Изобретение относится к медицине, в частности к методам лечения пациентов препаратами, содержащими генетический материал, и может быть использовано для лечения пациентов с онкологическими заболеваниями, причиной возникновения которых являются мутации в онкогенах, генах онкосупрессоров и тотальное огомозигочивание аллелей генов клетки, претерпевающей раковое перерождение.
Известен способ лечения, основанный на исправлении точечных мутаций в клетках [1].
Способ отличается относительно низкой эффективностью лечения и ограниченностью применения, поскольку согласно этому способу мутации должны быть точно определены еще до начала лечения.
Ввиду того, что ни для одной мутации нет достоверного доказательства того, что именно эта мутация является причиной рака, развитие этого метода требует скрупулезных и длительных исследований по выявлению конкретных мутаций, требующих коррекции, а следовательно, существует необходимость в создании методов, которые могли бы применяться на основе уже имеющихся знаний о причинах рака, не делая акцента на конкретных изменениях генома, приведших к злокачественному перерождению.
Известны также способы лечения, основанные на локальном применении ДНК фрагментов для лечения предраковых состояний в коже пациентов и стимуляции солнечного загара [2, 3].
Указанные способы также имеют ограниченную применимость, поскольку ни конкретные последовательности, ни источники ДНК в указанных патентах не определены. В них предлагается использовать как природную, так и синтетическую ДНК из "любых подходящих источников", например ДНК лосося длиной от 200 до мононуклеотидов и нуклеозидов, включая димеры, что в соответствии с тестами авторов является наиболее эффективным. Однако действие и последствия применения ДНК иной природы на человеческий организм пока не полностью изучены, что предопределяет определенную опасность применения способов.
Наиболее близким по сущности к предлагаемому является способ, основанный на активации репарации ДНК в клетках, в соответствии с которым производится доставка ферментов репарации в клетки кожи [4].
Недостатком наиболее близкого технического решения является ограниченная область применения, поскольку оно предназначено для лечения онкологических заболеваний только кожи и при этом не предполагает лечения уже имеющихся мутаций.
Из сказанного следует, что потребность в методах коррекции генетических мутаций по-прежнему является в высшей степени актуальной. В значительной мере это касается лечения индивидуумов с онкологическими заболеваниями, причиной которых являются как мутации в онкогенах и генах онкосупрессоров, так же как и тотальное огомозигочивание генов.
Требуемая задача заключается в расширении области применения и лечения пациентов с онкологическими заболеваниями, причиной которых являются как мутации в онкогенах и генах онкосупрессоров, так же как и тотальное огомозигочивание генов.
В более широком плане технической задачей настоящего изобретения является разработка экспериментально подтвержденного на культуре клеток аденокарциномы человека и искусственно индуцированных опухолях мышей способа воздействия на раковые клетки и опухоль в целом таким образом, что происходит изменение генного гомеостаза, когда клетки претерпевают реверсивное генетическое перерождение, при котором исчезает основное свойство раковой клетки - ее неограниченная пролиферативная активность.
Требуемый результат достигается тем, что по способу лечения онкологических заболеваний, основанному на введении в организм фрагментированной ДНК, используют фрагменты гомологической ДНК, составляющие полный геном физиологически и генетически здорового донора, при этом количество вводимой ДНК равно или превышает количество собственно ДНК плазмы крови и тканевых жидкостей пациента, но не более максимально допустимого количества, равного 30 мкг/мл.
В современной литературе отсутствуют указания на предлагаемый способ лечения онкологически больных пациентов с использованием фрагментированной генетически здоровой ДНК, естественного механизма ее доставки и депонирования в межхромосомном пространстве, и гомологического обмена доставленных фрагментов и соответствующих им участков хромосом, несущих мутации, приведшие к ораковлению данной клетки.
Следовательно, предложение отвечает критериям новизны и изобретательского уровня.
Ниже приводятся теоретические и экспериментальные данные, подтверждающие, что изобретение отвечает и критерию практической (промышленной) применимости.
На фиг.1 представлено распределение по клеточным компартментам клеток культуры клеток MCF-7 фрагментированной экстраклеточной ДНК человека в зависимости от времени ее нахождения в культуральной среде (нативные условия проведения фореза, логарифмическая фаза роста культуры). Справа блоки агарозы, окрашенные этидиумом бромидом. Слева рентгенограммы этих же блоков после высушивания. Цифры над блоками указывают время инкубации культуры клеток с меченой α32p экстраклеточной ДНК и αdATP*. Цифры слева и справа от блоков - маркеры молекулярных весов в т.п.о. α - исходный αdATP* предшественник.
На фиг.2 представлены здоровая последовательность и последовательность специфического участка мРНК гена каспазы 3, содержащего репарированный экзон 4, экзоны 5 и 6. На фигуре отмечены: набор праймеров и ожидаемый ПЦР фрагмент гена каспазы 3 размером 431 п.о. в РТ процедуре; набор праймеров и ожидаемые ПЦР фрагменты размером 236 и 304 п.о., подтверждающие принадлежность фрагмента 431 п.о.; амплификация фрагмента 431 п.о. свидетельствует о существовании мРНК, синтезирующейся с репарированного гена каспазы 3; амплификация ПЦР фрагментов 236 и 304 п.о., свидетельствующая о соответствии внутренней структуры фрагмента 431 п.о. району мРНК гена каспазы 3, содержащему репарированный экзон 4, экзоны 5 и 6; сравнение последовательности мРНК гена каспазы 3, содержащей экзоны 4, 5 и 6 и сиквенированных фрагментов 236 и 304 п.о. как внутренних сегментов, полученных с матрицы фрагмента 431 п.о.
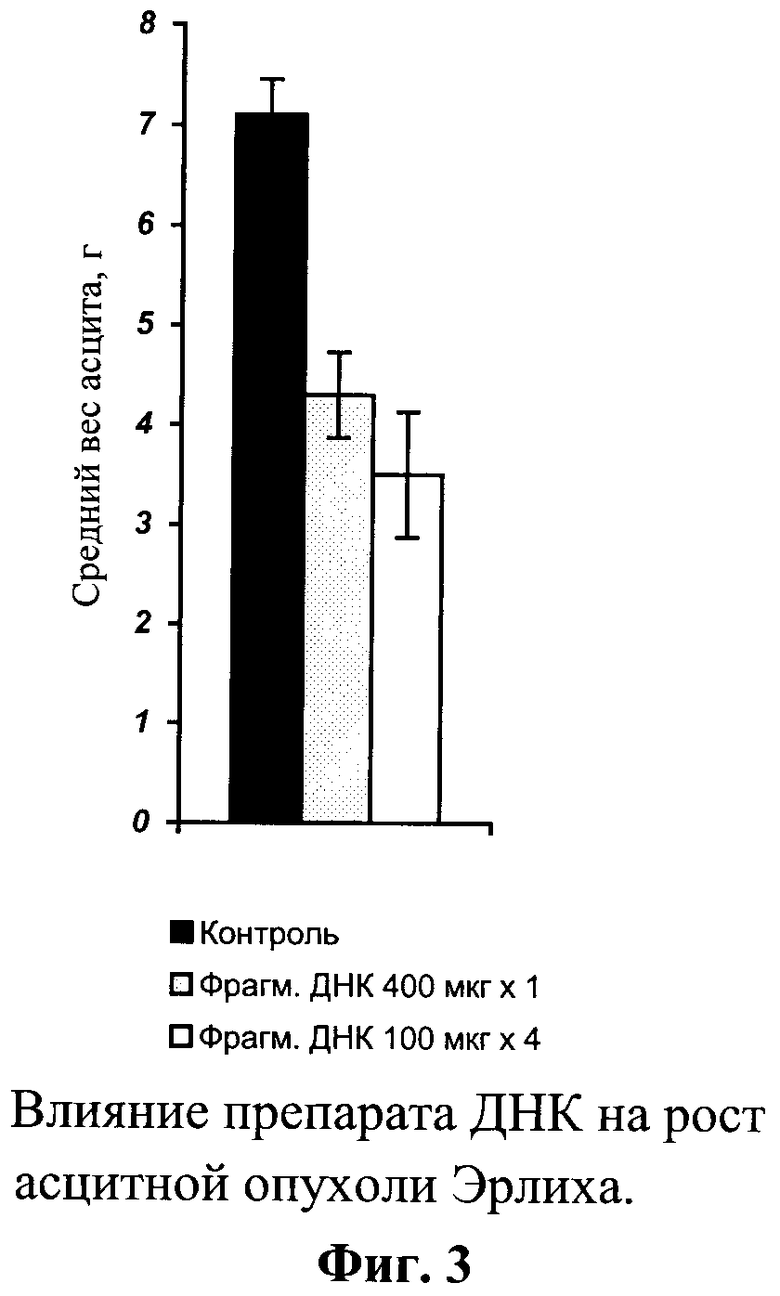
На фиг.3 продемонстрировано влияние препарата ДНК на рост асцитной опухоли Эрлиха.
В таблице 1 приводится общая характеристика количеств вещества, взятого во всех экспериментах.
В таблице 2 представлен абсолютный счет (AC cpm) αdATP* находящегося в различных клеточных компартментах в % к добавленному в культуральную среду меченому материалу. В эксперимент взята одна точка 180'.
В таблице 3 представлены количественные показатели содержания меченого материала (ДНК*) в различных компартментах клетки в зависимости от времени инкубации клеток с ним.
В таблице 4 приведены данные по продолжительности жизни мышей-носителей опухоли ГА-1 в контрольной группе и в группах, получавших ДНК.
В таблице 5 приведены данные о влиянии фрагментированной ДНК на рост и метастазирование карциномы Льюиса.
В таблице 6 приведены данные о влиянии различной концентрации экстраклеточной ДНК на скорость деления культуры клеток человека hTERT-BJ1.
В таблице 7 проанализирована частота структурных перестроек хромосом, возникающих в культуре диплоидных клеток человека hTERT-BJ1, обработанных в течение двух суток экстраклеточной ДНК в концентрации 300 мкг/мл.
Предложенный способ лечения онкологических заболеваний реализуется следующим образом.
Для его осуществления в организм пациента известными путями и способами (внутривенно, внутримышечно, путем внутрикожной инъекцией, подкожно, внутрибрюшинно, промыванием или нанесением на различные слизистые оболочки и/или кожу, приемом через рот (с или без предварительной нейтрализации кислой среды желудка либо с использованием специальной нерастворимой в желудке оболочки), через прямую кишку, интравагинально, интрокулярно, через нос, введением в опухоль, инрацеребрально, путем интроокулярной инъекции, ингаляцией или введением в спино-мозговой канал) вводится фрагментированная ДНК.
Для этого используются фрагменты гомологической ДНК, составляющие полный геном физиологически и генетически здорового донора.
Количество вводимой фрагментированной ДНК равно или превышает количество собственно ДНК плазмы крови и тканевых жидкостей пациента, которое многократно превышает количество ДНК плазмы крови и тканевых жидкостей здорового донора, но не более максимально допустимого количества, равного 30 мкг/мл.
При этом фрагменты ДНК, имея биологически активный размер, будучи введенные в организм пациента, доставляются в активно делящиеся клетки организма, в том числе и в клетки, подвергшиеся раковому перерождению при помощи естественного механизма доставки (кровоток, специфические рецепторы, расположенные на поверхности активно делящихся клеток, имеющие высокое сродство к указанным фрагментам ДНК).
Далее фрагменты ДНК, доставленные во внутриклеточное пространство, депонируются в межхромосомном компартменте ядра и подвергаются внутриядерному процессингу.
В этом случае фрагменты ДНК, будучи депонированными в межхромосомном компартменте ядра, вследствие естественного механизма гомологической рекомбинации (ГР) замещают мутантные участки генов, мутации в которых привели к раковому перерождению клетки на здоровые, а также замещают огомозигоченные аллели, что характерно для ораковевших клеток, приводя их к гетерозиготному состоянию, характерному для здоровой клетки. В результате указанных событий происходит реверсивное полое или частичное перерождение прежде раковой клетки, ведущее к выходу этой клетки из состояния неконтролируемой пролиферации.
Таким образом, при введении в организм гомологичной, фрагментированной ДНК от физиологически и генетически здорового донора происходит ее поглощение активно делящимися, претерпевшими раковое перерождение клетками посредством естественного механизма доставки, свойственного активно делящимся клеткам (рецептор-опосредованный пиноцитоз). Доставленная в ядерный компартмент - межхромосомное пространство - фрагментированная ДНК депонируется и вступает в процесс гомологического обмена с соответствующими локусами хромосом. Вследствие естественного механизма гомологической рекомбинации депонированные в межхромосомном пространстве фрагменты ДНК замещают мутантные участки генов, мутации в которых привели к раковому перерождению клетки на здоровые, а также замещают огомозигоченные аллели, что характерно для ораковевших клеток, приводя их к гетерозиготному состоянию, характерному для здоровой клетки. Вследствие указанных событий раковые клетки претерпевают частичное или полное реверсивное перерождение и вступают на путь развития дифференцированной клетки.
Эффект, достигаемый при использовании предлагаемого изобретения, может быть обоснован следующими теоретическими данными.
Каждая клетка организма имеет геномную ДНК, в которой закодирована информация как обо всех белках всех клеток организма, так и о пространственной организации генов в ядре, необходимой для корректной пространственно-временной экспрессии этих белков. В итоге геномная ДНК представляет собой матрицу жизни организма, включающую информацию как об организме в целом, так информацию о его развитии. Изначально геномная ДНК идентична во всех клетках организма. Однако по мере роста организма геномная ДНК каждой клетки подвергается мутационным воздействиям, вызванным факторами окружающей среды и ошибками, возникающими при клеточной репликации и репарации. Известно, что механизмы репарации замещают неправильные или ущербные основания ДНК и снова замыкают концы молекулы ДНК после одно- и двухзвенных разрывов (ДЦР) цепей сразу же после мутационных событий. Для этого в качестве матрицы они могут использовать вторую цепь ДНК. В случаях, когда участки поражения очень длинные и затрагивают обе цепи ДНК, репарация становится проблематичной, а результатом нарушения может явиться мутация. Таким образом, мутагенные факторы окружающей среды и ошибки клеточной репарации и репликации являются источниками соматических мутаций в клетках. В современном научном представлении о генезисе раков считается, что постепенное накопление мутаций в клетках вызывает многоступенчатую раковую трансформацию.
Генетическая мульти-мутационная природа раковых трансформаций клеток и самого рака говорит о том, что методы лечения рака должны быть направлены на устранение причины болезни и, прежде всего, на лечение (исправление) мутаций. Единственным принципиальным подходом к лечению заболеваний, связанных с генетическими нарушениями, каковым считается рак, является изменение тем или иным способом генома ораковевших клеток организма индивида и восстановление исходного, свойственного здоровой клетке генетического гомеостаза. В формате такого подхода были разработаны многочисленные методы генной терапии, однако все они имеют серьезные недостатки и малоэффективны в рамках сложного эукариотического организма.
Классические методы генной терапии (gene-targeting, ген-таргентинг), направленные на исправление хромосомных мутаций, с использованием вирусных конструкций способны эффективно ввести работающие копии генов в клеточный геном. Тем не менее, обеспечивая случайную встройку гена и регуляторных областей, эти подходы страдают рядом проблем, включающих зависимость генной экспрессии от места интеграции, сайленсинг введенного вирусного генома и инсерционный мутагенез, ассоциированный с карциногенезом [5, 6]. Привлекательной альтернативой этим методам могла бы стать коррекция или замещение мутантного гена в составе хромосомы путем естественной гомологичной рекомбинации с доставленной в клетку ДНК матрицей, не содержащей мутации, если бы не чрезвычайно низкая эффективность метода, обнаруженная в ранних попытках его использования [7, 8]. Исторически стратегия генного замещения (gene replacement), разработанная с целью преодоления проблемы инсерционного мутагенеза на дрожжах и позже на клетках млекопитающих, включала использование линейной рекомбинантной ДНК, в которой два конца гомологичны районам, фланкирующим замещаемый ген, а сам ген замещен селектируемым маркером [9]. Такой тип генного таргетинга называется "ends-out", поскольку концы конструкции соответствуют двум расходящимся (от целевого гена) последовательностям хромосомальной ДНК. При этом концы "ends-out" ген-таргетинг конструкции рекомбинагенны и облегчают замещение генной последовательности, заключенной между концевыми гомологиями. Недавно было показано, что у дрожжей при замещении целого гена гетерологичной последовательностью или замещении единственной мутантной пары оснований этим методом процесс инициируется двумя независимыми "strand invasions" (внедрение цепей), что подразумевает поиск гомологии и рекомбинационные события, осуществляемые двумя концевыми сегментами терапевтической конструкции [10]. Ранее подобный механизм был предложен для объяснения "ends-out" ген-таргетинга в клетках млекопитающих [11]. Соматическая ГР, судя по тому, что она сохранилась в эволюции миллиарды лет от простейших до млекопитающих, должна, очевидно, выполнять абсолютно необходимую и универсальную во всех царствах живого функцию и исполнять ее эффективно. Поскольку соматическая ГР у одноклеточных дрожжей является основным механизмом прецизионной репарации ДЦР, ее эффективность должна быть очень высокой, чтобы обеспечить спасение индивида. Ранние эксперименты с микроинъекциями плазмид в клеточное ядро [12] привели к открытию того, что плазмиды всегда интегрировали в клеточный геном в форме одного конкатемера, состоящего из всех (часто более ста) инъецированных плазмид, собранных в единую молекулу, хвост к голове, и что результатом этого была удивительно эффективная работа механизма ГР. Причиной же низкой эффективности классического ген-таргетинга, предполагающего использование ГР в клетках млекопитающих, может служить несоответствие между задачей, ставящейся перед ГР, и ее свойствами и возможностями, отобранными эволюцией для исполнения ее собственной, хотя и неизвестной функции. Данные, описанные в литературе, позволяют идентифицировать некоторые факторы, являющиеся критическими для работы ГР механизма.
Важным для эффективной работы механизма ГР является линейный размер доставленных к месту расположения рекомбинационного комплекса фрагментов ДНК. Так, описан 100-кратный экспоненциальный рост эффективности ген-таргетинга при увеличении длины гомологии в корректирующей конструкции с 2 до 14,5 т.п.о. и при этом достигнутая эффективность, 10-5 событий генной коррекции на клетку, оставалась все же недостаточной для применения метода без селекции [13]. Разительный контраст приведенным выше данным представляют результаты по использованию для коррекции мутантного гена коротких фрагментов гомологичной ДНК (SFHR, small fragments homologous replacement) с эффективностью исправления на клетку 1-20% [3, 14]. Полученная в этих экспериментах эффективность ген-таргетинга может означать, что ГР предпочитает работать либо с короткими фрагментами, либо формирует из коротких фрагментов при помощи лигирования фрагментов ДНК лигазой IV по типу голова-хвост мультимерные фрагменты, которые наиболее эффективны в использовании в рекомбинационных событиях.
Необходимым условием для получения рекомбинантного продукта в клетках млекопитающих является длина концевой гомологии фрагментов, которая составляет около 200 bp (пар оснований) [15, 16].
Принципиальным для осуществления ГР является линейная форма доставленных терапевтических ДНК как источника двуцепочечных концов. Кольцевые формы терапевтических плазмид практически не участвуют в гомологическом обмене. Генерация ДЦР и образование двуцепочечных концов молекулы ДНК локально активирует и усиливает хромосомальную и экстрахромосомальную ресомбинацию и ген-таргетинг [17]. В недавней публикации в Nature [18] этот принцип был использован для индукции коррекции гена IL2R, точечная мутация в 5-ом экзоне которого вызывает смертельную наследственную болезнь, связанный с Х-хромосомой тяжелый комбинированный иммунодефицит (severe combined immunodeficiency, SCID). Авторы путем трансфекции клетки генетическими конструкциями, экспрессирующими синтетические цинк-фингер нуклеазы, способные специфически расщепить последовательность вблизи мутации в гене IL2R в геномной ДНК клеток и плазмидой, содержащей немутантный фрагмент этого же гена, перекрывающий район SCID мутации, продемонстрировали высокий, до 20% уровень генной коррекции.
Можно полагать, что принципиальная способность ГР очень эффективно осуществлять генную коррекцию связана с инициирующим началом появления в ядерном пространстве свободных двуцепочечных концов ДНК, независимо будь то концы разорванной хромосомы, образовавшиеся в результате ДЦР, или двуцепочечные концы фрагментов ДНК, доставленных в ядро и являющихся типичными "ends-out" структурами. Любой немодифицированный фрагмент геномной ДНК, имея два конца гомологичных двум последовательностям хромосомальной ДНК и среднюю часть, способную заместить геномный фрагмент и соответственно исправить имеющуюся мутацию, представляет собой типичную "ends-out" конструкцию. Это означает, что концы фрагмента будут инициировать поиск гомологии и ГР с геномной ДНК, приводящую к замещению фрагмента геномной ДНК центральной частью фрагмента, доставленного из внешней по отношению к клетке среды. В условиях использования генетически корректной экзогенной ДНК эти события будут приводить к исправлению генетического дефекта [19].
В плазме крови и интерстициальных жидкостях млекопитающих и человека всегда присутствуют фрагменты геномной ДНК, образующиеся в результате фрагментации хроматина при естественной смерти клеток организма [20, 21]. Эти фрагменты ДНК участвуют в постоянном обороте ДНК, захватываясь активно делящимися клетками и доставляясь в клеточные компартменты. Доставка экстраклеточной ДНК осуществляется в результате активного рецептор-опосредованного пиноцитоза с участием специфических мембранных рецепторов [22, 23]. Существует множество методов генной трансфекции, однако эти методы лишь обеспечивают компактную упаковку терапевтических ДНК, необходимую для транспорта этой ДНК рассматриваемым рецептор-опосредованным механизмом [24, 25]. Создание высокой концентрации фрагментированной генетически корректной ДНК в плазме крови и интерстициальных жидкостях приведет к появлению конкурентного вытеснения этой ДНК из постоянного процесса гомологического обмена с соответствующими участками хромосом патологических фрагментов ДНК, циркулирующих в крови и появляющихся в результате гибели ораковевших клеток. Продолжительное применение такого воздействия приведет к восстановлению здорового генотипа клеток и реверсивной трансформации к норме [19, 26].
Поставленная техническая задача достигается тем, что в ее основе лежит разработанная теоретическая концепция оборота экстраклеточной ДНК в организме, подтвержденная многочисленными экспериментами. Ниже сформулированы основные положения концепции оборота экстраклеточной ДНК в эукариотическом организме.
Экстраклеточная фрагментированная геномная ДНК, содержащая мутантные последовательности в пропорциях, соответствующих встречаемости конкретных мутаций в клетках, образуется в результате программируемой клеточной смерти или, иначе говоря, апоптоза клеток организма и всегда присутствует в межклеточном пространстве и плазме крови [21, 27]. Клетки способны захватывать образующиеся фрагменты ДНК или хроматина рецептор-опосредованным механизмом и транспортировать их в клеточные ядра [22, 28]. Доставленные фрагменты рекомбинируют с геномной ДНК в ядрах по механизму гомологической рекомбинации (ГР) и сами способны инициировать этот процесс. Можно предполагать, что в совокупности эти три процесса составляют механизм, способный контролировать генетику соматических клеток на организменном уровне, исправляя мутации или индуцируя генетические изменения в клетках, используя экстраклеточную ДНК тканевых жидкостей и плазмы крови как внешний геномный стандарт. Работа такого механизма могла бы служить основой геномного единства клеток организма. Этот механизм мог бы элиминировать возникшие и установившиеся мутации, а также внедрять мутации в клетки, используя мутантные последовательности экстраклеточного стандарта ДНК. В том случае, если бы механизм мог использовать стандарт ДНК, практически лишенный вредных мутаций, его работой было бы исключительно устранение мутаций из клеток тела. Поскольку аллельные гены обычно обладают высокой степенью гомологии и часто отличаются лишь относительно небольшими участками измененных последовательностей, другим аспектом работы предполагаемого механизма могло бы стать замещение последовательностей аллельных генов в хромосоме на альтернативные, подобно замещению мутантных последовательностей на немутантные.
Поставленная техническая задача также достигается тем, что полный геном индивидуума вводится в организм в форме набора молекул полинуклеотидов, являющихся фрагментами геномной ДНК, циркулирует и захватывается клетками, попадает в ядро, где происходит его депонирование и гомологичная рекомбинация с геномной ДНК клетки. Таким образом, появляется возможность корректировать генетические дефекты путем введения в организм полного генома в форме молекул полинуклеотидов, являющихся фрагментами геномной ДНК, не имеющей генетических дефектов, а индивидуумам, страдающим заболеваниями и расстройствами, связанными с генетическими мутациями, может быть предоставлено эффективное лечение на основании способа, изложенного в настоящем изобретении. В заявленном способе лечения используются фрагменты гомологической ДНК, составляющие полный геном физиологически и генетически здорового индивида, находящихся в ассоциации с белками ядерного матрикса. Размер экзогенных фрагментов соответствует размеру фрагментов циркулирующих в плазме крови лечимого организма, возникающих в ходе апоптотического нуклеолиза (1-30 нуклеосомных единиц). В прототипах используются различные искусственные конструкции ДНК, не составляющие полный геном индивида. Эта ДНК не находится в ассоциации с белками ядерного матрикса и не соответствует размеру естественной апоптозной ДНК организма, составляющей 1-30 нуклеосомных единиц.
Приведенные отличительные свойства заявляемого способа лечения онкологических заболеваний, возникших как следствие мульти-мутантных событий, произошедших в клетке, были исследованы на культуре клеток аденокарциномы молочной железы человека (in culture). Заявляемый способ лечения онкологических заболеваний был исследован на экспериментально индуцированных опухолях мышей (in vivo). Предельно допустимое количество терапевтической ДНК, предлагаемое для введения и отраженное в заявленном способе лечения, было определено в тестах по цитотоксичности, выполненных на культуре клеток человека (in culture).
Доставка во внутренние клеточные компартменты фрагментированной геномной ДНК экзогенного происхождения, определенная в заявленном способе лечения, исследовалась на модели культуры клеток аденокарциномы молочной железы человека.
МАТЕРИАЛЫ И МЕТОДИКА.
Культура клеток.
Клетки культуры клеток аденокарциномы молочной железы человека MCF-7 культивировали в среде RPMI 1640 с 10мМ L-глутамина и 50 мкг/мл стрептамицина "Sigma" (США), при 37°С в присутствии 5% эмбриональной бычьей сыворотки (ФБС) "Биолот" (Россия), в атмосфере с 5% CO2 до плотности 0.7×107 клеток на 4 лунки в 24 луночном планшете.
Приготовление ДНК и предшественника.
10 мкг фрагментированной до размеров 500 п.о. ДНК плаценты человека метили ник-трансляцией в присутствии фрагмента Кленова, трех холодных и одного горячего dNTP. Для удаления не включившихся предшественников ДНК переосаждали 2 раза изопропанолом согласно процедуре, описанной у Гловера [29]. На выходе после очистки получали 7-15 мкг меченой ДНК. ДНК разводили в соответствующем объеме дистиллированной воды и добавляли в каждую лунку планшета (в объеме, не превышающем 20 мкл на лунку). В каждой лунке находилось 250-350 мл среды. Аликвоту 1 мкл из рабочего объема отбирали на счет радиоактивности.
Аликвоту αdNTP* разводили в соответствующем объеме Н2O. 1 мкл из разведенного трифосфата отбирали на счет радиоактивности. Ориентировочно одинаковое количество α по отношению к ДНК (по счету и объему) добавляли на точку.
Количество ДНК (мкг, cpm) и αdNTP* (cpm) для трех экспериментов указано в таблице 1.
РЕЗУЛЬТАТЫ.
Распределение по клеточным компартментам экстраклеточной ДНК размером 500 п.о., представляющей пул фрагментов, содержащий полный геном человека.
Необходимые цифры для количественной оценки результатов экспериментов.
Количество клеток культуры клеток аденокарциномы молочной железы человека MCF-7 в экспериментальной точке составляет 0.7×106.
Согласно протоколу для CF-7 107клеток содержат 60 мкг ДНК.
Одна клетка содержит 6 пикограмм ДНК.
Суммарное количество ДНК в клетках в экспериментальной точке составляло 4.2 мкг.
В экспериментальную точку было добавлено количество меченой ДНК, указанное в таблице 1. Счет радиоактивности добавленной ДНК составлял указанное в таблице 1 количество cpm.
Счет радиоактивности αdNTP* в точке составил количество cpm, указанное в таблице 1.
Гаплоидный геном клетки человека содержит 3.3×109 п.о.
Размер добавленных в среду меченых фрагментов геномной ДНК составлял исходно порядка 500 п.н.
Для анализа поведения экстраклеточной ДНК при ее взаимодействии с клеткой мы выбрали две стадии зрелости культуры (экспоненциальная фаза и фаза контактного торможения) и два типа электрофоретического анализа (нативные условия и денатурирующие условия). Были проанализированы качественные характеристики и количественные параметры поведения экстраклеточной ДНК при ее проникновении в клетку.
Качественные характеристики поведения меченого материала (нативные условия). Экспоненциальная фаза роста клеток в момент добавления ДНК и предшественника в культуральную среду (Фиг.1).
На чертеже представлены электорфореграмма (справа) и рентгеновский фильм после засветки этого же агарозного блока после высушивания (слева).
Цитоплазматическая фракция (верхняя группа блоков).
ДНК практически сразу проникает в цитоплазму клеток. В нулевой точке высвечивается низкомолекулярная фракция по подвижности, соответствующая исходному меченому материалу. На старте нулевой точки метка отсутствует. Во всех остальных образцах на старте присутствует значительное количество меченого материала. Это может означать, что ДНК формирует с компонентами цитоплазмы высокомолекулярные комплексы. Очевидной медузы осаждающейся ДНК не обнаруживалось. Во всех образцах наблюдается фракция ДНК с одинаковой подвижностью, совпадающей с подвижностью исходной меченой ДНК, добавленной в среду. Это означает, что со свободно мигрирующей частью ДНК, доставленной в цитоплазматичекую фракцию, не происходит метаболических превращений. Другая часть ДНК, обнаруживающаяся на старте, или претерпела некие превращения, или находится в составе высокомолекулярных комплексов. Межхромосомная фракция (средняя группа блоков).
ДНК практически сразу проникает в межхромосомную фракцию ядра. В нулевой точке высвечивается низкомолекулярная фракция, по подвижности соответствующая исходному меченому материалу. На старте нулевой точки и на старте всех остальных образцов метка отсутствует. Вся высокомолекулярная фракция ДНК при центрифугировании сформировала чечевидный осадок, который не обнаруживается в ядерном соке. Во всех образцах, за исключением нулевой точки, наблюдается изменение подвижности ДНК. ДНК формирует высокомолекулярный пул в виде "размытого облака", поднимающегося из нижней части в высокомолекулярную область агарозного блока. При этом на фоне равномерного облака можно наблюдать несколько дискретных, размер-зависимых бэндов. Особенно четко бандированность ДНК можно увидеть во фракции хроматина, где присутствует незначительная контаминация межхромосомным материалом (осадок хроматина не промывался). Такая картина распределения меченого материала может свидетельствовать о следующих событиях. Возможно, что попавшая в ядро фрагментированная ДНК выступает в качестве затравки-праймера для синтеза цепи ДНК. Это выражается в появлении разноразмерного набора меченых фрагментов ДНК, который на форезе выявляется как "размытое облако" меченого материала, достигающего размера порядка 10 т.п.о. Другая возможность заключается в том, что фрагменты ДНК лигируются по концам. Такое заключение можно сделать из картины дискретных меченых бэндов, по размеру кратных исходной ДНК. Также возможен комбинированный вариант увеличения линейного размера экстраклеточной ДНК в межхромосомальном пространстве ядра.
Хроматин (нижняя группа блоков).
ДНК (или метка в какой-то иной форме) практически сразу проникает в ядро и интегрирует во фракцию хроматина ядра. В области разделения геля наблюдается выраженная лестница фрагментов, по размеру кратных исходной ДНК. (Появление этого меченого материала связано с незначительной контаминацией межхромосомной фракцией, содержащей высокую удельную метку).
αdATP* (во всех блоках под обозначением 180α).
αdATP* в цитоплазматической фракции отсутствует. Однако присутствует меченый фрагмент, размером совпадающий с размером ДНК, которая использовалась в эксперименте. В межхромосомной фракции образует аналогичное, но менее интенсивное "размытое облако", морфологически полностью соответствующее таковому для аналогичной фракции с меченой ДНК. Через 180' метка обнаруживается в хроматине. Другие точки для αdATP* не контролировались. Количество метки, которое было взято в культуральной среде в точке 180 минут для меченой ДНК и для αdATP*, составляет ориентировочно равное количество (см. таблицы 1, 2). Однако количество меченого материала, выявляемого в указанной экспериментальной точке с αdATP*, приблизительно в 6 раз меньше, чем в случае с ДНК.
Количественные показатели поведения меченого материала.
Все количественные оценки, выполненные на основании сравнения полученных счетов в экспериментальных точках различных клеточных компартментов, приводятся в таблицах 1, 2. Следует отметить, что во всех процедурах материал присутствует в абсолютном количестве и поэтому уместно сравнение и его привязка к исходной, вносимой в культуральную среду метке.
В результате проведенных экспериментов неожиданным оказалось то, что, во-первых, αdATP* доставляется во все компартменты клетки не как мономер, а в составе фрагментов ДНК размером порядка 500 п.о. Далее эта ДНК участвует во всех процессах, как и ДНК, добавленная в среду. Во всех отношениях активность утилизации αdATP* в несколько раз менее интенсивна по сравнению с утилизацией меченой экстраклеточной ДНК. Далее было обнаружено, что экстраклеточная ДНК практически сразу (менее минуты, если клетки активно делятся) доставляется во все проанализированные компартменты клетки (цитоплазма, ядерное пространство) и интегрирует в хроматин. В цитоплазме присутствует приблизительно одинаковое количество меченой ДНК для всех точек за исключением нулевой точки. Наблюдается динамика накопления метки в компартментах ядра в зависимости от времени экспозиции с меченой ДНК, присутствующей в культуральной среде. ДНК присутствует в межхромосомном пространстве ядра в виде фрагментов ДНК различной длины. В нулевой точке в активно делящихся клетках размер фрагментов соответствует размеру экстраклеточной ДНК, добавленной в среду. Во всех остальных вариантах линейный размер ДНК увеличивается до размеров порядка 10 т.п.о. Причем, судя по "лестнице" выявляемых бандов, часть фрагментов лигируется по типу "голова к хвосту". В клетках культуры в стадии контактного торможения процессинг ДНК (денатурирующие условия, данные не приводятся) демонстрирует явно выраженную динамику. На данной стадии роста культуры клеток, по-видимому, метаболические процессы заторможены, и нам удалось зафиксировать динамическую стадию процесса формирования мультимеров исходных фрагментов ДНК. Увеличение линейных размеров молекул ДНК, доставленных в ядерное пространство, может быть связано с работой специфической ДНК лигазы IV, участвующей в репарационных событиях [см. 30].
Приведенные данные предполагают, что при заявленном способе лечения онкологических заболеваний в ядро раковой клетки доставляется, постоянно присутствует в межхромосомном пространстве и интегрирует в геном определенное количество ДНК. В межхромосомном пространстве ДНК претерпевает изменения, увеличивающие ее линейный размер до порядка 10 т.п.о.
Возможность исправления мутаций воздействием экстраклеточной ДНК на раковые клетки, рассматриваемая в заявленном способе лечения раков, была исследована на мутантном гене каспазы 3 аденокарциномы человека. В результате проведенных исследований было показано как восстановление активности гена каспазы 3 клеток аденокарциномы молочной железы человека MCF-7, так и апопттического пути развития прежде раковых клеток при воздействии экстраклеточной ДНК.
МАТЕРИАЛЫ И МЕТОДЫ. Культуры клеток и условия культивирования.
Клетки аденокарциномы молочной железы MCF-7 и саркомы мыши - L929 предоставлены НИИ клеточных культур ГНЦ ВБ "Вектор". Клетки культивировали в среде RPMI 1640 с 10мМ L-глутамина и 50 мкг/мл стрептамицина ("Sigma", США), при 37°C в присутствии 5% эмбриональной бычьей сыворотки (ФБС) ("Биолот", Санкт-Петербург) в атмосфере с 5% CO2. Для трансфекции клетки MCF-7 культивировали в среде RPMI 1640 с 10 мМ L- глутамина и 50 мкг/мл стрептамицина в присутствии 5% эмбриональной бычьей сыворотки (ЭБС) и 0.1-0.2 мг/мл фрагментированной ДНК плаценты человека. Время трансфекции варьировало от 5 до 40 дней, как это указано в подписях к чертежам.
ПЦР анализ.
ПЦР анализ проводили по стандартным протоколам, с использованием китов компании "Медиген" (Россия) и компании "BioRad" (США). Real time ПЦР анализ проводили согласно протоколу компании "BioRad" (США).
Для анализа мРНК гена каспазы 3 были синтезированы праймеры:
ПЦР-фрагменты ДНК разделяли в 2% агарозном геле и окрашивали бромистым этидием. Выделение РНК из культуры клеток.
Для выделения РНК использовали Total RNA Purification Kit (V-gene Biotechnology Limited). Процедуру выделения вели согласно прилагаемому протоколу.
ПЦР анализ мРНК клеток культуры клеток MCF 7, длительное время экспонированных с экстраклеточной человеческой ДНК.
Обнаружение в экстракте клеток, обработанных человеческой ДНК, либо фермента каспазы 3, либо соотвествующей ей мРНК с залеченной мутацией в экзоне 4, могло быть прямым доказательством гомологической интеграции терапевтического фрагмента в хромосому мутантных клеток. Мы выбрали подход, связанный с обнаружением и анализом мРНК, который позволил бы прямым сиквенированием показать места сплайсинга экзонов и присутствие в мРНК залеченной мутации. Используя РТ (reverse transcriptase) ПЦР процедуру, был амплифицирован участок мРНК, содержащий экзоны 4, 5 и 6. При этом в качестве прямого праймера использовался олигонуклеотид, гомологичный отсутствующему в геноме клеток MCF 7 участку 47 п.о. в 4 экзоне (праймер 3). В качестве обратного праймера использовался олигонуклеотид, комплементарный хвостовой части экзона 6, последний нуклеотид которого соответствовал последнему нуклеотиду экзона 6.
Первоначально из культуры клеток MCF 7, инкубированных с человеческой ДНК 15 суток, была выделена суммарная РНК и синтезирована кДНК. Для выявления участка мРНК, соответствующего гену каспазы 3 и содержащего репарированную делецию в 4-ом экзоне, были использованы праймеры 3 и 2 (пр.2, 3). В результате амплификации был получен искомый фрагмент размером 431 п.о., включающий сплайсированные экзоны 4 (без 20 начальных нуклеотидов), экзон 5 и экзон 6 (фиг.3). Выявление этого фрагмента во фракции суммарной РНК означило, что суммарная РНК содержит мРНК гена каспазы 3 с репарированной делецией 47 п.о. Для контроля правильности амплификации фрагмента 431 п.о. были получены 2 ожидаемых ПЦР продукта (фрагменты размером 236 и 304 п.о) с использованием двух внутренних праймеров (пр.1 и 4), двух концевых праймеров (пр.3 и 4) и ДНК фрагмента 431 п.о. в качестве матрицы (фиг.3). Сиквенс обоих фрагментов позволил сделать следующее заключение. Фрагмент 431 п.о. представляет собой кДНК копию участка мРНК гена каепазы 3, содержащего сплайсированные экзоны 4 (без 20 начальных нуклеотидов, что определено местом посадки праймера), экзон 5 и экзон 6 (фиг.3). Левый край фрагмента 431 п.о. является репарированным участком экзона 4, содержащим недостающие 47 нуклеотидов (область 320-367 мРНК каспазы 3), корректно объединенным с двумя следующими за ним экзонами, которые делегированы в исходной культуре MCF 7. Этот факт свидетельствует о том, что произошла гомологическая интеграция терапевтического фрагмента в дефектную область экзона 4. Интеграция произошла по механизму, описанному в работах [11, 10], когда срединная часть интегрируемого фрагмента не важна для процесса объединения. При сравнении сплайсированной последовательности фрагмента 431 п.о., содержащей экзоны 4, 5, и 6 с оригинальной последовательностью, были обнаружены две вставки 6 и 6 нуклеотидов (область 370 и 390 мРНК каспазы 3). Обе вставки не нарушают открытой рамки считывания. В экспериментальной последовательности отсутствующий в положении 370 гена каспазы 3 цистеин, по-видимому, компенсируется цистеином в положении 350 гена. Появление двух вставок связано либо с вариабельностью 4-го экзона гена каспазы 3, либо является следствием процесса интеграции экстрахромосомального фрагмента в хромосому. В остальной части последовательности имеется несколько не значимых замен. Практически полная гомология оставшейся части прочитанной матрицы, где места объединения экзонов без выпадения нуклеотидов соответствуют последовательности, содержащейся в архиве GeneBank, служат доказательством корректно прошедшего сплайсинга. Обнаружение репарированной мРНК и демонстрация активно протекающего апоптоза с характерной нуклеосомной фрагментацией свидетельствуют о появлении функционального фермента.
Известно, что ген каспазы расположен на хромосоме 4, имеет размер 21751 п.о. и состоит из 8 экзонов. В процессе клеточного метаболизма могут существовать два продукта экспрессии данного гена. Это транскрипты размером 17 и 19 т.п.о. и их белковые продукты [31]. Для появления функционального фермента каспазы 3 требуется сложный процесс считывания мРНК, при котором идет сплайсинг 8 экзонов. Делеция 47 п.о. в экзоне 4 приводит к пропуску 4-го экзона при сплайсинге и нарушению синтеза РНК и экспрессии активного фермента. ДНК, присутствующая в культуральной среде, фрагментирована до размера 500-1000 п.о. гидродинамическим способом. Это означает, что в эксперименте принципиально отсутствуют фрагменты, способные использоваться для синтеза полноразмерного продукта, дающего функциональный фермент. Ускользающе мала вероятность того, что произошло лигирование нужных фрагментов в правильной ориентации с восстановлением открытой рамки считывания каспазного гена и образование тем самым экстрахромосомальной матрицы, пригодной для синтеза фермента. На наш взгляд и согласно результатам проведенных экспериментов интеграция в геном MCF-7 посредством гомологической рекомбинации здорового фрагмента, реверсирующего ген каспазы к норме, представляется наиболее вероятным завершением событий перемещения экстраклеточной ДНК в проведенных опытах.
Таким образом, результаты проведенных исследований свидетельствуют о том, что в культуре клеток возникает новый признак, носителем которого являлась исключительно внеклеточная фрагментированная аллогенная ДНК.
Приведенные данные предполагают, что при заявленном способе лечения онкологических заболеваний доставленные естественным клеточным механизмом фрагменты экстраклеточной ДНК находят свои гомологичные сегменты на хромосомах и рекомбинируют с ними. При этом если участок хромосомы несет мутацию, то гомологичный ему экстраклеточный фрагмент способен исправить эту мутацию за счет гомологического замещения мутантного участка.
Заявленный способ лечения онкологических заболеваний был исследован в системе (in vivo) на экспериментальных опухолях мышей
МАТЕРИАЛЫ И МЕТОДЫ.
Эксперименты проведены на мышах линии A/Sn, CBA/Lac, C 57 BL/6 и ICR в возрасте 3-4 месяцев разводки вивария Института цитологии и генетики СО РАН. В исследованиях были использованы 3 штамма перевиваемых опухолей: опухоль Эрлиха, гепатома ГА-1 и опухоль Lewis. Асцитная опухоль Эрлиха и карцинома легкого Lewis получены из НИИ онкологии ТНЦ СО РАМН. Гепатома ГА-1 была первично индуцирована у мыши линии А/Не орто-аминоазотолуолом. Поддерживается в асцитной форме на мышах A/Sn. При подкожной или внутримышечной прививке растет на месте прививки в виде солидного узла и дает множественные метастазы в печень.
Источником ДНК человека служили плаценты здоровых рожениц, а ДНК мышей - смесь тканей органов (печень, почки, селезенка, тимус). ДНК выделяли бесфенольным методом (лабораторный регламент ИЦиГ СО РАН). Такой способ позволяет получить полноценную геномную ДНК с сохранением тех ее фрагментов, которые in vivo прочно ассоциированы с белками ядерного матрикса. ДНК фрагментировали ультразвуковым дезинтегратором, в результате чего получали смесь фрагментов размером 200-6000 п.н. Препараты хранили расфасованными небольшими порциями в морозильной камере при -18°С. Перед использованием необходимое количество ДНК размораживали и доводили до нужной концентрации физиологическим раствором.
Статистическую обработку данных проводили, используя компьютерную программу "Статистика". Достоверность различий между данными оценивали, используя t-критерий Стъюдента и критерий Уитни-Вилкоксона.
РЕЗУЛЬТАТЫ.
Проведенные исследования демонстрируют, что препараты фрагментированной геномной ДНК обладают способностью тормозить рост этих опухолей и оказывают умеренное ингибирующее влияние на развитие опухолевых метастазов (см. таблица 4, 5, фиг.3).
Влияние цитотоксического и абберативного действия ДНК в количестве, определенном в заявленном способе лечения, исследовалось на модели культуры клеток hTERT-BJ1 человека.
МАТЕРИАЛЫ И МЕТОДЫ.
В экспериментах использовали раствор фрагментированной ДНК 1,695 мг/мл. Препаратом сравнения являлась ДНК, выделенная из молок лосося в тех же условиях. Использовалась диплоидная линия культуры клеток hTERT-BJ1 CLONTECH Laboratories, Inc. Catalog# С 4001-1. Клетки в одинаковой концентрации были высажены на 6-и ячеечные плато. Через 24 часа в ячейки была добавлена ДНК человека или лосося в концентрации 300 или 30 мкг/мл. Подсчет клеток осуществлялся в камере Горяева через 48 и 72 часов после введения ДНК. Статистическая обработка проводилась с использованием программы "Origin".
Для оценки хромосомных перестроек, экспонированных с ДНК человека и лосося, клетки фиксировали на предметном стекле и окрашивали Гимза по стандартной методике.
РЕЗУЛЬТАТЫ.
Данные о цитотоксическом действии фрагментированной ДНК, выделенной из разных источников и аплицированной в разных дозах.
Анализ полученных данных показал, что введение ДНК как человека, так и лосося в концентрации 300 мкг/мл на третьи сутки культивирования угнетает рост культуры. Концентрация препаратов в 30 мкг/мл на третьи сутки культивирования не оказывает статистически значимого действия на рост клеток культуры hTERT-BJ1 (таблица 6).
Данные об аберративном действии фрагментированной ДНК, выделенной из разных источников. Анализ полученных данных свидетельствует о том, что при концентрации экзогенной ДНК человека или лосося в 300 мкг/мл наблюдается повышенный уровень хромосомных аберраций. При этом для ксеногенной ДНК наблюдается более ярко выраженное аберративное действие, чем для аллогенной человеческой ДНК (таблица 7).
Экспериментально установлено, что фрагментированная ДНК в концентрации, равной или превышающей 300 мкг/мл, может приводить к торможению роста клеток и к возникновению хромосомных нарушений. Концентрация препарата 30 мкг/мл не оказывает цитотоксического действия. Для препаратов ДНК, используемой в терапевтической практике, рекомендованы дозы 5 мг для однократного приема. Это соответствует 1 мг ДНК на литр крови или рабочей концентрации препарата в крови 1 мкг/мл, что в 300 раз меньше концентрации, при которой наблюдались нежелательные эффекты, и в 30 раз ниже концентраций экзогенной ДНК, не обладающих цитотоксичностью для клеток культуры клеток человека hTERT-BJ1.
Таким образом, в предложенном способе лечения онкологических заболеваний достигается требуемый результат, поскольку включается в расширение области применения и лечения пациентов с онкологическими заболеваниями, поскольку способ позволяет лечить причину заболевания, а именно исправлять мутации в онкогенах и генах онкосупрессоров, также вносить гетерозиготные аллели в огомозигоченные гены клетки.
Источники информации
1. 1.US, No. 5,795,972, 18.08.1998
2. US, No. 5,955,059, 21.09.1999
3. US, No. 5,470,577,28.11.1995
4. US, No. 5,302,389, 12.04.1994 (прототип)
5. Khon D. В., Sadelain M., Glorioso J. С. Occurrence of leukaemia following gene therapy of X-linked SCID. Nature Rev. Cancer. 477-88 (2003).
6. Persons D.A., Nienhuis A.W. Gene therapy of hemoglobin disorders. Hematol. Rep. 2, 348-355 (2003).
7. Yanez RJ. and Porter., ACG. Therapeutic gene targeting. Gene therapy. 5, 149-159, (1998).
8. Rothstein RJ. One-steplgene disruption in yeast. Methods Enzymol. 101:202-11 (1983).
9. Thomas KR, Capecchi MR. Site-directed mutagenesis by gene targeting in mouse embryo-derived stem cells. Cell. 51(3):503-12. (1987)
10. Langston LD, Symingtom LS. Gene targeting in yeast is initiated by two independent strand invasions. Proc NM Acad Sci USA. 101(43): 15392-7. (2004).
11. Li J, Read LR, Baker MD. The mechanism of mammalian gene replacement is consistent with the formation of long regions oi heteroduplex DNA associated with two crossing-over events. Mol. Cell Biol. 21(2):501-10 (2001).
12. Capecchi, M. Generating n\ice with targeted mutations. Nature Medicine 7, 1086-1090. (2001)
13. Deng, С. and Capecchi, M. К. Reexainination of gene targeting frequency as a function of the extent of homology between the targeted vector and the target locus. Mol. Cell Biol. 12.3365-3371 (1992).
14. Gruenert D., Bruscia E., Novelli G., Colosimo A., Dallapiccola В., Sangiuolo F., Goncz K. Sequence-specific modification of genomic DNA by small DNA fragments. Clin. Invest. 112, 637-641 (2003).
15. Lin Y., Lukacsovich Т., Waldman A.S. Multiple pathways for repair of DNA double-strand breaks in mammalian chromosomes. Mol. Cell Biol. 19, 8353-60 (1999).
16. Nassif N., Engels W. DNA homology requirements for mitotic gap repair in Drosophila. Proc. Nail, Acad. Sci. USA. 90 (4):1262-6 (1993).
17. Kucherlapati RS, Eves ЕМ, Song KY, Morse BS, Smithies 0. Homologous recombination between plasmids in mammalian cells can be enhanced by treatment of input DNA. Proc. Natl.Acad.Sci. USA. 81, 3153-7 (1984).
18. Urhov FD, Miller JC, Lee YL, Beausejour CM, Rock JM, Augustus S, Jamieson AC, Proteus MH, Gregory PD, Holmes MC. Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature. 435, 646-51 (2005).
19. Yakubov L., PopovaN., Nikolin V., Semenov D., Bogachev S., Oskina I. Extracellular genomic DNA protects mice against radiation and chemical mutagens. Genome Biology. 5:P3 (2003).
20. Anker P, Zajac V, Lyautey J, Lederrey C, Dunand C, Lefort F, Mulcahy H, Heinemann J, Stroun M. Transcession of DNA from bacteria to human cells in culture: a possible role in oncogenesis. Ann. N Y Acad Sci. 1022:195-201 (2004).
21. Anker P, Mulcahy H, Chen XQ, Stroun M. Detection of circulating tumour DNA in the blood (plasma/serum) of cancer patients. Cancer Metastasis Rev. 18,65-73 (1999).
22. Yakubov, L.A., Deeva, E.A., Zarytova V.F., Ivanova E.M., Ryte A.S., Yurchenko, L.Y., Vlassov V.V. Mechanisms of oligonucleotide uptake by cells: Involvement of specific receptors? Proc. Nail. Acad. Sci. USA. 86, 6454-6458, (1989).
23. Doerfler, W., Remus, R., Muller, K., Heller, H., Hohlweg, U., Schubbert, R. The fate of foreign DNA in mammalian cells and organisms. Dev. Biol. (Basel), 106, 89-97, (2001).
24. Budker V, Budker T, Zhang G, Subbotin V, Loomis A, Wolff JA. Hypothesis: naked plasmid DNA is taken up by cells in vivo by a receptor-mediated process.. / Gene Med. 2(2):76-88, (2000).
25. Satkauskas S, Bureau MF, Mahfoudi A, Mir LM. Slow accumulation of plasmid in muscle cells: supporting evidence for a mechanism of DNA uptake by receptor-mediated endocytosis. Mol. Ther. 4, 317-323 (2001).
26. Yakubov L.A., Petrova N.A., Popova K.A., Semenov D.V., Nikolin V.P., Oskina I.N. Role of extracellular DNA in maintaining of stability and variability of cellular genomes. Doklady Biochemistry and Biophysics. 382, 31 -34, (2002).
27. Giacona M.B. Ruben G.C., Iczkowski K. Roots Т., Porter D., Sorenson G. Cell-free DNA in human blood plasma: length measurements in patients with pancreatic cancer and healthy controls. Pancreas. 17, 89-97 (1998).
28. Шестова О.Е., Андреева А.Ю., Власов В.В., Якубов Л.А. Транспорт комплексов олигонуклеотидов с белками клеточной поверхности в клеточное ядро. ДАН. 368, 264-267, (1999).
29. Гловер Д. Клонирование ДНК. Методы. M.: Мир, С.538, (1988).
30. Lees-Miller S.P., Meek К. Repair of DNA double strand breaks by non-homologous end joining // Biochimie. 85, 1161-1173 (2003).
31. Wolf B.B., Schuler M., Echiverri F. and Green D.R.. Caspase-3 is the primary activator of apoptotic DNA fragmentation via DNA fragmentation factor-45/ingibitor of caspase-activated DNase inactivation. J. Biol. Chem. 274, 30651-30656(1999).
275481
Разница достоверна
100+\-17.55 и 66.46+\-18.99
100+\-17.55 и 51.27+\-16.55
83.11+\-23.16 и 51.27+\-16.55
Достоверного отличия нет
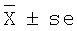
 t-test
t-test
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ЛЕЧЕНИЯ ОНКОЛОГИЧЕСКИХ ЗАБОЛЕВАНИЙ | 2008 |
|
RU2387456C1 |
| СПОСОБ ЛЕЧЕНИЯ ОНКОЛОГИЧЕСКИХ ЗАБОЛЕВАНИЙ | 2007 |
|
RU2345792C2 |
| СПОСОБ ПОЛУЧЕНИЯ СТИМУЛИРОВАННЫХ ДЕНДРИТНЫХ КЛЕТОК ДЛЯ ИНДУКЦИИ ИММУННОГО ОТВЕТА ПРОТИВ ТУБЕРКУЛЕЗА ЧЕЛОВЕКА | 2009 |
|
RU2401664C1 |
| СПОСОБ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ | 2006 |
|
RU2322264C1 |
| СПОСОБ ДЕТЕКЦИИ СТВОЛОВЫХ РАКОВЫХ КЛЕТОК | 2013 |
|
RU2530170C1 |
| СПОСОБ ЭРАДИКАЦИИ СТВОЛОВЫХ ИНИЦИИРУЮЩИХ РАКОВЫХ КЛЕТОК | 2013 |
|
RU2542410C1 |
| СПОСОБ ЛЕЧЕНИЯ ОНКОЛОГИЧЕСКИХ ЗАБОЛЕВАНИЙ | 2009 |
|
RU2429019C2 |
| Персонализированный способ реституции костного мозга для борьбы с прогрессированием и рецидивами различных болезней цивилизации, профилактики старения и внезапной смерти | 2023 |
|
RU2817892C1 |
| Биомедицинский клеточный продукт для лечения онкологических, нейродегенеративных, аутоимунных заболеваний и травм головного и спинного мозга | 2021 |
|
RU2798554C2 |
| СПОСОБ ЛЕЧЕНИЯ ОНКОЛОГИЧЕСКИХ ЗАБОЛЕВАНИЙ | 2010 |
|
RU2489169C2 |

Изобретение относится к медицине, в частности к способам лечения онкологических заболеваний животных препаратами, содержащими ДНК. При этом в качестве такой ДНК используют гомологическую ДНК, имеющую биологически активный размер, составляющий полный геном физиологически и генетически здорового донора. ДНК вводят в дозе, создающей такую концентрацию ДНК в плазме крови, которая равна или превышает концентрацию собственно ДНК плазмы крови, но не превышает максимально допустимую концентрацию, равную 30 мкг/мл. Изобретение обеспечивает естественную гомологическую репарацию ДНК с заменой участков хромосом, несущих мутации, приведшие к ораковлению клеток, без нежелательных эффектов при использовании ДНК в указанной дозе. 7 таб., 3 ил.
Способ лечения онкологических заболеваний у животных, основанный на введении в организм животного фрагментированной ДНК, отличающийся тем, что в качестве такой ДНК используют гомологическую ДНК, имеющую биологически активный размер, составляющий полный геном физиологически и генетически здорового донора, при этом ДНК вводят в дозе, создающей такую концентрацию ДНК в плазме крови, которая равна или превышает концентрацию собственно ДНК плазмы крови, но не превышает максимально допустимую концентрацию, равную 30 мкг/мл.
| ПРЕПАРАТ, ОБЛАДАЮЩИЙ ПРОТИВООПУХОЛЕВЫМ, АНТИТОКСИЧЕСКИМ И РАДИОПРОТЕКТОРНЫМ ДЕЙСТВИЕМ | 2003 |
|
RU2234323C1 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| WO 9604397 A1, 15.02.1996, реферат | |||
| Устройство для отображения информации на экране электронно-лучевой трубки | 1982 |
|
SU1156119A1 |
| YANEZ R.J | |||
| et al | |||
| Therapeutic gene targeting // Gene Ther | |||
| Способ и аппарат для получения гидразобензола или его гомологов | 1922 |
|
SU1998A1 |

