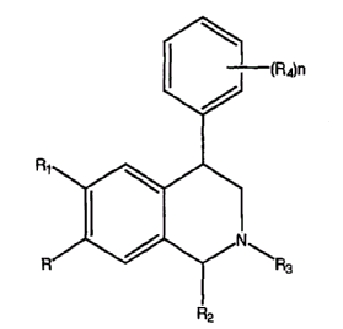
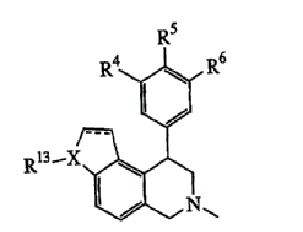
Область изобретения
Настоящее изобретение относится к соединениям, композициям и способам лечения различных неврологических и психологических нарушений. В частности, настоящее изобретение относится к таким соединениям, композициям и способам, где соединениями являются новые 4-фенилзамещенные производные тетрагидроизохинолина.
Предпосылки создания изобретения
Лечение различных неврологических и психиатрических нарушений, например дефицита внимания, связанного с гиперактивностью (ADHD), характеризуется рядом побочных действий, которые, как считают, являются результатом отсутствия подходящей селективности у соединений, используемых для лечения, например, неспособности соединений селективно блокировать некоторые нейрохимические соединения и не блокировать другие. ADHD, например, является заболеванием, поражающим 3-6% детей школьного возраста и обнаруживается также у одного процента взрослых людей. Помимо создания затруднений в выполнении функций в школе и на работе, ADHD является значительным фактором риска для последующего развития нарушений типа тревожных состояний, депрессии, кондуктивного расстройства и злоупотребления лекарственными средствами, поскольку современные схемы лечения требуют применения психостимуляторов и поскольку значительное число пациентов (30%) являются резистентными к стимуляторам или побочным действием. Кроме того, метилфенидат, современное лекарственное средство, выбранное для лечения ADHD, индуцирует ряд побочных действий; они включают анорексию, бессонницу и нервное состояние, тики, а также повышенное кровяное давление и частоту сердечных сокращений, вторичную к активации симпатической нервной системы. Метилфенидат имеет также высокую селективность в отношении белка-транспортера допамина по сравнению с белком-транспортером норэпинефрина (отношение DAT/NET Ki 0,1), что может привести к склонности к привыканию и требует многократных доз в день для оптимальной эффективности.
Данное изобретение относится к альтернативе таких известных режимов лечения с использованием новых производных 4-фенилтетрагидроизохинолина, причем указанные соединения никогда не были описаны в данной области. В патенте США № 3947456 (патент '456), например, описаны 4-фенилзамещенные тетрагидроизохинолины формулы:
где R представляет гидрокси или низший алкокси; в патенте '456 не описаны какие-либо другие группы в этом положении, не говоря уже о заместителях, имеющихся в положении (R4) в предложенных здесь соединениях. Фенилзамещенные тетрагидроизохинолины описаны также в Mondeshka et al. (Il Farmaco, 1994, 49 pp. 475-481). Однако соединения, описанные в ней, отличны от соединений, предложенных здесь.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
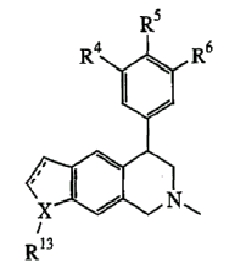
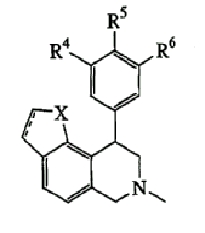
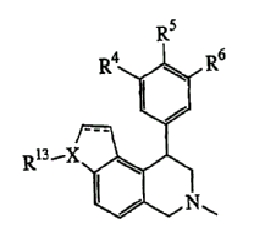
Данное изобретение относится к следующим соединениям формулы IA, IB, IIA, IIB, IIIA и IIIB
где R1-R13 имеют указанные здесь значения. В одном варианте осуществления R1 представляет С1-С6-алкил; R2 представляет Н, С1-С6-алкил, С3-С6-циклоалкил или С1-С6-галогеналкил; R3 в каждом случае представляет независимо Н, галоген, С1-С6-алкил или С1-С6-алкил, замещенный 1-3 OR8 или NR8R9; R4, R5 и R6 каждый, независимо, представляет Н или выбраны в каждом их случае из галогена, -OR10, -NR10R11, -NR10C(O)R11, -S(O)nR11, -CN, -C(O)R11, -C(O)2R11, -C(O)NR11R12, С1-С6-алкила, С3-С6-циклоалкила или С4-С7-циклоалкилалкила и где каждый С1-С6-алкил, С3-С6-циклоалкил и С4-С7-циклоалкилалкил необязательно замещен от 1 до 3 заместителями, независимо выбранными в каждом случае из С1-С3-алкила, галогена, -CN, -OR8, -NR8R9 и фенила, который необязательно замещен 1-3 раза галогеном, -CN, С1-С4-алкилом, С1-С4-галогеналкилом, -OR8 или -NR8R9; или R5 и R6 могут быть -О-С(R11)2-O-; и R7 - R13, n и Х имеют указанные здесь значения.
Соединения, предложенные здесь, блокируют повторное поглощение норэпинефрина, допамина и серотонина с определенными соотношениями селективности, например, являются более селективными для белка-транспортера норэпинефрина (NET), чем для белка-транспортера допамина (DAT), или белков-транспортеров серотонина (SERT). Следовательно, соединения полезны для селективного лечения различных неврологических и психологических нарушений.
Предложена также фармацевтическая композиция, включающая фармацевтически приемлемый носитель и терапевтически эффективное количество соединения формулы IA, IB, IIA, IIB, IIA, IIIB или IIIB. Далее предложен способ лечения млекопитающего, страдающего неврологическим или психологическим расстройством, выбранным из группы, включающей дефицит внимания, связанный с гиперактивностью, тревогу, депрессию, послетравматический стресс, надъядерный паралич, нарушения питания, обессивно-компульсивное заболевание, аналгезию, синдром прекращения курения, приступы паники, болезнь Паркинсона и фобию, включающий введение млекопитающему вышеуказанной фармацевтической композиции.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Данное изобретение относится к соединению формулы IA, IB, IIA, IIB, IIIA или IIIB:
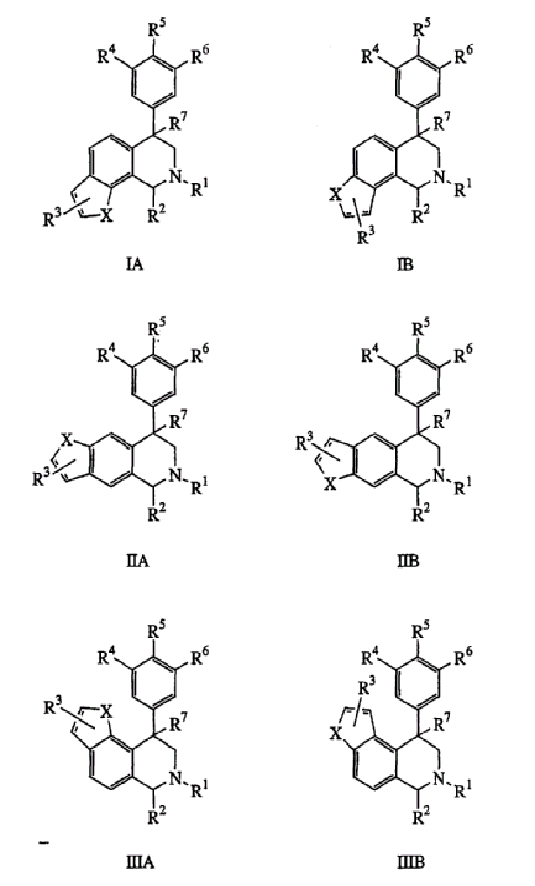
где:
R1 выбран из группы, состоящей из С1-С6-алкила, С2-С6-алкенила, С2-С6-алкинила, С3-С6-циклоалкила, С4-С7-циклоалкилалкила и бензила, каждый из которых необязательно замещен 1-3 заместителями, независимо выбранными в каждом случае из С1-С3-алкила, галогена, -CN, -OR8 и -NR8R9;
R2 выбран из группы, состоящей из Н, С1-С6-алкила, С2-С6-алкенила, С2-С6-алкинила, С3-С6-циклоалкила, С4-С7-циклоалкилалкила и С1-С6-галогеналкила;
R3 выбран из группы, состоящей из Н, галогена, С1-С6-алкила, С1-С6-галогеналкила и С3-С6-циклоалкила, где С1-С6-алкил, С1-С6-галогеналкил и С3-С6-циклоалкил необязательно замещены 1-3 заместителями, независимо выбранными в каждом случае из OR8 и NR8R9;
R4, R5 и R6, каждый независимо выбран в каждом случае из группы, состоящей из Н, галогена, -OR10, -NO2, -NR10R11, -NR10С(O)R11, -NR10С(О)NR11R12, -S(O)nR11, -CN, -C(O)R11, -C(O)2R11, -C(O)NR11R12, С1-С6-алкила, С2-С6-алкенила, С2-С6-алкинила, С3-С6-циклоалкила и С4-С7-циклоалкилалкила, где каждый С1-С6-алкил, С2-С6-алкенил, С2-С6-алкинил, С3-С6-циклоалкил и С4-С7-циклоалкилалкил необязательно замещен заместителями от 1 до 3, независимо выбранными в каждом случае из С1-С3-алкила, галогена, =О, -CN, -OR8, -NR8R9 и фенила, и где фенил необязательно замещен 1-3 заместителями, независимо выбранными в каждом случае из галогена, -CN, С1-С4-алкила, С1-С4-галогеналкила, -OR8 и -NR8R9;
в альтернативном случае R5 и R6 представляют -O-C(R11)2-O-;
R7 выбран из группы, состоящей из Н, галогена и OR10;
R8 и R9 каждый независимо выбран из группы, состоящей из Н, С1-С4-алкила, С1-С4-галогеналкила, С1-С4-алкоксиалкила, С1-С4-алкоксиалкилалкила, С3-С6-циклоалкила, С4-С7-циклоалкилалкила, -С(O)R12, фенила и бензила, где фенил и бензил необязательно замещены 1-3 заместителями, независимо выбранными в каждом случае из галогена, циано, С1-С4-алкила, С1-С4-галогеналкила, С1-С4-алкокси и С1-С4-галогеналкокси, или R8 и R9 взяты вместе с азотом, к которому они присоединены, c образованием кольца пиперидина, пирролидина, пиперазина, N-метилпиперазина, морфолина или тиоморфолина;
R10 выбран из группы, состоящей из Н, С1-С4-алкила, С1-С4-галогеналкила, С1-С4-алкоксиалкила, С3-С6-циклоалкила, С4-С7-циклоалкилалкила, -С(O)R12, фенила и бензила, где фенил и бензил необязательно замещены 1-3 заместителями, независимо выбранными в каждом случае из галогена, -NH2, -OH, циано, С1-С4-алкила, С1-С4-галогеналкила, С1-С4-алкокси и С1-С4-галогеналкокси;
R11 выбран из группы, состоящей из Н, С1-С4-алкила, С1-С4-галогеналкила, С1-С4-алкоксиалкила, С3-С6-циклоалкила, С4-С7-циклоалкилалкила, фенила и бензила, где фенил и бензил необязательно замещены 1-3 заместителями, независимо выбранными в каждом случае из галогена, -NH2, -OH, циано, С1-С4-алкила, С1-С4-галогеналкила, С1-С4-алкокси и С1-С4-галогеналкокси,
или R10 и R11 взятые вместе с азотом, к которому они присоединены, с образованием кольца пиперидина, пирролидина, N-метилпиперазина, морфолина или тиоморфолина; при условиии, что только один из R8 и R9 или R10 и R11 взяты вместе с азотом, к которому они присоединены, с образованием кольца пиперидина, пирролидина, пиперазина, N-метилпиперазина, морфолина или тиоморфолина.
R12 выбран из группы, состоящей из С1-С4-алкила, С1-С4-галогеналкила и фенила;
Х выбран из группы, состоящей из О, NR13 и S, при условии, что X не представляет NR13, когда соединение является соединением формулы (IA); кольцо, содержащее Х, выбрано из фурана, пиррола, тиофена, дигидрофурана, дигидропиррола и дигидротиофена;
n равно 0, 1 и 2 и
R13 выбран из группы, состоящей из Н, С1-С6-алкила, бензила и фенила, где С1-С6-алкил, бензил и фенил необязательно замещены 1-3 заместителями, независимо выбранными в каждом случае из галогена, -NH2, -OH, циано, С1-С4-алкила, С1-С4-галогеналкила, С1-С4-алкокси и С1-С4-галогеналкокси.
«Алкил» означает насыщенные углеводородные цепи, разветвленные или неразветвленные, имеющие определенное число атомов углерода. «Алкенил» означает углеводородные цепи либо неразветвленной, либо разветвленной конфигурации, и одну или несколько ненасыщенных углерод-углеродных связей, которые могут присутствовать в любом стабильном положении вдоль цепи, такие как этенил, пропенил и подобные. «Алкинил» означает углеводородные цепи либо неразветвленной, либо разветвленной конфигурации, и одну или несколько тройных углерод-углеродных связей, которые могут присутствовать в любом стабильном положении вдоль цепи, такие как этинил, пропинил и подобные. «Алкокси» означает алкильную группу с указанным числом атомов углерода, присоединенную через кислородный мостик. «Циклоалкил» означает группы насыщенных колец, включая системы моно-, би- или полициклических колец, такие как циклопропил, циклобутил, циклопентил и подобные. «Галоген» означает фтор, хлор, бром и иод. «Галогеналкил» означает алкилы как с разветвленными, так и неразветвленными цепями, имеющими определенное число атомов углерода, замещенные одним или несколькими галогенами. «Галогеналкокси» означает алкоксигруппу, замещенную, по меньшей мере, одним атомом галогена.
«Замещенный» или «замещение» атома означает, что один или несколько атомов водорода на указанном атоме заменены на указанную группу, с условием, что нормальная валентность указанных атомов не превышена. «Незамещенные» атомы имеют все атомы водорода, определенные их валентностью. Когда заместителем является кето (т.е. С=О), то на атоме заменены 2 водорода. Комбинации заместителей и/или символов являются допустимыми, только если такие комбинации приводят к стабильным соединениям; «стабильное соединение» или «стабильная структура» означает соединение, которое является достаточно устойчивым для того, чтобы выдерживать выделение из реакционной смеси до пригодной степени чистоты и превращение в эффективный терапевтический агент.
Одним вариантом осуществления данного изобретения являются соединения, в которых: R1 представляет С1-С6-алкил; R2 представляет Н, С1-С6-алкил, С3-С6-циклоалкил или С1-С6-галогеналкил; R3 представляет в каждом случае независимо Н, галоген, С1-С6-алкил или С1-С6-алкил, замещенный 1-3 OR8 или NR8R9; R4, R5 и R6, каждый независимо представляет Н, или выбраны в каждом случае из галогена, -OR10, -NR10R11, -NR10C(O)R11, -S(O)nR11, -CN, -C(O)R11, -C(O)2R11, -C(O)NR11R12, С1-С6-алкила, С3-С6-циклоалкила или С4-С7-циклоалкилалкила и где каждый С1-С6-алкил, С3-С6-циклоалкил и С4-С7-циклоалкилалкил необязательно замещен 1-3 заместителями, независимо выбранными в каждом случае из С1-С3-алкила, галогена, -CN, -OR8, -NR8R9 и фенила, который необязательно замещен 1-3 раза галогеном, -CN, С1-С4-алкилом, С1-С4-галогеналкилом, -OR8 или -NR8R9, или R5 и R6 могут быть -O-C(R11)2-O- и R7-R13, n и Х имеют указанные выше значения.
В этих вариантах осуществления выбор определенного заместителя в любом одном положении соединения необязательно влияет на выбор заместителя в другом положении того же самого соединения; то есть соединение, предложенное здесь, имеет любой из заместителей в любом из положений. Например, как описано выше, R1, предпочтительно, представляет, например, С1-С6-алкил, выбор R1 как любого одного из С1, С2, С3 С4, С5 или С6-алкила не ограничивает выбор R2, в частности, из любого одного из Н, С1-С6-алкила, С3-С6-циклоалкила или С1-С6-галогеналкила. Для R1 как любого из С1, С2, С3 С4, С5 или С6-алкила R2, предпочтительно, представляет любой из Н, С1, С2, С3 С4, С5, или С6-алкила, или С3 С4, С5, или С6-циклоалкила, или С1, С2, С3 С4, С5, или С6-галогеналкила. Аналогично, выбор R2, в частности, из любого одного из Н, С1, С2, С3 С4, С5, или С6-алкила, или С3 С4, С5, или С6-циклоалкила, или С1, С2, С3 С4, С5, или С6-галогеналкила не ограничивает выбор R3, в частности, из любого одного из его составляющих значений.
В другом варианте осуществления изобретения R1 представляет метил, этил, пропил или изопропил; R2 представляет Н или
С1-С6-алкил и R3 представляет Н, галоген или С1-С6-алкил, где С1-С6-алкил необязательно замещен 1-3 OR8; R4 и R5 и R6 каждый независимо представляет Н, галоген, -OR10, -S(O)nR11, -NR10R11,
-C(O)R11 или С1-С6-алкил, где С1-С6-алкил необязательно замещен, как указано выше; и R7-R13 и Х имеют указанные выше значения. Еще в одном варианте осуществления R1 представляет метил; R2 и R3 представляют Н; R4 и R5 и R6, каждый независимо, представляет Н, F, Cl, -OH, С1-С3-алкокси или С1-С3-алкил; R7 представляет H, F, -OH или -ОСН3 и R8-R13 и Х имеют указанные выше значения.
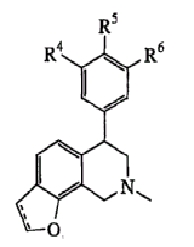
В одном варианте осуществления соединения включают, например и без ограничения, или соединения, которые приведены здесь ниже в таблицах I-VIA. То есть такие соединения включают соединения, имеющие следующую формулу (см. таблицы 1-1В),
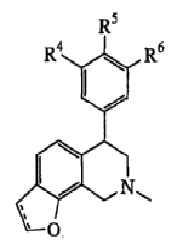
где кислородсодержащее кольцо является либо насыщенным, либо ненасыщенным, R4 представляет Н, Cl или F, R5 представляет Н, F или Ме и R6 представляет Н или F.
В другом варианте осуществления соединения включают соединения, имеющие следующую формулу (см. таблицы 2-2В).
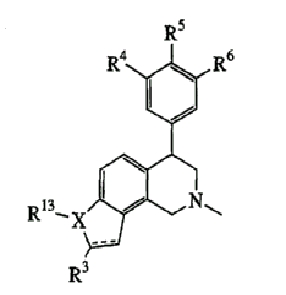
где Х представляет О, S или N, Х-содержащее кольцо является либо насыщенным, либо ненасыщенным, R3 представляет Н, Ме, Et или МеОН, R4 и R6 каждый представляет Н, F или Cl, R5 представляет H, F, Cl или OMe и R13, когда присутствует, представляет С1-С6-алкил. Еще в одном варианте осуществления соединения далее включают соединения, имеющие следующую формулу (см. таблицы 3-3А).
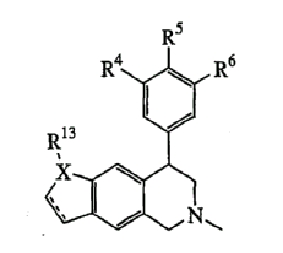
где Х представляет О или N, Х-содержащее кольцо является либо насыщенным, либо насыщенным, либо ненасыщенным, R4, R5 и R6 каждый представляет Н и R13, когда он присутствует, представляет Н или С1-С6-алкил.
Еще один вариант осуществления включает соединения, имеющие следующую формулу (см. таблицы 4-4В).
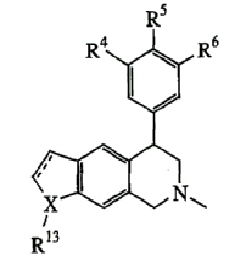
где Х представляет О или N, Х-содержащее кольцо является либо насыщенным, либо ненасыщенным, R4 представляет Н, R5 представляет Н, Cl, F или Bn, R6 представляет Н, Cl или F и R13 представляет Н или С1-С6-алкил. Следующие варианты осуществления включают соединения, имеющие следующую формулу (см. таблицу 5).
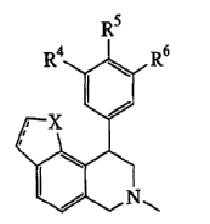
где Х представляет О или S, Х-содержащее кольцо является либо насыщенным, либо ненасыщенным, R4 представляет Н, R5 представляет Н, Cl, F или OMe, R6 представляет Н, Cl или F и R13 представляет С1-С6-алкил. Следующие соединения вариантов осуществления включают соединения, имеющие следующую формулу (см. таблицы 6-6A).
где Х представляет О, Х-содержащее кольцо является либо насыщенным, либо ненасыщенным, R4 представляет Н, R5 представляет Н или F, R6 представляет Н или F.
Предложена также каждая стереоизомерная форма соединений этого изобретения. То есть соединения могут иметь один или несколько асимметричных центров или плоскостей, и все хиральные (энантиомерные и диастереомерные) и рацемические формы соединений включены в настоящее изобретение. Многие геометрические изомеры олефинов, двойных связей C=N и подобных могут также присутствовать в соединениях, и все такие стабильные изомеры рассматриваются в настоящем изобретении. Соединения выделяют либо в рацемической форме, либо в оптически чистой форме, например, хиральной хроматографией или химическим разделением рацемической формы.
Предложены также фармацевтически приемлемые соли соединений данного изобретения. В этом отношении фразу «фармацевтически приемлемый» применяют для указания тех соединений, материалов, композиций и/или лекарственных форм, которые, с точки зрения медицинских структур являются подходящими для использования в контакте с тканями людей и животных без излишней токсичности, раздражения, аллергической реакции или другой проблемы или осложнения, соразмерных с приемлемым соотношением польза/риск. «Фармацевтически приемлемые соли» относятся к производным описанных соединений, где исходное (родительское) соединение модифицируют с образованием солей с кислотами или основаниями. Примеры фармацевтически приемлемых солей включают, но не ограничиваются ими, соли минеральных или органических кислот основных остатков, таких как амины; соли щелочных или органических основных соединений кислотных остатков, таких как карбоновые кислоты, и подобные. Фармацевтически приемлемые соли включают общепринятые нетоксичные соли или четвертичные аммониевые соли исходного соединения, образованные, например, из нетоксичных неорганических или органических кислот. Такие общепринятые нетоксичные соли включают соли, полученные из неорганических кислот, таких как хлористоводородная, бромистоводородная, серная, сульфаминовая, фосфорная, азотная и подобные; и соли, полученные из органических кислот, таких как уксусная, пропионовая, янтарная, гликолевая, стеариновая, молочная, яблочная, винная, лимонная, аскорбиновая, памовая, малеиновая, гидроксималеиновая, фенилуксусная, глутаминовая, бензойная, салициловая, сульфаниловая, 2-ацетоксибензойная, фумаровая, толуолсульфоновая, метансульфоновая, этандисульфоновая, щавелевая, изетионовая и подобные.
Предложены также пролекарственные формы соединений этого изобретения. Такие «пролекарства» являются соединениями, включающими соединения данного изобретения и части, ковалентно связанные с исходными соединениями, так что части исходного соединения наиболее вероятно связанные с токсичностью в организме субъектов, которым пролекарства вводят, блокированы от индуцирования таких действий. Однако, пролекарства также расщепляются в организме субъекта таким путем, чтобы высвободить исходное соединение без чрезмерной потери его терапевтического потенциала. Пролекарства включают соединения, у которых гидрокси, амино или сульфгидрильные группы связаны с любой группой, которая при введении субъекту-млекопитающему отщепляется с образованием свободной гидроксильной, амино или сульфгидрильной группы, соответственно. Примеры пролекарств включают, но не ограничиваются ими, ацетатные, формиатные и бензоатные производные функциональных групп спирта и амина в соединениях формул (I-III).
Предложены также соединения, меченные радиоактивными изотопами, т.е. соединения, у которых один или несколько описанных атомов заменены радиоактивным изотопом такого атома (например, С, замененный 14С или 11C, и Н, замененный 3Н или 18F). Такие соединения имеют различные потенциальные использования, например, в качестве стандартов и реагентов при определении способности потенциального фармацевтического средства связываться с белками-нейротрансмиттерами или для получения изображения соединений данного изобретения, связанных с биологическими рецепторами, in vivo или in vitro.
Данное изобретение относится к композициям, содержащим описанные здесь соединения, в том числе, в частности, фармацевтическим композициям, включающим терапевтически эффективные количества соединений и фармацевтически приемлемые носители. «Терапевтически эффективные количества» являются любыми количествами соединений, эффективными для облегчения, снижения симптомов, ингибирования или профилактики конкретного состояния, из-за наличия которого субъекта лечат. Такие количества обычно изменяются в соответствии с рядом факторов, находящихся вполне в пределах компетенции средних специалистов и приведенных здесь для определения и объяснения предложенного описания. Они включают, без ограничения: конкретного субъекта, а также его возраст, вес, рост, общее физическое состояние и историю болезни; конкретное используемое соединение, а также носитель, в котором его приготовляют, и выбранный для него путь введения, и природу и серьезность подвергаемого лечению состояния. Терапевтически эффективные количества включают оптимальные и субоптимальные дозы, их можно определить различными способами, известными обычно специалистам в данной области, например, введением различных количеств конкретного агента животному, страдающему от конкретного заболевания, с последующим определением относительной терапевтической пользы, полученной животным. Эти количества обычно составляют приблизительно от 0,001 мг на кг веса тела подвергаемого лечению субъекта до приблизительно 1000 мг на кг и, более типично, приблизительно от 0,1 до приблизительно 200 мг на кг. Эти количества можно вводить в соответствии с любой схемой приема лекарственного средства, приемлемой для средних специалистов, наблюдающих за лечением.
«Фармацевтически приемлемые носители» являются средами обычно приемлемыми в данной области для введения терапевтических соединений людям. Такие носители обычно изготавливают в соответствии с рядом факторов, находящихся вполне в пределах компетенции средних специалистов данной области и приведенных для определения и объяснения. Они включают, без ограничения, тип и природу активного агента, который вводят в композицию; субъект, которому нужно ввести содержащую агент композицию; заданный путь введения композиции и терапевтическое показание, на которое нацелено лечение. Фармацевтически приемлемые носители включают как водные, так и неводные жидкие среды, а также различные твердые и полутвердые формы. Такие носители могут включать, кроме активного ингредиента, ряд различных ингредиентов и добавок, причем такие дополнительные ингредиенты включают в препаративную форму по различным причинам, например, для стабилизации активного ингредиента, которые хорошо известны специалисту в данной области. Описания подходящих фармацевтически приемлемых носителей и факторов, учитываемых при их выборе, имеются в различных легко доступных источниках, например, в Remington's Pharmaceutical Sciences, 17th ed., Mack Publishing Company, Easton, PA, 1985, содержание которой включено сюда в качестве ссылки.
Соединения настоящего изобретения вводят, например, парентерально в различных водных средах, таких как водные растворы декстрозы и физиологические растворы; растворы гликолей также являются пригодными носителями. Растворы для парентерального введения, предпочтительно, содержат водорастворимую соль активного ингредиента, подходящие стабилизирующие агенты и, если необходимо, буферные вещества. Подходящими стабилизирующими агентами являются также антиоксиданты, такие как бисульфит натрия, сульфит натрия или аскорбиновая кислота, применяемые либо по отдельности, либо в комбинации. Используют также лимонную кислоту и ее соли и ЭДТУ. Кроме того, парентеральные растворы могут содержать консерванты, такие как хлорид бензалкония, метил- или пропилпарабен и хлорбутанол.
В альтернативном случае соединения вводят перорально в виде твердых лекарственных форм, таких как капсулы, таблетки и порошки, или жидких форм, таких как эликсиры, сиропы и/или суспензии. Можно использовать желатиновые капсулы, которые содержат активный ингредиент и подходящий носитель, включающий, но не ограничивающийся ими, лактозу, крахмал, стеарат магния, стеариновую кислоту или производные целлюлозы. Для получения прессованных таблеток можно использовать подобные разбавители. Как таблетки, так и капсулы можно изготовить в виде продуктов пролонгированного действия, чтобы обеспечить непрерывное высвобождение лекарственного средства на протяжении какого-либо периода времени. Прессованные таблетки можно покрыть сахаром или пленкой для маскирования неприятного вкуса или для защиты активных ингредиентов от действия атмосферы или для селективной дезинтеграции таблетки в желудочно-кишечном тракте.
Соединения данного изобретения обеспечивают особенно полезный терапевтических индекс по сравнению с другими соединениями, доступными для лечения подобных нарушений. Без намерения ограничиваться теорией считают, что это является результатом, по меньшей мере частично, способности соединений быть селективными в отношении белка-транспортера норэпинефрина (NET) по сравнению с селективностью в отношении транспортеров других нейротрансмиттеров. Сродство связывания демонстрируют различными способами, хорошо известными специалистам, включая, но не ограничиваясь ими, способы, описанные здесь в приведенных ниже Примерах.
Говоря кратко, например, содержащие белок экстракты из клеток, например, клеток НЕК293, экспрессирующих белки-транспортеры, инкубируют с мечеными радиоактивными изотопами лигандами для белков. Связывание радиолигандов с белками является обратимым в присутствии других лигандов белков, например, соединений настоящего изобретения; указанная обратимость, как описано ниже, обеспечивает средство измерения сродства связывания соединений к белкам (Ki). Более высокая величина Ki для соединения указывает на то, что соединение имеет меньшее сродство связывания к белку, чем соединение с более низким значением Ki, наоборот, более низкие величины Ki указывают на более высокое сродство связывания.
В соответствии с этим, более низкая величина Ki для белка, для которого соединение является более селективным, и более высокая величина Ki для белка, для которого соединение является менее селективным, указывают на различие в селективности соединения в отношении белков. Таким образом, чем выше отношение величины Ki соединения для белка А по сравнению с белком В, тем выше является селективность соединения в отношении последнего по сравнению с первым (причем, первый имеет более высокое значение Ki и последний более низкое значение Ki в отношении этого соединения). Предложенные здесь соединения вызывают меньшие побочные действия во время терапевтического использования вследствие их селективности в отношении белка-транспортера норэпинефрина, на что указывает соотношение их Ki для связывания с NET по сравнению с Ki для связывания других белков-транспортеров, например, транспортера допамина (DAT) и транспортера серотонина (SERT). Соединения настоящего изобретения обычно имеют соотношение Ki для DAT/NET приблизительно ≥ 2:1; соединения обычно имеют также соотношение SERT/NET ≥ 5:1.
Кроме того, оценку in vivo активности соединений в отношении транспортеров NE и DA проводят, например, определяя их способность предотвращать седативные действия тетрабеназина (TBZ) (см., например, G. Stille, Arzn. Forsch. 1964, 14, 534-537; содержание которой включено сюда в качестве ссылки). Взятые случайно и предварительно определенные дозы испытуемых соединений вводят мышам, как вводят затем дозу тетрабеназина. Животных затем оценивают на антагонизм индуцированной тетрабеназином потери исследовательской активности и птоза в определенные интервалы времени после введения лекарственного средства. Исследовательскую активность, например, оценивают, помещая животное в центр круга и затем оценивая время, которое требуется животному для пересечения периметра круга, - обычно, чем больше времени требуется животному для такого пересечения, тем больше потеря исследовательской активности. Кроме того, считается, что животное имеет птоз, если его веки закрыты, по меньшей мере, на 50%. Считают, что более чем 95% контрольных (обработанных наполнителем) мышей имеют потерю исследовательской активности и птоз; связанную с соединением активность затем вычисляют в виде процента повреждения мышей в ответ на дозу введения тетрабеназина, и считают, что терапевтически более эффективные соединения являются лучшими при снижении потери исследовательской активности и птоза.
В соответствии с этим, предложенные здесь фармацевтические композиции полезны для лечения субъектов, страдающих различными неврологическими и психиатрическими расстройствами при введении указанным субъектам предложенной здесь дозы фармацевтической композиции. Такие расстройства включают, без ограничения, дефицит внимания, связанный с гиперактивностью, тревогу, депрессию, послетравматическое стрессовое нарушение, надъядерный паралич, нарушения питания, обессивно-компульсивное расстройство, аналгезию, синдром прекращения курения, приступы паники, болезнь Паркинсона и фобии. Предложенные здесь соединения являются особенно полезными при лечении этих и других нарушений вследствие, по меньшей мере частично, их способности селективно связываться с белками-транспортерами некоторых нейрохимических веществ с большим сродством связывания по сравнению с белками-транспортерами других нейрохимических веществ.
Синтез
Соединения настоящего изобретения можно получить с использованием способов, описанных ниже, а также способы известных в области синтетической органической химии, или их вариантов, известных специалистам в данной области. Предпочтительные способы включают, но не ограничиваются ими, способы, описанные ниже.
Новые тетрагидроизохинолиновые ингибиторы повторного поглощения формул (I-IIIB) этого изобретения можно получить по общей схеме, приведенной ниже (схемы 1-4). R1-замещенные N-бензиламины формулы (V) схемы 1 можно приобрести из коммерческих источников или, в альтернативном случае, их можно получить простой реакцией восстановительного аминирования. Так, карбонилсодержащие соединения формулы (IV) можно обработать H2N-R1 в низших алкиловых спиртовых растворителях (предпочтительно, метаноле или этаноле) при комнатной или более низкой температуре. Образовавшийся имин можно восстановить, чаще всего борогидридами щелочных металлов (предпочтительно, борогидридом натрия), с образованием требуемых аминных промежуточных продуктов, восстановления оптимально проводят при комнатной или более низкой температуре. Обработка промежуточных бензиламинов формулы (V) электрофильными промежуточными соединениями формулы (VII) дает продукты алкилирования формулы (VIII). Реакции алкилирования можно проводить в различных условиях, известных специалисту в области органического синтеза. Типичные растворители включают ацетонитрил, толуол, диэтиловый эфир, тетрагидрофуран, диметилсульфоксид, диметилформамид, метиленхлорид и низшие алкиловые спирты, в том числе этанол. Реакции можно проводить при температурах, составляющих от 0°С до температуры кипения используемого растворителя. Ход реакции обычно контролируют стандартными хроматографическими и спектроскопическими методами. Реакцию алкилирования необязательно проводят с добавлением ненуклеофильного органического основания, включающего, но не ограничивающегося ими, пиридин, триэтиламин и диизопропилэтиламин, и время реакции до ее завершения может изменяться от 1 часа до нескольких дней.
Вышеуказанное электрофильное промежуточное соединение формулы (VII) обычно приобретают из коммерческих источников или получают обработкой необязательно замещенного ацетофенона формулы (VI) обычными бромирующими агентами, включающими, но не ограничивающимися ими, бром, NBS или трибромид тетрабутиламмония, такая обработка легко дает требуемые бромацетофеноны формулы (VII). Эти реакции оптимально проводят в уксусной кислоте или метиленхлориде с метанолом, используемым в качестве сорастворителя для трибромидного реагента, при комнатной или более низкой температуре. Другой вариант осуществления данной методики может включать использование хлорацетофеноновых соединений формулы (VII).
Ацетофеноны формулы (VI), в свою очередь, являются доступными также из коммерческих источников или их обычно получают с помощью нескольких хорошо известных способов, включающих обработку соответствующих промежуточных бензойной кислоты двумя стехиометрическими эквивалентами метиллития (см., например, Jorgenson, M.J. (Organic Reactions, 1970, 18, pg. 1)). В альтернативном случае соответствующие бензальдегиды можно обработать нуклеофильным алкилсодержащим реактивом Гриньяра (например, MeMgBr) или алкиллитием (например, MeLi) с последующим стандартным окислением в кетон (см., например, Larock, R.C. (Comprehensive Organic Transformations, VCH Publishers, New York, 1989, p. 604)).
Восстановления соединений формулы (VIII) в бензиловые спирты формулы (IX) проводят, используя многие восстанавливающие агенты, включающие, например, борогидрид натрия, борогидрид лития, боран, диизобутилалюминийгидрид и литийалюминийгидрид. Восстановления проводят в течение времени от 1 часа до 3 дней при комнатной температуре или повышенной температуре вплоть до температуры кипения используемого растворителя. Если используют боран, его можно применять в виде комплекса, включающего, например, но не ограничивающегося ими, комплекс боран-метилсульфид, комплекс боран-пиперидин или комплекс боран-тетрагидрофуран. Специалист в данной области должен знать оптимальную комбинацию восстанавливающих агентов и необходимых условий реакции или может использовать руководство из Larock, R.C. (см. выше).
Соединения формулы (IX) можно циклизовать в тетрагидроизохинолиновые соединения формулы (Ib), где R7=H, данного изобретения быстрой обработкой сильной кислотой. Подходящие кислоты включают, но не ограничиваются ими, концентрированную серную кислоту, полифосфорную кислоту, метансульфоновую кислоту и трифторуксусную кислоту. Реакции проводят без растворителя или необязательно, в присутствии сорастворителя, такого как, например, метиленхлорид или 1,2-дихлорэтан. Циклизации можно проводить при температуре от 0°С до температуры кипения используемого растворителя. Специалист в области химии гетероциклических соединений может легко выбрать эти условия или может принять во внимание указания в Mondeshka, et al. (Il Farmaco, 1994, 49, 475-480) или Venkov, et al. (Synthesis, 1990, 253-255). Циклизации можно также осуществить обработкой соединений формулы (IX) сильными кислотами Льюиса, такими как, например, трихлорид алюминия, обычно в галогенированных растворителях, таких как метиленхлорид. Специалист в данной области должен быть знаком со способом, описанным в Kaiser, et al. (J. Med. Chem., 1984, 27, 28-35) и Wyrick, et al. (J. Med. Chem., 1981, 24, 1013-1015).
Соединения формул (I-III) можно получить в энантиомерно чистой (R)- и (S)-форме кристаллизацией с хиральными солями, что хорошо известно специалисту в данной области, или, в альтернативном случае, можно выделить посредством хиральной ВЭЖХ с использованием коммерчески доступных хиральных колонок.
Соединения формул (I-III), где R7=ОН в схеме 1, 3 и 4 настоящего изобретения, можно получить в соответствии с Kihara, et al. (Tetrahedron, 1992, 48, 67-78) и Blomberg, et al. (Synthesis, 1977, p. 18-30). Так, кетонные соединения формулы (VIII), которые имеют орто-иод на ароматическом кольце и подвергаются циклизации, можно обработать сильными основаниями, включающими, но не ограничивающимися ими, низший алкил(С1-6)литиевые основания (предпочтительно, трет-BuLi или н-BuLi), для проведения ожидаемого обмена галоген-металл с последующей внутримолекулярной циклизацией Barbier с образованием соединений формул (I-III), где R7=OH. Необходимы инертные растворители, такие как диалкиловые простые эфира (предпочтительно, диэтиловый эфир), циклические простые эфиры (предпочтительно, тетрагидрофуран или 1,4-диоксан) и т.д., и поддерживают низкие температуры реакции (от -78°С до -25°С), чтобы избежать образования побочных продуктов. В альтернативном случае обмен галоген-металл можно также проводить в присутствии никеля с нулевой валентностью, в этом случае N,N-диалкилформамиды (предпочтительно, диметилформамид) служат в качестве идеальных растворителей. Эту циклизацию лучше всего проводить, когда Х=Br, чтобы избежать перевосстановления или межмолекулярной реакционной способности. Кроме этого, соединения формул (I-III), где R7=ОН, можно легко алкилировать (см. выше) для получения соединений формул (I-III), где R7=OR10. Наконец, дальнейшая обработка соединений формул (I-III), где R7=ОН, галогенирующим реагентом или, определенно, фторирующим реагентом, включающим, но не ограничивающимся им, трифторид диэтиламиносеры (DAST), легко дает соединения формулы (I-III), где R7=F. Следующую ссылку можно получить из Hudlicky (Organic Reactions, 1985, 35, p. 513-637).
Что касается предшественников соединений формулы (IV), для тех реагентов, которые могут быть коммерчески недоступными, существуют многочисленные синтетические пути получения из других коммерческих соединений или соединений, известных в данной области, и эти пути должны быть вполне очевидны любому специалисту в области органического синтеза. Без ограничения иллюстративный способ показан на схеме 2, где аллиловый спирт формулы (Х) легко подвергают озонолизу с последующей восстановительной обработкой реагентами, включающими, но не ограничивающимися им, диметилсульфид, получая лактол, который обрабатывают слабой кислотой в широком диапазоне условий с образованием бензофурана формулы (XI). Методика взаимного превращения функциональной группы сложного эфира в альдегид должна быть вполне знакома для специалиста в данной области для получения целевых соединений формулы (IV).
Кроме того, аминоспирты предварительной циклизации формулы (XII) схемы 3 и формул (XIII-XIV) схемы 4 синтезируют способом, полностью аналогичным способам, описанным выше для получения аминоспирта предварительной циклизации формулы (IX) схемы 1. Кроме того, как описано выше, аминоспирты предварительной циклизации формулы (XII) схемы 3 и формул (XIII-XIV) схемы 4 можно циклизовать, как описано, чтобы получить целевые тетрагидроизохинолины формулы (Ia) схемы 3 и формул (IIa, IIb, IIIa и IIIb) схемы 4. Должно быть также известно любому специалисту в данной области, что региомерные тетрагидроизохинолины получают циклизацией соединений формул (XIII-XIV).
В следующем варианте осуществления данного изобретения ненасыщенные фуран-, индол- и тиофентетрагидроизохинолины формул (I-III) можно частично восстановить в соответствующие дигидрофуран-, дигидроиндол- и дигидротиофентетрагидроизохинолины формул (I-III).
Восстановления проводят в присутствии водорода, либо при атмосферном давлении, либо при повышенном давлении и в присутствии растворителя из широкого диапазона растворителей, включающих, но не ограничивающихся ими, метанол, этанол и этилацетат. Реакции оптимально проводят в присутствии металлического катализатора, включающего, но не ограничивающегося ими, палладий, платину или родий. Оптимальные условия гидрирования должны быть вполне известны специалисту в данной области; в альтернативном случае он может найти их в Larock, R.C. (Comprehensive Organic Transformations, VCH Publishers, New York, 1989, p. 6.
В случаях, когда частичное восстановление вышеуказанных гетероциклов является невозможным (для соединения Ib, где R7=H) вследствие сопутствующего гидрогенолиза заместителей боковых арилов (например, Cl), необходимо восстановить гетероциклическую часть (т.е. бензофуран, индол или тиофен) на более ранней стадии синтеза (схема 1, промежуточный продукт (V)) и затем ввести боковой арил (VII) такими же способами, указанными на схеме 1.
Содержимое указанных выше публикаций включено сюда в качестве ссылки.
СХЕМА 1
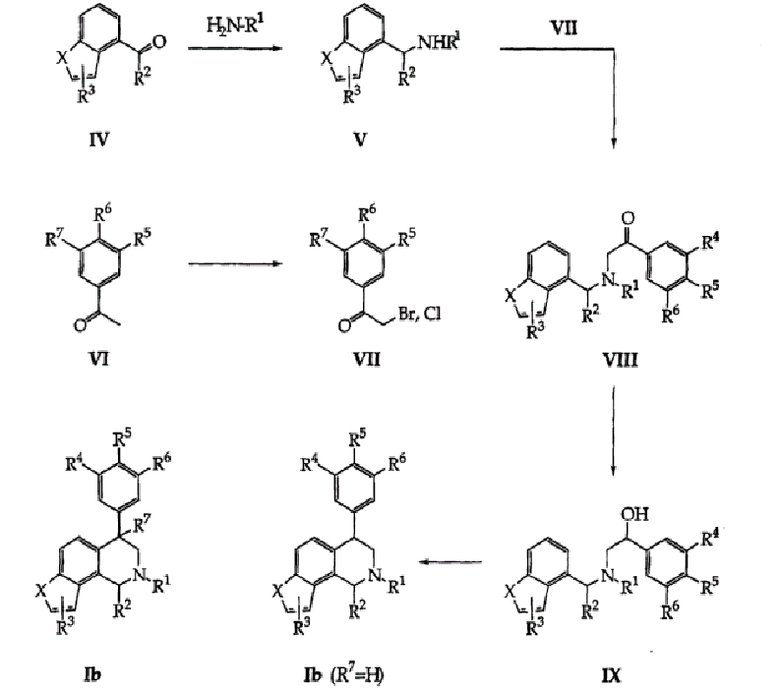
СХЕМА 2
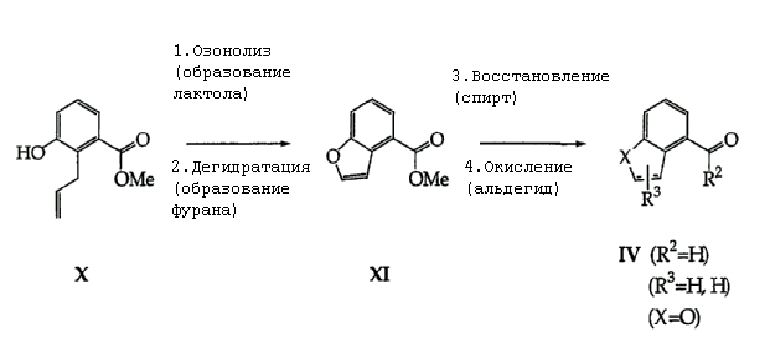
СХЕМА 3
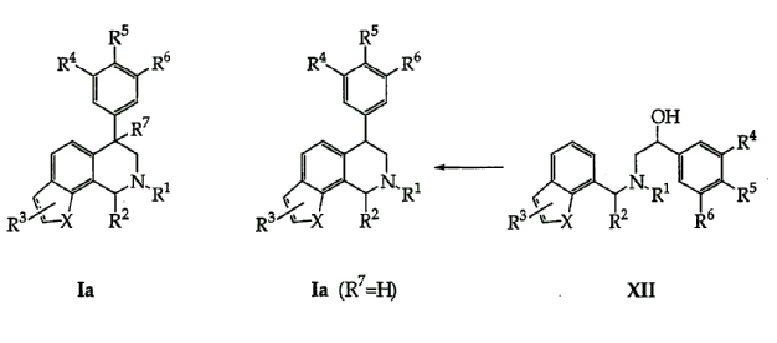
СХЕМА 4
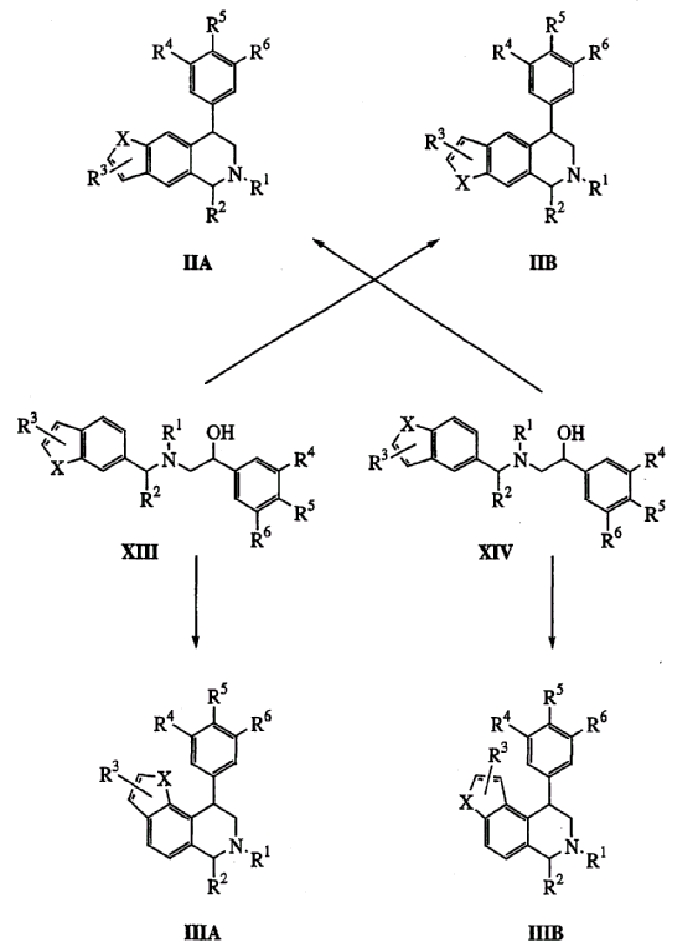
Данное изобретение можно лучше понять посредством ссылки на следующий раздел примеров. Однако средний специалист в данной области может легко понять, что примеры являются только иллюстрацией изобретения, как определено в формуле изобретения, которая приводится ниже.
ПРИМЕРЫ
Соединения, перечисленные ниже в таблицах I-VIA (примеры 1-131), получены в соответствии со схемами синтезов, приведенных выше, и имеют точки плавления, как указано в таблицах; когда соединение представляет собой масло или твердое вещество, оно указано здесь как таковое, и если оно является твердым, указана форма соли.
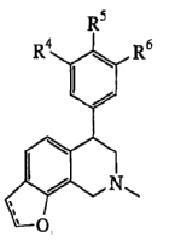
Энантиомерно чистые соединения (на основе общей структуры в таблице I)
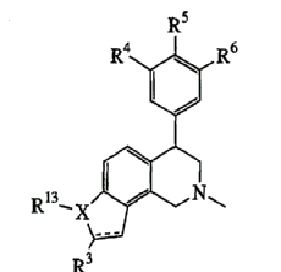
(идентичная общая структура, как показано в таблице II)
Энантиомерно чистые соединения (на основе общей структуры, показанной в таблице II, но с (R)- или (S)-абсолютной конфигурацией)
фумарат
малеат
фумарат
энантиомер В,
вращение +18,2°
(С=0,262, МеОН)
энантиомер В
вращение -55,2°
(С=0,372, МеОН)
вращение +55,5°
(С=0,384, МеОН)
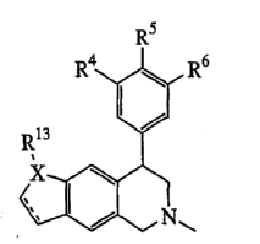
(идентичная общая структура, как показано в таблице III)
(идентичная общая структура, как показано в таблице IV)
энантиомерно чистые соединения (на основе общей структуры в таблице IV)
(идентичная общая структура, как показано в таблице VI)
Пример 5
Стадия А: Бензофуран-7-карбоксальдегид (4,44 г, 30,4 ммоль), водный метиламин (5,5 мл, 63 ммоль) и МеОН (35 мл) смешивают в колбе на 25 мл в атмосфере N2. Смесь охлаждают до 0°С при быстром перемешивании и порциями в течение 5 минут добавляют NaBH4 (0,61 г, 16 ммоль). Смесь нагревают до комнатной температуры при перемешивании в течение ночи. Смесь разбавляют водой (50 мл), перемешивают в течение 15 минут и экстрагируют (3х) СН2Cl2. Объединенные органические экстракты промывают (3х) 2 н. HCl. Эти кислотные экстракты делают основными твердым KOH, дополнительным количеством воды и концентрированным NH4OH. Основную смесь экстрагируют (3х) СН2Cl2. Эту вторую серию органических экстрактов объединяют и сушат над Na2SO4, фильтруют и концентрируют в вакууме, получая метиламинопродукт (3,51 г, 71%) в виде желтого масла: 1Н ЯМР (500 МГц, CDCl3) δ 7.66 (д, J=2.3 Гц,1Н); 7.53-7.55 (м,1Н); 7.22-7.29 (м,2Н); 6.80 (д, J=2.4 Гц,1Н); 4.10 (с,2Н); 2.51 (с,3Н).
Стадия В: Метиламин со стадии А (3,50 г, 21,7 ммоль) и 4'-хлорфенацилбромид (6,2 г, 23 ммоль) растворяют в СН2Cl2 (45 мл) в 250 мл колбе в атмосфере N2. Смесь быстро перемешивают, добавляют Et3N (3,0 мл, 22 ммоль) и смесь продолжают перемешивать в течение ночи. Смесь разбавляют водой, слои разделяют и водный слой экстрагируют дважды СН2Cl2. Объединенные органические экстракты сушат над Na2SO4, фильтруют и концентрируют в вакууме. Сырой остаток очищают хроматографией на силикагеле (20% EtOAc/гексаны), получая аминокетон (4,28 г, 63%) в виде желтого масла: 1Н ЯМР (300 МГц, CDCl3) δ 7.94-7.96 (м,1Н); 7.76-7.80 (м,1Н); 7.60 (д, J=2.3 Гц,1Н); 7.47-7.56 (м,2Н); 7.18-7.35 (м,3Н); 6.77 (д, J=2.3 Гц,1Н); 4.03 (с,2Н); 3.79 (с,2Н), 2.42 (с,3Н).
Стадия С: Аминокетон из стадии В (4,28 г, 13,6 ммоль) растворяют в МеОН (30 мл) в атмосфере N2. Смесь охлаждают до 0°С, добавляют по порциям NaBH4 (1,07 г, 28,2 ммоль) и смесь перемешивают в течение 5 часов при нагревании до комнатной температуры. Смесь разбавляют водой и экстрагируют (3 х) СН2Cl2. Объединенные органические экстракты сушат над Na2SO4, фильтруют и концентрируют в вакууме. Сырой остаток очищают хроматографией на силикагеле (20% EtOAc/гексаны), получая аминоспирт (3,14 г, 73%) в виде желтого масла: 1Н ЯМР (500 МГц, CDCl3) δ 7.61-7.69 (м,1Н); 7.53-7.56 (м,1Н); 7.32-7.40 (м,1Н); 7.18-7.29 (м,5Н); 6.78-6.83 (м,1Н); 4.75-4.81 (м,1Н); 4.35 (ушир.с,1Н); 4.06 (д, J=13.2 Гц,1Н); 3.87 (д,J=13.2Гц,1Н), 2.55-2.66 (м,1Н), 2.34 (с,3Н).
Стадия D: Аминоспирт со стадии С (580 мг, 1,83 ммоль) растворяют в СН2Cl2 (18 мл) в 100 мл колбе, снабженной холодильником, в атмосфере N2. Смесь охлаждают до 0°С при перемешивании и по каплям добавляют MeSO3H (6,0 мл, 92 ммоль). Смеси дают возможность нагреться до комнатной температуры, затем кипятят с обратным холодильником на протяжении ночи. Смесь охлаждают до комнатной температуры, медленно добавляют 2 н. NaOH, чтобы сделать смесь основной. Смесь экстрагируют (3х) СН2Cl2 и объединенные органические экстракты сушат над Na2SO4, фильтруют и концентрируют в вакууме. Сырой остаток очищают хроматографией на силикагеле (смесь 5% EtOAc/гексаны, содержащая 1% Et3N), получая соединение примера 5 (304 мг, 56%) в виде светло-желтого масла: 1Н ЯМР (300 МГц, CDCl3) δ 7.61 (д,J=2.1 Гц,1Н); 7.32 (д,J=8.1 Гц,1Н); 7.19-7.22 (м,3Н); 7.07-7.11 (м,1Н); 6.72-6.77 (м,2Н); 4.34 (т,J=6.2 Гц,1Н); 4.03 (д,J=15.5 Гц,1Н); 3.87 (д, J=15.3 Гц,1Н); 3.01-3.08 (м,1Н); 2.66 (дд,J=7.8, 11.5 Гц,1Н), 2.50 (с,3Н), 2.34 (с,3Н); CI MS m/z=298 [C18H16ClNO + H]+; Элементный анализ. Вычислено для С18Н16ClNO-0,25 Н2О: С, 71,52; Н, 5,50; N, 4,63. Найдено: С, 71,53; Н, 5,34; N, 4,42. Выделяют также исходный материал (115 мг, 20%).
Пример 6
Стадия А: Амин, полученный в примере 5, стадия А (1,24 г, 7,69 ммоль), растворяют в абсолютном EtOH (8 мл) в реакторе Парра. Добавляют 10% Pd/C (0,61 г, 50 масс.%) и смесь гидрируют при 30 фунт/кв. дюйм на протяжении ночи. Суспензию фильтруют через целит и слой промывают дважды МеОН. Фильтрат концентрируют в вакууме, получая дигидробензофуран 76 (1,27 г, выход количественный) в виде желтого масла: 1Н ЯМР (300 МГц, CDCl3) δ 7.07-7.13 (м,2Н); 6.81 (т,J=7.4 Гц,1Н); 4.58 (т,J=8.7 Гц,1Н); 3.78 (с,2Н); 3.18-3.27 (м,3Н); 2.45 (с,3Н).
Стадия В: Дигидробензофуранамин (1,27 г, 7,69 ммоль, получен на стадии А) 3'-хлорфенацилбромид 71 (1,9 г, 8,0 ммоль) и СН2Cl2 (15 мл) смешивают в 100 мл колбе в атмосфере N2. Смесь быстро перемешивают при добавлении Et3N (1,1 мл, 7,9 ммоль). После перемешивания в течение 2 часов смесь разбавляют водой и СН2Cl2 и слои разделяют. Водный слой экстрагируют дважды СН2Cl2 и объединенные органические экстракты сушат над Na2SO4, фильтруют и концентрируют в вакууме. Сырой остаток очищают хроматографией на силикагеле (20% EtOAc/гексаны), получая аминокетон (1,75 г, 72%) в виде желтого масла: 1Н ЯМР (300 МГц, CDCl3) δ 7.96 (т,J=1.7 Гц,1Н); 7.82-7.86 (м,1Н); 7.50 (дт,J=1.4, 8.3 Гц,1Н); 7.35 (т,J=7.9 Гц,1Н); 7.10 (дд,J=7.5, 15.6 Гц,2Н); 6.82 (т,J=7.4 Гц,1Н); 4.51 (т, J=8.7 Гц,2Н); 3.73 (с,2Н); 3.69 (с,2Н); 3.20 (т,J=8.7 Гц,2Н), 2.37 (с,3Н); CI MS m/z=316 [C18H18ClNO2 + H]+.
Стадия С: Аминокетон, который получен на стадии В (1,75 г, 5,54 ммоль), растворяют в МеОН (12 мл) в 100 мл колбе в атмосфере N2. Смесь охлаждают до 0°С и в виде одной порции добавляют NaBH4 (440 мг, 11,6 ммоль). Смеси дают возможность нагреться до комнатной температуры при перемешивании на протяжении ночи. Смесь разбавляют водой, затем экстрагируют (3 х) СН2Cl2. Объединенные органические экстракты сушат над Na2SO4, фильтруют и концентрируют в вакууме, получая аминоспирт (1,76 г, 99%) в виде желтого масла, которое отверждается при стоянии: 1Н ЯМР (300 МГц, CDCl3) δ 7.38 (с,1Н); 7.23-36 (м,3Н); 7.13-7.16 (м,1Н); 7.01 (д,J=7.4 Гц,1Н); 6.81 (т,J=7.4 Гц,1Н); 4.73 (дд, J=4.1, 9.8 Гц,1Н); 4.60 (т,J=9.0 Гц,2Н), 3.75 (д, J=12.9 Гц,1Н), 3.50 (д,J=12.9 Гц,1Н); 3.23 (т,J=8.7 Гц,2Н), 2.49-2.62 (м,2Н); 2.30 (с,3Н).
Стадия D: Аминоспирт, который получен на стадии С (814 мг, 2,56 ммоль), растворяют в СН2Cl2 (25 мл) в 100 мл колбе, снабженной холодильником, в атмосфере N2. Смесь охлаждают до 0°С при быстром перемешивании и по каплям добавляют MeSO3H (8,4 мл, 129 ммоль). Смеси дают возможность нагреться до комнатной температуры, затем кипятят с обратным холодильником в течение 48 часов. Смесь охлаждают до комнатной температуры и медленно гасят добавлением 2 н. NaOH. Слои разделяют и водный слой экстрагируют (3х) СН2Cl2. Объединенные органические экстракты сушат над Na2SO4, фильтруют и концентрируют в вакууме. Сырой остаток очищают хроматографией на силикагеле (1,5% МеОН/СН2Cl2), получая соединение примера 6 (603 мг, 75%) в виде светло-желтого масла: 1Н ЯМР (300 МГц, CDCl3) δ 7.17-7.22 (д,J=7.5 Гц,1Н); 4.57-4.64 (м,2Н); 4.19 (т,J=6.2 Гц,1Н); 3.69 (д,J=15.4 Гц,1Н); 3.50 (д,J=15.3 Гц,1Н); 3.18 (т,J=8.8 Гц,2Н); 2.91-2.98 (м, 1Н); 2.56 (дд,J=8.0, 11.4 Гц,1Н), 2.43 (с,3Н); API MS m/z=300 [C18H18ClNO + H]+; Элементный анализ. Вычислено для С18Н18ClNO-0,6 Н2О: С, 69,60; Н, 6,23; N, 4,51. Найдено: С, 69,53; Н, 5,88; N, 4,38.
Пример 12
Стадия А: Аллиловый спирт Х (2,0 г, 10,5 ммоль) растворяют в метаноле (90 мл), охлаждают до -78°С и озонируют до тех пор, пока не останется исходного материала (приблизительно 30 минут). Быстро добавляют диметилсульфид (4 мл) и образовавшейся смеси дают возможность нагреться до комнатной температуры на протяжении ночи. Растворитель удаляют в вакууме и остаток растворяют в диэтиловом эфире, затем промывают дважды водой и один раз насыщенным раствором соли. Органическую часть сушат над безводным сульфатом натрия, фильтруют и растворитель удаляют в вакууме, получая требуемый лактол, 1,18 г (58%) в виде вязкого желтого масла: 1Н ЯМР (300 МГц, CDCl3) δ 7.56-7.59 (м,1Н); 7.21-7.26 (м,1Н); 7.03 (д, 1Н, J=8.0 Гц); 6.12 (дд, 1Н, J=2.2, 6.5 Гц); 3.90 (с,3Н); 3.40-3.60 (м,2Н).
Стадия В: Продукт стадии А (8,0 г, 41,0 ммоль) перемешивают в Н3РО4 (85%, 50 мл) при комнатной температуре в течение 30 минут. Образовавшуюся мутную смесь разбавляют водой и экстрагируют (4х) диэтиловым эфиром. Объединенные органические экстракты промывают насыщенным раствором соли, сушат над безводным сульфатом натрия, фильтруют и растворитель удаляют в вакууме. Сырой материал очищают флэш-хроматографией на силикагеле (гексаны/этилацетат, 20:1), получая 3,87 г (53%) бензофуранметилового сложного эфира в виде светло-желтого масла: 1Н ЯМР (300 МГц, CDCl3) δ 7.99 (д, 1Н, J=7.1 Гц); 7.68-7.74 (м,2Н); 7.32-7.38 (м,2Н); 3.99 (м,3Н); CI MS m/z=177 [C10H8O3+H]+.
Стадия С: Продукт стадии В (4,67 г, 27,0 ммоль), растворенный в безводном тетрагидрофуране (60 мл), добавляют по каплям к перемешиваемой суспензии литийалюминийгидрида (2,5 г, 65,0 ммоль) в безводном тетрагидрофуране (50 мл) при 0°С в атмосфере азота. Серую суспензию перемешивают и оставляют для нагревания до комнатной температуры в течение двух часов. Смесь снова охлаждают до 0°С, затем гасят этилацетатом до прекращения выделения пузырьков и добавляют раствор насыщенного водного сульфата натрия до тех пор, пока не исчезнет серый цвет. Для удаления воды добавляют безводный сульфат натрия, раствор фильтруют и растворитель удаляют в вакууме. Остаток выдерживают при пониженном давлении в течение семи часов, получая 4,6 г (100%) требуемого спирта в виде желтого масла, который обычно используют без дополнительной очистки. Часть сырого продукта очищают флэш-хроматографией на силикагеле (гексан/этилацетат, 10:1, затем 2:1), получая чистый спирт в виде белого твердого вещества: 1Н ЯМР (300 МГц, CDCl3) δ 7.60 (д, 1Н, J=2.3 Гц); 7.43 (д, 1Н, J=8.1 Гц); 7.24 (т, 1Н, J=7.7 Гц); 7.16 (д, 1Н, J=7.4 Гц); 6.85-6.86 (м,1Н); 4.84 (с,3Н); 2.34 (ушир.с,1Н).
Стадия D: Раствор оксалилхлорида (2,9 мл, 33,0 ммоль) в метиленхлориде (75 мл) перемешивают в атмосфере азота при -78°С, и по каплям добавляют диметилсульфоксид (5,2 мл, 73,0 ммоль). Образовавшуюся смесь перемешивают при -78°С в течение 10 минут, затем по каплям в течение 20 минут добавляют раствор соединения стадии С (4,5 г, 30,0 ммоль) в метиленхлориде (75 мл). Смесь перемешивают при -78°С в течение более 20 минут, затем быстро добавляют триэтиламин (21,0 мл, 150 ммоль) и реакционной смеси дают возможность нагреться до комнатной температуры и перемешивают на протяжении ночи в атмосфере азота. Смесь разбавляют метиленхлоридом и водой. Слой метиленхлорида удаляют и водную часть экстрагируют дважды метиленхлоридом. Органические слои объединяют, промывают насыщенным раствором соли, сушат над безводным сульфатом натрия, фильтруют и растворитель удаляют в вакууме. Остаток очищают флэш-хроматографией на силикагеле (гексаны/этилацетат, 5:1, затем 1:1), получая требуемый бензофуранальдегид, 3,1 г (70%), в виде желтого масла: 1Н ЯМР (300 МГц, CDCl3) δ 10.19 (с,1Н); 7.79 (д, 1Н, J=2.1 Гц); 7.73 (т, 1Н, J=7.4 Гц); 7.51 (д, 1Н, J=1.7 Гц); 7.43 (т, 1Н, J=7.8 Гц).
Стадия Е: Продукт стадии D (2,91 г, 20 ммоль) в виде раствора в метаноле (30 мл) добавляют по каплям к 40% водному метиламину (3,4 мл, 40 ммоль) в метаноле. Реакционную смесь перемешивают в течение ночи при комнатной температуре в атмосфере азота, затем охлаждают до 0°С и небольшими порциями на протяжении двух минут добавляют борогидрид натрия (0,8 г, 20 ммоль). Образовавшуюся смесь перемешивают в течение 2,5 часов при комнатной температуре, затем гасят водой и экстрагируют (3х) 2 н. HCl. Водные экстракты делают основными 6 н. NaOH (рН 10) и продукт экстрагируют в метиленхлорид и сушат над безводным сульфатом натрия. Фильтрование и концентрирование дают требуемый метиламин, 2,88 г (89%), в виде светло-желтого масла: 1Н ЯМР (300 МГц, CDCl3) δ 7.62 (д, 1Н, J=2.3 Гц); 7.42 (д, 1Н, J=8.0 Гц); 7.25 (т, 1Н, J=7.7 Гц); 7.16-7.19 (м,1Н); 6.88-6.89 (м,1Н); 3.98 (ушир.с,2Н); 2.48 (ушир.с,3Н).
Стадия F: Продукт стадии Е (2,99 г, 19,0 ммоль), 2-бромацетофенон (3,7 г, 19,0 ммоль) и триэтиламин (2,7 мл, 19,6 ммоль) в метиленхлориде (40 мл) перемешивают при комнатной температуре в атмосфере азота на протяжении ночи. Смесь разбавляют метиленхлоридом, промывают водой и сушат над безводным сульфатом натрия. Фильтрование и концентрирование в вакууме дают продукт алкилирования, 5,3 г (99%), в виде желтовато-оранжевого масла: 1Н ЯМР (300 МГц, CDCl3) δ 7.89-7.93 (м,2Н); 7.38-7.61 (м,4Н); 7.17-7.27 (м,3Н); 6.99 (д, 1Н, J=1.9 Гц); 3.90 (с,2Н); 3.83 (с,2Н); 2.39 (с,3Н).
Стадия G: К раствору продукта стадии F (5,3 г, 18,8 ммоль) в метаноле (50 мл) при 0°С добавляют борогидрид натрия (1,4 г, 37,6 ммоль). После перемешивания в течение 1,5 часов при комнатной температуре реакционную смесь гасят водой, затем экстрагируют (3х) метиленхлоридом. Объединенные органические экстракты промывают насыщенным раствором соли, сушат над безводным сульфатом натрия, фильтруют и концентрируют в вакууме. Остаток очищают флэш-хроматографией на силикагеле (гексаны/этилацетат, медленный градиент от 10:1 до 1:1), получая аминоспирт, 3,22 г (61%), в виде вязкого желтого масла: 1Н ЯМР (300 МГц, CDCl3) δ 7.65 (д, 1Н, J=2.3 Гц); 7.46 (д, 1Н, J=8.2 Гц); 7.23-7.34 (м,6Н); 7.16 (д, 1Н, J=7.3 Гц); 6.93-6.94 (м,1Н); 4.74-4.78 (м,1Н); 3.91-4.00 (м,2Н); 3.75 (д, 1Н, J=12.9 Гц), 2.54-2.68 (м,2Н); 2.35 (с,3Н).
Стадия Н: Раствор продукта стадии G (3,2 г, 11,5 ммоль) в метиленхлориде перемешивают при комнатной температуре в атмосфере азота, и в течение 30 минут по каплям добавляют метансульфоновую кислоту (17 мл, 260,0 ммоль). Реакционный раствор перемешивают на протяжении ночи при комнатной температуре в атмосфере азота, затем охлаждают до 0°С и обрабатывают 2 н. NaOH до тех пор, пока рН водного слоя не будет 12, и затем разбавляют водой. Слой метиленхлорида удаляют и водную часть экстрагируют дважды метиленхлоридом. Объединенные органические слои промывают насыщенным раствором соли, сушат над безводным сульфатом натрия, фильтруют и растворитель удаляют в вакууме. Реакционную массу подщелачивают 10% водным гидроксидом аммония, образовавшуюся белую мутную смесь экстрагируют (3х) метиленхлоридом и органические слои объединяют, промывают насыщенным раствором соли, сушат над безводным сульфатом натрия, фильтруют и растворитель удаляют в вакууме, получая целевой циклизованный тетрагидроизохинолин, 2,0 г, в виде светло-коричневого масла: 1Н ЯМР (300 МГц, CDCl3) δ 7.62 (д, 1Н, J=2.3 Гц); 7.17-7.33 (м,6Н); 6.81 (д, 1Н, J=8.6 Гц); 6.73 (д, 1Н, J=2.2 Гц); 4.33-4.37 (м,1Н); 3.96 (д, 1Н, J=15.2 Гц); 3.79 (д, 1Н, J=14.5 Гц); 3.04-3.10 (м,1Н); 2.62-2.68 (м,1Н); 2.50 (с,3Н). Свободное основание (2,0 г, 7,6 ммоль) и малеиновую кислоту (0,88 г, 7,6 ммоль) очень быстро растворяют в абсолютном этаноле (70 мл) при кипячении с обратным холодильником. Раствору дают возможность охладиться до комнатной температуры, в течение этого времени образуется не совсем белый осадок. Выделение твердого вещества вакуумным фильтрованием дает требуемую малеатную соль, 1,45 г (33% от продукта стадии G) в виде не совсем белого твердого вещества; т. пл. 199-204°С; 1Н ЯМР (300 МГц, CD3OD) δ 7.88 (д, 1Н, J=2.3 Гц); 7.33-7.42 (м,4Н); 7.23-7.26 (м,2Н); 6.96-6.98 (м,1Н); 6.84 (д, 1Н, J=8.7 Гц); 6.22 (с,2Н); 4.82-4.88 (м,1Н); 4.64-4.74 (м,2Н); 3.84-3.90 (м,1Н); 3.55-3.63 (м,1Н); 3.13 (с,3Н); IR (KBr) 3448, 2363, 1700, 1578, 1456, 1354, 1049, 869, 748, 703, 652, 576 см-1; API MS m/z=264 [C18H17NO+H]+; Элементный анализ. Вычислено для С18Н17NO-С4Н4О4-0,25 Н2О: С, 72,11; Н, 6,05; N, 4,67. Найдено: С, 71,89; Н, 6,01; N, 4,59.
Пример 13
Свободное основание продукта примера 12, стадии Н (0,029 г), в абсолютном этаноле (6 мл) гидрируют над 5% Pd/C (0,030 г) при давлении незначительно выше атмосферного в течение 3 дней. Катализатор удаляют фильтрованием и растворитель удаляют в вакууме. Остаток подвергают колоночной хроматографии на силикагеле (гексаны/этилацетат, 2:1), получая дигидробензофурановое свободное основание, 0,015 г (52%), в виде бесцветной смолы: 1Н ЯМР (300 МГц, CDС13) δ 7.17-7.31 (м,5Н); 6.63 (д, 1Н, J=8.3 Гц); 6.54 (д, 1Н, J=8.3 Гц); 4.61 (т,2Н, J=8.7 Гц); 4.18-4.23 (м,1Н); 3.64 (д, 1Н, J=15.1 Гц); 3.47 (д, 1Н, J=15.3 Гц); 3.08 (т, 2Н, J=8.5 Гц); 2.97-3.03 (м,1Н); 2.56 (дд, 1Н, J=8.6, 11.5 Гц); 2.44 (с,3Н). Свободное основание (0,012, 0,045 ммоль) и малеиновую кислоту (0,005 г, 0,045 ммоль) в течение 10 минут растворяют в абсолютном этаноле (7 мл) и кипятят с обратным холодильником в атмосфере азота. Растворитель удаляют в вакууме и остаток перекристаллизовывают из смеси этанол/диэтиловый эфир, получая требуемую малеатную соль, 0,014 г (79%), в виде белого твердого вещества: т. пл. 168-169°С; 1Н ЯМР (300 МГц, CD3OD) δ 7.31-7.40 (м,3Н); 7.22-7.25 (м,2Н); 6.63 (с,3Н); 6.24 (с,3Н); 4.63 (т, 1Н, J=8.7 Гц); 4.40-4.51 (м,3Н); 3.73-3.79 (м,1Н); 3.43-3.53 (м,1Н); 3.12-3.27 (м,2Н); 3.06 (с,3Н); IR (KBr) 3448, 2923, 2364, 1578, 1484, 1355, 1258, 981, 868, 702, 574 см-1; CI MS m/z=266 [C18H19NO+H]+;
Пример 14
К перемешиваемому раствору подходящего аминного продукта, полученного с использованием методики стадии Н примера 12 (1,3 г, 4,3 ммоль), в безводном простом эфире (40 мл) в атмосфере азота добавляют 1 М раствор HCl в эфире (8,7 мл, 8,7 ммоль). Образовавшееся твердое вещество отфильтровывают, промывают эфиром и сушат, получая гидрохлоридную соль продукта в виде белого твердого вещества (1,4 г, 95%): т. пл. 240-243°С; 1Н ЯМР (500 МГц, CD3OD) δ 7.87 (д, J=2.2 Гц, 1Н); 7.45 (д, J=8.6 Гц, 1Н); 7.32-7.20 (м,2Н); 7.12 (с,1Н); 6.99 (дд, J=1.0, 2.23 Гц, 1Н); 4.88-4.74 (м,3Н); 3.90 (дд, J=12.2, 6.0 Гц,1Н); 3.62 (с,1Н); 3.15 (с,3Н); IR (KBr) 3423, 2935, 2547, 1610; CI MS m/z=300 [C18H15NO+H]+; Элементный анализ. Вычислено для С18Н15F2NO-HCl-0,20 Н2О: С, 63,70; Н, 4,87; N, 4,13. Найдено: С, 63,50; Н, 4,72; N, 4,06.
Пример 15
Подходящий ненасыщенный амин (320 г, 1,07 ммоль), полученный с использованием методики примера 12, стадия Н, обрабатывают в соответствии с условиями реакции, описанными в примере 13. Очисткой сырого остатка хроматографией (SiO2, EtOAc/гексаны, 1/1) выделяют свободный аминный продукт (230 мг, 71%) в виде белого твердого вещества: т. пл. 86-90°С; 1Н ЯМР (500 МГц, CDС13) δ 6.92-7.05 (м,3Н); 6.62 (д, J=8.3 Гц, 1Н); 6.55 (д, J=8.3 Гц, 1Н); 4.60 (т, J=8.6 Гц, 2Н); 4.12 (т, J=6.1 Гц, 1Н); 3.52 (с,2Н); 3.07 (т, J=8.6 Гц, 2Н); 2.90 (дд, J=11.4, 5.2 Гц, 1Н); 2.56 (дд, J=11.4, 7.4 Гц, 1Н); 2.42 (с,3Н); IR (KBr) 2940, 1609, 1517 см-1; CI MS m/z=302 [C18H17F2NO+H]+; Элементный анализ. Вычислено для С18Н17F2NO: С, 71,75; Н, 5,69; N, 4,65. Найдено: С, 71,50; Н, 5,61; N, 4,59.
Пример 16
К перемешиваемому раствору подходящего аминного продукта, полученного с использованием методики стадии Н примера 12 (1,6 г, 5,4 ммоль), в безводном эфире (50 мл) в атмосфере азота добавляют 1 М раствор HCl в эфире (10,7 мл, 10,7 ммоль). Образовавшееся твердое вещество фильтруют, промывают эфиром и перекристаллизовывают в метаноле, получая белое твердое вещество (950 мг, 50%): т. пл. 256-258°С; 1Н ЯМР (500 МГц, CD3OD) δ 7.90 (д, J=2.2 Гц, 1Н); 7.48 (д, J=8.6 Гц, 1Н); 6.91-6.99 (м,5Н); 4.75-4.84 (м,3Н); 3.92 (дд, J=12.5, 6.0 Гц, 1Н); 3.62 (ушир.с, 1Н); 3.15 (с,3Н); IR (KBr) 3424, 2467, 1624, 1597 см-1; MS (API) m/z=300 [C18H15F2NO+H]+; Элементный анализ. Вычислено для С18Н15F2NO-HCl-0,20 Н2О: С, 63,70; Н, 4,87; N, 4,13. Найдено: С, 63,50; Н, 4,72; N, 4,06.
Пример 17
Подходящий ненасыщенный амин (750 мг, 2,51 ммоль), полученный с использованием методики примера 12, стадия Н, обрабатывают в соответствии с условиями реакции, описанными в примере 13. Очищая сырой остаток хроматографией (SiO2, EtOAc/гексаны, 1/1), выделяют свободный аминный продукт (444 мг, 59%) в виде белого твердого вещества: т. пл. 107-109°С; 1Н ЯМР (500 МГц, CDС13) δ 6.56-6.75 (м,5Н); 4.61 (т, J=8.6 Гц, 2Н); 4.13 (т, J=5.9 Гц, 1Н); 3.54 (д, J=15.3 Гц, 1Н); 3.49 (д, J=15.3 Гц, 1Н); 3.08 (т, J=8.6 Гц, 2Н); 2.91 (дд, J=11.5, 5.5 Гц, 1Н); 2.60 (дд, J=11.5, 5.9 Гц, 1Н); 2.42 (с,3Н); IR (KBr) 3077, 1626, 1597 см-1; CI MS m/z=302 [C18H17F2NO+H]+; Элементный анализ. Вычислено для С18Н17F2NO: С, 71,75; Н, 5,69; N, 4,65. Найдено: С, 71,41; Н, 5,75; N, 4,42.Пример 20
Подходящий аминный продукт, полученный с использованием методики стадии Н примера 12 (2,8 г, 10,0 ммоль), растворяют в этиловом эфире (20 мл). Некоторое количество материала является нерастворимым, поэтому раствор декантируют от твердых веществ. Декантированный раствор обрабатывают 1 М HCl/Et2O (8,2 мл, 8,2 ммоль). Сразу образуется не совсем белый осадок. Твердое вещество отфильтровывают, получая 2,0 г вещества, которое перекристаллизовывают из смеси метанол/Et2O, получая гидрохлоридную соль (1,4 г, 56%): т. пл. 190-192°С; 1Н ЯМР (300 МГц, CD3OD) δ 7.87 (д, J=2.2 Гц, 1Н); 7.39 (д, J=8.7 Гц, 1Н); 7.35-7.26 (м,2Н); 7.12 (т, J=8.7 Гц, 2Н); 6.99 (д, J=1.4 Гц, 1Н); 6.79 (т, J=8.7 Гц, 1Н); 5.01-4.85 (м,1Н); 4.80-4.60 (м,1Н); 3.92-3.80 (м,1Н); 3.57 (т, J=12 Гц, 1Н); 3.34 (с,1Н); 3.17 (с,3Н); IR (KBr) 3422, 2926, 2550, 1580, 1224 см-1; CI MS m/z=282 [C18H16FNO+H]+; Элементный анализ. Вычислено для С18Н16FNO-HCl-0,75 Н2О: С, 65,26; Н, 5,63; N, 4,23. Найдено: С, 65,51; Н, 5,35; N, 4,14.
Пример 21
Подходящий ненасыщенный амин (512 г, 1,83 ммоль), полученный с использованием методики примера 12, стадия Н, обрабатывают в соответствии с условиями реакции, описанными в примере 13. Очисткой сырого остатка хроматографией (SiO2, EtOAc/гексаны, 1/1) продукт выделяют в виде свободного амина (200 мг, 38%) в виде светло-желтого твердого вещества: т. пл. 110-116°С; 1Н ЯМР (300 МГц, CDС13) δ 7.16-7.08 (м,2Н); 6.91 (т, J=8.7 Гц, 2Н); 6.60 (д, J=8.3 Гц, 1Н); 6.51 (д, J=8.3 Гц, 1Н); 4.59 (т, J=8.7 Гц, 2Н); 4.16 (т, J=6.9 Гц, 1Н); 3.59 (д, J=15.2 Гц, 1Н); 3.47 (д, J=15.2 Гц, 1Н); 3.08 (т, J=8.5 Гц, 2Н); 2.92 (дд, J=11.5, 5.5 Гц, 1Н); 2.50 (дд, J=11.5, 5.9 Гц, 1Н); 2.43 (с,3Н); IR (KBr) 2874, 2784, 1599, 1505, 1217 см-1; CI MS m/z=284 [C18H18FNO+H]+; Элементный анализ. Вычислено для С18Н18FNO2: С, 76,30; Н, 6,40; N, 4,94. Найдено: С, 75,96; Н, 6,43; N, 4,82.
Пример 22
К раствору подходящего продукта, аминоспирта, полученного с использованием методики стадии G примера 12 (2,5 г, 7,9 ммоль), в метиленхлориде (40 мл) при комнатной температуре в течение 10 мин добавляют метансульфоновую кислоту (10 мл, 150 ммоль). Реакционную смесь кипятят с обратным холодильником в атмосфере азота в течение ночи. После того, как смесь охладиться до комнатной температуры, добавляют 2 н. NaOH до рН ˜11 и образовавшийся раствор экстрагируют (3х) метиленхлоридом. Объединенные органические слои промывают насыщенным раствором соли, сушат над MgSO4 и концентрируют в вакууме. Остаток очищают хроматографией (SiO2, EtOAc/гексаны, 1/2), получая продукт в виде масла (1,2 г, 50%): 1Н ЯМР (500 МГц, CDС13) δ 7.63 (д, J=2.0 Гц, 1Н); 7.24 (д, J=6.0 Гц, 1Н); 7.20 (д, J=2.0 Гц, 1Н); 7.19 (с,2Н); 7.08 (д, J=5.6 Гц, 1Н); 6.80 (д, J=8.4 Гц, 1Н); 6.73 (д, J=1.4 Гц, 1Н); 4.30 (т, J=7.5 Гц, 1Н); 3.88 (д, J=13.0 Гц, 1Н); 3.85 (д, J=13.0 Гц, 1Н); 3.02 (дд, J=11.5, 7.5 Гц, 1Н); 2.66 (дд, J=11.5, 7.5 Гц, 1Н); 2.48 (с,3Н); IR (МеОН) 2950, 2778, 1593, 1432 см-1; CI MS m/z=298 [C18H16С1NO+H]+; Элементный анализ. Вычислено для С18Н16ClNO-HCl-0,1 H2O: С, 64,34; Н, 5,16; N, 4,17. Найдено: С, 63,98; Н, 5,07; N, 3,91.
Пример 23
Способ, описанный в примере 25, используют для получения соединения примера 23. Метансульфоновую кислоту (18 мл, 280 ммоль) добавляют при температуре окружающей среды к раствору подобного аминоспирта (3,6 г, 11,2 ммоль) в метиленхлориде (50 мл). Реакционную смесь кипятят с обратным холодильником в атмосфере азота. После охлаждения реакционной смеси до комнатной температуры ее делают основной (рН ˜11) 2 н. NaOH, смесь экстрагируют (3х) метиленхлоридом. Объединенные органические слои промывают насыщенным раствором соли, сушат над MgSO4 и концентрируют в вакууме. Остаток очищают хроматографией (SiO2, EtOAc/гексаны, 1/1), получая требуемый продукт, пример 21 (1,70 г, 51%) в виде белого порошка: т.пл. 78-80°С: 1Н ЯМР (500 МГц, CDС13) δ 7.12 (м,3Н); 7.08 (д, J=8.0 Гц, 1Н); 6.62 (д, J=8.2 Гц, 1Н); 6.56 (д, J=8.2 Гц, 1Н); 4.60 (т, J=8.6 Гц, 2Н); 4.16 (т, J=5.0 Гц, 1Н); 3.57 (д, J=15.3 Гц, 1Н); 3.50 (д, J=15.3 Гц, 1Н); 3.08 (т, J=8.6 Гц, 2Н); 2.94 (дд, J=11.4, 5.0 Гц, 1Н); 2.57 (дд, J=11.4, 7.8 Гц, 1Н); 2.43 (с,3Н); IR (СН2С12) 2940, 2784, 1594 см-1; CI MS m/z=300 [C18H18С1NO+H]+; Элементный анализ. Вычислено для С18Н18ClNO: С, 72,11; Н, 6,05; N, 4,67. Найдено: С, 71,87; Н, 6,09; N, 4,45, вместе с выделением 1,4 г исходного материала.
Пример 24
Подходящий аминный продукт, полученный с использованием методики стадии Н примера 12 (0,5 г, 3,0 ммоль), растворяют в этиловом эфире (10 мл) и обрабатывают раствором 1 М хлористоводородной кислоты в этиловом эфире (1,7 мл, 1,7 ммоль). Сразу образуется не совсем белый осадок, который фильтруют, получая продукт (320 мг, 60%): т. пл. 230-234°С; 1Н ЯМР (300 МГц, CD3OD) δ 7.87 (д, J=2.0 Гц, 1Н); 7.48-7.32 (м,3Н); 7.27 (д, J=8.4 Гц, 2Н); 6.99 (д, J=1.6 Гц, 1Н); 6.78 (д, J=8.7 Гц, 1Н); 4.95-4.67 (м,3Н); 3.93-3.78 (м,1Н); 3.58 (т, J=12.2 Гц, 1Н); 3.17 (с,3Н); IR (KBr) 3422, 2926, 2589, 1490, 1089 см-1; CI MS m/z=298 [C18H16С1NO+H]+.
Пример 25
Стадия А: К раствору N-метиламина (5,0 г, 31 ммоль, получен в примере 12, стадия Е) в этаноле (50 мл) добавляют 10% Pd/C (2,5 г) в атмосфере азота. Воздух из реакционного сосуда выкачивают и наполняют водородом, затем снова откачивают. Это повторяют более двух раз. Реакционный сосуд с водородом (45 фунт/кв. дюйм) помещают в вибратор Парра и встряхивают в течение 18 часов. Смесь фильтруют через слой целита и слой целита промывают метанолом. Фильтрат концентрируют в вакууме, получая N-метил-4-(2,3-дигидробензофуранил)амин (4,8 г, 94%) в виде желтого масла: 1Н ЯМР (300 МГц, CDС13) δ 7.10 (т, J=8.2 Гц, 1Н); 6.79 (д, J=8.3 Гц, 1Н); 6.67 (д, J=8.3 Гц, 1Н); 4.50 (т, J=8.6 Гц, 2Н); 3.65 (с,2Н); 3.07 (т, J=8.6 Гц, 2Н); 2.42 (с,3Н).
Стадия В: Раствор N-метил-4-(2,3-дигидробензофуранил)амин со стадии А (2,2 г, 13 ммоль) и триэтиламина (1,4 мл) в дихлорметане (25 мл) охлаждают на бане льда с водой. Добавляют 4'-хлорфенацилбромид (13,8 ммоль) и реакционной смеси дают нагреться до комнатной температуры. Реакционную смесь промывают водой и органический слой сушат над MgSO4, фильтруют и концентрируют, получая требуемый аминокетон в виде темно-оранжевого масла (3,7 г, 86% сырого продукта): 1Н ЯМР (300 МГц, CDС13) δ 7.82 (д, J=8.5 Гц, 2Н); 7.37 (д, J=8.4 Гц, 2Н); 7.07 (т, J=7.8 Гц, 1Н); 6.79 (д, J=7.6 Гц, 1Н); 6.72 (д, J=8.0 Гц, 1Н); 4.52 (т, J=8.8 Гц, 2Н); 3.74 (с,2Н); 3.61 (с,2Н); 3.16 (т, J=8.7 Гц, 2Н); 2.37 (с,3Н).
Стадия С: Аминокетон, полученный на стадии В (3,7 г, 12 ммоль), растворяют в метаноле (40 мл) и охлаждают на бане льда с водой. По порциям добавляют борогидрид натрия (0,44 г, 12 ммоль). Реакционную смесь перемешивают в течение 1 часа. Реакционную смесь концентрируют до половины первоначального объема. Добавляют воду (40 мл) и смесь экстрагируют (3х) дихлорметаном. Объединенные органические слои сушат над MgSO4, фильтруют и концентрируют, получая требуемый аминоспирт в виде светло-желтого масла (2,5 г, 67% сырого продукта): 1Н ЯМР (300 МГц, CDС13) δ 7.35-7.20 (м,4Н); 7.08 (т, J=7.8 Гц, 1Н); 6.78 (д, J=7.6 Гц, 1Н); 6.72 (д, J=8.0 Гц, 1Н); 4.70 (дд, J=8.6, 5.5 Гц, 1Н); 4.55 (т, J=8.6 Гц, 2Н); 3.65 (д, J=12.9 Гц, 1Н); 3.44 (д, J=12.9 Гц, 1Н); 3.18 (т, J=8.6 Гц, 2Н); 2.57-2.52 (м,2Н); 2.29 (с,3Н).
Стадия D: Аминоспирт (2,4 г, 7,5 ммоль, из стадии С) перемешивают в СН2Cl2 (40 мл) и в течение 5 минут добавляют CH3SO3H (9,8 мл). Реакционную смесь перемешивают при температуре окружающей среды до тех пор, пока исходный материал не будет обнаруживаться ЯМР-анализом (24 часа), затем раствор делают основным водным 2 н. NaOH. Слои разделяют и водный слой экстрагируют(2х) СН2Cl2. Органические экстракты объединяют, промывают насыщенным раствором соли, сушат над MgSO4, фильтруют и концентрируют в вакууме, получая коричневое твердое вещество, которое хроматографируют (SiO2, 20% EtOAc/гексаны) с получением требуемого продукта примера 23 (1,13 г, 50%): т. пл. 148-150°С; 1Н ЯМР (300 МГц, CDС13) δ 7.23 (д, J=8.5 Гц, 2Н); 7.11 (д, J=8.4 Гц, 2Н); 6.60 (д, J=8.3 Гц, 1Н); 6.54 (д, J=8.3 Гц, 1Н); 4.60 (т, J=8.7 Гц, 2Н); 4.16 (т, J=6.5 Гц, 1Н); 3.59 (д, J=15.2 Гц, 1Н); 3.47 (д, J=15.2 Гц, 1Н); 3.07 (т, J=9.0 Гц, 2Н); 2.93 (дд, J=11.3, 5.2 Гц, 1Н); 2.53 (дд, J=11.4, 8.0 Гц, 1Н); 2.42 (с,3Н); IR (KBr) 2944, 2788, 1480, 1253, 823 см-1; CI MS m/z=300 [C18H18С1NO+H]+; Элементный анализ. Вычислено для С18Н18ClNO: С, 72,11; Н, 6,05; N, 4,67. Найдено: С, 72,03; Н, 6,17; N, 4,56.
Пример 30
Охлажденный льдом раствор подходящего аминного продукта, полученного с использованием методики стадии Н примера 12 (450 мг, 1,44 ммоль), в СН2Cl2 (10 мл) обрабатывают 1 М HCl/Et2O (1,5 мл, 1,5 ммоль). Приблизительно через 30 минут образуется не совсем белый осадок. Раствор перемешивают при комнатной температуре в течение 1 часа и концентрируют в вакууме. Остаток растворяют в метаноле (10 мл) при 50°С, охлаждают до комнатной температуры и кристаллизацию начинают добавлением Et2O (20 мл). Раствор оставляют для кристаллизации на протяжении ночи. Эту процедуру повторяют несколько раз, получая гидрохлоридную соль в виде не совсем белого порошка (106 мг, 30%): т. пл. 279-284°С; 1Н ЯМР (300 МГц, CD3OD) δ 7.91-7.90 (м,1Н); 7.50-7.47 (м,1Н); 7.24-7.17 (м,2Н); 7.02-6.98 (м,2Н); 6.89-6.86 (м,1Н); 4.85-4.73 (м,3Н); 3.92 (дд, J=12.1, 6.1 Гц, 1Н); 3.70-3.60 (м,1Н); 3.15 (с,3Н); IR (KBr) 3424, 2933, 2466, 1606, 1590, 1443, 1137, 860 см-1; CI MS m/z=316 [C18H15С1FNO+H]+; Элементный анализ. Вычислено для С18Н15ClFNO-HCl-0,25 Н2О: С, 60,60; Н, 4,66; N, 3,93. Найдено: С, 60,30; Н, 4,79; N, 3,66.
Пример 32
Подходящий аминный продукт, полученный с использованием методики стадии Н примера 12 (0,88 г, 3,0 ммоль), растворяют в этиловом эфире (25 мл) и обрабатывают раствором 1 М хлористоводородной кислотой в этиловом эфире (3,4 мл, 3,4 ммоль). Сразу образуется не совсем белый осадок, который фильтруют, получая не совсем белое твердое вещество (795 мг, 80%): т. пл. 212-214°С; 1Н ЯМР (300 МГц, CD3OD) δ 7.88 (д, J=2.2 Гц, 1Н); 7.40 (д, J=8.5 Гц, 1Н); 7.16 (д, J=8.0 Гц, 2Н); 6.97-6.77 (м,4Н); 4.65-4.56 (м,1Н); 3.87-3.75 (м,2Н); 3.80 (с,3Н); 3.65-3.46 (м,2Н); 3.17 (с,3Н); IR (KBr) 3224, 2930, 2547, 1513, 1030 см-1; CI MS m/z=294 [C19H19NO+H]+; Элементный анализ. Вычислено для С19Н19CNO2-HCl-0,25 Н2О: С, 68,26; Н, 6,18; N, 4,19. Найдено: С, 68,01; Н, 6,20; N, 3,93.
Пример 33
Подходящий ненасыщенный амин (660 мг, 2,26 ммоль), полученный с использованием методики примера 12, стадия Н, обрабатывают в соответствии с условиями реакции, описанными в примере 13. При очистке сырого остатка хроматографией (SiO2, EtOAc/гексаны, 1/1) продукт выделяют в виде свободного амина (220 мг, 33%) в виде светло-желтого твердого вещества: т. пл. 119-121°С; 1Н ЯМР (300 МГц, CDС13) δ 7.08 (д, J=8.4 Гц, 5Н); 6.80 (д, J=8.5 Гц, 2Н); 6.62 (д, J=8.3 Гц, 1Н); 6.51 (д, J=8.3 Гц, 1Н); 4.59 (т, J=8.7 Гц, 2Н); 4.15 (т, J=6.9 Гц, 1Н); 3.78 (с, 3Н); 3.61 (д, J=15.2 Гц, 1Н); 3.43 (д, J=15.2 Гц, 1Н); 3.08 (т, J=8.5 Гц, 2Н); 2.92 (дд, J=11.5, 5.5 Гц, 1Н); 2.50 (дд, J=11.5, 5.9 Гц, 1Н); 2.42 (с,3Н); IR (KBr) 2785, 2762, 1610, 1509, 1251 см-1; CI MS m/z=296 [C19H21NO2+H]+; Элементный анализ. Вычислено для С19Н21NO2: С, 77,26; Н, 7,17; N, 4,74. Найдено: С, 76,93; Н, 7,31; N, 4,57.
Пример 34
Свободное основание продукта примера 12, стадия Н (0,3 г, 1,14 ммоль) в виде раствора в безводном тетрагидрофуране при -78°С обрабатывают раствором н-BuLi (0,91 мл, 2,5 М в гексанах, 2,3 ммоль) в атмосфере азота. После перемешивания в течение двух часов по каплям добавляют иодметан (0,17 мл, 2,7 ммоль). Образовавшуюся смесь перемешивают при -78°С в течение двух часов, затем позволяют ей нагреться до комнатной температуры. Смесь разбавляют водой и экстрагируют (3х) диэтиловым эфиром. Органические слои объединяют, промывают насыщенным раствором соли, сушат над безводным сульфатом натрия, фильтруют и растворитель удаляют в вакууме. Остаток очищают колоночной хроматографией на силикагеле с использованием медленного градиента от 0 до 10% метанола в метиленхлориде, получая метилзамещенный бензофуран, 203 мг (64%) в виде желтого масла: 1Н ЯМР (300 МГц, CDС13) δ 7.15-7.28 (м,5Н); 7.10 (д, 1Н, J=8.7 Гц); 6.70 (д, 1Н, J=8.5 Гц); 6.28-6.29 (м,1Н); 4.29-4.34 (м,1Н); 3.86 (д, 1Н, J=15.2 Гц); 3.70 (д, 1Н, J=15.2 Гц); 3.00-3.05 (м,1Н); 2.58-2.65 (м,1Н); 2.44 (с,3Н); 2.40 (с,3Н). Свободное основание (0,23 г, 0,73 ммоль) и малеиновую кислоту (0,085 г, 0,073 ммоль) в течение 5 минут растворяют в абсолютном этаноле (10 мл) и кипятят с обратным холодильником в атмосфере азота, затем дают охладиться до комнатной температуры. Смесь концентрируют в вакууме до объема приблизительно 2 мл, затем добавляют диэтиловый эфир, что вызывает образование кристаллов. Выделение твердого вещества вакуумным фильтрованием дает не совсем белое твердое вещество. Твердое вещество перекристаллизовывают из смеси этанол/диэтиловый эфир, затем из этанола, получая нужную малеатную соль, 0,043 г (15%), в виде белого кристаллического твердого вещества: т. пл. 187-192°С; 1Н ЯМР (300 МГц, CD3OD) δ 7.23-7.40 (м,6Н); 6.73 (д, 1Н, J=8.6 Гц); 6.57 (с,1Н); 6.21 (с,2Н); 4.63-4.80 (м,3Н); 3.83-3.88 (м,1Н); 3.53-3.61 (м,1Н); 3.12 (с,3Н); 2.48 (с,3Н); IR (KBr) 3448, 2548, 1584, 1495, 1354, 1270, 1195, 1078, 936, 866, 808, 704, 656, 583, 510 см-1; CI MS m/z=278 [C19H19NO+H]+; Элементный анализ. Вычислено для С19Н19NO-С4Н4О4-0,5 Н2О: С, 68,84; Н, 6,01; N, 3,48. Найдено: С, 68,49; Н, 5,84; N, 3,41.
Пример 36
Стадия А: Свободное основание продукта примера 12, стадия Н (1,0 г, 3,91 ммоль), в виде раствора в безводном тетрагидрофуране при -78°С обрабатывают раствором н-BuLi (3,3 мл, 2,5 М раствор в гексанах, 8,2 ммоль) в атмосфере азота. После перемешивания в течение одного часа по каплям добавляют диметилформамид (0,70 мл, 9,0 ммоль). Образовавшуюся смесь перемешивают при -78°С в течение двух часов, затем дают нагреться до комнатной температуры. Смесь разбавляют водой и экстрагируют (3х) диэтиловым эфиром. Органические слои объединяют, промывают насыщенным раствором соли, сушат над безводным сульфатом натрия, фильтруют и растворитель удаляют в вакууме. Остаток очищают колоночной хроматографией на силикагеле (гексаны/этилацетат, 1:1), получая ожидаемый альдегид, 430 мг (38%) в виде светло-желтого масла: 1Н ЯМР (300 МГц, CDС13) δ 9.86 (с,1Н); 7.54 (с,1Н); 7.17-7.33 (м,6Н); 7.05 (д, 1Н, J=8.7 Гц); 4.32-4.36 (м,1Н); 4.00 (д, 1Н, J=15.5 Гц); 3.83 (д, 1Н, J=15.5 Гц); 3.07-3.13 (м,1Н); 2.67 (дд, 1Н, J=8.2, 11.5 Гц); 2.51 (с,3Н); CI MS m/z=292 [C19H17NO2+H]+.
Стадия В: Продукт примера 36, стадия А (0,07 г, 0,23 ммоль) обрабатывают борогидридом натрия (0,02 г, 0,46 ммоль) в охлажденном метаноле (20 мл). Реакционной смеси дают нагреться до комнатной температуры и перемешивают в течение 1 часа, гасят водой и экстрагируют (3х) метиленхлоридом. Органические слои объединяют, промывают насыщенным раствором соли, сушат над безводным сульфатом натрия, фильтруют и растворитель удаляют в вакууме, получая спирт, 0,07 г (100%) в виде желтого масла; 1Н ЯМР (300 МГц, CDС13) δ 7.17-7.32 (м,6Н); 6.79 (д, 1Н, J=8.5 Гц); 6.57 (с,1Н); 4.76 (с,2Н); 4.33-4.38 (м,1Н); 3.89 (д, 1Н, J=15.2 Гц); 3.72 (д, 1Н, J=15.2 Гц); 3.06-3.11 (м,1Н); 2.63 (дд, 1Н, J=8.6, 11.4 Гц); 2.50 (с,3Н); CI MS m/z=294 [C19H19NO2+H]+. Свободное основание (0,03 г, 0,10 ммоль) и хлористоводородную кислоту (1 М раствор в диэтиловом эфире, 0,5 мл) растворяют в диэтиловом эфире (4 мл). Образовавшийся не совсем белый осадок выделяют вакуумным фильтрованием и сушат при пониженном давлении, получая нужную гидрохлоридную соль, 0,03 г (72%), т. пл. 149-162°С; 1Н ЯМР (300 МГц, CD3OD) δ 7.25-7.42 (м,6Н); 6.78-6.86 (м,2Н); 4.64-4.74 (м,5Н); 2.85-2.91 (м,1Н); 3.52-3.68 (м,1Н); 3.15 (с,3Н); IR (KBr) 3375, 2500, 1456, 1023, 811, 702 см-1; CI MS m/z=294 [C19H19NO2+H]+; Элементный анализ. Вычислено для С19Н19CNO2-HCl-0,75 Н2О: С, 66,47; Н, 6,31; N, 4,08. Найдено: С, 66,13; Н, 6,54; N, 3,82.
Пример 38
Стадия А: К смеси литийалюминийгидрида (1,3 г, 34 ммоль) в ТГФ (200 мл) добавляют при комнатной температуре метил-4-индолкарбоксилат (3,0 г, 17 ммоль) в ТГФ (100 мл). Реакционную смесь перемешивают при комнатной температуре в течение 2 часов и затем гасят этилацетатом. Смесь обрабатывают водой (1,3 мл), 15% NaOH (1,3 мл) и водой (3,9 мл) и затем фильтруют. Фильтрат концентрируют в вакууме, получая сырой 4-(гидроксиметил)индол (2,5 г, 99%): 1Н ЯМР (500 МГц, CDС13) δ 8.29 (ушир.с,1Н); 7.34 (д, J=9.0 Гц, 1Н); 7.16-7.22 (м,2Н); 7.12 (д, J=7.0 Гц, 1Н); 6.67 (т, J=1.0 Гц, 1Н); 4.98 (д, J=4.2 Гц, 2Н); CI MS m/z=147 [C9H9NO+H]+.
Стадия В: Перрутенат тетрапропиламмония (0,3 г, 0,85 ммоль) добавляют порциями к смеси продукта в виде спирта со стадии А (2,5 г, 17 ммоль), N-оксида N-метилморфолина (3,0 г, 25 ммоль) и молекулярных сит 4 А (3,0 г) в безводном метиленхлориде (30 мл) при комнатной температуре. Смесь перемешивают при комнатной температуре в атмосфере азота в течение 1 часа и затем фильтруют. Фильтрат концентрируют в вакууме и остаток очищают хроматографией (SiO2, СН2Cl2), получая индол-4-альдегид в виде белого порошка (2,0 г, 80%): 1Н ЯМР (300 МГц, CDС13) δ 10.2 (с,1Н); 8.52 (ушир.с,1Н); 7.64-7.69 (м,2Н); 7.31-7.44 (м,3Н); CI MS m/z=146 [C9H7NO+H]+.
Стадия С: К раствору альдегида из стадии В (2,0 г, 14 ммоль) в метаноле (100 мл) при комнатной температуре добавляют 40% метиламин в воде (2,27 мл, 27,6 ммоль) в течение 10 минут. Смесь перемешивают при комнатной температуре в атмосфере азота на протяжении ночи и затем охлаждают до 0°С. Добавляют борогидрид натрия (1,05 г, 27,6 ммоль). Реакционную смесь медленно нагревают до комнатной температуры в течение 2 часов. Большую часть метанола удаляют в вакууме и остаток разбавляют водой и экстрагируют (3х) эфиром. Объединенные органические слои экстрагируют 2 н. HCl (100 мл). Слой HCl делают основным (рН ˜11) 2 н. NaOH и экстрагируют (3х) метиленхлоридом. Объединенные органические слои промывают насыщенным раствором соли, сушат над Na2SO4 и концентрируют в вакууме, получая сырой
4-(аминометил)индол в виде белого порошка (1,95 г, 88%): 1Н ЯМР (300 МГц, CDС13) δ 8.29 (с,ушир,1Н); 7.31 (д, J=8.0 Гц, 1Н); 7.22 (т, J=2.7 Гц, 1Н); 7.16 (т, J=8.0, 7.3 Гц, 1Н); 7.08 (д, J=7.3 Гц, 1Н); 6.64 (т, J=2.0 Гц, 1Н); 4.06 (с,2Н); 2.51 (с,3Н); CI MS m/z=160 [C10H12N2+H]+.
Стадия D: К смеси аминного продукта со стадии С (1,0 г, 6,3 ммоль) и 2-бромацетофенона (1,2 г, 6,3 ммоль) в безводном метиленхлориде (20 мл) при комнатной температуре добавляют триэтиламин (0,96 мл, 6,9 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 4 часов и обрабатывают водой (20 мл). Органический слой отделяют и водный слой экстрагируют (2х) метиленхлоридом. Объединенные органические слои промывают насыщенным раствором соли, сушат над MgSO4 и концентрируют в вакууме. Остаток очищают хроматографией (SiO2, EtOAc/гексаны, 1:2), получая N-метил-α-аминокетон (1,5 г, 86%): 1Н ЯМР (300 МГц, CDС13) δ 8.32 (ушир.с,1Н); 7.90-7.93 (м,2Н); 7.52 (м,1Н); 7.31-7.39 (м,3Н); 7.08-7.19 (м,3Н); 6.72 (т, J=1.0 Гц, 1Н); 3.96 (с,2Н); 3.81 (с,2); 2.41 (с,3Н).
Стадия Е: К раствору продукта со стадии D, N-метил-α-аминокетона (1,5 г, 5,4 ммоль), в метаноле (50 мл) при 0°С в течение 5 минут добавляют борогидрид натрия (410 мг, 10,8 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 2 часов. Большую часть метанола удаляют в вакууме и остаток разбавляют водой (100 мл) и экстрагируют (3х) метиленхлоридом. Объединенные органические слои промывают насыщенным раствором соли, сушат над Na2SO4, фильтруют и концентрируют в вакууме, получая сырой аминоспирт в виде светло-желтого масла (1,5 г, 99%): 1Н ЯМР (500 МГц, CDС13) δ 8.23 (ушир.с,1Н); 7.21-7.36 (м,7Н); 7.15 (т, J=7.0 Гц, 1Н); 7.04 (д, J=7.0 Гц, 1Н); 6.71 (т, J=1.0 Гц, 1Н); 4.73 (дд, J=10.5, 3.4 Гц, 1Н); 4.03 (д, J=12.8 Гц, 1Н); 3.80 (д, J=12.8 Гц, 1Н); 2.65 (дд, J=12.4, 10.5 Гц, 1Н); 2.58 (дд, J=12.4, 3.4 Гц, 1Н); 2.38 (с, 3Н); CI MS m/z=281 [C18H20N2O+H]+.
Стадия F: К раствору аминоспирта со стадии Е (1,37 г, 4,89 ммоль), в метиленхлориде (40 мл) добавляют метансульфоновую кислоту (7,93 мл, 122 ммоль) при комнатной температуре в течение 10 минут. Реакционную смесь перемешивают при комнатной температуре в атмосфере азота в течение 24 часов и затем делают основной (рН ˜11) 2 н. NaOH. Органический слой отделяют и водный слой экстрагируют (2х) метиленхлоридом. Объединенные органические слои промывают насыщенным раствором соли, сушат над MgSO4 и концентрируют в вакууме. Остаток очищают хроматографией (SiO2, EtOAc/гексаны, 1:1), получая нужный продукт примера 36, пирролоконденсированный тетрагидроизохинолин, в виде белого порошка(450 мг, 35%): т. пл. 142-144°С; 1Н ЯМР (300 МГц, CDС13) δ 8.23 (ушир.с,1Н); 7.16-7.27 (м,6Н); 7.11 (д, J=8.5 Гц, 1Н); 6.70 (д, J=8.5 Гц, 1Н); 6.49 (т, J=1.5 Гц, 1Н); 4.37 (т, J=5.5 Гц, 1Н); 4.04 (д, J=15.2 Гц, 1Н); 3.85 (д, J=15.2 Гц, 1Н); 3.08 (дд, J=11.5, 5.5 Гц, 1Н); 2.66 (дд, J=11.5, 8.2 Гц, 1Н); 2.51 (с, 3Н); CI MS m/z=263 [C18H18N2O+H]+; IR (KBr) 3410, 3027, 2861, 2363, 1600, 1493 см-1; Элементный анализ. Вычислено для С18Н18N2-0,1 Н2О: С, 81,84; Н, 6,94; N, 10,60. Найдено: С, 81,94; Н, 7,10; N, 10,46.
Пример 39
К раствору индолового продукта примера 38, стадии F (182 мг, 0,694 ммоль), и диметилоксалата (90 мг, 0,76 ммоль) в ДМФ (5 мл) в виде одной порции при комнатной температуре в атмосфере азота добавляют трет-бутоксид калия (86 мг, 0,76 ммоль). Реакционную смесь кипятят с обратным холодильником в течение 30 мин и затем охлаждают до комнатной температуры. Смесь разбавляют водой (50 мл) и экстрагируют (3х) метиленхлоридом. Объединенные органические слои промывают насыщенным раствором соли, сушат над MgSO4 и концентрируют в вакууме. Остаток очищают хроматографией (SiO2, EtOAc/гексаны, 1:1), получая N-метилиндол примера 37 в виде белого твердого вещества (180 мг, 92%): т. пл. 106-108°С; 1Н ЯМР (500 МГц, CDС13) δ 7.19-7.28 (м,5Н); 7.05 (д, J=8.5 Гц, 1Н); 7.03 (д, J=3.0 Гц, 1Н); 6.73 (д, J=8.5 Гц, 1Н); 6.41 (дд, J=3.0, <1 Гц, 1Н); 4.37 (т, J=6.3 Гц, 1Н); 4.00 (д, J=15.1 Гц, 1Н); 3.85 (д, J=15.1 Гц, 1Н); 3.75 (с,3Н); 3.08 (дд, J=11.4, 5.4 Гц, 1Н); 2.66 (дд, J=11.4, 8.1 Гц, 1Н); 2.50 (с, 3Н); CI MS m/z=277 [C19H20N2+H]+; IR (KBr) 3050, 2939, 2783, 1487, 1451 см-1; Элементный анализ. Вычислено для С19Н20N2-0,1 Н2О: С, 82,04; Н, 7,32; N, 10,07. Найдено: С, 82,06; Н, 7,50; N, 9,85.
Пример 40
К раствору индолового продукта примера 38 (150 мг, 0,572 ммоль) и диэтилоксалата (92 мг, 0,63 ммоль) в ДМФ (5 мл) в виде одной порции при комнатной температуре в атмосфере азота добавляют трет-бутоксид калия (71 мг, 0,63 ммоль). Реакционную смесь кипятят с обратным холодильником в атмосфере азота в течение 1 часа и затем охлаждают до комнатной температуры. Смесь разбавляют водой (50 мл) и экстрагируют (3х) метиленхлоридом. Объединенные органические слои промывают насыщенным раствором соли, сушат над Na2SO4, фильтруют и концентрируют в вакууме. Остаток очищают хроматографией (SiO2, EtOAc/гексаны, 1:1), получая продукт примера 38, N-этилиндол, (144 мг, 86%): 1Н ЯМР (500 МГц, CDС13) δ 7.18-7.28 (м,5Н); 7.09 (д, J=3.2 Гц, 1Н); 7.07 (д, J=8.5 Гц, 1Н); 6.71 (д, J=8.5 Гц, 1Н); 6.42 (дд, J=3.2, <1 Гц, 1Н); 4.36 (т, J=8.0, 5.4 Гц, 1Н); 4.12 (кв, J=7.3 Гц, 2Н); 4.00 (д, J=15.1 Гц, 1Н); 3.85 (д, J=15.1 Гц, 1Н); 3.07 (дд, J=11.3, 5.4 Гц, 1Н); 2.66 (дд, J=11.3, 8.0 Гц, 1Н); 2.50 (с,3Н); 1.44 (т, J=7.3 Гц, 3Н).
Пример 41
К раствору индолового продукта примера 38 (150 мг, 0,572 ммоль) и дибензилоксалата (170 мг, 0,63 ммоль) в ДМФ (5 мл) в виде одной порции при комнатной температуре в атмосфере азота добавляют трет-бутоксид калия (71 мг, 0,63 ммоль). Реакционную смесь кипятят с обратным холодильником в атмосфере азота в течение 3 часов и затем охлаждают до комнатной температуры. Смесь разбавляют водой (50 мл) и экстрагируют (3х) метиленхлоридом. Объединенные органические слои промывают насыщенным раствором соли, сушат над Na2SO4, фильтруют и концентрируют в вакууме. Остаток очищают хроматографией (SiO2, EtOAc/гексаны, 1:1), получая N-бензилиндоловый продукт примера 39 (161 мг, 80%): 1Н ЯМР (500 МГц, CDС13) δ 7.09-7.29 (м,11Н); 7.01 (д, J=8.5 Гц, 1Н); 6.67 (д, J=8.5 Гц, 1Н); 6.48 (дд, J=3.1, <1 Гц, 1Н); 5.26 (с,2Н); 4.34 (т, J=8.1, 5.4 Гц, 1Н); 4.00 (д, J=15.1 Гц, 1Н); 3.86 (д, J=15.1 Гц, 1Н); 3.06 (дд, J=11.3, 5.4 Гц, 1Н); 2.67 (дд, J=11.3, 8.1 Гц, 1Н); 2.50 (с,3Н).
Пример 42
К раствору индолового продукта (200 мг, 0,763 ммоль) примера 38, стадия F, в уксусной кислоте (5 мл) добавляют по частям при комнатной температуре в течение 5 минут цианоборогидрид натрия (240 мг, 3,82 ммоль). Реакционную смесь перемешивают в атмосфере азота в течение 4 часов и затем большую часть уксусной кислоты удаляют в вакууме. Остаток разбавляют метиленхлоридом (100 мл), промывают 2 н. NaOH и насыщенным раствором соли, сушат над Na2SO4, фильтруют и концентрируют в вакууме. Остаток очищают хроматографией (SiO2, EtOAc/метанол, 8:1), получая индолин примера 40, в виде белого твердого вещества (176 мг, 87%): т. пл. 84-86°С: 1Н ЯМР (500 МГц, CDС13) δ 7.17-7.27 (м,5Н); 6.53 (д, J=8.0 Гц, 1Н); 6.40 (д, J=8.0 Гц, 1Н); 4.17 (дд, J=8.2, 5.4 Гц, 1Н); 3.62 (д, J=15.0 Гц, 1Н); 3.58 (т, J=8.4 Гц, 2Н); 3.44 (д, J=15.0 Гц, 1Н); 2.96 (дд, J=11.3, 5.4 Гц, 1Н); 2.91 (дт, J=8.4, 4.0 Гц, 2Н); 2.54 (дд, J=11.3, 8.2 Гц, 1Н); 2.42 (с,3Н); CI MS m/z=265 [C18H20N2+H]+; IR (KBr) 3241, 2924, 2873, 1611, 1486 см-1; Элементный анализ. Вычислено для С18Н20N2-0,1 Н2О: С, 81,23; Н, 7,65; N, 10,52. Найдено: С, 80,87; Н, 7,46; N, 10,48.
Пример 43
К раствору индолина примера 42 (110 мг, 0,420 ммоль) и уксусной кислоты (0,1 мл) в метаноле (4 мл) по каплям при комнатной температуре добавляют 37% водный формальдегид (0,04 мл, 0,5 ммоль). Реакционную смесь перемешивают при комнатной температуре в атмосфере азота в течение 1 часа и затем по порциям при комнатной температуре добавляют цианоборогидрид натрия (66 мг, 1,05 ммоль). Смесь перемешивают при комнатной температуре в атмосфере азота в течение 3 часов и затем гасят 2 н. NaOH и экстрагируют (3х) метиленхлоридом. Объединенные органические слои промывают насыщенным раствором соли, сушат над Na2SO4, фильтруют и концентрируют в вакууме. Остаток очищают хроматографией (SiO2, EtOAc/метанол, 10:1), получая N-метилиндолиновый продукт, пример 41, в виде белого твердого вещества (92 мг, 80%): т. пл. 88-90°С; 1Н ЯМР (500 МГц, CDС13) δ 7.17-7.28 (м,5Н); 6.59 (д, J=8.0 Гц, 1Н); 6.26 (д, J=8.0 Гц, 1Н); 4.17 (дд, J=8.5, 5.4 Гц, 1Н); 3.62 (д, J=15.0 Гц, 1Н); 3.43 (д, J=15.0 Гц, 1Н); 3.31 (м,2Н); 2.96 (дд, J=11.3, 5.4 Гц, 1Н); 2.82 (м,2Н); 2.70 (с,3Н); 2.54 (дд, J=11.3, 8.5 Гц, 1Н); 2.42 (с,3Н); CI MS m/z=279 [C19H22N2+H]+; IR (KBr) 3020, 2940, 2773, 1610, 1487 см-1; Элементный анализ. Вычислено для С19Н22N2-0,1 Н2О: С, 81,45; Н, 7,99; N, 10,00. Найдено: С, 81,21; Н, 8,00; N, 9,74.
Пример 45
К раствору подходящего аминоспирта, полученного с использованием методики стадии Е примера 38 (174 мг, 0,550 ммоль), растворяют в СН2Cl2 (11 мл) в 50 мл колбе, снабженной холодильником, в атмосфере азота, снабженной холодильником. Смесь охлаждают до 0°С при быстром перемешивании и по каплям добавляют MeSO3H (1,8 мл, 28 ммоль) и смесь перемешивают в течение 30 минут при нагревании до комнатной температуры, затем кипятят с обратным холодильником в течение 48 часов. Смесь охлаждают до комнатной температуры, нейтрализуют 2 н. NaOH, затем экстрагируют (3х) EtOAc. Объединенные органические экстракты сушат над Na2SO4, фильтруют и концентрируют в вакууме. Сырой остаток очищают хроматографией на силикагеле (градиент 30-45% EtOAc/гексаны), получая нужный индоловый продукт (19 мг, 12%) в виде оранжевого твердого вещества: т. пл. 164-169°С; 1Н ЯМР (300 МГц, CDС13) δ 8.18 (ушир.с,1Н); 7.15-7.24 (м,2Н); 6.92-7.10 (м,3Н); 6.70 (д, J=8.4 Гц, 1Н); 6.50-6.54 (м,1Н); 4.29 (т, J=5.8 Гц, 1Н); 3.92 (д, J=4.8 Гц, 1Н); 3.02 (дд, J=11.3, 5.1 Гц, 1Н); 2.67 (дд, J=11.3, 6.8 Гц, 1Н); 2.49 (с,3Н).
Следует отметить, что выделяют также 55 мг (32%) исходного материала. На основе выделенного исходного материала выход продукта примера 45 составляет 17%.
Пример 46
N-Метилиндолиновый продукт примера 47 (70 мг, 0,22 ммоль) растворяют в толуоле (9 мл) в 50 мл колбе, снабженной холодильником, в атмосфере N2. Добавляют MnO2 (199 мг, 2,3 ммоль) и смесь кипятят с обратным холодильником в течение 1,5 часа. Смесь охлаждают до комнатной температуры, пропускают через слой целита и слой несколько раз промывают обильными количествами МеОН. Фильтрат концентрируют в вакууме и остаток очищают хроматографией на силикагеле (градиент 25-35% EtOAc/гексан), получая N-метилиндолиновый продукт (39 мг, 57%) в виде оранжевого масла: 1Н ЯМР (300 МГц, CDС13) δ 6.93-7.11 (м,5Н); 6.72 (д, J=8.5 Гц, 1Н); 6.42 (д, J=3.1 Гц, 1Н); 4.29 (т, J=5.8 Гц, 1Н); 3.84-3.96 (м,2Н); 3.77 (с,3Н); 2.99 (дд, J=11.3, 5.0 Гц, 1Н); 2.67 (дд, J=11.3, 6.7 Гц, 1Н); 2.49 (с,3Н); ESI MS m/z=313 [C19H18F2N2+H]+.
Пример 47
Стадия А: Продукт, подходящий аминоспирт (730 мг, 2,31 ммоль), полученный с использованием методики примера 38, стадия Е, растворяют в ледяной НОАс (23 мл) в 100 мл колбе в атмосфере N2. В виде одной порции добавляют NaBH3CN (0,76 г, 12 ммоль) и смесь перемешивают в течение 2 часов. Смесь выливают в 200 мл быстро перемешиваемой смеси льда и воды и раствор делают основным концентрированным NH4OH. После перемешивания в течение 30 минут смесь экстрагируют (4х) СН2Cl2. Объединенные органические экстракты сушат над Na2SO4, фильтруют и концентрируют в вакууме. Сырой остаток очищают хроматографией на силикагеле (градиент 25-50% EtOAc/гексаны), получая нужный индолиновый продукт (434 мг, 59%) в виде не совсем белого твердого вещества: 1Н ЯМР (300 МГц, CDС13) δ 6.98-7.23 (м,4Н); 6.62 (дд, J=16.2, 7.7 Гц, 2Н); 4.67 (т, J=7.0 Гц, 1Н); 3.75-4.10 (ушир.с,2Н); 3.64 (д, J=12.8 Гц, 1Н); 3.58 (т, J=8.3 Гц, 2Н); 3.45 (д, J=12.7 Гц, 1Н); 3.04 (т, J=8.3 Гц, 2Н); 2.51 (д, J=7.0 Гц, 2Н); 2.30 (с,3Н); CI MS m/z=315 [C18H20N2O+H]+.
Следует отметить, что выделяют также 70 мг (10%) исходного материала. На основе выделенного исходного материала выход индолина составляет 65%.
Стадия В: Индолинаминоспирт стадии А данного примера (165 мг, 0,518 ммоль) растворяют в дихлорметане (5 мл) в 50 мл колбе с атмосферой N2. В виде одной порции добавляют MeSO3H (1,7 мл, 26 ммоль) и смесь быстро перемешивают при кипячении с обратным холодильником. Спустя 5 часов смесь охлаждают до комнатной температуры, выливают в 100 мл ледяной воды и делают основным 10% NaOH. После перемешивания в течение 30 минут смесь экстрагируют (4х) СН2Cl2. Объединенные органические экстракты сушат над Na2SO4, фильтруют и концентрируют в вакууме. Сырой остаток очищают хроматографией на силикагеле (градиент 1-4% МеОН/СН2Cl2), получая продукт примера 45 (81 мг, 52%) в виде не совсем белого твердого вещества: т. пл. 45-51°С; 1Н ЯМР (300 МГц, CDС13) δ 6.96-7.08 (м,2Н); 6.89-6.94 (м,1Н); 6.52 (д, J=8.0 Гц, 1Н); 6.43 (д, J=8.0 Гц, 1Н); 4.10 (т, J=6.0 Гц, 1Н); 3.68 (ушир.с,1Н); 3.59 (т, J=8.4 Гц, 2Н); 3.50 (с,2Н); 2.85-2.93 (м,3Н); 2.54 (дд, J=11.4, 7.2 Гц, 1Н); 2.41 (с,3Н); ESI MS m/z=301 [C18H18F2N2+H]+; Элементный анализ. Вычислено для С18Н18F2N2: С, 71,98; Н, 6,04; N, 9,33. Найдено: С, 72,14; Н, 6,69; N, 8,45.
Пример 48
Индолиновый продукт примера 47, стадия В (16 мг, 0,049 ммоль), растворяют в МеОН (2 мл) в 25 мл колбе в атмосфере N2. Добавляют каталитическое количество НОАс (1 капля) и водный формальдегид (15 мкл, 0,15 ммоль) и смесь перемешивают в течение 1 часа. Добавляют NaBH3CN (16 мг, 0,25 ммоль) и смесь перемешивают в течение дополнительного 1 часа. Смесь разбавляют СН2Cl2 (50 мл), затем промывают последовательно 0,5 н. NaOH (25 мл) и насыщенным водным NaCl (25 мл). Органический слой сушат над Na2SO4), фильтруют и концентрируют в вакууме, получая
N-метилиндолиновый продукт (14 мг, 87%) в виде оранжевого масла: 1Н ЯМР (300 МГц, CDС13) δ 6.93-7.09 (м,3Н); 6.59 (д, J=8.1 Гц, 1Н); 6.29 (д, J=8.1 Гц, 1Н); 4.12 (т, J=5.8 Гц, 1Н); 3.52 (с,2Н); 3.30-3.37 (м,2Н); 2.91 (дд, J=11.3, 5.1 Гц, 1Н); 2.82 (т, J=8.0 Гц, 2Н); 2.73 (с,3Н); 2.56 (дд, J=11.3, 7.3 Гц, 1Н); 2.42 (с,3Н); CI MS m/z=315 [C19H20F2N2+H]+.
Пример 49
Подходящий индоловый продукт, полученный с использованием методики примера 38, стадия F (41 мг, 0,137 ммоль), и диметилоксалат (21 мг, 0,17 ммоль) растворяют в ДМФ (2 мл) при быстром перемешивании в 25 мл колбе, снабженной холодильником, в атмосфере N2. Добавляют трет-бутоксид калия (22 мг, 0,19 ммоль) и смесь кипятят с обратным холодильником в течение 1 часа. Смесь охлаждают до комнатной температуры, разбавляют водой (100 мл) и экстрагируют (4х) смесью гексан/эфир, 1:1. Объединенные органические экстракты сушат над Na2SO4, фильтруют и концентрируют в вакууме. Сырой остаток очищают хроматографией на силикагеле (25% EtOAc/гексаны), получая N-метилиндоловый продукт, (20 мг, 47%) в виде желтого масла, которое отверждается при стоянии: т. пл. 110-112°С; 1Н ЯМР (300 МГц, CDС13) δ 7.11 (д, J=8.3 Гц, 1Н); 7.06 (д, J=3.1 Гц, 1Н); 6.72-6.80 (м,3Н); 6.59-6.67 (м,1Н); 6.42 (д, J=3.1 Гц, 1Н); 4.30 (т, J=5.9 Гц, 1Н); 3.95 (д, J=15.3 Гц, 1Н); 3.85 (д, J=15.2 Гц, 1Н); 3.77 (с,3Н); 3.00 (дд, J=11.3, 5.1 Гц, 1Н); 2.71 (дд, J=11.3, 6.6 Гц, 1Н); 2.49 (с,3Н); CI MS m/z=313 [C19H18F2N2+H]+; Элементный анализ. Вычислено для С19Н18F2N2: С, 73,06; Н, 5,81; N, 8,97. Найдено: С, 72,93; Н, 6,08; N, 8,13.
Пример 50
Аналогичный индолинаминоспирт, который получают по методике, описанной в примере 47, стадия А (199 мг, 0,625 ммоль), растворяют в дихлорэтане (6 мл) в 50 мл колбе, снабженной холодильником, с атмосферой N2. В виде одной порции добавляют MeSO3H ((2,0 мл, 31 ммоль) и смесь энергично перемешивают при кипячении с обратным холодильником на протяжении ночи. Смесь охлаждают до комнатной температуры, выливают в 100 мл быстро перемешиваемой смеси лед-вода и делают ее основной 2 н. NaOH. После перемешивания в течение 30 минут смесь экстрагируют (4х) СН2Cl2. Объединенные органические экстракты сушат над Na2SO4, фильтруют и концентрируют в вакууме. Сырой остаток очищают хроматографией на силикагеле (градиент 50-100% EtOAc/гексаны), получая индолиновый продукт, пример 48 (110 мг, 55%), в виде не совсем белого твердого вещества: т. пл. 71-75°С; 1Н ЯМР (300 МГц, CDС13) δ 6.73-6.77 (м, 2Н); 6.59-6.66 (м,1Н); 6.56 (д, J=8.0 Гц, 1Н); 6.45 (д, J=8.0 Гц, 1Н); 4.12 (т, J=6.0 Гц, 1Н); 3.72 (ушир.с,1Н); 3.61 (т, J=8.5 Гц, 2Н); 3.51 (с,2Н); 2.86-2.94 (м,3Н); 2.59 (дд, J=11.4, 7.0 Гц, 1Н); 2.42 (с,3Н); API MS m/z=301 [C18H18F2N2+H]+.
Следует отметить, что выделяют также 28 мг (14%) исходного материала. На основе выделенного исходного материала выход соединения примера 48 составляет 64%.
Пример 51
Продукт примера 50 (86 мг, 0,286 ммоль) растворяют в МеОН (3 мл) и каталитическом количестве НОАс (1 капля) в 25 мл колбе в атмосфере N2. Добавляют водный формальдегид (24 мкл, 0,32 ммоль) и смесь перемешивают в течение 2 часов. Добавляют NaBH3CN (29 мг, 0,46 ммоль) и смесь перемешивают в течение дополнительного 1 часа. Смесь разбавляют СН2Cl2 (50 мл), затем промывают последовательно 1 н. NaOH (40 мл) и насыщенным водным NaCl (40 мл). Органический слой сушат над Na2SO4, фильтруют и концентрируют в вакууме. Сырой остаток очищают хроматографией на силикагеле (50% EtOAc/гексаны), получая N-метилиндолин (72 мг, 80%) в виде бледно-желтого масла, которое отверждается при повторяющихся циклах замораживание/размораживание/промывка струей N2: т. пл. 111-116°С; 1Н ЯМР (300 МГц, CDС13) δ 6.73-6.77 (м,2Н); 6.59-6.67 (м,2Н); 6.30 (д, J=8.1 Гц, 1Н); 4.12 (т, J=5.9 Гц, 1Н); 3.50 (с,2Н); 3.30-3.36 (м,2Н); 2.89 (дд, J=11.4, 5.1 Гц, 1Н); 2.82 (т, J=8.2 Гц, 2Н); 2.73 (с,3Н); 2.59 (дд, J=11.3, 6.9 Гц, 1Н); 2.41 (с,3Н); API MS m/z=315 [C19H20F2N2+H]+; Элементный анализ. Вычислено для С19Н20F2N2-0,1 Н2О: С, 72,18; Н, 6,44; N, 8,86. Найдено: С, 72,04; Н, 6,46; N, 8,65.
Пример 52
К раствору подходящего аминоспирта, полученного с использованием методики стадии Е примера 38 (1,20 г, 3,81 ммоль) в метиленхлориде (20 мл) по каплям при 0°С в течение 2 минут добавляют 98% Н2SO4 (10 мл, 0,20 моль). Реакционную смесь перемешивают при 0°С в течение 15 минут и затем выливают в смесь льда и 2 н. NaOH (300 мл). Органический слой отделяют и водный слой экстрагируют (2х) метиленхлоридом. Объединенные органические слои промывают насыщенным раствором соли, сушат над MgSO4, фильтруют и концентрируют в вакууме. Остаток очищают хроматографией (SiO2, EtOAc/гексаны, 1:1), получая требуемый индоловый продукт в виде белого порошка (0,55 г, 48%): т. пл. 184-186°С; 1Н ЯМР (300 МГц, CDС13) δ 8.20 (ушир.с,1Н); 7.08-7.22 (м,6Н); 7.69 (д, J=8.5 Гц, 1Н); 6.50 (т, J=1.0 Гц, 1Н); 4.32 (т, J=7.5, 5.4 Гц, 1Н); 3.97 (д, J=15.2 Гц, 1Н); 3.88 (д, J=15.2 Гц, 1Н); 3.04 (дд, J=11.5, 5.4 Гц, 1Н); 2.66 (дд, J=11.5, 7.5 Гц, 1Н); 2.50 (с,3Н); CI MS m/z=297 [C18H17С1N2+H]+; IR (KBr) 3410, 2870, 2778, 1594, 1460, 1348 см-1; Элементный анализ. Вычислено для С18Н17ClN2: С, 72,84; Н, 5,77; N, 9,44. Найдено: С, 72,83; Н, 5,95; N, 9,28.Пример 53
К раствору индолового продукта примера 52, (160 мг, 0,539 ммоль) и диметилоксалата (70 мг, 0,59 ммоль) в ДМФ (5 мл) в виде одной порции при комнатной температуре в атмосфере азота добавляют трет-бутоксид калия (66 мг, 0,59 ммоль). Реакционную смесь кипятят с обратным холодильником в течение 3 минут и затем охлаждают до комнатной температуры. Смесь разбавляют водой (50 мл) и экстрагируют (3х) метиленхлоридом. Объединенные органические слои промывают насыщенным раствором соли, сушат над Na2SO4, фильтруют и концентрируют в вакууме. Остаток очищают хроматографией (SiO2, EtOAc/гексаны, 1:1), получая
N-метилиндоловый продукт в виде белого твердого вещества (160 мг, 96%): т. пл. 90-92°С; 1Н ЯМР (300 МГц, CDС13) δ 7.03-7.23 (м,6Н); 6.71 (д, J=8.5 Гц, 1Н); 6.41 (дд, J=3.0, <1 Гц, 1Н); 4.33 (т, J=7.5, 5.0 Гц, 1Н); 3.94 (д, J=15.1 Гц, 1Н); 3.88 (д, J=15.1 Гц, 1Н); 3.74 (с,3Н); 3.01 (дд, J=11.4, 5.0 Гц, 1Н); 2.66 (дд, J=11.4, 7.5 Гц, 1Н); 2.48 (с,3Н); CI MS m/z=311 [C19H19С1N2+H]+; IR (KBr) 2937, 2766, 1594, 1497, 1265 см-1; Элементный анализ. Вычислено для С19Н19ClN2-0,1 Н2О: С, 73,00; Н, 6,19; N, 8,96. Найдено: С, 72,78; Н, 6,09; N, 8,78.
Пример 54
Подходящий индоловый продукт (200 мг, 0,763 ммоль) восстанавливают по методике, описанной в примере 42. Продукт реакции выделяют и очищают, получая индолиновый продукт в виде свободного основания (239 мг, 82%): 1Н ЯМР (500 МГц, CDС13) δ 7.07-7.20 (м,4Н); 6.53 (д, J=8.0 Гц, 1Н); 6.43 (д, J=8.0 Гц, 1Н); 4.14 (т, J=8.4, 5.4 Гц, 1Н); 3.65 (ушир.с,1Н); 3.60 (т, J=8.3 Гц, 2Н); 3.58 (д, J=15.2 Гц, 1Н); 3.48 (д, J=15.2 Гц, 1Н); 2.96 (дд, J=11.4, 5.4 Гц, 1Н); 2.91 (т, J=8.3 Гц, 2Н); 2.55 (дд, J=11.4, 8.0 Гц, 1Н); 2.42 (с,3Н).
К перемешиваемому раствору индолинового свободного основания (239 мг, 0,80 ммоль) в метаноле (4 мл) при комнатной температуре в атмосфере азота по каплям добавляют 1н HCl (2,0 мл, 2,0 ммоль) в эфире. Реакционную смесь перемешивают при комнатной температуре в течение 10 мин и затем разбавляют эфиром (10 мл). Образовавшееся белое твердое вещество фильтруют, промывают безводным эфиром и сушат при 60°С в вакууме на протяжении ночи, получая дигидрохлоридную соль (210 мг, 70%): т. пл. 236-238°С; 1Н ЯМР (300 МГц, CD3OD) δ 7.25-7.43 (м,7Н); 6.95 (д, J=6.4 Гц, 1Н); 4.55-4.75 (м,4Н); 3.95 (т, J=7.7 Гц, 2Н); 3.86 (м,1Н); 3.61 (м,1Н); 3.35 (м, J=7.7 Гц, 2Н); 3.13 (с,3Н); CI MS m/z=299 [C18H19С1N2+H]+; IR (KBr) 3410, 2950, 2554, 1595, 1482 см-1; Элементный анализ. Вычислено для С18Н19ClN2-2 HCl-0,5 Н2О: С, 56,78; Н, 5,82; N, 7,36. Найдено: С, 56,74; Н, 5,92; N, 7,19.
Пример 55
Подходящий индоловый продукт получают по способу, описанному в примере 38, и затем восстанавливают в индолиновый продукт по методике, описанной в примере 42.
К раствору образовавшегося индолина (180 мг, 0,603 ммоль) и уксусной кислоты (0,1 мл) в метаноле (5 мл) по каплям при 0°С добавляют 37% водный формальдегид (0,054 мл, 0,723 ммоль). Реакционную смесь перемешивают при комнатной температуре в атмосфере азота в течение 1 часа и затем снова охлаждают до 0°С. Порциями при 0°С добавляют цианоборогидрид натрия (95 мг, 1,5 ммоль). Смесь перемешивают при комнатной температуре в атмосфере азота в течение 3 часов и затем гасят 2н NaOH и экстрагируют (3х) метиленхлоридом. Объединенные органические слои промывают насыщенным раствором соли, сушат над Na2SO4, фильтруют и концентрируют в вакууме. Остаток очищают хроматографией (SiO2, EtOAc/метанол, 10:1), получая N-метилиндолиновый продукт примера 53 в виде белого твердого вещества (140 мг, 74%): т. пл. 63-65°С; 1Н ЯМР (300 МГц, CDС13) δ 7.07-7.20 (м,4Н); 6.59 (д, J=8.0 Гц, 1Н); 6.28 (д, J=8.0 Гц, 1Н); 4.14 (т, J=7.9, 5.0 Гц, 1Н); 3.58 (д, J=15.0 Гц, 1Н); 3.46 (д, J=15.0 Гц, 1Н); 3.33 (т, J=8.2 Гц, 2Н); 2.93 (дд, J=11.3, 5.0 Гц, 1Н); 2.82 (т, J=8.2 Гц, 2Н); 2.72 (с,3Н); 2.54 (дд, J=11.3, 7.9 Гц, 1Н); 2.42 (с,3Н); CI MS m/z=313 [C19H21С1N2+H]+; IR (KBr) 2940, 2796, 1611, 1594, 1489, 1372, 1286 см-1; Элементный анализ. Вычислено для С19Н21ClN2: С, 72,95; Н, 6,77; N, 8,95. Найдено: С, 72,70; Н, 6,83; N, 8,78.
Пример 56
Серную кислоту (5,0 мл) добавляют к раствору подходящего аминоспирта, полученного с использованием методики стадии Е примера 38 (500 мг, 1,68 ммоль) в дихлорметане (25 мл) при 0°С. Реакционную смесь перемешивают при 0°С в атмосфере азота в течение 20 минут. После окончания реакции реакционную смесь делают основной (рН ˜11) 6 н. NaOH и экстрагируют (3х) метиленхлоридом. Объединенные органические слои промывают насыщенным раствором соли, сушат над Na2SO4, фильтруют и концентрируют в вакууме, получая коричневое масло, которое хроматографируют (SiO2, 20% EtOAc/гексаны), получая требуемый индол в виде не совсем белого порошка (120 мг, 34%): т. пл. 150-152°С; 1Н ЯМР (300 МГц, CDС13) δ 8.21 (ушир.с,1Н); 7.25-7.17 (м,2Н); 7.14 (д, J=8.6 Гц, 1Н); 7.01 (д, J=7.7 Гц, 1Н); 6.93-6.85 (м,2Н); 6.70 (д, J=8.5 Гц, 1Н); 6.50 (д, J=2.3 Гц, 1Н); 4.35 (т, J=6.3 Гц, 1Н); 3.97 (д, J=15.3 Гц, 2Н); 3.89 (д, J=15.3 Гц, 1Н); 3.05 (дд, J=5.2, 11.3 Гц, 1Н); 2.68 (дд, J=7.5, 11.3 Гц, 1Н); 2.50 (с,3Н); IR (KBr) 3427, 2921, 2473, 1617, 1590, 1484 см-1.; CI MS m/z=281 [C18H17FN2+H]+.
Пример 57
Индоловый продукт примера 56, (100 мг, 0,36 ммоль) и диметилоксалат (46 мг, 0,39 ммоль) в ДМФ (3 мл) обрабатывают трет-бутоксидом калия (44 мг, 0,39 ммоль). Реакционную смесь кипятят с обратным холодильником в течение 30 минут. Реакционную смесь охлаждают до комнатной температуры и разбавляют водой (25 мл). После экстракции (3х) этилацетатом органические слои промывают водой, насыщенным раствором соли, сушат над сульфатом натрия, фильтруют и концентрируют. Темный остаток хроматографируют (SiO2, 20% EtOAc/гексаны) и образовавшееся масло обрабатывают 1 М HCl (1 экв.) в диэтиловом эфире, получая
N-метилиндоловый продукт в виде белого твердого вещества (45 мг, 11%): т. пл. 255-258°С; 1Н ЯМР (300 МГц, CD3OD) δ 7.43-7.25 (м,3Н); 7.12-7.00 (м,2Н); 6.99 (д, J=10.0 Гц, 1Н); 6.70 (д, J=8.6 Гц, 1Н); 6.50 (д, J=3.1 Гц, 1Н); 4.8-4.67 (м,2Н); 3.89 (дд, J=5.6, 11.9 Гц, 1Н); 3.81 (с,3Н); 3.65-3.55 (м,1Н); 3.30-3.29 (м,1Н); 3.14 (с,3Н); IR (KBr) 3424, 2944, 2479, 1590, 1449 см-1.; CI MS m/z=295 [C19H19FN2+H]+.
Пример 59
1 М раствор HCl в эфире (2,0 мл, 2,0 ммоль) добавляют по каплям к раствору подходящего аминоспирта, полученного с использованием методики стадии Е примера 38 (129 мг, 0,459 ммоль) в метаноле (4 мл). Растворители и избыточный HCl удаляют в вакууме, после чего остается коричневое твердое вещество, которое перекристаллизовывают из EtOH-Et2O, получая требуемый индоловый продукт (64 мг, 42%) в виде коричневого твердого вещества: т. пл. 200-205°С (с разложением); 1Н ЯМР (300 МГц, CD3OD) δ 7.36-7.23 (м,4Н); 7.10 (т, J=8.7 Гц, 2Н); 6.62 (д, J=8.5 Гц, 1Н); 6.52 (д, J=3.0 Гц, 1Н); 4.82-4.69 (м,3Н); 3.85 (дд, J=11.3, 5.8 Гц, 1Н); 3.56 (т, J=11.5 Гц, 1Н); 3.14 (с,3Н); ); IR (KBr) 3238, 2954, 2588, 1605, 1590, 1463, 1348, 1224, 1160, 838, 740 см-1.; CI MS m/z=281 [C18H17FN2+H]+; Элементный анализ. Вычислено для С18Н17FN2-HCl-0,75 Н2О: С, 65,45; Н, 5,95; N, 8,48. Найдено: С, 65,75; Н, 5,94; N, 8,42.
Пример 61
Концентрированную серную кислоту (10,0 мл, 30,1 ммоль) добавляют к охлажденному льдом перемешиваемому раствору подходящего аминоспирта, полученного с использованием методики стадии Е примера 38 (1,00 г, 3,05 ммоль) в СН2Cl2 (50 мл). Эту смесь перемешивают при 0°С в течение 20 минут, затем перемешивают при комнатной температуре в течение 30 минут и охлаждают до -10°С. Небольшими порциями добавляют охлажденный льдом концентрированный водный гидроксид аммония (200 мл) до достижения рН раствора 12. Водный слой экстрагируют (2х) СН2Cl2. Органические экстракты объединяют, сушат над MgSO4/Na2SO4, фильтруют и концентрируют в вакууме. Очистка колоночной хроматографией (SiO2, 20 г, от гексанов до 10% EtOAc/гексаны) дает требуемый индоловый продукт (302 мг, 32%) в виде не совсем белого твердого вещества: т. пл. 149-153°С; 1Н ЯМР (500 МГц, CDС13) δ 8.16 (с,1Н); 7.28-7.21 (м,2Н); 7.16 (д, J=8.4 Гц, 1Н); 7.02 (д, J=10.4 Гц, 1Н); 6.97 (д, J=8.2 Гц, 1Н); 6.70 (д, J=8.4 Гц, 1Н); 6.51 (с,1Н); 4.29 (т, J=5.2 Гц, 1Н); 3.96 (д, J=15.2 Гц, 1Н); 3.87 (д, J=15.3 Гц, 1Н); 3.00 (дд, J=11.2, 5.0 Гц, 1Н); 2.69 (дд, J=11.3, 6.5 Гц, 1Н); 2.49 (с,3Н); IR (KBr) 3409, 2779, 1579, 1489, 1424, 1349, 1244, 1163, 1060 см-1.; CI MS m/z=315 [C18H16С1FN2+H]+; Элементный анализ. Вычислено для С18Н16ClFN2: С, 68,68; Н, 5,12; N, 8,90. Найдено: С, 68,36; Н, 5,13; N, 8,51.
Пример 62
Трет-бутоксид калия добавляют к раствору индолового продукта примера 61 (354 мг, 1,12 ммоль) и диметилоксалата (145 мг, 1,23 ммоль) в ДМФ (3 мл) и смесь кипятят с обратным холодильником в течение 1 часа. Смесь охлаждают до комнатной температуры и гасят водой (5 мл). После экстракции (2х) СН2Cl2 органический слой сушат над MgSO4/Na2SO4, фильтруют и концентрируют в вакууме. Очистка колоночной хроматографией (SiO2, 20 г, от гексанов до 10% EtOAc/гексаны) дает N-метилиндолиновый продукт (163 мг, 44%) в виде желтого порошка: т. пл. 120-124°С; 1Н ЯМР (500 МГц, CDС13) δ 7.27-7.24 (м,1Н); 7.08 (д, J=8.5 Гц, 1Н); 7.04 (д, J=3.1 Гц, 1Н); 7.01 (д, J=10.4 Гц, 1Н); 6.96 (д, J=8.2 Гц, 1Н); 6.71 (д, J=8.5 Гц, 1Н); 6.42 (д, J=3.1 Гц, 1Н); 4.29 (т, J=5.8 Гц, 1Н); 3.96 (д, J=15.2 Гц, 1Н); 3.85 (д, J=15.2 Гц, 1Н); 3.76 (с,3Н); 2.99 (дд, J=11.3, 5.2 Гц, 1Н); 2.68 (дд, J=11.4, 6.5 Гц, 1Н); 2.47 (с,3Н); IR (KBr) 3438, 2943, 2779, 1579, 1488, 1422, 1358, 1266, 1064 см-1.; ESI MS m/z=329 [C19H18С1FN2+H]+.
Пример 64
Аналогичный N-метилиндоловый продукт получали по способу, описанному в примере 39, и затем восстанавливали в индолиновый продукт по следующей методике.
Цианоборогидрид натрия (63 мг, 1,004 ммоль) добавляют к охлажденному льдом раствору N-метилиндола (110 мг, 0,335 ммоль) в ледяной уксусной кислоте (6 мл). Реакционной смеси дают нагреться до комнатной температуры, перемешивают в течение 2 часов, охлаждают на ледяной бане и разбавляют Н2О (10 мл). Добавляют охлажденный льдом концентрированный водный гидроксид аммония (30 мл) до тех пор, пока рН раствора не достигнет 12. После экстракции (2х) СН2Cl2 органический слой сушат над MgSO4/Na2SO4, фильтруют и концентрируют в вакууме. Очистка колоночной хроматографией (SiO2, 10 г, 10% EtOAc/гексаны) дает нужный N-метилиндолиновый продукт (15 мг, 15%) в виде коричневого масла. Продукт чувствителен к действию воздуха и требует хранения в атмосфере азота: 1Н ЯМР (500 МГц, CDС13) δ 7.28-7.25 (м,1Н); 7.01 (дд, J=10.4, 2.0 Гц, 1Н); 6.95 (дд, J=8.2, 1.9 Гц, 1Н); 6.59 (д, J=8.1 Гц, 1Н); 6.28 (д, J=8.1 Гц, 1Н); 4.11 (т, J=5.9 Гц, 1Н); 3.50 (дд, J=13, 2.0 Гц, 2Н); 3.36-3.32 (м,2Н); 2.88 (дд, J=11.3, 5.2 Гц, 1Н); 2.82 (т, J=8.2 Гц, 2Н); 2.73 (с,3Н); 2.56 (дд, J=11.4, 7.0 Гц, 1Н); 2.41 (с,3Н); IR (KBr) 3052, 2925, 2850, 2786, 1609, 1422, 1265, 739 см-1.; ESI MS m/z=331 [C19H20С1FN2+H]+.
Пример 65
Раствор подходящего аминоспирта, полученного с использованием методики стадии Е примера 38 (2,00 г, 6,01 ммоль), в СН2Cl2 (50 мл), охлажденный до 0°С, добавляют по каплям к концентрированной H2SO4 (20 мл), охлажденной до 0°С, в атмосфере азота. После перемешивания в течение 20 минут при 0°С реакционную смесь выливают в смесь лед-вода (400 мл). Водный слой гасят 6 н. NaOH до достижения рН ˜14, затем водный слой экстрагируют (3х) СН2Cl2. Объединенный экстракт в СН2Cl2 промывают смесью 1:5 6 н. NaOH и насыщенного NaCl, затем сушат над Na2SO4, фильтруют и концентрируют в вакууме. Хроматография на диоксиде кремния (60 г) и элюирование 66% EtOAc дает продукт (0,68 г, 36%). Перекристаллизация из смеси СН2Cl2/МеОН/гексаны дает требуемый индоловый продукт (0,12 г) в виде не совсем белого твердого вещества: т. пл. 189-195°С; 1Н ЯМР (300 МГц, 5% МеОН-D4/CDС13) δ 8.66 (ушир.с,1Н); 7.28-7.20 (м,2Н); 7.16 (д, J=8.6 Гц, 1Н); 7.11-6.97 (м,2Н); 6.66 (д, J=8.5 Гц, 1Н); 6.52-6.47 (м,1Н); 4.34 (т, J=6.5 Гц, 1Н); 4.00 (д, J=15.2 Гц, 1Н); 3.89 (д, J=15.2 Гц, 1Н); 3.05 (дд, J=11.1, 5.8 Гц, 1Н); 2.63 (дд, J=11.4, 8.0 Гц, 1Н); 2.51 (с,3Н); IR (KBr) 3410, 2780, 1498, 1461, 1347, 1247, 1132, 1059, 883, 824, 801, 736, 690, 560 см-1.; ESI MS m/z=315 [C18H16С1FN2+H]+; Элементный анализ. Вычислено для С18Н16ClFN2-0,10 Н2О: С, 68,29; Н, 5,16; N, 8,85. Найдено: С, 68,17; Н, 4,95; N, 8,68.
Пример 81
Стадия А: Метиламин (40 масс.% водный, 2,0 мл, 23 ммоль) добавляют к перемешиваемому раствору 5-формилбензофурана (8,2 г, 56 ммоль) в МеОН (55 мл). После перемешивания в течение 20 минут смесь охлаждают на бане лед-вода в течение 35 минут и затем порциями в течение 15 минут добавляют NaBH4 (1,3 г, 34 ммоль). После перемешивания в течение 30 минут добавляют Н2О (5 мл) для гашения любого оставшегося гидрида. После перемешивания в течение 15 минут МеОН удаляют в вакууме, остаток растворяют в 1 н. HCl и затем экстрагируют (2х) Et2O. Водную фазу делают сильно щелочной (рН 11) добавлением избыточного концентрированного NH4OH, затем экстрагируют (2х) Et2O. Органическую фазу промывают насыщенным NaCl, сушат над Na2SO4, фильтруют и растворитель удаляют в вакууме, получая соединение, являющееся продуктом восстановительного алкилирования (4,2 г, теоретический выход = 3,8 г) в виде прозрачной, желтой жидкости: 1Н ЯМР (300 МГц, CDС13) δ 7.61 (д, J=2.3 Гц, 1Н); 7.54 (с,1Н); 7.45 (с,1Н); 7.25 (дд, J=8.5, 1.7 Гц, 1Н); 6.74 (д, J=2.7 Гц, 1Н); 3.83 (с,2Н); 2.47 (с,3Н).
Стадия В: 2-Бромацетофенон (5,12 г, 26 ммоль) добавляют к перемешиваемому раствору метиламинного продукта со стадии А (4,08 г, 25 ммоль) и DIEA (5,5 мл, 31 ммоль) в безводном СН2Cl2 (50 мл) в атмосфере азота. После перемешивания в течение 20 часов смесь разбавляют Et2O и затем промывают (2х) 1 н. HCl. Водную фазу делают сильно щелочной (рН 12) добавлением избыточного концентрированного NH4OH, затем экстрагируют (2х) Et2O. Органическую фазу сушат над Na2SO4, фильтруют, растворитель удаляют в вакууме и остаток очищают флэш-хроматографией на силикагеле с использованием от 2% до 14% EtOAc/гексаны + 1% Et2O, получая аминокетон (4,32 г, 61%) в виде прозрачного, темно-желтого масла: 1Н ЯМР (300 МГц, CDС13) δ 7.94 (дд, J=8.5, 1.2 Гц, 2Н); 7.51-7.62 (м,3Н); 7.38-7.47 (м,3Н); 7.30 (дд, J=8.5, 1.7 Гц, 1Н); 6.72 (д, J=2.9 Гц, 1Н); 3.83 (с,2Н); 3.79 (с,2Н); 2.41 (с,3Н).
Стадия С: По методике, описанной в примере 10, стадия G, аминокетон, полученный на стадии В (4,31 г, 15,4 ммоль) используют для получения аминоспирта (3,61 г, 83%) в виде светло-желтого масла: 1Н ЯМР (300 МГц, CDС13) δ 7.62 (д, J=2.0 Гц, 1Н); 7.52 (д, J=0.9 Гц, 1Н); 7.47 (д, J=8.5 Гц, 1Н); 7.22-7.48 (м,6Н); 6.72-6.75 (м,1Н); 4.76 (дд, J=10.3, 3.7 Гц, 1Н); 3.83 (д, J=12.9 Гц, 1Н); 3.61 (д, J=12.9 Гц, 1Н); 2.63 (дд, J=12.4, 10.2 Гц, 1Н); 2.54 (дд, J=12.4, 3.7 Гц, 1Н); 2.33 (с,3Н).
Стадия D: Метансульфоновую кислоту (15,5 мл, 239 ммоль) добавляют к перемешиваемому раствору аминоспирта (3,45 г, 12 ммоль, получен на стадии С) в СН2Cl2 (60 мл) в атмосфере N2. Затем смесь кипятят с обратным холодильником в течение 6 часов и дают ей возможность охладиться до комнатной температуры. СН2Cl2 удаляют в вакууме и образовавшийся раствор CH3SO3H выливают на лед при перемешивании. Смесь делают сильно щелочной (рН 12) добавлением избыточного концентрированного NH4OH, затем экстрагируют (2х) Et2O. Органическую фазу промывают насыщенным NaCl, сушат над Na2SO4, фильтруют, растворитель удаляют в вакууме и остаток очищают флэш-хроматографией на силикагеле с использованием смеси от 5% до 15% EtOAc/гексаны + 1% Et3N, получая (в процессе элюирования) (i) соединение А (1,65 г, 51%) в виде прозрачного, бледно-желтого масла: 1Н ЯМР (300 МГц, CDС13) δ 7.16-7.39 (м,7Н); 7.04 (д, J=8.4 Гц, 1Н); 5.97-6.00 (м,1Н); 4.47 (т, J=6.6 Гц, 1Н); 3.75 (с,2Н); 3.09 (дд, J=11.6, 5.8 Гц, 1Н); 2.62 (дд, J=11.7, 7.5 Гц, 1Н); 2.44 (с,3Н); (ii) смесь 9:1 (0,44 г, 14%) соединений В и А; (iii) соединение А (0,54 г, 17%) в виде прозрачного, желтого масла: 1Н ЯМР (300 МГц, CDС13) δ 7.50 (д, J=2.3 Гц, 1Н); 7.19-7.37 (м,6Н); 6.99 (с,1Н); 6.66-6.69 (м,1Н); 4.40 (дд, J=8.8, 5.8 Гц, 1Н); 3.89 (д, J=14.3 Гц, 1Н); 3.70 (д, J=14.3 Гц, 1Н); 3.08 (ддд, J=11.6, 5.8, 1.5 Гц, 1Н); 2.59 (дд, J=11.6, 9.2 Гц, 1Н); 2.45 (с,3Н).
Стадия Е: Раствор HCl в эфире (1 М, 5 мл) добавляют к перемешиваемому раствору соединения В (0,53 г, 2,0 ммоль, со стадии D) в МеОН (20 мл). После перемешивания в течение 20 минут растворитель удаляют в вакууме, остаток снова растворяют в МеОН и растворитель снова удаляют в вакууме. Остаток перекристаллизовывают из EtOH-Et2O, получая соединение примера 67 (405 мг, 68%) в виде белого кристаллического твердого вещества: т. пл. 241-246°С; 1Н ЯМР (300 МГц, CD3OD) δ 7.75 (д, J=2.2 Гц, 1Н); 7.56 (с,1Н); 7.33-7.47 (м,3Н); 7.27-7.33 (м,2Н); 6.98 (с,1Н); 6.84-6.87 (м,1Н); 4.66-4.77 (м,3Н); 3.87 (дд, J=12.3, 6.4 Гц, 1Н); 3.60 (т, J=11.9 Гц, 1Н); 3.10 (с,3Н); IR (KBr) 3432, 2954, 2476, 1468, 1275, 1124, 701 см-1.; CI MS m/z=264 [C18H17NO+H]+; Элементный анализ. Вычислено для С18Н17NO-HCl-0,1 Н2О: С, 71,68; Н, 6,08; N, 4,64. Найдено: С, 71,53; Н, 6,04; N, 4,56.
Пример 123
По методике, описанной для получения соединения примера 81, стадии Е, соединение А (0,83 г, 3,2 ммоль, пример 81, стадия С) используют для получения соединения примера 123 (385 мг, 41%) в виде белого, аморфного твердого вещества: т. пл. 234-240°С; 1Н ЯМР (300 МГц, CD3OD) δ 7.50-7.60 (м,2Н); 7.29-7.42 (м,3Н); 7.20-7.28 (м,3Н); 5.95 (ушир.с,1Н); 4.83-4.91 (м,1Н); 4.69 (д, J=15.2 Гц, 1Н); 4.62 (д, J=15.2 Гц, 1Н); 3.97 (дд, J=12.4, 6.7 Гц, 1Н); 3.53 (ушир.т, J=11.4 Гц, 1Н); 3.09 (с,3Н); IR (KBr) 3424, 2936, 2588, 1466, 1431, 1268, 1148, 1039, 780, 705 см-1.; CI MS m/z=264 [C18H17NO+H]+; Элементный анализ. Вычислено для С18Н17NO-HCl-0,5 Н2О: С, 70,01; Н, 6,20; N, 4,54. Найдено: С, 70,05; Н, 6,06; N, 4,46.
АНАЛИЗЫ НА СВЯЗЫВАНИЕ
Анализы на первичное связывание
Для оценки относительного сродства связывания различных соединений с транспортерами NE, DA и 5НТ, были использованы линии клеток НЕК293Е для экспрессии каждого из трех транспортеров для человека. кДНК, содержащие полные области кодирования каждого транспортера, амплифицировали посредством PCR из библиотек мозга человека. кДНК, содержащиеся в pCRII-векторе, секвенировали для подтверждения их идентичности и затем субклонировали в экспрессирующую плазмиду на основе вируса Эпштейна-Барра (E. Shen, GM Cooke, RA Horlick, Gene 156:235-239, 1995). Эту плазмиду, содержащую кодирующую последовательность для одного из транспортеров человека, трансфицировали в клетки НЕК293Е. Успешную трансфекцию подтверждали способностью известных блокаторов повторного поглощения ингибировать поглощение меченых тритием NE, DA или 5НТ.
Для связывания клетки гомогенизировали, центрифугировали и затем повторно суспендировали в буфере для инкубации (50 мМ Трис, 120 мМ NaCl, 5 мМ KCl, рН 7,4). Затем добавляли подходящий радиолиганд. Для связывания NET добавляли [3H]-низоксетин (86,0 Ки/ммоль, NEN/DuPont) до конечной концентрации приблизительно 5 нМ. Для связывания DAT добавляли [3H]-WIN 35428 (84,5 Ки/ммоль) при 15 нМ. Для связывания 5НТТ добавляли [3H]-цитолапрам (85,0 Ки/ммоль) при 1 нМ. Затем для вытеснения радиолиганда добавляли различные концентрации (от 10∧-5 до 10∧-11 М) представляющего интерес соединения. Инкубацию проводили при комнатной температуре в течение 1 часа в 96-луночном планшете. После инкубации планшеты помещали на харвестер и быстро 4 раза промывали (50 мМ Трис, 0,9% NaCl, рН 7,4), где клеточные мембраны, содержащие связанную радиоактивную метку, улавливались на фильтрах Whatman GF/B. К фильтрам добавляли сцинтиляционный коктейль и затем подсчет фильтров проводили в Packard TopCount. Сродство связывания представляющих интерес соединений определяли методом нелинейной регрессии с построением кривой с использованием программного обеспечения GraphPad Prism 2.01. Неспецифическое связывание определяли вытеснением 10 микромолярным мазиндолом.
Анализ TBZ
Для определения in vivo активности соединений в отношении транспортеров NE и DA определяли их способность предотвращать седативные действия тетрабеназина (TNZ) (G. Stille, Arzn. Forsch 14:534-537, 1964). Самцов мышей CFI (Charles River Breeding Laboratories), весом 18-25 г во время испытания помещали минимум на 6 дней в тщательно контролируемые окружающие условия (22,2+1,1°С; 50% средняя влажность; 12-часовой световой цикл/24 часа). Мыши голодали на протяжении ночи (16-22 часов) перед испытанием. Мышей помещали в прозрачные «обувные» коробки из поликарбоната (17 см × 28,5 см × 12 см). Перорально вводили случайно выбранные и предварительно определенные дозы испытуемых соединений. Дозу тетрабеназина 45 мг/кг вводили внутрибрюшинно за 30 минут до начала отсчета времени. Все соединения вводили в объеме 0,1 мл/10 г веса тела. Животных оценивали на антагонизм индуцированной тетрабеназином потери исследовательской активности и птоз при определенных интервалах времени после введения лекарственного средства. В определенные интервалы времени мышей обследовали на симптомы исследовательской активности и птоз. Исследовательскую активность оценивали, помещая животное в центр круга 5 дюймов. Пятнадцать секунд давали животному для движения и исследования периметра. Это считали антагонизмом тетрабеназину и оценивали как 0. Неспособность выйти из круга считали как потерю исследовательской активности и оценивали как 4. Считали, что животное имеет птоз, если его веки закрыты, по меньшей мере, на 50% и оценивали как 4, если они полностью закрыты; отсутствие закрытия век оценивали как 0. Считали, что более чем 95% контрольных (обработанных наполнителем) мышей имели потерю исследовательской активности и птоз. Активность лекарственного средства вычисляли в виде процента мышей, у которых наблюдали отсутствие реакции на введенную дозу тетрабеназина.
Статистическая оценка
Средние эффективные дозы (ED50) и 95% доверительные пределы определяют количественно способами Thompson (1947) и Litchfield и Wilcoxon (1949).
Приложение I
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ТЕТРАГИДРОИЗОХИНОЛИНА, СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ИНГИБИРОВАНИЯ СИНАПТИЧЕСКОГО ЗАХВАТА ДОПАМИНА И СПОСОБ ЛЕЧЕНИЯ | 2000 |
|
RU2293728C2 |
| АКТИВИРУЮЩИЙ АГЕНТ ДЛЯ РЕЦЕПТОРА, АКТИВИРУЕМОГО СТИМУЛИРУЮЩИМИ РОСТ ПЕРОКСИСОМ АГЕНТАМИ | 2009 |
|
RU2501794C2 |
| ПЕСТИЦИДНЫЕ КОМПОЗИЦИИ И СВЯЗАННЫЕ С НИМИ СПОСОБЫ | 2013 |
|
RU2654327C2 |
| ЦИКЛИЧЕСКИЕ N, N'-ДИАРИЛТИОМОЧЕВИНЫ ИЛИ N, N'-ДИАРИЛМОЧЕВИНЫ - АНТАГОНИСТЫ АНДРОГЕННЫХ РЕЦЕПТОРОВ, ПРОТИВОРАКОВОЕ СРЕДСТВО, СПОСОБ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2010 |
|
RU2434851C1 |
| СОЕДИНЕНИЯ ДЛЯ ЛЕЧЕНИЯ РАКА | 2010 |
|
RU2581367C2 |
| ПРОИЗВОДНЫЕ 1,2,4-ТРИАЗОЛА В КАЧЕСТВЕ ИНГИБИТОРОВ СИГМА РЕЦЕПТОРА | 2007 |
|
RU2451015C2 |
| ПРОИЗВОДНЫЕ ПИПЕРИДИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ | 1997 |
|
RU2193029C2 |
| СПОСОБЫ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ БИЦИКЛО [3.1.0]ГЕКСАНА И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ДЛЯ ЭТОЙ ЦЕЛИ | 2004 |
|
RU2388747C2 |
| ПРОИЗВОДНЫЕ ПИРРОЛПИРИМИДИНОНА, СПОСОБ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2001 |
|
RU2263676C2 |
| ПРОИЗВОДНЫЕ ТАКСАНА, ФУНКЦИОНАЛИЗИРОВАННЫЕ ПО 14-ПОЛОЖЕНИЮ, И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2003 |
|
RU2320652C2 |
Изобретение относится к новым 4-фенилзамещенным тетрагидрохинолинам, имеющим формулу IA, IB, IIA, IIB, IIIA или IIIC, которые вследствие их селективного связывания нейротрансмиттеров пригодны для лечения различных неврологических или психологических нарушений, например, ADHD. Изобретение также относится к фармацевтической композиции на их основе и способу лечения.
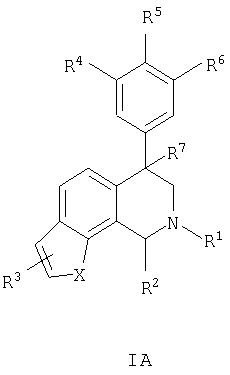
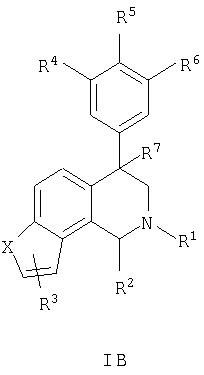
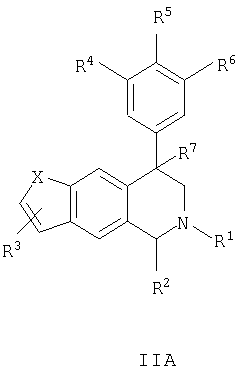
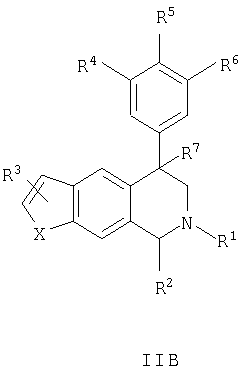
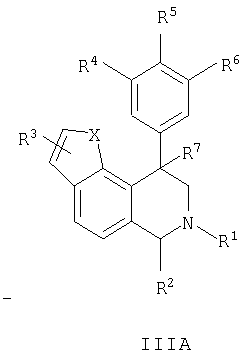
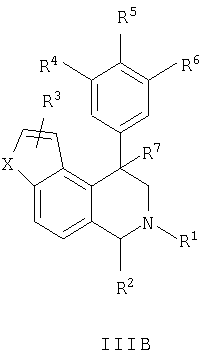
где значения X, R1-R7 указано в описании. 3 н. и 33 з.п. ф-лы, 1 ил., 16 табл.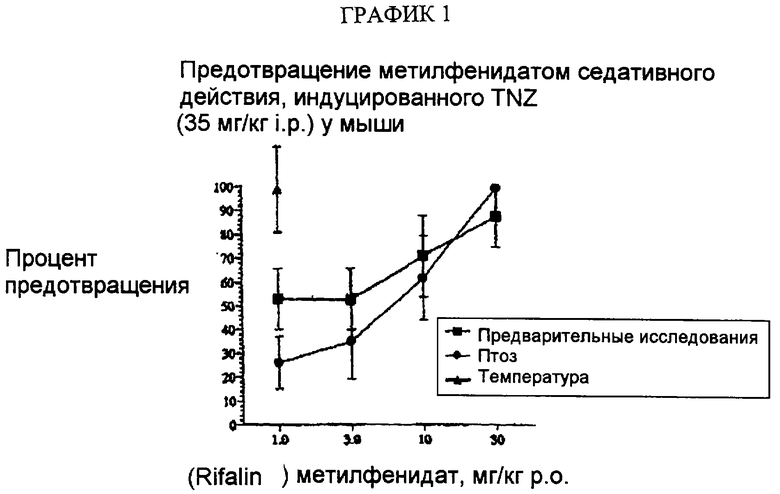
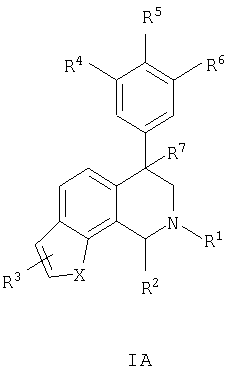
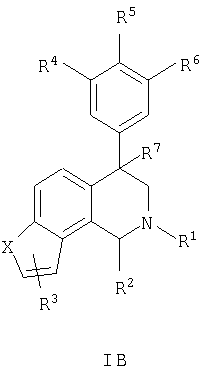
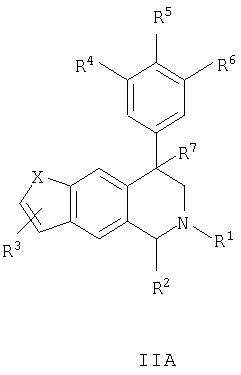
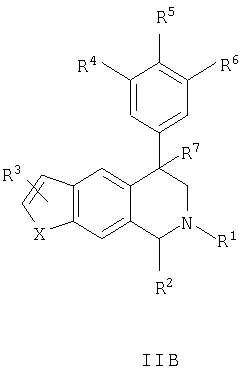
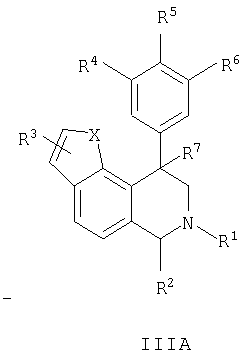
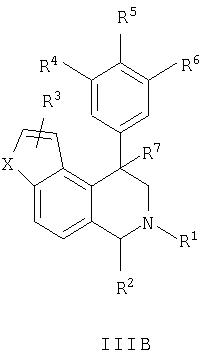
где R1 выбран из C1-С6-алкила;
R2 выбран из Н;
R3 выбран из Н, C1-С6-алкила, где C1-С6-алкил, необязательно замещен 1-3 заместителями, независимо выбранными в каждом случае из OR8;
R4, R5 и R6, каждый независимо выбран в каждом случае из группы, состоящей из Н, галогена, -OR10, C1-С6-алкила, где каждый C1-С6-алкил необязательно замещен заместителями от 1 до 3 независимо выбранными в каждом случае из фенила;
R7 выбран из Н;
R8 выбран из Н;
R10 выбран из группы, состоящей из Н, С1-С4-алкила
Х выбран из группы, состоящей из О, NR13 и S, при условии, что Х не представляет NR13, когда соединение является соединением формулы (IA);
R13 выбран из группы, состоящей из Н, C1-С6-алкила.
или C1-С6-алкил, замещенный 1-3 OR8.
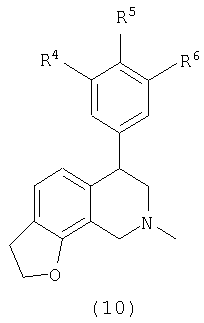
или его фармацевтически приемлемая соль, выбранное из группы, состоящей, по существу, из
соединения формулы (10), где R4 представляет Н, R5 представляет Н и R6 представляет Н;
соединения формулы (10), где R4 представляет Н, R5 представляет Me и R6 представляет Н;
соединения формулы (10), где R4 представляет Cl, R5 представляет Н и R6 представляет Н; и
соединения формулы (10), где R4 представляет Н, R5 представляет F и R6 представляет Н.
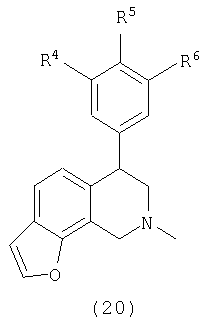
или его фармацевтически приемлемая соль, выбранное из группы, состоящей, по существу, из
соединения формулы (20), где R4 представляет H, R5 представляет Н и R6 представляет H;
соединения формулы (20), где R4 представляет Н, R5 представляет Me и R6 представляет Н;
соединения формулы (20), где R4 представляет Н, R3 представляет Cl и R6 представляет Н;
соединения формулы (20), где R4 представляет H, R5 представляет F и R6 представляет Н; и
соединения формулы (20), где R4 представляет F, R5 представляет Н и R6 представляет F.
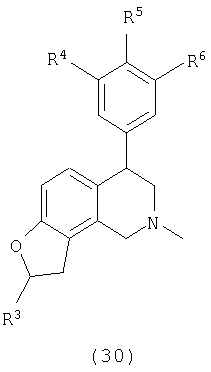
или его фармацевтически приемлемая соль, выбранное из группы, состоящей, по существу, из
соединения формулы (30), где R3 представляет H, R4 представляет Н, R5 представляет Н и R6 представляет Н;
соединения формулы (30), где R3 представляет Н, R4 представляет F, R5 представляет F и R6 представляет Н;
соединения формулы (30), где R представляет Н, R4 представляет F, R5 представляет Н и R6 представляет F;
соединения формулы (30), где R3 представляет Н, R4 представляет Н, R5 представляет F и R6 представляет Н;
соединения формулы (30), где R3 представляет Н, R4 представляет Cl, R5 представляет Н и R6 представляет Н;
соединения формулы (30), где R3 представляет Н, R4 представляет H, R5 представляет Cl и R6 представляет Н;
соединения формулы (30), где R3 представляет Н, R4 представляет Н, R5 представляет Cl и R6 представляет F;
соединения формулы (30), где R3 представляет H, R4 представляет Н, R5 представляет F и R6 представляет Cl;
соединения формулы (30), где R3 представляет Н, R4 представляет F, R5 представляет Н и R6 представляет Cl;
соединения формулы (30), где R3 представляет Н, R4 представляет Н, R5 представляет ОМе и R представляет Н; и
соединения формулы (30), где R3 представляет Н, R4 представляет F, R5 представляет Н и R6 представляет Н.
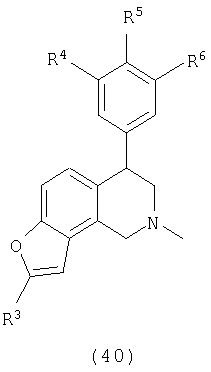
или его фармацевтически приемлемая соль, выбранное из группы, состоящей, по существу, из
соединения формулы (40), где R3 представляет Н, R4 представляет Н, R5 представляет Н и R6 представляет Н;
соединения формулы (40), где R3 представляет Н, R4 представляет F, R5 представляет F и R6 представляет Н;
соединения формулы (40), где R3 представляет Н, R4 представляет F, R5 представляет Н и R6 представляет F;
соединения формулы (40), где R3 представляет Н, R4 представляет F, R5 представляет Н и R6 представляет Н;
соединения формулы (40), где R3 представляет Н, R4 представляет Н, R5 представляет F и R6 представляет Н;
соединения формулы (40), где R3 представляет Н, R4 представляет Cl, R5 представляет Н и R6 представляет Н;
соединения формулы (40), где R3 представляет Н, R4 представляет Н, R5 представляет Cl и R6 представляет Н;
соединения формулы (40), где R3 представляет Н, R4 представляет Н, R5 представляет Cl и R6 представляет F;
соединения формулы (40), где R3 представляет Н, R4 представляет Н, R5 представляет F и R6 представляет Cl;
соединения формулы (40), где R3 представляет Н, R4 представляет F, R5 представляет Н и R6 представляет Cl;
соединения формулы (40), где R3 представляет Н, R4 представляет Н, R5 представляет ОМе и R6 представляет Н;
соединения формулы (40), где R3 представляет Me, R4 представляет Н, R5 представляет Н и R6 представляет Н;
соединения формулы (40), где R3 представляет Et, R4 представляет Н, R5 представляет Н и R6 представляет Н; и
соединения формулы (40), где R3 представляет CH2OH, R4 представляет Н, R5 представляет Н и R6 представляет Н.
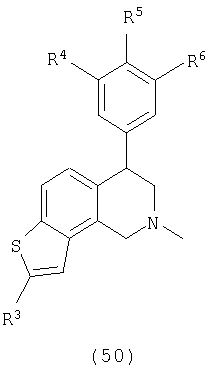
или его фармацевтически приемлемая соль, выбранное из группы, состоящей, по существу, из
соединения формулы (50), где R3 представляет H, R4 представляет Н, R5 представляет Н и R6 представляет Н.
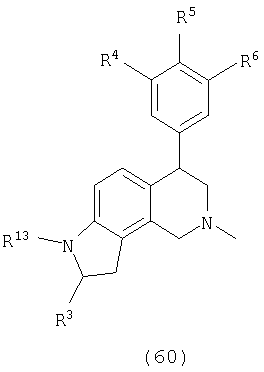
или его фармацевтически приемлемая соль, выбранное из группы, состоящей, по существу, из
соединения формулы (60), где R3 представляет Н, R4 представляет Н, R5 представляет Н, R6 представляет Н и R13 представляет Н;
соединения формулы (60), где R3 представляет Н, R4 представляет Н, R5 представляет Н, R6 представляет H и R13 представляет Me;
соединения формулы (60), где R3 представляет Н, R4 представляет Н, R5 представляет Н, R6 представляет Н и R13 представляет Et;
соединения формулы (60), где R3 представляет Н, R4 представляет Н, R5 представляет F, R6 представляет F и R13 представляет Н;
соединения формулы (60), где R3 представляет H, R4 представляет Н, R5 представляет F, R6 представляет F и R13 представляет Me;
соединения формулы (60), где R3 представляет Н, R4 представляет F, R5 представляет Н, R6 представляет F и R13 представляет Н;
соединение формулы (60), где R3 представляет H, R4 представляет F, R5 представляет Н, R6 представляет F и R13 представляет Me;
соединения формулы (60), где R3 представляет Н, R4 представляет Cl, R5 представляет Н, R6 представляет Н и R13 представляет Н;
соединения формулы (60), где R3 представляет Н, R4 представляет Cl, R5 представляет Н, R6 представляет H и R13 представляет Me;
соединения формулы (60), где R3 представляет Н, R4 представляет F, R5 представляет Н, R6 представляет Н и R13 представляет H;
соединения формулы (60), где R3 представляет Н, R4 представляет Н, R5 представляет F, R6 представляет Н и R13 представляет Н;
соединения формулы (60), где R3 представляет Н, R4 представляет F, R5 представляет Cl, R6 представляет Н и R13 представляет Н;
соединения формулы (60), где R представляет Н, R представляет F, R5 представляет Cl, R6 представляет H и R представляет Me;
соединения формулы (60), где R3 представляет Н, R4 представляет Cl, R5 представляет F, R6 представляет Н и R13 представляет H; и
соединения формулы (60), где R3 представляет Н, R4 представляет Cl, R5 представляет F, R6 представляет Н и R13 представляет Me.

или его фармацевтически приемлемая соль, выбранное из группы, состоящей, по существу, из
соединения формулы (70), где R3 представляет Н, R4 представляет Н, R5 представляет Н, R6 представляет Н и R13 представляет Н;
соединения формулы (70), где R3 представляет Н, R4 представляет Н, R5 представляет Н, R6 представляет Н и R13 представляет Me;
соединения формулы (70), где R3 представляет Н, R4 представляет Н, R5 представляет Н, R6 представляет H и R13 представляет Et;
соединения формулы (70), где R3 представляет Н, R4 представляет Н, R5 представляет H, R6 представляет Н и R13 представляет Bn;
соединения формулы (70), где R3 представляет Н, R4 представляет Н, R5 представляет F, R6 представляет F и R13 представляет Н;
соединения формулы (70), где R3 представляет Н, R4 представляет H, R5 представляет F, R6 представляет F и R13 представляет Me;
соединение формулы (70), где R3 представляет Н, R4 представляет F, R5 представляет Н, R6 представляет F и R13 представляет Me;
соединения формулы (70), где R3 представляет Н, R4 представляет Cl, R5 представляет Н, R6 представляет Н и R13 представляет Н;
соединения формулы (70), где R3 представляет Н, R4 представляет Cl, R5 представляет Н, R6 представляет H и R13 представляет Me;
соединения формулы (70), где R3 представляет Н, R4 представляет F, R5 представляет Н, R6 представляет Н и R13 представляет Н;
соединения формулы (70), где R3 представляет Н, R4 представляет F, R5 представляет Н, R6 представляет Н и R13 представляет Me;
соединения формулы (70), где R3 представляет Н, R4 представляет Н, R5 представляет F, R6 представляет H и R13 представляет Н;
соединения формулы (70), где R3 представляет Н, R4 представляет F, R5 представляет Cl, R6 представляет Н и R13 представляет Н;
соединения формулы (70), где R3 представляет Н, R4 представляет F, R5 представляет Cl, R6 представляет Н и R13 представляет Me;
соединения формулы (70), где R3 представляет Н, R4 представляет Cl, R5 представляет F, R представляет Н и R представляет Н; и
соединения формулы (70), где R3 представляет Н, R4 представляет Cl, R5 представляет F, R представляет H и R13 представляет Me.
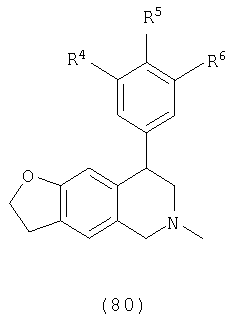
или его фармацевтически приемлемая соль, выбранное из группы, состоящей, по существу, из
соединения формулы (80), где R4 представляет Н, R5 представляет Н и R6 представляет Н;
соединения формулы (80), где R4 представляет Н, R5 представляет F и R6 представляет Н; и
соединения формулы (80), где R4 представляет Н, R5 представляет F и R6 представляет F.
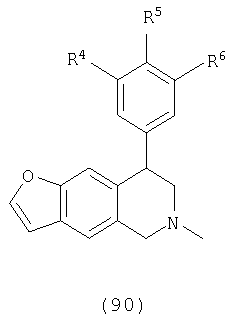
или его фармацевтически приемлемая соль, выбранное из группы, состоящей, по существу, из
соединения формулы (90), где R4 представляет Н, R5 представляет H и R6 представляет Н;
соединения формулы (90), где R4 представляет Н, R5 представляет F и R6 представляет F; и
соединения формулы (90), где R4 представляет Н, R5 представляет F и R6 представляет Н.
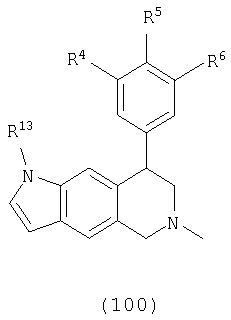
или его фармацевтически приемлемая соль, выбранное из группы, состоящей, по существу, из
соединения формулы (100), где R4 представляет Н, R5 представляет Н, R6 представляет Н и R13 представляет Н.
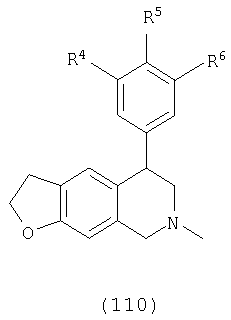
или его фармацевтически приемлемая соль, выбранное из группы, состоящей, по существу, из
соединения формулы (110), где R4 представляет Н, R5 представляет Н и R6 представляет Н;
соединения формулы (110), где R4 представляет Н, R5 представляет F и R6 представляет F;
соединения формулы (110), где R4 представляет Н, R5 представляет F и R6 представляет Н;
соединения формулы (110), где R представляет Н, R5 представляет Н и R6 представляет Cl;
соединения формулы (110), где R4 представляет Н, R5 представляет Cl и R6 представляет F;
соединения формулы (110), где R4 представляет Н, R5 представляет F и R6 представляет Cl; и
соединения формулы (110), где R4 представляет Н, R5 представляет ОМе и R6 представляет Н.
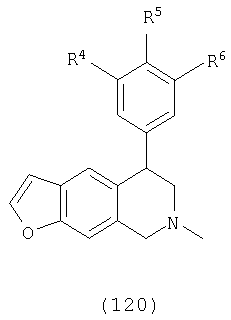
или его фармацевтически приемлемая соль, выбранное из группы, состоящей, по существу, из
соединения формулы (120), где R4 представляет H, R5 представляет Н и R6 представляет Н;
соединения формулы (120), где R4 представляет Н, R5 представляет F и R6 представляет F;
соединения формулы (120), где R4 представляет Н, R5 представляет F и R6 представляет Н;
соединения формулы (120), где R4 представляет Н, R5 представляет Н и R6 представляет Cl;
соединения формулы (120), где R4 представляет Н, R5 представляет Cl и R6 представляет F;
соединения формулы (120), где R4 представляет Н, R5 представляет ОМе и R6 представляет Н; и
соединения формулы (120), где R4 представляет Н, R5 представляет F и R6 представляет Cl.
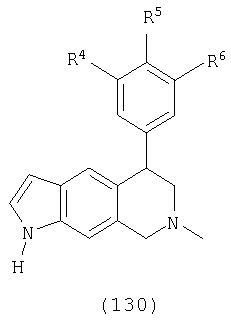
или его фармацевтически приемлемая соль, выбранное из группы, состоящей, по существу, из
соединения формулы (130), где R4 представляет Н, R5 представляет H и R6 представляет Н; и
соединения формулы (130), где R4 представляет H, R5 представляет Bn и R6 представляет Н.
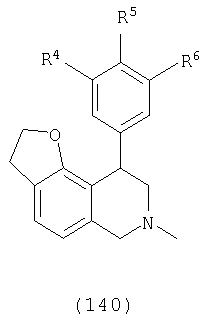
или его фармацевтически приемлемая соль, выбранное из группы, состоящей, по существу, из
соединения формулы (140), где R4 представляет Н, R5 представляет Н и R6 представляет Н;
соединения формулы (140), где R4 представляет Н, R5 представляет F и R6 представляет Н;
соединения формулы (140), где R4 представляет Н, R5 представляет F и R представляет Cl;
соединения формулы (140), где R4 представляет Н, R5 представляет Cl и R6 представляет F;
соединения формулы (140), где R4 представляет Н, R5 представляет Н и R6 представляет Cl;
соединения формулы (140), где R4 представляет Н, R5 представляет ОМе и R6 представляет Н; и
соединения формулы (140), где R4 представляет Н, R5 представляет F и R6 представляет F.
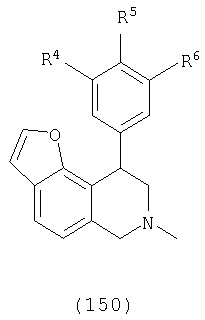
или его фармацевтически приемлемая соль, выбранное из группы, состоящей, по существу, из
соединения формулы (150), где R4 представляет Н, R3 представляет Н и R6 представляет Н;
соединения формулы (150), где R4 представляет H, R5 представляет F и R6 представляет Н;
соединения формулы (150), где R4 представляет Н, R5 представляет F и R6 представляет Cl;
соединения формулы (150), где R4 представляет Н, R5 представляет Cl и R6 представляет F;
соединения формулы (150), где R4 представляет Н, R5 представляет Н и R6 представляет Cl;
соединения формулы (150), где R4 представляет Н, R5 представляет ОМе и R6 представляет Н; и
соединения формулы (150), где R4 представляет Н, R5 представляет F и R6 представляет F.
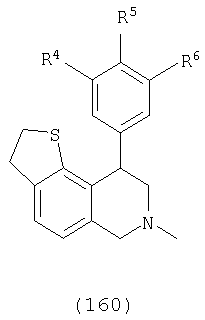
или его фармацевтически приемлемая соль, выбранное из группы, состоящей, по существу, из
соединения формулы (160), где R4 представляет Н, R5 представляет Н и R6 представляет Н.
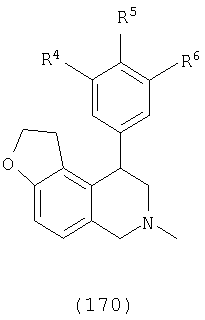
или его фармацевтически приемлемая соль, выбранное из группы, состоящей, по существу, из
соединения формулы (170), где R4 представляет Н, R5 представляет Н и R6 представляет Н;
соединения формулы (170), где R4 представляет Н, R5 представляет F и R6 представляет Н; и
соединения формулы (170), где R4 представляет Н, R5 представляет F и R6 представляет F.
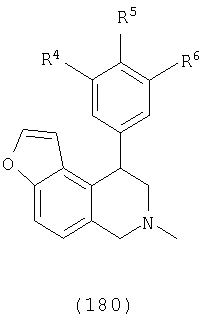
или его фармацевтически приемлемая соль, выбранное из группы, состоящей, по существу, из
соединения формулы (180), где R4 представляет Н, R5 представляет Н и R6 представляет Н;
соединения формулы (180), где R4 представляет Н, R5 представляет F и R6 представляет Н; и
соединения формулы (180), где R4 представляет Н, R5 представляет F и R6 представляет F.
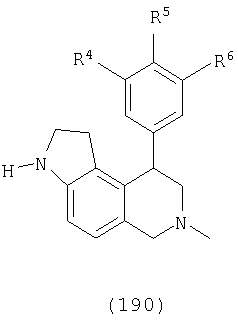
или его фармацевтически приемлемая соль, выбранное из группы, состоящей, по существу, из
соединения формулы (190), где R4 представляет Н, R5 представляет Н и R6 представляет Н.
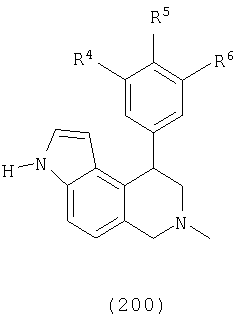
или его фармацевтически приемлемая соль, выбранное из группы, состоящей, по существу, из
соединения формулы (200), где R4 представляет Н, R5 представляет Н, R6 представляет Н и R13 представляет Н; и
соединения формулы (200), где R4 представляет Н, R5 представляет Н, R6 представляет Н и R13 представляет Me.
(R)-2-метил-4-фенил-1,2,3,4,8,9-гексагидрофуро[2,3-h]изохинолина;
(S)-2-метил-4-фенил-1,2,3,4,8,9-гексагидрофуро[2,3-h]изохинолина;
(R)-7-метил-5-фенил-5,6,7,8-тетрагидрофуро[3,2-g]изохинолина;
(S)-7-метил-5-фенил-5,6,7,8-тетрагидрофуро[3,2-g]изохинолина;
(R)-4-(3,4-дифторфенил)-2-метил-1,2,3,4-тетрагидрофуро[2,3-h]изохинолина;
(S)-4-(3,4-дифторфенил)-2-метил-1,2,3,4-тетрагидрофуро[2,3-h]изохинолина;
(R)-2-метил-4-фенил-1,2,3,4-тетрагидрофуро[2,3-h]изохинолина;
(S)-2-метил-4-фенил-1,2,3,4-тетрагидрофуро[2,3-h]изохинолина;
(R)-4-(4-хлорфенил)-2-метил-1,2,3,4-тетрагидрофуро[2,3-h]изохинолина;
(S)-4-(4-хлорфенил)2-метил-1,2,3,4-тетрагидрофуро[2,3-h]изохинолина;
(R)-8-метил-6-фенил-2,3,6,7,8,9-гексагидрофуро[3,2-h]изохинолина;
(S)-8-метил-6-фенил-2,3,6,7,8,9-гексагидрофуро[3,2-h]изохинолина;
(R)-4-(4-фторфенил)-2-метил-1,2,3,4-тетрагидрофуро[2,3-h]изохинолина;
(S)-4-(4-фторфенил)-2-метил-1,2,3,4-тетрагидрофуро[2,3-h]изохинолина;
(R)-4-(3,5-дифторфенил)-2-метил-1,2,3,4-тетрагидрофуро[2,3-h]изохинолина;
(S)-4-(3,5-дифторфенил)-2-метил-1,2,3,4-тетрагидрофуро[2,3-h]изохинолина;
(R)-2-метил-4-фенил-2,3,4,7-тетрагидро-1Н-пирроло[2,3-h]изохинолина; и
(S)-2-метил-4-фенил-2,3,4,7-тетрагидро-1Н-пирроло[2,3-h]изохинолина.
(+)-2-метил-4-фенил-1,2,3,4,8,9-гексагидрофуро[2,3-h]изохинолина;
(-)-2-метил-4-фенил-1,2,3,4,8,9-гексагидрофуро[2,3-h]изохинолина;
(+)-7-метил-5-фенил-5,6,7,8-тетрагидрофуро[3,2-g]изохинолина;
(-)-7-метил-5-фенил-5,6,7,8-тетрагидрофуро[3,2-g]изохинолина;
(+)-4-(4-фторфенил)-2-метил-1,2,3,4-тетрагидрофуро[2,3-h]изохинолина;
(-)-4-(4-фторфенил)-2-метил-1,2,3,4-тетрагидрофуро[2,3-h]изохинолина;
(+)-4-(3,4-дифторфенил)-2-метил-1,2,3,4-тетрагидрофуро[2,3-h]изохинолина;
(-)-4-(3,4-дифторфенил)-2-метил-1,2,3,4-тетрагидрофуро[2,3-h]изохинолина;
(+)-2-метил-4-фенил-1,2,3,4-тетрагидрофуро[2,3-h]изохинолина;
(-)-2-метил-4-фенил-1,2,3,4-тетрагидрофуро[2,3-h]изохинолина;
(+)-4-(4-хлорфенил)-2-метил-1,2,3,4-тетрагидрофуро[2,3-h]изохинолина;
(-)-4-(4-хлорфенил)2-метил-1,2,3,4-тетрагидрофуро[2,3-h]изохинолина;
(+)-8-метил-6-фенил-2,3,6,7,8,9-гексагидрофуро[3,2-h]изохинолина;
(-)-8-метил-6-фенил-2,3,6,7,8,9-гексагидрофуро[3,2-h]изохинолина;
(+)-4-(3,5-дифторфенил)-2-метил-1,2,3,4-тетрагидрофуро[2,3-h]изохинолина;
(-)-4-(3,5-дифторфенил)-2-метил-1,2,3,4-тетрагидрофуро[2,3-h]изохинолина;
(+)-2-метил-4-фенил-2,3,4,7-тетрагидро-1Н-пирроло[2,3-h]изохинолина и
(-)-2-метил-4-фенил-2,3,4,7-тетрагидро-1Н-пирроло[2,3-h]изохинолина.
| Способ изготовления металлокерамических фрикционных дисков | 1960 |
|
SU140070A1 |
| US 3947456 А, 30.03.1976 | |||
| US 3666763 A, 30.05.1972 | |||
| Способ получения октагидро- @ -пирроло-/2,3- @ /-изохинолинов в виде их рацемической смеси,цис- и транс-изомеров,в свободном виде или в виде соли | 1981 |
|
SU1014474A3 |
| Способ получения октагидро- @ -бензо-(4,5)-фуро-(3,2- @ )-изохинолинов или их солей | 1979 |
|
SU1060114A3 |

