. Yamanaka, ed.).
Производное бензимидазола настоящего изобретения, если это желательно, может быть превращено в медицински применимую, нетоксичную соль. Примеры рассматриваемых солей включают соли с такими ионами щелочных металлов, как Na+, и К+, ионами таких щелочноземельных металлов, как Mg2+ и Са2+ и такими ионами, как Al3+ и Zn2+, а также соли таких органических оснований, как аммоний, триэтиламин, этилендиамин, пропандиамин, пирролидин, пиперидин, пиперазин, пиридин, лизин, холин, этаноламин, N,N-диметилэтаноламин, 4-гидроксипиперидин, глюкозамин и N-метилглюкамин. Особенно предпочтительными являются соли Na+,К+, Са2+, лизин, холин, N,N-диметилэтаноламин, и N-метилглюкамин.
Производное бензимидазола настоящего изобретения сильно ингибируют активность человеческой химазы. Так, IC50 составляет 1000 нМ или менее, предпочтительно, от 0,01 нМ или более до менее 1000 нМ, и более предпочтительно, от 0,05 нМ или более до менее 500 нМ. Бензимидазольное производное настоящего изобретения, обладающее такой превосходной ингибирующей активностью в отношении человеческой химазы, может использоваться в качестве профилактического и/или терапевтического агента, применимого в клинических условиях для лечения разнообразных болезней.
Производное бензимидазола настоящего изобретения может применяться перорально или не-перорально, в виде фармацевтической композиции совместно с фармацевтически применимым носителем, который обеспечивает получение различных лекарственных форм указанной фармацевтической композиции.
Примерами не-перорального введения могут служить внутривенное, подкожное, внутримышечное, чрескожное, ректальное, назальное и внутриглазное применение.
Примерами лекарственных форм указанной фармацевтической композиции для пероральных препаратов могут служить таблетки, пилюли, гранулы, порошки, жидкости, суспензии, сиропы и капсулы.
В контексте изобретения таблетки могут формироваться обычным способом с использованием такого фармацевтически применимого носителя, как наполнитель, связующий агент или дезинтегрирующий агент. Пилюли, гранулы и порошки могут быть сформированы обычными способами, с использованием носителя и т.п. согласно тем же способам, что таблетки. Жидкости, суспензии и сиропы могут быть сформированы обычным способом, с использованием сложных эфиров глицерина, спиртов, воды и растительного масла. Капсулы из желатины и т.д. могут быть сформированы путем их заполнения гранулами, порошками или жидкостями и т.п.
Препараты не-перорального типа могут применяться в виде препаратов для инъекций в случае внутривенного, подкожного или внутримышечного применения. Примеры препаратов для инъекций включают вариант, в котором производное бензимидазола настоящего изобретения растворяют в таком водном жидком агенте, как физиологический раствор, или вариант, в котором его растворяют в неводной жидкости, состоящей из органического сложного эфира, например, растительного масла.
В случае чрескожного применения, можно использовать такую лекарственную форму, как мазь или крем. Мази могут формироваться смешиванием производного бензимидазола настоящего изобретения с маслом или вазелином и т.п., тогда как кремы можно формировать путем смешивания производного бензимидазола настоящего изобретения с эмульгатором.
В случае ректального введения, препараты могут применяться в виде свечей с использованием мягких желатиновых капсул и т.п.
При назальном введении может использоваться препарат, представляющий собой композицию из жидкости и порошка. Примерами основ для используемых жидких препаратов могут служить вода, физиологический раствор, фосфатный буфер и ацетатный буфер, причем указанные препараты могут также содержать поверхностно-активный агент, антиоксидант, стабилизатор, консервант и загуститель. Примерами основ для порошковых препаратов могут служить такие влагопоглощающие основы, как водно-растворимые полиакрилаты, низшие алкиловые эфиры целлюлозы, полиэтиленгликоль поливинилпирролидон, амилоза, и плюран, такие нерастворимые в воде основы, как целлюлоза, крахмалы, протеины, каучуки и сшитые виниловые полимеры, хотя водно-растворимые основы являются предпочтительными. Кроме этого, может использоваться смесь перечисленных веществ. В порошковые препараты могут добавляться антиоксидант, краситель, консервант, антисептический или полисапробный агенты. Указанные жидкие и порошковые препараты могут применяться с использованием распылителя и т.п.
Для внутриглазного введения могут использоваться водные или неводные глазные примочки. В водных глазных примочках в качестве растворителя могут использоваться стерильная очищенная вода и физиологический раствор. В том случае, когда растворитель представляет собой только стерильную очищенную воду, может использоваться водная суспензионная глазная примочка, получаемая в результате добавления поверхностно-активного агента, полимерного загустителя и т.п. Рассматриваемый препарат может применяться в виде растворимой глазной примочки в результате добавления такого солюбилизирующего агента, как неионный поверхностно-активный агент. В неводных глазных примочках могут использоваться неводные растворители для инъекций и такой препарат также может использоваться в виде неводной суспензионной глазной примочки.
Примерами лекарственных форм для глазного применения, отличных от глазных примочек, могут служить офтальмологические мази, аппликационные жидкости, спрэи и вставки.
При ингаляции через нос и ротовую полость, бензимидазольное производное настоящего изобретения вдыхается с использованием аэрозольного распылителя для ингаляции, при объединении основного ингредиента с типичным фармацевтическим носителем в виде раствора или суспензии. Кроме этого, бензимидазольное производное настоящего изобретения может применяться в виде сухого порошка при использовании ингалятора, находящегося в прямом контакте с легкими.
При необходимости, в различные перечисленные выше препараты могут добавляться разрешенные носители, например, изотонические агенты, консерванты, антисептики, увлажнители, буферы, эмульгаторы, диспергенты, и стабилизаторы.
Кроме этого, в случае необходимости, такие различные препараты могут быть подвергнуты стерилизации, включающей смешивание с дезинфицирующим агентом, фильтрование с применением противобактериального фильтра, нагревание или облучение. С другой стороны, может быть приготовлен стерильный твердый препарат, который используют после растворения или суспендирования в подходящей стерильной жидкости непосредственно перед применением.
Хотя дозировка бензимидазольного производного настоящего изобретения может изменяться в зависимости от природы заболевания, пути введения и симптомов, возраста, пола и веса тела пациента, в случае перорального применения, рассматриваемая дозировка составляет 1-500 мг/день/объект, предпочтительно, 10-300 мг/день/объект, и она составляет 0,1-100 мг/день/объект, предпочтительно 0,3-30 мг/день/объект в случае не-перорального применения, например, внутривенного, подкожного, внутримышечного, чрескожного, ректального, назального, внутриглазного или ингаляционного применения.
В случае применения бензимидазольного производного настоящего изобретения в качестве профилактического агента, препарат может применяться ранее известными методами в соответствие с каждым симптомом.
Примерами заболеваний, на которые нацелено действие профилактического агента и/или терапевтического агента настоящего изобретения, являются такие заболевания верхних дыхательных путей, как бронхиальная астма, такие воспалительные и аллергические заболевания, как аллергические риниты, атопические дерматиты, такие сердечно-сосудистые заболевания, как склеротические поражения сосудов, вазоконстрикция, нарушения периферического кровоснабжения, почечная и сердечная недостаточность, а также такие костные и хрящевые метаболические заболевания, как ревматоидный артрит, и остеоартрит.
Ниже приведено подробное разъяснение настоящего изобретения с помощью примеров, касающихся получения соединений и тестовых примеров. Однако область настоящего изобретения никоим образом не ограничивается этими примерами.
[Справочный пример 1]
Получение 5,6-диметилбензимидазол-2-тиола
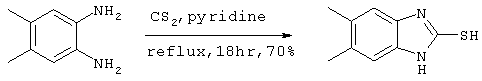
40 мл (0,66 ммоля) сероуглерода добавляли в пиридиновый раствор (40 мл) 4,5 г (33 ммоля) 5,6-диметилортофенилендиамина. После перемешивания, полученный в результате раствор в течение 18 часов нагревали с обратным холодильником, добавляли воду и полученную смесь экстрагировали этилацетатом. После сушки этилацетатной фазы над безводным сульфатом магния, ее концентрировали при пониженном давлении и сушили в течение 6 часов при 80°С и пониженном давлении с образованием 4,1 г целевого соединения (выход: 70%).
Спектр 1Н-ЯМР (270 Мгц, ДМСО-d6) (ч/млн): 12,30 (широкий, 1Н), 6,91 (с, 2Н), 2,21 (с, 6Н).
[Справочный пример 2]
Получение этилового эфира 4-(5,6-диметилбензимидазол-2-илтио)бутаноата
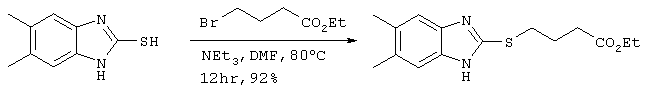
35 мкл (0,25 ммоля) триэтиламина и 36 мкл (0,25 ммоля) этилового эфира 4-бромбутаноата добавляли к 36 мг (0,20 ммоля) 5,6-диметилбензимидазол-2-тиола. После перемешивания полученного в результате раствора в течение 12 часов при 80°С, добавляли воду и проводили экстракцию диэтиловым эфиром. После сушки диэтилэфирной фазы над безводным сульфатом магния, раствор концентрировали и остаток очищали методом хроматографии на колонке с силикагелем (гексан:этилацетат =4:1) с получением 54 мг целевого соединения (выход: 92%). Подтверждение структуры полученного соединения осуществляли путем идентификации по молекулярному весу с использованием метода ЖХ-МС.
Вычисленное значение М=292,12, измеренное значение (М+Н)+=293,40.
[Справочный пример 3]
Следующие ниже соединения синтезировали по методике Справочного примера 2. Подтверждение структур полученных соединений осуществляли путем идентификации по молекулярному весу с использованием метода ЖХ-МС.
Этиловый эфир 4-(бензимидазол-2-илтио)бутаноата
Вычисленное значение М=264,09, измеренное значение (М+Н)+=293,40.
Этиловый эфир 4-(5,6-дифторбензимидазол-2-илтио)бутаноата
Вычисленное значение М=300,07, измеренное значение (М+Н)+=301,3.
[Справочный пример 4]
Получение 3-бромметил-5-метилбензо[b]тиофена
Стадия 1
Получение 3-гидроксиметил-п-нитротолуола
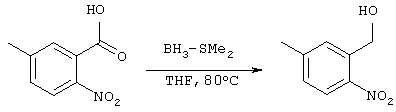
5,02 г (27,7 ммоля) 5-метил-2-нитробензойной кислоты растворяли в 20 мл ТГФ, после чего раствор прикапывали в 11,1 мл 10,2 М диметилсульфидного комплекса борана и нагревали при 80°С. Через 1,5 часа в реакционную систему, при охлаждении льдом и перемешивании, прикапывали 30 мл 1М раствора хлористоводородной кислоты. Затем полученную систему концентрировали при пониженном давлении с получением 100 мл водной фазы, которую экстрагировали этилацетатом (100 мл ×2). После промывки этилацетатной фазы насыщенным рассолом, органическую фазу сушили над сульфатом магния, концентрировали при пониженном давлении и сушили с образованием 3,91 г целевого соединения (выход: 85%).
Стадия 2
Получение 3-формил-п-нитротолуола
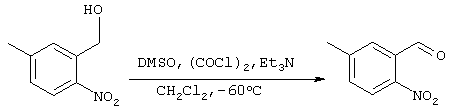
5,5 мл (63,2 ммоля) хлористого оксалила добавляли к 50 мл дихлорметана и полученную смесь охлаждали до -60°С. Через 20 минут добавляли 9,13 мл (138,6 ммоля) ДМСО, смесь перемешивали при -60°С и через 15 минут, при -60°С и перемешивании, добавляли 3,91 г (23,3 ммоля) 3-гидроксиметил-п-нитротолуола, полученного на стадии 1. Через 30 минут, при температуре -60°С, прикапывали 45 мл триэтиламина и системе давали нагреваться до комнатной температуры. После концентрирования при пониженном давлении, к остатку добавляли 0,1М хлористоводородной кислоты, после чего проводили экстракцию этилацетатом (150 мл ×2). Затем органическую фазу сушили над сульфатом магния и концентрировали при пониженном давлении с получением 5,02 г целевого соединения (выход сырого продукта: 130%).
Стадия 3
Получение 2-карбоксиэтил-5-метилбензо[b]тиофена
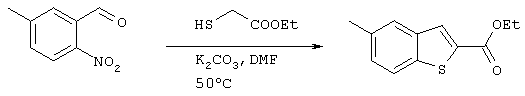
5,02 г (63,2 ммоля) 3-формил-п-нитротолуола, полученного на стадии 2, растворяли в 50 мл ДМФ, после чего добавляли 3,06 мл (28,1 ммоля) этилмеркаптоацетата и 4,85 г (35,1 ммоля) карбоната калия и полученную смесь перемешивали при 50°С. Через 9,5 часов температуру повышали до 80°С и в течение 100 минут осуществляли дополнительное нагревание. После завершения реакции в реакционный раствор добавляли 250 мл воды, после чего осуществляли экстракцию этилацетатом (100 мл ×3) и систему сушили над сульфатом магния. После концентрирования растворителя при пониженном давлении, остаток очищали методом хроматографии на колонке с силикагелем (гексан:этилацетат =8:1), после чего проводили дополнительную хроматографическую очистку на колонке с силикагелем (гексан:этилацетат =10:1) с получением 2,48 г (11,2 ммоля) целевого соединения (выход: 48%).
Спектр 1H-ЯМР (400 Мгц, CDCl3) (ч/млн): 7,98 (с, 1Н), 7,73 (д, 1Н, J=8,28 Гц), 7,65 (с, 1Н), 7,27 (д, 1Н, J=8,32 Гц), 4,39 (квартет, 2Н), 2,47 (с, 3Н), 1,41 (с, 3Н).
Стадия 4
Получение 2-карбокси-5-метилбензо[b]тиофена
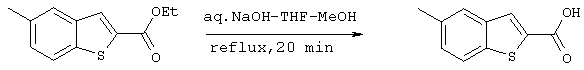
30 мл раствора метанола, ТГФ и 2М водного раствора гидроксида натрия (1:1:1) добавляли к 2,17 г (9,87 ммоля) 2-карбоксиэтил-5-метилбензо[b]тиофена, полученного на стадии 3, и полученную смесь нагревали с обратным холодильником. Через 20 минут раствор нейтрализовали кислотой и затем концентрировали при пониженном давлении и выделяли осадок. Осадок промывали 50 мл воды и высушивали с образованием 2,03 г (10,5 ммоля) целевого соединения (выход сырого продукта: 107%).
Спектр 1Н-ЯМР (400 Мгц, CDCl3) (ч/млн): 7,94 (с, 1Н), 7,74 (д, 1Н, J=8,56 Гц), 7,69 (с, 1Н), 7,27 (д, 1Н, J=8,30 Гц), 2,47 (с, 3Н).
Стадия 5
Получение 5-метилбензо[b]тиофена

2,03 г (9,87 ммоля) 2-карбокси-5-метилбензо[b]тиофена, полученного на стадии 4, растворяли в 10 мл хинолина, после чего добавляли 799,2 мг медного порошка и смесь нагревали до 190°С. Через 100 минут, полученный раствор охлаждали с последующим добавлением 40 мл 0,5М хлористоводородной кислоты и экстракцией этилацетатом (40 мл ×2). Органическую фазу промывали 40 мл воды и затем сушили над сульфатом магния. После концентрирования растворителя при пониженном давлении, смесь очищали методом хроматографии на колонке с силикагелем (гексан:этилацетат =20:1) с получением 1,41 г (9,51 ммоля) целевого соединения (выход за две стадии, начиная со стадии 4: 96%).
Спектр 1Н-ЯМР (270 Мгц, CDCl3) (ч/млн): 7,76 (д, 1Н, J=8,24 Гц), 7,62 (с, 1Н), 7,40 (д, 1Н, J=5,44 Гц), 7,24 (м, 1Н), 7,17 (д, 1Н, J=8,24 Гц), 2,47 (с, 3Н).
Стадия 6
Получение 3-хлорметилкарбонил-5-метилбензо[b]тиофена
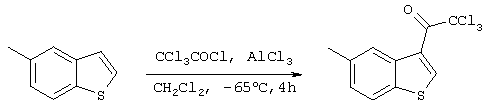
10 мл дихлорметана добавляли к 1,33 г (9,97 ммоль) треххлористого алюминия, после чего смесь охлаждали до -65°С сухим льдом и ацетоном. Через 10 минут прикапывали 1,12 мл (10,0 ммоль) хлористого трихлорацетила. Еще через 20 минут прикапывали 10 мл дихлорметанового раствора, содержащего 1,41 г (9,51 ммоль) 5-метилбензо[b]тиофена, полученного на стадии 5, после чего смесь перемешивали при -65°С. Через 1 час 40 минут температура повышалась до -40°С. Еще через 1 час 10 минут температура повышалась до 0°С. Еще через 1 час 40 минут добавляли 10 мл 1М хлористоводородной кислоты и полученную смесь перемешивали. После добавления в реакционную систему 20 мл воды, удаления дихлорметановой фазы методом сепарации жидкости и дополнительной экстракции водной фазы этилацетатом, водную фазу объединяли с дихлорметановой фазой и концентрировали при пониженном давлении. 3,2 г полученного остатка очищали методом хроматографии на колонке с силикагелем (силикагель; 120 г, гексан) с получением 686,7 мг (2,34 ммоля) целевого соединения (выход: 24%).
Спектр 1Н-ЯМР (400 Мгц, CDCl3) (ч/млн): 8,89 (с, 1Н), 8,51 (с, 1Н), 7,78 (д, 1Н, J=8,28 Гц), 7,30 (д, 1Н, J=8,32 Гц), 2,53 (с, 3Н).
Стадия 7
Получение 3-карбокси-5-метилбензо[b]тиофена
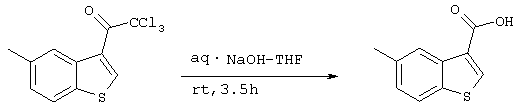
686,7 мг (2,34 ммоль) 3-хлорметилкарбонил-5-метилбензо-[b]тиофена, полученного на стадии 6 растворяли в 2,0 мл ТГФ и 3,0 мл МеОН, после чего добавляли 2 мл 2М водного раствора гидроксида натрия и смесь перемешивали при комнатной температуре. Через 2 часа 45 минут добавляли 5 мл 2М водного раствора гидроксида натрия и полученную смесь нагревали до 60°С. Через 30 минут смесь охлаждали и добавляли 10 мл 2М раствора хлористоводородной кислоты и 30 мл воды, раствор экстрагировали этилацетатом с последующим концентрированием при пониженном давлении и сушили с получением 438,9 мг (2,28 ммоля) целевого соединения (выход: 97%).
Спектр 1H-ЯМР (400 Мгц, CDCl3) (ч/млн): 8,44 (с, 1Н), 8,36 (с, 1Н), 7,74 (д, 1Н, J=8,04 Гц), 7,22 (д, 1Н, J=8,28 Гц), 2,50 (с, 3Н).
Стадия 8
Получение 3-гидроксиметил-5-метилбензо[b]тиофена
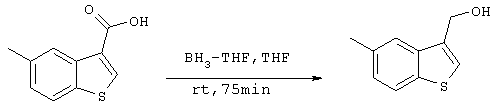
438,9 мг (2,28 ммоля) 3-карбокси-5-метилбензо[b]тиофена, полученного на стадии 7, растворяли в 5 мл ТГФ с последующим добавлением раствора комплекса ВН3·ТГФ и перемешиванием при комнатной температуре. Через 1 час 15 минут добавляли 4 мл 2М раствора хлористоводородной кислоты и смесь перемешивали, после чего добавляли 50 мл этилацетата. Органическую фазу промывали 30 мл воды и сушили над сульфатом магния с последующей концентрацией при пониженном давлении. Полученный остаток очищали с помощью Biotage (гексан:этилацетат =4:1) с образованием 347,6 мг (1,95 ммоля) целевого соединения (выход: 86%).
Спектр 1H-ЯМР (400 Мгц, CDCl3) (ч/млн): 7,74 (д, 1Н, J=8,04 Гц), 7,65 (с, 1Н), 7,34 (с, 1Н), 7,19 (д, 1H, J=8,28 Гц), 4,89 (с, 2Н), 2,48 (с, 3Н).
Стадия 9
Получение 3-бромометил-5-метилбензо[b]тиофена
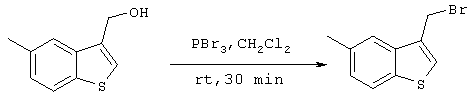
326 мг (1,83 ммоля) 3-гидроксиметил-5-метилбензо[b]тиофена, полученного на стадии 8, растворяли в 10 мл дихлорметана с последующим добавлением 0,262 мл трехбромистого фосфора и перемешиванием при комнатной температуре. Через 30 минут добавляли 30 мл воды с последующим перемешиванием в течение 10 минут и экстракцией дихлорметаном (30 мл ×2). Затем органическую фазу концентрировали при пониженном давлении и сушили с получением 397,5 мг (1,65 ммоль) целевого соединения (выход: 90%).
Спектр 1H-ЯМР (270 Мгц, CDCl3) (ч/млн): 7,74-7,67 (м, 2Н), 7,46 (с, 1Н), 7,22 (д, 1H, J=8,24 Гц), 4,74 (с, 2Н), 2,51 (с, 3Н).
[Справочный пример 5]
Получение йодистого ((4-метилбензо[b]тиофен-3-ил)метил)-триметиламмония
Стадия 1
Получение 2-циано-3-нитротолуола
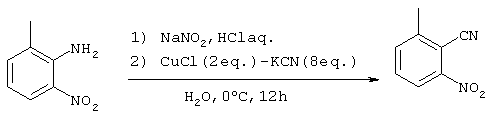
76,07 г (500 ммоль) 2-амино-3-нитротолуола добавляли к 100 г (990 ммоль) 36% хлористоводородной кислоты и 500 г льда с последующим энергичным перемешиванием при 0°С. Затем медленно прикапывали 80 мл водного раствора, содержащего 37,95 г (550 ммоль) нитрата натрия, поддерживая температуру в интервале 0-5°С. После завершения прикапывания в систему добавляли 100 мл толуола с последующим перемешиванием в течение 30 минут при 0°С. Реакционный раствор помещали на баню с охлаждающей системой лед-NaCl, после чего медленно добавляли бикарбонат натрия, интенсивно перемешивая реакционную систему для нейтрализации рН до, примерно, 6 (раствор соли диазония (1)).
Водный раствор (550 мл), содержащий 260,5 г (4000 ммоль) цианида калия медленно добавляли при 0°С к водному раствору (650 мл), содержащему 99,0 г (1000 ммоль) хлористой меди (I) с последующим перемешиванием в течение 90 минут и добавлением 200 мл этилацетата. Затем полученный выше раствор соли диазония (1) прикапывали к полученному раствору в течение 30 минут, поддерживая при этом температуру в интервале 0-5°С. Полученный раствор перемешивали в течение 12 часов на бане со льдом, после чего смеси давали нагреваться до комнатной температуры. После экстракции реакционного раствора этилацетатом и промывания органической фазы водой, систему сушили над сульфатом магния с последующим концентрированием растворителя при пониженном давлении. Полученный остаток очищали методом хроматографии на колонке с силикагелем (гексан:этилацетат  с получением 58,63 г (362 ммоль) целевого соединения (выход: 72%).
с получением 58,63 г (362 ммоль) целевого соединения (выход: 72%).
Спектр 1Н-ЯМР (270 Мгц, CDCl3) (ч/млн): 7,68 (м, 2Н), 8,13 (1Н, м), 2,715 (3Н, с).
Стадия 2
Получение 3-амино-2-этоксикарбонил-4-метилбензо[b]тиофена
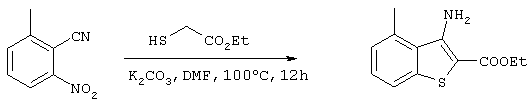
ДМФ раствор (250 мл), содержащий 58,63 г (362 ммоль) 2-циано-3-нитротолуола, полученного на стадии 1, 47,5 г (395 ммоль) этил 2-меркаптоацетата и 57,5 г (416 ммоль) карбоната калия перемешивали в течение 12 часов при 100°С. Затем реакционный раствор концентрировали при пониженном давлении для удаления некоторого количества ДМФ. Воду добавляли для растворения неорганических веществ, после чего проводили экстракцию этилацетатом. После промывки органической фазы водой, ее сушили над сульфатом магния, после чего концентрировали растворитель при пониженном давлении. Затем остаток очищали методом хроматографии на колонке с силикагелем (гексан:этилацетат =10:1) с получением 62,86 г (267 ммоль) целевого соединения (выход: 74%).
Спектр 1Н-ЯМР (270 Мгц, CDCl3) (ч/млн): 7,54 (д, 2Н), 7,29 (т, 1Н), 7,03 (д, 1Н), 6,28 (с, 2Н), 4,35 (кв., 2Н), 2,82 (с, 3Н), 1,39 (т, 3Н).
Стадия 3
Получение 3-циано-2-этоксикарбонил-4-метилбензо[b]тиофена
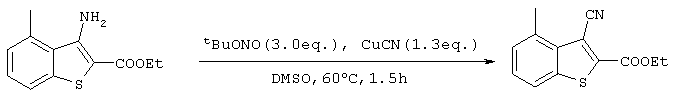
После продувки реакционной системы азотом, 82,0 г (795 ммоль) трет-бутил нитрита и 30,9 г (345 ммоль) цианида меди добавляли к 250 мл ДМСО и осуществляли растворение в результате перемешивания при 55°С в течение 30 минут. Кроме этого, раствор, содержащий 62,2 г (265 ммоль) 3-амино-2-этоксикарбонил-4-метилбензо-[b]тиофена, полученного на стадии 2, в ДМСО (100 мл), медленно прикапывали в течение 2 часов, поддерживая температуру, равной 55°С. После нагревания реакционного раствора до 60°С и перемешивания в течение 140 минут, раствор охлаждали до 0°С с последующим медленным добавлением воды и перемешиванием в течение 1 часа при 0°С. После этого реакционный раствор фильтровали через Целит для удаления примесей и после экстракции дихлорметаном и промывки органической фазы водой, реакционную смесь сушили над сульфатом магния с последующим концентрированном растворителя при пониженном давлении. Затем остаток очищали методом хроматографии на колонке с силикагелем (гексан:этилацетат  с получением 15,59 г (63,6 ммоль) целевого соединения (выход: 24%).
с получением 15,59 г (63,6 ммоль) целевого соединения (выход: 24%).
Спектр 1Н-ЯМР (270 Мгц, CDCl3) (ч/млн): 7,73 (д, 1Н), 7,44 (т, 1Н), 7,30 (д, 1Н), 4,50 (кв., 2Н), 2,95 (с, 3Н), 1,47 (т, 3Н).
Стадия 4
Получение 3-циано-4-метилбензо[b]тиофена
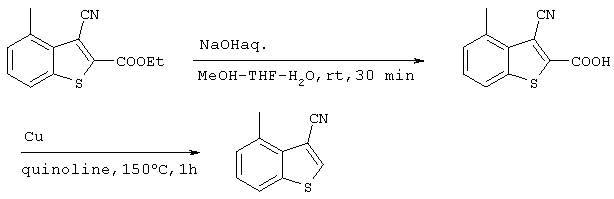
15,59 г (63,6 ммоль) 3-циано-2-этоксикарбонил-4-метилбензо[b]тиофена, полученного на стадии 3 растворяли в смеси метанола (150 мл), ТГФ (150 мл) и воды (150 мл) после чего добавляли 30 мл 5М водного раствора гидроксида натрия и полученную смесь перемешивали в течение 2 часов при комнатной температуре. После концентрирования растворителя при пониженном давлении, рН понижали до 4 путем добавления 1М раствора хлористоводородной кислоты и, после экстракции этилацетатом и промывкой органической фазы водой, ее сушили над сульфатом магния. Затем растворитель концентрировали при пониженном давлении с получением 3-циано-2-карбокси-4-метилбензо[b]тиофена. Этот продукт и 1,27 г (20 ммоль) медного порошка добавляли к 18 мл хинолина с последующим перемешиванием в течение 2 часов при 150°С. После охлаждения реакционного раствора его фильтровали через целит и рН фильтрата понижали до 3 добавлением хлористоводородной кислоты с целью переноса хинолина, как растворителя, в водную фазу с последующей экстракцией этилацетатом. После промывки органической фазы водой ее сушили над сульфатом магния, и растворитель концентрировали при пониженном давлении. Затем остаток очищали методом хроматографии на колонке с силикагелем (гексан:этилацетат =20:1) с получением 9,10 г (52,6 ммоль) целевого соединения (выход за две стадии: 83%).
Спектр 1Н-ЯМР (270 Мгц, CDCl3) (ч/млн): 8,15 (с, 1Н) и 7,74 (д, 1Н), 7,36 (т, 1Н), 7,25 (д, 1Н), 2,91 (с, 3Н).
Стадия 5
Получение 3-((N,N-диметиламино)метил)-4-метилбензо[b]тиофена
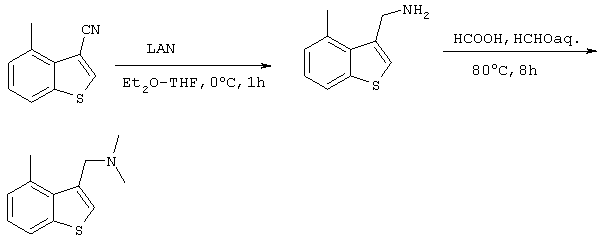
После прикапывания раствора из диэтилового эфира (20 мл) и ТГФ (20 мл), содержащего 9,10 г (52,6 ммоль) 3-циано-4-метилбензо[b]тиофена, полученного на стадии 4, в суспензию из 50 мл диэтилового эфира и 2,0 г (53 ммоль) литийалюминий гидрида в течение 15 минут при 0°С, полученный раствор перемешивали в течение 30 минут при комнатной температуре. После завершения реакции избыток LAH в реакционном растворе обрабатывали хлористоводородной кислотой с последующим добавлением водного раствора гидроксида натрия с целью подщелачивания системы. После насыщения водной фазы карбонатом калия, экстракции дихлорметаном и промывания органической фазы водой, ее сушили над сульфатом магния. После этого растворитель концентрировали при пониженном давлении с получением 3-аминометил-4-метилбензо[b]тиофена. К полученному веществу последовательно добавляли 11,5 г (250 ммоль) муравьиной кислоты и 10,0 г (123 ммоль) 37% водного раствора формальдегида, после чего систему перемешивали в течение 5 часов при 80°С. После завершения реакции и последующего добавления в реакционный раствор водного раствора хлористоводородной кислоты, реакционный раствор концентрировали при пониженном давлении с целью удаления муравьиной кислоты и формальдегида. Для подщелачивания полученного раствора добавляли водный раствор гидроксида натрия, после чего проводили экстракцию дихлорметаном. После промывания органической фазы водой, ее сушили над сульфатом магния и растворитель концентрировали при пониженном давлении. Остаток подвергали хроматографической очистке на колонке с силикагелем (гексан:этилацетат =10:1) с получением 2,61 г (12,8 ммоль) целевого соединения (выход за две стадии: 24%). Подтверждение структуры полученного соединения проводили по данным 1Н-ЯМР-анализа.
Спектр 1H-ЯМР (270 Мгц, CDCl3) (ч/млн): 7,66 (с, 1Н), 7,26-7,09 (м, 3Н), 3,65 (с, 2Н), 2,85 (с, 3Н), 2,27 (с, 6Н)
Стадия 6
Получение иодистого ((4-метилбензо[b]тиофен-3-ил)метил)-триметиламмония
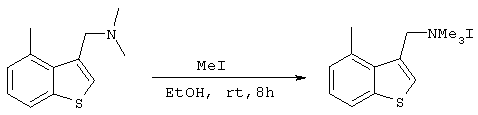
3,69 г (26 ммоль) иодистого метила добавляли к 20 мл этанольного раствора, содержащего 2,61 г (12,8 ммоль) 3-((N,N-диметиламино)метил-4-метилбензо[b]тиофена, полученного на стадии 5, с последующим перемешиванием в течение 18 часов при комнатной температуре. Полученную в результате белую суспензию, после фильтрации от избытка иодистого метила и растворителя, промывали этанолом (10 мл ×2) и диэтиловым эфиром (10 мл ×3) с получением 3,08 г (8,88 ммоль) целевого соединения в виде белого твердого вещества (выход: 69%).
Спектр 1Н-ЯМР (270 Мгц, ДМСО) (ч/млн): 8,19 (с, 1Н), 7,93 (д, 1Н), 7,36-7,25 (м, 2Н), 4,91 (с, 2Н), 3,05 (с, 9Н), 2,77 (с, 3Н).
[Справочный пример 6]
Получение иодистого ((1,4-диметилиндол-3-ил)метил)метиламмония
Стадия 1
Получение 4-метилиндола
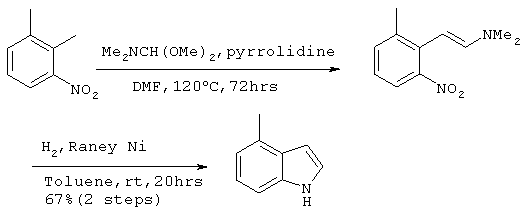
30,5 г (256 ммоль) диметилацетали N,N-диметилформамида и 10,9 г (153 ммоль) пирролидина добавляли к 150 мл N,N-диметилформамидного раствора, содержащего 19,4 г (128 ммоль) 2,3-диметилнитробензола. После перемешивания полученного раствора в течение 72 часов при 120°С его концентрировали. К полученному в результате коричневому маслянистому продукту добавляли 100 мл толуола с последующим добавлением 11 г никеля Рэнея (50%, водная суспензия, рН>9) и перемешиванием. Реактор продували газообразным водородом и содержимое перемешивали в течение 20 часов при комнатной температуре в атмосфере водорода. После фильтрации реакционного раствора через целит, фильтрат концентрировали с получением 30 г темного раствора. Полученный раствор подвергали хроматографической очистке на колонке с силикагелем (гексан:этилацетат =10:1) с получением 11,33 г (86 ммоль) целевого соединения (выход за две стадии: 67%). Подтверждение структуры полученного соединения проводили по его идентификации методом 1Н-ЯМР.
Спектр 1Н-ЯМР (270 Мгц, CDCl3) (ч/млн): 7,28-7,07 (м, 3Н), 6,93 (м, 1Н), 6,57 (м, 1Н), 2,57 (с, 3Н)
Стадия 2
Получение 1,4-диметилиндола

В предварительно высушенный реакционный сосуд вводили 12,7 г (134 ммоль) трет-бутокси калия и 80 мл N,N-диметилформамида. Добавляли 8,9 г (67,9 ммоль) 4-метилиндола, полученного на стадии 1 с последующим перемешиванием в течение 35 минут при комнатной температуре. К полученной смеси добавляли 15,8 г (134 ммоль) диметилоксалата с последующим перемешиванием в течение 5 часов 30 минут при 120°С. После концентрирования при пониженном давлении добавляли 200 мл воды, после чего проводили обработку 1М раствором хлористоводородной кислоты для подкисления реакционной системы (рН 3), смесь экстрагировали этилацетатом (200 мл ×2) и сушили над безводным сульфатом магния. После отгонки растворителя при пониженном давлении, остаток подвергали хроматографической очистке на колонке с силикагелем (гексан:этилацетат =5:1) с получением 9,24 г (53 ммоля) целевого соединения (выход: 94%). Подтверждение структуры полученного соединения проводили путем его идентификации методом 1H-ЯМР.
Спектр 1Н-ЯМР (270 Мгц, CDCl3) (ч/млн): 7,25-7,09 (м, 2Н), 7,03 (м, 1Н), 6,90 (м, 1Н), 6,49 (м, 1Н), 3,78 (с, 3Н), 2,55 (с, 3Н)
Стадия 3
Получение 1,4-диметил-3-(N,N-диметиламинометил)индола
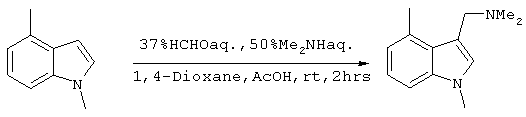
5,9 мл (72,0 ммоль) 37% водного раствора формальдегида и 7,08 мл (78 ммоль) 50% водного раствора диметиламина последовательно добавляли к смешанной системе, содержащей по 25 мл 1,4-диоксана и уксусной кислоты. После охлаждения до комнатной температуры, поскольку в ходе реакции выделяется тепло, добавляли 10 мл 1,4-диоксанового раствора, содержащего 9,24 г (63,6 ммоля) 1,4-диметилиндола, полученного на стадии 2, с последующим перемешиванием в течение 2 часов при комнатной температуре. Затем реакционный раствор концентрировали. К полученному остатку, с целью его подщелачивания (рН 12), добавляли 5М водный раствор гидроксида натрия, общий объем системы доводили до 100 мл, после чего проводили экстракцию этилацетатом (100 мл ×2). Затем органическую фазу сушили над безводным сульфатом магния и концентрировали при пониженном давлении с получением 12,93 г (63,9 ммоль) целевого соединения (выход сырого продукта: 100,4%). Подтверждение структуры полученного соединения проводили путем его идентификации методом 1Н-ЯМР.
Спектр 1Н-ЯМР (270 Мгц, CDCl3) (ч/млн): 7,15-7,06 (м, 2Н), 6,91 (м, 1Н), 6,85 (м, 1Н), 3,71 (с, 3Н), 3,59 (с, 2Н), 2,74 (с, 3Н), 2,27 (с, 6Н).
Стадия 4
Получение иодистого ((1,4-диметилиндол-3-ил)метил)триметиламмония
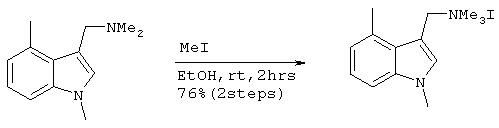
12,93 г (63,6 ммоль) 1,4-диметил-3-(N,N-диметиламинометил)-индола, полученного на стадии 3, растворяли в 60 мл этанола с последующим добавлением 4,36 мл (70 ммоль) иодистого метила. После перемешивания в течение 2 часов при комнатной температуре образовывался белый осадок. Этот осадок фильтровали, дважды промывали 10 мл этанола и сушили в вакууме с получением 16,66 г (48,4 ммоль) целевого соединения (выход за две стадии: 76%.) Подтверждение структуры полученного соединения проводили путем его идентификации методом 1H-ЯМР.
Спектр 1H-ЯМР (270 Мгц, ДМСО) (ч/млн): 7,65 (с, 1Н), 7,36 (д, 1Н), 7,13 (т, 1Н), 6,91 (д, 1Н), 4,74 (с, 2Н), 3,82 (с, 3Н), 3,01 (с, 9Н), 2,65 (с, 3Н),
[Справочный пример 7]
Получение бромистоводородной соли 4-(5-метоксибензимидазол-2-илтио)бутаноата
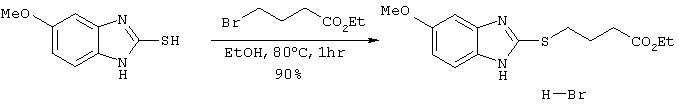
6,48 г (33,2 ммоль) этилового эфира 4-бромбутаноата добавляли к 10 мл этанольного раствора, содержащего 5,0 г (27,7 ммоль) 5-метоксибензимидазол-2-тиола с последующим перемешиванием в течение 1 часа при 80°С и добавлением 90 мл этилацетата. Реакционному раствору давали охлаждаться до комнатной температуры и образовавшиеся кристаллы фильтровали и сушили с получением 9,34 г целевого продукта (выход: 90%).
Спектр 1Н-ЯМР (270 Мгц, CDCl3) (ч/млн): 7,65 (д, 1Н, J=8,91 Гц), 7,24 (с, 1Н), 7,00 (дд, 1Н, J=2,43, 8,91 Гц), 4,21 (кв., 2Н, J=7,29 Гц), 3,88 (с, 3Н), 3,74 (м, 2Н), 2,61 (м, 2Н), 2,10 (м, 2Н), 1,30 (т, 3Н, J=7,29 Гц).
[Пример 1]
Получение соединения №39
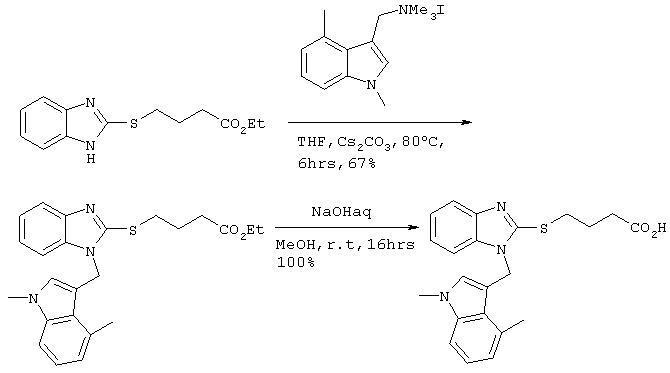
480 мг (2,49 ммоль) Cs2СО3 и 10 мл тетрагидрофурана вводили в заранее высушенный реакционный сосуд. Добавляли 505 мг (1,91 ммоль) этилового эфира 4-(бензимидазол-2-илтио)бутаноата, полученного по методике справочного примера 3, и 724 мг (2,10 ммоль) иодистого ((1,4-диметилиндол-3-ил)метил)триметиламмония с последующим перемешиванием в течение 6 часов при 80°С. После фильтрации полученного раствора через целит, его концентрировали при пониженном давлении. Остаток подвергали хроматографической очистке на колонке с силикагелем (дихлорметан:этилацетат =8:1) с получением 540 мг (1,28 ммоль) этилового эфира 4-(1-((1,4-диметилиндол-3-ил)метил)бензимидазол-2-илтио)бутаноата (выход: 67%).
Затем 2,0 мл 2М водного раствора гидроксида натрия добавляли к 6 мл метанольного раствора, содержащего 540 мг (1,28 ммоль), полученного в результате этилового эфира 4-(1-((1,4-диметилиндол-3-ил)метил)бензимидазол-2-илтио)бутаноата. После перемешивания в течение 16 часов при комнатной температуре для прекращения реакции в систему добавляли 6М хлористоводородную кислоту. Растворитель удаляли до некоторого остаточного количества путем концентрирования при пониженном давлении, после чего проводили экстракцию этилацетатом. После промывки этилацетатной фазы насыщенным рассолом, ее сушили над безводным сульфатом магния. После отгонки растворителя при пониженном давлении, продукт подвергали хроматографической очистке на колонке с силикагелем (дихлорметан:метанол =8:1) с получением 502 мг (1,28 ммоль) целевого соединения (выход: 100%). Подтверждение структуры полученного соединения осуществляли определением его молекулярного веса методом ЖХ-МС.
Рассчитанное значение М=393,15; измеренное значение (М+Н)+=394,2.
[Пример 2]
Следующие ниже соединения и соединения, указанные в следующей таблице, синтезировали по методике Примера 1 с использованием соединений, указанных в справочных примерах 2 или 3, а также различных солей четвертичного аммония или галоидных производных, синтезированных по методикам справочных примеров 4-6 и другим методикам, указанным в тексте. Подтверждение структур полученных соединений осуществляли определением их молекулярных масс методом ЖХ-МС. Однако некоторые соединения были синтезированы в условиях, несколько отличных от условий Примера 1, включая использование ДМФ и т.п. в качестве растворителя и использование карбоната калия в качестве основного агента реакции сочетания, использования ТГФ и EtOH в качестве растворителя для реакции гидролиза и использование температуры в интервале от комнатной до 50°С.
Кроме того, следующие ниже соединения были синтезированы по одинаковым методикам.
4-(1-(2-(1-метилиндол-3-ил)этил)бензимидазол-2-илтио)бутановая кислота (Соединение №1153)
Однако, в этом случае, вместо соли четвертичного аммония и галоидного производного использовали метансульфонатный эфир 2-(1-метилиндол-3-ил)этанола. Идентификацию этого соединения проводили с использованием ЖХ-МС анализа. Выход составил 19% (две стадии N-алкилирования и гидролиз сложного эфира).
Вычисленное значение М=393,15; измеренное значение (М+Н)+=394,0.
4-(1-(4-метил-7-хлорбензо[b]тиофен-3-ил)метил)бензимидазол-2-илтио)бутановая кислота (Соединение №1154)
Выход: 15% (две стадии N-алкилирования и гидролиза сложного эфира).
Вычисленное значение М=430,6; Измеренное значение (M+H)+=431,2.
Спектр 1H-ЯМР (270 Мгц, ДМСО-d6) (ч/млн): 12,17 (широкий, 1Н), 7,63 (д, 1Н, J=7,83 Гц), 7,47-7,40 (м, 2Н), 7,26 (д, 1Н, J=8,10 Гц), 7,22-7,11 (м, 2Н), 6,46 (с, 1Н), 5,86 (с, 2Н), 3,34 (т, 2Н, J=7,29 Гц), 2,84 (с, 3Н), 2,34 (т, 2Н, J=7,29 Гц), 1,94 (м, 2Н).
4-(1-(4-метил-7-бромбензо[b]тиофен-3-ил)метил)бензоимидазол-2-илтио)бутановая кислота (Соединение №1155)
Выход: 56% (две стадии N-алкилирования и гидролиза сложного эфира).
Вычисленное значение М=474,01; Измеренное значение (М+Н)+=477,0.
Спектр 1Н-ЯМР (270 Мгц, ДМСО-d6) (ч/млн): 12,18 (широкий, 1H), 7,63 (д, 1Н, J=7,56 Гц), 7,53 (д, 1Н, J=7,56 Гц), 7,46 (д, 1Н, J=7,56 Гц), 7,22-7,11 (м, 3Н), 6,46 (с, 1Н), 5,85 (с, 2Н), 3,34 (т, 2Н, J=7,29 Гц), 2,83 (с, 3Н), 2,34 (т, 2Н, J=7,29 Гц), 1,97 (м, 2Н).
[Пример 3]
Получение соединения №148
Стадия 1
Получение ((бензотиофен-3-ил)метил)(4-метокси-2-нитрофенил)амина
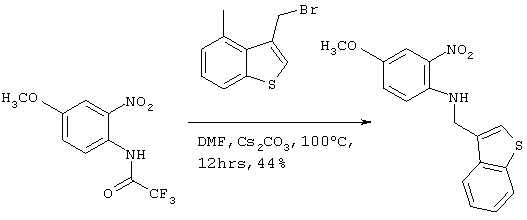
740 мг (2,8 ммоль) 4-метокси-2-нитротрифторанилида растворяли в 5 мл диметилформамида, после чего последовательно добавляли 503 мг (3,64 ммоль) карбоната калия и 773 мг (3,4 ммоль) 3-бромометилбензотиофена и смесь нагревали до 100°С. Через 12 часов добавляли 5 мл 5М водного раствора гидроксида натрия и полученный раствор нагревали с обратным холодильником в течение 1 часа. Через 15 минут раствор охлаждали до комнатной температуры с последующим добавлением 10 мл воды и экстракцией хлороформом. После двукратной промывки органической фазы 25 мл насыщенного рассола и сушки над сульфатом магния, полученный раствор концентрировали и сушили при пониженном давлении. Полученный остаток очищали методом хроматографии на колонке с силикагелем (гексан:этилацетат =60:1) с получением 400 мг ((бензотиофен-3-ил)метил)(4-метокси-2-нитрофенил)амина в виде оранжевого порошка (выход: 44%).
Стадия 2
Получение 1-((бензотиофен-3-ил)метил)-5-метоксибензо-имидазол-2-тиола
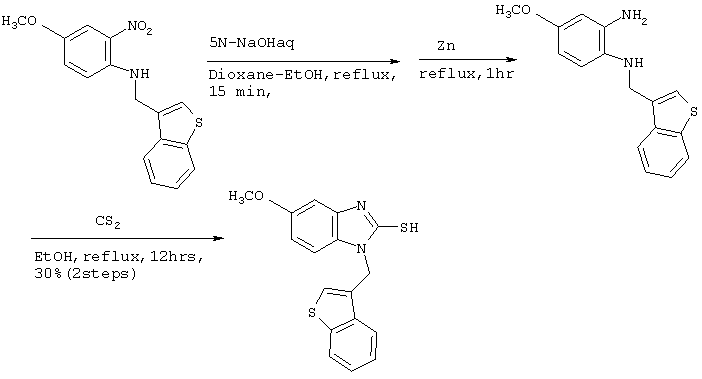 4 мл этанола и 4 мл 1,4-диоксана добавляли к 400 мг (1,23 ммоль) ((бензотиофен-3-ил)метил)(4-метокси-2-нитрофенил)амина с последующим добавлением 0,34 мл 5М водного раствора гидроксида натрия и нагреванием с обратным холодильником. Через 15 минут, реакционный раствор снимали с масляной бани и в систему добавляли 320 мг (4,9 ммоль) цинкового порошка. Реакционный раствор снова нагревали с обратным холодильником в течение 1 часа. После охлаждения до комнатной температуры цинк отфильтровывали и фильтрат концентрировали при пониженном давлении, после чего проводили экстракцию хлороформом. Органическую фазу дважды промывали 5 мл насыщенного рассола, после чего сушили над сульфатом магния, концентрировали при пониженном давлении и сушили с образованием 309 мг коричневого масла.
4 мл этанола и 4 мл 1,4-диоксана добавляли к 400 мг (1,23 ммоль) ((бензотиофен-3-ил)метил)(4-метокси-2-нитрофенил)амина с последующим добавлением 0,34 мл 5М водного раствора гидроксида натрия и нагреванием с обратным холодильником. Через 15 минут, реакционный раствор снимали с масляной бани и в систему добавляли 320 мг (4,9 ммоль) цинкового порошка. Реакционный раствор снова нагревали с обратным холодильником в течение 1 часа. После охлаждения до комнатной температуры цинк отфильтровывали и фильтрат концентрировали при пониженном давлении, после чего проводили экстракцию хлороформом. Органическую фазу дважды промывали 5 мл насыщенного рассола, после чего сушили над сульфатом магния, концентрировали при пониженном давлении и сушили с образованием 309 мг коричневого масла.
Затем полученное в результате коричневое масло растворяли в 10 мл этанола с последующим добавлением 2,5 мл (42 ммол) сероуглерода и нагреванием с обратным холодильником. Через 12 часов температура реакционного раствора возвращалась к комнатной, и раствор концентрировали при пониженном давлении, после чего добавляли 2 мл этанола и проводили ультразвуковую обработку с целью измельчения продукта на мелкие фрагменты, которые далее отфильтровывали. Полученный в результате порошок дважды промывали этанолом и затем сушили с получением 120 мг (0,37 ммоль) 1-((бензотиофен-3-ил)метил)-5-метоксибензимидазол-2-тиола (выход за две стадии: 30%).
Стадия 3
Получение этилового эфира 4-(1-((бензотиофен-3-ил)метил)-5-метоксибензимидазол-2-илтио)бутаноата.
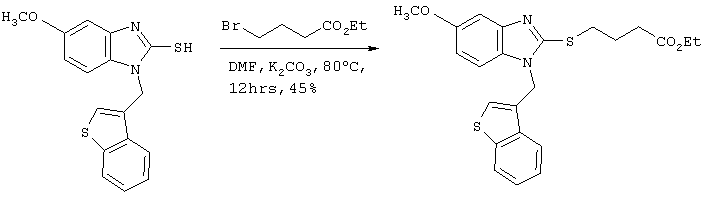
101 мг (0,30 ммоль) 1-((бензотиофен-3-ил)метил)-5-метокси-бензимидазол-2-тиола растворяли в 2 мл диметилформамида с последующим добавлением 62 мг (0,45 ммоль) карбоната калия и 53 мг (0,40 ммоль) этилового эфира 4-бромбутаноата и нагреванием до 80°С. Через 12 часов реакционный раствор концентрировали при пониженном давлении и экстрагировали диэтиловым эфиром, после чего дважды промывали 10 мл насыщенного рассола и сушили над сульфатом магния. Затем растворитель концентрировали при пониженном давлении, и остаток очищали методом хроматографии на колонке с силикагелем (гексан:этилацетат =1:1) с получением 60 мг (0,136 ммоль) этилового эфира 4-(1-((бензотиофен-3-ил)метил)-5-метоксибензимидазол-2-илтио)бутаноата (выход: 45%).
Стадия 4
Получение 4-(1-((бензотиофен-3-ил)метил)-5-метоксибенз-имидазол-2-илтио)бутановой кислоты
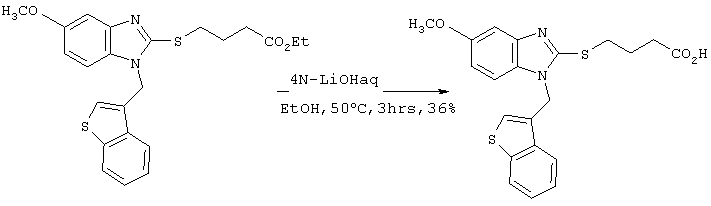
60 мг (0,136 ммоль) этилового эфира 4-(1-((бензотиофен-3-ил)метил)-5-метоксибензимидазол-2-илтио)бутаноата растворяли в 2 мл метанола с последующим добавлением 0,5 мл 4М водного раствора гидроксида натрия. После перемешивания в течение 3 часов при 50°С для завершения реакции добавляли 6М хлористоводородную кислоту, после чего проводили концентрирование при пониженном давлении и экстракцию хлороформом. После промывания органической фазы насыщенным рассолом, ее сушили над безводным сульфатом магния. Растворитель концентрировали при пониженном давлении, и остаток подвергали хроматографической очистке на колонке с силикагелем (этилацетат) с получением 20 мг (0,048 ммоль) целевого соединения (выход: 36%). Подтверждение структуры полученного соединения осуществляли путем идентификации молекулярного веса с помощью ЖХ-МС анализа.
Рассчитанное значение М=412,09; Измеренное значение (М+Н)+=413,1
[Пример 4]
Получение соединения №135
Целевое соединение получали согласно методике Примера 3.
Однако в реакции, соответствующей стадии 1, использовали иодистый ((1,4-диметилиндол-3-ил)метил)триметиламмоний.
Подтверждение структуры полученного соединения осуществляли путем идентификации молекулярного веса с помощью ЖХ-МС анализа.
Рассчитанное значение М=423,16; Измеренное значение (М+Н)+=424,3
Получение соединения №137
Целевое соединение получали по методике Примера 3.
Однако в реакции, соответствующей стадии 1, использовали иодистый ((1-метил-4-хлориндол-3-ил)метил)триметиламмоний.
Подтверждение структуры полученного соединения осуществляли путем идентификации молекулярного веса с помощью ЖХ-МС анализа.
Рассчитанное значение М=443,11; Измеренное значение (М+Н)+=444,3
[Пример 5]
Получение соединения №244
Целевое соединение получали с использованием методики Примера 3. В качестве реагента, используемого на стадии 1, применяли 4-циано-2-нитротрифторацетонитрил. Кроме этого, стадию, на которой производное 2-нитроанилина восстанавливали в производное ортофенилендиамина, и стадию, на которой это соединение циклизовали в производное бензимидазол-2-тиола, проводили с использованием описанных ниже методик.
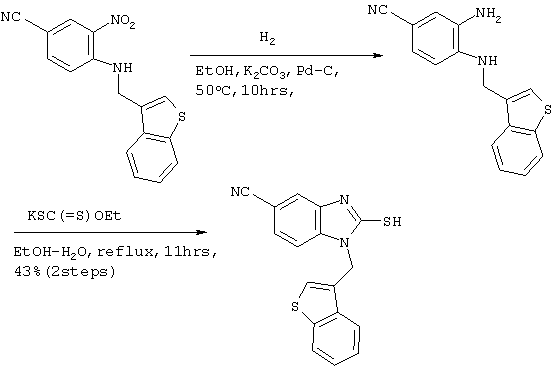
10 мл этанола добавляли к 1,1 г (3,56 ммоль) ((3-бензотиофенил)метил)(4-циано-2-нитрофенил)амина с последующим добавлением 2,4 г (17,8 ммоля) карбоната калия. После продувки реакционной системы азотом, добавляли 220 мг 10% палладия на угле, после чего реакционную систему заполняли водородом и нагревали до 60°С.
Через 4 часа 30 минут добавляли еще 220 мг 10% палладия на угле, после чего реакционную систему заполняли водородом и нагревали до 60°С. Через 5 часов 10 минут после начала реакции, реакционную систему охлаждали до комнатной температуры. Реакционный раствор фильтровали через целит, и концентрировали при пониженном давлении с получением 0,93 г жидкого остатка. Затем 0,93 г (2,63 ммоль) ((2-бензотиофенил)метил)(2-амино-4-метилфенил)амина растворяли в 10 мл этанола и 2 мл воды с последующим нагреванием с обратным холодильником после добавления 2,1 г (13,3 ммоль) этилксантата калия. Через 11 часов прикапывали 12,5 мл 40% водного раствора уксусной кислоты. После охлаждения до комнатной температуры и концентрирования при пониженном давлении, остаток подвергали хроматографической очистке на колонке с силикагелем (гексан:ацетон =2:1) с получением 491,7 мг 1-((2-бензотиофенил)метил)-6-цианобензимидазол-2-тиола (выход за две стадии: 43%). Подтверждение структуры соединения №244 осуществляли путем идентификации молекулярного веса с использованием методов 1H-ЯМР и ЖХ-МС.
Вычисленное значение М=407,08; Измеренное значение (М+Н)+=408,2.
Спектр 1Н-ЯМР (270 Мгц, CDCl3) (ч/млн): 7,94 (с, 1Н), 7,76 (дд, 1Н), 7,52 (дд, 1Н), 7,42 (м, 3Н), 7,31 (д, 1Н), 7,00 (с, 1Н), 5,56 (с, 2Н), 3,35 (т, 2Н), 2,47 (т, 2Н), 2,15 (п, 2Н)
[Пример 6]
Следующие целевые соединения получали с использованием методики Примера 5.
Получение соединения №340
В качестве реагента, соответствующего Стадии 1, использовали 4-метил-2-нитротрифторацетоанилид.
Подтверждение структуры соединения №340 осуществляли идентификацией по молекулярному весу с использованием ЖХ-МС анализа. Вычисленное значение М=396,10; Измеренное значение (М+Н)+=397,0.
Получение соединения №436
В качестве реагента, соответствующего Стадии 1, использовали 5-метил-2-нитротрифторацетоанилид.
Подтверждение структуры соединения №436 осуществляли идентификацией по молекулярному весу с использованием ЖХ-МС анализа.
Вычисленное значение М=396,10; Измеренное значение (М+Н)+=397,0.
[Пример 7]
Получение соединения №34
Стадия 1
Получение ((1-метилиндол-3-ил)метил)(2-аминофенил)амина
829 мг (6 ммоль) 2-нитроанилина и 1242 мг (7,8 ммоль) 1-метилиндол карбоксиальдегида растворяли в 20 мл тетрагидрофурана с последующим последовательным добавлением 200 мкл уксусной кислоты и 5087 мг (24 ммоль) NaBH(OAc)3 и перемешиванием в течение ночи при комнатной температуре. После добавления насыщенного водного раствора бикарбоната натрия, экстракции этилацетатом и сушки над безводным сульфатом магния, растворитель отгоняли и остаток подвергали хроматографической очистке на колонке с силикагелем (гексан:этилацетат =95:5) с получением 264 мг ((1-метилиндол-3-ил)метил)(2-нитрофенил)амина (выход: 18%). Затем 264 мг (0,939 ммоль) ((1-метилиндол-3-ил)метил)(2-нитрофенил)амина растворяли в 10 мл этанола с последующим добавлением 50 мг (0,047 ммоль) 10% Pd-C и перемешиванием системы в течение 6 часов при комнатной температуре в атмосфере водорода. После завершения реакции, Pd-C отфильтровывали и растворитель отгоняли при пониженном давлении с получением 212 мг ((1-метилиндол-3-ил)метил)(2-аминофенил)амина (выход: 90%).
Стадия 2
Получение 1-((1-метилиндол-3-ил)метил)бензимидазол-2-тиола
212 мг (0,845 ммоль) ((1-метилиндол-3-ил)метил)(2-аминофенил)амина растворяли в 1мл пиридина с последующим добавлением 1 мл (16,9 ммоль) сероуглерода и нагреванием с обратным холодильником в течение 1 часа в атмосфере азота. Растворитель отгоняли и остаток подвергали хроматографической очистке на колонке с силикагелем (гексан:этилацетат =2:1) с получением 96 мг 1-((1-метилиндол-3-ил)метил)бензимидазол-2-тиола (выход: 39%).
Стадия 3
Получение 4-(1-((1-метилиндол-3-ил)метил)бензимидазол-2-илтио)бутановой кислоты
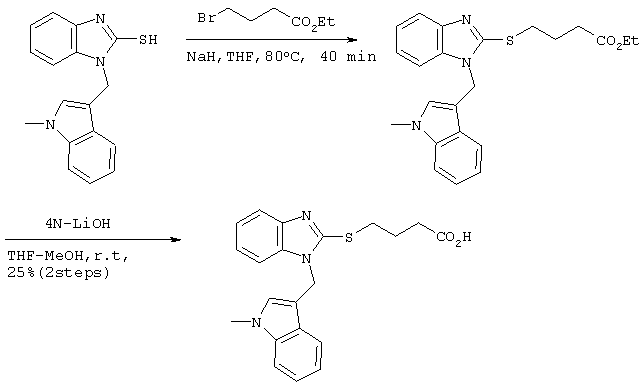
12 мг (0,342 ммоль) гидрида натрия и 2 мл тетрагидрофурана загружали в предварительно высушенный реакционный сосуд. Далее в реакционный сосуд добавляли 50 мг (0,171 ммоль) 1-((1-метилиндол-3-ил)метил)бензимидазол-2-тиола и 34 мкл (0,23 ммоль) этилового эфира 4-бромбутаноата, после чего смесь перемешивали в течение 40 минут при 60°С. Затем добавляли воду, и смесь экстрагировали этилацетатом. После сушки этилацетатной фазы над безводным сульфатом магния, реакционный раствор концентрировали при пониженном давлении и полученный в результате остаток подвергали хроматографической очистке на колонке с силикагелем (гексан:этилацетат =3:1) с получением этилового эфира 4-(1-((1-метилиндол-3-ил)метил)(бензимидазол-2-илтио)бутаноата. Далее 0,25 мл 4М водного раствора гидроксида лития добавляли к 1 мл тетрагидрофурана, содержащего указанный этиловый эфир 4-(1-((1-метилиндол-3-ил)метил)(бензимидазол-2-илтио)бутаноата и 0,5 мл метанола. После перемешивания в течение ночи при комнатной температуре, для завершения реакции добавляли 6М раствор хлористоводородной кислоты, после чего смесь экстрагировали этилацетатом. После промывания этилацетатной фазы насыщенным рассолом, ее сушили над безводным сульфатом магния. Растворитель отгоняли при пониженном давлении с получением 16 мг (0,042 ммоль) целевого соединения (выход: 25%).
Подтверждение структуры соединения осуществляли идентификацией по молекулярному весу с использованием ЖХ-МС анализа.
Вычисленное значение М=379,14; Измеренное значение (М+Н)+=380,2.
[Пример 8]
Получение 5-(1-((1,4-диметилиндол-3-ил)метил)бензимидазол-2-ил)пентановой кислоты
Стадия 1
Получение этилового эфира 5-(бензимидазол-2-ил)пентаноата
696 мкл (5,0 ммоль) триэтиламина и 893 мг (5,0 ммоль) метиладипохлорида прикапывали к 10 мл хлороформного раствора, содержащего 540 мг (5,0 ммоль) ортофенилендиамина с последующим перемешиванием в течение 12 часов при комнатной температуре. Затем добавляли 20 мл этанола и 4 мл концентрированной хлористоводородной кислоты, после чего смесь перемешивали в течение 10 часов при нагревании с обратным холодильником. После этого, реакционный раствор нейтрализовали с использованием 5М водного раствора гидроксида натрия и раствор экстрагировали этилацетатом. После промывания водой и концентрирования при пониженном давлении, остаток подвергали хроматографической очистке на колонке с силикагелем (только этилацетат) с получением 359 мг этилового эфира 5-(бензимидазол-2-ил)пентаноата (выход: 30%).
Стадия 2
Получение 5-(1-((1,4-диметилиндол-3-ил)метил)бензимидазол-2-ил)пентановой кислоты
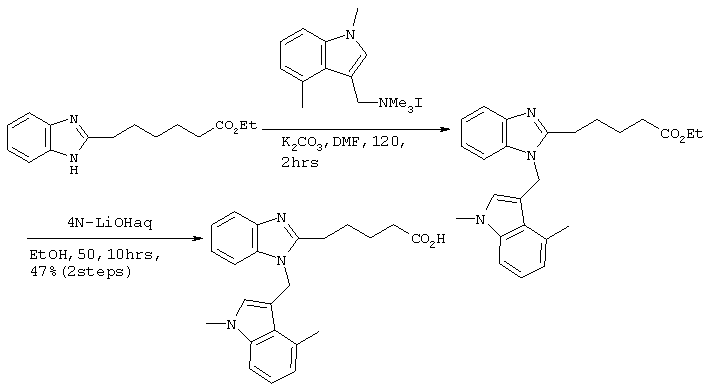
42 мг (0,3 ммоль) карбоната калия и 103 мг (0,3 ммоль) иодистого ((1,4-диметилиндол-3-ил)метил)триметиламмония добавляли к 2 мл ДМФ раствора, содержащего 50 мг (0,2 ммоль) этилового эфира 5-(бензимидазол-2-ил)пентаноата с последующим перемешиванием в течение 2 часов при 120°С. Полученный в результате раствор экстрагировали дихлорметаном, промывали водой и концентрировали, после чего полученный остаток подвергали очистке методом колоночной хроматографии (гексан:этилацетат =1:2). В полученную систему добавляли 5 мл этанола и 0,5 мл 4М водного раствора гидроксида натрия, после чего смесь перемешивали в течение 10 часов при 50°С и затем для прекращения реакции, добавляли 6М хлористоводородную кислоту. Полученный раствор экстрагировали хлороформом и, после промывки водой и концентрирования при пониженном давлении, остаток подвергали хроматографической очистке на колонке с силикагелем с получением 35 мг целевого соединения (выход за две стадии: 47%). Подтверждение структуры соединения осуществляли идентификацией по молекулярному весу с использованием ЖХ-МС анализа.
Вычисленное значение М=375,19; Измеренное значение (М+Н)+=376,5.
[Пример 9]
Получение натриевой соли соединения №519
11,9 мл (1,19 ммоль) 0,1М водного раствора гидроксида натрия добавляли к 100 мл водного раствора, содержащего 503 мг (1,19 ммоль) указанного выше соединения №519, после чего раствор перемешивали при комнатной температуре. После этого, реакционный раствор сушили вымораживанием с получением 470 мг (1,05 ммоль) натриевой соли (выход: 89%).
Спектр 1H-ЯМР (400 Мгц, ДМСО-d6) (ч/млн): 7,37 (с, 1Н), 7,19 (д, 1Н, J=8,24 Гц), 7,09-7,01 (м, 2Н), 6,80 (д, 1Н, J=7,09 Гц), 6,32 (с, 1Н), 5,66 (с, 2Н), 3,59 (с, 3Н), 3,26 (м, 2Н), 2,66 (с, 3Н), 2,27 (с, 3Н), 2,21 (с, 3Н), 1,95 (м, 2Н), 1,81 (м, 2Н).
[Пример 10]
Указанные ниже соединения синтезировали по методике Примера 9 с использованием соответствующих субстратов.
Натриевая соль соединения №39
Спектр 1Н-ЯМР (270 Мгц, ДМСО-d6) (ч/млн): 7,57 (д, 1Н,), 7,28 (д, 1Н, J=7 Гц), 7,20 (д, 1Н, J=8 Гц), 7,15-7,00 (м, 3Н), 6,77 (д, 1Н, J=7 Гц), 6,47 (с, 1Н), 5,69 (с, 2Н), 3,60 (с, 3Н), 3,31 (т, 2Н, J=7 Гц), 2,61 (с, 3Н), 1,99 (т, 2Н, J=7 Гц), 1,84 (п, 2Н, J=7 Гц)
Натриевая соль соединения №52
Спектр 1H-ЯМР (400 Мгц, ДМСО-d6) (ч/млн): 7,97 (д, 1Н), 7,91 (д, 1Н, J=6,76 Гц), 7,57 (д, 1Н, J=7,75 Гц), 7,44-7,38 (м, 3Н), 7,30 (с, 1Н), 7,12 (м, 2Н), 5,63 (с, 2Н), 3,33 (м, 2Н), 2,03 (м, 2Н), 1,87 (м, 2Н).
Натриевая соль соединения №135
Спектр 1H-ЯМР (400 Мгц, ДМСО-d6) (ч/млн): 7,21-7,00 (м, 4Н), 6,79 (д, 1Н, J=7,29 Гц), 6,67 (дд, 1Н, J=2,43, 8,91 Гц), 6,51 (с, 1Н), 5,65 (с, 2Н), 3,75 (с, 3Н), 3,62 (с, 3Н), 3,31 (м, 2Н), 2,59 (с, 3Н), 1,95 (м, 2Н), 1,82 (м, 2Н).
Натриевая соль соединения №532
Спектр 1Н-ЯМР (400 Мгц, ДМСО-d6) (ч/млн): 7,98 (д, 1Н, J=7,42 Гц), 7,90 (д, 1Н, J=6,43 Гц), 7,44-7,39 (м, 2Н), 7,35 (с, 1Н), 7,18 (м, 2Н), 5,57 (с, 2Н), 3,28 (м, 2Н), 2,26 (с, 3Н), 2,23 (с, 3Н), 1,99 (м, 2Н), 1,84 (м, 2Н).
[Пример 10]
Получение этилового эфира 4-(1-((4-метилбензотиофен-3-ил)метил)-5-метоксибензимидазол-2-илтио)бутаноата и этилового эфира 4-(1-((4-метилбензотиофен-3-ил)метил)-6-метоксибензимидазол-2-илтио)бутаноата
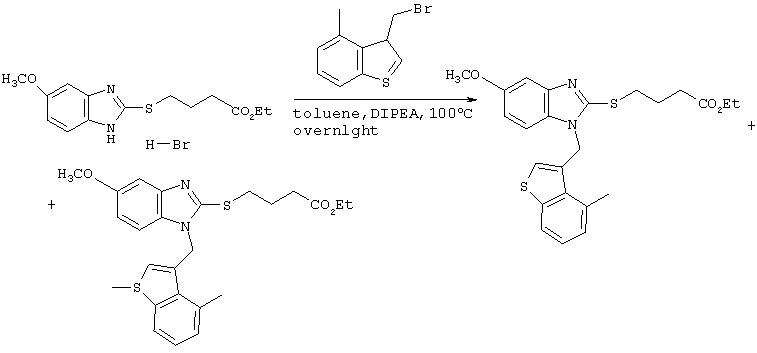
539 мг (1,44 ммоль) этилового эфира 4-(5-метоксибензимидазол-2-илтио)бутаноата суспендировали в 4 мл толуола, после чего добавляли 616 мкл (3,60 ммоль) диизопропилэтиламина и 384 мг (1,59 ммоль) 4-метил-3-(бромометил)бензо[b]тиофена и смесь нагревали при 100°С. После проведения реакции в течение ночи добавляли насыщенный раствор бикарбоната натрия, после чего проводили экстракцию этилацетатом. Органическую фазу промывали водой, после чего сушили над сульфатом магния и концентрировали растворитель при пониженном давлении. Полученный в результате остаток подвергали хроматографической очистке на колонке с силикагелем (гексан:этилацетат =4:1) с получением 114 мг этилового эфира 4-(1-((4-метилбензотиофен-3-ил)метил)-5-метоксибенз-имидазол-2-илтио)бутаноата и 68 мг этилового эфира 4-(1-((4-метилбензотиофен-3-ил)метил)-6-метоксибензимидазол-2-илтио)-бутаноата (выход: 10%).
Этиловый эфир 4-(1-((4-метилбензотиофен-3-ил)метил)-5-метоксибензимидазол-2-илтио)бутаноата
Спектр 1Н-ЯМР (270 Мгц, CDCl3) (ч/млн): 7,71 (д, 1Н, J=7,56 Гц), 7,62 (д, 1Н, J=8,64 Гц), 7,30-7,18 (м, 2Н), 6,87 (дд, 1Н, J=2,43, 8,64 Гц), 6,61 (д, 1Н, J=2,43 Гц), 6,42 (с, 2Н), 5,74 (с, 2Н), 4,10 (кв, 2Н, J=7,29 Гц), 3,75 (с, 2Н), 3,38 (т, 2Н, J=7,29 Гц), 2,89 (с, 3Н), 2,45 (т, 2Н, J=7,29 Гц), 2,11 (м, 2Н), 1,23 (т, 3Н, J=7,29 Гц)
Этиловый эфир 4-(1-((4-метилбензотиофен-3-ил)метил)-6-метоксибензимидазол-2-илтио)бутаноата
Спектр 1Н-ЯМР (270 Мгц, CDCl3) (ч/млн): 7,70 (д, 1Н, J=8,10 Гц), 7,29-7,17 (м, 3Н), 7,02 (д, 1Н, J=8,91 Гц), 6,80 (дд, 1Н, J=2,43, 8,91 Гц), 6,40 (с, 1Н), 5,74 (с, 2Н), 4,11 (кв, 2Н, J=7,29 Гц), 3,87 (с, 3Н), 3,42 (т, 2Н, J=7,02 Гц), 2,88 (с, 3Н), 2,46 (т, 2Н, J=7,29 Гц), 2,10 (м, 2Н), 1,23 (т, 3Н, J=7,29 Гц)
[Пример 11]
Следующие ниже соединения получали по методике Примера 10.
Этиловый эфир 4-(1-((5-метилбензотиофен-3-ил)метил)-5-метоксибензимидазол-2-илтио)бутаноата
(Выход: 24%)
Спектр 1Н-ЯМР (270 Мгц, CDCl3) (ч/млн): 7,76 (д, 1Н, J=8,10 Гц), 7,62 (с, 1Н), 7,58 (д, 1Н, J=8,64 Гц), 7,25 (1Н), 6,84 (дд, 1Н, J=2,43, 8,91 Гц), 6,81 (с, 1Н), 6,65 (д, 1Н, J=2,16 Гц), 5,47 (с, 2Н), 4,11 (кв, 2Н, J=7,02 Гц), 3,74 (с, 3Н), 3,39 (т, 2Н, J=7,02 Гц), 2,51 (с, 3Н), 2,47 (т, 2Н, J=7,56 Гц), 2,11 (м, 2Н), 1,24 (т, 3Н, J=7,02 Гц)
Этиловый эфир 4-(1-((5-метилбензотиофен-3-ил)метил)-6-метоксибензимидазол-2-илтио)бутаноата
(Выход: 18%)
Спектр 1Н-ЯМР (270 Мгц, CDCl3) (ч/млн): 7,75 (д, 1Н, J=8,10 Гц), 7,60 (с, 1Н), 7,26-7,22 (м, 2Н), 7,04 (д, 1Н, J=8,91 Гц), 6,83 (с, 1Н), 6,78 (дд, 1Н, J=2,43, 8,91 Гц), 5,47 (с, 2Н), 4,12 (кв, 2Н, J=7,02 Гц), 3,84 (с, 3Н), 3,43 (т, 2Н, J=7,29 Гц), 2,50 (с, 3Н), 2,48 (т, 2Н, J=7,29 Гц), 2,12 (м, 2Н), 1,24 (т, 3Н, J=7,02 Гц)
[Пример 12]
Получение 4-(1-((4-метилбензотиофен-3-ил)метил)-5-метоксибензимидазол-2-илтио)бутановой кислоты (Соединение №154)
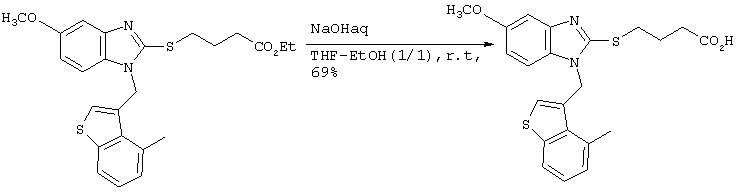
84,7 мг (0,186 ммоль) этилового эфира 4-(1-((4-метилбензотиофен-3-ил)метил)-5-метоксибензимидазол-2-илтио)-бутаноата, полученного в Примере 10, растворяли в смешанном растворителе из 1 мл ТГФ и 1 мл этанола, после чего добавляли 1 мл 1М водного раствора гидроксида натрия и полученную смесь перемешивали в течение 1 часа при 40°0. После завершения реакции добавляли 1,5 мл 1М хлористоводородной кислоты с последующим перемешиванием в течение 30 минут при комнатной температуре. Полученный в результате осадок отфильтровывали, промывали водой, этанолом и сушили с получением 54,9 мг целевого соединения (выход: 69%). ЖХ-МС.
Вычисленное значение М=426,11; Измеренное значение (М+Н)+=427,2
Спектр 1Н-ЯМР (270 Мгц, ДМСО-d6) (ч/млн): 7,80 (д, 1Н, J=7,29 Гц), 7,60 (д, 1Н, J=8,91 Гц), 7,31-7,20 (м, 3Н), 6,95 (дд, 1H, J=2,16, 8,91 Гц), 6,53 (с, 1Н), 5,94 (с, 2Н), 3,73 (с, 3Н), 3,37 (т, 2Н, J=7,29 Гц), 2,86 (с, 3Н), 2,34 (т, 2Н, J=7,29 Гц), 1,90 (м, 2Н)
[Пример 13]
Следующие соединения синтезировали по методике Примера 12.
4-(1-((4-метилбензотиофен-3-ил)метил)-6-метоксибензимидазол-2-илтио)бутановая кислота (Соединение №1114)
Выход: 60%. ЖХ-МС: Вычисленное значение М=426,11; Измеренное значение (М+Н)+=427,2
Спектр 1Н-ЯМР (270 Мгц, ДМСО-d6) (ч/млн): 7,78 (д, 1Н, J=7,83 Гц), 7,52 (д, 1Н, J=8,91 Гц), 7,34-7,17 (м, 3Н), 6,77 (дд, 1Н, J=2,34, 8,91 Гц), 6,37 (с, 1Н), 5,83 (с, 2Н), 3,78 (с, 3Н), 3,32 (т, 2H, J=7,29 Гц), 2,82 (с, 3Н), 2,34 (т, 2Н, J=7,56 Гц), 1,93 (м, 2Н)
Однако в этом случае, в реакционную систему после завершения реакции добавляли 1М хлористоводородную кислоту, после чего проводили экстракцию хлороформом и промывание водой. Затем систему сушили над сульфатом магния, растворитель концентрировали при пониженном давлении и остаток сушили с получением целевого соединения.
4-(1-((5-метилбензотиофен-3-ил)метил)-5-метоксибензимидазол-2-илтио)бутановая кислота (Соединение №152)
Выход: 63%. ЖХ-МС: Вычисленное значение М=426,11; Измеренное значение (М+Н)+=426,8
Спектр 1Н-ЯМР (270 Мгц, ДМСО-d6) (ч/млн): 7,88 (д, 1Н, J=8,64 Гц), 7,76 (с, 1Н), 7,58 (д, 1Н, J=8,64 Гц), 7,28-7,24 (м, 3Н), 6,94 (дд, 1Н, J=2,16, 8,64 Гц), 5,72 (с, 2Н), 3,74 (с, 3Н), 3,40 (т, 2Н, J=7,29 Гц), 2,42 (с, 3Н), 2,36 (т, 2Н, J=7,29 Гц), 1,92 (м, 2Н)
4-(1-((5-метилбензотиофен-3-ил)метил)-6-метоксибензимидазол-2-илтио)бутановая кислота (Соединение №1112)
Выход: 79%. ЖХ-МС: Вычисленное значение М=426,11; Измеренное значение (М+Н)+=427,0
Спектр 1Н-ЯМР (270 МГц, ДМСО-d6) (ч/млн): 7,87 (д, 1Н, J=8,10 Гц), 7,71 (с, 1Н), 7,47 (д, 1Н, J=8,91 Гц), 7,24 (м, 2Н), 7,17 (д, 1Н, J=2,16 Гц), 6,84 (дд, 1Н), 5,64 (с, 2Н), 3,77 (с, 3Н), 3,38 (т, 2Н, J=7,02 Гц), 2,41 (с, 3Н), 2,37 (т, 2Н, J=7,56 Гц), 1,95 (м, 2Н)
[Пример 14]
Получение HCl соли соединения №532

1,5 мл 4М раствора хлористоводородной кислоты в диоксане добавляли к 50 мг (0,122 ммоль) соединения №532 с последующим перемешиванием при 100°С. После завершения реакции реакционный раствор концентрировали при пониженном давлении с получением 53 мг (1,05 ммоль) целевого соединения (выход: 97%).
Спектр 1Н-ЯМР (270 Мгц, ДМСО-d6) (ч/млн): 8,00 (м, 1Н), 7,89 (м, 1Н), 7,52 (м, 2Н), 7,45-7,42 (м, 2Н), 7,32 (с, 1Н), 5,78 (с, 2Н), 3,48 (т, 2Н, J=7,42 Гц), 2,37 (м, 2Н), 2,34 (с, 3Н), 2,30 (с, 3Н), 1,92 (т, 2Н, J=7,09 Гц)
[Пример 15]
Получение HCl соли соединения №56
Целевое соединение получали по методике Примера 14.
Спектр 1Н-ЯМР (270 Мгц, ДМСО-d6) (ч/млн): 7,87 (д, 1Н, J=8,08 Гц), 7,74 (с, 1Н), 7,66 (д, 1Н, J=6,76 Гц), 7,58 (д, 1Н, J=8,74 Гц), 7,26 (м, 4Н), 5,70 (с, 2Н), 3,45 (т, 2Н, J=7,26 Гц), 2,42 (с, 3Н), 2,39 (т, 2Н, J=7,26 Гц), 1,98 (м, 2Н)
[Пример 16]
Получение химазы рекомбинантных человеческих мастоцитов
Химазу рекомбинантных человеческих мастоцитов получали в соответствие с сообщением Urata с сотр., (Journal of Biological Chemistry, v.266, p.17173 (1991)). Химазу человеческих мастоцитов очищали гепарин сефарозой (Pharmacia) от клеток насекомых (Th5), инфицированных рекомбинантным бациловирусом, содержащим кДНК, кодирующую химазу человеческих мастоцитов. Более того, после активации в соответствие с сообщением Murakami с сотр. (Journal of Biological Chemistry, vol.270, p.2218 (1995)), химазу человеческих мастоцитов очищали гепарин сефарозой для получения активной химазы человеческих мастоцитов.
[Пример 17]
Измерение ингибирования энзимной активности химазы рекомбинантных человеческих мастоцитов.
После добавления 2 мкл ДМСО раствора, содержащего соединение настоящего изобретения, к 50 мкл буфера А (0,5-3,0 М NaCl, 50 мМ Трис-HCl, рН 8,0), содержащего 1-5 нг активной химазы человеческих мастоцитов, полученной в Примере 16, добавляли 50 мкл Буфера А, содержащего в качестве субстрата 0,5 мМ сукцинил-аланил-гистидил-пролил-фенилаланилпаранитроанилид (Bacchem) и смеси давали реагировать в течение 5 минут при комнатной температуре. Для исследования ингибиторной активности снимали зависимость оптического поглощения при 405 нм от времени.
В результате было установлено, что соединения №№39, 56, 58, 59, 63, 148, 154, 519, 532, 534, 536, 538, 615, 1112 и 1114 проявляют ингибирующую активность IC50= от 1 нМ до менее 10 нМ, тогда как соединения №№34, 38, 41, 42, 52, 54, 135, 137, 152, 244, 340,436, 514, 521 и 628 проявляют ингибирующую активность IC50=10-100 нМ.
Как показано выше, производные бензимидазола настоящего изобретения обладают мощной ингибирующей активностью по отношению к химазе. Таким образом, продемонстрировано, что бензимидазольные производные настоящего изобретения являются ингибиторами активности человеческой химазы, которые могут применяться в клинических условиях для профилактики и/или лечения различных заболеваний, связанных с действием человеческой химазы.
[Пример 18]
Производство таблеток
Таблетки производили из индивидуального состава, представленного ниже.
Соединение настоящего изобретения (соединение из примеров), лактозу и картофельный крахмал смешивали друг с другом, затем однородно смачивали 20% этанольным раствором поливинилпирролидона, пропускали через сито с отверстиями 20 меш, сушили при 45°С и снова пропускали через сито с отверстиями 15 меш. После этого, полученные таким образом гранулы смешивали со стеаратом магния и прессовали в таблетки.
[Пример 19]
Измерение концентраций в крови крыс в ходе применения путем внутрижелудочного принудительного питания
Соединения, обозначенные выше номерами 39, 52 и 244, применяли путем внутрижелудочного принудительного питания голодных самцов SD крыс с дозировкой 30 мг/кг, после чего отбирали образцы крови сразу после применения и через 30 минут, 1, 2 и 4 часа после применения. После отбора образцов крови их немедленно разделяли на сывороточные компоненты, соединение настоящего изобретения экстрагировали обычными методами твердофазной экстракции и полученные в результате образцы анализировали методом ЖХВР с использованием колонки ODS (в качестве подвижной фазы для анализа соединений №№52 и 244 использовали фазу 32% ацетонитрила-вода - 0,05% TFA, тогда как для анализа соединения №39, в качестве подвижной фазы использовали систему 47% ацетонитрила-вода - 10 мМ аммоний ацетатный буфер (рН 4,0)) после чего измеряли количество неизменной формы. Полученные результаты приведены в следующей таблице 2.
Из приведенных выше результатов можно заключить, что соединения настоящего изобретения быстро абсорбируются после применения и указанные в таблице концентрации неизменной формы в крови были измерены через 30 минут. Более того, хотя концентрации в крови постепенно уменьшаются к 4 часу после применения, значительные количества неизменных форм сохраняются в крови даже через 4 часа после применения. Таким образом, установлено, что соединения настоящего изобретения представляют собой группу соединений с превосходными фармакокинетическими свойствами. Особенно хорошими фармакокинетическими свойствами обладает группа веществ, в которой А представляет собой -СН2СН2СН2-.
[Пример 20]
In vitro тест на метаболизм с использованием микросом печени (Ms)
Метод измерения:
* Состав реакционного раствора и условия проведения реакции
*Метод расчета MR
Скорость метаболизма определяли по уменьшению количества неизмененной формы в каждое из указанных времен реакции и во время реакции, соответствующее величине в 100%, приписанной количеству неизменной формы при исходной концентрации (время реакции: 0 минут), причем скорость метаболизма во время достижения максимального значения оценивали, как величину MR.
MR = (концентрация субстрата во время реакции: 0 мин - концентрация субстрата после реакции)÷время реакции÷концентрация протеина (нмоль/мин/мг протеина)
Указанные методы использовали для получения результатов, представленных в таблице 3.
В соответствие с приведенными выше результатами, соединения настоящего изобретения представляют собой группу метаболически устойчивых соединений. Группа соединений, в которых А представляет собой -СН2СН2СН2-, является группой метаболически особенно устойчивых соединений.
Промышленная применимость
Бензимидазольные производные настоящего изобретения или их медицински применимые соли демонстрируют мощную ингибиторную активность в отношении человеческой химазы. Вследствие этого, бензимидазольные производные настоящего изобретения или их медицински применимые соли могут использоваться в качестве профилактических и/или терапевтических агентов, которые могут применяться в клинических условиях в качестве ингибиторов человеческой химазы при воспалительных заболеваниях, аллергических заболеваниях, респираторных заболеваниях, сердечно-сосудистых заболеваниях или костных и хрящевых метаболических заболеваниях.
Дополнительные примеры соединений по изобретению и их некоторые физико-химические и биологические свойства
4-(5-трифторметил-1((4-метилбензо[b]тиофен-3-ил)метил)бензимидазол-2-илтио)бутановая кислота формулы
C22H19 F3N2O2S2
Точно масс.: 464,08; М. вес: 464,52.
Рассчитанная величина М+1=465,2 IC50: 1,02Е-08 (М)
4-(5-гидрокси-1-((4-метилбензо[b]тиофен-3-ил)метил)бензимидазол-2-илтио)бутановая кислота формулы
C21H20N2O3S2
Точно масс.: 412,09; М. вес: 412,09.
Рассчитанная величина М+1=413,2 IC50: 1,09Е-08 (М)
4-(6-гидрокси-1-((4-метилбензо[b]тиофен-3-ил)метил)бензимидазол-2-илтио)бутановая кислота формулы
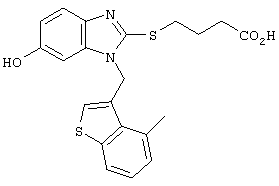
C21H20N2O3S2
Точно масс.: 412,09; М. вес: 412,53.
Рассчитанная величина М+1=413,2 IC50: 1,58Е-08 (М)
4-(1-((4-трифторметоксибензо[b]тиофен-3-ил)метил)бензимидазол-2-илтио)бутановая кислота формулы
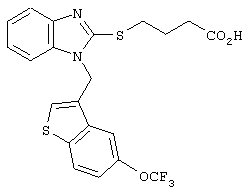
С21Н17F3N2O3S2
Точно масс.: 466,06; М. вес: 466,5.
Рассчитанная величина М+1=467,0 IC50: 5,20Е-07 (М)
4-(1-((5-трифторметилбензо[b]тиофен-3-ил)метил)бензимидазол-2-илтио)бутановая кислота формулы
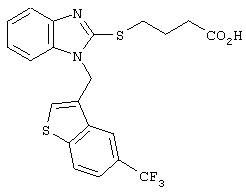
C21H17F3N2O2S2
Точно масс.: 450,07; М. вес: 450,5.
Рассчитанная величина М+1=451,2 IC50: 7,52Е-08 (М)
4-(1-((4-метилбензо[b]тиофен-3-ил)метил)бензимидазол-2-илтио)бутановая кислота формулы
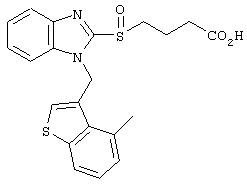
C21H20N2O3S2
Точно масс.: 412,09; М. вес: 412,53.
Рассчитанная величина М+1=413,0 IC50: 8,33Е-08 (М).
4-(1-((4-метилбензо[b]тиофен-3-ил)метил)бензимидазол-2-илсульфонил)бутановая кислота формулы
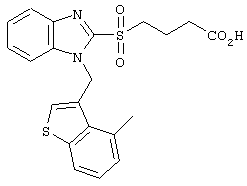
C21H20N2O4S2
Точно масс.: 428,09; М. вес: 428,52.
Рассчитанная величина М+1=429,2 IC50: 6,10Е-08 (М).
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ ПРОИЗВОДНЫЕ АЦЕТАМИДА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКИЙ СОСТАВ И ИНГИБИТОРЫ ПРОТЕАЗ НА ИХ ОСНОВЕ | 1997 |
|
RU2181360C2 |
| ПРОИЗВОДНОЕ ТИОБЕНЗИМИДАЗОЛА И СОДЕРЖАЩИЕ ЕГО ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 1999 |
|
RU2237663C2 |
| ПРОИЗВОДНЫЕ ТИОФЕНА | 2003 |
|
RU2296758C2 |
| ПРОИЗВОДНЫЕ ПИРИДИНА | 1992 |
|
RU2035461C1 |
| МАКРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ ПИРИДАЗИНОНА | 2014 |
|
RU2660435C2 |
| ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1998 |
|
RU2243968C2 |
| ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛА, ПОЛЕЗНЫЕ ПРИ ЛЕЧЕНИИ РЕСПИРАТОРНО-СИНЦИТИАЛЬНОЙ ВИРУСНОЙ ИНФЕКЦИИ | 2012 |
|
RU2612530C2 |
| АНТАГОНИСТЫ РЕЦЕПТОРА FLT3 | 2015 |
|
RU2710928C2 |
| ОМЕГА-АМИНОАЛКИЛАМИДЫ R-2-АРИЛПРОПИОНОВЫХ КИСЛОТ В КАЧЕСТВЕ ИНГИБИТОРОВ ХЕМОТАКСИСА ПОЛИМОРФНОЯДЕРНЫХ И ОДНОЯДЕРНЫХ КЛЕТОК, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2002 |
|
RU2272024C2 |
| ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛА, СОДЕРЖАЩИЕ ИХ КОМПОЗИЦИИ, ИХ ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ | 2004 |
|
RU2346938C2 |
Изобретение относится к бензимидазольным производным или их применимым в медицине солям, общей формулы (1)

где R1 и R2 могут иметь одинаковые или различные значения и, независимо друг от друга, представляют собой атом водорода, атом галогена, циано группу, гидроксильную группу, алкильную группу, содержащую 1-4 углеродных атома, алкокси группу, содержащую 1-4 углеродных атома, трифторметильную группу; А представляет собой незамещенную, линейную алкиленовую группу, содержащую 1-7 углеродных атомов; Е представляет собой группу -COOR3, где R3 представляет собой атом водорода или линейную алкильную группу, содержащую 1-6 углеродных атома; G представляет собой незамещенную, линейную алкиленовую группу, содержащую 1-6 углеродных атомов; М представляет собой простую связь или -S(O)m-, где m представляет собой целое число в интервале 0, 1 или 2; J представляет собой замещенную или незамещенную гетероциклическую группу, содержащую 4-10 углеродных атомов и один гетероатом в кольце, выбранный из группы, состоящей из атома азота или атома серы, исключая незамещенное пиридиновое кольцо; заместитель в указанной ароматической гетероциклической группе выбирают из атома галогена, циано группы, линейной алкильной группы, содержащей 1-6 углеродных атомов, линейной алкокси группы, содержащей 1-6 углеродных атомов, трифторметильной группы и трифторметокси группы; причем один или более из указанных заместителей могут быть замещены по произвольным положениям кольца; и Х представляет собой метиновую группу (-СН=). Изобретение также относится к фармацевтической композиции для ингибирования активности химазы человека, на основе этих соединений. Технический результат - получение новых соединений и фармацевтической композиции на их основе в целях профилактики и/или лечения воспалительного заболевания, сердечно-сосудистого заболевания, аллергического заболевания, респираторное заболевание или костного либо хрящевого метаболического заболевания. 4 н. и 10 з.п. ф-лы, 3 табл.
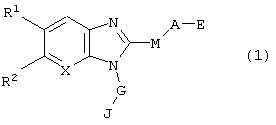
где R1 и R2 могут иметь одинаковые или различные значения и независимо друг от друга представляют собой атом водорода, атом галогена, цианогруппу, гидроксильную группу, алкильную группу, содержащую 1-4 углеродных атома, алкоксигруппу, содержащую 1-4 углеродных атома, трифторметильную группу;
А представляет собой незамещенную линейную алкиленовую группу, содержащую 1-7 углеродных атомов;
Е представляет собой группу -COOR3, где R3 представляет собой атом водорода или линейную алкильную группу, содержащую 1-6 углеродных атома;
G представляет собой незамещенную линейную алкиленовую группу, содержащую 1-6 углеродных атомов;
М представляет собой простую связь или -S(O)m-, где m представляет собой целое число в интервале 0, 1 или 2;
J представляет собой замещенную или незамещенную гетероциклическую группу, содержащую 4-10 углеродных атомов и один гетероатом в кольце, выбранный из группы, состоящей из атома азота или атома серы, исключая незамещенное пиридиновое кольцо; заместитель в указанной ароматической гетероциклической группе выбирают из атома галогена, цианогруппы, линейной алкильной группы, содержащей 1-6 углеродных атомов, линейной алкоксигруппы, содержащей 1-6 углеродных атомов, трифторметильной группы и трифторметоксигруппы; причем один или более из указанных заместителей могут быть замещены по произвольным положениям кольца; и
Х представляет собой метиновую группу (-СН=).
| JP 62212386 А, 18.09.1987 | |||
| Устройство для изготовления изделий из термопластичной пленки | 1984 |
|
SU1265089A1 |
| WO 9926932 А1, 03.06.1999 | |||
| СПОСОБ ПОВЫШЕНИЯ ВОДОСТОЙКОСТИ СТРУКТУРЫ ДЕРНОВО-ПОДЗОЛИСТОЙ ПОЧВЫ | 2009 |
|
RU2430950C2 |
| ПРОИЗВОДНЫЕ ХИНОЛИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ПРОТИВОВОСПАЛИТЕЛЬНОЕ СРЕДСТВО | 1994 |
|
RU2132330C1 |

