ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к новому способу получения некоторых эфиров циклопропилкарбоновой кислоты и других производных циклопропилкарбоновой кислоты; новому способу получения диметилсульфоксония метилида и диметилсульфония метилида; применению некоторых эфиров циклопропилкарбоновой кислоты в способе получения промежуточных соединений, которые можно использовать в синтезе фармацевтически активных соединений, и к некоторым промежуточным соединениям, получаемым с помощью этих способов. Упомянутые фармацевтически активные соединения могут обладать полезными фармакологическими свойствами в качестве, например, сердечно-сосудистых препаратов. Конкретно, из данных промежуточных соединений могут быть получены соединения, которые являются антагонистами рецепторов Р2Т и полезны в качестве антитромботических агентов.
ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Таким образом, в первом аспекте изобретения предлагается способ получения соединения формулы (I):
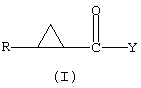
в которой:
R представляет фенил, замещенный одним или несколькими галогенами;
Y представляет OR1, где R1 представляет нормальный алкил, разветвленный алкил, циклоалкил или замещенный бициклогептил (например, борнил), который включает взаимодействие соединения формулы (II):
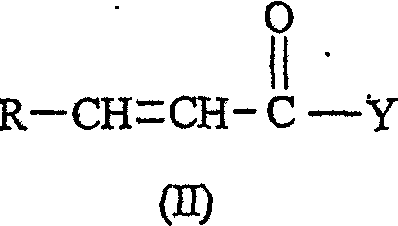
где R и Y имеют значения, определенные выше, с диметилсульфоксонием метилидом в присутствии растворителя.
В удобном случае растворитель является полярным растворителем, предпочтительно диметилсульфоксидом. Соответственно реакцию проводят при -10°С - 90°С, предпочтительно при 25°С.
Диметилсульфоксоний метилид можно получить взаимодействием соли триметилсульфоксония с твердым сильным основанием, предпочтительно в твердом виде, в диметилсульфоксиде при комнатной или повышенной температуре. В удобном случае основание является гидроокисью металла, например NaOH, LiOH, или гидридом щелочного металла, например NaH. Предпочтительно основание представляет гидроокись натрия.
Предпочтительно иодид триметилсульфоксония перемешивают с порошкообразной гидроокисью натрия в диметилсульфоксиде (в отсутствие межфазного катализатора), необязательно в атмосфере азота, при 20 - 25°С в течение 90 мин. Альтернативно диметилсульфоксоний метилид можно получить из соли триметилсульфоксония (предпочтительно иодида или хлорида) с использованием гидроокиси натрия в диметилсульфоксиде с межфазным катализатором, например бромидом тетрабутил-н-аммония или с другими сильными основаниями такими, как гидриды щелочного металла, в диметилсульфоксиде.
Соединение формулы (II) можно получить взаимодействием соединения формулы (III):
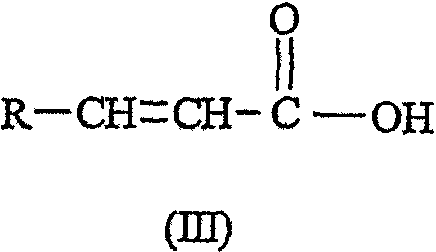
в которой R имеет значения, определенные выше, с подходящим хлорирующим реагентом в присутствии инертного растворителя и необязательного катализатора при температуре 0-200°С. Предпочтительно Y представляет OR1, хлорирующим реагентом является тионилхлорид, инертным растворителем является толуол, и катализатор представляет собой пиридин. В удобном случае температура реакции составляет 70°С. Затем полученный хлорангидрид кислоты взаимодействует с YH или Y- (где Y- представляет анионные частицы Y), Y имеет значения, определенные выше, необязательно при повышенной температуре такой, как 100°С.
Соединение формулы (III) можно получить с использованием обычных химических операций, например взаимодействием соединения формулы (IV):
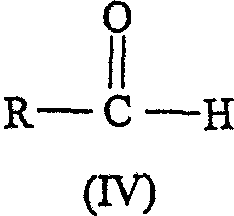
где R имеет значения, определенные выше, с малоновой кислотой в присутствии пиридина и пиперидина при повышенной температуре, предпочтительно 50-90°С.
Соединение формулы (I) можно гидролизовать с использованием основного гидролиза с получением соединения формулы (V):
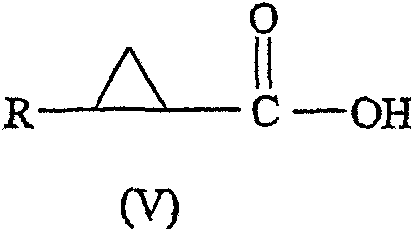
где R имеет значения, определенные выше. Например, эфирные группы предпочтительно удаляют основным гидролизом с использованием гидроокиси щелочного металла, такой как гидроокись натрия или гидроокись лития, или гидроокиси четвертичного аммония в растворителе, таком как вода, водный спирт или водный тетрагидрофуран, при температуре в пределах 10 - 100°С. Наиболее предпочтительно основанием является гидроокись натрия, растворителем является этанол и температура реакции составляет 50°С.
Соединение формулы (V) можно использовать для получения соединения формулы (VI):
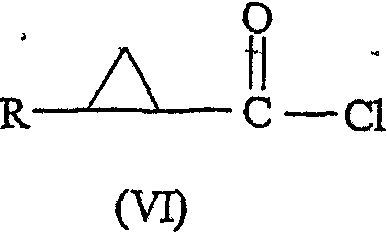
где R имеет значения, определенные выше, взаимодействием с тионилхлоридом или другим подходящим хлорирующим реагентом в присутствии толуола или другого подходящего растворителя и, необязательно, катализатора, предпочтительно пиридина, при 0 - 200°С. Предпочтительно температура составляет 65 - 70°С.
Соединение формулы (VI) можно использовать в синтезе соединения формулы (VII):
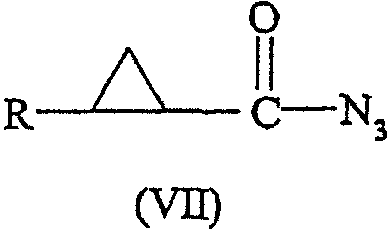
где R имеет значения, определенные выше, взаимодействием азида щелочного металла (предпочтительно азида натрия) в присутствии межфазного катализатора (предпочтительно бромида тетра-н-бутиламмония), водного раствора карбоната калия и инертного растворителя (предпочтительно толуола). Предпочтительно температура реакции составляет 0 - 10°С.
Соединение формулы (VII) можно использовать в синтезе соединения формулы (VIII):
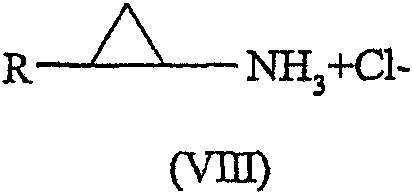
где R имеет значения, определенные выше, перегруппировкой в толуоле при температуре в пределах от 0°С до 200°С, предпочтительно при температуре реакции 90 - 100°С, после чего изоцианатное промежуточное соединение взаимодействует с соляной кислотой при повышенной температуре, предпочтительно 85 - 90°С.
Непротонированный исходный амин (свободное основание) формулы (IX):
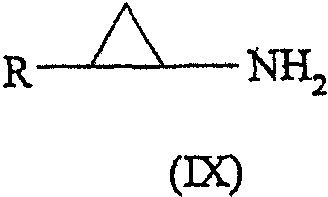
где R имеет значения, определенные выше, можно выделить доведением значения рН водного раствора соли соединения формулы (VIII) до 10 или выше. Затем его можно превратить в другие соли органических кислот или неорганических кислот, предпочтительно миндальной кислоты. Соль R-(-)-миндальной кислоты и соединения формулы (IX) можно получить добавлением R-(-)-миндальной кислоты при комнатной или повышенной температуре к раствору соединения формулы (IX) в растворителе, предпочтительно этилацетате. Предпочтительно температура составляет 20°С.
В удобном случае R представляет фенил, необязательно, замещенный одним или несколькими атомами галогена. Предпочтительно R представляет фенил, замещенный одним или несколькими атомами фтора. Более предпочтительно R является 4-фторфенилом или 3,4-дифторфенилом.
Предпочтительно Y представляет D-ментокси или более предпочтительно L-ментокси.
Соединения формул (I)-(IX) могут существовать в различных изомерных формах (таких как цис/транс, энантиомеры или диастереоизомеры). Способ по данному изобретению включает все такие изомерные формы и их смеси во всех соотношениях.
Там, где Y является хиральным, соединение формулы (I) будет представлять смесь диастереоизомеров, и ее можно разделить кристаллизацией или хроматографическими методами с получением диастереоизомерно обогащенного соединения формулы (Ia):
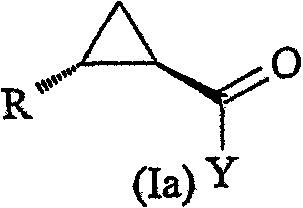
где R и Y имеют значения, определенные выше. Предпочтительно кристаллизацию проводят in situ после синтеза соединения формулы (I), описанной выше, нагреванием неочищенной реакционной смеси, пока не будет достигнуто полного или почти полного растворения, затем охлаждением с соответствующей скоростью, пока не образуется достаточное количество кристаллов желаемого качества. Затем кристаллы собирают фильтрованием. Альтернативно разделение можно провести в любом другом подходящем растворителе таком, как углеводород, например гептан, экстракцией соединения формулы (I) подходящим количеством растворителя, нагреванием экстрактов, пока не будет достигнуто полного растворения, затем охлаждением с соответствующей скоростью, пока не образуется достаточное количество кристаллов желаемого качества. Необязательно органические экстракты можно промыть водой, высушить над сульфатом магния и профильтровать перед кристаллизаций, описанной выше.
Соединение формулы (Ia) можно гидролизовать с получением соединения формулы (Va):
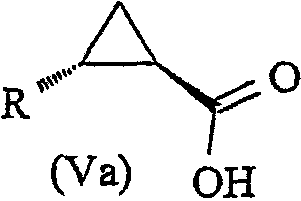
где R имеет значения, определенные выше, с использованием способа, описанного выше для гидролиза соединения формулы (I), с получением соединения формулы (V).
Соединение формулы (Va) можно использовать для получения соединения формулы (VIa):
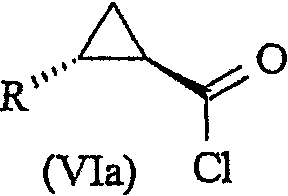
где R имеет значения, определенные выше, с использованием способа, описанного выше для превращения соединения формулы (V), с получением соединения формулы (VI).
Соединение формулы (VIa) можно использовать в синтезе соединения формулы (VIIa):
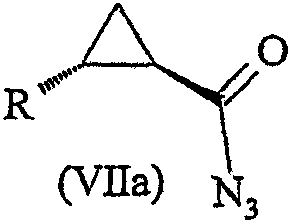
где R имеет значения, определенные выше, с использованием способа, описанного выше для превращения соединения формулы (VI), с получением соединения формулы (VII).
Соединение формулы (VIIa) можно использовать в синтезе соединения формулы (VIIIa):
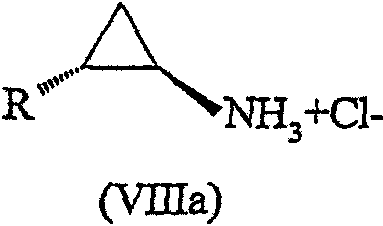
где R имеет значения, определенные выше, с использованием способа, описанного выше для превращения соединения формулы (VII), с получением соединения формулы (VIII).
Соединение формулы (VIIIa) можно использовать в синтезе соединения формулы (IXa):
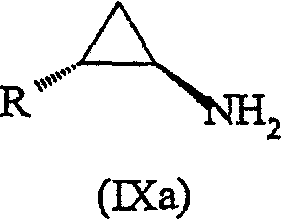
где R имеет значения, определенные выше, с использованием способа, описанного выше для превращения соединения формулы (VIII), с получением соединения формулы (IX).
Соль R-(-)-миндальной кислоты и соединения формулы (IXa) можно получить с использованием способа, описанного выше для получения соли миндальной кислоты и соединения формулы (IX).
Новые соединения образуют дополнительный аспект изобретения. Следовательно, в дополнительном аспекте изобретения предлагаются соединения формул (I), (Ia), (II), (III), (V), (Va), (VI), (VIa), (VII), (VIIa), (VIII), (VIIIa), (IX) и (IXa), определенные выше.
Особенно предпочтительные соединения включают:
((1R,2S,5R)-2-изопропил-5-метилциклогексил)транс-2-(3,4-дифторфенил)циклопропанкарбоксилат;
((1R,2S,5R)-2-изопропил-5-метилциклогексил)транс-(1R,2R)-2-(3,4-дифторфенил)циклопропанкарбоксилат;
((1R,2S,5R)-2-изопропил-5-метилциклогексил)-(Е)-3-(3,4-дифторфенил)-2-пропеноат;
(Е)-3-(3,4-дифторфенил)-2-пропеновая кислота;
(Е)-3-(3,4-дифторфенил)-2-пропеноилхлорид;
транс-(1R,2R)-2-(3,4-дифторфенил)циклопропанкарбоновая кислота;
транс-(1R,2R)-2-(3,4-дифторфенил)циклопропанкарбонилхлорид;
транс-(1R,2R)-2-(3,4-дифторфенил)циклопропанкарбонилазид;
транс-(1R,2S)-2-(3,4-дифторфенил)циклопропиламин;
и транс-(1R,2S)-2-(3,4-дифторфенил)циклопропанаминий (2R)-2-гидрокси-2-фенилэтаноат.
Примеры
Изобретение иллюстрируется следующими неограничивающими примерами.
Пример 1
Данный пример показывает получение (Е)-3-(3,4-дифторфенил)-2-пропеновой кислоты.
Перемешиваемую смесь пиридина (15,5 кг) и пиперидина (0,72 кг) нагревали до 90°С. Добавляли малоновую кислоту (17,6 кг) с последующим медленным добавлением в течение 50 мин 3,4-дифторбензальдегида (12,0 кг). Реакционную смесь перемешивали при 90°С еще в течение 4 ч 36 мин. Добавляли воду (58,5 кг) и затем 32 л смеси пиридин/вода упаривали из реактора при пониженном давлении. Реакционную смесь подкисляли до рН 1 37% соляной кислотой (6,4 кг) в течение 40 мин, затем охлаждали до 25°С при энергичном перемешивании. Твердые частицы собирали фильтрованием, два раза промывали 1% соляной кислотой (34,8 л на порцию), один раз водой (61 л) и затем тщательно удаляли жидкость пропусканием через фильтр. Затем продукт высушивали под вакуумом при 40°С в течение 24 ч 40 мин с получением 13,7 кг кристаллического продукта.
Пример 2
Пример показывает получение (Е)-3-(3,4-дифторфенил)-2-пропеноилхлорида.
Перемешиваемую смесь (Е)-3-(3,4-дифторфенил)-2-пропеновой кислоты (8,2 кг), толуола (7,4 кг) и пиридина (0,18 кг) нагревали при 65°С и затем добавляли тионилхлорид (7,4 кг) в течение 30 мин. Реакционную смесь перемешивали еще в течение 2 ч 15 мин после завершения добавления, затем разбавляли толуолом (8,7 кг). Затем отгоняли избыток тионилхлорида, двуокиси серы и хлористого водорода вместе с толуолом (10 л) при пониженном давлении с получением раствора (Е)-3-(3,4-дифторфенил)-2-пропеноилхлорида (примерно 9 кг) в толуоле.
Пример 3
Данный пример показывает получение (1R,2S,5R)-2-изопропил-5-метилциклогексил (Е)-3-(3,4-дифторфенил)-2-пропеноата.
Раствор L-ментола (7,1 кг) в толуоле (8,5 кг) добавляли в течение 20 мин к раствору (Е)-3-(3,4-дифторфенил)-2-пропеноилхлорида (полученного в примере 2) и пиридина (0,18 кг, 2,28 ммоль), перемешивая при 65°С. Реакционную смесь перемешивали при 65°С еще в течение 4 ч 40 мин после завершения добавления, затем охлаждали до 25°С и перемешивали в течение 14 ч. Раствор разбавляли толуолом (16 кг), промывали 5% водным раствором хлорида натрия (6,4 кг), затем 6% бикарбонатом натрия (6,47 кг), затем водой (6,1 кг). Раствор азеотропно высушивали отгонкой растворителя (20 л) при пониженном давлении. Добавляли диметилсульфоксид (33,9 кг) и оставшийся толуол отгоняли при пониженном давлении с получением 47,3 кг раствора (1R,2S,5R)-2-изопропил-5-метилциклогексил (Е)-3-(3,4-дифторфенил)-2-пропеноата (примерно 13,3 кг) в диметилсульфоксиде.
Пример 4
Данный пример показывает способ получения диметилсульфоксония метилида (диметил(метилен)оксо-λ6-сульфана).
Порошкообразную гидроокись натрия (1,2 кг), полученную измельчением гранул гидроокиси натрия в роторной мельнице с металлическим ситом 1 мм, и иодид триметилсульфоксония (6,2 кг) перемешивали в диметилсульфоксиде (25,2 кг) в атмосфере азота при 25°С в течение 90 мин. Раствор непосредственно использовали для получения (1R,2S,5R)-2-изопропил-5-метилциклогексил-транс-2-(3,4-дифторфенил)циклопропанкарбоксилата.
Пример 5
Данный пример показывает способ получения диметилсульфония метилида (диметил(метилен)-λ4-сульфана).
Порошкообразную гидроокись натрия (970 мг), полученную измельчением гранул гидроокиси натрия в роторной мельнице с металлическим ситом 1 мм, и иодид триметилсульфония (4,66 г) перемешивали в диметилсульфоксиде (17 мл) в атмосфере азота при 20 - 25°С в течение 10 мин. Раствор непосредственно использовали для получения (1R,2S,5R)-2-изопропил-5-метилциклогексил транс-2-(3,4-дифторфенил)циклопропанкарбоксилата.
Пример 6
Данный пример показывает получение (1R,2S,5R)-2-изопропил-5-метилциклогексил-транс-2-(3,4-дифторфенил)циклопропанкарбоксилата.
Раствор (1R,2S,5R)-2-изопропил-5-метилциклогексил (3,4-дифторфенил)-2-пропеноата (примерно 8,6 кг) в диметилсульфоксиде (примерно 27,9 кг) добавляли при перемешивании в течение 20 мин к смеси диметилсульфоксония метилида (примерно 2,6 кг, полученного, как описано выше), иодида натрия ((Е-3) примерно 4,2 кг), воды (примерно 500 г) и гидроокиси натрия (примерно 56 г) в диметилсульфоксиде (27,7 кг) при 25°С. Реакционную смесь перемешивали еще в течение 2 ч 50 мин при 25°С, затем непосредственно использовали для получения (1R,2S,5R)-2-изопропил-5-метилциклогексил-транс-(1R,2R)-2-(3,4-дифторфенил)циклопропанкарбоксилата.
Пример 7
Данный пример показывает получение (1R,2S,5R)-2-изопропил-5-метилциклогексил-транс-(1R,2R)-2-(3,4-дифторфенил)циклопропанкарбоксилата.
Неочищенный раствор (1R,2S,5R)-2-изопропил-5-метилциклогексил-транс-2-(3,4-дифторфенил)циклопропанкарбоксилата, полученного, как описано в примере 6, нагревали при перемешивании от 25°С до 50°С в течение 1 ч и температуру поддерживали еще в течение часа. Затем смесь охлаждали при перемешивании от 50°С до 35°С в течение 4 ч, выдерживали при 35°С в течение 1 ч, затем охлаждали до 26°С в течение 4 ч, выдерживали при 26°С в течение 1 ч, затем охлаждали до 19°С в течение 3 ч и выдерживали при 19°С в течение 5 ч 10 мин. Продукт кристаллизовали и собирали фильтрованием с получением кристаллического твердого вещества (2,7 кг), которое, как было показано, включает смесь (1R,2S,5R)-2-изопропил-5-метилциклогексил-транс-(1R,2R)-2-(3,4-дифторфенил)циклопропанкарбоксилата (1,99 кг) и (1R,2S,5R)-2-изопропил-5-метилциклогексил-транс-(1S,2S)-2-(3,4-дифторфенил)циклопропанкарбоксилата (85 г).
Пример 8
Данный пример показывает альтернативный способ получения (1R,2S,5R)-2-изопропил-5-метилциклогексил-транс-(1R,2R)-2-(3,4-дифторфенил)циклопропанкарбоксилата.
н-Гептан (82,5 л) отгоняли при пониженном давлении из раствора (1R,2S,5R)-2-изопропил-5-метилциклогексил-транс-2-(3,4-дифторфенил)циклопропанкарбоксилата (14,3 кг, 44,4 моль) в гептане (128,6 л). Затем смесь охлаждали от 34°С до 24°С в течение 3 ч 20 мин. Затем вносили затравочные кристаллы (1R,2S,5R)-2-изопропил-5-метилциклогексил-транс-(1R,2R)-2-(3,4-дифторфенил)циклопропанкарбоксилата и смесь охлаждали до 0°С в течение 5 ч 50 мин. Фильтрование давало продукт в виде кристаллического, содержащего растворитель твердого вещества (7,05 кг), которое, как было показано, содержало смесь (1R,2S,5R)-2-изопропил-5-метилциклогексил- транс-(1R,2R)-2-(3,4-дифторфенил)циклопропанкарбоксилата (4,7 кг) и (1R,2S,5R)-2-изопропил-5-метилциклогексил-транс-(1S,2S)-2-(3,4-дифторфенил)циклопропанкарбоксилата (1,1 кг).
Пример 9
Данный пример показывает способ получения транс-(1R,2R)-2-(3,4-дифторфенил)циклопропанкарбоновой кислоты.
(1R,2S,5R)-2-Изопропил-5-метилциклогексил-транс-(1R,2R)-2-(3,4-дифторфенил)циклопропанкарбоксилат (9,6 кг, 91,8% избыток диастереоизомера) растворяли в этаноле (13,8 кг) и нагревали при перемешивании до 46°С. Добавляли 45% водный раствор гидроокиси натрия (3,1 кг) в течение 20 мин и смесь перемешивали еще в течение 2 ч 27 мин. Растворитель (28 л) отгоняли из смеси при пониженном давлении, затем смесь охлаждали до 24°С и разбавляли водой (29,3 кг), после чего выделившийся ментол экстрагировали толуолом (3 порции по 3,3 кг каждая). Оставшееся водное вещество подкисляли до рН 2 37% соляной кислотой (3,3 л) и продукт экстрагировали толуолом (8,6 кг, 2 порции по 4,2 кг и 4,3 кг). Объединенные толуольные экстракты промывали 1% соляной кислотой (4,9 л), затем разбавляли еще толуолом (4,2 кг) и высушивали азеотропной отгонкой растворителя (25 л) при пониженном давлении. После конечного разбавления толуолом (24,2 кг) растворитель отгоняли при пониженном давлении (10 л) с получением раствора, содержащего транс-(1R,2R)-2-(3,4-дифторфенил)циклопропанкарбониловой кислоты (примерно 3,45 кг), подходящую для получения транс-(1R,2R)-2-(3,4-дифторфенил)циклопропанкарбонилхлорида.
Пример 10
Данный пример показывает способ получения транс-(1R,2R)-2-(3,4-дифторфенил)циклопропанкарбонилхлорида.
Пиридин (70 мл) добавляли к раствору транс-(1R,2R)-2-(3,4-дифторфенил)циклопропанкарбоновой кислоты (примерно 3,45 кг) в толуоле (примерно 12-15 кг), полученной, как описано выше, и затем смесь нагревали до 65°С. Добавляли тионилхлорид (2,3 кг) в течение 1 ч и смесь перемешивали при 70°С в течение 3 ч. Добавляли тионилхлорид (0,5 кг) и смесь перемешивали еще в течение 2 ч при 70°С. Добавляли конечную порцию тионилхлорида (0,5 кг) и реакционную смесь перемешивали в течение 1 ч при 70°С, затем охлаждали до 40°С. Во время отгонки растворителя (примерно 60 л) из смеси при пониженном давлении проводили периодические добавления толуола (45 кг, 3 внесения по 15 кг каждое), затем раствор транс-(1R,2R)-2-(3,4-дифторфенил)циклопропанкарбонилхлорида (примерно 3,8 кг) в толуоле (примерно 6-9 л) охлаждали до 20°С.
Пример 11
Данный пример показывает способ получения транс-(1R,2R)-2-(3,4-дифторфенил)циклопропанкарбонилазида.
Раствор транс-(1R,2R)-2-(3,4-дифторфенил)циклопропанкарбонилхлорида (примерно 3,8 кг) в толуоле (примерно 6-9 л) при 1°С добавляли в течение 74 мин к смеси азида натрия (1,24 кг), бромида тетрабутиламмония (56 г) и карбоната натрия (922 г) в воде (6,2 кг) при перемешивании при 1,5°С. Смесь перемешивали при 0°С в течение 1 ч 55 мин, затем водный слой разбавляли холодной водой (3,8 кг), быстро перемешивали, затем разделяли. Толуольный слой промывали еще раз при 0°С водой (3,8 кг), затем 20% водным раствором хлорида натрия (3,8 л), затем хранили при 3°С для дальнейшего применения.
Пример 12
Данный пример показывает способ получения транс-(1R,2S)-2-(3,4-дифторфенил)циклопропиламина.
Холодный раствор транс-(1R,2R)-2-(3,4-дифторфенил)циклопропанкарбонилазида, полученного, как описано в примере 11, добавляли в течение 41 мин к толуолу (6,0 кг), перемешивая при 100°С. Смесь перемешивали еще 55 мин при 100°С, затем охлаждали до 20°С и добавляли в течение 2 ч 15 мин к соляной кислоте (3 М, 18,2 кг), перемешивая при 80°С. Через 65 мин раствор разбавляли водой (34 кг) и охлаждали до 25°С. Толуольный слой удаляли и водный слой подщелачивали до рН 12 45% раствором гидроокиси натрия (3,8 кг) и затем продукт экстрагировали этилацетатом (31 кг) и дважды промывали водой (13,7 кг на порцию) с получением раствора, содержащего транс-(1R,2S)-2-(3,4-дифторфенил)циклопропиламин (2,6 кг, 91,8% избыток этантиомера) в этилацетате (29,5 л).
Пример 13
Данный пример показывает способ получения транс-(1R,2S)-2-(3,4-дифторфенил)циклопропанаминия (2R)-2-гидрокси-2-фенилэтаноата.
R-(-)миндальную кислоту (2,26 кг) добавляли к раствору, содержащему транс-(1R,2S)-2-(3,4-дифторфенил)циклопропиламин (2,6 кг, более 91,8% избыток энантиомера), перемешивая при 17°С в этилацетате (45,3 л). Смесь перемешивали при 25°С в течение 3 ч 8 мин, затем фильтровали и дважды промывали этилацетатом (в целом 13,8 кг). Кристаллический продукт высушивали при 40°С при пониженном давлении в течение 23 ч с получением транс-(1R,2S)-2-(3,4-дифторфенил)циклопропанаминия (2R)-2-гидрокси-2-фенилэтаноата (4,45 кг).
| название | год | авторы | номер документа |
|---|---|---|---|
| ПИРИМИДИНОВЫЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ), ПРОМЕЖУТОЧНЫЕ ПРОДУКТЫ (ВАРИАНТЫ) И СПОСОБЫ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ), СПОСОБ ПОЛУЧЕНИЯ ТРИАЗОЛОПИРИМИДИНОВЫХ СОЕДИНЕНИЙ | 2001 |
|
RU2295526C2 |
| СПОСОБ ПОЛУЧЕНИЯ (R)-5-(3,4-ДИФТОРФЕНИЛ)-5-[(3-МЕТИЛ-2-ОКСОПИРИДИН-1(2H)-ИЛ)МЕТИЛ]ИМИДАЗОЛИДИН-2,4-ДИОНА И ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ ДЛЯ ЕГО ПОЛУЧЕНИЯ | 2017 |
|
RU2735600C2 |
| ПРОЛЕКАРСТВО ПРОИЗВОДНОГО АМИНОКИСЛОТЫ | 2017 |
|
RU2739318C2 |
| ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛА, ПОЛЕЗНЫЕ В КАЧЕСТВЕ БЛОКАТОРОВ T/L КАНАЛОВ | 2009 |
|
RU2478095C2 |
| АРИЛ-ЗАМЕЩЕННЫЕ КАРБОКСАМИДНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ БЛОКАТОРОВ КАЛЬЦИЕВЫХ ИЛИ НАТРИЕВЫХ КАНАЛОВ | 2010 |
|
RU2575168C2 |
| ПРОИЗВОДНОЕ ЦИННАМИДА ТИПА МОРФОЛИНА | 2006 |
|
RU2381225C1 |
| СЛОЖНОЭФИРНОЕ ПРОИЗВОДНОЕ 2-АМИНО-БИЦИКЛО[3.1.0]ГЕКСАН-2,6-ДИКАРБОНОВОЙ КИСЛОТЫ | 2004 |
|
RU2409557C2 |
| НОВЫЕ ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ДЛЯ ПОЛУЧЕНИЯ ТРИАЗОЛО(4,5-d)ПИРИМИДИНА | 2012 |
|
RU2593201C2 |
| ЧАСТИЧНЫЕ И ПОЛНЫЕ АГОНИСТЫ АДЕНОЗИНОВЫХ РЕЦЕПТОРОВ A | 2003 |
|
RU2340623C2 |
| ПРОЛЕКАРСТВО ФТОРСОДЕРЖАЩЕЙ АМИНОКИСЛОТЫ | 2013 |
|
RU2639868C1 |
Изобретение относится к новому способу получения некоторых эфиров циклопропилкарбоновой кислоты и других производных циклопропилкарбоновой кислоты общей формулы I
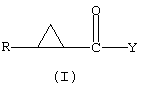
Изобретение также относится к новому способу получения диметилсульфоксония метилида и диметилсульфония метилида, применению некоторых эфиров циклопропилкарбоновой кислоты в способе получения промежуточных соединений, которые можно использовать в синтезе фармацевтически активных соединений, и некоторым промежуточным соединениям, получаемым данными способами. 5 н. и 10 з.п. ф-лы.
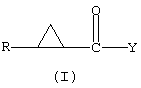
в которой
R представляет фенил, замещенный одним или несколькими галогенами;
Y представляет OR1, где R1 представляет линейный алкил, разветвленный алкил, циклоалкил или замещенный бициклогептил, который включает
взаимодействие соли триметилсульфоксония с твердой гидроокисью металла в диметилсульфоксиде при комнатной или повышенной температуре с получением диметилсульфоксония метилида;
взаимодействие соединения формулы (II)
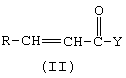
где R и Y имеют значения, определенные выше,
с диметилсульфоксонием метилидом в присутствии растворителя при температуре в пределах 10-90°С.
в которой
R представляет фенил, замещенный одним или несколькими галогенами;
Y представляет OR1, где R1 представляет линейный алкил, разветвленный алкил, циклоалкил или замещенный бициклогептил, который включает
а) хлорирование соединения формулы (III)
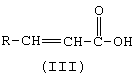
где R и Y имеют значения, определенные выше, взаимодействием с хлорирующим реагентом в присутствии инертного растворителя и катализатора при температуре в пределах 0-200°С, и затем взаимодействие полученного раствора с YH или Y-, где Y имеет значения, определенные выше, при повышенной температуре, с получением соединения формулы (II)
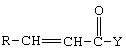
где R и Y имеют значения, определенные выше;
б) взаимодействие соли триметилсульфоксония с твердой гидроокисью металла в диметилсульфоксиде при комнатной или повышенной температуре с получением диметилсульфоксония метилида; и
в) взаимодействие соединения формулы (II) с диметилсульфоксонием метилидом в присутствии растворителя при температуре в пределах 10-90°С.
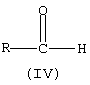
где R имеет значения, определенные в п.1, с малоновой кислотой в присутствии пиридина и пиперидина при повышенной температуре.
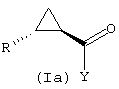
где R и Y имеют значения, определенные в п.1, кристаллизацией или хроматографическими методами.
| ПРОИЗВОДНЫЕ БИЦИКЛИЧЕСКИХ АМИДОВ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЛЕЧЕНИЯ ПАТОЛОГИЧЕСКИХ И ВОСПАЛИТЕЛЬНЫХ СОСТОЯНИЙ | 1994 |
|
RU2143422C1 |
| WO 200021910 A2, 20.04.2000 | |||
| Journal of the American Chemicals Society | |||
| Торфодобывающая машина с вращающимся измельчающим орудием | 1922 |
|
SU87A1 |
| E.COREY et al | |||
| Устройство для автоматического выключения оборванного трамвайного провода | 1923 |
|
SU1353A1 |
| Organic Chemistry | |||
| Fifth Edition | |||
| T.W.GRAHAM SOLOMONS | |||
| Пуговица для прикрепления ее к материи без пришивки | 1921 |
|
SU1992A1 |
| US 5929291 A, 27.07.1999. | |||

