Перекрестная ссылка на родственные заявки
В данной заявке испрашивается приоритет по 35 U.S.C. § 119(е) на основании предварительной заявки US 60/518391, поданной 7 ноября 2003 года.
Область техники, к которой относится изобретение
Настоящее изобретение относится к способам получения производных бицикло[3.1.0]гексана, которые используются в качестве модуляторов метаботропных глутаматных рецепторов. Данное изобретение относится также к новым промежуточным соединениям, получаемым в ходе данных способов, и к гидрохлоридной соли (+)-(1R,2S,5S,6S)-2-амино-6-фтор-4-оксобицикло[3.1.0]гексан-2,6-дикарбоновой кислоты, и к ее полиморфам.
Уровень техники
Возбуждающие аминокислоты, включая глутамат, модулируют множество физиологических процессов в центральной нервной системе млекопитающих (ЦНС), например долговременное потенцирование (обучение и память), развитие синаптической пластичности, регулирование двигательной активности, дыхание, регулирование сердечно-сосудистой деятельности и сенсорное восприятие.
Глутамат действует посредством рецепторов, по крайней мере, двух различных классов. Первый класс включает в себя ионотропные глутаматные рецепторы (iGlu), действующие в качестве лиганд-регулируемых ионных каналов. Второй класс представляет собой G белковый или второй мессенджер-связанный «метаботропный» глутаматный (mGluR) рецептор. По-видимому, рецепторы обоих классов опосредуют нормальную синаптическую трансмиссию по путям возбуждения, а также принимают участие в модификации синаптических связей во время развития и на протяжении жизни. Schoepp, Bockaert and Sladeczek, Trends in Pharmacol. Sci., 11, 508 (1990); McDonald and Johnson, Brain Research Reviews, 15, 41 (1990).
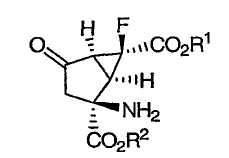
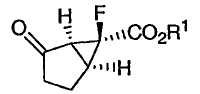
В качестве модуляторов mGluR были известны различные соединения функционализированных производных бицикло[3.1.0]гексанов. Модуляторы mGluR терапевтически применимы для лечения или предупреждения психических расстройств, шизофрении, беспокойства и сопутствующих заболеваний, депрессии, биполярного расстройства и эпилепсии, и неврологических заболеваний, таких как лекарственная зависимость, нарушение познавательной деятельности, болезнь Альцгеймера, хорея Хантингтона, болезнь Паркинсона, дискинезия, сопровождаемая мышечным окоченением, ишемия головного мозга, церебральная недостаточность, миелопатия и травмы головы. Например, в патенте США № 6333428, выданном 25 декабря 2001 года, описаны некоторые mGluR агонисты, которые представляют собой производные 2-амино-6-фторбицикло[3.1.0]гексана приведенной ниже формулы:
в которой каждый из R1 и R2 выбирают из группы, включающей в себя
(1) водород,
(2) С1-10алкил,
(3) С3-8циклоалкил и
(4) С3-8циклоалкил-С1-5алкил,
и их фармацевтически приемлемые соли. В патенте '428 заявлено, что данные соединения могут находиться в форме рацемата или могут быть в виде энантиомера. В патенте '428 описаны также некоторые новые промежуточные соединения приведенной ниже формулы:
в которой R1 определен выше.
В патенте США № 6160009, выданном 12 декабря 2000 года, раскрыт класс функционализированных производных бицикло[3.1.0]гексана приведенной ниже формулы, являющихся терапевтически применимыми в качестве mGluR агонистов:
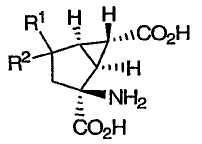
в которой R1 и R2 вместе могут представлять собой =О.
В патенте США № 5750566, выданном 12 мая 1998 года, описан mGluR агонист приведенной ниже формулы:
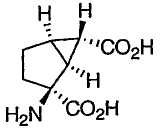
известный как LY 354740.
Получение описанных выше модуляторов mGluR и промежуточных соединений было описано в выше упомянутых патентах, у Nakazato et al., J. Med. Chem., 2000, 43, 4893-4909 и в WO 02/00595 (который опубликован на английском языке как ЕР 1295862). Однако описанные синтезы имеют недостатки, которые делают их непригодными для крупномасштабного производства. Например, в синтезах, описанных в патенте '428 и у Nakazato, требуется получение рацемических промежуточных соединений, которые затем нужно подвергнуть сложным методам разделения, включая ВЭЖХ, что приводит к низкой производительности. Обычно в известных способах синтеза требуется также применение дорогостоящих и опасных реагентов, таких как Pd(OAc)2 и (PhSe)2, которые должны присутствовать в стехиометрических количествах, и CH2N2. Кроме того, в способе синтеза Nakazato в качестве последней стадии синтеза необходим жесткий гидролиз с применением H2SO4 при высокой температуре (145°С) в течение пяти дней, что приводит к низкому выходу и требует трудоемкого выделения конечного продукта из предшествующего производного гидантоина.
Понятно, что модуляторы mGluR, описанные в патентах США №№ 6333428, 6160009 и 5570566, применимы в качестве терапевтических агентов. По существу, есть необходимость в разработке способа получения данных соединений, легко поддающегося масштабированию, в котором используются рентабельные и относительно безопасные реагенты и который, таким образом, может быть использован на практике в крупномасштабном производстве.
В настоящее время заявителями разработан новый способ синтеза класса модуляторов mGluR на основе энантиомерно чистых функционализированных производных бицикло[3.1.0]гексана и энантиомерно чистых промежуточных соединений.
Сущность изобретения
Настоящее изобретение относится к новому способу синтеза класса модуляторов mGluR на основе энантиомерно чистых функционализированных производных бицикло[3.1.0]гексана формулы (I)
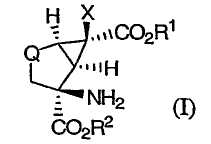
в которой R1 и R2 независимо выбирают из группы, включающей в себя
(1) водород,
(2) С1-10алкил,
(3) С3-8циклоалкил и
(4) -(СН2)n-фенил
где n равно 1 или 2, а указанные алкил, циклоалкил и фенил не замещены или замещены одним или более галогеном, гидрокси, С1-6алкилом или С1-6алкокси,
Х выбирают из группы, включающей в себя
(1) галоген и
(2) водород, а
Q представляет собой -СН2- или -С(=О),
и их фармацевтически приемлемых солей.
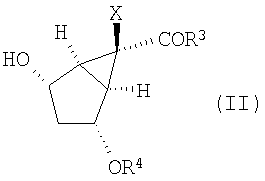
Кроме того, изобретение относится к новым способам получения соединений формулы (II)
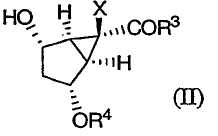
в которой R3 выбирают из группы, включающей в себя
(1) -ОН,
(2) -O-Ra и
(3) -NRbRc,
где Ra выбирают из группы, включающей в себя
(a) С1-10алкил и
(b) С3-8циклоалкил,
и Ra не замещен или замещен одним или более
(i) С1-10алкокси,
(ii) гидрокси,
(iii) галогеном,
(iv) SRd,
(v) арилом, не замещенным или замещенным одним или более гидрокси, С1-10алкокси, С1-10алкилом или галогеном,
(vi) гетероарилом, не замещенным или замещенным одним или более гидрокси, С1-10алкокси, С1-10алкилом или галогеном, и
(vii) NReRf;
Rb, Rc, Re и Rf выбирают из группы, включающей в себя
(а) водород,
(b) С1-10алкил и
(c) С3-8циклоалкил,
и когда Rb, Rc, Re или Rf представляют собой С1-10алкил или С3-8циклоалкил, указанные С1-10алкил или С3-8циклоалкил не замещены или замещены одним или более
(i) гидрокси,
(ii) С1-10алкокси,
(iii) SRd,
(iv) арилом, не замещенным или замещенным одним или более гидрокси, С1-10алкокси, С1-10алкилом или галогеном, и
(v) гетероарилом, не замещенным или замещенным одним или более гидрокси, С1-10алкокси, С1-10алкилом или галогеном, и
(vi) NRgRh;
где Rg или Rh представляют собой водород, С1-10алкил или С3-8циклоалкил,
или Rb или Rc вместе с атомом N, с которым они связаны, образуют группу

в которой r равно 1 или 2, а группа NRbRc может быть незамещенной или замещенной по атомам углерода цикла одним или более
(i) гидрокси,
(ii) С1-10алкокси,
(iii) SRd,
(iv) арилом, не замещенным или замещенным одним или более гидрокси, С1-10алкокси, С1-10алкилом или галогеном, и
(v) гетероарилом, не замещенным или замещенным одним или более гидрокси, С1-10алкокси, С1-10алкилом или галогеном, и
(vi) NRgRh;
Rd представляет собой водород или С1-10алкил;
Х выбирают из группы, включающей в себя
(1) галоген и
(2) водород, а
R4 выбирают из группы, включающей в себя
(1) водород,
(2) С1-10алкил,
(3) Si-(R9)(R10)(R11),
(4) С(=О)-R12,
(5) СН2-фенил, где указанный фенил не замещен или замещен одним или более заместителями, выбранными из группы, включающей в себя нитро, галоген, С1-10алкил и С1-10алкокси,
(6) (СН2)р-О-(СН2)q-X'-R14,
(7) тетрагидропиранил,
где каждый из R9, R10 и R11 представляет собой С1-10алкил или фенил, а R14 выбирают из группы, включающей в себя
(а) водород,
(b) С1-10алкил,
р равно 1 или 2,
q является целым числом от 1-10, а
Х' представляет собой О или связь,
и их солей.
Изобретение относится также к новым способам получения соединений формулы (XII)
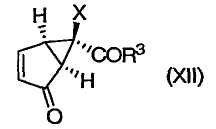
или их энантиомеров (XII')
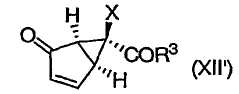
в которых R3 и Х определены выше, и их солей.
Соединения формул (II), (XII) и (XII') представляют собой промежуточные соединения, получаемые в процессе синтеза модуляторов mGluR формулы (I). Способы применения соединения (XII) или (XII') для получения модуляторов mGluR формулы (I) описаны в упомянутых выше патентах '566, '428 и '009 и у Nakazato et al., J. Med. Chem., 2000, 43, 4893-4909. Изобретение относится также к некоторым новым промежуточным соединениям, получаемым в процессе синтеза в соответствии с данным изобретением.
Краткое описание чертежей
Изобретение описано в связи с прилагаемыми чертежами, где:
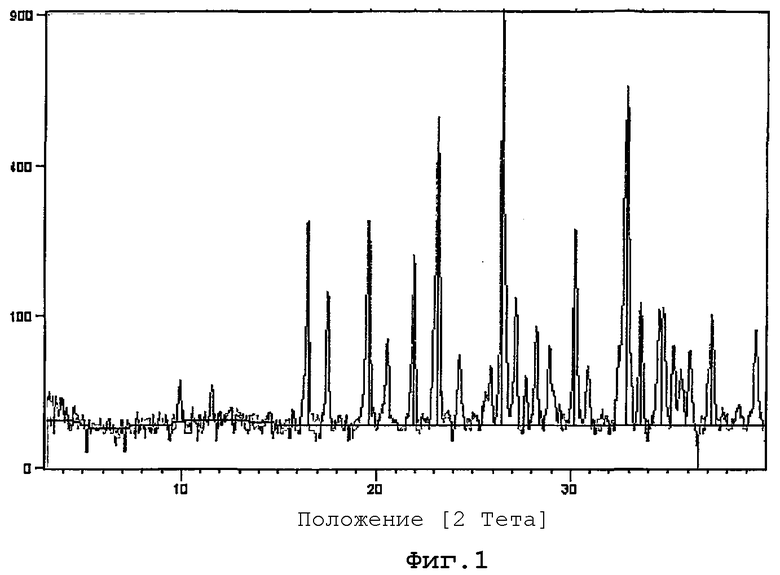
Фиг.1 представляет собой порошковую рентгенограмму (XPRD) кристаллической формы гидрохлоридной соли (+)-(1R,2S,5S,6S)-2-амино-6-фтор-4-оксобицикло[3.1.0]гексан-2,6-дикарбоновой кислоты, и
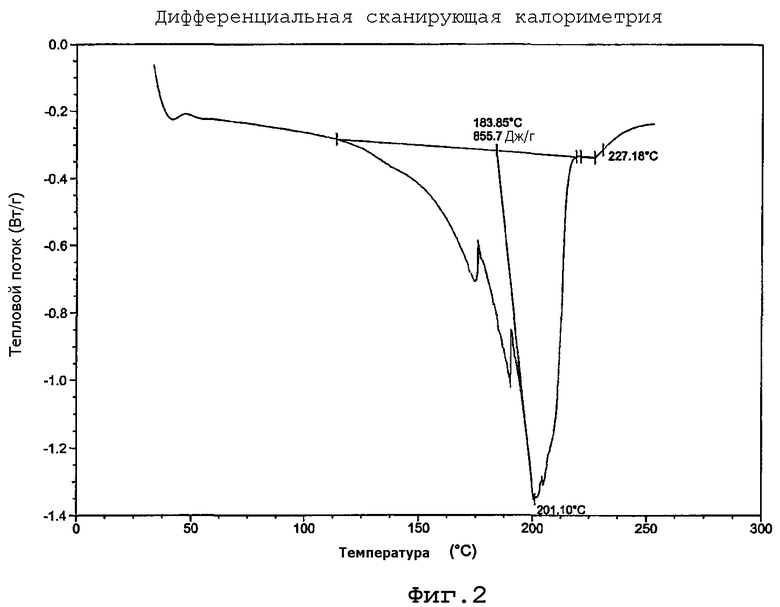
на Фиг. 2 изображена кривая дифференциальной сканирующей калориметрии для кристаллической формы гидрохлоридной соли (+)-(1R,2S,5S,6S)-2-амино-6-фтор-4-оксобицикло[3.1.0]гексан-2,6-дикарбоновой кислоты.
Подробное описание изобретения
Настоящее изобретение направлено на способы получения функционализированных производных бицикло[3.1.0]гексана формулы (I)
в которой R1 и R2 независимо выбирают из группы, включающей в себя
(1) водород,
(2) С1-10алкил,
(3) С3-8циклоалкил и
(4) (СН2)n-фенил,
где n равно 1 или 2, а указанные алкил, циклоалкил и фенил не замещены или замещены одним или более галогеном, гидрокси, С1-6алкилом или С1-6алкокси,
Х выбирают из группы, включающей в себя
(1) галоген и
(2) водород, а
Q представляет собой -СН2- или -С(=О),
и их фармацевтически приемлемых солей.
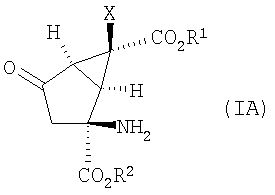
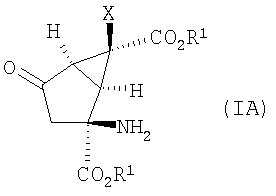
В одном из вариантов осуществления изобретение направлено на способ получения соединений формулы (IA):
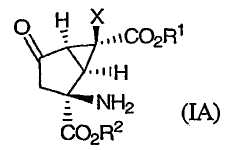
в которой Х, R1 и R2 определены выше.
В данном варианте осуществления изобретение включает в себя окисление промежуточного соединения формулы (II):
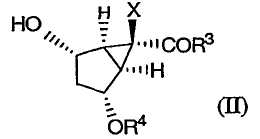
в которой Х, R3 и R4 определены выше,
с получением соединения формулы (IV):
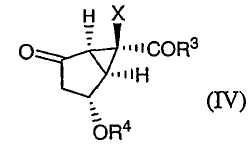
снятие защиты с гидроксильной группы соединения формулы (IV) с получением соединения формулы (V):
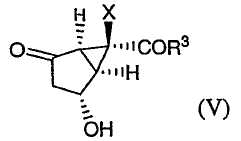
и взаимодействие соединения формулы (V) с соединением формулы (VI)
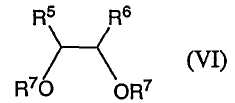
в которой каждый из R5 и R6 независимо выбирают из группы, включающей в себя
(1) водород,
(2) С1-10алкил,
(3) С3-8циклоалкил и
(4) (СН2)m-фенил,
где m равно 0, 1 или 2, а
R7 выбирают из группы, включающей в себя
(1) водород и
(2) Si-(R9)(R10)(R11), где каждый из R9, R10 и R11 представляет собой С1-10алкил или фенил,
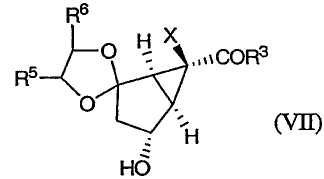
с получением соединения формулы (VII):
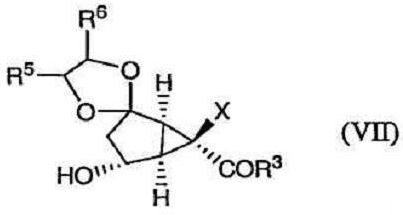
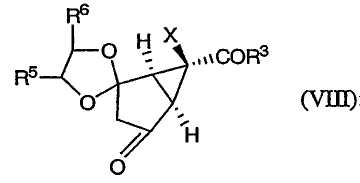
После этого соединение формулы (VII) окисляют, получая соединение формулы (VIII):
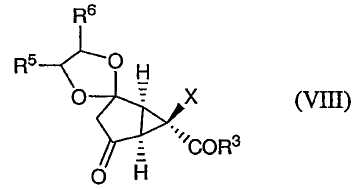
которое превращают в соединение формулы (IX):
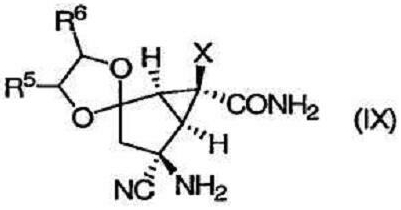
Затем соединение формулы (IX) превращают в требуемое соединение формулы (IA):
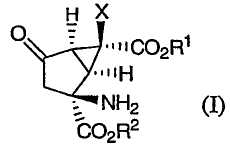
в которой Х, R1 и R2 определены выше.
В предпочтительных вариантах осуществления способа получения соединений формулы (IA) Х представляет собой фтор. В других предпочтительных вариантах осуществления Х представляет собой водород.
В предпочтительных вариантах осуществления способа получения соединений формулы (IA) R1 и R2 представляют собой водород.
В предпочтительных вариантах осуществления способа получения соединений формулы (IA) R3 является метокси, этокси или бензилокси.
В способе получения соединений формулы (IA) предпочтительными группами R4 являются TBS, TMS и TES. Предпочтительной группой R7 является TMS.
В предпочтительных вариантах осуществления способа получения соединений формулы (IA) R5 и R6 выбирают из группы, включающей в себя метил и фенил. Предпочтительным является, чтобы R5=R6.
В предпочтительных вариантах осуществления способа получения соединений формулы (IA) стадия превращения соединения (IX) в соединение (I) включает в себя гидролиз соединения (IX).
Изобретение направлено также на новые промежуточные соединения формул (VII), (VIII) и (IX):
 и
и
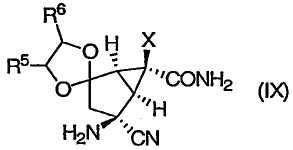
которые образуются в ходе синтеза модуляторов mGluR формулы (I), и их соли. В соединениях формул (VII), (VIII) и (IX) R3, R4, R5 и Х определены выше.
Настоящее изобретение направлено также на способы получения промежуточных соединений формулы (II):
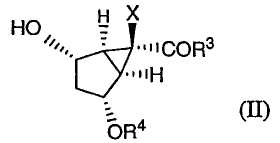
в которой R3, Х и R4 определены выше, и их солей. В данном способе соединение формулы (Х):
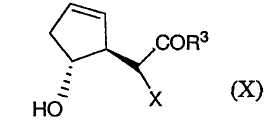
в котором Х является водородом, а R3 определен выше, подвергают эпоксидированию, например, в результате реакции с пероксидом, таким как гидропероксид трет-бутила, или с другими окислителями (включая надкислоты, такие как надбензойная кислота и надуксусная кислота), предпочтительно в присутствии металлического катализатора, такого как VO(acac)2. После этого гидроксильную группу соединения (Х) можно защитить, например, при помощи TBS или TMS, получая в результате соединение формулы (XI):
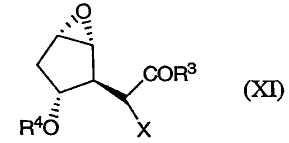
в которой Х является водородом, а R4 определен выше. После этого данное соединение можно фторировать (где Х является фтором). Альтернативным образом, соединение (Х) можно сперва фторировать (где Х является F). После этого фторированное соединение можно подвергнуть описанному выше эпоксидированию.
Альтернативным образом получение эпоксидного производного можно осуществить через галогенгидрин реакцией с источником галогена. Например, соединение формулы (Х) можно ввести во взаимодействие с N-бромсукцинимидом с последующей обработкой основанием, а затем выделить эпоксидный продукт.
Затем защищенное эпоксидное производное (XI) вводят во взаимодействие с подходящим основанием в присутствии кислоты Льюиса, получая соединение формулы (II):
в которой Х, R3 и R4 определены выше. Затем соединение формулы (II) может быть окислено с получением соединения (IV):
которое можно потом окислить в соответствии с описанными выше стадиями процесса, получая соединения формулы (IA).
Альтернативным образом соединения формулы (IV) можно перевести в соединения формулы (IA) при помощи методов, описанных в предшествующем уровне техники. Например, у Nakazato, J. Med. Chem. 2000, 43, 4893-4909, описано применение соединения формулы (IV) для получения соединения формулы (IA) на схеме 5 страницы 4898. В способе, предложенном Nakazato, требуется получение дитиокеталя, а затем производного гидантоина.
В патенте США № 6160009 в колонках 8-13 описано применение соединения формулы (IV) для получения соединения формулы (IA). Данная реакция протекает через производное гидантоина.
В предпочтительных вариантах осуществления способа получения соединений формулы (II) R3 является метокси, этокси или бензилокси.
В предпочтительных вариантах осуществления способа получения соединений формулы (II) Х является фтором. В других предпочтительных вариантах осуществления Х представляет собой водород.
В способе получения соединений формулы (II) предпочтительными группами R4 являются TBS, TMS и TES.
В других предпочтительных вариантах осуществления данного способа окисление соединения (II) включает в себя контактирование соединения (II) с RuCl3 и окислителем. Предпочтительными окислителями являются отбеливатели. Предпочтительным отбеливателем является NaClO.
Изобретение направлено также на новые промежуточные соединения формул (ХА), (XI), (IVA) и (II), изображенные ниже:
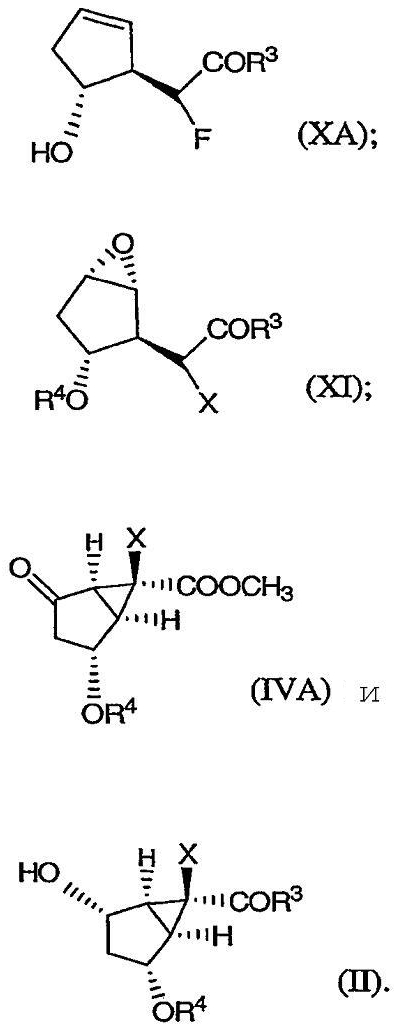
В соединениях (ХА), (XI), (IVA) и (II) R3, Х и R4 определены выше.
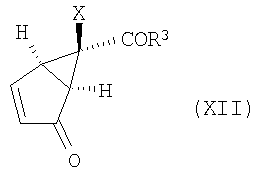
Изобретение направлено также на способ получения промежуточных еноновых соединений формулы (XII):
и их энантиомеров (XII'):
в которых R3 и Х определены выше, и их солей.
В одном из вариантов осуществления данного способа получения соединений формулы (XII) соединение формулы (II)
в котором Х, R3 и R4 определены выше, вводят в реакцию для получения соединения формулы (XIII), содержащего следующую уходящую группу R8:
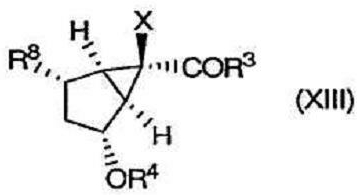
где R8 выбирают из группы, включающей в себя
(1) галоген и
(2) O-SO2-R12, где R12 выбирают из группы, включающей в себя
(а) С1-10алкил,
(b) С1-10перфторалкил,
(с) фенил, замещенный или не замещенный одним или более заместителями, выбранными из группы, включающей в себя нитро, галоген, С1-10алкил или С1-10алкокси.
После этого группу R4 удаляют, получая приведенное ниже гидроксильное сложноэфирное производное (XIV):
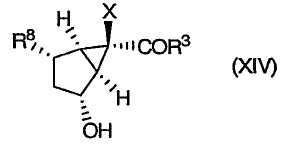
которое затем окисляют, получая требуемый [3.1.0]бициклический-α,β-ненасыщенный кетон формулы (XII):
В одном из вариантов осуществления данного способа получения соединения формулы (XII') соединение формулы (II) окисляют, получая соединение формулы (IV):
в котором Х, R3 и R4 определены выше. Затем соединение (IV) вводят в реакцию элиминирования, например, при взаимодействии с основанием, таким как DBU, получая соединение формулы (XII')
представляющее собой энантиомер соответствующего соединения формулы (XII).
Еноновое соединение формулы (XII) или (XII') можно перевести в соединение формулы (I) при помощи способов, известных в данной области техники. Например, у Nakazato, J. Med. Chem. 2000, 43, 4893-4909, описано применение соединения формулы (XII) для получения соединения формулы (IA) на схеме 5 страницы 4898.
В патенте США № 5750566 описано применение соединения формулы (XII) для получения соединений формулы (I), где Q является СН2, в колонке 12 на схеме IV.
У Dominguez et al, Tetrahedron: Asymmetry, 1997, 8, 511-514, описано применение соединения формулы (XII) для получения соединений формулы (I), где Q является СН2, на схеме 2 страницы 513. В данном способе требуется получение производного гидантоина.
В предпочтительных вариантах осуществления синтеза соединений формулы (XII) или (XII') R3 представляет собой метокси, этокси или бензилокси.
В предпочтительных вариантах осуществления синтеза соединений формулы (XII) или (XII') Х представляет собой фтор. В других предпочтительных вариантах осуществления Х представляет собой водород.
В синтезе соединений формулы (XII) и (XII') предпочтительные защитные группы R4 представляют собой TBS, TMS и TES.
В синтезе соединений формулы (XII) и (XII') предпочтительные группы R8 включают в себя О-тозил (пара-толуолсульфонил), О-мезил и О-трифлат.
Изобретение направлено также на гидрохлоридные соли соединений формулы (I). В предпочтительных вариантах осуществления данная гидрохлоридная соль является солью соединения формулы (I), в которой Х представляет собой фтор, а как R1, так и R2 являются водородом, что обозначено как соединение (I'):
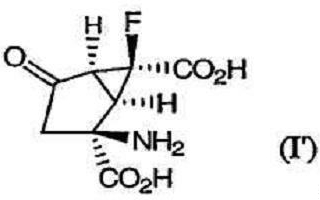
которое представляет собой (+)-(1R,2S,5S,6S)-2-амино-6-фтор-4-оксобицикло[3.1.0]гексан-2,6-дикарбоновую кислоту. Изобретение направлено также на новый кристаллический полиморф гидрохлоридной соли соединения (I').
Определения
Использованный здесь термин «синтез по Штрекеру» относится к реакции, известной специалистам в области органического синтеза, для получения альфа-аминонитрилов.
Использованный здесь термин «по существу энантиомерно чистая форма» означает, что требуемый энантиомер присутствует в количестве, по меньшей мере, 50% е/е (энантиомерного избытка) по отношению к нежелательному энантиомеру.
Использованный здесь термин «кислота Льюиса» относится к соединению, способному принимать электроны.
Использованный здесь термин «арил» относится к полиненасыщенному ароматическому углеводородному заместителю, который может представлять собой одно кольцо или несколько колец (предпочтительно от 1 до 3 колец), которые могут быть конденсированными или ковалентно связанными. Не ограничивающие примеры арильных групп включают в себя фенильную, нафтильную и дифенильную.
Использованный здесь термин «гетероарил» относится к полиненасыщенному ароматическому кольцу, содержащему в цикле, по меньшей мере, один гетероатом (азот, кислород или серу). Гетероарильная группа может представлять собой одно кольцо или несколько колец (предпочтительно от 1 до 3 колец), которые могут быть конденсированными или ковалентно связанными. Не ограничивающие примеры гетероарильных групп включают в себя пиррол, пиразол, имидазол, пиридин, пиразин, пиримидин, фуран, пиран, оксазол, изоксазол, пурин, бензимидазол, хинолин, изохинолин, индол и так далее.
В том случае, когда определенная здесь гетероарильная группа замещена, заместитель может быть связан с атомом углерода цикла данной гетероарильной группы или с гетероатомом цикла (то есть азотом, кислородом или серой), которые обладают валентностями, допускающими замещение. Предпочтительно заместитель связан с атомом углерода цикла.
Использованный здесь термин «галоген» относится к фтору, хлору и брому. Предпочтительным галогеном является фтор.
Использованный здесь термин «алкил», сам по себе или как часть другого заместителя, означает углеводородный радикал с линейной или разветвленной цепочкой, содержащей обозначенное число атомов углерода (например, С1-10алкил означает алкильную группу, содержащую от одного до десяти атомов углерода). Примеры алкильных групп включают в себя метил, этил, н-пропил, изопропил, н-бутил, трет-бутил, изобутил, втор-бутил, пентил, гексил и так далее.
Использованный здесь термин «алкокси», сам по себе или как часть другого заместителя, означает группу О-алкил, в которой алкил определен выше, включая линейные или разветвленные алкильные группы.
Использованный здесь термин «циклоалкил», сам по себе, или как часть другого заместителя, означает насыщенный циклический углеводородный радикал, содержащий обозначенное число атомов углерода (например, С3-8циклоалкил означает циклоалкильную группу, содержащую от трех до восьми атомов углерода).
Использованный здесь термин «фармацевтически приемлемый» относится к молекулярным объектам и композициям, которые «в общем, считаются безопасными», например, которые физиологически переносимы и при введении человеку, как правило, не вызывают аллергической или аналогичной неблагоприятной реакции, такой как расстройство желудка, головокружение и так далее. Предпочтительно, использованный здесь термин «фармацевтически приемлемый» означает одобренный органом государственного регулирования федерального или государственного правительства или упомянутый в Фармакопее США или другой общепринятой фармакопее для использования для животных и, в частности, для людей.
В одном из вариантов осуществления способ изобретения представлен на приведенной ниже схеме 1.
Схема 1
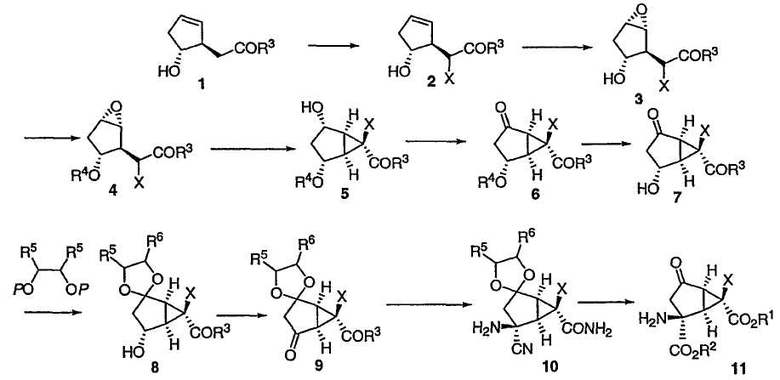
где R1, R2, R3, R4, R5, R6, R7, R8 и Х определены выше.
Оптически активный транс-гидрокси сложный эфир 1 можно получить в соответствии с указаниями Partridge et al., Org. Synth., 1985, 83, 44. Смотри также Tolstikov et. al, J. Org. Chem. USSR, 1989, 25(1.2) и 1990, 26(7.1, 1274). Транс-гидрокси сложный эфир 1 предпочтительно имеет более 90% е/е, более предпочтительно, более 95% е/е и еще более предпочтительно, свыше 96% е/е.
Транс-гидрокси сложный эфир 1 можно фторировать, не защищая вторичную спиртовую группу, получая соединение 2.
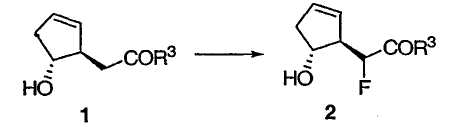
Один из способов достижения требуемого фторирования включает в себя реакцию с фторирующим агентом, таким как N-фторбензолсульфонимид (NFSI) с сильным основанием в подходящем растворителе, например в тетрагидрофуране. Предпочтительно, чтобы данная реакция протекала при температуре ниже -65°С, предпочтительно ниже -75°С, наиболее предпочтительно ниже -78°С. Подходящие сильные основания включают в себя диизопропиламид лития (LDA), тетраметилпиперазид лития, гексаметилдисилазид лития (LHMDS) или соответствующие соли калия или натрия.
После этого можно осуществить стереоселективное эпоксидирование 2 реакцией в толуоле с окислителем, таким как производное перекиси (например, гидроперекись трет-бутила), и катализатором (например, каталитическим количеством ацетилацетоната ванадила (VO(acac)2). Предпочтительно, чтобы реакция протекала при температуре примерно от около 0°С до около 40°С.
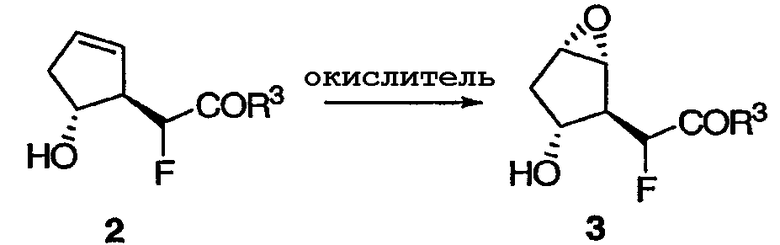
Альтернативные окислители включают в себя мета-хлорпероксибензойную кислоту (mCPBA). Конечный эпоксид 3 получают виде транс-изомера.
Альтернативным образом, транс-гидрокси сложный эфир 1 можно сначала подвергнуть стереоселективному эпоксидированию, а полученный эпоксид 2' фторировать, получая соединение 3.
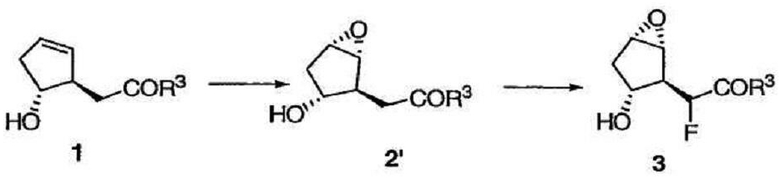
Эпоксидирование можно также осуществить в результате обработки 1 (или фторированного соединения 2) галогенирующим агентом, например NBS или NIS, в подходящем растворителе (например, в смеси ДМСО и воды). После этого соединение 1 образует производное галогенгидрина, которое подвергают циклизации под действием основания (такого, как DBU) с образованием эпоксида.
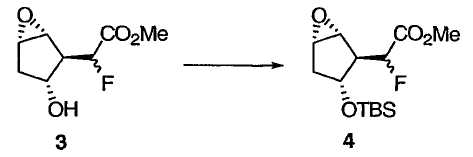
При защите гидроксильной группы 3 защитным агентом R4, например силильным защитным агентом, таким как трет-бутилдиметилсилилхлорид (TBSCl) в подходящих условиях, например в имидазоле и ДМФА, получают защищенное эпоксидное соединение 4, приведенное ниже:
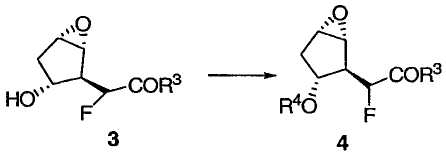
Затем защищенный эпоксид 4 можно ввести во внутримолекулярную реакцию циклопропанирования с раскрытием эпоксида. Данная реакция протекает при добавлении основания в присутствии кислоты Льюиса, такой как Et3Al. Предпочтительно реакция протекает примерно при -50°С.
В предпочтительном варианте осуществления соединение 4 сначала обрабатывают Et3Al, а затем по каплям прибавляют LiHMDS. Данная реакция может протекать в течение от 0,5 до 6 часов при температуре от -20°С до -80°С. Предпочтительное время составляет около 1 часа. Предпочтительная температура составляет примерно -60°С. Альтернативные кислоты Льюиса, которые могут быть использованы в данной реакции, включают в себя RTi(OR)3, R2Ti(OR)2, RAlX2 или R2AlX, где Х является галогеном или неорганическим радикалом, а каждый R представляет собой углеводородную группу. Примеры кислот Льюиса включают в себя Al(OiPr)3, Ti(OiPr)4, эфират BF3, Et2Zn, Et3Al и Sc(OTf)3. Соединение 5 получают в виде требуемой стереоизомерной формы.
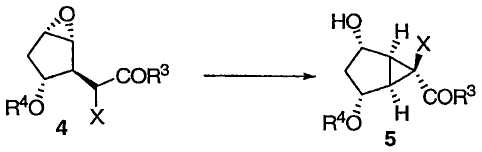
Окисление полученного свободного спирта и снятие защитной группы приводит к бициклическому кетону 7 (соединение II). Предпочтительные окислители включают в себя химически чистый раствор гипохлорита натрия или промышленный отбеливатель. Реакция может протекать в присутствии каталитического количества RuCl3 и в присутствии уксусной кислоты (1,5 эквивалентов) при 0°С в ацетонитриле. Затем избыток гипохлорита натрия следует удалить (например, путем гашения изопропиловым спиртом). Добавление любой кислоты (например, 20 мол.% 1М HCl) к раствору ацетонитрила приводит к снятию защитной группы R4.
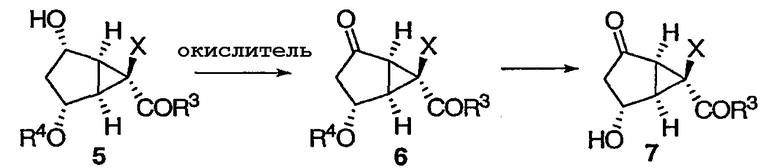
Соединение 7 можно защитить в виде кеталя 8 реакцией с производными диола. Предпочтительной группой R7 является TMS.
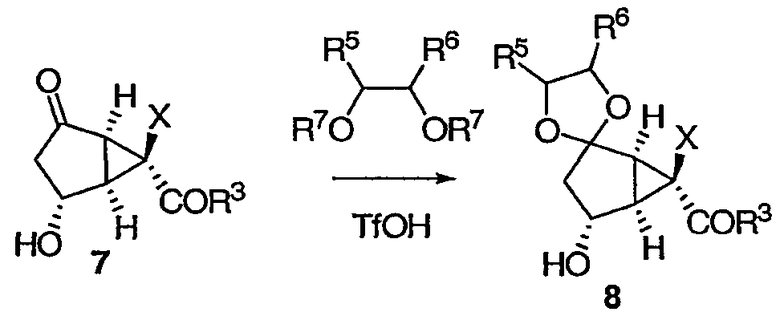
Реакция протекает в присутствии кислоты (например, 0,1 эквивалента) при температуре от около 0°С до около -10°С. Предпочтительной кислотой является TfOH или TfOTMS.
В результате окисления вторичного спирта 8 получают кетон 9.
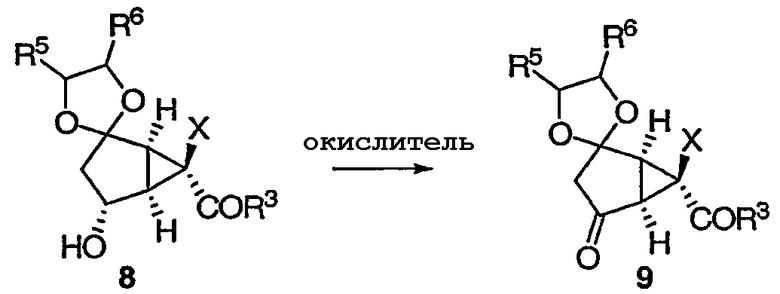
Реакция окисления может протекать при любых условиях окисления, таких как условия Сверна. Альтернативным образом окисление может происходить в присутствии RuCl3 (0,5 мол%) с NaClO в ацетонитриле и уксусной кислоте при температуре от 0°С до комнатной.
Затем соединение 9 вводят в реакцию Штрекера с аммиаком. Данная реакция может протекать в спиртовом растворителе (например, метаноле) с аммиаком при комнатной температуре.
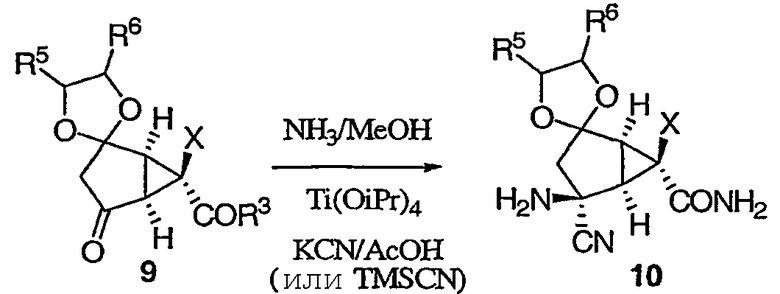
После этого можно добавить TMSCN при температуре от -10°С до 0°С. TMSCN можно заменить на KCN/NaCN в присутствии кислот. Реакция приводит к требуемому аминонитрилу 10 с высокой диастереоселективностью.
Затем соединение 10 подвергают гидролизу, получая желаемый 2-амино-6-фторбицикло[3.1.0]гексан (соединение 11).
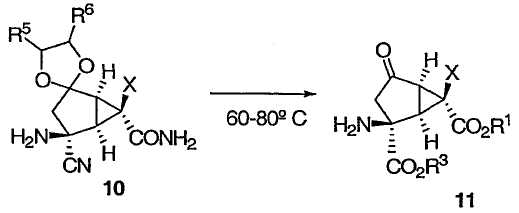
Реакция гидролиза может протекать в течение 5 часов с применением 1:3 смеси уксусной кислоты и 8М HCl при 75°С. Альтернативным образом реакция может протекать в присутствии 60% H2SO4 приблизительно при 100°С в течение примерно 2 часов или альтернативным образом при обработке смесью уксусной кислоты/H2SO4 при 60°С в течение примерно 2 часов.
После этого желаемое соединение 11 можно выделить в виде гидрохлоридной соли при помощи методов, известных специалистам в данной области техники.
В другом варианте осуществления способ изобретения представлен на приведенной ниже схеме 2.
Схема 2
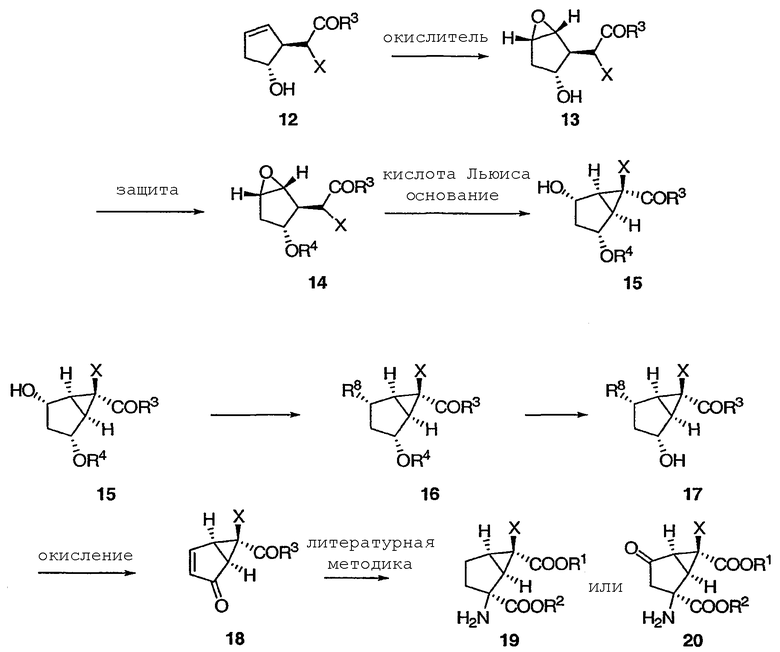
где Х, R3, R4 и R8 определены выше.
На схеме 2 оптически активный транс-гидрокси сложный эфир 12 получали в соответствии с приведенными выше указаниями при описании схемы 1. Эпоксидирование 12 протекает диастереоселективно, приводя к эпоксиду 13, защита гидроксильной группы в 13 приводит к 14, а при обработке 14 кислотой Льюиса с последующей обработкой основанием получают бицикло[3.1.0]соединение 15. Применение энантиомера 12, описанного у Partridge et al., Org. Synth., 1985, 83, 44, приведет к получению энантиомеров 13, 14 и 15.
Монозащищенный [3.1.0]бициклический диол 15 (который идентичен 5 из схемы 1) переводят в [3.1.0]бициклический α,β-ненасыщенный кетон. На этой схеме гидроксильную группу в спирте 15 превращают в уходящую группу R8, а защитную группу R4 удаляют, получая гидроксильный сложный эфир 17. Пригодные уходящие группы R8 включают в себя сульфонат (например, пара-толуолсульфонат) и галогениды. Окисление 17 вызывают элиминированием уходящей группы R8, получая в результате [3.1.0]бициклический α,β-ненасыщенный кетон 18, который можно использовать для синтеза mGluR агонистов 19 (который идентичен 11 из схемы 1) и 20, в соответствии с указаниями патентов США №№ 5750566, 6333428 и 6160009 и Nakazato et al., J. Med. Chem., 2000, 43, 4893-4909.
В другом варианте осуществления способ изобретения изображен на приведенной ниже схеме 3:
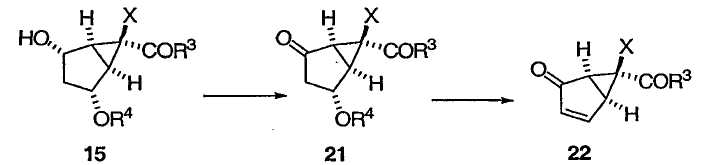
где Х, R3 и R4 определены выше. На схеме 3 представлен синтез энантиомера енона 18 (из схемы 2).
Описанные выше химические структуры включают в себя каждый энантиомер либо в энантиомерно чистой форме, либо в виде смеси.
Исходные вещества и реактивы для описанного здесь способа являются либо коммерчески доступными, либо известны из литературы, либо могут быть получены по литературным методам, описанным для аналогичных соединений. Навыки, необходимые для проведения реакций и очистки полученных продуктов реакции, известны специалистам в данной области техники. Методы очистки включают в себя кристаллизацию, перегонку, хроматографию с нормальной фазой или обращенной фазой.
Следующие примеры приведены только в целях дополнительной иллюстрации и не подразумевают ограничения описываемого изобретения. Примеры 1-10 иллюстрируют способ по схеме 1. Примеры 11-15 иллюстрируют способ по схеме 2. Примеры 16 и 17 иллюстрируют способ по схеме 3.
ПРИМЕР 1
Метилфтор[(1R,5R)-5-гидроксициклопент-2-ен-1-ил]ацетат 2
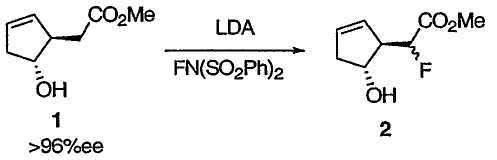
К раствору диизопропиламина (10,8 мл, 76,8 ммоль) в ТГФ (28 мл) прибавляли раствор бутиллития (28,2 мл, 70,4 ммоль, 2,5 М в гексане) в течение 40 мин, поддерживая температуру реакционной массы в интервале от 0 до 5°С. Полученный раствор перемешивали при 0°С в течение 3 мин перед тем, как охладить его до -78°С на бане с сухим льдом и ацетоном. К раствору LDA прибавляли по каплям раствор сложного эфира 1 (5,00 г, 32,0 ммоль) в ТГФ (41,3 мл) в течение 45 мин, поддерживая температуру реакционной массы ниже -73°С, и перемешивали полученный раствор при -78°С в течение 20 мин, получая раствор дианиона оранжевого (или темно-оранжевого) цвета. В отдельную колбу загружали N-фторбензолсульфонимид (14,1 г, 44,8 ммоль) и ТГФ (62 мл) и охлаждали полученный раствор до -96°С на бане с жидким азотом-ацетоном. К суспензии фторирующего реагента прибавляли из капельной воронки раствор дианиона в течение 1 ч, поддерживая температуру реакционной массы примерно при -95°С. Воронку и колбу промывали 2,5 мл ТГФ и сливали его в реакционную смесь. Полученную смесь перемешивали при -96°С в течение 1 ч перед нагреванием до -80°С в течение 30 мин. В течение 7 мин медленно прибавляли уксусную кислоту (11 мл) в ТГФ (5 мл). Смеси давали нагреться до температуры окружающей среды после прибавления МТВЕ (100 мл). Полученный осадок удаляли фильтрованием и тщательно промывали МТВЕ (70 мл × 6). Объединенные фильтрат и промывные фракции снова фильтровали и анализировали методом ВЭЖХ. Химический выход составлял 86%. Фильтрат пропускали через небольшой слой силикагеля (30 г) и промывали этот слой МТВЕ (200 мл). Объединенные растворы МТВЕ концентрировали при пониженном давлении. Остаток растворяли в EtOAc (250 мл) и промывали насыщенным раствором NaHCO3 (170 мл). Водный слой снова экстрагировали EtOAc (60 мл × 2). Объединенные органические растворы промывали насыщенным раствором соли (60 мл) и сушили над Na2SO4. При упаривании растворителя получали неочищенный сложный эфир, который подвергали перегонке из колбы в колбу (1,6 мм рт. ст.), получая сложный эфир в виде желтого масла.
Аналитически чистый образец получали в виде бесцветного масла в результате последующей флэш-хроматографии на колонке с силикагелем.
1Н ЯМР (400 МГц, CDCl3): δ 5,84 (м, 1Н), 5,55 (м, 1Н), 4,95 (дд, J = 48,8, 5,5 Гц, 1Н), 4,49 (дт, J = 7,2, 4,6 Гц, 1Н), 3,82 (с, 3Н), 3,11 (дм, J = 24,4 Гц, 1Н), 2,75 (м, 1Н), 2,51 (с, 1Н), 2,33 (м, 1Н); 13С ЯМР (101 МГц, CDCl3): δ 170,02 (д, J = 24,1 Гц), 132,27, 126,13 (д, J = 5,0 Гц), 89,52 (д, J = 188,0 Гц), 73,92 (д, J = 4,0 Гц), 57,12 (д, J = 20,1 Гц), 52,64, 41,85; 19F ЯМР (376 МГц, CDCl3): -196,5; ИК (пленка) 3409, 3059, 1744, 1439, 1288, 1209, 1153, 1099, 1048, 951, 733 см-1; 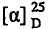 = -123,5 (c 1,02, CHCl3).
= -123,5 (c 1,02, CHCl3).
ПРИМЕР 2
Метилфтор[(1R,2S,3R,5S)-3-гидрокси-6-оксабицикло[3.1.0]гекс-2-ил]ацетат 3
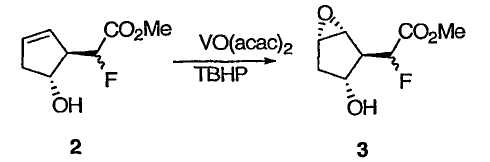
К раствору олефина 2 (1,92 кг, 11,0 моль) в толуоле (4,83 л) прибавляли ацетилацетонат ванадила (VO(acac)2, 58,3 г, 0,22 моль) при 0°С. После прибавления к раствору раствора ТВНР (5,7 М в декане, 38,6 мл) при 0°С полученной суспензии давали нагреться до 14°С. К реакционной смеси медленно прибавляли дополнительное количество раствора ТВНР (5,7 М в декане, 4,36 л) в течение 50 мин, поддерживая температуру смеси в интервале 14-28°С. Полученную суспензию перемешивали еще в течение 2 ч, а затем нагревали при 40°С в течение 8 ч. Избыток ТВНР гасили водным раствором Na2S2O3 (2,95 кг Na2S2O3 и 4,71 кг Н2О), который медленно прибавляли при 0°С. Полученную смесь перемешивали при 20°С в течение 1,5 ч. Исчезновение перекисей подтверждали при помощи индикаторной бумаги. Водный слой отделяли и экстрагировали EtOAc (9,42 л × 2). Объединенные органические растворы промывали насыщенным раствором соли (6,33 л). Солевой слой повторно экстрагировали EtOAc (3,42 л × 4). ГХ проба объединенных органических растворов указывала на присутствие продукта 3. Объединенные органические растворы концентрировали, а полученный остаток очищали хроматографией на силикагеле в чашке фильтра (сначала элюировали смесью гексан/EtOAc (4/1), затем чистым EtOAc). Аналитически чистый образец получали в результате флэш-хроматографии на колонке с силикагелем (гексан/МТВЕ) с последующей перекристаллизацией (EtOAc) в виде бледно-желтых кристаллов: Т.пл. 31-33°С; 1Н ЯМР (400 МГц, CDCl3): δ 5,01 (дд, J = 48,3, 3,9 Гц, 1Н), 4,13 (ушир. с, 1Н), 3,86 (с, 3Н), 3,71 (м, 1Н), 3,59 (м, 1Н), 2,77 (дд, J = 32,8, 3,9 Гц, 1Н), 2,30 (ушир. с, 1Н), 2,11 (м, 2Н); 13С ЯМР (101 МГц,): δ 168,4 (д, J = 24,1 Гц), 88,1 (д, J = 186,1 Гц), 73,2 (д, J = 1,6 Гц), 58,4, 57,1 (д, J = 5,6 Гц), 52,8, 51,6 (д, J = 19,3 Гц), 37,7 (д, J = 1,6 Гц); 19F ЯМР (376 МГц, CDCl3): δ -200,8 (дд, J = 48,3, 32,8 Гц); МСНР m/z 191 (М+1), 189 (М-1), 172 ([М-Н2О]+), 59 ([СООСН3]+, основной пик); = -56 (c 1,0, CHCl3).
C, 50,53; H, 5,83; F, 9,99
ПРИМЕР 2А
Метил[(1R,2S,3R,5S)-3-гидрокси-6-оксабицикло[3.1.0]гекс-2-ил]ацетат
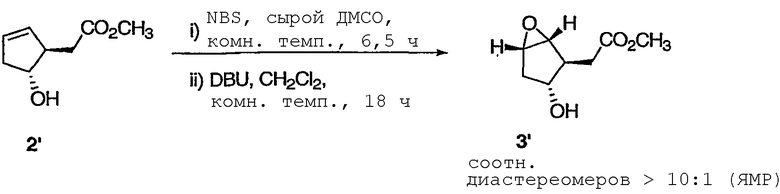
К раствору олефина 2' (50,0 мг, 0,320 ммоль) в сыром ДМСО (6,4 мкл Н2О в 1,2 мл ДМСО) при комнатной температуре прибавляли NBS (68,4 мг, 0,384 ммоль). После перемешивания полученного раствора при комнатной температуре в течение 4,5 ч добавляли еще 10 мг NBS. Реакционную смесь перемешивали еще в течение 2 ч, разбавляли EtOAc и промывали Н2О. Водный слой экстрагировали EtOAc (дважды) и сушили объединенные органические слои над Na2SO4. Растворитель удаляли при пониженном давлении, а полученный остаток растворяли в CH2Cl2 (1,2 мл). Прибавляли DBU (57,4 мкл, 0,384 ммоль) к данному раствору, который перемешивали при комнатной температуре в течение 18 ч. Растворитель выпаривали, а полученный остаток очищали флэш-хроматографией на колонке с силикагелем, получая эпоксид 3' в виде смеси диастереомеров, которая не разделялась хроматографически. Основной диастереомер имеет следующие спектральные данные: 1Н ЯМР (400 МГц, CDCl3): δ 3,80 (дд, J = 11,6, 5,6 Гц, 1Н), 3,72 (с, 3Н), 3,65 (м, 1Н), 3,61 (м, 1Н), 2,68 (дд, J = 8,4, 7,2 Гц, 1Н), 2,36 (д, J = 11,6 Гц, 1Н), 2,26 (дд, J = 15,7, 7,2 Гц, 1Н), 2,20 (дд, J = 15,7, 8,4 Гц, 1Н), 2,11 (д, J = 15,3 Гц, 1Н), 2,02 (дд, J = 15,3, 5,6 Гц, 1Н).
В аналогичных условиях реакции получали также следующие эпоксиды:
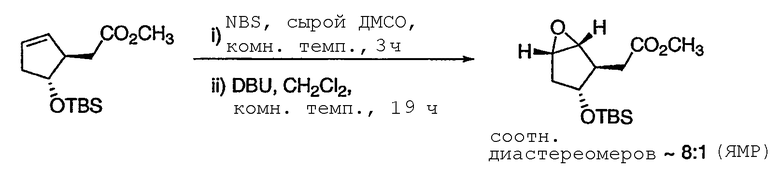
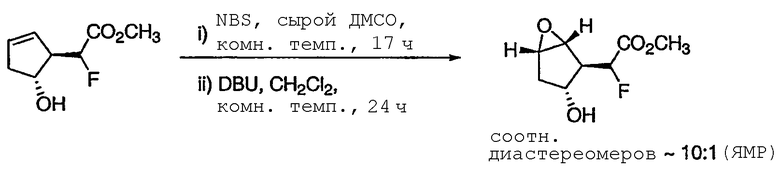
ПРИМЕР 3
Метил[(1R,2R,3R,5S)-3-{[трет-бутил(диметил)силил]окси}-6-оксабицикло[3.1.0]гекс-2-ил)фторацетат 4
К раствору эпоксиспирта 3 (1,60 кг, 8,40 моль) и ДМФА (3,40 л) прибавляли имидазол (1,26 кг, 18,5 моль) при 10°С. К реакционной смеси добавляли TBSCl (1,52 кг, 10,1 моль), поддерживая температуру смеси ниже 8°С. Полученный раствор перемешивали при 5°С в течение 10 мин, затем давали нагреться до 20°С в течение 30 мин и перемешивали 2 ч при той же температуре. Расходование исходного спирта контролировали методом ГХ и разбавляли реакционную смесь холодным толуолом (17,0 л, 5°С). Полученную смесь промывали Н2О (5,67 л), насыщенным водным раствором NaHCO3 (5,67 л), Н2О (5,67 л × 2) и насыщенным раствором соли (5,67 л). Проба органического раствора указывала на присутствие 4. В результате концентрирования раствора получали 4 в виде желтой жидкости, которую использовали на следующей стадии без дополнительной очистки. Аналитически чистый образец получали колоночной флэш-хроматографией на силикагеле (гексан/МТВЕ) в виде бесцветных кристаллов: Т.пл. 28-30°С; 1Н ЯМР (400 МГц, CDCl3): δ 5,00 (дд, J = 48,2, 3,5 Гц, 1Н), 4,45 (м, 1Н), 3,85 (с, 3Н), 3,51 (м, 1Н), 3,42 (м, 1Н), 2,64-2,52 (дм, J = 34,5 Гц, 1Н), 2,14 (м, 1Н), 1,91 (м, 1Н), 0,88 (с, 9Н), 0,054 (с, 3Н) и 0,051 (с, 3Н); 13С ЯМР (101 МГц, CDCl3): δ 168,8 (д, J = 24,1 Гц), 88,3 (д, J = 186,1 Гц), 75,4 (д, J = 1,6 Гц), 58,3, 57,2 (д, J = 7,2 Гц), 52,8 (д, J = 19,3 Гц), 52,7, 38,3, 25,9, 18,0, -4,5 и -4,7; 19F ЯМР (376 МГц, CDCl3): δ -199,9 (дд, J = 48,2, 34,5 Гц); МСНР m/z 305 (М+1), 121 (основной пик); = -27 (c 1,0, CHCl3).
C, 55,23; H, 8,28; F, 6,24
ПРИМЕР 4
Метил(1R,2R,4S,5S,6R)-2-{[трет-бутил(диметил)силил]окси}-6-фтор-4-гидроксибицикло[3.1.0]гексан-6-карбоксилат 5
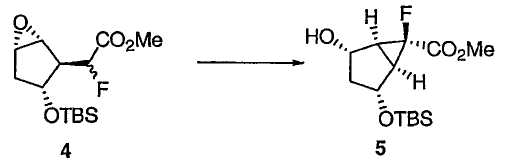
К раствору эпоксида TBS-эфира 4 (масса пробы 1,60 кг, 5,24 моль) в ТГФ (16,1 л) прибавляли раствор Et3Al (1,0 М в гексане, 6,81 л, 6,81 моль), поддерживая температуру смеси при -60°С в течение 1 ч, и перемешивали полученный раствор при -60°С в течение 20 мин. К реакционной смеси прибавляли LHMDS (1,0 М раствор в гексане, 7,86 л, 7,86 моль) в течение 1 ч, поддерживая температуру смеси при -60°С, и реакционную смесь выдерживали при -60°С. Протекание реакции контролировали методом ГХ. После полного израсходования эпоксида (6 ч) прибавляли водный раствор лимонной кислоты (3 М, 10,5 л) в течение 1 ч, поддерживая температуру смеси при -50°С. После добавления МТВЕ (12,4 л) полученной суспензии давали постепенно нагреться до 15°С при перемешивании. После добавления Н2О (4,93 л) смесь превращалась в двухфазный раствор. Органический слой отделяли и дважды промывали насыщенным раствором NaHCO3 (11,1 л, затем 5,6 л). ГХ проба органического раствора указывала на присутствие соединения 5. В результате концентрирования органического слоя получали сырой спирт в виде желтого масла, которое использовали в следующей реакции без дополнительной очистки.
Аналитически чистый образец получали колоночной флэш-хроматографией на силикагеле в виде бесцветного аморфного твердого вещества: 1Н ЯМР (400 МГц, CDCl3): δ 4,47 (д, J = 4,4 Гц, 1Н), 4,34 (м, 1Н), 3,83 (с, 3Н), 2,44 (д, J = 6,8 Гц, 1Н), 2,37 (д, J = 11,2 Гц, 1Н), 2,25 (д, J = 6,8 Гц, 1Н), 2,07 (м, 1Н), 1,84 (м, 1Н), 0,91 (с, 9Н), 0,131 (с, 3Н) и 0,128 (с, 3Н); 13С ЯМР (101 МГц, CDCl3): δ 169,2 (д, J = 26,5 Гц), 79,7 (д, J = 244,3 Гц), 74,1, 74,0, 52,9, 44,6 (д, J = 10,4 Гц), 37,9 (д, J = 11,2 Гц), 25,8, 18,0, -4,8, -4,9; 19F ЯМР (376 МГц, CDCl3): δ -217,1 (м); МСНР m/z 305 (М+1), 304 (М), 303 (М-1), 75 (основной пик); = +7 (c 1,1, CHCl3).
C, 55,23; H, 8,28; F, 6,24
ПРИМЕР 5
Метил(1R,2R,5S,6S)-2-{[трет-бутил(диметил)силил]окси}-6-фтор-4-оксобицикло[3.1.0]гексан-6-карбоксилат 6
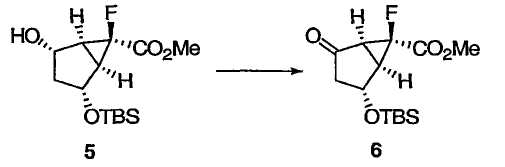
К раствору бициклического моно-TBS-диола 5 (2,08 кг; 6,83 моль) в ацетонитриле (8,0 л) при -5°С прибавляли уксусную кислоту (0,70 л) и воду (2,5 л), затем гидрат RuCl3 (14,20 г). К этой смеси прибавляли водный раствор гипохлорита натрия (~13%; 7,0 л), в течение 2 ч, поддерживая температуру около 0°С. Полученную смесь перемешивали при 0°С еще 1 ч до тех пор, пока весь бициклический моно-TBS-диол 5 не исчез, что контролировали методами ТСХ и ЯМР. Избыток водного гипохлорита натрия разлагали добавлением изопропанола (0,70 л), выдерживали при 0°С в течение 15 мин. Два слоя разделяли и водный слой отбрасывали. Раствор использовали на следующей стадии без дополнительной очистки. Аналитически чистый образец получали колоночной флэш-хроматографией на силикагеле (МТВЕ/гексан) в виде бесцветных кристаллов: Т.пл. 70-71°С; 1Н ЯМР (400 МГц, CDCl3): δ 4,66 (д, J = 5,4 Гц, 1Н), 3,86 (с, 3Н), 3,73 (с, 3Н), 2,73 (м, 2Н), 2,54 (дт, J = 19,1, 5,7 Гц, 1Н), 2,22 (дд, J = 19,1, 3,8 Гц, 1Н), 0,91 (с, 9Н), 0,13 (с, 3Н), 0,11 (с, 3Н); 13С ЯМР (101 МГц, CDCl3): δ 206,2, 167,1 (д, J = 26,1 Гц), 78,9 (д, J = 246,4 Гц), 67,6 (д, J = 2,8 Гц), 53,4, 47,5 (д, J = 3,9 Гц), 42,0 (д, J = 11,4 Гц), 39,6 (д, J = 13,3 Гц), 25,7, 18,0, -4,76, -4,78; 19F ЯМР (376 МГц, CDCl3): δ -210,7 (м); = +58,2 (c 0,50, CH3OH).
C, 55,60; H, 7,67; F, 6,28
ПРИМЕР 6
Метил(1R,2R,5S,6S)-6-фтор-2-гидрокси-4-оксобицикло[3.1.0]гексан-6-карбоксилат 7

Упомянутый выше органический слой, содержащий TBS-кетон 6 (6,83 моль), нагревали до 22°С и добавляли 1 М HCl (1,37 л). Смесь перемешивали при 22-24°С в течение 3,5 ч до удаления всех TBS групп. К данной смеси прибавляли насыщенный раствор гидрокарбоната натрия (4,8 л). Смесь перемешивали в течение 15 мин, разбавляли изопропилацетатом (20 л) и отделяли органический слой. Водный слой снова экстрагировали изопропилацетатом (6 л). Объединенные органические слои концентрировали досуха и очищали соединение хроматографией на силикагеле в чашке фильтра (сначала элюировали 30% МТВЕ в гексане, затем только МТВЕ), получая соединение 7 в виде почти белых кристаллов. Аналитически чистый образец получали дополнительной колоночной флэш-хроматографией на силикагеле в виде бесцветных кристаллов: Т.пл. 61-62°С; 1Н ЯМР (400 МГц, CDCl3): δ 4,92 (ушир. с, 1Н), 3,85 (с, 3Н), 2,86 (дд, J = 6,2, 2,1 Гц, 1Н), 2,71 (д, J = 6,2 Гц, 1Н), 2,61 (дт, J = 19,4, 5,7 Гц, 1Н), 2,59 (ушир. с, 1Н), 2,30 (дд, J = 19,4, 3,7 Гц, 1Н); 13С ЯМР (100 МГц, CDCl3): δ 206,9, 167,0 (д, J = 26,2 Гц), 79,0 (д, J = 246,6 Гц), 67,0 (д, J = 3,1 Гц), 53,5, 46,8 (д, J = 4,2 Гц), 41,6 (д, J = 11,8 Гц), 39,4 (д, J = 13,1 Гц); 19F ЯМР (376 МГц, CDCl3): δ -210,6; = +77 (c 0,50, CH3OH).
C, 51,07; H, 4,82; F, 10,10
ПРИМЕР 6А
Метил(1R,2R,5R,6R)-2-гидрокси-4-оксобицикло[3.1.0]гексан-6-карбоксилат
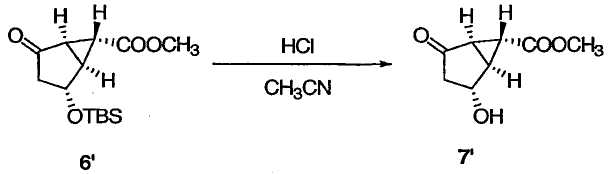
TBS-эфир 6' (150 мг, 0,528 ммоль) обрабатывали 1М HCl (0,106 мл) в ацетонитриле (0,8 мл) при комнатной температуре в течение 2 ч. Реакционную смесь разбавляли EtOAc, гасили добавлением небольшого количества насыщенного водн. NaHCO3, промывали Н2О и насыщенным водным раствором соли (дважды) и сушили над Na2SO4. Растворители удаляли при пониженном давлении, а полученный остаток очищали колоночной флэш-хроматографией на силикагеле, получая соединение 7' в виде бесцветного твердого вещества: 1Н ЯМР (400 МГц, CDCl3): δ 4,60 (д, J = 5,2 Гц, 1Н), 3,72 (с, 3Н), 2,67 (дд, J = 5,2, 3,6 Гц, 1Н), 2,42 (дд, J = 5,2, 2,4 Гц, 1Н), 2,34 (дд, J = 18,9, 5,27 Гц, 1Н), 2,22 (ушир. с, 1Н), 2,08 (д, J = 18,9 Гц, 1Н), 1,93 (дд, J = 3,6, 2,4 Гц, 1Н); 13С ЯМР (101 МГц, CDCl3): δ 208,8, 169,8, 68,3, 52,5, 42,7, 36,2, 34,2, 25,2.
ПРИМЕР 7
Метил(1S,4R,4'S,5R,5'S,6S)-6-фтор-4-гидрокси-4',5'-дифенилспиро[бицикло[3.1.0]гексан-2,2'-[1.3]диоксолан]-6-карбоксилат 8
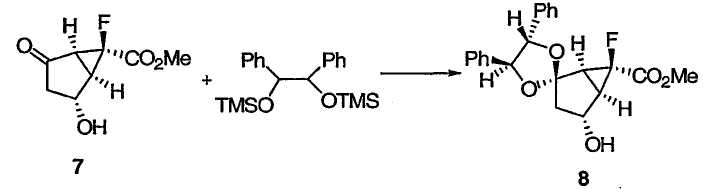
К раствору гидроксикетона 7 (1,09 кг; 5,76 моль) и CH2Cl2 (7,7 л) прибавляли раствор (S,S)-бис-О-TMS-гидробензоина (2,01 кг по анализу; 5,60 моль) и CH2Cl2 (2,55 л). Раствор охлаждали до -20°С. Через капельную воронку загружали TfOH (50,9 мл; 0,576 моль) в течение 4 мин при -15~-20°С. Раствор нагревали до -10°С и выдерживали при -10°С в течение 1,5 ч. К реакционной смеси при -10°С прибавляли дополнительное количество раствора (S,S)-бис-О-TMS-гидробензоина (107 г по анализу; 0,298 моль) в CH2Cl2 (188 г). Реакция завершалась после 30-минутной дополнительногй выдержки при -10°С. Реакционную смесь гасили добавлением пиридина (46,9 мл; 0,576 моль) при <-15°С. Раствор нагревали до -10°С, промывали по очереди 5 мас.% холодным водным раствором NaHCO3 (3,75 л), 1 М холодным водным HCl (8,6 л), 5 мас.% холодным водным раствором NaHCO3 (3,75 л) и 10 мас.% холодным водным NaCl (5,0 л), сушили над Na2SO4 (1,5 кг). Растворитель органического слоя заменяли на ацетонитрил и использовали в следующей реакции без дополнительной очистки. Анализ данного раствора методом ВЭЖХ на этой стадии указывал на наличие кетального спирта 8. Аналитически чистый образец получали колоночной флэш-хроматографией на силикагеле в виде бесцветных кристаллов: Т.пл. 118-120°С; 1Н ЯМР (401 МГц, CDCl3): δ 7,38-7,21 (м, 10Н), 4,89 (д, J = 8,3 Гц, 1Н), 4,83 (д, J = 8,3 Гц, 1Н), 4,51 (ушир. с, 1Н), 3,89 (с, 3Н), 2,54-2,51 (м, 2Н), 2,43-2,37 (м, 2Н), 2,18 (ушир. с, 1Н); 13С ЯМР (101 МГц, CDCl3): δ 168,7 (д, J = 26,2 Гц), 136,6, 135,8, 128,7, 128,6, 128,5, 128,4, 126,9, 126,3, 117,7, 86,2, 86,1, 77,6 (д, J = 247,1 Гц), 71,1, 53,0, 45,7 (д, J = 7,8 Гц), 37,5 (д, J = 12,1 Гц), 36,7 (д, J = 11,9 Гц); 19F ЯМР (377 МГц, CDCl3): δ -216,3.
ПРИМЕР 8
Метил(1S,4'S,5R,5'S,6S)-6-фтор-4-оксо-4',5'-дифенилспиро[бицикло[3.1.0]гексан-2,2'-[1.3]диоксолан]-6-карбоксилат 9
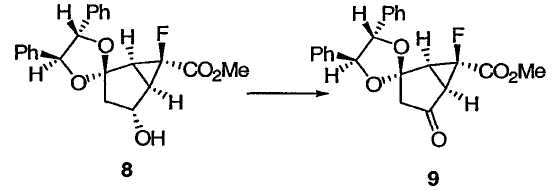
К раствору гидроксикеталя 8 (2,04 кг по анализу, 5,31 моль) в ацетонитриле (36,7 л) прибавляли гидрат RuCl3 (8,25 г), затем воду (2,0 л) и уксусную кислоту (0,41 л) при 0°С. К реакционному раствору медленно в течение 19 мин прибавляли водный раствор гипохлорита натрия (~13%; 5,37 л), поддерживая температуру реакционной смеси ниже 4°С. Раствор выдерживали при 0-3,5°С в течение 2 ч. Реакционную смесь гасили добавлением изопропанола (2,2 л) при 3,5°С. Через 30 мин выдерживания при той же температуре к смеси добавляли холодный водный NaHCO3 (5 мас.%, 10,7 л) в течение 12 мин в интервале от 0,4 до 3,3°С. Полученную взвесь перемешивали в течение 30 мин при 3°С и отфильтровывали продукт 9. Сырой остаток на фильтре промывали холодной водой (2 л × 2) и сушили, получая первую порцию кетального кетона 9. Фильтрат и промывные воды объединяли и разделяли слои. Органический слой концентрировали в вакууме. Полученную взвесь фильтровали. Остаток на фильтре промывали водой (0,48 л × 2) и перекристаллизовывали из ацетонитрила (1,8 л) и воды (1,08 л), получая вторую порцию кетального кетона 9. Аналитически чистый образец получали колоночной флэш-хроматографией на силикагеле в виде бесцветных кристаллов: Т.пл. 58,5-59,5°С; 1Н ЯМР (401 МГц, CDCl3): δ 7,40-7,34 (м, 6Н), 7,28-7,25 (м, 4Н), 4,97 (д, J = 8,4 Гц, 1Н), 4,88 (д, J = 8,4 Гц, 1Н), 3,93 (с, 3Н), 3,10 (дд, J = 6,4, 2,0 Гц, 1Н), 2,94 (д, J = 4,0 Гц, 2Н), 2,87 (д, J = 6,4 Гц, 1Н); 13С ЯМР (101 МГц, CDCl3): δ 201,6, 166,9 (д, J = 25,7 Гц), 136,1, 135,3, 129,0, 128,8, 128,72, 128,69, 126,8, 126,5, 110,8, 86,3, 85,8, 78,9 (д, J = 251,6 Гц), 53,6, 48,3 (д, J = 3,3 Гц), 42,2 (д, J = 13,2 Гц), 41,7 (д, J = 12,0 Гц); 19F ЯМР (376 МГц, CDCl3): δ -208,5.
ПРИМЕР 9
(1S,4'S,5R,5'S,6S)-4-Амино-4-циано-6-фтор-4',5'-дифенилспиро[бицикло[3.1.0]гексан-2,2'-[1.3]диоксолан]-6-карбоксамид 10
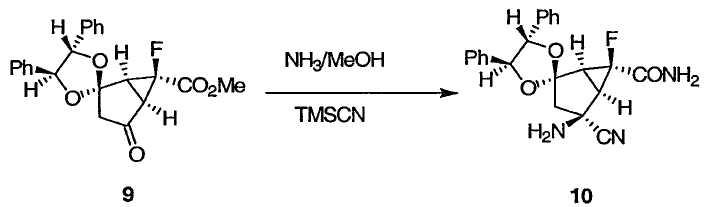
К раствору 7М аммиака в метаноле (7,4 л, 47,8 моль) и Ti(OiPr)4 (1,77 л, 5,93 моль) при 23°С прибавляли кетальный кетон 9 (2,11 кг, 1,89 кг в виде чистого 9, 4,94 моль). Смесь перемешивали в течение 4 ч при 20-23°С. Смесь охлаждали до -12°С и добавляли TMSCN (505 г, 5,09 моль). Смесь нагревали до -4,5°С и перемешивали при этой температуре в течение 16 ч. Смесь фильтровали и промывали кристаллы холодным МеОН (7,0 л) и сушили при 20-25°С при пониженном давлении, получая аминонитрил 10 в виде бесцветного твердого вещества. Аналитически чистый образец получали колоночной хроматографией на силикагеле в виде бесцветных кристаллов: Т.пл. 196,9-197,4°С; 1Н ЯМР (400 МГц, ДМСО-d6): δ 8,04 (с, 1Н), 7,78 (с, 1Н), 7,38-7,25 (м, 10Н), 5,15 (д, J = 8,8 Гц, 1Н), 4,81 (д, J = 8,8 Гц, 1Н), 2,86 (с, 2Н), 2,78 (дд, J = 14,5, 3,2 Гц, 1Н), 2,63 (д, J = 6,8 Гц, 1Н), 2,46 (д, J = 6,8 Гц, 1Н), 2,23 (дд, J = 14,5, 4,4 Гц, 1Н); 13С ЯМР (101 МГц, ДМСО-d6): δ 168,7 (д, J = 23,3 Гц), 136,5, 135,9, 128,6, 128,5, 128,5, 127,1, 126,9, 123,4, 115,1, 84,7, 84,3, 81,1 (д, J = 255,4 Гц), 54,6, 48,3 (д, J = 7,2 Гц), 36,6 (д, J = 11,2 Гц) и 35,9 (д, J = 10,4 Гц); 19F ЯМР (377 МГц, ДМСО-d6): δ -211,6.
ПРИМЕР 10
(1R,2S,5S,6S)-2-Амино-6-фтор-4-оксобицикло[3.1.0]гексан-2,6-дикарбоновая кислота 11
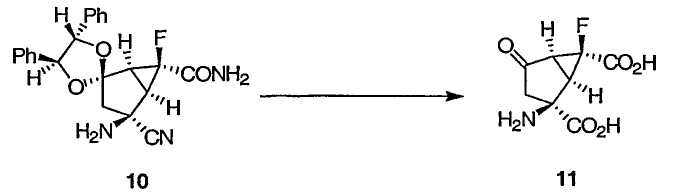
Смесь аминонитрила 10 (1,63 кг неочищенного, 1,55 кг из расчета чистого), НОАс (3,25 л), Н2О (3,25 л) и конц. HCl (6,50 л) нагревали до 75±2°С в течение 4 ч. По данным ЯМР 19F реакция завершалась. Раствор охлаждали до 18°С и экстрагировали CH2Cl2 (1 × 9 л и 2 × 5 л). Водный слой концентрировали при 10-25 мм рт. ст. и температуре смеси 50°С до ~2л. Полученную взвесь охлаждали до 0°С и перемешивали 1 ч. Охлажденную взвесь фильтровали, а остаток с фильтра, содержащий соль HCl продукта 11, выдерживали в условиях фильтрования под вакуумом в течение 5-10 мин для удаления наибольшего возможного количества фильтрата. Упомянутый выше остаток с фильтра соли HCl добавляли в воду (5,0 л) при 65°С и промывали горячей водой (300 мл). Раствору давали остыть до 17°С в течение 45 мин. Доводили рН до 1,25 50%-ным NaOH (230 мл). Суспензию охлаждали до 0°С и перемешивали в течение 45 мин. Взвесь фильтровали, промывали Н2О (2 × 1 л) и сушили в атмосфере азота, получая почти белый кристаллический продукт 11 в виде моногидрата. Аналитически чистую соль HCl 11 получали из 20%-ного HCl: Т.пл. 195-220°С (разл); 1Н ЯМР (401 МГц, ДМСО-d6): δ 8,99 (с, 2Н), 3,08 (дд, J = 6,4, 1,6 Гц, 1Н), 3,02 (д, J = 6,4 Гц, 1Н), 2,86 (дд, J = 18,5, 3,6 Гц, 1Н), 2,57 (дд, J = 18,5, 4,8 Гц, 1Н); 13С ЯМР (101 МГц, ДМСО-d6): δ 201,3 (д, J = 2,7 Гц), 170,4, 166,3 (д, J = 25,7 Гц), 78,9 (д, J = 247,0 Гц), 58,1 (д, J = 1,5 Гц), 40,6 (д, J = 13,1 Гц), 36,8 (д, J = 11,1 Гц); 19F ЯМР (377 МГц, ДМСО-d6): δ -204,8; Титрование Cl 13,96% (Теория 13,98%).
ПРИМЕР 11
Метил((1R,2R,3R,5S)-3-{[трет-бутил(диметил)силил]окси}-6-оксабицикло[3.1.0]гекс-2-ил)ацетат
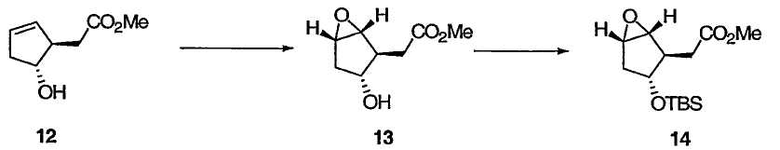
К раствору олефина 12 (4,25 г, 27,2 ммоль) в толуоле (10,8 мл) прибавляли ацетилацетонат ванадила (VO(acac)2, 289 мг, 1,09 ммоль, 4 мол%). В течение 30 мин добавляли раствор ТВНР (14,3 мл, 81,6 ммоль, 5,7 М в декане), поддерживая температуру реакционной массы ниже 28°С. Полученную смесь перемешивали при комнатной температуре в течение 5,5 ч и гасили добавлением насыщенного водн. Na2S2O3. Водный слой отделяли и экстрагировали этилацетатом (х 5). Объединенные органические слои промывали насыщенным раствором соли и сушили над Na2SO4. Растворители выпаривали, а полученный остаток очищали флэш-хроматографией на силикагеле, получая эпокси-спирт 13 в виде бесцветной жидкости, содержащей неотделяемые побочные продукты. Данный спирт (3,21 г) обрабатывали имидазолом (2,78 г, 40,9 ммоль) и TBSCl (3,36 г, 22,3 ммоль) в ДМФА (7,2 мл) при температуре окружающей среды для того, чтобы перевести гидроксильную группу в TBS-эфир. Реакционную смесь перемешивали при комнатной температуре в течение 2,5 ч, а затем обрабатывали МТВЕ (36 мл) и Н2О (12 мл). Органический слой отделяли, промывали насыщенным водн. NaHCO3, Н2О и насыщенным раствором соли и сушили над Na2SO4. Растворитель выпаривали, а полученный остаток очищали флэш-хроматографией на силикагеле, получая TBS-эфир 14 в виде бесцветной жидкости: 1Н ЯМР (400 МГц, CDCl3): δ 4,08 (м, 1Н), 3,72 (с, 3Н), 3,49 (м, 1Н), 3,37 (м, 1Н), 2,49 (м, 1Н), 2,31 (д, J = 7,2 Гц, 1Н), 2,31 (м, 1Н), 2,09 (м, 1Н), 1,93 (м, 1Н), 0,88 (с, 9Н), 0,04 (с, 3Н), 0,03 (с, 3Н); 13С ЯМР (101 МГц, CDCl3): δ 171,9, 77,0, 60,4, 57,4, 51,7, 46,4, 37,2, 34,6, 25,8, 18,0, -4,7; МСНР m/z 287 (М+1), 286 (М), 285 (М-1), 169 (основной пик).
C, 58,70; H, 9,15
ПРИМЕР 12
Метил(1S,2R,4S,5R,6S)-2-{[трет-бутил(диметил)силил]окси}-4-гидроксибицикло[3.1.0]гексан-6-карбоксилат
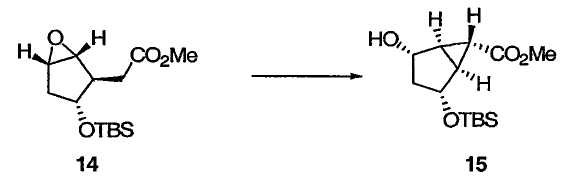
К раствору эпоксида 14 (3,52 г, 12,3 ммоль) в ТГФ (37,8 мл) при -70°С прибавляли раствор Et3Al (16,0 мл, 16,0 ммоль, 1 М в гексане). После перемешивания полученного раствора при -70°С в течение 10 мин медленно, в течение 30 мин, прибавляли раствор LHMDS (18,4 мл, 18,4 ммоль, 1 М в гексане). Полученный раствор перемешивали при -70°С в течение 100 мин и гасили добавлением водн. лимонной кислоты (24,9 мл, 3 М). После добавления толуола (24,9 мл) полученной смеси давали нагреться до температуры окружающей среды и добавляли Н2О (11,7 мл). Водный слой отделяли и экстрагировали МТВЕ (20 мл). Объединенные органические слои промывали насыщенным водн. NaHCO3 (36 мл × 2) и насыщенным раствором соли и сушили над Na2SO4. Растворитель выпаривали, а полученный остаток очищали колоночной флэш-хроматографией на силикагеле, получая бициклический спирт 15 в виде бесцветного масла: 1Н ЯМР (400 МГц, CDCl3): δ 4,34 (д, J = 4,4 Гц, 1Н), 4,18 (дд, J = 11,6, 4,4 Гц, 1Н), 3,68 (с, 3Н), 2,46 (д, J = 11,6 Гц, 1Н), 2,26 (дд, J = 6,0, 2,8 Гц, 1Н), 2,10 (дд, J = 6,0, 2,8 Гц, 1Н), 1,67 (д, J = 15,3 Гц, 1Н), 1,49 (дт, J = 15,3, 4,4 Гц, 1Н), 1,16 (т, J = 2,8 Гц, 1Н), 0,90 (с, 9Н), 0,13 (с, 3Н), 0,11 (с, 3Н); 13С ЯМР (101 МГц, CDCl3): δ 172,2, 73,8, 73,6, 51,9, 40,3, 33,3, 33,0, 25,7, 21,8, 17,9, -4,7, -5,0; МСНР m/z 287 (М+1), 286 (М), 285 (М-1), 169 (основной пик).
C, 58,70; H, 9,15
ПРИМЕР 13
Метил(1S,2R,4S,5R,6R)-2-{[трет-бутил(диметил)силил]окси}-4-{[(4-метилфенил)сульфонил]окси}бицикло[3.1.0]гексан-6-карбоксилат

К перемешиваемому раствору спирта 15 (929 мг, 3,24 ммоль) в CH2Cl2 (3,8 мл) при 0°С прибавляли пиридин (2,62 мл, 32,4 ммоль) и п-толуолсульфонилхлорид (1,24 г, 6,49 ммоль). Полученной смеси давали нагреться до температуры окружающей среды и перемешивали при той же температуре в течение 15 ч. К реакционной смеси прибавляли насыщенный водн. NaHCO3 (5 мл) и перемешивали полученную смесь при комнатной температуре в течение 1 ч. Водный слой отделяли и экстрагировали МТВЕ (10 мл × 2). Объединенный органический слой промывали 1 М HCl (40 мл), насыщенным водн. NaHCO3 (10 мл) и насыщенным раствором соли (10 мл) и сушили над Na2SO4. Растворитель выпаривали, а полученный остаток очищали колоночной флэш-хроматографией на силикагеле, получая п-толуолсульфонатный эфир 16 в виде бесцветного твердого вещества: 1Н ЯМР (400 МГц, CDCl3): δ 7,81 (д, J = 8,0 Гц, 2Н), 7,33 (д, J = 8,0 Гц, 2Н), 5,02 (д, J = 5,2 Гц, 1Н), 4,27 (д, J = 4,8 Гц, 1Н), 3,65 (с, 3Н), 2,45 (с, 3Н), 2,30 (дд, J = 5,6, 2,8 Гц, 1Н), 2,15 (дд, J = 5,6, 3,2 Гц, 1Н), 1,85 (д, J = 16,5 Гц, 1Н), 1,64 (ддд, J = 16,5, 5,2, 4,8 Гц, 1Н), 1,06 (дд, J = 3,2, 2,8 Гц, 1Н), 0,86 (с, 9Н), 0,07 (с, 3Н), 0,04 (с, 3Н); 13С ЯМР (101 МГц, CDCl3): δ 171,4, 144,5, 134,5, 129,7, 127,6, 82,4, 72,7, 52,0, 40,0, 34,8, 31,3, 25,7, 21,6, 21,1, 17,9, -4,7, -4.
ПРИМЕР 14
Метил(1R,2R,4S,5R,6R)-2-гидрокси-4-{[(4-метилфенилсульфонил]окси}бицикло[3.1.0]гексан-6-карбоксилат
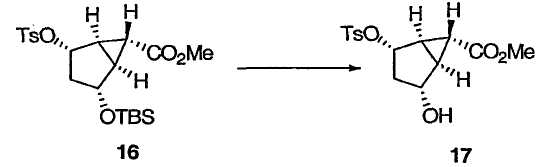
TBS-эфир 16 (1,86 г, 4,22 ммоль) обрабатывали 0,84 мл водн. HCl (1 М) в ацетонитриле (9,4 мл) при комнатной температуре в течение 4 ч. Реакционную смесь гасили добавлением насыщенного водн. NaHCO3 (8,7 мл) и МТВЕ (20 мл). Водный слой отделяли и экстрагировали МТВЕ (10 мл × 2). Объединенный органический слой сушили над Na2SO4 и концентрировали при пониженном давлении. В результате обработки полученного остатка гексаном получали кристаллы, которые фильтровали и перекристаллизовывали из смеси гексан/EtOAc, получая чистый спирт 17 в виде бесцветных кристаллов: 1Н ЯМР (400 МГц, CDCl3): δ 7,82 (д, J = 8,0 Гц, 2Н), 7,38 (д, J = 8,0 Гц, 2Н), 5,01 (д, J = 5,2 Гц, 1Н), 4,24 (д, J = 5,2 Гц, 1Н), 3,67 (с, 3Н), 2,47 (с, 3Н), 2,33-2,28 (м, 2Н), 1,93 (д, J = 16,5 Гц, 1Н), 1,67 (дт, J = 16,5, 5,2 Гц, 1Н), 1,16 (т, J = 3,0 Гц, 1Н); 13С ЯМР (101 МГц, CDCl3): δ 171,1, 145,1, 133,9, 130,1, 127,8, 83,2, 72,7, 52,2, 39,3, 33,9, 30,8, 21,8, 21,7.
ПРИМЕР 15
Метил(1R,5S,6S)-4-оксобицикло[3.1.0]гекс-2-ен-6-карбоксилат
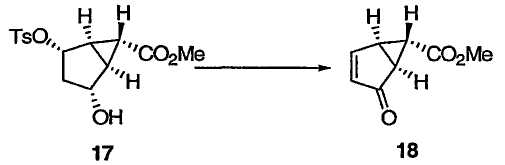
К раствору ДМСО (0,404 мл, 5,70 ммоль) в CH2Cl2 (2,6 мл) прибавляли раствор трифторуксусного ангидрида (0,604 мл, 4,28 ммоль) в CH2Cl2 (1,5 мл) при -78°С. Полученный раствор перемешивали при -78°С в течение 30 мин и добавляли раствор спирта 17 (0,885 г, 2,85 ммоль) в CH2Cl2 (4,1 мл) (колбу промывали 1,0 мл CH2Cl2). После перемешивания полученного раствора при -78°С в течение 30 мин медленно прибавляли Et3N (1,59 мл, 11,4 ммоль). Полученную смесь перемешивали при -78°С в течение 2,5 ч и гасили реакционную смесь добавлением Н2О (5 мл). После добавления МТВЕ (10 мл) полученной смеси давали нагреться до комнатной температуры, а водный слой отделяли и экстрагировали МТВЕ (10 мл). Объединенный органический слой промывали 1М HCl (15 мл), насыщенным водным NaHCO3 (10 мл), Н2О (10 мл) и насыщенным раствором соли (10 мл) и сушили над Na2SO4. Растворитель выпаривали и очищали полученный остаток колоночной флэш-хроматографией на силикагеле, получая α,β-ненасыщенный кетон 18 в виде бледно-желтых кристаллов: 1Н ЯМР (400 МГц, CDCl3): δ 7,61 (ддд, J = 5,6, 2,4, 0,8 Гц, 1Н), 5,74 (д, J = 5,6 Гц, 1Н), 3,71 (с, 3Н), 2,96 (м, 1Н), 2,62, (м, 1Н), 2,27 (м, 1Н); 13С ЯМР (101 МГц, CDCl3): δ 203,1, 168,4, 159,5, 129,7, 52,3, 45,4, 30,0, 28,9.
ПРИМЕР 16
Метил(1S,2R,5R,6R)-2-{[трет-бутил(диметил)силил]окси}-4-оксобицикло[3.1.0]гексан-6-карбоксилат (21)
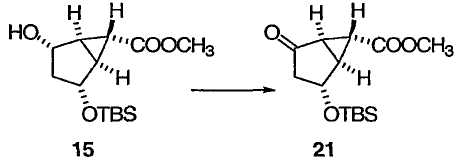
К раствору ДМСО (0,358 мл, 5,04 ммоль) в CH2Cl2 (2,5 мл) прибавляли по каплям раствор трифторуксусного ангидрида (0,534 мл, 3,78 ммоль) в CH2Cl2 (1,3 мл), поддерживая температуру реакционной смеси ниже -70°С. Полученный раствор перемешивали при -78°С в течение 55 мин. Прибавляли по каплям раствор спирта 15 (722 мг, 2,52 ммоль) в CH2Cl2 (3,7 мл + 1,0 мл промывки), поддерживая температуру смеси ниже -75°С. После перемешивания при -78°С в течение 30 мин медленно прибавляли триэтиламин (1,05 мл, 7,56 ммоль) в течение 15 мин, поддерживая температуру реакционной смеси ниже -74,5°С. Полученную смесь перемешивали при -78°С в течение 30 мин и давали нагреться до -20°С в течение 20 мин. Реакционную смесь перемешивали еще при -20°С в течение 30 мин и гасили добавлением Н2О. Органический слой отделяли, разбавляли МТВЕ, промывали 0,5 М HCl, Н2О, насыщенным водным NaHCO3 и насыщенным раствором соли и сушили над Na2SO4. Растворитель выпаривали при пониженном давлении и очищали полученный остаток колоночной флэш-хроматографией на силикагеле, получая бесцветное твердое вещество 21 (673 мг, выход 94%): 1Н ЯМР (400 МГц, CDCl3): δ 4,52 (д, J = 5,2 Гц, 1Н), 3,72 (с, 3Н), 2,57 (дд, J = 5,2, 3,6 Гц, 1Н), 2,40 (м, 1Н), 2,28 (дд, J = 18,5, 5,2 Гц, 1Н), 1,99 (д, J = 18,5 Гц, 1Н), 1,87 (дд, J = 3,6, 2,8 Гц, 1Н), 0,89 (с, 9Н), 0,11 (с, 3Н), 0,09 (с, 3Н); 13С ЯМР (101 МГц, CDCl3): δ 209,2, 170,0, 68,8, 52,4, 43,2, 36,8, 34,5, 25,7, 25,0, 18,0, -4,7, -4,8.
ПРИМЕР 17
Метил(1S,5R,6R)-4-оксобицикло[3.1.0]гекс-2-ен-6-карбоксилат (22)
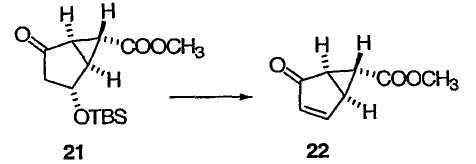
TBS-эфир 21 (50,0 мг, 0,176 ммоль) обрабатывали DBU (0,0789 мл, 0,528 ммоль) в CH2Cl2 (0,9 мл) при комнатной температуре в течение 1 ч. Реакционную смесь разбавляли МТВЕ, промывали 1 М HCl и насыщенным водным раствором соли (дважды) и сушили над Na2SO4. Растворитель удаляли при пониженном давлении и очищали полученный остаток колоночной флэш-хроматографией на силикагеле, получая бесцветное твердое вещество 22: +272,2 (c 1,1, CHCl3). Другие спектральные данные идентичны данным для α,β-ненасыщенного кетона 18, полученного в примере 15.
Характеристика полиморфа гидрохлоридной соли (1R,2S,5S,6S)-2-амино-6-фтор-4-оксобицикло[3.1.0]гексан-2,6-дикарбоновой кислоты 11.
Исследование методом рентгеноструктурного анализа широко используется для выяснения молекулярной структуры, кристалличности и полиморфизма. Рентгенограммы (XRPD) получали для кристаллической формы образца соли HCl, синтезированной в примере 10, с использованием дифрактометра Philips. Измерения проводили в интервале от 3,0080 градусов до 39,9830 градусов (2 Тета).
XRPD приведена на фигуре 1. Для идентификации кристаллической формы можно использовать следующие отражения:
Список пиков XRPD приведен далее в таблице.
Таким образом, в одном из вариантов осуществления полиморфная форма (1R,2S,5S,6S)-2-амино-6-фтор-4-оксобицикло[3.1.0]гексан-2,6-дикарбоновой кислоты HCl имеет d-расстояние, определенное при помощи рентгеновской дифракции на порошке, CuK альфа, составляющее около 5,37 ангстрем. В другом варианте осуществления полиморфная форма (1R,2S,5S,6S)-2-амино-6-фтор-4-оксобицикло[3.1.0]гексан-2,6-дикарбоновой кислоты HCl имеет, по меньшей мере, одно d-расстояние, определенное при помощи рентгеновской дифракции на порошке, CuK альфа, составляющее около 4,52, 4,05, 3,84, 3,37, 2,96, 2,73, 2,67, 2,59 или 2,42 ангстрем.
Дифференциальную сканирующую калориметрию (DSC) соли HCl, полученной в примере 10, проводили на приборе ТА Instruments DSC 2910 при скорости нагревания 10°С/мин от 20 до 175°С и при 2°С/мин от 175 до 255°С в атмосфере азота в открытом тигле. Результаты представлены на фигуре 2. Из данных результатов виден широкий интервал температуры плавления с начальной температурой примерно 184°С с последующим экзотермическим разложением выше 227°С.
Таким образом, в одном из вариантов осуществления полиморфная форма (1R,2S,5S,6S)-2-амино-6-фтор-4-оксобицикло[3.1.0]гексан-2,6-дикарбоновой кислоты HCl имеет начальную температуру плавления, экстраполированную дифференциальной сканирующей калориметрией, свыше 184°С.
В тексте использованы следующие сокращения:
Ме: метил;
Et: этил;
iPr: изопропил;
Bu: бутил;
Ac: ацетил;
DBU: 1,8-диазабицикло[5.4.0]ундец-7-ен;
NBS: N-бромсукцинимид;
NIS: N-йодсукцинимид;
ДМФА: N,N'-диметилформамид;
ТГФ: тетрагидрофуран;
TBHP: гидроперекись трет-бутила;
MTBE: метил-трет-бутиловый эфир;
LDA: диизопропиламид лития;
TBS: трет-бутилдиметилсилил;
TMS: триметилсилил;
TES: триэтилсилил;
ДМСО: диметилсульфоксид;
TfOH: трифторметансульфокислота;
LHMDS: гексаметилдисилазид лития;
Ts: пара-толуолсульфонил (тозил);
ВЭЖХ: высокоэффективная жидкостная хроматография;
ГХ: газовая хроматография;
ЯМР: ядерный магнитный резонанс;
DSC: дифференциальная сканирующая калориметрия;
ТСХ: тонкослойная хроматография;
XRPD: рентгеновская дифракция на порошке;
Комн. темп.: комнатная температура.
Несмотря на то что данное изобретение было описано и иллюстрировано со ссылкой на некоторые конкретные варианты его осуществления, специалистам в данной области будет понятно, что можно осуществлять различные адаптации, изменения, модификации, замены, исключения или дополнения методик, не отступая от сути и не выходя за рамки изобретения. Например, можно применять условия реакции, отличающиеся от конкретных условий, приведенных здесь выше, вследствие изменения реагентов или методики для получения соединений по указанным выше способам изобретения. Аналогичным образом индивидуальная реакционная способность исходных веществ может различаться в соответствии и исходя из конкретных имеющихся заместителей или условий производства, и подобные ожидаемые изменения или различия в результатах рассматривают в соответствии с целями и практическим осуществлением настоящего изобретения. Поэтому подразумевается, что изобретение определено последующей формулой изобретения и что данную формулу изобретения следует интерпретировать настолько широко, насколько это возможно.
Кроме того, нужно понимать, что все величины являются приблизительными и приведены для описания. В данной заявке цитированы патенты, патентные заявки, публикации, описания продуктов и протоколы, раскрытие которых включено здесь ссылкой во всей полноте для любых целей.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРЕДШЕСТВЕННИКИ ВИТАМИНА D, СПОСОБ ИХ ПОЛУЧЕНИЯ И ПРОМЕЖУТОЧНЫЕ ПРОДУКТЫ | 2000 |
|
RU2247710C2 |
| БИСЧЕТВЕРТИЧНЫЕ СОЛИ АЛКАЛОИДА ХИННОГО ДЕРЕВА В КАЧЕСТВЕ АСИММЕТРИЧЕСКИХ МЕЖФАЗНЫХ КАТАЛИЗАТОРОВ | 2013 |
|
RU2667909C2 |
| 4-ФЕНИЛЗАМЕЩЕННЫЕ ТЕТРАГИДРОИЗОХИНОЛИНЫ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЛЕЧЕНИЯ НА ИХ ОСНОВЕ | 2001 |
|
RU2301808C2 |
| ПРОИЗВОДНЫЕ ТЕТРАГИДРОИЗОХИНОЛИНА, СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ИНГИБИРОВАНИЯ СИНАПТИЧЕСКОГО ЗАХВАТА ДОПАМИНА И СПОСОБ ЛЕЧЕНИЯ | 2000 |
|
RU2293728C2 |
| КАРБАМИДЗАМЕЩЕННЫЕ ИМИДАЗОХИНОЛИНОВЫЕ ЭФИРЫ | 2001 |
|
RU2302418C2 |
| ПРОИЗВОДНЫЕ 6-ФТОРБИЦИКЛО[3.1.0]ГЕКСАНА | 2002 |
|
RU2315622C2 |
| АМИДНОЕ ПРОИЗВОДНОЕ ПИРАЗОЛА | 2015 |
|
RU2658827C2 |
| ПРОИЗВОДНЫЕ ТАКСАНА, ФУНКЦИОНАЛИЗИРОВАННЫЕ ПО 14-ПОЛОЖЕНИЮ, И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2003 |
|
RU2320652C2 |
| ЗАМЕЩЕННЫЕ БИАРИЛЬНЫЕ СОЕДИНЕНИЯ ИЛИ ЗАМЕЩЕННЫЕ ПИРИДИНЫ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ НА ИХ ОСНОВЕ | 1997 |
|
RU2195443C2 |
| АКТИВИРУЮЩИЙ АГЕНТ ДЛЯ РЕЦЕПТОРА, АКТИВИРУЕМОГО СТИМУЛИРУЮЩИМИ РОСТ ПЕРОКСИСОМ АГЕНТАМИ | 2009 |
|
RU2501794C2 |
Настоящее изобретение относится к способам получения производных бицикло[3.1.0]гексана, применимых в качестве агонистов mGluR, выраженных формулами (IA), (XII), где R1 и R2 представляют собой водород, Х представляет собой галоген, R3 представляет собой -O-Ra, Ra представляет собой C1-10алкил и R4 представляет собой (1) водород или (2) Si-(R9)(R10)(R11),
где каждый R9, R10 и R11 представляют собой С1-10алкил, а также к промежуточным соединениям, полученным в ходе осуществления этих способов. 12 н. и 14 з.п. ф-лы, 2 ил., 1 табл.
 ,
, 
1. Способ получения соединения формулы (IA):
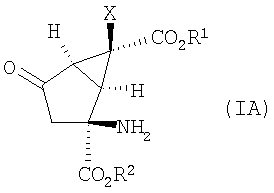 ,
,
в которой R1 и R2 представляют собой водород,
Х представляет собой галоген,
и его фармацевтически приемлемых солей,
включающий в себя:
(А) окисление соединения формулы (II):
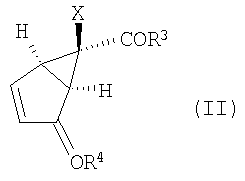 ,
,
в которой R3 представляет собой -O-Ra,
где Rа представляет собой С1-10алкил, и
R4 представляет собой
(1) водород или
(2) Si-(R9)(R10)(Rll),
где каждый R9, R10 и R11 представляют собой С1-10алкил,
с получением соединения формулы (IV):
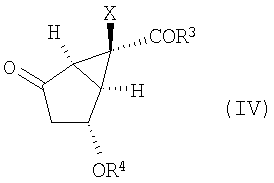 ,
,
(В) снятие защиты с гидроксильной группы соединения формулы (IV) с получением соединения формулы (V):
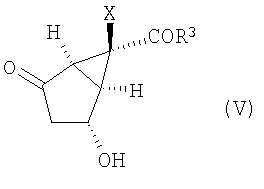 ,
,
(С) проведение реакции соединения формулы (V) с соединением формулы (VI) в присутствии кислоты:
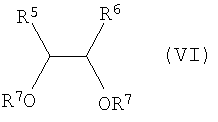 ,
,
в которой каждый из R5 и R6 независимо выбирают из
(1) C1-10алкила и
(2) фенила,
R7 представляет собой
(1) водород или
(2) Si-(R9)(R10)(R11), где каждый из R9, R10 и R11 представляют собой С1-10алкил,
с получением соединения формулы (VII):
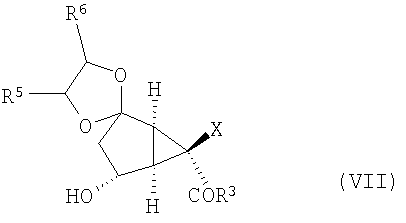 ,
,
(D) окисление соединения формулы (VII) с получением соединения формулы (VIII):
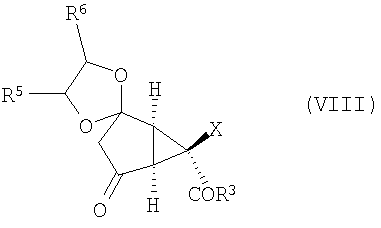 ,
,
(Е) проведение реакции Штрекера с аммиаком в спиртовом растворителе соединения формулы (VIII) в соединение формулы (IX):
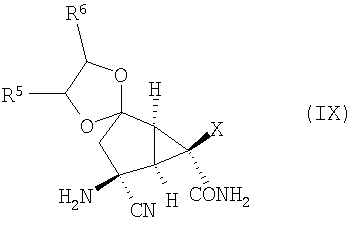
и (F) гидролиз соединения формулы (IX) с получением соединения формулы (IA).
2. Способ по п.1, где R5 и R6 представляют собой метил.
3. Способ по п.1, где R5 и R6 представляют собой фенил.
4. Способ по п.1, где R3 представляет собой метокси.
5. Способ по п.1, где R7 представляет собой триметилсилил.
6. Способ по п.1, где Х представляет собой фтор.
7. Способ по п.1, где R4 представляет собой трет-бутилдиметилсилил.
8. Способ получения соединения формулы (IA):
 ,
,
в которой R1 и R2 представляют собой водород,
Х представляет собой галоген, и
его фармацевтически приемлемых солей,
включающий гидролиз соединения формулы (IX):
,
в которой R5 и R6 независимо выбирают из
(1) С1-10алкила и
(2) фенила,
с получением соединения формулы (IA).
9. Способ по п.8, где R5 и R6 представляют собой метил.
10. Способ по п.8, где R5 и R6 представляют собой фенил.
11. Способ по п..8, где Х представляет собой фтор.
12. Способ получения соединения формулы (II):
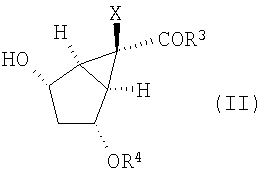 ,
,
в которой R3 представляет -O-Ra,
где Ra представляет собой С1-10алкил,
Х представляет собой галоген;
R4 представляет собой Si-(R9)(R10)(R11),
где R9, R10 и R11 представляют собой С1-10алкил,
включающий в себя:
(А) окисление соединения формулы (X):
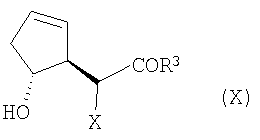
с последующей защитой гидроксильной группы защитным агентом R4 с получением соединения формулы (XI):
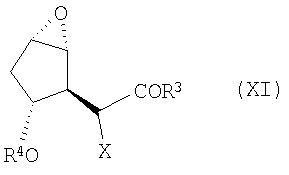
и (В) проведение реакции полученного защищенного эпоксида с основанием в присутствии кислоты Льюиса с получением соединения формулы (II).
13. Способ по п.12, в котором превращение соединения формулы (X) в соединение формулы (XI) включает в себя стадию эпоксидирования соединения формулы (X) в присутствии источника пероксида и каталитического количества VO(acac)2.
14. Способ по п.12, в котором превращение соединения формулы (X) в соединение формулы (XI) включает в себя обработку соединения формулы (X) галогенирующим агентом с последующей обработкой основанием.
15. Способ по п.12, в котором Х представляет собой фтор.
16. Способ получения соединения формулы (XII)
,
в которой R3 представляет собой -O-Ra,
где Ra представляет собой C1-10алкил, и
R4 представляет собой Si-(R9)(R10)(R11),
где R9, R10 и R11 представляют собой С1-10алкил,
Х представляет собой галоген;
включающий в себя:
(А) превращение соединения формулы (II)
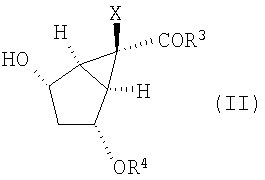 ,
,
в которой R4 представляет собой Si-(R9)(R10)(R11),
где R9, R10 и R11 представляют собой С1-10алкил,
в соединение формулы (XIII) введением уходящей группы R8
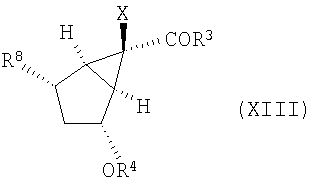 ,
,
в которой R8 представляет собой O-SO2-R12, где R12 выбирают из группы, включающей фенил, замещенный одним заместителем, выбранным из группы, включающей С1-10алкил,
(В) удаление R4 с получением соединения формулы (XIV)
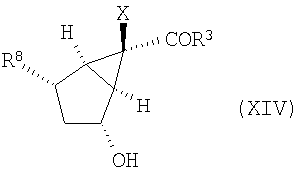
и (С) окисление соединения формулы (XIV) с получением соединения формулы (XII).
17. Способ по п.16, где R3 представляет собой метокси.
18. Способ получения соединения формулы (ХII')
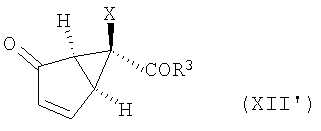 ,
,
в которой R3 представляет собой -O-Ra,
где Ra представляет собой С1-10алкил,
Х представляет собой галоген,
R4 представляет собой Si(R9)(R10)(R11),
где R9, R10 и R11 представляют собой С1-10алкил,
включающий в себя обработку соединения формулы (IV) DBU в CH2Cl2
в соединение формулы (XII').
19. Соединение формулы (VII):
в которой R3 представляет -О-Ra,
где Ra представляет собой С1-10алкил,
каждый из R5 и R6 независимо выбирают из
(1) C1-10алкила и
(2) фенила,
Х представляет собой галоген;
и их солей.
20. Соединение формулы (VIII):
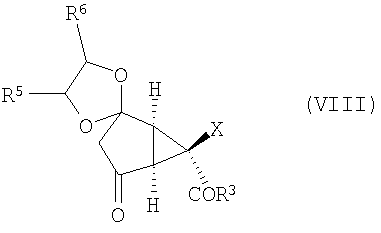 ,
,
в которой R3 представляет собой -O-Ra,
где Ra представляет собой С1-10алкил,
каждый из R5 и R6 независимо выбирают из
(1) C1-10алкила и
(2) фенила,
Х представляет собой галоген;
и его соли.
21. Соединение формулы (IX):
,
в которой каждый из R5 и R6 независимо выбирают из
(1) C1-10алкила и
(4) фенила,
Х представляет собой галоген;
и его соли.
22. Соединение формулы (ХА):
 ,
,
в которой R3 представляет собой -O-Ra,
где Ra представляет собой С1-10алкил,
и его соли.
23. Соединение формулы (XI):
,
в которой R3 представляет собой -O-Ra,
где Ra представляет собой С1-10алкил,
R4 представляет собой
(1) водород,
(2) Si-(R9)(R10)(R11),
где R9, R10 и R11 представляют собой С1-10алкил,
Х представляет собой галоген;
и его соли.
24. Соединение формулы (II):
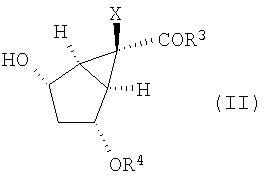 ,
,
в которой R3 представляет собой -O-Ra,
где Ra представляет собой С1-10алкил,
и R4 представляет собой
(1)водород,
(2) Si-(R9)(R10)(R11),
где R9, R10 и R11 представляют собой С1-10алкил, и
Х представляет собой галоген;
и его соли.
25. Способ получения соединения формулы (IV):
где R3 представляет собой -ORa,
где Ra представляет собой С1-10алкил,
Х представляет собой галоген;
R4 представляет собой Si-(R9)(R10)(R11),
где R9, R10 и R11 представляют собой С1-10алкил,
включающий в себя:
(А) окисление соединения формулы (X):
с последующей защитой гидроксильной группы защитным агентом R4 с получением соединения формулы (XI):
и (В) проведение реакции соединения формулы (XI) с основанием в присутствии кислоты Льюиса с получением соединения формулы (II):
 ,
,
и (С) окисление соединения формулы (II) с получением соединения формулы (IV).
26. Способ по п.25, в котором Х представляет собой фтор.
| US 6160009 А, 12.12.2000 | |||
| US 6333428 B1, 25.12.2001 | |||
| ПРОИЗВОДНЫЕ АМИНОБИЦИКЛО[3.1.0]ГЕКСАН-2,6-ДИКАРБОНОВОЙ КИСЛОТЫ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1995 |
|
RU2152925C1 |

