Область техники
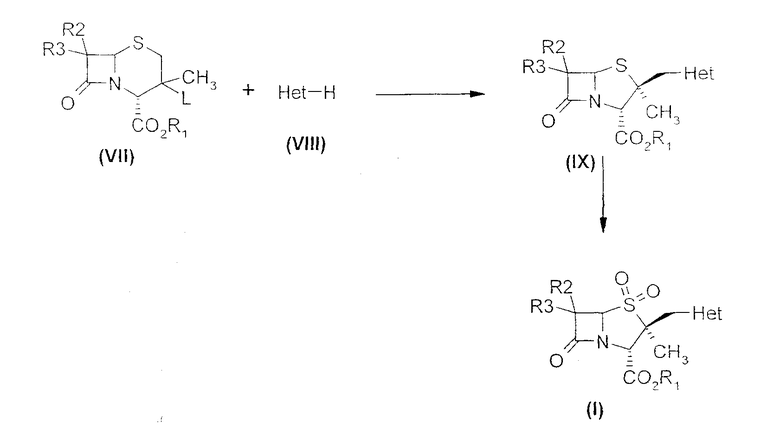
Настоящее изобретение относится к способу получения производных 2α-метил-2β-замещенный метил пенама из производных цефама. Более конкретно, настоящее изобретение предлагает новый способ получения производных 2β-гетероциклилметил пенама формулы (I)
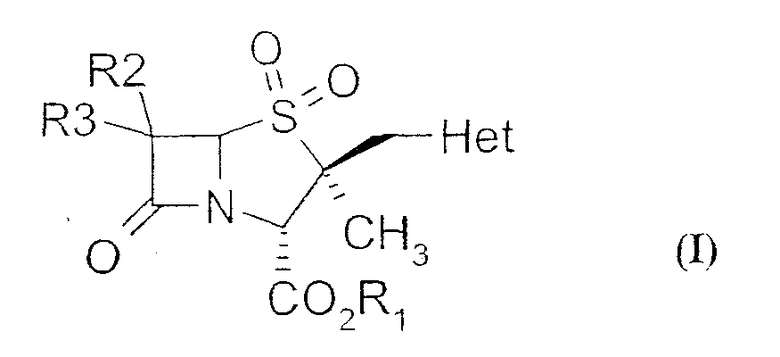
где R1 обозначает водород, защитную группу для карбоксильной группы, которая образует, например, сложный эфир или фармацевтически приемлемую соль; R2 и R3 могут быть одинаковыми или разными и независимо обозначают водород, галоген, NH2, ациламино, фталимидо, при условии, что оба R2 и R3 не обозначают NH2, ациламино, фталимидо; Het обозначает 5- или 6-членную гетероциклическую систему, содержащую NH и гетероциклическую систему, в состав которой входят один или несколько гетероатомов, выбранных из O, S или N.
Производные 2β-гетероциклилметил пенама формулы (I) используются как β-лактамовые антибиотики.
Применение β-лактамовых антибиотиков ограничено из-за устойчивости, проявляемой микроорганизмами благодаря действию фермента β-лактамазы. Действие данного фермента заключается в том, что он расщепляет β-лактамовый цикл указанных антибиотиков, в результате чего лекарственное средство разрушается и теряет активность. Следовательно, существует потребность в ингибиторах β-лактамазы, которые нейтрализуют активность фермента и устраняют устойчивость к антибиотикам. Для обеспечения антибиотической активности β-лактамазные ингибиторы используют вместе с β-лактамовыми антибиотиками. Таким образом, продолжается поиск новых производных пенама и новых способов их получения.
Предпосылки создания изобретения
В нескольких патентах раскрыты разные способы получения производных 2β-замещенный метил пенама. Например, патенты США 4529592, 4562073 и 4668514, а также EP 97446, раскрывают способ, который включает в себя обработку производных 2β-азидометилпенама формулы (II):
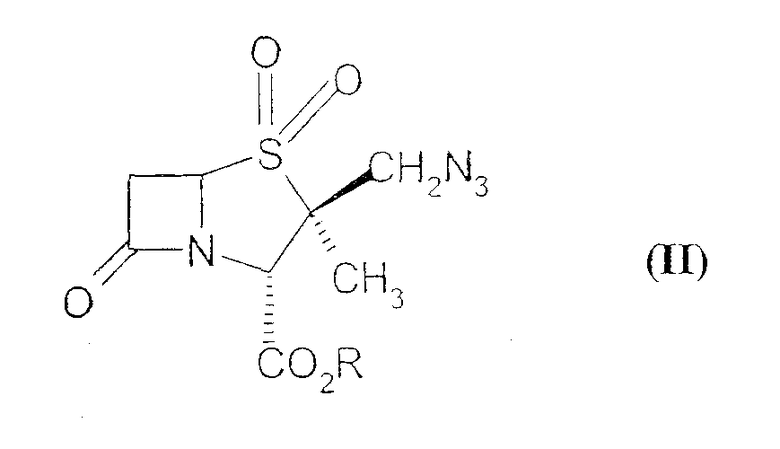
где R обозначает карбоксизащитную группу, ацетиленом/ацетиленовым производным или виниловым производным при высоком давлении в герметично закрытом реакторе и при повышенной температуре и затем удаление защитных групп при помощи подходящего реагента с получением ингибитора β-лактамазы формулы (I).
В свою очередь производное 2β-азидометилпенама формулы (II) получают из производных 2β-замещенный метил пенама формулы (III)
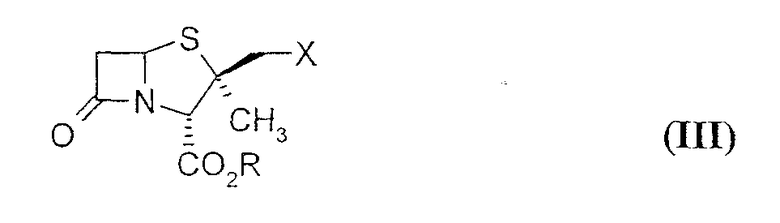
где R = карбоксизащитная группа; X = хлор или бром, путем обработки азидом натрия в водных полярных апротонных растворителях с последующим окислением.
Недостаток вышеуказанного способа заключается в том, что он не позволяет вводить широкий диапазон гетероциклов, а только небольшое количество таких гетероциклов, как 1,2,3-триазольная группа. Кроме того, при проведении данного способа приходится иметь дело с газообразным ацетиленом при высоком давлении и высокой температуре, что связано с большим риском из-за присущей ему высокой скорости детонации, следовательно, данный способ не является промышленным и экологически безвредным. В данном способе также используется избыток азида натрия, который остается в слишком больших количествах для экологической характеристики технологии (ETP); азид натрия опасен из-за высвобождения азотистоводородной кислоты, которая потенциально взрывоопасна и вредна для здоровья.
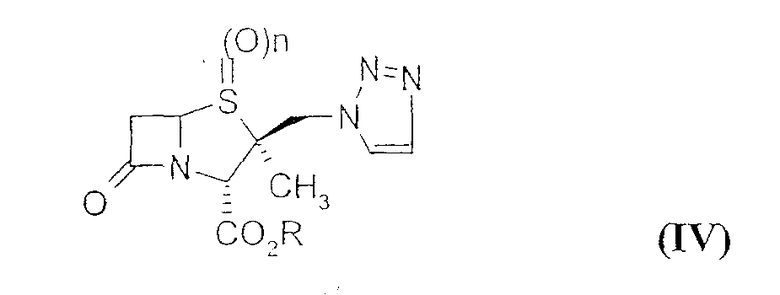
В EP 0273699 раскрывается другой подход, который включает в себя получение 2β-триазолилметилпенамовых производных формулы (IV)
где R = карбоксизащитная группа, n=0, путем обработки производного 2β-галогенметилпенама формулы (III)
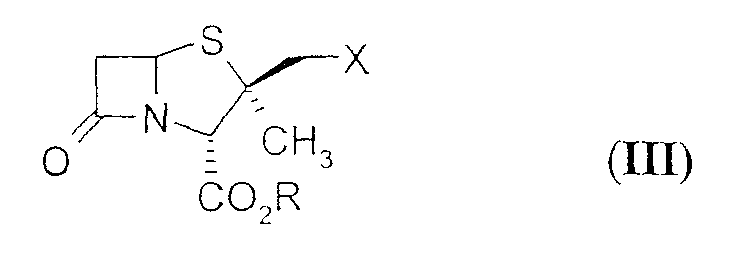
где X = хлор или бром; R обозначает карбоксизащитную группу, 1H-1,2,3-триазолом. Полученный продукт можно подвергнуть окислению и удалению защитных групп с получением производных 2β-замещенный метил пенама формулы (I).
В EP 306924 раскрывается способ восстановления с использованием таких соединений свинца, как хлорид свинца или бромид свинца; с помощью данного способа получают 2β-триазолилметилпенамовое производное формулы (IV) (n=0-2) из 6,6-дибром-2β-триазолилметилпенамового производного формулы (V).
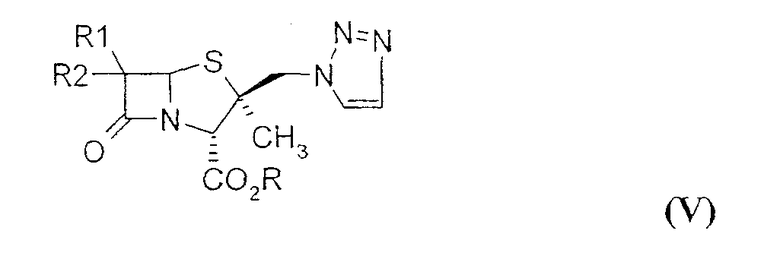
где R1, R2 могут обозначать H, Br; Br, Br; R обозначает карбоксизащитную группу.
В другом способе, раскрытом в патенте США 4895941, сульфоксид пенама формулы (VI)
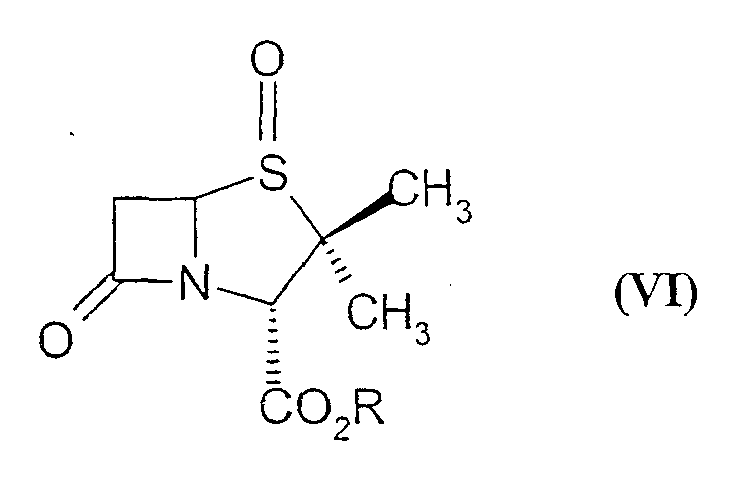
где R обозначает карбоксизащитную группу, обрабатывают 2-триметилсилил-1,2,3-триазолом в герметично закрытой пробирке при повышенной температуре с получением смеси, из которой можно выделить 2β-триазолилметилпенамовое производное формулы (IV) (n=0) с помощью колоночной хроматографии.
В большинстве включенных в данное описание способов в качестве ключевого промежуточного соединения используют 2β-галогенметилпенам формулы (III). Его используют как в описанном выше азидном способе, так и в описанном выше триазольном способе. Однако сам пятичленный 2-галогенметилпенам формулы (III) является нестабильным промежуточным соединением, поэтому его всегда трудно хранить и получать в больших количествах. Обнаружено, что данное соединение разлагается при хранении даже при низкой температуре, как в чистом виде, так и в растворителе, из которого его выделяют. Следовательно, все операции, связанные с получением данного промежуточного соединения, нужно выполнять быстро, и выделенное соединение должно быть сразу превращено в конечный продукт. Вследствие указанных недостатков масштабирование производства всегда приводит к уменьшению выхода, снижению качества и, в результате, к снижению уровня воспроизводимости.
Все вышеописанные способы связаны с одним или несколькими из следующих недостатков: (i) нестабильность ключевого промежуточного соединения; (ii) использование вредных и взрывоопасных реагентов; (iii) необходимость применения высокого давления вместе с высокой температурой, особенно в случае использования ацетилена; (iv) использование большого избытка азида натрия и вытекающие отсюда проблемы, связанные с тем, что он является взрывоопасным и вредным для окружающей среды; (v) использование высокотоксичных и загрязняющих окружающую среду соединений тяжелых металлов, таких как свинец, особенно на предпоследних стадиях получения фармацевтических средств. Данные факторы оказывают влияние на единообразие качества и выхода промежуточных соединений и конечного продукта, а также на безопасность при получении в промышленном масштабе.
Чтобы преодолеть вышеуказанные недостатки, авторы настоящего изобретения разработали новый способ промышленного получения 2β-замещенных метилпенамов, в котором используются стабильные промежуточные соединения и безопасные реагенты/условия реакций. Чтобы найти новое решение для промышленного получения упомянутого пенама, авторы в своей лаборатории провели экстенсивные исследования и изучили ряд схем и методов синтеза.
Цель изобретения
Основной целью настоящего изобретения является предоставление способа получения производных 2β-гетероциклилметил пенама формулы (I), который включает в себя превращение шестичленного цефамового фрагмента.
Другой целью настоящего изобретения является предоставление способа получения производных 2β-гетероциклилметил пенама формулы (I) с хорошим выходом и высокой степенью чистоты.
Следующей целью настоящего изобретения является предоставление способа получения производных 2β-гетероциклилметил пенама формулы (I) в чистом виде, без примесей других изомеров.
После продолжительных исследований авторы настоящего изобретения смогли найти новый способ, в котором используется цефамовый фрагмент, а не производные пенама, использующиеся до настоящего времени. Преимущество применения шестичленного цефамового фрагмента заключается в том, что он, в отличие от используемых до настоящего времени пенамов, представляет собой стабильное промежуточное соединение, и, следовательно, его применение поможет преодолеть указанные выше недостатки.
Во всех доступных публикациях описано применение 2β-хлорметилпенамов формулы (III) для получения 2β-триазолилметилзамещенных пенамов формулы (IV), тогда как в основе настоящего изобретения лежит явление сокращения цикла, имеющее место при превращении шестичленных 3-галогенметилцефамов формулы (VII) в 2β-гетероциклилметилпенамы формулы (I).
Краткое описание изобретения
Соответственно, настоящее изобретение предоставляет способ получения производных 2β-гетероциклилметил пенама формулы (I),
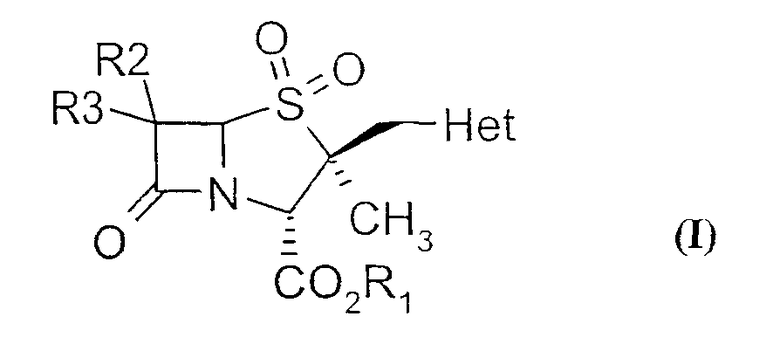
где R1 обозначает водород, защитную группу для карбоксильной группы, которая образует, например, сложный эфир или фармацевтически приемлемую соль; R2 и R3 могут быть одинаковыми или разными и независимо обозначают водород, галоген, NH2, ациламино, фталимидо, при условии, что оба R2 и R3 не обозначают NH2, ациламино, фталимидо; Het обозначает 5- или 6-членную гетероциклическую систему, содержащую NH и гетероциклическую систему, в состав которой входят один или несколько гетероатомов, выбранных из O, S или N, который включает в себя:
Способ приведен на схеме 1
Подробное описание изобретения
В одном воплощении настоящего изобретения карбоксизащитная группа, например образующая сложный эфир, выбрана из п-нитробензила, п-метоксифенила, дифенилметила и т.п.
В другом воплощении настоящего изобретения L обозначает уходящую группу, выбранную из галогена, например хлора, брома, иода; п-толуолсульфонилокси, метансульфонилокси.
В следующем воплощении настоящего изобретения группа, обозначаемая Het, выбрана из пирролила, пирролидинила, пиперидинила, имидазолила, оксазолидинила, 1,2,3-триазолила, 1,2,4-триазолила и др.
В очередном воплощении настоящего изобретения группа, обозначаемая ациламино, выбрана из фенацетиламино, феноксиацетиламино или бензоиламино.
В следующем воплощении настоящего изобретения реакцию, в которой 3-замещенное производное цефама формулы (VII) взаимодействует с соединением формулы (VIII), проводят в подходящем растворителе, в присутствии или в отсутствие катализатора межфазного переноса и в присутствии или в отсутствие основания. Молярное соотношение соединения формулы (VIII) к цефамовому соединению формулы (VII) находится в интервале приблизительно от 1 до 30, предпочтительно приблизительно от 1 до 10. Используемый гетероциклический амин может находится или в свободном виде, или в виде соли минеральной кислоты или органической сульфоновой или карбоновой кислоты.
Растворители не имеют решающего значения и, следовательно, можно использовать широкий ряд растворителей, например, эфирные растворители, такие как ТГФ, диоксан, диглим, моноглим и др., полярные апротонные растворители, такие как ДМФ, DMAC, ДМСО, ацетон, этилацетат, сульфолан, ацетонитрил и др., протонные растворители, такие как н-бутанол, изопропанол, метанол, этанол, циклогексанол и др., ароматические растворители, такие как толуол, анизол и др., хлорсодержащие растворители, такие как дихлорэтан, дихлорметан, четыреххлористый углерод, хлорбензол и др. Данные органические растворители можно использовать по отдельности или в сочетании с некоторым количеством воды как дополнительного компонента. В случае несмешиваемых с водой растворителей реакцию проводят в двухфазной среде в присутствии катализатора межфазного переноса при энергичном перемешивании. Катализатором межфазного переноса может служить соль четвертичного аммония, например, бромид тетрабутиламмония, бромид бензилтрибутиламмония, бромид бензилтриоктиламмония и др., или соль фосфония, например, бромид бензилтрифенилфосфония и др. Основание может быть неорганическим или органическим, предпочтительно, оно представляет собой неорганический оксид или карбонат щелочного или щелочноземельного металла, например, карбонат магния, карбонат кальция, карбонат цезия, карбонат бария, гидрокарбонат калия, карбонат натрия, гидрокарбонат натрия, оксид меди, карбонат меди, карбонат калия и др. Температура реакции обычно находится в интервале от -10 до 110°C, предпочтительно от 30 до 65°C.
Продукт вышеуказанной реакции можно очистить, удалив нежелательные изомеры, или его можно непосредственно использовать на следующей стадии без очистки, так как продукт, полученный на следующей стадии, обрабатывают, получая чистое соединение. Полученный таким образом продукт выделяют в пастообразном виде и окисляют окисляющим агентом в кислой водной среде. Окисляющим реагентом может быть обычный окислитель для серы, например перманганат калия, перуксусная кислота, трифторперуксусная кислота, м-хлорпербензойная кислота, оксон и др., предпочтительно перманганат калия. Окисление можно проводить в присутствии органической кислоты, например алифатической карбоновой кислоты, алифатической сульфоновой кислоты и др., предпочтительно, уксусной кислоты, метансульфоновой кислоты и др. Температуру реакции можно варьировать от -30 до +50°C, предпочтительно от -10 до +30°C. Время реакции может варьировать приблизительно от 15 мин до 8 часов, предпочтительно, от 15 мин до 2 часов. В конце реакции реакционную смесь гасят подходящим реагентом, чтобы разрушить избыток окисляющего реагента, после чего реакционную среду нейтрализуют неорганическим основанием, таким как бикарбонат натрия. На данной стадии продукт очищают в этилацетате, при этом другие изомеры, образовавшиеся в результате реакции, растворяются в данном растворителе. Избирательность удаления нежелательных изомеров в других растворителях меньше, поэтому этилацетат является предпочтительным растворителем для получения изомера требуемой степени чистоты.
Полученный таким образом 2β-триазолилметилзамещенный пенам формулы (IX) превращают в 2β-триазолилметилпенамовое производное формулы (I), используя подходящий метод деэтерификации, в зависимости от типа защиты карбоксильной группы. Например, в случае п-нитробензильной защитной группы используют следующий метод удаления защитной группы с получением β-лактамового ингибитора формулы (I). 2β-Триазолилметилзамещенный пенам формулы (I) (n=2; R обозначает карбоксизащитную группу), превращают в соединение формулы (I) (n=2; R=H) в присутствии катализатора на основе благородного металла, неорганического основания и источника водорода; реакцию проводят в двухфазной среде при повышенном давлении. Катализатором на основе благородного металла может быть 5-10% Pd/C, 5% Pt, катализатор Адама и др., предпочтительно 10% Pd. Реакцию проводят в присутствие или отсутствии органического или неорганического основания, предпочтительно в присутствии неорганического основания. Неорганическим основанием является карбонат щелочного или щелочноземельного металла, выбранный из карбоната магния, карбоната кальция, карбоната цезия, карбоната бария, гидрокарбоната калия, карбоната натрия, гидрокарбоната натрия, карбоната калия или бикарбоната натрия, предпочтительно, бикарбонат натрия. Хотя реакцию можно проводить и в однофазной, и в двухфазной среде, предпочтительно используют водно-органическую среду, содержащую несмешиваемый с водой растворитель, такой как толуол, этилацетат, метилацетат и др., предпочтительно этилацетат. После обработки продукт выделяют из водной среды кристаллизацией.
Способ получения 2β-триазолилметилпенамового производного формулы (I) подробно описан в приведенных ниже ссылочных примерах, которые даны только для иллюстрации и не должны рассматриваться как ограничивающие объем настоящего изобретения.
Интересно отметить, что при получении производного пенама, содержащего пятичленный цикл, из производного цефама, содержащего шестичленный цикл, стереохимически предпочтительным направлением реакции является образование β-изомера. Кроме того, в процессе сокращения цикла 3-замещенных производных цефама формулы (VII) с образованием 2β-гетероциклилметилпенамовых производных формулы (I) конфигурация карбоксильной группы остается неизменной. Карбоксильная группа находится в трансположении по отношению к 2β-триазолилметильной группе. Относительную стереохимию однозначно подтверждают в эксперименте, основанном на ядерном эффекте Оверхаузера (NOE).
ПРИМЕРЫ
Пример 1:
Получение 4-нитробензил-2β-(1H-1,2,3-триазол-1-илметил)-2α-метилпенам-3α-карбоксилата формулы (IX):
К раствору 4-нитробензил-3-бром-3-метилцефам-4-карбоксилата (50 г) в ацетоне (250 мл), который находится в 2-литровой круглодонной колбе, добавляют воду (65 мл) и 1H-1,2,3-триазол (100 мл) при комнатной температуре. К прозрачному раствору добавляют карбонат кальция (25 г) при энергичном перемешивании. Реакционную смесь нагревают до 50-60°C в течение 15 мин и оставляют стоять 9 часов, энергично перемешивая при указанной температуре. Протекание реакции отслеживают с помощью ТСХ. После завершения реакции реакционную смесь фильтруют для удаления неорганических солей и фильтр промывают ацетоном (50 мл). Прозрачный раствор перегоняют в вакууме для удаления ацетона при температуре ниже 30°C. После удаления ацетона раствор выливают в дихлорметан (250 мл) и хорошо перемешивают при 26-28°C. Органический слой отделяют и промывают очищенной водой (200 мл) четыре раза. Органический слой концентрируют в вакууме для удаления дихлорметана, вначале при <25°C и затем при 35-40°C. Полученную пасту используют на следующей стадии без очистки.
Пример 2:
Получение 4-нитробензил 2β-(1H-1,2,3-триазол-1-илметил)-2α-метилпенам-3α-карбоксилата 1,1-диоксида формулы (I):
К уксусной кислоте (350 мл), которая находится в 2-литровой круглодонной колбе, при 20°С добавляют 4-нитробензил-2β-(1H-1,2,3-триазол-1-илметил)-2α-метилпенам-3α-карбоксилат (полученный в вышеописанном примере) и очищенную воду (35 мл). Гомогенную реакционную смесь охлаждают до 5-10°C при перемешивании. К гомогенной реакционной смеси добавляют порошкообразный перманганат калия (30 г) 12 порциями в течение 1,5-2,0 часов, поддерживая температуру при 5-10°C. Перемешивание продолжают еще 0,5 часа и протекание реакции отслеживают с помощью ТСХ. После завершения реакции реакционную смесь помещают в измельченный лед (500 г) при энергичном перемешивании в течение 0,5-1,0 часов. К полученной массе добавляют холодный этилацетат (500 мл), поддерживая температуру при 0-5°C. Разбавленный раствор пероксида водорода (25%; 40 мл) медленно добавляют в течение 1 часа с такой скоростью, чтобы температура оставалась в интервале 0-5°. После полного обесцвечивания добавляют этилацетат (200 мл). К совершенно бесцветному раствору добавляют хлорид натрия (100 г) и хорошо перемешивают в течение 15 мин. Этилацетатный слой отделяют и дважды промывают водой (250 мл). К этилацетатному слою медленно добавляют 8% раствор бикарбоната натрия (˜400 мл), пока pH водного слоя не достигнет 7,2. Реакционную смесь перемешивают еще 15 мин и снова проверяют pH. После стабилизации pH при 7,2 перемешивание останавливают и слои разделяют. Органический слой дважды промывают водой (250 мл) и обрабатывают активированным углем (10 г). Органический слой концентрируют в вакууме для удаления этилацетата, пока объем смеси не уменьшится до 150 мл, когда продукт будет выделяться из смеси. Оставляют перемешиваться в течение 5 часов, вещество фильтруют и промывают этилацетатом (30 мл). После сушки в вакууме получают бесцветный 4-нитробензил-2β-(lH-1,2,3-триазол-1-илметил)-2α-метилпенам-3α-карбоксилат-1,1-диоксид в чистом виде с выходом 50-75%.
Масс-спектр m/e: M+l пик 436,3; данные Н1 ЯМР (CDCl3): δ 1,29 (3H, с, 2α-Me), 3,53 (1H, дд, J=1,9 & 16,3 Гц, 7H-транс), 3,61 (1H, дд, J=4,3 & 16,3 Гц, 7H-цис), 4,63 (1H, с, CH-CO2), 4,66 (1H, дд, J=1,9 & 4,2 Гц, 6H), 5,07 (2H, Abкв., J=15,1 Гц, 2β-CH2), 5,35 (2H, Abкв., J=14 Гц, COO-CH2), 7,61 (2H, д, J=8,7 Гц, протоны в ортоположении ароматического цикла), 8,30 (2H, д, J=8,7 Гц, протоны в метаположении ароматического цикла), и 7,75 & 7,79 (2H, протоны триазола).
Пример 3:
Получение 2β-(1H-1,2,3-триазол-1-илметил)-2α-метилпенам-3α-карбоновой кислоты 1,1-диоксида формулы (I):
В 2-литровый автоклав высокого давления для гидрирования добавляют этилацетат (500 мл), 10% Pd/C (2,5 г) и 4-нитробензил-2β-(1H-1,2,3-триазол-1-илметил)-2α-метилпенам-3a-карбоксилата 1,1-диоксид (25 г). Гетерогенную реакционную смесь при перемешивании охлаждают до 20-22°C. Раствор бикарбоната натрия (24 г в 375 мл очищенной воды) добавляют в течение 10-15 мин при 20-22°C. Автоклав для гидрирования продувают азотом и создают давление водорода 200 фунтов/кв. дюйм (1379 кПа) в течение 10 мин при 20-22°C. В течение 1,5-2,0 часов поддерживают указанное давление водорода и отслеживают протекание реакции. После завершения реакции давление водорода снимают и автоклав продувают азотом. Реакционную массу охлаждают до 0-5°C. Катализатор Pd/C отделяют фильтрацией и фильтр промывают охлажденной очищенной водой (50 мл). Водный слой отделяют и промывают этилацетатом (150 мл) три раза. pH доводят до 6,4-6,6 с помощью 6N HCl (требуется ˜37 мл) и водный слой промывают этилацетатом (150 мл). Водный слой обрабатывают активированным углем (4 г) в течение 15 мин и подложку промывают очищенной водой (50 мл). pH доводят до 3,2 с помощью 6N HCl (˜60 мл) и оставляют на 15 мин. Происходит кристаллизация. Перемешивание продолжают 30 мин при указанном pH. Затем pH доводят до 2,5-2,6 с помощью 6N HCl (˜15 мл) и оставляют на 2 часа. Кристаллы отфильтровывают и промывают водой и этилацетатом (40 мл). Вещество сушат в вакууме в течение 5 часов при 26-30°C. Выход продукта 2β-(1H-1,2,3-триазол-1-илметил)-2α-метилпенам-3α-карбоновой кислоты 1,1-диоксида составляет приблизительно 85-90%.
Масс-спектр m/e: M-l пик 299,1; данные Н1 ЯМР (ДМСО-d6): δ 1,33 (3H, с, 2α-Me), 3,31 (1Н, дд, J=1,4 & 16,5 Гц, 7H-транс), 3,71 (1H, дд, J=4,5 & 16,5 Гц, 7H-цис), 4,80 (1H, с, CH-CO2), 4,91 (1H, д, J=15,3 Гц, H' из 2β-CH2), 5,19 (1H, дд, J=1,5 & 4,4 Гц, H6), 5,24 (1Н, д, J=15,3 Гц, H" из 2β-CH2), и 7,8 & 8,1 (2H, протоны триазола).
Стереохимию групп 2α-метил и 2β-метилен подтверждают в эксперименте, основанном на ядерном эффекте Оверхаузера (NOE).
Способ получения 2β-гетероциклилметилпенамовых производных формулы (I)
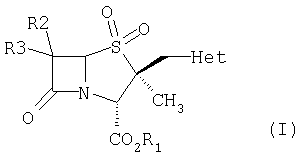
где R1 обозначает водород, защитную группу для карбоксильной группы, которая образует, например, сложный эфир или фармацевтически приемлемую соль; R2 и R3 могут быть одинаковыми или разными и независимо обозначают водород, галоген, NH2, ациламино, фталимидо, при условии, что оба R2 и R3 не обозначают NH2, ациламино, фталимидо; Het обозначает 5-членную гетероциклическую систему, содержащую NH, в состав которой входят один или несколько гетероатомов N, который включает в себя: (i) взаимодействие соединения формулы (VII), 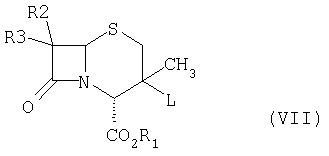
где L обозначает уходящую группу, а все другие заместители определены выше, с соединением формулы (VIII),  где Het такой, как определено выше, в присутствии растворителя и основания, при температуре в интервале от -10 до 110°С, с получением соединения формулы (IX)
где Het такой, как определено выше, в присутствии растворителя и основания, при температуре в интервале от -10 до 110°С, с получением соединения формулы (IX)
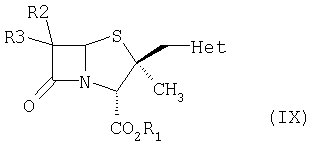
где все заместители определены выше; (ii) окисление соединения формулы (IX) традиционными окисляющими агентами в присутствии смешивающегося с водой растворителя и органической кислоты при температуре от -30 до +50°С, с получением соединения формулы (I); и, при необходимости, (iii) деэтерификацию соединения формулы (I), где R1 обозначает карбоксизащитную группу, с получением соединения формулы (I), где R1 обозначает водород, с использованием металлического катализатора, в присутствии основания и смешивающегося с водой растворителя. 12 з.п. ф-лы.
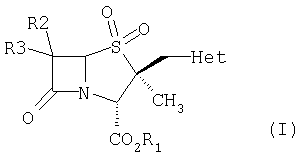
где R1 обозначает водород, защитную группу для карбоксильной группы, которая образует, например, сложный эфир или фармацевтически приемлемую соль; R2 и R3 могут быть одинаковыми или разными и независимо обозначают водород, галоген, NH2, ациламино, фталимидо, при условии, что оба R2 и R3 не обозначают NH2, ациламино, фталимидо; Het обозначает 5-членную гетероциклическую систему, содержащую NH, в состав которой входят один или несколько гетероатомов N, который включает в себя:
(i) взаимодействие соединения формулы (VII),
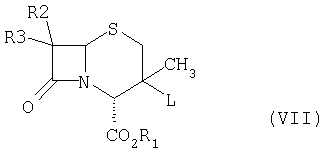
где L обозначает уходящую группу, а все другие заместители определены выше, с соединением формулы (VIII),

где Het такой, как определено выше, в присутствии растворителя и основания, при температуре в интервале от -10 до 110°С, с получением соединения формулы (IX)
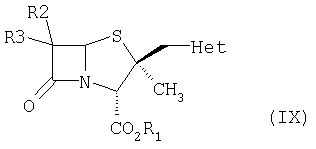
где все заместители определены выше;
(ii) окисление соединения формулы (IX) традиционными окисляющими агентами в присутствии смешивающегося с водой растворителя и органической кислоты при температуре от -30 до 50°С, с получением соединения формулы (I); и, при необходимости,
(iii) деэтерификацию соединения формулы (I), где R1 обозначает карбоксизащитную группу, с получением соединения формулы (I), где R1 обозначает водород, с использованием металлического катализатора, в присутствии основания и смешивающегося с водой растворителя.
| ALDO BALSAMO et all, EUROPEAN JOURNAL OF MEDICINAL CHEMISTRY, vol.24, №6, 1989, p.573-577 | |||
| Способ получения 5(R) пенемовых производных | 1983 |
|
SU1375139A3 |
| Способ нарезания цилиндрических зубчатых колес с бочкообразным зубом | 1953 |
|
SU97446A1 |
| US 4912213 А, 27.03.1990. | |||

