ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к трансгенным организмам, которые способны разрушать гербицид - дикамбу, включая трансгенные растения, которые могут быть толерантными к дикамбе. Также изобретение касается дикамба-разрушающих ферментов и молекул ДНК и ДНК-конструкций, кодирующих дикамба-разрушающие ферменты. Также изобретение относится к способу борьбы с сорняками на плантациях толерантных к дикамбе трансгенных растений и к способу удаления дикамбы из загрязненного им материала (биологическая нейтрализация). Наконец, изобретение относится к способам отбора трансформантов, основанным на толерантности к дикамбе или на выявлении флуоресценции 3,6-дихлорсалициловой кислоты, образующейся в процессе разрушения дикамбы.
ПРЕДПОСЫЛКИ
Гербициды широко применяются в сельскохозяйственном производстве. Их эффективность часто определяется способностью подавлять рост сорняков в посевах культурных растений и толерантностью культурных растений к такому гербициду. Если же культурное растение не толерантно к гербициду, то гербицид обусловливает либо снижение продуктивности данной культуры, либо вообще приводит к ее полному уничтожению. С другой стороны, если гербицид не является достаточно мощным, то он может сохранять такой уровень роста сорняков на плантации культурного растения, который, со своей стороны, обусловит снижение продуктивности культурного растения. Соответственно, с экономической точки зрения желательным является создание сельскохозяйственных культурных растений, которые бы были толерантными к гербицидам. С точки зрения защиты вод и параметров внешней среды в районах расположения сельскохозяйственных угодий также желательно развитие технологий, которые бы обеспечивали разрушение гербицидов в случае аварийных выбросов гербицидов или в случаях неприемлемо высокого уровня загрязнения почвы и вод.
Гены, кодирующие ферменты, которые инактивируют гербициды и другие соединения-ксенобиотики, ранее были выделены у различных прокариотических и эукариотических организмов. В некоторых случаях эти гены были успешно экспрессированы у растений с применением методов генетической инженерии. С использованием такого подхода были разработаны растения, которые являются толерантными к гербицидам: 2,4-дихлорфеноксиуксусной кислоте (Streber & Willmitzer, 1989, Bio/Technol., 7, 811-816: 2,4-D), бромоксинилу (Stalker et al., 1988, Science, 242, 419-423: торговая марка Buctril), глифозату (Comai et al., 1985, Nature, 317, 741-744: торговая марка Round-Up) и фосфинотрицину (De Block et al., 1987, EMBO J., 6, 2513-2518: торговая марка Basta).
Дикамба (торговая марка Banvel) используется в качестве предвсходового и послевсходового гербицида для борьбы с одно- и многолетними широколистными сорняками и некоторыми злаковыми сорняками в посевах кукурузы, сорго, зерновых культур, кормовых культур, сенокосных трав, пастбищных культур, сахарного тростника, спаржи, дерновых и семенных травяных культур: см. Crop Protect-on Ref., pp.1803-1821 (Chemical & Phamaceutical Press Inc., New York, NY, 11th ed., 1995). К сожалению, дикамба способна повреждать многие сельскохозяйственные культуры (включая бобы, сою, хлопчатник, горох, картофель, подсолнечник, помидор, табак и фруктовые деревья), декоративные растения и деревья, а также другие широколистные растения при контакте с данным гербицидом. Дикамба характеризуется химической стабильностью и в ряде случаев может сохраняться во внешней среде.
Дикамба относится к классу гербицидов на основе бензойной кислоты. Предполагается, что растения, толерантные к гербицидам, основанным на бензойной кислоте, включая дикамбу, могут быть созданы путем внесения в растения антисмыслового гена синтазы 1-аминоциклопропан-1-карбоновой кислоты (АСС), антисмыслового гена АСС-оксидазы, гена АСС-деаминазы или их сочетаний. См. заявку на патент под №2165036 (опубликована 16 июня 1996 г.). Однако в этой заявке не представлено никаких экспериментальных данных по анализу такой толерантности.
Известны бактерии, которые способны метаболизировать дикамбу. См. патент США №5445962; Krueger et al., 1989, J. Agricult. Food Chem., 37, 534-538; Cork & Krueger, 1991, Adv. Appl. Microbiol., 38, 1-66; Cork & Khalil, 1995, Adv. Appi. Microbiol., 40, 289-320. Было выдвинуто предположение, что конкретные гены, участвующие в метаболизме дикамбы, осуществляемом этими бактериями, могут быть выделены и использованы для создания резистентных к дикамбе растений и других организмов. См. цитированное выше и Yang et al., 1994, Anal. Biochem., 219, 37-42. Однако до настоящего изобретения такие гены не были идентифицированы и выделены.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение предоставляет выделенную и по крайней мере частично очищенную дикамба-разрушающую О-деметилазу, выделенную и по крайней мере частично очищенную дикамба-разрушающую оксигеназу, выделенный и по крайней мере частично очищенный дикамба-разрушающий ферредоксин и выделенную и по крайней мере частично очищенную дикамба-разрушающую редуктазу, причем все они определены и описаны ниже.
Также настоящее изобретение предоставляет выделенную молекулу ДНК, включающую последовательность ДНК, кодирующую дикамба-разрушающую оксигеназу, и выделенную молекулу ДНК, включающую последовательность ДНК, кодирующую дикамба-разрушающий ферредоксин и выделенную молекулу ДНК, включающую последовательность ДНК, кодирующую дикамба-разрушающую редуктазу. Также настоящее изобретение предоставляет ДНК-конструкцию, включающую последовательность ДНК, кодирующую дикамба-разрушающую оксигеназу, последовательность ДНК, кодирующую дикамба-разрушающий ферредоксин, или последовательность ДНК, кодирующую дикамба-разрушающую редуктазу, причем каждая кодирующая последовательность ДНК функционально связана с последовательностями, регулирующими экспрессию. В дополнение, изобретение предоставляет ДНК-конструкцию, включающую последовательность ДНК, кодирующую дикамба-разрушающую оксигеназу, последовательность ДНК, кодирующую дикамба-разрушающий ферредоксин, или последовательность ДНК, кодирующую дикамба-разрушающую редуктазу, причем каждая последовательность ДНК функционально связана с последовательностями, регулирующими экспрессию.
Кроме того, изобретение относится к любой из указанных выше ДНК-конструкций, которые также включают последовательность ДНК, кодирующую сигнальный пептид, который направляет дикамба-разрушающий фермент(-ы), в органеллы клеток растения или микроорганизма (хлоропласт и/или митохондрию).
Также изобретение предоставляет трансгенную клетку-хозяин, содержащую ДНК, кодирующую дикамба-разрушающую оксигеназу, ДНК, кодирующую дикамба-разрушающий ферредоксин, или ДНК, кодирующую дикамба-разрушающую редуктазу, причем каждая ДНК функционально связана с последовательностями, регулирующими экспрессию. Кроме того, изобретение предоставляет трансгенную клетку-хозяин, несущую ДНК, кодирующую дикамба-разрушающую оксигеназу, и ДНК, кодирующую дикамба-разрушающий ферредоксин, ДНК, кодирующую дикамба-разрушающую редуктазу, или ДНК, кодирующую дикамба-разрушающий ферредоксин, и ДНК, кодирующую дикамба-разрушающую редуктазу, причем каждая ДНК функционально сзязана с последовательностями, регулирующими экспрессию. ДНК в некоторых вариантах осуществления также может включать последовательность ДНК, кодирующую сигнальный пептид, который направляет дикамба-разрушающий(-е) фермент(-ы) в органеллы клеток растения или микроорганизма (хлоропласт и/или митохондрию). Трансгенная клетка-хозяин может быть растительной клеткой или прокариотическим или эукариотическим микроорганизмом.
Настоящее изобретение также предоставляет трансгенное растение или часть растения, включающую одну или более клеток, содержащих ДНК, кодирующую дикамба-разрушающую окскгеназу, ДНК, кодирующую дикамба-разрушающий ферредоксин, или ДНК, кодирующую дикамба-разрушающую редуктазу, причем каждая ДНК функционально связана с последовательностями, регулирующими экспрессию. Кроме того, изобретение также предоставляет трансгенное растение или часть растения, включающую одну или более клеток, содержащих ДНК, кодирующую дикамба-разрушающую оксигеназу, и ДНК, кодирующую дикамба-разрушающий ферредоксин, ДНК, кодирующую дикамба-разрушающую редуктазу, или ДНК, кодирующую дикамба-разрушающий ферредоксин, и ДНК, кодирующую дикамба-разрушающую редуктазу, причем каждая ДНК функционально связана с последовательностями, регулирующими экспрессию. В некоторых осуществлениях данная ДНК также включает последовательность ДНК, кодирующую сигнальный пептид, который направляет дикамба-разрушающий(-е) фермент(-ы) в органеллы клеток растения. Трансгенное растение или часть растения толерантно к дикамбе или характеризуется усилением имеющейся толерантности к дикамбе в результате экспрессии дикамба-разрушающих ферментов.
Также настоящее изобретение предоставляет способ борьбы с сорняками на плантации трансгенных растений, толерантных к дикамбе. Способ включает нанесение такого количества дикамбы на плантацию, которое будет эффективно с точки зрения борьбы с сорняками.
Также настоящее изобретение представляет способы очистки материала, содержащего дикамбу. В одном из вариантов такой способ включает нанесение эффективного количества трансгенного дикамба-разрушающего микроорганизма на материал. В другом осуществлении способ включает нанесение эффективного количества дикамба-разрушающей О-деметилазы или сочетания дикамба-разрушающей оксигеназы, дикамба-разрушающего ферредоксина и дикамба-разрушающей редуктазы на материал.
Также настоящее изобретение предоставляет способ отбора клеток трансформированного растения и трансформированных растений с использованием толерантности к дикамбе в качестве селективного маркера. В одном из осуществлений способ включает трансформацию по крайней мере нескольких растительных клеток в популяции растительных клеток, так что они становятся толерантными к дикамбе, и выращивание полученной в результате популяции растительных клеток в культуральной среде, содержащей дикамбу в концентрации, выбранной таким образом, чтобы трансформированные растительные клетки росли в ней, а нетрансформированные растительные клетки не росли. В другом варианте осуществления способ включает нанесение дикамбы на популяцию растений, предположительно включающей растение, трансформированное таким образом, что они становятся толерантными к дикамбе, причем дикамбу наносят в таком количестве, чтобы трансформированные растения росли, а рост нетрансформированных растений подавлялся.
Наконец, настоящее изобретение предоставляет способ отбора или скрининга трансформированных клеток-хозяев, целых организмов и частей организмов. Способ включает получение популяции клеток-хозяев, целых организмов или частей организмов, которая предположительно включает клетки-хозяева, целые организмы или части организмов, трансформированные таким образом, что они приобретают способность разрушать дикамбу, контактирование популяции клеток-хозяев, целых организмов или частей организма с дикамбой и оценку наличия или уровня флуоресценции вследствие присутствия 3,6-дихлорсалициловая кислоты. 3,6-Дихлорсалициловая кислота образуется в трансформированных клетках-хозяевах, целых организмах или частях организмов в результате разрушения дикамбы, но не может образовываться в нетрансформированных клетках хозяина, целых организмах или частях организмов.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
Фигура 1. Диаграмма схемы предполагаемого транспорта электронов для дикамбы. Электроны от НАД последовательно переносятся с редуктазыDIC на ферредоксинDIC и затем на оксигеназуDIC. Реакция кислорода с субстратом дикамбы c образованием 3,6-дихлорсалициловой кислоты катализируется оксигеназойDIC. Обозначения: "окис" - окисленный; "восст" - восстановленный.
Фигура 2. Сравнение производной аминокислотной последовательности ферредоксинового компонента дикамбы-O-деметилазы с аминокислотными последовательностями членов адренодоксинового семейства ферредоксинов. На фигуре 2: ferr di6 = ферредоксиновый компонент дикамбы-O-деметилазы Pseudomonas maltophilia DI-6 [SEQ ID NO:5]; fer6 rhoca = ферредоксин Rhodobacter capsulatus [SEQ ID NO:10]; fer caucr = ферредоксин Caulobacter crescentus [SEQ ID NO:11]; thcc rhocr = ферредоксин Rhodococcus erythropolis [SEQ ID NO:12]; putx psepu = ферредоксин Pseudomonas putida [SEQ ID NO:13]; terp psesp = ферредоксин Pseudomonas sp. [SEQ ID NO:14]. Также на фигуре 1 номера 1-3 обозначают три консервативных мотива адренодоксинового семейства бактериальных ферредоксинов.
Фигура 3. Сравнение производной аминокислотной последовательности двух редуктазных компонентов дикамбы-O-деметилазы с аминокислотными последовательностями членов семейства FAD-зависимых пиридиннуклеотидредуктаз. На фигуре 3: red1 di6 = редуктазный компонент дикамбы-O-деметилазы Р. maltophilia DI-6 [SEQ ID NO:7]; red2 di6 = редуктазный компонент дикамбы-O-деметилазы Р. maltophilia DI-6 [SEQ ID NO:9]; AJ002606 = редуктаза Sphingomonas sp. [SEQ ID NO:15]; thcd rhoer = редуктаза R. erythropolis [SEQ ID NO:16]; cama psepu = редуктаза P. putida [SEQ ID NO:17]; tera pscsp = редуктаза Pseudomonas sp. [SEQ ID NO:18]. Также на фигуре 3 номера 1-5 обозначают пять консервативных мотивов FAD-зависимых пиридиннуклеотидредуктаз.
Подробное описание предпочтительных вариантов осуществление изобретения
Предыдущие исследования (Cork & Krueger, Adv. Appi. Microbiol., 36, 1-56 и Yang et al., 1994, Anal Biochem., 219, 37-42) показали, что почвенная бактерия Pseudomonas maltophilia штамма DI-6 способна нарушать гербицидную активность дикамбы в результате одностадийной реакции, в которой дикамба (3,6-дихлор-2-метоксибензойная кислота) превращается в 3,6-дихлорсалициловую кислоту (3,6-DCSA). 3,6-DCSA не обладает гербицидной активностью и для нее не было ранее установлено наличия какого-либо отрицательного действия на растения. Кроме того, 3,6-DCSA легко разрушается нормальной бактериальной флорой, присутствующей в почве.
Описанные здесь эксперименты подтвердили гипотезу Янга с соавт. (Yang et al., цит. выше), согласно которой O-деметилаза контролирует превращение дикамбы с образованием 3,6-DCSA с участием P.maltophilia штамма DI-6, и позволили установить, что О-деметилаза является трехкомпонентной ферментной системой, включающей редуктазу, ферредоксин и оксигеназу. См. примеры 1 и 3, в которых подробно описаны выделение, очистка и характеристика О-деметилазы Р. maltophilia и трех ее компонентов. Схема реакции, катализируемой этими тремя компонентами дикамбы-О-деметилазы, представлена на фигуре 1. Как показано на фигуре 1, электроны от НАД переносятся по короткой электронной цепочке, состоящей из редуктазы и ферредоксина, на конечную оксигеназу, которая катализирует окисление дикамбы с образованием 3,6-DCSA.
В первом осуществлении настоящее изобретение представляет выделенные и по крайней мере частично очищенные дикамба-разрушающие ферменты. Понятие "выделенные" при использовании здесь означает то, что эти ферменты по меньшей мере удалены из клеток, в которых они вырабатываются (т.е. они находятся в составе клеточного лизата). Понятие "по крайней мере частично очищенные" при использовании в данном тексте означает то, что эти ферменты по крайней мере частично были отделены от других компонентов клеточного лизата. Предпочтительно, чтобы ферменты были очищены до такой степени, чтобы полученные ферментные препараты были гомогенными по крайней мере примерно на 70%.
В частности, настоящее изобретение предоставляет выделенную и по крайней мере частично очищенную дикамба-разрушающую О-деметилазу. По использованию в данном тексте "дикамба-разрушающая О-деметилаза" обозначает комбинацию дикамба-разрушающей оксигеназы, дикамба-разрушающего ферредоксина и дикамба-разрушающей редуктазы, все, как определено ниже.
Также настоящее изобретение представляет выделенную и по крайней мере частично очищенную дикамба-разрушающую оксигеназу. При использовании здесь термин "дикамба-разрушающая оксигеназа" обозначает оксигеназу, очищенную из P.maltophilia штамма DI-6, и оксигеназы, аминокислотные последовательности которых гомологичны на уровне по крайней мере примерно 65% (и предпочтительно идентичны), и, более предпочтительно, гомологичны на уровне по крайней мере примерно 75% (и предпочтительно идентичны), и, более предпочтительно, гомологичны на уровне по крайней мере примерно 85% (и предпочтительно идентичны), и, даже более предпочтительно, гомологичны на уровне по крайней мере примерно 95% (и предпочтительно идентичны) по отношению к последовательности оксигеназы P.maltophilia и которые могут участвовать в разрушении дикамбы. "Дикамба-разрушающие оксигеназы" включают мутантные оксигеназы, характеризующиеся аминокислотной последовательностью оксигеназы P.maltophilia, где одна или более аминокислот добавлены, дилетированы или замещены в составе последовательности оксигеназы P.maltophilia. Такие оксигеназы включают ферментативно активные фрагменты оксигеназы, а также оксигеназы, обладающие определенным процентом гомологии или идентичности по отношению к последовательности оксигеназы P.maltophilia. Активность дикамба-разрушающих оксигеказ может быть определена так, как это описано в примерах 1 и 3.
Также настоящее изобретение предоставляет выделенный по крайней мере частично очищенный дикамба-разрушающий ферредоксин. При использовании здесь термин "дикамба-разрушающий ферредоксин" обозначает ферредоксин, очищенный из P.maltophilia штамма DI-6, и ферредоксины, аминокислотные последовательности которых гомологичны на уровне по крайней мере примерно 65% (и предпочтительно идентичны), и, более предпочтительно, гомологичны на уровне по крайней мере примерно 75% (и предпочтительно идентичны), и, более предпочтительно, гомологичны на уровне по крайней мере примерно 85% (и предпочтительно идентичны), и, даже более предпочтительно, гомологичны на уровне по крайней мере примерно 95% (и предпочтительно идентичны) по отношению к ферредоксину Р.maltophilia, которые могут участвовать в разрушении дикамбы. "Дикамба-разрушающие ферредоксины" включают мутантные ферредоксины, характеризующиеся аминокислотной последовательностью ферредоксина P.maltophilia, где одна или более аминокислот добавлены, делетированы или замещены в составе последовательности ферредоксина P.maltophilia. Такие ферредоксины включают ферментативно активные фрагменты ферредоксина, а также ферредоксины, обладающие определенным процентом гомологии или идентичности по отношению к последовательности ферредоксина P.maltophilia. Активность дикамба-разрушающих ферредоксинов может быть определена так, как это описано в примерах 1 и 3.
Наконец, настоящее изобретение предоставляет выделенную и по крайней мере частично очищенную дикамба-разрушающую редуктазу. По использованию в данном тексте термин "дикамба-разрушающая редуктаза" обозначает редуктазу, очищенную из Р.maltophilia штамма DI-6, и редуктазы, аминокислотные последовательности которых гомологичны на уровне по крайней мере примерно 65% (и предпочтительно идентичны), и, более предпочтительно, гомологичны на уровне по крайней мере примерно 75% (и предпочтительно идентичны), и, более предпочтительно, гомологичны на уровне по крайней мере примерно 85% (и предпочтительно идентичны), и, даже более предпочтительно, гомологичны на уровне по крайней мере примерно 95% (и предпочтительно идентичны) по отношению к редуктазе P.maltophilia, которые могут участвовать в разрушении дикамбы. "Дикамба-разрушающие редуктазы" включают мутантные редуктазы, характеризующиеся аминокислотной последовательностью редуктазы P.maltophilia, где одна или более аминокислот добавлены, делетированы или замещены в составе последовательности редуктазы P.maltophilia. Такие редуктазы включают обладающие ферментативной активностью фрагменты редуктазы, а также редуктазы, обладающие определенным процентом гомологии или идентичностью по отношению к последовательности редуктазы P.maltophilia. Активность дикамба-разрушающих редуктаз может быть определена так, как это описано в примерах 1 и 3.
Способы определения степени гомологии аминокислотных последовательностей хорошо известны в данной области. Например, компьютерная программа FASTA из программного пакета Genetics Computing Group (GCG; University of Wisconsin, Madison, Висконсин) может быть использована для сравнение последовательностей из различных белковых баз данных, например, Swiss Protein Database.
Дикамба-разрушающие ферменты по настоящему изобретению могут быть выделены и очищены так, как это описано в примерах 1 и 3, из P.maltophilia или других организмов, которые разрушают дикамбу. Другие подходящие организмы включают другие, помимо P.maltophilia штамма DI-6, бактерии, которые разрушают дикамбу. Некоторые штаммы таких бактерий известны и включают многие почвенные бактерии, а также пресноводные бактерии. См. патент США №5445962; Krueger et al., 1989, J. Agricult. Food Chem., 37, 534-538; Cork & Krueger, 1991, Adv. Appi. Microbiol., 38, 1-66; Cork & Khalil, 1995, Adv. Appi. Microbiol, 40, 289-32C. Дикамба-разрушающие бактерии (т.е. бактерии, которые используют дикамбу в качестве единственного источника углерода) обнаружены во многих родах бактерий, включая в качестве неограничивающих примеров Pseudomonas, Sphingomonas, Aeromonas, Xanthomonas, Alcaligenes и Moraxella. Другие дикамба-разрушающие бактерии, могут быть выделены, как были выделены данные штаммы, способами, хорошо известными в данной области.
Предпочтительно, однако, дикамба-разрушающие ферменты по настоящему изобретению получают с применением технологий рекомбинантной ДНК (см. ниже). В частности, мутантные ферменты, характеризующиеся аминокислотной последовательностью фермента P.maltophilia, где одна или более аминокислот добавлены, делетирована или заменены по отношению к аминокислотной последовательности P.maltophilia, получают данным способом с использованием, например, опосредованного олигонуклеотидами мутагенеза, мутагенеза на основе "сканирования линкером", мутагенеза с использованием полимеразной цепной реакции и тому подобного. См. Ausubel et al. (eds.), "Current Protocols in Molecular Biology", Wiley Interscience, 1990 и McPherson (ed.), "Directed Mutagenesis: A Practical Approach", IRL Press, 1991.
Во втором осуществлении настоящее изобретение представляет выделенные молекулы ДНК, кодирующие дикамба-разрушающие ферменты по изобретению. Термин "выделенные" при использовании здесь означает, что молекула ДНК была изъята из ее естественной среды или же не является встречающейся в природе молекулой ДНК. Способы получения таких молекул ДНК хорошо известны в данной области. См., например, Maniatis et al., Molecular Cloning: A Laboratory Manual", Cold Spring Harbor, NY, 1982; Sambrook et al., "Molecular Cloning: A Laboratory Manual", Cold Spring Harbor, NY, 1989.
Например, молекулы ДНК по настоящему изобретению могут быть выделенными кДНК или геномными клонами и могут также включать молекулы РНК. Идентификация и выделение клонов, кодирующих дикамба-разрушающие ферменты P.maltophilia штамма DI-6, описаны в примерах 2 и 4-5. Дополнительные клоны, кодирующие дикамба-разрушающие ферменты, могут быть получены сходным образом. Выделенные клоны или их части могут быть использованы в качестве зондов, предназначенных для идентификации и выделения дополнительных клонов из тех организмов, которые отличаются от организмов, из которых такие клоны были исходно выделены. Подходящие организмы включают бактерии, которые разрушают дикамбу. Как отмечалось выше, в дополнение к P.maltophilia штамма DI-6 известны некоторые другие бактериальные штаммы, способные разрушать дикамбу. См. патент США №5445962; Kruger et al., 1989, J. Agricult. Food Chem., 37, 534-538; Cork & Krueger, 1991, Adv. Appl. MicrobioL, 38, 1-66; Cork & Khalil, 1995, Adv. Appi. Microbiol., 40, 289-320,
Молекулы ДНК по настоящему изобретению также могут быть химически синтезированы с использованием последовательностей выделенных клонов. Такие способы хорошо известны в данной области. Например, последовательности ДНК могут быть синтезированы с помощью метода фосфоамидитного синтеза с применением автоматического ДНК-синтезатора. Химический синтез имеет ряд достоинств. В частности, химический синтез является предпочтительным, поскольку для оптимизации экспрессии могут быть использованы кодоны, которые являются предпочтительными для хозяина, в котором будет экспрессироваться последовательность ДНК. Улучшение параметров экспрессии не требует изменения всех кодонов, однако предпочтительным является изменение тех кодонов, которые редко используются в геноме организма-хозяина. Высокий уровень экспрессии может быть достигнут путем изменения более чем примерно 50% кодонов, более предпочтительно, по крайней мере, примерно 80% кодонов на кодоны, предпочитаемые организмом-хозяином. Предпочтительность использования кодонов в различных клетках-хозяев известна. См., например, "Maximizing Gene Expression", pp.225-285, Reznikoff & Gold (eds.), 1986, PCT WO 97/31115, PCT WO 97/11086, EP 646643, EP 553494 и патенты США W 5689052, 5567862, 5567600, 5552299 и 5017692. Предпочтительности использования кодонов у других клеток-хозяев могут быть определены способами, известными в данной области. Также, с использованием химического синтеза последовательности молекулы ДНК или кодируемого ею белка могут быть легко изменены, например, с целью оптимизации экспрессии (например, путем удаления тех вторичных структур мРНК, которые могут являться помехами для транскрипции и трансляции), добавления уникальных рестрикционных сайтов по стандартным положениям, удаления сайтов расщепления протеазами, и т.д.
В третьем варианте осуществления настоящее изобретение предоставляет ДНК-конструкции, включающие ДНК, кодирующую дикамба-разрушающий фермент, функционально присоединенную к последовательностям, регулирующим экспрессию. "ДНК-конструкции" определены здесь как конструируемые (не встречающиеся в природе) молекулы ДНК, которые могут использоваться для введения ДНК внутрь клеток-хозяев, и при этом данный термин включает химерные гены, экспрессионные кассеты и векторы.
При использовании здесь термин "функционально присоединенный" означает связывание последовательностей ДНК (включая порядок расположения последовательностей, ориентацию последовательностей и относительный размер отдельных последовательностей) таким образом, чтобы обеспечить экспрессию кодируемых ими белков. Способы функционального присоединения контролирующих экспрессию последовательностей к кодирующим последовательностям хорошо известны в данной области техники. См., например, Maniatis et al., "Molecular Cloning: A Laboratory Manual", Cold Spring Harbor, NY, 1982; Sambrook et al., "Molecular Cloning: A Laboratory Manual", Cold Spring Harbor, NY, 1989.
"Последовательностями, регулирующими экспрессию" являются последовательности ДНК, участвующие в осуществляемой любым путем регуляции транскрипции или трансляции у прокариот и эукариот. Подходящие последовательности, регулирующие экспрессию, и способы их получения и использования хорошо известны в данной области.
Последовательности, регулирующие экспрессию, должны включать промотор. Промотором может быть любая последовательность ДНК, которая проявляет транскрипционную активность в выбранных клетке-хозяине или организме. Промотор может быть индуцибельным или конститутивным. Он может быть встречающимся в природе (нативным), может включать фрагменты различных встречающихся в природе промоторов или может быть частично или полностью синтетическим. Руководство для конструирования промоторов предоставлено на основании исследований структуры промоторов, таких как работа Harley & Reynolds, 1987, Nucl. Acids Res., 15, 2343-2361. Также может быть оптимизировано расположение промотора по отношению к сайту инициации транскрипции: см., например, Roberts et al., 1979, Proc. Natl. Acad. Sci. USA, 76, 760-764. Много промоторов, подходящих для использования у прокариот и эукариот, хорошо известны в данной области.
Например, конститутивные промоторы, подходящие для использования у растений, включают: промоторы растительных вирусов, такие как промотор 35S вируса мозаики цветной капусты (Odell et al., 1985, Nature, 313, 810-812), полноразмерный транскрипционный промотор с дуплицированными энхансерными доменами колимовируса хлоротичной полосатости арахиса (Maiti Shepherd, BBRC 244:440-444 (1998)), промоторы генов метилтрансферазы вируса хлореллы (патент США №5563328); полноразмерный транскрипционный промотор вируса мозаики листьев инжира (патент США №5378619); промоторы таких генов, как ген актина риса (McElroy et al., 1990, Plant Cell, 2, 163-171), ген убиквитина (Christensen et al., 198-9, Plant Mol. Biol., 12, 619-632 и Christensen et al., 1992, Plant Mol. Biol., 18, 675-689), pEMU (Last et al., 1991, Theor. Appl. Genet., 81, 581-588), MAS (Velten et al., 1984, EMBO J., 3, 2723-2730), ген гистона Н3 кукурузы (Lepetit et al., 1992, Mol. Gen. Genet., 231, 276-285; и Atanassova et al., 1992, Plant J., 2 (3), 291-300), ALS3 Brassica napus (заявка PCT WO 97/41228); промоторы различных генов Agrooacterium (см. патенты США №№4771002, 5102796, 5182200, 5428147).
Индуцибельные промоторы, подходящие для использования в растениях, включают: промотор системы АСЕ1, реагирующий на медь (Mett et al., 1993, PNAS, 90, 4567-4571), промотор гена In2 кукурузы, реагирующий на бензолсульфонамидный гербицидный сафенерс (Hershey et al., 1991, Mol. Gen. Genetics, 227, 229-237; и Gatz et al., 1994, Mol. Gen. Genetics, 243, 32-38), и промотор Tet-репрессорной системы Tn10 (Gatz et al., 1991, Mol. Gen. Genet., 227, 229-237). Особенно предпочтительным индуцибельным промотором для использования в растениях является один из тех промоторов, которые реагируют на индуцирующий агент, на который в норме растения не реагируют. Примером индуцибельного промотора такого типа является индуцибельный промотор гена сфероидного гормона, транскрипционная активность которого индуцируется глюкокортикостероидным гормоном (Schena et al., 1991, Proc. Natl. Acad. Sci. USA, 88, 10421). Другие индуцибельные промоторы для использования в растениях описаны в ЕР 332104, РСТ WO 93/21334 и РСТ WO 97/02269.
Промоторы, подходящие для использования в бактериях, включают промотор гена мальтогенной амилазы Bacillus stearothermophilus, гена α-амилазы Bacillus licheniformis, гена BAN-амилазы Bacillus amyloliquefaciens, гена щелочной протеазы Bacillus subtilis, гена ксилозидазы Bacillus pumilis, промоторы PR и PL фага λ и промоторы оперонов lac, trp и tac Escherichia coli. См. РСТ WO 96/23898 и РСТ WO 97/42320.
Промоторы, подходящие для использования в дрожжевых клетках-хозяевах, включают промоторы генов гликолитического пути дрожжей, промоторы генов алкогольдегидрогеназы, промотор ТРI1 и промотор ADH2-4C. См. РСТ WO 96/23898.
Промоторы, подходящие для использования в нитчатых грибах, включают промотор ADH3, промотор tpiA, промоторы генов, кодирующих ТАКА-амилазу Aspergillus oryzae, аспартопротеазу Rhizomucor miehei, нейтральную α-амилазу Aspergillus niger, кислую стабильную α-амилазу А.niger, глюкоамилазу А.niger или Aspergillus awamori, липазу R. miehei, щелочную протеазу А. oryzae, триозофосфатизомеразу A.oryzae и ацетамидазу Aspergillus nidulans. См. РСТ WO 96/23898.
Промоторы, подходящие для использования в клетках млекопитающих, включают промотор SV40, промотор гена металлотионеина, промотор вируса опухоли молочной железы мышей, промотор вируса саркомы Рауша и главный поздний промотор аденовируса - 2. См. РСТ WO 97/42320 и РСТ WO 96/23898.
Промоторы, подходящие для использования в клетках насекомых, включают промотор полиэдрина, промотор Р10, промотор гена основного белка вируса полиэдроза ядер Autographa californica, бакуловирусный промотор предраннего гена-1 и бакуловирусный промотор постраннего гена 39К. См. РСТ WO 96/23898.
Наконец, могут быть использованы промоторы, составленные из частей других промоторов, а также частично или полностью синтетические промоторы. См., например, Ni et al., 1995, Plant J., 7, 661-676 и РСТ WO 95/14098, описывающие такие промоторы, использующиеся в растениях.
Промотор может включать или быть так модифицированным, чтобы включать один или более энхансерных элементов. Предпочтительно промотор включает множество энхансерных элементов. Промоторы, несущие энхансерные элементы, обеспечивают более высокий уровень транскрипции по сравнению с промоторами, в которых таких элементов нет. Энхансерные элементы, подходящие для использования в растениях, включают энхансерный элемент 353 генома вируса мозаики цветной капусты (патенты США №№5106739 и 5164316) и энхансерный элемент вируса мозаики инжира (Maiti et al., 1997, Transgenic Res., 6, 143-156). Известны и другие энхансеры, подходящие для использования в других типах клеток. См. PCT WO 96/23898 и "Enhancers and Eukaryotic Expression" (Cold Spring Harbor Press, Cold Spring Harbor, NY, 1983).
Для обеспечения эффективной экспрессии кодирующие последовательности предпочтительно функционально соединяют также с 3'-нетранслируемой последовательностью. 3'-Нетранслируемая последовательность включает последовательности терминации транскрипции и/или трансляции. 3'-Нетранслируемые участки могут быть выделены из фланкирующих сегментов генов бактериальных, растительных или других эукариотических клеток. Для использования в прокариотах 3'-нетранслируемый сегмент должен включать последовательность терминации транскрипции. Для использования в растениях и других эукариотах 3'-нетранслируемая последовательность должна включать последовательность терминации транскрипции и последовательность полиаденилирования. 3'-Нетранслируемые последовательности, подходящие для использование в растениях, включают последовательности гена 35S вируса мозаики цветной капусты, гена запасающего белка семян фасоли фазеолина, гена малой субъединицы Е9 фермента рибулозо-бисфосфаткарбоксилазы гороха, гена запасающего белка 7S сои, гена октопинсинтазы и гена нопалинсинтазы.
В растениях и других эукариотах также используется 5'-нетранслируемая последовательность. 5'-Нетранслируемая последовательность является частью молекулы мРНК, которая простирается от 5'-"кэп"-сайта до кодона инициации трансляции. Этот участок мРНК необходим для инициации трансляции у эукариот и играет роль в регуляции генной экспрессии. Подходящие для использования в растениях 5'-нетранслируемые сегменты включают последовательности вируса мозаики люцерны, гена капсидного белка вируса мозаики огурца и вируса мозаики табака.
Как отмечалось выше, ДНК-конструкция может представлять собой вектор. Вектор может включать одну или более реплицирующихся систем, которые обеспечивают его репликацию в клетках-хозяевах. Самореплицирующиеся векторы включают плазмиды, космиды и вирусные векторы. С другой стороны, вектор может являться интегрирующим вектором, который обеспечивает интегрирование в хромосому клетки-хозяина последовательности, кодирующей дикамба-разрушающий фермент по настоящему изобретению. Также желательно, чтобы вектор включал уникальные рестрикционные сайты для встраивания последовательностей ДНК. Если вектор не имеет уникальных рестрикционных сайтов, то он может быть модифицирован с целью введения или удаления рестрикционных сайтов так, чтобы сделать его более пригодным для дальнейших манипуляций.
ДНК-конструкции по настоящему изобретению могут быть использованы для трансформации различных клеток-хозяев (см. ниже). Генетический маркер должен быть использован для отбора трансформированных клеток-хозяев ("селективный маркер"). Селективные маркеры обычно позволяют получать трансформированные клетки путем негативной селекции (т.е. происходит подавление роста тех клеток, которые не несут селективный маркер) или путем скрининга продукта, кодируемого селективным маркером.
Наиболее часто используемым в трансформации растений геном, являющимся селективным маркером, является ген неомицинфосфотрансферазы-II (nptII), выделенный из Тп5, который, в случае помещения его под контроль растительных регуляторных сигналов, обеспечивает резистентность к канамицину (Fraley et. al., 1983, Proc. Natl. Acad. Sci. USA, 80, 4803). Другим обычно используемым в качестве селективного маркера геном является ген гигромицинфосфотрансферазы, который обеспечивает резистентность к антибиотику гигромицину (Vanden Elzen et al., 1985, Plant Mol. Biol., 5, 299).
Другие рассматриваемые в качестве селективных маркеров гены бактериального происхождения, которые обусловливают резистентность к антибиотикам, включают ген гентамицинацетилтрансферазы, ген стрептомицинфосфотрансферазы, ген аминогликозид-3'-аденилтрансферазы и ген резистентности блеомицину (Hayford et al., 1988, Plant Physiol, 86, 1216, 1988; Jones et al., 1987, Mol. Gen. Genet., 210, 86; Svab et al., 1990, Plant Mol. BioL, 14, 197; Hille et al., 1986, Plant Mol. BioL, 7, 171). Другие являющиеся селективными маркерами гены обеспечивают резистентность к гербицидам, таким как глифозат, глюфозинат или бромксинил (Comai et al., 1985, Nature, 317, 741-744; Gordon-Kamm et al., 1990, Plant Cell, 2, 603-618; и Stalker et al., 1988, Science, 242, 419-423).
Другие используемые в качестве селективных маркеров гены, пригодные для трансформации растений, имеют небактериальное происхождение. Эти гены включают, например, ген дигидрофолатредуктазы мыши, ген растительной 5-енолпирувилшикимат-3-фосфатсинтазы и растительной ацетолактатсинтазы (Eichholtz et al., 1987, Somatic Cell Mol. Genet., 13, 67; Shah et al., 1986, Science, 233, 478; Charest et al., 1990, Plant Cell Rep., 8, 643).
Гены, обычно используемые для скрининга предположительно трансформированных клеток, включают ген β-глюкуронидазы (GUS), β-галактозидазы, люциферазы и хлорамфениколацетилтрансферазы (Jefferson R.A., 1987, Plant Mol. Biol. Rep., 5, 387; Teeri. et al., 1959, EMBO J., 8, 343; Koncz et al., 1987, Proc. Natl. Acad. Sci. USA, 84, 131; De Block et al., 1984, EMBO J., 3, 1681), ген зеленого флуоресцентного белка (GFP) (Chalfie et al., 1994, Science, 263, 802; Haseloff et al., 1995, TIG, 11, 328-329 и заявка РСТ WO 97/41228). Другой подход к идентификации относительно редких случаев трансформации основан на использовании гена, который кодирует доминантный конститутивный регулятор метаболического пути антоциановой пигментации у Zea mays (Ludwig et al., 1990, Science, 247, 449).
Также хорошо известны селективные маркеры, подходящие для использования в прокариотах и эукариотах, помимо растений. См., например, РСТ WO 96/23898 и РСТ WO 97/42320. Например, в качестве селективного маркера может быть использована резистентность к антибиотикам (ампициллину, какамицину, тетрациклину, хлорамфениколу, неомицину или гигромицину).
В соответствии с другим аспектом настоящего изобретения, толерантность к дикамбе может быть использована в качестве селективного маркера для растений и растительных клеток. Понятие "толерантности" означает, что трансформированные растительные клетки способны расти (выживать и регенерировать в растения) тогда, когда их помещают в культуральную среду, содержащую такое количество дикамбы, которое предотвращает рост нетрансформированных клеток. Также понятие "толерантности" означает, что трансформированные растения способны расти после нанесения на них такого количества дикамбы, которое подавляет рост нетрансформированных растений.
Способы отбора трансформированных растительных клеток хорошо известны в данной области. В кратком изложении, по крайней мере некоторые растительные клетки в популяции растительных клеток (например, эксплантата или зародышевой суспензионной культуры) трансформируют ДНК-конструкцией или комбинацией ДНК-конструкций для обеспечения разрушения дикамбы. Полученную в результате популяцию растительных клеток помещают в культуральную среду, содержащую дикамбу в концентрации, выбранную таким образом, чтобы трансформированные растительные клетки могли расти, а нетрансформированные растительные клетки нет. Подходящие концентрации дикамбы могут быть определены эмпирически способами, известными в данной области.
Способы отбора трансформированных растений также известны в данной области. В кратком изложении, дикамбу наносят на популяцию растений, которые предположительно несут ДНК-конструкцию или комбинацию ДНК-конструкций, обеспечивающую разрушение дикамбы. Количество дикамбы выбирают таким образом, чтобы трансформированные растения могли расти, а рост нетрансформировакных растений подавлялся. Степень подавления должна быть достаточной для того, чтобы трансформированные и нетрансформированные растения могли быть легко различимы (т.е. подавление должно быть статистически значимым). Такие количества могут быть определены эмпирически способами, известными в данной области.
Далее, образование 3,6-DCSA в результате разрушения дикамбы может быть использовано для проведения отбора и скрининга. Образование 3,6-DCSA может быть легко выявлено путем наблюдения флуоресценции этого соединения, что позволяет осуществлять отбор и скрининг трансформированных клеток-хозяев, целых организмов и частей организмов (например, микроорганизмов, растений, частей растений и растительных клеток). В этом отношении настоящее изобретение обеспечивает проведение отбора и скрининга трансформированных клеток-хозяев, целых организмов и частей организмов тем же путем, что и в случае с зеленым флуоресцентным белком (GFP). См. патенты США №№5162227 и 5491084 и заявки РСТ WO 96/27675, WO 97/11094, WO 97/41228 и WO 97/42320: все они включены сюда в качестве ссылки. В частности, 3,6-DCSA может быть выявлено в трансформированных клетках-хозяевах, целых организмах частях организмов с использованием стандартных спектрофотокетрических методов. Например, микроскопы могут быть оснащены подходящими наборами фильтров, необходимых для возбуждения и детекции флуоресценции. Для работы в полевых условиях можно использовать лампы-переноски (см. пример 1). Может быть также применена сортировка активированных по флуоресценции клеток. Возбуждение 3,6-DCSA происходит при длинах волн 312-313 нм при максимальной эмиссионной длине волны 424 нм.
"Части" организмов включают органы, ткани или любые другие части. "Части растений" включают семена, пыльцу, зародыши, цветки, плоды, побеги, листья, корни, стебли, эксплантаты и т.п.
Отбор, основанный на толерантности к дикамбе или разрушении дикамбы, может быть использован для получения толератных к дикамбе растений или дикамба-разрушающих микроорганизмов, причем в этом случае использование другого селективного маркера может не быть необходимым. Отбор, основанный на толерантности дикамбе или разрушении дикамбы, также может быть использован для получения трансгенных клеток или организмов, которые экспрессируют другие представляющие интерес гены. Многие из таких генов известны и включают гены, кодирующие белки, имеющие коммерческое значение, и гены, которые обуславливают улучшение агрономических признаков растений (см., например, РСТ WO 97/41228, полное описание которой включено в данный текст в качестве ссылки).
ДНК-конструкции по настоящему изобретению могут быть использованы для трансформации различных клеток-хозяев, включая прокариоты и эукариоты. Последовательности ДНК, кодирующие дикамба-разрушающий(-ие) фермент(-ы) и селективный маркер, если используется отдельный селективный маркер, могут входить в состав одной и той же или разных ДНК-конструкций. Предпочтительно, чтобы они были расположены в одной ДНК-конструкции в общей транскрипционной единице так, чтобы все кодирующие последовательности экспрессировались вместе. Также представляющий интерес ген(-ы) и последовательности ДНК, кодирующие разрушающий(-ие) дикамбу фермент(-ы), в случае когда толерантность к дикамбе или разрушение дикамбы используется в качестве селективного признака, могут находиться в составе одной и той же или разных ДНК-конструкций. Такие конструкции получают тем же образом, что и описанные выше конструкции.
Подходящие клетки-хозяева включают прокариотические и эукариотические микроорганизмы (например, бактерии (включая Agrobacterium tumefaciens и Eschenchia coli), дрожжи (включая Saccharomyces cerevisiae) и другие грибы (включая Aspergillus sp.), растительные клетки, клетки насекомых и клетки млекопитающих. Предпочтительно клеткой-хозяином должна быть такая клетка, которая в норме не способна разрушать дикамбу. Однако настоящее изобретение также может быть использовано для увеличения уровня разрушения дикамбы в клетках-хозяевах, которые в норме разрушают дикамбу. Таким образом, "трансгенные" клетки организмы по изобретению включают клетки и организмы, которые в норме не разрушают дикамбу, но которые были трансформированы в соответствии с настоящим изобретением так, чтобы они приобрели способность разрушать данный гербицид. "Трансгенные" клетки и организмы по настоящему изобретению также включают клетки организмы, которые в норме разрушают дикамбу, но которые были трансформированы в соответствии с настоящим изобретением так, чтобы они приобрели способность разрушать данный гербицид в больших количествах или разрушать данный гербицид с большей эффективностью.
Способы трансформации прокариотических и эукариотических клеток-хозяев хорошо известны в данной области. См., например, Maniatis et al., "Molecular Cloning: A Laboratory Manual", Cold Spring Harbor, NY, 1982; Sambrook et al., "Molecular Cloning: A Laboratory Manual", Cold Spring Harbor, NY, 1989; PCT WO 96/23898 и PCT WO 97/42320.
Например, были разработаны многочисленные способы трансформации растений, включая протоколы трансформации биологическими и физическими путями. См., например, Miki et al., 1993, "Procedures for introducing foreign DNA into Plants", in "Methods in Plant Molecular Biology & Biotechnology", B.R.Glick & J.E.Thompson (Eds.), CRC Press Inc., Boca Raton, pp.1993, 67-88. Кроме того, известны векторы и способы культивирования in vitro для растительных клеток или трансформации тканей и регенерации растений. См., например, Gruber et al., "Vectors for Plant Transformation", In "Methods in Plant Molecular Biology & Biotechnology", B.R.Glick & J.E.Thompson (Eds.), CRC Press Inc., Boca Raton, pp.1993, 89-119. В примере 6 представлены конкретные примеры трансформации в соответствии с настоящим изобретением.
Наиболее широко распространенный метод введения экспрессирующего вектора в растения основан на естественной трансформационной системе Agrobacterium. См., например, Horsch et al., 1985, Science, 227, 1229. Виды A.tumefaciens и A.rhizogenes являются патогенными почвенными бактериями, которые генетически трансформируют растительные клетки. Плазмиды Ti и Ri из A.tumefaciens и A.rhizogenes, соответственно, несут гены, ответственные за генетическую трансформацию растения. См., например, Kado C.J., 1991, Crit. Rev. Plant. Sci., 10, 1. Описания векторных систем Agrobacterium и методы опосредованного Agrobacterium переноса генов представлены многочисленными публикациями, включая Gruber et al. (цит. выше), Miki et al. (цит. выше), Moloney et al., 1989, Plant Cell Rep., 8, 238 и патенты США №№4940838 и 5464763.
Основным применяющимся способом трансформации растений является трансформация, основанная на бомбардировке микрочастицами, при том, что ДНК находится на поверхности этих микрочастиц. Экспрессирующий вектор вносится в растительные ткани с помощью биобаллистического устройства, которое обеспечивает ускорение микрочастиц до скоростей, достаточных для проникновения через клеточные стенки и мембраны. Sanford et al., 1987, Part. Sci. Technol., 5, 27; Sanford J.C., 1988, Trends Biotechnol., 6, 299; Sanford J.C., 1990, Physiol. Plant, 79, 206; Klein et al., 1992, Biotechnology, 10, 268.
Другим способом физической доставки ДНК в растения явлется обработка клеток-мишеней ультразвуком. Zhang et al., 1991, Biotechnology, 9, 996. Альтернативно, слияние липосом и сферопластов может быть использовано для введения экспрессирующих векторов в растения. Deshayes et al., 1985, ЕМВС J., 4, 2731; Christou et al., 1987, Proc. Natl. Acad. Sci. USA, 84, 3962. Также имеются сообщения о прямом введении ДНК в протопласты с использованием преципитации CaCl2, обработки поливиниловым спиртом или поли-L-орнитином. См. Hain et al., 1985, Mol. Gen. Genet., 199, 161; и Draper et al., 1982, Plant Cell Physiol, 23, 451. Также была описана электропорация протопластов и целых клеток и тканей. Donn et al., 1990, In "Abstr. VII Intern. Congr. Plant Cell и Tissue Culture IAPTC, A2-38, p.53; D'Halluin et al., 1992, Plant Cell, 4, 1495-1505; и Spencer et al., 1994, Plant Mol. Biol., 24, 51-61.
В соответствии с настоящим изобретением могут быть получены трансгенные толерантные к дикамбе растения любого типа. В частности, широколистные растения (включая бобы, сою, хлопчатник, горох, картофель, подсолнечник, помидор, табак, фруктовые деревья и декоративные растения и деревья), которые, как хорошо известно, поражаются дикамбой, могут быть трансформированы так, чтобы они стали толерантными к гербициду. Другие растения (такие, как кукуруза, сорго, хлебные злаки, сахарный тростник, спаржа и травы), которые обычно являются толерантными к дикамбе, могут быть трансформированы так, чтобы стать более толерантными к этому гербициду. В частности, двудольное и однодольное растения можно трансформировать конструкцией ДНК по настоящему изобретению. "Толерантность" означает то, что трансформированные растения могут расти в присутствии такого количества дикамбы, которое подавляет рост нетрансформированных растений.
Предполагается, что дикамба-разрушающие оксигеназы по настоящему изобретению могут функционировать в разрушении дикамбы с участием эндогенных редуктаз и ферредоксинов, обнаруживаемых в трансгенных клетках хозяина и организмах. Особенно богатыми редуктазами и ферредоксинами являются хлоропласты растений. Соответственно, предпочтительный вариант осуществления изобретения получения трансгенных толерантных к дикамбе растений основано на использовании последовательности, кодирующей пептид (например, сигнальный пептид), который обеспечит доставку дикамба-разрушающей оксигеназы в хлоропласты ("последовательность, направленная на хлоропласты"). ДНК, кодирующую последовательность, направленную на хлоропласты, предпочтительно располагают выше (в 5'-направлении) от последовательности, кодирующей дикамба-разрушающую охсигеназу, однако она может располагаться и ниже (в 3'-направлении) кодирующей последовательности, или одновременно выше и ниже от кодирующей последовательности. Может использоваться любая последовательность, направленная на хлоропласты. Примеры последовательностей, направленных на хлоропласты, включают сигнальную последовательность cab-m7 кукурузы (см. Backer et al., 1992, Plant Mol. Biol., 20, 49; и РСТ WO 97/41228), сигнальную последовательность глутатионредуктазы гороха (Creissen et al., 1991, Plant J., 2, 129; и РСТ WO 97/41228) и сигнальную последовательность малой субъединицы рибулозо-1,5-бисфосфаткарбоксилазы гороха (нуклеиновая кислота, представленная SEQ ID NO:19) (Fluhr et al, EMBO J., 5, 2063-2071, (1986)). Альтернативным предпочтительным вариантом осуществления является прямая трансформация хлоропластов использованием конструкции, включающей промотор, функционирующий в хлоропластах, для получения экспрессии оксигеназы в хлоропластах. См., например, заявку WO 95/24492 и патент США №5545818. Конечно, если выбранная трансгенная клетка-хозяин или организм не продуцирует достаточно эндогенной редуктазы, ферредоксина или их обоих, клетка-хозяин или организм может трансформироваться так, чтобы они продуцировали один из данных ферментов или оба, а также оксигеназу.
Еще в одном варианте осуществления настоящее изобретение представляет способ борьбы с сорняками на плантации, где выращиваются трансгенные толерантные к дикамбе растения. Способ включает нанесение эффективного количества дикамбы на плантацию для борьбы с сорняками. Способы нанесения дикамбы и определение эффективного количества дикамбы для борьбы с различными видами сорняков известны. См. Crop Protection References (Chemical & Pharmaceutical Press Inc., New York, KY, llth ed, 1555, pp.1503-1621).
В другом осуществлении настоящее изобретение предоставляет способ разрушения дикамбы, присутствующей в материале, таком, как почва, вода или отходы предприятия по производству дикамбы. Такое разрушение может быть осуществлено с применением дикамба-разрушающих ферментов по настоящему изобретению. Ферменты могут быть очищены из микроорганизмов, которые их экспрессируют в природе (см. примеры 1 и 3), или могут быть очищены из вырабатывающих их трансренных клеток-хозяев. Если в таких способах используются ферменты, то также могут быть предоставлены подходящие кофакторы (см. пример 1). Эффективные количества могут быть определены эмпирически, что хорошо известно в данной области (см. пример 1). Альтернативно, трансгенные прокариотические и эукариотические микроорганизмы могут быть использованы для разрушения дикамбы в таких материалах. Трансгенные прокариотические и эукариотические микроорганизмы могут быть продуцированы так, как это описано выше, а эффективные количества могут быть определены эмпирически так, как это известно в данной области.
Дикамба постоянно попадает в окружающую среду в больших количествах. Удаление дикамбы в значительной степени зависит от действия ферментных систем, которые обнаруживаются в микроорганизмах, обитающих в почвах и водах планеты. Расшифровка этих ферментных систем, включая дикамба-разрушающие О-деметилазы и их три компонента, является важной составляющей для использования естественных и генетически модифицированных микроорганизмов для целей биологического восстановления и восстановления загрязненных почв, вод и других материалов. Таким образом, дикамба-разрушающие ферменты, молекулы ДНК, ДНК-конструкции и т.д. по настоящему изобретению могут быть использованы в качестве исследовательских инструментов для изучения разрушения дикамбы и биологического восстановления.
Наконец, дикамба-разрушающие ферменты по настоящему изобретению могут быть использованы в тесте на выявление дикамбы. Образец, предположительно содержащий дикамбу, смешивают с дикамба-разрушающей О-деметилазой или с комбинацией дикамба-разрушающей оксигеназы, дикамба-разрушающего ферредоксина и дикамба-разрушающей редуктазы. Подходящие тесты описаны в примерах 1 и 3. В частности, пригодным для стандартного теста является выявление или количественный анализ флуоресценции, связанной с образованием 3,6-DCSA.
ПРИМЕРЫ
ПРИМЕР 1: Очистка и характеристика компонентов дикамба-О-деметилазы Pseudomonas maltophilia штамма DI-6
Методы и материалы
Бактерии и условия выращивания. Pseudomonas maltophilia, штамм DI-6 (Kreuger J.P., et al., 1989, J. Agricult. Food Chem., 37, 534-538) были выделены из участка почвы, постоянно подвергавшегося загрязнению дикамбой. Бактерии были предоставлены д-ром Douglas Cork из Illinois Institute of Technology (Чикаго, Иллинойс) и поддерживались в восстановленной хлоридной среде (Kreuger J.P., 1989, Ph. D.thesis, Illinois Inst. Technol., Чикаго, Иллинойс) с добавлением либо дикамбы (2 мг/мл), либо смеси глюкозы (2 мг/мл) и казаминовых кислот (2 мг/мл). Источники углерода были стерилизованы фильтрованием и добавлены в культуральную среду после ее автоклавирования. Твердые среды получали путем добавления 1% (масс./об.) Gelrite (Scott Laboratories, West Warwick, Род-Айленд).
Химикаты и реагенты. Дикамба, 3,6-DCSA и [14С]-дикамба (U-фенил-14С: 42,4 мКи/моль, радиохимическая чистота более 98%, были поставлены Sandoz Agro Inc. (Des Plaines, Иллинойс). Для повышения растворимости маточные растворы дикамбы и 3,6-DCSA получали путем титрования КОН до рН 7,0. Все химикаты были приобретены в Sigma Chemical Co. (St. Louis, Миссури), помимо специально оговоренных. Superose-12, Mono-Q, Q-сефароза (быстрый поток) и фенилсефароза (CL-4B) для забивки колонок в аппарате FPLC (жидкостная хроматография быстрого разделения) были получены от Pharmacia (Milwaukee, Висконсин). Амфолиты с рН 4-6 и амфолиты с рН 4-9 были приобретены у Serva (Heidelberg, Германия). Акриламид, β-меркаптоэтанол, N,N,N',N'-тетраметилэтилендиамин (TEMED) и персульфат аммония (APS) получены от Bio-Rad Laboratories (Hercules, Калифорния). Пластины для тонкослойной хроматографии (TLC) были изготовлены из силикагеля (толщиной 250 мкм) с индикатором УФ-254 и приобретены у J.Т.Baker Chem. Co. (Phillipsburg, Нью-Джерси).
Тесты на активность ферментов. Активность дикамба-O-деметилазы оценивали путем измерения уровня образования [14С]-3,6-DCSA из [14С]-дикамбы. В кратком изложении, активность смесей компонентов фермента определяли при 30°С в стандартной реакционной смеси в объеме 300 мкл, включающей 25 мМ калий-фосфатного буфера (рН=7,0), 10 мМ MgCl2, 0,5 мМ NADH (восстановленная форма бета-никотинамидадениндинуклеотида), 0,5 мМ сульфата железа (II), 50 мкМ холодной дикамбы, 2,5 мкМ [14С]-дикамбы (конечная специфическая активность меченой дикамбы составляла 1/9 мКи/ммоль) и различное количество клеточного лизата или частично очищенного фермента. Все тесты на ферментную активность в своих конечных этапах проводили в фосфатном буфере, поскольку было установлено, что оптимальная величина рН для дикамба-О-деметилазы находится в средней части диапазона, характерного для фосфатных буферов, и что более высокая активность этого фермента наблюдается именно в фосфатном буфере в сравнении с буфером Tns-HCl [Трис (гидроксиметил)аминометангидрохлорид] при рН=7,0. Реакции инициировали добавлением соответствующего субстрата, дикамбы. В определенное время реакции останавливали добавлением 50 мкл 5%-ного (об./об.) раствора серной кислоты. Затем экстракцию дикамбы и метаболитов дикамбы осуществляли дважды одним объемом простого эфира и экстракты выпаривали досуха. Эффективность выделения (средняя величина ± стандартное отклонение) для процедуры экстракции составила 87±2% для дикамбы и 85±3% для 3,6-DCSA (Yang et al., 1994, Anal. Biochem., 219, 37-42).
[14С]-дикамбу и меченные 14С метаболиты дикамбы разделяли с помощью тонкослойной хроматографии (TLC). Экстрагированную эфиром дикамбу и ее метаболиты вновь растворяли в 50 мкл эфира с последующим раскапыванием на пластинку для TLC. Для проведения TLC использовали систему растворителей, представленную смесью хлороформ-этанол-уксусная кислоты при их объемном соотношению 85:10:5. Полученные при повторном растворении продукты визуализировали и оценивали количественно путем помещение пластины для TLC на фосфорный экран на 24 часа с последующим сканированием полученной на экране картины с помощью Phosphorlmager SF (Molecular Dynamics, Sunnyvale, Калифорния). Оценки уровня радиоактивности в конкретном пятне на пластине TLC определяли путем сравнения общего числа пикселей данного пятна по сравнению с пятном, расположенным на той же пластине и характеризующимся известным количеством [14С]-дикамбы. Единицу активности определяли, как такое количество фермента, которое катализирует образование 1 нмоль 3,6-DCSA из дикамбы за минуту при 30°С. Специфические активности определяли по отношению к общей концентрации белка в тестируемой смеси.
Активность редуктазного компонента дикамба-деметилазы оценивали путем измерения уровня восстановления 2,6-дихлорфенолиндофенола (DCIP) с использованием спектрофотометра Hitachi U-2000. Реакционная смесь включала 0,5 мМ NADH, 0,2 мМ FAD (флавинадениндинуклеотид), 50 мкМ DCIP, 20 мМ буфера Tris (рН 8,0) и 10-100 мкл образца фермента при общем объеме 1 мл. Изменение поглощения во времени при 600 нм определяли при комнатной температуре. Специфическую активность рассчитывали с использованием коэффициента экстинкции при 600 нм, равного 21,0 мМ-1см-1 для восстановленного DCIP. Специфическую активность выражали в нмоль восстановленного DCIP в минуту на 1 мг белка.
Кроме того, тест с DCIP in situ использовали для выявления и картирования редуктазной активности в белковых препаратах, разделенных в гелях для изоэлектрического фокусирования (IEF). После электрофореза белков в IEF-геле вырезанные из геля дорожки промывали в 20 мл холодного 20 мМ буфера Tris-HCl (pH=8,0). Низкоплавкую агарозу растворяли с нагреванием в 10 мл 20 мМ буфера Tris-HCl (pH=8,0) при конечной концентрации 1,5% (мас./об.). Когда агароза охлаждалась примерно до комнатной температуры, к ней добавляли 0,2 мМ FAD, 50 мкМ DCIP и 0,5 мМ NADH. Полученную для тестирования смесь переносили на стеклянную пластину и оставляли до застывания. Пластинку геля помещали на верхушку застывшей реакционной смеси и оставляли при комнатной температуре на 15 минут. Если пластинка геля содержит белок, обладающий редуктазной активностью, то образуются бесцветные полосы восстановленного DCIP при сохранении голубого фона исходного DCIP.
Клеточные лизаты. Клетки выращивали до оптической плотности 1,3-1,5 при 550 нм в жидкой редуцированной хлоридной среде, включающей смесь глюкозы и казаминовых кислот, с использованием роторного шейкера (250 об./мин при 30°С). Клетки собирали центрифугированием, дважды промывали холодным раствором 100 мМ хлорида магния и снова центрифугировали. Клеточные центрифугаты либо сразу использовали, либо быстро замораживали на жидком азоте и хранили при -80°С. Во время проведения очистки фермента 25 г замороженных клеток растаивали и ресуспендировали в 50 мл буфера для выделения, включающего 25 мМ буфера Tris (pH 7,0), 10 мМ MgCl2 и 0,5 мМ EDTA. Фенилметилсульфонилфторид и дитиотреит добавляли до конечных концентраций 0,5 мМ и 1 мМ, соответственно. После добавления 10 мг лизоцима и 1 мг ДНКазы клетки перемешивали в течение 10 минут на льду и разрушали ультразвуком (ультразвуковая установка XL2020; Heat Systems) на льду в среднем установочном положении прибора (положение 5) при 20-секундных импульсах и 40-секундных промежутках без обработки. Полученные в результате клеточные лизаты разводили в 90 мл буфера выделения и центрифугировали при 76000×g в течение 1 часа при 4°С. Надосадочную фракцию использовали в качестве источника осветленного клеточного лизата.
Очистка ферментов. Помимо специально оговоренных случаев, все процедуры были выполнены при 4°С. Твердый сульфат аммония медленно добавляли в объем, равный 90 мл, осветленного клеточного лизата до достижения 40%-ного насыщения (масс./об.) при постоянном перемешивании. После 15 минут перемешивания смеси центрифугировали при 15400×g в течение 15 минут и удаляли образовавшийся осадок. Вновь добавляли твердый сульфат аммония до достижения 70%-ного насыщения (масс./об.) при постоянном перемешивании надосадочной фракции. После 15 минут перемешивания смеси центрифугировали в тех же условиях, что были описаны выше. Надосадсчную фракцию удаляли, а осадок ресуспендировали в минимальном объеме буфера А (20 мМ Tris, pH 8,0; 2,5 мМ MgCl2, 0,5 мМ EDTA, 5% (об./об.) глицерина и 1 мМ дитиотреита).
Фракцию сульфата аммония 40-70% затем загружали в фенил-сефарозную колонку (2,5×10 см), связанную с аппаратом для FPLC (Pharmacia), и элюировали в нисходящем линейном градиенте (NH4)2SO4 от 10% (мас./об.) до 0% (мас./об.). Колонку предварительно уравновешивали буфером-А, включающим 10% (мас./об.) сульфата аммония. Скорость потока составила 1 мл/мин. Концентрация белка непрерывано контролировали при A280 в ходе элюции колонки. Колонку промывали 120 мл буфера-А, включающего 10% (мас./об.) сульфата аммония, до достижения исходной линии считывания A280. Связанные белки элюировали нисходящим градиентом (NH4)2SO4 в буфере А [от 10 до 0% сульфата аммония (мас./об.) при общем объеме 210 мл]. Собирали фракции объемом 2 мл. Аликвоты по 10 мкл были отобраны из каждой фракции и добавлены к стандартней смеси, изготовленной для тестирования дикамба-О-деметилазы так, как указано выше, за исключением того, что в качестве субстрата использовали нерадиоактивную дикамбу. Активность дикамба-О-деметилазы определяли путем контроля превращения дикамбы в продукт 3,6-DCSA с интенсивным уровнем флуоресцентной активности, с помощью переносной ультрафиолетовой лампы (312 нм, Fotodyne) в темной комнате.
Данная процедура позволила разделить дикамба-О-деметилазу на три пула, включающих отдельные компоненты (обозначенные как компоненты I, II и III). Каждый из этих компонентов был необходим для проявления активности дикамба-О-деметилазы (см. ниже). Когда проводилось тестирование отдельного компонента, то другие два компонента включались в избытке. Были отобраны фракции, включающие единственный тип активности (компонент I, фракции 128-145; компонент II, несвязавшиеся фракции 12-33; компонент III, фракции 62-92).
(i) Очистка компонента I. Фракции, включающие активность компонента I (элюированные из фенил-сефарозной колонки при 0 М (NH4)2SO4, фракции 128-145), объединяли так, чтобы их общий объем составил 34 мл. Объединенные образцы концентрировали до 10 мл центрифугированием с использованием прибора Centriprep-10 (Amicon) и затем загружали в Q-сефарозную колонку (Fast Flow) для FPLC (Pharmscia) (2,5×6 см), уравновешенную буфером А, и промывали 80 мл буфера А. Белки, связанные с колонкой, элюировали 100 мл линейного градиента от 0 до 0,6 М KCl в буфере А при скорости потока 1 мл/мин. Фракции отбирали с интервалом в 1,5 минуты. Те фракции, которые проявляли активность компонента I (фракции 29-37), отбирали, подвергали диализу против буфера А в течение ночи при 4°С и загружали в анионообменную колонку Mono-Q HR-5/5 для FPLC в буфере А. Белки элюировали при скорости 1 мл/мин с использованием 50 мл градиента с увеличением концентрации KCl (от 0 до 0,5 М). Фракции, проявлявшие активность компонента I (фракции 19-25), объединяли и концентрировали до 0,4 мл центрифугированием в приборе Centricon-10. Затем концентрированный образец подвергали хроматографическому анализу в колонке с суперозой-12 для FPLC (1,6×50 см) при скорости потока 0,2 мл/мин с буфером А, включающим 100 мМ KCl. Фракции 7-10, проявлявшие активность компонента I, объединяли и концентрировали центрифугированием с использованием прибора Centncon-10.
Частично очищенный компонент I растворяли с холодным 1%-ным глицином (мас./об.) и концентрировали центрифугированием в приборе Centricon-10 еще трижды с целью обессоливания для анализа с помощью гель-электрофореза на основе изоэлектрического фокусирования (IEF). Обессоленный концентрированный образец затем загружали в 6%-ный (мас./об.) гель для IEF (рН 4-6) и подвергали электрофорезу в течение 1,5 часов при 4°С (см. ниже). После проведения электрофореза гель промывали холодным 25 мН фосфатным буфером (рН 7,0) в течение 5 минут, и затем каждый отрезок дорожки геля нарезали маленькими кусочками (6×4 мм). Белок элюировали из нарезанных фрагментов геля путем измельчения их кончиком пипетки в присутствии 10 мкл 25 мМ фосфатного буфера (рН 7,0). Белок из каждого сегмента смешивали с избытком компонентов II и III и тестировали на активность дикамба-О-деметилазы. Гелевый сегмент, который проявляет активность компонента I (который также имел красновато-коричневое окрашивание), загружали на 12,5%-ный (мас./об.) полиакриламидный гель с додецилсульфатом натрия (SDS-PAGE) для проверки чистоты образца.
(ii) Очистка компонента II. Компонент II, выделенный из хроматографической фенил-сефарозной колонки, подвергали диализу против буфера А в течение ночи при 4°С и загружали в Q-сефарозную колонку для FPLC (2,5×6 см). Условия элюции образца были идентичны тем условиям, которые были описаны выше для компонента I, за исключением того, что градиент элюции представлял собой 0-1 М KCl в буфере А. Фракции, проявлявшие активность компонента II (фракции 30-37), объединяли, диализовали против буфера А, концентрировали до 0,4 мл и загружали в колонку с суперозой-12 для FPLC (1,6×50 см). Процедуры загрузки и элюции образцов были идентичны тем, которые были описаны выше для компонента I. Фракции, проявлявшие активность компонента II (фракции 3-6), объединяли, растворяли разным объемом буфера А и загружали в колонку Mono-Q для FPLC. Белки элюировали из колонки с использованием того же градиента хлористого калия, который использовался для компонента I. Фракции 20-25 проявили активность компонента II. Частично очищенный компонент II далее был подвергнут очистке с помощью IEF (рН 4-6) с использованием тех же условий, которые были описаны для компонента I. Вырезанные сегменты геля, которые проявляли активность компонента II, загружали на 12,5%-ный (мас./об.) SDS-PAGE для дальнейшего анализа.
(iii) Очистка компонента III. Компонент III, выделенный из хроматографической фенил-сефарозной, колонки, подвергали диализу против буфера А в течение ночи при 4°С и загружали в Q-сефарозную колонку для FPLC (2,5×6 см). Условия были идентичными тем, которые были описаны выше для компонента I. Фракции, проявлявшие активность компонента III (фракции 26-38), подвергали диализу против буфера В [10 мМ Tris-HCl (pH 7,5), 2,5 мМ MgCl2, 5% (об./об.) глицерина, 1 мМ дитиотреита] и концентрировали до 5 мл. Окрашенный в голубой цвет аффинный матрикс [Cibacron Blue 3GA 3000 (Sigma)] упаковывали в колонку для FPLC (1×5 см) и предварительно уравновешивали 20 мл буфера В. Концентрированный компонент III загружали в окрашенную голубой краской колонку и промывали 20 мл буфера В при скорости потока 0,2 мл/мин до тех пор, когда будет достигнут уровень базовой линии колонки А280. Затем связанный белок элюировали с использованием 5 мМ NADH в буфере В. Фракции, обладающие редуктазной активностью, были выявлены путем тестирования активности дикамба-О-деметилазы в присутствии избытка компонентов I и II, а также путем оценки способности каждой фракции восстанавливать DCIP в присутствии NADH. Фракции, характеризующиеся сильной редуктазной активностью в обоих тестах, объединяли и диализовали против буфера А, включающего 100 мМ KCl, концентрировали до 0,2 мл и загружали в колонку с суперозой-12 для FPLC. Для прохождения суперозной колонки были применены те же условия, которые были описаны для компонента I. Фракции, включающие белки, которые катализируют восстановление DCIP, объединяли, диализовали против буфера-А и загружали в колонку Mono-Q для FPLC. Белки элюировали с использованием тех же условий, которые были описаны для компонента I. Частично очищенный компонент III далее очищали с использованием IEF-гель-электрофореза (рН 4-6). Редуктазную активность белков в составе IEF-геля выявляли путем тестирования по восстановлению DCIP в агарозном геле, полученном так же, как это было описано выше. Гелевый сегмент, который проявлял активность компонента II, загружали в 12,5%-ный (мас./об.) SDS-PAGE для дальнейшего анализа.
Определение NH2-концевых аминокислотных последовательностей. Белковые полосы вырезали из IEF-гелей и помещали в лунки 12,5%-ного (мас./об.) SDS-полиакриламидного геля. После проведения электрофореза вырезанные пластинки геля, содержащие очищенные белки, подвергали трансблоттингу на PVDF-(поливинилидендифторидную) мембрану (Millipore) в транс-блот-камеру (Bio-Rad, Richmond, Калифорния) при 25 В на 16 часов. В качестве буфера блоттинга использовали раствор 20% (об./об.) метанола с 10 мМ CAPS [3-(циклогексиламино)-1-пропансульфоновая кислота], рН 10,0. Секвенирование осуществляли с использованием аппарата Applied Biosystems Inc., модель 420-Н по методу расщепления Эдмана (Edman & Henschen, 1975, in "Protein Sequence Determination", S.B.Needleman (ed.), 2d ed., Springer-Verlage, New York).
Определение концентрации белка. Концентрации белков были определены с помощью метода Брэдфорда (Bradford, 1976, Anal. Biochem., 72, 248-254) с использованием сывороточного бычьего альбумина в качестве стандарта.
SDS-PAGE. Электрофорез в полиакриламидном геле с додецилсульфатом натрия (SDS-PAGE) был проведен в соответствии с модифицированным методом Лэммли (Laemmli, 1970, Nature, 227, 680-685). Пластины 12,5%-ного SDS-геля размером 85×65×0,75 мм получали следующим образом: основной разделяющий гель - 2,5 мл 40%-ного (мас./об.) акриламид/бис (37:5:1), 1 мл раствора проточного буфера [3М Tris-HCl (рН 8,8), 0,8% SDS (мас./об.)], 4,5 мл воды, 5 мкл TEMED и 40 мкл 10% (мас./об.) APS; концентрирующий гель - 0,5 мл 40%-ного (мас./об.) раствора акриламида/бис, 0/5 мл раствора концентрирующего буфера [1 М Tns-HCl (рН 6,8), 0,8% SDS (мас./об.)], 3 мл воды, 5 мкл TEMED и 12,5 мкл 10% (мас./об.) APS. В состав проточного буфера входят 25 мМ Tris-HCl (рН 8,3), 0,2 М глицина и 0,1% (мас./об.) SDS. Буфер для образца включал 0,25 мл концентрирующего буфера, 0,6 мл 20% (мас./об.) SDS, 0,2 мл β-меркаптоэтанола и 0,95 мл 0,1%-ного бромфенолового синего (мас./об.) в 50%-ном (об./об.) глицерине. Электрофорез проводили при напряжении 80 В в устройстве BioRad Mini-Gel до тех пор, пока след красителя не достигал участка в 0,5 см от анодного края геля. Белки окрашивали 0,1%-ным (мас./об.) кумасси бриллиантовым голубым R-250 в смеси изопропанола, воды, уксусной кислоты в объемной соотношении 3:6:1. Отмывку от красителя проводили смесью метанола, воды и уксусной кислоты в объемном соотношении 7:83:10. В качестве стандартов использовали следующие белки (Gibco BRL): миозин (214,2 кДа), фосфорилазу В (111,4 кДа), бычий сывороточный альбумин (74,25 кДа), овальбумин (45,5 кДа), карбоангидразу (29,5 кДа), β-лактоглобулин (18/3 кДа) и лизоцим (15,4 кДа).
Определение молекулярной массы. Молекулярную массу (Mr) пептидов, помещаемых в денатурирующие условия, определяли с использованием анализа SDS-PAGE. Молекулярные. массы нативных компонентов I, II и III оценивали с помощью гель-фильтрации через колонку с суперозой-12 HR-10/30 для FPLC (Pharmacia), скорость потока 0,2 мл/мин, в буфере А, включающем 100 мМ KCl. В качестве белков для проведения гель-фильтрации использовали следующие стандарты производства фирмы BioRad: бычий тиреоглобулин (670 кДа), бычий γ-глобулин (158 кДа), куриный овальбумин (44 кДа), миоглобин лошади (17 кДа) и витамин В12 (1,35 кДа). Свободный объем хроматографической колонки с суперозой-12 определяли с использованием голубого декстрана (молекулярная масса 2000000, Sigma).
IEF. Гель-электрофорез на основе изоэлектрического фокусирования (IEF) проводили в вертикальном аппарате для минигелей (модель № MGV-100) производства C.B.S. Scientific Co. (Del Mar, Калифорния). Полиакриламидный 6%-ный (мас./об.) гель для IEF размером 70×90×1 мм получали путем смешивания следующих компонентов: 1,6 мл 30% (мас./об.) акриламида/бис (37:5:1), 0,8 г глицерина, 0,32 мл амфолита рН 4-6 (Serva), 0,08 мл амфолита pH 4-9 (Serva), 5,2 мл воды, 10 мкл TEMED и 80 мкл 10% (мас./сб.) APS. Катодным буфером являлся 100 мМ β-аланин, а анодный буфер представлял собой 100 мМ уксусную кислоту. Образцы белка приблизительно в 1-10 мкл 1%-ного глицина (мас./об.) смешивали с равным количеством буфера для образца [50% (об./об.) глицерина, 1,6% (об./об.) амфолита рН 4-9, 2,4% (об./об.) амфолита рН 4-6]. Образцы загружали на катодный конец геля и позволяли им мигрировать при напряжении 200 В в течение 1,5 часов и при 400 В в течение следующих 1,5 часов. Белки окрашивали кумасси бриллиантовым голубым R-250 с применением процедуры, описанной выше для SDS-полиакриламидных гелей. Были использованы следующие маркеры (Sigma) для IEF: амилогликозидаза (pI 3,6), глюкозооксидаза (pI 4,2), ингибитор трипсина (pI 4,6), β-лактоглобулин А (pI 5,1), карбоангидраза II (pI 5,4), карбоангидраза II (pI 5,9) и карбоангидраза I (pI 6,6).
Кинетический анализ. Кинетику реакции деметилирования, катализируемой дикамба-O-деметилазой, анализировали с помощью определения исходных скоростей реакции в присутствии постоянной концентрации фермента и увеличивающейся концентрации субстрата, которым является дикамба. Реакционные смеси включали 25 мМ калий-фосфатного буфера (рН 7,0), 10 мМ MgCl2, 0,5 мМ NADH, 0,5 мМ FeSO4, 25 мкг частично очищенной О-деметилазы [фракция 40-70% (NH4)2SO4 (мас./об.), выделенная из очищенного клеточного лизата], различные концентрации дикамбы (0,5-50 мкМ) и различные концентрации (0,025-2,5 мкМ) меченой [14С]-дикамбы (U-фенил-14С, 42,4 мКи/ммоль) при общем объеме 300 мкл. Для проведения теста в диапазоне концентрации дикамбы от 0,5 мкМ до 1 мкМ объем реакционной смеси увеличивали до 900 мкл для того, чтобы гарантировать присутствие количеств меченой дикамбы и ее метаболитов, достаточных для их выявления. В этих реакционных смесях количество всех остальных компонентов реакции утраивали. Превращение [14С]-дикамбы в [14С]-3,6-DCSA определяли в различные моменты времени для каждой концентрации дикамбы с использованием PhosphorImager SF, считывающего уровень радиоактивности фосфорных экранов, которые соприкасали с хроматографическими пластинами TLC в течение 24 часов. Единицу активности определяли как количество фермента, которое образует 1 нмоль 3,6-DCSA за минуту при 30°С. Исходные скорости каждой из реакций определяли путем построения графика зависимости скоростей реакций от времени для каждой концентрации субстрата. Данные анализировали по параметрам кинетики Михаэлиса-Ментена, а величины Km и Vmax определяли путем построения кривой по графику Лайнуивера-Берка с использованием SigmaPlot® (Jandel Scientific, Corte Madera, Калифорния).
Потребность в кислороде. Предварительные эксперименты с использованием кислородного электрода Кларка продемонстрировали потребление кислорода в ходе стандартного теста по активности дикамба-О-деметилазы при использовании дикамбы в качестве субстрата. Для проверки потребности в кислороде в реакции О-деметилирования дикамбы с участием дикамба-О-деметилазы реакции проводили в анаэробной камере, содержание кислорода в которой составляет менее 1 м.д. Процедуры замещения кислорода в реакционной смеси осуществляли при 4°С. Лишенные фермента реакционные смеси помещали в сосуд и закрывали резиновой крышкой. Для замещения кислорода из сосуда дважды откачивали газ вакуумом и каждый раз заполняли его азотом. После третьего откачивания сосуд заполняли смесью 90% азота и 10% водорода. Раствор фермента также был освобожден от кислорода (следя за тем, чтобы раствор фермента не закипал). И реакционные смеси, и растворы фермента переносили в анаэробную камеру (атмосфера которого состояла из 95% азота и 5% водорода). 240 мкл очищенного клеточного лизата инъецировали через резиновую пробку с помощью микрошприца и осторожно смешивали с 960 мкл бескислородной реакционной смеси. Реакции проводили при 30°с.
Оценка продуктов реакции на пластинах TLC показала, что скорость образования [14С]-3,6-DCSA из [14С]-дикамбы в анаэробных условиях оказалась существенно меньшей, чем скорость реакций с тем же количеством фермента, проводившихся в аэробных условиях. В анаэробных условиях превращение дикамбы в 3,6-DCSA в течение 1 часа практически не происходило. Однако, когда в параллельном эксперименте реакционную смесь брали из анаэробной камеры через 30 минут и инкубировали в воздухе, то существенное количество одного из компонентов дикамба-О-деметилазного каталитического комплекса оказалось представлено оксигеназой.
Следует заметить, что превращение [14С]-дикамбы в [14C]-3,6-DCSA in vitro имитирует in vivo превращение, описанное ранее (Cork & Kreuger, 1991, Adv. Appl. MicrobioL, 36, 1-66; Yang et al., 1994, Anal. Biochem., 219, 37-42). В данных исследованиях DCSA идентифицировали как продукт реакции и при TLC, и при капиллярном электрофорезе. Точная идентификация первого основного компонента разрушения дикамбы как DCSA и in vivo, и in vitro была установлена путем анализа, включающего газовую хроматографию с масс-спектрометрической детекцией.
Требования к компонентам и кофакторам. После исходного разделения трех компонентов дикамба-О-деметилазы путем хроматографии в фенил-сефарозных колонках частично очищенные препараты были взяты индивидуально благодаря дополнительной очистке, осуществлявшейся в Q-сефарозной колонке (2,5×6 см). Образцы загружали на Q-сефарозу (быстрый поток) в колонке для скоростной жидкостной хроматографии белков (Pharmacia) в буфере А и элюировали 100 мл линейного градиента от 0 до 0,6 М KCl (для оксигеназного компонента) или от 0 до 1,0 М KCl (для ферредоксинового и редуктазного компонентов) в фракциях объемом 1,5 мл. Подходящие объединенные фракции из отдельных колонок для очистки оксигеназы (фракции 29-37), для очистки ферредоксина (фракции 30-37) или для очистки редуктазы (фракции 26-38) использовали для определения требований к компонентам и кофакторам.
Три компонента тестировали на О-деметилазную активность в различных сочетаниях с целью определения потребности в разных компонентах.
Для определения потребности в кофакторах О-деметилазную активность определяли с использованием смеси трех компонентов с [14С]дикамбой в течение 30 минут при 30°С. Количества частично очищенного белка (представленные в оптимизированном отношении) в реакционных смесях составили 85 мкг оксигеназы, 55 мкг ферредоксина и 50 мкг редуктазы. Концентрация кофакторов, использовавшихся в реакционных смесях, составляли 0,5 мМ NADH, 0,2 мМ FAD, 3,5 мМ FeSO3, 10 мМ MgCl2, 0,5 мМ NADPH и 0,2 мМ FMN.
РЕЗУЛЬТАТЫ
Компонент I. Компонент дикамба-O-деметилазы, который наиболее тесно связывается с фенил-сефарозной колонкой (исходно обозначенный как компонент I и, соответственно, определенный как оксигеназа), характеризовался отчетливым красновато-коричневым цветом. Это соответствует вероятному присутствию белка(-ов), имеющему железосерный(-ые) кластер(-ы) или группу(-ы) гема. Фракции из фенил-сефарозной колонки, характеризующиеся активностью компонента I, подвергали дальнейшей очистке с помощью хроматографии в Q-сефарозе (быстрый поток) и хромотографии на Mono-Q, и затем разделяли на колонке для гель-фильтрации с суперозой-12. Затем белок, идентифицированный как компонент I, был дополнительно очищен в геле для IEF.
Белок из основной полосы IEF-геля (соответствующей величине pI на уровне приблизительно 4,6) вырезали и отделяли от любых остальных минорных загрязнителей с помощью электрофореза в SDS-PAGE. Белок, соответствующий основному компоненту I, полученный после очистки с помощью IEF, проявил более чем 90%-ную чистоту, что подтверждается при денситометрическом анализе данного SDS-полиакриламидного геля, окрашенного кумасси голубым.
Определяли N-концевую аминокислотную последовательность основного белка, характеризующегося молекулярной массой около 40000 Да. Результаты аминокислотного секвенирования показали, что первые 29 аминокислот N-концевого сегмента присутствуют в следующей последовательности (аминокислотные остатки, взятые в скобки, является наиболее вероятными):
Thr Phe Val Arg Asn Ala Trp Tyr Val Ala Ala Leu Pro Glu Glu Leu Ser Glu Lys Pro Leu Gly Arg Thr Ile Leu Asp (Asp or Thr) (Pro)
[SEQ ID NO:1].
Сравнение с аминокислотными последовательностями из различных баз данных, показало незначительную гомологию или вообще отсутствие гомологии с NH2-концевыми последовательностями, приведенными для других монооксигеназ или диоксигеназ.
Компонент II. Белковая фракция, которая не связывалась с фенил-сефарозной колонкой, была обозначена как компонент II. Поскольку эта фракция, характеризующася желтоватой окраской, может быть замещена ферредоксином из Clostridium pasteurianum (но с меньшими скоростями реакции) при проведении тестов в комбинации с компонентами I и III, ее обозначали в эксперименте как ферредоксин-содержащую фракцию. Ферредоксин Clostridium явно функционировал вместо компонента II, но, с учетом очень высокой степени неочищенности естественного компонента II, использованного в этих экспериментах, эффективность фермента Clostridium была существенно ниже, чем эффективность предполагаемого ферредоксина штамма DI-6. В частности, 55 мкг частично очищенного компонента II, смешанного с избыточными количествами компонентов I и III, катализировали превращение дикамбы в 3,6-DCSA со скоростью приблизительно 5 нмоль в минуту на 1 мг, в то время как 100 мкг высокоочищенного ферредоксина Clostridium обусловливает активность на уровне лишь 0,6 нмоль мин-1 мг-1.
Этапы очистки, связанные с применением Q-сефарозной хроматографии (быстрого потока), гель-фильтрации с использованием суперозы-12 и хроматографии в Mono-Q, приводили к получению приблизительно 1 мг очищенного белка из исходных 25 г клеточной массы. Данную фракцию очищали способом, сходным со способом очистки оксигеназного компонента, с помощью электрофореза в IEF-геле с последующим электрофорезом активной IEF-фракции в SDS-полиакриламидном геле.
Анализ активности компонента II в белках, элюированных из сегментов IEF-геля, показал, что фракция, характеризующаяся изоэлектрической точкой приблизительно 3,0, содержит активный белок, соответствующий компоненту II. Белок из этого вырезанного геля элюировали и подвергали электрофорезу в SCS-PAGE. Окрашивание геля с использованием кумасси голубого показало наличие одной отчетливой белковой полосы (молекулярная масса приблизительно 28000 Да) наряду с размазыванием низкомолекулярных белков.
Компонент III. Компонент III дикамба-О-деметилазы задерживался на фенил-сефарозной колонке при высокой концентрации и его элюировали приблизительно при 4% (NH4)2SO4 (мас./об.). Данную светло-желтую фракцию экспериментально определяли как содержащую редуктазу фракцию, основываясь на ее способности восстанавливать окисленный цитохром-с и DCIP в присутствии NADH, а также благодаря тому, что она может быть замещена редуктазой цитохрома-с из сердца свиньи (тип I, Sigma) в тестах, в которых анализировали компоненты I и II. В данном наборе реакций использование 50 мкг частично очищенного компонента III обусловливало скорость реакции на уровне приблизительно 5 нмоль мин-1 мг-1, в случае смешивания с избытком компонентов I и II. Высокоочищенная редуктаза цитохрома-с показала специфическую активность на уровне приблизительно 2,5 нмоль мин-1 мг-1, т.е. активность, отчетливо уступающую таковой для компонента III, причем даже с учетом недостаточной чистоты сырого компонента III, использовавшегося в этих тестах. Также компонент III проявлял редуктазную активность в случае инкубирования его с цитохромом-с или с 2,6-дихлорфенолиндофенолом (DCIP) в присутствии NADH. В обоих этих тестах на редуктазную активность ни компонент I, ни компонент II активности не проявлял.
Дополнительная очистка данной фракции с помощью хроматографии на колонках, содержащих Q-сефарозу (быстрый поток), окрашенный голубым аффинный матрикс, набивки суперозы-12 и Mono-Q, приводила к получению малого количества белка во фракциях, проявляющих редуктазную активность. Белок-компонент III характеризовался чистотой на уровне примерно 70%, что подтверждалось денситометрическим анализом активного белка фракции после разделения путем SDS-PAGE и окрашивания кумасси голубым.
В связи с дальнейшим усилением очистки компонента III было обнаружено, что две различные белковые фракции, выделенные на колонке Mono Q, включали редуктазную активность, выявляемую при тестировании с добавлением ферредоксинового и оксигеназного компонентов. Дальнейшая очистка этих двух фракций с помощью электрофореза в IEF-геле показала, что редуктазные активности этих двух фракций характеризуются отчетливо различающимися изоэлектрическими точками (pI). Это было продемонстрировано путем вырезания дорожек, содержащих каждую из двух редуктазных фракций, из IEF-геля и путем помещения срезов в верхнюю часть подушечки из легкоплавкой агарозы, содержащей реакционную смесь DCIP. Редуктазная активность обоих гелевых срезов была идентифицирована с помощью NADH-зависимого восстановления DCIP до его бесцветной, восстановленной формы. Редуктаза в составе фракции 35 характеризовалась значением pI, приблизительно равным 5,6, в то время как редуктаза в составе фракции 27 обладала значением pI, приблизительно равным 4,8.
Обе редуктазные активности, выделенные из срезов IEF-геля, были нестабильными и присутствовали в очень небольшом количестве. Действительно, только редуктаза из состава фракции 35, выделенной из колонки Mono-Q, сохраняла достаточную концентрацию белка и активность, позволяющие проводить дальнейшие очистку и характеристику. Срез IEF-геля, содержащий редуктазную активность, элюировали и отделяли от загрязняющих белков путем SDS-PAGE. Доминирующим белковым компонентом этого геля оказался белок, характеризующийся молекулярной массой приблизительно 45000 Да. Путем гель-фильтрации была выявлена молекулярная масса для компонента III, приблизительно равная 50 кДа в его нативном состоянии.
Биохимическая характеристика дикамба-О-деметилазы. Активность дикамба-О-деметилазы измеряли в ходе инкубации in vitro в температурном диапазоне от 20°С до 50°С и в диапазоне значений рН от 6 до 9. Отчетливый пик активности наблюдался при 30°С, а более размытый пик активности был выявлен в диапазоне рН 6,5-7,5. Каталитическая активность оказалась зависимой от используемого для поддержания рН буфера. При рН 7, например, активность была приблизительно на 40% меньше в буферах на основе Tris в сравнении с фосфатсодержащими буферами.
Значения Km и Vmax для дикамба-O-деметилазы были определены с использованием SigmaPlot® с целью создания кривых лучшего соответствия, обобщающих данные графиков Михаэлиса-Ментена и Лайнуивера-Берка по экспериментам, проведенным в двух параллелях. Величина константы Михаэлиса Кm для дикамбы была оценена приблизительно в 9,9±3,9 мкМ, а величина Vmax для реакции была оценена приблизительно в 108±12 нмоль/мин/мг.
Три компонента тестировали по их дикамба-О-деметилазной активности в различных сочетаниях. Ни один из этих компонентов не проявил ожидаемой каталитической активности при их тестировании по отдельности. Действительно, существенный уровень дикамба-О-деметилазной активности мог обнаруживаться только тогда, когда все три компонента комбинировали друг с другом. Смесь компонентов I и II проявляла очень слабый уровень каталитической активности, что может объясняться наличием следовых количеств компонента III во фракции компонента I.
И NADH, и NADPH поддерживают каталитическую активность, при том, что NADH оказывается существенно более эффективным в сравнении с NADPH. Mg2+ является необходимым для проявления каталитической активности. Fe2+, флавинадениндинуклеотид (FAD) и флавинмононуклеотид (FMN) либо проявляли слабый стимулирующий эффект в отношении каталитической активности, либо не проявляли его вовсе при анализе частично очищенных белковых препаратов в описанных экспериментах. Наивысший уровень активности был выявлен при использовании сочетания NADH, Fe2+, Mg2+ и FAD.
Обсуждение
Дикамба-O-деметилаза Pseudomonas maltophilia штамма DI-6 является трехкомпонентной оксигеназой (Wang X.-Z., 1996, Ph. D. thesis, Univ. Nebrasca-Lincoln, Lincoln, Небраска), ответственной за превращение гербицида дикамбы (2-метокси-3,6-дихлорбензойной кислоты) в 3,6-дихлорсалициловую кислоту (3,6-DCSA: 2-гидрокси-3,6-дихлорбензойная кислота). Были разработаны такие схемы очистки, которые бы позволили выделить каждый из трех компонентов до гомогенного или до почти гомогенного состояния.
Исходное разделение трех компонентов было достигнуто хроматографией на фенил-сефарозной колонке. Каталитические активности и другие характеристики частично очищенных компонентов позволили осуществить точную идентификацию компонентов как редуктазу, ферредоксин и оксигеназу - состав, сходный с таковым, который ранее был обнаружен в ряде других исследованных гемсодержащих и лишенных гема мультикомпонентных оксигеназ (Batie et al., 1992, In "Chemistry & Biochemistry of Flavoenzymes", F.Muller (ed.). Vol.Ill, CRC Press, Boca Raton, pp.543-565; Harayama et al., 1992, Annu. Rev. Microbiol., 46, 565-601; Mason & Cammack, 1992, Annu. Rev. Microbiol, 46, 277-305; Rosche et al., 1-995, Biochem. Biophys. Acta, 1252, 177-179). Компонент III, выделенный из фенил-сефарозной колонки, катализировал NADH-зависимое восстановление цитохрома-с и красителя DCIP. Кроме того, его способность обеспечивать превращение дикамбы в 3,6-DCSA в случае комбинирования с компонентами I и II может быть отчасти замещена цитохром-с редуктазой. Компонент II может быть замещен добавлением ферредоксина Clostridium pasteurianum в реакционных смесях, содержащих компоненты I и III. Абсолютная необходимость в присутствии молекулярного кислорода для поддержания реакции О-деметилирования показала, что оставшимся компонентом являлась оксигеназа.
Оксигеназаdic. Компонент I дикамба-О-деметилазы (обозначенный как оксигеназаdic) очищали до гомогенного состояния и подвергали N-концевому аминокислотному секвенированию. Полученная в результате последовательность из 29 аминокислотных остатков не позволила выявить сколько-нибудь существенной гомологии с аминокислотными последовательностями других белков, присутствующих в различных банках данных. Однако информация, полученная в анализе этой аминокислотной последовательности, позволяет конструировать вырожденные олигонуклеотидные зонды, которые могут быть успешно использованы для выявления и клонирования гена, кодирующего компонент I (см. пример 2). Более того, сравнение аминокислотной последовательности, расшифрованной по нуклеотидной последовательности в составе такого клона, с аминокислотными последовательностями белка в базе данных показал наличие существенной гомологии с другими оксигеназами (см. пример 2).
Вероятная молекулярная масса оксигеназыdic, оцененная с помощью электрофореза в SDS-полиакриламидном геле, составила приблизительно 40000 Да. Очищенные оксигеназные препараты проявляли только одну основную полосу в SDS-полиакриламидном геле, окрашенном кумасси голубым, а расщепление по Эдману белка, находящегося в составе этой полосы, показало присутствие только одной N-концевой формы. Оценки, полученные в анализе поведения нативной оксигеназыdic с помощью гель-фильтрации, подтвердили наличие молекулярного размера на уровне приблизительно 90000 Да. Все эти данные подтверждают, что нативная оксигеназа существует в виде гомодимера.
Оксигеназно-гидроксилазный компонент ряда мультикомпонентных систем составлен комплексом субъединиц (αβ)n-типа, в котором большая α-субъединица имеет молекулярную массу приблизительно 50000 Да, а меньшая β-субъединица имеет молекулярную массу приблизительно 20 Да (Harayama et al., 1992, Annu. Rev. Microbiol., 46, 565-601). С другой стороны, оказалось, что оксигеназный компонент дикамба-О-деметилазы включает единственную субъединицу, имеющую молекулярную массу приблизительно 40 кДа, которая в нативной форме может существовать в виде димера. Этот комплекс субъединиц (α)n-типа сходен с таким комплексом, который ранее был хорошо изучен и охарактеризован у других оксигеназ, таких как 4-хлорфенилацетат-3,4-диоксигеназа Pseudomonas sp. штамма CBS (Markus et al., 1986, J. Biol. Chem., 261, 12883-12888), фталатдиоксигеназы Pseudomonas cepacia (Batie et al., 1987, J. Biol. Chem., 262, 1510-1518), 4-сульфобензоат-3,4-диоксигеназы Comamonas testosteroni (Locher et al., 1991, Biochem. J., 274, 833-842), 2-оксо-1,2-дигидрохинолин-8-монооксигеназы Pseudomonas putida 86 (Rosche et al., 1995, Biochem. Biophys. Acta, 1252, 177-179), диоксигеназы 4-карбоксифенилового простого эфира Pseudomonas pseudoalcaligenes (Dehmel et al., 1995, Arch. Microbiol., 163, 35-41) и 3-хлорбензоат-3,4-диоксигеназы Pseudomonas putida (Nakatsu et al., 1995, Microbiology [Reading], 141, 485-495).
Ферредоксинdic. Компонент II дикамба-О-деметилазы (обозначенный как ферредоксинdic) очищали до практически полной гомогенности путем колоночной хроматографии и изоэлектрического фокусирования, проводившихся в несколько этапов. Окончательная очистка с помощью SDS-PAGE позволила получить единственную белковую полосу (Mr приблизительно 28000 Да) и размытый след белков чуть меньшей массы.
Определяли N-концевую последовательность белка с очевидной молекулярной массой, составляющей примерно 28000 Да. Наличие данной аминокислотной последовательности позволило получить вырожденные олигонуклеотидные зонды, и данные зонды применяли для выделения геномного клона, кодирующего данный белок. Хотя белок, продуцируемый данным клоном, представлял собой ферредоксин (ферредоксин28кДа), он, как впоследствии было определено, не был активным в разрушении дикамбы при сочетании с двумя другими компонентами дикамбы-О-деметилазы (данные не показаны).
Другое доказательство поддерживает вывод, что ферредоксин28кДа не является ферредоксиновым компонентом дикамбы-О-деметилазы. Во-первых, молекулярная масса данного белка (28 кДа) существенно выше, чем величины, характерные для ферредоксинов в составе других мультикомпонентных оксигеназ бактерий (т.е. 8-13 кДа) (Batie et al., 1992, In "Chemistry & Biochemistry of Flavoenzymes", F.Muller (ed.). Vol.Ill, CRC Press, Boca Raton, pp.543-565; Harayama et al., 1992, Annu. Rev. Microbiol, 46, 565-601).
Во вторых, сравнение N-концевой последовательности, включающей 20 аминокислотных остатков, полученной из ферредоксина28кДа, с другими аминокислотными последовательностями, входящими в состав различных баз данных белков, с использованием пакета программ Genetics Computer Group (GCG) (University of Wisconsin, Madison, Висконсин), показало наличие высокого уровня гомологии (80-85% идентичности по сравнению с более вероятной N-концевой последовательностью ферредоксина28кДа) с рядом дикластерных бактериальных ферредоксинов (таких, как Pseudomonas stutzeri, Pseudomonas putida, Rhodobacter capsulatus и Azotobacter vinelandii). Четыре дикластерных ферредоксина, которые проявляют высокий уровень гомологии с ферредоксином28кДа включают кластер [3Fe-4S], за которым расположен кластер [4Fe-4S], на N-конце белка. Это подтверждает, что ферредоксин28кДа четко отличается от ферредоксиновых компонентов, включающих кластер [2Fe-2S], который обычно связан с негемовыми мультикомпонентными оксигеназами (Harayama et al., 1992, Annu. Rev. Microbiol., 46, 565-601; Mason & Cammack, 1992, Annu. Rev. Microbiol., 46, 277-305; Rosche et al., 1995, Biochem. Biophys. Acta, 1252, 177-179).
РедуктазаDIC. Компонент III дикамба-О-деметилазы (обозначенный как редуктазаDIC) является наименее поддающимся очистке среди трех компонентов. Это обусловлено отчасти тем, что этот компонент является нестабильным и содержится в малом количестве в лизатах клеток штамма DI-6. Несмотря на это, белок был очищен с определением точной молекулярной массы, составившей 45000 Да. Эта величина сходна с молекулярной массой приблизительно 50000 Да, полученной при тестировании с помощью гель-фильтрации, и подтверждает, что редуктазаDIC существует в своей нативной форме в виде мономера. Очистка редуктазного компонента также затруднена из-за того, что хроматография с применением колонок Mono Q и IEF разделяют очищенные препараты редуктазы на две активности, характеризующиеся отчетливо различающимися величинами pI. Обе фракции, выделенные с колонки Mono Q, в сочетании с очищенными ферредоксиномDIC и оксигеназойDIC фунциональны в обеспечение дикамба-O-деметилазной активности. Присутствие у Sphingomonas sp. штамма RW1 двух сходных флавопротеинов, которые равно функционируют как редуктазные компоненты в составе трехкомпонентной дибензофуран-4,4а-диоксигеназы, было недавно сообщено Бюнцом и Куком (Bunz & Cook, 1993, J. Bacteriol., 175, 6467-6475). Интересно, что обе редуктазы характеризуются молекулярной массой по 44000 Да, что довольно сходно с молекулярной массой редуктазыDIC равной 45000 Да. Множественные компоненты леггемоглобинредуктазы также были выявлены в корневых клубеньках люпина при использовании метода изоэлектрического фокусирования (Topunov, et al. (1982) Biokhimiya (English edition) 162:378-379). В этом случае IEF показало наличие четырех раздельных компонентов, проявляющих NADH-зависимую редуктазную активность. Решение проблемы, связанной с тем, имеется ли только одна редуктазаDIC, которая существует в двух формах, или у штамма DI-6 имеются две редуктазы, позволит разработать улучшенные процедуры выделения увеличенного количества белков и/или клонировать и секвенировать вовлеченный(-ые) ген(-ы) (см. примеры 3 и 5).
Характеристики дикамбы-О-деметилазы. В дополнение к физическим и биохимическим свойствам отдельных компонентов, обозначенных выше, анализ каталитических активностей показал, что О-деметилазная система характеризуется высоким уровнем аффинности (Km˜10 мкМ) в отношении ее субстрата, а величина Vmax составляет приблизительно 100-110 нмоль/мин/мг. Как и ожидается для почвенной бактерии, обитающей в умеренной климатической зоне, максимальная каталитическая активность была выявлена при температуре около 30°С. В то время как оптимумы рН для ферментной системы находились в диапазоне рН 6,5-7,5, активность, измеренная для данного ферментного препарата, в значительной степени зависела от типа использовавшейся для поддержания рН буферной системы. Активность в присутствии Tris-буферов была по крайней мере на 40% ниже, чем в фосфатных буферах, при сравнении при одинаковых величинах рН.
Схема реакции, катализируемой тремя компонентами дикамба-О-деметилазы, показана на фиг.1. Электроны от NADH переносятся по короткой электронной цепочке, включающей редуктазу и ферредоксин, к конечной оксигеназе, которая и катализирует окисление дикамбы. Сходство между дикамбой-О-деметилазой и некоторыми многокомпонентными диоксигеназами предполагает, что дикамба-О-деметилаза может проявлять скрытую диоксигеназную активность. Ясно, однако, что этот фермент не входит в класс диоксигеназ, которые расщепляют молекулярный кислород О2 и включают один атом кислорода в состав основного субстрата, а другой атом - в малый органический субстрат, такой как α-кетоглутарат (Fukumori & Hausinger, 1993, J. Biol. Chem., 268, 24311-24317). Действительно, комбинирование высокоочищенных редуктазыDIC, ферредоксинаDIC и оксигеназыDIC для проявления своей активности нуждается только в О2, NADH, Mg2+, Fe2+ и дикамбе.
ПРИМЕР 2. Идентификация и секвенирование клона, кодирующего оксигеназу дикамба-О-деметилазы Pseudomonas maltophilia DI-6
Как отмечалось в примере 1, были определены первые 29 аминокислот N-концевой аминокислотной последовательности оксигеназыDIC (остатки в скобках приведены как наиболее вероятные):
Thr Phe Val Arg Asn Ala Trp Tyr Val Ala Ala Leu Pro Glu Glu Leu Ser Glu Lys Pro Leu Gly Arg Thr Ile Leu Asp (Asp or Thr) (Pro)
[SEQ ID NO:1].
Данная последовательность позволяет конструировать олигонуклеотидные зонды, которые были синтезированы Operon (Alameda, Калифорния). В частности, провели смешение 32 зондов, длина каждого из которых составляла 17 нуклеотидов, и они содержали все возможные нуклеотидные последовательности, которые могли кодировать аминокислотную последовательность, выделенную в вышеприведенной последовательности жирным шрифтом. Олигонуклеотидные зонды были помечены по 3'-концу с использованием дигоксигенина (DIG) в соответствии с инструкциями фирмы Boehringer Mannheim (Indianapolis, Индиана).
Меченные DIG зонды, во-первых, гибридизовали с геномной ДНК P.maltophilia штамма DI-6, которую расщепляли различными сочетаниями ректриктаз, разгоняли в 1%-ном агарозном геле и подвергали блоттингу на нейлоновый фильтр. Основываясь на полученных результатах, конструировали фракционированную по размеру геномную библиотеку с использованием вектора pBluescript-II KS+ и трансформировали компетентные клетки DH5α E.coli. Геномная библиотека включала фрагменты длиной 1-2 тыс.н.п., вырезанные XhoI/HindIII. Олигонуклеотидные меченные DIG зонды гибридизовали с серией бактериальных колоний, нанесенными логосами на нейлоновые фильтры. Плазмидную ДНК выделяли из позитивных колоний и субклонировали. Обе цепи каждого субклона подвергали секвенированию с применением DNA Sequencing Facility в University of Nebraska-Lincoln. Гибридизация и выявление меченных DIG зондов осуществляли в соответствии с протоколами, предоставленными Boehringer Mannheim.
Идентифицировали клоны геномной ДНК, кодирующие оксигеназуDIC. Нуклеотидная последовательность и выведенная аминокислотная последовательность полной оксигеназыDIC показаны в списке последовательностей, приведенном ниже, как SEQ ID NO: 2 и SEQ ID NO: 3, соответственно.
Сравнение аминокислотной последовательности, расшифрованной по нуклеотидной последовательности данного клона, с последовательностями белков, включенными в швейцарскую базу данных Swiss Protein Database, показали наличие гомологии с другими оксигеназами. Гомология была установлена с использованием программы FASTA, разработанной группой GCG. Наиболее значительная гомология отмечена при сравнении с оксигеназным компонентом ваниллатдеметилазы (от Pseudomonas sp. штамм АТСС 19151), с уровнем идентичности 33,8%.
ПРИМЕР 3: Очистка и характеристика других компонентов дикамбы-О-деметилазы Pseudomonas maltophilia PI-6
Бактериальные культуры и получение очищенных клеточных лизатов. Штаммом DI-6 Pseudomonas maltophilia инокулировали шесть двухлитровых флаконов Erlenmeyer, содержащих один литр восстановленной хлоридной среды (Kreuger, J.P. 1989, Ph. D. thesis, Illinois Institute of Technology, Чикаго), дополненной глюкозой (2,0 мг/мл) и казаминовыми кислотами (2,0 мг/кл) в качестве источников углерода. Культуры инкубировали на круговом шейкере (225 об./мин при 30°С). Клетки собирали при A600, изменяющем от 1,5 до 2,0, с использованием ротора JLA-10.500 в центрифуге Beckman Avanti J-25I при 4000×g в течение 20 минут. Осажденные клетки хранили при -80°С. Замороженные клетки ресуспендировали в 40 мл 100 мМ MgCl2, опять осаждали с использований тех же условий, как указанные выше, и ресуспендировали в разрушающем буфере (100 мМ 3-[N-морфолино]пропансульфоновой кислоты (MOPS), (pH 7,2), 1 мМ дитиотреит, 5% глицерин) в отношении 2 мл разрушающего буфера на грамм влажного веса клеток. Лизоцим добавляли в отношении 80 мкл на грамм клеток вместе с коктейлем ингибиторов протеаз для бактериальных экстрактов (Sigma, P 8465) в отношении 5 мл на 20 грамм сырого веса клеток. Наконец, добавляли фенилметилсульфонилфторид (0,1 М маточный раствор в 100% этаноле) в отношении 250 мкл на 50 мл разрушающего буфере. Клетки разрушали ультразвуковым устройством (Sonics and Materials Inc., Model VCX 600) импульсами длительностью 9,3 секунд, с промежутками длительностью 3,0 секунд, в течение 33 минут с амплитудой, равной 50%. Лизированные клетки центрифугировали в течение 75 минут при 56000×g в роторе JA-25.50 в центрифуге Beckman Avanti J-25I при 4°С. Надосадочнузэ жидкость (очищенный клеточный лизат) декантировали и добавляли глицерин до конечной концентрации 15% перед хранением при -80°С.
Начальная очистка компонентов дикамбы-О-деметилазы. Аликвоту осветвленного клеточного лизата, содержащую примерно 2,7 грамм белка, наносили на колонку Pharmacia ХК 26/60, содержащую 25 мл DEAE-сефарозы быстрого потока, уравновешенную 50 мМ MOPS (pH 7,2), 1 мМ дитиотреита и 15% (об./об) глицерина (буфер А). Колонку соединяли с Bio-Cad Perfusion Chromatography Workstation (USDA NRICGP GRANT # 9504266) и запускали со скоростью потока, равной 5,0 мл/мин. После загрузки колонки ее промывали буфером А до того, как считываемое при 280 нм поглощение не снижалось до значения ниже 0,1. Все три компонента дикамбы-О-деметилазы связывались с DEAE-колонкой при данных условиях. Колонку обрабатывали в линейном градиенте от 0 до 500 мМ NaCl в буфере А. Это приводило к элюции ферредоксина при 400 мМ NaCl и совместной элюции редуктазного и оксигеназного компонентов при 250 мМ NaCl.
Очистка ферредоксинаDIC. Фракции, содержащие ферредоксинDIC, элюированный из колонки с DEAE-сефарозой, объединяли и меняли буфер на 50 мМ MOPS (рН 7,2), 5% глицерин (об./об.) и 200 мМ NaCl (буфер В). Затем их концентрировали примерно до 2 мл с использованием концентратора клеток Amicon с мембраной YM10 и концентратором Centricon 10. Затем данный образец наносили на предварительно упакованную колонку сефакрила S-100 Pharmacia HiPrep 26/60, уравновешенную буфером В, и запускали со скоростью потока 0,5 мл/мин в аппарат FPLC Pharmacia. Фракции, проявившие активность, объединяли и меняли буфер на 50 мМ MOPS (рН 7,2), 1 мМ дитиотреит и 5% глицерин (об./об.) (буфер С). Фракции концентрировали примерно до 2 мл и затем загружали на колонку Pharmacia Mono Q HP 5/5, уравновешенную буфером С. Колонку обрабатывали в линейном градиенте 0-2,0 М NaCl в буфере С. Фракции, проявляющие ферредоксиновую активность, анализировали на предмет содержания белка и хранили при -80°С.
Очистка редуктазыDIC. Фракции из начальной колонки с DEAE-сефарозой, содержащей компоненты оксигеназы/редуктазы, объединяли и затем добавляли сульфат аммония до конечной концентрации, равной 1,5 М. После инкубации при 4°С в течение 1,5 часов образцы центрифугировали в течение 75 минут при 56000×g при 4°С в роторе JA-25.50 в центрифуге Beckman Avanti J-251. Надосадочную фракцию сохраняли и загружали со скоростью потока 5,0 мл/мин в колонку Pharmacia XK 26/20, содержащую 25 мл фенил-сефарозы 6 с быстрым потоком (high sub), которую уравновешивали буфером А, содержащим 1,5 М (NH4)2SO4. Фракции, которые содержали редуктазный компонент, объединяли, буфер меняли на буфер В и концентрировали примерно до 2 мл с использованием концентратора Amicon с мембраной YM30 и концентратором Centricon 10. 2 мл образца наносили на предварительно упакованную колонку сефакрила S-100 Pharmacia HiPrep 26/60, уравновешенную буфером В, и запускали со скоростью потока 0,5 мл/мин. Фракции, проявляющие редуктазную активность, объединяли, меняли буфер на буфер С и концентрировали примерно до 2 мл. 2 мл образца загружали на колонку Pharmacia Mono Q HP 5/5, уравновешенную буфером С. Колонку обрабатывали в линейное градиенте 0-0,2 М NaCl в буфере С при скорости потока 0,5 мл/мин. Фракции, проявлявшие редуктазную активность, анализировали на предмет содержания белка и хранили при -80°С.
Быстрый ферментативный анализ. Активность каждого из трех индивидуальных компонентов отслеживали в реакциях с использованием меченной [14С] дикамбы в реакционной смеси, содержащей избыток двух остальных компонентов. Для каждой реакции буферный раствор, содержащий 24 мМ калий-фосфатный буфер (KPi) (рН 7,0), 0,48 мМ NADH, 0,48 мМ FeSO4 и 9,6 мМ MgCl2, добавляли к 200 мкл образца белка вместе с 30 мкл специальной смеси (562,4 мкл стерильной воды, 12,0 мкл 50 мМ дикамбы и 25,6 мкл маточного раствора [14С]-дикамбы [1,28 мкКи]) до общего объема 311 мкл и специфической активности [14С]-дикамбы, равной 4,12 мкКи/мл. Через 60 мин каждую реакцию останавливали добавлением 50 мкл 5% H2SO4 (об./об.) и 500 мкл простого эфира. Реакционные пробирки встряхивали на вортексе и центрифугировали в микроцентрифуге (Eppendorf, Model 5415C) при 14000×g в течение 2 минут. Для быстрой визуальной оценки ферментативной активности каждую реакционную пробирку помещали под ручной источник УФ-излучения (Fotodyne Inc., Model 3-6000) для детекции флуоресценции реакционного продукта DCSA. Для более аккуратных полуколичественных измерений ферментативных активностей продукты реакции разделяли с использованием тонкослойной хроматографии, как описано в примере 1.
Определения концентрации белка. Фракции, полученные на различных стадиях протокола очистки, анализировали на предмет концентрации белка с использованием анализа Брэдфорд (стандартный протокол, Bio Rad; см. пример 1).
ПРИМЕР 4: Идентификация и секвенирование клона, кодирующего ферредоксинDIC
N-концевая последовательность, полученная из очищенного белка ферредоксина (очистка описана в примере 3), составляла 29 аминокислот в длину (определение последовательности белков проводили на Protein Core Facility в University of Nebraska-Lincoln с использованием стандартной процедуры деградации по Эдману; см. пример 1). Сравнение данной последовательности с банком данных Genbank показало, что она характеризовалась 35% идентичностью в 26 аминокислотах перекрывания с терпредоксином Pseudomonas sp., бактериальным [2Fe-2S]-ферредоксином семейства адренодоксинов. Информацию о последовательности использовали для конструирования трех вырожденных олигонуклеотидных праймеров (двух прямых и одного обратного). Последовательность двух 17-членных прямых праймеров основывалась на N-концевой аминокислотной последовательности, полученной из очищенного белка ферредоксина. Последовательность 17-членного обратного праймера основывалась на консервативной последовательности из шести аминокислот вблизи С-конца шести секвенированных ранее бактериальных ферредоксинов адренодоксинового типа. Праймеры использовали в гнездовой реакции PCR с целью амплификации продукта длиной 191 н.п. из общей ДНК Р. maltophilia. Продукт клонировали в вектор pGEM-T Easy (Promega, Madison, Висконсин) и определяли его последовательность. Определение последовательности ДНК проводили на DNA Sequencing Core Facility в University of Nebraska-Lincoln с использованием стандартной процедуры опосредованной дидезоксинуклеотидами терминации цепи. Анализ предсказанной аминокислотной последовательности данного клона подтверждал ее соответствие N-концевой и внутренней аминокислотной последовательности, полученной ранее из очищенного белка ферредоксина. Более того, выделенная аминокислотная последовательность обладает 48%-ной идентичностью по всей ее длине с [2Fe-2S]-ферредоксином из Rhodobacter capsulatus. Клонированный фрагмент метили дигоксигенином (DIG) (Roche Diagnostics) с использованием стандартного протокола ПЦР (руководство пользователей системой DIG/Genius) и гибридизовали путем саузерн-блоттинга с общей ДНК Р. maltophilia, которую расщепляли различными ферментами рестрикции. Карту сайтов рестрикции, окружающих ген, конструировали, основываясь на размерах фрагментов рестрикции, которые гибридизовались с зондом. Данный начальный эксперимент показал, что ген содержится в фрагменте XhoI/PstI, который по длине был примерно равен 1 тыс.н.п. Впоследствии общую ДНК Р. maltophilia расщепляли XhoI и PstI и разделяли на геле фрагменты рестрикции. Фрагменты длиной от 0,5 до 1,5 тыс.н.п. вырезали из геля, лигировали в вектор pBluescript II KS+ (Stratagene, Inc.) и трансформировали в клетки DH5α (Gibco BRL, Inc.). Бактериальные колонии, содержащие фракционированную по размеру библиотеку, подвергали скринингу меченным DIG зондом и идентифицировали фрагмент XhoI/PstI длиной 900 н.п. Анализ последовательности показал, что данный клон содержал полноразмерный ген ферредоксина длиной 315 н.п. [SEQ ID NO:4], кодирующий белок 11,4 кДа, состоящий из 105 аминокислотных остатков [SEQ ID NO:5]. Аминокислотная последовательность, предсказанная из клонированного гена, соответствовала N-концевой и внутренней аминокислотным последовательностям, полученным ранее из очищенного белка ферредоксина. Более того, предсказанная аминокислотная последовательность была гомологична по всей ее длине пяти другим членам адренодоксинового семейства бактериальных [2Fe-2S]-ферредоксинов, в интервале от 42%-ной идентичности с ферредоксином Rhodobacter capsulatus до 35%-ной идентичности с ферредоксином Pseudomonas (см. фиг.2). Три других ферредоксина происходили из Caulobacter crescentus, Rhodococcus erhytropolis и Pseudomonas putida. Белки данного семейства образуют единственный 2Fe-2S-железосерный кластер и содержат три консервативных мотива. Мотив 1 включает три консервативных цистеина, которые являются лигандами 2Fe-2S. Мотив 2 содержит кластер отрицательно заряженных остатков. Мотив 3 включает четвертый консервативный цистеин 2Fe-23-кластера.
ПРИМЕР 5: Идентификация и секвенирование клонов, кодирующих редуктазуDIC
Два гена редуктазы клонировали тем же способом, который использовали в примере 4 для клонирования гена ферредоксина. N-концевая последовательность, полученная из очищенного белка редуктазы (очистка описана в примере 3), в длину составляла 25 аминокислот. Сравнение данной последовательности с банком данных Genbank показало, что она характеризовалась 90% идентичностью в 20 аминокислотах перекрывания с редуктазным компонентом относящегося к цитохрому Р450 типа диоксиндиоксигеназы, трехкомпонентного фермента, выделенного ранее из Sphingomonas sp. RW1. Внутренняя последовательность из 10 аминокислотных остатков также была получена после расщепления трипсинов очищенного белка. Внутренняя последовательность обладала 87,5%-ной идентичностью с остатками 62-69 той же редуктазы цитохрома Р450 типа Sphingomonas sp. RW1. Данную информацию о последовательности использовали для конструирования трех вырожденных олигонуклеотидных праймеров (двух прямых и одного обратного). Последовательность двух 17-членных прямых праймеров основывалась на N-концевой аминокислотной последовательности, а последовательность 17-членного обратного праймера основывалась на внутренней аминокислотной последовательности. Праймеры использовали в гнездовой реакции ПЦР с целью амплификации продукта длиной 180 н.п.из общей ДНК Р. maltophilia. Продукт клонировали в вектор pGEM-T Easy и секвенировали. Анализ предсказанной аминокислотной последовательности данного клона подтвердил, что она кодирует белок, соответствующий N-концевой и внутренней аминокислотной последовательности, полученной ранее из очищенного белка редуктазы. Более того, предсказанная последовательность обладала 80%-ной идентичностью по всей длине с редуктазным компонентом относящегося к цитохрому Р450 типа диоксиндиоксигеназы Sphingomonas sp. RW1.
Клонированный фрагмент метили DIG и гибридизовали путем саузерн-блоттинга с общей ДНК Р. maltophilia, которую до этого расщепляли несколькими ферментами рестрикции. Саузерн-блот показал, что меченный DIG зонд распознавал два различных локуса в различных продуктах расщепления рестриктазами общей ДНК Р. maltophilia. Данное наблюдение указывает на то, что имеются два редуктазных гена, локализованных в различных положениях в геноме Р. maltophilia. Возможно, что два данных гена представляют собой идентичные дупликации, которые кодируют идентичные редуктазные белки. Альтернативно, один из этих генов мог бы кодировать укороченный белок без активности или полноразмерный белок с низкой активностью в анализе дикамбы-О-деметилазы, проводимом авторами изобретения. Поскольку было необходимо клонировать ген с оптимальной активностью, для получения обоих генов редуктазы использовали меченный DIG зонд.
При расщеплении ДНК Р. maltophilia KpnI и EcoRI меченный DIG зонд гибридизовался с одним рестрикционным фрагментом, который составлял примерно 4,0 тыс.н.п. в длину, и другим более крупным фрагментом размером примерно 20 тыс.н.п. Карта числа сайтов рестрикции, окружающих ген, локализованный на фрагменте KpnI/EcoRI длиной 4,0 тыс.н.п., конструировали, основываясь на размерах различных фрагментов рестрикции, которые гибридизовались с зондом. Рестрикционная карта показала, что целый ген должен располагаться на данном фрагменте длиной 4,0 тыс.н.п. Впоследствии общую ДНК Р. maltophilia расщепляли KpnI и EcoRI и разделяли на геле фрагменты рестрикции. Фрагменты длиной от 3,0 до 5,0 тыс.н.п. вырезали из геля, лигировали в вектор pBluescript II KS+ и трансформировали в клетки DH5α. Бактериальные колонии, содержащие фракционированную по размеру библиотеку, подвергали скринингу меченным DIG зондом и идентифицировали фрагмент KpnI/EcoRI длиной 4,3 тыс.н.п. Анализ последовательности показал, что данный клон содержал полноразмерный ген редуктазы длиной 1224 н.п. [SEQ ID NO:6], кодирующий белок длиной 43,7 кДа, состоящий из 403 аминокислотных остатков [SEQ ID NO:7]. Аминокислотная последовательность, предсказанная из клонированного гена соответствовала N-концевой и внутренней аминокислотным последовательностям, полученным ранее из очищенного белка редуктазы. Более того, предсказанная аминокислотная последовательность была гомологична по всей ее длине по крайней мере четырем другим FAD-зависимым пиридиннуклеотидредуктазам, в интервале от 69% идентичности с редуктазным компонентом относящегося к цитохрому Р450 типа диоксиндиоксигеназы Sphingomonas sp. RW1 до 36% идентичности с терпредоксинредуктазой вида Pseudomonas (см. фиг.3). Две другие FAD-зависимые пиридиннуклеотидредуктазы происходили из R. erythropolis и Р. putida. Белки в данном семействе FAD-зависимых пиридиннуклеотидредуктаз содержат пять консервативных мотивов. Мотивы 1 и 3 содержат три консервативных остатка глицина и соответствуют АДФ-связывающему участку для FAD и NAD(P), соответственно. Мотив 5 соответствует участку, связывающемуся с флавиновой группой FAD.
Для клонирования второго гена общую ДНК Р. maltophilia расщепляли KpnI и EcoRI и разделяли на агарозном геле полученные в результате фрагменты рестрикции. Фрагменты размером примерно 20 тыс.н.п. вырезали из геля, расщепляли несколькими ферментами рестрикции и затем гибридизовали путем саузерн-блоттинга с меченным DIG зондом. Карту сайтов рестрикции, окружающих второй ген, конструировали, основываясь на размерах фрагментов рестрикции, которые гибридизовались с зондом. Данные эксперименты показали, что полноразмерный второй редуктазный ген содержался в фрагменте ApaI, который по длине составлял примерно 3,0 тыс.н.п. Впоследствии общую ДНК Р. maltophilia расщепляли ApaI и разделяли на геле фрагменты рестрикции. Фрагменты длиной от 2,0 до 4,0 тыс.н.п. вырезали из геля, лигировали в вектор pBluescript II KS+ и трансформировали в клетки DН5α. Бактериальные колонии, содержащие фракционированную по размеру библиотеку, подвергали скринингу меченным DIG зондом и идентифицировали фрагмент ApaI длиной 3,0 тыс.н.п. Анализ последовательности показал, что клон длиной 3,0 тыс.н.п. содержал открытую рамку считывания длиной 1227 н.п. [SEQ ID NO:8] и кодировал белок 43,9 кДа, состоящий из 409 аминокислотных остатков [SEQ ID NO:9]. Аминокислотная последовательность, кодируемая данным вторым редуктазным геном, является почти идентичной (98,8% идентичности) к последовательности первого гена.
ПРИМЕР 6: Трансформация растений
Для помещения каждого из трех генов, которые кодируют компоненты дикамбы-O-деметилазы индивидуально в кассеты, подходящие для экспрессии в растениях, осуществляли следующие стадии. Конструировали олигонуклеотидные праймеры для образования сайта NcoI с 5'-конца и сайта XbaI с 3'-конца каждого из трех генов путем амплификации ПЦР. Аутентичность полученных продуктов ПЦР подтверждали секвенированием и каждый ген клонировали индивидуально в вектор pRTL2 (предоставленный д-ром Torn Clemente из Plant Transformation Core Research Facility, University of Nebraska, Lincoln, Небраска). Данный вектор содержит последовательность-энхансер трансляции длиной 144 н.п. из вируса гравировки табака (TEV) (Carrington and Freed, J. Virology, 64:1590-1597 (1990)) с 5'-конца полилинкера. Гены оксигеназы, редуктазы и ферредоксина, каждый с 5'-концевым энхансером трансляции, затем клонировали индивидуально в виде фрагментов XhoI/XbaI в экспрессирующий вектор для растений pKLP36 (полученный от Indu Maiti, University of Kentucky, Lexington, Kentucky) (Maiti and Sheppard, Biochem. Biophys. Res. Commun., 244: 440-444 (1998)). Данный бинарный вектор содержит полноразмерный промотор вируса хлороза арахиса (PCISV FLt36) с дуплицированным энхансерным доменом для конститутивной экспрессии в растениях и 3'-концевая последовательность гороха rbcS для эффективной терминации транскрипции (Maiti and Sheppard, Biochem Biophys Res Commun, 244: 440-444 (1998)). Конструкции со всеми тремя генами на одном бинарном векторе может продуцироваться путем комбинации трех генов в некотором количестве разных порядков и ориентации.
Следующие способы использовались для направления конструкций оксигеназы, ферредоксина и редуктазы индивидуально в Agrobacterium tumifaciens и для трансформации каждой конструкции в Arabidopsis и табак. Три конструкции перемещали в А. tumefaciens штамма С58С1 путем модифицированной процедуры три-родительского спаривания, обычно используемой в Plant Transformation Core Facility. Процедура включала инкубацию клеток Escherichia coli, несущих каждую конструкцию со смесью клеток A. tumefaciens и клеток Е. coli, несущих хелперную плазмиду pRK2013 (для мобилизации плазмиды). Клетки А. tumefaciens, содержащие каждую конструкцию, затем использовали для трансформации табака и Arabidopsis с помощью Plant Transformation Core Research Facility. Для табака эксплантаты листьев инкубировали с суспензией клеток A. tumefaciens, содержащих каждую конструкцию, и побеги регенерировали на твердой среде, содержащей канамицин (Horsch et al., Science, 227: 1229-1231 (1985)). Детали протокола трансформации табака следующие.
Получение эксплантатов табака:
3-4 дюймовых листа выбирали от растений в возрасте 1 месяца (верхние 2 листа). Этого было достаточно для получения 30-40 эксплантатов. Листья помещали в большую мензурку и покрывали на 20-30 минут дист. Н2О. Воду сливали и листья покрывали 10% Chlorox™ с Tween20™ (1 капля на 50 мл). Листья высушивали в течение 15 минут и промывали 3Х-5Х стерильной дист. Н2О. Внешний край листа вместе с кончиком и черешком отрезали, срединную жилку отсекали и оставшуюся ткань нарезали на квадраты 0,5×0,5 см. Эксплантаты повергали предварительному культивированию по 30 на чашку кверху осевой стороной в течение 1 суток перед инокуляцией.
Препарат для инокуляции Agrobacterium
2 мл культуры Agrobacterium инициировали в среде YEP (10 г/л пептона, 10 г/л NaCl и 5 г/л дрожжевого экстракта, pH 7,0) с добавлением подходящих антибиотиков. Культуру доводили до насыщения. 2 мл культуры субкультивировали в 50 мл среды YEP и позволяли расти в течение 6-8 часов (28°С при постоянном встряхивании). Клетки собирали центрифугированием (3000-4000 об./мин). Бактериальные осадки ресуспендировали до конечного OD660=0,3-1,0 в среде для совместного культивирования.
Препарат для инокуляции Agrobacterium переносили на чашки Петри. Предварительно культивированные эксплантаты инокулировали в течение 30 минут. Эксплантаты подвергали блоттингу на стерильную фильтровальную бумагу и помещали на среду для совместного культивирования (10 эксплантатов на чашку), которую сделали более твердой 0,8% агаром, накрытую стерильной фильтровальной бумагой (эксплантаты для совместного культивирования осевой стороной кверху). Эксплантатам позволяли совместно культивироваться в течение 3 суток. После периода совместного культивирования эксплантаты в течение краткого времени промывали в среде регенерации.
Промытые эксплантаты подвергали блоттингу на стерильную фильтровальную бумагу и ткань (10 эксплантатов на чашку) переносили на среду регенерации, которую сделали более твердой 0,8% агаром, с добавлением подходящего средства селекции (канамицин 150 мг/л). Эксплантаты культивировали осевой стороной кверху. Ткань переносили каждые 2 недели на свежую среду регенерации и побеги извлекали (>3 см) и субкультивировали в среде для укоренения. Укоренившиеся побеги затем акклиматизировали на почву.
Реагенты:
Среда предварительного культивирования (РМ):
Соли MS с витаминами В5, 3% сахарозы с добавлением 1 мг/л ВАР, 0,1 мг/л NAA и 8 мкг/л рСРА (п-хлорфеноксиуксусная кислота), рН 5,7 (все регуляторы роста стерилизуют фильтрованием).
Среда совместного культивирования (СМ);
1/10 среда MS/B5, дополненная 3% сахарозой, 1 мг/л ВАР, 0,1 мг/л NAA и 8 мкг/л рСРА
Среда регенерации (RM):
Среда MS/B5, дополненная 1 мг/л ВАР, 0,1 мг/л NAA и 3% сахарозой, рН 5,7. Используют антибиотики: карбенициллин (500 мг/л) и цефотаксим (100 мг/л).
Корневая среда:
Соли MS с витаминами В5 полноценного действия, дополненные 0,1 мг/л NAA и 1% сахарозой. Среде придают твердость 0,8% агаром. Поддерживают концентрацию антибиотиков (при использовании npt II 75 мг/л) карбенициллина (500 мг/л) и цефотаксима (100 мг/л).
Десять побегов выбирали из каждого из трех экспериментов по трансформации, помещали на корневую среду в течение нескольких недель и затем помещали в горшки в теплице. Для Arabidopsis горшок с цветущими растениями инкубировали с суспензией клеток A. tumefaciens, содержащих каждую из конструкций, и затем на растениях позволяли завязаться семенам (Bechtold et al., С.R. Acad. Sci. Paris, Sciences la vie/Life sciences, 316. 1194-1199 (1993); Clough and Bent, Plant J., 16 (6): 735-743 (1598)). Семена собирали и проращивали на среде с канамицином. После развития у саженцев адекватной корневой системы некоторые растения выбирали из каждого эксперимента по трансформации и помещали в горшки в ростовой камере.
Для оценки экспрессии каждого гена в трансформированных растениях получали вестерн-блоты лизатов листьев некоторых трансформированных растений и тестировали их поликлональными антителами, детектирующими три компонента дикамбы-О-деметилазы. Для определения ферментативных активностей каждого фермента в растительных экстрактах применяли подход комбинирования лизатов листьев из трансформированных растений, экспрессирующих белки, оксигеназу, ферредоксин или редуктазу с избыточными количествами других компонентов O-деметилазы, очищенных из Р. maltophila штамма DI-6. Данные смеси тестировали на предмет активности дикамбы-О-деметилазы как в стандартном анализе с меченной 14С дикамбой (см. примеры 1 и 3), так и в анализе HPLC, использующим в качестве субстрата немеченую дикамбу (дикамба и 3,6-DCSA элюируются в разных точках при HPLC и могут подвергаться количественному анализу).
Для тестирования экспрессии генных продуктов в компартменте хлоропласта получали конструкции с сигнальной пептидной последовательностью с 5'-конца генов оксигеназы, ферредоксина и редуктазы. Такой подход был успешным для придания толерантности к гербицидам на основе сульфонилмочевины в растения табака (O'Keefe at al., Plant. Physiol., 105:473-4778 (1994)). Более конкретно, последовательность сигнального пептида гена малой субъединицы рибулозо-1,5-бисфосфаткарбоксилазы гороха (последовательность нуклеиновой кислоты, представленная SSQ 12 NO:19) была путем генной инженерии встроена в промежуточный вектор pRTL2, содержащий гены оксигеназы, ферредоксина или редуктазы, каждый из которых с своей собственной последовательностью, регулирующей экспрессию (см. выше). Конструирование завершали созданием сайтов Ncol с обоих концов сигнального пептида с использованием олигонуклеотидных праймеров и протокола ПЦР. Участки Ncol способствовали введение сигнального пептида между лидерной последовательностью TEV к репортерным геном (оксигеназы, редуктазы или ферредоксина) вектора pRTL2. Всю кассету (лидер TEV, сигнальный пептид и репортерный ген) затем вырезали из вектора pRTL2 в виде фрагмента XhoI/XbaI и вводили в бинарный вектор pKLP36, содержащий промотор PC1SV FLt36 и 3'-концевой терминатор rbcS.
Для оценки возможности того, что использование кодонов в гене бактериального ферредоксина не вполне оптимально для эффективной трансляции в растительной клетке, на Entelechon GmbH (Регенсберг, Германия) синтезировали синтетический ген ферредоксина, кодирующий ту же аминокислотную последовательность, что и у ферредоксина Р. maltophila штамма DI-6, но с кодонным смещением, оптимизированным для двудольных растений. Подробно описано, что изменения в использовании кодонов являются существенными для оптимальной экспрессии генов бактериального токсина B.t. в растительных клетках (см., например, Diehn et al., Genetic Engineering, 18: 83-99 (1996)).
ПРИМЕР 7: Опрыскивание растений
Для тестирования устойчивости растений трансгенного табака к дикамбе семена от поколения Т0, описанные выше в примере 6, проращивали и выращивали до стадии 10-12 листьев. С помощью Department of Agronomy данные растения подвергали «распылению с постоянной скоростью» с использованием изготовленного на заказ распылителя (Burnside, Weed Science Vol.17, No.1, 102-104, 1969), произведенном ISCO. Распылитель имеет насадку TEEJET и экран и использует скорость доставки 1,87 миль в час и давление 42 фунта на кв.дюйм. Высота насадки установлена на 8'' над пологом. Дикамбу (коммерческий продукт - Clarity) в различных концентрациях распыляли на растения в смеси с неионным поверхностно-активным веществом в отношении 20 галлонов на акр с помощью сжатого воздуха.
Для установления инициального графика гибели возрастающими концентрациями дикамбы, изменяющимися от 0 до 0,5 фунтов/акр, обрабатывали растения табака дикого типа. После установки 100%чувствительности растений дикого типа к 0,5 фунтов/акр дикамбы трансгенные растения, содержащие только ген оксигеназы с сигнальным пептидом или без него, обрабатывали возрастающими концентрациями дикамбы, изменяющимися от 0,5 до 5 фунтов/акр. Трансгенные растения, несущие конструкции с геном оксигеназы, но в отсутствие последовательности, кодирующей сигнальный пептид, проявляли устойчивость (т.е. росли и были здоровыми) к обработке дикамбой по крайней мере при 2,5 фунтов/акр. Трансгенные растения табака, несущие генные конструкции оксигеназы с сигнальным пептидом росли и были здоровыми при распылении с концентрацией дикамбы вплоть до 5 фунтов/акр. То есть трансгенные растения, содержащие ген оксигеназы с сигнальным пептидом или без него, оказывались сходными с необработанными контрольными растениями по крайней мере до указанных количеств дикамбы, тогда как нетрансгенные, обработанные теми же количествами дикамбы, характеризовались сниженным ростом, засыхали и подвергались сильному некрозу.
В сходном эксперименте трансгенные растения, содержащие три генные конструкции (гены оксигеназы, ферредоксина и редуктазы), опылялись 0,125 фунтами/акр дикамбы. Трансгенные растения табака, несущие три генные конструкции, росли и были здоровыми при данной концентрации дикамбы, тогда как нетрансгенные, обработанные теми же количествами дикамбы, характеризовались сниженным ростом, засыхали и подвергались сильному некрозу. Поскольку трансгенные растения только с конструкцией оксигеназы характеризовались устойчивостью к дикамбе вплоть до 2,5 фунтов/акр, предполагается, что трансгенные растения, содержащие три генные конструкции будут проявлять сходную устойчивость вплоть до 2,5 фунтов/акр или даже более.
Данные результаты ясно демонстрируют, что трансгенные растения, трансформированные дикамба-разрушающей оксигеназой или тремя генными конструкциями по настоящему изобретению характеризуются приобретенной устойчивостью к дикамбе.
Эксперимент 1
Авторами изобретения были проведены эксперименты по идентификации дополнительных генов, которые кодируют дикамба-разрушающие оксигеназы. В частности, авторами изобретения были выделены дополнительные дикамба-разрушающие бактерии и идентифицированы несколько копий генов, кодирующих дикамба-разрушающие оксигеназы. Эти эксперименты выполняли следующим образом.
а. В указанных исследованиях были использованы пять штаммов бактерий:
(1) Штамм Pseudomonas maltophilia DI-6 был получен от Дугласа Корка, Иллинойский технологический университет, Чикаго, Иллинойс (Douglas Cork, Illinois Institute of Technology, Chicago, Illinois).
(2) Три штамма были выделены авторами изобретения из почвы, взятой из разных участков пруда-отстойника при заводе по производству дикамбы в г.Бомоне, Техас (Beaumont, Texas) (Pseudomonas sp. BRW1, Sphingomonas sp. BRW2, Pseudomonas sp.BRWS). Указанные штаммы были выделены при посеве с серийными разведениями на чашках Петри (в стерильной воде) образцов почвы на среде с пониженным содержанием хлорида ("RCl"; Krueger et al., J.Agric. Food Chem., 37, 534-538(1989)), содержащей 5 мМ дикамбы в качестве единственного источника углерода с последующим инкубированием чашек при 30°С. Было выделено несколько десятков штаммов, способных утилизировать дикамбу в качестве единственного источника углерода; однако некоторые изоляты, которые образуют крупные колонии на агаровой среде, но не растут в жидкой культуре, не использовались в дальнейших исследованиях. Три штамма, которые использовались в исследованиях (Pseudomonas sp. BRW1, Sphingomonas sp. BRW2, Pseudomonas sp. BRW3), быстро растут в жидкой среде.
(3) Штамм 3МР представляет собой штамм DH5α Escherichia coli, трансформированный ДНК, кодирующей дикамба-разрушающую оксигеназу штамма DI-6 P.maltophilia (получение описано в эксперименте 2а ниже).
(4) Все пять указанных штаммов бактерий представляют собой дикамба-разрушающие бактерии, которые способны утилизировать дикамбу в качестве единственного источника углерода.
b. Бактерии выращивали на чашках Петри, содержащих гелрит (10 г/л) и RCl-среду с 5 мМ дикамбы. Единичные колонии инокулировали в 5 мл жидкой RCl-среды, содержащей 5 мМ дикамбы (культуры выращивали в двух повторах). Культуры выращивали в стерильных тест-пробирках на 15 мл при встряхивании со скоростью 225 об/мин при 30°С до насыщенной плотности, для чего требуется от 48 до 72 часов. 2 мл каждой культуры инокулировали в 25 мл RCl-среды, содержащей 5 мМ дикамбы в колбе на 125 мл. Культуры выращивали до насыщенной плотности, для чего вновь требовалось от 48 до 72 часов. В этот момент становится видимым розовый побочный продукт. Десять мл каждой из указанных культур инокулировали в 500 мл RCl-среды, содержащей 5 мМ дикамбы в литровых колбах. Культуры выращивали при 30°С со встряхиванием при 225 об/мин до насыщенной плотности, примерно 48 часов. Далее культуры центрифугировали при 4°С в центрифуге Beckman J-251 со скоростью 5000×G в течение 10 мин с соблюдением условий стерильности. Супернатант сливали и осадок ресуспендировали в 10 мл RCl. После ресуспендирования 10 мл помещали в литровую колбу с 500 мл RCl, содержащей 5 мМ дикамбы, и инкубировали, как описано выше, в течение примерно 48-72 часов. Затем клетки осаждали на центрифуге Beckman J-251 при 4°С со скоростью 5000×G в течение 15 мин.
с. Набор для очистки плазмид Qiagen® (Qiagen, Inc.) использовали для выделения плазмид по методике получения плазмид с очень низким числом копий (Qiagen® tip-100). Плазмидную ДНК осаждали, распределяя плазмидную ДНК каждого штамма по двум пробиркам. В каждую пробирку добавляли одну десятую объема 3М ацетата натрия и два объема 100% этанола. Пробирки инкубировали в течение ночи при -20°С. Затем проводили центрифугирование в микроцентрифуге при 4°С в течение 15 минут. Супернатант сливали. В каждую пробирку добавляли 500 мкл 70% холодного этанола. Пробирки центрифугировали в микроцентрифуге при 4°С в течение 15 минут при 4000×G. Супернатант сливали и осадок сушили в течение одного часа. ДНК суспендировали в 50 мкл ТЕ буфера (0,01 М Tris, 0,001 М EDTA), рН 8,0.
d. Для получения образцов для анализа гель-электрофорезом по методу CHEF (в электрическом гомогенном поле с контурными зажимами) к образцам добавляли ТЕ буфер, рН 8,0 вместе с IxSALB красителем (1,5 мг/мл бромфенолового синего, 0,5×ТВЕ (Трис-борат-ЭДТА буфер, 90 мМ Трис-борат, 2 мМ ЭДТА), 0,6 г/мл сахарозы, 4 мкл прокипяченой РНКазы/мл (10 мг/мл)). Используемые маркеры молекулярного размера представляли собой высокомолекулярные маркеры (Gibco BRL) (15 мкл маркера, 5 мкл ТЕ, рН 8,0, 5 мкл SALE красителя, который непосредственно перед использованием выдерживали в течение 10 минут при 55°С), маркеры Low Range PFG (Biolabs) (0,25 мкг маркера запечатанного с расплавленной агарозой) и маркер DIG Marker II (Boehringer Mannheim GmbH) (15 мкл маркера, 5 мкл Н2О). Полученные образцы вносили в ячейки 0,7% агарозного геля в аппарат для проведения гель-электрофореза по методу CHEF (BioRad). Для проведения CHEF были установлены следующие контрольные параметры: исходное время включения (2 секунды), конечное время выключения (2 секунды), время разделения (10 часов) и напряжение (6 В). В аппарат для проведения гель-электрофореза по методу CHEF вносили два литра 1×TAE (0,04 М Трис-ацетат, 0,001 М ЭДТА). По окончании времени разделения гель погружали в раствор этидиума бромида (1 мкг/мл) примерно на 30 минут. Окрашенный гель фотографировали в УФ свете с использованием аппарата Fotodyne Gel Documentation.
е. Зонды, меченные дигоксигенином, получали путем амплификации участка гена оксигеназы методом ПЦР (Genius System User's Guide For Membrane Hybridization). Полученный зонд составлял примерно 900 п.н. в длину. Продукты ПЦР проверяли внесением образцов в 1% агарозный гель. Для гена оксигеназы использовали следующие ПЦР праймеры:
прямой: - 5'-GCTGCCCGAGGAACTGTCCGAAAAG-3';
обратный: - 5'-CGACGACGACCTTGTCCTCCTTGAC-3'.
f. Плазмидную ДНК переносили на нейлоновую мембрану с использованием методики Саузерн-блоттинга. Саузерн-блоттинг проводили с использованием меченной дигоксигенином оксигеназы в качестве зонда. Использовали жесткие условия гибридизации и промывки мембраны. Для обнаружения гибридизации использовали набор для хемилюминесценции (Roche, Genius Nouradioactive Detection Kit). Проводили 5-минутную, часовую и 24-часовую экспозиции.
д. Из полученных результатов было видно, что зонд оксигеназы интенсивно гибридизуется со многими полосами, относящимися к каждому бактериальному штамму, указывая на наличие близкого сходства между зондом и несколькими бактериальными генами (с более чем 90% идентичностью, обеспечиваемой жесткими условиями гибридизации и размером зонда) и между различными бактериальными генами. На основании этих результатов был сделан вывод о том, что каждый из трех впервые обнаруженных штаммов (Pseudomonas sp. BRW1, Sphingomonas sp. BRW2, Pseudomonas sp. BRW3) содержит, по меньшей мере, один ген дикамба-разрушающей оксигеназы.
Экспиремент 2
Получение трансгенного штамма E.coli 3MP, экспресирующего оксигеназу штамма P.maltophilia.
а. Как отмечалось выше в эксперименте 1, трансгенный штамм E.coli 3MP получали путем трансформации штамма E.coli DH5α ДНК, кодирующей дикамба-разрушающую оксигеназу (SEQ ID NO:3). Штамм E.coli DH5α трансформировали плазмидной ДНК, выделенной из штамма DI-6 P.maltophilia способом, описанным в эксперименте 1. Культуры штамма E.coli DH5α делали компетентными к трансформации согласно метидикам, описанным в работе Самбрука и соавт. (Sambrook at al., Molecular Cloning, стр.1,74-1,84 (2-е изд., Cold Spring Harbor Press, 1989)).
b. Как уже отмечалось выше в эксперименте 1, штамм E.coli 3МР способен утилизировать дикамбу в качестве единственного источника углерода. Штамм E.coli DH5α - родительский штамм E.coli 3МР - не может расти на дикамбе в тех же условиях, что и трансгенный штамм 3МР. Таким образом, именно наличие оксигеназы штамма DI-6 P.maltophilia и ассоциированных диамба-разрушающих генов штамма DI-6 P.maltophilia позволяет штамму E.coli 3МР разрушать дикамбу и утилизировать ее в качестве источника углерода.
с. Результаты эксперимента, описанного в эксперименте 1, показывают, что зонд штамма DI-6 P.maltophilia (из SEQ ID NO:3) гибридизовался с плазмидной ДНК из штамма E.coli 3МР, указывая на присутствие в штамме ЗМР ДНК, кодирующей оксигеназу DI-6.
d. Оксигеназа, продуцируемая трансгенным штаммом E.coli 3МР, очищали и смешивали с очищенными ферредоксиновым и редуктазным компонентами (очищенными из штамма DI-6 P.maltophilia, как описано в примере 1). Указанное сочетание определяет дикамба-O-деметилазную активность. Сочетание только ферредоксина и редуктазы не приводит к дикамба-O-деметилазной активности. Анализ проводили по методике, описанной в примере 1.





















| название | год | авторы | номер документа |
|---|---|---|---|
| МОЛЕКУЛА ДНК, КОДИРУЮЩАЯ ДИКАМБА-РАЗРУШАЮЩУЮ ОКСИГЕНАЗУ, И ЕЕ ПРИМЕНЕНИЕ | 1998 |
|
RU2226215C2 |
| ТРАНСГЕННЫЕ РАСТЕНИЯ С УЛУЧШЕННЫМИ ХАРАКТЕРИСТИКАМИ РОСТА | 2009 |
|
RU2582260C2 |
| ПОЛИПЕПТИД, ИМЕЮЩИЙ NADH-ЗАВИСИМУЮ HMF-РЕДУКТАЗНУЮ АКТИВНОСТЬ | 2008 |
|
RU2483107C2 |
| СОЗДАНИЕ ГАПЛОИДНЫХ РАСТЕНИЙ И УСОВЕРШЕНСТВОВАНИЕ СЕЛЕКЦИИ РАСТЕНИЙ | 2010 |
|
RU2571927C2 |
| РАСТЕНИЯ ПШЕНИЦЫ С ПОВЫШЕННОЙ УСТОЙЧИВОСТЬЮ К ИМИДАЗОЛИНОНОВЫМ ГЕРБИЦИДАМ | 2002 |
|
RU2337532C2 |
| СПОСОБЫ И ВЕЩЕСТВА ДЛЯ ОСНОВАННОГО НА РЕКОМБИНАЦИИ ПОЛУЧЕНИЯ СОЕДИНЕНИЙ ШАФРАНА | 2012 |
|
RU2676730C2 |
| СПОСОБ И КОНСТРУКТ ДЛЯ СИНТЕТИЧЕСКОГО ДВУНАПРАВЛЕННОГО РАСТИТЕЛЬНОГО ПРОМОТОРА SCBV | 2012 |
|
RU2627595C2 |
| СПОСОБ ПОЛУЧЕНИЯ 2,4-ДИГИДРОКСИБУТИРАТА | 2013 |
|
RU2645260C2 |
| СПОСОБЫ И ВЕЩЕСТВА ДЛЯ ПРИДАНИЯ РАСТЕНИЯМ УСТОЙЧИВОСТИ К ЗАБОЛЕВАНИЯМ | 1996 |
|
RU2203320C2 |
| РАСТЕНИЯ ПШЕНИЦЫ С ПОВЫШЕННОЙ УСТОЙЧИВОСТЬЮ К ИМИДАЗОЛИНОНОВЫМ ГЕРБИЦИДАМ | 2002 |
|
RU2337531C2 |
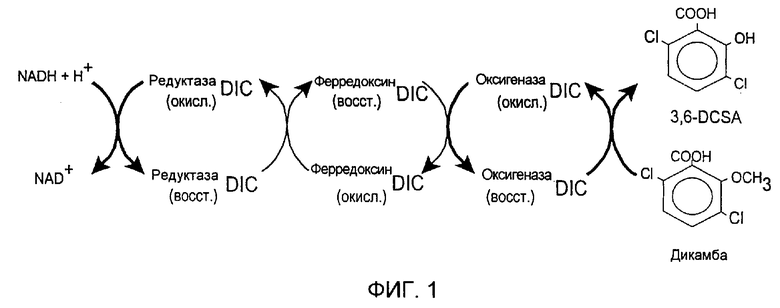


Изобретение относится к биотехнологии, в частности к дикамба-разрушающим ферментам, выделенным молекулам ДНК, трансгенным организмам, способам борьбы с сорняками и способам очистки материала. Выделяют последовательности ДНК, кодирующие дикамба-разрушающий ферредоксин и редуктазу. Конструируют конструкции ДНК, содержащие, по меньшей мере, одну последовательность, регулирующую экспрессию, и последовательность, кодирующую один из вышеуказанных протеинов или O-деметилазу. Получают трансгенные растения устойчивые к дикамбе, а также микроорганизм, разрушающий дикамбу. Способ борьбы с сорняками проводят путем нанесения дикамбы на плантацию вышеуказанных растений. Способ очистки материала заключается в нанесении дикамба-разрушающего трансгенного микроорганизма на материал, содержащий дикамбу. Способ селекции трансформированных клеток проводят путем выращивания полученной популяции клеток в культуральной среде, содержащей дикамбу в такой концентрации, чтобы трансформированные растительные клетки росли, а нетрансформированные не росли. Изобретение позволяет эффективно бороться с сорняками, а также проводить очистку материала, загрязненного дикамбой. 14 н. и 17 з.п. ф-лы, 3 ил.
a) аминокислотной последовательности, содержащей SEQ ID NO: 5;
b) аминокислотной последовательности, которая представляет собой ферментативно активный фрагмент SEQ ID NO: 5; и
c) аминокислотной последовательности, которая по меньшей мере приблизительно на 65% идентична SEQ ID NO: 5 и которая обладает дикамба-разрушающей активностью ферредоксина.
а) аминокислотной последовательности, выбранной из группы, состоящей из SEQ ID NO: 7 и SEQ ID NO: 9;
b) аминокислотной последовательности, которая представляет собой ферментативно активный фрагмент аминокислотной последовательности, указанной в (а); и
c) аминокислотной последовательности, которая по меньшей мере приблизительно на 75% идентична аминокислотной последовательности, указанной в (а), и которая обладает дикамба-разрушающей активностью редуктазы.
a) дикамба-разрушающую оксигеназу, содержащую аминокислотную последовательность, выбранную из группы, состоящей из:
i) SEQ ID NO:3;
ii) аминокислотной последовательности, которая представляет собой ферментативно активный фрагмент SEQ ID NO: 3; и
iii) аминокислотной последовательности, которая по меньшей мере приблизительно на 65% идентична SEQ ID NO: 3 и которая обладает дикамба-разрушающей активностью оксигеназы;
b) дикамба-разрущающий ферредоксин, содержащий аминокислотную последовательность, выбранную из группы, состоящей из:
i) аминокислотной последовательности, содержащей SEQ ID NO: 5;
ii) аминокислотной последовательности, которая представляет собой биологически активный фрагмент SEQ ID NO: 5; и
iii) аминокислотной последовательности, которая по меньшей мере приблизительно на 65% идентична SEQ ID NO: 5 и которая обладает дикамба-разрушающей активностью ферредоксина; и
c) дикамба-разрушающую редуктазу, содержащую аминокислотную последовательность, выбранную из группы, состоящей из:
i) аминокислотной последовательности, выбранной из группы, состоящей из SEQ ID NO: 7 и SEQ ID NO: 9;
ii) аминокислотной последовательности, которая представляет собой ферментативно активный фрагмент аминокислотной последовательности, указанной в (а); и
iii) аминокислотной последовательности, которая по меньшей мере приблизительно на 75% идентична аминокислотной последовательности, указанной в (а), и которая обладает дикамба-разрушающей активностью редуктазы.
получение популяции растительных клеток;
трансформацию по меньшей мере некоторых растительных клеток из популяции растительных клеток ДНК-конструкцией по любому из пп.8-13 таким образом, чтобы они стали устойчивыми к дикамбе; и
выращивание полученной популяции растительных клеток в культуральной среде, содержащей дикамбу в концентрации, выбранной так, чтобы трансформированные растительные клетки росли, а нетрансформированные растительные клетки не росли.
получение популяции растений, которая может содержать растения, которые были трансформированы ДНК-конструкцией по любому из пп.8-13 таким образом, чтобы они стали устойчивыми к дикамбе; и
нанесение дикамбы на популяцию растений в количестве дикамбы, выбранном так, чтобы трансформированные растения росли, а рост нетрансформированных растений был ингибирован.
a) аминокислотной последовательности, содержащей SEQ ID NO: 5;
b) аминокислотной последовательности, которая представляет собой ферментативно активный фрагмент SEQ ID NO: 5; и
c) аминокислотной последовательности, которая по меньшей мере приблизительно на 65% идентична в отношении SEQ ID NO: 5;
причем указанная аминокислотная последовательность обладает ферментативной активностью дикамба-разрушающего ферредоксина.
a) аминокислотной последовательности, выбранной из группы, состоящей из SEQ ID NO: 7 и SEQ ID NO: 9;
b) аминокислотной последовательности, которая представляет собой ферментативно активный фрагмент аминокислотной последовательности, указанной в (а); и
c) аминокислотной последовательности, которая по меньшей мере приблизительно на 75% идентична аминокислотной последовательности, указанной в (а);
причем указанная аминокислотная последовательность обладает ферментативной активностью дикамба-разрушающей редуктазы.
| WO 9845424, 15.10.1998 | |||
| US 5445962, 29.08.1995 | |||
| СПОСОБ ПОЛУЧЕНИЯ РАСТЕНИЙ ZEA MAYS L., УСТОЙЧИВЫХ К ПОВРЕЖДЕНИЯМ, ВЫЗЫВАЕМЫМ НАСЕКОМЫМИ | 1988 |
|
RU2126047C1 |

