1
Изобретение относится к способу получения тетрапептидов или их солей - новых биологически активных соединений, которые могут найти применение в медицине.
Известен способ деблокирования соответственно защищенных пептидов в кислой среде. Использование известного способа позволяет получить новые соединения, обладающие интересными фармакологическими свойствами 1 1 .
Цель изобретения - получение новых производных тетрапептидов или их солей, расширяющих арсенал средств воздействия на живой организм.
Поставленная цель достигается способом получения производных тетрапептидов формулы
О ( о oWR,
Нч 1 п I N-CH-C-NH-CH-C-NW-СН,-C-W - C-Z
I
/., сн,
R.
где L и D означают хиральность;
R. - первичный алкил С..,- C-j; , К„ - водород или первичный
алкил С - Сз ; RI - водород или первичный
алкил С ,
1 CONH. (при условии, когда один из радикалов Rj и первичный алкил, второй - водород),
или их солей, заключающийся в том, что защищенные соединения, соответствующие производным тетрапептидов формулы 1, деблокируют обработкой в кислой среде.
Причем деблокирование осуществляют в трифторуксусной кислоте или в ледяной уксусной кислоте, насыщенной газообразным НСК.
Фармацевтически приемлемые нетоксичные кислые соли присоединения включают соли органических и неорганических кисл-от, например хлористоводородной, серной, сульфокислоты, винной, фумаровой, бромистоводородной, гликолевой, лимонной, малеиновой, фосфорной, янтарной, уксусной, азотной, бензойной, аскорбиновой кислот, паратолуолсульфокислоты, бензолсуль30;фокислоты, нафталинсульфокислоты и .
пропионовой кислоты. Предпочтительно для образования кислых солей выбирать хлористоводородную, уксусную или янтарную кисло.ту. Любую из указанных солей получают известными методами.
Соединения формулы 1 являются первичнымгГ амидами, первичными спиртами или производными нитрилов специально определенных тетрапептидов. Хиральность остатков аминокислот, при рассмотрении от положения 1 до 4 - oL, D, отсутствует. Остаток в положении 3 - глициновая группа, следовательно он не может обладать хиральностью. Остаток в положении 4 может быть первичным амидом, первичным спиртом или нитрилом, его Хиральность согласуется с соответствующим предполагаемым остатком L аминокислоты.
Соединения формулы Г получают с помощью известных способов синтеза пептидов. Возможно, что в процессе синтеза и некоторых соединений формулы I происходит частичная рацемизация. Однако степень рацемизации, если таковая происходит, недостаточна, чтобы существенно изменить анестезирующую активность соединений формулы I.
Способы получения -оединений формулы I включают соче ание аминокислот или фрагментов пептида посредством взаимодействия функционалной группой с другой молекулой с образованием амидного связывания. Для обеспечения.эффективного сочетания желательно, чтобы все реакционноспособные функциональные группы, непосредственно не участвующие в реакции, были дезактивированы посредством использования соответствущих блокирующих групп и карбоксильная функциональная группа, которая подлежит сочетанию, была соответствующим образом активирована для обеспечения протекания сочетания.
Все это требует тщательного выбора как последовательности превращений, так и условий реакции, а также использования, специальных блокирующих групп. Каждую из аминокислот которые используют для получения содинений формулы I и которые имеют специально подобранные защитные групы и/или активированные функциональные группы, получают при использовании известных методик.
На каждой стадии общего синтеза соединений формулы I применяют выбранные комбинации блокирующих групп, в синтезе соединений формулы I могут работать и другие комбинации, хотя, возможно, с меньшей степенью эффективности. Так, например, бензилоксикарбонил, трет.-бутилоксикарбонил, трет.-амилоксикарбонил, 1-меток сибензилоксикарбонил, а дама и тилоксикарбонил и изоборнилоксикарбонил могут в различных случаях использоваться в качестве групп блокирующих аминогруппы в синтезе соединений формулы. Более того, в качестве защищающего оксигруппу для остатка тирозила обычно используют радикал бензил. (Бзл), даже если могут быть использованы другие агенты, например п-нитробензил, п-метоксибензил.
Применяемые при синтезе соединений формулы 1 группы, блокирующие карбоксил, могут представлять собой любые из типичных эфирообразующих групп, например, включающие метил,, этил,бензил, п-нитробензил,п-метоксибензил и 2,2,2-трихлорэтил.
.Сочетание соответствующим образом защищенных N-блокированных аминокислот или пептидных фрагментов с соответственно защищенными карбоксиблокированными аминокислотами или фрагментами пептида при получении соединений формулы I заключается в превращении свободных функциональных карбоксильных групп аминокислот или пептидных фрагментов, активных в соотношении реакции сочетания. Это может быть осуществлено посредством использования любой из известных методик, одна из которых заключается в превращении функциональной группы карбоксила в смешанный ангидрид. Свободные функциональные карбоксильные группы активируют посредством взаиг 1одействия с другой кислотой, обычно производной карбоновой кислоты, например хлорангидрид кислот. Примерами хлорангидридов кислот, применяемых для образования смешанных ангидридов, являются этилхлороформиа фенилхлороформиат, втор.-бутилхлороформиат, изобутилхлороформиат и пивалоилхлорид. Предпочтительно использовать изобутилхлороформиат.
Другим способом активирования карбоксильных групп для целей проведения реакции сочетания является превращение их в соответствующие активные эфирные производные. Такие активные эфиры включают, например, эфир 2,4,5-трихлорфенила, эфир пентахлорфенила и эфир п-нитрофенила. Другим способом сочетания является метод азидного сочетания.
Предпочтительный способ сочетания при получении соединений формулы I включает использование N ,Ы-дициклогексилкарОодиимида (ДСС) для активирования свободной карбоксильной группы и тем самым способствует протеканию реакции сочетания. Этот способ проводится при использовании эквимолярного количества ДСС относительно аминокислоты или пептидного фрагмента и осуществляетс я в присутствии эквимолярног-о количества 1-.окси6ензотриазола frOBT) . Наличие ОБТ подавляет нежелательные побочные реакции, включая вероятную рацемизацию. Отщепление выбранных блокирующих групп необходимо на определенных стадиях в последовательности синтеза при получении соединений формулы Г. Отщепление карбоксилэащищающих групп может быть достигнуто посредством щелочного омыления. Обычно для деэтерификации защищенного карбоксила применяют относительно сильные щелочи, типичным является испольэование гидроокисей щелочных металлов (гидроокись натрия,гидроокись калия и гидроокись лития).Блокирующие карбоксил группы, кроме того, могут быть удалены каталитическим гидрогенолизом, например, включающим гидрогенолиз в присутствии ка тализатора - палладий на угле. Более того, в тех случа.ях, когда блокирующая карбоксилгруппа является н-нитробензилом или 2,2,2трихлорэтилом, деблокирование може осуществляться посредством восстановления в присутствии цинка и соляной кислоты. Группы, блокирующие аминную функ цию, отщепляются посредством обработки защищенной аминокислоты или пептида кислотой, например, муравьи ной кислотой, трифторуксусной кислотой (ТФУ), п-толуолсульфокислотой (ТСК), бензолсульфокислотой (БСК) и нафталинсульфокислотой с образованием в качестве продукта соли присоединения соответствующей кисло ты. Отщепление аминоблокирующей гру пы также может осуществляться посре ством обработки блокированной амино кислоты или пептида смесью НВ-г. или НС и уксусной кислоты, для того чтобы получить соответствующую кисл соль присоединения, гидробромид или гидрохлорид. Конкретный способ или используемые реагенты зависят от химических или физических характеристик применяемых материалов в специальной реакции деблокирования. В тех случаях, когда группа R, отличается о водорода и пептид содержит, по край ней мере, три остатка аминокислоты, которые следует деблокировать, наиболее предпочтительно деблокировать пептид ТФУ или муравьиной кислотой, получая соответствующую кислую соль присоединения. Эту соль можно превратить в более приемлемую фармацевт чески нейтральную фор1му посредством обработки соответствующей ионообменной смолой, например DEAE, Сефадект А25 и Амберлит А27. Присутствующая в тирозильном остатке оксиэащищающая группа может быть сохранена в пептиде на протяжении последовательных стадий его получения, причем ее удаляют на последней стадии синтеза в сочетании с отщеплением аминоблокирующих групп. Однако, 1, зависимости от применяемых условий для удаления карбоксилблокирующих групп, она может быть удалена ранее, в процессе получения. Когда карбоксильную группу отщепляют посредством.щелочного огиолления, оксизащищакидая группа остается, однако при использовании каталитического гидрогенолиза для удаления карбоксилзащищающей группы океизащищающая группа также отщепляется. Но получение соединений формулы I может осуществляться в присутствии тирозильного остатка, имеющего свободную гидроксильную группу. Пример 1. Получение L-тирозил-0-аналин-глицил-№ -метил-1.-фенилаланинамида (уксуснокислой.соли). А. Бензил-В-алинат-п-толуолсульфонат. К смеси 100 мл бензилового спирта и 200 мл бензола, содержащей 55,1 г (0,29 моль) моногидрата п-толуолсульфокислоты, добавляют 25 г (0,201 моль) D-аланина. Смесь доводят до кипения, причем воду удаляют в виде азеотропа с помощью ловушки Дина-Старка. Эту смесь нагревают в течение 15 ч и затем охлаждают до комнатной температуры и разбавляют эфиром. Полученный осадок отделяют и перекристаллизовывают из смеси метанола и эфира, получая 55,3 г (56 %) соединения, т. пл. 112-115°С. Молекулярный вес 351,42. Элементный анализ: Бычислено,%:С 58,10;Н 6,02;N 3,99. С И,;, N05Найдено,%: С 58,19;Н 6,06;N 3,82. Б. Бeнзил-N --тpeт.-бyтилo cикapбонил-О-бензил-L- тирозил-П-алинат. К 200 мл сухого диметилформамида (ДМФ) добавляют 35,1 г (0,1 моль) диазабициклооктана (ДАБЦО). Смесь перемешивают в течение 10 мин при 0°С и добавляют 37,1 -г (0,1 моль) N -Tpet.-бутилоксикарбонил-о-бензил1.-тирозина и затем 13,5 г (0,1 моль) 1-оксибензотриазола (НВТ) и 20,6 г (0,1 моль)Ы ,Н-дициклогексилкарбодиимида (ДС.С) . Образовавшуюся смесь перемешивают при 0°С в течение 3 ч и затем при комнатной температуре в течение 1 сут. Затем эту смесь охлаждают до , полученную суспензию фильтруют и фильтрат концентрируют в вакууме. Полученный осадок снова растворяют в этилацетате и последовательно промывают 1 н. раствором NaHCO3f водой, холодным 0,75 н. раствором лимонной кислоты и водой. Органический слой сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Полученный остаток растворяют в горячем этаноле. При охлаждении происходит кристаллизания. После первой кристаллизации из этанола получают 41,5 г (80 %) чистого вещества, т. пл. 121-123 С. Молекулярный ес 520,63,
Элементный анализ:
Вычислено, %: С 69,21;Н 6,97;N 5,38 СзоНзбНгОб
Найдено, % :С 68,99;Н 6,75:N 5,17
В. №-трет.-бутилоксикарбонил-0бензил-Ь-тирозил-В-аланин.
К смеси 200 мл тетрагидрофурана (ТГФ) и 20 мл воды добавляют 31,2 г (0,06 моль) полученного продукта. Полученный раствор охлаждают до 0°С и медленно добавляют к нему 13,2 мл (1,1.эквивалента} 5 н, раствора гидроокиси натрия. Полученную смесь перемешивают и оставляют медленно нагреваться до комнатной температуры. Черзз 5 ч смесь распределяют между эфирным и водным слоями. Водный слой отделяют и охлаждают, поддерживая рН посредством добавления лимонной кислоты, и смесь экстрагируют этилацетатом. Этилацетатный экстракт промывают водой, сушат над сульфатом магния, фильтруют и разбавляют эфиром. Полученный осадок собирают, получая 17,7 г (67 %) соединения, т. пл. 1бО-1б2°С. Молекулярный вес 442,51.
Элементный анализ:
Вычислено,%:С 65, 6,B3;N 6,6 C24H3oN2.0b
Найдено, % :С 64,73;Н 6,70;N 6,20
Г. Бензил-М -трет . -бутилоксикарбонил-О-бензил-L тирозил-0--аланилглицинат.
К 70 мл сухого ДМФ добавляют 6,74 г (0,02 моль) кислой п-толуолсульфокислотной соли бензилглицината Полученную смесь охлаждают до 0°С и добавляют 2,24 г (0,020 моль) DABCO. Смесь перемешивают в течение нескольких минут и добавляют 8,84 г (0,020 моль) полученного продукта и затем 2,7 г (0,020 моль) НВТ и 4,12 г (0,020 моль) ДСС. Реакционную смесь перемешивают в течение 2 ч при 0°С и затем в течение 1 сут при комнатной температуре. Полученную суспензию охлаждают до ос, фильтруют и фильтрат концентрируют в вакууме. Полученный осадок растворяют в этилацетате и последовательно промывают 1 н. раствором бикарбоната натрия, водой, холодным 0,75 н. раствором лимонной кислоты и водой. Органическую фазу сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Полученный остаток кристаллизуют из этанола, получая 10,8 г (92,8 %) чистого соединения, т.пл. 145 147С. Молекулярный вес 589,69.
Элементный анализ:
Вычислено,%:С 67,22;Н 6,67;N 7,1 Cji Нзэы О,
Найдено, % :С 67,32;Н 6,83:N 6,9
Д. .-бутилоксикарбонил-Обензил-1.-тирозил-В-аланил глицин.
К 150 мл смеси тетрагидрофурана и воды (9:1) добавляют 15,95 г (27 моль) полученного продукта.Смесь охлаждают до Ос при перемешивании и к полученной смеси- по каплям добавляют 30 мл 1 н. раствора гидроокиси натрия. Смесь перемешивают в течение 2 ч до завершения прика-
пывания и затем ее дважды экстрагируют эфиром. Выделенный водный слой подкисляют до рН 2,5 добавлением 30 мл 1 н. раствора соляной кислоты. Вещество кристаллизуют, отделяют фильтрацией и перекристаллизовывают
5 один раз из смеси метанола и воды и дважды из этилацетата, получая 11,43г (85% от теории) , т. пл. 104-107°С.
31,4° (,5; метанол). Молекулярный вес 499,54.
Элементный анализ:
Вычислено,%:С 62,51;Н 6,66;N 8,41
NjO
Найдено, % :С 62,31;Н 6,83;N 8,12. 5 Е. .-бутилоксикарбонил-№ метил-1-фенил-аланин, соль d(+)- метилбензиламина.
К 75 мл тетрагидрофурана добавляют 13,26 г (0,05 моль) .0 бутилоксикарбонил-с -фенилаланина и полученную смесь по каплям в течение 30 мин прибавляют при перемешивании к суспензии 0,15 моль гидрида калия и 0,5 г 18-крау-6 эфира при
5 в атмосфере азота. Перемешивание продолжают при 0°С еще 1 ч. По каплям добавляют раствор 6,23 мл (0,1 моль) йодистого метила в 15 мл тетрагидрофурана в течение 15 мин.
0 Смесь выдерживают в течение 2 ч и по каплям добавляют смесь 10 мл уксусной кислоты и 10 мл тетрагидрофурана и затем добавляют 20 мл этанола.- Затем смесь вливают в 400 мл льда. рН полученной водной фазы доводятдо 12-13 путем добавления 2 н.раствора гидроокиси натрия. Водную смесь дважды экстрагируют эфиром и затем подкисляют до рН 3,0, добавляя твердую лимонную кислоту. Затем водную смесь экстрагируют эфиром-(3 X 200 мл) . Эфирные экстракты объединяют, экстрагируют водой, высушивают над сульфатом магния и упаривают в вакууме до сиропообразного состояния. Этот сироп растворяют в 50 мл .эфира и добавляют 6,44мл (0,05 моль) d(+)-о -метилбензиламина. Продукт осаждают добавлением 350 мл гексана, отфильтровывают, получают 15,83 г (79% от теории) соединения. При перекристаллизации из этилацетата получают 13,70 г (68 % от теории) соединения, т. пл; 136 .139°С; ct - 28,2(С-1, этанол).
5 Молекулярный вес 400,50, Элементный анализ: Вычислено,%:С 68,97;Н 8,05;N 6,9 q2i Найдено, % :С 68,75;Н 7,81;N 6,7 Ж. .-бутилоксикарбонил-Ы метил-Ь-фенилаланиламид. , N -трет.-бутилоксикарбонил-N -ме тил-i-фенилаланин (4 г, 0,01 моль) полученный посредством подкисления d (t) -метилбензиламинной соли и последукндей экстракцией эфиром, растворяют в 20 мл ДМФ. Смесь охлаж дают до -5С и добавляют 1,56 мл (0,012 моль) изобутилхлороформиата и затем 1,32 мл (0,012 моль) N-метилморфолина. Реакционную смесь пер мешивают в течени,е 10 мин при и барботируют в нее безводный сшмиа в течение 1,5 ч. Полученную смесь п ремешивают в течение 1 ч при и затем вливают в сосуд, содержащий 200 мл льда. Водный раствор экстр агируют этилацетатом. Органический слой отделяют и промывают последовательно 1,5 н. раствором лимонной кислоты, водой, 1 н. раствором бика боната натрия и водой. Затем этилацетатный раствор высушивают над сульфатом магния и упаривают в вакууме до сиропообразного состояния. Сироп кристаллизуется из смеси эфира и петролейного эфира, получают 2,12 г (76 % от теории) соединения т. пл. 91 - 92°C,laL:i - 111,2 (С 0,5 CHC6j ). Молекулярный вес 278,33. Элеме н т н ый а н ализ: Вычислено,%:С 64,73;Н 7,97;N 10, C 5-%CN203 Найдено, % :С 64,95;Н 7,8l;N 9,7 3. .-бутилоксикарбонил-Обензил-1-тирозил-О-аланилглицил-№метил- -фенилаланиламид. К 20 мл свежеприготовленной ледя ной, уксусной кислоты, содержащей безводный хлористый водород (1н.рас вор) и 2 мл анизола, до&авляют 1,95 (0,007 моль) №-ткет.-бутилоксикарбонил-Н метил-Ь-фенилаланиламида. Полученную смесь перемешивают при комнатной температуре в течение 30 мин. Затем вливают в эфир, выпавший осадок отделяют, высушивают получают 1,5 г хлористоводородной соли, которую растворяют в 30 мл ДМФ. Раствор охлаждают до 0°С и добавляют к нему 1,4 мл (0,007 мол дициклогексиламина. Смесь перемешивают в течение нескольких минут ипобавляют 3,5 г (0,007 моль) .-бутилоксикарбонил-0-бензилЬ-тирозил-П-аланилглицина, 950 мг (0,007 моль) НВТ и 1,4 г (0,007моль ДОС. Затем реакционную смесь перемешивают при в течение 2 ч и затем при .в течение 1 сут, внов охлаждают до и фильтруют. Филь.т рат концентрируют в вакууме до маслообразного состояния; масло повторно растворяют в этилацетате, Этилацетатннй раствор последовательно экстрагируют 1 н. раствором бикарбонату натрия, водой, холодным раствором 0,75 н. лимонной кислоты и водой. Органическую фазу высушивают над сульфатом магния и концентрируют в вакууме до масла. Масло подвергают хроматографической очистке на колонке 40 см х 3 см с силйкагелем марки Грейс и Дэвисон 62 в хлороформе. Продукт элюиругот, используя ступенчатый градиент концентрации хлороформа в смеси 10 % метанола в хлороформе. Контроль за ходом очистки ведут с помощью тонкослойной хроматографии отбираемых фракций. Получают 3,55 г (77 % от теории) соединения, ,- 9,2 (С О, 5; метанол). Молекулярный вес 659,8. Элементный анализ: Вычислено,%:С 65,54;Н 6,57;il 10,61 С Н N О Найдено, % :С 65,46;Н 6,58;N 10,36 И. 1 Р -трет.-бутилоксикарбонил1.-тирозил-П-аланил-глицил-К -метилL-фенилаланиламид. 3,2 г полученного продукта (0,0485 моль) растворяют в 60 мл этанола и добавляют к смеси в виде водной суспензии 1,5 г палладия (5%) на угле. Через реакционную смесь барботируют азот с помощью газораспределительной трубки в течение 5 мин и затем пропускают водород в течение 6 ч. Затем реакционную смесь продувают азотом и палладиевый катализатор отделяют фильтрованием. Смесь концетрируют в вакууме до сиропообразного состояния. Сироп растворяют в хлороформе и пропускают через хломатографическую колонку 40 X 3 см, содержащую силикагель марки Грейс и Дэвисон 62. Продукт элюируют, используя ступенчатый градиент концентрации хлороформа в смеси 10 % метанола в хлороформе, и выделяют в соответствии с тонкослойным профилем отобранных фракций, получая 2,0 г соединения (74% от теории), 9,9 (,5 метанол) . Аминокислотный состав-гССу 1,01, Аба 0,99; Туг 0,99; ННз 1,14. .К; .4j-тирозил-1)-аланил-глицилN -метил-Л-фенилаланиламид, ацетатная соль. 1,6 г полученного продукта (0,00281 моль) растворяют в 10 мл ТФУ, содержащей 0,5 мл анизола. Смесь перемешивают при 0°С в течение 30 мин, затем выливают в эфир и полученный осадок собирают и высушивают (1,1 г). Твердое вещество растворяют в соответствуюЕдем количестве водного буферного раствора (1 % пиридина и 0,05 % уксусной кислоты) до объема 15 мл и раствор пропускают через колонку DEAEСефа.цекс А-25 (ацетат) размером 2,5 X 99 см,уравновешенную тем же буфером. Элюат просматривают при 280 нм и соответствующие фракции оСъединяют и лиофилизуют.При повтор ной лиoфиJy зaции из 10 %-ной уксусНОЙ кислоты, с последующей лиофилизацией из смеси воды и ацетонитрила (3:1), получают 0,84 г соединения. + -27,8 (С 1,1 н. раствор HC Аминокислотный состав: Туг 0,98 1,03; сеу 1,00; ЫНз 1,05. II р и м е р 2. Получение L-тир зил-0-аланил-глицил-Ь-с --метилфенил аланиламида, ацетатная соль. А. L-o rмeтилфeнилaлaнин, бензило вый эфир, тозилатная соль. К 100 мл бензола добавляют 3,0 (0,0168 моль) L-bt-метилфенилалани К полученной суспензии затем добав ляют 3,5 г (1,1 эквивалента) гидра п-толуолсульфокислоты и 10 мл бензилового спирта. Смесь кипятят с обратным холодильником и ловушкой Дина-Старка для воды в течение 4 су Затем смесь охлаждают до комнатной температуры и добавляют к ней эфир чтобы осадить соль тозилата. Полученный осадок собирают и высушивают получая 7,0 г (94 %) соединения, т. пл. 129 - 131°С. bU|f- 10,7° (С 0,5; 1 н. раствор в метиловом спирте). Молекулярный вес 441,5. Элементный анализ: Вычислено,%: N 3,17 N GSНайдено, % : N 2,87. Б. N-трет.-бутилоксикарбонил-Обензил-Ь-тирозил-В-аланил-глицил-ЬоС-метилфенилаланин, бензиловый эфир К 80 Nm ДМФ добавляют 5,74 г (0,013 моль) полученного продукта. Полученную смесь охлаждают до в течение 5 мин и добавляют 6,5 г (13 моль). № -трет-бутилоксикарбони 0-бен:зил-Ь-тирозил-0-аланил-глицина (полученного как в примере 1), 1,8 г (13 моль) НВТ и 2,7 г (13 мо ДСС. Смесьперемешивают при течение 2 ч и затем при комнатной температуре в течение 1 сут. Затем смесь охлаждают до 0°С и полученный осадок отделяют фильтрацией. Фильтрат упаривают в вакууме. Полученный остаток растворяют в этилацетате и этилацетатный раствор последовательно экстрагируют 1 н. раствором бикарбоната натрия, водой, 0,75 н. раствором лиманной кислоты и водой Органический слой затем высушивают над сульфатом магния и упаривают в вакууме до маслообразного состояния Масло кристаллизуют из эфира и лерекристаллизуют из смеси этилацеТ1та и эфира. Получают 7,0 г (72 % соедине: ия. У. +7,9 (С 0,5 метанол). Молекулярный вес 750,86, Элементный анализ: Вычислено,%:С 68,78;;; 6,7l;N 7,46 Найдено.. % :С 68,75;Н 6,46;N 7,21. В. .-бутилоксик-арбонилЬ-тирозил-В-аланил-глицил-Ь-о1-метилфенилаланин, соль дицигслогексиламина. К 50 мл этанола добавляют 4,0 г (0,053 моль) полученного продукта. Затем добавляют суспензию 2,0 г палладия (5%) на угле в ДМФ. Через смесь пропускают азот, подаваемый через газораспределительную трубку в течение 5 мин, и затем подают газообразный водород в течение 4. Затем смесь обильно продувают азотом и отделяют палладиевый катализатор фильтрацией. Фильтрат концетрируют в вакууме до сиропообразного состояния . Сироп в растворе хлороформа подают на колонку размером 10 х 2см, родержащую силикагель марки Грейс и Дэвисон 62. Колонку элюируют при ступенчатом градиенте концентрации хлороформа в смеси хлороформ метанол (9 у 5: О, 5). Основные фракции объединяют и растворитель упаривают. Полученное масло растворяют в этилацетате и добавляют к раст.вору 1 мл дициклогексиламина. Выпавший в осадок продукт отфильтровывают, высушивают, получая 2,6 г (65 %) соединения,т. пл. 142 - d +46,3 (С 0,5, метанол). Г. N -трет.-бутилоксикарбонилЬ-тирозил-О-аланил-глицил-Ь-о -метилфенилаланиламид. 2,0 г полученного продукта (0,0227 моль) нейтрализуют смесью этилацетата и 0,75 н. раствора лимонной кислоты. Выделившийся органический слой отделяют, экстрагируют водой, высушивают над сульфатом магния и упаривают в вакууме до масла (1,5 г). Полученную свободную кислоту растворяют в 30 мл ДМФ и раствор охлаждают до 0°С в бутыли, выдерживающей давление. Добавляют 560 мг ДСС (0,0027 моль) и смесь перемешивают 4 ч при 0°С и затем в течение 3 ч при комнатной температуре. Затем бутыль охлаждают до -78С и добавляют 30 мл безводного аммиака. Бутыль снова закупоривают и смесь оставляют перемешиваться при комнатной температуре в течение 48 ч. Смесь охлаждают до -78°С, бутыль открывают и непрореагировавший аммиак дегазируют в процессе нагревания бутыли до комнатной температуры. Затем растворитель выпаривают в вакууме. Полученный остаток растворяют в этилацетате и этилацетатный раствор экстрагируют сначала 0,75 н. раствором лимонной кислоты и затем водой. Раствор высушлвают над сульфатом магния, и растворитель выпаривают в вакууме. Остаток растворяют в хлороформе и подают на колонку размером 3 х 45 с с силикагелем. Колонку элюируют при постепенном градиенте, включающем добавление хлороформа к смеси хлоро форм - метанол (9:1). Контроль за ходом очистки ведут с помощью хромотографии в тонком слое. Фракции, содержащие продукт, объединяют, упа ривают и получают 1,1 г (72%) -соеди нения, 26° (С 4, метанол) Анализ аминокислот: G€y 0,99J АСа 1,00; Туг 0,99; ЫНз 1,12. Д. Ь-тирозил-О-аланил-глицилL-ci-метилфенилаланиламид, ацетатная соль. К 20 мл смеси 1 н. раствора хлористого водорода в ледяной уксусной кислоте, содержащей 0,3 мл анизола, добавляют 0,9 г (0,0016 моль) полученного продукта. Смесь перемешиваю при комнатной температуре в течение 30 мин и затем вливают в эфир. Полу ченный осадоК собирают и высушивают (720 мг). Твердое вещество рас творяют в таком количестве буферного раствора (1 % пиридина и 0,05 % уксусной кислоты), чтобы конечный объем был 5 мл, и раствор подают на колонку размером 2,5 см х 99 см с DEAE-Сефадекс А-25 (ацетата) , предварительно доведенным до равновесия с тем же самым буфером. Элюат просматривают при 2ЬО им и соответству щие фракции объединяют и лиофи лизируют. Повторную лиофилизацию проводят из 10 %-ной уксусной кислоты и затем из смеси воды и ацето нитрила (3:1). Получают 400 мг соединения 23,9° (С 0,5; 1 н. раствор неб). Молекулярный вес 529,60. Элементный анализ; Вычислено,%: С 58,97; Н 6,66; N 13,22; О 21,15. С2бНз5%07 Найдено, % : С 59,02) Н 6,36, N 12,99; О 21,41. Анализ аминокислот: Туг 0,96) Аба l,0i; Ggy 1,00; NHj 1,03. Примерз. Получение Ь-тиро зил-В-аланил-глицил-Ы н-пропил-Ьфенилаланиламида, ацетата. А. N -трет.-бутилоксикарбонилN -н-пропил-Ь-фенилаланин. К 70 мл тетрагидрофурана добавляют 10,6 г (0,04 моль) .-бу тилоксикарбонил-Ь-фенилаланина и по лученную смесь по каплям в течение 30 мин прибавляют при сильном перемешивании к суспензии 0,12 моль гид рида калия в 220 мл тетрагидрофурана и 0,5 г 18-краун-6 эфира при . Процесс ведут в атмосфере азота. Смесь перемешивают еще 10 мин п ОС. По каплям добавляют раствор 23,3 мл (0,24-моль.) 1-иодопропана 40 мл тетрагидрофурана в течение 20 мин. Смесь выдерживают .в течени 2,5 ч при и вводят в нее по каплям 11,5 мл (0,12 моль) 1-иодопропана. Перемешивание продолжают еще 2 ч при , затем вносят 10мл ледяной уксусной кислоты и вновь перемешивают в течение-10 мин. Затем смесь выливают на раздробленный лед. рН полученной водной фазы доводят до 8,0, добавляя 2 н.раствор гидроокиси натрия. Водную фазу дважды экстрагируют эфиром и затем подкисляют до рН 2,5 добавлением холодного 2 н. раствора НСб. Затем водную фазу экстрагируют этилацетатом. Этилацетатный экстракт промывают один раз водой, высушивают нгщ сульфатом маг- ния и упаривают в вакууме до сиропообразного состояния. Сироп растворяют в 200 мл эфира и добавляют 8 мл ,(0,04 моль) DCHA. Осадок о1-фильтровывают и фильтрат промывают 1,. 5 н. раствором лимонной кислоты и волы. Эфирный слой высушивают над сульфатом магния и упаривают в вакууме до маслообразного состояния. Масло очищают хроматографически на колонке размером 3 х 40 см с силикагелем в хлороформе-. Продукт элюируют, используя ступенчатый градиент хлороформа в смеси 5 % метанола в хлороформе. За ходом очистки следят с помощью хроматографии в тонком слое. Фракции, содержащие продукт, собирают, упаривают и получают 3,6г (31 % от теории) соединения, aL - 153,3 (С - 1 метанол). ЯМР-спектр: сГ () 10,47; сГ(МезС-) 1,50. Молекулярный вес 295,4. Элементный анализ: Вычислено,%:С 65,06;Н 8,53; 4,74 C. найдено, % :С 65,2б;Н 8,29; N4,69 Б..-бутилоксикарбоксилN -н-пропил-Ь-фенилаланиламид. К раствору м-трет.-бутилоксикарбонил-к н-пропил-Ь-февилаланинав ДМФ, охлажденному до -15°С, добавляют один эквивалент изобутилхлороформиата и затем один эквивалент N-метилморфолина. Смесь перемешивают в течение 10 мин при -15 С и затем пропускают через нее в течение 30 мин безводный аммиак. Полученную смесь перемешивают 1 ч при -15 С и затем смеь вливают в сосуд, содержащий 200 мл льда. Водный раствор экстрагируют этилацетатом. Органический слой отделяют и промывают последовательно 1,5 н. раствором лимонной кислоты, водой, 1 н. раствором бикарбоната натрия и водой. Слой этилацетата отделяют и высушивают над сульфатом магния, упаривают в вакууме, получая указанное соединение. В. ы -трет. -бутилоксикарбрнилЬ-тирозил-В-аланил-глицил-Ы -н-пропил-Ь-фенилаланиламид. К 20 мл свежеприготовленного 1 н раствора безводного НСБ в ледяной уксусной кислоте/ содержащего 2 мл анизола, добагляют один эквивалент .трет.-бутилоксикарбонил-Ы -н-про пил-Ь-фёнилаланиламида. Полученную смесь перевешивают при комнатной температуре 30 мин, затем вливают в эфир и образовавшийся осадок собирают и высушивают. Затем хлористо водородную соль растворяют.в 30 мл ДМФ. Раствор охлаждают до и добавляют один эквивалент N -трет,бутилоксикарбонил-О-бензил-Ь-тирозил-О-аланил-глицина {полученного как в примере 1 Д),один эквивалент HAT 1 экв ивалент ДСС.Затем реакционную смесь перемешивают при в течение 2 ч и при 4°С в течение 1 сут.Смесь охлаждают до и фильт руют. Фильтрат концентрируют в ва кууме до масла, которое снова растворяют в этилацетате, Этилац Татный раствор промывают посЛедо шательно 1 н, раствором бикарбо ната натрия, водой, холодным 0,7 н. раствором лимонной кислоты водой. Органическую фазу высушивают над сульфатом магния и концен трируют в вакууме до масла. Масло хроматографируют на колонке размером 3 X 40 см с силикагелем в хлороформе. Продукт элюируют, используя ступенчатый градиент хлороформа в смеси 10 % метанола в хлороформе , За ходом хроматографии следят с помощью хроматографии в тонком слое отобранных фракций, получают Ы -трет, -бутилоксикарбонил-0бензил-Ь-тирозил-Ь-аланил-глицилМ -н-пропил-Ь-фенилаланиламид. Полученный продукт растворяют в 60 мл этанола и к смеси добавляют 1,5 г палладия (5%) на угле в виде водной суспензии, В реакционную смесь через газораспределительную трубку пропускают азот в течение 5мин и затем водород в течение 6ч. Затем реакционную смесь проду вают i3OTOM и палладиевый катализа тор удаляют фильтрованием. Смесь концентрируют в вакууме до сиропообразного состояния. Сироп растворяют в хлороформе и чистят хромат графией на колонке 40 х 3 см, содержащей силикагель. Продукт элюир ют, используя ступенчатый градиен хлороформа в смеси 10 % метанола в хлороформе, и выделяют в соответст с тонкослойным- профилем отобранных фракций, получая соединение, молекулярный вес 597,7, oi- - 34,8 (С 0,5 метанол). Элементный анализ: Вычислено,%:С 62,29;Н 7,25;N 11 Сэ1 Н43 ЩО-, Найдено, % :С 62,13;Н 7,24;N 11 Г. Ь-тирозил-О-аланил-глицил-К 1-пропил-1|-фенилаланиламид, ацетат 800 мг полученного продукта (1,34 ммоль) растворяют в 10 мл трифторуксусной кислоты, содержащей 0,5 мл анизола. Смесь перемешивают при в течение 30 мин. Реакционную смесь сушат лиофилизацией. Твердое вещество растворяют в достаточном количестве водного буферного раствора (1% пиридина и 0,5 % уксусной кислоты), объем доводят до 10 мл; раствор педают на колонку размером 2,5 х 99 см с DEAE-Сефадексом А-25 (ацетат), который уравновешен тем же самым буфером, Элюат просматривают при длине волны 280 нм и соответствующие фракции объединяют и лиофилизуют. Повторная лиофилизация из 1 М раствора уксусной кислоты дает 655 мг соединения, - 11,0° (С 0,5, 1 н, раствор НСР), Молекулярный вес 557,6, Элементный анализ: Вычислено,%:С 60,31;Н 7,05: N12,56 C2gH39%07 Найдено, % :С 60,23;Н 6,98;N 12,49 Анализ аминокислот. Туг 0,99J Аеа 1,00, сеу 1,01; NH 0,95. П р и м е р 4, Получение L-тиpoзил-D-aлaнил-глицил-№ этил-L-фeнилаланиламида, ацетатная соль, А, N -бутилоксикарбонил-N-этилL-фенилаланйн, К 70 мл тетрагидрофурана добавляют 10,6 г (0,04 моль) № бутилоксикарбонил-Ь-фенилаланина, Полученную смесь добавляют по каплям в течение 30 мин к механически перемешиваемой суспензии 0,12 моль гидрида калия в 220 мл тетрагидрофурана и 0,5 г 18-краун-б эфира при Ос в атмосфере азота. Смесь перемешивают дополнительно в течение 10 мин при , Затем по каплям в течение 20 мин добавляют раствор 19,4 мл (0,24 моль) йодистого этила в 40 мл. тетрагидрофурана. Смесь выдерживают в течение 4 ч при 0°С, Добавляют еще 19,4 мл (0,24 моль) йодистого этила к смеси двумя равными порциями. Смесь перемешивают дополнительно 2 ч при и затем добавляют 10 мл ледяной уксусной кислоты. После перемешивания в течение 10 мин смесь вливают на 400 мл измельченного льда, рН полученной водной фазы увеличивают до 8,0 добавлением 2 н, раствора гидроокиси натрия. Водную смесь дважды экстрагируют эфиром и затем подкисляют до рН 2,5 добавлением холодного 2 н. раствора соляной кислоты. Затем в.рдную смесь экстрагируют этилацетатом, экстракт промывают водой, высушивают над сульфатом магния и выпаривают в вакууме до сиропообразного состояния. Сироп растворяют в 200 мл эфира и добавляют 8 мл (0,04 моль) DCHA, Осадок отфильтровывают и фильтрат экстрагируют 1/5 н. раствором лимонной кислаты и водой,. Эфирный слой высушивают .над сульфатом магния и упаривают в вакууме, получают 4,6 г (39 % от теории) соединения. Спект ЯМР: cf (фенил) 7,2; {f (Ме,СОС-)1,4. Б. N -трет.-бутилоксикарбонилN -этил-Ь-фенилаланиламид. 4,3 г полученного ,-бутилоксикарбонил-Ы этил-Ь-фенилаланила (0,0416 моль) растворяют в 60 мл ДМФ. Смесь охлаждают до 0°С и добавляют к ней 3,0 г (0,0146 мол ДСС. Реакционную смесь перемешивают в течение 2 ч при и затем 72 ч при комнатной температуре. Затем смесь охлаждают до и фильтруют Фильтрат концентрируют в вакууме до масла, которое повторно растворяют в этилацетате. Раствор экстрагируют 1 и. раствором бикарбоната натрия, водой, холодным 1,5 н. раствором ли монной кислоты и водой. Органическую фазу высушивают над сульфатом магния и концентрируют, получая 3,33 г (91 % от теории) соединения - 101,51 (С 1, метанол). Молекулярный вес 292,4. Элементный анализ: Вычислено,%:С 65,73;Н 8,27;N 9, С еН24«2.С3 Найдено, % :С 66,03;Н 8,13;N 9, В. Н этил-Ь-фенилаланиламид, хл ристоводородная соль. 3,5 г полученного соединения (11,95 моль) растворяют в 40 мл свежеприготовленной уксусной кисло ты, содержащей безводныйхлористый водород (1 н. раствор) и 1,5 мл анизола и (С,-) SiH. Реакционную смесь перемешивают при комнатной температуре 30 мин и затем вливают в эфир. Выпавший в осадок продукт отфильтровывают и высушивают, полу чают 2,6 г (96 % от теории) соединения, т. пл. 276 - 211°С. Молекулярный вес 227,7. Элементный Вычислено,%:С 58,02;Н 7,08;N 12 С И-f N2.C2 Найдено, % :С 57,97;Н 7,26;N 12 Г. N-трет.-бутилоксикарбонилL-тиpoзил-D-aлaнил-глицил-N этилL-фенилаланинамид. К 50 мл ДМФ добавляют 1,14 г (0,005 моль) М этил-Ь-фенилалан амида в виде хлористоводородной со ли. Смесь охлаждают до 0°С и затем добавляют 2,95 г (0,005 моль) соли DCHA . -бутилоксикарбонилЬ-тирсзил-О-аланил-глицина. Реакционную мяссу перемешивают при 5 мин и затем добавляют к ней 675 (0,005 моль) МВТ и 1,03 г (0,005мо ДСС. Перемешивание продолжают при 6,5 ч и аатем при комнатной те пературе в течение 20 ч. Смесь ох.лаждают до ОС и фильтруют. Фильтр концентрируют в вакууме до масла, которое снова растворяют в этиладетате, экстрагируют 1 н. раствором бикарбоната натрия, водой, холодным 1,5 fj. раствором лимонной кислоты и водой. Органическую фазу высушивают над сульфатом магния и концентрируют в вакууме до масла. Масло очищают хроматографически на колонке 3 х 40 см с силикагелем в хлороформе. Продукт элюируют, используя ступенчатый градиент хлороформа в смеси 15 % метанола с хлороформом. Продукт выделяют согласно тонкослойному профилю отобранных фракций, получают 1,13 г (39 % от теории) соединения, ot 21,0 (С 0,5, метанол). Молекулярный вес 583,7. Элементный анализ: Вычислено,%:С 61,73;Н 7,08;N 12,0 С30 4 N507 Найдено, % :С 60,35;Н 7v26;N 11,25 Д. Ь-Гирозил-О-аланил-глицил,N -зтил-Ь-фенилаланиламид, ацетат. 1 г полученного продукта (1,71 ммоль) растворяют в 20 мл трифторуксусной кислоты, содержащей 3 мл анизола и 3 мл (С.}-,, SiH. Смесь перемешивают при 0°С в течение ЗОмин и вливают в эфир, выпавший в осадок, продукт отфильтровывают и высушивают (660 мг) . Твердое веидество растворяют в небольшом количестве буферного водного раствора (1 % пиридина и 0,05 % уксусной кислоты), объем доводят до 10 мл и этот раствэр подают на колонку 2,5 х 90 см с DEAE-Сефадексом А-25 (ацетат), который уравновешивают предварительно с тем же самым буфером. Элюат просматривают при длине волны 280 нм и соответствующие фракции собирают и лиофилизуют. Твердое ве1цество растворяют в 0. ,2М растворе уксусной кислоты (10 мл) и. раствор чистят хроматографически на колонке 2,5х99см с Сефадексом G-10, который предварительно уравновешивают с тем же самым растворителем. Элюат просматривают при длине волны 280 нм и соответствующие фракции объединяют и лиофилизуют, получая 448 мг (48 % от теории) соединения 10,6 (С 0,5, 1 н. раствор нее). Молекулярный вес 543,6. Элементный анализ: Вычислено,%:С 59,65;Н 6,86;N 12,88 Са7Нэ7М5-07 найдено, % :С 59,41;Н 7,26;N 13,18 Анализ аминокислот :Туг 1,03; Аба 0,99; СЕу. 0,97; NH 0,98. Анестезирующая активность соединений формулы 1 изучена в опыте с мышами на горячей пластине. Мышь помещают в вертикальный акриловый цилиндр, в основании которого расположена горячая пластина, температура которой поддерживается равной 52С. Мышь получает подкожную
иньекцйю заданного количества испытуемого соединения, растворенного или суспендированного в соответствущем носителе. Выдерживают заданный промежуток времени после введения испытуемого соединения и затем мышь помещают tj поверхность горячей платины. Затем записывают латентности двух отдельных явлений в секундах. Сначала измеряется латентность до тех пор, покамышь не начнет лизать задние лапы, и затем измеряется латентность до момента, когда мышь прыгнет с горячей пластины. Вещество, которое проявляет анестезирующую активность, обеспечивает увеличение этих латентных периодов по сравнению с контрольными мышами, которым при инъекции вводили только
носитель. Это должно происходить при величине дозы, которая не вызывает нарушения моторной координации или выводит мышь из строя.
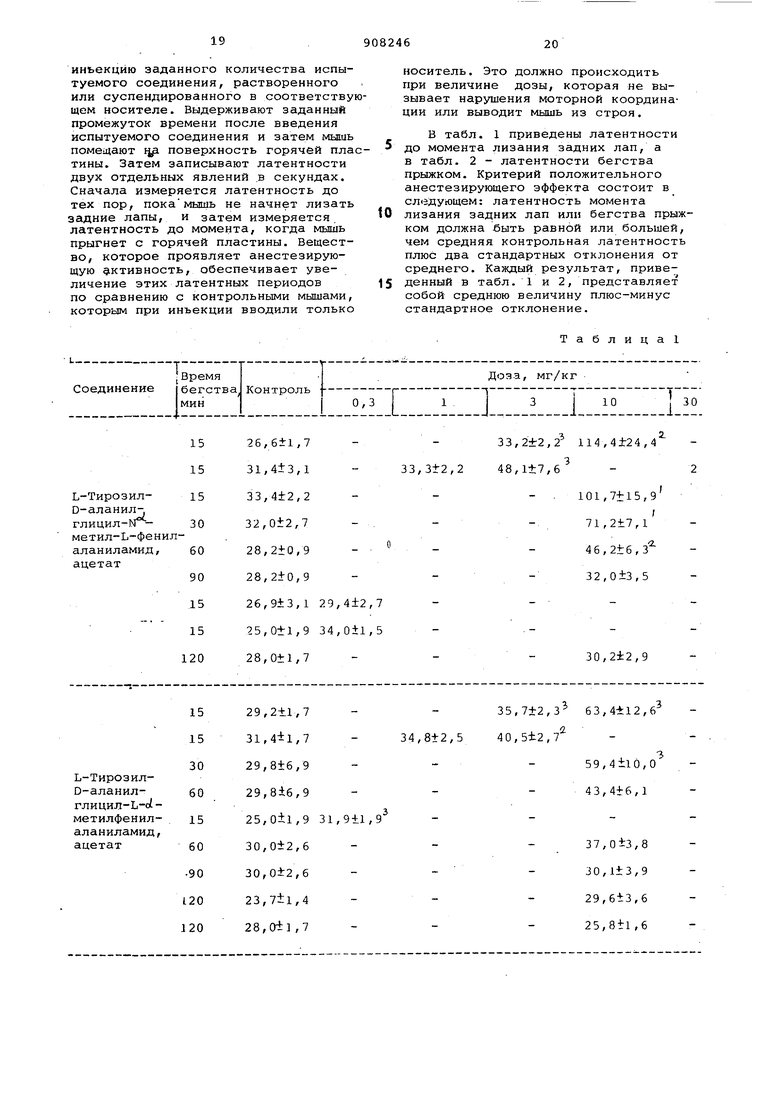
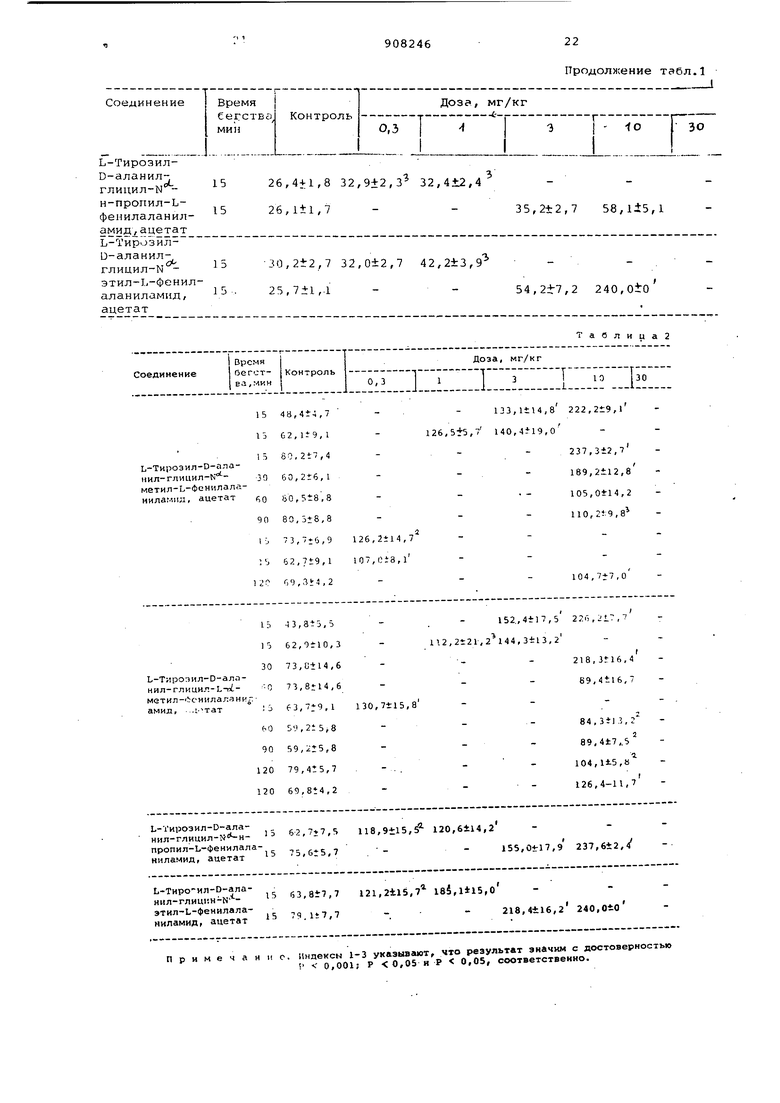
В табл. 1 приведены латентности 5 до момента лизания задних лап, а в табл. 2 - латентности бегства прыжком. Критерий положительного анестезирующего зффекта состоит в следующем: латентность момента
0 лизания задних лап или бегства прыжком должна быть равной или большей, чем средняя контрольная латентность плюс два стандартных отклонения от среднего. Каждый результат, приведенный в табл. 1 и 2, представляет собой среднюю величину плюс-минус стандартное отклонение.
Таблица
15 26,4il,8 32,9±2,3 32,4t2,4
26,,7
30,2±2,7 32,0±2,7 42,2±3,9 15 .25,7±1,.1
Продолжение табл.1
РО
35,2±2,7 58,li5,l
54,2i:7,2 240,OtO
Тавлица2









| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения тетрапептидов или их кислотно-аддитивных солей | 1981 |
|
SU1082319A3 |
| Способ получения пептидов | 1977 |
|
SU753358A3 |
| Способ получения пептидов с последовательностью актг-человека,содержащих в -конечном положении аминооксикислоту | 1973 |
|
SU490284A3 |
| Способ получения сульфата октапептидамида-терминального октапетида холецистокининпанкреазимина | 1977 |
|
SU659083A3 |
| Аналог Энкефалина,обладающий пролонгированной анальгетической активностью | 1981 |
|
SU1048705A1 |
| Способ получения аминоацильных и пептидных производных фосфоновой или фосфиновой кислоты | 1976 |
|
SU619100A3 |
| Способ получения пентапептидов или их эфиров или их амидов или их солей | 1977 |
|
SU772481A3 |
| Способ получения -сульфоната в-цепи инсулина человека | 1977 |
|
SU696011A1 |
| Циклический аналог энкефалина, обладающий пролонгированной анальгетической активностью | 1981 |
|
SU1048702A1 |
| Способ получения солей мурамилпептидов | 1983 |
|
SU1299516A3 |
Ь-Тирозил-О-аланил-гляцил-К метил-Ь-феннлаланилампл, ацетат
9080,5±8,8
1-J73,,9 126,2il4,7
:.62,7i9,l lC7,ei8,l
,,2
L:. 13,,5 Ii 62,9i-10,3
30 73,i;tl4,6
-0 73,,6
63,,1 130,7±15,
60 59,
90 59,,8 120 79,4t5,7 . - .. 120 69,8i4,2
;;л- л «и;-; н: s 62,7,7,. 118,, i2o,6tu.2
пропил-Ь-Фенилала15 75,Gt5,7
ниламид, ацетат
:;:л:: лйци;°« - ,8.7,7 121,2115,7-X85,,o
этил-Ь-фенилала15 7q.li7,7
ниламид, ацетат Примечай. с.
lJ3,ltl4,8 222,2±9,l 140,,0
237,,7 189,2112,8 105,,2 110,219,8
104, ,0
112,2t21,2 144,,2
I
218,ЗГ16,4 69,4tl6,
89,4t7,.5 104,Its,« 126,4-11,7
I55,0tl7,9 237,,4
218,,2 240,Oio Индексн 1-3 указывают, что результат значим с лостоверностью V 0,001; Р 0,05 и Р 0,05, соответственно.
Формула изобретения
I. Способ получения производных тетрапептидов формулы I
„ о , oW«,
Нч Л п I
; H-CH-t-NH-CH c-NH- сн.- с-н - с- z
Н
L и D означают хиральность; R - первичный алкил R/j - водород или первичный
алкил Сз;
Ra - водород или первичный алкил СМ ;
Z - CONH (при условии, когда один из радикалов Ку и Rjпервичный алкил, второй водород),
или их солей, отличающийс я тем, что защищенные соединения, соответствующие производным тетрапептидов формулы I, деблокируют обработкой в кислой среде.
Источники информации, принятые во внимание при экспертизе 1. Шредер Э., Любке К. Пептиды,
Мир
ч. 1, 1967, М,,
с, 112.
