Настоящее изобретение относится к пиримидиновому соединению, пригодному в качестве фармацевтического промежуточного соединения, способу получения указанного пиримидинового соединения, промежуточным соединениям, используемым в указанном способе, и применению указанного пиримидинового соединения при получении фармацевтических препаратов.
В настоящем изобретении предлагается соединение формулы (I):
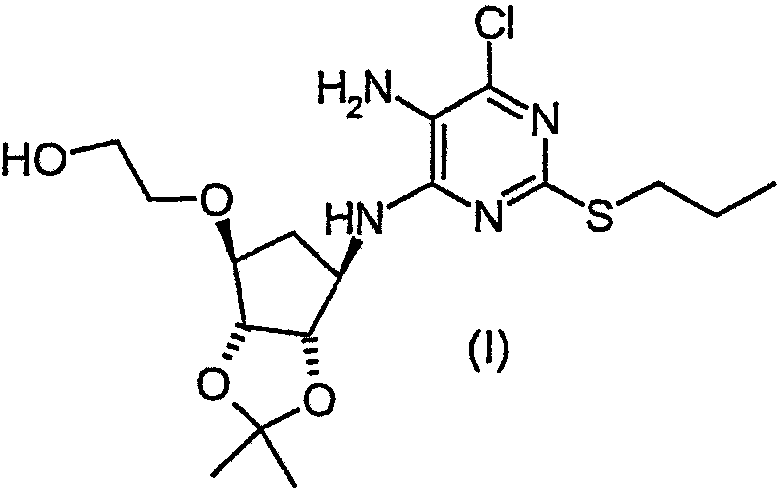
В настоящем изобретении предлагается также способ получения соединения формулы (I), включающий взаимодействие соединения формулы (II):
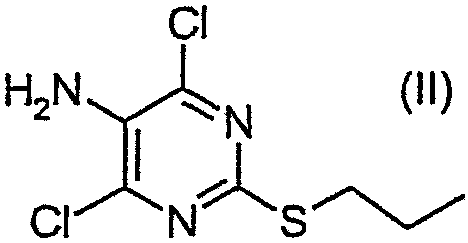
с солью соединения формулы (III):

в присутствии подходящего основания (такого как гидроокись щелочного металла (как, например, гидроокись натрия или калия), третичный амин (такой как три(С1-6алкил)амин, например, триэтиламин)), подходящего растворителя (такого как спирт, как, например, алифатический спирт, содержащий 1-6 атомов углерода, например этанол), предпочтительно при температуре в пределах 100 - 150°С, и, где это необходимо (например, когда температура превышает точку кипения растворителя), в герметичной системе под автогенным давлением.
Подходящей солью соединения формулы (III) является соль минеральной или органической кислоты. Подходящие минеральные кислоты включают соляную, бромистоводородную, иодистоводородную, азотную или серную кислоту. Подходящей органической кислотой является, например, органическая ахиральная кислота, такая как уксусная, трифторуксусная, щавелевая или п-толуолсульфоновая кислота, или органическая хиральная кислота, такая как L-винная кислота, дибензоил-L-винная кислота или ди-п-толуоил-L-винная кислота.
В другом аспекте настоящего изобретения предлагается способ получения соединения формулы (I), включающий гидрирование соединения формулы (IV):
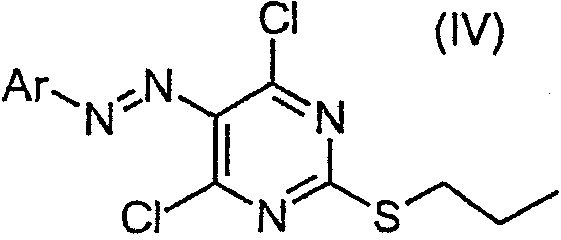
в которой Ar представляет фенил, необязательно замещенный галогеном, С1-4алкилом или С1-4алкокси, с получением соединения формулы (II), и взаимодействие соединения формулы (II) с соединением формулы (III) (как описано выше) с получением соединения формулы (I).
Предпочтительно гидрирование проводят с использованием катализатора на основе тяжелого металла (такого как платина на угле) в подходящем растворителе (таком как С1-6алифатический спирт, например, 2-пропанол (изопропанол)) при подходящей температуре (такой, как 10 - 70°С, например, 20 - 50°С) и при подходящем давлении (таком как 1-5 бар, например, примерно 3 бар).
Соединение формулы (IV) можно получить хлорированием соединения формулы (VIII):
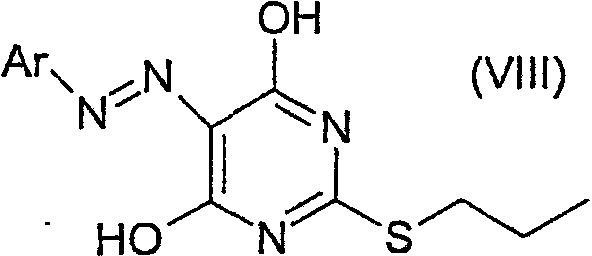
где Ar имеет значения, определенные выше, с помощью подходящего хлорирующего реагента (такого как хлорокись фосфора) в присутствии подходящего азотсодержащего основания (такого как триэтиламин, особенно пиридин) и при подходящей температуре (такой как в пределах от 50°С до точки кипения хлорокиси фосфора, например, 70 - 90°С). Соединение формулы (VIII) можно получить обычным применением способов, описанных в литературе.
В дополнительном аспекте настоящего изобретения предлагается способ, как описано выше, получения соединения формулы (II).
Соединение формулы (I) можно использовать для получения фармацевтического соединения формулы (А):

как описано ниже.
Так, соединение формулы (А) можно получить снятием защиты с соединения формулы (V):
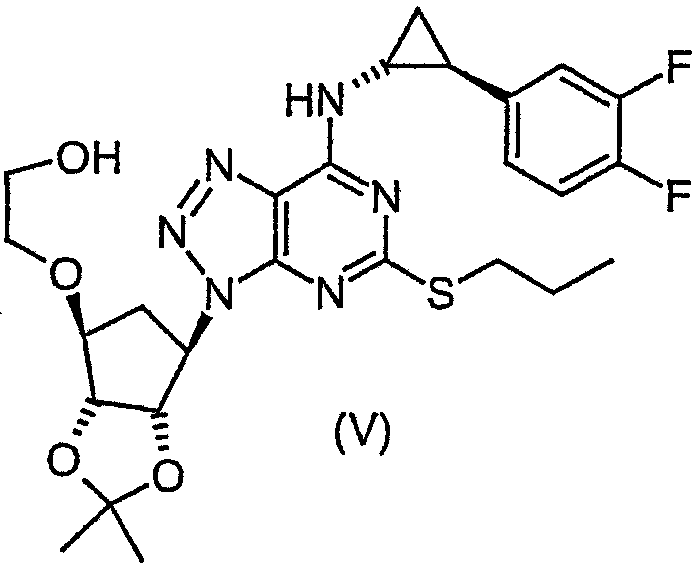
например, с использованием сильной минеральной кислоты (такой как соляная кислота) в подходящем растворителе (таком как метанол или этанол).
Соединение формулы (V) можно получить сочетанием соединения формулы (VI) {или его соли (такой как манделат), из которой соединение формулы (VI) получают in situ} с соединением формулы (VII):

например, в присутствии подходящего основания (такого как третичный амин, такой как три(С1-6алкил)амин, например, триэтиламин) и подходящего растворителя (например, полярного растворителя, такого как спирт (такого как алифатический спирт, содержащий 1-6 атомов углерода, например этанол) или нитрил (такого как ацетонитрил) и при подходящей температуре (такой как температура в пределах 10 - 40°С, например комнатной температуре).
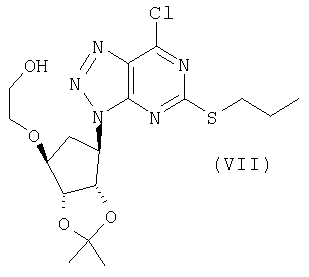
Соединение формулы (VII) можно получить взаимодействием соединения формулы (I) с нитритом щелочного металла (такого как NaNO2) или органическим нитритом (например, изоамилнитритом) в присутствии подходящей кислоты (такой как уксусная кислота) и подходящего растворителя (такого как вода или смесь воды и уксусной кислоты) и при подходящей температуре (такой как температура в пределах от -10 до 15°С, например от -10 до 10°С).
Таким образом, в дополнительном аспекте настоящего изобретения предлагается применение соединения формулы (I) в способе получения соединения формулы (А).
Соль соединения формулы (III) можно получить взаимодействием соединения формулы (III) с необходимой кислотой в подходящем растворителе (таком как вода, алифатический спирт, содержащий 1-4 атомов углерода (например, этанол), или обычном сложном эфире (таком как этилацетат) при подходящей температуре (такой как от 10 до 60°С, например, от 30 до 50°С).
Соединение формулы (III) можно получить снятием защиты с соединения формулы (IX):

например, гидрированием (как, например, с катализатором на основе тяжелого металла (такого как палладий на угле) в присутствии растворителя (такого как алифатический спирт, содержащий 1-4 атомов углерода, например этанол) при комнатной температуре при подходящем давлении (таком как 1-3 бар, например 1,0-1,5 бар)).
Соединение формулы (IX) можно получить восстановлением соединения формулы (X):

в которой R* представляет С1-4алкил (предпочтительно этил), подходящим боргидридом (например, боргидридом щелочного металла, таким как боргидрид лития), алюмогидридом лития или DIBAL-H в подходящем полярном растворителе (таком как тетрагидрофуран).
Соединение формулы (X) можно получить взаимодействием соединения формулы (XI):

с подходящим соединением L-CH2CO2R* {где R* представляет С1-4алкил (особенно этил); и L представляет уходящую группу, особенно галоген (например, бром)} в присутствии подходящего полярного растворителя (такого как тетрагидрофуран) и в присутствии подходящего основания (такого как трет-бутилат калия, гидрид натрия или разновидности С1-6алкиллития).
Соединение формулы (XI) можно получить взаимодействием соединения формулы (XII):

с бензилхлорформиатом в присутствии подходящего основания (такого как карбонат калия) и подходящего растворителя (такого как кетон (например, 4-метил-2-пентанон) или углеводорода (например, толуола)).
В еще одном дополнительном аспекте настоящего изобретения предлагается способ получения соли соединения формулы (III), описанной ранее.
В дополнительных аспектах настоящего изобретения предлагается промежуточное соединение формул (II), (IV), (VII), (VIII), (X) или (XI) или соль соединения формулы (III).
Последующие примеры иллюстрируют изобретение.
ПРИМЕР 1
Данный пример показывает получение 2-{[(3aR,4S,6R,6aS)-6-амино-2,2-диметилтетрагидро-3аН-циклопента[d][1,3]диоксол-4-ил]окси}-1-этанола, соль с L-винной кислотой (1:1).
Стадия а: получение фенилметилового эфира [3aS-(3aα,4α,6α,6aα)]-[тетрагидро-6-гидрокси-2,2-диметил-4Н-циклопента-1,3-диоксол-4-ил]карбаминовой кислоты.
Карбонат калия (39,3 г) добавляли к суспензии [3aR-(3aα,4α,6α,6aα)]-6-амино-тетрагидро-2,2-диметил-4Н-циклопента-1,3-диоксол-4-ола гидрохлорида (полученного, как описано в WO 9905142) (27,1 г) в 4-метил-2-пентаноне (500 мл). Затем добавляли воду (150 мл) с последующим добавлением по каплям бензилхлорформиата (23,1 г). Реакционную смесь перемешивали при комнатной температуре в течение 4 ч перед отделением органической фазы. Водную фазу экстрагировали 4-метил-2-пентаноном (2×50 мл). Объединенные органические фазы концентрировали и осадок очищали (SiO2, смесь дихлорметан:метанол, от 95:5 до 90:10 в качестве элюента) с получением указанного в подзаголовке соединения (39,23 г).
1Н ЯМР (CDCl3) δ 7,32 (5H, м), 5,65 (1H, шир. с), 5,10 (2H, шир. с), 4,59 (1H, д), 4,48 (1H, д), 4,27 (1H, м), 4,19 (1H, шир. м), 2,24 (1H, шир. с), 1,69 (1H, д), 1,41 (3H, с), 1,26 (3H, с).
Стадия b: получение фенилметилового эфира [3aS-(3aα,4α,6α,6aα)]-[2,2-диметил-6-(2-гидроксиэтокси)тетрагидро-4Н-циклопента-1,3-диоксол-4-ил]карбаминовой кислоты.
Трет-бутилат калия (3,6 г) в тетрагидрофуране (20 мл) добавляли в течение 5 мин к раствору продукта со стадии (а) (39,23 г) в тетрагидрофуране (200 мл). Через 15 мин добавляли по каплям этилбромацетат (3,7 мл) в тетрагидрофуране (10 мл). Смесь перемешивали при 0°С в течение 10 мин, затем вновь добавляли этилбромацетат (3,7 мл × 4). Реакционную смесь перемешивали при 0°С в течение еще 2 ч.
Затем к реакционной смеси добавляли порциями боргидрид лития (2,79 г), которую перемешивали при <5°С в течение 16 ч. К холодной смеси добавляли ледяную уксусную кислоту (23 г). Через 30 мин перемешивания добавляли по каплям воду (100 мл) и полученную смесь перемешивали в течение 30 мин. Затем фазы разделяли и водную фазу экстрагировали этилацетатом. Объединенные органические фазы промывали насыщенным раствором бикарбоната натрия и рассолом, высушивали и концентрировали. Остаток очищали (SiO2, смесь этилацетат:гексан, от 25:75 до 50:50 в качестве элюента) с получением указанного в подзаголовке соединения (38,6 г).
МС (химическая ионизация при атмосферном давлении) (APCI) 218 (М+Н+, 100%).
Стадия с: получение [3aR-(3aα,4α,6α,6aα)]-2-[[6-амино-2,2-диметил-тетрагидро-4Н-циклопента-1,3-диоксол-4-ил]окси]этанола (альтернативно называемого: 2-{[(3aR,4S,6R,6aS)-6-амино-2,2-диметилтетрагидро-3аН-циклопента[d][1,3]диоксол-4-ил]окси}-1-этанол).
Суспензию 5% палладия на угле (4 г) в этаноле добавляли к раствору продукта со стадии (b) (39,96 г) в этаноле (250 мл) и смесь гидрировали под давлением 1,2 бар в течение 20 ч. Катализатор отфильтровывали и фильтрат концентрировали с получением указанного в подзаголовке соединения (23,65 г).
МС (APCI) 160 (М+Н+, 100%).
Стадия d: получение [3aR-(3аα,4α,6α,6aα)]-2-[[6-амино-2,2-диметил-тетрагидро-4Н-циклопента-1,3-диоксол-4-ил]окси]этанол-L тартрата (альтернативно называемого: 2-{[(3aR,4S,6R,6aS)-6-амино-2,2-диметилтетрагидро-3аН-циклопента[d][1,3]диоксол-4-ил]окси}-1-этанол, соль с L-винной кислотой (1:1)).
Перемешиваемый раствор продукта, полученного на стадии (с) (545 г) в этаноле (3,8 л), нагревали до 35°С. Добавляли L-винную кислоту (352 г) (температуру поднимали до 45°С) и смесь перемешивали при 40 - 45°С в течение 1 ч. Смесь охлаждали до 20°С и полученную густую суспензию перемешивали в течение 16 ч, затем фильтровали. Собранное твердое вещество промывали двумя порциями 2-пропанола (300 мл, затем 500 мл), отсасывали досуха, затем высушивали в вакууме при 40°С с получением продукта в виде белых кристаллов (728 г).
ПРИМЕР 2
Данный пример показывает получение транс-(1R,2S)-2-(3,4-дифторфенил)-2-циклопропиламина R-манделата (альтернативно называемого транс-(1R,2S)-2-(3,4-дифторфенил)циклопропанаминий (2R)-2-гидрокси-2-фенилэтаноатом).
Стадия 1: получение (Е)-3-(3,4-дифторфенил)-2-пропеновой кислоты.
Перемешиваемую смесь пиридина (15,5 кг) и пиперидина (0,72 кг) нагревали до 90°С. Добавляли малоновую кислоту (17,6 кг), затем медленно в течение 50 мин 3,4-дифторбензальдегид (12,0 кг). Реакционную смесь перемешивали при 90°С в течение еще 4 ч 36 мин. Добавляли воду (58,5 кг) и затем 32 л смеси пиридин/вода упаривали из реактора при пониженном давлении. Реакционную смесь подкисляли до рН 1 37% соляной кислотой (6,4 кг) в течение 40 мин, затем охлаждали до 25°С при энергичном перемешивании. Твердые частицы собирали фильтрованием, дважды промывали 1% раствором соляной кислоты (34,8 л на порцию), один раз водой (61 л) и затем тщательно удаляли жидкость пропусканием через фильтр. Продукт высушивали под вакуумом при 40°С в течение 24 ч 40 мин с получением 13,7 кг кристаллического продукта.
Стадия 2: получение (Е)-3-(3,4-дифторфенил)-2-пропеноилхлорида.
Перемешиваемую смесь (Е)-3-(3,4-дифторфенил)-2-пропеновой кислоты (8,2 кг), толуола (7,4 кг) и пиридина (0,18 кг) нагревали до 65°С и затем в течение 30 мин добавляли тионилхлорид (7,4 кг). Реакционную смесь перемешивали в течение еще 2 ч 15 мин после полного добавления, затем разбавляли толуолом (8,7 кг). Избыток тионилхлорида, двуокиси серы и хлористого водорода затем отгоняли вместе с толуолом (10 л) при пониженном давлении с получением раствора (Е)-3-(3,4-дифторфенил)-2-пропеноилхлорида (примерно 9 кг) в толуоле.
Стадия 3: получение (1R,2S,5R)-2-изопропил-5-метилциклогексил (Е)-3-(3,4-дифторфенил)-2-пропеноата.
Раствор L-ментола (7,1 кг) в толуоле (8,5 кг) добавляли в течение 20 мин к раствору (Е)-3-(3,4-дифторфенил)-2-пропеноилхлорида (полученного на стадии 2) и пиридина (0,18 кг, 2,28 моль), перемешивая при 65°С. Реакционную смесь перемешивали при 65°С в течение 4 ч 40 мин после полного добавления, затем охлаждали до 25°С и перемешивали в течение еще 14 ч. Раствор разбавляли толуолом (16 кг), промывали 5% водным раствором хлорида натрия (6,4 кг), затем 6% раствором бикарбоната натрия (6,47 кг), затем водой (6,1 кг). Раствор высушивали азеотропной отгонкой растворителя (20 л) при пониженном давлении. Добавляли диметилсульфоксид (33,9 кг) и оставшийся толуол отгоняли при пониженном давлении с получением 47,3 кг раствора (1R,2S,5R)-2-изопропил-5-метилциклогексил (Е)-3-(3,4-дифторфенил)-2-пропеноата (примерно 13,3 кг) в диметилсульфоксиде.
Стадия 4: получение диметилсульфоксония метилида (диметил(метилен)оксо)-λ6-сульфана.
Порошкообразную гидроокись натрия (1,2 кг), полученную измельчением гранул гидроокиси натрия в роторной мельнице с металлическим ситом 1 мм, и триметилсульфоксоний иодид (6,2 кг) перемешивали в диметилсульфоксиде (25,2 кг) в атмосфере азота при 25°С в течение 90 мин. Раствор непосредственно использовали для получения (1R,2S,5R)-2-изопропил-5-метилциклогексил-транс-2-(3,4-дифторфенил)циклопропанкарбоксилата.
Стадия 5: получение (1R,2S,5R)-2-изопропил-5-метилциклогексил-транс-2-(3,4-дифторфенил)циклопропанкарбоксилата.
Раствор (1R,2S,5R)-2-изопропил-5-метилциклогексил (Е)-3-(3,4-дифторфенил)-2-пропеноата (примерно 8,6 кг) в диметилсульфоксиде (примерно 27,9 кг) добавляли при перемешивании в течение 20 мин к смеси диметилсульфоксония метилида (примерно 2,6 кг полученного, как описано выше), иодида натрия (примерно 4,2 кг), воды (примерно 500 г) и гидроокиси натрия (примерно 56 г) в диметилсульфоксиде (27,7 кг) при 25°С. Реакционную смесь перемешивали в течение еще 2 ч 50 мин при 25°С, затем непосредственно использовали для получения (1R,2S,5R)-2-изопропил-5-метилциклогексил-транс-(1R,2R)-2-(3,4-дифторфенил)циклопропанкарбоксилата.
Стадия 6: получение (1R,2S,5R)-2-изопропил-5-метилциклогексил-транс-(1R,2R)-2-(3,4-дифторфенил)циклопропанкарбоксилата.
Неочищенный раствор (1R,2S,5R)-2-изопропил-5-метилциклогексил-транс-2-(3,4-дифторфенил)циклопропанкарбоксилата, полученный, как описано на стадии 5, нагревали при перемешивании от 25°С до 50°С в течение 1 ч и температуру поддерживали в течение еще одного часа. Затем смесь охлаждали при перемешивании от 50°С до 35°С в течение 4 ч, выдерживали при 35°С в течение 1 ч, затем охлаждали до 26°С в течение 4 ч, выдерживали при 26°С в течение 1 ч, затем охлаждали до 19°С в течение 3 ч и выдерживали при 19°С в течение 5 ч 10 мин. Продукт собирали фильтрованием с получением кристаллического твердого вещества (2,7 кг), которое, как было показано, включало смесь (1R,2S,5R)-2-изопропил-5-метилциклогексил-транс-(1R,2R)-2-(3,4-дифторфенил)циклопропанкарбоксилата (1,99 кг) и (1R,2S,5R)-2-изопропил-5-метилциклогексил-транс-(1S,2S)-2-(3,4-дифторфенил)циклопропанкарбоксилата (85 г).
Стадия 7: получение транс-(1R,2R)-2-(3,4-дифторфенил)циклопропанкарбоновой кислоты.
(1R,2S,5R)-2-Изопропил-5-метилциклогексил-транс-(1R,2R)-2-(3,4-дифторфенил)циклопропанкарбоксилат (9,6 кг, 91,8% избыток диастереоизомера) растворяли в этаноле (13,8 кг) и нагревали при перемешивании до 46°С. В течение 20 мин добавляли 45% водный раствор гидроокиси натрия (3,1 кг) и смесь перемешивали в течение еще 2 ч 27 мин. Растворитель (28 л) отгоняли из смеси при пониженном давлении, затем смесь охлаждали до 24°С и разбавляли водой (29,3 кг), после чего выделенный ментол экстрагировали толуолом (3 порции по 3,3 кг каждая). Оставшееся водное вещество подкисляли до рН 2 37% соляной кислотой (3,3 л) и продукт экстрагировали толуолом (8,6 кг, затем еще 2 порции по 4,2 кг и 4,3 кг). Объединенные толуольные экстракты промывали 1% соляной кислотой (4,9 л), затем разбавляли еще толуолом (4,2 кг) и высушивали азеотропной отгонкой растворителя (25 л) под пониженном давлении. После конечного разбавления толуолом (24,2 кг) растворитель выпаривали при пониженном давлении (10 л) с получением раствора, содержащего транс-(1R,2R)-2-(3,4-дифторфенил)циклопропанкарбоновую кислоту (примерно 3,45 кг), подходящую для получения транс-(1R,2R)-2-(3,4-дифторфенил)циклопропанкарбонилхлорида.
Стадия 8: получение транс-(1R,2R)-2-(3,4-дифторфенил)циклопропанкарбонилхлорида.
Пиридин (70 мл) добавляли к раствору транс-(1R,2R)-2-(3,4-дифторфенил)циклопропанкарбоновой кислоты (примерно 3,45 кг) в толуоле (примерно 12-15 кг), полученному, как описано выше, и смесь затем нагревали до 65°С. Тионилхлорид (2,3 кг) добавляли в течение 1 ч и смесь перемешивали при 70°С в течение 3 ч. Добавляли тионилхлорид (0,5 кг) и смесь перемешивали в течение еще 2 ч при 70°С. Добавляли последнюю порцию тионилхлорида (0,5 кг) и реакционную смесь перемешивали в течение 1 ч при 70°С, затем охлаждали до 40°С. Во время отгонки растворителя (примерно 60 л) из смеси при пониженном давлении периодически добавляли толуол (45 кг, 3 внесения по 15 кг каждое), затем раствор транс-(1R,2R)-2-(3,4-дифторфенил)циклопропанкарбонилхлорида (примерно 3,8 кг) в толуоле (примерно 6-9 л) охлаждали до 20°С.
Стадия 9: получение транс-(1R,2R)-2-(3,4-дифторфенил)циклопропанкарбонилазида.
Раствор транс-(1R,2R)-2-(3,4-дифторфенил)циклопропанкарбонилхлорида (примерно 3,8 кг) в толуоле (примерно 6-9 л), полученный на стадии 8, при 1°С добавляли в течение 74 мин к смеси азида натрия (1,24 кг), бромида тетрабутиламмония (56 г) и карбоната натрия (922 г) в воде (6,2 кг), перемешивая при 1,5°С. Смесь перемешивали при 0°С в течение 1 ч 55 мин, затем водный слой разбавляли холодной водой (3,8 кг), быстро перемешивали, затем разделяли. Толуоловый слой еще раз промывали при 0°С водой (3,8 кг), затем 20% водным раствором хлорида натрия (3,8 л), затем хранили при 3°С для последующего использования.
Стадия 10: получение транс-(1R,2S)-2-(3,4-дифторфенил)циклопропиламина.
Холодный раствор транс-(1R,2R)-2-(3,4-дифторфенил)циклопропанкарбонилазида, полученный, как описано на стадии 9, добавляли в течение 41 мин к толуолу (6,0 кг), перемешивая при 100°С. Смесь перемешивали в течение 55 мин при 100°С, затем охлаждали до 20°С и добавляли в течение 2 ч 15 мин к соляной кислоте (3 М, 18,2 кг), перемешивая при 80°С. Через 65 мин раствор разбавляли водой (34 кг) и охлаждали до 25°С. Толуоловый слой удаляли и водный слой подщелачивали до рН 12 45% водным раствором гидроокиси натрия (3,8 кг) и затем продукт экстрагировали этилацетатом (31 кг) и дважды промывали водой (13,7 кг на порцию) с получением раствора, содержащего транс-(1R,2S)-2-(3,4-дифторфенил)циклопропиламин (2,6 кг, 91,8% избыток энантиомера) в этилацетате (29,5 л).
Стадия 11: получение транс-(1R,2S)-2-(3,4-дифторфенил)циклопропанаминия (2R)-2-гидрокси-2-фенилэтаноата.
R-(-)-миндальную кислоту (2,26 кг) добавляли к раствору, содержащему транс-(1R,2S)-2-(3,4-дифторфенил)циклопропиламин (2,6 кг, 91,8% избыток энантиомера), перемешивая при 17°С в этилацетате (45,3 л). Смесь перемешивали при 25°С в течение 3 ч 8 мин, затем фильтровали и дважды промывали этилацетатом (в целом 13,8 кг). Кристаллический продукт высушивали при 40°С при пониженном давлении в течение 23 ч с получением транс-(1R,2S)-2-(3,4-дифторфенил)циклопропанаминия (2R)-2-гидрокси-2-фенилэтаноата (4,45 кг).
ПРИМЕР 3
Данный пример показывает получение 4,6-дихлор-2-(пропилсульфанил)-5-пиримидинамина.
Стадия 1: 4,6-Дигирокси-2-(пропилсульфанил)пиримидин.
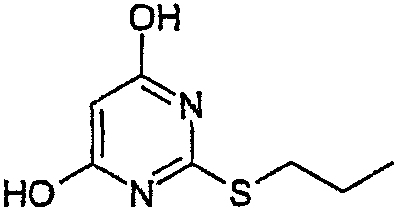
Воду (670 мл) добавляли к 2-тиобарбитуровой кислоте (200 г). Полученную смесь перемешивали и добавляли порциями гидроокись натрия (126,3 г). Смесь перемешивали в течение 40 мин, затем разбавляли водой. Добавляли 1-метил-2-пирролидинон (400 мл) и 1-иодпропан (140,9 мл). Полученную суспензию перемешивали при 20°С в течение 22 ч. Значение рН смеси доводили до 6,5 добавлением 1 М HCl (600 мл) в течение 30 мин, затем до рН 2,5 добавлением 6 М HCl (180 мл) в течение еще 30 мин. Полученную суспензию перемешивали в течение 18 ч и продукт выделяли фильтрованием и последовательно промывали водой (4×100 мл), этанолом (200 мл) и водой (2×200 мл). Продукт высушивали при пониженном давлении в течение ночи при 50°С с получением указанного в заголовке продукта в виде белого порошка (185 г).
Стадия 2: 4,6-Дигидрокси-5-[(E)-2-(4-метилфенил)диазенил]-2-(пропилсульфанил)пиримидин.
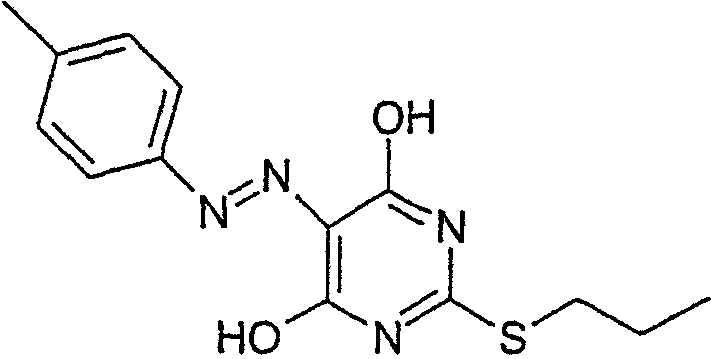
Этанол (25 мл), 4,6-дигидрокси-2-(пропилсульфанил)пиримидин (стадия 1, 5 г) и воду (25 мл) перемешивали вместе при комнатной температуре. Добавляли гидроокись натрия (1,02 г) и получали прозрачный раствор. Полученный раствор охлаждали до 0°С и затем добавляли ацетат натрия (9,42 г) с получением раствора А.
В отдельной емкости готовили раствор п-толуидина (3,01 г) в воде (10 мл). К нему добавляли концентрированную соляную кислоту (37% мас./мас. водный раствор, 8,45 мл). Полученную смесь охлаждали до 0°С, и раствор нитрита натрия (2,16 г) в воде (10 мл) охлаждали до 0°С, и добавляли по каплям к содержащей толуидин реакционной смеси в течение 30 мин. Температуру во время добавления поддерживали между 0 и 5°С. Полученную смесь охлаждали до 0°С и добавляли к холодному (0°С) раствору А (температура поднималась до 8°С). Полученную желтую суспензию перемешивали в течение ночи и рН смеси доводили до рН 1 добавлением 6 М HCl. Смесь фильтровали и собранный продукт промывали водой (25 мл) и этанолом (10 мл). Продукт высушивали при пониженном давлении при 50°С в течение 24 ч с получением продукта в виде желтого твердого вещества (6,97 г).
Стадия 3: 4,6-Дихлор-5-[(E)-2-(4-метилфенил)диазенил]-2-(пропилсульфанил)пиримидин.

Пиридин (2,58 мл) добавляли к перемешиваемой, нагретой (70°С) суспензии 4,6-дигидрокси-5-[(E)-2-(4-метилфенил)диазенил]-2-(пропилсульфанил)пиримидина (стадия 2, 5 г) в толуоле (15 мл). К смеси добавляли по каплям хлорокись фосфора (18,7 мл) в течение 15 мин (повышение температуры до 94°С). Реакционную смесь нагревали в течение еще 4,5 ч, затем упаривали. Остаток дважды подвергали азеотропной отгонке с толуолом (2×30 мл). Остаток растворяли в толуоле (50 мл) и фильтровали для удаления твердых частиц. Собранное твердое вещество растворяли в толуоле (50 мл) и фильтровали для удаления некоторых твердых частиц. Собранное твердое вещество промывали толуолом и объединенные фильтраты промывали водой (30 мл) и насыщенным водным раствором бикарбоната натрия (30 мл). Выпаривание давало указанный в заголовке продукт (4,98 г) в виде красного масла, которое медленно кристаллизовалось при стоянии.
Стадия 4: получение 4,6-дихлор-2-(пропилсульфанил)-5-пиримидинамина.

Перемешиваемый раствор 4,6-дихлор-5-[(E)-2-(4-метилфенил)диазенил]-2-(пропилсульфанил)пиримидина (стадия 3, 1,1 кг) в 2-пропаноле (16,6 кг) гидрировали в течение 1 ч при 40°С/3,2 бар над катализатором платиной на угле (0,81 кг, 50% мас./мас. Pt/С). Находящийся под давлением газообразный водород выпускали и реактор наполняли азотом. Реакционную смесь фильтровали. Собранное твердое вещество промывали 2-пропанолом (1,7 кг) и объединенные фильтраты концентрировали при пониженном давлении. Оставшееся масло охлаждали до 20°С и растворяли в этилацетате (5 кг) и добавляли воду (5,5 л). Значение рН перемешанной смеси доводили до рН 2 добавлением 3 М водного раствора соляной кислоты (800 мл). Фазам давали разделиться и водную фазу отбрасывали. Добавляли воду (2,75 л) к органической фазе и рН доводили до 2 добавлением небольшого количества 3 М HCl (45 мл). Водную фазу отделяли и органическую фазу концентрировали при пониженном давлении при 30 - 50°С с получением 4,6-дихлор-2-(пропилсульфанил)-5-пиримидинамина в виде красноватого вязкого масла, содержащего этилацетат, которое растворяли в этаноле (8,5 кг). Затем растворитель (6,5 л этанола/этилацетата) удаляли выпариванием при пониженном давлении. К остатку добавляли еще порцию этанола (4,5 кг) и выпаривание повторяли для удаления 6,5 л растворителя. Этанольный раствор продукта использовали без дополнительной очистки на следующей стадии.
ПРИМЕР 4
Данный пример показывает получение [1S-[1α,2α,3β-(1S*,2R*),5β]]-3-[7-[2-(3,4-дифторфенил)циклопропиламино]-5-(пропилтио)-3Н-1,2,3-триазоло[4,5-d]пиримидин-3-ил]-5-(2-гидроксиэтокси)циклопентан-1,2-диола (альтернативно называемого (1S,2S,3R,5S)-3-[7-{[(1R,2S)-2-(3,4-дифторфенил)циклопропил]амино}-5-(пропилсульфанил)-3Н-[1,2,3]триазоло[4,5-d]пиримидин-3-ил]-5-(2-гидроксиэтокси)-1,2-циклопентандиолом).
Стадия 1: получение [(3aR-(3аα,4α,6α,6aα)]-2-[[6-[[5-амино-6-хлор-2-(пропилтио)-4-пиримидинил]амино]тетрагидро-2,2-диметил-3аН-циклопента[d][1,3]-диоксол-4-ил]окси]этанола (альтернативно называемого 2-[((3aR,4S,6R,6aS)-6-{[5-амино-6-хлор-2-(пропилсульфанил)-4-пиримидинил]амино}-2,2-диметилтетрагидро-3аН-циклопента[d][1,3]диоксол-4-ил)окси]-1-этанолом.

Этанольный раствор 4,6-дихлор-2-(пропилсульфанил)-5-пиримидинамина (полученного как в примере 3, стадия 4) добавляли к 2-{[(3aR,4S,6R,6aS)-6-амино-2,2-диметилтетрагидро-3аН-циклопента[d][1,3]диоксол-4-ил]окси}-1-этанолу, соль L-винной кислоты (1:1) (1,18 кг). К полученной перемешиваемой густой суспензии добавляли триэтиламин (0,95 кг), поддерживая температуру между 20 и 25°С. Реактор герметизировали и температуру повышали до 120 - 125°С. Реакционную смесь выдерживали при данном интервале температуры в течение 30 ч, затем охлаждали до 75°С и давление сбрасывали. Температуру смеси доводили до 50°С и растворитель отгоняли при пониженном давлении при 30 - 40°С. Добавляли этилацетат (4,95 кг) и воду (5,5 л), рН смеси доводили до рН 5 добавлением 3 М соляной кислоты (100 мл) и фазы разделяли. Органическую фазу промывали 15% рассолом мас./мас. (5,5 л), затем отделяли. Органическую фазу концентрировали при пониженном давлении (4,81 л растворителя удаляется) с получением 2-[((3aR,4S,6R,6aS)-6-{[5-амино-6-хлор-2-(пропилсульфанил)-4-пиримидинил]амино}-2,2-диметилтетрагидро-3аН-циклопента[d][1,3]диоксол-4-ил)окси]-1-этанола в виде коричнево-красного вязкого масла, содержащего этилацетат. Продукт использовали без дополнительной очистки на следующей стадии.
Стадия 2: получение [3aR-(3aα,4α,6α,6aα)]-2-[[6-[7-хлор-5-(пропилтио)-3Н-[1,2,3]триазоло[4,5-d]пиримидин-3-ил]тетрагидро-2,2-диметил-3аН-циклопента[d][1,3]диоксол-4-ил]окси]этанола (альтернативно называемого 2-({(3aR,4S,6R,6aS)-6-[7-хлор-5-(пропилсульфанил)-3Н-[1,2,3]триазоло[4,5-d]пиримидин-3-ил]-2,2-диметилтетрагидро-3аН-циклопента[d][1,3]диоксол-4-ил}окси)-1-этанолом).
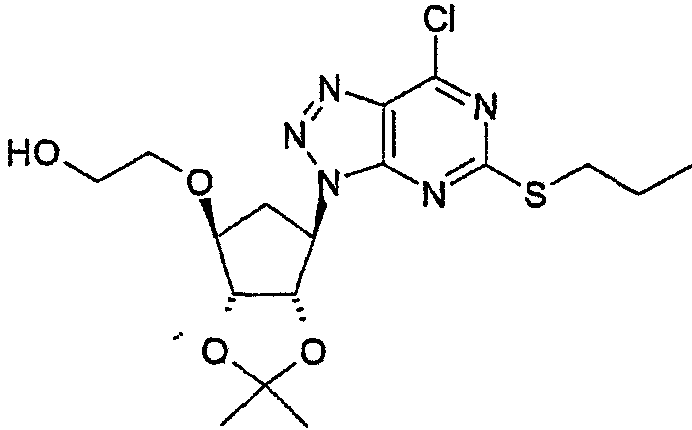
2-[((3aR,4S,6R,6aS)-6-{[5-Амино-6-хлор-2-(пропилсульфанил)-4-пиримидинил]амино}-2,2-диметилтетрагидро-3аН-циклопента[d][1,3]диоксол-4-ил)окси]-1-этанол, полученный на стадии 1, растворяли в уксусной кислоте (5,75 кг) и воде (650 мл). Полученный раствор охлаждали до 2°С (при перемешивании) и добавляли раствор нитрита натрия (232 г) в воде (1,25 л) таким образом, чтобы температура смеси поддерживалась ниже 7°С. Затем смеси давали подогреться до 7°С, затем добавляли этилацетат (8,9 кг). Добавляли водный раствор карбоната калия (4 л, 37% мас./мас.). Смесь разделяли и органическую фазу еще раз промывали водным раствором карбоната калия (3,8 кг, 21% мас./мас.). Водную фазу отбрасывали и органическую фазу концентрировали при пониженном давлении с получением указанного в подзаголовке соединения в виде красно-коричневого вязкого масла, используемого без дополнительной очистки на следующей стадии.
Стадия 3: получение {3aR-[3aα,4α,6α(1R*,2S*),6aα]}-2-[6-({7-[2-(3,4-дифторфенил)циклопропил]амино-5-(пропилтио)-3Н-1,2,3-триазоло[4,5-d]пиримидин-3-ил}тетрагидро-2,2-диметил-4Н-циклопента-1,3-диоксол-4-ил)окси]этанола (альтернативно называемого 2-({(3aR,4S,6R,6aS)-6-[7-{[(1R,2S)-2-(3,4-дифторфенил)циклопропил]амино}-5-(пропилсульфанил)-3Н-[1,2,3]триазоло[4,5-d]пиримидин-3-ил]-2,2-диметилтетрагидро-3аН-циклопента[d][1,3]диоксол-4-ил}окси)-1-этанолом).

Транс-(1R,2S)-2-(3,4-дифторфенил)циклопропанаминий(2R)-2-гидрокси-2-фенилэтаноат (0,77 кг) загружали в емкость с последующим внесением раствора 2-({(3aR,4S,6R,6aS)-6-[7-хлор-5-(пропилсульфанил)-3Н-[1,2,3]триазоло[4,5-d]пиримидин-3-ил]-2,2-диметилтетрагидро-3аН-циклопента[d][1,3]диоксол-4-ил}окси)-1-этанола (полученного на стадии 2), растворенного в ацетонитриле (3,85 кг). К полученной перемешиваемой смеси добавляли триэтиламин (0,81 кг) с такой скоростью, чтобы температура реакционной смеси сохранялась в пределах 20 - 25°С. Реакционную смесь перемешивали в течение 13 ч, затем концентрировали при пониженном давлении при 30°С. К остатку добавляли этилацетат (8,1 кг) и воду (4,6 л). Значение рН перемешиваемой двухфазной смеси доводили до рН 4 добавлением 3 М HCl (450 мл). Затем смеси давали осесть и разделиться. Водную фазу отделяли и оставшуюся органическую фазу промывали 15% мас./мас. водным раствором хлорида натрия (4,15 кг), органическую фазу концентрировали при пониженном давлении при 30 - 50°С с получением сырого указанного в заголовке соединения в виде красного масла, которое непосредственно использовали на следующей стадии.
Стадия 4: получение [1S-[1α,2α,3β(1S*,2R*),5β]]-3-[7-[2-(3,4-дифторфенил)циклопропиламино]-5-(пропилтио)-3Н-1,2,3-триазоло[4,5-d]пиримидин-3-ил]-5-(2-гидроксиэтокси)циклопентан-1,2-диола (альтернативно называемого (1S,2S,3R,5S)-3-[7-{[(1R,2S)-2-(3,4-дифторфенил)циклопропил]амино}-5-(пропилсульфанил)-3Н-[1,2,3]триазоло[4,5-d]пиримидин-3-ил]-5-(2-гидроксиэтокси)-1,2-циклопентандиолом).

Водный раствор соляной кислоты (3 М, 4,8 л) добавляли к перемешиваемому раствору 2-({(3aR,4S,6R,6aS)-6-[7-{[(1R,2S)-2-(3,4-дифторфенил)циклопропил]амино}-5-(пропилсульфанил)-3Н-[1,2,3]триазоло[4,5-d]пиримидин-3-ил]-2,2-диметилтетрагидро-3аН-циклопента[d][1,3]диоксол-4-ил}окси)-1-этанола (1,931 кг) в метаноле (13,4 кг), поддерживая температуру во время добавления в пределах 20 - 25°С. Затем смесь перемешивали в течение 24 ч при 20°С. Затем добавляли гидроокись натрия (45% мас./мас. водный раствор, 780 мл) для доведения рН смеси до рН 7,2. Затем метанол удаляли отгонкой при пониженном давлении и добавляли этилацетат (14,3 кг). Смесь нагревали до 45°С и водный слой отделяли. Затем органическую фазу промывали 15% мас./мас. водным раствором хлорида натрия (7,2 кг). Этилацетат (10 л) удаляли отгонкой при пониженном давлении. Добавляли свежую порцию этилацетата (7,2 кг) и смесь фильтровали. Фильтр промывали этилацетатом (1,5 кг). Объединенные фильтраты высушивали повторным добавлением/отгонкой этилацетата. Когда раствор был сухой, определяли содержание продукта в этилацетатном растворе анализом хроматографией и установили, что он содержит 1016 г продукта, концентрацию этилацетата доводили, пока не достигали концентрации 5 мл этилацетата/г сырого продукта. Этилацетатный раствор нагревали до 47°С и затем добавляли изооктан (2,5 мл/г продукта, 2540 мл) в течение 15 мин. Полученную суспензию перемешивали в течение 30 мин и затем добавляли еще изооктан (2540 мл) в течение 5 мин. Полученную смесь перемешивали при 48 - 50°С в течение 30 мин, затем охлаждали до 20°С в течение 3 ч. Суспензию перемешивали при 20°С в течение 6,5 ч, затем фильтровали и промывали смесью, состоящей из изооктана (1,25 кг) и этилацетата (1,6 кг). Собранное твердое вещество высушивали в вакууме с получением указанного в заголовке соединения (920 г).
Если желательно, сырой продукт можно очистить дополнительно с использованием одного из трех последующих способов.
Перекристаллизация из этилацетат/изооктана
Сырой продукт растворяют в этилацетате (4,8 мл/г) при 55°С, затем фильтруют для удаления частиц. Прозрачный раствор переносят обратно в реактор для перекристаллизации и температуру устанавливают при 50°С. Затем добавляют изооктан (4,8 мл/г) в течение 10 мин. Суспензии дают постоять в течение 30 мин, после чего ее охлаждают до 20°С в течение 2-3 ч и в конце температуру поддерживают при 20°С в течение примерно 30 мин. Затем продукт фильтруют и промывают изооктаном (2×1,5 мл/г). Продукт высушивают при пониженном давлении при 50°С с получением чистого продукта (чистота >98% по данным ВЭЖХ).
Суспензия с н-бутилацетатом
Сырой продукт суспендируют в н-бутилацетате из расчета 4 мл/г и перемешивают при комнатной температуре в течение 10 ч. Суспензию охлаждают до 0°С в течение 3-4 ч и выдерживают при 0°С в течение 1 ч. Продукт фильтруют и промывают холодным н-бутилацетатом из расчета 2 мл/г (<0°С). Затем продукт высушивают в вакууме при 50°С с получением чистого продукта (чистота >98% по данным ВЭЖХ).
Суспензия с изопропанолом
Сырой продукт суспендируют в изопропаноле из расчета 3 мл/г и перемешивают при 50°С в течение 72 ч. Затем суспензию охлаждают до 20°С в течение 3 ч и температуру сохраняют при 20°С в течение примерно 30 мин. Затем продукт фильтруют и промывают холодным изопропанолом из расчета 1 мл/г (<0°С). Наконец, продукт высушивают при пониженном давлении при 50°С с получением чистого продукта (чистота >98% по данным ВЭЖХ).
Изобретение относится к улучшенному способу получения фармакологически активного соединения формулы (А):
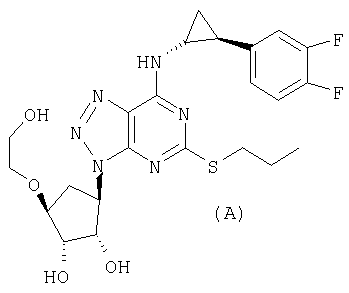
включающему: а) взаимодействие соединения формулы (I)

с нитритом щелочного металла в присутствии подходящей кислоты с получением соединения формулы (VII):

b) сочетание соединения формулы (VII) с соединением формулы (VI):

с получением соединения формулы (V):

и
с) снятие защиты с соединения формулы (V) с получением соединения формулы А. Соединение формулы А обладает свойствами антагонистов рецепторов Р2T и обладает высокой стабильностью к метаболизму и биодоступностью. Изобретение также относится к новому промежуточному продукту формулы (I) и способам его получения, а также к новым промежуточным продуктам для его получения. 11 н. и 1 з.п. ф-лы.


с солью соединения формулы (III)


в которой Ar представляет фенил, необязательно замещенный галогеном, С1-4алкилом или С1-4алкокси.

включающий
а) взаимодействие соединения формулы (I) с нитритом щелочного металла в присутствии подходящей кислоты с получением соединения формулы (VII)

b) сочетание соединения формулы (VII) с соединением формулы (VI)

с получением соединения формулы (V)
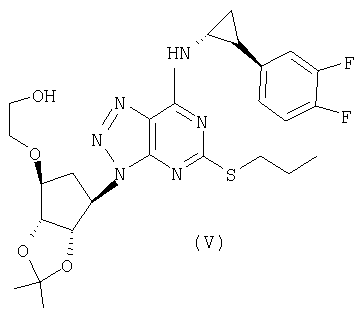
и
с) снятие защиты с соединения формулы (V) с получением соединения формулы А.
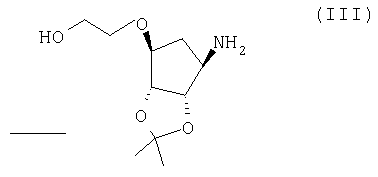
включающий взаимодействие соединения формулы (III) с необходимой кислотой.
включающий стадии
а) получения соединения формулы (XI)
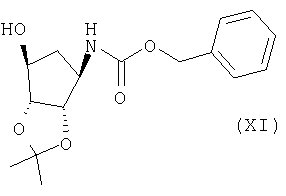
взаимодействием соединения формулы (XII)
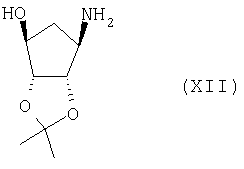
с бензилхлорформиатом в присутствии подходящего основания и подходящего растворителя;
b) получения соединения формулы (X)
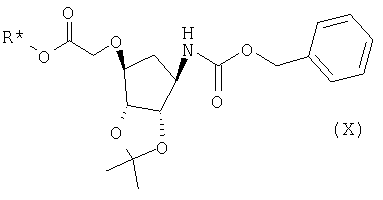
взаимодействием соединения формулы (XI) с соединением L-CH2CO2R* (где R* представляет С1-4алкил и L представляет уходящую группу);
с) получения соединения формулы (IX)

восстановлением соединения формулы (X);
d) получения соединения формулы (III)
снятием защиты с соединения формулы (IX) и
е) получения соли соединения формулы (III) взаимодействием соединения формулы (III) с необходимой кислотой.
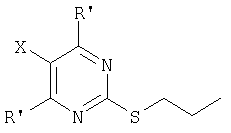
где оба R' одновременно представляют собой Cl или оба R' одновременно представляют собой ОН, а Х означает группу Ar-N=N-, где Ar является фенилом, который необязательно замещен галогеном, C1-4-алкилом или C1-4-алкокси.
где Ar определен, как указано в п.7.
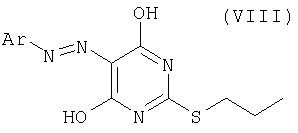
где Ar определен, как указано в п.7.
где R* является C1-4-алкилом.
| Преобразователь температуры вчастоту | 1974 |
|
SU508687A1 |
| Н.Н.ВОРОЖЦОВ «Основы синтеза промежуточных продуктов и красителей», М.: Государственное научно-техническое издательство химической литературы, 1955, стр.255 | |||
| ВЕЙГАНД-ХИЛЬГЕТАГ «Методы эксперимента в органической химии, М.: Химия, 1968, стр.202 | |||
| JUNG MICHAEL et al | |||
| "Total synthesis of neplanocin A", Helvetica Chimica Acta, | |||

