В настоящем изобретении предлагаются производные пиридазинон-амидов формулы (I) в качестве ингибиторов IRAK и их применение в лечении рака и других заболеваний, связанных с повышенной экспрессией IRAK, таких как ревматоидный артрит, системная красная волчанка или волчаночный нефрит.
Предпосылки создания изобретения
Киназы катализируют фосфорилирование белков, липидов, сахаров, нуклеозидов и других клеточных метаболитов и играют главную роль во всех аспектах эукариотической физиологии клеток. В особенности протеинкиназы и липидкиназы участвуют в сигнальных событиях, контролирующих активацию, рост, дифференциацию и выживаемость клеток под влиянием внеклеточных медиаторов или стимулов, таких как факторы роста, цитокины или хемокины. В целом, протеинкиназы подразделены на две группы - те, которые преимущественно фосфорилируют остатки тирозина и те, которые преимущественно фосфорилируют остатки серина и/или треонина.
Киназы являются важными терапевтическими мишенями для разработки противовоспалительных лекарственных препаратов (Cohen, 2009 Current Opinion in Cell Biology 21, 1-8), например, киназы, которые участвуют в управлении адаптивными и врожденными иммунными реакциями. Представляющие особый интерес киназные мишени являются членами семейства IRAK.
Киназы, связанные с рецептором интерлейкина-1 (IRAKs), принимают непосредственное участие в регуляции внутриклеточной сети сигналов, контролирующих воспаление (Ringwood и Li, 2008. Cytokine 42, 1-7). IRAKs экспрессируются во многих типах клеток и могут опосредовать сигналы от различных клеточных рецепторов, включая толл-подобные рецепторы (TLRs). IRAK4 считается инициальной протеинкиназой активированной ниже рецептора интерлейкина-1 (IL-1) и всех толл-подобных рецепторов (TLRs), за исключением TLR3, и инициирует передачу сигналов во врожденную иммунную систему через быструю активацию IRAKI и более медленную активацию IRAK2. IRAK1 была впервые выявлена вследствие биохимической очистки IL-1 зависимой киназной активности, которая подвергается коиммунопреципитации с IL-1 рецептором типа 1 (Cao et al., 1996. Science 271(5252): 1128-31). IRAK2 была идентифицирована во время поиска маркера экспрессируемой последовательности человека (EST) в базе данных для последовательностей, гомологичных IRAKI (Muzio et al., 1997. Science 278(5343): 1612-5). IRAK3 (также называемая IRAKM) была идентифицирована с использованием мышиной EST последовательности, кодирующей полипептид со значительной гомологией к IRAK1 для скрининга человеческого активированного фитогемагглютинином лейкоцита периферической крови (PBL) библиотеки кДНК (Wesche et al., 1999. J. Biol. Chem. 274(27): 19403-10). IRAK4 была идентифицирована с помощью поиска в базе данных для IRAK-подобных последовательностей и ПЦР универсальной библиотеки кДНК (Li et al., 2002. Proc. Natl. Acad. Sci. USA 99(8): 5567-5572).
Мыши, которые экспрессируют каталитически неактивный мутант IRAK4 вместо киназы дикого типа полностью устойчивы к септическому шоку, вызванному несколькими агонистами TLR, и ослаблены в своей реакции на IL-1. Дети с дефицитом активности IRAK4 вследствие генетического дефекта, страдают от повторяющихся инфекций, вызванных пиогенными бактериями. Получается, что IRAK-зависимые TLRs и IL-1 Rs жизненно необходимы для детского иммунитета против некоторых пиогенных бактерий, но в иммунной защите от большинства инфекций у взрослых играют резервную роль. Поэтому ингибиторы IRAK4 могут быть пригодными для лечения хронических воспалительных заболеваний у взрослых, не делая их слишком чувствительными к бактериальным и вирусным инфекциям (Cohen, 2009. Current Opinion in Cell Biology 21, 1-8). Были разработаны сильные ингибиторы IRAK4 (Buckley et al., 2008. Bioorg Med Chem Lett. 18(12): 3656-60). IRAK1 незаменим для TLR7-опосредованной и TLR9-опосредованной активации IRF7 и получения интерферона-альфа (IFN-α), давая основание предполагать, что ингибиторы IRAK1 могут быть пригодны для лечения системной красной волчанки (СКВ). IRAK2 активируется ниже от IRAK4 и имеет важное значение для выработки противовоспалительного цитокина. Поэтому ингибиторы IRAK2 могут быть пригодны для лечения воспалительных заболеваний.
Сущность изобретения
В соответствии с одним аспектом изобретения предлагаются соединения формулы (I).
В соответствии с другим аспектом изобретения предлагаются соединения формулы (I), которые пригодны для лечения и/или предупреждения заболеваний, связанных с IRAK.
В соответствии с другим аспектом изобретения предлагаются соединения, способные модулировать, в частности ингибировать активность или действие IRAK при болезненных состояниях у млекопитающих, особенно у людей.
В соответствии с другим аспектом изобретения предлагаются способы лечения и/или предупреждения расстройств, выбранных из аутоиммунных, воспалительных заболеваний, сердечно-сосудистых заболеваний, нейродегенеративных расстройств, бактериальных и вирусных инфекций, аллергии, астмы, панкреатита, полиорганной недостаточности, заболеваний почек, агрегации тромбоцитов, рака, трансплантации органов, подвижности сперматозоидов, дефицита эритроцитов, отторжения трансплантата, повреждения легких, респираторных заболеваний и ишемических болезней.
В соответствии с другим аспектом в настоящем изобретении предлагаются соединения формулы (I), являющиеся селективными IRAK-4 и/или IRAK-1 по сравнению с другими изоформами.
В соответствии с другим аспектом изобретения предлагается набор или комплект, содержащий, по меньшей мере, одно соединение формулы (I), предпочтительно в комбинации с иммуномодулирующими средствами. Предпочтительно, набор состоит из отдельных упаковок:
(а) эффективного количества соединения формулы (I) и/или его фармацевтически приемлемых производных, сольватов, солей, гидратов и стереоизомеров, включая их смеси во всех соотношениях, и
(б) эффективного количества дополнительного действующего вещества лекарственного средства.
В соответствии с другим аспектом изобретения, предлагается способ синтеза соединений формул (I) и связанных формул.
Подробное описание изобретения:
В одном варианте осуществления, настоящее изобретение обеспечивает соединение формулы (I)
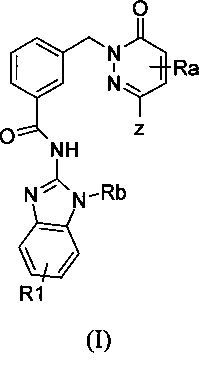
в которой
Z означает группу
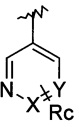
в которой
X означает CH или N,
Y означает CH или N,
Ra, Rc, R1 каждый независимо означают H, Hal или A1,
Rb означает H или алкил
А1 означает разветвленный или линейный алкил, имеющий от 1 до 12 атомов углерода, в котором один или больше, например, от 1 до 7, атомов Н могут быть заменены посредством Hal, ORb, COORb, CN или N(Rb)2 и причем одна или больше, предпочтительно от 1 до 5 CH2-группы могут быть заменены посредством О, СО, NRb или S, SO, SO2, 1,2-, 1,3- или 1,4-фенилен, -СН=СН- или -С≡С-,
и
Hal означает F, Cl, Br, I
и его фармацевтически приемлемые производные, сольваты, таутомеры, соли, гидраты и стереоизомеры, включая их смеси во всех соотношениях.
Настоящее изобретение в частности охватывает таутомерную форму (I'):
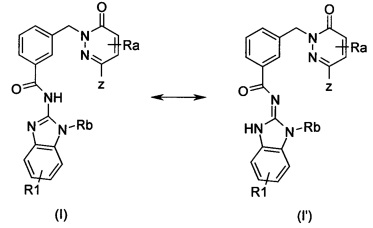
Если не указано иное, то алкил означает углеродную цепь, имеющую от 1 до 12 атомов углерода, предпочтительно от 1 до 8 атомов углерода и наиболее предпочтительно от 1 до 6 атомов углерода. Алкил весьма предпочтительно означает метил, кроме того этил, пропил, изопропил, бутил, изобутил, втор-бутил или трет-бутил, кроме того также пентил, 1, 2 или 3 метилбутил, 1,1, 1,2- или 2,2-диметилпропил, 1-этилпропил, гексил, 1,2,3 или 4 метилпентил, 1,1, 1,2, 1,3, 2,2, 2,3- или 3,3-диметилбутил, 1 или 2 этилбутил, 1 этил-1-метилпропил, 1 этил-2-метилпропил, 1,1,2- или 1,2,2-триметилпропил.
Группа Оалкил предпочтительно означает метокси и этокси.
R означает предпочтительно метил, этил, н-пропил или н-бутил.
Ra означает предпочтительно Н, Hal ORd или алкил, причем Rd означает Н, алкил или CORb.
R1 означает предпочтительно Н, алкил, Hal, Оалкил, ORd, или (CH2)nCONHRb или (CH2)nCOORb, причем n означает 0, 1, 2, 3, 4, 5, или 6 и Rb является таким, как определено выше и, причем Rd означает Н, алкил или CORb.
Rb предпочтительно означает Н, метил или этил.
Z предпочтительно означает пиридинил или пиримидинил.
Выше и ниже, все радикалы и индексы имеют значения, указанные под формулой (I), если специально не указано иное.
Как правило, соединения формулы I являются более предпочтительными, если более предпочтительны заместители, которые они несут.
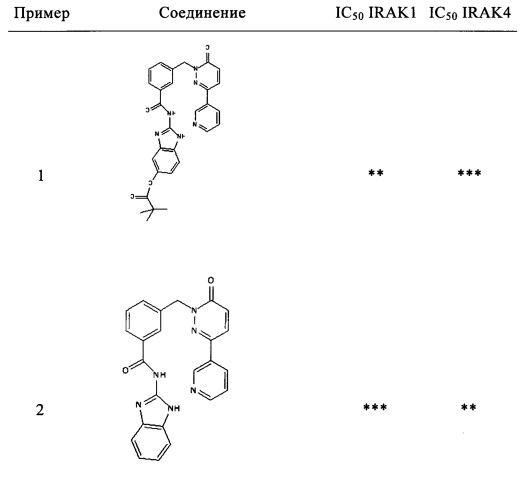
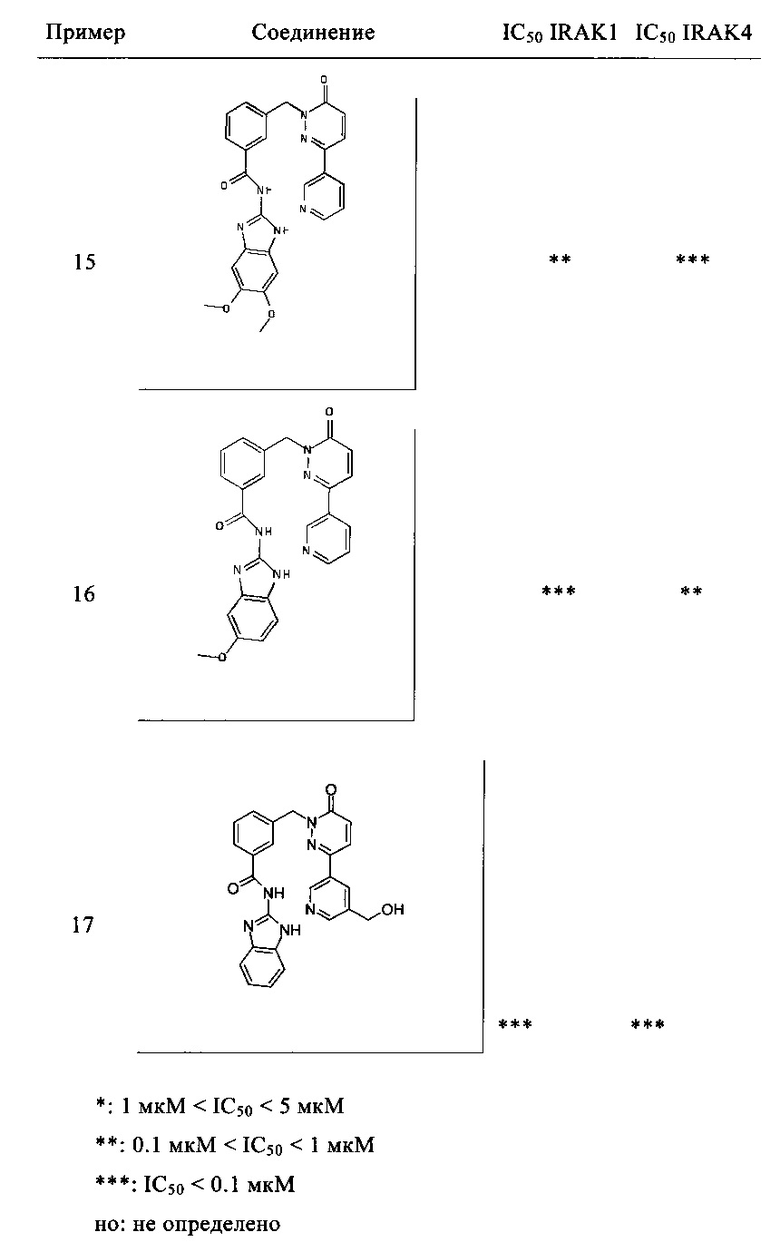
Предпочтительные соединения от 1 до 17 формулы I приведены ниже вместе с их активностями (значения IC50 были получены в соответствии с ферментативными анализами IRAK 1 и IRAK 4, описанными в Примере 18):
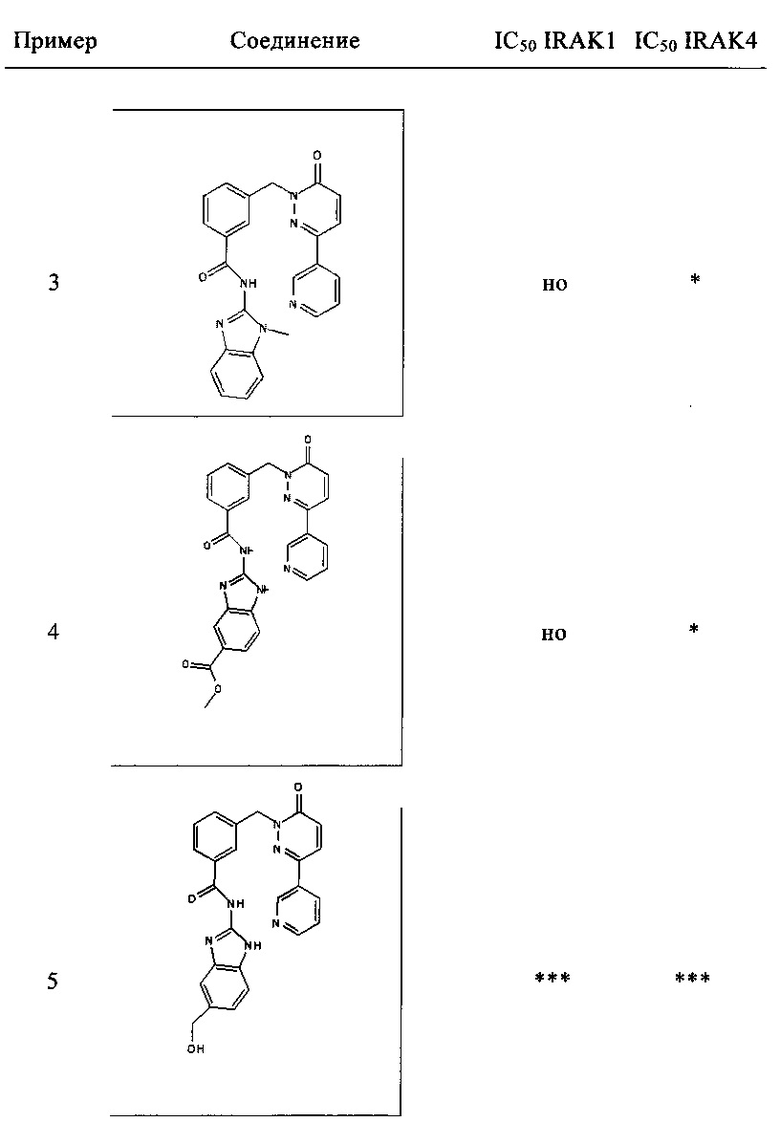
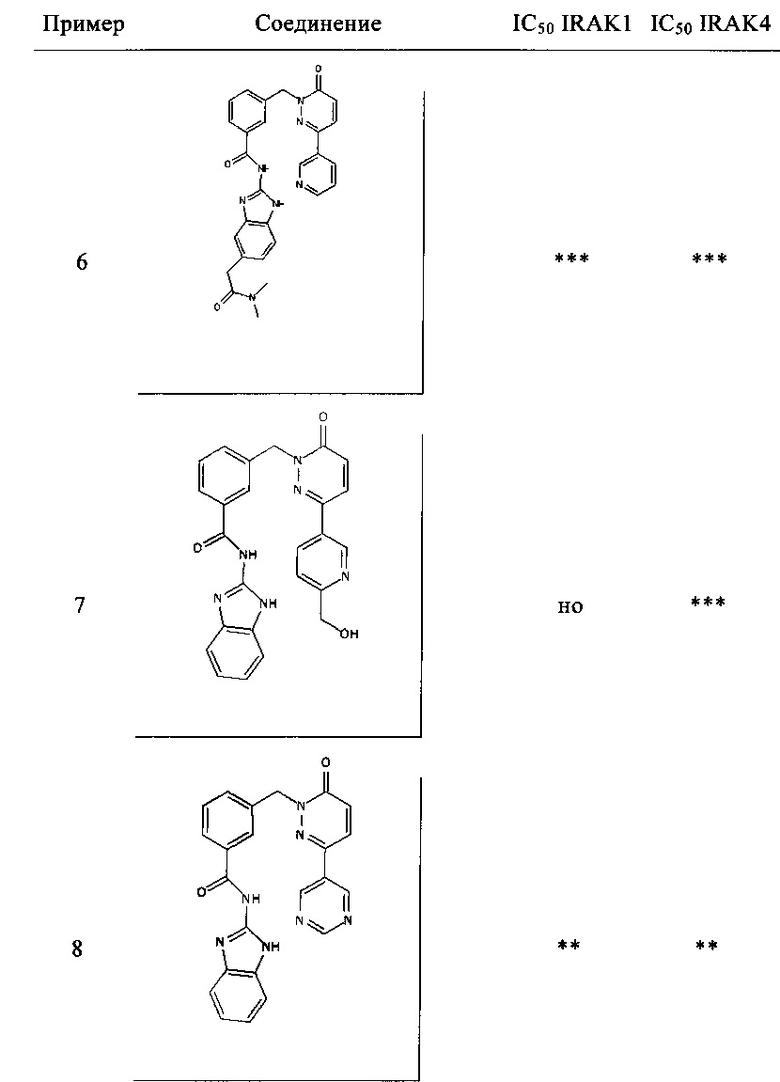
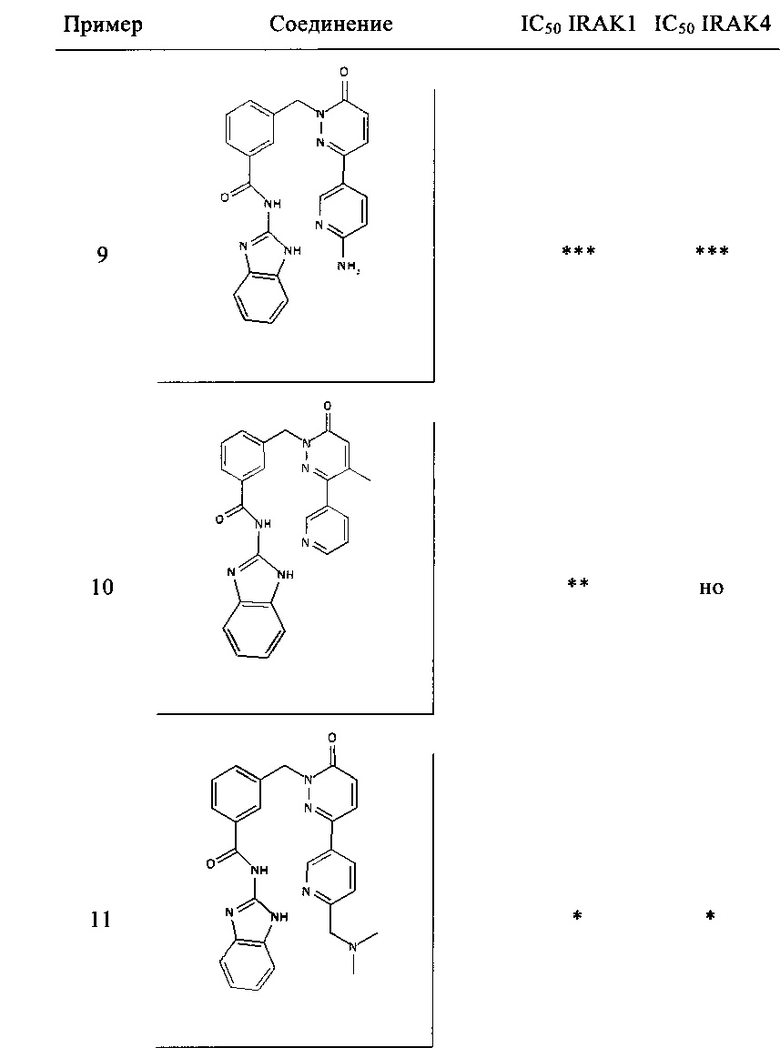
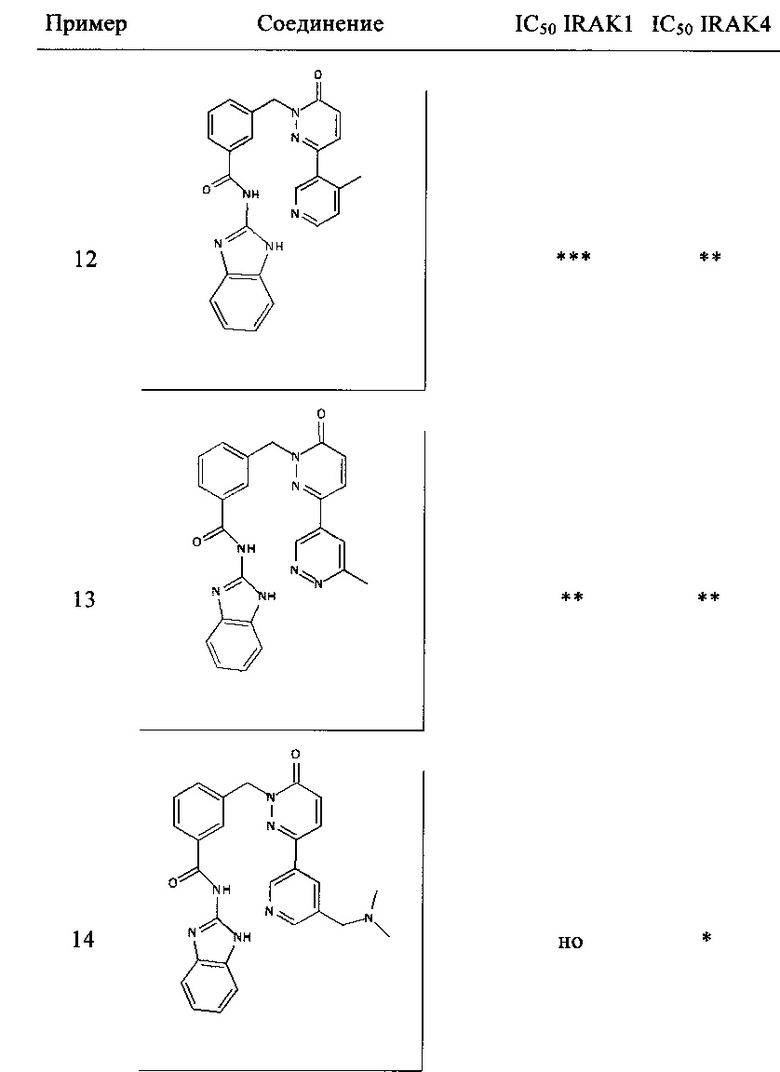
Нижеследующие сокращения относятся к применяемым ниже сокращениям:
Ас (ацетил), BINAP (2,2'-бис(дисфенилфосфино)-1,1'-бинафталин), dba (дибензилиден ацетон), Bu (бутил), tBu (трет-бутил), ДХЭ (дихлорэтан), ДХМ (дихлорметан), DIEA (диизопропилэтиламин), ДМА (диметилацетамид), ДМСО (диметилсульфоксид), ДМФА (N,N-диметилформамид), Dppf (1,1'-бис(дифенил фосфин ферроцен)), EtOAc (этилацетат), EtOH (этанол), г (грамм), сНех (циклогексан), HATU (N-[(диметиламино)(3Н-[1,2,3]триазоло[4,5-b]пиридин-3-илокси)метилен]-N-метилметанаминия гексафторфосфат), HBTU (N,N,N',N'-тетраметил-O-(1Н-бензотриазол-1-ил)урония гексафторфосфат), ВЭЖХ (высокоэффективная жидкостная хроматография), ч. (час), ЖХ (жидкостная хроматография), LDA (диизопропиламин лития), LiHMDS (бис(триметилсилил)амид лития), МГц (мегагерц), МеОН (метанол), мин. (минута), мл (миллилитр), ммоль (миллимоль), мМ (милимолярный), т.п. (точка плавления кипения), МС (масс-спектрометрия), MB (микроволны), NMM (N-метилморфолин), ЯМР (ядерный магнитный резонанс), О/N (в течение ночи), PBS (фосфатнобуфферный солевой раствор), PPh3 (трифенилфосфин), КТ (комнатная температура), ТЭА (триэтиламин), ТФУ (трифторуксусная кислота), ТГФ (тетрагидрофуран), ТСХ (тонкослойная хроматография), oTol (орто-толил), Т3Р (ангидрид пропилфосфоновой кислоты), УФ (ультрафиолет).
В общем, соединения в соответствии с формулой (I) и связанных формул настоящего изобретения могут быть получены из легкодоступных исходных веществ. Если такие исходные вещества не являются коммерчески доступными, они могут быть получены стандартными методами синтеза. Как правило, пути синтеза для любого отдельного соединения формулы (I) и связанных формул будут зависеть от конкретных заместителей каждой молекулы, такие факторы являются понятными обычным специалистам в данной области. Следующие общие способы и методики, описанные далее в примерах, могут быть использованы для получения соединений формулы (I) и связанных формул.
Условия реакций, показанные на следующих схемах, такие как температуры, растворители или совместные реагенты, приведены только в качестве примеров и не являются ограничивающими. Следует понимать, что там, где приведены типичные или предпочтительные экспериментальные условия (т.е. температуры реакции, время, моли реагентов, растворители и т.д.), также могут быть использованы другие экспериментальные условия, если не указано иное. Оптимальные реакционные условия могут варьироваться в зависимости от конкретных реагентов или растворителей, которые применяют, и такие условия могут быть определены специалистом в данной области техники, с использованием типовых методик оптимизации. Для всех методик защиты и снятия защиты, см. Philip J. Kocienski, in "Protecting Groups", Georg Thieme Verlag Stuttgart, New York, 1994 and, Theodora W. Greene and Peter G.M. Wuts in "Protective Groups in Organic Synthesis", Wiley Interscience, 3-е издание 1999.
В зависимости от природы R1, Ra, Rb, X, Y и Z для синтеза соединений формулы (I) могут быть выбраны различные синтетические стратегии. В способе, показанном на следующих схемах R1, Ra, Rb, X, Y и Z являются такими, как определено выше в описании, если не указано иное.
Соединения формулы (I) могут быть получены сочетанием соединения карбоновой кислоты общей формулы (II), в которой А означает Н, Li, Na или K и амино-бензимидазола общей формулы (III), в которой R1 и Rb являются такими, как определены выше, как приведено на схеме 1. Общие протоколы для такой реакции приведены ниже в примерах, с применением условий и способов, хорошо известных специалисту в данной области техники. Стандартный агент реакции сочетания, такой как HBTU, EDC, Т3Р или изобутилхлороформиат можно использовать в присутствии или без добавки, такой как HOBt и основания, такого как DIEA, TEA или NMM в приемлемом растворителе, таком как ДМФ, ацетонитрил, ТГФ или ДХМ при температуре, повышающейся от приблизительно 0°C до 50°C. В качестве альтернативы, производное карбоновой кислоты (такое как хлорангидрид) может вступать в реакцию с применением условий и способов, хорошо известных специалисту в данной области техники, в присутствии основания, такого как пиридин или DIEA в приемлемом растворителе, таком как толуол, ДХМ, ТГФ или ДМФ, при температуре, повышающейся от приблизительно 0°C до КТ, предпочтительно при КТ, в течение нескольких часов.
Схема 1
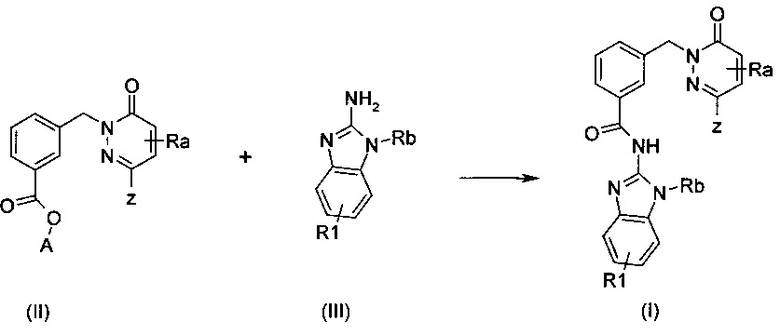
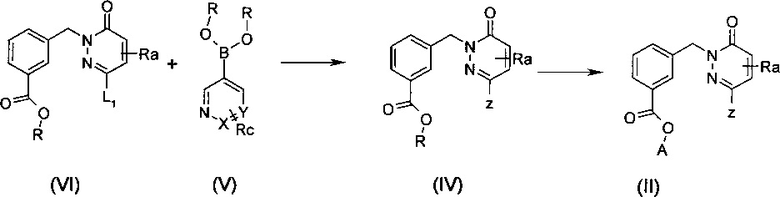
Соединения формулы (II), в которой А означает Н или Li, Na или K и Ra и Z являются такими, как определены выше могут быть получены в две стадии при помощи реакции сочетания Сузуки-Мияуры между пиридазиноном общей формулы (VI), в которой L1 означает галоген или группу трифторметансульфоната и R означает алкильную группу и бороновой кислотой или сложным эфиром формулы (V), в которой R означает алкильную группу до получения сложного эфира общей формулы (IV), в которой R означает алкильную группу, за которой следует гидролиз сложного эфира (IV) в кислоту или кислотную соль (II), как изображено на схеме 2. Общие протоколы для реакции сочетания Сузуки-Мияура приведены ниже в примерах, с применением условий и способов, хорошо известных специалисту в данной области техники, чтобы осуществить такое сочетание (см. например Miyaura, N.; Suzuki, A. Chem. Rev. 1995, 95, 2457; Takahiro I. и Toshiaki M., Tetrahedron Lett. 2005, 46, 3573-3577). При стандартной процедуре, пиридазинон общей формулы (VI) и бороновую кислоту или сложный эфир формулы (V) нагревают в приемлемом растворителе, таком как ТГФ, толуол, ДМФ или диоксан, в присутствии или отсутствии воды в качестве сорастворителя, в присутствии основания, такого как CS2CO3, Na2CO3, K2CO3, CsF, и с приемлемым катализатором, таким как, но не ограничиваясь только ним, дихлорбис(трифенилфосфин)палладий(II), Pd(PPh3)4 или 1,1'-бис(дифенилфосфино)ферроцендихлор палладий(II), Pd(OAc)2, Pd2(dba)3, Pd(Cl)2(PPh3)2 или Pd/C в присутствии или отсутствии дополнительного лиганда, такого как, но не ограничиваясь только ним Р(tBu)3, Р(oTol)3, PPh3, BINAP. Эта реакция сочетания может быть осуществлена при температуре от приблизительно 20°C до приблизительно 150°C, предпочтительно приблизительно при 120°C, в течение нескольких минут до нескольких часов, возможно под действием микроволнового излучения. Гидролиз сложного эфира (IV) может быть осуществлен, например, с применением HCl, H2SO4, или с применением LiOH, NaOH или KОН в воде, вода/ТГФ, вода/ТГФ/этанол или вода/диоксан, при температурах между 0 и 100°C. Кислотную или соляную форму получают в зависимости от выбранной реакционной обработки (основные или кислотные условия).
Схема 2
Аминобензимидазолы общей формулы (III) могут быть получены из коммерческих источников или могут быть синтезированы следуя методикам, хорошо известным специалисту в данной области техники таким как, но не ограничиваясь только ним, описаны в J. Org. Chem. 1977, 42, 542  Bioorganic & Medicinal Chemistry Letters 2006, 16, 2842-2845.
Bioorganic & Medicinal Chemistry Letters 2006, 16, 2842-2845.
Соединения формулы (VI), в которой Ra, L1 и R являются такими, как определены выше могут быть получены алкилированием пиридазинона общей формулы (VIII), в которой Ra и L1 являются такими, как определены выше с соединением общей формулы (VII), в которой R является таким, как определено выше и L2 означает уходящую группу, такую как бром, хлор, йод, алкилсульфонат или любую другую пригодную уходящую группу, известную специалисту в данной области техники или группу ОН, как показано на схеме 3. Общие протоколы для такого превращения приведены ниже в примерах, с применением условий и способов, хорошо известных специалисту в данной области техники. При стандартной процедуре, соединение формулы (VII), в которой L2 означает уходящую группу, обрабатывают основанием, таким как, но не ограничиваясь только ним, NaH, K2CO3, CS2CO3, LDA, LiHMDS, предпочтительно NaH, и с пиридазиноном формулы (VIII), в приемлемом растворителе, таком как ТГФ, диоксан, ДМФ, ДМА, при температуре между -20°C до приблизительно 150°C, в течение периода времени от нескольких минут до нескольких часов. В качестве альтернативы, соединения формулы (VI), в которой Ra, L1 и R являются такими, как определены выше могут быть получены в ходе реакции соединения формулы (VII), в которой L2 означает группу ОН с пиридазиноном формулы (VIII) с применением условий, хорошо известных специалисту в данной области техники для реакции Митсунобу (см. например Hughes, D.L. Organic Reactions (New York), 1992, 42, 335-656; Reynolds, A.J.; Kassiou, M. Current Organic Chemistry, 2009, 13 (16); 1610-1632). Как правило, реакция происходит в присутствии фосфина, такого как, но не ограничиваясь только ним P(tBu)3, PPBu3, Р(oTol)3, PPh3, в присутствии азадикарбоксилата, такого как, но не ограничиваясь только ним, диэтилазадикарбоксилат, диизопропилазадикарбоксилат, тетраметилазодикарбоксамид, в растворителе, таком как ТГФ, диоксан, ДХМ, ДХЭ, при температуре между -20°C до приблизительно 150°C, предпочтительно при комнатной температуре, в течение периода времени от нескольких минут до нескольких часов.
Схема 3
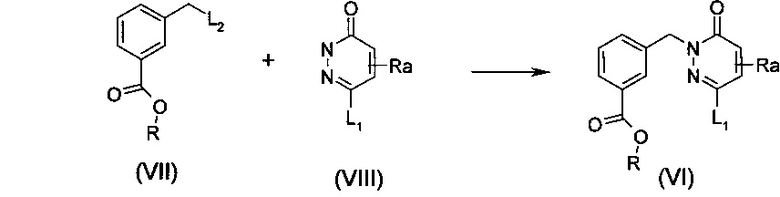
В качестве альтернативы, соединение общей формулы (I) может быть получено с применением подобных химических стадий, но в другом порядке, например, как приведено на схеме 4. После гидролиза соединения общей формулы (VI) в кислоту или кислотную соль общей формулы (X), сочетание с аминобензимидазолом общей формулы (II) может обеспечить пиридазинон общей формулы (XI), который в заключение может вступить в реакцию с бороновой кислотой или сложным эфиром общей формулы (V) посредством реакции сочетания Сузуки-Мияура с получением соединения общей формулы (I). Общие протоколы для такого превращения приведены ниже в примерах, с применением условий и способов, хорошо известных специалисту в данной области техники. Стандартные условия для таких превращений являются такими же, как и описано выше.
Схема 4
Соединения согласно настоящему изобретению могут быть выделены совместно с молекулами растворителей путем кристаллизации из соответствующего растворителя или выпариванием соответствующего растворителя.
Фармацевтически приемлемые анионные соли соединений формулы (I), содержащие основный центр, могут быть получены стандартным способом. Например, раствор свободного основания может быть обработан с помощью приемлемой кислоты или в чистом виде, или в приемлемом растворе, и полученную соль выделяют или фильтрацией, или выпариванием под вакуумом реакционного растворителя.
Фармацевтически приемлемые катионные соли соединений формулы (I), содержащие кислотный центр, могут быть получены традиционным способом. Например, раствор свободной кислоты может быть обработан с помощью приемлемого основания или в чистом виде, или в пригодном растворе, и полученную соль выделяют или фильтрацией, или упариванием под вакуумом реакционного растворителя. В некоторых случаях, соли могут быть получены смешиванием раствора кислоты с раствором соли щелочного или щелочноземельного металла (такого как, этилгексаноат натрия, олеат магния), используя растворитель, в котором требуемая соль щелочного или щелочноземельного металла соединения формулы (I) осаждается, или может быть иным образом выделена путем концентрации и добавления осадителя.
Оба типа солей могут быть образованы или взаимопревращены с применением методик ионообменной смолы.
В зависимости от применяемых условий время реакции, как правило, составляет от нескольких минут до 14 дней. Температура реакций находится в диапазоне от приблизительно -30°C до приблизительно 140°C, обычно от -10°C до 90°C, в частности от приблизительно 0°C до 70°C.
Формула (I) и связанные формулы также охватывают оптически активные формы (стереоизомеры), энантиомеры, рацематы, диастереомеры и гидраты, и сольваты этих соединений. Понятие "сольваты соединений" следует понимать как аддукции молекул инертного растворителя на соединениях, которые образуются благодаря их взаимной силе притяжения. Сольваты, например, представляют собой моно- или дигидраты или алкоголяты.
Понятие «фармацевтически приемлемые производные» означает, например, соли соединений формулы I и так называемых про лекарственных соединений.
Понятие «пролекарственные производные» следует понимать как соединения формулы I, которые были модифицированы с помощью, например, алкильных или ацильных групп, Сахаров или олигопептидов и которые быстро расщепляются в организме с образованием действующих соединений. Предпочтительно «пролекарство», в отношении соединений формулы I, относится к производным соединений, которые быстро превращаются in vivo с получением исходного соединения формулы I, как например, путем гидролиза в крови. У Т. Higuchi and V. Stella представлено исчерпывающее обсуждение концепции пролекарств в "Prodrugs as Novel Delivery Systems", том 14 Symposium Series, American Chemical Society (1975). Примеры эфиров, пригодных в качестве пролекарств для соединений, содержащих карбоксильные группы можно найти на страницах 14-21 в "Bioreversible Carriers in Drug Design: Theory and Application", под редакцией E.В. Roche, Pergamon Press: New York (1987). Предполагается, что эти ссылки, и любые другие приведенные в данном описании, включены в данное описание посредством ссылки.
Они также охватывают производные поддающихся биологическому разложению полимеров соединений в соответствии с изобретением, как описано, например, в Int. J. Pharm. 115, 61-67 (1995).
Формула (I) и связанные формулы также охватывают смеси соединений формулы I, например, смеси двух диастереомеров, например, в соотношении 1:1, 1:2, 1:3, 1:4, 1:5, 1:10, 1:100 или 1:1000.
В особенности предпочтительно они представляют собой смеси стереоизомерных соединений.
Фармацевтические составы могут быть введены в виде лекарственных дозированных форм, содержащих предопределенное количество действующего вещества на дозированную единицу. Такая дозированная единица может содержать, например, от 0,5 мг до 1 г, предпочтительно от 1 мг до 700 мг, предпочтительно в особенности от 5 мг до 100 мг соединения в соответствии с изобретением, в зависимости от состояния заболевания, которое подвергают лечению, способа введения и возраста, веса и состояния пациента, или же фармацевтические композиции можно вводить в виде дозированных единиц, которые содержат предопределенное количество действующего вещества на дозированную единицу. Предпочтительными композициями дозированных единиц являются составы, содержащие суточную дозу или часть суточной дозы, как указано выше, или соответствующую ей порцию действующего вещества. Кроме того, фармацевтические композиции этого типа могут быть получены с применением способа, который, в общем, известен в области фармацевтики.
Фармацевтические составы могут быть адаптированы для введения посредством любого пригодного способа, например, путем перорального (включая буккальное или подъязычное), ректального, назального, местного (включая буккальное, подъязычное или трансдермальное), вагинального или парентерального (включая подкожное, внутримышечное, внутривенное или внутрикожное) введения. Такие составы могут быть приготовлены с помощью любого способа, известного в области фармацевтики, например, путем соединения действующего вещества с наполнителем(ями) или вспомогательным(ыми) веществом(ами).
Фармацевтические составы, приспособленные для перорального введения, могут быть введены в виде отдельных единиц, таких как, например, капсулы или таблетки; порошки или гранулы; растворы или суспензии в водных или неводных жидкостях; съедобных пенок или пенистых пищевых продуктов; или жидких эмульсий типа масло-в-воде или жидких эмульсий типа вода-в-масле.
Так, например, в случае перорального введения в виде таблетки или капсулы, действующее вещество может быть соединено с пероральным, нетоксичным и фармацевтически приемлемым инертным наполнителем, таким как, например, этанол, глицерин, вода и т.п. Порошки получают путем измельчения соединения до приемлемого мелкого размера и смешивания его с фармацевтическим наполнителем, измельченным аналогичным способом, таким как, например, пищевой углевод, такой как, например, крахмал или маннит. Равным образом можно добавлять ароматизатор, консервант, диспергирующее вещество и краситель.
Капсулы получают путем приготовления порошковой смеси, как описано выше и заполнения ею желатиновых оболочек. Вещества, способствующие скольжению и смазывающие вещества, такие как, например, высокодисперсная кремниевая кислота, тальк, стеарат магния, стеарат кальция или полиэтиленгликоль в твердой форме, могут быть добавлены к порошковой смеси перед процессом наполнения. В целях улучшения доступности лекарственного средства после того, как капсула была принята, также может быть добавлен разрыхлитель или солюбилизатор, такой как, например, агар-агар, карбонат кальция или карбонат натрия.
К тому же, при желании или необходимости, в смесь также могут быть добавлены пригодные связующие вещества, смазывающие средства и дезинтеграторы, а также красители. Пригодные связующие вещества охватывают крахмал, желатин, природные сахара, такие как, например, глюкоза или бета-лактоза, подсластители, полученные из кукурузы, природных и синтетических смол, такие как, например, аравийская камедь, трагакантовая камедь или альгинат натрия, карбоксиметилцеллюлоза, полиэтиленгликоль, воски и т.п. Смазывающие средства, которые могут быть применены в таких дозированных формах, включают олеат натрия, стеарат натрия, стеарат магния, бензоат натрия, ацетат натрия, хлорид натрия и т.п. Дезинтеграторы включают, но не ограничиваются только ними, крахмал, метилцеллюлозу, агар, бентонит, ксантановую камедь и т.п. Таблетки изготавливают, например, путем приготовления порошковой смеси, гранулирования или сухого прессования смеси, добавления смазывающего вещества и дезинтегратора и прессования полученной смеси в таблетки. Порошковую смесь готовят путем смешивания соединения, измельченного пригодным образом, с разбавителем или основанием, как описано выше, и необязательно со связующим веществом, таким как, например, карбоксиметилцеллюлоза, альгинат, желатин или поливинилпирролидон, замедлителем растворения, таким как, например, парафин, усилителем поглощения, таким как, например, четвертичная соль, и/или абсорбентом, таким как, например, бентонит, каолин или фосфат дикальция. Порошковую смесь можно гранулировать путем ее смачивания связующим веществом, таким как, например, сироп, крахмальная паста, клейкое вещество акации или растворы целлюлозы или полимерных веществ, и ее прессования через сито. В качестве альтернативы грануляции, порошковую смесь можно пропускать через таблетировочную машину, получая крупные куски неправильной формы, которые разрушаются, образуя гранулы. Гранулы можно замасливать путем добавления стеариновой кислоты, стеарата, талька или минерального масла для предотвращения слипания в таблетировочной литейной форме. После этого смазанную смесь спрессовывают, получая таблетки. Соединения в соответствии с изобретением также можно объединять с сыпучим инертным наполнителем и затем подвергать прямому прессованию, получая таблетки без осуществления стадий грануляции или сухого прессования. Таблетки также можно покрывать прозрачным или светонепроницаемым защитным слоем, состоящим из шеллакового герметизирующего слоя, слоя сахара или полимерного вещества и глянцевого слоя воска. В эти покрытия также можно добавлять красители для возможности различения между разными дозированными единицами.
Жидкости для перорального введения, такие как, например, раствор, сиропы и эликсиры, могут быть приготовлены в виде дозированных единиц таким образом, чтобы они содержали заранее предопределенное количество соединения. Сиропы могут быть получены путем растворения соединений в водном растворе с пригодным ароматизатором, тогда как эликсиры готовят с применением нетоксичного спиртового наполнителя. Суспензии могут быть приготовлены путем диспергирования соединений в нетоксичном наполнителе. Равным образом можно добавлять солюбилизаторы и эмульгирующие вещества, такие как, например, этоксилированные изостеариловые спирты и полиоксиэтиленовые эфиры сорбита, консерванты, ароматические добавки, такие как, например, масло мяты перечной, или натуральные заменители сахара или сахарин, или другие искусственные заменители сахара и т.п.
Составы для перорального введения в виде дозированных единиц, при желании, могут быть инкапсулированы в микрокапсулы. Также состав может быть приготовлен таким образом, чтобы пролонгировать или замедлить высвобождение, например, путем применения покрытий или заделывания требуемого вещества в полимеры, воск и т.п.
Соединения формулы (I) и связанных формул и их соли, сольваты, таутомеры и стереоизомеры также могут быть введены в виде липосомных систем доставки, таких как, например, небольшие однослойные пузырьки, большие однослойные пузырьки и многослойные пузырьки. Липосомы могут быть образованы с помощью различных фосфолипидов, таких как, например, холестерин, стеариламин или фосфатидилхолины.
Соединения формулы (I) и связанных формул и их соли, сольваты, таутомеры и стереоизомеры также могут быть доставлены с применением моноклональных антител в качестве индивидуальных носителей, с которым соединены молекулы соединения. Соединения также могут быть соединены с растворимыми полимерами в качестве нацеливающих носителей лекарственных средств. Такие полимеры могут охватывать поливинилпирролидон, сополимер пирана, полигидроксипропилметакриламидофенол, полигидроксиэтиласпартамидофенол или полиэтиленоксидполилизин, замещенный пальмитоиловыми радикалами. Кроме того, соединения могут быть соединены с классом способных к биологическому разложению полимеров, которые пригодны для обеспечения контролируемого высвобождения лекарственного средства, например полимолочной кислотой, поли-эпсилон-капролактоном, полигидроксимасляной кислотой, полиортоэфирами, полиацеталями, полидигидроксипиранами, полицианоакрилатами и перекрестно-сшитыми или амфипатическими блок-сополимерами гидрогелей.
Фармацевтические составы, приспособленные для трансдермального введения, могут быть введены в виде независимых пластырей для пролонгированного, непосредственного контакта с эпидермисом реципиента. Таким образом, например, действующее вещество может быть доставлено из пластыря посредством ионтофореза, как, в общем, описано в Pharmaceutical Research, 3(6), 318 (1986).
Фармацевтические соединения, приспособленные для местного введения, могут быть приготовлены в виде мазей, кремов, суспензий, лосьонов, порошков, растворов, паст, гелей, спреев, аэрозолей или масел.
Для лечения глаз или других наружных тканей, например рта и кожи, предпочтительно применяются составы в виде местной мази или крема. Для приготовления состава в виде мази, действующее вещество можно применять с парафиновым или смешиваемым с водой основанием для крема. В качестве альтернативы для получения крема действующее вещество может быть приготовлено с основой для крема типа масло-в-воде или основой вода-в-масле.
Фармацевтические составы, приспособленные для местного введения в глаза, включают глазные капли, в которых действующее вещество растворено или суспендировано в пригодном носителе, предпочтительно в водном растворителе.
Фармацевтические составы, приспособленные для местного введения в полость рта, включают лепешки, пастилки и жидкости для полоскания рта.
Фармацевтические составы, приспособленные для ректального введения, могут быть введены в виде суппозиториев или клизм.
Фармацевтические составы, приспособленные для назального введения, в которых носитель представляет собой твердое вещество, включают крупный порошок, имеющий размер частичек, например, в диапазоне 20-500 микрон, который вводится путем вдыхания, то есть путем быстрого вдоха через нос из контейнера, содержащего порошок, который придерживают возле носа. Пригодные составы для введения в виде назального спрея или носовых капель с жидкостью в качестве носителя включают растворы активного вещества в воде или в масле.
Фармацевтические составы, приспособленные для введения путем ингаляции, включают тонкоизмельченные частички в виде пыли или аэрозоля, которые могут быть получены с помощью различных диспергирующих устройств под давлением с аэрозолями, распылителей или инсуффляторов.
Фармацевтические составы, адаптированные для вагинального введения, могут вводиться в виде пессариев, тампонов, кремов, гелей, паст, пен или композиций в виде спреев.
Фармацевтические составы, приспособленные для парентерального введения, включают водные или неводные стерильные растворы для инъекций, содержащие антиоксиданты, буферы, бактериостатические вещества и растворенные вещества, с помощью которых состав поддерживается изотоническим по отношению к крови реципиента, подвергаемого лечению; и водные или неводные стерильные суспензии, которые могут содержать суспензионную среду и загустители. Составы могут быть введены с помощью емкостей для однократного или многократного введения, например, запечатанных ампул и флаконов, и храниться в вымороженном (лиофилизированном) состоянии, при этом непосредственно перед введением необходимо только добавить стерильную жидкость-носитель, например, воду для инъекций.
Растворы и суспензии для инъекций, приготовленные согласно рецептуре, могут быть приготовлены из стерильных порошков, гранул и таблеток.
Само собой разумеется, что дополнительно к предпочтительным вышеописанным составляющим, составы также могут содержать другие вещества, которые используются в данной области для конкретных типов составов; например, составы, пригодные для перорального введения, могут содержать ароматизаторы.
Терапевтически эффективное количество соединения формулы (I) и связанных формул зависит от многих факторов, включая, например, возраст и вес животного, определенное состояние, которое необходимо лечить, и его тяжесть, природу лекарственного средства и способ введения, и в конечном счете оно может быть определено лечащим врачом или ветеринаром. Тем не менее, эффективное количество соединения, как правило, находится в пределах от 0,1 до 100 мг/кг веса тела реципиента (млекопитающего) в сутки и предпочтительно обычно находится в пределах от 1 до 10 мг/кг веса тела в сутки. Таким образом, действующее суточное количество для взрослого млекопитающего весом 70 кг обычно может составлять от 70 до 700 мг, причем это количество может быть введено в виде отдельной дозы один раз в день или обычно в виде циклов частичных доз (таких как, например, два, три, четыре, пять или шесть раз) в сутки, таким образом, что общая суточная доза является одинаковой. Эффективное количество его соли, сольвата, таутомера и стереоизомера может быть определено в виде доли эффективного количества соединения как такового.
Кроме того, настоящее изобретение относится к способу лечения пациента, страдающего от связанных с IRAK расстройств, включающий введение указанному субъекту эффективного количества соединения формулы I и связанных формул. Настоящее изобретение предпочтительно относится к способу, где связанное с IRAK расстройство представляет собой аутоиммунное заболевание или состояние, связанное с гиперактивностью иммунной реакции или раком. Настоящее изобретение, кроме того, относится к способу лечения пациента, страдающего от иммунорегуляторного нарушения, включающему введение указанному субъекту соединения формулы (I), и связанных формул в количестве, эффективном для лечения указанного иммунорегуляторного нарушения. Настоящее изобретение предпочтительно относится к способу, в котором иммунорегуляторное нарушение представляет собой аутоиммунное или хроническое воспалительное заболевание, выбранное из группы, состоящей из: аллергических заболеваний, бокового амиотрофического склероза (ALS), системной красной волчанки, хронического ревматоидного артрита, сахарного диабета I типа, воспалительного заболевания кишечника, билиарного цирроза печени, увеита, множественного склероза, болезни Крона, язвенного колита, буллезного пемфигоида, саркоидоза, псориаза, аутоиммунного миозита, гранулематоза Вегенера, ихтиоза, офтальмопатии Грейвса и астмы. Настоящее изобретение, кроме того, относится к способу, в котором иммунорегуляторное нарушение представляет собой патологию костного мозга или отторжение пересаженного органа или реакцию «трансплантат против хозяина».
Помимо этого, настоящее изобретение относится к способу, в котором иммунорегуляторное нарушение выбрано из группы, которая включает: трансплантацию органов или тканей, реакцию «трансплантат против хозяина», вызванную пересадкой, аутоиммунные синдромы, в том числе ревматоидный артрит, системную красную волчанку, тиреоидит Хашимото, рассеянный склероз, системную склеродермию, миастению, сахарный диабет I типа, увеит, задний увеит, аллергический энцефаломиелит, гломерулонефрит, послеинфекционные аутоиммунные заболевания, включая ревматизм и послеинфекционный гломерулонефрит, воспалительные и гиперпролиферативные кожные заболевания, псориаз, атонический дерматит, контактный дерматит, экзематозный дерматит, себорейный дерматит, красный плоский лишай, пузырчатка, буллезный пемфигоид, буллезный эпидермолиз, крапивницу, ангионевротический отек, васкулит, эритему, кожную эозинофилию, красную волчанку, акне, алопецию, кератоконъюнктивит, весенний конъюнктивит, увеит, связанный с болезнью Бехчета, кератит, герпетический кератит, коническую роговицу, дистрофию эпителия роговицы, роговичную лейкому, глазную пузырчатку, язву Мурена, склерит, офтальмопатию Грейвса, синдром Фогта-Коянаги-Харада, саркоидоз, аллергию на пыльцу, обратимые обструктивные заболевания дыхательных путей, бронхиальную астму, аллергическая астма, внутренняя астма, внешняя астма и астма из-за пыли, хроническая или запущенная астма, поздняя астма и гиперчувствительность дыхательных путей, бронхит, язва желудка, повреждение сосудов, вызванное ишемическими заболеваниями и тромбозами, ишемические заболевания кишечника, воспалительные заболевания кишечника, некротический энтероколит, кишечные повреждения, связанные с термическими ожогами, целиакические заболевания, проктит, эозинофильный гастроэнтерит, мастоцитоз, болезнь Крона, язвенный колит, мигрень, ринит, экзема, интерстициальный нефрит, синдром Гудпасчера, гемолитико-уремический синдром, диабетическая нефропатия, дерматомиозит, синдрома Гийена-Барре, болезнь Меньера, полиневрит, множественный неврит, мононеврит, радикулопатию, гипертиреоз, базедову болезнь, чистую красноклеточную аплазию, апластическую анемию, гипопластическую анемию, идиопатическую тромбоцитопеническую пурпуру, аутоиммунную гемолитическую анемию, агранулоцитоза, злокачественной анемии, мегалобластной анемии, анэритроплазии, остеопороза, саркоидоза, миомы легких, идиопатической интерстициальной пневмонии, дерматомиозита, обычной лейкодермы, обычного ихтиоза, фотоаллергической чувствительности, кожной Т-клеточной лимфомы, хронического лимфоцитарного лейкоза, артериосклероза, атеросклероза, синдрома аортита, узелкового полиартериита, миокардоза, склеродермии, гранулематоза Вегенера, синдрома Шегрена, ожирения, эозинофильного фасцита, поражения десен, периодонта, альвеолярной кости, костного вещества зуба, гломерулонефрита, алопеции по мужскому типу или старческой плешивости, посредством предотвращения выпадения волос или обеспечивая прорастание волос и/или стимулирование восстановления волос и роста волос, мышечной дистрофии, пиодермии и синдрома Сезари, болезни Аддисона, ишемически-реперфузионного повреждения органов, которое происходит при консервировании, трансплантации или ишемической болезни, эндотоксического шока, псевдомембранозного колита, колита, вызванного применением лекарственных средств или радиацией, ишемической острой почечной недостаточности, хронической почечной недостаточности, токсичности, вызванной кислородным отравлением или лекарственными средствами, рака легких, эмфиземы легких, катаракты, сидероза, пигментного ретинита, старческой дегенерации желтого пятна, витреального рубцевания, ожога роговицы щелочью, дерматита, многоформной эритемы, линейного IgA буллезного дерматита и цементного дерматита, гингивита, периодонтита, сепсиса, панкреатита, заболеваний, вызванных загрязнением окружающей среды, старения, канцерогенеза, метастазов рака и гипобаропатии, заболевания, вызванного высвобождением гистамина или лейкотриена-С4, болезни Бехчета, аутоиммунного гепатита, первичного билиарного цирроза печени, склерозирующего холангита, частичной резекции печени, острого некроза печени, некроза, вызванного токсинами, вирусного гепатита, шока или гипоксии, гепатита В, гепатита С, цирроза печени, алкогольного цирроза, печеночной недостаточности, молниеносной печеночной недостаточности, печеночной недостаточности с поздним началом, обострения хронической печеночной недостаточности, увеличения химиотерапевтического эффекта, цитомегаловирусной инфекции, ЦМВ инфекции, СПИДа, рака, старческого слабоумия, болезни Паркинсона, травм, а также хронических бактериальных инфекций.
Предпочтительно, расстройства, связанные с IRAK, выбраны из ревматоидного артрита, псориатического артрита, остеоартрита, системной красной волчанки, волчаночного нефрита, анкилозирующего спондилоартрита, остеопороза, системного склероза, множественного склероза, псориаза, сахарного диабета I типа, сахарного диабета II типа, воспалительного заболевания кишечника (болезнь Крона и язвенный колит), гипериммуноглобулинемии D и синдрома периодической лихорадки, криопирин-связанных периодических синдромов, синдрома Шницлера, системного ювенильного идиопатического артрита, приобретенной болезни Стилла, подагры, псевдоподагры, синдрома САПГО, болезни Кастлемана, сепсиса, инсульта, атеросклероза, целиакии, DIRA (дефицит антагониста рецептора IL-1), болезнь Альцгеймера, болезни Паркинсона, рака.
Предпочтительные соединения формулы (I) и связанных формул проявляют IC50 для связывания с IRAK менее чем приблизительно 5 мкМ, предпочтительно менее чем приблизительно 1 мкМ и еще более предпочтительно менее чем приблизительно 0,100 мкМ.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
В дальнейшем настоящее изобретение будет проиллюстрировано посредством некоторых примеров, которые не следует истолковывать как ограничивающие объем притязаний изобретения.
Общая часть:
Предоставленные данные ВЭЖХ в примерах, описанных ниже, были получены следующим образом.
Метод А: колонка Waters XbridgeTM С8 50 мм × 4.6 мм при производительности 2 мл/мин.; 8 мин. градиент Н2О : CH3CN : ТФУ от 100:0:0.1% до 0:100:0.05%
УФ обнаружение: макс, площадь или определенная длина волны.
Предоставленные данные ЖХ/МС в примерах, описанных ниже были получены следующим образом:
ЖХ:
Метод А: колонка Waters XbridgeTM С8 50 мм × 4.6 мм при производительности 2 мл/мин.; 8 мин. градиент Н2О : CH3CN : ТФУ от 100:0:0.1% до 0:100:0.05%
Метод В: колонка Waters XbridgeTM С8 50 мм × 4.6 мм при производительности 1 мл/мин.; 8 мин. градиент H2O : CH3CN : NH4HCO3 от 100:0:0.1% до 0:100:0.05%
УФ обнаружение: макс, площадь или определенная длина волны.
Масс-спектр: МС Waters ZMD (ESI).
Представленные данные ЯМР в примерах, описанных ниже были получены с применением Bruker AV-400 Мгц.
Предлагаемые в изобретении соединения получили названия в соответствии со стандартами, применяемыми в программе Autonom.
Соединения в соответствии с формулой (I) могут быть получены из легко доступных исходных веществ при помощи некоторых синтетических подходов, с применением химических протоколов, как в растворах, так и в твердых фазах, или смешанных химических протоколов в растворах и в твердых фазах. Примеры способов синтеза описаны в примерах ниже. Если не указано иное, соединения формулы (I), и связанных формул, полученные в виде рацемической смеси могут быть выделены с получением энантиомерно обогащенной смеси или чистого энантиомера.
Коммерчески доступные исходные вещества, используемые в следующем экспериментальном описании, были приобретены у Aldrich или Sigma или ABCR, если не указано иное.
Промежуточное соединение 1: 3-(6-оксо-3-пиридин-3-ил-6Н-пиридазин-1-илметил)-бензоат лития
Стадия 1: Образование метилового эфира 3-(6-оксо-3-пиридин-3-ил-6Н-пиридазин-1 -илметил)-бензойной кислоты
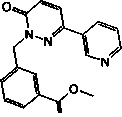
Трибромфосфан (1.7 г, 6.6 ммоль) добавляли к раствору из метилового эфира 3-гидроксиметил-бензойной кислоты (1 г, 6.0 ммоль) в диэтиловом эфире (20 мл) при 0°C. Реакционную смесь оставляли нагреваться до КТ и перемешивали в течение 2 ч. Затем ее обрабатывали водой. Водную фазу подщелачивали насыщенным раствором NaHCO3 (15 мл) и экстрагировали при помощи дихлорметана. Объединенные органические фазы высушивали над безводным Na2SO4, фильтровали и концентрировали с получением метилового эфира 3-бромметил-бензойной кислоты, который растворяли в НМП. Затем к этому раствору добавляли 6-пиридин-3-ил-2Н-пиридазин-3-он (1.1 г, 6.5 ммоль) и карбонат цезия (2.1 г, 6.5 ммоль), и реакционную смесь перемешивали при КТ в течение 12 ч. Ее обрабатывали водой, водную фазу экстрагировали при помощи этилацетата (3×15 мл) и объединенные органические фазы высушивали над безводным Na2SO4, фильтровали и концентрировали под сниженным давлением. Очищение флэш-хроматографией на силикагеле обеспечило указанный в заголовке продукт в виде твердого вещества желтого цвета (0.8 г, 57%). ЖХ/МС: (метод А) 322.2 (М+Н), КТ. 2.51 мин., 66.4% (макс.).
Стадия 2: Образование 3-(6-оксо-3-пиридин-3-ил-6Н-пиридазин-1-илметил)-бензоата лития
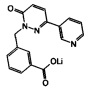
Раствор из лития гидроксида моногидрата (0.35 г, 8.7 ммоль) и метилового эфира 3-(6-оксо-3-пиридин-3-ил-6Н-пиридазин-1-илметил)-бензойной кислоты (1.4 г, 4.35 ммоль) в ТГФ: вода (1:2, 10 мл) перемешивали при КТ в течение 12 ч. Реакционную смесь концентрировали, и подвергали азеотропной перегонке с толуолом с получением указанного в заголовке соединения в виде твердого вещества желтого цвета (0.5 г, 52%). 1HNMR (400 Мгц, ДМСО-d6): δ 13.01 (brs, 1Н), 9.09-9.08 (m, 1Н), 8.65-8.63 (m, 1Н), 8.27-8.24 (m, 1Н), 8.15 (d, J=9.8 Гц, 1Н), 7.95 (s, 1Н), 7.86 (t, J=1.3 Гц, 1Н), 7.63 (d, J=7.8 Гц, 1Н), 7.54-7.47 (m, 2Н), 7.15 (d, J=9.8 Гц, 1Н), 5.41 (s, 2Н). ЖХ/МС: (метод А) 308.0 (М+Н), КТ. 2.0 мин., 90.1% (макс.).
Промежуточное соединение 2: метиловый эфир 3-(3-Хлор-6-оксо-6Н-пиридазин-1-илметил)-бензойной кислоты
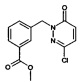
Указанное в заголовке соединение получали, следуя методике, описанной для промежуточного соединения 1, стадия 1 из метилового эфира 3-гидроксиметил-бензойной кислоты и 6-хлор-3-ил-2Н-пиридазин-3-она в виде твердого вещества не совсем белого цвета (9.8 г, 94%). ЖХ/МС: (метод А) 279.0 (М+Н), КТ. 3.7 мин., 93.8% (макс.), 93.9% (254 нм).
Промежуточное соединение 3: 3-(3-Хлор-6-оксо-6Н-пиридазин-1-илметил)-бензойная кислота
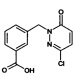
Раствор из лития гидроксида моногидрата (0.307 г, 7.5 ммоль) и метилового эфира 3-(3-хлор-6-оксо-6Н-пиридазин-1-илметил)-бензойной кислоты (1.2 г, 4.30 ммоль) в ТГФ : вода (2:1, 30 мл) перемешивали при КТ в течение 12 ч. Реакционную смесь затем концентрировали, подкисляли насыщенным раствором лимонной кислоты (15 мл) и экстрагировали при помощи дихлорметана (3×10 мл). Объединенные органические фазы промывали рассолом, высушивали над безводным Na2SO4, фильтровали, и концентрировали с получением указанного в заголовке соединения в виде твердого вещества не совсем белого цвета (1.5 г, 83%). ЖХ/МС: (метод А) 265.0 (М+Н), КТ. 2.9 мин., 94.1% (макс.).
Промежуточное соединение 4: 2-(2-амино-1Н-бенз[d]имидазол-5-ил)-N,N-диметилацетамид
Стадия 1: Образование 2-(бенз[с][1,2,5]тиадиазол-5-ил)-N,N-диметилацетамида
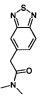
К раствору из бензо[1,2,5]тиадиазол-5-ил-уксусной кислоты (получено, как описано в Bioorg. Med. Chem. Lett. (1998) стр. 17-22, 5 г, 25.7 ммоль) в ТГФ добавляли N,N-диметиламин (15.4 мл, 30.8 ммоль) и триэтиламин (0.1 мл, 0.8 ммоль) при 0°C. К этой реакционной смеси добавляли Т3Р (50% вес/объем раствор в этилацетате, 49 мл, 77.2 ммоль) и перемешивали при комнатной температуре в течение 12 ч. Реакционную смесь промывали при помощи 10% двууглекислого натрия (15 мл) и экстрагировали при помощи дихлорметана (3×10 мл). Объединенные органические фазы промывали 10%-ым раствором лимонной кислоты, высушивали над безводным Na2SO4, фильтровали и концентрировали с получением указанного в заголовке соединения в виде твердого вещества желтого цвета (3 г, 53%). 1Н ЯМР (400 Мгц, ДМСО-d6): δ 8.00 (d, J=9.0 Гц, 1Н), 7.88 (d, J=0.7 Гц, 1Н), 7.58-7.56 (m, 1Н), 3.93 (s, 2Н), 3.06 (s, 3Н), 2.85 (s, 3Н). ЖХ/МС: (метод А) 222.0 (М+Н), КТ. 2.4 мин., 96.1% (макс.), 96.5% (220 нм).
Стадия 2: Образование 2-(3,4-диаминофенил)-N,N-диметилацетамид
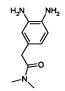
Никель Ренея (9 г, 40.5 ммоль) добавляли к раствору из 2-(бензо[с][1,2,5]тиадиазол-5-ил)-N,N-диметилацетамида (3 г, 13.5 ммоль) в метаноле (100 мл). Затем реакционную смесь нагревали в автоклаве при 45°C в течение 12 ч. Затем ее фильтровали через прокладку из целита, и фильтрат концентрировали под сниженным давлением с получением указанного в заголовке соединения в виде твердого вещества коричневого цвета (2.0 г, 76%). 1Н ЯМР (400 Мгц, ДМСО-d6): δ 6.72-6.35 (m, 2Н), 6.29-6.16 (m, 1Н), 4.39 (brs, 2Н), 4.29 (brs, 2Н), 3.32 (s, 2Н), 2.92 (s, 3Н), 2.78 (s, 3Н). ЖХ/МС: (метод В) 194.3 (М+Н), КТ. 2.4 мин., 92.9% (макс.).
Стадия 3: Образование 2-(2-амино-1Н-бензо[d]имидазол-5-ил)-N,N-диметилацетамида
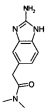
Раствор из 2-(3,4-диаминофенил)-N,N-диметилацетамида (3.0 г, 15.5 ммоль) в этаноле (15 мл) добавляли в течение периода времени в 30 мин. К перемешанному раствору из бромистого циана (1.8 г, 17.0 ммоль) в воде (100 мл). Реакционную смесь перемешивали при КТ в течение 20 ч. Этанол удаляли под сниженным давлением. Полученную водную фазу подщелачивали насыщенным раствором NaHCO3 и экстрагировали при помощи этилацетата. Объединенные органические фазы высушивали над безводным Na2SO4, фильтровали и концентрировали с получением указанного в заголовке соединения в виде твердого вещества коричневого цвета (1.0 г, 45%). 1Н ЯМР (400 Мгц, ДМСО-d6): δ 10.58 (s, 1Н), 6.99-6.95 (m, 1Н), 6.95-6.90 (m, 1Н), 6.70 (d, J=16.3 Гц, 1Н), 6.05 (brs, 2Н), 3.62 (s, 2Н), 2.95 (s, 3Н), 2.81 (s, 3Н). ЖХ/МС: (метод А) 219.2 (М+Н), КТ. 1.5 мин., 97.0% (макс.), 97.0% (220 нм).
Промежуточное соединение 5: 3-[3-(6-Гидроксиметил-пиридин-3-ил)-6-оксо-6Н-пиридазин-1-илметил]-бензойная кислота
Стадия 1: Образование метилового эфира 3-[3-(6-гидроксиметил-пиридин-3-ил)-6-оксо-6Н-пиридазин-1-илметил]-бензойной кислоты
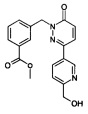
Смесь из метилового эфира 3-(3-хлор-6-оксо-6Н-пиридазин-1-илметил)-бензойной кислоты (0.5 г, 1.79 ммоль) и [5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-2-пиридил]метанола (0.831 г, 3.53 ммоль) в ДМФ/Н2О (9 мл/1 мл) дегазировали под атмосферой N2 в течение 10 мин., Na2CO3 (2.6 мл, 2 М раствор, 5.39 ммоль) добавляли к указанной выше смеси, после чего следовал бис(трифенилфосфин)палладия(II) дихлорид (0.063 г, 0.089 ммоль). Затем реакционную смесь нагревали при 100°C в течение 3 ч., разбавляли с водой и экстрагировали при помощи EtOAc. Объединенные органические слои затем промывали водой, рассолом, высушивали над безводным Na2SO4, фильтровали и концентрировали. Очищение сырого продукта флэш-хроматографией на силикагеле (н-гексан : EtOAc, 80:20) обеспечивало указанное в заголовке соединение в виде твердого вещества желтого цвета (380 мг, 52%). ЖХ/МС: (метод А) 352.0 (М+Н), КТ. 2.4 мин., 94.8% (макс.).
Стадия 2: Образование 3-[3-(6-гидроксиметил-пиридин-3-ил)-6-оксо-6Н-пиридазин-1-илметил]-бензойной кислоты
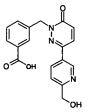
Указанное в заголовке соединение получали, следуя методике, описанной для промежуточного соединения 3 из метилового эфира 3-[3-(6-гидроксиметил-пиридин-3-ил)-6-оксо-6Н-пиридазин-1-илметил]-бензойной кислоты в виде твердого вещества не совсем белого цвета (230 мг, 63%). ЖХ/МС: (метод А) 338.2 (М+Н), КТ. 1.9 мин., 95.1% (макс.), 93.4% (254 нм).
Промежуточное соединение 6: N-(1Н-Бензимидазол-2-ил)-3-(3-хлор-6-оксо-6Н-пиридазин-1-илметил)-бензамид
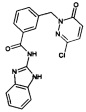
Раствор из 1Н-бензимидазол-2-иламина (0.55 г, 4.17 ммоль), 3-(3-хлор-6-оксо-6Н-пиридазин-1-илметил)-бензойной кислоты (0.85 г, 3.21 ммоль), N-метилморфолина (0.4 мл, 3.38 ммоль), 1-гидроксибензотриазола (47 мг, 3.53 ммоль) и HBTU (1.4 г, 3.69 ммоль) в ДМФ (5 мл) перемешивали при КТ в течение 12 ч. Реакционную смесь затем гасили водой и концентрировали под сниженным давлением. Сырой продукт очищали флэш-хроматографией на силикагеле с получением указанного в заголовке соединения в виде твердого вещества желтого цвета (0.7 г, 58%). 1Н ЯМР (400 Мгц, ДМСО-d6): δ 12.31 (s, 2Н), 8.07 (dd, J1=2.0 Гц, 9.8 Гц, 1Н), 8.03 (s, 1Н), 7.61 (d, J=9.8 Гц, 1Н), 7.51 (d, J=6.7 Гц, 2Н), 7.44-7.42 (m, 2Н), 7.14-7.11 (m, 3Н), 5.28 (s, 2Н). ЖХ/МС: (метод А) 380.0 (М+Н), КТ. 3.0 мин., 98.8% (макс.).
Промежуточное соединение 7: метиловый эфир 3-(3-хлор-4-метил-6-оксо-6Н-пиридазин-1-илметил)-бензойной кислоты
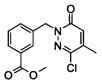
Смесь из метилового эфира 3-бромметил-бензойной кислоты (3.0 г, 13.1 ммоль), 6-хлор-5-метил-2Н-пиридазин-3-она (1.9 г, 13.1 ммоль) и карбоната цезия (4.25 г, 13.1 ммоль) в N-метилпирролидине (15 мл) перемешивали при КТ в течение 14 ч. Реакционную смесь затем выливали на лед и экстрагировали при помощи ДХМ (3 раза). Объединенные органические фазы промывали рассолом, высушивали над Na2SO4 фильтровали и концентрировали. Очищение колоночной флэш-хроматографией на силикагеле обеспечивало указанное в заголовке соединение в виде твердого вещества коричневого цвета (1.5 г, 39%). ЖХ/МС: (метод А) 293.0 (М+Н), КТ. 4.4 мин., 90.1% (макс.). 1Н ЯМР (400 Мгц, ДМСО-d6): δ 7.88 (dd, J=1.2, 7.7 Гц, 2Н), 7.57 (t, J=6.4 Гц, 1H), 7.50 (t, J=7.7 Гц, 1H), 7.04 (d, J=1.2 Гц, 1H), 5.25 (s, 2H), 3.84 (s, 3H), 2.19 (s, 3H).
Промежуточное соединение 8: 3-(4-Метил-6-оксо-3-пиридин-3-ил-6Н-пиридазин-1-илметил)-бензоат лития
Стадия 1: Образование метилового эфира 3-(4-метил-6-оксо-3-пиридин-3-ил-6Н-пиридазин-1-илметил)-бензойной кислоты
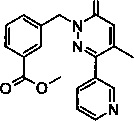
Смесь из метилового эфира 3-(3-хлор-4-метил-6-оксо-6Н-пиридазин-1-илметил)-бензойной кислоты (1.5 г, 5.13 ммоль) и пиридин-3-бороновой кислоты (0.94 г, 7.7 ммоль) в ДМФ/Н2О (9:1; 30 мл) дегазировали под атмосферой N2 в течение 10 мин., Na2CO3 (7.7 мл, 15.4 ммоль) добавляли к указанной выше смеси, после чего следовал бис(трифенилфосфин)палладия(II) дихлорид (0.18 г, 0.25 ммоль). Реакционную смесь затем нагревали при 100°C в течение 4 ч. и фильтровали через прокладку из целита. Целитную прокладку промывали при помощи дихлорметан/метанола, и фильтрат концентрировали под сниженным давлением. Очищение сырого продукта флэш-хроматографией на силикагеле обеспечивало указанное в заголовке соединение в виде твердого вещества коричневого цвета (1 г, 59%). 1Н ЯМР (400 Мгц, ДМСО-d6): δ 8.69-8.64 (m, 2Н), 7.98-7.95 (m, 2Н), 7.95-7.86 (m, 1Н), 7.62-7.60 (m, 1H), 7.52-7.48 (m, 2H), 6.98 (d, J=1.2 Гц, 1H), 5.35 (s, 2H), 3.84 (s, 3H), 2.15 (s, 3Н). ЖХ/МС: (метод A) 336.2 (М+Н), KT. 2.5 мин., 89.5% (макс.), 89.1% (254 нм).
Стадия 2: Образование 3-(4-Метил-6-оксо-3-пиридин-3-ил-6Н-пиридазин-1-илметил)-бензоата лития
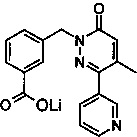
Указанное в заголовке соединение получали, следуя методике, описанной для промежуточного соединения 1, стадия 2 из метилового эфира 3-(4-метил-6-оксо-3-пиридин-3-ил-6Н-пиридазин-1-илметил)-бензойной кислоты в виде твердого вещества коричневого цвета (0.5 г, 65%). 1H ЯМР (400 Мгц, ДМСО-d6): δ 8.69-8.64 (m, 2Н), 7.98-7.95 (m, 1H), 7.80-7.74 (m, 2H), 7.51-7.47 (m, 1H), 7.24-7.18 (m, 2H), 6.97 (d, J=1.2 Гц, 1H), 5.27 (s, 2H), 2.16 (s, 3Н). ЖХ/МС: (метод A) 322.2 (M+H), KT. 2.0 мин., 88.7% (макс.), 90.2% (254 нм).
Промежуточное соединение 9: Диметил-[5-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-пиридин-2-илметил]-амин
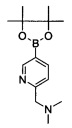
Смесь из (5-бром-пиридин-2-илметил)-диметил-амина (приобретено у rare Chemicals, 3 г, 13.94 ммоль) и бис(пинаколато)диборона (3.9 г, 15.34 ммоль) в диоксане (40 мл) дегазировали под атмосферой N2 в течение 10 мин., к указанной выше смеси добавляли ацетат калия (2.8 г, 27.89 ммоль), после чего следовал 1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий(II). CH2Cl2 (0.50 г, 0.69 ммоль). Реакционную смесь затем нагревали при 60°C в течение 14 ч. и фильтровали через прокладку из целита. Целитную прокладку промывали при помощи дихлорметан/метанола, и фильтрат концентрировали под сниженным давлением с получением указанного в заголовке соединения в виде коричневой смолы (1.5 г, 41%). ЖХ/МС: (метод А) 181.0 (М-82, бороновой кислоты), КТ. 0.42 мин., 97.09% (макс.).
Пример 1: 2-[3-(6-оксо-3-пиридин-3-ил-6Н-пиридазин-1-илметил)-бензоиламино]-1Н-бензимидазол-5-иловый эфир 2,2-диметил-пропионовой кислоты
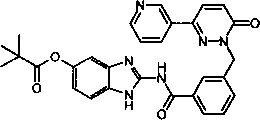
Раствор из 2-амино-1Н-бензимидазол-5-илового эфира 2,2-диметил-пропионовой кислоты (приобретен у Ambinter Stock Screening Collection, 0.15 г, 0.6 ммоль), 3-(6-оксо-3-пиридин-3-ил-6Н-пиридазин-1-илметил)бензоата лития (0.1 г, 0.3 ммоль), N-метилморфолина (0.1 мл, 0.9 ммоль), 1-гидрокси бензотриазола (80 мг, 0.6 ммоль) и HBTU (200 мг, 0.6 ммоль) в ДМФ (5 мл) перемешивали при комнатной температуре в течение 12 ч. Реакционную смесь затем разбавляли 10%-ым раствором двууглекислого натрия (15 мл) и экстрагировали при помощи дихлорметана (3×10 мл). Объединенные органические фазы промывали 10%-ым раствором лимонной кислоты, высушивали над безводным Na2SO4, фильтровали и концентрировали. Полученное твердое вещество перемешивали с метанолом (5 мл), фильтровали и высушивали под вакуумом с получением указанного в заголовке соединения в виде твердого вещества коричневого цвета (29 мг, 10%). 1HNMR (400 Мгц, ДМСО-d6): δ 12.37 (brs, 2Н), 9.12 (t, J=1.6 Гц, 1Н), 8.65 (dd, J=4.8, 1.6 Гц, 1Н), 8.28 (dt, J=4.9, 2.3 Гц, 1Н), 8.17-8.12 (m, 2Н), 8.07 (d, J=7.8 Гц, 1Н), 7.62 (d, J=7.6 Гц, 1Н), 7.54-7.49 (m, 2Н), 7.42 (d, J=8.4 Гц, 1Н), 7.18-7.13 (m, 2Н), 6.81 (dd, J=8.6, 1.9 Гц, 1Н), 5.44 (s, 2Н), 1.31 (s, 9Н). ЖХ/МС: (метод А) 523.3 (М+Н), КТ. 3.2 min, 91.9% (макс.), 90.3% (254 нм). ВЭЖХ: (метод А) КТ 3.5 мин., 92.1% (макс.), 93.1% (254 нм).
Пример 2: N-(1Н-Бензимидазол-2-ил)-3-(6-оксо-3-пиридин-3-ил-6Н-пиридазин-1-илметил)-бензамид
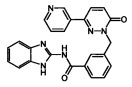
Указанное в заголовке соединение получали, следуя методике, описанной для примера 1 из 3-(6-оксо-3-пиридин-3-ил-6Н-пиридазин-1-илметил)-бензоата лития и 1Н-бензимидазол-2-амина в виде твердого вещества коричневого цвета (135 мг, 12%). 1HNMR (400 Мгц, ДМСО-d6): δ 12.39 (brs, 2Н), 9.12 (t, J=1.7 Гц, 1Н), 8.65 (dd, J=4.76, 1.6 Гц, 1H), 8.29-8.26 (m, 1H), 8.17-8.14 (m, 2H), 8.07 (d, J=7.8 Гц, 1H), 7.60 (d, J=7.5 Гц, 1H), 7.54-7.50 (m, 2H), 7.44-7.42 (m, 2H), 7.15-7.11 (m, 3H), 5.44 (s, 2H). ЖХ/МС: (метод A) 423.0 (M+H), KT. 2.4 мин., 97.8% (макс.), 98.3% (254 нм). ВЭЖХ: (метод А) КТ 2.4 мин., 98.3% (макс.), 97.8% (254 нм).
Пример 3: N-(1-Метил-1Н-бензимидазол-2-ил)-3-(6-оксо-3-пиридин-3-ил-6Н-пиридазин-1-илметил)-бензамид
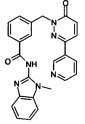
Указанное в заголовке соединение получали, следуя методике, описанной для примера 1 из 3-(6-оксо-3-пиридин-3-ил-6Н-пиридазин-1-илметил)-бензоата лития и 1-метилбензимидазол-2-амина в виде твердого вещества не совсем белого цвета (11 мг, 18%). 1HNMR (400 Мгц, ДМСО-d6): δ 12.70 (s, 1Н), 9.13 (d, J=1.8 Hz, 1Н), 8.65 (dd, J=4.7, 1.5 Гц, 1H), 8.31-8.26 (m, 2H), 8.18-8.16 (m, 2H), 7.55-7.43 (m, 5H), 7.27-7.20 (m, 3H), 5.44 (s, 2H), 3.64 (s, 3Н). ЖХ/МС: (метод A) 437.2 (M+H), KT. 2.5 мин., 96.3% (макс.), 96.7% (254 нм). ВЭЖХ: (метод А) КТ 2.4 мин., 96.1% (макс.), 95.6% (254 нм).
Пример 4: метиловый эфир 2-[3-(6-оксо-3-пиридин-3-ил-6Н-пиридазин-1-илметил)-бензоиламино]-1Н-бензимидазол-5-карбоновой кислоты
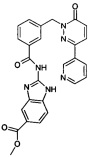
Указанное в заголовке соединение получали, следуя методике, описанной для примера 1 из 3-(6-оксо-3-пиридин-3-ил-6Н-пиридазин-1-илметил)-бензоата лития и метил 2-амино-1Н-бензимидазол-5-карбоксилата (приобретен у PharmaCore inc.) в виде твердого вещества коричневого цвета (13 мг, 19%). 1HNMR (400 Мгц, HMCO-d6): δ 12.65 (brs, 1Н), 12.25 (brs, 1H), 9.12 (d, J=1.8 Гц, 1H), 8.65 (dd, J=4.7, 1.3 Гц, 1H), 8.28 (d, J=7.8 Гц, 1H), 8.17 (d, J=9.7 Гц, 1H), 8.12-7.98 (m, 3H), 7.84-7.78 (m, 1H), 7.65 (d, J=6.8 Гц, 1H), 7.55-7.51 (m, 3H), 7.17 (d, J=9.72 Гц, 1H), 5.45 (s, 2H), 3.85 (s, 3Н). ЖХ/МС: (метод A) 481.2 (M+H), KT. 2.6 мин., 94.9% (макс.), 93.7% (254 нм). ВЭЖХ: (метод А) КТ 2.5 мин., 92.6% (макс.), 92.5% (254 нм).
Пример 5: N-(5-Гидроксиметил-1Н-бензимидазол-2-ил)-3-(6-оксо-3-пиридин-3-ил-6Н-пиридазин-1-илметил)-бензамид
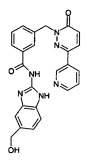
Указанное в заголовке соединение получали, следуя методике, описанной для примера 1 из 3-(6-оксо-3-пиридин-3-ил-6Н-пиридазин-1-илметил)-бензоата лития и (2-амино-1Н-бензимидазол-5-ил)метанола (приобретен у FCH Group Reagents for Synthesis) в виде твердого вещества желтого цвета (6 мг, 11%). 1HNMR (400 Мгц, ДМСО-d6): δ 12.60 (brs, 1Н), 9.12 (s, 1Н), 8.65 (d, J=3.2 Гц, 1Н), 8.28 (d, J=7.2 Гц, 1Н), 8.17-8.14 (m, 2Н), 8.07 (d, J=7.2 Гц, 1Н), 7.61-7.48 (m, 3Н), 7.40 (s, 1Н), 7.36 (d, J=8.0 Гц, 1Н), 7.18-7.15 (m, 2Н), 5.44 (s, 2Н), 5.15 (brs, 1Н), 4.54 (s, 2Н), 2.53 (s, 1Н). ЖХ/МС: (метод А) 453.3 (М+Н), КТ. 2.0 мин., 94.5% (макс.), 96.0% (254 нм). ВЭЖХ: (метод А) КТ 2.0 мин., 98.1% (макс.), 97.3% (254 нм).
Пример 6: N-(5-Диметилкарбамоилметил-1Н-бензимидазол-2-ил)-3-(6-оксо-3-пиридин-3-ил-6Н-пиридазин-1-илметил)-бензамид
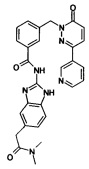
Указанное в заголовке соединение получали, следуя методике, описанной для примера 1 из 3-(6-оксо-3-пиридин-3-ил-6Н-пиридазин-1-илметил)-бензоата лития и 2-(2-амино-1Н-бенз[d]имидазол-5-ил)-N,N-диметилацетамида в виде твердого вещества не совсем белого цвета (49 мг, 19%). 1HNMR (400 Мгц, ДМСО-d6): δ 12.26 (brs, 2Н), 9.11 (d, J=1.8 Гц, 1Н), 8.64 (dd, J=4.7, 1.5 Гц, 1H), 8.28 (td, J=8.1, 1.9, Гц, 1H), 8.17 (s, 1H), 8.15 (s, 1H), 8.07 (d, J=7.8 Гц, 1H), 7.55-7.51 (m, 2H), 7.45 (t, J=7.7 Гц, 1H), 7.30-7.28 (m, 1H), 7.23 (s, 1H), 7.16 (d, J=9.7 Гц, 1H), 6.94-6.92 (m, 1H), 5.42 (s, 2H), 3.71 (s, 2H), 3.0 (s, 3H), 2.8 (s, 3H). ЖХ/МС: (метод A) 508.2 (M+H), KT. 2.2 мин., 97.1% (макс.), 98.9% (254 нм). ВЭЖХ: (метод А) КТ 2.1 мин., 96.5% (макс.), 95.4% (254 нм).
Пример 7: N-(1Н-Бензимидазол-2-ил)-3-[3-(6-гидроксиметил-пиридин-3-ил)-6-оксо-6Н-пиридазин-1-илметил]-бензамид
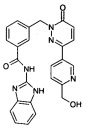
Указанное в заголовке соединение получали, следуя методике, описанной для примера 1 из 3-[3-(6-гидроксиметил-пиридин-3-ил)-6-оксо-6Н-пиридазин-1-илметил]-бензойной кислоты и 1Н-бензимидазол-2-амина в виде твердого вещества не совсем белого цвета (28 мг, 12%). 1HNMR 400 Мгц, ДМСО-d6: δ 12.29 (brs, 2Н), 9.02 (d, J=2.0 Гц, 1Н), 8.28 (dd, J=8.2, 2.2, Гц, 1Н), 8.16 (s, 1Н), 8.14 (s, 1Н), 8.07 (d, J=7.8 Гц, 1Н), 7.61-7.59 (m, 2Н), 7.50 (t, J=7.6 Гц, 1Н), 7.44-7.42 (m, 2Н), 7.16-7.12 (m, 3Н), 5.52 (s, 1Н), 5.43 (s, 2Н), 4.61 (d, J=5.32 Гц, 2Н). ЖХ/МС: (метод А) 453.3 (М+Н), КТ. 2.2 мин., 97.8% (макс.), 98.2% (254 нм). ВЭЖХ: (метод А) КТ 2.4 мин., 98.3% (макс.), 97.8% (254 нм).
Пример 8: N-(1Н-Бензимидазол-2-ил)-3-(6-оксо-3-пиримидин-5-ил-6Н-пиридазин-1-илметил)-бензамид
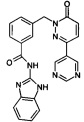
Указанное в заголовке соединение получали, следуя методике, описанной для промежуточного соединения 5, стадия 1 из N-(1Н-бензимидазол-2-ил)-3-(3-хлор-6-оксо-6Н-пиридазин-1-илметил)-бензамида и 5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиримидина в виде твердого вещества не совсем белого цвета (12 мг, 9%). 1HNMR (400 Мгц, ДМСО-d6): δ 12.30 (brs, 2Н), 9.31 (s, 2Н), 9.26 (s, 1Н), 8.22-8.07 (m, 3Н), 7.62 (s, 1Н), 7.49 (t, J=6.6 Гц, 3Н), 7.18 (t, J=9.6 Гц, 3Н), 5.44 (s, 2Н). ЖХ/МС: (метод А) 424.2 (М+Н), КТ. 2.6 мин., 95.6% (макс.), 97.9% (254 нм). ВЭЖХ: (метод А) КТ 2.6 мин., 98.7% (макс.), 98.4% (254 нм).
Пример 9: 3-[3-(6-Амино-пиридин-3-ил)-6-оксо-6Н-пиридазин-1-илметил]-N-(1Н-бензимидазол-2-ил)-бензамид
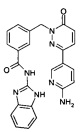
Указанное в заголовке соединение получали, следуя методике, описанной для промежуточного соединения 5, стадия 1 из N-(1Н-Бензимидазол-2-ил)-3-(3-хлор-6-оксо-6Н-пиридазин-1-илметил)-бензамида и 5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-амина в виде твердого вещества желтого цвета (55 мг, 36%). 1HNMR (400 Мгц, ДМСО-d6): δ 12.32 (s, 2Н), 8.47 (d, J=2.3 Гц, 1Н), 8.10 (s, 1Н), 8.06 (d, J=7.8 Гц, 1Н), 8.00 (d, J=9.8 Гц, 1Н), 7.88 (dd, J=8.7, 2.4 Гц, 1Н), 7.56 (d, J=7.5 Гц, 1Н), 7.49 (t, J=7.6 Гц, 1Н), 7.44-7.42 (m, 2Н), 7.14-7.12 (m, 2Н), 7.04 (d, J=9.7 Гц, 1Н), 6.50 (d, J=8.8 Гц, 1Н), 6.39 (s, 2Н), 5.36 (s, 2Н). ЖХ/МС: (метод А) 438.2 (М+Н), КТ. 2.3 мин., 94.9% (макс.), 98.3% (254 нм). ВЭЖХ: (метод А) КТ 2.5 мин., 99.5% (макс.), 99.2% (254 нм).
Пример 10: N-(1Н-бенз[d]имидазол-2-ил)-3-(2-циано-5-(пиридин-3-ил)фенокси)бензамид
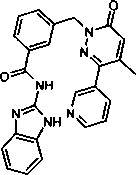
Указанное в заголовке соединение получали, следуя методике, описанной для примера 1 из 3-(4-Метил-6-оксо-3-пиридин-3-ил-6Н-пиридазин-1-илметил)-бензоат лития и 1Н-бензимидазол-2-амина с дополнительной стадией очищения флэш-хроматографией на силикагеле в виде твердого вещества не совсем белого цвета (26 мг, 20%). 1Н ЯМР (400 Мгц, ДМСО-d6): δ 12.32 (s, 2Н), 8.73-8.66 (m, 2Н), 8.11-7.98 (m, 3Н), 7.52 (t, J=9.5 Гц, 5Н), 7.13 (s, 2Н), 6.99 (s, 1Н), 5.36 (s, 2Н), 2.16 (s, 3Н). ЖХ/МС: (метод В) 437.3 (М+Н), КТ. 4.73 мин., 96.41% (макс.), 93.83% (254 нм). ВЭЖХ: (метод А) КТ. 2.37 мин., 94.94% (макс.), 95.06% (254 нм).
Пример 11: N-(1Н-бенз[d]имидазол-2-ил)-3-((3-(6-((диметиламино)метил)пиридин-3-ил)-6-оксопиридазин-1(6Н)-ил)метил)бензамид
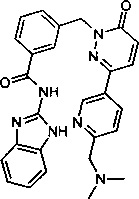
Указанное в заголовке соединение получали, следуя методике, описанной для промежуточного соединения 5, стадия 1 из N-(1Н-бензимидазол-2-ил)-3-(3-хлор-6-оксо-6Н-пиридазин-1-илметил)-бензамида и диметил-[5-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-пиридин-2-илметил]-амина в виде твердого вещества не совсем белого цвета (103 мг, 32%). 1Н ЯМР (400 Мгц, ДМСО-d6): δ 12.29 (s, 2Н), 9.01 (d, J=2.0 Гц, 1Н), 8.26 (dd, J=2.3, 9.1 Гц, 1Н), 8.15 (d, J=9.8 Гц, 2Н), 8.07 (d, J=7.7 Гц, 1H), 7.60 (d, J=7.6 Гц, 1Н), 7.54-7.47 (m, 2Н), 7.44-7.41 (m, 2Н), 7.16-7.11 (m, 3Н), 5.42 (s, 2Н), 3.56 (s, 2Н), 2.19 (s, 6Н). ЖХ/МС: (метод А) 437.3 (М+Н), КТ. 2.34 мин., 98.15% (макс.), 98.58% (254 нм). ВЭЖХ: (метод А) КТ. 2.38 мин., 98.34% (макс.), 98.69% (254 нм).
Пример 12: N-(1Н-бенз[d]имидазол-2-ил)-3-((3-(4-метилпиридин-3-ил)-6-оксопиридазин-1(6Н)-ил)метил)бензамид
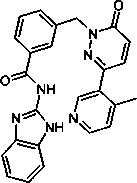
Указанное в заголовке соединение получали, следуя методике, описанной для промежуточного соединения 5, стадия 1 из N-(1Н-бензимидазол-2-ил)-3-(3-хлор-6-оксо-6Н-пиридазин-1-илметил)-бензамида и 4-метил-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридина (приобретен у Boron Molecular) в виде твердого вещества бежевого цвета (12 мг, 5%). 1Н ЯМР (400 Мгц, ДМСО-d6): δ 12.35 (s, 2Н), 8.59 (s, 1Н), 8.49 (d, J=5.0 Гц, 1H), 8.12 (s, 1H), 8.08 (d, J=7.6 Гц, 1H), 7.80 (d, J=9.6 Гц, 1H), 7.58 (d, J=7.8 Гц, 1H), 7.51 (t, J=7.9 Гц, 1H), 7.44 (t, J=1.9 Гц, 2H), 7.35 (d, J=5.0 Гц, 1H), 7.14 (dd, J=1.9, 4.9 Гц, 3Н), 5.40 (s, 2H), 2.31 (s, 3Н). ЖХ/МС: (метод A) 480.3 (M+H), KT. 2.32 мин., 98.90% (макс.), 99.16% (254 нм). ВЭЖХ: (метод А) КТ. 2.41 мин., 96.97% (макс.), 97.22% (254 нм).
Пример 13: N-(1Н-бенз[d]имидазол-2-ил)-3-((3-(6-метилпиридазин-4-ил)-6-оксопиридазин-1(6Н)-ил)метил)бензамид
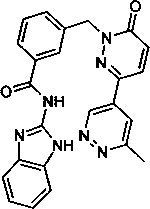
Указанное в заголовке соединение получали, следуя методике, описанной для промежуточного соединения 5, стадия 1 из N-(1Н-Бензимидазол-2-ил)-3-(3-хлор-6-оксо-6Н-пиридазин-1-илметил)-бензамида и 3-метил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридазина (приобретен у Combi-Blocks) в виде твердого вещества белого цвета (35 мг, 12%). 1Н ЯМР (400 Мгц, ДМСО-d6): δ 12.30 (s, 2Н), 9.58 (d, J=1.9 Гц, 1Н), 8.24 (d, J=9.7 Гц, 1Н), 8.14 (brs, 1Н), 8.08 (d, J=7.7 Гц, 1Н), 8.04 (d, J=1.9 Гц, 1Н), 7.63 (d, J=7.6 Гц, 1Н), 7.52 (t, J=7.6 Гц, 1Н), 7.44 (m, 2H), 7.23 (d, J=9.8 Гц, 1H), 7.13-7.11 (m, 2H), 5.46 (s, 2H), 2.68 (s, 3Н). ЖХ/МС: (метод A) 438.3 (M+H), KT. 2.45 мин., 96.32% (макс.), 93.47% (254 нм). ВЭЖХ: (метод А) КТ. 2.49 мин., 94.55% (макс.), 94.92% (254 нм).
Пример 14: N-(1Н-бенз[d]имидазол-2-ил)-3-((3-(5-((диметиламино)метил)пиридин-3-ил)-6-оксопиридазин-1(6Н)-ил)метил)бензамид
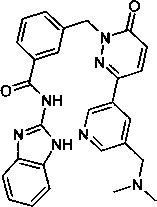
Указанное в заголовке соединение получали, следуя методике, описанной для промежуточного соединения 5, стадия 1 из N-(1Н-бензимидазол-2-ил)-3-(3-хлор-6-оксо-6Н-пиридазин-1-илметил)-бензамида и диметил-[5-(4,4,5,5-тетраметил-[1,3,21диоксаборолан-2-ил)-пиридин-3-илметил]-амина (приобретен у Small Molecules, inc.) в виде твердого вещества белого цвета (94 мг, 32%). 1Н ЯМР (400 Мгц, ДМСО-d6): δ 12.30 (s, 2Н), 9.01 (d, J=1.8 Гц, 1Н), 8.54 (s, 1Н), 8.18-8.12 (m, 3Н), 8.07 (d, J=7.7 Гц, 1Н), 7.58 (d, J=7.5 Гц, 1Н), 7.51 (t, J=7.6 Гц, 1Н), 7.44 (m, 2Н), 7.16-7.11 (m, 3Н), 5.44 (s, 2Н), 3.47 (s, 2Н), 2.13 (s, 6Н). ЖХ/МС: (метод А) 480.2 (М+Н), КТ. 2.31 мин., 98.46% (макс.), 99.18% (254 нм). ВЭЖХ: (метод А) КТ. 2.41 мин., 99.04% (макс.), 98.82% (254 нм).
Пример 15: N-(5,6-диметокси-1Н-бенз[d]имидазол-2-ил)-3-((6-оксо-3-(пиридин-3-ил)пиридазин-1(6Н)-ил)метил)бензамид
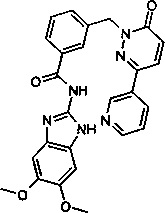
Указанное в заголовке соединение получали, следуя методике, описанной для промежуточного соединения 6 из 5,6-Диметокси-1Н-бензимидазол-2-иламин (приобретен у Enamine) и 3-(6-оксо-3-пиридин-3-ил-6Н-пиридазин-1-илметил)бензоата лития в виде твердого вещества желтого цвета (46 мг, 8%). 1Н ЯМР (400 Мгц, ДМСО-d6): δ 12.16 (s, 2Н), 9.12 (d, J=1.7 Гц, 1Н), 8.65 (d, J=6.3 Гц, 1Н), 8.29-8.26 (m, 1Н), 8.17 (d, J=9.8 Гц, 1Н), 8.11 (s, 1Н), 8.05 (d, J=7.8 Гц, 1Н), 7.60 (d, J=6.7 Гц, 1Н), 7.54-7.46 (m, 2Н), 7.17 (d, J=9.7 Гц, 1Н), 7.03 (s, 2Н), 5.43 (s, 2Н), 3.75 (s, 6Н). ЖХ/МС: (метод А) 483.3 (М+Н), КТ. 2.34 мин., 96.83% (макс.), 96.63% (254 нм). ВЭЖХ: (метод А) КТ. 2.37 мин., 96.48% (макс.), 97.91% (254 нм).
Пример 16: N-(5-метокси-1Н-бенз[d]имидазол-2-ил)-3-((6-оксо-3-(пиридин-3-ил)пиридазин-1(6Н)-ил)метил)бензамид
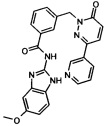
Указанное в заголовке соединение получали, следуя методике, описанной для промежуточного соединения 6 из 5-метокси-1Н-бензимидазол-2-иламин (приобретен у Anichem) и 3-(6-оксо-3-пиридин-3-ил-6Н-пиридазин-1-илметил)бензоата лития в виде твердого вещества желтого цвета (101 мг, 11%). 1Н ЯМР (400 Мгц, HMCO-d6): δ 12.20 (s, 2Н), 9.12 (s, 1Н), 8.65 (d, J=4.5 Гц, 1H), 8.28 (d, J=8.0 Гц, 1H), 8.16 (t, J=9.8 Гц, 2H), 8.06 (d, J=7.7 Гц, 1H), 7.60 (d, J=7.5 Гц, 1H), 7.54-7.47 (m, 2H), 7.32 (d, J=8.6 Гц, 1H), 7.17 (d, J=9.8 Гц, 1H), 6.98 (d, J=1.5 Гц, 1H), 6.76 (m, 1H), 5.43 (s, 2H), 3.75 (s, 3Н). ЖХ/МС: (метод A) 453.3 (M+H), KT. 2.44 мин., 97.51% (макс.), 99.01% (254 нм). ВЭЖХ: (метод А) КТ. 2.45 мин., 99.09% (макс.), 98.82% (254 нм).
Пример 17: N-(1Н-Бензимидазол-2-ил)-3-[3-(5-гидроксиметил-пиридин-3-ил)-6-оксо-6Н-пиридазин-1-илметил]-бензамид
Указанное в заголовке соединение получали следуя процедуре, описанной для примера 1 из 3-[3-(5-гидроксиметил-пиридин-3-ил)-6-оксо-6Н-пиридазин-1-илметил]-бензоата лития и 1Н-бензимидазол-2-амина в виде твердого вещества не совсем белого цвета (119 мг, 41%). 1HNMR (400 Мгц, ДМСО-d6): δ 12.30 (brs, 2Н), 8.99 (d, J=2.1 Гц, 1Н), 8.59 (d, J=1.8 Гц, 1Н), 8.20 (s, 1Н), 8.16 (d, J=9.8 Гц, 1Н), 8.11 (s, 1Н), 8.08 (d, J=7.8 Гц, 1Н), 7.58 (d, J=7.6 Гц, 1Н), 7.44-7.51 (m, 2Н), 7.42 (t, J=3.2 Гц, 1Н), 7.17-7.12 (m, 2Н), 7.11 (s, 1Н), 5.43 (d, J=6. Гц, 2Н), 4.60 (d, J=5.2 Гц, 3Н). ЖХ/МС: (метод А) 453.0 (М+Н), КТ. 2.2 мин., 98.7% (макс.), 99.5% (254 нм). ВЭЖХ: (метод А) КТ 2.2 мин., 98.2% (макс.), 97.6% (254 нм).
Пример 18: ферментативные анализы IRAK1 и IRAK4
Ферментативный анализ IRAK1:
IRAK1 является очищенным рекомбинантным ферментом человека (His-TEV-IRAK1 (194-712))
В этом анализе IRAK-1 гидролизует АТФ и аутофосфорилирует.
Измерения ингибирования IRAK-1 осуществляют на покрытом стрептавидином 384-луночном планшете FlashPlate (PerkinElmer #SMP410A).
His-TEV-IRAK-1 (15 нг/лунку), АТФ (1 мкМ, [33Р] АТФ 0.25 мкСi/лунку) и соединения в ДМСО (диапазон концентраций от 20 мкМ до 1 нМ) или контрольные образцы (2% ДМСО) инкубировали в течение 3 часов при 30°C в буфере для анализа: Hepes рН 7.0 50 мМ, не содержащий жирных кислот БСА 0,1%, дитиотреитол DTT 2 мМ, MgCl2, 10 мМ, EGTA 0,5 мМ, Triton-X-100 0,01%. Киназную реакцию останавливали путем добавления EDTA. Супернатант отбрасывали, планшеты промывали три раза с помощью 150 мМ NaCl и затем измеряли радиоактивность в считывающем устройстве Microbeta Trilux.
Ферментативный анализ IRAK4:
IRAK4 является очищенным рекомбинантным ферментом человека (His-TEV-IRAK1 (194-712)
IRAK4 гидролизует АТФ, аутофосфорилирует и фосфорилирует генерический пептидный субстрат серин/треонин (STK: 61ST1BLC от CisBio International находящийся в  ).
).
Измерения ингибирования IRAK-4 осуществляют на покрытом стрептавидином 384-луночном планшете FlashPlate (PerkinElmer #SMP410A). His-TEV-IRAK4 (20 нг/лунку), АТФ (2 мкМ, [33Р] АТФ 0.25 мкСi/лунку), STK1-биотин пептид (300 нМ) и соединения в ДМСО (диапазон концентраций от 20 мкМ до 1 нМ) или контрольные образцы (2% ДМСО) инкубировали в течение 3 часов при 30°C в буфере для анализа: Hepes рН7.0 50 мМ, не содержащий жирных кислот БСА 0.1%, дитиотреитол DTT 2 мМ, MgCl2 10 мМ, EGTA 0.5 мМ, Tween-20 0.01%, MnCl2 5 мМ.
Киназную реакцию останавливали добавлением EDTA. Супернатант отбрасывали, планшеты промывали три раза с помощью 150 мМ NaCl и затем измеряли радиоактивность в считывающем устройстве Microbeta Trilux.
Пример 19: Получение фармацевтического состава
Состав 1 - Таблетки
Соединение формулы (I) смешивают в виде сухого порошка с сухим желатиновым связующим веществом в весовом соотношении приблизительно 1:2. В качестве смазывающего вещества добавляют незначительное количество стеарата магния. В таблеточном прессе смеси придают форму таблеток массой 240-270 мг (80-90 мг действующего соединения в соответствии с изобретением на таблетку).
Состав 2 - Капсулы
Соединение формулы (I) смешивают в виде сухого порошка с крахмальным разбавителем в весовом соотношении приблизительно 1:1. Смесь наполняют в 250 мг капсулы (125 мг действующего соединения в соответствии с изобретением на капсулу).
Состав 3 - Жидкость
Смешивают соединение формулы (I) (1250 мг), сахарозу (1.75 г) и ксантановую камедь (4 мг), пропускают через сито №10 меш U.S., и затем смешивают в воде с заранее подготовленным раствором микрокристаллической целлюлозы и натрий-карбоксиметилцеллюлозы (11:89, 50 мг). Бензоат натрия (10 мг), ароматизатор и краситель разбавляют с водой и добавляют при помешивании. Затем добавляют необходимое количество воды для получения общего объема в 5 мл.
Состав 4 - Таблетки
Соединение формулы (I) смешивают в виде сухого порошка с сухим желатиновым связующим веществом в весовом соотношении приблизительно 1:2. В качестве смазывающего вещества добавляют незначительное количество стеарата магния. В таблеточном прессе смеси придают форму таблеток массой 450-900 мг (150-300 мг действующего соединения в соответствии с изобретением на таблетку).
Состав 5 - Инъекционный раствор
Соединение формулы (I) растворяют в стерильной буферизованной солевой водной среде для инъекций до концентрации приблизительно 5 мг/мл.
| название | год | авторы | номер документа |
|---|---|---|---|
| МАКРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ ПИРИДАЗИНОНА | 2014 |
|
RU2660435C2 |
| КЛАСС БИФУНКЦИОНАЛЬНЫХ ХИМЕРНЫХ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ ДЛЯ НАПРАВЛЕННОГО РАЗРУШЕНИЯ АНДРОГЕННЫХ РЕЦЕПТОРОВ И ИХ ПРИМЕНЕНИЕ | 2020 |
|
RU2825000C2 |
| БЕНЗОКСЕПИНОВЫЕ ИНГИБИТОРЫ PI3 И СПОСОБЫ ПРИМЕНЕНИЯ | 2010 |
|
RU2557658C2 |
| ПРОИЗВОДНЫЕ 4-ГИДРОКСИ-1,2,3,4-ТЕТРАГИДРОНАФТАЛИН-1-ИЛ-МОЧЕВИНЫ И ИХ ПРИМЕНЕНИЕ В ЛЕЧЕНИИ, СРЕДИ ПРОЧЕГО, ЗАБОЛЕВАНИЙ ДЫХАТЕЛЬНОГО ТРАКТА | 2011 |
|
RU2586333C1 |
| ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛА, СОДЕРЖАЩИЕ ИХ КОМПОЗИЦИИ, ИХ ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ | 2004 |
|
RU2346938C2 |
| ИНГИБИТОРЫ КИНАЗЫ | 2012 |
|
RU2623734C9 |
| НОВЫЕ ПРОИЗВОДНЫЕ ПИРРОЛО[3,2-d]ПИРИМИДИН-4-ОНА И ИХ ПРИМЕНЕНИЕ В ТЕРАПИИ | 2005 |
|
RU2577858C2 |
| ТИЕНОПИРИМИДИНДИОНЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ) И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2002 |
|
RU2294937C9 |
| ФЕНИКОЛОВЫЕ ПРОТИВОБАКТЕРИАЛЬНЫЕ СРЕДСТВА | 2013 |
|
RU2593204C2 |
| 1-(2-ИЗОПРОПОКСИЭТИЛ)-2-ТИОКСО-1,2,3,5-ТЕТРАГИДРО-ПИРРОЛО[3,2-d]ПИРИМИДИН-4-ОН И ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ | 2005 |
|
RU2409578C2 |
Настоящее изобретение относится к соединениям формулы I и к их фармацевтически приемлемым солям, в которой R1, Ra, Rb и Z имеют значение, приведенное в п. 1. Изобретение также относится к фармацевтической композиции их содержащей и к их применению в профилактике и лечении заболеваний, связанных с повышенной экспрессией IRAK, таких как воспалительные заболевания, аутоиммунные заболевания, рак, множественный склероз. 4 н. и 8 з.п. ф-лы, 18 пр.
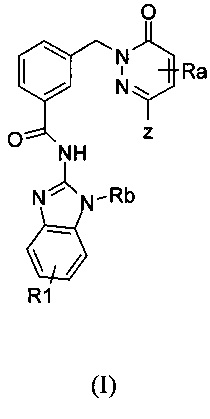
1. Соединение формулы (I)
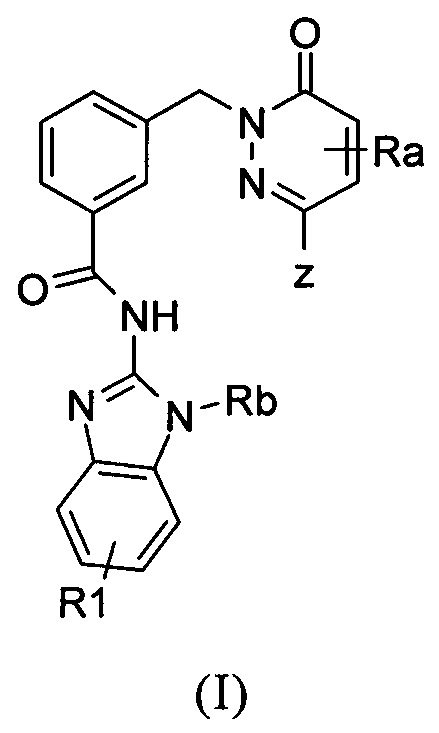
в которой
Z означает группу
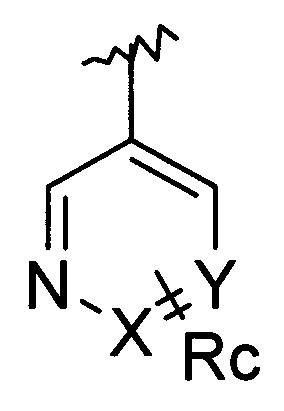
в которой
X означает CH или N,
Y означает CH или N,
причем X и Y одновременно не могут означать N,
Ra означает Н, Hal или разветвленный или линейный алкил, имеющий от 1 до 6 атомов углерода,
Rb означает Н или разветвленный или линейный алкил, имеющий от 1 до 12 атомов углерода,
Rc означает Н, NH2 или разветвленный или линейный алкил, имеющий от 1 до 12 атомов углерода, в котором один или больше, например от 1 до 7, атомов Н могут быть заменены посредством ОН, N(Rb)2 или Hal,
R1 означает Н или разветвленный или линейный алкил, имеющий от 1 до 12 атомов углерода, необязательно замещенный Hal или ORb; ORd, (CH2)nCON(Rb)2 или (CH2)nCOORb, где n означает 0, 1, 2, 3, 4, 5 или 6 и Rb является таким, как определено выше, и где Rd означает Н, алкил, имеющий от 1 до 12 атомов углерода, или CORb, и
Hal означает F, Cl, Br, I
и его фармацевтически приемлемые соли.
2. Соединение формулы (I) по п. 1, где Ra означает Hal или алкил.
3. Соединение формулы (I) по п. 1 или 2, где R1 означает Н, алкил, Оалкил, ORd, или (CH2)nCONHRb, или (CH2)nCOORb, где n означает 0, 1, 2, 3, 4, 5 или 6 и Rb является таким, как определено выше, и где Rd означает Н, алкил, или CORb.
4. Соединение формулы (I) по п. 1 или 2, где Z означает пиридинил или пиримидинил.
5. Соединение формулы (I), выбранное из следующей группы:
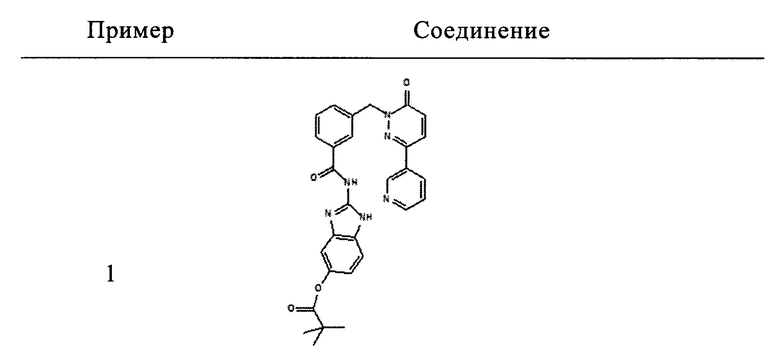
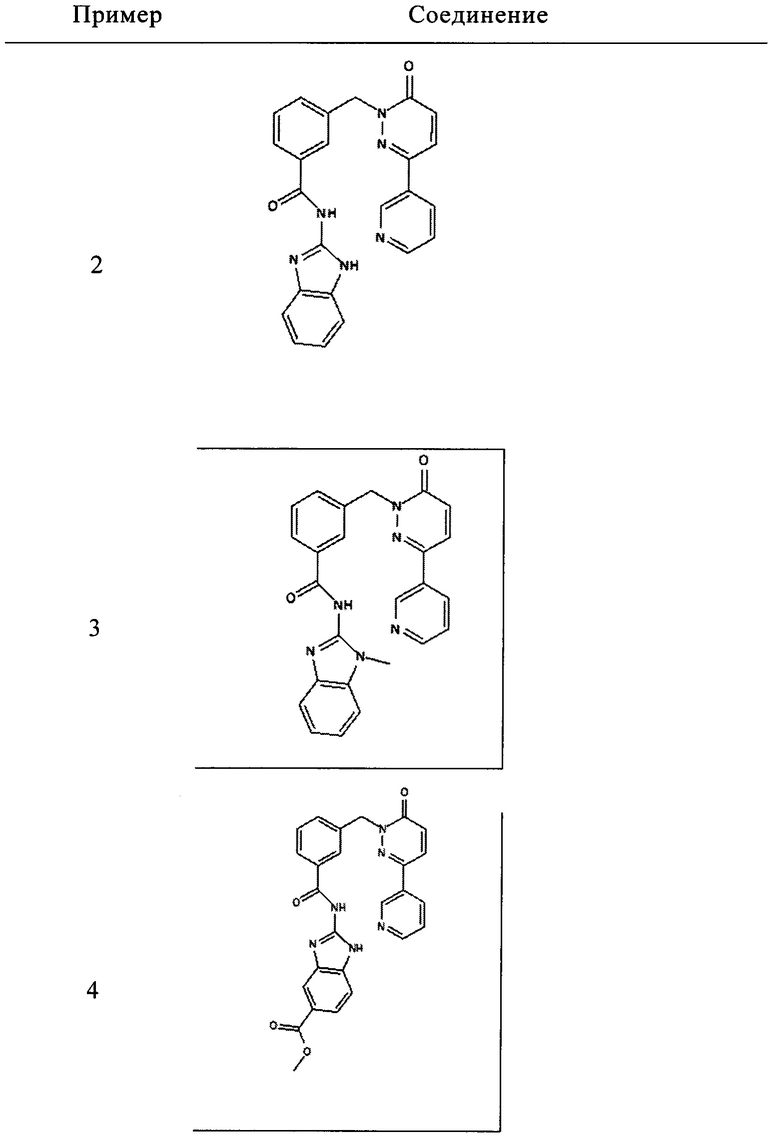
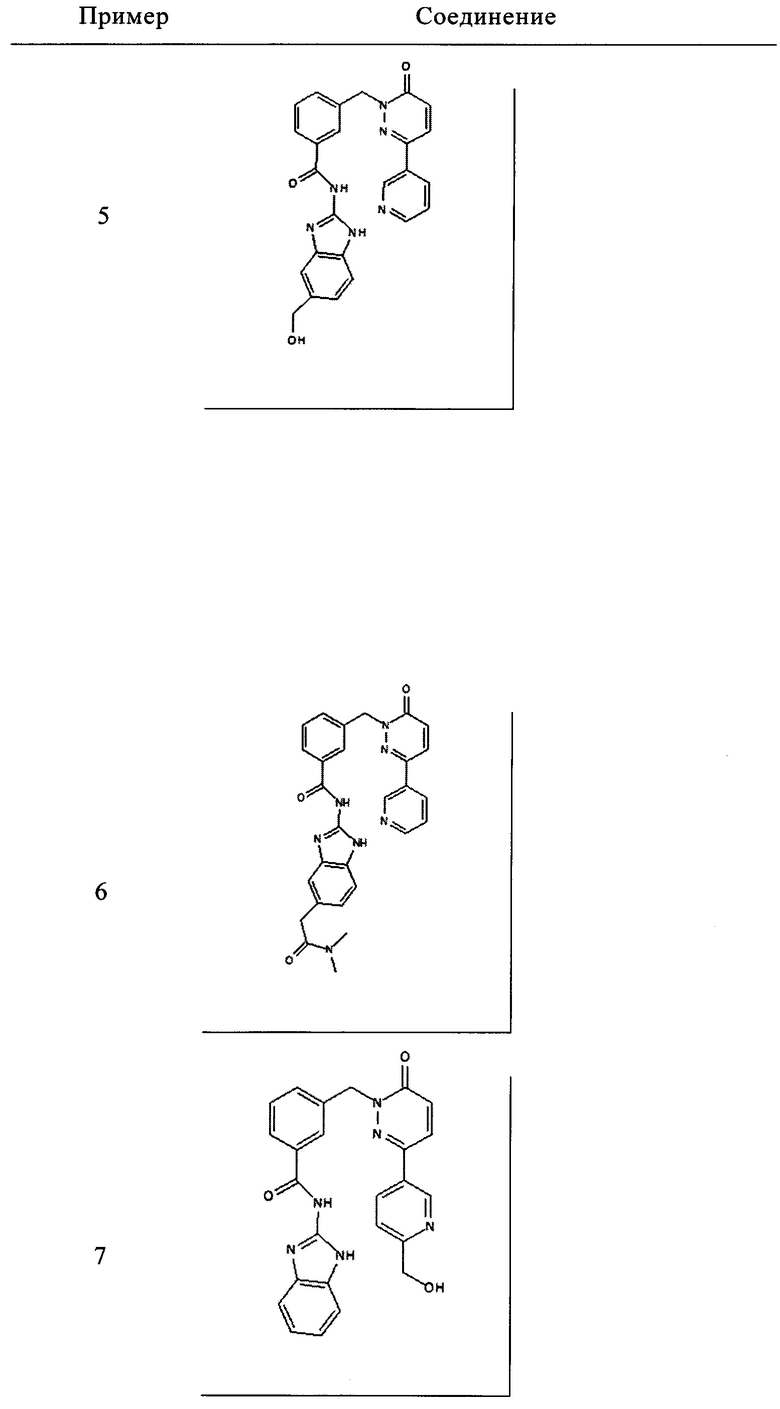
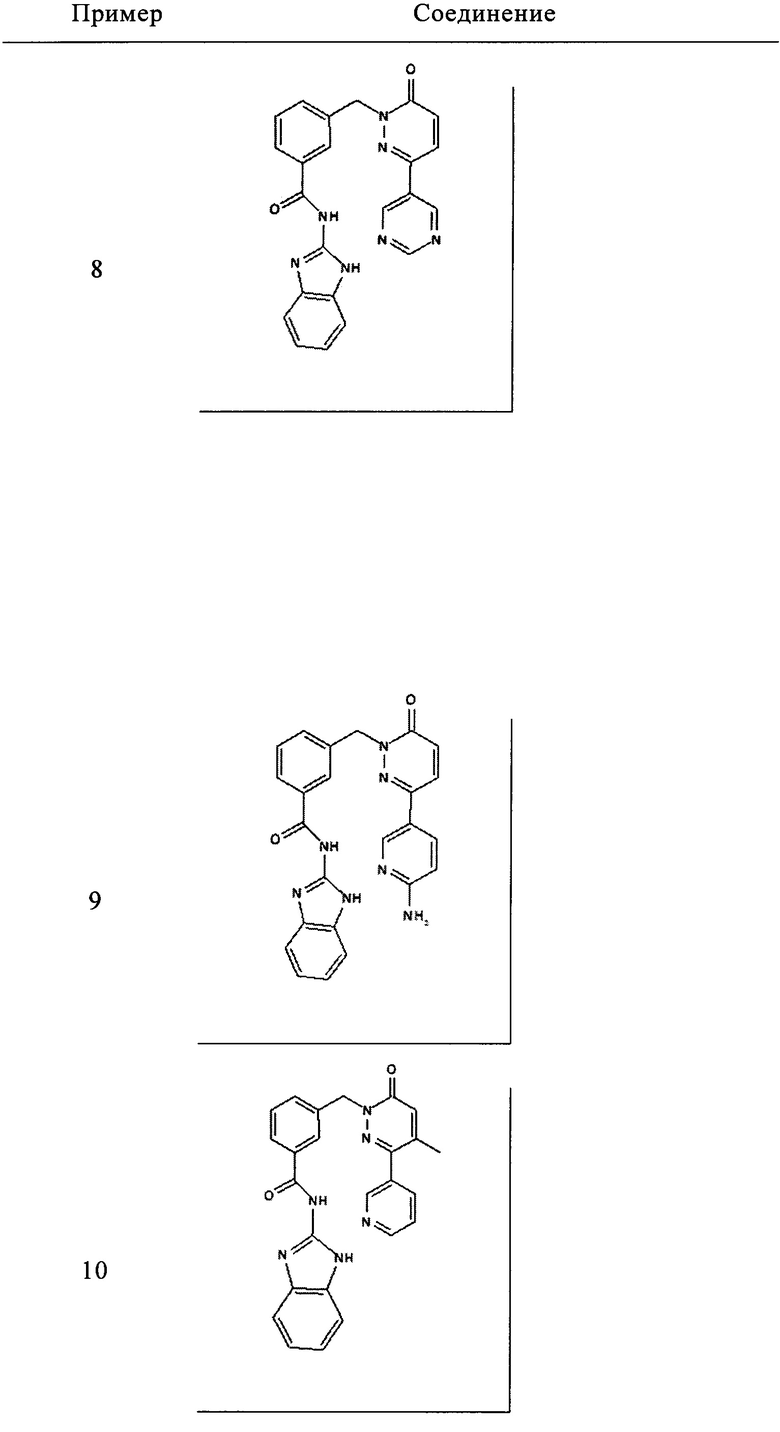
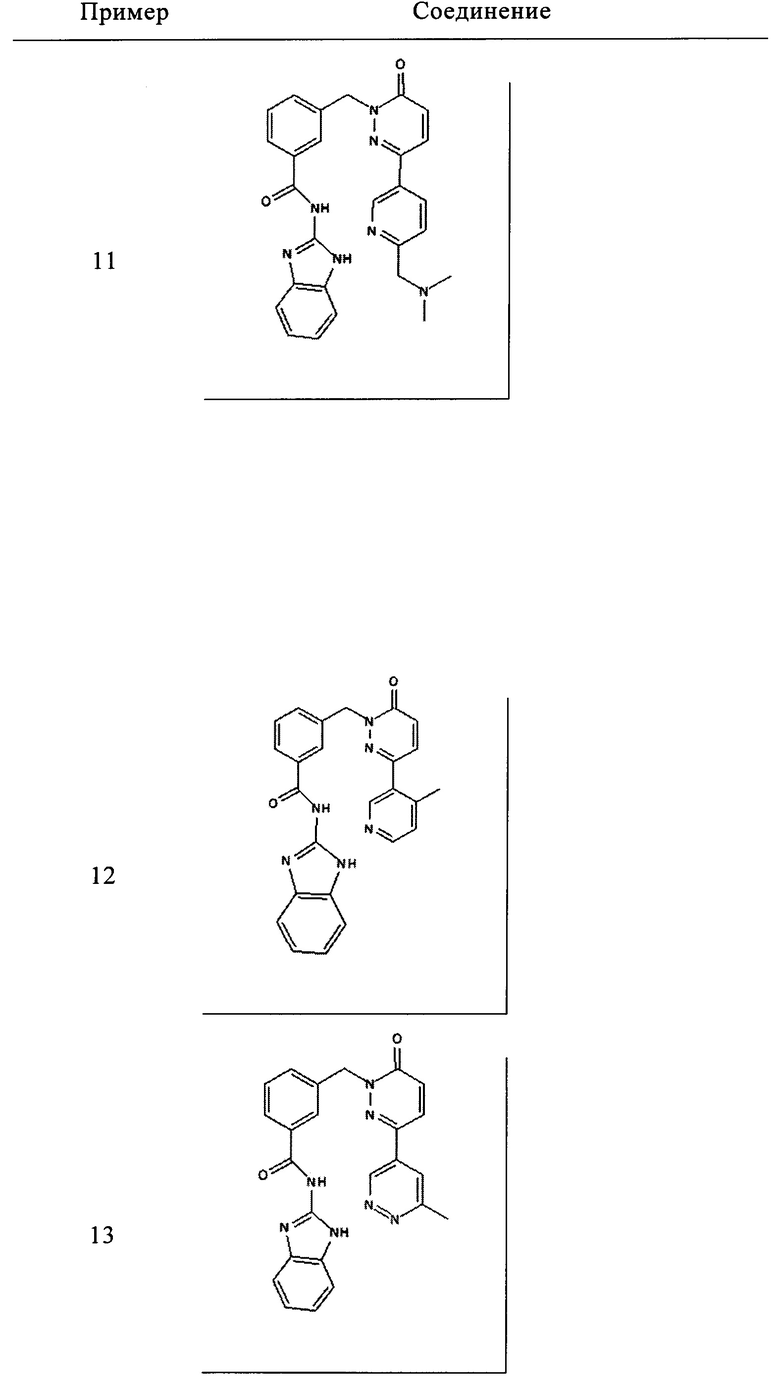
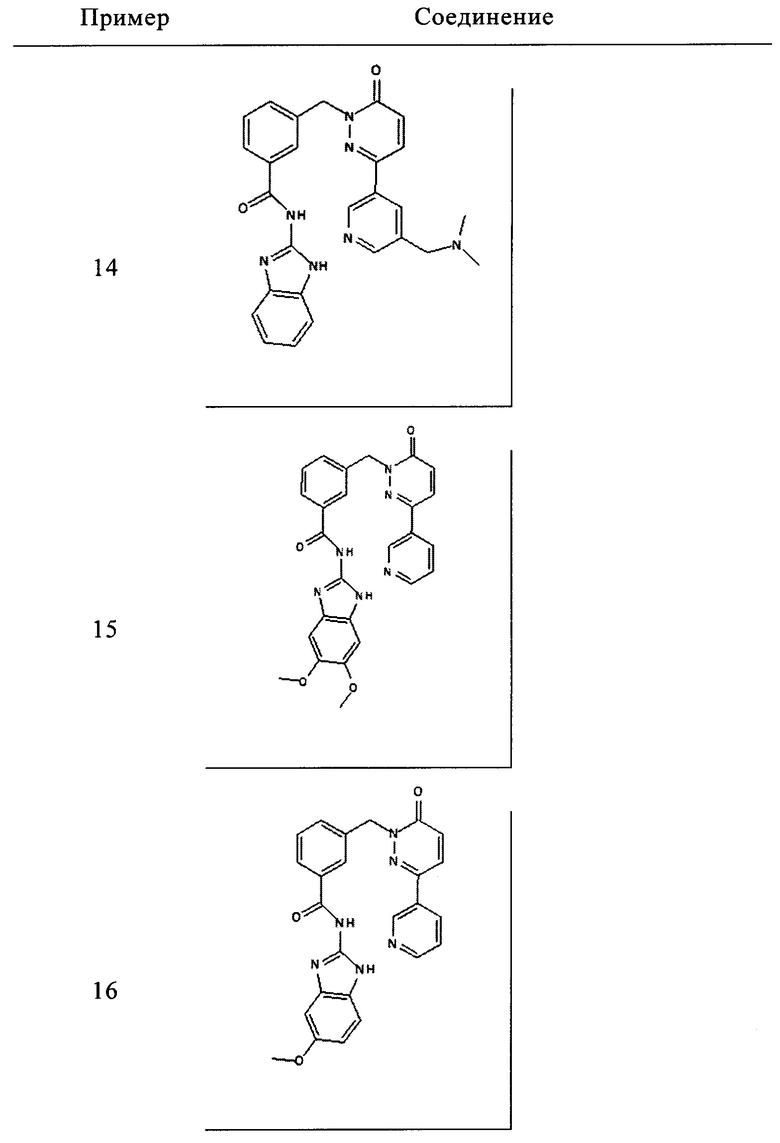
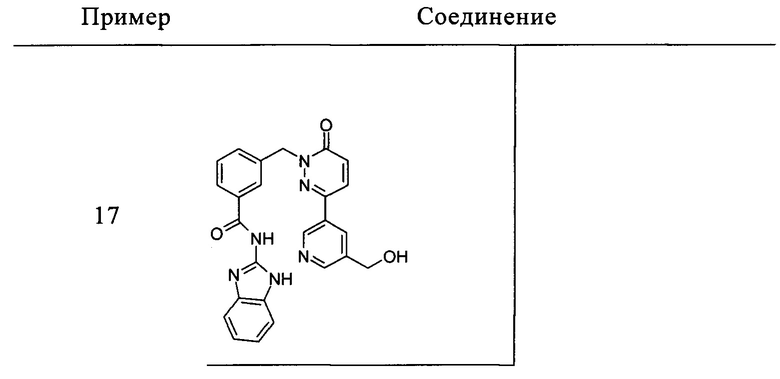
и его фармацевтически приемлемые соли
6. Применение соединения формулы (I) по предыдущим пунктам и его фармацевтически приемлемых солей в качестве лекарственного средства, ингибирующего IRAK.
7. Применение по п. 6, для лечения или предупреждения воспалительного заболевания, аутоиммунного заболевания, рака или множественного склероза и связанных заболеваний.
8. Применение по п. 7, где аутоиммунное заболевание выбирают из группы, состоящей из астмы, ревматоидного артрита, острого множественного энцефаломиелита (ОМЭМ), болезни Аддисона, очаговой алопеции, анкилозирующего спондилита, синдрома антифосфолипидных антител (АФС), аутоиммунной гемолитической анемии, аутоиммунного гепатита, аутоиммунного заболевания внутреннего уха, буллезного пемфигоида, болезни Бехчета, целиакии, антитрансглутаминазы, болезни Чагаса, хронического обструктивного заболевания легких, болезни Крона, дерматомиозита, сахарного диабета 1 типа, эндометриоза, синдрома Гудпасчера, болезни Грейвса, синдрома Гийена-Барре (СГБ), тиреоидита Хашимото, гнойного гидраденита, синдрома Кавасаки, нефропатии IgA, идиопатической тромбоцитопенической пурпуры, интерстициального цистита, эритематозной волчанки, смешанной болезни соединительной ткани, кольцевидной склеродермии, множественного склероза (МС), тяжелой миастении, нарколепсии, нейромиотомии, обыкновенной пузырчатки, злокачественной анемии, псориаза, псориатического артрита, полимиозита, билиарного первичного цирроза печени, ревматоидного артрита, шизофрении, склеродермии, синдрома Шегрена, синдрома скованного человека, системного склероза, височного артериита, язвенного колита, васкулита, витилиго, гранулематоза Вегенера.
9. Применение по п. 7, где заболевание выбирают из ревматоидного артрита, псориатического артрита, остеоартрита, системной красной волчанки, волчаночного нефрита, анкилозирующего спондилоартрита, остеопороза, системного склероза, множественного склероза, псориаза, сахарного диабета I типа, сахарного диабета II типа, воспалительного заболевания кишечника (болезнь Крона и язвенный колит), гипериммуноглобулинемии D и синдрома периодической лихорадки, криопирин-связанных периодических синдромов, синдрома Шницлера, системного ювенильного идиопатического артрита, приобретенной болезни Стилла, подагры, псевдоподагры, синдрома САПГО, болезни Кастлемана, сепсиса, инсульта, атеросклероза, целиакии, DIRA (дефицит антагониста рецептора IL-1), болезни Альцгеймера, болезни Паркинсона, рака.
10. Применение по п. 6, где заболевание выбирают из ревматоидного артрита, волчаночного нефрита, системной красной волчанки.
11. Соединение формулы (I) по п. 1 для предупреждения и/или лечения заболеваний, связанных с повышенной экспрессией IRAK.
12. Фармацевтическая композиция, ингибирующая IRAK, содержащая по меньшей мере одно из соединений формулы (I) по любому из пп. 1-5 и/или любую его фармацевтически приемлемую соль.
| Способ удаления снега с надувной конструкции | 1988 |
|
SU1604984A1 |
| Изложница с суживающимся книзу сечением и с вертикально перемещающимся днищем | 1924 |
|
SU2012A1 |
| ПРОИЗВОДНЫЕ ПИРИДАЗИН-3-ОНА И СОДЕРЖАЩИЕ ИХ ЛЕКАРСТВЕННЫЕ СРЕДСТВА | 2000 |
|
RU2232755C2 |
| МОДУЛЯТОРЫ КИНАЗЫ, АССОЦИИРОВАННОЙ С РЕЦЕПТОРОМ ИНТЕРЛЕЙКИНА-1 | 2007 |
|
RU2459821C2 |

