Настоящее изобретение касается новых соединений формулы (I), фармацевтических композиций, включающих указанные соединения, их получения, а также применения в качестве лекарственных препаратов при лечении гиперлипидемии.
За последние несколько лет широкое признание приобрела причинная связь между гиперхолестеринемией, в особенности связанной с повышенной концентрацией остатков липопротеинов низкой плотности (LDL) и очень низкой плотности (VLDL) в плазме, и преждевременным атеросклерозом. Врачами и общественностью единодушно признано, что лечение гиперхолестеринемии приносит терапевтическую пользу. Имеется ограниченное количество лекарств для лечения гиперлипидемии. Важнейшие агенты, используемые для лечения гиперлипидемии, включают секвестранты желчной кислоты, фибраты (fibrates), никотиновую кислоту и ингибиторы HMG-CoA-редуктазы (гидроксиметилглютарил-CoA-редуктазы). Неудобство введения и желудочно-кишечные побочные эффекты имеющихся доступных секвестрантов желчной кислоты, вызывают много проблем. Фибраты имеют только ограниченную применимость при лечении гиперхолестеринемии определенных типов. Применение никотиновой кислоты вызывает побочные эффекты и проблемы, связанные с токсичностью. Ингибиторы HMG-CoA-редуктазы уже используют для первоочередной терапии обычной гиперхолестеринемии. Однако по-прежнему существует потребность в новых агентах, понижающих уровень липидов, которые предпочтительно действовали бы по механизму, отличному от действия упомянутых выше лекарств. Европейский патент EP-0,006,711-A, опубликованный 9 сентября 1980 г., раскрывает гетероциклические производные (4-фенилпиперазин-1- ил-арилоксиметил-1,3-диоксолан-2-ил)-метил-1H-имидазолов и 1H-1,2,4-триазолов, обладающие противогрибковыми свойствами. Заявленные здесь соединения отличаются от них присутствием атома серы, соседнего с гетероциклом (Het) и фармакологическим профилем, в частности их активностью по ингибированию синтеза аполипопротеина В. Настоящее изобретение предлагает новые соединения формулы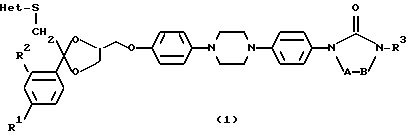
где A и B, взятые вместе, образуют бивалентный радикал формулы
-N = CH- (a)
-CH= N - (b)
-CH= CH - (c)
-CH2-CH2 - (d)
-С(=O) - CH2) - (e)
-CH2 - С(=0) - (f) причем в бивалентных радикалах (а) и (b) атом водорода может быть замещен C1-6-алкилом; в бивалентных радикалах (е) и (f) два атома водорода могут быть замещены C1-6-алкилом;
R1 является водородом, C1-6-алкилом или галогеном;
R2 является водородом или галогеном;
R3 является C1-6-алкилом;
C3-6-циклоалкилом; или C1-8-алкилом, замещенным гидроксилом, оксогруппой; Het является гетероциклом, выбранным из группы, включающей пиридинил; пиримидинил; пиримидинил, замещенный двумя заместителями, выбранными из C1-6-алкила, аминогруппы; тетразолил, замещенный C1-6-алкилом или арилом; триазолил; триазолил, замещенный одним или двумя заместителями, выбранными из C1-6-алкила, тригалогенметила, гидроксила, аминогруппы; тиадиазолил, замещенный C1-6-алкилом; тиазолил; оксазолил, замещенный C1-6-алкилом; имидазолил; имидазолил, замещенный C1-6-алкилом; его N - оксид, стереохимически изомерная форма или фармацевтически приемлемая соль присоединения кислоты.
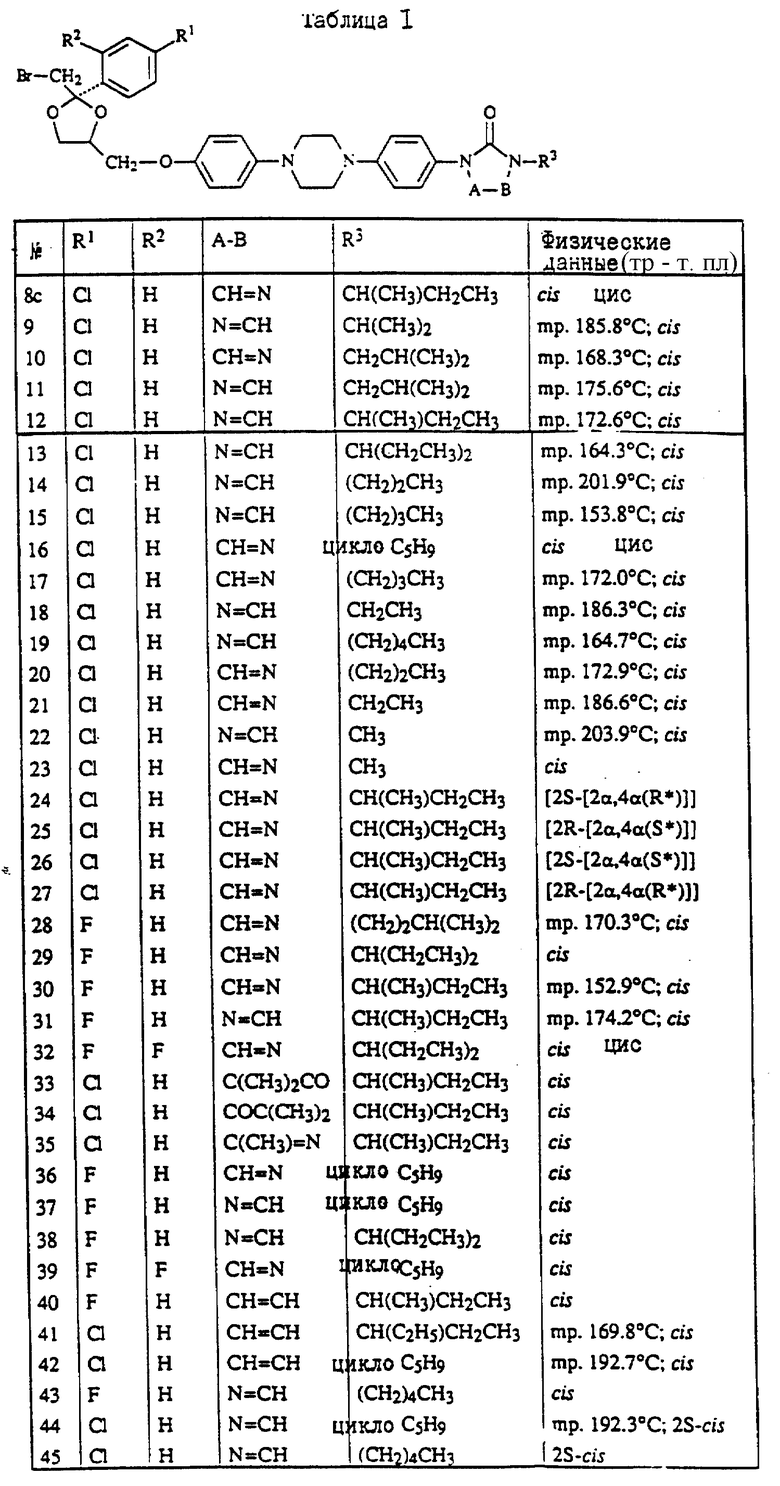
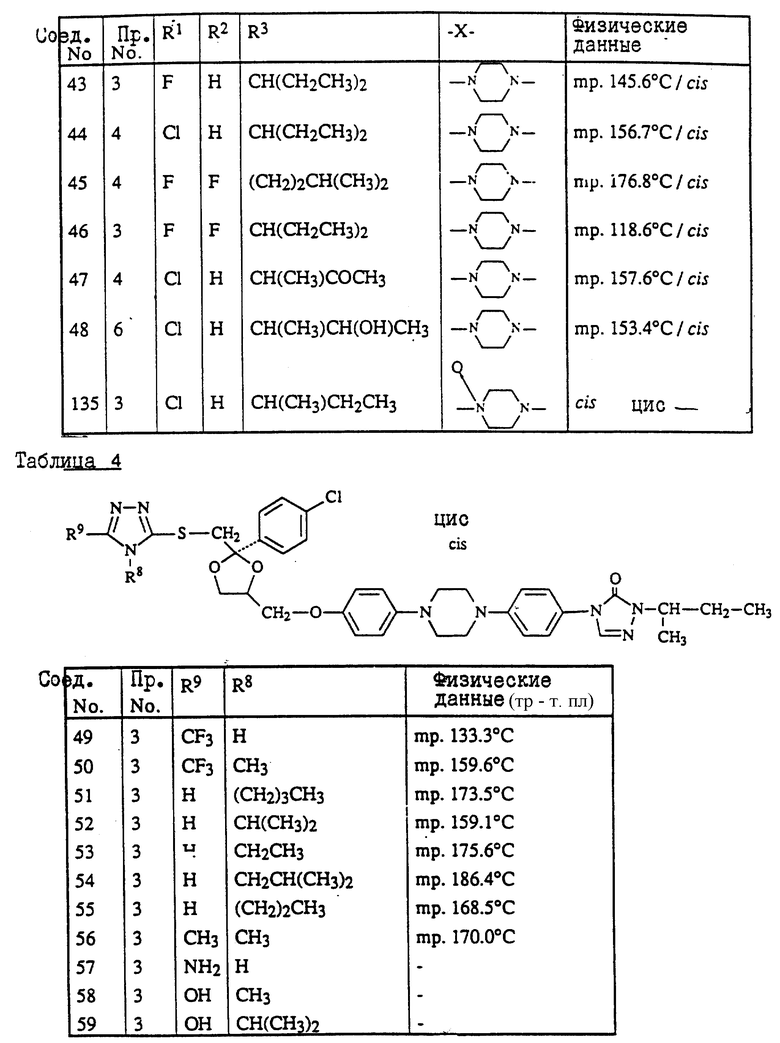
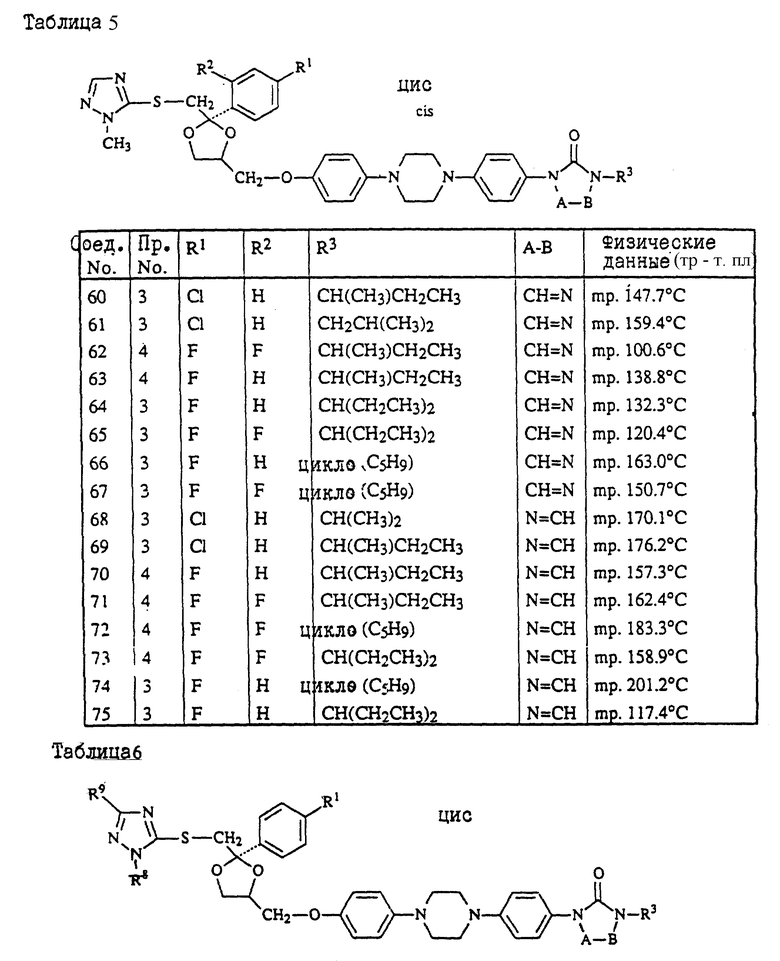
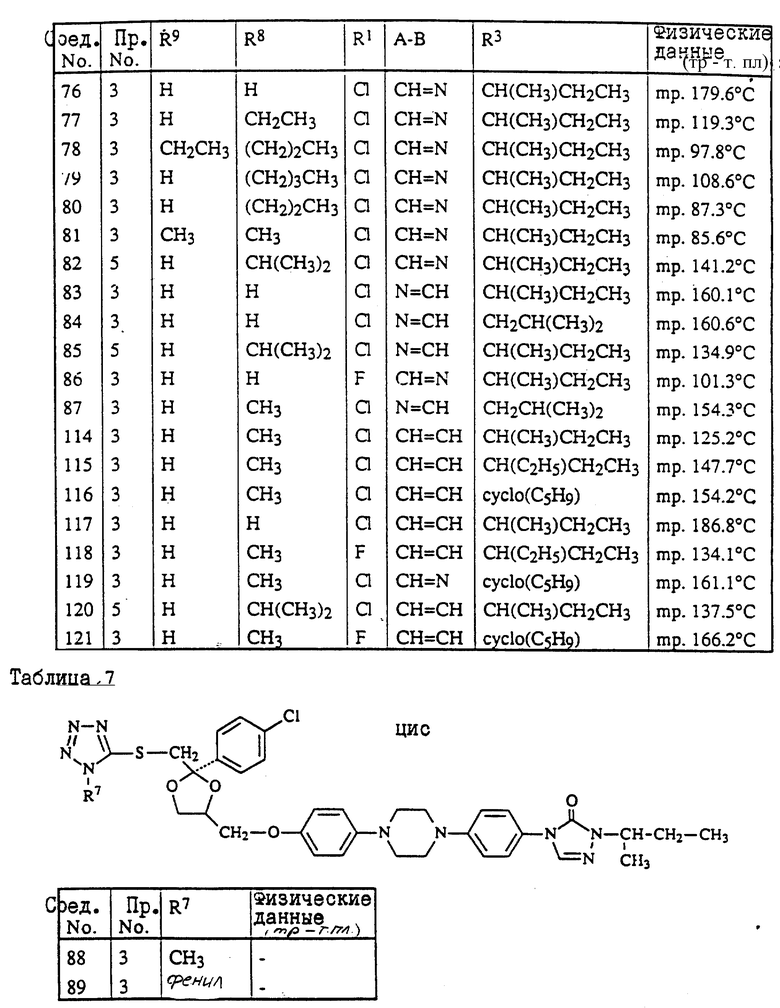
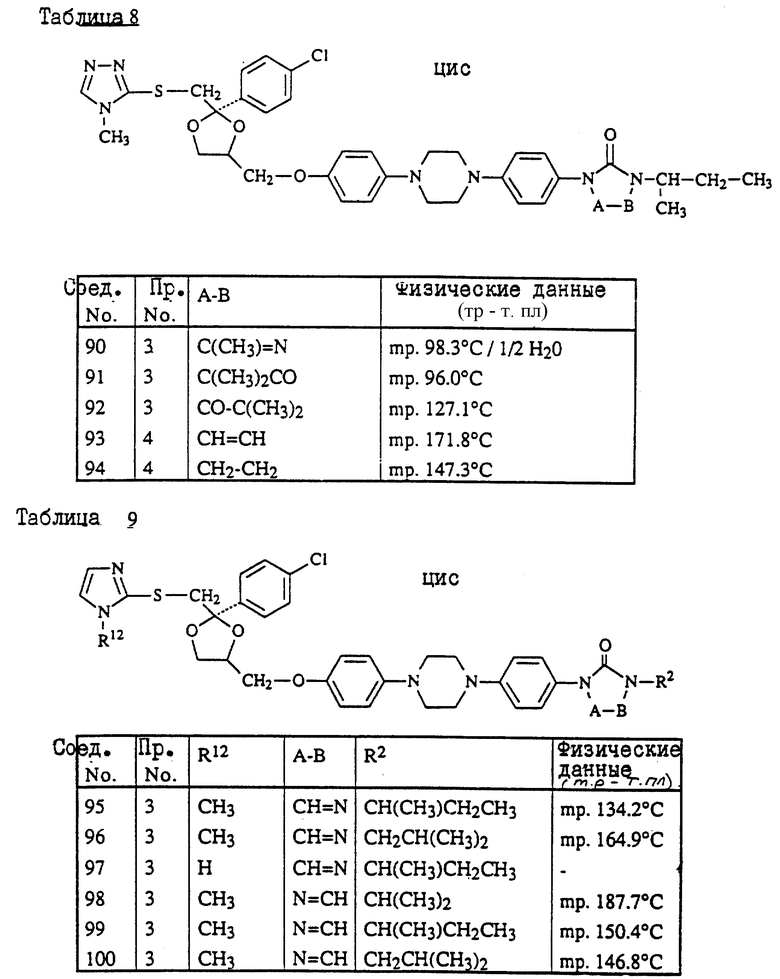
Предпочтительны соединения, где R1 является хлором или фтором, а также соединения, в которых R1 является метилом. Еще одной предпочтительной группой являются соединения в которых у бивалентного радикала -A-B-, являющегося -N = CH- или -CH=N-, атом водорода может быть замещен C1-6- алкилом.
Следующая предпочтительная группа включает соединения, в которых R3 является бутилом, пентилом или циклопентилом.
Группа наиболее предпочтительных соединений включает
цис-4-[4-[4-[4-[[2-(4-хлорфенил)-2-[[(4-метил-4H-1,2,4-триазол-3-ил)тио] метил] -1,3-диоксолан-4-ил] метокси] фенил]-1-пиперазинил]фенил]-2,4- дигидро-2-(1-метилпропил)-3H-1,2,4-триазол-3-он;
цис-2-[4-[4-[4-[[2-(4-хлорфенил)-2-[[4-метил-4H-1,2,4-триазол- 3-ил)тио] метил] -1,3-диоксолан-4-ил] метокси] фенил]-1- пиперазинил]фенил]-2,4-дигидро-4-(1-метилпропил)-3H-1,2,4- триазол-3-он;
цис-2-[4-[4-[4-[[2-(4-фторфенил)-2-[[(4-метил-4H-1,2,4-триазол-3-ил)тио] метил] -1,3-диоксолан-4-ил] метокси]фенил]-1-пиперазинил]фенил] -4-циклопентил-2,4-дигидро-3H-1,2,4-триазол-3-он;
цис-2-[4-[4-[4-[[2-(4-хлорфенил)-2-[[(4-метил-4H-1,2,4-триазол-3-ил) тио]метил]-1,3-диоксолан-4-ил]метокси]фенил]-1-пиперазинил]фенил] -2,4-дигидро-4-пентил-3H-1,2,4-триазол-3-он;
цис-4-(1-этилпропил)-2-[4-[4-[4-[[2-(4-фторфенил)-2-[[(4-метил-4H- 1,2,4-триазол-3-ил)тио] метил] -1,3-диоксолан-4-ил]метокси]фенил]-1- пиперазинил]-фенил]-2,4-дигидро-3H-1,2,4-триазол-3-он;
и их фармацевтически приемлемые соли присоединения кислот или стереохимически изомерные формы.
Вышеуказанные соединения обладают способностью ингибировать аполипопротеин B.
Таким образом, следующим аспектом изобретения является фармацевтическая композиция, обладающая способностью ингибировать аполипопротеин B, включающая активный ингредиент и фармацевтически приемлемый носитель, а в качестве активного ингредиента содержащая соединение 1 в терапевтически эффективном количестве.
Фармацевтическая композиция может быть получена обычным способом.
Способ получения фармацевтической композиции является следующим объектом изобретения и заключается в том, что терапевтически эффективное количество соединения 1 тщательно смешивают с фармацевтически приемлемым носителем.
Предложенные в изобретении соединения могут быть получены из промежуточных продуктов общей формулы (III)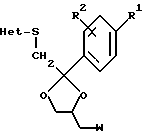
или их солей присоединения кислоты или стереохимически изомерных форм, где R1, R2 и Het являются такими, как определено для соединения 1, a W является подходящей удаляемой группой, такой как галоген или сульфонилоксигруппа.
Еще одним аспектом изобретения является способ получения гетероциклических производных формулы (I), заключающийся в том, что: O - алкилируют промежуточный продукт формулы (II), где A-B и R3 являются такими, как определено выше промежуточным продуктом формулы (III):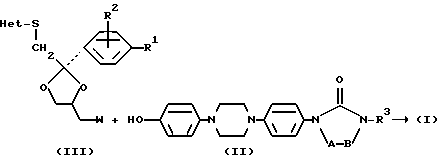
в котором R1, R2 и Het являются такими, как определено выше и W является подходящей уходящей группой, такой как галоген или сульфонилоксигруппа, и необязательно переводят соединение формулы (I) в другое соединение 1 посредством трансформации функциональных групп; и/или, если желательно, переводят соединение формулы (I) в терапевтически активную нетоксичную соль присоединения кислоты или наоборот, с помощью щелочи переводят присоединения кислоты или наоборот, с помощью щелочи переводят соль присоединения кислоты в форму свободного основания; и/или получают N-оксид или стереохимически изомерные формы.
Другим вариантом способа получения соединений 1 является способ получения, заключающийся в том, что проводят реакцию промежуточного продукта формулы (V), в котором Het является таким, как определено выше с промежуточным продуктом формулы (IV), в котором R1, R2, R3, -A-B- являются такими, как определено в пункте 1, a W является подходящей удаляемой группой, такой как галоген или сульфонилоксигруппа;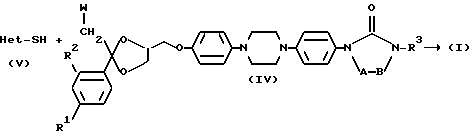
и необязательно переводят соединение формулы (I) в другое соединение (1) посредством трансформации функциональных групп; и/или, если желательно, переводят соединение формулы (I) в терапевтически активную нетоксичную соль присоединения кислоты или наоборот, с помощью щелочи переводят соль присоединения кислоты в форму свободного основания; и/или получают N-оксид или стереохимически изомерные формы.
Указанные выше фармацевтически приемлемые соли присоединения кислот включают терапевтически активные нетоксические формы - соли присоединения кислот, которые способны образовывать соединения формулы (I). Соли можно получить обычным способом путем обработки основной формы подходящей кислотой.
Подходящие кислоты включают, например, неорганические кислоты, такие как галогенводородные кислоты, например, хлористоводородная или бромистоводородная кислота; серная; азотная; фосфорная кислота и подобные; или такие органические кислоты как, например, уксусная, пропионовая, гидроксиуксусная, молочная, пировиноградная, щавелевая, малоновая, янтарная, малеиновая, фумаровая, яблочная, винная, лимонная, метансульфоновая, этансульфоновая, бензолсульфоновая, паратолуолсульфоновая, цикламиновая, салициловая, парааминосалициловая, памовая и подобные кислоты. Использованный выше термин "соли присоединения" также включает сольваты, которые могут образовывать соединения формулы (I), а также их соли. Такими сольватами являются, например, гидраты, алкоголяты и тому подобное. Путем обработки щелочью форму соли можно перевести обратно в форму свободного основания.
Использованный здесь выше термин "стереохимически изомерные формы" определяет все возможные изомерные формы, которые может давать соединение формулы (I). Если специально не упомянуто или не указано, химическое название соединений обозначает смесь всех возможных стереохимически изомерных форм, причем указанные смеси содержат все диастереомеры и энантиомеры основной молекулярной структуры. Более подробно, стереоспецифические центры могут иметь R- или S-конфигурацию; заместители на бивалентных циклических насыщенных радикалах могут иметь либо цис-, либо транс-конфигурацию. Очевидно имеется в виду, что область данного изобретения охватывает стереохимически изомерные формы соединений формулы (I).
Подразумевается, что формы N-оксидов соединений формулы (I) включают те соединения формулы (I), в которых один или несколько атомов азота окислены до так называемого N-оксида, в частности, те N-оксиды, в которых N-окисленными являются один или более пиперазиновых азотов.
Заместители на диоксолановой части соединений формулы (I) могут иметь цис- или транс-конфигурацию. Предпочтительными являются соединения формулы (I), имеющие цис-конфигурацию.
Предпочтительными являются также соединения формулы (I), в которых стереоспецифический углерод во 2-м положении диоксолановой части имеет S-конфигурацию.
Соединения формулы (I) могут также существовать в таутомерных формах. Например, в таутомерных формах могут существовать такие гетероциклы как пиридин, пиримидин, триазол, тиадиазол, оксадиазол, имидазол, тиазол и оксазол, которые замещены гидроксилом, аминогруппой или C1-6-алкиламиногруппой. Хотя в приведенной выше формуле конкретно не указано, имеется в виду, что область настоящего изобретения охватывает такие формы.
Как указано выше соединения формулы (I) можно получить путем О-алкилирования фенолов формулы (II) производными 1,3-диоксолана (III), в которых W обозначает подходящую уходящую группу, такую как, например, хлор или бром или уходящую сульфонилоксигруппу, например, 4-метилбензолсульфонилокси-группу (тозилат) или метансульфонилоксигруппу (мезилат).
Указанную реакцию О-алкилирования обычно можно проводить следуя известным в практике процедурам, например, посредством перемешивания и нагревания реагентов в подходящем растворителе, таком как биполярный апротонный растворитель, например, N,N-диметилформамид, N,N-диметилацетамид в присутствии такого основания, как гидроксид или карбонат щелочного металла, например, гидроксид натрия или калия или карбонат натрия или калия.
Промежуточные продукты формулы (II) можно получить аналогичным образом, как раскрыто в упомянутом выше Европейском патенте EP-0006711. EP-0331232, опубликованный 6 сентября 1989 года, и WO 93/19061, опубликованный 30 сентября 1993 года, также раскрывают пути получения промежуточных продуктов формулы (II).
Соединения формулы (I) можно также получить по реакции промежуточного продукта формулы (IV), в котором, как определено здесь выше, W является подходящей уходящей группой, с гетероциклическим производным формулы (V).
Указанную реакцию можно проводить при перемешивании и нагревании промежуточных продуктов в подходящем растворителе, таком как биполярный апротонный растворитель, например, N,N-диметилформамид, N,N-диметилацетамид, диметилсульфоксид в присутствии такого основания как карбонат или гидроксид щелочного металла, например, карбонат натрия или калия или гидроксид натрия или калия.
Соединения формулы (I) можно также переводить одно в другое. Например, соединения, в которых R3 является С1-8-алкилом, замещенным гидроксилом, можно получить путем восстановления соответствующих соединений формулы (I), в которых R3 является C1-8-алкилом, замещенным оксогруппой. Соединения формулы (I), в которых эндоциклический или экзоциклический атом азота не замещен. Соединения формулы (I), в которых R3 отличен от водорода, можно получить известной специалистам реакцией N-алкилирования из соединений формулы (I), в которых R3 является водородом.
Соединения формулы (I) можно также превратить в соответствующие формы N-оксидов, осуществляя известные специалистам процедуры для перевода трехвалентного азота в форму N-оксида. Указанное N-оксилирование в основном можно осуществлять посредством реакции исходного вещества формулы (I) с подходящей органической или неорганической перекисью. Подходящие неорганические перекиси включают, например, перекись водорода, перекиси щелочных или щелочноземельных металлов, например, пероксид натрия, пероксид калия; подходящие органические перекиси могут включать пероксо-кислоты, такие как, например, бензолкарбопероксо-кислота или галогензамещенная бензолкарбопероксо-кислота, например, 3-хлорбензолкарбопероксо-кислота, пероксоалкановые кислоты, например, пероксоуксусная кислота, алкилгидропероксиды, например, трет-бутилгидропероксид. Подходящими растворителями являются, например, вода; низшие спирты, например, этанол и подобные; углеводороды, например, толуол; кетоны, например, 2-бутанон; галогенированные углеводороды, например, дихлорметан; и смеси подобных растворителей.
Промежуточные продукты формулы (III), которые считаются новыми, можно получить путем следующей последовательности реакций. Осуществляют S-алкилирование гетероциклического реагента (V) промежуточным соединением формулы (VI), где W является подходящей уходящей группой, как определено здесь выше, при перемешивании и нагревании промежуточных продуктов в подходящем реакционно-инертном растворителе, таком как кетон, например, ацетон, в присутствии такого основания как карбонат или гидроксид щелочного металла, например, карбонат натрия или калия, гидроксид натрия или калия. Затем полученный таким образом кетон формулы (VII) превращают в соответствующий кеталь формулы (VIII) при перемешивании и нагревании промежуточного продукта формулы (VII) с глицерином в присутствии кислоты, такой как, например, паратолуолсульфокислота, в реакционно-инертном растворителе, таком как толуол. В завершение гидроксифункцию промежуточного продукта формулы (VIII) превращают в подходящую уходящую группу посредством известных в практике реакций трансформирования функциональных групп, таких как, например, превращение гидроксила в тозилат путем реакции с паратолуолсульфонилхлоридом.
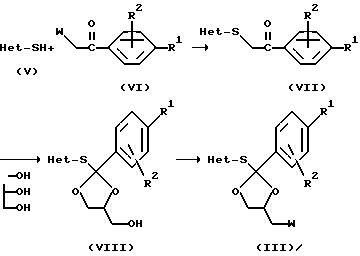
Промежуточные продукты формулы (IV) можно получить аналогичным образом.
Промежуточный продукт формулы (VI) превращают в кеталь, как описано выше. После этого гидроксильную функцию превращают в подходящую уходящую группу, например, сульфонилоксигруппу. Реакция полученного таким образом промежуточного продукта (IX) с промежуточным продуктом (II) дает в результате промежуточный продукт (IV).
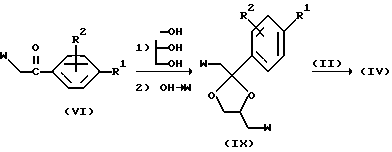
Применяя известные специалистам процедуры, можно получить чистые стереохимически изомерные формы соединений формулы (I). Диастереомеры можно разделить физическими методами деления, такими как селективная кристаллизация или хроматография, например, жидкостная хроматография". Энантиомеры можно отделить друг от друга путем образования диастереомерных солей с оптически чистыми хиральными кислотами и последующей селективной кристаллизации. Указанные чистые стереохимически изомерные формы можно также получить из соответствующих стереохимически изомерных форм подходящих исходных материалов, обеспечивающих стереспецифическое протекание реакции. Если требуется конкретная стереохимически изомерная форма, предпочтительно синтезировать указанную форму стереоспецифическими способами получения. В качестве исходных материалов полезно использовать в этих способах чистые энантиомеры.
Настоящие соединения ингибируют синтез аполипопротеина B, что очевидно из результатов, полученных в "Тесте на ингибирование аполипопротеина B (аро B)", как описано здесь далее. Аполипопротеин B является основным протеиновым компонентом липопротеинов очень низкой плотности (VLDL) и липопротеинов низкой плотности (LDL). Примерно от 60 до 70% общего количества холестерина сыворотки транспортируется в LDL. Увеличенная концентрация LDL холестерина в сыворотке является причиной атеросклероза. При ингибировании синтеза аполипопротеина В снижается количество вредных липопротеинов низкой плотности.
Настоящие соединения не оказывают вовсе или оказывают небольшие нежелательные побочные действия, такие как, например, активность по ингибированию альбумина, активность по ингибированию биосинтеза андрогена или активность по ингибированию биосинтеза холестерина.
С точки зрения своей активности по ингибированию синтеза аполипопротеина B и активности по снижению синтеза сопутствующих липидов настоящие соединения полезны в качестве лекарственных препаратов, особенно в способе лечения пациентов, страдающих от гиперлипидемии. В частности, настоящие соединения можно использовать для производства лекарственных препаратов для лечения нарушений, вызванных избытком липопротеинов очень низкой плотности (VLDL) или липопротеинов низкой плотности (LDL), и особенно нарушений, вызванных холестерином, связанным с указанными VLDL и LDL. Гиперлипидемия может быть вызвана большим количеством генетических и приобретенных заболеваний. Их можно классифицировать на первичные и вторичные гиперлипидемические состояния. Наиболее общими случаями вторичной гиперлипидемии являются сахарный диабет, злоупотребление алкоголем, лекарства, гипотиреоз, хроническая почечная недостаточность, нефротический синдром, холестаз и булимия. Первичной гиперлипидемией являются обычная гиперхолестеринемия, семейная объединенная гиперлипидемия, семейная гиперхолестеринемия, остаточная гиперлипидемия, хиломикронемический синдром (chylomicronaemai syndrome), семейная гипертриглицеридемия (hypertriglyceridaemia). Настоящие соединения можно также применять для профилактики или лечения пациентов, страдающих от атеросклероза, особенно, коронарного атеросклероза, а в более общем случае, от заболеваний, связанных с атеросклерозом, таких как ишемическая болезнь сердца, периферийные сосудистые заболевания, церебральные сосудистые заболевания. Данные соединения могут вызвать регрессию атеросклероза и ингибировать клинические последствия атеросклероза, в частности, болезненность и смертность.
С точки зрения своей активности по ингибированию синтеза аполипопротеина В рассматриваемые соединения можно включать в различные фармацевтические формы для введения пациентам. Для приготовления таких фармацевтических композиций смешивают эффективное количество индивидуального соединения в виде основания или соли присоединения кислоты в качестве активного ингредиента с фармацевтически приемлемым носителем. Указанный носитель может находиться в разнообразных формах в зависимости от формы препарата, необходимого для введения. Желательно, чтобы такие фармацевтические композиции представляли собой единообразную лекарственную форму, предпочтительно удобную для орального приема, ректального введения и парентеральной инъекции. Например, при получении композиций в виде лекарственной формы для орального приема можно использовать любую фармацевтическую среду (такую как, например, вода, гликоли, масла, спирты и подобное), обычную для орально применяемых жидких препаратов, таких как суспензии, сиропы, эликсиры и растворы; или твердые носители (такие как крахмалы, сахара, каолин, лубриканты, связующие, разрыхляющие агенты и подобное) для случая получения порошков, пилюль, капсул и таблеток. Вследствие легкости приема таблетки и капсулы представляют собой наиболее преимущественные лекарственные формы для орального приема, в которых, как очевидно, используют твердые фармацевтические носители. В композициях для парентерального введения носитель обычно будет включать стерильную воду, по крайней мере в большей части, хотя может включать другие ингредиенты, например, способствующие растворимости. Например, можно приготовить растворы для инъекций, в которых носитель включает физиологический раствор, раствор глюкозы или смесь физиологического раствора и раствора глюкозы. Можно также приготовить суспензии для инъекций, в этом случае можно использовать подходящие жидкие носители, суспендирующие агенты и подобное. В композициях, подходящих для введения через кожу, носители обычно включают агент для повышения проницаемости и/или подходящий смачивающий агент, как правило, объединенный с подходящими добавками любой природы в меньших количествах, указанные добавки не оказывают существенного вредного воздействия на кожу. Указанные добавки могут облегчать нанесение на кожу и/или могут быть полезными в приготовлении требуемых композиций. Такие композиции можно применять различными способами, например, в виде бляшек, пятен, мазей для трансдермального введения. Очевидно, что соли присоединения кислот соединений формулы (I), благодаря повышенной растворимости по сравнению с соответствующей основной формой, больше подходят для приготовления водных композиций. Для простоты использования и единообразия дозировки предпочтительно составлять указанные выше фармацевтические композиции в виде стандартных лекарственных форм. Использованный в данном описании термин "стандартная лекарственная форма" обозначает дискретные единицы, подходящие для унифицированной дозировки, причем каждая единица содержит заранее определенное количество активного ингредиента, рассчитанное с целью произведения требуемого терапевтического действия, в ассоциации с подходящим фармацевтическим носителем. Примерами таких стандартных лекарственных форм являются таблетки (включая таблетки с насечкой или в оболочке), капсулы, пилюли, пакетики с порошками, облатки, растворы или суспензии для инъекций, объемы, равные чайной ложке, объемы, равные столовой ложке, и подобное и отдельные их множества.
Специалисты в области терапии гиперлипидемии могут легко определить ежедневное эффективное количество по представленным здесь результатам тестов. В основном предполагается, что терапевтически эффективная доза должна быть от 0,001 до 5 мг/кг веса тела, более предпочтительно от 0,01 мг/кг до 0,5 мг/кг веса. Подходящим вариантом может быть введение терапевтически эффективной дозы за два, три, четыре и более приемов в день (субдозы) с подходящими временными интервалами. Указанные субдозы могут представлять собой стандартные лекарственные формы, содержащие, например, от 0,05 до 250 мг (в особенности, от 0,5 до 5 мг) активного ингредиента на стандартную лекарственную форму.
Как хорошо известно специалистам, точная дозировка и частота приема зависят от конкретного использованного соединения формулы (I), конкретных условий лечения, тяжести состояния, подлежащего лечению, возраста, веса и общего физического состояния конкретного пациента, а также от возможного приема других лекарственных препаратов. Кроме того, очевидно, что указанное эффективное дневное количество можно снизить или увеличить в зависимости от реакции пациента и/или в зависимости от оценки врача, прописывающего соединения настоящего изобретения. Следовательно, указанный здесь выше интервал для эффективного дневного количества является только ориентировочным.
Экспериментальная часть
Здесь далее обозначение "ДИПЭ" относится к диизопропиловому эфиру, "МИК" обозначает метилизопропилкетон и "ДМФ" обозначает N,N-диметилформамид.
A. Получение промежуточных продуктов
Пример 1.
а) Смесь 1-метил-1H-1,2,4-триазол-5-тиола (35 г), 2-хлор-1-(фторфенил)-этанона (51,4 г) и карбоната натрия (32,5 г) в 2-пропаноле (500 мл) перемешивают и нагревают с обратным холодильником в течение 4 часов. Растворитель выпаривают, остаток растворяют в CH2Cl2, фильтруют и фильтрат выпаривают. Остаток кристаллизуют из ДИПЭ, получая выход продукта 25 г (33%). Образец (3 г) чистят колоночной хроматографией на силикагеле (элюент CH2Cl2/CH3OH = 99/1). Чистые фракции собирают и выпаривают. Остаток кристаллизуют из ДИПЭ, получая 1-(4-фторфенил)-2-[(2-метил-2H-1,2,4-триазол-3-ил)тио]этанон (промежуточный продукт 1).
б) Смесь промежуточного продукта (1) (22 г), глицерина (39,6 г) и паратолуолсульфокислоты (20 г) в толуоле (200 мл) перемешивают при нагревании с обратным холодильником в течение ночи. Смесь охлаждают и добавляют воду. Смесь экстрагируют толуолом и промывают водой. Органический слой сушат, фильтруют и выпаривают растворитель. Остаток чистят способом высокоэффективной жидкостной хроматографии на силикагеле (элюент CH2Cl2/CH3OH=98/2). Чистые фракции собирают и выпаривают, получая 9 г (31,6%) (±)цис-2-(4-фторфенил) -2-[[(2-метил-2H-1,2,4-триазол-3-ил)тио] метил] -1,3-диоксолан-4-метанол (промежуточный продукт 2).
в) Смесь промежуточного продукта 2 (9 г), паратолуолсульфохлорида (6,3 г) и N,N-диметил-4-пиридинамина (1 г) в CH2Cl2 (150 мл) и N,N-диэтилэтанамине (5 мл) перемешивают при комнатной температуре в течение 4 часов. Добавляют воду и разделяют слои. Органический слой промывают водой, сушат, фильтруют и выпаривают растворитель. Остаток чистят колоночной хроматографией на силикагеле (элюент CH2Cl2/CH3OH=99/1). Чистые фракции собирают и выпаривают при температуре < 35oC. Остаток растворяют в МИК и переводят в соль паратолуолсульфокислоты (1:1). Добавляют немного ДИПЭ и продукт высаживается в виде кристаллов. Осадок отфильтровывают и сушат, получая 6,8 г (37.8%) {(±)-цис-2-(4-фторфенил)-2-[[(2-мeтил-2H-1,2,4-тpиaзoл-3-ил)тио] метил]-1,3-диоксолан-4-метанол}-4-метилбензолсульфоната 4-метилбензолсульфонат (1:1) (промежуточный продукт 3). Аналогичным образом получают также:
{(±)-цис-2-(4-фторфенил)-2-[[(4-метил-4H-1,2,4-триазол-3-ил)- тио]метил] -1,3-диоксолан-4-метанол} -4-метилбензолсульфонат (эфир) 4-метилбензолсульфонат (1:1) т. пл. 136,4oC (промежуточный продукт 4);
{(±)-цис-2-(2,4-дифторфенил)-2-[[(4-метил-4H-1,2,4-триазол-3-ил) тио]метил] -1,3-диоксолан-4-метанол} -4-метилбензолсульфонат (эфир) 4-метилбензолсульфонат (1:1) (промежуточный продукт 5);
{ (±)-транс-2-(4-хлорфенил)-2-[[(4-мeтил-4H-1,2,4-тpиaзoл-3-ил) тио]метил] -1,3-диоксолан-4-метанол} -4-метилбензолсульфонат (эфир) 4-метилбензолсульфонат (1:1) т. пл. 151,9oC (промежуточный продукт 6);
{(±)-цис-2-(2,4-дифторфенил)-2-[[(2-мeтил-2H-1,2,4-триазол-3-ил) тио]метил] -1,3-диоксолан-4-метанол} -4-метилбензолсульфонат (эфир) (промежуточный продукт 7); и
{ (±)-цис-[2-(бромметил)-2-(2,4-дифторфенил)-1,3-диоксолан-4-ил] метил} -2-нафталинсульфонат (промежуточный продукт 40).
Пример 2.
а) Смесь 2-бром-1-(4-хлорфенил)этанона (350 г), глицерина (322 г) и паратолуолсульфокислоты (35 г) в толуоле (3000 мл) перемешивают и нагревают с обратным холодильником в течение 24 часов, используя водный сепаратор. Реакционную смесь выливают в водный раствор NaHCO3 перемешивают в течение некоторого времени. Органический слой отделяют, сушат, фильтруют и выпаривают растворитель, получая 485 г (93%; масло) (цис+транс)-2-(бромметил)-2-(4-хлорфенил)-1,3-диоксолан-4-метанол (промежуточный продукт 8a). К смеси промежуточного продукта 8a (25 г) и N,N-диметил-4-пиридинамина (1 г) в N, N-диэтилэтанамине (25 мл) и CH2Cl2 (250 мл) порциями добавляют 2-нафталинсульфонилхлорид (21 г) и смесь перемешивают при комнатной температуре в течение 2 часов. Смесь выливают в воду и промывают. Органический слой сушат, фильтруют и выпаривают растворитель. Остаток чистят колоночной хроматографией на силикагеле (элюент CH2Cl2/CH3OH=99/1). Чистые фракции собирают и выпаривают. Остаток чистят колоночной хроматографией (элюент CH2Cl2, /гексан от 40/60 до 60/40). Чистые фракции собирают и выпаривают, получая выход 21,8 г (55%) {(±)-цис-[2-(бромметил) -2-(4-хлорфенил)-1,3-диоксолан-4-ил]метил} 2-нафталинсульфоната (промежуточный продукт 8b).
в) 2,4-дигидро-4-[4-[4-(4-гидроксифенил)-1-пиперазинил] фенил]-2-(1-метилпропил)-3H-1,2,4-триазол-3-он (206,9 г) добавляют к раствору промежуточного продукта (8b) (250 г) в диметилсульфоксиде (2000 мл). Добавляют гидроксид калия (67 г) и реакционную смесь перемешивают в течение ночи при комнатной температуре. Смесь выливают в воду (3000 мл) и перемешивают в течение 30 минут. Осадок отфильтровывают, промывают 2-пропанолом (1000 мл) и ДИПЭ (1000 мл), затем сушат, получая выход 316 г (92,2%) (±)-циc-4-[4-[4-[4-[[2-(бромметил)-2-(4-хлорфенил)-1,3- диоксолан-4-ил] метокси]фенил]-1-пиперазинил]фенил]-2,4-дигидро- 2-(1-метилпропил)-3H-1,2,4-триазол-3-она (промежуточный продукт 8c).
Аналогичным образом получают:
Б. Получение целевых соединений
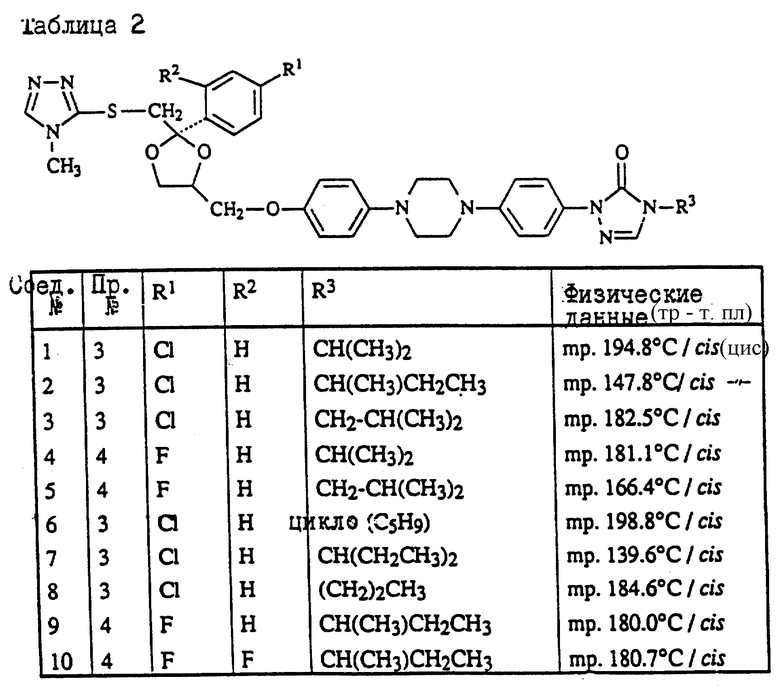
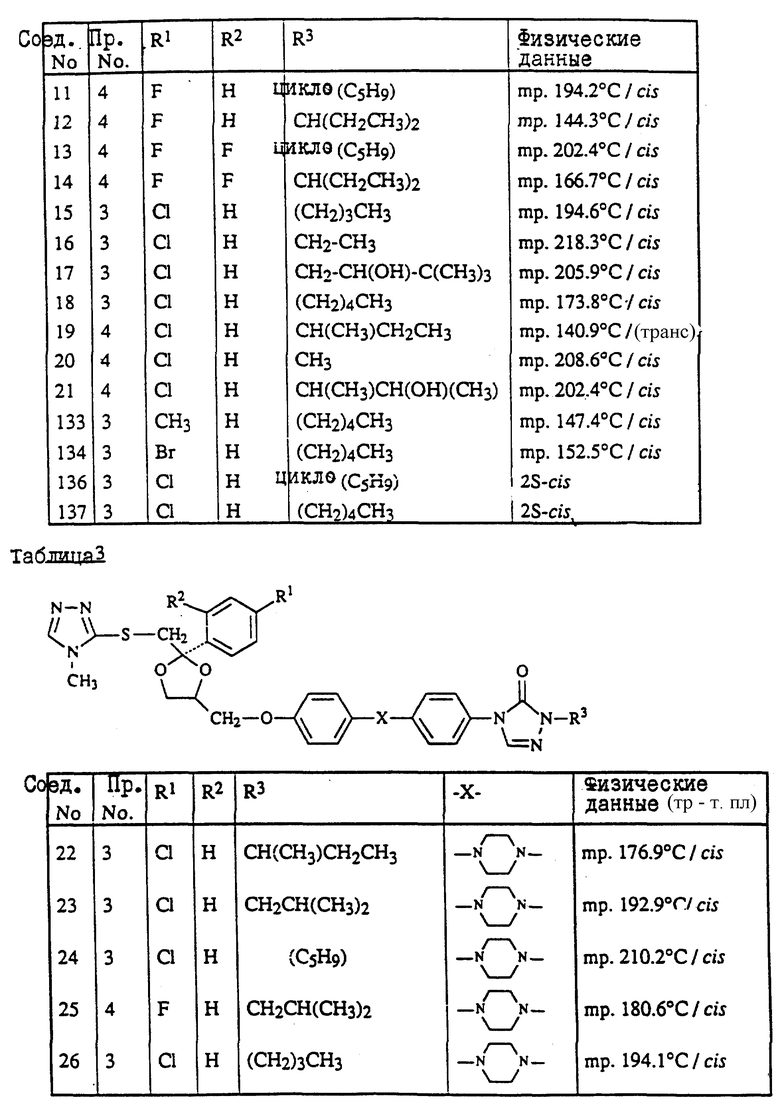
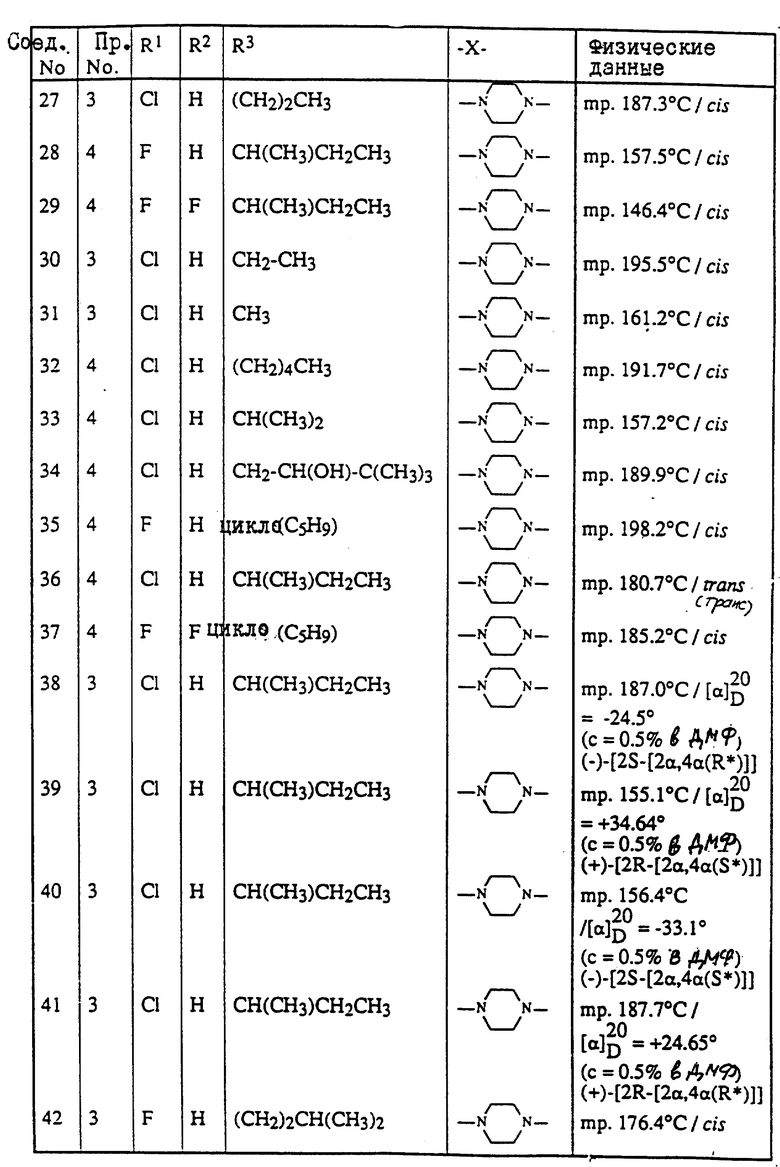
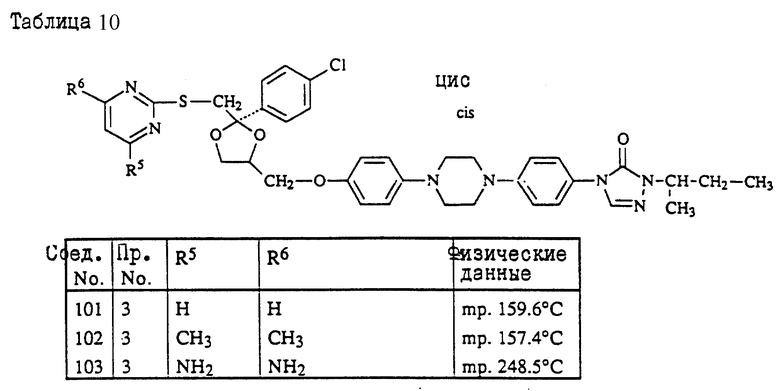
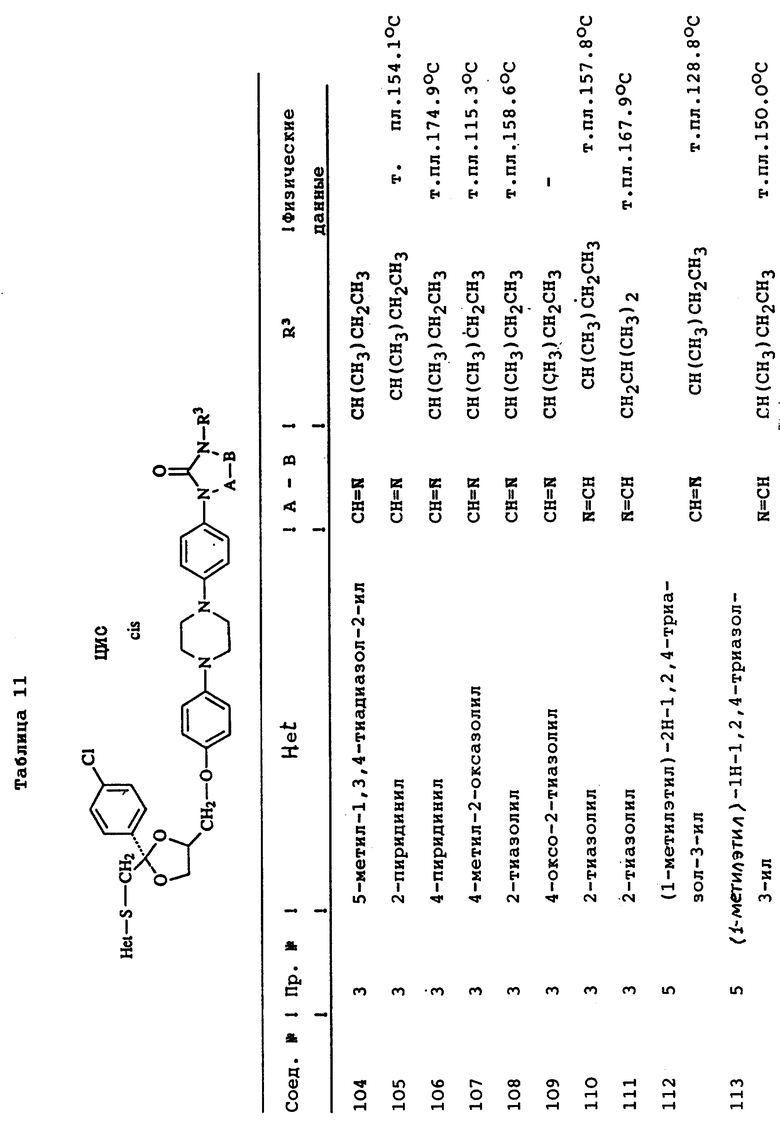
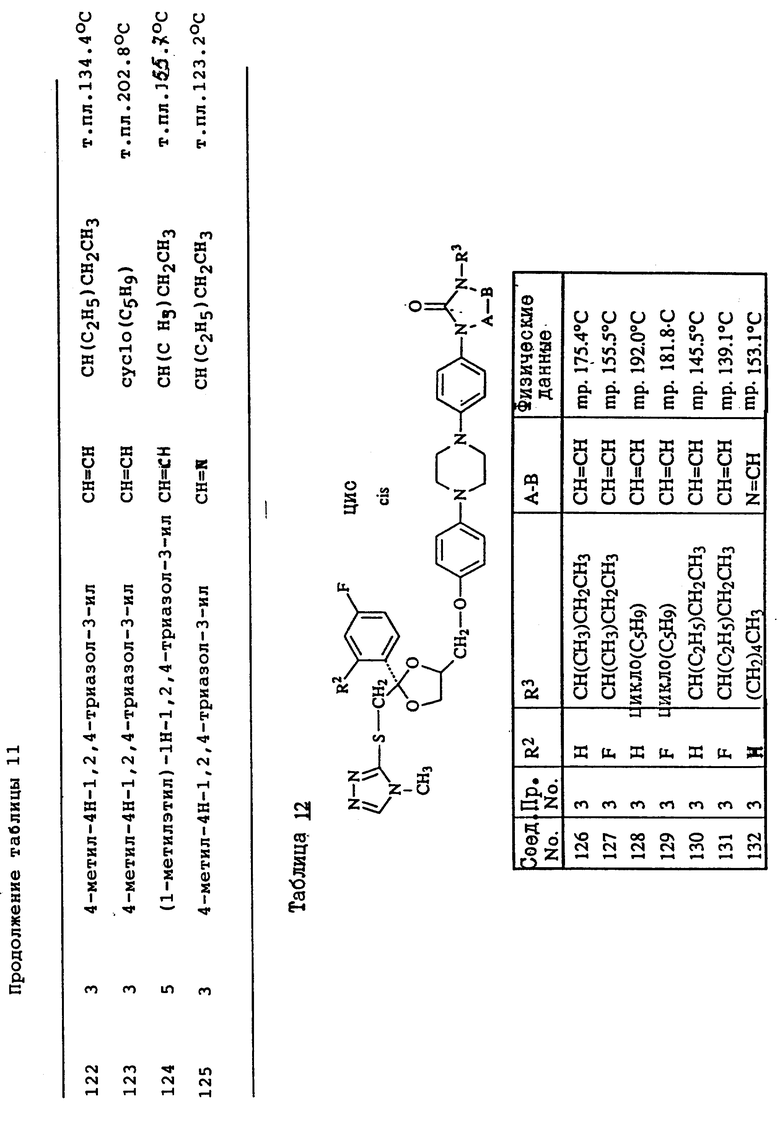
Пример 3. Смесь 4-метил-4H-1,2,4-триазол-3-тиола (1,9 г), промежуточного продукта (8с) (9 г) и карбоната натрия (3 г) в ДМФ (150 мл) перемешивают под N2 при 120oC в течение ночи. Смесь охлаждают, разбавляют водой, продукт выпадает в виде кристаллического осадка. Осадок отфильтровывают и чистят колоночной хроматографией на силикагеле (элюент CH2Cl2/н-гексан/EtOAc/ /CH3OH= 500/250/250/2). Чистят фракции, собирают и выпаривают. Остаток растирают в CH3ОН и перекристаллизовывают из н-C4H9OH, получая выход 6,3 г (±)-циc-4-[4-[4-[4-[[2-(4-xлopфeнил)-2-[[4-мeтил- 4H-1,2,4-триазол-3-ил)тио]метил]-1,3-диоксолан-4- ил] метокси] фенил]-1-пиперазинил]фенил]-2,4-дигидро-2- (1-метилпропил)-3H-1,2,4-триазол-3-она (68%); т. пл. 173oC (соединение 22).
Пример 4. Смесь промежуточного продукта (3) (3,3 г), 2,4-дигидро-2-[4-[4-(4-гидроксифенил)-1-пиперазинил] фенил] -4-(1-метилпропил)-3H-1,2,4-триазол-3-она (2 г), и гидроксида калия (1 г) в ДМФ (100 мл) перемешивают при комнатной температуре под N2 в течение 6 часов. В смесь еще добавляют промежуточный продукт (3) (1 г) и перемешивают в течение 1 часа. Смесь выливают в воду и фильтруют. Осадок чистят колоночной хроматографией на силикагеле (элюент CH2Cl2/CH3OH= 99/1). Чистые фракции собирают и выпаривают. Остаток кристаллизуют из МИК, получая выход 1,6 г (±)-циc-2-[4-[4-[4-[[2-(4-фтopфeнил)-2-[[2-метил-2H-1,2,4-триазол-3-ил) тио] метил] -1,3-диоксолан-4-ил] метокси] -фенил] -1-пиперазинил] фенил] -2,4- дигидро-4-(1-метилпропил)-3H-1,2,4-триазол-3-она (45.7%); т. пл. 157,3oC (соединение 70).
Пример 5. Гидрид натрия, 50% дисперсию в минеральном масле (0,31 г), добавляют к смеси соединения (76) (4,3 г) в ДМФ (100 мл), и смесь перемешивают при комнатной температуре в течение 30 минут. Добавляют 2-бромпропан (0,86 г) и смесь перемешивают при комнатной температуре в течение 48 часов. Снова добавляют гидрид натрия, 50% дисперсию в минеральном масле, и 2-бромпропан и смесь перемешивают в течение 4 часов. Смесь выливают в воду, экстрагируют CH2Cl2 и промывают водой. Органический слой сушат, фильтруют и выпаривают растворитель. Остаток чистят колоночной хроматографией на силикагеле (элюент CH2Cl2/CH3OH= 99/1). Чистые фракции собирают и выпаривают. Остаток кристаллизуют из CH3ОН. Остаток чистят способом высокоэффективной жидкостной хроматографии. Чистые фракции собирают и выпаривают. Фракцию 1 кристаллизуют из н-C4H9OH, получая выход 0,4 г (±)-циc-4-[4-[4-[4-[[2-(4-xлopфeнил)-2-[[[1-(1-метилэтил) -1H-1,2,4-триазол-3-ил]тио]метил]-1,3-диоксолан-4-ил]метокси] фенил]-1- пиперазинил]фенил]-2,4-дигидро-2-(1-метилпропил)-3H-1,2,4-триазол- 3-она; т. пл. 128,8oC (соединение 112). Фракцию 2 растирают в CH3ОН, получая выход 1.4 г (±)-циc-4-[4-[4-[4-[[2-(4-xлopфeнил)-2-[[[2- (1-метилэтил)-2H-1,2,4-триазол-3-ил] тио] метил]-1,3- диоксолан-4-ил]метокси]фенил]-1-пиперазинил] фенил] -2,4-дигидро-2- (1-метилпропил)-3H-1,2,4-триазол-3-она; т. пл. 141,2oC (соединение 82).
Пример 6. Раствор боргидрида натрия (1 г) в воде (20 мл) добавляют по капле к раствору соединения (47) (3,6 г) в ДМФ (100 мл). Реакционную смесь перемешивают в течение ночи при комнатной температуре. Добавляют уксусную кислоту (1 мл). Добавляют воду (750 мл), в результате чего происходит кристаллизация продукта. Остаток чистят способом колоночной хроматографии на силикагеле (элюент CH2Cl2/CH3OH=90/10). Чистые фракции собирают и выпаривают растворитель. Остаток растирают в 2-пропаноле. Осадок отфильтровывают и сушат, получая выход 2,9 г (±)-цис-4-[4-[4-[4-[[2-(4-xлopфeнил)-2-метил-4H-1,2,4-триазол-3-ил) тио] метил]-1,3-диоксолан-4-ил]метокси]фенил]-1-пиперазинил] фенил]-2,4- дигидро-2-(2-гидрокси-1-метилпропил)-3H-1,2,4-триазол-3-она; т. пл. 153,4oC (соединение 48).
В. Фармакологический пример
Пример 7:
Тест на ингибирование аполипопротеина B (apo B)
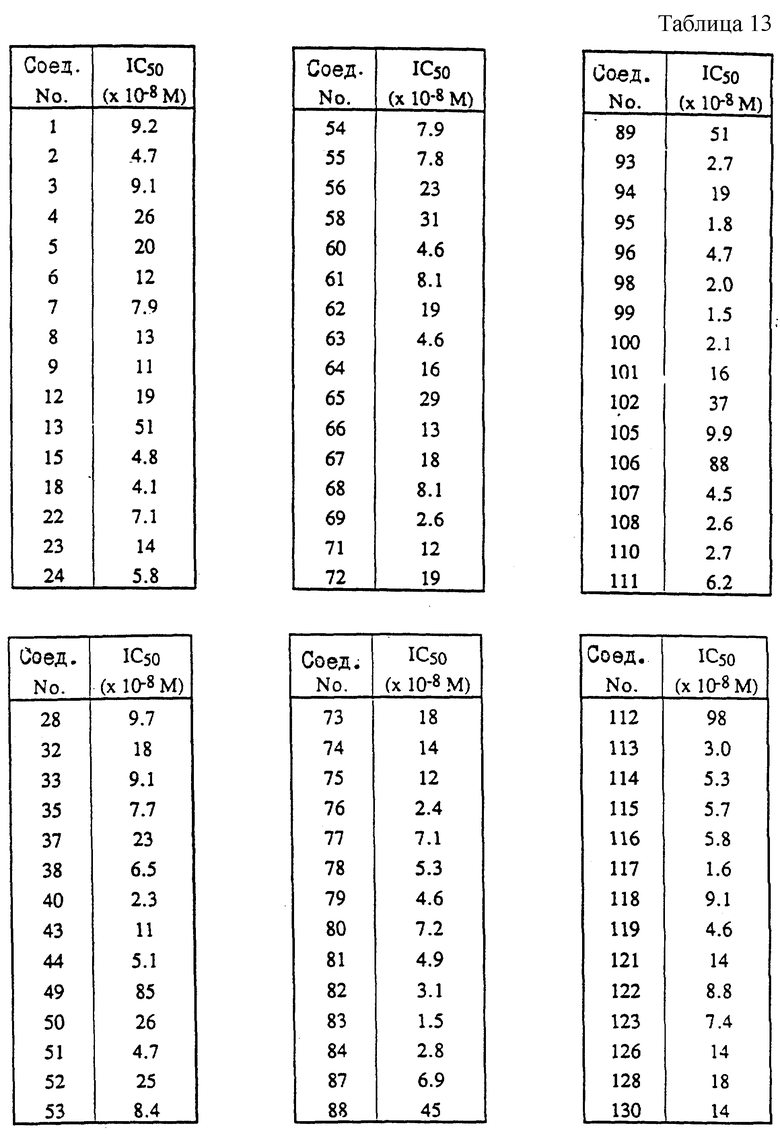
Культивированные клетки печени человека (НерG2-клетки), которые синтезируют и выделяют липопротеины низкой плотности, инкубируют в течение ночи при температуре 37oC в жидкой среде, содержащей радиоактивно-меченый лецитин. Таким образом радиоактивно-меченый лецитин включают в аполипопротеин B. Жидкую среду декантируют и выделяют аполипопротеин B путем двойного иммуноосаждения, а именно, сначала к жидкой среде добавляют специфические по отношению к аполипопротеину B антитела (антитело1), а потом добавляют вторые антитела (антитело2), которые специфически присоединяются к комплексу apoB-антитело1. Образованный таким образом комплекс apoB-антитело1-антитело2 осаждается, и его выделяют посредством центрифугирования. Количество синтезированного в течение ночи аполипопротеина B получают измеряя радиоактивность выделенного комплекса. Для измерения ингибирующей активности исследуемого соединения это исследуемое соединение добавляют к жидкой среде в различных концентрациях и сравнивают концентрацию аполипопротеина B, синтезированного в присутствии исследуемого соединения (концентрация apoB (после)), с концентрацией аполипопротеина B, синтезированного в отсутствие исследуемого соединения (концентрация apoB (контроль)). Для каждого эксперимента ингибирование образования аполипопротеина B выражают следующим образом: %ингибирования=100•(1-конц.apoB (после)/конц.apoB (контроль)).
После проведения нескольких экспериментов с одинаковой концентрацией рассчитывают среднее значение ингибирования для данных экспериментов. Рассчитывают также величины IC50 (концентрация лекарства, необходимая для снижения выделения apoB на 50% от контрольного значения).
В табл. 13 представлены IC50-величины для некоторых соединений формулы (I). Соединения формулы (I), не перечисленные в таблице 13, но для которых существуют соответствующие данные, имеют IC50-величины 1•10-6 М или более.
Г. Примеры композиций
Следующие препаративные формы представляют типичные фармацевтические композиции в виде стандартной (единичной) дозы, подходящие для общего или локального применения для теплокровных животных в соответствии с настоящим изобретением.
Использованный в этих примерах термин "активный ингредиент" (АИ) относится к соединению формулы (I), форме N-оксида, его фармацевтически приемлемой соли присоединения кислоты или его стереохимически изомерной форме.
Пример 8:
Растворы для орального применения
9 г метил 4-гидроксибензоата и 1 г пропил-4-гидроксибензоата растворяют в 4 л кипящей очищенной воды. В 3 л этого раствора растворяют сначала 10 г 2,3-гидроксибутандионовой кислоты и затем 20 г АИ. Последний раствор объединяют с оставшейся частью предшествующего раствора и добавляют туда 12 л 1,2,3-пропантриола и 3 л 70% раствора сорбитола. 40 г натрийсахарина (sodium saccharin) растворяют в 0,5 л воды и добавляют 2 мл малиновой эссенции и 2 мл эссенции крыжовника. Последний раствор объединяют с предыдущим, добавляют воды q.s. до объема 20 л, получая раствор для орального приема, включающий 5 мг АИ на чайную ложку (5 мл). Полученным раствором заполняют подходящие контейнеры (емкости).
Пример 9:
Капсулы
Энергично смешивают 20 г АИ, 6 г лаурилсульфата натрия, 56 г крахмала, 56 г лактозы, 0,8 г коллоидного диоксида кремния и 1,2 г стеарата магния. Затем полученной смесью наполняют 1000 подходящих отвержденных желатиновых капсул, каждая из которых включает 20 мг АИ.
Пример 10:
Таблетки в оболочке
Приготовление сердцевины таблеток. Хорошо перемешивают 100 г АИ, 570 г лактозы и 200 г крахмала и после этого смесь смачивают раствором 5 г додецилсульфата натрия и 10 г поливинилпирролидона (Kollidon - K90) примерно в 200 мл воды. Влажную порошкообразную смесь просеивают, сушат и просеивают снова. Затем добавляют 100 г микрокристаллической целлюлозы (Avicel) и 15 г гидрированного овощного масла (Sterotex). Все это хорошо перемешивают и прессуют таблетки, получая 10000 таблеток, каждая из которых включает 10 мг активного ингредиента.
Оболочка. К раствору 10 г метилцеллюлозы (Methocel 60HG) в 75 мл денатурированного этанола добавляют раствор 5 г этилцеллюлозы (Ethocel 22 cps) в 150 мл дихлорметана. Затем добавляют 75 мл дихлорметана и 2,5 мл 1,2,3-пропантриола. 10 г полиэтиленгликоля расплавляют и растворяют в 75 мл дихлорметана. Последний раствор добавляют к предыдущему и затем добавляют 2,5 г октадеканоата магния, 5 г поливинилпирролидона и 30 мл концентрированной цветной суспензии (Opaspray К-1-2109), полученную смесь гомогенизируют. Сердцевину таблеток покрывают полученной таким образом смесью в установке для нанесения покрытия.
Пример 11:
Растворы для инъекций
1,8 г метил-4-гидроксибензоата и 0,2 г пропил-4-гидроксибензоата растворяют примерно в 0,5 л кипящей воды для инъекций. После охлаждения примерно до 50oC добавляют при перемешивании 4 г молочной кислоты, 0,05 г пропиленгликоля и 4 г АИ. Раствор охлаждают до комнатной температуры и разбавляют водой q. s. для инъекций до объема 1 л, получая раствор, содержащий 4 мг АИ на мл. Раствор стерилизуют посредством фильтрации (Фармакопея США U.S.P.) XVII p. 811) и наполняют им стерильные контейнеры.
Приложение: дополнительная экспериментальная информация
Соединение 138 получают способом, описанным в примере 3. R1 в соединении 138, которое имеет следующую структуру, является водородом.
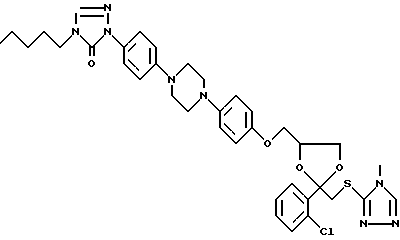
Соединение 139 (0,0027 моль) перемешивают в кипящем 2-пропаноле (20 мл). HCl добавляют в 2-пропанол (5 мл) по каплям. Происходит полное растворение. Затем смеси дают охладиться до комнатной температуры, и происходит кристаллизация. Осадок отфильтровывают, перемешивают в 2-пропаноле, отфильтровывают, затем сушат, получают 1,25 г соединения 137 в форме его HCl (1: 3)•H2O (1:1) соли, представляющее соединение 139, имеющее структуру: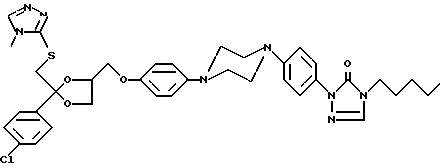
Стереохимия: (2S - ClS)
Форма соли: гидрохлорид (1:3)- гидрат (1:1)
В табл. 14 и 15 представлены физико-химические константы соединений, которые ранее не были охарактеризованы.
В табл. 14 представлены результаты элементного анализа; представлены как теоретические (колонка "теоретич. "), так и экспериментальные (колонка "эксперим. ") данные. В табл. 15 представлены данные по масс-спектрометрии; представлены молекулярный ион и базовый пик.
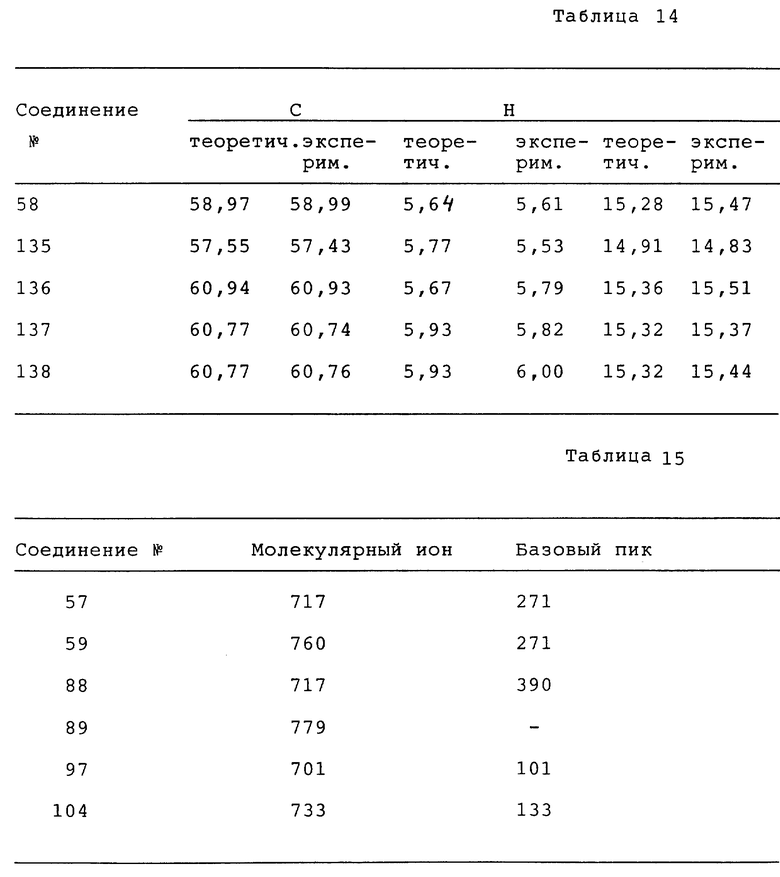
Изобретение описывает новые соединения формулы I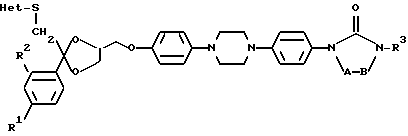
их N-оксиды, стереохимически изомерные формы и фармацевтически пригодные соли присоединения кислот, где А и В вместе взятые образуют бивалентный радикал формулы: -N=CH-(a), -CH=N-(b), -CH=CH-(c), CH2-CH2-(d), -C(= O)-CH2-(e), -CH2-C(=O)-(f); R1 является водородом; С1-6-алкилом или галогеном; R2 является водородом или галогеном; R3 является С1-8-алкилом; С3-6-циклоалкилом или С1-8-алкилом, замещенным гидроксилом, оксогруппой; Het является пяти- или дестичленным гетероциклическим кольцом. Раскрывается применение в качестве лекарственных препаратов, в частности в качестве агентов для снижения уровня липидов, а также фармацевтические композиции и способы получения соединений и композиций. 6 с. и 7 з.п.ф-лы, 15 табл.
где А и В, взятые вместе, образуют бивалентный радикал формулы:
-N=CH- (a)
-CH=N- (в)
-CH-CH- (c)
-CH2=CH2- (d)
-C(=O)-CH2) (e)
-CH2-C(=O)- (f)
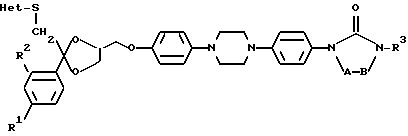
причем в бивалентных радикалах (a) и (в) атом водорода может быть замещен C1-6-алкилом; в бивалентных радикалах (e) и (f) два атома водорода могут быть замещены C1-6-алкилом; R1 является водородом, C1-6-алкилом или галогеном; R2 является водородом или галогеном; R3 является C1-8-алкилом; C3-6-циклоалкилом; или C1-8-алкилом, замещенным гидроксилом, оксогруппой; Het является гетероциклом, выбранным из группы, включающей пиридинил; пиримидинил; пиримидинил, замещенный двумя заместителями, выбранными из C1-6-алкила, аминогруппы; тетразолил, замещенный C1-6-алкилом или арилом; триазолил; триазолил, замещенный одним или двумя заместителями, выбранными из C1-6-алкила, тригалогенметила, гидроксила, аминогруппы; тиадиазолил, замещенный C1-6-алкилом; тиазолил; оксазолил, замещенный C1-6-алкилом; имидазолил, имидазолил, замещенный C1-6-алкилом; его N-оксид, стереохимически изомерная форма или фармацевтически приемлемая соль присоединения кислоты.
его соль присоединения кислоты или стереохимически изомерная форма, где R1, R2 и Het являются такими, как определено в п.1, а W является подходящей уходящей группой, такой как галоген или сульфонилоксигруппа.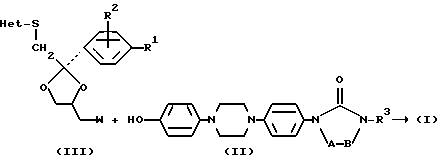
в котором R1, R2 и Het являются такими, как определено в п.1 и W является подходящей уходящей группой, такой как галоген или сульфонилоксигруппа, и необязательно переводят соединение формулы I в другое соединение 1 посредством трансформации функциональных групп; и/или, если желательно, переводят соединение формулы I в терапевтически активную нетоксичную соль присоединения кислоты или наоборот, с помощью щелочи переводят соль присоединения кислоты в форму свободного основания; и/или получают N-оксид или стереохимически изомерные формы.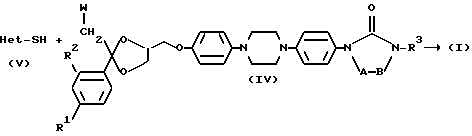
и необязательно переводят соединение формулы I в другое соединение I посредством трансформации функциональных групп; и/или, если желательно, переводят соединение формы I в терапевтически активную нетоксичную соль присоединения кислоты или наоборот, с помощью щелочи переводят соль присоединения кислоты в форму свободного основания; и/или получают N-оксид или стереохимически изомерные формы.
Приоритет по пунктам и признакам:
19.10.95 по п.3 и N-оксид соединения по п.1;
27.10.94 по п.1, кроме N-оксида, пп.2, 4-13.
| Приспособление в автоматическом оружии для скрепления при помощи сцепляющихся выступов ствола с боевой личинкой на время выстрела и последующего короткого отката | 1925 |
|
SU6711A1 |
| Киноэкран с изменяющимся радиусом кривизны | 1958 |
|
SU118138A1 |
| EP 228125 A1, 08.07.87 | |||
| 0 |
|
SU402989A1 | |
| Машковский М.Д | |||
| Лекарственные средства | |||
| - М.: Медицина, 1972, ч.II, с.137-143. | |||

