Настоящее изобретение относится к способу получения имидазолильных соединений.
1,2,3,9-Тетрагидро-9-метил-3-[2-метил]-1H-имидазол-1-ил)метил]-4Н-карбазол-4-он (ондансетрон) известен из EP191562 и US 4695578. В этих патентных публикациях описан основной класс соединений, включая ондансетрон и гомологичные соединения, их получение и применение в качестве сильных селективных антагонистов "нейрональных" 5-гидрокситриптаминовых рецепторов и их применение для лечения мигрени и психотических расстройств.
Из EP-B-0297651, EP-B-0601345 и EP-B-768309 известен (10R)-5,6,9,10-тетрагидро-10-[(2-метил-1Н-имидазол-1-ил)метил]-4Н-пиридо[3,2,1-jk]-карбазол-11(8H)-он (цилансетрон) (известный также как (R)-(-)-4,5,6,8,9,10-гексагидро-10-[(2-метил-1H-имидазол-1-ил)метил]-11Н-пиридо-[3,2,1-jk]-карбазол-11-он). В первой патентной публикации описан общий класс соединений, включая цилансетрон и гомологичные соединения, их получение и применение в качестве 5-HT антагонистов. Во второй патентной публикации описано применение выбранных соединений этого типа для лечения некоторых заболеваний и в третьей описано получение энантиомерно чистых соединений и их гидрохлорида моногидрата.
Общим признаком вышеуказанных соединений является то, что они содержат замещенную имидазолильную группу, присоединенную в α-положении относительно кетогруппы карбазольной системы с метиленовым мостиком. Некоторые варианты синтеза этих соединений описаны в указанных патентных публикациях. Общим признаком в данных синтезах является то, что замещенная имидазолильная группа вводится посредством реакции Манниха с последующим дезаминированием и получением промежуточного экзометиленового соединения, которое взаимодействует с замещенной имидазолильной группой (смотри схему 1, например).
Недостатком этого пути синтеза является то, что выход продукта в этой последовательности реакционных стадий довольно низкий. В US 4695578 первая стадия, которая обычно дает самый низкий выход продукта, не описана, а вторая стадия (пример 7 по US 4695578) дает выход 68%. В EP-B-0297651 на первой стадии (пример 1c EP-B-0297651) выход продукта равен 53%, и на второй стадии (пример 1d EP-B-0297651) выход продукта равен 87%. При широкомасштабном получении данный способ приводит к образования значительного количества дегтеобразных побочных продуктов.
Схема 1
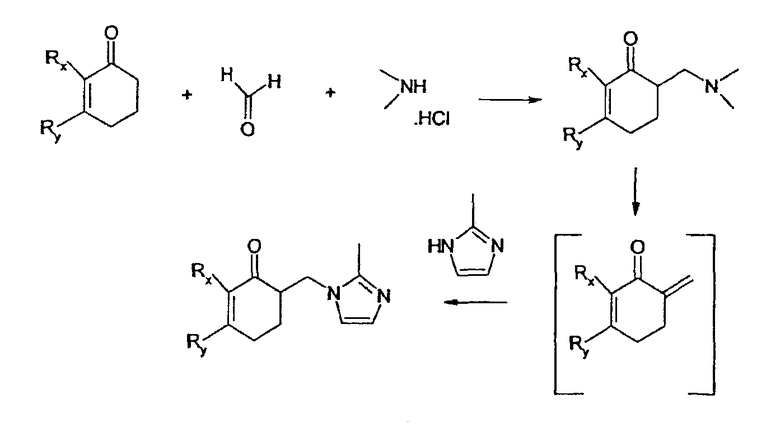
Объектом настоящего изобретения является альтернативный способ получения имидазолилсоединений, который является экономически эффективным и отвечает одному или нескольким следующим требованиям: a) сравнительно высокий выход продукта, b) короткое время реакции по сравнению с предшествующими способами, c) небольшое количество побочных реакций, d) более высокое качество конечного продукта и e) использование реакционных условий, не требующих разбавителя, с экологически приемлемым растворителем.
Неожиданно было обнаружено, что этот тип имидазолильных соединений может быть легко получен, используя замещенное оксазолидиновое соединение для введения метиленового мостика.
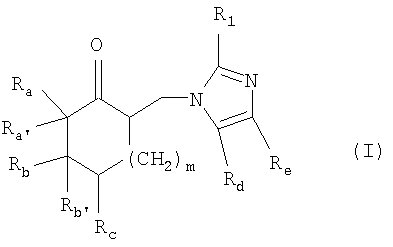
Таким образом, настоящее изобретение относится к способу получения имидазолильного соединения общей формулы
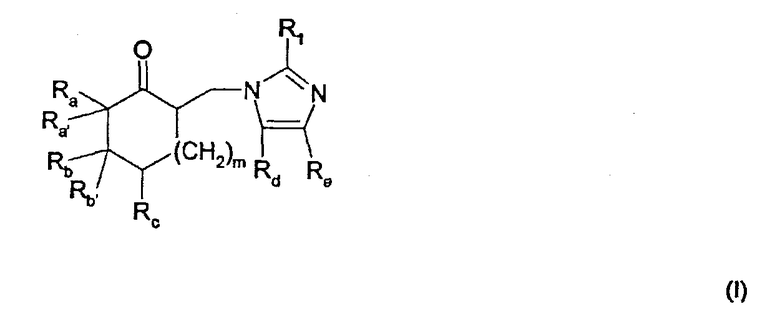
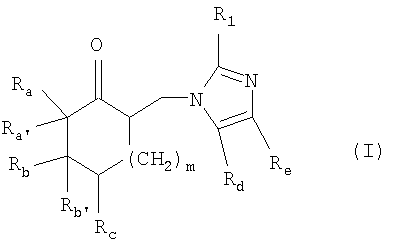
где Ra и Rb, каждый, независимо, представляют собой (C1-C6)алкил, (C1-C6)алкоксиалкил, необязательно замещенный арил или гетероарил;
или где Rа и Rb вместе образуют дополнительную гомоциклическую или гетероциклическую систему, содержащую одно или несколько колец;
Ra' и Rb', каждый, представляют собой водород, или вместе образуют углерод-углеродную двойную связь, указанная углерод-углеродная двойная связь необязательно является частью ароматической системы;
Rс представляет собой водород, (C1-C6)алкил, (C1-C6)алкокси, (C1-C6)алкоксиалкил или галоген;
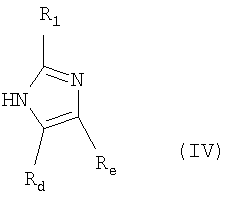
Rd представляет собой водород или (C1-С4)алкил;
Re представляет собой водород или (C1-C4)алкил;
m равно 1 или 2; и
R1 представляет собой водород или (С1-C4)алкил;
а также его соли добавления кислот;
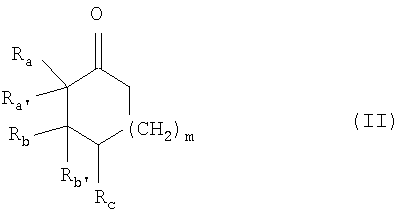
отличающегося тем, что соединение общей формулы
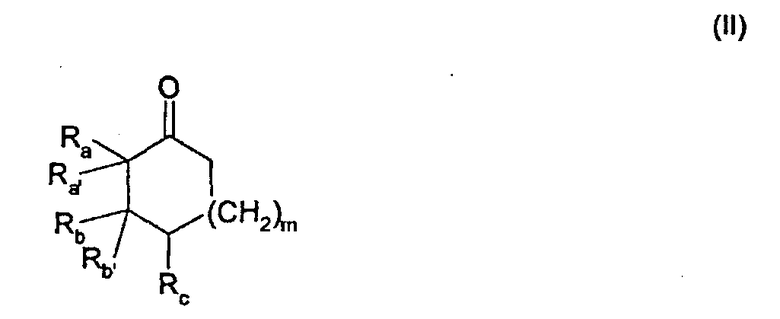
подвергают взаимодействию с соединением формулы
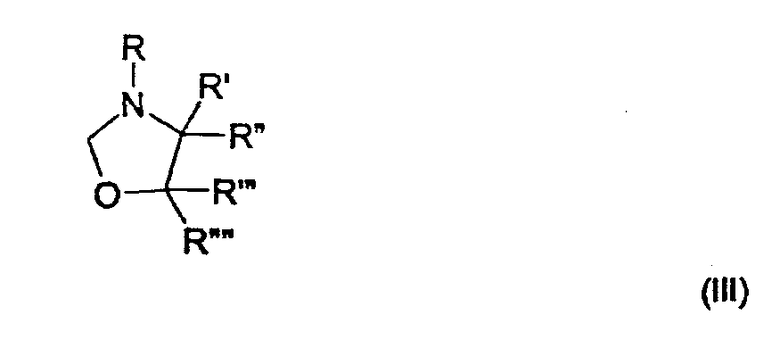
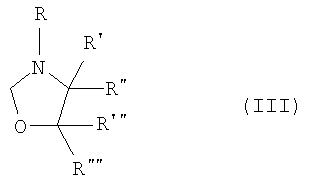
где R представляет собой водород, (C1-C4)алкильную группу, необязательно замещенную гидроксигруппой или необязательно замещенную арильную группу,
R', R", R"' и R"", каждый, независимо, представляют собой водород или (C1-C4)алкильную группу;
затем подвергают взаимодействию с соединением формулы

где R1, Rd и Re имеют значения, определенные выше;
и затем, необязательно, подвергают взаимодействию с соответствующей кислотой.
Алкильные группы по настоящему изобретению включают алкильные радикалы с прямой, разветвленной цепью и циклические алкильные радикалы, содержащие до 6 атомов углерода. Соответствующие алкильные группы могут быть насыщенными или ненасыщенными. Более того, алкильная группа также может быть замещена один или несколько раз заместителями, выбранными из группы, включающей арил, галоген, гидрокси, циано или моно- или диалкилзамещенный амино.
Арильные группы по данному изобретению включают арильные радикалы, которые могут содержать до 6 гетероатомов. Арильная группа может также быть необязательно замещена один или несколько раз заместителем, выбранным из группы, состоящей из арила, (C1-C6)алкила, галогена, гидрокси, циано или моно- или диалкилзамещенного амино и также может быть конденсирована с арильной группой или циклоалкильными кольцами. Соответствующие арильные группы включают, например, фенил, нафтил, толил, имидазолил, пиридил, пирроил, тиенил, пиримидил, тиазолил и фурильные группы.
Под гомоциклической системой подразумевается система, содержащая, по крайней мере, одну насыщенную или ненасыщенную циклическую группу, содержащую только атомы углерода и атомы водорода.
Под гетероциклической системой подразумевается система, содержащая, по крайней мере, одну насыщенную или ненасыщенную циклическую группу, также содержащую один или несколько гетероатомов, таких как N, О или S. И гомоциклическая. и гетероциклическая система могут быть необязательно замещены заместителем, выбранным из группы, состоящей из алкила, арила, циано, галогена, гидрокси или моно- или диалкилзамещенного амино.
В предпочтительном варианте осуществления изобретения Rс представляет собой водород или (C1-C6)алкил, Rd представляет собой водород или (C1-C4)алкил; Re представляет собой водород или (C1-C4)алкил; и R1 представляет собой водород, метил или этил.
Способ по настоящему изобретению особенно эффективен для получения соединений общей формулы
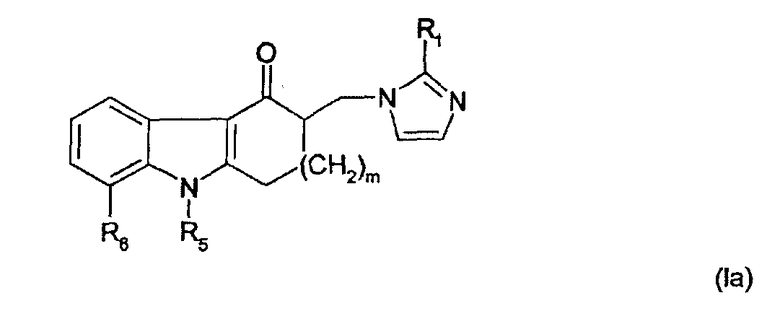
где m равно 1 или 2;
R1 представляет собой водород, метил или этил;
R5 представляет собой (C1-C4)алкил;
R6 представляет собой водород или (С1-C4)алкил, или
R5 и R6 вместе с промежуточными атомами образуют 5-, 6- или 7-членное кольцо, необязательно замещенное одним или двумя заместителями, выбранными из группы, включающей галоген, гидрокси, (C1-C4)алкил, (С1-C4)алкоксиалкил и (С1-C4)алкокси.
В данном случае исходное соединение представляет собой соединение общей формулы
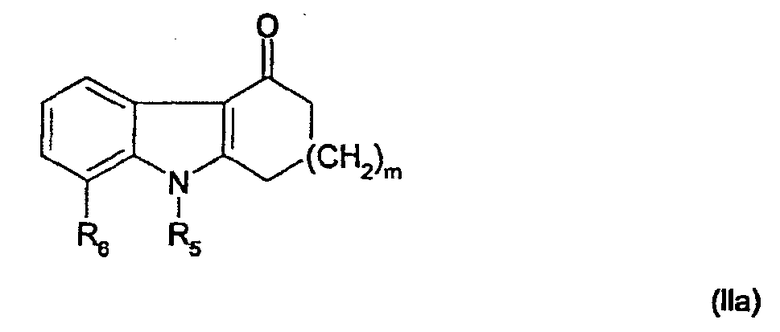
Это соединение далее называется карбазолоновым соединением.
Предпочтительные соединения общей формулы Ia представляют собой соединение, где m = 1, и R5 и R6 вместе с промежуточными атомами образуют 6-членное кольцо, и соединение, где m = 1, R5 представляет собой метил и R6 представляет собой водород. Для первого соединения выход продукта в способе, использующем в качестве исходного продукта 5,6,9,10-тетрагидро-4H-пиридо[3,2,1-jk]карбазол-11(8Н)-он и 3-оксазолидинэтанол составляет 77% (смотри пример 2), по сравнению с общим выходом в способе по EP0297651, равным 46% (примеры Ic и Id). Наибольший выход продукта может быть получен при широкомасштабном производстве.
В замещенном оксазолидине, предпочтительно, один из R' и R" и один из R"' и R"" представляет собой водород, так как оксазолидин, дизамещенный по одному и тому же атому углерода, например 4,4-диметилоксазолидин, дает меньший выход продукта в этой реакции. Предпочтительные оксазолидины представляют собой 3-оксазолидинэтанол и 3-этилоксазолидин. Наиболее предпочтительный оксазолидин представляет собой 3-оксазолидинэтанол.
Взаимодействие проводят в кислой среде, и степень кислотности зависит от реакционной активности взаимодействующей системы. В случае карбазолоновых систем, среда должна быть очень кислой. Примерами соответствующих кислот в последнем случае являются метансульфоновая кислота, трифторметансульфоновая кислота, п-толуолсульфоновая кислота и газообразный HCl в спиртовой среде.
Для получения высокого выхода продукта реакционный раствор должен содержать лишь небольшое количество воды. Количество воды предпочтительно должно быть ниже 0,6% (об./об.), более предпочтительно ниже 0,3% об./об. и наиболее предпочтительно ниже 0,1% об./об.
Оптимальная температура взаимодействия зависит от исходного продукта и растворителя и отличается на этих двух реакционных стадиях. Первая стадия взаимодействия может проводиться при температуре между 40°C и 110°C. Для карбазолоновых систем предпочтительная реакционная температура в первой стадии составляет между 50°C и 90°C, и наиболее предпочтительная температура равняется около 70°C. Вторая стадия обычно может проводиться при температуре, равной между 100°C и 140°C. Для карбазолоновых систем предпочтительная реакционная температура на второй стадии составляет между 110°C и 130%, и наиболее предпочтительная температура равна около 120°C.
Взаимодействие может проводиться в различных растворителях, например, диполярных апротонных растворителях, таких как ДМФ или в спиртах. Предпочтительными растворителями являются C4-C7 спирты, и выбор может зависеть от желаемой реакционной температуры. Примерами соответствующих спиртов являются 1-бутанол, 1-гексанол и изоамиловый спирт. Предпочтительным спиртом является 1-бутанол. Также подходят смеси ароматических углеводородов и спиртов, например смеси толуола и спирта и монохлорбензола и спирта. Предпочтительная смесь представляет собой смесь монохлорбензола и метанола. При использовании смесей растворителей низкокипящий растворитель может быть отогнан перед второй стадией для того, чтобы достичь высоких температур кипения с обратным холодильником системы растворителя на второй стадии.
Отношение объема растворителя к количеству реагентов в смеси может изменяться в относительно широком диапазоне и зависит от растворимости реагентов. Обычно отношение количества растворителя к количеству реагентов может составлять от около 1:1 до около 15:1, где отношение выражается как отношение объема растворителя к массе реагентов в растворителе (в мл/г). Предпочтительно отношение составляет от около 1:1 до около 10:1. В случае с карбазолоновыми системами предпочтительное отношение объема растворителя к массе реагентов равно около 4:1.
Получаемые продукты могут быть кристаллизованы из различных растворителей. Примерами растворителей для кристаллизации свободных оснований являются ароматические углеводороды, такие как толуол. Соль добавления хлористоводородной кислоты может. например, быть кристаллизована из спиртовых растворителей, таких как изопропанол или 1-бутанол.
Нижеследующие примеры предназначены лишь для более полной иллюстрации изобретения и поэтому эти примеры не следует рассматривать как ограничивающие каким-либо образом объем изобретения.
Пример 1: Материалы и методы
5,6,9,10-Тетрагидро-4H-пиридо[3,2,1-jk]карбазол-11(8H)-он был получен по EP0375045. 3,4-Дигидро-1(2H)-нафталенон приобретали из коммерческого источника. 1,2,3,9-Тетрагидро-9-метил-4H-карбазол-4-он был получен в соответствии с патентом США 3892766 от фирмы Warner-Lambert Company и Elz, S. и Heil, W., Bioorganic & Medicinal Chemistry Letters 1995, 5, 667-672. Метансульфоновую кислоту приобретали из коммерческого источника.
Спектр ЯМР измеряли на Varian VXR 200, а спектр MS на Finnigan TSQ 7000. Исследование с помощью ВЭЖХ проводили на системе HP1050 с детектором Separations 757 (250 нм) и колоночной печи Separations Marathon XT при 35°C. Использовали колонку Zorbax XDB C8, 15×0,3 см. Элюенты готовили следующим способом: смесь 2 л воды, 2 мл триэтиламина и 5 мл 25% аммония делали буферным при pH 4 с помощью муравьиной кислоты и добавляли 0,5 л ацетонитрил. Скорость потока составляла 1 мл/мин.
Пример 1a: Получение оксазолидинов
3-Оксазолидинэтанол получали следующим способом:
Эквимолярные количества диэтаноламина и параформальдегида в 1-бутаноле нагревали до 70°C. Через 1 час реакционного времени образовавшуюся воду удаляли путем азеотропной дистилляции с 1-бутанолом. 3-Этилоксазолидин получали в соответствии с Heany, H. et al., Tetrahedron 1997, 53, 14381-96.
4,4-Диметилоксазолидин является коммерчески доступным и его приобретали в виде 75% масс/масс раствора в воде. Из водного слоя экстрагировали 4,4-диметилоксазолидин, промывая раствором дихлорметана/насыщенного NaCl. Дихлорметановый слой сушили над безводным сульфатом натрия и затем упаривали.
Пример 2. Взаимодействие 5,6,9,10-тетрагидро-4H-пиридо[3,2,1-jk]карбазол-11(8H)-она с 3-оксазолидинэтанолом
5,6,9,10-Тетрагидро-4H-пиридо[3,2,1-jk]карбазол-11(8H)-он (25,00 г ≡ 111,0 ммоль) и метансульфоновую кислоту (17,06 г ≡ 177,5 ммоль) в 1-бутаноле (100 мл) нагревали до 70°C. В течение 3 минут к раствору добавляли 3-оксазолидинэтанол (19,49 г 166,4 ммоль) в 1-бутаноле (39 мл).
Через 50 минут при 80°C добавляли 2-метилимидазол (45,55 г = 554,8 ммоль) и 1-бутанол (10 мл). Через 1,5 часа при 120°C реакционную смесь частично упаривали до 30 мл 1-бутанола.
При 70°C к остатку добавляли 75 мл толуола и 50 мл воды. Слои разделяли. Водный слой экстрагировали 75 мл толуола. Объединенные толуольные слои промывали три раза 100 мл воды.
Органический слой упаривали досуха и затем добавляли 125 мл 1-бутанола. К полученному раствору добавляли 12,5 мл 36% масс/масс соляной кислоты. После перемешивания в течение 2 часов при комнатной температуре образовавшееся твердое вещество отфильтровывали и промывали 1-бутанолом и МТБЭ. Выход продукта после сушки составлял: 30,40 г (77,0%) гидрохлорид 5,6,9,10-тетрагидро-10-[(2-метил-1H-имидазол-1-ил)метил]-4H-пиридо[3,2,1-jk]карбазол-11(8H)-она (77,0%). ВЭЖХ: ≥ 95%.
1Н ЯМР [200 МГц, ДМСО-d6:CDCl3 4:1] δ 1,97 (1Н, м), 2,18 (3Н, м), 2,68 (3Н, с), 2,95 (2Н, т), 3,00 (1Н, дд), 3,12 (2Н, м), 4,13 (2Н, м), 4,29 (1Н, дд), 4,66 (1Н, дд), 6,97 (1Н, д), 7,09 (1Н, т), 7,55 (1Н, д), 7,68 (1Н, д) и 7,71 (1Н, д).
МС [ESI] МН+ = 320.
Пример 3. Взаимодействие 5,6,9,10-тетрагидро-4H-пиридо[3,2,1-jk]карбазол-11(8H)-она c 4,4-диметилоксазолидином.
5,6,9,10-Тетрагидро-4H-пиридо[3,2,1-jk]карбазол-11(8H)-он (20,00 г 88,8 ммоль) и метансульфоновую кислоту (13,65 г 142,0 ммоль) в 1-бутаноле (60 мл) нагревали до 70°C. В течение 2 минут добавляли 4,4-диметилоксазолидин (13,47 г 133,2 ммоль) в 1-бутаноле (10 мл).
Через 50 минут при 80°C добавляли 2-метилимидазол (36,45 г 444,0 ммоль) и бутанол (10 мл). Через 2 часа при 120°C реакционную смесь частично упаривали до 20 мл 1-бутанола.
К остатку при 70°C добавляли 60 мл толуола и 40 мл воды. Слои разделяли. Водный слой экстрагировали 60 мл толуола. Объединенные толуольные слои промывали три раза 80 мл воды.
Органический слой упаривали досуха и затем добавляли 100 мл 1-бутанола. К полученному раствору добавляли 10,0 мл 36% масс/масс соляной кислоты. После перемешивания в течение 2 часов при комнатной температуре образовавшееся твердое вещество отфильтровывали и промывали 1-бутанолом и МТБЭ. Выход продукта после сушки составлял: 12,38 г гидрохлорида 5,6,9,10-тетрагидро-10-[(2-метил-1H-имидазол-1-ил)метил]-4H-пиридо[3,2,1-jk]карбазол-11(8H)-она (39,2%). ВЭЖХ: ≥ 95%. 1НЯМР и MS: смотри пример 2. Маточный раствор содержал 3,45 г (10,9%) продукта.
Пример 4. Взаимодействие 5,6,9,10-тетрагидро-4H-пиридо[3,2,1-jk]карбазол-11(8H)-она с 3-этилоксазолидином.
5,6,9,10-Тетрагидро-4H-пиридо[3,2,1-jk]карбазол-11(8H)-он (20,00 г ≡ 88,8 ммоль) и метансульфоновую кислоту (13,65 г 142,0 ммоль) в 1-бутаноле (60 мл) нагревали до 70°C. В течение 2 минут добавляли 3-этилоксазолидин (13,46 г 133,2 ммоль) в 1-бутаноле (10 мл).
Через 50 минут при 80°C добавляли 2-метилимидазол (36,45 г 444,0 ммоль) и 1-бутанол (10 мл). Через 2 часа при 120°C реакционную смесь частично упаривали до 20 мл 1-бутанола.
При 70°C к остатку добавляли 60 мл толуола и 40 мл воды. Слои разделяли. Водный слой экстрагировали 60 мл толуола. Объединенные толуольные слои промывали три раза 80 мл воды.
Органический слой упаривали досуха и затем добавляли 100 мл 1-бутанола. К полученному раствору добавляли 10,0 мл 36% масс/масс соляной кислоты. После перемешивания в течение 2 часов при комнатной температуре, образовавшееся твердое вещество отфильтровывали и промывали 1-бутанолом и МТБЭ. Выход продукта после сушки составлял: 22,10 г (70,0%) гидрохлорида 5,6,9,10-тетрагидро-10-[(2-метил-1H-имидазол-1-ил)метил]-4H-пиридо[3,2,1-jk]карбазол-11(8H)-она (70,0%). ВЭЖХ: ≥ 95%. 1НЯМР и MS: смотри пример 2.
Пример 5. Взаимодействие 3,4-дигидро-1(2H)-нафталинона с 3-оксалидинэтанолом.
3,4-Дигидро-1(2H)-нафталинон (12,98 г 88,8 ммоль) и метансульфоновую кислоту (13,65 г 142,0 ммоль) в 1-бутаноле (60 мл) нагревали до 50°C. В течение 2 минут к раствору добавляли 3-оксазолидинэтанол (15,59 г 133,1 ммоль) в 1-бутаноле (14 мл).
Через 50 минут при 80°С добавляли 2-метилимидазол (36,45 г 444,0 ммоль) и 1-бутанол (10 мл). Через 2 часа при 120°C реакционную смесь частично упаривали до тех пор, пока не осталось 20 мл 1-бутанола.
При 70°C к остатку добавляли 60 мл толуола и 40 мл воды. Слои разделяли. Водный слой экстрагировали 60 мл толуола. Объединенные толуольные слои промывали три раза 80 мл воды.
Органический слой упаривали досуха и затем добавляли 100 мл 1-бутанола. К полученному раствору добавляли 10,0 мл 36% мас/мас соляной кислоты. Полученный раствор упаривали до объема 60 мл. После перемешивания в течение 2 часов при комнатной температуре образовавшееся твердое вещество отфильтровывали и промывали 1-бутанолом и МТБЭ. Выход продукта после сушки составлял: 15,28 г (62,2%) гидрохлорида 3,4-дигидро-2-[(2-метил-1H-имидазол-1-ил)метил]-1(2H)-нафталинона. ВЭЖХ: ≥ 95%.
1Н ЯМР [200 МГц, ДМСО-d6:CDCl3 4:1] δ 2,00 (2Н, м), 2,73 (3Н, с), 3,20 (3Н, м), 4,27 (1Н, дд), 4,68 (1Н, дд), 7,35 (2Н, т), 7,55 (1Н, м), 7,70 (1Н, д) и 7,90 (1Н, д).
МС [ESI] МН+ = 241.
Маточный раствор содержит 3,28 г (13,3%) продукта
Пример 6. Взаимодействие 3,4-дигидро-1(2H)-нафталинона с 4,4-диметилоксазолидином.
3,4-Дигидро-1(2H)-нафталинон (12,98 г 88,8 ммоль) и метансульфоновую кислоту (13,65 г = 142,0 ммоль) в 1-бутаноле (60 мл) нагревали до 70°C. В течение 2 минут добавляли 4,4-диметилоксазолидин (13,46 г = 133,1 ммоль) в 1-бутаноле (10 мл). Через 50 минут при 80°C добавляли 2-метилимидазол (36,45 г 444,0 ммоль) и 1-бутанол (10 мл). Через 2 часа при 120°C реакционную смесь частично упаривали до тех пор, пока не останется 20 мл 1-бутанола.
К остатку при 70°C добавляли 60 мл толуола и 40 мл воды. Слои разделяли. Водный слой экстрагировали 60 мл толуола. Объединенные толуольные слои промывали три раза 80 мл воды.
Органический слой упаривали досуха и затем добавляли 100 мл 1-бутанола. К полученному раствору добавляли 10,0 мл 36% масс/масс соляной кислоты. Полученный раствор упаривали до объема 50 мл. После перемешивания в течение 2 часов при 0°C образовавшееся твердое вещество отфильтровывали и промывали 1-бутанолом и МТБЭ. Выход продукта после сушки: 14,13 г (57,5%) гидрохлорида 3,4-дигидро-2-[(2-метил-1H-имидазол-1-ил)метил-1(2H)-нафталинона. ВЭЖХ: ≥95%. 1H ЯМР и MS: смотри пример 5. Маточный раствор содержит 2,33 г (9,5%) продукта.
Пример 7. Взаимодействие 3,4-дигидро-1(2Н)-нафталинона с 3-этилоксазолидином
3,4-Дигидро-1(2H)-нафталенон (12,98 г ≡88,8 ммоль) и метансульфоновую кислоту (13,65 г ≡142,0 ммоль) в 1-бутаноле (60 мл) нагревали до 50°C. В течение 2 минут добавляли 3-этилоксазолидин (13,46 г ≡ 133,1 ммоль) в 1-бутаноле (10 мл). Через 50 минут при 80°C добавляли 2-метилимидазол (36,45 г ≡ 444,0 ммоль) и 1-бутанол (10 мл). Через 2 часа при 120°C реакционную смесь частично упаривали, оставляя 20 мл 1-бутанола.
К остатку добавляли при 70°C 60 мл толуола и 40 мл воды. Слои разделяли. Водный слой экстрагировали 60 мл толуола. Объединенные слои толуола промывали три раза 80 мл воды.
Органический слой упаривали досуха и затем добавляли 100 мл 1-бутанола. К полученному раствору добавляли 10,0 мл 36% масс/масс соляной кислоты. Полученный раствор упаривали до окончательного объема 50 мл. После перемешивания в течение 2 часов при 0°C полученный твердый продукт отфильтровывали и промывали 1-бутанолом и MTBE. Выход после высушивания: 17,30 г гидрохлорида 3,4-дигидро-2-[(2-метил-1H-имидазол-1-ил)метил]-1(2H)-нафталинона (70,4%). ВЭЖХ: ≥ 95%. 1H ЯМР и MS: смотри пример 5.
Пример 8. Взаимодействие 1,2,3,9-тетрагидро-9-метил-4H-карбазол-4-он с 3-оксазолидинэтанолом
1,2,3,9-Тетрагидро-9-метил-4H-карбазол-4-он (13,26 г ≡ 66,5 ммоль) и метансульфоновую кислоту (10,23 г ≡ 106,4 ммоль) в 1-бутаноле (45 мл) нагревали при 90°C. В течение 2 минут добавляли 11,68 г (99,8 ммоль) 3-оксазолидинэтанол в 1-бутанол (11 мл).
Через 50 минут при 80°C добавляли 2-метилимидазол (27,32 г ≡ 332,5 ммоль) и 1-бутанол (8 мл). Через 2 часа при 120°C добавляли 180 мл толуола и 120 мл воды при 80°C. Слои разделяли. Водный слой экстрагировали 180 мл толуола и 60 мл 1-бутанола. Объединенные органические слои дважды промывали 240 мл воды. Органический слой упаривали досуха. К остатку добавляли 150 мл 1-бутанол и 10 мл 36% масс/масс соляной кислоты. При 0°С вскоре происходила кристаллизация. Через 1 час при 0°C полученные кристаллы отфильтровывали, промывали 1-бутанолом и MTBE и затем сушили: получали 15,39 г (70,1%) гидрохлорид 1,2,3,9-тетрагидро-9-метил-3-[(2-метил-1Н-имидазол-1-ил)метил]-4Н-карбазол-4-она. ВЭЖХ: ≥ 95%.
1Н ЯМР [200 МГц, ДМСО-d6:CDCl3 4:1] δ 2,00 (1Н, м), 2,20 (1Н, м), 3,69 (3Н, с), 3,09 (3Н, м), 3,75 (3Н, с), 4,30 (1Н, дд), 4,67 (1Н, дд), 7,23 (2Н, м), 7,53 (2Н, м), 7,69 (1Н, д), 8,01 (1Н, д).
МС [ESI] МН+ = 294.
Маточный раствор содержал 3,19 г (14,5%) продукта.
Пример 9. Взаимодействие 1,2,3,9-тетрагидро-9-метил-4H-карбазол-4-она с 4,4-диметилоксазолидином.
1,2,3,9-Тетрагидро-9-метил-4H-карбазол-4-он (13,26 г 66,5 ммоль) и метансульфоновую кислоту (10,23 г 106,4 ммоль) в 1-бутаноле (45 мл) нагревали до 90°C. В течение 2 минут добавляли 4,4-диметилоксазолидин (10,09 г 99,9 ммоль) в 1-бутаноле (8 мл).
Через 50 минут при 80°C добавляли 2-метилимидазол (27,32 г 332,5 ммоль) и 1-бутанол (8 мл). Через 2 часа при 120°C добавляли 180 мл толуола и при 80°C 120 мл воды. Слои разделяли. Водный слой экстрагировали 180 мл толуола и 60 мл 1-бутанола. Объединенные органические слои промывали дважды 240 мл воды. Органический слой упаривали досуха. К остатку добавляли 150 мл 1-бутанола и 10 мл 36% масс/масс соляной кислоты. При 0°C вскоре происходила кристаллизация. Через 1 час при 0°C образовавшиеся кристаллы отфильтровывали, промывали 1-бутанолом и МТБЭ и затем сушили: 10,02 г (45,7%) гидрохлорид 1,2,3,9-тетрагидро-9-метил-3-[(2-метил-1H-имидазол-1-ил)метил]-4H-карбазол-4-она. Маточный раствор содержал 2,70 г (12,3%) продукта ВЭЖХ: ≥ 95%. ЯМР и MS: смотри пример 6.
Пример 10. Взаимодействие 1,2,3,9-тетрагидро-9-метил-4H-карбазол-4-она с 3-этилоксазолидином.
1,2,3,9-Тетрагидро-9-метил-4H-карбазол-4-он (13,26 г 66,5 ммоль) и метансульфоновую кислоту (10,23 г 106,4 ммоль) в 1-бутаноле (45 мл) нагревали до 90°C. В течение 2 минут добавляли 3-этилоксазолидин (10,09 г 99,9 ммоль) в 1-бутаноле (8 мл). Через 50 минут при 80°C добавляли 2-метилимидазол (27,32 г 332,5 ммоль) и 1-бутанол (8 мл). Через 2 часа при 120°C добавляли 180 мл толуола и 120 мл воды при 80°C. Слои разделяли. Водный слой экстрагировали 180 мл толуола и 60 мл 1-бутанола. Объединенные органические слои промывали дважды 240 мл воды. Органический слой упаривали досуха. К остатку добавляли 150 мл 1-бутанола и 10 мл 36% масс/масс соляной кислоты. При 0°C вскоре происходила кристаллизация. Через 1 час при 0°C образовавшиеся кристаллы отфильтровывали, промывали 1-бутанолом и МТБЭ и затем сушили: отделяли 15,67 г (71,4%) гидрохлорида 1,2,3,9-тетрагидро-9-метил-3-[(2-метил-1Н-имидазол-1-ил)метил]-4Н-карбазол-4-она. ВЭЖХ: 95%. ЯМР и MS: смотри пример 8. Маточный раствор содержал 2,06 г (9,4%) продукта.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ 1,2,3,9-ТЕТРАГИДРО-9-МЕТИЛ-3-[(2-МЕТИЛ-1Н-ИМИДАЗОЛ-1-ИЛ)-МЕТИЛ]-4Н-КАРБАЗОЛ-4-ОНА | 1993 |
|
RU2109741C1 |
| Производные тетрагидротриазолопиримидина в качестве ингибиторов нейтрофильной эластазы человека | 2012 |
|
RU2622643C2 |
| ПРОИЗВОДНЫЕ ТЕТРАГИДРОКАРБАЗОЛА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2005 |
|
RU2430088C2 |
| ЛЕКАРСТВЕННЫЕ СРЕДСТВА, СОДЕРЖАЩИЕ ПРОИЗВОДНЫЕ ИМИДАЗОЛ-1-ИЛА | 1993 |
|
RU2125877C1 |
| ПРОИЗВОДНЫЕ КАРБАЗОЛОНА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1993 |
|
RU2119914C1 |
| СПОСОБЫ ПОЛУЧЕНИЯ АПОПТОЗ АГЕНТА | 2014 |
|
RU2660424C2 |
| ТРИЦИКЛИЧЕСКИЕ АЗОТСОДЕРЖАЩИЕ СОЕДИНЕНИЯ И ПРОИЗВОДНЫЕ ТЕТРАГИДРОХИНОЛИНА ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ | 1991 |
|
RU2023712C1 |
| ПРОИЗВОДНЫЕ 3-(ГЕТЕРОАРИЛАМИНО)-1,2,3,4-ТЕТРАГИДРО-9Н-КАРБАЗОЛА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ МОДУЛЯТОРОВ РЕЦЕПТОРОВ ПРОСТАГЛАНДИНА D2 | 2011 |
|
RU2562255C2 |
| СОЕДИНЕНИЯ, СПОСОБ ПОЛУЧЕНИЯ СОЕДИНЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2000 |
|
RU2244709C2 |
| 1,7-АНЕЛЛИРОВАННЫЕ ПРОИЗВОДНЫЕ 3-(ПИПЕРАЗИНОАЛКИЛ)-ИНДОЛА, СПОСОБ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ), СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ПРОМЕЖУТОЧНЫЙ ПРОДУКТ ДЛЯ ИХ ПОЛУЧЕНИЯ | 1992 |
|
RU2083580C1 |
Настоящее изобретение относится к способу получения производного имидазолила общей формулы I, где Ra и Rb, каждый, независимо, представляют собой (С1-С6)алкил, (С1-С6)алкоксиалкил, необязательно замещенный арил или гетероарил; или где Ra и Rb вместе образуют дополнительную гомоциклическую или гетероциклическую систему, содержащую одно или несколько колец; Ra' и Rb', каждый, представляют собой водород, или вместе образуют углерод-углеродную двойную связь, указанная углерод-углеродная двойная связь необязательно является частью ароматической системы; Rc представляет собой водород, (С1-С6)алкил, (C1-С6)алкокси, (С1-С6)алкоксиалкил или галоген; Rd представляет собой водород или (С1-С4)алкил; Re представляет собой водород или (С1-С4)алкил; m равно 1 или 2; и R1 представляет собой водород или (С1-С4)алкил; а также его соли добавления кислоты; отличающийся тем, что соединение общей формулы II, где значения Ra, Ra', Rb, Rb' такие, как указано выше; подвергают взаимодействию с соединением формулы III, где R представляет собой водород, (С1-С4)алкильную группу, необязательно замещенную гидроксигруппой или необязательно замещенную арильную группу, R', R", R" и R'''', каждый, независимо, представляют собой водород или (C1-С4)алкильную группу; затем подвергают взаимодействию с соединением формулы IV, где R1, Rd и Re имеют значения, указанные выше; и затем, необязательно, подвергают взаимодействию с соответствующей кислотой. Технический результат - способ по настоящему изобретению особенно эффективен для получения ондансетрона и цилансетрона. 9 з.п. ф-лы/
где Ra и Rb каждый независимо представляет собой (С1-С6)алкил, (С1-С6)алкоксиалкил, необязательно замещенный арил или гетероарил;
или где Ra и Rb вместе образуют дополнительную гомоциклическую или гетероциклическую систему, содержащую одно или несколько колец;
Ra' и Rb' каждый представляет собой водород, или вместе образуют углерод-углеродную двойную связь, указанная углерод-углеродная двойная связь необязательно является частью ароматической системы;
Rc представляет собой водород, (С1-С6)алкил, (С1-С6)алкокси, (С1-С6)алкоксиалкил или галоген;
Rd представляет собой водород или (С1-С4)алкил;
Re представляет собой водород или (С1-С4)алкил;
m равно 1 или 2;
R1 представляет собой водород или (С1-С4)алкил;
а также его соли добавления кислоты;
отличающийся тем, что соединение общей формулы
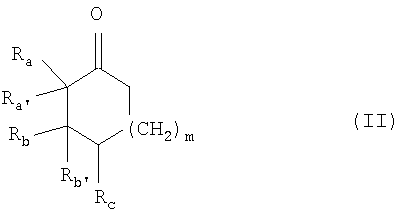
где значения Ra, Ra', Rb, Rb' такие, как указано выше;
подвергают взаимодействию с соединением формулы
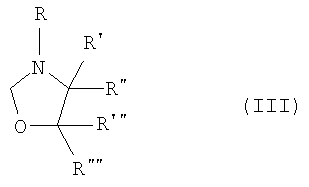
где R представляет собой водород, (С1-С4)алкильную группу, необязательно замещенную гидроксигруппой или необязательно замещенную арильную группу,
R', R", R" и R"" каждый независимо представляет собой водород или (С1-С4)алкильную группу;
затем подвергают взаимодействию с соединением формулы
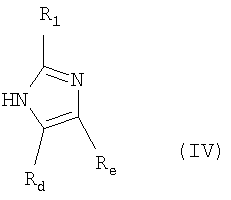
где R1, Rd и Re имеют значения, указанные выше;
и затем, необязательно, подвергают взаимодействию с соответствующей кислотой.
где Rc представляет собой водород, (С1-С6)алкил; Rd представляет собой водород или (С1-С4)алкил; Re представляет собой водород или (С1-С4)алкил; m равно 1 или 2; R1 представляет собой водород, метил или этил.
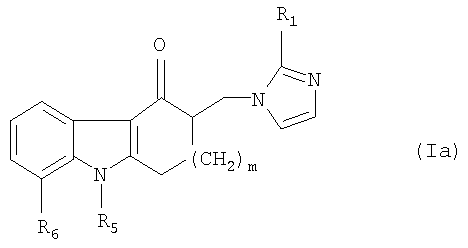
где m равно 1 или 2;
R1 представляет собой водород, метил или этил;
R5 представляет собой (С1-С4)алкил;
R6 представляет собой водород или (С1-С4)алкил, или
R5 и R6 вместе с промежуточными атомами образуют 5-, 6- или 7-членное кольцо, необязательно замещенное одним или двумя заместителями, выбранными из группы, включающей галоген, гидроксил, (С1-С4)алкил, (С1-С4)алкоксиалкил и (С1-С4)алкокси, а также его фармацевтически приемлемой соли добавления кислоты;
отличающийся тем, что соединение общей формулы
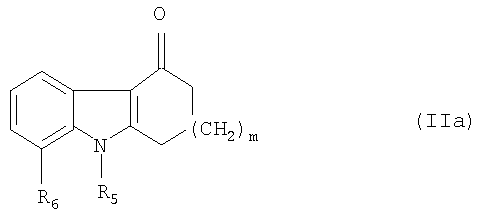
где R5, R6 и m имеют значения, указанные выше,
подвергают взаимодействию с соединением формулы
где R, R', R", R'" и R"" имеют значения, указанные в п.1;
а затем подвергают взаимодействию с соединением формулы
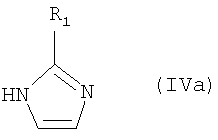
где R1 имеет значение, указанное в п.1.
| Способ получения термостойкого связующего на основе ароматических дималеимидов | 1969 |
|
SU297651A1 |
| Способ получения 1-этил-имидазоловили их солей | 1974 |
|
SU509225A3 |

