В данной заявке испрошен приоритет по заявкам США №60/096,078, поданной 11 августа 1998 года и №60/133,970, поданной 13 мая 1999 года.
Правительство США имеет в данном изобретении оплаченную лицензию и право, в ограниченных обстоятельствах, требовать от владельца патента выдачи на разумных условиях лицензии другим лицам, как это предусмотрено условиями контракта №HD-34362, заключенного с National Institute of Child's Health and Human Development.
Трансгенные животные важны для целей науки, фармацевтики и сельского хозяйства. Скот, полученный методами генной инженерии и используемый для получения чужеродных белков в молоке, считают подходящей системой для получения терапевтически активных рекомбинантных протеинов. Более того, введение человеческих генов в геном животных, таких как свиньи, может позволить таким животным играть роль живого органа или клеточных "фабрик" для органов или клеток человека, которые не будут отторгнуты иммунной системой человека.
Существует ряд документированных способов получения трансгенных млекопитающих путем введения чужеродной ДНК в их соматические и зародышевые клетки. Один из этих способов, нашедший широкое применение, пронуклеарная микроинъекция, впервые был разработан на модели мыши в начале 1980-х. Пронуклеарная микроинъекция предусматривает инъекцию трансгенной (тг) ДНК в пронуклеус одноклеточного эмбриона {(J.W.Gordon, et al., Proc. Natl. Acad. Sci. USA. 77,7380 (1980); J.W.Gordon and F.H.Ruddle, Science 214, 1244 (1981); R.D.Palmiter and R.L.Brinster, Ann.Rev.Genet.20, 465 (1986); and J.W.Gordon Int. Rev. Cytol. 115, 171 (1989)}. Хотя получение пронуклеарных зигот оказалось простым у мышей, это не всегда справедливо для видов - представителей крупных коммерчески разводимых животных. Например, зиготы представляют собой сложные объекты для инъекции пронуклеуса, если насыщенность липидами делает их непрозрачными, как это имеет место у скота и свиней; хотя в отличие от этого зиготы мышей прозрачны.
Трансгенные эмбриональные стволовые клетки (ЭС), полученные трансфекцией конструкциями ДНК, были использованы для получения животных-химер у скота, овец и им подобных. Этот способ предусматривает инъекцию полученных методами генной инженерии ЭС клеток, несущих желаемую мутацию в оплодотворенные эмбрионы, находящиеся в эмбриональном развитии на стадии морулы (примерно от 20 до 50 клеток) или на стадии бластоциста (примерно 100 клеток). После имплантации такие эмбрионы часто дают животные-химеры, последующее скрещивание которых с животными дикого типа с различной частотой (часто равной нулю), приводит к передаче зародышевым путем генома, происходящего из ЭС клеток. Поскольку эффективность передачи гена низка, а также вследствие того, что для пересадки эмбрионов требуется большое количество животных-реципиентов, получение крупных трансгенных животных этим способом оказалось сложным.
Ни описанный выше способ пронуклеарной микроинъекции, ни способ трансфекции ЭС клеток до сих пор не позволяют контролировать или предсказывать результат трансгенного введения, поскольку введение гетерологической ДНК в клетку часто приводит к эффектам нежелательного "положения" или числа копий, вызванным квази-случайным характером, с которым трансген или его множественные копии встраиваются в геном хозяина (J.W.Gordon, supra). Поэтому эффективность этих способов при получении крупных трансгенных животных оказалась низкой.
Есть данные, свидетельствующие о том, что более строгий контроль за результатом трансгенной интеграции можно осуществить при использовании линии ЭС клеток мыши, трансфецированных конструкциями ДНК, способными к гомологической рекомбинации {(M.J.Evans and M.H.Kaufman, Natura 292, 154 (1981); M.Kuehn, et.al., ibid. 326, 295 (1987)}. Этими "генетически нацеленными" ЭС клетками являются такие клетки, в которых один или более специфических генов удалены или модифицированы строго определенным способом, который не влияет ни на какой другой локус во всем геноме. "Бессмертные" трансгенные линии ЭС клеток были созданы и хорошо изучены in vitro для того, чтобы определить место внедрения конструкции. Однако генетическое нацеливание в настоящее время ограничено одним видом, для которого существуют зародышевые линии ЭС клеток, - мышью.
Недостатки существующих способов модификации генетических линий млекопитающих послужили толчком к поиску альтернативных способов, включая использование рекомбинантных ретровирусов для инфицирования ооцитов или предимплантационных эмбрионов {D.Jahner, et al., Proc. Natl. Acad. Sci. USA, 82, 6927 (1985): A.W.S.Chan et al., ibid., 95, 14028 (1998)} и использование репликационно-недостаточных, передаваемых аденовирусами, систем доставки {Y.Kanegal et al., Nucleic Acids Res. 23, 3816 (1995)}. Однако методика с применением вирусов подразумевает необходимость проведения дополнительных операций при клонировании, что влечет за собой потребность в специализированном оборудовании для тех рекомбинантных аденовирусов и ретровирусов, которые должны быть созданы. Доставка вируса этими способами все еще требует либо оборудования для микроинъекций, либо удаления зоны пеллюцида ооцита.
Также есть данные о том, что в качестве средств доставки ДИК во время оплодотворения in vitro (in vitro fertilization, IVF), можно использовать сперматозоиды {M.Lavitrano et al., Cell 57, 717 (1998)}. В данном случае в качестве вектора in vitro введения рекомбинантной ДНК в ооцит используют живые сперматозоиды. Несмотря на то, что опосредованный сперматозоидами перенос ДНК потомкам потенциально может значительно упростить получение трансгенных животных, существуют серьезные разногласия относительно эффективности способов с использованием живых сперматозоидов при осуществлении трансгенеза вследствие его ненадежности с точки зрения стабильности воспроизведения трансгенных животных {М. Lavitrano, et al., Mol. Reprod. Dev.50, 406 (1998)}. Так, в одной из работ было показано, что экзогенная ДНК связывается с интактным сперматозоидом обратимым образом {М. Lavitrano, et al., Mol. Reprod. Dev.31, 161 (1992)}, что указывает на то, что мембранные структуры могут служить барьером для стабильной ассоциации головок спермиев с чужеродной рекомбиногенной ДНК. В другой работе живые сперматозоиды мыши, инкубированные in vitro в течение двух часов с плазмидной ДНК, показали незначительное включение экзогенной ДНК в ядро, так же как и в плазменную мембрану. Сперма vas deferens, в которую за шесть часов до этого инъецировали плазмидную ДНК, также показала незначительное включение ее в ядро. Однако ни один из этих сперматозоидов не был использован для оплодотворения ооцитов {Е. Huguet and P.Esponda, Mol. Reprod. Dev.51, 42 (1998)}.
Поэтому все еще существует потребность в способе эффективного трансгенного переноса, который можно надежно использовать для получения трансгенных животных. В частности, существует потребность в эффективном способе получения методами генной инженерии скота или других крупных животных для использования в качестве фармацевтических "фабрик" и как источник человеческих органов или клеток для ксенотрансплантации.
Согласно изобретению предложен способ получения трансгенного эмбриона, включающий стадии совместного введения экзогенной нуклеиновой кислоты и головки спермия с разрушенной мембраной или головки спермия с удаленной мембраной в цитоплазму неоплодотворенного ооцита для образования трансгенного оплодотворенного ооцита и предоставление возможности трансгенному оплодотворенному ооциту развиться в трансгенный эмбрион и, при желании, в живого потомка. Стадия совместного введения предпочтительно включает подстадию преинкубации головки спермия с разрушенной мембраной или удаленной мембраной с экзогенной нуклеиновой кислотой в течение времени от примерно 30 с до примерно 5 мин, обычно примерно от 45 с до примерно 3 мин, чаще от примерно 1 мин до примерно 2 мин. Совместное введение головки спермия и экзогенной нуклеиновой кислоты в ооцит проводят микроинъекцией, предпочтительно микроинъекцией, проводимой пьезоэлектрически. Экзогенная нуклеиновая кислота в смеси с головками спермиев с нарушенной мембраной или с удаленной мембраной может нести более одного трансгена для получения эмбриона, который будет трансгенным более чем по одному трансгену.
Головки спермиев с разрушенными мембранами, которые подходят для использования в изобретении, можно получить из замороженных/размороженных сперматозоидов или регидратированных лиофилизированных сперматозоидов. Способ сохранения сперматозоидов лиофилизированием и использование получающихся в результате этого восстановленных лиофилизированных сперматозоидов для оплодотворения ооцитов in vitro с целью получения эмбрионов и живых потомков является предметом нашей заявки на патент США №09/177,391 от 23 октября 1998 года, описание которой, таким образом, включено здесь в виде ссылки. Головки спермиев с удаленной мембраной, включающие ядро и перинуклеарные материалы и пригодные для использования в изобретении, можно получить, обрабатывая свежие сперматозоиды детергентами, как это описано ниже.
Способ изобретения можно использовать для получения трансгенных эмбрионов или живых потомков млекопитающих, таких как приматы, овцы, быки, свиньи, медведи, кошачьи, псовые, лошади и грызуны. Способ также можно использовать для получения трансгенных беспозвоночных, таких как, но не ограничиваясь, морские ежи, лобстер, оболочник или моллюски. Способ также можно использовать для получения трансгенных рыб, амфибий, рептилий и птиц. При этом было обнаружено, что живые трансгенные потомки (животные-родоначальники), полученные в процессе реализации изобретения, сами способны производить трансгенных потомков, демонстрируя стабильную интеграцию тг в первоначальный геном и плодовитость родоначальника.
Описанный здесь способ трансгенеза млекопитающих отличается от предыдущих in vitro способов, предусматривающих пронуклеарнуто инъекцию экзогенной ДНК в оплодотворенные ооциты или смешивание живых интактных сперматозоидов с экзогенной ДНК, и использование этих обработанных сперматозоидов для оплодотворения ооцитов для образования трансгенных эмбрионов. Использование неоплодотворенных Мет II ооцитов по способу изобретения представляет сильно упрощенный и ускоренный способ по сравнению со способами, которые требуют использования зигот. Более того, трансгенез посредством интрацитоплазматической инъекции спермия (ВЦИС), может преодолеть некоторые недостатки пронуклеарной микроинъекции. Например, применение микроинъекционных пипеток, имеющих примерно в 100 раз большее отверстие на конце (т.е. примерно 0,78 мкм для пронуклеарной микроинъекции при кончике диаметром 1 мкм, по сравнению с примерно 78 мкм2 для ВЦИС, при кончике диаметром 10 мкм) ускорит обработку крупных конструкций, таких как дрожжи или искусственные хромосомы млекопитающих. Более того, по способу изобретения ассоциация тг ДНК с головками спермиев с разрушенными мембранами или удаленными мембранами предполагает дальнейшую стабилизацию и защиту ее первичной и вторичных структур.
Фиг.1 представляет собой микрофотографию, показывающую сагиттальное сечение через головки сперматозоидов мыши, которые были либо интактными (свежими) (А), либо сперматозоидами, мембраны которых были разрушены Тритоном Х-100 (В), замораживанием-размораживанием (С), или лиофилизированием (D): ас - акросомальная шапочка, eq - экваториальный сегмент, pa - постакросомальный район.
Фиг.2 представляет собой микрофотографию, показывающую трансгенные эмбрионы, полученные двойным трансгенезом при однократной инъекции. Ооциты микроинъецировали сперматозоидами, которые были проинкубированы со смесью pCX-LacZ и pCX-EGFP тг ДНК. Те же самые эмбрионы (х 400) показаны через 3,5 суток под Hoffman modulation контрастным микроскопом неокрашенными (Фиг.2А), под длинноволновым (480 нм) ультрафиолетовым (УФ) светом для экспрессии GFP (Фиг.2В); и окрашенными X-gal для экспрессии β-галактозидазы (Фиг.2С).
Фиг.3 представляет собой микрофотографию, показывающую результат анализа биопсий, взятых из кончика хвоста трансгенных родоначальников и нетрансгенных контрольных животных. (Фиг.3 А) Флуоресцентная стерео микроскопия (х 40) кончиков хвостов нетрансгенной (а) (мышь 16) и тг, зеленая флюоресценция (в) (мышь 3) линий. Флюоресцирующую зеленым кожу можно рассмотреть через незеленые шерстинки. (Фиг.3В) Анализ по методу Southern blot общей ДКН контроля B6D2F1 (wt, дикий тип, дт) (0) и мыши номер 3 (5-9), 19 (>50), 28 (5-9) и 41 (2) с применением фрагмента рСХ-EGFP в качестве пробы. Результаты определения числа копий тг на геном приведены в скобках. (Фиг.3 С) PCR анализ общей ДНК мыши номер 16, 17, 30, 36, 47, 49, контроля B6D2F1 (дт), мыши номер 3, 19, 28 и 41.
В соответствии с изобретением предложен способ получения трансгенного эмбриона путем совместного введения экзогенной нуклеиновой кислоты и головки спермия с разрушенной мембраной или головки спермия, лишенной мембраны в неоплодотворенный ооцит. Способ изобретения включает следующие стадии: (1) получение сперматозоида, имеющего разрушенную мембрану, или головки сперматозоида, лишенной мембраны, (2) смешивание сперматозоида, имеющего разрушенную мембрану, или головки сперматозоида, лишенной мембраны, с экзогенной нуклеиновой кислотой, несущей желаемый ген, и (3) совместное введение экзогенной нуклеиновой кислоты и головки спермия с разрушенной мембраной или лишенной мембраны головки спермия в изолированный неоплодотворенный ооцит для получения трансгенного эмбриона, экспрессирующего желаемый трансген. Способ далее может включать стадию имплантации трансгенного эмбриона в матку суррогатной матери и предоставление возможности эмбриону развиться до живого трансгенного потомка.
Примеры реализации отдельных стадий и подстадий способа изобретения представлены далее более подробно.
Приготовление свежих сперматозоидов.
Свежие сперматозоиды беспозвоночных и позвоночных животных получают способами, хорошо известными специалистам. Например, зрелые сперматозоиды грызунов, таких как мышь, золотой (Сирийский) хомяк, морская свинка, кролик и им подобных, можно получить из каудального эпидидимиса, тогда как у других видов, таких как люди, свиньи, лошади, быки, козлы, петухи и им подобные, зрелые сперматозоиды можно выделить из свежеэякулированного семени плодовитых самцов. Сперматозоиды рыб (например, мечехвоста Xiphophorus helleri) и беспозвоночных, таких как морские ежи (Tripneustis gratilla), можно получить из яичек зрелых самцов.
Далее следует пример способа получения сперматозоидов из каудального эпидидимиса. У зрелого (примерно 8 педель после рождения или старше) самца мыши удаляют каудальный эпидидимис. С поверхности каудального эпидидимиса убирают кровь и жировую ткань. Затем его сдавливают, чтобы высвободить плотную массу сперматозоидов. Каплю (примерно 2 мкл, μл) массы спермы помещают на дно 1,5 миллилитровой (мл) полипропиленовой центрифужной пробирки и заливают 0,5 мл теплой физиологической среды, такой как среда CZB (состав которой описан ниже), фосфатно забуференный физраствор или изотонический физраствор. По прошествии примерно 10-20 мин при 37°С, из надосадочной жидкости отбирают подвижные сперматозоиды.
Далее следует пример способа получения сперматозоидов из семенной жидкости. Свежеэякулированной семенной жидкости человека позволяют разжижиться в течение примерно 30 мин при комнатной температуре (около 25°С). Затем семенную жидкость разбавляют примерно 10 мл физраствора и фильтруют примерно через два слоя фильтровальной бумаги для удаления загрязнений. Затем фильтрат центрифугируют при 400 × g в течение примерно 10 мин и осадок сперматозоидов ресуспензируют в физиологическом растворе в желаемой концентрации.
Далее следует пример получения сперматозоидов из яичек. Удаленные яички помещают в эритроцит-лизирующий буфер (например, 155 миллимолярный (мМ) NH4Cl, 10 мМ КНСО3, 2 мМ EDTA, рН 7,2-7,4), измельчают при помощи острых ножниц и, для того, чтобы удалить загрязнения, фильтруют через примерно два слоя фильтровальной бумаги. Затем фильтрат центрифугируют (например, 700 × g, 5 мин) и осадок ресуспензируют в физиологическом растворе в желаемой концентрации.
Полученные таким способом сперматозоиды мыши, имеющие интактные плазменные и акросомальные мембраны, показаны на Фиг.1 (А), которая является микрофотографией типичного сагиттального сечения через головку сперматозоида мыши, где "ас" это акросомальная шапочка, "eq" - экваториальный сегмент и "pa" - постакросомальный район. Сперматозоиды суспензированы в физиологической среде, описанной ниже, в момент подготовки к процессам замораживания-размораживания или лиофилизации.
Альтернативно, сперматозоиды можно подвергнуть дальнейшей обработке для получения лишенных мембран головок спермиев.
Приготовление сперматозоидов с разрушенными мембранами
Свежие сперматозоиды с разрушенными мембранами
Мембраны свежих сперматозоидов, полученных, как описано выше, можно разрушить механическим способом, таким как отделение головок спермиев от хвостов в микроинъекционной пипетке приложением единичного импульса от прибора для пьезоэлектрически проводимых микроинъекций, как это будет описано ниже. Используемый в данном случае термин "свежие" сперматозоиды относится к сперматозоидам с разрушенной мембраной, которые используют для микроинъекции в неоплодотворенные ооциты, и каковые отличают от, и каковые имеют отличие от "живых" сперматозоидов, используемых как носитель для доставки ДНК в предыдущих работах по IVF.
Замороженные - размороженные сперматозоиды
Замораживание и размораживание сперматозоидов приводит к разрушению плазменной мембраны, как это подтверждают методиками прижизненного окрашивания, которые способны различать клетки с интактной плазменной мембраной (живые) и с разрушенной плазменной мембраной (мертвые), как это детально описано ниже. Такие замороженные - размороженные сперматозоиды с разрушенной плазменной мембраной в обычном смысле считаются "мертвыми". Замороженные-размороженные сперматозоиды можно приготовить в соответствии со способами, описанными у T.Wakayama, et al., J. Reprod. Fert. 112, 11 (1996) и у S. Kuretake, et al., Biology of Reproduction 55, 789 (1996). В частности, сперматозоиды эпидидимиса мыши, суспензированные перед охлаждением до минус 20°С или минус 50°С, или минус 196°С в среде CZB с добавлением или без добавления криопротекторов, таких как 18% (масс./об.) раффиноза, и хранившиеся в замороженном состоянии до размораживания в течение от одного до 28 дней, поддерживали развитие нормальных плодовитых живых потомков после того, как их головки были микроинъецированы в неоплодотворенные ооциты.
В типичном способе замораживания сперматозоидов эпидидимиса мыши концентрация сперматозоидов в среде CZB составляет примерно от 3 до 10×106/мл. Порции суспензии спермиев, по 100 микролитров каждая, переносят в 1,5 мл полипропиленовые микроцентрифужные пробирки (Fisher Scintific, Pittsburg, PA) и тщательно перемешивают с рапным объемом среды CZB с добавлением или без добавления 36% (мас./об.) D (+) - раффинозы (задавая конечную концентрацию раффинозы 18%). Порции этой суспензии, по 50 микролитров каждая, помещают в снабженные этикетками 1 мл криогенные флаконы (A/S NUNS, Copenhagen). Флаконы плотно закупоривают и помещают непосредственно в минус 20°С или минус 50°С, морозильник или жидкий азот (минус 196°С). Образцы можно хранить в течение времени от одного дня до четырех недель.
Для размораживания флаконы вынимают из морозильника или жидкого азота и помещают в воду или оставляют на воздухе при температуре от 24°С до 26°С примерно на 10 мин. Размороженная суспензия спермиев теперь готова для интрацитоплазматической инъекции спермиев (ВЦИС), как это описано ниже.
Хотя способ получения замороженной - размороженной спермы был в данном случае описан для сперматозоидов эпидидимиса мыши, любой специалист сможет приспособить способ к сперматозоидам других позвоночных и беспозвоночных без проведения дополнительных экспериментов.
Регидратированные лиофилизированпые сперматозоиды
Лиофилизирование сперматозоидов приводит к разрушению плазменной мембраны, как это подтверждают методиками прижизненного окрашивания, которые способны различать клетки с интактной плазменной мембраной (живые) и с разрушенной плазменной мембраной, которые в обычном смысле считаются "мертвыми". Лиофилизированные сперматозоиды можно приготовить в соответствии со способами, описанными у T.Wakayama and R. Yanagimachi, Nature Biotechnology 16, 639, (1998) и в нашей заявке на патент США №09/177,391 поданной 23 октября 1998. В частности, заявка на патент раскрывает основные способы, которые можно использовать для лиофилизирования сперматозоидов позвоночных и беспозвоночных животных. В типичном способе сперматозоиды эпидидимиса мыши перед замораживанием в жидком азоте и высушиванием до содержания воды около нуля процентов, суспензированные в (1) среде CZB без этилендиаминтетрауксусной кислоты (EDTA), содержащей 4 мг/мл BSA, или (2) в модифицированной Dulbecco среде Eagle (DMEM), с добавкой 10% (об./об.) фетальной сыворотки быка (Hyclone, Logan, UT) и хранившиеся до регидратации в лиофилизироваином состоянии вплоть до 6 месяцев, поддерживали развитие нормальных плодовитых живых потомков после того, как их регидратировали и их головки вводили способом микроинъекции в неоплодотворештые ооциты.
В типичном способе замораживания эпидимисных сперматозоидов мыши концентрация спермиев в средах CZB или DMEM составляет примерно от 3 до 10×10 на мл. Порцию (100 мкл) суспензии спермиев помещают в 2 мл ампулу (Wheaton Scientific, Millville, NJ, Catalogue №651506), которую погружают непосредственно в жидкий азот. Через 10 мин ампулы помещают в предварительно охлажденную (минус 50°С) морозильную колбу, подсоединенную к лиофилизирующей системе (модель 10-020, VicTis Co., Gardner, NY). Давление на выходе приблизительно равно 1 млТорр. Примерно через 12 ч колбу после того, как ее заполняют аргоном, подаваемым через систему осушки газа, вынимают из системы (Fisher Scientific, Pittsburg, PA Catalogue №09-204). Каждую ампулу подсоединяют к вакуумному насосу и после того, как из нее откачивают более 99% газа, запаивают. Ампулы по отдельности заворачивают в алюминиевую фольгу и, до использования, хранят в темноте при комнатной температуре (примерно 25°С) или при 4°С до года.
Для регидратации упомянутой выше лиофилизировапной спермы ампулу, содержащую лиофилизированную сперму, приготовленную, как описано выше, вскрывают и, для получения восстановленной суспензии спермиев, добавляют 100 мкл дистиллированной воды.
Хотя способ получения регидратироваппой лиофилизированной спермы был описан здесь на примере эпидидимисных сперматозоидов мыши, любой специалист может применить способ к сперматозоидам других позвоночных и беспозвоночных животных без проведения дополнительных экспериментов, как описано в заявке на патент США №09/177,391.
Было отмечено, что процент активации ооцита и нормального оплодотворения вслед за инъекцией головки спермия, как оказалось, уменьшается с увеличением времени после регидратации лиофилизированпой спермы. Допустимый период времени между регидратацией и инъекцией может варьировать у разных видов, однако, в качестве примера, это время для сперматозоидов мыши предпочтительно составляет один час или менее.
Приготовление лишенных мембран головок спермиев
Лишенные мембран головки спермиев - это извлеченные детергентом головки, у которых отсутствуют все мембраны, включая плазменную мембрану и внутреннюю и наружную акросомальные мембраны, но сохранено ядро и перинуклеарный материал. Например, головки спермиев можно лишить мембран обработкой Тритоном Х-100 с добавлением или без добавления SDS (sodium dodecyi sylfate, додецилсульфата натрия). Тритон Х-100 - это хорошо известный неионный сурфактант, который широко используют для удаления мембранных компонентов в условиях без денатурации. SDS является анионным детергентом, используемым для растворения различных протеинов, включая мембранные протеины. Было показано, что у мыши головки спермиев, лишенные мембран при помощи Тритона Х-100, способны активировать ооциты, что ведет к нормальному эмбриональному развитию.
Далее следует типичный способ удаления мембран головок спермиев. Часть суспензии спермиев, приготовленной как описано выше, сонифицируют. Например, сперматозоиды, полученные из каудального эпидидимиса, яичек или семени, как это описано выше, можно суспензировать в 5 мл ВМ буфера (75 мМ NaCl, 24 мМ EDTA и 50 мМ Трис-HCl, рН 7.2) и сонифицировать в течение 30 секунд при 70%-80% мощности соникатора Biosonic (Bronwill Scientific, Rochester, NY). При такой обработке обезглавливается более 95% сперматозоидов. Для того чтобы удалить мембрану с головок спермиев, сонифицированную суспензию спермиев центрифугируют при 700 × g в течение 5 мин и осадок промывают ВМ буфером и затем, при комнатной температуре, обрабатывают в течение 5 мин 1% Тритоном Х-100 в среде NIM (среда NIM состоит из 123,0 мМ KCl, 2,6 мМ NaCl, 7,8 мМ КН2РО4, 3 мМ Na2EDTA и имеет рН 7.2). Затем головки тщательно промывают средой NIM и ресуспендируют в среде для суспензирования спермы.
Определение жизнеспособности спермы
Микрофотография на Фиг.1 (В), (С) и (D), изображающая продольное сечение через переднюю часть головок спермиев, показывает, что плазменная и акросомальные мембраны, кроме расположенных в районе экватора, в сперматозоидах, обработанных Тритоном Х-100 (детергент) (В), замороженых/размороженных (С) или лиофилизированных (D) отсутствуют или разрушены. Жизнеспособность сперматозоидов можно определить при помощи любого метода окрашивания, который способен различать сперматозоиды, которые в обычном смысле являются живыми или мертвыми. Коммерчески доступным подходящим для использования в изобретении тестом на жизнеспособность является Live/Dead FertiLight, который можно приобрести у Molecular Probes, Eugene, Oregon, и который позволяет различать клетки с интактными плазменными мембранами (живые) и нарушенными плазменными мембранами (мертвые) по характеру флюоресценции под ультрафиолетовым (УФ) микроскопом после окрашивания иодидом пропидия/SYBR 14. Ядра "живых" сперматозоидов с интактной плазменной мембраной флюоресцируют зеленым, тогда как ядра "мертвых" сперматозоидов флюоресцируют ярким оранжево-красным. Ожидается, что все сперматозоиды, приготовленные способом физического разрушения мембраны или замораживанием/размораживанием, лиофилизированием и процедурами удаления мембран, описанными выше, будут "мертвыми" в обычном смысле этого слова.
Отбор и подготовка экзогенной нуклеиновой кислоты, содержащей трансген
Генетическая трансформация в соответствии с изобретением представляет собой стабильную интеграцию экзогенной чужеродной ДНК в геном зиготы и включает интеграцию чужеродной ДНК в ядерную ДНК клетки хозяина и/или в экстрапуклеарную ДНК митохондрии. Чужеродная ДНК представляет собой генетический материал, которые не является врожденным (т.е. который в норме отсутствует) в зиготе до трансформации, или в норме представлен не более чем одной копией. Однако "чужеродная" ДНК может включать добавочные копии врожденного гена или генетической последовательности, которые вводят с целью взаимоподавления.
Чужеродный генетический материал может включать в себя ДНК из любого источника, включая, но, не ограничиваясь, растениями, бактериями, вирусами, бактериофагами, плазмидами, пластидами, млекопитающими и синтетическими конструкциями ДНК. ДНК может быть в кольцевой или линейной форме и может быть одинарной цепочкой или двойной спиралью. ДНК может быть введена в ДНК клетки хозяина в смысловой или антисмысловой конфигурации и в форме одинарной или двойной спирали. Вся или часть ДНК, вводимая в клетку-хозяина, может интегрироваться в геном хозяина.
Отбор и/или синтетическое построение плазмид и других векторов клонирования, содержащих специфические гены, хорошо известны в науке. Синтетические конструкции химерных плазмид содержат ген или интересующий нас ген и часто, для ускорения внедрения в геном хозяина, включают промоутер и/или лидерную последовательность, полученные из различных источников. Хотя последовательности прокариотического вектора клонирования не оказывают видимого влияния на частоту интеграции микроинъецированных генов, было отмечено, что они могут значительно подавлять экспрессию эукариотических генов, внедренных в зародышевую линию млекопитающих, таких как мышь {см. B.Hogan et al., Manipulating the Mouse Embryo, Section E, Второе изд., Cold Spring Harbor Laboratory Press, p.22 (1994)}. Вследствие этого, если желательна оптимальная экспрессия гена, можно порекомендовать удалять большей частью все векторные последовательности из клонируемого гена до внедрения его в зародышевую линию млекопитающих, таких как мышь. Векторные последовательности можно удалять в соответствии с сайтами ограничения, представленными на векторе, путем использования рестриктаз, известными методами, с получением фрагментов, несущих желаемый ген, промоутеров, усиливающих агентов и тому подобное. Уровень экспрессии интродуцированного гена в основном зависит от силы промоутера и количества копий интегрированной ДНК в трансфектированных клетках. Поэтому в векторах экспрессии применяют очень сильные промоутеры, такие как передний или задний промоутер SV40 (SV40 early или late), непосредственно передний(СМУ-ГЕ) промоутер цитомегаловируса, промоутер цитоплазматического β-актина и основной задний промоутер аденовируса.
Успешность доставки ДНК в клетку можно предварительно оценить по экспрессии гена "репортера". Ген репортер является компонентом ДНК, используемым для трансформации, и может быть таким же или иным, чем трансген, придающий другое желаемое свойство. Свойство, придаваемое трансформированной клетке или ткани геном репортером, обычно легко прослеживается (выявляется) при помощи гистохимических или флуоресцентных анализов. Для количественного определения эффективности транссфекции существует ряд часто используемых in vitro генов-репортеров, а также большое число плазмид и векторов клонирования, содержащих репортерные гены, коммерчески доступных от хорошо известных специалистам источников, таких как Stratogene, Inc., LaJolla, СА и Clontech Laboratories, Inc., Palo Alto, СА. Образцы генов-репортеров для использования в настоящем изобретении включают, но не ограничены секретируемой щелочной фосфатозой (secreted alkaline phosphatase, SEAP), β-галактозидазой (β-gal), люциферазой светлячка и хлорамфеникол ацетилтрансферазой (chloramphenicol acetyltransferase GUS), а люциферазные тесты также пригодны для in situ обнаружения переноса гена либо в фиксированных клетках либо на гистологических срезах. Эти методики позволяют, после окрашивания энзиматическими субстратами или антителами, визуализировать трансфектные клетки. Из этих in situ методик широко используемым способом, благодаря его простоте и чувствительности, является окрашивание β-gal после экспрессии гена LacZ в Escherichia coli. В данной методике реакция β-gal с X-gal субстратом дает яркий голубой цвет, который легко заметен под оптическим микроскопом и обеспечивает прямое определение эффективности переноса.
Флуоресцирующий зеленым белок (GFP) медузы Aequorea victoria стал важным репортером для мониторинга экспрессии генов и локализации белков в самых разных клетках и организмах {R.Y.Tsien, Annu. Rev. Biochem., 67, 509 (1998); G.Zhang et al., Biochemical and Biotechnology 15, 458-460 (1997)}. Поскольку для выявления GFP не нужно никакого субстрата, он может быть походящим маркером для отбора трансгенных эмбрионов. GFP, экспрессированный в эукариотических клетках, проявляет зеленую флюоресценцию, когда клетки возбуждают УФ или синим светом. Хромофор в GFP присущ первичной структуре протеина, и флюоресценция GFP не требует дополнительных кофакторов, субстратов или дополнительных генетических продуктов. Флуоресценция GFP стабильна, не зависит от вида и может отслеживаться без внутреннего вмешательства, с применением методик флуоресцентной микроскопии, поточной цитометрии и визуально под микроскопом. Для увеличения интенсивности флуоресценции GFP при возбуждении синим светом был разработан усиленный вариант GFP (EGFP) (pEGFP-Cl, можно приобрести в Clontech Laboratories), который, для запуска экспрессии EGFP гена в клетках млекопитающих, содержит немедленный ранний промоутер сигналов полиадениляции CMW и SV40 человека.
Смешивание сперматозоидов с векторным фрагментом, содержащим желаемый трансген
Приготовленные, как описано выше, сперматозоиды можно смешать с фрагментом вектора без дальнейшей подготовки (свежие) или после того, как они были подвергнуты одному из трех описанных выше способов разрушения мембраны. В типичном способе смешивания, 1 мкл раствора ДНК, содержащей фрагмент вектора (примерно 2,5 нг/мкл) смешивают с 9 мкл суспензии, содержащей примерно от 2 до 5×105 сперматозоидов в физиологической среде, такой как CZB или NIM и перемешивают пипеттированием для получения конечной концентрации фрагментов ДНК, равной 7 нг/мкл. Смесь инкубируют при комнатной температуре (примерно 25°С) или на льду в течение от примерно 30 с до примерно 5 мин, обычно от примерно 45 с до примерно 3 мин, чаще от примерно одной до примерно 3 мин, предпочтительно около 1 мин. Концентрацию спермиев и фрагментов ДНК можно варьировать, так же как и время инкубации и температуры, в зависимости от размера фрагмента или размера спермия и тому подобного, как это известно специалистам.
Микроинъекцию смеси сперматозоидов и фрагментов ДНК обычно проводят при комнатной температуре в течение одного часа после смешивания спермиев и ДИК или в течение одного часа после удаления мембран спермиев.
Ооциты-реципиенты
Ооциты - реципиенты, которые можно использовать по способу изобретения, включают как незрелые (т.е. стадия GV), которые впоследствии дозревают in vitro, так и зрелые (т.е. стадия Мет II) ооциты, которые отбирают у животных. Зрелые ооциты можно получить, например, вызвав суперовуляцию у животных путем инъекции гонадотропных или других гормонов (например, последовательным назначением лошадиного и человеческого хорионального гонадотропина) и хирургическим отбором яиц вскоре после овуляции (т.е. через 80-84 часа после начала эструса у домашней кошки, через 72-96 часов после начала эструса у коровы и через 13-15 часов после начала эструса у мыши). Там, где возможно получение только незрелых ооцитов, их культивируют в ускоряющей созревание среде до тех пор, пока они не достигнут стадии Метафазы II, этот процесс известен как вызревание in vitro (in vitro maturation, "TVM"). Способы для IVM незрелых ооцитов коровы описаны в W098/07841, а для незрелых ооцитов мыши - Eppig and Telfer (Methods in Embriology 225, 77-84, Academic Press, 1993).
Ранее было известно, что стадия созревания ооцитов in vitro очень важна для успешного применения способов in vitro пересадки ядер для получения эмбрионов. Известно, что химизм цитоплазмы ооцита меняется в процессе созревания. Например, активность цитоплазмы, связанная с созреванием, фактор ускорения метафазы (metaphase promoting factor, "MPF"), высок в незрелых ооцитах на стадии метафазы первого мейотического деления, снижается с образованием и выталкиванием первого полярного тельца и снова достигает высоких значений на стадии Метафазы II. Активность MPF остается высокой у ооцитов, задержанных на стадии Метафазы II, и быстро снижается после активации ооцитов. В общем, работы по пересадке ядер у млекопитающих описывают использование в качестве реципиентов ооцитов на стадии Метафазы II. Ооциты на стадии Метафазы II относятся к типу, готовому быть активированными оплодотворяющими сперматозоидами. Когда ядро вводят в цитоплазму неоплодотворенного ооцита на стадии Метафазы II (т.е. ооцита с высокой активностью MPF), оболочка ядра (если она имеется) клетки разрушается и хроматин конденсируется, что приводит к образованию метафазных хромосом.
Ооциты реципиенты хирургически отбирают из яйцеводов в виде комплексов ооцитов и клеток кумулюса и помещают в забуференную среду, такую как среда Hepez-CZB (приведена ниже). Клетки кумулгоса диспергируют при помощи диспергирующих 4 ферментов, таких как 0,1% гиалуронидаза яичек быка (например, 300 USP единиц/мг, ICN Pharmaceuticals, Costa Mesa, CA). Предпочтительно хранить освобожденные от клеток кумулюса ооциты при 37°С под минеральным маслом (таким, какое можно приобрести у E.R.Squibb and Sons, Princeton, NJ) в среде, такой как среда CZB, уравновешенная при 5% (об./об.) СО2 в воздухе, в течение времени не более одного часа до дальнейшей обработки.
Компоненты спермия, необходимые для успешного оплодотворения in vitro
Известно, что нормального оплодотворения у мыши можно добиться путем инъекции изолированных головок сперматозоидов в ооциты и что плазменные и акросомальные мембраны и все компоненты хвостовой части сперматозоида не важны для нормального развития эмбриона. Мышь и, возможно, большинство обычных лабораторных грызунов являются "исключениями" в том смысле, что им для нормального оплодотворения не требуется центросома спермия, расположенная в шейном отделе сперматозоида, которая, таким образом, дегенерирует внутри ооцита во время нормального оплодотворения.
В противоположность этому у большинства остальных наземных животных, включая скот и человека, центросома спермия играет важную роль в образовании микротрубочек, которые необходимы для объединения мужского и женского пронуклеусов, так же как и для последующего дробления во время эмбрионального развития. Поэтому оказалось, что у этих видов для получения нормальных потомков важно введение в ооцит как ядра спермия (головка), так и центросомы. В настоящее время не известно, могут ли центросомы всех видов пережить замораживание-размораживание, лиофилизирование или удаление мембран детергентами. Если нет, то для обеспечения нормального эмбрионального развития необходимо вводить центросому незамороженного спермия в ооцит совместно с замороженной-размороженной, лиофилизировапной или лишенной мембраны головкой спермия. Введение излишнего количества центросом, однако, приведет к абнормальному пронуклеарному развитию и абнормальному эмбриональному развитию.
В норме центросома остается прикрепленной либо к заднему концу головки спермия или к переднему концу хвоста спермия, в том случае, если головка и хвост разделены. Таким образом, центросому спермия можно вести в ооцит одновременно с головкой спермия, или ее можно ввести способами одновременного или последующего введения хвоста спермия.
Введение ядра сперматозоида в ооцит-реципиент
Совместно с экзогенной нуклеиновой кислотой в цитоплазму ооцита реципиента можно ввести и целый сперматозоид, но у видов, у которых сперматозоиды крупные, предпочтительнее инъецировать головку сперматозоида (ядро) в цитоплазму ооцита реципиента методами микроинъекции. В предпочтительном способе совместной микроинъекции экзогенной нуклеиновой кислоты с замороженной - размороженной, регидратированной лиофилизированной головкой спермия или литейной мембраны головкой спермия в ооцит реципиент используют микропипетку с пьезоэлектрическим приводом.
В качестве подходящего пьезоэлектрического привода можно использовать Piezo Micromanipulator/Piezo Impact Drive Unit от Prime Tech Ltd. (Tsukuba, Ibaraki-ken, Japan). Для продвижения держателя (инъекционной) пипетки вперед быстро, строго контролируемым образом, на короткое расстояние (примерно 0,5 микрон), в приборе используется пьезоэлектрический эффект. Интенсивность и продолжительность каждого импульса можно изменять, и она регулируется контрольным прибором.
Для инъекции в ооцит единичный сперматозоид в смеси со спермиями/головками спермиев и экзогенной аминокислотой отбирают хвостом вперед (если у него имеется хвост) в инъекционную пипетку с коротким, плоским кончиком, с диаметром внутреннего отверстия примерно 5 мкм, помещенную в прибор с пьезоэлектрическим приводом в соответствии с инструкциями изготовителя. Головку и хвост спермия отделяют друг от друга приложением одного или нескольких пьезоимпульсов к району шейки. Затем головку засасывают вглубь пипетки. Альтернативно, в инъекционную пипетку для инъекции в ооцит можно засосать единичную головку спермия в смеси спермии/головки спермиев с экзогенной аминокислотой.
Во время совместной инъекции головки спермия (ядра) и экзогенной нуклеиновой кислоты ооцит удерживают подходящей удерживающей пипеткой. Кончик инъекционной пипетки с отобранной головкой спермия приводят в тесный контакт с зоной пеллюцида ооцита и, для того, чтобы продвинуть пипетку, прикладывают несколько пьезоимпульсов, установив на контрольной шкале интенсивность 1-5, скорость 4-6, одновременно поддерживая небольшое отрицательное давление внутри. Когда кончик пипетки проходит зону пеллюцида, получающуюся пробочку зоны выталкивают в перивителлиповое пространство и головку спермия продвигают вперед до тех пор, пока она не окажется вблизи кончика пипетки. Кончик пипетки затем прикладывают к плазменной мембране и нажимают (в направлении противоположной стороны ооцита), так что удерживающая пипетка почти достигает противоположной стороны кортекса ооцита. Теперь плазменная мембрана глубоко инвагинирована вокруг кончика инъекционной иглы. При приложении одного или двух пьезоимпульсов (интенсивностью 1-2, скоростью 1), оолемма на кончике пипетки прорывается, на что указывает четко видимое быстрое расслабление оолеммы. Затем головку спермия выталкивают в ооплазму с минимальным количеством (около 6 пл) сопровождающей среды, содержащей экзогенную нуклеиновую кислоту. Затем пипетку осторожно убирают, оставляя вновь интродуцированную головку внутри цитоплазмы ооцита. Эту операцию проводят быстро, обычно на группе из 10-15 ооцитов, которые все остальное время содержат в условиях культуры.
Для процедуры совместной инъекции можно применять и альтернативные варианты микроинъекции, в которых используют обычные инъекционные пипетки. Пример подходящего способа микроинъекции с применением обычной пипетки для инъекции головки спермия в ооцит хомяка описан у Yanigida, К., Yanagimachi, R., Perreault, S.D. and R.G.Klinfeld, Biology of Reproduction 44, 440-447 (1991), описание которого в части способа включено в виде ссылки.
Совместная микроинъекция экзогенной аминокислоты и системы "сперматозоид/головка спермия/толовка спермия, лишенная мембраны" обладает рядом преимуществ. Во-первых, доставка сперматозоида/головки спермия микроинъекцией применима для широкого спектра типов сперматозоидов, вне зависимости от размера, морфологии и тому подобного. Во-вторых, микроинъекция позволяет четко контролировать совместное (с донорским сперматозоидом/головкой спермия) введение в ооцит во время инъекции других агентов, в дополнение к описанной выше экзогенной аминокислоте. Это продемонстрировано на приведенных ниже примерах. В-третьих, в способе реализации изобретения, в котором введение сперматозоида/головки спермия осуществляется пьезоэлектрически проводимой микроинъекцией, достигается быстрая и эффективная обработка проб, что уменьшает травмирование спермиев и ооцитов, подвергнутых манипуляциям. Ооциты некоторых видов (например, мыши), невозможно микроинъецировать с использованием обычных иголок, тогда как пьезоэлектрически проводимая микроинъекция часто оказывается успешной.
Активация оплодотворенных ооцитов
Известно, что ооцит мытой можно активировать введением единичного интактного сперматозоида мыши или его изолированной головки. Изолированные хвосты спермиев не способны активировать ооцит. Активные фактор(ы) активации ооцита, происходящие от спермия, обычно появляются во время трансформации круглой сперматиды в сперматозоид. Действие этих факторов не является строго видоспецифичным, поскольку ооциты мыши активируют инъекцией сперматозоидов других видов, таких как хомяк, кролик, свинья, человек и даже рыб. Есть данные, что один из таких факторов активации является белком в 33 кДа, располагающимся в районе экваториального сегмента акросомы. Этот белок, называемый осциллином, легко выделить из зрелых сперматозоидов (хомяка) простым замораживанием и размораживанием. Оказалось, что зрелые сперматозоиды помимо осциллина, несут другой фактор активации, который трудно выделить, но который можно получить последовательной обработкой сперматозоидов Тритоном Х-100 и SDS. Неизвестно, являются ли легко выделяемый осциллин и факторы, устойчивые к экстракции при замораживании-размораживании, биологически и химически идентичными.
Известно, что головки спермиев, сонифицированные в присутствии Тритона X-100, теряют все компоненты, кроме ядра и перинуклеарных материалов. Однако после микроинъекционого введения в ооциты, такие, обработанные Тритоном Х-100, головки спермиев (имеющие ядра и перинуклеарпые материалы, но не имеющие плазменных мембран) могут активировать ооциты с такой же эффективностью, как и интактные сперматозоиды.
Как описано в нашей заявке на патент США номер 09/177,391 и T.Wakayama et al., 1997, supra, по крайней мере, у мыши, происходящие от сперматозоидов молекулы, активирующие ооциты, должны быть устойчивы к замораживанию-размораживанию и лиофилизации, поскольку большинство ооцитов, перенесших инъекцию замороженных - размороженных или лиофилизированных головок спермиев были нормально активированы и оплодотворены.
Если у других видов инъекция головки спермия не приводит к активации ооцита, может иметь место партеногенетическая активация, например, путем электроактивации, инъекции одного или более ооцит-активирующих веществ, или переноса ооцитов в среду, содержащую одно или более ооцит-активирующих веществ. Реагенты, способные обеспечивать активирующий стимул (или комбинацию активирующих стимулов) включают, но не ограничиваются активирующим фактором цитоплазмы спермия и некоторыми фармакологическими соединениями (например, Са2+ и другими модуляторами сигнальной трансдукции), которые можно ввести микроинъекцией после или одновременно с совместной инъекцией головки спермия и экзогенной аминокислоты. Некоторые активирующие стимулы можно получить при переносе оплодотворенных ооцитов в среду, содержащую один из членов подгруппы активирующих соединений, включающей стимуляторы высвобождения Са2+ (например, кофеин, Са2+ иопофоры, такие как А23187, иономицин и этанол), сигнальные фосфопротеидные модуляторы (например, 2-аминопурин, стауроспурин и сфингозин), ингибиторы протеинового синтеза (например, А23187, циклогексимид (cycloheximide)), 6-диметиламинопурин или их комбинации (например, 6-диметиламинопурин и иономицин). В типичном способе активацию ооцитов мыши получают культивированием в течение 1-6 ч в среде CZB без Са2+, содержащей от 2 до 10 мМ Sr2+.
Развитие эмбрионов для получения жизнеспособных плодов и потомков
После образования пронуклеусов, эмбрионы можно культивировать in vitro до достижения ими стадии в 2-8 клеток или стадии морула/бластоцист, во время которых эмбрионы можно перенести в яйцеводы пли матки суррогатных матерей.
Инъекция биологически значимых веществ одновременно с головками спермиев
В одном способе реализации изобретения совместная микроинъекция головки спермия и экзогенной нуклеиновой кислоты в ооцит позволяет осуществить введение до, во время или после инъекции головки спермия в ооцит одного или более агентов, могущих изменить выход развития эмбрионов. Например, способом микроинъекции в ооцит может быть введена дополнительная рибонуклеиновая кислота (РНК) или ДНК. Например, инъекция рекомбинантной ДНК, включающей cis-активные сигналы, может привести к транскрипции последовательностей, присутствующих на рекомбинантной ДНК резидентным или совместно инъецированным фактором транскрртции и последующей экспрессии закодированных протеинов с антагонистическим действием на факторы, ингибирующие развитие, или с положительным влиянием на эмбриональное развитие. Более того, транскрипт может обладать антисмысловой активностью против мРНК, кодирующих протеины, ингибирующие развитие. Альтернативно, антисмысловая регуляция может быть достигнута инъекцией аминокислот (или их производных), которые способны влиять на эффект ингибирования, взаимодействуя с их аминокислотными мишенью (мишенями) напрямую, без предварительной транскрипции внутри ооцита.
Рекомбинантная ДНК (линейная или иная), введенная способами изобретения, может включать функциональный репликон, содержащий один или более экспрессированных функциональных генов под контролем промоутера, проявляющего любые, от узкого до широкого, профили экспрессии развития. Например, промоутер может вызвать мгновенную, но краткую экспрессию там, где промоутер активен только в ранней зиготе. Введенная ДНК может либо теряться в некоторый момент эмбрионального развития, либо интегрироваться в один или более геномный локус, чтобы стабильно реплинироваться во время жизни получившегося трансгенного индивидуума. В одном способе реализации микроинъекцией в ооцит могут быть введены конструкции ДНК, кодирующие протеины, предположительно "направленные против старения", такие как теломераза или супероксидная дисмутаза. Альтернативно, такие протеины можно вводить напрямую.
ПРИМЕРЫ
Для того чтобы проиллюстрировать способ изобретения, была проведена оценка способности сперматозоидов с разрушенными и/или удаленными мембранами вносить в неоплодотворенный ооцит репликационно недостаточный фрагмент плазмиды, содержащий экспрессируемый ген репортер, а также способности вызывать развитие трансгенных эмбрионов мыши и живых трансгенных потомков. Использование гена-репортера позволило непосредственно идентифицировать эмбрионы и живых потомков, экспрессирующих трансген.
Примеры, описанные здесь, не являются ограничивающими, так как любой специалист может понять, что согласно способу изобретения могут быть использованы и другие трансгены, сперматозоиды и ооциты из источников, отличных от мыши.
Среда и реагенты
Все неорганические и органические соединения были приобретены у Sigma Chemicals Co (St. Louis МО), если только это не оговорено особо.
Собранные ооциты, до совместного введения в них экзогенной ДНК и головок спермиев с разрушенной мембраной или лишенных мембраны, хранили в среде CZB (Chatot et al., 1989. J. Reprod. Fert. 86, 679-688). Среда CZB содержит 81,6 мМ NaCl, 4,8 мМ KCl, 1,7 мМ CaCl2, 1,2 мМ MgSO4, 1,8 мМ КН2РО4, 25,1 мМ NaHCO3, 0,1 мМ NaEDTA, 31 мМ лактата натрия, 0,3 мМ пирувата натрия, 7 Ед./мл пенициллина G, 5 Ед./мл сульфата стрептомицина и 4 мг/мл альбумина сыворотки быка (BSA). Средой для сбора ооцитов из яйцеводов, последующей обработки и микроманипуляций служила модифицированная CZB, содержащая 20 мМ Hepes, уменьшенное количество NaHCO3 (5 мМ) и BSA (3 мг/мл). Здесь мы называем эту среду Hepez-SZB. Для целей микроинъекции предпочтительно заменить BSA в Hepez-SZB на 0,1 мг/мл поливинилового спирта (ПВС, растворим в холодной воде, средняя молекулярная масса 10×103), поскольку ПВС гораздо дольше сохраняет стенки инъекционной пипетки менее липкими, чем BSA, и удобен при неоднократном применении одной и той же пипетки во время множественных переносов головка спермия/ооцит. рН обеих сред приблизительно равен 7,4. Все манипуляции с ооцитами проводили в Hepez, забуференной CZB (Hepez-SZB) под минеральным маслом при комнатной температуре (от 23 до 25°С), на воздухе.
Среда, которую использовали для выделения свежих сперматозоидов, была средой SZB. Замороженные-размороженные и регидратированные лиофилизированные сперматозоиды суспендировали либо в среде CSB, либо в среде для выделения ядер (NIM), состоящей из 123,0 мМ KCl, 2,6 мМ NaCl, 7,8 мМ NaH2РO4, 1,4 мМ KHPO4, 3 мМ Na2EDTA. Значение ее рН устанавливали на уровне 7,2 путем добавления небольшого количества 1 М HCl. Свежие сперматозоиды для удаления мембран отбирали в NIM и также в NIM обрабатывали экстрактом Тритона Х-100. После промывания лишенные мембран головки спермиев суспензировали в среде NIM или CZB. После инкубации спермы с экзогенной ДНК (в CZB или NIM) в смесь добавляли PVP (средняя молекулярная масса 360,000б, ICN Biochemicals, Costa Mesa, CA).
Животные
Животных, использованных в данных примерах, содержали в соответствии с рекомендациями лаборатории Службы животных Гавайского Университета и таковыми, приготовленными Комитетом по уходу и использованию лабораторных животных Института лабораторных ресурсов национального совета по исследованиям (издание DNEW №(NIH) 80-23, пересмотренное в 1985). Методика содержания и обработки животных пересмотрена и одобрена Комитетом по уходу и использованию животных Гавайского университета.
ПРИМЕР 1
Подготовка ооцита
У зрелых B6D2F1 (C57BL/6 × DBA/2) самок мышей вызывали сверховуляцию двумя, следующими через 48 ч друг за другом, инъекциями 7,5 международных единиц (international units IU) гоиадотропина сыворотки беременной кобылы и 7,5 IU человеческого хорионального гонадотропина (hGT). Через 14 ч после инъекции hGT комплексы ооцит-кумулюс отбирали из яйцеводов и, для удаления клеток кумулюса, обрабатывали в течение 3 мин гиалорунидазой яичек быка (300 USP ед/мл, ICN Biochemicals, Costa Mesa, СА) в среде Hepez-SZB. До инъекции ядра спермия ооциты промывали и хранили в среде SZB под минеральным маслом, уравновешенным при 5% (об./об.) СО2 в воздухе при 37°С в течение времени до 4 ч.
ПРИМЕР 2
Подготовка свежих сперматозоидов
Свежие сперматозоиды получали из каудальных эпидидимисов B6D2F1. Одновременно со сдавливанием каждого эпидидимиса пальцами, его дистальную часть протыкали острым пинцетом. Плотную массу сперматозоидов, вытекающую из эпидидимиса переносили в чашку Петри. Капли (примерно 2 мкл) сперматозоидов помещали на дно 1,5 мл полипропиленовых микроцентрифужных пробирок (Fisher Scientific, Pittsburg, PA) и сверху заливали 0,2-0,5 мл среды CZB. После инкубации в течение примерно 20 мин верхние 0,4 мл среды отбирали и исследовали. Более 90% сперматозоидов в суспензии (примерно от 3 до 10×106 на мл) были активно подвижными.
ПРИМЕР 3
Подготовка замороженых - размороженных сперматозоидов
Замороженные - размороженные сперматозоиды готовили в соответствии со способом, описанным у T.Wakayama, D.G. Whittingham and R. Yanaghnachi, J. Reprod. Fert., 112, 11-17, 1998. Вкратце, капли сперматозоидов, полученные из каудального эпидидимиса, помещают на дно 1,5 мл полипропиленовых центрифужных пробирок и заливают 0,5 мл теплой среды CZB. После примерно 20 мин при 37°С, отбирают верхние 0,2 мл среды. Суспензия, содержит приблизительно от 3 до 10×106 сперматозоидов на мл. Аликвотные доли суспензии сперматозоидов (по 100 мкл) переносят в 1,5 мл полипропиленовые микроцентрифужпые пробирки и тщательно смешивают с равным объемом среды CZB с добавлением или без добавления 36% (мас./об.) D (+) - раффинозы. Конечная концентрация раффинозы составляет 18% или 0% (мас./об.) соответственно. Аликвотные доли каждой суспензии (по 50 мкл) распределяют по снабженным этикетками 1 мл криогенным колбам (A/S NUNS, Copenhagen). Каждую колбу плотно закрывают и помещают непосредственно в морозильник при минус 20°С, или минус 50°С, или в жидкий азот (при минус 196°С). Все образцы хранятся в течение времени от одного дня до четырех недель.
Для размораживания колбы вынимают из морозильника или жидкого азота и оставляют в воде или на воздухе при 24-26°С в течение примерно 10 мин. Образец размороженной суспензии спермиев тестируют на подвижность и "жизнеспособность" при помощи коммерчески доступного набора для определения живучести спермиев (Live/dead FertiLight, Molecular Probes, Inc., Eugene, OR), как это было описано выше. Все сперматозоиды, замороженные в отсутствие раффинозы, были неподвижными и "мертвыми" (мембрана разрушена). По меньшей мере, 97% сперматозоидов, замороженных в присутствии раффинозы при любой температуре, были неподвижными и "мертвыми".
Размороженную суспензию спермиев до смешивания с экзогенной ДНК однократно промывают и ресуспензируют в 400 мкл среды CZB.
Разрушение мембран было подтверждено методами электронной микроскопии, как это проиллюстрировано на Фиг.1 (С). Разрушение наиболее четко заметно на мембранах акросомальной шапочки.
ПРИМЕР 4
Подготовка регидратированных лиофилизированных сперматозоидов
Регидратированные лиофилизированные сперматозоиды были приготовлены в соответствии со способами, описанными у T.Wakayama and R. Yanagimachi, Nature Biotechnology 16, 638-640, 1998 и в нашей одновременно поданной заявке на патент США серийный №09/177,391. Вкратце, части (но 100 мкл) суспензии сперматозоидов мыши, подготовленной как описано выше, переносят в 1,5 мл полипропиленовые микроцентрифужные пробирки и тщательно смешивают с одним мл либо среды CZB без EDTA, содержащей 4 мг/мл BSA либо с модифицированной Dulbecco средой Eagle (DMEM), подкрепленной 10% (об./об.) фетальной сыворотки быка (Hyclone, Logan, UT). После инкубации в течение 30 мин при 37,5°С, из пробирки отбирают верхние 0,3-0,5 мл среды. Суспензия содержит примерно от 3 до 10×106 спермиев на мл.
Равные части (по 100 мкл) суспензии спермиев помешают в 2 мл ампулы (Weaton Scientific, Millville, NJ), которые непосредственно погружают в жидкий азот. Десять минут спустя ампулы переносят в предварительно охлажденные (до минус 50°С) сосуды для замораживания, подсоединенные к системе лиофилизации (Model 10-020, VirTis, Gardner, NY). Давление на выходе приблизительно составляет 1 млТорр. Спустя примерно 12 ч сосуд после того, как его заполняют аргоном, проходящим через систему осушки газа, из системы вынимают (Fisher Scientific, Pittsburg, PA, Catalogue №09-204). Каждую ампулу подсоединяют к вакуумному насосу и, после вакуумирования на 99%, запаивают. Ампулы по отдельности заворачивают в алюминиевую фольгу и хранят в темноте при комнатной температуре (примерно при 25°С) или при 4°С.
Для регидратации ампулы с лиофилизированными спермиями, приготовленные как описано выше, вскрывают и, для получения суспензии спермиев, добавляют в каждую ампулу по 100 мкл дистиллированной воды.
Разрушение мембран было подтверждено методами электронной микроскопии, как это проиллюстрировано на Фиг.1 (D). Разрушение наиболее четко заметно на мембранах акросомальной шапочки.
ПРИМЕР 5
Приготовление головок сперматозоидов с удаленными мембранами
Сперматозоиды для экстракции Тритоном Х-100 были выделены путем тщательного измельчения двух каудальных эпидидимисов при температуре от 0°С до 1°С в среде NIM и фильтрацией полученной суспензии спермиев с получением окончательного объема в 900 мкл. Для экстракции Тритоном Х-100, 100 мкл 0,5% (об./об. в NIM) Тритона Х-100 добавляют к 900 мкл суспензии спермиев в NIM и смешивают растиранием на льду в течение 30 с. Клетки концентрируют центрифугированием в течение 1 мин при 20000 g при 2°С. Окончательный осадок ресуспензируют в 400 мкл среды CZB или NIM.
Удаление мембран с головок спермиев было подтверждено методами электронной микроскопии, как это проиллюстрировано на Фиг.1 (В). Разрушение наиболее четко заметно на мембранах акросомальной шапочки.
ПРИМЕР 6
Приготовление трансгена
Усиленный трансген (EGFP) протеина зеленой флуоресценции был крупным (3,5 kb) SalGI-Bam фрагментом плазмиды pCX-EGFP. Фрагмент включает ген EGFP, выделенный из сильной комбинации усиливающий агент - промоутер β-актина цитомегаловируса IE цыпленка, у которого отсутствует эукариотическое начало репликации {H.Niwa, К.Yamamura, J.Miyazaki, Gene 108, 193 (1991); G.Zhang, G. Vanessa, S.R. Kain, Biochem. Biophys. Res. Commun. 227, 707 (1996); T.Takada et al., Nature Biotechnol. 15, 458 (1997)}. Фрагмент размером 3,5 kb, включающий ген EGFP, получали расщеплением плазмиды pCX-EGFP ферментами рестрикции Sal GI и BamIII и очищали хорошо известными специалистам способами.
Очищенные включающие lac-Z линеаризованные фрагменты px-CANLacZ получили расщеплением ее при помощи либо Sal GT, либо Xho I и Sal GI. У фрагментов Xho I -Sal GI плазмиды px-CANLacZ отсутствует репликационное начало. Кодируемая px-CANLacZ β-галактозидаза несет сигнал локализации в ядре.
В некоторых описанных ниже экспериментах фрагмент плазмиды pCX-LacZ получали расщеплением ее при помощи Sal GI и PstI для того, чтобы получить фрагмент pCX-LacZ Sal GI-Pst I. pCX- LacZ является производным рСХ- EGFP, в котором ген EGFP замещен на ген, кодирующий β-галактозидазу
ПРИМЕР 7
Приготовление смеси фрагментов ДНК и сперматозоидов
Подготовленные описанным выше способом сперматозоиды либо свежие, либо после того, как они были подвергнуты одной из трех методик разрушения мембран: замораживанию - размораживанию, лиофилизации или экстракции Тритоном Х-100, смешивали с фрагментами ДНК, как это описано ниже.
Объем, равный 1 мкл раствора вышеописанных фрагментов, содержащих ген GFP, пипеттированием смешивают с 9 мкл предварительно приготовленной суспензии спермиев (содержащей от 2 до 5×105 сперматозоидов) до получения окончательной концентрации фрагментов GFP ДНК, равной 7 нг/мкл. Аналогично, объемы, равные 1 мкл раствора фрагментов Sal GI или Xho I/Sal GI, каждый по отдельности, смешивают с частями суспензии спермиев, по 9 мкл каждая, для получения окончательных концентраций фрагментов Sal GI px-CANLacZ, равных 4,5 нг/мкл и 9 нг/мкл соответственно.
В некоторых экспериментах, в которых проводили двойной трансгенез при единичном введении с получением эмбрионов, экспрессирующих совместно, два трансгена в результате единичной совместной микроинъекции, головки спермиев до микроинъекции были смешаны с двумя трансгенами, например с фрагментами рСХ-EGFP Sal Gi-Bam HI (конечная концентрация 2.5 нг/мкл) и фрагментами pCX-LacZ Sal GI-Pst I (конечная концентрация 2.5 нг/мкл), содержащимися в одном растворе ДНК.
Описанные выше смеси ДНК - спермии инкубируют при комнатной температуре (около 25°С) или на льду в течение 1 мин и затем смешивают с раствором поливинилпирролидона (polyvinilpyrrolidone, PVP, средняя молекулярная масса 360,000) до получения окончательной концентрации PVP, примерно равной 10% (мас./об.).
В экспериментах по определению действия промывания сперматозоидов после инкубации с фрагментами плазмид смесь ДНК - спермии сразу после смешивания и инкубации в течение 1 мин с ДНК рСХ- EGFP разделяют па две равные части по 5 мкл каждая. Одну часть (промытые спермии) разбавляют и промывают путем тщательного перемешивания с 50 мкл ледяной, свежеприготовленной CZB или NIM. Затем обе части центрифугируют в течение 2 мин при 20000 g при 2°С. Надосадочную жидкость из варианта с промытыми спермиями осторожно удаляют и замещают 5 мкл свежей CZB или NIM. Надосадочную жидкость от второй части используют для ресуспензирования ее собственного осадка (таким образом, этот образец оказался непромытым).
В некоторых экспериментах для того, чтобы оценить влияние инъекции одних только фрагментов ДНК, без одновременной инъекции сперматозоидов, свежий раствор фрагментов Sal G I-Bam HI плазмиды рСХ- EGFP (7 нг/мкл) до инъекции смешивают с равным объемом PVP 20% (об./об.).
Во всех экспериментах смеси, полученные описанными выше способами, затем помещают на столик микроскопа для микроинъекции, как это будет описано ниже. Все инъекции проводят в среде Hepez-CZB при комнатной температуре в течение одного часа после смешивания спермиев и ДНК или в течение одного часа после смешивания спермиев с Тритоном Х-100.
ПРИМЕР 8
Микроинъекция ядер спермиев в ооциты
Для совместной инъекции головок спермиев и экзогенной ДНК в подготовленные ооциты готовили микроинъекционную камеру, используя для пластиковой чашки (100 мм × 15 мм. Falcon Plastics, Oxnard, CA, Catalogue №1001) крышечку (глубиной 10 мм). Вдоль центральной линии чашки размещают ряд, состоящий из двух круглых капель и одной продолговатой капли. Первая капля (2 мкл, диаметром 2 мм) - для промывания пипетки (Hepez-CZB с добавлением 12% (мас./об.) PVP, средняя молекулярная масса 360,000 Дальтон). Вторая капля (2 мкл, диаметром 2 мм) - с приготовленной, как описано выше, смесью сперматозоидов и фрагментов ДНК. Третья, продолговатая капля (6 мкл, 2 мм шириной и 6 мм длиной) со средой Hepez-CZB для ооцитов. Каждую из этих капель покрывают минеральным маслом (E.R.Squibb and Sons, Princeton, NJ). Чашку помещают на столик инверсионного микроскопа с интерференционной контрастной оптикой.
Совместную микроинъекцию ядер спермиев и экзогенной ДНК в ооциты проводили описанным выше способом пьезоэлектрической микроинъекции с применением Пьезомикроманипулятора модели MB-U от Prime Tech Ltd. (Tsulcuba, Ibaraki-ken, Japan). Этот прибор использует пьезоэлектрический эффект для продвижения держателя пипетки единовременно на очень короткие расстояния (например, 0,5 мкм) с очень большой скоростью. Интенсивность и скорость импульсов устанавливают при помощи регулятора.
Для инъекции в ооцит, подготовленный описанным выше способом, единичную головку спермия в смеси с экзогенной ДНК засасывают в инъекционную пипетку (внутренний диаметр кончика около 5 мкм), прикрепленную к пьезоэлектрическому приводу. В том случае, когда используют целые сперматозоиды, единичный сперматозоид в инъекционную пипетку засасывают хвостом вперед. Головку спермия и хвост разделяют приложением одного или нескольких пьезоимпульсов в районе шейки. Интенсивность и скорость (частоту) импульсов регулируют регулятором PMAS-CTО1 (показания шкал регулятора: интенсивность 2, скорость 1). Затем головки засасывают вглубь пипетки и в проксимальный конец инъекционной пипетки забирают небольшой объем (около 0,5 мкл) ртути. Отделение головок от хвостов сперматозоидов разрушает мембраны, что, таким образом, составляет отличие от свежих сперматозоидов, использованных в этих примерах и предыдущих работах по трансгенезу, проводимому путем IVF с использованием живых сперматозоидов.
Одновременно зрелый неоплодотворенный ооцит помещают па столик в среде Hepez-CZB. Ооцит удерживают удерживающей пипеткой, а кончик инъекционной пипетки приводят в тесный контакт с зоной пеллюцида на 3 часа. Для продвижения пипетки подают несколько пьезоимпульсов (интенсивность 1-2, скорость 1-2), одновременно создавая небольшое отрицательное давление. Когда кончик пипетки проходит через зону пеллюцида, цилиндрическую пробку зоны пеллюцида выталкивают из пипетки в перивителлиновое пространство. После того как головку сперматозоида проталкивают вперед до того, как она достигает конца пипетки, последнюю механически продвигают вперед до тех пор, пока ее кончик почти не достигнет противоположной стороны кортекса ооцита. Оолемму прокалывают приложением 1 или 2 пьезоимпульсов (интенсивность 1-2, скорость 1) и головку сперматозоида выталкивают в ооплазму. Было подсчитано, что во время одной инъекции из пипетки вытесняют около 1 пкл смеси, содержащей экзогенную ДНК. Затем пипетку осторожно вынимают, оставляя головку сперматозоида внутри ооплазмы.
Все инъекции проводят в среде Hepez-CZB при комнатной температуре. Каждый ооцит инъецируют одной головкой спермия. Таким способом в течение 10-15 мин микроинъецируют приблизительно от 5 до 20 ооцитов. Ооциты, которые вскоре после инъекции подверглись лизису, выбрасывают.
В экспериментах по определению влияния одних фрагментов ДНК, без одновременной инъекции сперматозоидов, около 1 пкл фрагментов Sal GI-BamHI плазмиды рСХ - EGFP и PVP, как описано выше, инъецировали в ооцит. После восстановления в течение времени от 5 до 10 мин при комнатной температуре инъецированные ооциты переносили в среду CZB, свободную от Са2+, содержащую 10 мМоль S2Cl2 и агент, блокирующий цитокинез - цитохалазин В в концентрации 5 мкг/мл, и инкубировали в течение 6 ч при 37°С. Ооциты, которые не были активированы сперматозоидами или головками сперматозоидов для того, чтобы имело место нормальное эмбриональное развитие, должны быть активированы другими способами. Активация ионами стронция является одним из множества способов партеногенетической активации, известных специалистам, и подробно описана в нашей заявке на патент США №09/132,104 от 10 августа 1998 года, описание которой в отношении активации ооцита в данном случае включено в виде ссылки. Использование агентов, блокирующих цитокинез для предотвращения выталкивания хромосом, хорошо известно специалистам. Описание заявки на патент США №09/132,104 в отношении блокирования цитокинеза в ооците также включено здесь в виде ссылки.
Партеногенетически активированные ооциты затем переносят в среду CZB и продолжают инкубацию в условиях стандартной культуры эмбрионов, описанной ниже. Экспрессию GFP эмбрионами определяют после культивирования в течение 3,5 суток описанными ниже способами.
ПРИМЕР 9
Исследование ооцитов, культура эмбрионов и пересадка суррогатным матерям
Ооциты, инъецированные головками спермиев и экзогенной ДНК, инкубировали в среде CZB при 37°С под минеральным маслом, уравновешенным при 5% (об./об.) СО2 в воздухе, и исследовали под инвертирующим микроскопом через 5-6 ч. Ооциты с двумя четко видимыми пронуклеусами и вторым полярным тельцем считали оплодотворенными нормально и культивировали в течение 4 суток в среде CZB. Те, которые достигли стадий морулы или бластоцисты, переносили в маточные трубы реципиентных самок (обычно CD-1 самки альбиносы), которые за три дня до этого были спарены с вазектомированными (CD-1) самцами, для синхронизации эмбрионального развития с развитием эндометрия матки. В каждую трубу в среднем переносили по 8 морул/бластоцист. Самкам предоставляли возможность родить и вырастить суррогатных потомков. Для того чтобы проверить их плодовитость, случайным образом отбирали и скрещивали несколько получившихся зрелых самцов и самок.
ПРИМЕР 10
Проверка эмбрионов на экспрессию трансгена
Через 3-3,5 суток после совместной микроинъекции эмбрионы проверяли на экспрессию GFP при помощи флуоресцентной микроскопии с источником УФ света (480 нм) и флуоресциновыми изотиоцианатными фильтрами. Это позволяло четко выделить нефлуоресциругощие (неэкспрессирующие GFP), слабо флуоресцирующие и сильно флуоресцирующие эмбрионы и мозаики, которые, соответственно, подсчитывают.
Экспрессию px-CANLacZ β-галактозидазы проверяли у трехдневных эмбрионов способом, описанным у T.Tsukul, et al., Nature Biotechnology 14, 982 (1996) после 5-минутной фиксации при комнатной температуре в фосфатно-забуференном физиологическом растворе (phosphate-buffered saline, PBS) (pH 7,6), содержащем 1% (об./об.) формальдегида, 0,2% (об./об.) глютаральдегида и BSA (5 мг/мл) и окрашивания путем инкубации в течение 5 часов при 37°С в PBS, содержащей BSA (5 мг/мл), 4 мМ калий феррицианида, 4 мМ калий ферроцианида, 2 мМ MgCl2 и 5-бромо-4-хлоро-3индолил β-D-галактопиранозида (X-gal) (1 мг/мл). Эмбрионы исследовали и подсчитывали под световым микроскопом.
В тех экспериментах, где совместно с головками спермиев одновременно инъецировали два трансгена, 3-3,5 дневные эмбрионы сначала просчитывали с точки зрения экспрессии GFP, а затем - экспрессии β-галактозидазы описанным выше способом. Для фотографирования эмбрионы помешали между столиком микроскопа и покровным стеклом и делали ряд фотографий для того, чтобы выявить развитие и экспрессию гена GFP перед фиксацией и окрашиванием, которые использовали для выявления экспрессии LacZ.
ПРИМЕР 11
Исследование живых потомков на экспрессию трансгена
Живых потомков, полученных из эмбрионов, имплантированных в суррогатных матерей описанным выше способом, исследовали с точки зрения экспрессии постороннего GFP через 1-4 суток после рождения. Экспрессия GFP четко заметна в виде зеленого цвета кожи при боковом освещении УФ источником света (480 нм).
ПРИМЕР 12
Анализ интеграции трансгепа в геном
Методом Southern Blotting или реакцией цепочной полимеразы (polymerase chain reaction, PCR) был проведен физический анализ взятой из кончиков хвостов геномной ДНК. Биопсии кончиков хвостов отбирали у животных одного помета. Способами, хорошо известными специалистам, для получения общей геномной ДНК используют ткани копчика хвоста. Фотографируют хвосты под флуоресцентным стереомикроскопом, оборудованным фильтром 480/440 нм.
Для теста Southern blot no 10 мкг геномной ДНК на пробу энзиматически разрушают при помощи Eco RI и тестируют, используя 733 -пар-оснований Eco RI фрагмента pCX-EGFP. Для обнаружения гена GFP проводят PCR анализ с 1 мкг геномной ДНК на реакцию, используя передний (TTGAATTCGCCACCATGGTGAGC) и задний (TTGAATTCTTACTTGTACAGCTCG-TCC) олигонуклеотидные праймеры. Параметры реакции были следующими: 95°С в течение 9 мин (1 цикл) и 94°С в течение 45 с, 60°С в течение 30 с, 72°С в течение 45 с (40 циклов). Продукты PCR разделяли электрофорезом и визуализировали после окрашивания бромидом этидиума (ethidium bromide).
Экспрессия трансгена у эмбрионов, полученных после микроинъекции экзогенной кодирующей репортер ДНК или головками спермиев, или тем и другим одновременно в ооциты на стадии метафазы II
Экспрессия GFP и β-галактозидазы была отмечена у эмбрионов, которых культивировали in vitro в течение 3,5 суток после того, как спермий и ДНК предварительно инкубировали в течение 1 мин, а затем совместно инъецировали. GFP выявляли эпифлуоресцентной микроскопией, а β-галактозидазу выявляли окрашиванием, как это описано выше. Результаты представлены в Таблице 1. Доля эмбрионов, имеющих флуоресцирующие бластомеры, была наименьшей (26%), когда pCX-EGFP ДНК вводили совместно со сперматозоидами, подвергнутыми разрушению мембран при помощи Тритона Х-100 (64%), замораживанию-размораживанию (82%) или лиофилизации (87%). Совместное введение в неоплодотворенные ооциты фрагментов линеаризованной px-CANLacZ ДНК и либо замороженых-размороженных, либо лиофилизированных спермиев также породило высокий процент (от 92% до 94%) эмбрионов, экспрессирующих продукт lacZ тг-β-галактозидазу. Более того, совместное введение головки спермия в смеси с двумя различными ДНК (соответственно кодирующими GFP и LacZ), дало возможность после однократной инъекции получить эмбрионы, экспрессирующие оба тг. Фиг.2 показывает трансгенные эмбрионы, полученные таким двойным трансгенезом при однократной инъекции. Ооциты были микроинъецированы сперматозоидами, которые до этого преинкубировали в смеси pCX-LacZ и pCX-EGFP тг ДНК. Те же самые эмбрионы (х 400) показаны через 3,5 суток при помощи контрастной модулирующей микроскопии Гоффмана неокрашенпыми (Фиг.2А) под динноволновым (480 нм) УФ светом для экспрессии GFP (Фиг.2В) и окрашенными X-gal для экспрессии β-галоктозидазы (Фиг.2С).
В целом вышеприведенные данные свидетельствуют о том, что совместное введение головок спермиев с разрушенными мембранами и экзогенной нуклеиновой кислоты в неоплодотворенный ооцит может эффективно производить трансгенные эмбрионы.
Сперматозоиды, которые после смешивания с pCS-EGFP ДНК были промыты свежей средой, сохранили способность производить флуоресцентные бластоцисты, хотя и с несколько меньшей эффективностью (63% против 80%) по сравнению с их непромытыми двойниками (Таблица 1). Это предполагает быстрое образование связи между экзогенной ДНК и сперматозоидом во время смешивания (до инъекции).
Для проверки, может ли подобное же взаимодействие происходить внутри ооцитов (после инъекции), мы инъецировали головки спермиев и pCX-EGFP ДНК по очереди, без смешивания перед инъекцией. Нам ни разу не удалось выявить экспрессии экзогенной (GFP) ДНК, даже если в контроле флуоресцентными были 75% позитивных эмбрионов (замороженные-размороженные головки спермиев совместно введенные с pCX-EGFP, как в Таблице 1). Замороженные-размороженные головки спермиев, инъецированные совместно с pCX-EGFP в концентрации 500 пкг/мкл (но не в концентрации 50 пкг/мкл) производили бластоцисты, экспрессирующие видимый GFP. Эта граница обнаружения GFP (соответствует от 50 до 500 пкг pCX-EGFP ДНК на мкл) представляет собой в среднем от 15 до 150 молекул на внесенный инъекнией пкл.
В противоположность инъекции совместно с головкой спермия инъекция такого же количества одной только GFP тг ДНК не мешает хорошему партеногенетическому развитию (98% ооцитов, переживших инъекцию, развились до стадии морулы-бластоцисты) (Таблица 1). Более того, ни один из получившихся эмбрионов не проявлял наблюдаемой экспрессии тг. Однако и в отсутствие головок спермиев может быть незначительная экспрессия тг или эпихромосомальное сохранение транскрипционно активной ДНК.
Данные Таблицы 1 подтверждвают идею прединъекционной ассоциации экзогенной ДНК и субмембранных структур головки спермия, по-видимому, задействующей преимущественно основные протеины перинуклеарного матрикса {F.J.Longo et al., J.Cell Biol. 105, 1105 (1987)}. В других экспериментах нашей лаборатории мы обнаружили, что ядра спермиев содержат по меньшей мере одну эндонуклеазу и при 25°С лишенные мембран сперматозоиды быстро теряют способность поддерживать полное эмбриональное развитие {B.Maione et al., DNA Cell Biol., 16, 1087 (1997)}, Поэтому маловероятно, что использованная здесь геномная ДНК спермия не имела повреждений, соответствующих разрывам одной цепочки, которые и будут ускорять опосредованную ооцитом интеграцию тг.
С удивлением мы наблюдали мозаичные эмбрионы, имеющие после совместной инъекции головки спермия и pCX-EGFP, но не после инъекции одной ДНК pCX-EGFP, как GFP положительные, так и отрицательные бластомеры (+/-морула-бластописта) (Таблица 1). Частота появления таких +/- мозаик означает, что интеграция тг ДНК иногда откладывается до прохождения первой S фазы клеточного цикла после ICSI. Такая отложенная интеграция, очевидно, не происходит, если только тг ДНК не инъецируется совместно с головкой спермия. Одно из объяснений этому таково, что происходящий от сперматозоида материал стабилизирует экзогенную ДНК внутри раннего эмбриона, таким образом ускоряя отложенную интеграцию, в отсутствие такого материала (например, при партеногенезе) экзогенная ДНК деградирует до того, как она сможет интегрироваться.
После совместной инъекции головки спермия и pCX-EGFP потенциал развития эмбрионов при возрастании доли эмбрионов, имеющих флуоресцентные бластомеры, уменьшается (Таблица 1). В противоположность этому экспрессия тг после совместной инъекции головки спермия и pxCANLacZ, не ингибирует эмбриональное развитие (Таблица 1). Без теоретических ограничений, возможно, это отражает вредное воздействие экспрессии GFP. То есть раннее эмбриональное развитие может быть особенно чувствительно к выделению Н2О2, которое сопровождает созревание хроматофора GFP (R.Y.Tsien, supra).
Экспрессия трансгена у живых потомков, полученных после микроинъекции экзогенной ДНК, кодирующей репортер, или головок спермиев, или того и другого в ооцит на стадии метафазы II
Для того чтобы определить, можно ли продемонстрировать интеграцию тг конструкций ДНК на живых потомках (мыши родоначальники), головки спермиев, которые были подвергнуты одной их трех разрушающих мембраны процедур, инъецировали совместно с pCX-EGFP ДНК. Полученных эмбрионов культивировали in vitro, как это описано выше в течение 3,5-4 суток (до стадии морула-бластоциста), а затем случайным образом, то есть не на основе флюоресценции, переносили в суррогатных матерей. Результаты фенотипического анализа интеграции тг, проведенного путем исследования биопсий кончика хвоста трансгенных мышей и нетрансгенных контрольных мышей, показаны на Фиг.3А (а) и Фиг.3А (в) соответственно под длинноволновым УФ светом. Флуоресцирующую зеленым кожу трансгенных мышей можно видеть сквозь незеленые шерстинки. Значительная часть (от 17% до 21%) потомков была трансгенной в отношении наблюдаемой экспрессии GFP в коже (Таблица 2). Такая эффективность экспрессии не зависит от способа разрушения мембран, который использовали при подготовке сперматозоидов. Уровень полного развития зигот сравним для каждой из трех групп головок сперматозоидов с разрушенными мембранами (от 12% до 14%), но относительно мало похож на уровень, полученный после микроинъекции обработанных сходным образом головок в отсутствие экзогенной ДНК.
Эти данные соотносятся с результатами, приведенными на Таблице 1. Данные указывают на то, что эмбрионы, имеющие GFP отрицательные клетки, более вероятно разовьются полностью, чем эмбрионы, у которых все клетки позитивны. Кроме того, некоторые живые потомки, отнесенные при подсчетах к отрицательным, вероятно развились из мозаичных эмбрионов, которые на 3,5 сутки культивирования имели как GFP положительные, так и отрицательные клетки. Без теоретического подтверждения нам кажется, что совместная инъекция pCX-FGFP ДНК может имеет отрицательное влияние как на пре- так и на постимплантационное эмбриональное развитие. Однако неизвестно, является ли ингибирование постимплантационного развития следствием экспрессии тг, или присутствием экзогенной ДНК per се.
Физический анализ полной геномной ДНК кончиков хвостов методом Southern blotting (Фиг.3В) или методом PCR (Фиг.3С) показал, что все линии мышей родоначальников, которые проявляли зеленую флуоресценцию, имели тг, включая и тех, которых первоначально отнесли как фенотипически отрицательным, но биопсии кончиков хвостов которых демонстрировали экспрессию GFP. В трех случаях тг был выявлен методом PCR у родоначальников, не имеющих видимой зеленой флуоресценции. Без теоретического подтверждения мы считаем, что отсутствие экспрессии тг объясняется локальными cis-активными элементами в локусе интеграции тг. Результаты анализа по методу Southern blot общей ДНК контроля D6D2F (дт) (0) и мышей родоначальников №3 (5-9), 19 (> 50), 28 (5-9) и 41 (2) с использованием фрагментов pCX-EGFP в качестве пробы показаны на Фиг.3В, где в скобках указано вычисленное число копий тг на геном. Анализ по методу Southern blot показал, что число копий тг у родоначальников варьирует от <= 1 до >50. Этот результат напоминает характер интеграции тг после пронуклеарной микроинъекции. Как физическая характеристика геномной pCX-EGFP ДНК, так и эффективность экспрессии GFP указывают на то, что в момент интеграции тг ДНК не претерпевает сильных перестроек.
Результаты анализа методом PCR общей ДНК мышей 16, 17, 30, 36, 47, 49, контроля B6D2F1 (дт), 3, 19, 28 и 41 показаны на Фиг.3С и подтверждают наличие трансгена у мышей 17, 3, 19 и 28.
Двенадцать отобранных случайным образом экспрессирующих GFP основателей (8 самок, 4 самца из Таблицы 2 и аналогичных серий) скрестили с не трансгенными животными и получили пометы во всех, кроме одного (самка), случаях. Из 11 плодовитых родоначальников 8 произвели мышат, экспрессирующих GFP наружно, в коже, с частотой от 27% до 50% (в среднем 40%). Характер наследования тг в большинстве случаев согласуется с прямой гаметной передачей одиночного GFP гена по Менделю.
Хотя изобретение было описано в отношении предпочтительного способа реализации, следует понимать, что это не предполагает ограничения изобретения только теми формами, которые были раскрыты. Наоборот, мы предпочитаем охватить все модификации и альтернативные формы, подпадающие под дух и букву изобретения.
+ Когда сравнивали значения в одной колонке с надпечатками "а" и "в", они различались достоверно (Р<0,05)
Значения в той же колонке с надпечаткой "с" не различались достоверно
$ Экспрессия тг: - отрицательная, + положительная; +/-, м-б содержали как + клетки, так и - клетки (мозаики).
+ м-б, Морулы - бластоцисты. Значения в скобках показывают число суррогатных матерей, использованных в качестве реципиентов при пересадке эмбрионов.
$ Экспрессия тг: + - позитивные мышата - те, которые внешне в коже экспрессируют GFP
**Величины различаются недостоверно
| название | год | авторы | номер документа |
|---|---|---|---|
| РАЗВИТИЕ НОРМАЛЬНОГО ПОТОМСТВА ИЗ ООЦИТОВ, ИНЪЕЦИРОВАННЫХ ОБРАБОТАННЫМИ СУШКОЙ ВЫМОРАЖИВАНИЕМ СПЕРМАТОЗОИДАМИ | 1999 |
|
RU2244525C2 |
| СПОСОБ ПОЛУЧЕНИЯ ТРАНСГЕННЫХ МЫШЕЙ | 2009 |
|
RU2425880C2 |
| СПОСОБ ПОЛУЧЕНИЯ ТРАНСГЕННЫХ КУР ОПОСРЕДОВАННЫМ ПЕРЕНОСОМ ГЕНА ПУТЕМ ОБЫЧНОГО ИСКУССТВЕННОГО ОСЕМЕНЕНИЯ | 2011 |
|
RU2517731C2 |
| СПОСОБ ВВЕДЕНИЯ ЧУЖЕРОДНОЙ ДНК В СПЕРМАТОЗОИДЫ | 1994 |
|
RU2081914C1 |
| ТРАНСГЕННЫЕ КОПЫТНЫЕ ЖИВОТНЫЕ, ИМЕЮЩИЕ ПОНИЖЕННУЮ АКТИВНОСТЬ ПРИОННОГО БЕЛКА, И ИХ ПРИМЕНЕНИЯ | 2003 |
|
RU2384059C2 |
| СПОСОБ ПОЛУЧЕНИЯ ТРАНСГЕННЫХ МЛЕКОПИТАЮЩИХ, КОТОРЫЕ ПРОДУЦИРУЮТ ЭКЗОГЕННЫЕ БЕЛКИ В МОЛОКЕ, И ТРАНСГЕННЫЕ МЛЕКОПИТАЮЩИЕ, ПОЛУЧЕННЫЕ ТАКИМ СПОСОБОМ | 2004 |
|
RU2390562C2 |
| ПРИМЕНЕНИЕ ЦИКЛИЧЕСКОГО ТРИПЕПТИДА ДЛЯ УЛУЧШЕНИЯ КЛЕТОЧНОГО ЭНЕРГЕТИЧЕСКОГО МЕТАБОЛИЗМА | 2016 |
|
RU2737116C2 |
| СПОСОБ ПОЛУЧЕНИЯ ТРАНСГЕННЫХ КРОЛИКОВ, ПРОДУЦИРУЮЩИХ БЕЛКИ В МОЛОЧНУЮ ЖЕЛЕЗУ | 2007 |
|
RU2402211C2 |
| СПОСОБ ПРОДУЦИРОВАНИЯ ЭКЗОГЕННОГО БЕЛКА В МОЛОКЕ ТРАНСГЕННЫХ МЛЕКОПИТАЮЩИХ И СПОСОБ ОЧИСТКИ БЕЛКОВ ИЗ МОЛОКА | 2004 |
|
RU2360002C2 |
| ИЗОЦИТРАТДЕГИДРОГЕНАЗА, ЕЕ ГЕН И ИХ ПРИМЕНЕНИЕ В ЛЕЧЕНИИ ОЖИРЕНИЯ, ГИПЕРЛИПИДЕМИИ И ЖИРОВОЙ ИНФИЛЬТРАЦИИ ПЕЧЕНИ ПРИ БИОСИНТЕЗЕ ЛИПИДОВ | 2001 |
|
RU2266131C2 |
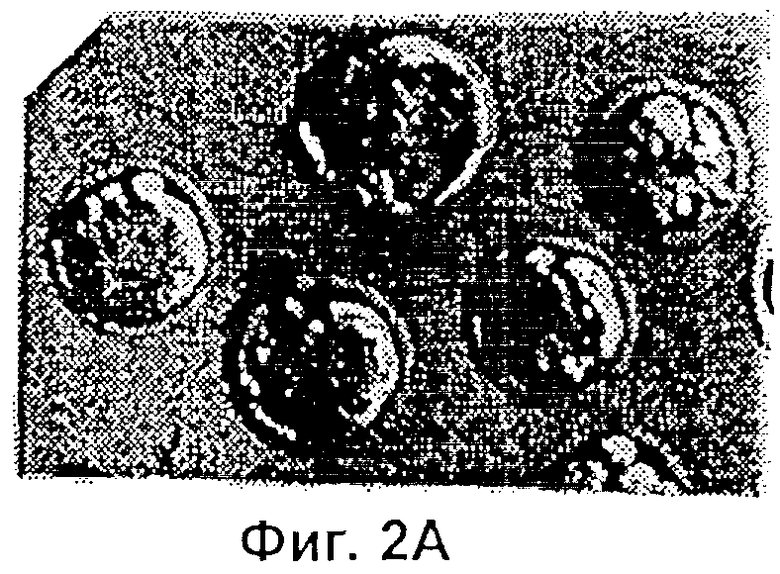
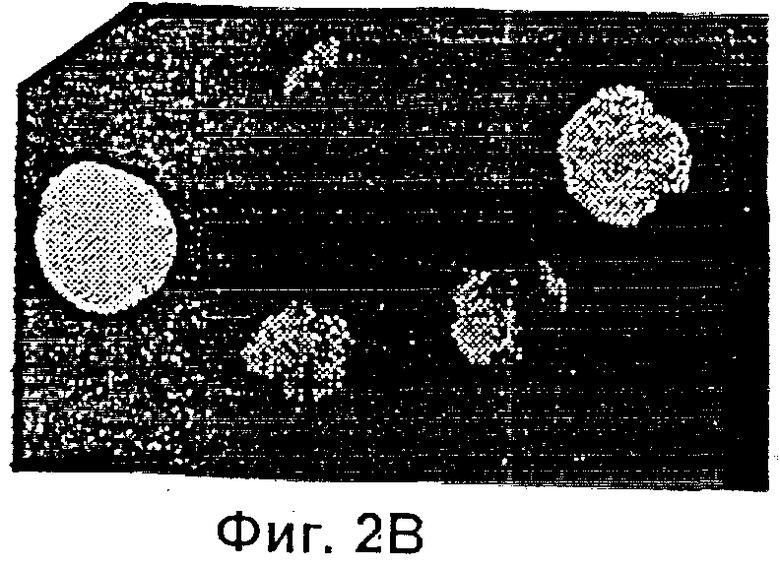


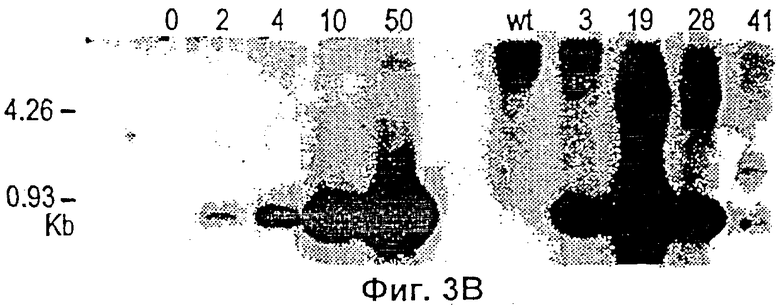
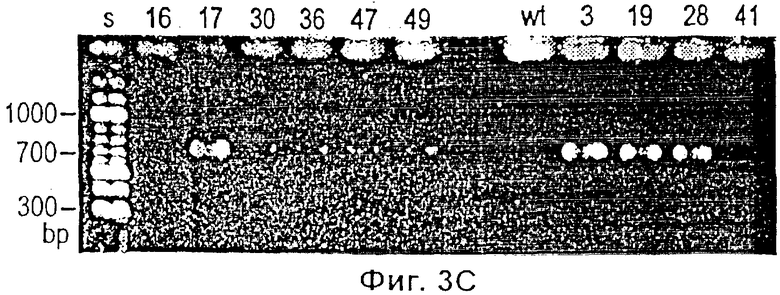
Изобретение относится к области генетической инженерии, в частности к способам получения трансгенного эмбриона. Способ предусматривает инкубирование экзогенной нуклеиновой кислоты и головки спермия с разрушенной или удаленной мембраной. Далее получают трансгенный оплодотворенный ооцит путем совместного введения экзогенной нуклеиновой кислоты и головки спермия в неоплодотворенный ооцит. В заключение развивают оплодотворенный ооцит в трансгенный эмбрион. Другой вариант способа предусматривает предварительное получение головок спермиев с разрушенной или удаленной мембраной. Предложенные способы позволяют повысить эффективность трансгенного переноса и повысить выход трансгенных животных. Изобретение может быть использовано для получения методами генетической инженерии скота и других крупных животных, которые могут быть использованы для получения фармацевтических «фабрик» и как источник органов или клеток для ксенотрансплантации. 2 н. и 18 з.п. ф-лы, 7 ил., 2 табл.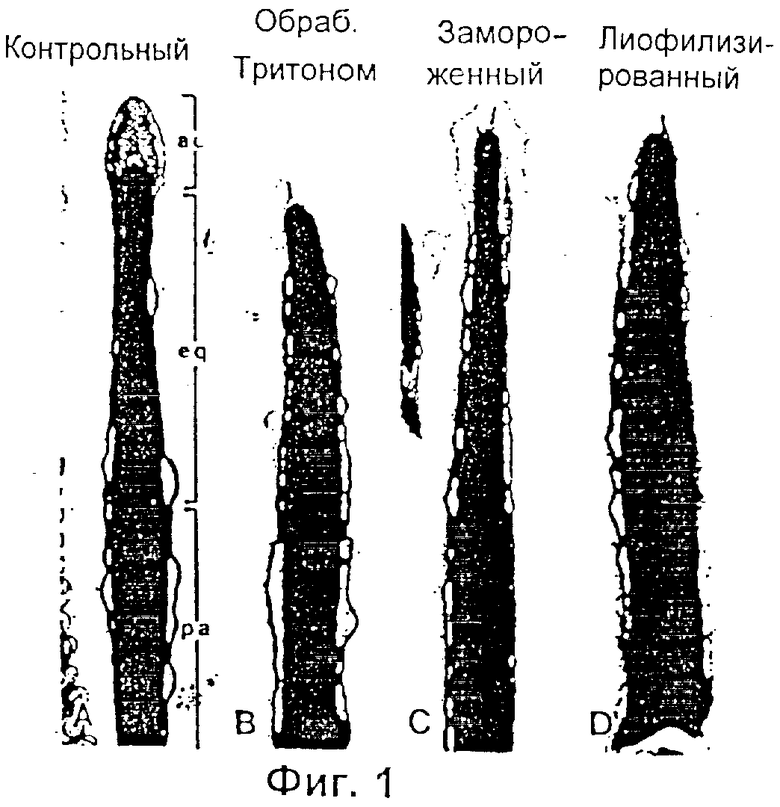
инкубирования экзогенной по отношению к эмбриону нуклеиновой кислоты и головки спермия с разрушенной мембраной или головки спермия с удаленной мембраной в течение промежутка времени;
совместного введения экзогенной нуклеиновой кислоты и указанной головки спермия в неоплодотворенный ооцит с образованием трансгенного оплодотворенного ооцита и
предоставления трансгенному оплодотворенному ооциту возможности развиться в трансгенный эмбрион.
получения головки спермия с разрушенной мембраной или головки спермия с удаленной мембраной;
смешивания головки спермия с разрушенной мембраной или головки спермия с удаленной мембраной с экзогенной по отношению к эмбриону нуклеиновой кислотой, несущей желаемый ген;
совместного введения смеси в изолированный неоплодотворенный ооцит на стадии метафазы II с образованием трансгенного оплодотворенного ооцита и
предоставление трансгенному оплодотворенному ооциту возможности развиться в трансгенный эмбрион.
| Lavitrano M | |||
| et al., «Sperm cells as vectors for introducing foreign DNA into eggs: genetic transformation of mice.», Cell 1989 Jun 2; 57(5):717-23 | |||
| Kimura Y | |||
| et al, «Analysis of mouse oocyte activation suggests the involvement of sperm perinuclear material.», Biol Reprod 1998 Jun; 58(6): 1407-15 | |||
| Kuretake S, et al., «Fertilization and development |

