ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение касается новых производных тетрациклина, способов получения новых производных и способов применения этих производных.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Соединение тетрациклин имеет следующую общую структуру:
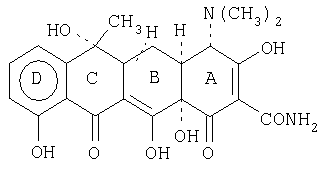
Система нумерации циклических ядер представляет собой следующее:
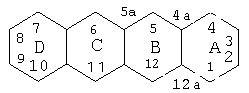
Тетрациклин, а также его производные 5-ОН (террамицин) и 7-Cl (ауреомицин) существуют в природе и являются хорошо известными антибиотиками. Природный тетрациклин может быть модифицирован без потери его антибиотических свойств, хотя определенные элементы структуры должны сохраняться. Модификации, которые могут и не могут быть сделаны в основной тетрациклиновой структуре, были представлены в обзоре Mitscher в The Chemistry of Tetracyclines, Chapter 6, Marcel Dekker, Publishers, New York (1978). Согласно Mitscher, заместители в положениях 5-9 тетрациклиновой кольцевой системы могут быть модифицированы без полной потери антибиотических свойств. Однако изменения в основной кольцевой системе или замена заместителей в положениях 1-4 и 10-12 обычно приводят к образованию синтетических тетрациклинов со значительно меньшей антимикробной активностью или фактически неактивных. Некоторые примеры химически модифицированных неантимикробных тетрациклинов (в дальнейшем ХМТ) представляют собой 4-дедиметиламино-тетрациклин, 4-дедиметиламиносанциклин(6-деметил-6-дезокси-4-дедиметиламинотетрациклин), 4-дедиметиламиноминоциклин(7-диметиламино-4-дедиметиламинотетрациклин) и 4-дедиметиламинодоксициклин(5-гидрокси-6-дезокси-4-дедиметиламинотетрациклин).
Некоторые производные 4-дедиметиламинотетрациклина раскрыты в Патентах US 3029284 и US 5122519. Они включают 6-деметил-6-дезокси-4-дедиметиламинотетрациклин и 5-гидрокси-6-дезокси-4-дедиметиламинотетрациклин с водородом и другими заместителями в С7- и С9-положениях в D-кольце. Эти заместители включают амино, нитро, ди-(низший алкил)амино и моно-(низший алкил)амино или галоген. Авторы упоминают, что производные 6-деметил-6-дезокси-4-дедиметиламинотетрациклина и производные 5-гидрокси-6-дезокси-4-дедиметиламинотетрациклина полезны в качестве антимикробных агентов.
Другие производные 4-дедиметиламинотетрациклина с оксим-группой в С4-положении в А-кольце раскрыты в Патентах US 3622627 и US 3824285. Эти оксим-производные имеют водород и галоген в качестве заместителей в С7-положении и включают 7-галоген-6-деметил-6-дезокси-4-дедиметиламино-4-оксиминотетра-циклин и 7-галоген-5-гидрокси-6-дезокси-4-дедиметиламино-4-оксиминотетрациклин.
Группы алкиламино (МН-алкил) и алкилгидразон (N-NH-алкил) были замещены в А-кольце в С4-положении 4-дедиметиламинотетрациклина. Эти соединения известны своими антимикробными свойствами. См. Патенты US 3345370, US 3609188, US 3622627, US 3502660, US 3509184, US 3502696, US 3515731, US 3265732, US 5122519, US 3849493, US 3772363 и US 3829453.
Было описано, что кроме антимикробных свойств тетрациклины имеют и некоторые другие применения. Например, известно также, что тетрациклины ингибируют активность ферментов, разрушающих коллаген, таких как матриксные металлопротеиназы (ММР), включая коллагеназы (ММР-1), желатиназу (ММР-2) и стромелизин (ММР-3) (Golub et al., J. Periodont. Res. 20:12-23 (1985); Golub et al. Crit. Revs. Oral Biol. Med. 2:297-322 (1991); Патенты US 4666897, US 4704383, US 4935411, US 4935412). Известно также, что тетрациклины ингибируют атрофию и деградацию белков в скелетных мышцах млекопитающих (Патент US 5045538) и увеличивают продукцию IL-10 в клетках млекопитающих.
Кроме того, сообщалось, что тетрациклины увеличивают белковый синтез в костях (Патент US Re. 34656) и уменьшают резорбцию кости в органной культуре (Патент US 4704383).
Аналогично, Golub с соавт. в Патенте US 5532227 раскрывают, что тетрациклины могут воздействовать на избыточное гликозилирование белков. В частности, тетрациклины ингибируют избыточное перекрестное связывание коллагена, которое является результатом избыточного гликозилирования коллагена при диабете.
Известно, что тетрациклины ингибируют избыточную активность фосфолипазы А2, вовлеченной в воспалительные состояния, такие как псориаз, как раскрыто в Патенте US 5532227. Кроме того, также известно, что тетрациклины ингибируют циклооксигеназу-2 (СОХ-2), фактор некроза опухоли (TNF), окись азота и IL-1 (интерлейкин-1).
Эти свойства делают тетрациклины полезными в лечении множества заболеваний. Например, существует множество предположений, что тетрациклины, включая неантимикробные тетрациклины, эффективны в лечении артрита. См, например, Greenwald et al. "Tetracyclines Suppress Metalloproteinase Activity in Adjuvant Arthritis and, in Combination with Flurbioprofen, Ameliorate Bone Damage," Journal of Rheumatology 19:927-938 (1992); Greenwald et al. "Treatment of Destructive Arthritic Disorders with MMP Inhibitors; Potential Role of Tetracyclines in Inhibition of Matrix Metalloproteinases: Therapeutic Potential", Annals of the New York Academy of Sciences 732:181-198 (1994); Kloppenburg et al. "Minocycline in Active Rheumatoid Arthritis," Arthritis Rheum 37:629-636 (1994); Ryan et al. "Potential of Tetracycline to Modify Cartilage Breakdown in Osteoarthritis", Current Opinion in Rheumatology 8:238-247 (1996); O'Dell et al. "Treatment of Early Rheumatoid Arthritis with Minocycline or Placebo," Arthritis Rheum 40:842-848 (1997).
Предположили, что тетрациклины могут быть использованы для лечения кожных заболеваний. Например, White с соавт. (Lancet, Apr. 29, р.966 (1989)) сообщают, что тетрациклин миноциклин эффективен в лечении дистрофического врожденного буллезного эпидермолиза, который является жезнеопасным кожным заболеванием, связанным, как полагают, с избытком коллагеназы.
Кроме того, исследования также позволили предположить, что тетрациклины и ингибиторы металлопротеиназ ингибируют развитие опухоли (DeClerck et al., Annals N.Y. Acad. Sci, 732:222-232, 1994), резорбцию кости (Rifkin et al., Annals N.Y. Acad. Sci., 732:165-180, 1994), ангиогенез (Maragoudakis et al., Br. J. Pharmacol, 111:894-902, (1994) и могут обладать противовоспалительными свойствами (Ramamurthy et al., Annals N.Y. Acad. Sci., 732, 427-430, 1994).
На основе вышесказанного можно сделать вывод, что тетрациклины эффективны в лечении многочисленных заболеваний и состояний. Поэтому существует потребность в новых и еще более полезных производных тетрациклина.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Соединения, раскрытые здесь, являются производными тетрациклина, которые проявляют антимикробную и/или противораковую активность.
В предпочтительной форме осуществления настоящее изобретение относится к способу лечения, направленного на ингибирование микробного или опухолевого роста, включающему введение субъекту, нуждающемуся в таком лечении, эффективного количества тетрациклинового соединения или его соли общей формулы:
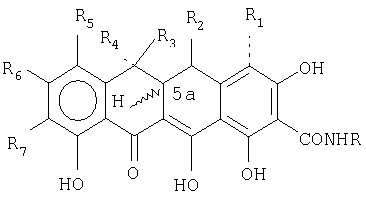
в которой R выбран из СН2N(RаRb) или 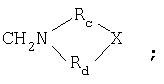
RaRb представляют собой С1-С6-алкил;
Rc и Rd представляют собой (CH2)nCHRe, n означает 0 или 1, и Re представляет собой Н, С1-С6-алкил или NH2, и Х представляет собой NH, S или СН2;
R1 представляет собой Н или ОН;
R2 представляет собой Н, ОН, =O или OCOR8, где R8 представляет собой С1-С6-алкил, или альтернативно, R2 представляет собой N(R9)2, где R9 представляет собой водород или С1-С6-низший алкил, или R9CO, при условии, что если R2 представляет собой кетогруппу или N(R9)2, то R2 и R4 представляют собой Н или СН3, и R3 и R4 не являются одновременно СН3 или Н;
5а водород является α- или β-;
R3 и R4 представляют собой Н, СН3 или F, при условии, что если R3 представляет собой СН3, то R4 представляет собой Н или F, и если R4 представляет собой СН3, то R3 представляет собой Н или F, и R4 и R5 не являются одновременно F;
R5 и R7 представляют собой галоген, Н, NO2, N3, N2 +, C2H5OC(S)S-, CN, NR10R11, где R10 и R11 представляют собой Н, C1-С10-алкил и R12(CH2)nCO-, где n означает от 0 до 5, и R9 представляет собой Н, NH2, моно- или ди-замещенный амин, выбранный из прямой, разветвленной или циклической С1-С10-алкильной группы, при условии, что R10 и R11 не являются одновременно R12(СН2)nСО-;
R7, альтернативно, представляет собой четвертичный бутил; и
R6 представляет собой галоген, ацетилен, R13-C6H5, где R13 представляет собой NO2, галоген, ацетиламино, амино, фенил, алкил или алкокси.
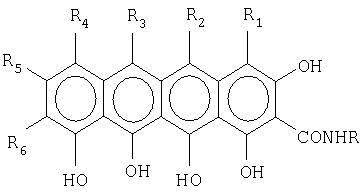
В еще одной предпочтительной форме осуществления, настоящее изобретение относится к способу лечения, направленного на ингибирование микробного или опухолевого роста, включающему введение субъекту, нуждающемуся в таком лечении, эффективного количества тетрациклинового соединения или его соли общей формулы:
в которой R выбран из CH2N(RaRb) или 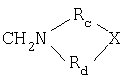 ;
;
где RaRb представляют собой C1-C6-алкил;
Rc и Rd представляют собой (CH2)nCHRe, n означает 0 или 1, и Re представляет собой Н, C1-С6-алкил или NH2, и Х представляет собой NH, S или СН2;
R1 представляет собой Н или ОН;
R2 представляет собой Н или ОН;
R3 представляет собой Н или СН3;
R4 и R6 представляют собой галоген, Н, NO2, N3, N2 +, C2H5OC(S)S-, CN, NR7R8, где R7 и R8 представляют собой Н, C1-С10-алкил и R9(CH2)nCO-, где n означает от 0 до 5, и R9 представляет собой Н, NH2, моно- или дизамещенный амин, выбранный из прямого, разветвленного или циклического C1-С10-алкила, при условии, что R7 и R8 не являются одновременно R9(CH2)nCO-; и
R6 представляет собой Н или галоген, ацетилен, R10-C6H5, где R10 представляет собой NO2, галоген, ацетиламино, амино, фенил, алкил или алкокси.
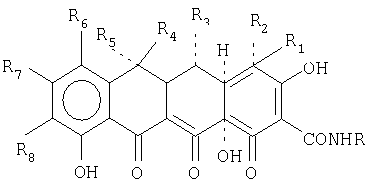
В еще одной предпочтительной форме осуществления настоящее изобретение относится к способу лечения, направленного на ингибирование микробного или опухолевого роста, включающему введение субъекту, нуждающемуся в таком лечении, эффективного количества тетрациклинового соединения или его соли общей формулы:
в которой R выбран из CH2N(RaRb) или ;
где RaRb представляют собой C1-C6-алкил;
Rc и Rd представляют собой (CH2)nCHRe, n означает 0 или 1, и Re представляет собой Н, C1-C6-алкил или NH2, и Х представляет собой NH, S или СН2;
R1 и R2 представляют собой Н или N(R9)2, где R9 представляет собой Н или С1-С6-алкил при условии, что R1 и R2 не являются одновременно N(R9)2, R1 и R2 представляют собой также N(R10)3l, где R10 представляет собой C1-С6-алкил, при условии, что R1 и R2 не являются одновременно N(R10)3l;
R3 представляет собой Н;
R4 и R5 представляют собой Н, СН3 или F, при условии, что если R4 представляет собой СН3, то R5 представляет собой Н или F, и если R5 представляет собой СН3, то R4 представляет собой Н или F, и R4 и R5 не являются одновременно F;
R6 и R8 представляют собой галоген, Н, NO2, N3, N2 +, C2H5OC(S)S-, CN, NR11R12, где R11 и R12 представляют собой Н, C1-С10-алкил и R13(CH2)nCO-, где n означает от 0 до 5, и R13 представляет собой Н, NH2, моно- или дизамещенный амин, выбранный из прямых, разветвленных или циклических C1-С10-алкильных групп, при условии, что R10 и R11 не являются одновременно R12(СН2)nСО-; и R8 может быть также четвертичным бутилом, и R7 представляет собой Н или галоген.
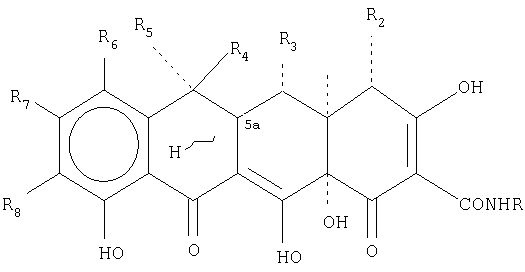
В дополнительной предпочтительной форме осуществления настоящее изобретение относится к способу лечения, направленного на ингибирование микробного или опухолевого роста, включающему введение субъекту, нуждающемуся в таком лечении, эффективного количества тетрациклинового соединения или его соли общей формулы:
в которой R выбран из CH2N(RaRb) или ;
RaRb представляют собой C1-C6;
Rc и Rd представляют собой (СН2)nCHRе, n означает 0 или 1, Re представляет собой Н, алкил(C1-C6), NH2, и Х представляет собой NH, S или СН2;
R1 и R2 представляют собой Н или N(R9)2, где R9 представляет собой водород или низший алкил (C1-C6), при условии, что R1 и R2 не могут одновременно представлять собой N(R9)2.
R3 представляет собой Н, ОН, =O, OCOR10, где R10 представляет собой C1-C6, или альтернативно, R3 представляет собой N(R9)2, где R9 представляет собой водород или C1-C6, при условии, что если R3 представляет собой =O или N(R9)2, то R4 и R5 представляют собой Н или СН3, и R4 и R5 не являются одновременно СН3 или Н;
5а водород является α- или β-;
R4 и R5 представляют собой Н, СН3 или F, при условии, что если R4 представляет собой СН3, то R5 представляет собой Н или F, и если R5 представляет собой СН3, то R4 представляет собой Н или F, и R4 и R5 не являются одновременно F;
R6 и R8 представляют собой галоген, Н, NO2, N3, N2 +, C2H5OC(S)S-, CN, NR10R11, где R10 и R11 представляют собой Н, алкил(C1-С10) и R12(СН2)nСО-, где n означает от 0 до 5, и R12 представляет собой Н, NH2, моно- или дизамещенный амин, выбранный из прямых, разветвленных или циклических C1-C10-групп, при условии, что R10 и R11 одновременно представляют собой R12(CH2)nCO-, или, альтернативно, R8 представляет собой четвертичный бутил;
R7 представляет собой Н или галоген, ацетилен, R12-C6H5, где R12 представляет собой NO2, галоген, ацетиламино, амино, фенил, алкил или алкокси.
В еще одной предпочтительной форме осуществления настоящее изобретение относится к способу лечения, направленного на ингибирование микробного или опухолевого роста, который включает введение субъекту, нуждающемуся в таком лечении, эффективного количества тетрациклинового соединения или его соли общей формулы:
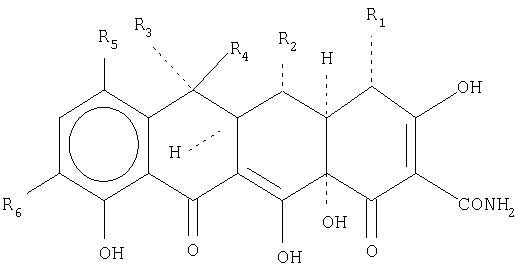
в которой R1 представляет собой Н или N(СН3)2; R2 представляет собой Н, СН3 или ОСОСН3; R3 представляет собой Н или СН3; R4 представляет собой Н; R5 представляет собой Н, N(CH3)2, NH2, N(C4H9)2, N(C6H13)2, N(3,3-диметилбутил)2, N3, N2 +, NO2, NHCOCH3, NH(н-пропил)2, NH-изобутил, NH-изобутилметил, NH(циклобутил), NH(циклобутилметил); и R6 представляет собой Н, NO2, NH2, (CH3)2CHNH, N3, NHCOCH3, N2 +, N3 или (СН3)3С.
В еще одной предпочтительной форме осуществления настоящее изобретение относится к способу лечения, направленного на ингибирование микробного или опухолевого роста, который включает введение субъекту, нуждающемуся в таком лечении, эффективного количества тетрациклинового соединения или его соли общей формулы:
в которой R1 представляет собой Н или N(СН3)2; R2 представляет собой Н, ОН или ОСОСН3; R3 представляет собой Н или СН3; R4 представляет собой Н; R5 представляет собой Н, N(СН3)2; N(C4H9)2, N(C6H13)2, N(3,3-диметилбутил)2; N3, N2 +, NO2, NHCOCH3, NH(н-пропил)2, NH-изобутил, NH-изобутилметил, N(СН3СО)(изобутил), NH(циклобутил), NH(циклобутилметил) или NH2; и R6 представляет собой Н, NO3, NH2, (СН3)2CHNH, N3, NHCOCH3, N2 + или (СН3)3С.
СВЕДЕНИЯ, ПОДТВЕРЖДАЮЩИЕ ВОЗМОЖНОСТЬ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
Следующие примеры описывают получение различных веществ согласно настоящему изобретению.
ВЭЖХ-анализы выполняли на колонках C18-symmetry фирмы Waters с обращенными фазами, используя воду, содержащую 0,1% ТФУ, и ацетонитрил в качестве элюентов.
Аналитические ЖХ/МС-измерения выполняли на жидкостном хроматографе Hewlett Packard Series 1100, снабженном детектором Series 1100 MSD, с ионизацией электрораспылением (ЭР) и УФ-детекцией при 270 нм, с использованием колонки размером 4,6×100 мм со скоростью протока 0,4 мл/мин. Препаративную ВЭЖХ выполняли на Gilson serial HPLC с УФ-детекцией при 270 нм, с использованием колонки размером 19×150 мм и скоростью потока 15 мл/мин. 1H-ЯМР регистрировали на спектрометре Bruker 300 МГц в дейтерированном метаноле.
9-Нитро-доксициклина сульфат
К перемешиваемому раствору доксициклина гидрохлорида (1 г, 2,08 ммоль) в 30 мл концентрированной серной кислоты при 0°С добавляли нитрат калия (252 мг, 2,49 ммоль). Полученную смесь перемешивали в течение 20 мин при 0°С, затем вливали в 1,2 л холодного эфира при перемешивании. Твердое вещество, которое осаждалось, отфильтровывали, промывали холодным эфиром и высушивали под вакуумом, и получали 1,22 г продукта кремового цвета. При необходимости продукт очищали, растворяя в метаноле, фильтруя и вливая фильтрат в эфир. Выделившееся твердое вещество отфильтровывали и высушивали.
9-Амино-доксициклина сульфат
К перемешиваемому раствору 9-нитро-доксициклина сульфата (1 г, 1,7 ммоль) в 35 мл монометилового эфира этиленгликоля и 5 мл метанола добавляли 700 мг 10%-ного Pd/C. Реакционную смесь гидрировали при атмосферном давлении в течение 6 ч. Катализатор фильтровали через целит, промывая метанолом. Фильтрат концентрировали, затем добавляли по каплям к 800 мл холодного эфира при перемешивании. Выделившееся твердое вещество отфильтровывали, промывали холодным эфиром и высушивали под вакуумом, и получали 843 мг продукта кремового цвета.
9-Изопропиламино-доксициклина сульфат
К перемешиваемому раствору 9-амино-доксициклина сульфата (100 мг, 0,18 ммоль) в 10 мл монометилового эфира этиленгликоля добавляли 0,05 мл концентрированной серной кислоты, 0,5 мл ацетона и 100 мг 10%-ного Pd/C. Полученную смесь гидрировали при атмосферном давлении в течение 1 ч 15 мин, затем катализатор фильтровали через целит, промывая метанолом. Фильтрат концентрировали, затем вливали в 150 мл холодного эфира при перемешивании. Выделившееся твердое вещество отфильтровывали, промывали холодным эфиром и высушивали под вакуумом, и получали 109 мг продукта кремового цвета. Этот продукт очищали, растворяя в метаноле, фильтруя и вливая фильтрат в эфир. Образовавшееся твердое вещество отфильтровывали и высушивали.
9-Ацетамидо-доксициклина сульфат
К перемешиваемому раствору 9-амино-доксициклина сульфата (72 мг, 0,13 ммоль) в 1,5 мл 1,3-диметил-2-имидазолидинона добавляли 70 мг бикарбоната натрия, затем 0,05 мл ацетилхлорида. Смесь перемешивали при комнатной температуре в течение 1 часа и затем фильтровали. Фильтрат концентрировали и вливали в 100 мл холодного эфира при перемешивании. Выделившееся твердое вещество отфильтровывали и высушивали под вакуумом, и получали 92 мг продукта.
9-Диазоний-доксициклина сульфата гидрохлорид
К перемешиваемому раствору 9-амино-доксициклина сульфата (300 мг, 0,54 ммоль) в 9 мл 0,1 н. соляной кислоты в метаноле, охлажденной на ледяной бане, добавляли 0,3 мл н-бутилнитрита. Раствор перемешивали при 0°С в течение 30 минут, затем вливали в 300 мл холодного эфира при перемешивании. Выделившееся твердое вещество отфильтровывали и высушивали под вакуумом, и получали 256 мг оранжево-коричневого продукта.
9-Азидо-доксициклина сульфат
К перемешиваемому раствору 9-диазоний-доксициклина сульфата гидрохлорида (80 мг, 0,14 ммоль) в 4,5 мл 0,1 н. соляной кислоты в метаноле добавляли 10 мг азида натрия. Раствор перемешивали при комнатной температуре в течение 2 часов, затем вливали в 100 мл холодного эфира при перемешивании. Выделившееся твердое вещество отфильтровывали и высушивали под вакуумом, и получали 43 мг продукта.
9-(4-фторфенил)доксициклин
К раствору 9-диазоний-доксициклина сульфата гидрохлорида (358 мг, 0,59 ммоль) и 4-фторфенилбороновой кислоты (107 мг, 0,76 ммоль) в 4 мл метанола добавляли 18 мг ацетата палладия(II) и полученную смесь перемешивали при комнатной температуре в течение 16 часов. Катализатор отфильтровывали и остаток концентрировали и очищали с помощью препаративной ВЭЖХ. Собранные после ВЭЖХ фракции концентрировали и вливали в 20 мл холодного эфира. Выделившееся твердое вещество отфильтровывали и высушивали под вакуумом и получали 32 мг продукта.
7-[1,2-бис(карбобензилокси)гидразино]доксициклин
К перемешиваемому раствору доксициклина гидрохлорида (300 мг, 0,62 ммоль) в 2,5 мл ТГФ и 3,2 мл метансульфокислоты при 0°С добавляли 230 мг дибензилазодикарбоксилата. Полученную смесь перемешивали при 0°С в течение 2 часов, затем вливали в 400 мл холодного эфира. Выделившееся твердое вещество отфильтровывали и высушивали под вакуумом, и получали 479 мг продукта не совсем белого цвета.
7-Амино-доксициклин
К раствору 7-[1,2-бис(карбобензилокси)гидразино]доксициклина (478 мг) в 30 мл монометилового эфира этиленгликоля добавляли 200 мг 10%-ного Pd/C и полученную смесь гидрировали в течение 3 часов при комнатной температуре. Катализатор фильтровали через целит, промывая метанолом, и фильтрат концентрировали и вливали в 500 мл холодного эфира. Выделившееся твердое вещество отфильтровывали и высушивали под вакуумом, и получали 268 мг продукта.
7-Диметиламино-доксициклина сульфат
К раствору 7-амино-доксициклина (100 мг) в 8 мл монометилового эфира этиленгликоля добавляли 1 мл 37%-ного водного раствора формальдегида, 0,05 мл концентрированной серной кислоты и 100 мг 10%-ного Pd/C. Полученную смесь гидрировали при атмосферном давлении в течение 6 часов, затем катализатор фильтровали через целит, промывая метанолом. Остаток концентрировали и вливали в 100 мл холодного эфира при перемешивании. Выделившееся очень липкое вещество с трудом отфильтровывали. Остаток сразу же снова растворяли в метаноле и вливали в 100 мл холодного эфира при перемешивании. Выделившееся твердое вещество отфильтровывали и высушивали под вакуумом, и получали 89 мг продукта.
7-Дипропил-доксициклина сульфат
К раствору 7-амино-доксициклина (90 мг) в 8 мл монометилового эфира этиленгликоля добавляли 0,5 мл пропионового альдегида, 0,05 мл концентрированной серной кислоты и 80 мг 10%-ного Pd/C. Полученную смесь гидрировали при атмосферном давлении в течение 5 часов, затем катализатор фильтровали через целит, промывая метанолом. Остаток концентрировали и вливали в 100 мл холодного эфира при перемешивании. Выделившееся твердое вещество отфильтровывали и высушивали под вакуумом, и получали 415 мг продукта.
7-Дибутиламино-доксициклина сульфат
К раствору 7-амино-доксициклина (100 мг, 0,2 ммоль) в 6 мл монометилового эфира этиленгликоля добавляли 0,6 мл масляного альдегида, 0,05 мл концентрированной серной кислоты и 100 мг 10%-ного Pd/C. Полученную смесь гидрировали при атмосферном давлении в течение 4 часов, затем катализатор фильтровали через целит, промывая метанолом. Остаток концентрировали и вливали в 100 мл холодного эфира при перемешивании. Выделившееся твердое вещество отфильтровывали и высушивали под вакуумом, и получали 103 мг продукта.
7-Ди-(3,3-диметил)бутиламино-доксициклина сульфат
К раствору 7-амино-доксициклина (100 мг, 0,2 ммоль) в 6 мл монометилового эфира этиленгликоля добавляли 0,5 мл 3,3-диметилмасляного альдегида, 0,05 мл концентрированной серной кислоты и 100 мг 10%-ного Pd/C. Полученную смесь гидрировали при атмосферном давлении в течение 4 ч, затем катализатор фильтровали через целит, промывая метанолом. Остаток концентрировали и вливали в 100 мл холодного эфира при перемешивании. Выделившееся твердое вещество отфильтровывали и высушивали под вакуумом, и получали 122 мг продукта.
7-Дигексиламино-доксициклина сульфат
К раствору 7-амино-доксициклина (100 мг, 0,2 ммоль) в 6 мл монометилового эфира этиленгликоля добавляли 0,6 мл гексаналя, 0,05 мл концентрированной серной кислоты и 100 мг 10%-ного Pd/C. Полученную смесь гидрировали при атмосферном давлении в течение 4 часов, затем катализатор фильтровали через целит, промывая метанолом. Остаток концентрировали и вливали в 100 мл холодного эфира при перемешивании. Выделившееся твердое вещество отфильтровывали и высушивали под вакуумом, и получали 129 мг продукта.
7-Изопропиламино-доксициклина сульфат
К раствору 7-амино-доксициклина (90 мг, 0,2 ммоль) в 8 мл монометилового эфира этиленгликоля добавляли 0,5 мл изомасляного альдегида, 0,05 мл концентрированной серной кислоты и 90 мг 10%-ного Pd/C. Полученную смесь гидрировали при атмосферном давлении в течение 6 часов, затем катализатор фильтровали через целит, промывая метанолом. Остаток концентрировали и вливали в 100 мл холодного эфира при перемешивании. Выделившееся твердое вещество отфильтровывали и высушивали под вакуумом, и получали 142 мг продукта.
7-Метил-7-изопропиламино-доксициклина сульфат
К раствору 7-изопропиламино-доксициклина (60 мг) в 4 мл монометилового эфира этиленгликоля добавляли 0,4 мл 37%-ного водного раствора формальдегида, 0,05 мл концентрированной серной кислоты и 50 мг 10%-ного Pd/C. Полученную смесь гидрировали при атмосферном давлении в течение 2 часов, затем катализатор фильтровали через целит, промывая метанолом. Остаток концентрировали и вливали в 80 мл холодного эфира при перемешивании. Выделившееся твердое вещество отфильтровывали и высушивали под вакуумом, и получали 52 мг продукта.
7-Циклобутиламино-доксициклина сульфат
К раствору 7-амино-доксициклина (80 мг) в 7 мл монометилового эфира этиленгликоля добавляли 0,3 мл циклобутанона, 0,04 мл концентрированной серной кислоты и 60 мг 10%-ного Pd/C. Полученную смесь гидрировали при атмосферном давлении в течение 24 часов, затем катализатор фильтровали через целит, промывая метанолом. Остаток концентрировали и вливали в 100 мл холодного эфира при перемешивании. Выделившееся твердое вещество отфильтровывали и высушивали под вакуумом, и получали 85 мг продукта.
7-Метил-7-циклобутиламино-доксициклина сульфат
К раствору 7-циклобутиламино-доксициклина (40 мг) в 3,5 мл монометилового эфира этиленгликоля добавляли 0,3 мл 37%-ного водного раствора формальдегида, 0,03 мл концентрированной серной кислоты и 40 мг 10%-ного Pd/C. Полученную смесь гидрировали при атмосферном давлении в течение 6 ч, затем катализатор фильтровали через целит, промывая метанолом. Остаток концентрировали и вливали в 80 мл холодного эфира при перемешивании. Выделившееся твердое вещество отфильтровывали и высушивали под вакуумом, и получали 35 мг продукта.
7-Ацетамидо-доксициклина сульфат
К перемешиваемому раствору 7-амино-доксициклина (200 мг) в 3 мл 1,3-диметил-2-имидазолидинона добавляли 200 мг бикарбоната натрия, затем 0,09 мл ацетилхлорида. Смесь перемешивали при комнатной температуре в течение 1 ч и затем фильтровали. Фильтрат концентрировали и вливали в 200 мл холодного эфира при перемешивании. Продукт, который осаждался, отфильтровывали и высушивали под вакуумом. Получали 202 мг продукта.
7-Ацетамидо-7-изопропил-доксициклина сульфат
К перемешиваемому раствору 7-изобутиламино-доксициклина (60 мг) в 1,5 мл 1,3-диметил-2-имидазолидинона добавляли 60 мг бикарбоната натрия, затем 0,05 мл ацетилхлорида. Смесь перемешивали при комнатной температуре в течение 1 ч 30 мин и затем фильтровали. Фильтрат концентрировали и вливали в 80 мл холодного эфира при перемешивании. Продукт, который осаждался, отфильтровывали и высушивали под вакуумом.
7-Диазоний-доксициклина гидрохлорид или тетрафторборат
К перемешиваемому раствору 7-амино-доксициклина (200 мг, 0,4 ммоль) в 4 мл 0,1 н. соляной кислоты в метаноле, охлажденной на ледяной бане, добавляли 0,2 мл н-бутилнитрита. Раствор перемешивали при 0°С в течение 30 минут, затем вливали в 200 мл холодного эфира при перемешивании. Выделившееся твердое вещество отфильтровывали и высушивали под вакуумом, и получали 173 мг продукта. Диазоний-тетрафторборатную соль приготавливали с помощью тетрафторборной кислоты (54%-ный раствор в эфире) вместо соляной кислоты.
7-Азидо-доксициклин
К перемешиваемому раствору 7-диазоний-доксициклина гидрохлорида (80 мг, 0,14 ммоль) в 4,5 мл 0,1 н. соляной кислоты в метаноле, охлажденной на ледяной бане, добавляли 12 мг азида натрия. Раствор перемешивали при 0°С в течение 2 часов, затем вливали в 100 мл холодного эфира при перемешивании. Выделившееся твердое вещество отфильтровывали и высушивали под вакуумом, и получали 43 мг продукта.
7-Амино-9-нитро-доксициклина сульфат
К перемешиваемому раствору 7-амино-доксициклина (220 г, 0,44 ммоль) в 4 мл концентрированной серной кислоты при 0°С добавляли нитрат калия (52 мг, 0,51 ммоль). Полученную смесь перемешивали в течение 15 минут при 0°С, затем вливали в 300 мл холодного эфира при перемешивании. Выделившееся твердое вещество отфильтровывали, промывали холодным эфиром и высушивали под вакуумом. Продукт снова растворяли в метаноле, фильтровали, и фильтрат вливали в эфир. Выделившееся твердое вещество отфильтровывали и высушивали под вакуумом, и получали 222 г продукта.
7-Диметиламино-9-нитро-доксициклина сульфат
К перемешиваемому раствору 7-диметиламино-доксициклина (40 г, 0,07 ммоль) в 1,5 мл концентрированной серной кислоты при 0°С добавляли нитрат калия (12 мг, 0,12 ммоль). Полученную смесь перемешивали в течение 30 мин при 0°С, затем вливали в 60 мл холодного эфира при перемешивании. Выделившееся твердое вещество отфильтровывали, промывали холодным эфиром и высушивали под вакуумом. ЖХ/МС демонстрирует, главным образом, один продукт, содержащий две нитрогруппы (возможно, азотный эфир желаемого продукта).
7-Диметиламино-9-диазоний-доксициклина сульфата гидрохлорид
К перемешиваемому раствору 7-диметиламино-9-амино-доксициклина (50 мг) в 1,5 мл 0,1 н. соляной кислоты в метаноле, охлажденной на ледяной бане, добавляли 0,05 мл н-бутилнитрита. Раствор перемешивали при 0°С в течение 30 минут, затем вливали в 80 мл холодного эфира при перемешивании. Выделившееся твердое вещество отфильтровывали и высушивали под вакуумом, и получали 38 мг продукта.
7-Ацетамидо-9-нитро-доксициклина сульфат
К перемешиваемому раствору 7-ацетамино-доксициклина (60 г, 0,095 ммоль) в 2 мл концентрированной серной кислоты при 0°С добавляли нитрат калия (11 мг, 0,11 ммоль). Полученную смесь перемешивали в течение 5 минут при 0°С, затем вливали в 80 мл холодного эфира при перемешивании. Выделившееся твердое вещество отфильтровывали, промывали холодным эфиром и высушивали под вакуумом.
7-Ацетамидо-9-амино-доксициклина сульфат
К перемешиваемому раствору 7-ацетамидо-9-нитро-доксициклина сульфата (77 мг, 0,12 ммоль) в 7 мл монометилового эфира этиленгликоля добавляли 60 мг 10%-ного Pd/C. Реакционную смесь гидрировали при атмосферном давлении в течение 7 часов. Катализатор фильтровали через целит, промывая метанолом. Фильтрат концентрировали, затем добавляли по каплям к 100 мл холодного эфира при перемешивании. Выделившееся твердое вещество отфильтровывали, промывали холодным эфиром и высушивали под вакуумом, и получали 62 мг продукта.
7-Ацетамидо-9-диазоний-доксициклина сульфата гидрохлорид
К перемешиваемому раствору 7-ацетамидо-9-амино-доксициклина (100 мг) в 3 мл 0,1 н. соляной кислоты в метаноле, охлажденной на ледяной бане, добавляли 0,1 мл н-бутилнитрита. Раствор перемешивали при 0°С в течение 30 минут, затем вливали в 100 мл холодного эфира при перемешивании. Выделившееся твердое вещество отфильтровывали и высушивали под вакуумом, и получали 92 мг продукта.
7-Ацетамидо-9-азидо-доксициклина сульфат
К перемешиваемому раствору 7-ацетамидо-9-диазоний-доксициклина (112 мг, 0,19 ммоль) в 6 мл 0,1 н. соляной кислоты в метаноле, охлажденной на ледяной бане, добавляли 14 мг (0,21 ммоль) азида натрия. Раствор перемешивали при 0°С в течение 35 минут, затем вливали в 150 мл холодного эфира при перемешивании. Выделившееся твердое вещество отфильтровывали и высушивали под вакуумом, и получали 94 мг продукта.
Доксициклина метилиодид
Доксициклина свободное основание (315 мг) растворяли в 2 мл метанола и 10 мл ТГФ, затем добавляли 1 мл метилиодида. Реакционную смесь выдерживали в течение 5 дней; никакой кристаллизации продукта не наблюдалось. Реакционную смесь концентрировали и вливали в 300 мл холодного эфира при перемешивании. Выделившийся продукт отфильтровывали и высушивали под вакуумом, и получали 269 мг; ЖХ/МС демонстрирует чистый продукт.
5-Ацетокси-доксициклин
Раствор доксициклина (100 мг) в 3 мл 30%-ного раствора HBr в уксусной кислоте перемешивали при комнатной температуре в течение 3,5 дней; реакционную смесь затем вливали в 100 мл холодного эфира при перемешивании, и выделившийся продукт отфильтровывали. ЖХ/МС показала неполное превращение исходного материала, поэтому продукт (86 мг) снова подвергали реакционным условиям в течение 9 ч и затем выделяли тем же способом.
9-трет-бутил-доксициклин
Раствор доксициклина (100 мг) в 2 мл трет-бутанола и 3 мл метансульфокислоты перемешивали при комнатной температуре в течение 18 часов, затем вливали в 100 мл холодной воды и экстрагировали н-бутанолом. Органические экстракты подщелачивали 1 н. раствором гидроксида натрия и рН быстро доводили до 6,5-7 концентрированной соляной кислотой. Твердое вещество, осажденное при фильтрации, затем растворяли в метаноле, концентрировали и добавляли по каплям к 30 мл холодного раствора эфир/петролейный эфир при перемешивании. Выделившееся твердое вещество отфильтровывали и высушивали под вакуумом.
9-трет-бутил-7-нитро-доксициклин
Раствор доксициклина гидрохлорида (2 г) в 10 мл трет-бутанола и 30 мл метансульфокислоты перемешивали при комнатной температуре в течение 3 часов, затем добавляли 500 мг нитрата калия и полученную смесь перемешивали еще час. Реакционную смесь вливали в 100 мл холодной раствора воды, содержащей 23 г гидроксида натрия; по каплям добавляли более разбавленный раствор NaOH до тех пор, пока реакционная смесь не станет щелочной. рН быстро доводили до 6,5-7 концентрированной соляной кислотой. Выделившееся липкое темное вещество отфильтровывали. Водный фильтрат экстрагировали еще раз н-бутанолом для получения большего количества продукта. Неочищенный продукт растворяли в минимальном количестве метанола и очищали с помощью ВЭЖХ. Объединенные после ВЭЖХ фракции концентрировали досуха, растворяли в метаноле и добавляли по каплям к 50 мл смеси эфир/петролейный эфир (3:1). Выделившееся твердое вещество отфильтровывали и получали 788 мг бледно-желтого продукта.
9-трет-бутил-7-амино-доксициклин
К раствору 9-трет-бутил-7-доксициклина (500 мг) в 25 мл монометилового эфира этиленгликоля добавляли 350 мг 10%-ного Pd/C и полученную смесь гидрировали при атмосферном давлении в течение 17 часов. Катализатор фильтровали через целит, промывая метанолом. Остаток концентрировали, растворяли в ТГФ и добавляли по каплям к холодному раствору эфир/петролейный эфир (1:1) при перемешивании. Выпавший в осадок продукт отфильтровывали и высушивали под вакуумом, и получали 452 мг продукта.
9-трет-бутил-7-диазоний-доксициклин
К перемешиваемому раствору 9-трет-бутил-7-амино-доксициклина (93 мг, 0,18 ммоль) в 2,5 мл 0,1 н. соляной кислоты в метаноле, охлажденной на ледяной бане, добавляли 0,1 мл н-бутилнитрита. Раствор перемешивали при 0°С в течение 30 минут и затем половину раствора вливали в 30 мл холодной смеси эфир/петролейный эфир (3:1) при перемешивании. Продукт приклеивался к дну лабораторного стакана в виде смолы. Его снова растворяли в метаноле и концентрировали, остаток вливали в 20 мл холодного эфира, и выделившееся твердое вещество сразу же отфильтровывали и высушивали под вакуумом; получали 42 мг продукта. Другую половину реакционной смеси использовали непосредственно для получения 9-трет-бутилового соединения, описанного ниже ниже.
9-трет-бутил-7-азидо-доксициклин
К перемешиваемому раствору реакционной смеси 9-трет-бутил-7-диазоний-доксициклина (1,5 мл) при 0°С добавляли 8 мг (0,12 ммоль) азида натрия. Раствор перемешивали в течение 3 часов, позволяя нагреться до комнатной температуры. После концентрирования реакционной смеси остаток вливали в холодную смесь эфир/петролейный эфир (3:1) при перемешивании. Твердое вещество не выделялось, поэтому раствор концентрировали досуха с получением 35 мг продукта.
9-трет-бутил-7-диметиламино-доксициклин
К перемешиваемой реакционной смеси из 9-трет-бутил-7-амино-доксициклина, содержащей 167 мг 9-трет-бутил-7-амино-доксициклина в 10 мл монометилового эфира этиленгликоля и 115 мг 10%-ного Pd/C, добавляли 1,2 мл 37%-ного водного раствора формальдегида. Полученную смесь гидрировали в течение 3,5 часов при атмосферном давлении, затем катализатор фильтровали через целит, промывая метанолом. Фильтрат концентрировали и остаток вливали в 10 мл холодного эфира при перемешивании. Выделившееся твердое вещество отфильтровывали и высушивали под вакуумом. ЖХ/МС показала ожидаемый продукт, но он был загрязнен основной примесью.
9-трет-бутил-7-ацетамидо-доксициклин
К перемешиваемому раствору 9-трет-бутил-7-амино-доксициклина (100 мг) в 2 мл 1,3-диметил-2-имидазолидинона добавляли 100 мг бикарбоната натрия, затем 0,07 мл ацетилхлорида. Смесь перемешивали при комнатной температуре в течение 1,5 часов и затем фильтровали. Фильтрат концентрировали и вливали в 100 мл холодного эфира при перемешивании. Выделившийся продукт отфильтровывали. Неочищенный продукт снова растворяли в метаноле, фильтровали и добавляли по каплям в холодную смесь эфир/петролейный эфир (3:1) при перемешивании. Продукт собирали фильтрацией и высушивали под вакуумом и получали 76 мг продукта.
4-Дедиметиламино-доксициклин
К перемешиваемому раствору тетрациклина метилиодида (860 мг) в 12 мл уксусной кислоты и 12 мл воды добавляли 432 мг цинковой пыли. После перемешивания в течение 15 мин цинковый порошок отфильтровывали. Фильтрат разбавляли 86 мл воды, содержащей 0,86 мл концентрированной соляной кислоты. Выделившийся продукт отфильтровывали и высушивали под вакуумом, и получали 419 мг продукта.
4-Дедиметиламино-9-нитро-доксициклин
К перемешиваемому раствору 4-дедиметиламино-доксициклина (402 мг, 1 ммоль) в 20 мл концентрированной серной кислоты при 0°С добавляли 111 мг нитрата калия. После перемешивания в течение 20 минут реакционную смесь вливали в 100 мл холодной воды. В воду, содержащую продукт, добавляли н-бутанол и отделяли органический слой. Экстракцию н-бутанолом повторяли дважды. Объединенные органические экстракты концентрировали и добавляли в холодную воду при перемешивании. Выделившийся продукт отфильтровывали; большее количество продукта получали путем повторной экстракции водного фильтрата н-бутанолом, концентрирования, вливания в холодную воду и фильтрации. После высушивания под вакуумом получали 353 мг продукта.
4-Дедиметиламино-9-амино-доксициклин
К перемешиваемому раствору 4-дедиметиламино-9-нитро-доксициклина (353 мг) в 17 мл монометилового эфира этиленгликоля добавляли 0,1 мл концентрированной серной кислоты и 200 мг 10%-ного Pd/C, и полученную смесь гидрировали при атмосферном давлении в течение 10 часов. Катализатор фильтровали через целит, промывая метанолом. Фильтрат концентрировали и вливали в 100 мл эфира. Выделившееся твердое вещество отфильтровывали и высушивали под вакуумом, и получали 292 мг продукта.
4-Дедиметиламино-9-диазоний-доксициклингидрохлорид
К перемешиваемому раствору 4-дедиметиламино-9-амино-доксициклина (220 мг) в 5 мл 0,1 н. соляной кислоты в метаноле, охлажденной на ледяной бане, добавляли 0,2 мл н-бутилнитрита. Раствор перемешивали при 0°С в течение 30 минут, затем вливали в 150 мл холодной смеси эфир/петролейный эфир (2:1) при перемешивании. Выделившееся твердое вещество отфильтровывали и высушивали под вакуумом, и получали 191 мг продукта.
4-Дедиметиламино-9-азидо-доксициклин
К перемешиваемому раствору 4-дедиметиламино-9-диазоний-доксициклина (150 мг, 0,32 ммоль) в 6,5 мл 0,1 н. соляной кислоты в метаноле, охлажденной на ледяной бане, добавляли 23 мг азида натрия, и полученную смесь перемешивали в течение 1 часа при 0°С. Затем смесь вливали в 120 мл холодного эфира при перемешивании. Некоторое количество образовавшегося твердого вещества фильтровали и обнаружили, что оно не является ожидаемым продуктом. Фильтрат концентрировали досуха, растворяли в метаноле и вливали в 50 мл холодной воды при перемешивании. Выделившееся твердое вещество отфильтровывали и высушивали под вакуумом; большее количество продукта получали путем экстракции фильтрата н-бутанолом, концентрирования, вливания в воду и фильтрации твердого вещества; получали 84 мг продукта.
4-Дедиметиламино-7-нитро-9-трет-бутил-доксициклин
Раствор 4-дедиметиламино-доксициклина (500 мг 1,25 ммоль) в 3 мл трет-бутанола и 15 мл метансульфокислоты перемешивали при комнатной температуре в течение 15 часов, затем добавляли 140 мг нитрата калия и полученную смесь перемешивали еще час. Реакционную смесь вливали в 200 мл холодной воды при перемешивании. Выделившееся твердое вещество отфильтровывали и высушивали на воздухе. Неочищенный продукт растворяли в минимальном количестве метанола и очищали с помощью ВЭЖХ. Объединенные после ВЭЖХ фракции концентрировали досуха, растворяли в метаноле и добавляли по каплям к 50 мл холодной воды. Выделившееся твердое вещество отфильтровывали и высушивали под вакуумом, и получали 217 мг продукта.
4-Дедиметиламино-7-амино-9-трет-бутил-доксициклин
К перемешиваемому раствору 4-дедиметиламино-7-нитро-9-трет-бутил-доксициклина (217 мг, 0,42 ммоль) в 12 мл монометилового эфира этиленгликоля добавляли 170 мг 10%-ного Pd/C и полученную смесь гидрировали при атмосферном давлении в течение 13 часов. Катализатор фильтровали через целит, промывая метанолом. Фильтрат концентрировали и вливали в 20 мл смеси эфир/петролейный эфир (3:1), и получали 40 мг продукта. Оранжевый фильтрат концентрировали досуха с получением еще 138 мг продукта.
4-Дедиметиламино-7-диазоний-9-трет-бутил-доксициклин
К перемешиваемому раствору 4-дедиметиламино-7-амино-9-трет-бутил-доксициклина (165 мг) в 5 мл 0,1 н. соляной кислоты в метаноле, охлажденной на ледяной бане, добавляли 0,15 мл н-бутилнитрита. Раствор перемешивали при 0°С в течение 30 минут; половину раствора вливали в 50 мл холодного эфира при перемешивании. Выделившееся твердое вещество отфильтровывали и высушивали под вакуумом, и получали 50 мг продукта.
4-Дедиметиламино-7-амино-доксициклин
К перемешиваемому раствору 4-дедиметиламино-доксициклина (400 мг) в 4 мл ТГФ и 4,5 мл метансульфокислоты при 0°С добавляли 418 мг (1,4 ммоль) дибензилазодикарбоксилата, и полученную смесь перемешивали в течение 4 часов, нагревая до комнатной температуры. Добавляли воду и н-бутанол, и два слоя разделяли. Водный слой экстрагировали смесью эфир/н-бутанол. Объединенные органические экстракты концентрировали досуха и добавляли 10 мл монометилового эфира этиленгликоля. Затем добавляли 430 мг 10%-ного Pd/C и полученную смесь гидрировали при атмосферном давлении в течение 12 часов. Катализатор фильтровали через целит, промывая метанолом. Фильтрат концентрировали и вливали в холодную смесь эфир/петролейный эфир (3:1) при перемешивании. Выделившийся продукт отфильтровывали и высушивали под вакуумом, и получали 114 мг продукта.
9-Нитро-миноциклина сульфат
К перемешиваемому раствору миноциклина гидрохлорида (1 г, 2,02 ммоль) в 30 мл концентрированной серной кислоты при 0°С добавляли нитрат калия (246 мг). Полученную смесь перемешивали в течение 45 минут при 0°С, затем вливали в 1,2 л холодного эфира при перемешивании. Выделившееся твердое вещество отфильтровывали, промывали холодным эфиром и высушивали под вакуумом, и получали 1,33 г продукта.
9-Амино-миноциклина сульфат
К перемешиваемому раствору 9-нитро-миноциклина сульфата (500 мг, 0,83 ммоль) в 10 мл монометилового эфира этиленгликоля и 7 мл метанола добавляли 250 мг 10%-ного Pd/C. Реакционную смесь гидрировали при атмосферном давлении в течение 4 часов. Катализатор фильтровали через целит, промывая метанолом. Фильтрат концентрировали, затем добавляли по каплям к 600 мл холодного эфира при перемешивании. Выделившееся твердое вещество отфильтровывали, промывали холодным эфиром и высушивали под вакуумом, и получали 465 мг продукта.
9-Диазоний-миноциклина сульфата гидрохлорид
К перемешиваемому раствору 9-амино-миноциклина сульфата (100 мг) в 3 мл 0,1 н. соляной кислоты в метаноле, охлажденной на ледяной бане, добавляли 0,1 мл трет-бутил нитрита (или н-бутилнитрита). Раствор перемешивали при 0°С в течение 30 минут, затем вливали в 100 мл холодного эфира при перемешивании. Выделившееся твердое вещество отфильтровывали и высушивали под вакуумом, и получали 87 мг оранжево-коричневого продукта.
9-Азидо-миноциклина сульфат
К перемешиваемому раствору 9-диазоний-миноциклина сульфата гидрохлорида (485 мг, 0,83 ммоль) в 18 мл 0,1 н. соляной кислоты в метаноле добавляли 59 мг азида натрия. Раствор перемешивали при комнатной температуре в течение 2 часов, затем фильтровали и фильтрат вливали в 500 мл холодного эфира при перемешивании. Выделившееся твердое вещество отфильтровывали и высушивали под вакуумом, и получали 480 мг бежевого продукта.
9-Ацетамидо-миноциклин
К перемешиваемому раствору 9-амино-миноциклина сульфата (100 мг, 0,175 ммоль) в 1,5 мл 1,3-диметил-2-имидазолидинона добавляли 100 мг бикарбоната натрия, затем 0,05 мл ацетилхлорида. Смесь перемешивали при комнатной температуре в течение 45 минут и затем вливали в 150 мл холодного эфира при перемешивании. Выделившееся твердое вещество отфильтровывали и высушивали под вакуумом, и получали 165 мг продукта.
4-Дедиметиламино-миноциклин
Миноциклина свободное основание (175 мг, 0,38 ммоль) растворяли в 2 мл ТГФ, затем добавляли 0,43 мл метилиодида. Кристаллизации продукта не наблюдалось. Реакционную смесь концентрировали и вливали в 200 мл смеси холодной смеси эфир/петролейный эфир (40:60) при перемешивании. Выделившийся продукт отфильтровывали и высушивали под вакуумом; продукт снова подвергали реакционным условиям еще в течение 3 дней, затем осаждали из эфира, как описано выше, и получали 123 мг миноциклина метилиодида. Неочищенный продукт растворяли в 2 мл уксусной кислоты и добавляли 2 мл воды и 60 мг цинковой пыли при перемешивании. Через 15 минут цинковый порошок отфильтровывали. Фильтрат разбавляли 15 мл воды, содержащей концентрированную соляную кислоту. Объединенные органические экстракты высушивали над сульфатом магния и концентрировали. Выделившееся твердое вещество отфильтровывали и высушивали под вакуумом, и получали 150 мг желтого продукта.
4-Дедиметиламино-9-нитро-миноциклин
4-Дедиметиламиноминоциклин (415 мг, 1 ммоль) растворяли в 30 мл концентрированной серной кислоты при комнатной температуре. Раствор охлаждали на ледяной бане и добавляли нитрат калия (110 мг, 1,09 ммоль) при перемешивании. Полученную смесь перемешивали в течение 15 минут, затем вливали в 300 мл холодного эфира при перемешивании. Выделившееся твердое вещество отфильтровывали, промывали холодным эфиром и высушивали под вакуумом. ЖХ/МС показала неполное превращение исходного материала, поэтому продукт снова растворяли в 15 мл концентрированной серной кислоты, добавляли 50 мг нитрата калия при 0°С и полученную смесь перемешивали в течение 45 минут. Продукт выделяли, как описано выше, и получали 542 мг продукта.
7,9-Дибромсанциклин
К перемешиваемому раствору санциклина (50 мг, 0,12 ммоль) в 1,5 мл концентрированной серной кислоты при 0°С добавляли 42 мг N-бромсукцинимида. Через 30 минут реакционную смесь вливали в 60 мл холодного эфира при перемешивании. Выделившееся твердое вещество отфильтровывали и высушивали под вакуумом, и получали 70 мг желтого продукта.
Смесь 7-нитро- и 9-нитро-санциклина
К перемешиваемому раствору санциклина (40 мг, 0,09 ммоль) в 1,5 мл концентрированной серной кислоты при 0°С добавляли 10 мг нитрата калия. Через 20 минут реакционную смесь вливали в 50 мл холодного эфира при перемешивании. Выделившееся твердое вещество отфильтровывали и высушивали под вакуумом, и получали 59 мг продукта. ЖХ/МС демонстрирует два моно-нитрированных продукта в соотношении 1,6:1.
При дополнительном тестировании настоящего изобретения оценивали Минимальные Ингибирующие Концентрации (МИК) пяти исследуемых продуктов при введении в организм десяти разных штаммов микроорганизмов. Исследование представляло собой определение МИК для пяти исследуемых продуктов, выполненное с использованием модификации Способа Макроразведений Бульона, изложенного в документе Национального комитета по Клиническим Лабораторным Стандартам США (NCCLS) M7-A5 (Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically, 5th Edition). Каждый исследуемый продукт оценивали в двойной повторности относительно провокационных суспензий десяти штаммов микроорганизмов. Перед тестированием каждого продукта его разбавляли (масса/объем) в стерильном бульоне Мюллера-Хинтона (МНВ от англ. Mueller-Hinton Broth) для достижения начальной концентрации 160 мкг/мл, основываясь на эффективности 1000 мкг/мг.
Приблизительно за 48 часов до тестирования отдельные стерильные пробирки с триптическим соевым бульоном (Tryptic Soy Broth) были инокулированы из виал, каждая из которых содержала лиофилизированные провокационные микроорганизмы, как указано в Таблице I ниже. Бульонные культуры инкубировали при 35±2°С в течение приблизительно 24 часов. Эти бульонные культуры, приготовленные, как описано выше, инокулировали на поверхность триптического соевого агара, содержащегося в чашках Петри, и инкубировали при 35±2°С в течение приблизительно 24 часов. Это приводило к образованию "газонов" микроорганизмов на поверхности чашек с агаром, которые использовали для приготовления провокационных суспензий.
До начала тестирования для каждого микроорганизма приготавливали исходную провокационную суспензию, содержащую приблизительно 1,0×109 КОЕ/мл, путем инокуляции тест-пробирки микроорганизмами, взятыми из чашек с твердой средой после орошения хлоридом натрия, приготовленных, как описано выше. Конечные провокационные суспензии, содержащие приблизительно 1,0×106 КОЕ/мл, приготавливали для каждого вида микроорганизмов, помещая аликвоту 0,2 мл суспензии с концентрацией 1,0×109 КОЕ/мл в стерильный полипропиленовый флакон объемом 250 мл, содержащий 200 мл бульона Мюллера-Хинтона. Конечные провокационные суспензии тщательно перемешивали перед тем, как использовать в тестировании. Конечные разведения на чашках составляли 10-3, 10-4 и 10-5. Эти чашки инкубировали при 35±2°С до тех пор, пока наблюдался достаточный рост (Таблица I).
После инкубации колонии на чашках подсчитывали вручную, используя счетчик с ручным подсчетом (hand-tally). При расчете данных использовали подсчеты в диапазоне от 30 до 300 КОЕ.
Исходную популяцию (КОЕ/мл) рассчитывали для каждой провокационной суспензии следующим образом:
КОЕ/мл=(Сi×10-D)
где:
Ci=Среднее для 2 подсчитанных чашек
D=Фактор разведения использующихся для подсчета чашек.
Популяцию (КОЕ/мл) в пробирке, содержащей продукт/бульон, рассчитывали после инокуляции для каждой провокационной суспензии следующим образом:
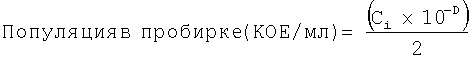
где:
Ci=Среднее для двух (2) подсчитанных чашек
D=Фактор разведения использующихся для подсчета чашек
2=Общий объем (мл) в каждой пробирке, содержащей продукт/бульон, после инокуляции.
Процедуру тестирования проводили следующим образом: аликвоты по 30 мл бульона Мюллера-Хинтона переносили в стерильные флаконы. Аликвоту продукта объемом 30 мл с исходной концентрацией 160 мкг/мл переносили в первый из 11 стерильных флаконов, каждый из которых содержит 30 мл бульона Мюллера-Хинтона, и тщательно перемешивали, для того чтобы получить разведение продукта 1:2 (об/об). Аликвоту 30 мл отбирали из флакона, приготовленного, как описано выше, и использовали для последовательных разведении 1:2 (об/об) в оставшихся 10 флаконах (разведения продукта - 1:4, 1:8, 1:16, 1:32, 1:64, 1:128, 1:256, 1:512, 1:1024 и 1:2048), каждый из которых содержит 30 мл бульона Мюллера-Хинтона. Каждый флакон тщательно перемешивали перед тем, как отобрать аликвоту 30 мл, необходимую для следующего в этой последовательности разведения. Аликвоты по 1,0 мл из каждого приготовленного разведения продукта и аликвоты по 1,0 мл из исходного препарата продукта (концентрация продукта 160 мкг/мл) переносили в отдельные стерильные тест-пробирки. Для каждого микроорганизма, оцениваемого относительно каждого продукта, приготавливали серию из 12 пробирок, каждая из которых содержит 1,0 мл соответствующего разведения продукта (1:1, 1:2, 1:4, 1:8, 1:16, 1:32, 1:64, 1:128, 1:256, 1:512, 1:1024 и 1:2048) (Таблица I). Эта процедура давала в итоге концентрации продукта, варьирующие от 160 мкг/мл до 0,078 мкг/мл.
Аликвоту суспензии 1,0 мл, содержащую приблизительно 1,0×106 КОЕ/мл, вносили в каждую пробирку для разведения продукта в этой серии, получая таким образом конечную последовательность разведении продукта 1:2, 1:4, 1:8, 1:16, 1:32, 1:64, 1:128, 1:256, 1:512, 1:1024, 1:2048 и 1:4096, с каждой пробиркой для разведения, содержащей приблизительно 5,0×105 КОЕ/мл провокационного микроорганизма. Это давало конечные концентрации продукта, варьирующие от 80 мкг/мл до 0,039 мкг/мл. Описанную выше процедуру тестирования выполняли в двойных повторностях для каждого тестируемого вида микроорганизма (Таблица I) относительно каждого исследуемого продукта. Для каждого микроорганизма приготавливали положительную контрольную пробирку (контроль роста), содержащую аликвоту 1,0 мл бульона Мюллера-Хинтона и аликвоту 1,0 мл провокационной суспензии (Таблица I). Приготавливали также отрицательную (среда) контрольную пробирку (контроль стерильности; без инокуляции микробов) с бульоном Мюллера-Хинтона.
Контроли плотности продукта приготавливали для каждого продукта путем переноса аликвот по 1,0 мл из каждого разведения продукта в отдельные стерильные тест-пробирки. Аликвоту 1,0 мл стерильного бульона Мюллера-Хинтона вносили в каждую из полученных пробирок для разведения продукта и тщательно перемешивали, используя вихревой смеситель и получая, таким образом, конечную последовательность разведений продукта, идентичную той, что описана выше. Пробирки, содержащие провокационную суспензию/разведение продукта, и контроли инкубировали при 35±2°С в течение 20 часов, до тех пор, пока в положительных контрольных пробирках наблюдался хороший рост.
После инкубации пробирки проверяли на рост микроорганизмов, определяемый визуально на основании плотности.
Минимальная Ингибирующая Концентрация (МИК) для каждого исследуемого продукта в зависимости от каждого провокационного микроорганизма регистрировалась как наибольшее разведение исследуемого продукта, которое полностью ингибировало рост этого микроорганизма, что определялось невооруженным глазом. Результаты дублированных измерений для каждого исследуемого продукта в зависимости от каждого микроорганизма регистрировали и затем усредняли одновременно с получением конечных представленных значений.
Таблица II представляет Минимальные Ингибирующие Концентрации, выраженные как разведение продукта для каждого исследуемого продукта в отношении каждого из 10 тестируемых микроорганизмов. Таблица III представляет Минимальные Ингибирующие Концентрации, выраженные как концентрация продукта (мкг/мл) для каждого исследуемого продукта в отношении каждого из 10 тестируемых микроорганизмов. Исходный раствор, приготовленный для каждого продукта, имел концентрацию 160 мкг/мл, основанную на эффективности 1000 мкг/мл.
В дополнительных исследованиях изучали активность производных тетрациклина по настоящему изобретению в качестве ингибиторов матрикс-разрушающих металлопротеиназ (ММР), которые вовлекаются в злокачественный опухолевый рост и метастазы. В этих исследованиях использовали опухолевую модель Dunning MAT LyLu, которая развивалась в крысах Copenhagen из трансплантируемой опухоли, перенесенной из первичной опухоли простаты из дорзальной простаты старых самцов крыс Copenhagen. Опухоль MAT LyLu является спонтанно метастазирующей в лимфатические узлы и легкие при инъекции и дает метастазы в кости приблизительно у 80-100% реципиентных животных. Свежие дозированные растворы, содержащие 10 мг/мл каждого исследуемого вещества в 2%-ной карбоксиметилцеллюлозе, приготавливали ежедневно и использовали при тестировании.
Сначала в исследованиях оценивали токсичность в двух группах из пяти не несущих опухоль животных, каждое из которых получало два аналога тетрациклина (9-амино-доксициклин и 9-нитро-доксициклин) в дозе 40 мг/кг ежедневно в течение семи дней.
Трем группам животных (10/группу) вводили либо носитель, либо один из двух аналогов ежедневно в течение трех недель и затем трижды в неделю до смерти или до забоя. Введение доз начинали за семь дней до имплантации опухолевых клеток MAT LyLu инъекцией в хвостовую вену. Приблизительно 0,1 мл суспензии опухолевых клеток инъецировали в одну из боковых хвостовых вен. Доза, которую нужно выбрать, основывалась на двух факторах: (1) эффективности и (2) заболеваемости, связанной с введением аналога, если таковая имеется. Следующая таблица (Таблица IV ниже) демонстрирует время жизни и еженедельный вес тела животных в контрольных и обработанных группах. Как показано в Таблице, 9-амино-доксициклин и 9-нитро-доксициклин продемонстрировали значительное увеличение коэффициента выживания после опухолевой имплантации, хотя и дающее лишь незначительные изменения в весе тела, по сравнению с контрольной группой. Это последнее открытие является особенно важным, так как обычные способы лечения рака часто приводят к значительной потере веса.
В дополнительных исследованиях оценивали ингибирующую активность ММР, а также процентное ингибирование разных злокачественных опухолей различными производными тетрациклина. Результаты представлены в Таблице V ниже и демонстрируют эффективность способов лечения по настоящему изобретению. Одна цель исследований состояла в том, чтобы оценить ряд производных тетрациклина в качестве ингибиторов очищенных матриксных металлопротеиназ человека, включая ММР1, 2, 3, 7, 9 и 14. Производные тетрациклина были эффективными ингибиторами в диапазоне от мМ до мкМ.
ПРОТОКОЛЫ ЭКСПЕРИМЕНТОВ
1. Протокол измерения ингибирования ММР1 (коллагеназы-1) производными тетрациклина
Активность ММР1 определяли по высвобождению растворимых 14С-меченых фрагментов коллагена из 14С-ацетилированного коллагена I типа из кожи крысы (Methods in Enzymology 80, 711, 1981).
Составляющие анализа:
100 мкг 14С-меченного коллагена I типа из кожи крысы (5-6000 dpm)
50 мМ Трис-HCl, рН 7,9
15 мМ CaCl2, 0,02% азида
Рекомбинантная ММР1 человека, 3-4 нМ
± Тетрациклин (до 1 мМ) в общем объеме 300 мкл.
Инкубации проводили при 35°С в течение 5 часов. Нерасщепленные фибриллы коллагена, которые образуются в течение первых 10 минут, удаляли центрифугированием при 10000 g, 4°С, 10 минут. 200 мкл супернатанта просчитывали в Packard Opti Phase Supermix сцинтилляционной жидкости с помощью сцинтилляционного счетчика. Использовали только данные, полученные в пределах линейной части анализа (10-70% лизиса коллагена). На основании процентного ингибирования активности только ММР1 диапазоном концентраций каждого образца рассчитывали величину IC50 для каждого тетрациклина с помощью линейного регрессивного анализа.
2. Протокол измерения ингибирования ММР2 (желатиназы А)
Активность ММР2 определяли по высвобождению ТХУ (трихлоруксусная кислота)-растворимых фрагментов из 14С-ацетилированного желатина I типа из кожи крысы (Methods in Emymology 248, 470, 1995; Biochem. J. 195. 1981)
Составляющие анализа:
100 мкг 14С-меченного желатина (коллагена I типа, денатурированного при 60°С в течение 20 мин)
50 мМ Трис-HCl, рН 7,9
15 мМ CaCl2, 0,02% азида
Рекомбинантная ММР1 человека, 0,1-0,5 нМ
± Тетрациклин (до 1 MM) в общем объеме 250 мкл.
Инкубации проводили при 37°С в течение 16 часов. Реакцию останавливали охлаждением инкубации и добавлением 50 мкл холодной 90% (масса/объем) трихлоруксусной кислоты при осторожном перемешивании. ТХУ-преципитацию продолжали в течение 15 минут при 4°С перед центрифугированием при 10000 g в течение 10 минут при 4°С. 200 мкл ТХУ-супернатанта брали для определения содержания радиоактивности с помощью сцинтилляционного счетчика (сцинтилляционная жидкость Packard Opti Phase Supermix). Использовали только данные, полученные в пределах линейной части анализа (10-70% лизиса). Процентное ингибирование ММР2 каждой концентрацией тетрациклина наносили на график, для того чтобы получить величину IC50, используя линейный регрессивный анализ.
3. Протокол измерения ингибирования ММРЗ (стромелизина 1)
Активность ММР3 определяли по высвобождению ТХУ-растворимых фрагментов из 14С-ацетилированного β-казеина (Sigma), как описано в Methods in Enzymology 248, 451 (1995).
Составляющие анализа:
100 мкг 14С-казеина (7000 dpm)
50 мМ Трис-HCl, рН 7,9
15 мМ CaCl2, 0,02% азида
Рекомбинантная ММР3 человека, 0,25-1,0 нМ
±тетрациклин (до 1 мМ) в общем объеме 250 мкл.
Инкубации проводили при 37°С в течение 16 часов. Реакцию останавливали добавлением 500 мкл холодной 18%-ной ТХУ и выдерживанием на льду в течение 15 минут. ТХУ-преципитаты удаляли центрифугированием при 10000 g в течение 10 минут при 4°С, и 200 мкл супернатанта брали для сцинтилляционного подсчета. Использовали только данные, полученные в пределах линейной части анализа (10-60% лизиса). Процентное ингибирование ММР3 каждой концентрацией тетрациклина-кандидата наносили на график, для того чтобы получить величину IC50, используя линейный регрессивный анализ.
4. Протокол измерения ингибирования ММР7 (матрилизина)
Активность ММР7 определяли по высвобождению ТХУ-растворимых фрагментов из 14С ацетилированного β-казеина (Sigma), как описано в Methods in Enzymology 248, 451 (1995).
Составляющие анализа:
100 мкг 14С-казеина (7000 dpm)
50 мМ Трис-HCl, рН 7,9
15 мМ CaCl2, 0,02% азида
Рекомбинантная ММР7 человека, 1,5 нМ
± тетрациклин (до 1 мМ) в общем объеме 250 мкл.
Инкубации проводили при 37°С в течение 16 часов. Инкубацию останавливали добавлением 500 мкл холодной 18%-ной ТХУ и выдерживанием на льду в течение 15 минут. ТХУ-преципитаты удаляли центрифугированием при 10000 g в течение 10 минут при 4°С, и 200 мкл супернатанта брали для сцинтилляционного подсчета. Использовали только данные, полученные в пределах линейной части анализа (10-60% лизиса). Процентное ингибирование ММР7 каждой концентрацией тетрациклина-кандидата наносили на график, для того чтобы получить величину IC50.
5. Протокол измерения ингибирования ММР9 (желатиназы В)
Активность ММР9 определяли по высвобождению ТХУ-растворимых фрагментов из 14С-ацетилированного коллагена I типа из кожи крысы (Methods in Enzymology 248, 470, 1995; Biochem. J. 195, 1981)
Составляющие анализа:
100 мкг 14С-меченного желатина (коллагена, денатурированного при 60°С в течение 20 мин)
50 мМ Трис-HCl, рН 7,9
15 мМ CaCl2, 0,02% азида
Рекомбинантная ММР9 человека, 1,2-2 нМ
±Тетрациклин (до 1 мМ) в общем объеме 250 мкл.
Инкубации проводили при 37°С в течение 16 часов. Инкубации останавливали охлаждением инкубации и добавлением 50 мкл холодной 90%-ной (масса/объем) трихлоруксусной кислоты при осторожном перемешивании. ТХУ-преципитацию продолжали в течение 15 минут при 4°С перед центрифугированием при 10000 g в течение 10 минут при 4°С. 200 мкл ТХУ-супернатанта брали для определения содержания радиоактивности с помощью сцинтилляционного счетчика (сцинтилляционная жидкость Packard Opti Phase Supermix). Использовали только данные, полученные в пределах линейной части анализа (10-70% лизиса). Процентное ингибирование ММР9 каждой концентрацией тетрациклина наносили на график, для того чтобы получить величину IC50.
6. Протокол измерения ингибирования ММР14 (MTI-MMP) производными тетрациклина
Активность ММР14 определяли по высвобождению растворимых 14С-меченых фрагментов коллагена из 14С-ацетилированного коллагена I типа из кожи крысы (Methods in Emymology 80, 711, 1981).
Составляющие анализа:
100 мкг 14С-меченого коллагена (5-6000 dpm)
50 мМ Трис-HCl, рН 7,9
15 мМ CaCl2 0,02% азида
Человеческий рекомбинантный ММР14 50-100 нМ
± Тетрациклин (до 1 мМ) в общем объеме 300 мкл.
Инкубации проводили при 35°С в течение 15 часов. Нерасщепленные фибриллы коллагена, которые образуются в течение первых 10 минут, удаляли центрифугированием при 10000 g, 4°С, 10 минут. 200 мкл супернатанта просчитывали в сцинтилляционной жидкости Packard Opti Phase Supermix с помощью сцинтилляционного счетчика. Использовали только данные, полученные в пределах линейной части анализа (10-70% лизиса коллагена). На основании процентного ингибирования активности только ММР1 диапазоном концентраций каждого образца рассчитывали величину IC50 для каждого тетрациклина.
Другая цель состояла в том, чтобы проанализировать in vitro эффекты специфических, тетрациклин-производных соединений на хемоинвазивный потенциал следующих клеточных линий:
С8161, клетки меланомы человека
MDA-MB-231, клетки рака молочной железы человека
РС3, клетки рака простаты человека
RPMI-8226, клетки миеломы человека
Ark, клетки миеломы человека
Arp-1, клетки миеломы человека
Хемоинвазивный анализ in vitro с мембранной инвазивной культуральной системы (МИКС) был выбран, для того чтобы измерить изменения в инвазивном потенциале специфических раковых клеток человека в ответ на тетрациклин-производные соединения. Маточные растворы каждого соединения гидратировали в воде, содержащей 2% ДМСО, с рН 10,0. После солюбилизации к раствору добавляли HCl для установления рН 7,5-8,0. Затем этот раствор заворачивали в фольгу и хранили при 4°С в течение 24 часов анализа. Для каждого анализа приготавливали свежее соединение. Анализы по хемоинвазии in vitro для определения инвазивности опухолевых клеток выполняют с помощью мембранной инвазивной культуральной системы (МИКС), как описано ранее.
МИКС-система представляет собой термически обработанную пластиковую разветвленную (коллекторную) систему, состоящую из двух подогнанных комплектов чашек, содержащих четырнадцать лунок с диаметром 13 мм. Между этими чашками был размещен поликарбонатный фильтр, содержащий 10 мкм поры и покрытый определенным матриксом, состоящим из человеческого ламинина, коллагена IV, желатина, образующих барьер толщиной 35 мкм между верхней и нижней частями лунок в МИКС. До помещения этого барьера на место нижние лунки заполняли бессывороточной культуральной средой RPMI, сделанной на 50% из кондиционированной среды, полученной от 2-дневных культур фибробластов человека, которая была очищена от клеток и клеточного дебриса либо центрифугированием, либо фильтрацией через 0,45 мкм стерильный фильтр. Затем в нижние лунки помещали барьер, и после сборки системы в верхние лунки добавляли свежую бессывороточную среду. Затем в лунки добавляли пятьдесят тысяч опухолевых клеток либо в присутствии соединения, либо в присутствии носителя ДМСО (контроли). Из 12 аналитических лунок в каждом коллекторе 3 случайным образом выбранные лунки служили в качестве контролей, 3 лунки содержали 1 мкг/мл соединения, 3 лунки - 10 мкг/мл и 3 лунки - 25 мкг/мл. Через 24 часа из нижних лунок удаляли клетки и среду и заменяли фосфатно-солевым буфером плюс 2 мМ ЭДТА. Этот раствор использовали для удаления всех клеток из нижней лунки, и эту промывку добавляли к полученным клеткам плюс среда из той же лунки. Эти образцы затем собирали на полилизин-содержащую поликарбонатную мембрану, содержащую 3 мкм поры, используя дот-блот коллекторную систему. Эти коллектор-фильтры затем фиксировали в метаноле и окрашивали in situ с помощью красителя Райта (набор для окрашивания Leukostat). Затем фильтры помещали на предметные стекла для микроскопа с иммерсионным маслом для микроскопии, которое уменьшало преломление света от пор, и просчитывали 5 микроскопических полей для расчета количества клеток, которые проникли через мембрану в течение 24 часов. Эти числа затем статистически анализировали, как описано ниже.
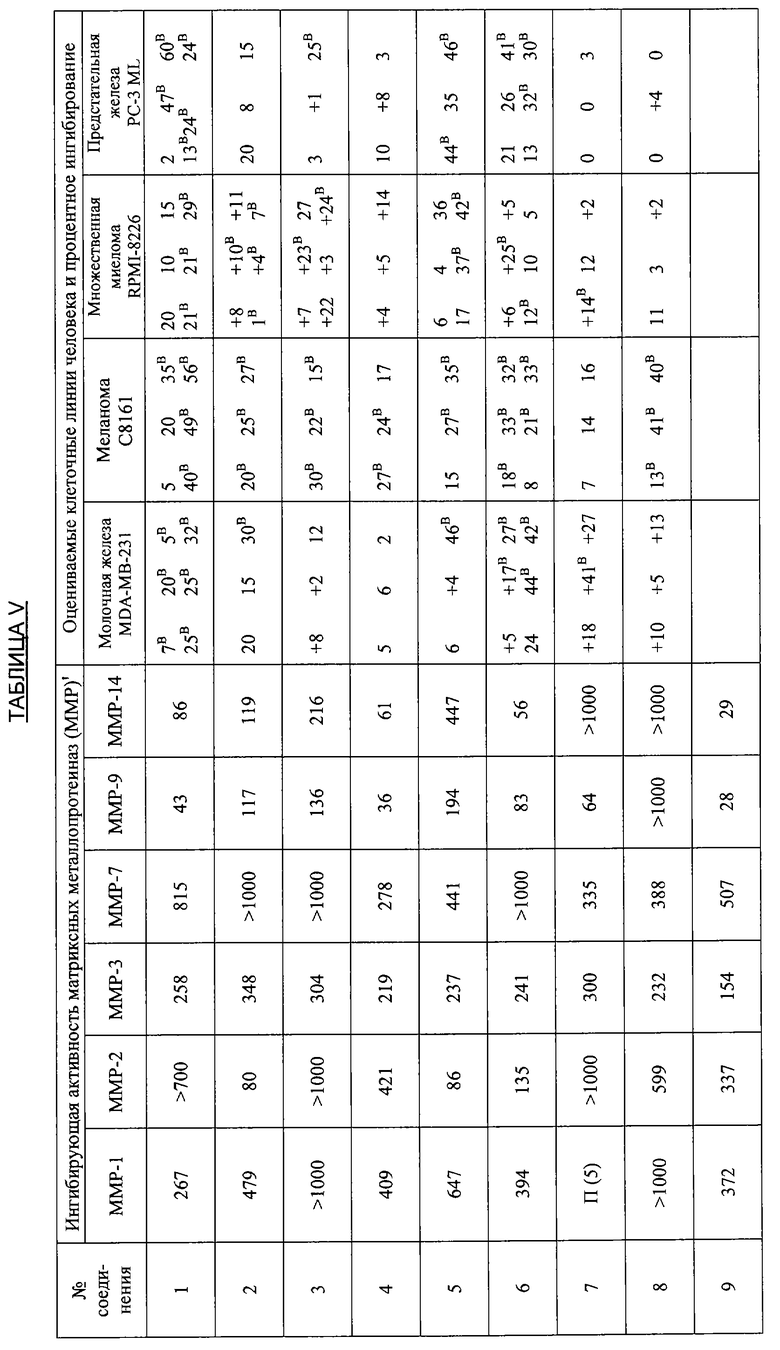
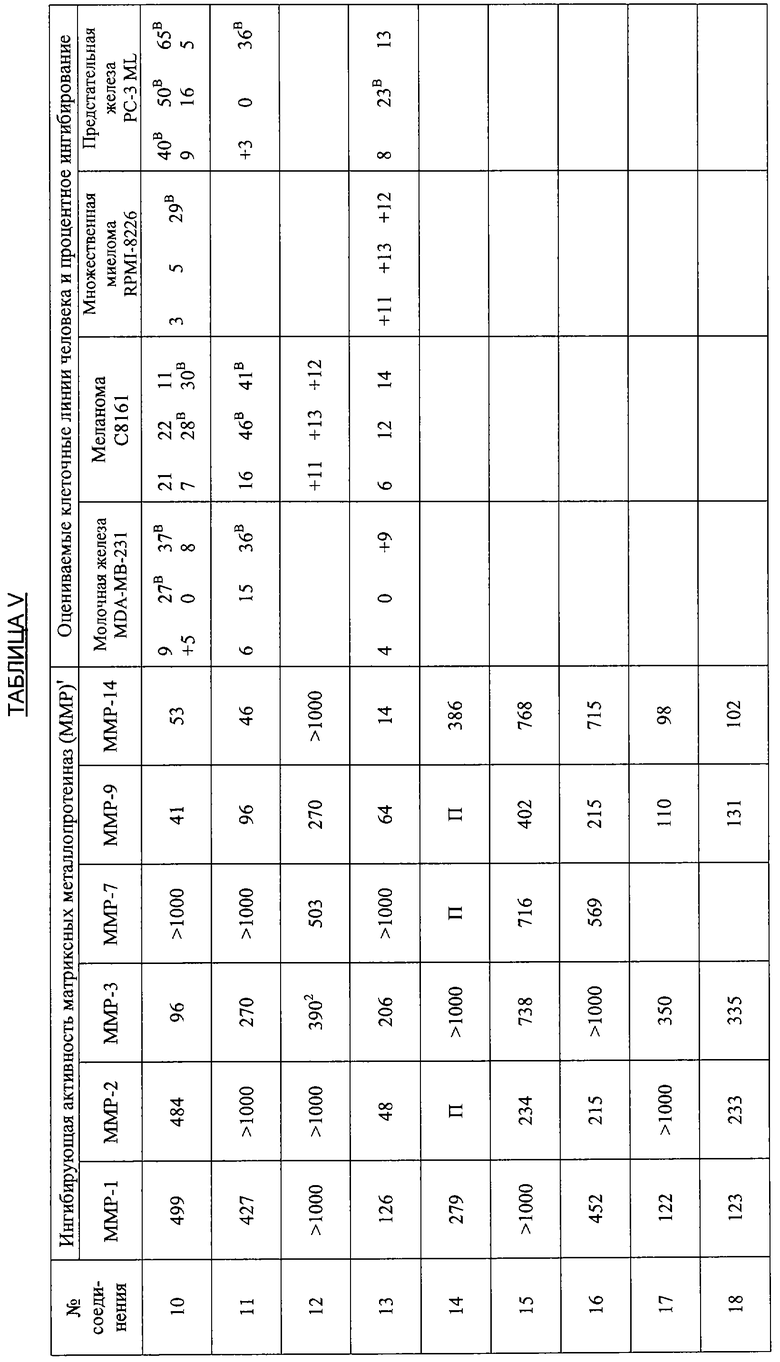
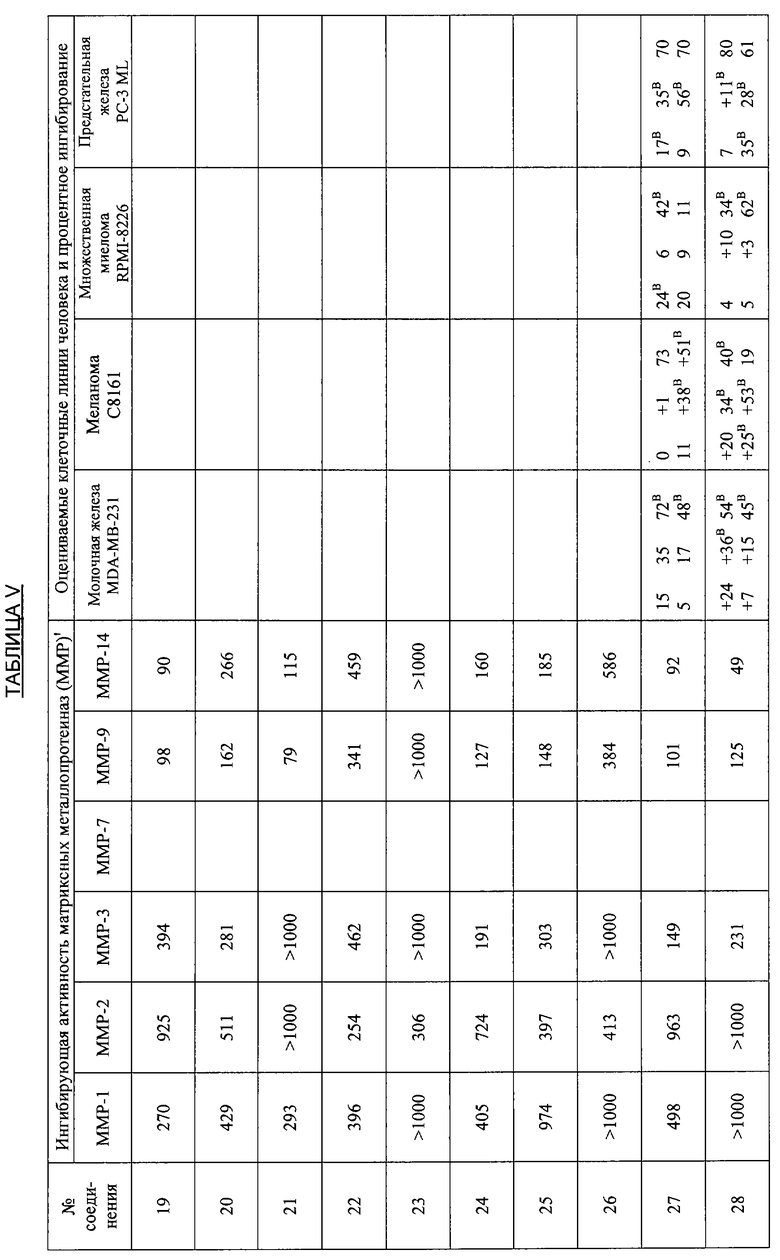
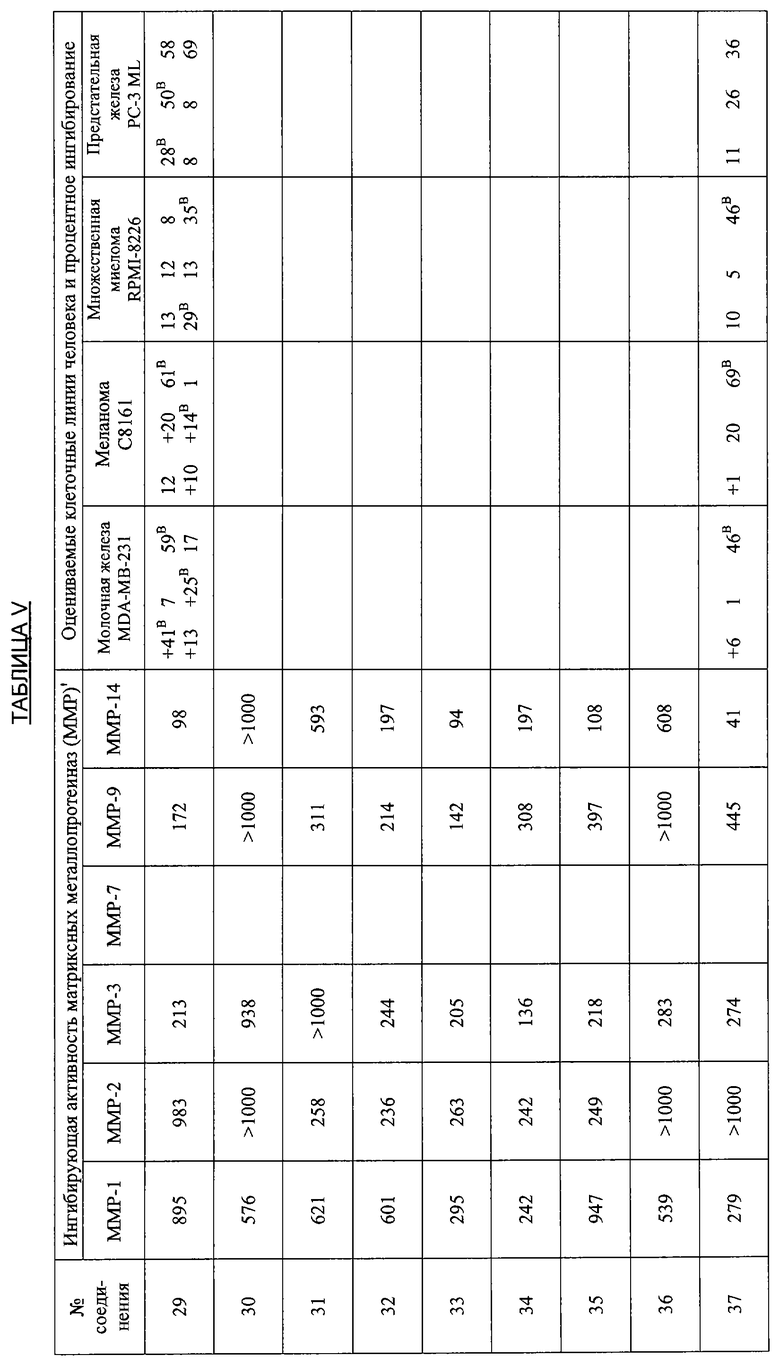
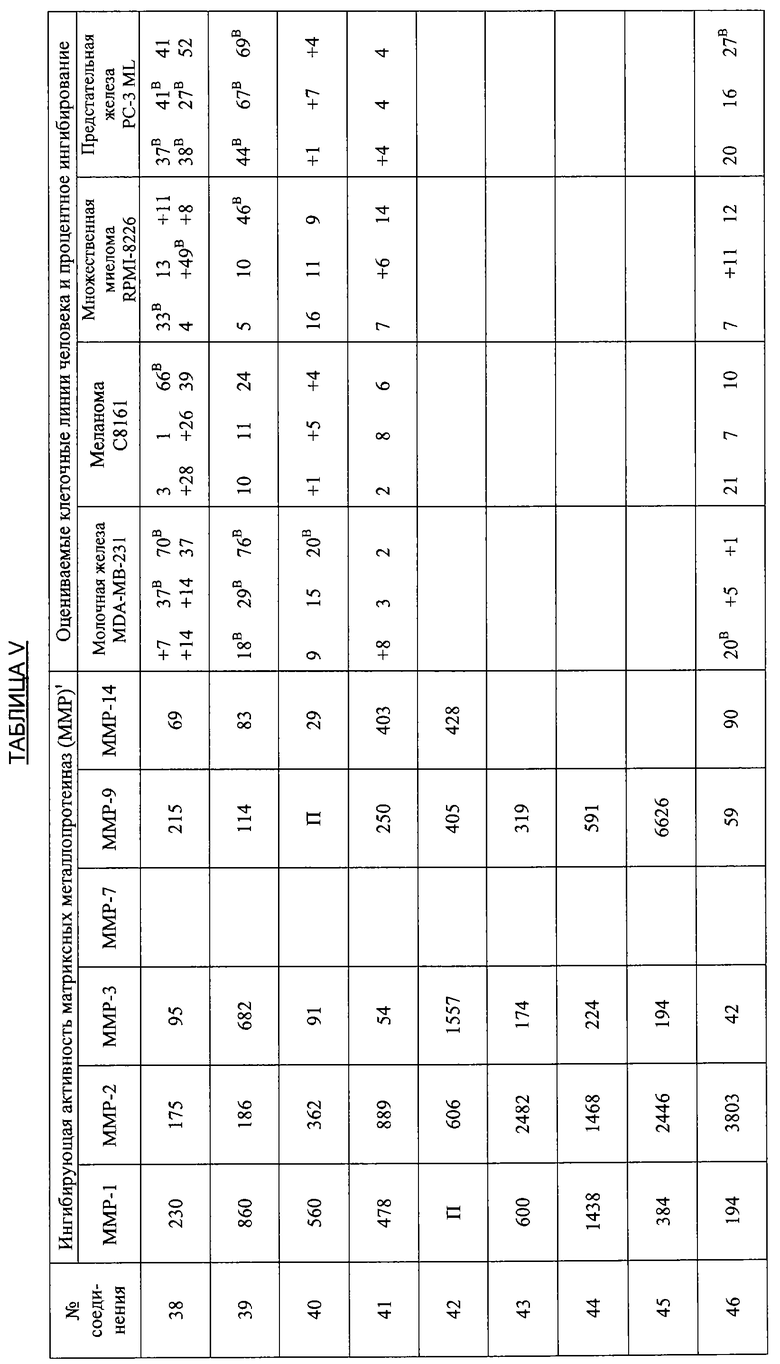
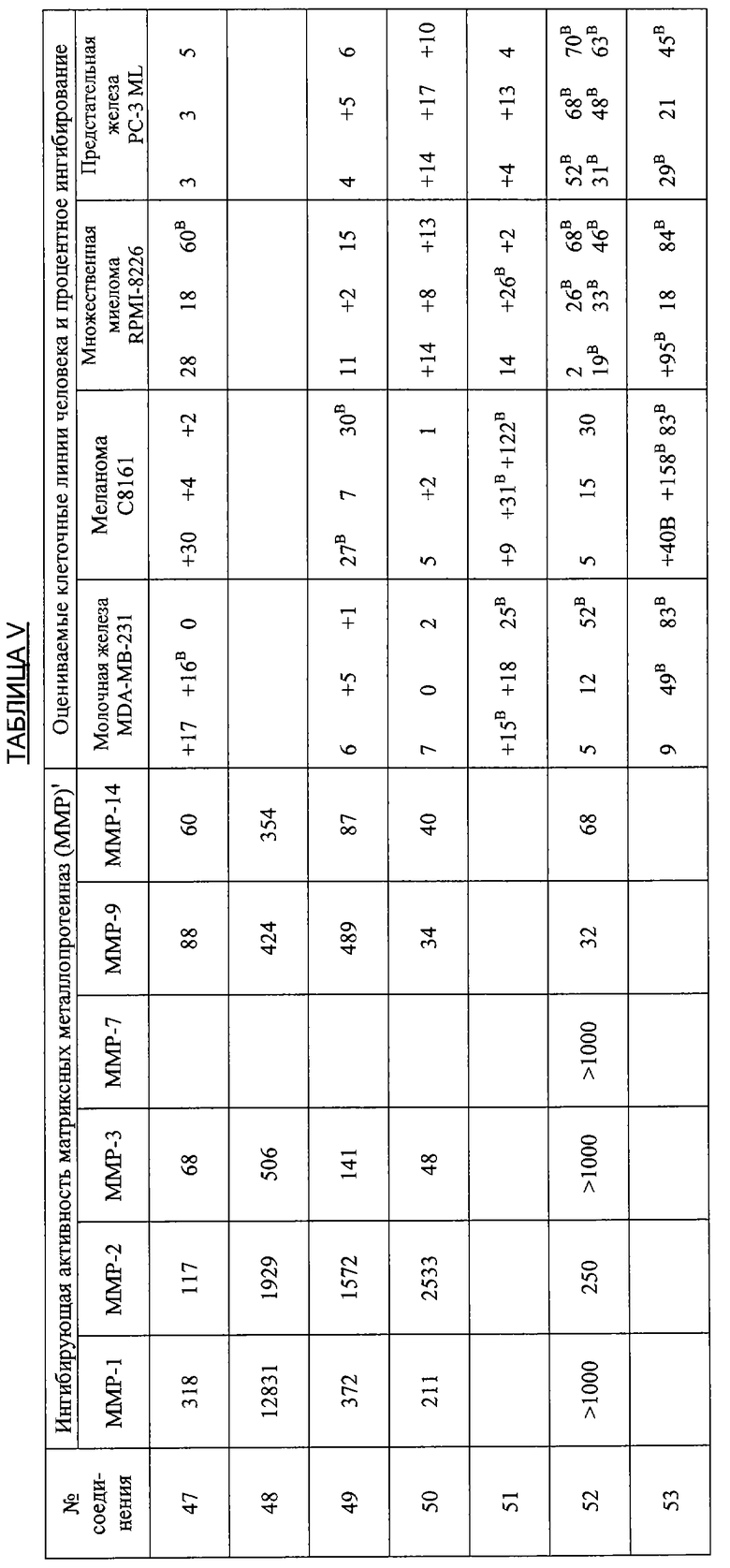
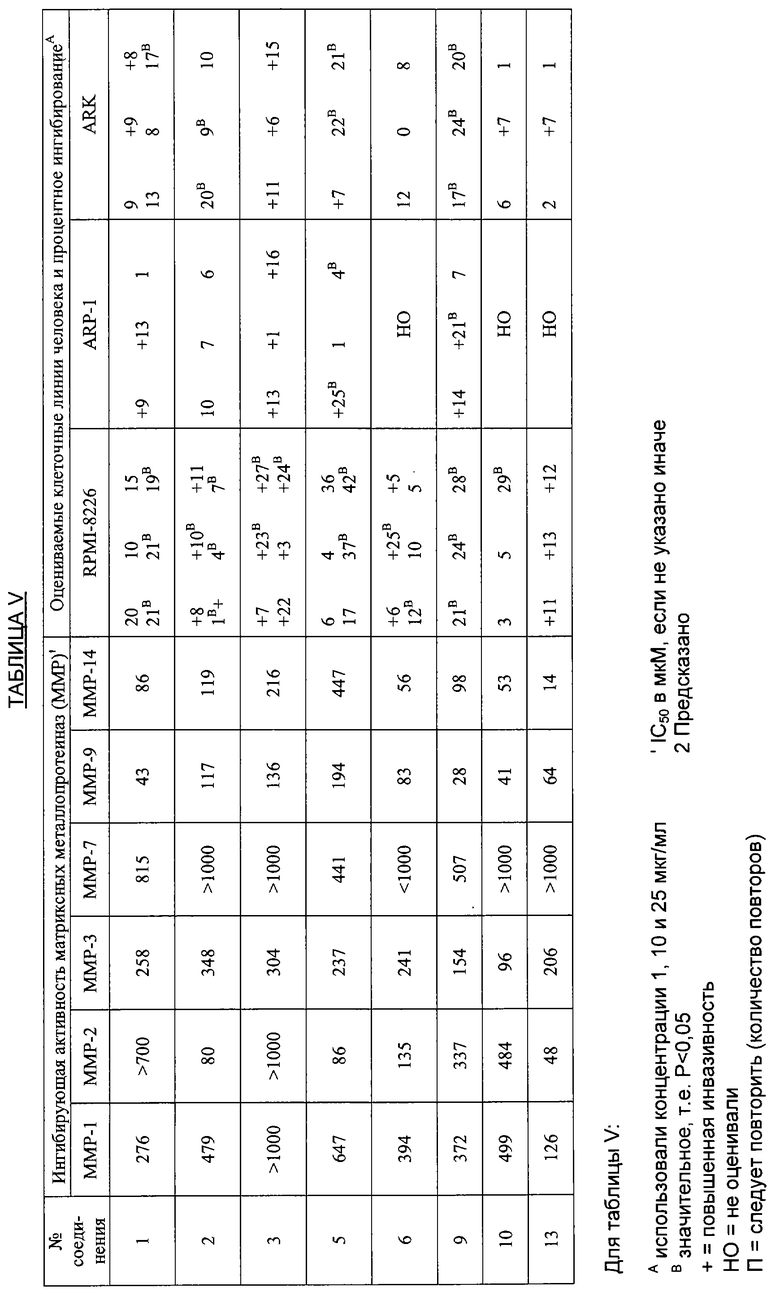
Ключ к Таблице V:
Соединение Вещество
1: 9-Нитро-6-дезокси-5-гидрокситетрациклин H2SO4
2: 9-Амино-6-дезокси-5-гидрокситетрациклин H2SO4
3: 9-Изопропиламино-6-дезокси-5-гидрокситетрациклин H2SO4
4: 7-Диметиламино-6-деметил-6-дезокси-9-нитротетрациклин H2SO4
5: То же, что и 2
6: 9-Азидо-6-дезокси-5-гидрокситетрациклин H2SO4
7: 9-Амино-7-диметиламино-6-деметил-6-дезокситетрациклин H2SO4
8: 9-Ацетамидо-7-диметиламино-6-деметил-6-дезокситетрациклин H2SO4
9: 7-Диметиламино-6-деметил-6-дезокситетрациклин-9-диазоний H2SO4 HCl
10: 9-Азидо-7-деметиламино-6-деметил-6-дезокситетрациклин H2SO4
11: 6-Дезокси-5-гидрокситетрациклин-9-диазоний H2SO4
12: 7-Амино-6-дезокси-5-гидрокситетрациклин H2SO4
13: 7,9-Дибром-6-деметил-6-дезокситетрациклин H2SO4
14: 7-Амино-6-дезокси-5-гидрокси-9-нитротетрациклин H2SO4
15: 7-Диметиламино-6-дезокси-5-гидрокситетрациклин H2SO4
16: 9-Ацетамидо-6-дезокси-5-гидрокситетрациклин H2SO4
17: 7-Ди-н-Бутиламино-6-дезокси-5-гидрокситетрациклин H2SO4
18: 7-Ди-н-Гексиламино-6-дезокси-5-гидрокситетрациклин H2SO4
19: 7-Ди-(3,3-диметилбутил)амино-6-дезокси-5-гидрокситетрациклин H2SO4
20: 7-Азидо-6-дезокси-5-гидрокси-тетрациклин H2SO4
21: 6-Дезокси-5-гидрокситетрациклин-7-диазоний H2SO4
22: 7-Ацетамидо-9-нитро-6-дезокси-5-гидрокситетрациклин H2SO4
23: 7-Ацетамидо-6-дезокси-5-гидрокситетрациклин H2SO4
24: 7-Ди-н-пропиламино-6-дезокси-5-гидрокситетрациклин H2SO4
25: 7-Изобутиламино-6-дезокси-5-гидрокситетрациклин H2SO4
26: 7-Ацетамидо-9-амино-6-дезокси-5-гидрокситетрациклин H2SO4
27: 7-Изобутилметиламино-6-дезокси-5-гидрокситетрациклин H2SO4
28: 6-Дезокси-5-ацетокси-тетрациклин H2SO4
29: 7-Ацетилизобутиламино-6-дезокси-5-гидрокситетрациклин H2SO4
30: 7-Ацетамидо-6-дезокси-5-гидрокситетрациклин-9-диазоний H2SO4
31: 7-Диметиламино-6-дезокси-5-гидрокситетрациклин-9-диазоний H2SO4
32: 7-Цикпобутиламино-6-дезокси-5-гидрокситетрациклин H2SO4
33: 7-Циклобутилметиламино-6-дезокси-5-гидрокситетрациклин H2SO4
34: 4-Дедиметиламино-6-дезокси-5-гидрокситетрациклин
35: 4-Дедиметиламино-6-дезокси-5-гидрокси-9-нитротетрациклин
36: 9-Амино-4-дедиметиламино-6-дезокси-5-гидрокситетрациклинсульфат
37: 4-Дедиметиламино-6-дезокси-5-гидрокситетрациклин-9-диазонийсульфат
38: 7-Нитро-9-трет-бутил-6-дезокси-5-гидрокситетрациклин H2SO4
39: 9-трет-Бутил-6-дезокси-5-гидрокситетрациклин H2SO4
40: 7-Нитро-9-трет-бутил-4-дедиметиламино-6-дезокси-5-гидрокситетрациклин H2SO4
41: 4-Дедиметиламино-7-диметиламино-6-деметил-6-дезокситетрациклин H2SO4
42: 7-Ацетиламино-9-азидо-6-дезокси-5-гидрокситетрациклин
43: 9-трет-Бутил-6-дезокси-5-гидрокситетрациклин-7-диазоний H2SO4
44: 7-Амино-9-трет-бутил-6-дезокси-5-гидрокситетрациклин H2SO4
45: 7-Азидо-9-трет-бутил-6-дезокси-5-гидрокситетрациклин H2SO4
46: 6-Дезокси-5-гидрокситетрациклин-4-метилиодид
47: 7-(1,2-Бензилкарбоксигидразин)-6-дезокси-5-гидрокситетрациклин HCl
48: 4-Дедиметиламино-7-амино-6-дезокси-5-гидрокситетрациклин H2SO4
49: 7-Амино-9-трет-бутил-4-дедиметил-6-дезокси-5-гидрокситетрациклин-H2SO4
50: 4-Дедиметиламино-9-трет-бутил-6-дезокси-5-гидрокситетрациклин-7-диазоний H2SO4
51: 9-(4-Фторфенил)-6-дезокси-5-гидрокситетрациклин
52: 4-Эпи-7-хлортетрациклин гидрохлорид
53: 6-Деметил-6-дезокситетрациклин HCl
Хотя настоящее изобретение было описано со ссылкой на его частную форму осуществления, понятно, что другие многочисленные формы и модификации этого изобретения будут очевидны специалистам в данной области. Приложенную формулу изобретения и само изобретение следует истолковывать в общем смысле, для того чтобы охватить все очевидные формы и модификации, которые соответствуют сущности и находятся в рамках настоящего изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ 6-МЕТИЛЕНТЕТРАЦИКЛИНА | 1972 |
|
SU341225A1 |
| МОНОЦИКЛИЧЕСКИЕ L-НУКЛЕОЗИДЫ, ИХ АНАЛОГИ И ПРИМЕНЕНИЯ | 1997 |
|
RU2188828C2 |
| Способ получения пиримидиновых нуклеозидов | 1987 |
|
SU1731064A3 |
| ПЕНТАСАХАРИДНЫЙ КОНЪЮГАТ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ | 2000 |
|
RU2266913C2 |
| 3,7-ДИАЗАБИЦИКЛО[3.3.1]-ПРЕПАРАТЫ КАК АНТИАРИТМИЧЕСКИЕ СОЕДИНЕНИЯ | 2002 |
|
RU2286993C2 |
| ТАКСАНЫ С БОКОВОЙ ЦЕПЬЮ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ, СПОСОБЫ ПОЛУЧЕНИЯ | 1993 |
|
RU2125042C1 |
| РАЦЕМИЧЕСКИЕ ИЛИ ОПТИЧЕСКИ АКТИВНЫЕ ПЕРГИДРО-1H-ПИРИДО(1,2-А) ПИРАЗИНЫ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1991 |
|
RU2090565C1 |
| НИТРАТНЫЕ СОЛИ АНТИМИКРОБНЫХ СОЕДИНЕНИЙ | 2001 |
|
RU2288231C2 |
| СУЛЬФОНАМИДНОЕ ПРОИЗВОДНОЕ ИЗОКСАЗОЛА ИЛИ ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМАЯ СОЛЬ | 1993 |
|
RU2116301C1 |
| Способ получения производных тетрапептидов или их солей | 1978 |
|
SU908246A3 |
Изобретение относится к медицине, в частности к онкологии, и касается вариантов способа лечения рака, включающего введение субъекту, нуждающемуся в таком лечении, эффективного количества 9-амино-6-дезокси-5-гидрокситетрациклина или его соли, либо 9-нитро-6-дезокси-5-гидрокситетрациклина или его соли. При этом рак выбран из группы, включающей рак молочной железы, меланому, миелому и рак простаты. Изобретение обеспечивает противоопухолевый, антиметастатический эффект и увеличение коэффициента выживаемости за счет установленного ингибирования указанными производными тетрациклина матриксразрушающих металлопротеиназ, причем без значительной потери веса тела. 2 н. и 2 з.п. ф-лы, 5 табл.
| US 5494903 А, 27.02.1996 | |||
| НОВЫЕ 7-(ЗАМЕЩЕННЫЕ)-8-(ЗАМЕЩЕННЫЕ)-9-[(ЗАМЕЩЕННЫЕ ГЛИЦИЛ)АМИДО]-6-ДЕМЕТИЛ- 6-ДЕЗОКСИТЕТРАЦИКЛИНЫ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ ИЛИ КОМПЛЕКСЫ С МЕТАЛЛАМИ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ АНТИБИОТИЧЕСКОЙ АКТИВНОСТЬЮ IN VIVO | 1993 |
|
RU2125985C1 |
| US 5834450, 10.11.1998 | |||
| US 6277061 B1, 21.08.2001 | |||
| US 5837696, 17.11.1998 | |||
| Беликов В.Г | |||
| Фармацевтическая химия, М., Высшая школа, 1993, т.1, с.43-47. | |||

