Настоящее изобретение относится к новым производным бензазепин, бензоксазепин- и бензотиазепин-N-уксусной кислоты, которые в α-положении к атому азота содержат оксо-группу и в положении 3 замещены 1-(карбоксиалкил)-циклопентилкарбонил- амино-остатком, и к их солям и биолабильным (бионестабильным) сложным эфирам, а также к содержащим эти соединения фармацевтическим композициям и способу получения этих соединений.
В основу настоящего изобретения положена задача нахождения новых бензазепиновых, бензоксазепиновых и бензотиазепиновых соединений с ценными фармакологическими свойствами. Далее, в основу изобретения положена задача нахождения новых, используемых для лечения сердечной недостаточности, фармацевтических биологически активных веществ.
В настоящее время найдено, что предлагаемые согласно изобретению новые, содержащие в положении 3, в случае необходимости, этерифицированный до сложного эфира 1-(карбоксиалкил) циклопентилкарбониламино-остаток, производные бензазепин-, бензоксазепин- и бензотиазепин-N-уксусной кислоты обладают ценными, эффективными в отношении воздействия на сердце, фармакологическими свойствами и проявляют отчетливо выраженное подавляющее воздействие на нейтральную эндопептидазу с благоприятным профилем действия, на основании которого они уменьшают наступающее при сердечной недостаточности высокое сердечное давление наполнения и таким образом разгружают сердце и могут вызывать усиление лиуреза.
Изобретение относится поэтому к новым соединениям общей формулы (I)
(см. формулу (I) в приложении)
где R1 обозначает низший алкокси - низшую алкильную группу, низший алкоксильный остаток которой замещен низшей алкоксильной группой;
фенил (низшую) алкильную группу или фенилокси (низшую) алкильную группу, которая в случае необходимости в фенильном кольце может быть замещена низшим алкилом, низшим алкоксилом или галогеном; или нафтил (низшую) алкильную группу;
A обозначает CH2, O или S;
R2 обозначает водород или галоген;
R3 обозначает водород или галоген;
R4 обозначает водород или образующую биолабильный сложный эфир группу, и
R5 обозначает водород или образующую биолабильный сложный эфир группу и к физиологически переносимым солям кислот формулы (I).
Если в соединениях формулы (I) заместители обозначают или содержат низшие алкильные или алкоксильные группы, то они могут быть линейными или разветвленными и содержат в особенности 1-4, предпочтительно 1-2, атома углерода и предпочтительно представляют собой метил или метоксигруппу. Если заместители обозначают галоген или содержат галогенные заместители, то принимают во внимание в особенности фтор, хлор или бром, предпочтительно фтор или хлор.
В соединениях формулы (I) A может обозначать метиленовую группу, кислород или серу и предпочтительно представляет собой метилен.
Соединения формулы (I) в фенильном кольце могут содержать заместители R2 и R3. Предпочтительно оба заместителя R2 и R3 или по меньшей мере, однако, один из этих заместителей обозначают водород.
R1 представляет собой предпочтительно содержащий ароматическое кольцо остаток, например, в случае необходимости замещенный фенил-(низший) алкильный или фенилокси (низший) алкильный остаток, в котором низшая алкиленовая цепь может содержать 1-4, предпочтительно 1-2, атома углерода. В особенности R1 представляет собой в случае необходимости замещенную фенетильную группу, которая в случае необходимости может быть замещена однократно или многократно галогеном, низшим алкоксилом или низшим алкилом, или нафтилэтильную группу. Если R1 обозначает замещенную низшим алкоксилом (низший) алкокси (низшую) алкильную группу, то она предпочтительно представляет собой (низший) алкокси-метильную группу, где низший алкоксильный остаток содержит 1-4, предпочтительно 1-2, атома углерода и замещен низшим алкоксилом, в особенности метоксигруппой.
Соединения формулы (I) в случае необходимости представляют собой этерифицированные до сложных эфиров производные дикарбоновой кислоты. В зависимости от формы применения - это биолабильные сложные моноэфиры, в частности соединения, где R4 обозначает образующую биолабильный сложный эфир группу и R5 обозначает водород, или дикарбоновые кислоты, причем последние пригодны в особенности для внутривенного введения.
В качестве образующих биолабильные сложные эфиры групп R4 и R5 пригодны низшие алкильные группы, в случае необходимости замещенные в фенильном кольце низшим алкилом или связанной с двумя соседними атомами углерода низшей алкиленовой цепью фенильные или фенил (низшие) алкильные группы; в случае необходимости замещенные в диоксолановом кольце низшим алкилом диоксоланилметильные группы или в случае необходимости замещенные в оксиметильной группе низшим алкилом (C2-C6)-алканоилоксиметильные группы. Если образующая биолабильный сложный эфир группа R4 или R5 обозначает низший алкил, то она может представлять собой предпочтительно неразветвленную алкильную группу с 1-4, предпочтительно 2-мя, атомами углерода. Если образующая биолабильный сложный эфир группа представляет собой, в случае необходимости, замещенную фенил (низшую) алкильную группу, то ее алкиленовая цепь может содержать 1-3, предпочтительно 1, атома углерода. Если фенильное кольцо замещено низшей алкиленовой цепью, то она может содержать 3-4, в особенности 3, атома углерода. В качестве фенилсодержащих заместителей R4 и/или R5 пригодны в особенности фенил, бензил или инданил. Если R4 и/или R5 представляют собой в случае необходимости замещенную алканоилоксиметильную группу, то ее алканоилокси-группа может содержать 2-6, предпочтительно 3-5, атомов углерода и предпочтительно разветвлена и может, например, представлять собой пивалоилоксиметильный остаток (= трет.- бутилкарбонилоксиметильный остаток).
Согласно изобретению, новые соединения формулы (I) и их соли получают тем, что само по себе известным образом кислоты общей формулы (II):
(см. формулу (II) в приложении)
где R1 имеет вышеуказанное значение и R4a обозначает защитную для кислотной функции группу, или их реакционноспособные производные вводят во взаимодействие с аминами общей формулы (III);
(см. формулу (III) в приложении)
где R2, R3 и A имеют вышеуказанное значение, a R5a обозначает защитную для кислотной функции группу, с получением амидов общей формулы (IV):
(см. формулу (IV) в приложении)
где R1, R2, R3, R4a, R5a, и A имеют вышеуказанное значение, и в соединениях формулы (IV) одновременно или в любой последовательности друг после друга отщепляют защитные для кислотной функции группы R4a и R5a, если они не представляют собой желательную, образующую биолабильный сложный эфир, группу; и в желательном случае, смотря по обстоятельствам, высвободившуюся кислотную группу этерифицируют до сложного эфира с помощью спирта общей формулы (V) или с помощью соответствующего реакционноспособного производного общей формулы (Va):
(см. формулы (V) и (Va) в приложении)
где R6 обозначает образующую биолабильный сложный эфир группу и X обозначает отщепляемую реакционноспособную группу;
и в желательном случае полученные кислоты формулы (I) переводят в их физиологически переносимые соли или соли кислот формулы (I) переводят в свободные кислоты.
В качестве физиологически переносимых солей дикарбоновых кислот или сложных моноэфиров формулы (I) принимают во внимание соли щелочных металлов, щелочноземельных металлов или аммония, например, соли натрия или кальция, или соли с физиологически приемлемыми, фармакологически нейтральными органическими аминами, как, например, диэтиламин или трет.-бутиламин.
Соединения формулы (I) содержат два хиральных атома углерода, а именно несущий амидную боковую цепь атом углерода в положении 3 циклического скелета и несущий остаток R1 атом углерода амидной боковой цепи. Таким образом соединения могут находиться в нескольких оптически активных стереоизомерных формах или в виде рацемата. Настоящее изобретение охватывает как рацемические смеси, так и также чистые изомерные соединения формулы (I).
Взаимодействие кислот формулы (II) с аминами формулы (III) с получением амидов формулы (IV) можно осуществлять само по себе обычными для получения амидных группировок путем амино-ацилирования способами. В качестве ацилирующих средств можно использовать кислоты формулы (II) или их реакционноспособные производные. В качестве реакционноспособных производных принимают во внимание в особенности смешанные ангидриды или галоидангидриды кислот. Так, например, можно использовать хлорангидриды или бромангидриды кислот формулы (II) или смешанные сложные эфиры кислот формулы (II) с органическими сульфокислотами, например, с (низшей) алкансульфокислотой, как например, метансульфокислота, или ароматическими сульфокислотами, как, например, бензолсульфокислота, или замещенными низшим алкилом или галогеном бензолсульфокислотами, как, например, толуолсульфокислоты или бромбензолсульфокислоты. Ацилирование можно осуществлять в инертном в реакционных условиях органическом растворителе, предпочтительно при температурах от -20oC до комнатной температуры. В качестве растворителей в особенности пригодны галогенированные углеводороды, как дихлорметан, или ароматические углеводороды, как бензол или толуол, или циклические простые эфиры, как тетрагидрофуран или диоксан, или смеси этих растворителей.
Ацилирование, в особенности когда в качестве ацилирующего средства применяют смешанный ангидрид кислоты формулы (II) с сульфокислотой, целесообразно осуществлять в присутствии кислотосвязующего реагента. В качестве кислотосвязующих средств пригодны растворимые в реакционной смеси основания, в частности органические основания, такие как третичные (низший)алкиламины и пиридины, как, например, триэтиламин, трипропиламин, пиридин, 4-диметиламинопиридин, 4-диэтиламинопиридин или 4-пирролидинопиридин. Используемые в избытке органические основания одновременно могут служить также в качестве растворителей.
Смешанные ангидриды кислот формулы (II) с органическими сульфокислотами предпочтительно получать in situ путем взаимодействия кислот формулы (II) с галоидангидридом, в особенности хлорангидридом, органической сульфокислоты и далее без выделения непосредственно сразу вводить во взаимодействие с аминосоединением формулы (III).
В случае, если в качестве ацилирующих средств используют сами кислоты формулы (II), взаимодействие аминосоединений формулы (III) с кислотами формулы (II) также целесообразно осуществлять в присутствии известного из химии пептидов, пригодного для образования амида, реагента связывания. В качестве примера реагентов связывания, которые способствуют образованию амида со свободными кислотами благодаря тому, что они реагируют с кислотой, in situ при образовании реакционноспособного производного кислоты, следует в особенности назвать алкилкарбодиимиды, например, циклоалкилкарбодиимиды, как дицикло- гексилкарбодиимид или 1-этил-3-/3-(диметиламино)пропил/карбодиимид, карбонилдиимидазол и N-(низший) алкил-2-галогенпиридиниевые соли, в особенности галогениды и тозилаты, предпочтительно N-метил-2-хлорпиридиний-иодид (см., например, Mukaijama в Angewandte Chemie, 91, с. 789-812). Взаимодействие в присутствии реагента связывания целесообразно можно осуществлять при температурах от -30oC до +50oC, при использовании растворителей, как галогенированные углеводороды и/или ароматические растворители, в случае необходимости в присутствии кислотосвязующего амина.
От полученных путем взаимодействия соединений формулы (II) с соединениями формулы (III) соединений формулы (IV) само по себе известным образом можно отщеплять защитные группы R4a и R5a, если они не представляют собой никаких желательных в соединениях формулы (I), образующих биолабильный сложный эфир групп.
В качестве защитных групп R4a и R5a можно выбирать сами по себе обычные для защиты кислотных функций защитные группы, которые затем снова отщепляют согласно сами по себе известным способам. Пригодные защитные для кислотной функции группы известны, например, из McOmie "Protective Groups in Organic Chemistry", Plenum Press и Greene, "Protective groups in Organic Synthesis", Wiley, Intersience Publication.
Если нужно получить соединения формулы (I), в которых R4 и R5 идентичны, то целесообразнее в исходных соединениях формул (II) и (III) выбирать идентичные защитные группы R4a и R5a.
Если нужно получить соединения формулы (I), где R4 и R5 имеют различное значение, то целесообразнее в исходных соединениях формул (II) и (III) выбирать разные защитные группы, которые селективно можно снова отщеплять сами по себе известным образом при различных условиях. В качестве примеров трех, отщепляющихся при различных условиях защитных групп следует назвать:
1. сложные метиловые или этиловые эфиры, которые легко расщепляются в основных условиях, однако значительно устойчивее по отношению к кислым условиям или гидрогенолизу;
2. трет.-бутиловые сложные эфиры, которые могут легко расщепляться с помощью кислот, однако, значительно стабильнее по отношению к основным условиям или гидрогенолизу; и
3. сложные бензиловые эфиры, которые могут легко расщепляться гидрогенолитически или также в основных условиях, однако, значительно стабильнее по отношению к кислым условиям.
Если, например, нужно получить дикарбоновые кислоты формулы (I), где R4 и R5 оба обозначают водород, то в качестве защитных групп R4a и R5a предпочтительно используют отщепляемые в кислых условиях защитные группы, например, трет.-бутильная группа, и полученные путем взаимодействия соединений формулы (II) с соединениями формулы (III) сложные трет.-бутиловые эфиры формулы (IV) затем расщепляют путем обработки кислотой. Расщепление можно осуществлять, например, путем обработки с помощью трифторуксусной кислоты как таковой или раствора трифторуксусной кислоты в галогенированном углеводороде, например, в дихлорметане, или путем обработки с помощью газообразного HCl в инертном в условиях реакции органическом растворителе, например, как этилацетат. Реакцию можно проводить при температурах от -25oC до комнатной температуры.
Если, например, нужно получить монокарбоновые кислоты формулы (I), где R4 обозначает образующую биолабильный сложный эфир группу и R5 обозначает водород, то в качестве исходных соединений формулы (II) можно использовать соединения, в которых R4a представляет собой уже желательную, образующую биолабильный сложный эфир, группу, например, этильную группу, и в качестве защитной группы R5a в соединениях формулы (III) используют защитные группы, которые отщепляются в условиях, в которых не расщепляется R4-OCO-группа. В случае, если R4-OCO-группа представляет собой относительно кислотоустойчивую группу сложного этилового эфира, то в качестве защитной группы R5a пригодны, например, отщепляемая с помощью кислоты трет.-бутильная группа или гидрогенолитически отщепляемая группа, как бензил.
В случае, если R4a в соединениях формулы (II) представляет собой чувствительную к кислоте, образующую биолабильный сложный эфир группу, то в качестве защитной группы R5a в соединениях формулы (III) целесообразно выбирать гидрогенолитически отщепляемую группу, как бензил, и отщеплять ее от получаемых путем взаимодействия соединений формулы (II) с соединениями формулы (III) исходных из соединений формулы (IV) гидрогенолитически. Гидрогенолиз можно осуществлять путем каталитического гидрирования в присутствии катализатора, предпочтительно Pd/C - катализатора, в органическом инертном в реакционных условиях растворителе, например, низшем спирте, как этанол, или низшем алкиловом сложном эфире, как этилацетат. Целесообразно осуществлять каталитическое гидрирование при давлении водорода 4-5 бар при комнатной температуре.
Для получения соединений формулы (I), где R4 обозначает образующую биолабильный сложный эфир группу и R5 обозначает водород, также можно однако выбирать исходные соединения формулы (II) и (III) с различными защитными группами R4a и R5a с различной реакционноспособностью и от полученных путем взаимодействия соединений формулы (II) с соединениями формулы (III) соединений формулы (IV) отщеплять сначала защитную группу R4a при сохранении защитной группы R5a, затем в реакционный продукт общей формулы (IV'):
(см. формулу (IV') в приложении)
где R1, R2, R3, R5a и A имеют вышеуказанное значение, вводить желательную, образующую биолабильный сложный эфир группу R4 путем взаимодействия свободной кислотной группы соединения формулы (IV') с соединением формулы (V) или формулы (VI) и после этого от полученных соединений формулы (IV) отщеплять защитную группу R5a.
Так, например, от соединений формулы (IV), где R4a обозначает отщепляемую с помощью кислоты защитную группу, в особенности трет-бутильную группу, и R5a обозначает кислотостабильную защитную группу, например, бензил, сначала можно отщеплять с помощью кислоты только защитную группу R4a. Полученную монокарбоновую кислоту формулы (IV') затем само по себе обычными для образования сложных эфиров способами можно этерифицировать с помощью спирта формулы (V) или соответствующего соединения формулы (Va). В качестве отщепляемых реакционноспособных групп X в соединениях формулы (Va) пригодны галогены, в особенности хлор или бром, или органический сульфокислотный остаток, например, остаток (низший) алкансульфокислоты, как, например, метансульфокислота, или остаток ароматических сульфокислот, как бензолсульфокислота, или замещенных низшим алкилом или галогеном бензолсульфокислот, как толуолсульфокислоты. Для этерификации до сложного эфира, спирты формулы (V) можно вводить во взаимодействие, например, с кислотой формулы (IV') или реакционноспособным производным этой кислоты само по себе известным для ацилирования спиртов образом. Взаимодействие можно осуществлять, например, при условиях, указанных для взаимодействия соединений формулы (II) с соединениями формулы (III).
Аналогичным образом, путем выбора соответствующих разных защитных групп также можно получать соединения формулы (I), где R5 обозначает образующую биолабильный сложный эфир группу и R4 обозначает водород или отличную от R5, образующую биолабильный сложный эфир группу.
В случае вышеописанных взаимодействий хиральные центры в исходных соединениях формул (II) и (III) не изменяются, так что в зависимости от рода исходных соединений можно получать чистые изомерные соединения формулы (I) или смеси изомеров. Для получения чистых изомерных соединений и таким образом оптически единообразных соединений формулы (I) целесообразнее чистые энантиомерные соединения формулы (II) вводить во взаимодействие с чистыми энантиомерными соединениями формулы (III). В случае, если чистое энантиомерное соединение формулы (II) вводят во взаимодействие с рацемическим соединением формулы (III) или рацемическое соединение формулы (II) вводят во взаимодействие с чистым энантиомерным соединением формулы (III), то, смотря по обстоятельствам, получают смесь из двух диастереомеров, которую в желательном случае можно разделять само по себе известным образом. Взаимодействие рацемических соединений формулы (II) с рацемическими соединениями формулы (III) приводит к соответствующим смесям из 4-х изомеров, которые в желательном случае само по себе известным образом можно разделять.
Исходные соединения формулы (II) можно получать само по себе известными способами.
Например, соединения общей формулы (IIa):
(см. формулу (IIa) в приложении)
где R4a имеет вышеуказанное значение, а R1a имеет указанное для R1 значение за исключением (низший) алкокси (низший) алкоксиметильного остатка, получают тем, что производные акриловой кислоты общей формулы (VI):
см. формулу (VI) в приложении
где R4a и R1a имеют вышеуказанное значение, вводят во взаимодействие с циклопентанкарбоновой кислотой формулы (VII)
см. формулу (VII) в приложении
Реакцию можно проводить само по себе известным образом в условиях присоединения по Михаэлю в инертном в реакционных условиях органическом растворителе путем взаимодействия циклопентанкарбоновой кислоты с сильным, способным образовывать дианион циклопентакарбоновой кислоты основанием и последующего взаимодействия с производным сложного акрилового эфира формулы (VI). В качестве растворителей пригодны простые эфиры, в особенности циклические простые эфиры, как, например, тетрагидрофуран. В качестве сильных оснований пригодны не нуклеофильные органические амиды щелочных металлов, как, например, диизопропиламид лития. Целесообразно циклопентанкарбоновую кислоту в тетрагидрофуране вводить во взаимодействие с двумя эквивалентами диизопропиламида лития и затем реакционную смесь вводят во взаимодействие с соединением формулы (VI). Температура реакции может составлять от -70oC до 0oC.
Соединения общей формулы (IIb):
см. формулу (IIb) в приложении
где R4a имеет вышеуказанное значение и R1b обозначает (низший) алкокси-(низший) алкоксиметильный остаток, можно получать тем, что сложный эфир галогенкарбоновой кислоты общей формулы (VIII):
см. формулу (VIII) в приложении
где R4a имеет вышеуказанное значение и V обозначает галоген, вводят во взаимодействие с циклопентанкарбоновой кислотой формулы (VII) и полученный продукт реакции общей формулы (IX):
см. формулу (IX) в приложении
где R4a имеет вышеуказанное значение, вводят во взаимодействие с соединениями общей формулы (Xb):
см. формулу (Xb) в приложении
где R1b и X имеют вышеуказанное значение. Взаимодействие сложного эфира галогенкарбоновой кислоты формулы (VIII) с циклопентанкарбоновой кислотой формулы (VII) можно осуществлять само по себе известным образом в инертном при реакционных условиях растворителе в присутствии сильного, способного образовывать дианион циклопентанкарбоновой кислоты основания. Например, взаимодействие можно осуществлять в условиях, указанных для взаимодействия циклопентанкарбоновой кислоты с соединениями формулы (VI). Последующее взаимодействие кислот формулы (IX) с соединениями формулы (Xb) можно осуществлять само по себе известным образом в пригодных для α -алкилирования сложных эфиров карбоновых кислот условиях в инертном при реакционных условиях органическом растворителе в присутствии сильного основания. Предпочтительно вводят соединения формулы (Xb), в которых X обозначает хлор или бром. В качестве растворителей пригодны простые эфиры, в особенности циклические простые эфиры, как тетрагидрофуран или диоксан. В качестве сильных оснований можно применять гидриды или амиды щелочных металлов, например, диизопропиламид лития.
Соединения формулы (II) на несущем остаток R1 атоме углерода имеют центр хиральности и при синтезе получаются в виде их рацематов. Оптически активные соединения можно получать из рацемических смесей само по себе известным образом, например, путем хроматографического разделения на хиральных разделительных средствах или путем введения во взаимодействие с пригодными оптически активными основаниями, например, с α--метилбензиламином или псевдоэфедрином, и последующего разделения на их оптические антиподы путем фракционной кристаллизации полученных солей.
Производные сложных эфиров акриловой кислоты формулы (VI) можно получать само по себе известным образом тем, что производные сложных эфиров (ди/низший/алкилфосфоно)-уксусной кислоты общей формулы (XI):
(см. формулу (XI) в приложении)
где R4a и R1a имеют вышеуказанное значение и R7 и R8, каждый, обозначают низший алкил, предпочтительно метил или этил, вводят во взаимодействие с формальдегидом в инертном при реакционных условиях органическом растворителе в основных условиях. Например, соединения формулы (XI) можно вводить во взаимодействие с параформальдегидом в простом эфире, предпочтительно в циклическом простом эфире, как тетрагидрофуран, в присутствии основания, предпочтительно не нуклеофильного алкоголята щелочного металла, как трет.-бутилат калия, при температурах от -20oC до +30oC.
Соединения формулы (XI) можно получать само по себе известным образом тем, что производные фосфоноуксусной кислоты общей формулы (XII):
(см. формулу (XII) в приложении)
где R4a, R7 и R8 имеют вышеуказанное значение, вводят во взаимодействие с соединениями формулы (Xa):
(см. формулу (Xa) в приложении)
где Ra и X имеют вышеуказанное значение. Взаимодействие можно осуществлять в обычных для алкилирования условиях в инертном при реакционных условиях полярном апротонном органическом растворителе в присутствии основания при температурах в пределах 0 - 80oC. Предпочтительно вводят соединения формулы (Xa), где X обозначает галоген, в особенности бром или иод, или тозилат. В качестве растворителей пригодны, например, амиды, как диметилформамид, или также простые эфиры. В качестве оснований пригодны не нуклеофильные алкоголяты щелочных металлов, как, например, трет.-бутилат калия.
Соединения формулы (VI) также можно получать тем, что производные малоновой кислоты общей формулы (XIII):
(см. формулу (XIII) в приложении)
где R4a и R1a имеют вышеуказанное значение, само по себе известным образом обрабатывают формальдегидом в основных условиях. Так, производные малоновой кислоты формулы (XIII), например, можно вводить во взаимодействие с водным раствором формальдегида в присутствии вторичного органического амина, в особенности пиперидина, при температурах 0-30oC, предпочтительно при температурах ниже комнатной. Производные малоновой кислоты формулы (XIII) также можно вводить во взаимодействие с параформальдегидом в пиридине при температурах 40-60oC.
Сложные моноэфиры малоновой кислоты формулы (XIII) можно получать тем, что сложные диэфиры малоновой кислоты общей формулы (XIV):
(см. формулу (XIV) в приложении)
где R4a имеет вышеуказанное значение и R9 обозначает низший алкил, в особенности метил, или бензил, вводят во взаимодействие с соединениями формулы (Xa) и полученные производные сложных диэфиров малоновой кислоты общей формулы (XV):
(см. формулу (XV) в приложении)
где R1a, R4a и R9 имеют вышеуказанное значение, путем частичного гидролиза переводят в производные сложных моноэфиров малоновой кислоты формулы (XIII).
Введение остатка R1a в сложный диэфир малоновой кислоты формулы (XIV) можно осуществлять само по себе известным образом путем взаимодействия сложного эфира формулы (XIV) с соединением формулы (Xa) в полярном апротонном органическом растворителе, предпочтительно в диметилформамиде, в присутствии основания, например, не нуклеофильного алкоголята щелочного металла, как трет.-бутилат калия, при температурах в пределах 0-80oC. Взаимодействие можно осуществлять, например, при условиях, указанных для взаимодействия соединений формулы (XI) с соединениями формулы (Xa).
Полученные замещенные сложные диэфиры малоновой кислоты формулы (XV) путем отщепления остатка R9 само по себе известным образом можно переводить в соответствующие сложные моноэфиры малоновой кислоты формулы (XIII). Если защитная группа R4a и остаток R9 представляют собой разные остатки с различной реакционноспособностью, то для отщепления остатка R9 целесообразнее выбирать такие условия, при которых остаток R4a не затрагивается. Если R9 обозначает бензил, то отщепление можно осуществлять само по себе известным образом гидрогенолитически. Низшие сложные алкиловые эфиры R9 расщепляют само по себе известным образом гидролитически, в зависимости от рода алкильного остатка в кислых или щелочных условиях. Предпочтительно R9 представляет собой этил, который можно отщеплять путем щелочного гидролиза. Для этой цели сложные алкиловые эфиры формулы (XV) в низшем спирте или смеси из низшего спирта с водой можно обрабатывать с помощью гидроксида щелочного металла, например, гидроксида калия. Если остатки R4a и R9 идентичны, то при этом количество гидроксида щелочного металла берется настолько незначительным, чтобы имел место только частичный гидролиз.
Соединения формулы (III) можно получать само по себе известным образом тем, что соединения общей формулы (XVI):
(см. формулу (XVI) в приложении)
где R2, R3 и A имеют вышеуказанное значение и R10R11N-группа представляет собой защищенную с помощью защитной для амино-функции группы амино-группу, вводят во взаимодействие с соединениями общей формулы (XVII):
(см. формулу (XVII) в приложении)
где R5a и X имеют вышеуказанное значение, и в полученном продукте реакции общей формулы (XVIII):
(см. формулу (XVIII) в приложении)
где R2, R3, R5a, A и R10R11N - группа имеют вышеуказанное значение, из R10R11N-группы высвобождают свободную амино-группу. Взаимодействие соединений формулы (XVI) с соединениями формулы (XVII) можно осуществлять способами, само по себе обычными для алкилирования амидов. Предпочтительно использовать соединения формулы (XVII), в которых X обозначает галоген, предпочтительно бром или иод. Взаимодействие можно осуществлять в полярном апротонном органическом растворителе, например, в диметилформамиде, или в циклическом простом эфире, как тетрагидрофуран, в присутствии основания. В качестве оснований пригодны не нуклеофильные основания, как, например, трет.-бутилат калия. В желательном случае взаимодействие также можно осуществлять в присутствии гидроксида щелочного металла, например, гидроксида калия, в двухфазной системе, в присутствии катализатора переноса фаз, например, тетра(низший) алкиламмонийгалогенида, как тетрабутиламмонийбромид.
Затем в полученных соединениях формулы (XVIII) путем отщепления защитной группы само по себе известным образом можно высвобождать аминогруппу. Для защиты амино-группы можно использовать само по себе известные для защиты амино-групп, снова легко отщепляемые защитные группы, например, известные из химии пептидов защитные группы. Пригодные защитные группы, например, известны из E. McOmie "Protective groups in Оrganic chemistry" Plenum Press, 1971. Например, в качестве защитных групп пригодны фталимидная группа или трет. -бутоксикарбонильная группа или также бензилоксикарбонильная группа. В зависимости от значения R5a, смотря по обстоятельствам, нужно выбирать защитные группы, которые затем отщепляются при условиях, при которых не затрагивается группа R5a. В качестве отщепляющейся в основной среде защитной группы пригодны, например, фталимидная группа, которую можно отщеплять путем обработки этаноламином или гидразином при повышенных температурах, например, при температурах 70-90oC. Фталимидная группа пригодна, например, в качестве защитной группы для соединений, где A обозначает серу. В качестве отщепляемой путем воздействия кислоты защитной группы пригодна, например, трет.-бутоксикарбонильная группа, которую можно снова отщеплять путем обработки кислотой, например, путем обработки трифторуксусной кислотой или газообразным хлороводородом в этилацетате. Трет.-Бутоксикарбонильная группа пригодна, например, в качестве защитной группы для соединений, где A обозначает кислород. В качестве гидрогенолитически отщепляемой защитной группы пригодна, например, бензилоксикарбонильная группа, которую можно отщеплять путем гидрирования с помощью водорода в присутствии катализатора палладия-на-угле.
Соединения формулы (III) содержат центр хиральности на атоме углерода, несущем амино- группу. Если исходят из оптически чистых исходных соединений формулы (XVI), то получают оптически чистые соединения формулы (III). Это относится в особенности к таким соединениям, где A обозначает кислород или серу. Если исходят из рацемических соединений формулы (XVI), то также получают рацемические соединения формулы (III). Это имеет место, в общем, в случае соединений, где A обозначает метиленовую группу. Рацемические смеси соединений формулы (III) само по себе известным образом можно разделять на их оптические изомеры, например, путем хроматографического разделения на хиральных разделительных материалах или путем введения во взаимодействие с пригодными оптически активными кислотами, например, с винной кислотой, и последующего разделения оптических антиподов путем фракционной кристаллизации полученных солей. Для повышения выхода желательного оптического изомера при взаимодействии с пригодными оптически активными кислотами одновременно с или после последующего осаждения соли одного изомера с помощью оптически активной кислоты в реакционной смеси можно осуществлять вторичную рацемизацию остающегося в растворе изомера путем добавки предпочтительно ароматического альдегида, как, например, бензальдегид. При этом рацемизация центра хиральности вызывается путем образования имина с помощью альдегида.
Соединения формулы (XVI) можно получать само по себе известным образом. Например, соединения общей формулы (XVIa):
(см. формулу (XVIa) в приложении)
где R2, R3 и R10R11N-группа имеют вышеуказанное значение, можно получать тем, что в соединениях общей формулы (XIX):
(см. формулу (XIX) в приложении)
где R2, R3 и Y имеют вышеуказанное значение, галоген Y само по себе известным образом заменяют R10R11N-группой. Например, соединение формулы (XIX) можно вводить во взаимодействие с солью щелочного металла амида R10R11NH, предпочтительно с фталимидом калия. Взаимодействие можно осуществлять в инертном при реакционных условиях апротонном органическом растворителе, предпочтительно в диметилформамиде, при температурах 40-80oC.
Соединения формулы (XIX) можно получать само по себе известным образом путем перегруппировки Бекмана оксимных соединений общей формулы (XX):
(см. формулу (XX) в приложении)
где R2, R3 и Y имеют вышеуказанное значение, тем, что соединения формулы (XX) в условиях перегруппировки Бекмана обрабатывают кислотой. Соединения формулы (XX) целесообразнее перегруппировывать в соединения формулы (XIX) путем обработки полифосфорной кислотой при температурах 60-90oC.
Оксимы формулы (XX) можно получать исходя из циклических кетонов общей формулы (XXI):
(см. формулу (XXI) в приложении)
где R2 и R3 имеют вышеуказанное значение, тем, что кетоны формулы (XXI) для введения остатка Y сначала обрабатывают галогеном и полученные галогенированные кетоны затем вводят во взаимодействие с гидроксиламином. Целесообразнее всего αгалогенирование кетона и последующее образование оксима можно осуществлять по одностадийному способу, причем кетон формулы (XXI) в инертном органическом растворителе, например, в низшем спирте, как метанол, сначала обрабатывают галогеном и затем к реакционной смеси добавляют гидроксиламин. Гидроксиламин целесообразнее использовать в форме соли гидроксиламина, например, в виде гидрохлорида, и к реакционной смеси добавлять немного воды. Способ можно осуществлять при температурах 0-40oC, предпочтительно при комнатной температуре.
Соединения общей формулы (XVIb):
(см. формулу (XVIb) в приложении)
где R2, R3 и R10R11N-группа имеют вышеуказанное значение и Aa обозначает кислород или серу, можно получать само по себе известным образом путем циклизации ароматических аминокислот общей формулы (XXII):
(см. формулу (XXII) в приложении)
где R2, R3, Aa и R10R11N-группа имеют вышеуказанное значение. Циклизация соединений формулы (XXII) протекает с отщеплением воды и ее можно осуществлять само по себе известными для получения лактамов способами. Так, циклизацию можно осуществлять, например, в присутствии активирующего кислотную группу, известного из химии пептидов для получения амидов, реагента связывания, например, карбодиимида, в инертном при реакционных условиях полярном органическом растворителе, например, диметилформамиде. Взаимодействие можно осуществлять, например, при условиях, указанных для взаимодействия соединений формулы (II) с соединениями формулы (III). В качестве активирующего кислотную группу агента можно использовать также диэтилфосфоноцианид и взаимодействие осуществляют в присутствии органического основания, например, три (низший) алкиламина, как триэтиламин.
Соединения общей формулы (XXII) можно получать само по себе известным образом путем восстановления соответствующих нитросоединений общей формулы (XXIII):
(см. формулу (XXIII) в приложении)
где R2, R3, Aa и R10R11N-группа имеют вышеуказаноное значение. Восстановление нитро-группы можно осуществлять по само по себе известными для восстановления нитро-бензольных соединений до анилиновых соединений способами, например, путем каталитического гидрирования в присутствии палладия-на-угле в качестве катализатора. Восстановление также можно осуществлять при применении других восстановителей, которые выделяют водород in situ, например, таких как смеси металлическое железо/соляная кислота или металлический цинк/соляная кислота.
Соединения формулы (XXIII) можно получать само по себе известным образом путем взаимодействия о-фтор-нитробензольных соединений общей формулы (XXIV):
(см. формулу (XXIV) в приложении)
где R2 и R3 имеют вышеуказанное значение, с кислотами общей формулы (XXV):
(см. формулу (XXV) в приложении)
где Aa и R10R11N-группа имеют вышеуказанное значение. Соединения формулы (XXV) представляют собой производные серина, соответственно, цистеина, амино-группа которых защищена. Взаимодействие осуществляют в инертном при реакционных условиях органическом растворителе в присутствии основания. Взаимодействие фторнитробензолов с сильно нуклеофильным производным цистеина можно осуществлять в низшем спирте или смеси спирта с водой в присутствии слабого основания, как гидрокарбонат натрия. Для взаимодействия со сравнительно более слабым нуклеофильным производным серина целесообразнее всего применять сильное основание, например, гидрид щелочного металла, в полярном органическом растворителе, как диметилформамид.
В желательном случае, после образования соединений формулы (XXIII) первоначально имеющуюся в соединениях формулы (XXV) защитную для амино-функции группу само по себе известным образом можно обменивать на другую защитную для амино-функции группу, которая по своей реакционноспособности лучше отличается от остатка R5a и таким образом лучше пригодна для дальнейшей обработки соединений формулы (XXIII).
Соединения формулы (I) и их фармакологически приемлемые соли отличаются интересными фармакологическими свойствами. В особенности вещества оказывают ингибирующее воздействие на нейтральную эндопептидазу (= NEP). NEP представляет собой фермент, который вызывает деструкцию эндогенных натриуретических пептидов, например, атриального натриуретического пептида (= ANP). Благодаря своему подавляющему воздействию на активность NEP вещества могут улучшать биологическую активность и продолжительность жизни уязвимых на счет NEP натриуретических пептидов, в особенности ANP, и поэтому пригодны для лечения болезненных состояний, на которые оказывает благоприятное влияние воздействие такого рода гормонов, в особенности сердечной недостаточности.
В случае сердечной недостаточности, благодаря обусловленной заболеванием пониженной сердечной пропускной деятельности приходят к рефлекторно повышенному периферическому сопротивлению и таким образом к застою крови в круге кровообращения легких и самого сердца. В дальнейшем возникает высокое давление за счет наполнения сердца, которое вызывает растяжение стенки желудочка в предсердии и в желудочках сердца. При этих обстоятельствах сердце функционирует как эндокринный орган, т.е. оно в состоянии выделять в кровеносное русло ANP, который обладает выраженными сосудорасширяющей и диуретически/натриуретической активностями. ANP действует понижающе на повышенное давление за счет наполнения сердца. Это происходит за счет диуреза/натриуреза (снижение циркулирующей общей массы крови) и за счет уменьшения периферического сопротивления (снижение предварительной и последующей нагрузки). Это разгрузочное в отношении сердца действие ANP рассматривается как эндогенный кардиозащитный механизм. Действие ANP, однако, только кратковременное, так как гормон быстро расщепляется под действием NEP.
На основании своих подавляющих NEP свойств предлагаемые согласно изобретению соединения могут улучшать кардиозащитный механизм действия ANP и обладают в особенности высокой эффективностью в отношении усиления диуретически/натриуретических активностей.
Предлагаемые согласно изобретению соединения отличаются благоприятным профилем действия с хорошей переносимостью и при этом обладают значительной селективностью в отношении ингибирующего NEP действия и дополнительно еще могут оказывать незначительные ингибиторные воздействия на конвертирующий эндотелин фермент (= ECE). При чрезвычайно высоких стадиях сердечной недостаточности приходят рефлекторно к высоким уровням в крови ангиотензина-II, эндотелина и катехоламинов и вместе с этим к дальнейшему повышению периферического сопротивления и давления за счет наполнения сердца, вследствие чего гипертрофируется и расширяется миокард (сердечная мышца). Ингибирующие ECE дополнительные свойства при этом могут усиливать понижающее воздействие предлагаемых согласно изобретению веществ на периферическое сопротивление.
Ингибирующие NEP и ECE и усиливающие диурез/натриурез свойства веществ обнаруживаются стандартными фармакологическими методами испытания in vitro и in vivo.
Описание фармакологических методов исследования:
1. Определение минимальной токсичной дозы
Самцам мышей весом 20-25 г перорально вводят максимальные дозы 300 мг/кг испытуемого вещества. В течение 3-х часов животных тщательно наблюдают в отношении симптомов токсичности. В течение промежутка времени 72 часа после введения дополнительно регистрируют все симптомы и случаи смерти. Сопутствующие симптомы также наблюдают и регистрируют. Если наблюдают гибель или сильные токсичные симптомы, то следующим мышам вводят в возрастающей степени меньшие дозы, пока более не будут появляться токсичные симптомы. Самая низкая доза, которая вызывает гибель или сильные токсичные симптомы, указывается в нижеприводимой таблице A как минимальная токсичная доза. Указанные в таблице A номера примеров относятся к нижеследующим примерам получения.
2. Исследование in vitro подавляющего NEP действия веществ и определение сродства молекул веществ к молекуле фермента
Для обнаружения подавляющего действия предлагаемых согласно изобретению веществ на нейтральную эндопептидазу ( =NEP) в стандартном тесте in vitro изучают ингибирующее воздействие веществ на происходящее благодаря ферментативной активности NЕР гидролитическое разрушение метионин-энкефалина (= Met-энкефалин). При этом в качестве меры ингибирующей эффективности веществ определяют их Ki - значение (= константа ингибирования). Ki - Значение действующего ингибирующе на фермент испытуемого вещества представляет собой константу диссоциации комплекса фермент - испытуемое вещество,
соответственно, комплекса (фермент - субстрат) - испытуемое вещество и имеет размерность концентрации.
Осуществление теста
Для осуществления теста, смотря по обстоятельствам, готовят по 100 мкл образцов различных инкубационных растворов, содержащих 10 нг очищенной NEP (E. C. 3.4.24.11) и, смотря по обстоятельствам, различные количества тест-вещества и субстрата (Met-энкефалин) и 50 ммоль Трис-буфера (= трис (гидроксиметил)аминометан/HCl, pH 7,4).
На одно тест-вещество, смотря по обстоятельствам, готовят 24 инкубационных раствора с 3-мя различными концентрациями тест- вещества (испытуемого вещества), смотря по обстоятельствам, в комбинации с содержанием Met-энкефалина 2, 5, 7, 10, 12, 15, 40 и 100 мкмоль.
В каждом тесте, смотря по обстоятельствам, совместно также обрабатывают два различных контрольных инкубационных растворов, во-первых для контролирования фермента, которые не содержат никакого тест-вещества, и во-вторых для контролирования субстрата, которые не содержат ни фермента, ни тест-вещества.
Инкубационные растворы инкубируют в течение 45 минут при 37oC на водяной бане при встряхивании. При этом реакцию фермента спустя 15 минут инициируют путем добавки субстрата (Met-энкефалин) и по окончании времени инкубации останавливают путем нагревания в течение 5 минут при 95oC. Затем инкубационный раствор, в котором прекращена реакция, центрифугируют в течение 3 минут при 12000 g, и в надосадочной жидкости определяют концентрации непровзаимодействовавшего субстрата и образовавшихся за счет ферментативной реакции продуктов гидролиза. Для этой цели, смотря по обстоятельствам, образцы надосадочных жидкостей разделяют с помощью ВЭЖХ (= высокоэффективная жидкостная хроматография) на гидрофобизированном силикагеле, и продукты ферментативной реакции и непровзаимодействовавший субстрат определяют фотометрически при длине волны 205 нм. Для ВЭЖХ-разделения используют разделительную колонку (4,6 х 250 мм), которая заполнена в качестве разделительного материала обращенной фазы с помощью Encapharm® 100 RR 18,5 микрон. Поток растворителя составляет 1,4 мл/мин, колонка нагревается до 40oC. Элюирующее средство A представляет собой 5 мМ H3PO4, pH 2,5, а элюирующее средство В представляет собой ацетонитрил +1% 5 мМ H3PO4, pH 2,5.
Из измеренных в различных пробах концентраций продуктов гидролиза и непровзаимодействовавшего субстрата для каждого тест- вещества рассчитывают само по себе известным образом Ki - значение. В нижеследующей таблице В приводятся найденные для тест- веществ Ki-значения. Указанные в таблице В номера примеров относятся к нижеследующим примерам получения.
3. Исследования in vitro подавляющего ECE действия веществ
Для обнаружения подавляющего действия предлагаемых согласно изобретению веществ на конвертирующий эндотелии фермент ( =ECE) в стандартном тесте in vitro исследуют ингибирующее действие веществ на происходящее благодаря ферментной активности ECE гидролитическое разрушение Big-эндотелина 1 (Big ET-1). При этом в качестве меры ингибиторной эффективности веществ определяют их IC50-значение. IC50 - Значение активного в отношении ингибирования фермента испытуемого (тест-) вещества представляет собой концентрацию испытуемого вещества, при которой ингибируются 50% ферментативной активности ECE.
Получение содержащей ECE мембранной фракции эндотелиальных клеток
Яйцеклетки китайского хомяка (= Chiense hamster ovarian cells, далее сокращенно называют как CHO-клетки), в которые рекомбинантно экспримирован человеческий ECE (см. Schmidt и др. Federation of European Biochemical Soсieties Letters, 356 (1994), 238-243), лизируют и клеточные мембраны отделяют путем центрифугирования при 10000 g в течение 10 минут. Клеточные мембраны промывают путем трехкратного повторного суспендирования и повторного центрифугирования. Содержащую ECE клеточную мембранную фракцию снова суспендируют в 100 мМ Трис/HCl - буфер (=трис (гидроксиметил)аминометан/HCl, pH 7,0, содержащий 250 мМ NaCl) и вплоть до ферментного теста хранят замороженной при -70oC.
Осуществление теста
Для осуществления теста готовят, смотря по обстоятельствам, пробы по 100 мкл каждая различных инкубационных растворов, содержащие 5 мкг протеина содержащего ECE препарата эндотелиальных клеточных мембран и, смотря по обстоятельствам, различные количества испытуемого вещества и 24 мкмоль субстрата (синтетический пептид: H2N-Asp-Ile-Ala-Trp-Phe-Asn-Thr-Pro-Glu-His-Val-Val-Pro-Tyr- Gly-Leu-Gly-COOH) и 100 ммоль трис-буфера (= трис(гидроксиметил)аминометан/HCl, pH 7,0, содержащий 250 ммоль хлорида натрия). Сверх того, в каждом инкубационном растворе содержатся 100 мкмоль тиорфана и 10 мкмоль каптоприла и 1 ммоль фенилсульфонилфторида, 100 мкмоль пепстатина A (= ингибитор протеазы) и 100 мкмоль амастатина (= ингибитор протеазы).
На одно испытуемое вещество готовят, смотря по обстоятельствам, 6 различных инкубационных растворов с 3-мя различными концентрациями испытуемого вещества, смотря по обстоятельствам, для двойных определений.
В случае каждого теста, смотря по обстоятельствам, также совместно обрабатывают контроль, который не содержит никакого фермента.
Инкубационные растворы предварительно инкубируют в течение 15 минут при 37oC, прежде, чем добавляют субстрат. Ферментную реакцию инициируют путем добавки субстрата; она продолжается в течение 60 минут при 37oC и ее прекращают путем нагревания инкубационных растворов в течение 5 минут при 95oC. Образовавшиеся благодаря ферментативной реакции из субстрата продукты гидролиза H2N-Asp-Ile-Ala-Trp- COOH и H2N-Phe-Asn-Thr-Pro-Glu-His-Val-Val-Pro-Tyr- Gly-Leu-Gly-COOH определяют с помощью высокоэффективной жидкостной хроматографии (ВЭЖХ). Осуществление определения с помощью ВЭЖХ проводят как в случае вышеописанного ин витро исследования подавляющего NEP действия.
Из измеренных в различных пробах концентраций продуктов гидролиза для каждого испытуемого вещества рассчитывают само по себе известным образом IC50. В нижеследующей таблице C приводятся найденные для испытуемых веществ IC50-значения.
4. In vivo определение влияния веществ на диурез/натриурез в случае крыс со связанной с увеличением объема нагрузкой
Исследуют in vivo активность в случае крыс со связанной с увеличением объема нагрузкой. В этом эксперименте путем инфузии изотонического раствора хлорида натрия вызывают высокое сердечное давление наполнения, вследствие которого приходят к высвобождению ANP и благодаря этому к диурезу/натриурезу.
Осуществление теста:
Опыты проводят при использовании самцов крыс Wistar с весом тела 200-400 г. При нейролептоанальгезии (фентанил; Hypnorm®, изготовитель: фирма Janssen) в правую феморальную вену вставляют катетер для периферической инфузии (Hintegrundinfusion) и объемной нагрузки с помощью изотонического раствора хлорида натрия. После раскрытия брюшной полости в мочевой пузырь вводят второй катетер и перевязывают уретру так, чтобы можно было измерять объем мочи, натриурез и калиурез.
Брюшную полость снова закрывают и животные получают длительную инфузию раствора хлорида натрия (0,5 мл /100 г веса тела) в течение всего промежутка опыта 2 часа. После периода уравновешивания 30 минут в предварительной фазе до введения испытуемого вещества, смотря по обстоятельствам, трижды через промежуток времени 10 минут собирают пробы мочи. Эти предварительные значения (= "Predrug"-значения) определяют для того, чтобы дополнительно проверить, что у подопытных животных происходит непрерывное мочеиспускание.
Затем содержащие испытуемые вещества растворы вводят внутривенно (инъекция лекарства в феморальную вену) или орально (с помощью желудочного зонда) группам, смотря по обстоятельствам, из 10 крыс. В случае обеих форм введения, смотря по обстоятельствам, контрольная группа получает лишь растворы плацебо, которые не содержат никакого биологически активного вещества. Спустя 5 минут после внутривенного введения, соответственно, спустя 120 минут после орального введения веществ крыс нагружают повышенным объемом раствора хлорида натрия внутривенно (2 мл /100 г веса тела в течение 2-х минут) и через промежуток времени 60 минут собирают мочу. Определяют выделившиеся за этот промежуток времени количества мочи и измеряют содержащиеся в ней количества натрия и калия. Из выделившихся количеств мочи определяют происходящее при объемной нагрузке повышение выделения по сравнению с предварительными значениями.
В нижеследующей таблице D указываются наступающие при объемной нагрузке после введения испытуемого вещества повышения мочеиспускания в % в расчете на происходящие при объемной нагрузке после введения плацебо увеличения мочевыделения. Далее, также указываются выделяющиеся при объемной нагрузке после введения испытуемого вещества количества натрия и калия в % в расчете на количества натрия и калия, выделяющиеся при объемной нагрузке после введения плацебо.
Вышеприведенные результаты испытаний показывают, что соединения формулы (I) обладают большим сродством к NEP и благодаря подавлению этого, разрушающего ANP фермента способствуют повышению уровня ANP в крови и благодаря этому повышают вызываемые ANP диуретически/натриуретические эффекты, не вызывая значительной потери калия.
На основании своего вышеописанного действия соединения формулы (I) пригодны в качестве лекарственных средств для более крупных млекопитающих, в особенности людей, с целью лечения сердечной недостаточности и для способствования диурезу/натриурезу, в особенности в случае страдающих сердечной недостаточностью пациентов. При этом дикарбоновые кислоты формулы (I) и их соли целесообразно использовать в парентерально, в особенности внутривенно, вводимых лекарственных формах, а сложные моно- или диэфиры формулы (I) целесообразно используют в орально вводимых лекарственных формах. Применяемые дозы индивидуально могут быть различны, и, естественно, изменяются в зависимости от вида излечиваемого состояния, используемого вещества и формы введения. Например, парентеральные формулировки в общем содержат меньше биологически активного вещества, чем оральные препараты. В общем, однако, для введений более крупным млекопитающим, в особенности людям, пригодны лекарственные формы с содержанием биологически активного вещества 1-200 мг на разовую дозу.
В качестве лекарственных средств, соединения формулы (I) могут содержаться вместе с обычными фармацевтическими вспомогательными веществами в галеновых композициях, как, например, таблетки, капсулы, свечи или растворы. Эти галеновые композиции можно приготовлять само по себе известными методами при применении обычных твердых или жидких носителей, как, например, молочный сахар, крахмал или тальк, или жидкие парафины, и/или при применении обычных фармацевтических вспомогательных веществ, например, как наполнители таблеток (Sprengmittel), агенты растворения или консерванты.
Нижеследующие примеры должны подробнее пояснить изобретение, однако никоим образом не ограничивая его объема охраны.
Структуры новых соединений гарантированы путем спектроскопических исследований, в особенности путем анализа ЯМР-, масс-, ИК-/или УФ-спектров и в случае необходимости путем определения величин оптического вращения.
Пример 1: трет.-Бутиловый эфир 3-{1-/2'-(этоксикарбонил)-4'- фенил-бутил/циклопентан-1-карбониламино} -2,3, 4,5-тетрагидро-2-оксо-1H-1-бензазепин-1-уксусной кислоты.
A). К раствору 160,1 г диэтилового эфира малоновой кислоты в 1 л диметилформамида при температуре 15oC порциями добавляют 123,4 г трет.-бутилата калия. Реакционную смесь перемешивают в течение 30 минут, затем при комнатной температуре прикапывают раствор из 207,7 г фенетилбромида в 200 мл диметилформамида. Затем реакционную смесь нагревают в течение часа при 60oC и оставляют снова охлаждаться. Диметилформамид испаряют при пониженном давлении, остающийся остаток обрабатывают смесью простого метил- трет, бутилового эфира с водой. Органическую фазу отделяют, промывают водой, сушат над сульфатом натрия и выпаривают. Остающийся в виде маслянистого остатка сырой продукт очищают путем перегонки при пониженном давлении. Получают 202,5 г этилового эфира 2-этоксикарбонил-4-фенил-бутановой кислоты. Т. кип. = 148- 153oC (1,5 мм рт. ст.).
Б.). К раствору 23,6 г вышеполученного сложного диэфира в 285 мл этанола добавляют раствор 6,17 г гидроксида калия в 76 мл воды при охлаждении льдом. Реакционную смесь перемешивают несколько часов при комнатной температуре. Затем этанол испаряют при пониженном давлении и остаток обрабатывают смесью простого метил-трет.-бутилового эфира с водой. Органическую фазу отделяют и отбрасывают, а водную фазу при охлаждении льдом подкисляют с помощью разбавленной водной соляной кислоты и после этого многократно экстрагируют метил-трет. - бутиловым простым эфиром. Объединенные фазы в метил-трет.- бутиловом эфире промывают водой, сушат над сульфатом натрия и выпаривают при пониженном давлении. Получают 20,1 г сырого маслянистого этилового эфира 2-карбокси-4-фенил-бутановой кислоты, который без дальнейшей очистки вводят во взаимодействие далее.
В). К 20,2 г вышеполученного продукта при охлаждении льдом последовательно добавляют 11 мл 35%-ного водного раствора формальдегида и 9,23 мл пиперидина. Реакционную смесь перемешивают в течение нескольких часов при комнатной температуре, затем разбавляют метил-трет.-бутиловым простым эфиром, промывают водным раствором гидросульфата калия и водой, сушат над сульфатом натрия и выпаривают. Остаток высушивают при пониженном давлении. Получают 14,8 г этилового эфира α- (2-фенилэтил)акриловой кислоты.
Г). В атмосфере азота 25,2 мл диизопропиламина растворяют в 150 мл абсолютного тетрагидрофурана и охлаждают до -35oC. К раствору прикапывают 100 мл 1,6 н. раствора бутиллития в н-гексане. После этого реакционную смесь перемешивают в течение 30 минут при 0oC и затем прикапывают раствор 8,1 мл циклопентанкарбоновой кислоты в 20 мл абсолютного тетрагидрофурана. Реакционную смесь перемешивают 2 часа при 0oC. После этого прикапывают раствор 16,8 г полученного в п. В) сложного акрилового эфира в 20 мл абсолютного тетрагидрофурана, реакционную смесь оставляют стоять в течение 2 часов при 0oC и затем в течение нескольких часов при -15oC. Для обработки реакционную смесь подкисляют с помощью 10%-ного водного раствора соляной кислоты и экстрагируют н-гексаном. Органическую фазу промывают 7 раз с помощью полунасыщенного водного раствора бикарбоната натрия и один раз водой, сушат над сульфатом натрия и выпаривают при пониженном давлении. Полученный в виде остатка сырой продукт очищают путем флэш-хроматографии на силикагеле при применении смеси н-гексана с этилацетатом (8:2). Получают 19,6 г чистой 1-/2'-(этоксикарбонил)-4'-фенил-бутил/циклопентан-1- карбоновой кислоты с т.пл. 68-69oC.
Д). К раствору 100 г α- тетралона в 820 мл метанола при охлаждении льдом медленно прикапывают 108,3 г брома. Затем реакционную смесь перемешивают в течение 30 минут при комнатной температуре и после этого при комнатной температуре добавляют сначала 122,4 г гидроксиламин-гидрохлорида и затем 110 мл воды. Смесь перемешивают в течение 3-х дней при комнатной температуре. После этого добавляют следующие 493 мл воды, причем спустя 1 час выпадает белого цвета осадок. Реакционную смесь перемешивают в течение следующих 3-х дней и затем охлаждают до 5oC. Осадок отсасывают, промывают водой и высушивают при 40oC при пониженном давлении. Получают 136,7 г 2-бром-3,4-дигидронафталин- 1(2H)-он-оксима с т.пл. 130-132oC.
Е). 79,5 г Вышеполученного оксима порциями добавляют к 452 г нагретой до 80oC полифосфорной кислоты и реакционную смесь перемешивают в течение 18 часов при 80oC. Затем осторожно разбавляют с помощью 710 мл воды и смесь перемешивают в течение 2 часов при комнатной температуре. Образовавшийся осадок отсасывают, промывают водой, водным раствором бикарбоната натрия, еще раз водой и затем метил-трет.- бутиловым простым эфиром и высушивают над гидроксидом калия при температуре 60oC. Получают 66,6 г 3-бром-4,5-дигидро-1H-1- бензазепин-2-(3H)-она с т.пл. 168-170oC.
Ж). 80 г Вышеполученного продукта суспендируют в 140 мл диметилформамида. Суспензию смешивают с раствором 72,6 г фталимида калия в 205 мл диметилформамида и затем перемешивают в течение 16 часов при 60oC. Для обработки, охлаждают до комнатной температуры и медленно прикапывают 800 мл воды и смесь перемешивают в течение 2 часов при охлаждении льдом. Образовавшуюся кашицу кристаллов отсасывают и промывают сначала смесью воды с диметилформамидом и затем метил- трет.-бутиловым простым эфиром и после этого высушивают в течение 2-х дней при 60oC и пониженном давлении. Получают 73,3 г 4,5- дигидро-3-фталимидо-1H-1-бензазепин-2(3H)-она с областью плавления 185-195oC.
З). К суспензии из 27 г вышеполученного продукта в 90 мл диметилформамида при охлаждении льдом добавляют раствор 12,3 г трет.-битулата калия в 40 мл диметилформамида. После перемешивания в течение 30 минут при охлаждении льдом в течение часа при 0-5oC прикапывают 20,7 г трет.-бутилового эфира бромуксусной кислоты. Перемешивают в течение 1 часа при 0oC. После этого реакционную смесь нагревают до 40oC и в течение 3 часов прикапывают 164 мл воды и перемешивают еще в течение часа при 30oC. Затем водный раствор отделяют путем декантации от образовавшегося осадка и остающийся твердый остаток кристаллизуют из метил-трет.-бутилового эфира. Образовавшиеся кристаллы отсасывают, промывают водой и метил- трет.бутиловым эфиром и высушивают при пониженном давлении при 60oC. Получают 26,3 г трет. -бутилового эфира 2,3,4,5-тетрагидро-2- оксо-3-фталимидо-1H-1-бензазепин-1-уксусной кислоты с т.пл. 194- 197oC.
И). 7 г вышеполученного сложного эфира в течение 5 минут добавляют к 13,8 мл нагретого до 80oC этаноламина. Спустя 5 минут образуется прозрачный раствор, который охлаждают до комнатной температуры и разбавляют с помощью 105 мл толуола. Раствор встряхивают со 140 мл 5%-ного водного раствора хлорида натрия, органическую фазу отделяют, сушат над сульфатом натрия и выпаривают. Остающийся остаток кристаллизуют из метил-трет.- бутилового эфира. Получают 4,0 г трет.-бутилового эфира 3-амино- 2,3,4,5-тетрагидро-2-оксо-1H-1-бензазепин-1-уксусной кислоты с т.пл. 117-118oC.
К). 2,9 г вышеполученного амина и 3,2 г вышеполученной в п. Г) кислоты растворяют в 100 мл дихлорметана. К реакционной смеси при охлаждении льдом добавляют 2,2 мл N- метилморфолина, 1,27 г гидрокси-бензотриазола и 3,81 г N-этил-N- (3-диметиламинопропил)карбодиимидгидрохлорида. После этого реакционную смесь перемешивают в течение 1 часа при комнатной температуре. Для обработки, реакционную смесь разбавляют дихлорметаном и промывают последовательно водой, водным раствором гидросульфата калия, водой, водным раствором бикарбоната натрия и снова водой. Органическую фазу затем сушат над сульфатом натрия и растворитель выпаривают при пониженном давлении. Таким образом полученный сырой продукт очищают с помощью колоночной хроматографии на силикагеле при слегка повышенном давлении ( = флэш-хроматография) при применении смеси н-гексана с этилацетатом, причем этилацетатная доля элюирующего средства во время элюирования повышается от первоначальной 1:9 до 3:7. Получают 5,4 г чистого целевого соединения в виде маслянистого продукта.
ИК-спектр (в виде пленки): 3400 см-1, 1725 см-1, 1660 см-1.
Пример 2: 3-{1-/2'-(этоксикарбонил-4'-фенил-бутил/циклопентан- 1-карбониламино}-2,3,4,5-тетрагидро-2-оксо-1H-1-бензазепин-1-уксусная кислота
5 г трет. -Бутилового эфира 3-{ 1-/2'-(этоксикарбонил)-4'- фенилбутил/циклопентан-1-карбониламино} -2,3,4,5-тетрагидро-2- оксо-1H-1-бензазепин-1-уксусной кислоты (получение см. в примере 1) растворяют в 16 мл трифтороуксусной кислоты. Раствор перемешивают 3 часа при комнатной температуре. Для обработки, трифторуксусную кислоту выпаривают при пониженном давлении. Остающийся остаток растворяют в дихлорметане и раствор промывают водой до нейтральной реакции. Затем органическую фазу сушат над сульфатом натрия и выпаривают при пониженном давлении. Остающийся остаток многократно перемешивают с н-гексаном и, смотря по обстоятельствам, снова выпаривают досуха. Получают 3,4 г целевого соединения в виде твердой пены с областью плавления 81-104oC.
Пример 3: Трет.-Бутиловый сложный эфир (3S,2'R)-3- {1-/2'-(этоксикарбонил)-4'-фенил-бутил/циклопентан-1-карбониламино- 2,3,4,5-тетрагидро-2-оксо-1H-1-бензазепин-1-уксусной кислоты.
A). 30,5 г 1-/2'-(этоксикарбонил)-4'-фенил-бутил/циклопентан- 1-карбоновой кислоты (получение см. пример 1 Г) и 11,6 г L-(-)- α-метилбензиламина при нагревании растворяют в этаноле. Реакционную смесь в течение 12 часов охлаждают в холодильнике, затем образовавшуюся кашицу кристаллов отсасывают, высушивают и многократно (вплоть до постоянства величины вращения) перекристаллизуют из этанола и затем высушивают при пониженном давлении. Получают 17,7 г α-метилбензиламмониевой соли вышеуказанной кислоты с т.пл. 118-121oC, величина оптического вращения [α]
Для высвобождения кислоты эту соль обрабатывают смесью воды с дихлорметаном и смесь подкисляют водным раствором гидросульфата калия. Органическую фазу отделяют, а водную фазу еще трижды экстрагируют дихлорметаном. Объединенные органические экстракты промывают водой, сушат над сульфатом натрия и выпаривают при пониженном давлении. Остающийся остаток высушивают. Получают 11,2 г чистой (2'R)-1-/2'- (этоксикарбонил)-4'-фенил-бутил/циклопентан-1-карбоновой кислоты; величина оптического вращения [α]
Б). К нагретому до 65oC раствору 24,5 г рацемического трет.-бутилового эфира 3-амино-2,3,4,5-тетрагидро-2- оксо-1H-1-бензазепин-1-уксусной кислоты (получение см. пример 1 И) добавляют раствор 12,65 г L-(+)-винной кислоты в 54 мл нагретого до 65oC этанола. Реакционную смесь перемешивают в течение часа при комнатной температуре. Затем прикапывают раствор 1,72 мл бензальдегида в 1,3 мл этанола. Полученную суспензию кипятят с обратным холодильником в течение 14 часов при 80oC и после этого охлаждают до комнатной температуры. Образовавшийся кристаллический осадок отсасывают, обрабатывают с помощью 80 мл этанола и снова в течение 8 часов кипятят с обратным холодильником. Затем охлаждают до комнатной температуры и кристаллы отсасывают и высушивают при 50oC и при пониженном давлении. Получают 23,6 г виннокислой соли с т.пл. 195-196oC и величиной оптического вращения [α]
Для высвобождения основания, 23,6 г виннокислой соли в смеси из 250 мл воды и 108 мл дихлорметана при перемешивании охлаждают до 0oC и путем добавки водного раствора аммиака устанавливают pH 9,6. Органическую фазу отделяют, водную фазу еще раз экстрагируют с помощью 30 мл дихлорметана и органические фазы объединяют, сушат над сульфатом натрия и концентрируют при пониженном давлении. Остающийся остаток кристаллизуют из метил- трет.-бутилового эфира и высушивают при пониженном давлении. Получают 12,2 г трет.-бутилового эфира (3S)-3-амино-2,3,4,5- тетрагидро-2-оксо-1H-1-бензазепин-1-уксусной кислоты с т.пл. 113-115oC и величиной оптического вращения [α]
В). 5,4 г Полученной выше в п. A) кислоты растворяют в 60 мл безводного дихлорметана. Раствор смешивают с 2,33 мл триэтиламина и охлаждают до -20oC. Затем медленно прикапывают раствор 1,31 мл хлорангидрида метансульфокислоты в 5 мл безводного дихлорметана. После перемешивания в течение 15 минут прикапывают раствор из 4,8 г полученного выше в п.Б). амина и 2,33 мл триэтиламина в 60 мл дихлорметана. Затем реакционную смесь перемешивают в течение 1 часа при комнатной температуре. Для обработки, реакционную смесь добавляют в воду, органическую фазу отделяют и промывают с помощью водного раствора гидросульфата калия и после этого водой, сушат над сульфатом натрия, отфильтровывают и концентрируют при пониженном давлении. Остающийся сырой продукт очищают путем флэш-хроматографии на 500 г силикагеля при применении смеси н-гексана с этилацетатом (7: 3). После высушивания при пониженном давлении получают 9,5 г чистого целевого соединения в виде масла; величина оптического вращения [α]
Пример 4: (3S, 2'R)-3-{ 1-/2'-(Этоксикарбонил)-4'- фенил-бутил/-циклопентан-1-карбониламино} -2,3,4,5-тетрагидро-2- оксо-1H-1-бензазепин-1-уксусная кислота
9,4 г трет. -Бутилового эфира (3S,2'R)-3-{1-/2'-(этоксикарбонил)- 4'-фенил-бутил/-циклопентан-1-карбониламино} -2,3,4,5-тетрагидро-2-оксо-1H-1-бензазепин-1-уксусной кислоты (получение см. пример 3) при охлаждении льдом растворяют в 15 мл дихлорметана. Раствор смешивают с 31 мл трифторуксусной кислоты и реакционную смесь выдерживают в течение примерно 12 часов при 4oC в холодильнике. Для обработки дихлорметан и трифторуксусную кислоту выпаривают при пониженном давлении. Полученный сырой продукт обрабатывают этилацетатом, полученный раствор промывают водой, разбавленным водным раствором бикарбоната натрия и снова водой. Органическую фазу отделяют, сушат над сульфатом натрия и выпаривают при пониженном давлении. Остающийся остаток очищают путем флэш-хроматографии на силикагеле, причем в качестве элюирующего средства применяют сначала дихлорметан и затем смесь дихлорметана с метанолом (95: 5). Полученный продукт высушивают в течение 2-х дней при 80oC и при пониженном давлении. Получают 7,3 г чистого целевого соединения в виде твердой пены с т.пл. 71-74oC; величина оптического вращения [α]
Пример 5: трет.-Бутиловый сложный эфир 3-{1-/2'-{трет.- бутоксикарбонил)-4'-фенил-бутил/циклопентан-1-карбониламино} -2,3,4,5-тетрагидро-2-оксо-1H-1-бензазепин-1-уксусной кислоты.
A). 118 г трет.-Бутилового эфира диметилфосфоноуксусной кислоты в атмосфере азота растворяют в 875 мл безводного диметилформамида. К раствору при охлаждении льдом добавляют 58,9 г трет.-бутилата калия. Затем реакционную смесь кратковременно нагревают при 60oC и затем оставляют охлаждаться до комнатной температуры. К реакционной смеси прикапывают раствор 104,9 г фенетилбромида в 110 мл диметилформамида. После этого реакционную смесь нагревают в течение 2 часов при 60oC. Для обработки, диметилформамид далее выпаривают при пониженном давлении и остающийся остаток растворяют в метил-трет. -бутиловом эфире. Раствор подкисляют с помощью водного раствора гидросульфата калия. Затем органическую фазу отделяют, промывают водой, сушат над сульфатом натрия и выпаривают при пониженном давлении. Полученный сырой продукт очищают путем флэш-хроматографии на 3 кг силикагеля при применении смеси дихлорметана с метил-трет.-бутиловым эфиром (4:1) в качестве элюирующего средства. Получают 105,1 г чистого трет.-бутилового эфира 2-(диметилфосфоно)-4-фенил-н-масляной кислоты в виде маслянистого продукта.
В). 105,1 г Вышеполученного продукта в атмосфере азота растворяют в 705 мл безводного тетрагидрофурана. К раствору добавляют 28,4 г параформальдегида. Затем медленно прикапывают раствор 32,5 г трет.-бутилата калия в 100 мл тетрагидрофурана. После этого реакционную смесь перемешивают в течение 1 часа. Для обработки, реакционную смесь подкисляют с помощью холодного водного раствора гидросульфата калия и разбавляют метил-трет.-бутиловым эфиром. Затем органическую фазу отделяют, промывают водой, сушат над сульфатом натрия и концентрируют при пониженном давлении. Полученный сырой продукт очищают путем флэш-хроматографии на 700 г силикагеля при применении смеси н-гексана с этилацетатом (9: 1). Получают 47,0 г трет.-бутилового эфира α- (фенетил)акриловой кислоты в виде бесцветного масла.
В). К охлажденному до -50oC раствору из 50,2 мл диизопропиламина в 450 мл абсолютного тетрагидрофурана прикапывают 200 мл 1,6 М раствора бутиллития в н-гексане и реакционную смесь выдерживают следующие 30 минут при 0oC. Затем при этой температуре прикапывают раствор 16,2 мл циклопентанкарбоновой кислоты в 40 мл абсолютного тетрагидрофурана. Реакционную смесь перемешивают следующие 2 часа при 0oC. После этого к смеси медленно добавляют раствор 38 г полученного выше в п. Б). продукта в 50 мл абсолютного тетрагидрофурана. Реакционную смесь перемешивают следующие 2 часа при 0oC и затем еще в течение нескольких часов оставляют стоять при -15oC. Для обработки реакционную смесь при охлаждении льдом подкисляют с помощью насыщенного водного раствора гидросульфата калия и трижды экстрагируют н-гексаном. Объединенные органические фазы промывают 7 раз полунасыщенным водным раствором бикарбоната натрия и затем водой, после этого сушат над сульфатом натрия и выпаривают при пониженном давлении. Полученный маслянистый сырой продукт кристаллизуют из охлажденного льдом н-гексана. Получают 41,9 г чистой кристаллической 1-/2'-(трет. -бутоксикарбонил)- 4'-фенил-бутил/циклопентан-1-карбоновой кислоты с т.пл. 75- 77oC.
Г). 3,3 г Вышеполученного продукта, 2,7 г трет.-бутилового эфира 3-амино-2,3,4,5-тетрагидро-2-оксо-1H-1-бензазепин-1-уксусной кислоты (получение см. пример 1 И)), 1,53 мл N-метилморфолина и 1,18 г гидроксибензотриазола в атмосфере азота растворяют в 93 мл абсолютного дихлорметана. К этому раствору при охлаждении льдом добавляют 3,52 г N-этил-N'-(3-диметиламинопропил)карбодиимидгидрохлорида. Затем реакционную смесь перемешивают в течение 2 часов при охлаждении льдом. Для обработки, реакционную смесь промывают последовательно водой, водным раствором гидросульфата калия, водой, водным раствором бикарбоната натрия и снова водой. Органическую фазу сушат над сульфатом натрия и концентрируют при пониженном давлении. Остающийся сырой продукт очищают путем флэш- хроматографии на 200 г силикагеля при применении смеси н-гексана с этилацетатом (7:3) в качестве элюирующего средства и кристаллизуют из метил-трет.-бутилового эфира. Получают 4,2 г чистого целевого соединения с т.пл. 110-114oC.
Пример 6: 3-/1-(2'-Карбокси-4'-фенил-бутил)циклопентан- 1-карбониламино/-2,3,4,5-тетрагидро-2-оксо-1H-1- бензазепин-1-уксусная кислота
4,1 г трет. -Бутилового эфира 3-{1- /2'-(трет.-бутоксикарбонил)-4'-фенил-бутил/циклопентан-1- карбониламино} -2,3,4,5-тетрагидро-2-оксо-1H-1-бензазепин-1- уксусной кислоты (получение см. пример 5), при температуре 4oC и при исключении влаги растворяют в 13 мл трифторуксусной кислоты. Полученный раствор перемешивают следующие 3 часа при этой температуре. Для обработки реакционную смесь концентрируют при пониженном давлении. Для полного удаления трифтороуксусной кислоты остаток многократно смешивают с дихлорметаном и снова выпаривают. Полученный остаток затем растворяют в дихлорметане, раствор промывают водой, сушат над сульфатом натрия и концентрируют при пониженном давлении. Остающийся в виде остатка сырой продукт кристаллизуют из дихлорметана. Получают 2,7 г чистого целевого соединения с т.пл. 178-183oC.
Пример 7: трет. -Бутиловый сложный эфир (3S,2'R)-2-{1- /2'-(трет.-бутоксикарбонил)-4'-фенил-бутил/циклопентан-1- карбониламино} -2,3,4,5-тетрагидро-2-оксо-1H-1-бензазепин-1- уксусной кислоты.
A). 68 г 1-/2'-(трет.-Бутоксикарбонил)-4'-фенил-бутил/ циклопентан-1-карбоновой кислоты (получение см. пример 5 В)) и 23,5 мл 1-(-) -α- метилбензиламина при нагревании растворяют в 200 мл этанола. Реакционную смесь обрабатывают как описано в примере 3 A). Получают 32,2 г α- метилбензиламмониевой соли вышеуказанной кислоты с т.пл. 118-119oC; величина оптического вращения составляет:
[α]
Б). 60,1 г Вышеполученной кислоты вводят во взаимодействие с 50,3 г трет. - бутилового эфира (3S)-3-амино-2,3,4,5-тетрагидро-2-оксо-1H-1- бензазепин-1-уксусной кислоты (получение см. пример 3 Б)), согласно описанному в примере 3 В) способу, и полученную реакционную смесь обрабатывают как описано в примере 3 В). Получают 94,3 г целевого соединения в виде пены; величина оптического вращения [α]
Пример 8: (3S,2'R)-3-/1-(2'-Карбокси-4'-фенил-бутилциклопентан- 1-карбониламино/-2,3,4,5-тетрагидро-2-оксо-1H-1-бензазепин-1-уксусная кислота.
93 г трет. -Бутилового эфира (3S,2'R)-3-{1-/2'-(трет.- бутоксикарбонил)-4'-фенил-бутил/циклопентан-1-карбониламино} - 2,3,4,5-тетрагидро-2-оксо-1H-1-бензазепин-1-уксусной кислоты (получение см. пример 7) согласно описанному в примере 6 способу гидролизуют с помощью трифторуксусной кислоты и реакционную смесь обрабатывают как описано в примере 6. Получают 63,5 г целевого соединения в виде твердой пены с областью плавления 97-122oC; величина оптического вращения [α]
Пример 9: Бензиловый сложный эфир (3S,2'S)-3- {1-/2'-(трет-бутоксикарбонил)-3'-(2-метоксиэтокси) пропил/циклопентан-1-карбониламино}-3,4-дигидро-4-оксо-1,5- бензоксазепин-5(2H)-уксусной кислоты.
A). К раствору из 100 г 3-бромпропионовой кислоты в 200 мл диэтилового эфира добавляют 7 мл серной кислоты и реакционную смесь охлаждают до -20oC. Затем добавляют 123 мл сжиженного изобутена. Реакционную смесь перемешивают в течение нескольких часов при комнатной температуре в сосуде высокого давления. Затем реакционную смесь, с целью обработки, вносят в разбавленный, охлажденный льдом водный раствор гидроксида натрия. Эфирную фазу отделяют и водную фазу еще раз экстрагируют эфиром. Объединенные органические фазы промывают водным раствором хлорида натрия, сушат над сульфатом натрия и выпаривают при пониженном давлении. Полученный сырой продукт перегоняют при пониженном давлении. Получают 100 г трет.-бутилового эфира 3-бром-пропионовой кислоты. Т. кип. 75-77oC (20 мм рт.ст.).
Б). 50,4 мл Диизопропиламина в атмосфере азота растворяют в 300 мл абсолютного тетрагидрофурана и раствор охлаждают до -70oC. К раствору при этой температуре медленно прикапывают 200 мл 1,6 М раствора бутиллития в н-гексане. Реакционную смесь оставляют нагреваться до 0oC, перемешивают 30 минут при этой температуре и снова охлаждают до -20oC. При этой температуре прикапывают раствор 16,2 мл циклопентанкарбоновой кислоты в 30 мл абсолютного тетрагидрофурана. Затем реакционную смесь в течение 2 часов перемешивают при комнатной температуре. После этого смесь охлаждают до -10oC и медленно прикапывают к охлажденному до -10oC раствору 35 г трет.-бутилового эфира 3-бром-пропионовой кислоты в 100 мл тетрагидрофурана. Реакционную смесь перемешивают в течение нескольких часов при комнатной температуре. С целью обработки, смесь подкисляют с помощью разбавленного водного раствора соляной кислоты и разбавляют с помощью 375 мл диэтилового эфира. Органическую фазу отделяют и водную фазу экстрагируют еще три раза по 100 мл диэтиловым эфиром. Объединенные органические экстракты промывают водным раствором хлорида натрия, сушат над сульфатом натрия и выпаривают при пониженном давлении. Остающийся остаток растворяют в 300 мл диэтилового эфира. Раствор встряхивают 6 раз с водным раствором бикарбоната натрия и затем 4 раза с 10%-ным водным раствором карбоната натрия. Объединенные растворы карбоната натрия подкисляют при охлаждении льдом и экстрагируют трижды по 150 мл эфиром. Эти эфирные экстракты объединяют с эфирным раствором и полученный раствор промывают водным раствором хлорида натрия, сушат над сульфатом натрия и концентрируют при пониженном давлении. Полученный сырой продукт кристаллизуют из охлажденного льдом н-гексана. Получают 7,7 г чистой 1-/2'-(трет.- бутоксикарбонил)этил/-1-циклопентанкарбоновой кислоты с т.пл. 78- 81oC.
В). 30 мл Диизопропиламина в атмосфере азота растворяют в 100 мл абсолютного тетрагидрофурана и раствор охлаждают до -70oC. К раствору прикапывают 132 мл 1,6 М раствора бутиллития в н-гексане и реакционную смесь перемешивают следующие 30 минут при 0oC и затем снова охлаждают до -70oC. После этого к реакционной смеси последовательно прикапывают раствор 24,2 г полученного выше в п. Б) продукта в 100 мл абсолютного тетрагидрофурана и после этого раствор 14,2 мл метоксиэтоксиметилхлорида в 20 мл абсолютного тетрагидрофурана. Реакционную смесь перемешивают еще в течение 16 часов при комнатной температуре. Для обработки реакционную смесь вносят в смесь льда с водой, подкисляют водным раствором гидросульфата калия и экстрагируют трижды по 300 мл этилацетатом. Органические фазы объединяют, промывают водным раствором хлорида натрия, сушат над сульфатом натрия и концентрируют при пониженном давлении. Остающийся сырой продукт очищают путем флэш-хроматографии на 500 г силикагеля при применении смеси дихлорметана с эфиром (8:2) в качестве элюирующего средства. Получают 26,5 г чистой 1-/2'-(трет.-бутоксикарбонил)-3'-(2-метоксиэтокси)-пропил/-циклопентан-1-карбоновой кислоты в виде масла.
Г). 36,7 г Вышеполученной рацемической кислоты растворяют в 184 мл н-гексана и раствор смешивают с 18,4 г (+)-псевдоэфедрина. Выпавший осадок путем кратковременного кипячения с обратным холодильником снова растворяют. Затем раствор охлаждают и оставляют стоять в течение нескольких часов в холодильнике. Образовавшийся кристаллический осадок отсасывают, промывают охлажденным льдом н-гексаном и еще 4 раза перекристаллизуют из н-гексана. Получают 16,2 г соли псевдоэфедрина вышеуказанной кислоты с т.пл. 89 - 91oC. Величина оптического вращения [α]
Для высвобождения кислоты 16 г этой соли суспендируют в н-гексане, и реакционную смесь подкисляют с помощью охлажденного льдом водного раствора гидросульфата калия. Органическую фазу отделяют, а водную фазу еще 2 раза экстрагируют н-гексаном. Объединенные органические фазы промывают с помощью водного раствора хлорида натрия, сушат над сульфатом натрия и выпаривают при пониженном давлении. Остаток высушивают при пониженном давлении при 50oC. Получают 9,9 г (2'S)-1-/2'- (трет.-бутоксикарбонил)-3'-(2-метоксиэтокси)-пропил/циклопентан- 1-карбоновой кислоты в виде масла; величина оптического вращения [α]
Д). 17,2 г Гидрида натрия (80%-ного), в атмосфере азота и при исключении влаги, растворяют в 400 мл безводного диметилформамида. К этому раствору при 0oC медленно прикапывают раствор из 50 г L-БОК-серина ( =N-(трет.-бутоксикарбонил)-серин) в 50 мл безводного диметилформамида. Реакционную смесь оставляют медленно нагреваться до 15oC, затем прикапывают раствор 37,4 г о-нитрофенола в 50 мл диметилформамида, и реакционную смесь перемешивают еще несколько часов при комнатной температуре. С целью обработки, реакционную смесь выливают в охлажденный льдом водный раствор гидросульфата калия. Затем многократно экстрагируют этилацетатом и объединенные органические фазы смешивают с водным раствором бикарбоната натрия. Водную фазу отделяют, промывают эфиром и затем подкисляют с помощью раствора гидросульфата калия и экстрагируют этилацетатом. Этилацетатный экстракт промывают водой, сушат над сульфатом натрия и выпаривают при пониженном давлении. Получают 54,2 г сырой (2S)-3-(2-нитрофенокси)-2-(трет.- бутоксикарбониламино)-пропионовой кислоты, которую без дальнейшей очистки вводят во взаимодействие далее.
Е). 54,2 г Вышеполученной кислоты растворяют в 600 мл метанола. Раствор смешивают с 1,8 г палладиевого катализатора (5% палладия-на-угле). Затем в течение 1 часа гидрируют при давлении водорода 5 бар. После этого катализатор отфильтровывают и отфильтрованный раствор концентрируют при пониженном давлении. Полученный сырой продукт кристаллизуют из смеси метил-трет.- бутилового эфира с н-гексаном при охлаждении льдом. Получают 30,1 г (2S)-3-(2-аминофенокси)-2-(трет. -бутоксикарбониламино)- пропионовой кислоты с т.пл. 87-91oC, величина оптического вращения [α]
Ж). 13,3 г Вышеполученной кислоты, при исключении влаги, растворяют в 71 мл безводного диметилформамида. К раствору, при охлаждении льдом, добавляют 7,8 мл диэтилфосфоноцианида в 6 мл диметилформамида. Спустя 10 минут прикапывают 5,7 мл триэтиламина и реакционную смесь перемешивают в течение 1 часа при комнатной температуре. Затем реакционную смесь, с целью обработки, выливают в воду со льдом и многократно экстрагируют метил-трет.-бутиловым эфиром. Объединенные органические фазы сушат и выпаривают при пониженном давлении. Остающийся в виде остатка сырой продукт кристаллизуют из этанола. Получают 1,3 г (3S)-3-(трет. -бутоксикарбониламино)- 2,3-дигидро-1,5-бензоксазепин-4(5H)-она; величина оптического вращения [α]
З). 16 г Вышеполученного продукта, при исключении влаги, растворяют в 313 мл тетрагидрофурана. К раствору последовательно прикапывают раствор 7,1 г трет.-бутилата калия в 30 мл тетрагидрофурана и раствор 10,9 мл бензилового эфира бромуксусной кислоты в 10 мл тетрагидрофурана. Реакционную смесь перемешивают в течение 1 часа при комнатной температуре. Затем, для обработки, разбавляют метил-трет.-бутиловым эфиром, промывают водой, органическую фазу сушат над сульфатом натрия и концентрируют при пониженном давлении. Полученный сырой продукт очищают путем флэш-хроматографии на 500 г силикагеля при применении смеси н-гексана с этилацетатом (3:2) в качестве элюирующего средства. Получают 20,5 г чистого бензилового эфира (3S)-3-(трет.-бутоксикарбониламино)-4-оксо-3,4-дигидро-1,5- бензоксазипен-5(2H)-уксусной кислоты в виде масла; величина оптического вращения [α]
И). 20 г Вышеполученного продукта растворяют в 137 мл дихлорметана. Раствор смешивают с 77 мл трифторуксусной кислоты и перемешивают в течение 1 часа. Затем концентрируют при пониженном давлении, остаток растворяют в дихлорметане и смешивают с водным раствором бикарбоната натрия вплоть до щелочной реакции. Органическую фазу отделяют, промывают водой, сушат над сульфатом натрия и концентрируют при пониженном давлении. Получают 15,7 г чистого бензилового эфира (3S)-3-амино-4-оксо-3,4-дигидро-1,5- бензоксазепин-5(2H)-уксусной кислоты; величина оптического вращения [α]
К). 15,7 г Вышеполученного продукта растворяют в 48 мл безводного дихлорметана и раствор при комнатной температуре последовательно смешивают с 1,6 г полученной выше в п.Г) кислоты, 0,79 мл N-метилморфолина и 1,83 г N-этил-N'-(3-диметиламинопропил)- карбодиимид-гидрохлорида. Реакционную смесь перемешивают еще 1 час при комнатной температуре. Затем, с целью обработки, последовательно промывают водой, водным раствором гидросульфата калия, водой, водным раствором бикарбоната натрия и снова водой, сушат над сульфатом натрия и выпаривают при пониженном давлении. Полученный в качестве остатка сырой продукт очищают путем флэш-хроматографии на силикагеле при применении смеси н-гексана с этилацетатом (7:3) в качестве элюирующего средства. Получают 1,8 г целевого соединения в виде масла; величина оптического вращения [α]
Пример 10: Бензиловый сложный эфир (3S,2'S)-3-{1-/2'- карбокси-3'-(2-метоксиэтокси)пропил/циклопентан-1-карбониламино} - 4-оксо-3,4-дигидро-1,5-бензоксазепин-5(2H)-уксусной кислоты.
1,6 г Бензилового эфира (3S,2'S)-3-{1-/2'-(трет.- бутоксикарбонил)-3'-(2-метоксиэтокси)пропил/циклопентан-1- карбониламино} -4-оксо-3,4-дигидро-1,5-бензоксазепин-5(2H)- уксусной кислоты (получение см. пример 9) при охлаждении льдом растворяют в 5 мл трифторуксусной кислоты. Раствор оставляют стоять в течение нескольких часов при температуре 4oC. Затем, для обработки, выпаривают при пониженном давлении и остающийся в качестве остатка сырой продукт очищают путем флэш-хроматографии на силикагеле при применении смеси дихлорметана с метил-трет.-бутиловым эфиром и метанолом (85:15:5). После высушивания получают 1,0 г целевого соединения в виде масла; величина оптического вращения [α]
Пример 11: (3S, 2'S)-3-(1-/2'-Карбокси-3'-(2- метoкcэтокси)-пропил/циклопентан-1-карбониламино)-4- оксо-3,4-дигидро-1,5-бензоксазепин-5(2H)-уксусная кислота.
0,95 г Бензилового эфира (3S,2'S)-3-(1-/2'-карбокси- 3'-(2-метоксиэтокси)пропил/циклопентан-1-карбониламино)-4- оксо-3,4-дигидро-1,5-бензоксазепин-5(2H)-уксусной кислоты (получение см. пример 10) растворяют в 50 мл этанола. К раствору добавляют 0,2 г палладиевого катализатора (5% палладия-на- угле). После этого гидрируют в течение 2 часов при давлении водорода 5 бар. Для обработки, отфильтровывают от катализатора, отфильтрованный раствор выпаривают при пониженном давлении и остающийся остаток высушивают. Получают 0,7 г целевого соединения в виде пенообразного продукта: величина оптического вращения [α]
Пример 12: трет.-Бутиловый сложный эфир (3R)-3-(1-/2'- (трет.-бутоксикарбонил)-4'-(4-фторфенокси)-бутил/циклопентан- 1-карбониламино)-4-оксо-3,4-дигидро-1,5-бензотиазепин-5 (2H)-уксусной кислоты.
A). 20,5 г трет.-Бутилового эфира диметилфосфоноуксусной кислоты вводят во взаимодействие с 25 г 4-фторфеноксиэтилбромида согласно описанному в примере 5 A) способу. Реакционную смесь обрабатывают как в примере 5 A). Получают 20,4 г трет.-бутилового сложного эфира 4-(4-фторфенокси)-2-(диметилфосфоно)-н-масляной кислоты.
Б). 20,4 г Вышеполученного продукта вводят во взаимодействие с 4,8 г параформальдегида согласно описанному в примере 5 Б) способу. Реакционную смесь обрабатывают как описано в примере 5 Б). В качестве сырого продукта получают 15,3 г маслянистого трет.-бутилового эфира α- /2-(4-фторфенокси) этил/-акриловой кислоты. Этот эфир без дальнейшей хроматографической очистки используют в самой ближайшей стадии.
В). 15,2 г Вышеполученного продукта вводят во взаимодействие с 5,1 мл циклопентанкарбоновой кислоты согласно описанному в примере 5 В) способу. Реакционную смесь обрабатывают как описано в примере 5 В). Получают 6,0 г 1-/2'-(трет. -бутоксикарбонил)-4'-(4- фторфенокси)-бутил/циклопентан-1-карбоновой кислоты с т.пл. 58- 63oC и дальнейшие 7,6 г маслянистого, еще слегка загрязненного продукта.
Г). К раствору 100 мл 1-фтор-2-нитро-бензола в 1800 мл этанола добавляют раствор 122,4 г N-ацетил-L-цистеина и 181,9 г гидрокарбоната натрия в 550 мл воды. Реакционную смесь кипятят с обратным холодильником в течение 3 часов, затем охлаждают до комнатной температуры и отфильтровывают от осадка. Фильтрат концентрируют примерно до объема 700 мл и остающийся остаток обрабатывают с помощью 1,8 л воды. Водную фазу экстрагируют диэтиловым эфиром и затем устанавливают pH 1 путем добавки концентрированного водного раствора соляной кислоты. Выпадает желтого цвета твердое вещество, которое отсасывают. Получают 253,6 г сырого R-(2-нитрофенил)-N-ацетил-L-цистеина, который без дальнейшей очистки вводят во взаимодействие далее.
Д). 253,6 г Вышеполученного продукта смешивают с 825 мл 18 М серной кислоты и 3,3 л воды. Реакционную смесь в течение 40 минут кипятят с обратным холодильником и затем охлаждают до 0oC. Добавляют 1925 мл водного концентрированного раствора аммиака. Выпадающее в осадок при этом твердое вещество отсасывают и перекристаллизуют из воды. Получают 143 г R-(2-нитрофенил)-L-цистеина.
Е). 100 г Вышеполученного продукта и 62,2 г карбоната калия растворяют в 7 л воды. Затем в течение 3 часов порциями добавляют 120 г карбэтоксифталимида и реакционную смесь перемешивают следующие 5 часов и еще в течение нескольких часов оставляют стоять. Затем выпавшее в осадок твердое вещество отсасывают и фильтрат доводят до pH-значения 2-3 с помощью концентрированного водного раствора соляной кислоты. Выпадающий при этом осадок отсасывают, промывают многократно водой и затем взмучивают примерно с 1 л этанола при легком нагревании (примерно 40oC). После охлаждения твердое вещество отсасывают и высушивают на воздухе. Получают 100 г (2R)-3-(2-нитрофенилтио)-2-фталимидо- пропионовой кислоты, которую без дальнейшей очистки вводят во взаимодействие далее.
Ж). 100 г Вышеполученного продукта суспендируют в 1,5 л метанола. К полученной суспензии добавляют 0,8 г 5%-ного палладия-на-угле и реакционную смесь гидрируют в течение 5 часов. После этого катализатор отделяют и растворитель выпаривают при пониженном давлении. Получают 71,6 г сырой (2R)-3- (2-аминофенилтио)-2-фталимидопропионовой кислоты в виде желто-коричневого масла, которую без дальнейшей очистки вводят во взаимодействие далее.
З). 71,6 г Вышеполученного продукта растворяют в диметилформамиде. К раствору добавляют 38,0 г 1-/3- (диметиламино)пропил/-3-этил-карбодиимид-гидрохлорида и реакционную смесь перемешивают в течение 3 часов при комнатной температуре. Затем реакционную смесь разбавляют с помощью 1,5 л этилацетата и экстрагируют многократно по 1,5 л 1 н. водным раствором гидрокарбоната натрия. Органическую фазу затем промывают дважды по 200 мл водой, сушат над сульфатом магния и выпаривают досуха при пониженном давлении. Остаток очищают путем флэш- хроматографии на колонке при применении смеси этилацетата с циклогексаном (1:1) в качестве элюирующего средства. Получают 46,3 г (3R)-2,3-дигидро-3-фталимидо-1,5-бензотиазепин-4(5H)-она.
И). К раствору из 46,3 г вышеполученного продукта в 300 мл тетрагидрофурана добавляют 10,6 г порошкообразного гидроксида калия и 4,8 г тетрабутиламмонийбромида. Реакционную смесь охлаждают до 0oC и затем медленно прикапывают 23,2 мл трет.-бутилового эфира бромуксусной кислоты. Реакционную смесь перемешивают еще следующие 3 часа при комнатной температуре. Затем отфильтровывают и фильтрат выпаривают досуха при пониженном давлении. Остаток обрабатывают диэтиловым эфиром и эфирную фазу промывают водой и 1 М раствором гидросульфата калия, сушат над сульфатом магния и затем концентрируют при пониженном давлении. Остающийся маслянистый продукт смешивают с этилацетатом и диэтиловым эфиром. Выделившийся осадок отсасывают. Получают 34 г (3R)-5-(трет.-бутоксикарбонилметил)-2,3-дигидро-3- фталимидо-1,5-бензотиазепин-4(5H)-она в виде твердого вещества. После концентрирования маточного раствора при пониженном давлении получают еще 25 г слегка загрязненного маслянистого продукта. Величина оптического вращения [α]
К). 2 г Вышеполученного продукта смешивают с 7,5 мл этаноламина и смесь перемешивают в течение 10 минут при 80oC. Затем источник нагрева удаляют и перемешивают еще следующие 30 минут. После этого реакционную смесь, с целью обработки, смешивают с 70 мл 5%-ного водного раствора хлорида натрия и полученную смесь экстрагируют толуолом. Органическую фазу отделяют, сушат над сульфатом натрия и выпаривают досуха при пониженном давлении. Получают 1,46 г сырого, еще содержащего толуол трет.-бутилового эфира (3R)-3-амино-4-оксо-3,4-дигидро- 1,5-бензотиазепин-5(2H)-уксусной кислоты в виде содержащего толуол твердого вещества.
Л). К 100 мл сухого дихлорметана добавляют 1,45 г вышеполученного продукта, 1,75 г полученного в п. В) производного циклопентанкарбоновой кислоты, 0,70 г гидроксибензотриазола и 1,50 мл N-метилморфолина. После этого реакционную смесь охлаждают до 0oC и добавляют 1,76 г 1-этил-3-(3- диметиламинопропил)-карбодиимид-гидрохлорида и реакционную смесь перемешивают при комнатной температуре в течение следующих 5 часов. Для обработки, реакционную смесь смешивают с 1 М раствором гидросульфата калия, органическую фазу отделяют и промывают 1 М раствором гидрокарбоната калия и насыщенным раствором хлорида натрия, сушат над сульфатом натрия и выпаривают досуха при пониженном давлении. Полученный сырой продукт очищают путем колоночной флэш-хроматографии при применении смеси н-гексана с этилацетатом (4:1) в качестве элюирующего средства. Получают 1,9 г целевого соединения в виде масла.
ИК-спектр (в виде пленки): 3366 см, 3059 см, 2969 см, 2874 см, 1727 см, 1657 см, 1505 см-1.
Пример 13: (3R)-3-(1-/2'-Карбокси-4'-(4-фторфенокси) бутил/циклопентан-1-карбониламино)-4-оксо-3,4- дигидро-1,5-бензотиазепин-5(2H)-уксусная кислота.
1,9 г трет. -Бутилового эфира (3R)-3-(1-/2'-(трет.-бутоксикарбонил)- 4'-(4-фторфенокси)бутил/циклопентан-1-карбониламино)-4-оксо-3,4-дигидро- 1,5-бензотиазепин-5(2H)-уксусной кислоты (получение см. пример 12) описанным в примере 6 способом гидролизуют с помощью трифторуксусной кислоты. Реакционную смесь обрабатывают как описано в примере 6. Получают 0,56 г целевого соединения в виде аморфного твердого вещества. Т.пл. 90-94oC.
Пример 14: Трет.-Бутиловый сложный эфир (3R)-3-(1-/2'- (трет.-бутоксикарбонил)-5'-(3,4-диметоксифенил)пентил/циклопентан- 1-карбониламино)-4-оксо-3,4-дигидро-1,5 -бензотиазепин-5(2H)-уксусной кислоты.
A). 6,7 г Трифенилфосфина растворяют в 200 мл ацетонитрила. После охлаждения раствора до 0oC прикапывают 1,3 мл брома. Затем охлаждающую баню удаляют и прикапывают раствор 5 г 3-(3,4- диметоксифенил)-1-пропанола в 80 мл ацетонитрила. Реакционную смесь затем кипятят с обратным холодильником, причем с помощью водоотделителя многократно в течение 6 часов, смотря по обстоятельствам, отбирают по 10 мл дистиллята и отобранное количество заменяют свежим ацетонитрилом. Для обработки, растворитель выпаривают при пониженном давлении и остающийся остаток обрабатывают диэтиловым эфиром и фильтруют. Фильтрат концентрируют при пониженном давлении и остаток очищают путем колоночной флэш-хроматографии при применении смеси циклогексана с метил-трет. -бутиловым эфиром (7:2). Получают 5,5 г 3-(3,4-диметоксифенил)-1-бром-пропана в виде бесцветного масла.
Б). 5,5 г Вышеполученного продукта описанным в примере 5 A) способом вводят во взаимодействие с 3,8 мл трет.-бутилового эфира диметилфосфоноуксусной кислоты. Реакционную смесь обрабатывают как описано в примере 5 A). Получают 6,1 г трет.-бутилового эфира 4- (3,4-диметоксифенил)-2-(диметил-фосфоно)-валериановой кислоты в виде бесцветного масла.
В). 6 г Вышеполученного продукта описанным в примере 5 Б) способом вводят во взаимодействие с параформальдегидом. Реакционную смесь обрабатывают описанным в примере 5Б) образом. Полученный сырой продукт очищают путем колоночной флэш-хроматографии при применении смеси метил-трет.- бутилового эфира с циклогексаном (1:3) в качестве элюирующего средства. Получают 3,4 маслянистого трет.-бутилового эфира 1-/3- (3,4-диметоксифенил)пропил/акриловой кислоты.
Г). 3,4 г Вышеполученного продукта вводят во взаимодействие с 1,3 мл циклопентанкарбоновой кислоты согласно описанному в примере 5 В) способу. Реакционную смесь обрабатывают как описано в примере 5 В). Сырой продукт очищают путем колоночной флэш-хроматографии при применении смеси этилацетата с циклогексаном (1:3) в качестве элюирующего средства. Получают 2,5 г маслянистой 1-/2-(трет.- бутоксикарбонил)-5-(3,4-диметоксифенил)пентил/циклопентанкарбоновой кислоты.
Д). 2,5 г Вышеполученного продукта растворяют в 50 мл ацетонитрила. При температуре 0oC, при исключении влаги, к раствору добавляют последовательно 4,2 мл диизопропилэтиламина, 1,7 г 2-хлор-1-метилпиридинийиодида и 2,5 г трет.-бутилового сложного эфира (3R)-3-амино-4-оксо-3,4-дигидро-1,5-бензодиазепин- 5(2H)-уксусной кислоты (получение см. пример 12 К)). Реакционную смесь перемешивают еще 30 минут при 0oC и 2 часа при комнатной температуре. Для обработки, реакционную смесь концентрируют досуха при пониженном давлении и остающийся остаток растворяют в дихлорметане. Раствор встряхивают сначала с разбавленным водным раствором соляной кислоты и затем с водой. Органическую фазу отделяют, вводную фазу экстрагируют еще 2 раза дихлорметаном. Затем объединенные органические фазы сушат над сульфатом натрия и концентрируют при пониженном давлении. В виде остатка получают 3 г целевого соединения в виде масла. Тонкослойная хроматография на силикагеле: Rf - значение = 0,4 (элюирующее средство циклогексан/этилацетат = 1:1).
Пример 15: (3R)-3-{ 1-/2'-Карбокси-5'-(3,4-диметоксифенил)- пентил/-циклопентан-1-карбониламино} -4-оксо-3,4-дигидро-1,5- бензотиазепин-5(2H)-уксусная кислота.
3 г трет.-Бутилового сложного эфира (3R)-3-(1-/2'-(трет.- -бутоксикарбонил)-5-(3,4-диметоксифенил)пентил/циклопентан-1- карбониламино)-4-оксо-3,4-дигидро-1,5-бензотиазепин-5(2H)- уксусной кислоты (получение см. пример 14) растворяют в 20 мл дихлорметана. К раствору добавляют 3 мл трифторуксусной кислоты и реакционную смесь перемешивают в течение 2-х дней при комнатной температуре. Для обработки, реакционную смесь концентрируют при пониженном давлении. Для полного удаления трифторуксусной кислоты остаток многократно смешивают, смотря по обстоятельствам, с толуолом в количестве по 2 мл и снова выпаривают. Таким образом полученный сырой продукт очищают путем флэш-хроматографии на силикагеле, причем в качестве элюирующего средства применяют сначала смесь дихлорметана с этилацетатом в соотношении 1:1 и затем чистый этилацетат. После концентрирования элюата при пониженном давлении получают 1,26 г целевого соединения в виде аморфного твердого вещества. ИК-спектр (в виде таблетки с KBr): 3365, 2942, 1726, 1652 см-1.
Пример 16: Бензиловый сложный эфир 3-(1-/2'-)трет.-бутоксикарбонил)- 4'-фенил-бутил/циклопентан-1-карбониламино)-2,3,4,5-тетрагидро-2-оксо- 1H-1-бензазепин-1-уксусной кислоты.
A). 10,5 г трет.-Бутилового сложного эфира 3-амино-2,3,4,5- тетрагидро-2-оксо-1H-1-бензазепин-1-уксусной кислоты (получение см. пример 1 И)), 8,25 г гидрата п-толуолсульфокислоты и 20,1 мл бензилового спирта вносят в 174 мл толуола. Реакционную смесь кипятят в течение 4 часов с водоотделителем, причем первоначально выпавший осадок медленно растворяется. Затем толуол удаляют при пониженном давлении и остающийся остаток перемешивают с метил-трет.-бутиловым эфиром и затем отфильтровывают. Таким образом полученный твердый остаток растворяют в дихлорметане и раствор подщелачивают путем добавки водного раствора карбоната натрия при охлаждении льдом. После этого отделяют дихлорметановую фазу, промывают водой, сушат над сульфатом натрия и выпаривают. Полученный сырой продукт для очистки перекристаллизуют из метил-трет. - бутилового эфира. Получают 8,2 г бензилового эфира 3-амино- 2,3,4,5-тетрагидро-2-оксо-1H-1-бензазепин-1-уксусной кислоты с т.пл. 105-107oC.
Б). 12,8 г Вышеполученного продукта вводят во взаимодействие с 13,7 г 1-/2'-(трет. -бутоксикарбонил)-4'- фенил-бутил/-циклопентан-1-карбоновой кислоты (получение см. пример 5 B)) согласно описанному в примере 3 В) способу. Реакционную смесь обрабатывают как описано в примере 3 В). Получают 19,3 г целевого соединения с т.пл. 118-123oC.
Пример 17: Бензиловый сложный эфир 3-/1-(2'-карбокси-4- фенил-бутил)циклопентан-1-карбониламино/-1,3,4,5-тетрагидро-2-оксо- 1H-1-бензазепин-1-уксусной кислоты.
15 г Бензилового сложного эфира 3-(1-/2'-(трет-бутоксикарбонил)- 4'-фенилбутил/циклопентан-1-карбониламино)-2,3,4,5- тетрагидро-2-оксо-1H-1-бензазепин-1-уксусной кислоты (получение см. пример 16) вводят во взаимодействие с 56 мл трифтор-уксусной кислоты согласно описанному в примере 6 способу. Реакционную смесь обрабатывают как описано в примере 6, и полученный сырой продукт кристаллизуют из метил-трет.бутилового эфира. Получают 13,1 г целевого соединения с т.пл. 86-90oC.
Пример 18: Бензиловый сложный эфир 3-(1-/2'-(трет.-бутил- карбонилоксиметоксикарбонил)-4'-фенилбутил/циклопентан-1- карлбониламино)-2,3,4,5-тетрагидро-2-оксо-1H-1-бензазепин-1- уксусной кислоты.
2 г Бензилового сложного эфира 3-/1-(2'-карбокси-4'- фенил-бутил) циклопентан-1-карбониламино/-2,3,4,5-тетрагидро- 2-оксо-1H-1-бензазепин-1-уксусной кислоты (получение см. пример 17) при исключении влаги растворяют в 20 мл безводного дихлорметана. К раствору добавляют 0,46 мл триэтиламина и 0,1 г диметиламинопиридина. Затем, при охлаждении льдом, прикапывают раствор 0,5 г хлорметилового эфира пивалиновой кислоты в 3 мл безводного дихлорметана. Реакционную смесь затем перемешивают в течение 2-х дней при комнатной температуре. Для обработки, реакционную смесь вносят в воду, органическую фазу отделяют, промывают водным раствором бикарбоната натрия и затем водой, сушат над сульфатом натрия и концентрируют при пониженном давлении. Остающийся в качестве остатка сырой продукт очищают путем флэш- хроматографии на 150 г силикагеля, причем в качестве элюирующего средства используют смесь н-гексана с этилацетатом состава: сначала 7:3 и затем 1:1. Получают 1,1 г чистого бензилового эфира 3-(1-/2' - (пивалоилоксиметоксикарбонил)-4'-фенил-бутил/ циклопентан-1-карбониламино)-2,3,4,5-тетрагидро-2-оксо-1H-1- бензазепин-1-уксусной кислоты в виде твердой пены с областью плавления 71-78oC.
Пример 19: 3-(1-/2'-(Пивалоилоксиметоксикарбонил)-4'- фенил-бутил/циклопентан-1-карбониламино)-2,3,4,5-тетрагидро-2- оксо-1H-1-бензазепин-1-уксусная кислота.
1,0 г Бензилового сложного эфира 3-(1-/2'- (пивалоилоксиметоксикарбонил)-4'-фенил-бутил/циклопентан-1-карбониламино) -2,3,4,5-тетрагидро-2-оксо-1H-1-бензазепин-1-уксусной кислоты (получение см. пример 18) растворяют в 100 мл этанола. Раствор смешивают с 0,5 г катализатора 5%-ного палладия-на-угле. Затем гидрируют в течение 3 часов при давлении водорода 5 бар. Для обработки, отфильтровывают от катализатора, и отфильтрованный раствор выпаривают. Полученный остаток высушивают при 80oC и пониженном давлении. Получают 0,7 г целевого соединения в виде стеклообразного продукта.
ИК-спектр (в виде таблетки с KBr): 3410, 1750, 1660 см-1.
Согласно описанным в вышеприведенных примерах способам также можно получать указанные в последующей таблице 1 соединения формулы (I):
Пример I: Таблетки, содержащие (3S,2'R)-3-(1-/2'- (этоксикарбонил)-4'-фенил-бутил/циклопентан-1-карбониламино) -2,3,4,5-тетрагидро-2-оксо-1H-1-бензазепин-1-уксусную кислоту.
Готовят таблетки следующего состава, в расчете на одну таблетку, мг:
(3S, 2'R)-3-(1-/2'-(Этоксикарбонил)-4'-фенил-бутил/циклопентан- 1-карбониламино)-2,3,4,5-тетрагидро-2-оксо-1H-1-бензазепин-1- уксусная кислота - 20
Кукурузный крахмал - 60
Молочный сахар - 135
Желатина (в виде 10%-ного раствора) - 6
Биологически активное вещество, кукурузный крахмал и молочный сахар концентрируют вместе с 10%-ным раствором желатины. Пасту размельчают и образовавшийся гранулят наносят на пригодный металлический лист и высушивают при 45oC. Высушенный гранулят пропускают через машину для размельчения и в миксере смешивают со следующими другими вспомогательными веществами, мг:
Тальк - 5
Стеарат магния - 5
Кукурузный крахмал - 9
и затем прессуют в таблетки весом 240 мг
Пример II: Раствор для инъекции, содержащий (3S,2'R)-3-/1- (2'-карбокси-4'-фенил-бутил)циклопентан-1-карбониламино-2,3,4,5-тетрагидро-2-оксо-1H-1-бензазепин-1-уксусную кислоту.
Готовят раствор для инъекции следующего состава, в расчете на 5 мл, мг:
(3S,2'R)-3-/1-(2'-Карбокси-4'-фенил-бутил)-циклопентан-1- карбониламино/-2,3,4,5-тетрагидро-2-оксо-1H-1-бензазепин-1-уксусная кислота - 10
Na2HPO4•7H2O - 43,24
NaH2PO4•2H2O - 7,72
NaCl - 30,0
Очищенная вода - 4948,0
Твердые вещества растворяют в воде, раствор стерилизуют и порциями, смотря по обстоятельствам, по 5 мл разливают в ампулы.


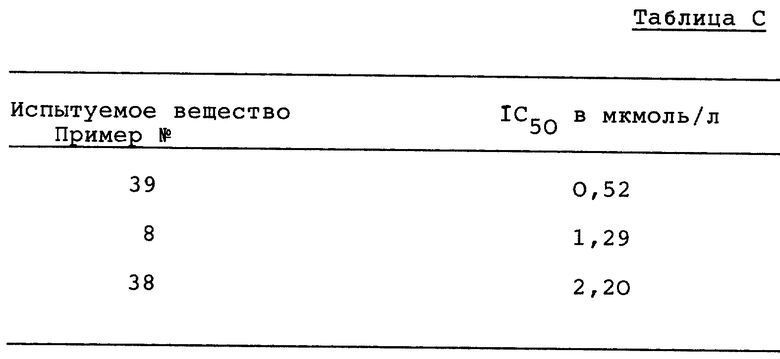
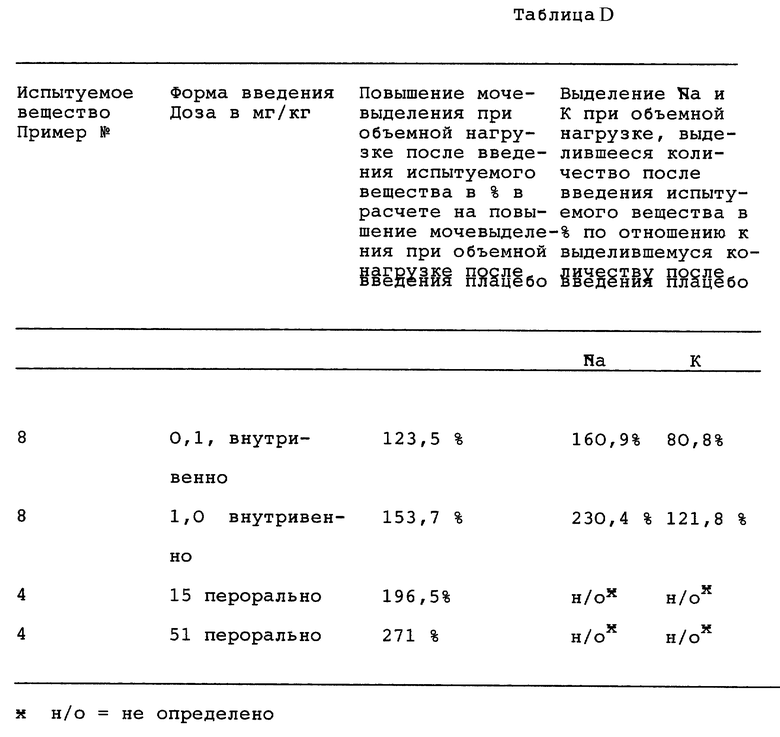
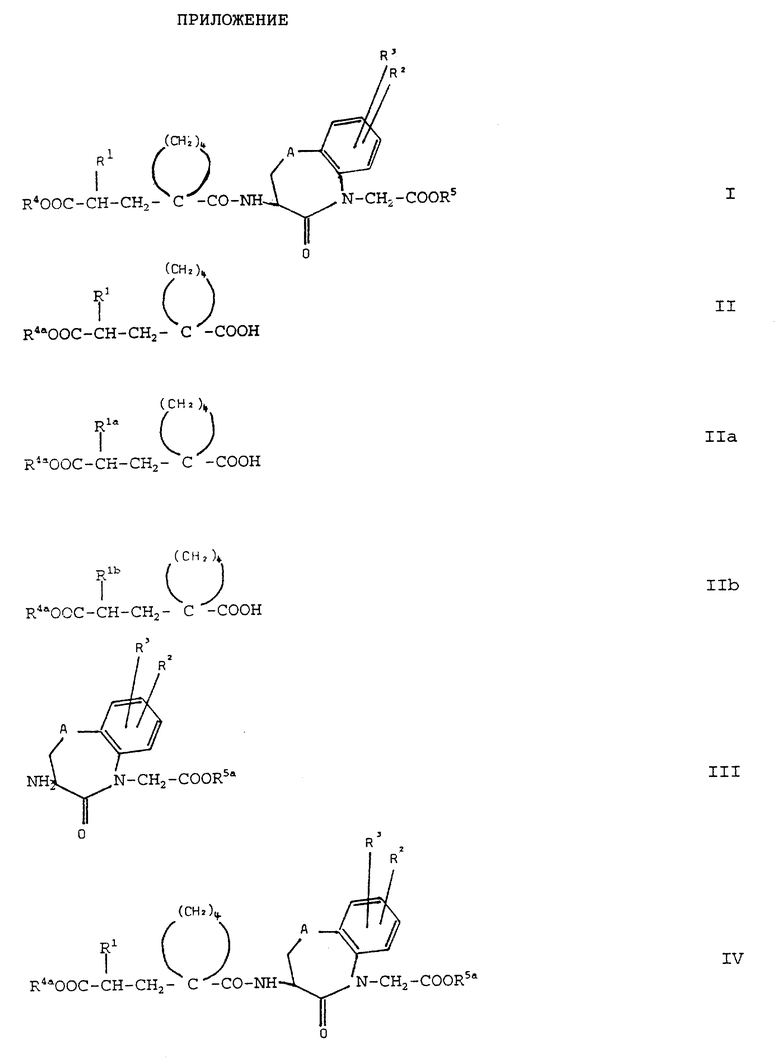
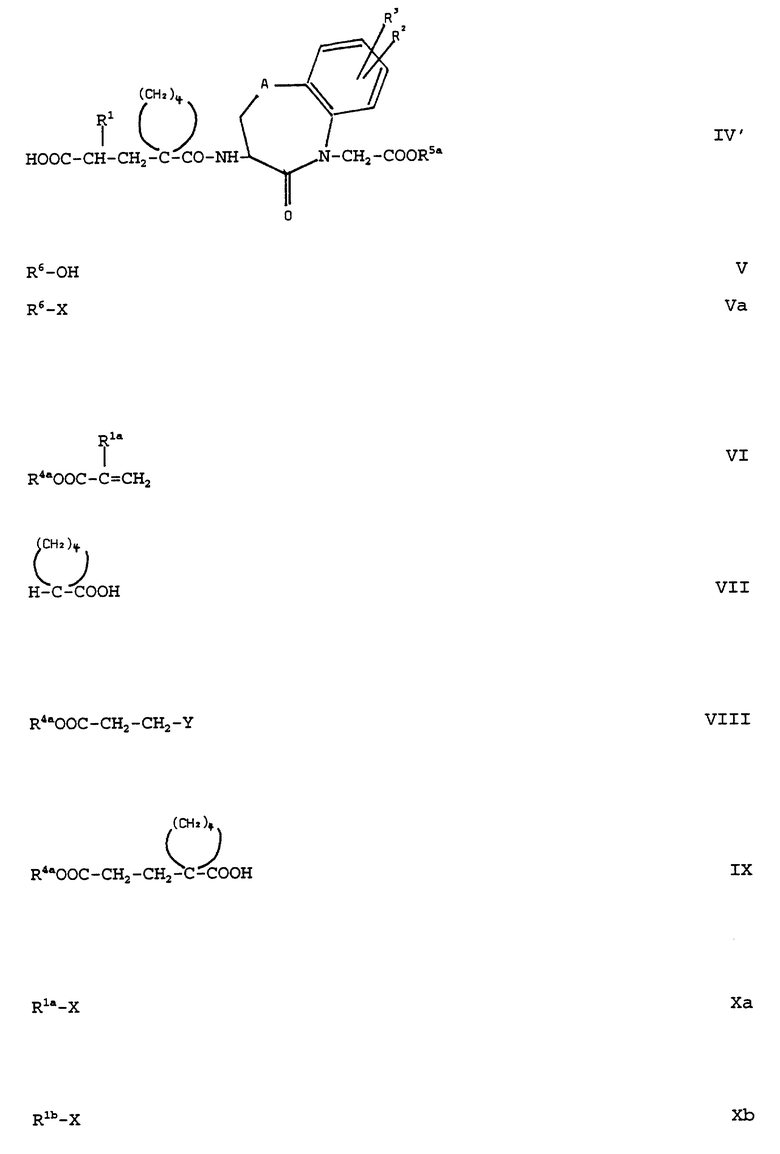
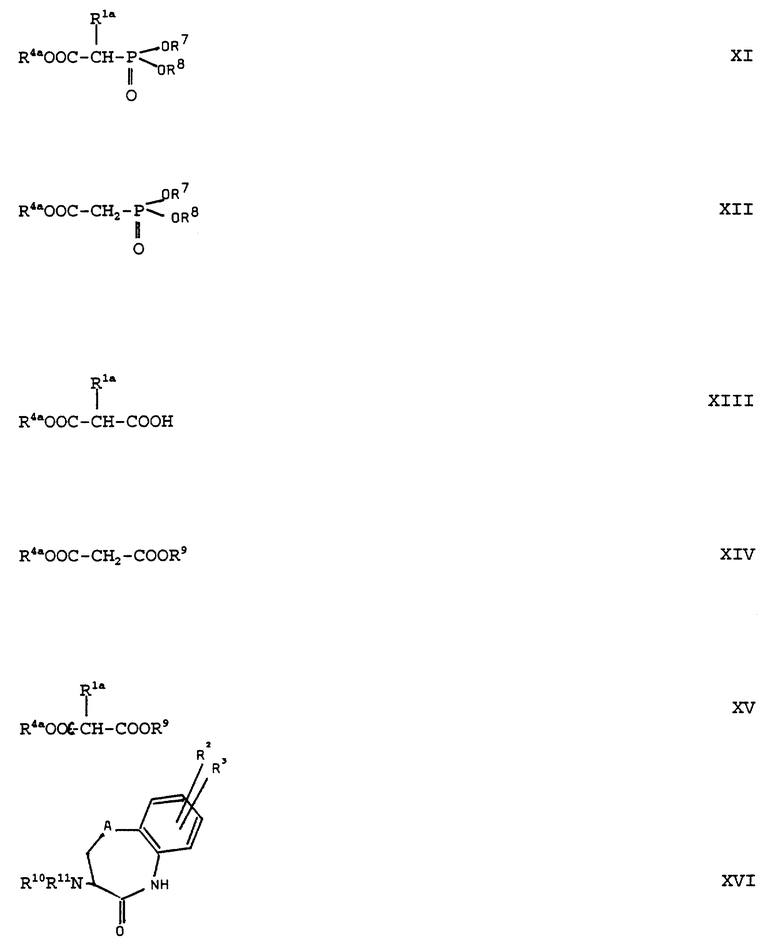
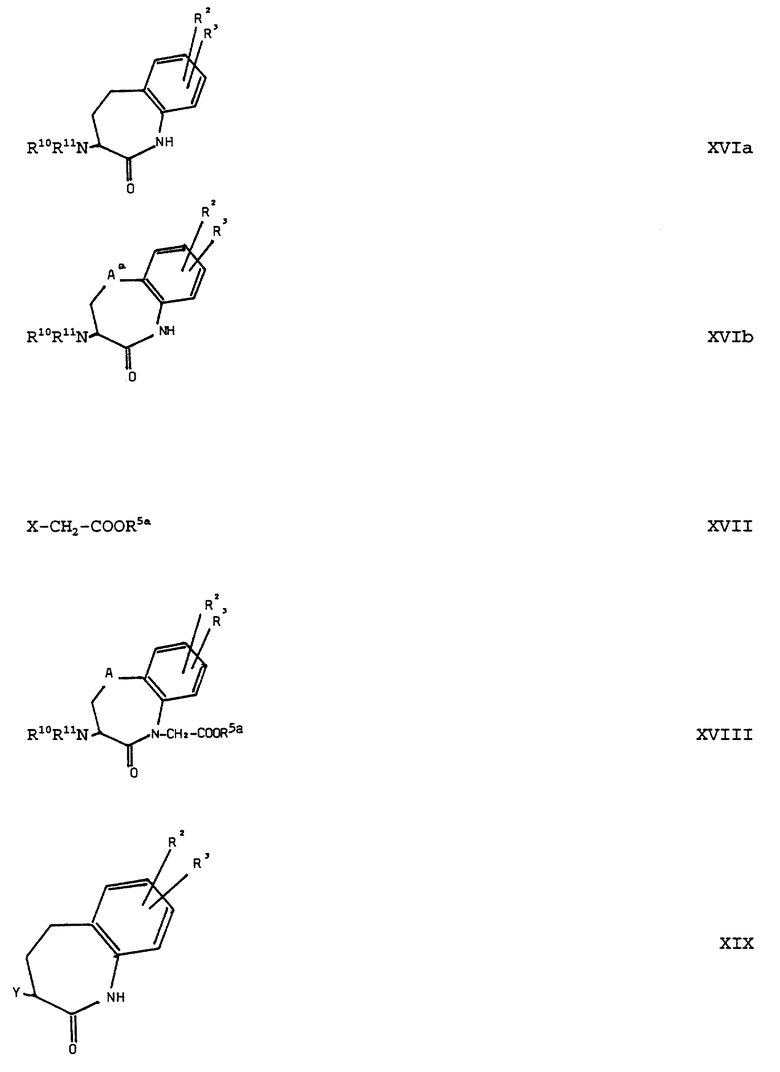
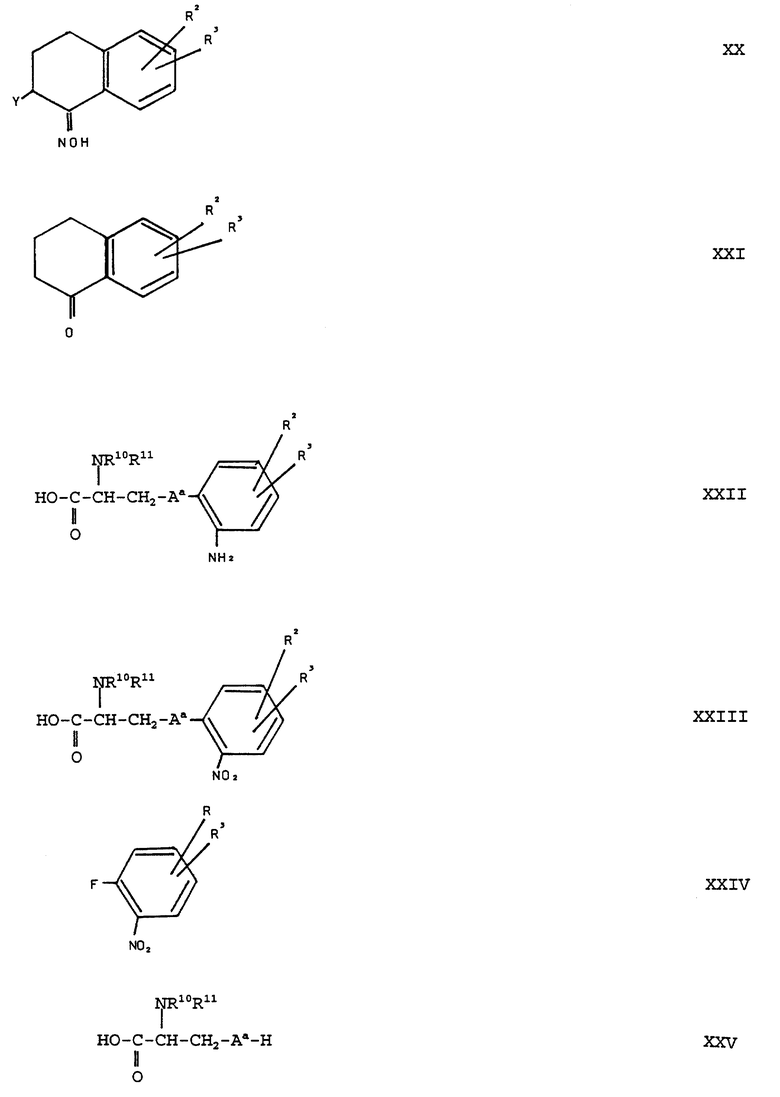
Описываются новые производные бензазепин-, бензоксазепин-, бензотиазепин- N-уксусной кислоты общей формулы I, где R1 обозначает (низший) алкокси (низшую) алкильную группу, низший алкоксильный остаток, который замещен низшей алкоксильной группой, фенил(низшую)алкильную группу или фенилокси(низшую)алкильную группу, которая в случае необходимости в фенильном кольце может быть замещена низшим алкилом, низшим алкоксилом или галогеном, или нафтил(низшую)алкильную группу, A обозначает CH2, O или S; R2 обозначает водород или галоген, R3 обозначает водород или галоген, R4 обозначает водород или образующую биолабильный сложный эфир группу, и R5 обозначает водород или образующую биолабильный сложный эфир группу, и физиологически переносимые соли кислот формулы I. Они обладают ценными, эффективными в отношении воздействия на сердце, фармакологическими свойствами и проявляют отчетливо выраженное подавляющее воздействие на нейтральную эндопептидазу с благоприятным профилем действия, на основании которого они уменьшают наступающее при сердечной недостаточности высокое сердечное давление наполнения и таким образом разгружают сердце и могут вызывать усиление лиуреза. Описываются также способ получения соединений формулы I и лекарственное средство, их содержащее. 3 с. и 5 з.п. ф-лы, 5 табл.
где R1 - обозначает (низший)алкокси(низшую)алкильную группу, низший алкоксильный остаток которой замещен низшей алкоксильной группой, фенил(низшую)алкильную группу или фенилокси(низшую)алкильную группу, которая в случае необходимости в фенильном кольце может быть замещена низшим алкилом, низшим алкоксилом или галогеном, или нафтил(низшую)алкильную группу;
А - обозначает CH2, О или S;
R2 - обозначает водород или галоген;
R3 - обозначает водород или галоген;
R4 - обозначает водород или образующую биолабильный сложный эфир группу;
R5 - обозначает водород или образующую биолабильный сложный эфир группу,
и физиологически переносимые соли кислот формулы I.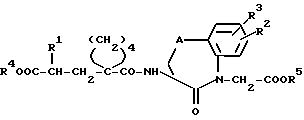
где R1 - обозначает (низший)алкокси(низшую)алкильную группу, низший алкоксильный остаток которой замещен низшей алкоксильной группой, фенил(низшую)алкильную группу или фенилокси(низшую)алкильную группу, которая в случае необходимости может быть замещена в фенильном кольце низшим алкилом, низшим алкоксилом или галогеном, или нафтил(низшую)алкильную группу;
А - обозначает CH2, О или S;
R2 - обозначает водород или галоген;
R3 - обозначает водород или галоген;
R4 - обозначает водород или образующую биолабильный сложный эфир группу;
R5 - обозначает водород или образующую биолабильный сложный эфир группу,
и физиологически переносимых солей кислот формулы I, отличающийся тем, что кислоты общей формулы II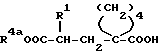
где R1 имеет вышеуказанное значение,
R4a обозначает защитную для кислотной функции группу,
или их реакционноспособные производные вводят во взаимодействие с аминами общей формулы III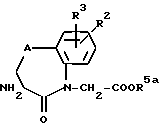
где R2, R3 и А имеют вышеуказанное значение,
R5a обозначает защитную для кислотной функции группу,
с получением амидов общей формулы IV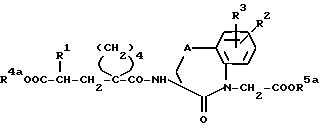
где R1, R2, R3, R4a, R5a и А имеют вышеуказанное значение,
и в соединениях формулы IV, одновременно или в любой последовательности друг за другом, отщепляют защитные для кислотной функции группы R4a и R5a, если они не представляют собой желательную, образующую сложный биолабильный эфир группу, и при желании, смотря по обстоятельствам, высвобождающуюся кислотную группу этерифицируют с помощью спирта общей формулы V или соответствующего реакционноспособного производного общей формулы Va
R6 - ОН (Y); R6 - X
где R6 представляет собой образующую биолабильный сложный эфир группу;
Х обозначает отщепляемую реакционноспособную группу,
и полученные кислоты формулы I переводят в их физиологически переносимые соли или соли кислот формулы I переводят в свободные кислоты.
| Способ получения производных 1,5-бензотиазепина или их фармацевтически приемлемых солей | 1985 |
|
SU1435152A3 |
| EP, 0487207 A1, 27.05.1992 | |||
| EP, 0135349 A1, 27.03.1985 | |||
| EP, 0125056 A3, 14.11.1984 | |||
| SU, 1454252 A1, 23.01.1989. | |||

