Настоящее изобретение относится к способу получения эфиров гидроксифенилкарбоновой кислоты с использованием определенных катализаторов. Настоящее изобретение относится также к новым эфирам гидроксифенилкарбоновой кислоты, которые можно получать предлагаемым в изобретении способом.
Эфиры гидроксифенилкарбоновой кислоты, прежде всего общей формулы (I), применяются предпочтительно в качестве антиоксидантов. Известны многочисленные соединения формулы (I). Эти соединения можно получать, например, путем переэтерификации с использованием пригодных для этой цели катализаторов. Такие способы переэтерификации описаны, например, в US 4716244, US 5481023, US 5563291.
Некоторые соединения формулы (I) являются важными коммерческими продуктами. Так, например, их используют для защиты органических материалов, таких как пластмассы и смазочные вещества, от деструкции при термическом воздействии, окислительном воздействии и/или воздействии актиничного излучения. Кроме того, существует потребность в новых соединениях такого типа для их применения в качестве антиоксидантов, а также в усовершенствованных способах их получения.
При создании изобретения было установлено, что при осуществлении известного способа переэтерификации определенные используемые в качестве катализаторов щелочнометаллические соли карбоновых кислот, такие, например, как ацетат лития, ацетат натрия или ацетат калия, можно применять в практически нейтральной среде и без растворителей, при этом продукты получают в виде бесцветных расплавов, которые можно использовать непосредственно в предусмотренных целях без их последующей очистки. При этом, что также является неожиданным фактом, отпадает необходимость в нейтрализации катализатора, а реакция обратной этерификации не наблюдается даже при контакте прореагировавшей реакционной массы со спиртами. Следует отметить и тот факт, что описанные выше катализаторы при указанных условиях, прежде всего при нейтральных значениях рН и без использования растворителей, катализируют реакции переэтерификации, при которых n может равняться 1, 2, 3, 4 или иметь большее значение.
Отличительные особенности предлагаемого в изобретении решения охарактеризованы в формуле изобретения.
В соответствии с этим настоящее изобретение относится прежде всего к способу получения соединений формулы (I)
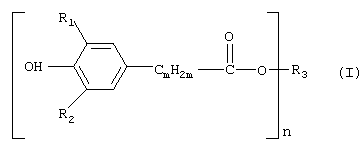
в которой
R1 И R2 независимо друг от друга обозначают С1-С8алкил, циклопентил или циклогексил,
m обозначает 1, 2 или 3, предпочтительно 2,
n обозначает целое число от 1 до 30, предпочтительно целое число от 1 до 10, прежде всего 1, 2, 3, 4, 5 или 6, более предпочтительно 1, 2, 3 или 4,
R3 обозначает n-валентный линейный или разветвленный С4-С30алкильный остаток, который необязательно прерван одним или несколькими атомами кислорода, либо обозначает (при n=1-12) n-валентный С5-С12диклоалкильный остаток, либо остаток R4-[NR5-CmH2m-]p,
R4 обозначает водород, n-валентный линейный или разветвленный С4-С30алкильный остаток, который необязательно прерван одной или несколькими группами -NR5-, либо обозначает (при m=1-12) m-валентный С5-С12пиклоалкильный остаток,
R5 в каждом случае независимо обозначает водород, метил или -CmН2m-, предпочтительно водород, а
p соответствует тому числу групп -[NR5-CmH2m-], которое равняется количеству n остатков -CmН2m-, приходящихся на одну молекулу, заключающемуся во взаимодействии соединения формулы (II)
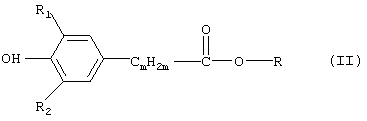
в которой R обозначает C1-С3алкил, с соединением формулы (III)

в которой R3 и n имеют вышеуказанные значения, и отличающемуся тем, что взаимодействие указанных соединений проводят при практически нейтральном значении водородного показателя (рН) в присутствии по крайней мере одной растворенной или суспендированной в реакционной смеси щелочнометаллической соли органической карбоновой кислоты либо в присутствии смеси таких щелочнометаллических солей, при этом (I) указанная щелочнометаллическая соль образована катионом щелочного металла и анионом органической карбоновой кислоты и (II) органическая карбоновая кислота в используемых для проведения реакции условиях является по меньшей мере частично летучей.
При этом под используемыми для проведения взаимодействия между указанными соединениями условиями реакции подразумеваются температура, предпочтительно составляющая от 50 до 250°С, более предпочтительно от 80 до 220°С, наиболее предпочтительно от 140 до 220°С, и давление, составляющее от 0,1 мбара до 1 атм (нормальное давление), предпочтительно от 0,1 мбара до 100 мбар, более предпочтительно от 0,1 мбара до 50 мбар, наиболее предпочтительно от 0,1 мбара до 20 мбар. Температура кипения солеобразующей карбоновой кислоты предпочтительно находится в указанных интервалах значений температуры и давления.
В качестве солеобразующих катионов щелочных металлов могут рассматриваться прежде всего катионы лития, натрия или калия. Предпочтительными являются катионы натрия, калия и/или лития, соответственно натриевые, калиевые и/или литиевые соли органических карбоновых кислот.
В качестве примера карбоновых кислот, которые в используемых при проведении реакции условиях по меньшей мере частично являются летучими в их кислой форме, можно назвать алифатические насыщенные или ненасыщенные карбоновые кислоты предпочтительно с 2-10 С-атомами, наиболее предпочтительно с 2-6 С-атомами, такие, например, как муравьиная кислота, уксусная кислота, пропионовая кислота, н-масляная кислота, изомасляная кислота, н-валериановая кислота, триметилуксусная кислота, капроновая кислота, н-гептиловая кислота или пеларгоновая кислота. Другими примерами указанных кислот являются малоновая кислота, малеиновая кислота, фумаровая кислота или же монометиловый эфир малоновой кислоты. Пригодными для использования являются также галогенированные кислоты, такие, например, как фторуксусная кислота, хлоруксусная кислота, бромуксусная кислота, дифторуксусная кислота, дихлоруксусная кислота, трифторуксусная кислота, трихлоруксусная кислота, α-хлорпропионовая кислота или β-хлорпропионовая кислота. Предпочтительны ацетат натрия, ацетат калия, ацетат лития, формиат натрия, формиат калия или формиат лития либо смесь этих соединений.
Катализатор добавляют предпочтительно в количестве от 0,05 до 5 мол.% в пересчете на превращаемое молярной количество соединения формулы (I).
R1 предпочтительно обозначает С1-С8алкил, при этом такой алкильный остаток может быть линейным или разветвленным. Более предпочтительно R1 обозначает алкильный остаток с 1-4 С-атомами, наиболее предпочтительно представляет собой метил, этил, пропил или бутил. Особенно предпочтительно R1 обозначает разветвленный остаток, наиболее предпочтительно метил или трет-бутил.
R2 обозначает предпочтительно С1-С8алкил, при этом такой алкильный остаток может быть линейным или разветвленным. Более предпочтительно R2 обозначает алкильный остаток с 1-4 С-атомами, наиболее предпочтительно представляет собой метил, этил, пропил или бутил. Предпочтительно R2 обозначает разветвленный остаток, наиболее предпочтительно метил или трет-бутил, особенно предпочтительно трет-бутл.
Соединения формулы (I) могут содержать в одной и той же молекуле различным образом замещенные радикалами R1 и R2 фенильные остатки, а R1 и R2 в одном и том же фенильном остатке в свою очередь могут быть идентичными или различными. В соответствии с этим оба R1 и R2 могут, например, обозначать метил или трет-бутил либо R1 может обозначать метил, а R2 может обозначать трет-бутил.
R3 в качестве n-валентного алкильного остатка с 4-30 С-атомами, который необязательно прерван кислородом, может быть линейным или разветвленным либо может быть представлен в виде смеси. Подобные смеси могут состоять из соединений, в которых R3 представляет собой смесь преимущественно линейных или разветвленных алкильных остатков, например, с 14, 16, 18 или 20 С-атомами, причем такие алкильные остатки частично могут быть также разветвленными.
Если R3 обозначает остаток R4-[NR5-CmH2m-]p, то этот остаток предпочтительно обозначает R4-[NH-CmH2m-]p или R4-[N(CmH2m-)2]p, наиболее предпочтительно R4-[NH-CmH2m-]p. В этом остатке R4 предпочтительно обозначает алкильный остаток с 4-30 С-атомами, который необязательно прерван группой -NR5-, более предпочтительно группой -NH-, и может быть линейным или разветвленным, например, с 14, 16, 18 и 20 С-атомами. Аналогичным образом R4 может представлять собой производное (при n=2) алкилендиамина, такого как этилендиамин или пропилендиамин, либо полиалкилендиамина, предпочтительно диалкилендиамина, такого как диэтилендиамин или дипропилендиамин.
Если n=1, то R3 предпочтительно обозначает одновалентный остаток, более предпочтительно н-бутил, изобутил, трет-бутил, пентил, изопентил, гексил, гептил, октил, 2-этилгексил, нонил, децил, ундецил, додецил, тридецил, тетрадецил, пентадецил, гексадецил, гептадецил, октадецил, эйкозил, а также соответствующие гомологические остатки с увеличивающимся до 30 числом атомов углерода или смесь таких С4-С30остатков. При n=1 R3 также предпочтительно обозначает одновалентный остаток, такой как циклоалкил с 5-12 С-атомами, предпочтительно циклопентил и циклогексил, прежде всего циклогексил. R3 предпочтительно обозначает (при n=1) одновалентный C8-С30алкильный остаток, наиболее предпочтительно изооктил, 2-этилгексил, н-гексадецил и н-октадецил либо смесь таких алкильных остатков. Указанные значения R3 являются также предпочтительными значениями R4.
Если n=2, то R3 обозначает предпочтительно С3-С8алкилен или прерванный кислородом С4-С13алкилен. В соответствии с этим R3 представляет собой (при n=2) дериватизированную из двухвалентного спирта при исключении обеих ОН-групп алкиленовую группу, которая может быть прервана кислородом. Примерами таких двухвалентных спиртов являются алкиленгликоли, такие как 1,2-этандиол, 1,3-пропандиол, 1,4-бутандиол, 1,5-пентандиол, 1,6-гександиол, 1,7-гептандиол и 1,8-октандиол, полиалкиленгликоли, такие как диэтиленгликоль, триэтиленгликоль и тетраэтиленгликоль, дипропиленгликоль и трипропиленгликоль, или (при n=1) глицерин, пентаэритрит [С(СН2OH)4] либо аналогичные многоатомные спирты. Равным образом прерванные кислородом n-валентные остатки R3 могут представлять собой производные этерифицированных полигидроксисоединений, предпочтительно полиглицерина или полипентаэритрита.
Аналогичным образом R4 (при n=2) предпочтительно представляет собой дериватизированную из диамина при исключении обеих H2N-групп алкиленовую группу, которая может быть прервана группой -NR5-, предпочтительно группой -NH-. Примерами таких диаминов являются алкилендиамины, такие как 1,2-этилендиамин или 1,2-пропилендиамин, либо диалкилендиамины, такие как ди(этилендиамин) или ди(пропилендиамин). При определенных условиях ди(этилендиамин) может содержать, например, у среднего атома азота группу [-NH-CmH2m-].
Если n=3, то R3 представляет собой дериватизированную из трехвалентного спирта при исключении ОН-групп разветвленную алкилалкиленовую группу. В этом случае R3 обозначает предпочтительно метан-C1-С6алкилтриметилен. Примерами таких трехвалентных спиртов являются 1,1,1-трис-гидроксиметилэтан (триметилолэтан), 1,1,1-трис-гидроксиметилпропан (триметилолпропан).
Если n=4, то R3 представляет собой дериватизированную из четырехвалентного или многоатомного спирта при исключении ОН-групп разветвленную алкилалкиленовую группу. В этом случае R3 обозначает предпочтительно метантетраметилен, например тетракисгидроксиметилметан (пентаэритрит).
В алкиленовом остатке -(CmН2m)-, присутствующем в соединении формулы (I), соответственно формулы (II), m предпочтительно равен 2, а алкиленовый остаток обозначает группу -СН2-СН2-.
Предлагаемым в изобретении способом особенно предпочтительно получать соединения формулы (I), в которых R1 представляет собой трет-бутил, R2 представляет собой метил или трет-бутил, R3 представляет собой н-октадецил, смесь высших алкильных остатков с 8-30 С-атомами (C8-С30арильные остатки) или остаток, представляющий собой производное 1,6-гександиола, триэтиленгликоля, пентаэритрита, 1,1,1-трис-гидроксиметилэтана, 1,1,1-трис-гидроксиметилпропана, либо R4 обозначает остаток, представляющий собой производное 1,2-этилендиамина, либо 1,2-пропилендиамина, или ди(этилендиамина), либо ди(пропилендиамина).
Настоящее изобретение относится также к применению щелочнометаллической соли органической карбоновой кислоты или смеси таких щелочнометаллических солей, описанных выше, в качестве катализатора при получении соединений формулы (I), исходя из приведенных выше соединений формулы (II) и (III), при этом предпочтительно применяют ацетат натрия, ацетат калия, ацетат лития, формиат натрия, формиат калия, или формиат лития, либо смесь этих соединений.
Катализатор добавляют к реакционной смеси в количестве от 0,05 до 5 мол.%, предпочтительно от 0,05 до 3 мол.%, наиболее предпочтительно от 0,1 до 1 мол.%, в пересчете на превращаемое молярной количество соединения формулы (I).
Согласно изобретению предпочтительно работать без растворителей. Вместе с тем изобретение не исключает возможности проведения реакции переэтерификации в инертном органическом алифатическом и/или ароматическом растворителе либо в смеси таких растворителей.
Согласно изобретению соединения формул (II) и (III) вместе с катализатором нагревают в инертных условиях предпочтительно при температуре порядка 90-120°С и при перемешивании до образования расплава. Затем с целью сдвинуть химическое равновесие в реакции переэтерификации давление понижают, а температуру повышают, т.е. реакцию предпочтительно проводят при давлении от 0,1 до 200 мбар и при температуре от 140 до 220°С. При этом предпочтительно работать при давлении в интервале от 0,1 до 50 мбар, наиболее предпочтительно от 0,1 до 20 мбар. Температуру реакции в свою очередь предпочтительно повышать до 160-220°С, наиболее предпочтительно до 165-185°С.
Продолжительность реакции зависит от давления и температуры и в целом составляет от 1 до 12 ч, предпочтительно от 1 до 10 ч, наиболее предпочтительно от 2 до 6 ч.
Сложный эфир формулы (II) обычно применяют в небольшом избытке в пересчете на функциональные гидроксигруппы спирта формулы (III). Количественное соотношение между соединением формулы (II) и соединением формулы (III) предпочтительно составляет [в пересчете на мольные эквиваленты] от 0,8:1 до 1,5:1, предпочтительно от 1:1 до 1,2:1, наиболее предпочтительно от 1,05:1 до 1,15:1.
При использовании соединения формулы (II) в избытке это избыточное количество предпочтительно отгонять в конце реакции, при этом такой избыток одновременно служит азеотропом для образующихся при любых условиях нежелательных побочных компонентов, которые отрицательно влияют на цвет продукта формулы (I).
Продукт формулы (I) можно непосредственно путем охлаждения, с затравкой или без нее подвергать кристаллизации или отверждению, после чего его можно непосредственно подвергать дальнейшей переработке для придания соответствующего товарного вида без дополнительной очистки, например, перекристаллизацией. Очевидно, что согласно изобретению расплав можно также растворять в пригодном для этой цели растворителе, охлаждать и кристаллизовать с затравкой или без нее. В качестве примера соответствующих растворителей можно назвать алифатические углеводороды, такие как гептан, или циклогексан, либо их смеси, ароматические углеводороды, такие как толуол и/или ксилол, спирты, такие как метанол, этанол, пропанол и/или изопропанол, а также соответствующие смеси спирт/вода (с содержанием спирта 50-100%). Предпочтительны метанол и/или изопропанол и их смеси с водой.
Остаточное содержание катализатора в продукте, например, до или после проводимой при необходимости фильтрации прореагировавшего реакционного расплава обычно не создает никаких помех при их использовании в качестве стабилизатора. Вместе с тем присутствующие в продукте щелочнометаллические соли можно удалять путем простой фильтрации, например через пористую керамическую пластину с размером пор 20 мкм при температуре от 90 до 130°С. После фильтрации содержание катализатора обычно лежит в диапазоне миллионных долей.
Особые преимущества предлагаемого в изобретении способа заключаются в получении продукта в чистом для анализа виде без нежелательного изменения его цвета, т.е. без цветных или изменяющих цвет побочных компонентов в реакционном расплаве, соответственно в продуктах реакции, благодаря чему отпадает необходимость проведения дальнейших операций очистки, а самому продукту реакции можно придавать готовый для его применения товарный вид без введения добавок лишь путем его переработки простыми в осуществлении физическими методами, например путем измельчения или таблетирования.
Используемые в предлагаемом в изобретении способе соединения формул (II) и (III) как таковые известны. Вместе с тем соединения формулы (I), в которых n=3, а R3 обозначает трехвалентный остаток, который представляет собой производное 1,1,1-трис-гидроксиметилэтана (триметилолэтана) или в которых R3 обозначает 1,1,1-трис-метиленэтан, являются новыми. Равным образом новыми являются и соединения формулы (I), в которых n=3, а R3 обозначает трехвалентный остаток, представляющий собой производное 1,1,1-трис-гидроксиметилпропана (триметилолпропана), или в которых R3 обозначает 1,1,1-трис-метиленпропан.
Новыми являются также соединения формулы (I), в которых R3 обозначает остаток R4-[NR5-CmH2m-]p, предпочтительно R4-[NH-CmHm-]p, где R4 обозначает остаток, представляющий собой производное 1,2-этилендиамина, или 1,2-пропилендиамина, либо ди(этилендиамина), или ди(пропилендиамина), a R1, R2 и m имеют указанные выше значения.
Все такие соединения также являются объектом настоящего изобретения. Эти соединения можно получать и по другим известным методам, и поэтому они не "привязаны" конкретно к предлагаемому в изобретении способу их получения. Такие соединения в основном ацетоновом растворе проявляют, как неожиданно было установлено, лишь незначительную тенденцию к пожелтению, что является важным подтверждением преимуществ их практического применения с учетом степени их пожелтения, являющейся показателем изменения окраски.
Ниже изобретение проиллюстрировано на примерах, которые не ограничивают его объем.
Пример 1. Октадециловый эфир β-(3,5-ди-трет-бутил-4-гидроксифенил)пропионовой кислоты (соединение формулы (I), в которой R1 и R2 означают трет-бутил, n=1, m=2, R3 обозначает н=C18H37)
В реакционный сосуд помещают 109 г (0,374 моля) метилового эфира β-(3,5-ди-трет-бутил-4-гидроксифенил)пропионовой кислоты, 92,5 г (0,34 моля) стеарилового спирта и 0,1 г ацетата лития в виде дигидрата (0,001 моля) помещают, расплавляют при температуре 100°С и перемешивают в атмосфере азота. Сразу же после образования расплава систему осторожно вакуумируют и одновременно нагревают до внутренней температуры (ВТ) 150-160°С. Образующийся метанол отгоняют и конденсируют в вымораживающей ловушке. Сразу же после снижения внутреннего давления до уровня менее 10 мбар внутреннюю температуру повышают до 170-180°С и затем вакуумируют до давления менее 1 мбара. Сразу же после повышения внутренней температуры до 180°С и снижения давления до уровня менее 1 мбара смесь выдерживают в течение 1 ч, после чего в течение 1-2 ч при ВТ 200-210°С отгоняют избыток метилового эфира β-(3,5-ди-трет-бутил-4-гидроксифенил)пропионовой кислоты. Содержание эдуктов в реакционном расплаве составляет менее 0,2%, а содержание октадецилового эфира β-(3,5-ди-трет-бутил-4-гидроксифенил)пропионовой кислоты составляет более 98%. Реакционный расплав охлаждают и оставляют кристаллизоваться.
Выход: 96,5%.
Температура плавления: 51°С.
Пример 2. Октадециловый эфир β-(3,5-ди-трет-бутил-4-гидроксифенил)пропионовой кислоты (соединение формулы (I), в которой R1 и R2 обозначают трет-бутил, n=1, m=2, R3 обозначает н-C18H37).
В реакционный сосуд помещают 109 г (0,374 моля) метилового эфира β-(3,5-ди-трет-бутил-4-гидроксифенил)пропионовой кислоты, 92,5 г (0,34 моля) стеарилового спирта и 0,2 г формиата натрия (0,003 моля), расплавляют при температуре 100°С и перемешивают в атмосфере азота. Сразу же после образования расплава систему осторожно вакуумируют и одновременно нагревают до внутренней температуры 150-160°С. Образующийся метанол отгоняют и конденсируют в вымораживающей ловушке. Сразу после снижения внутреннего давления до уровня менее 10 мбар внутреннюю температуру повышают до 170-180°С и затем вакуумируют до давления менее 1 мбара. Сразу после повышения внутренней температуры до 180°С и снижении давления до уровня менее 1 мбара смесь выдерживают в течение 1 ч, после чего в течение 1-2 ч при ВТ 200-210°С отгоняют избыток метилового эфира β-(3,5-ди-трет-бутип-4-гидроксифенил)пропионовой кислоты. Содержание эдуктов в реакционном расплаве составляет менее 0,2%, а содержание октадецилового эфира β-(3,5-ди-трет-бутил-4-гидроксифенил)пропионовой кислоты составляет более 98%. Реакционный расплав охлаждают и оставляют кристаллизоваться.
Выход: 96,5%.
Температура плавления: 51°С.
Пример 3. Эфир триэтиленглицил-бис[β-(3,5-ди-трет-бутил-4-гидроксифенил)пропионовой кислоты] (соединение формулы (I), в которой R1 и R2 обозначают трет-бутил, n=2, m=2, R3 обозначает -(СН2СН2O)2СН2СН2)-)
В реакционный сосуд помещают 76 г (0,26 моля) метилового эфира β-(3,5-ди-трет-бутил-4-гидроксифенил)пропионовой кислоты, 15 г (0,1 моля) триэтиленгликоля и 0,17 г ацетата лития в виде дигидрата (0,0015 моля), расплавляют при температуре 100°С и перемешивают в атмосфере азота. Сразу же после образования расплава систему осторожно вакуумируют и одновременно нагревают до внутренней температуры 160-180°С. Образующийся метанол отгоняют и конденсируют в вымораживающей ловушке. Сразу после снижения внутреннего давления до уровня менее 10 мбар внутреннюю температуру повышают до 180-190°С и затем вакуумируют до давления менее 1 мбара. Сразу же после повышения внутренней температуры до 190°С и снижении давления до уровня менее 1 мбара смесь выдерживают в течение 1 ч, после чего в течение 1-2 ч при ВТ 200-220°С отгоняют избыток метилового эфира β-(3,5-ди-трет-бутил-4-гидроксифенил)пропионовой кислоты. Содержание эдуктов в реакционном расплаве составляет менее 0,2%, а содержание эфира триэтиленглицил-бис[β-(3,5-ди-трет-бутил-4-гидроксифенил)пропионовой кислоты] составляет более 97%. Реакционный расплав охлаждают и оставляют кристаллизоваться.
Выход: 94%.
Температура плавления: 113-115°С.
Пример 4. Эфир триэтиленглицил-бис[β-(3-трет-бутил-5-метил-4-гидроксифенил)пропионовой кислоты] (соединение формулы (I), в которой R1 обозначает трет-бутил, R2 обозначает метил, n=2, m=2, R3 обозначает -(СН2СН2O)2СН2СН2)-)
В реакционный сосуд помещают 65 г (0,26 моля) метилового эфира β-(3-трет-бутил-5-метил-4-гидроксифенил)пропионовой кислоты, 15 г (0,1 моля) триэтиленгликоля и 0,17 г ацетата лития в виде дигидрата (0,0015 моля), расплавляют при температуре 100°С и перемешивают в атмосфере азота. Сразу же после образования расплава систему осторожно вакуумируют и одновременно нагревают до внутренней температуры 160-170°С. Образующийся метанол отгоняют и конденсируют в вымораживающей ловушке. Сразу после снижения внутреннего давления до уровня менее 10 мбар внутреннюю температуру повышают до 170-180°С и затем вакуумируют до давления менее 1 мбара. Сразу же после повышения внутренней температуры до 180°С и снижении давления до уровня менее 1 мбара смесь выдерживают в течение 1 ч, после чего в течение 1-2 ч при ВТ 190-200°С отгоняют избыток метилового эфира β-(3-трет-бутил-5-метил-4-гидроксифенил)пропионовой кислоты. Содержание эдуктов в реакционном расплаве составляет менее 0,2%, а содержание эфира триэтиленглицил-бис[β-(3-трет-бутил-5-метил-4-гидроксифенил)пропионовой кислоты] составляет более 97%. Реакционный расплав охлаждают и оставляют кристаллизоваться.
Выход: 94,5%.
Температура плавления: 74-77°С.
Пример 5. 1,1,1-трис-[β-(3,5-ди-трет-бутил-4-гидроксифенил)пропионилоксиметил]пропан (соединение формулы (I), в которой R1 и R2 обозначают трет-бутил, n=3, m=2, R3 обозначает СН3СН2С(СН2)3)
В реакционный сосуд помещают 85,5 г (0,293 моля) метилового эфира β-(3,5-ди-трет-бутил-4-гидроксифенил)пропионовой кислоты, 10,1 г (0,075 моля) 1,1,1-трет-(гидроксиметил)пропана и 0,23 г ацетата лития в виде дигидрата (0,002 моля), расплавляют при температуре 120°С и перемешивают в атмосфере азота. Сразу же после образования расплава систему осторожно вакуумируют и одновременно нагревают до внутренней температуры 160°С. Образующийся метанол отгоняют и конденсируют в вымораживающей ловушке. Сразу после снижения внутреннего давления до уровня менее 10 мбар внутреннюю температуру повышают до 180°С и затем вакуумируют до давления менее 1 мбара. Сразу после повышения внутренней температуры до 180°С и снижении давления до уровня менее 1 мбара смесь выдерживают в течение 1 ч, после чего в течение 1-2 ч при ВТ 200-210°С отгоняют избыток метилового эфира β-(3,5-ди-трет-бутил-4-гидроксифенил)пропионовой кислоты. Содержание метилового эфира β-(3,5-ди-трет-бутил-4-гидроксифенил)пропионовой кислоты в реакционном расплаве составляет менее 0,2%, а содержание 1,1,1-трис-[β-(3,5-ди-трет-бутил-4-гидроксифенил)пропионилоксиметил]пропана составляет менее 97%. Бесцветный реакционный расплав фильтруют, охлаждают и отверждают/кристаллизуют.
Выход: 95%.
Температура плавления: 66-79°С (аморфная форма).
Пример 6. 1,1,1-трис-[β-(3,5-ли-трет-бутцл-4-гидроксифенил)пропионилоксиметил1пропан (соединение формулы (I), в которой R1 и R2 обозначают трет-бутил., n=3, m=2, R3 обозначает СН3СН2С(СН2)3)
В реакционный сосуд помещают 85,5 г (0,293 моля) метилового эфира β-(3,5-ди-трет-бутил-4-гидроксифенил)пропионовой кислоты, 10,1 г (0,075 моля) 1,1,1-трис-(гидроксиметил)пропана и 0,19 г ацетата натрия (0,002 моля), расплавляют при температуре 120°С и перемешивают в атмосфере азота. Сразу же после образования расплава систему осторожно вакуумируют и одновременно нагревают до внутренней температуры 160°С. Образующийся метанол отгоняют и конденсируют в вымораживающей ловушке. Сразу после снижения внутреннего давления до уровня менее 10 мбар внутреннюю температуру повышают до 180°С и затем вакуумируют до давления менее 1 мбара. Сразу же после повышения внутренней температуры до 180°С и снижении давления до уровня менее 1 мбара смесь выдерживают в течение 1 ч, после чего в течение 1-2 ч при ВТ 200-210°С отгоняют избыток метилового эфира β-(3,5-ди-трет-бутил-4-гидроксифенил)пропионовой кислоты. Содержание метилового эфира β-(3,5-ли-трет-бутил-4-гидроксифенил)пропионовой кислоты в реакционном расплаве составляет менее 0,2%, а содержание 1,1,1-трис-[β-(3,5-ди-трет-бутил-4-гидроксифенил)пропионилоксиметил]пропана составляет более 97%. Бесцветный реакционный расплав фильтруют, охлаждают и отверждают/кристаллизуют.
Выход: 95,5%.
Температура плавления: 66-79°С (аморфная форма).
Пример 7. 1,1,1-трис-[β-(3,5-ди-трет-бутил-4-гидроксифенил)пропионилоксиметил]этан (соединение формулы (I), в которой R1 и R2 обозначают трет-бутил, n=3, m=2, R3 обозначает СН3С(СН2)3)
В реакционный сосуд помещают 85,5 г (0,293 моля) метилового эфира β-(3,5-ди-трет-бутил-4-гидроксифенил)пропионовой кислоты, 9 г (0,075 моля) 1,1,1-трис-(гидроксиметил)этана и 0,23 г ацетата лития в виде дигидрата (0,002 моля), расплавляют при температуре 120°С и перемешивают в атмосфере азота. Сразу же после образования расплава систему осторожно вакуумируют и одновременно нагревают до внутренней температуры 160°С. Образующийся метанол отгоняют и конденсируют в вымораживающей ловушке. Сразу после снижения внутреннего давления до уровня менее 10 мбар внутреннюю температуру повышают до 180°С и затем вакуумируют до давления менее 1 мбара. Сразу же после повышения внутренней температуры до 180°С и снижении давления до уровня менее 1 мбара смесь выдерживают в течение 1 ч, после чего в течение 1-2 ч при ВТ 200-210°С отгоняют избыток метилового эфира β-(3,5-ди-трет-бутил-4-гидроксифенил)пропионовой кислоты. Содержание метилового эфира β-(3,5-ди-трет-бутил-4-гидроксифенил)пропионовой кислоты в реакционном расплаве составляет менее 0,2%, а содержание 1,1,1-трис-[β-(3,5-ди-трет-бутил-4-гидроксифенил)пропионилоксиметил]этана составляет более 97%. Бесцветный реакционный расплав фильтруют, охлаждают и отверждают/кристаллизуют.
Выход: 95%.
Температура плавления: 55-78°С (аморфная форма).
Пример 8. Тетракис[β-(3,5-ди-трет-бутил-4-гидроксифенил)пропионилоксиметил]метан (соединение формулы (I), в которой R1 и R2 обозначают трет-бутил, n=4, m=2, R3 обозначает С(СН2)4)
В реакционный сосуд помещают 75,9 г (0,26 моля) метилового эфира β-(3,5-ди-трет-бутил-4-гидроксифенил)пропионовой кислоты, 6,8 г (0,05 моля) пентаэритрита и 0,16 г ацетата натрия (0,002 моля), расплавляют при температуре 120°С и перемешивают в атмосфере азота. Сразу же после образования расплава систему осторожно вакуумируют и одновременно нагревают до внутренней температуры 160-170°С. Образующийся метанол отгоняют и конденсируют в вымораживающей ловушке. Сразу после снижения внутреннего давления до уровня менее 10 мбар внутреннюю температуру повышают до 180°С и затем вакуумируют до давления менее 1 мбара. Сразу же после повышения внутренней температуры до 180°С и снижении давления до уровня менее 1 мбара смесь выдерживают в течение 1 ч, после чего в течение 1-2 ч при ВТ 200-210°С отгоняют избыток метилового эфира β-(3,5-ди-трет-бутил-4-гидроксифенил)пропионовой кислоты. Содержание метилового эфира (β-(3,5-ди-трет-бутил-4-гидроксифенил)пропионовой кислоты в реакционном расплаве составляет менее 0,2%, а содержание тетракис[β-(3,5-ди-трет-бутил-4-гидроксифенил)пропионилоксиметил]метана составляет более 96%. Бесцветный реакционный расплав фильтруют, охлаждают и отверждают/кристаллизуют.
Выход: 96%.
Температура плавления: 55-85°С (аморфная форма).
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ БЕНЗОФУРАН-2-ОНОВ, СТАБИЛИЗИРОВАННАЯ КОМПОЗИЦИЯ, СПОСОБ СТАБИЛИЗАЦИИ, ПРОИЗВОДНЫЕ МИНДАЛЬНОЙ КИСЛОТЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1993 |
|
RU2130931C1 |
| КОМПОЗИЦИЯ ДЛЯ ПОКРЫТИЯ | 1992 |
|
RU2107704C1 |
| ЖИДКИЕ СМЕСИ ФОСФИТОВ В КАЧЕСТВЕ СТАБИЛИЗАТОРОВ | 2007 |
|
RU2455325C2 |
| 3-АРИЛБЕНЗОФУРАНОНЫ, СТАБИЛИЗИРОВАННАЯ КОМПОЗИЦИЯ, СПОСОБ СТАБИЛИЗАЦИИ | 1994 |
|
RU2134689C1 |
| СПОСОБ СИНТЕЗА ПРОСТЫХ ЭФИРОВ АМИНОКСИЛОВ ИЗ ВТОРИЧНЫХ АМИНООКСИДОВ | 2001 |
|
RU2273634C2 |
| ОЛИГОМЕРНЫЕ СОЕДИНЕНИЯ, КОМПОЗИЦИЯ, СПОСОБ СТАБИЛИЗАЦИИ И ОЛИГОМЕРНЫЕ ПРОДУКТЫ | 1993 |
|
RU2130461C1 |
| ЗАМЕЩЕННЫЕ БЕНЗОТРИАЗОЛЫ, СТАБИЛИЗИРОВАННЫЕ КОМПОЗИЦИИ И ПОКРЫВНАЯ СИСТЕМА | 1992 |
|
RU2127264C1 |
| ПРОИЗВОДНЫЕ ЭФИРОВ БИСФЕНОЛА, СТАБИЛИЗИРОВАННАЯ КОМПОЗИЦИЯ И СПОСОБ СТАБИЛИЗАЦИИ ПОЛИМЕРА | 1995 |
|
RU2141469C1 |
| ПОЛИМЕРНЫЕ КОМПОЗИЦИИ | 2004 |
|
RU2358990C2 |
| ПОЛИМЕРНАЯ КОМПОЗИЦИЯ И СПОСОБ СТАБИЛИЗАЦИИ ПОЛИМЕРОВ | 1990 |
|
RU2068424C1 |
Изобретение относится к усовершенствованному способу получения соединений формулы (I)
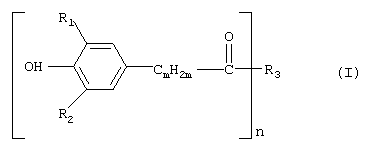 которые применяются предпочтительно в качестве антиоксидантов. В формуле (I) R1 и R2 независимо друг от друга обозначают С1-С8алкил, циклопентил или циклогексил, m обозначает 1, 2 или 3, предпочтительно 2, n обозначает целое число от 1 до 4 и R3 обозначает n-валентный линейный или разветвленный С4-С30алкильный остаток, который необязательно прерван кислородом, заключающийся во взаимодействии соединения формулы (II)
которые применяются предпочтительно в качестве антиоксидантов. В формуле (I) R1 и R2 независимо друг от друга обозначают С1-С8алкил, циклопентил или циклогексил, m обозначает 1, 2 или 3, предпочтительно 2, n обозначает целое число от 1 до 4 и R3 обозначает n-валентный линейный или разветвленный С4-С30алкильный остаток, который необязательно прерван кислородом, заключающийся во взаимодействии соединения формулы (II)

в которой R обозначает C1-С3алкил, с соединением формулы R3(ОН)n (III), в которой R3 и n имеют вышеуказанные значения, отличающийся тем, что взаимодействие указанных соединений проводят при практически нейтральном значении водородного показателя (рН) в присутствии по крайней мере одной растворенной или суспендированной в реакционной смеси щелочнометаллической соли органической карбоновой кислоты либо в присутствии смеси таких щелочнометаллических солей, при этом (I) указанная щелочнометаллическая соль образована катионом щелочного металла и анионом органической карбоновой кислоты и (II) органическая карбоновая кислота в используемых для проведения реакции условиях является по меньшей мере частично летучей. Предпочтительными щелочнометаллическими солями при этом являются формиат щелочного металла и ацетат щелочного металла. Способ осуществляют не только в отсутствие носителя, но и в практически нейтральной среде, что позволяет избежать ряд недостатков предыдущего уровня техники. 15 з.п. ф-лы.
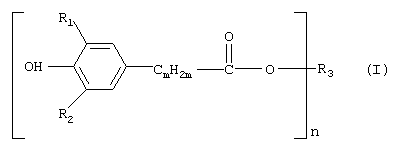
в которой R1 и R2 независимо друг от друга обозначают С1-С8алкил, циклопентил или циклогексил;
m обозначает 1, 2 или 3, предпочтительно 2;
n обозначает целое число от 1 до 4 и
R3 обозначает n-валентный линейный или разветвленный С4-С30алкильный остаток, который необязательно прерван кислородом, заключающийся во взаимодействии соединения формулы (II)
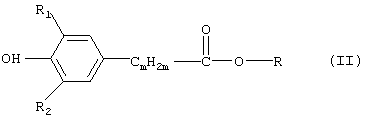
в которой R обозначает C1-С3алкил, с соединением формулы (III)
R3(ОН)n (III)
в которой R3 и n имеют вышеуказанные значения,
отличающийся тем, что взаимодействие указанных соединений проводят при практически нейтральном значении водородного показателя (рН) в присутствии по крайней мере одной растворенной или суспендированной в реакционной смеси щелочнометаллической соли органической карбоновой кислоты либо в присутствии смеси таких щелочнометаллических солей, при этом (I) указанная щелочнометаллическая соль образована катионом щелочного металла и анионом органической карбоновой кислоты и (II) органическая карбоновая кислота в используемых для проведения реакции условиях является по меньшей мере частично летучей.
Приоритет по пунктам и признакам:
| US 5563291 А, 08.10.1996 | |||
| US 5206414 A, 27.04.1993 | |||
| US 4716244 А, 29.12.1987 | |||
| Способ получения октадецил-3-(3,5-ди-трет-бутил-4-гидроксифенил)пропионата | 1986 |
|
SU1553010A3 |
| SU 1014213 A1, 30.07.1991. | |||

