УРОВЕНЬ ТЕХНИКИ
В Международных патентных заявках US03/12929, поданной 25 апреля 2003 г. и US03/13042, поданной 25 апреля 2003 г., описаны соединения тетрагидропиранилциклопентил-тетрагидропиридопиридина. Данные соединения пригодны для лечения заболеваний или состояний людей или других видов, которые могут поддаваться лечению ингибиторами, модуляторами или промоторами функции хемокинового рецептора. Подобные заболевания или состояния включают заболевания или состояния, перечисленные в упомянутых заявках.
((1R,3S)-3-Изопропил-3-{[3-(трифторметил)-7,8-дигидро-1,6-нафтиридин-6(5Н)-ил]карбонил}циклопентил)[(3S,4S)-3-метокситетрагидро-2Н-пиран-4-ил]амин, 1:
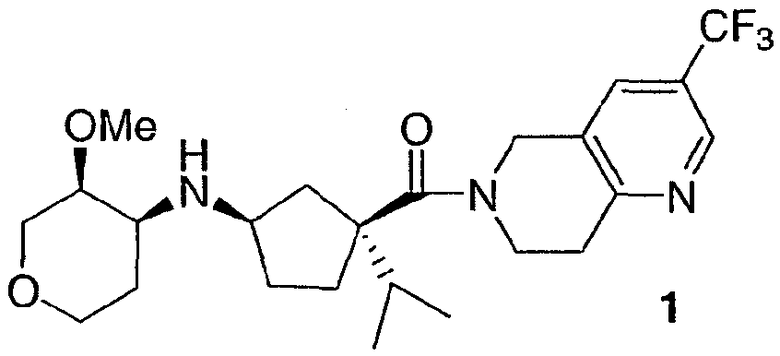
является сильным ингибитором CCR-2. Лабораторное получение соединения 1, включая лабораторное получение некоторых промежуточных соединений и предшественников, используемых в синтезе соединения 1, описано в Международных патентных заявках US03/12929 и US03/13042.
Известные способы синтеза соединения 1 неэффективны, требуют много времени и, таким образом, являются дорогостоящими с точки зрения производства. Например, синтез нафтиридинового структурного блока включает девять отдельных стадий и требует использования нескольких дорогостоящих реагентов, что снижает общую эффективность процесса синтеза. Подобным образом синтез циклопентенового структурного блока приводит к высокому уровню содержания нежелательных стереоизомеров. Другие аспекты синтеза также являются неэффективными, дорогостоящими и/или неподходящими для промышленного производства. Более того, синтезированные ранее гидрохлориды соединения 1 проявляют далеко не идеальные свойства при их растворении, обработке (например, гидроскопичность) и т.п. Таким образом, сохраняется потребность в разработке усовершенствованного синтетического способа получения соединения 1, пригодного для крупномасштабного производства, хранения и распространения. Также сохраняется потребность в разработке усовершенствованных солевых форм соединения 1.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретения касается эффективного синтеза для получения ((1R,3S)-3-изопропил-3-{[3-(трифторметил)-7,8-дигидро-1,6-нафтиридин-6(5Н)-ил]карбонил}циклопентил)[(3S,4S)-3-метокситетрагидро-2Н-пиран-4-ил]амина и его сукцината. Настоящее изобретение также касается эффективного синтеза для получения промежуточных соединений (3R)-3-метокситетрагидро-4Н-пиран-4-она; (1S,4S)-4-(2,5-диметил-1Н-пиррол-1-ил)-1-изопропилциклопент-2-ен-1-карбоновой кислоты и 3-(трифторметил)-5,6,7,8-тетрагидро-1,6-нафтиридина; а также для получения предшественника (3S,4S)-N-((1S,4S)-4-изопропил-4-{[3-(трифторметил)-7,8-дигидро-1,6-нафтиридин-6(5Н)-ил]карбонил}циклопент-2-ен-1-ил)-3-метокситетрагидро-2Н-пиран-4-амина. Кроме того, данное изобретение основано на превосходных свойствах сукцината ((1R,3S)-3-изопропил-3-{[3-(трифторметил)-7,8-дигидро-1,6-нафтиридин-6(5Н)-ил]карбонил}циклопентил)[(3S,4S)-3-метокситетрагидро-2Н-пиран-4-ил]амина.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Согласно одному из аспектов настоящее изобретение касается способа получения (3S,4S)-N-((1S,4S)-4-изопропил-4-{[3-(трифторметил)-7,8-дигидро-1,6-нафтиридин-6(5Н)-ил]карбонил}циклопент-2-ен-1-ил)-3-метокситетрагидро-2Н-пиран-4-амина, 2:
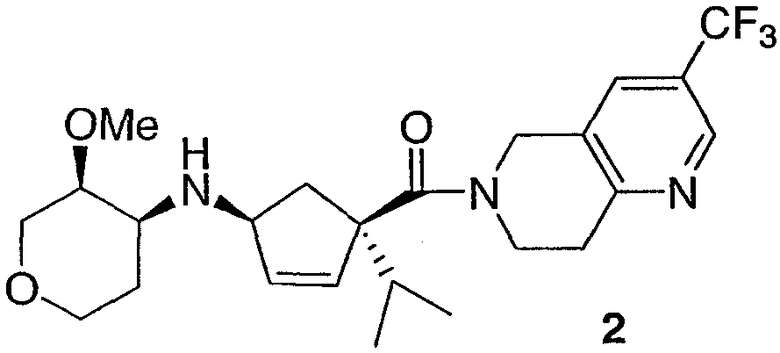
включающего следующие стадии:
(1) взаимодействие (1S,4S)-4-(2,5-диметил-1Н-пиррол-1-ил)-1-изопропилциклопент-2-ен-1-карбоновой кислоты с 3-(трифторметил)-5,6,7,8-тетрагидро-1,6-нафтиридином для получения 6-{[(1S,4S)-4-(2,5-диметил-1Н-пиррол-1-ил)-1-изопропилциклопент-2-ен-1-ил]карбонил}-3-(трифторметил)-5,6,7,8-тетрагидро-1,6-нафтиридина;
(2) обработку 6-{[(1S,4S)-4-(2,5-диметил-1Н-пиррол-1-ил)-1-изопропилциклопент-2-ен-1-ил]карбонил}-3-(трифторметил)-5,6,7,8-тетрагидро-1,6-нафтиридина гидроксиламином для получения (1S,4S)-4-изопропил-4-{[3-(трифторметил)-7,8-дигидро-1,6-нафтиридин-6(5Н)-ил]карбонил}циклопент-2-ен-1-амина; и
(3) сочетание (1S,4S)-4-изопропил-4-{[3-(трифторметил)-7,8-дигидро-1,6-нафтиридин-6(5Н)-ил]карбонил}циклопент-2-ен-1-амина с (3R)-3-метокситетрагидро-4Н-пиран-4-оном путем восстановительного аминирования.
Согласно следующему аспекту настоящее изобретение касается способа получения ((1R,3S)-3-изопропил-3-{[3-(трифторметил)-7,8-дигидро-1,6-нафтиридин-6(5Н)-ил]карбонил}циклопентил)[(3S,4S)-3-метокситетрагидро-2Н-пиран-4-ил]амина, 1:
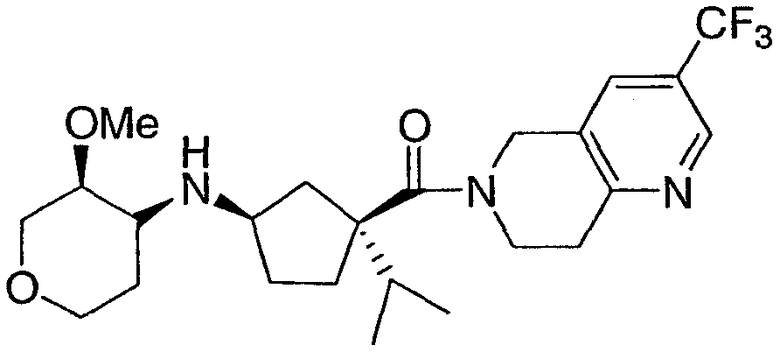
включающего следующие стадии:
(4) гидрогенизации (3S,4S)-N-((1S,4S)-4-изопропил-4-{[3-(трифторметил)-7,8-дигидро-1,6-нафтиридин-6(5Н)-ил]карбонил}циклопент-2-ен-1-ил)-3-метокситетрагидро-2Н-пиран-4-амина.
Согласно другому аспекту данного изобретения, более подробно описываемому ниже, стадию гидрогенизации осуществляют перед получением соединения 2.
Синтезы (3S,4S)-N-((1S,4S)-4-изопропил-4-{[3-(трифторметил)-7,8-дигидро-1,6-нафтиридин-6(5Н)-ил]карбонил}циклопент-2-ен-1-ил)-3-метокситетрагидро-2Н-пиран-4-амина, 2, и ((1R,3S)-3-изопропил-3-{[3-(трифторметил)-7,8-дигидро-1,6-нафтиридин-6(5Н)-ил]карбонил}циклопентил)[(3S,4S)-3-метокситетрагидро-2Н-пиран-4-ил]амина, 1, представлены на схеме 1:
Схема 1
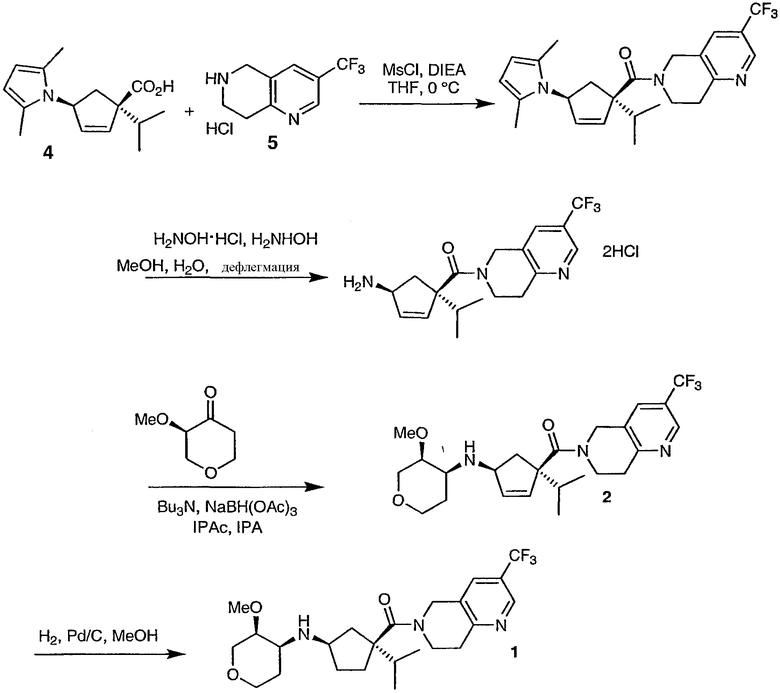
N-((1R,3S)-3-изопропил-3-{[3-(трифторметил)-7,8-дигидро-1,6-нафтиридин-6(5Н)-ил]карбонил}циклопентил)-N-[(цис-3S,4S)-3-метокситетрагидро-2Н-пиран-4-ил]амин, 1, синтезируют, последовательно сочетая три соединения-структурных блока, 4, 5 и 6 (представленные ниже схемы 3, 4 и 5). Реакцию амидирования между 3-(трифторметил)-5,6,7,8-тетрагидро-1,6-нафтиридином, 5, и (1S,4S)-4-(2,5-диметил-1Н-пиррол-1-ил)-1-изопропилциклопент-2-ен-1-карбоновой кислотой, 4, осуществляют в присутствии метансульфонилхлорида, получая 6-{[(1S,4S)-4-(2,5-диметил-1Н-пиррол-1-ил)-1-изопропилциклопент-2-ен-1-ил]карбонил}-3-(трифторметил)-5,6,7,8-тетрагидро-1,6-нафтиридин. Снятие защиты с группы пиррола осуществляют на данной стадии, используя гидроксиламин в качестве прямого процесса для получения соли амина. Соль амина и (3R)-3-метокситетрагидро-4Н-пиран-4-он, 6, сочетают путем восстановительного аминирования в присутствии трибутиламинового буфера, и с NaBH(OAc)3, получая (3S,4S)-N-((1S,4S)-4-изопропил-4-{[3-(трифторметил)-7,8-дигидро-1,6-нафтиридин-6(5Н)-ил]карбонил}циклопент-2-ен-1-ил)-3-метокситетрагидро-2Н-пиран-4-амин, 2. Циклопентеновый фрагмент соединения 2 затем гидрогенизируют, получая соединение 1 в виде свободного основания.
Другой аспект данного изобретения включает способ получения сукцината ((1R,3S)-3-изопропил-3-{[3-(трифторметил)-7,8-дигидро-1,6-нафтиридин-6(5Н)-ил]карбонил}циклопентил)[(3S,4S)-3-метокситетрагидро-2Н-пиран-4-ил]амина, 3:
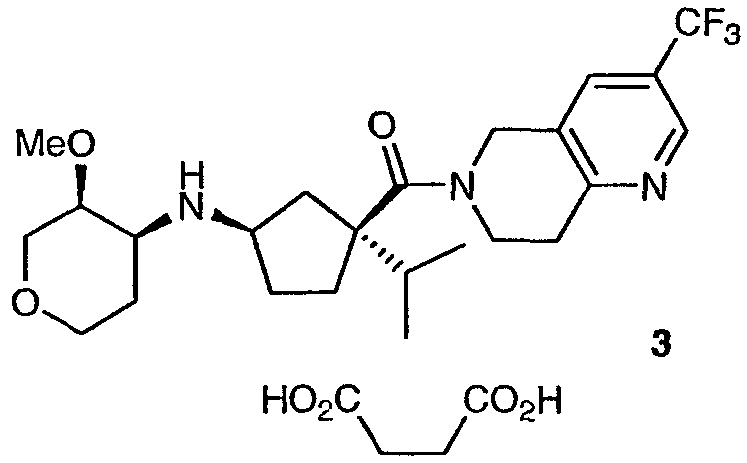
включающий дополнительную стадию:
(5) контактирования ((1R,3S)-3-изопропил-3-{[3-(трифторметил)-7,8-дигидро-1,6-нафтиридин-6(5Н)-ил]карбонил}циклопентил)[(3S,4S)-3-метокситетрагидро-2Н-пиран-4-ил]амина, 1, с янтарной кислотой.
Перед стадией (5) соединение 1 может быть, необязательно, очищено при помощи кристаллизации либо в виде сукцината, либо иной соли, такой как бензолсульфонат, с последующим разрушением соли. Очистка таким способом описана в следующих примерах.
Синтез соединения 3, сукцината, представлен на схеме 2:
Схема 2
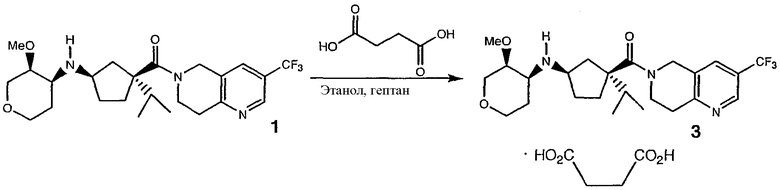
Другой аспект данного изобретения касается способа получения промежуточной (1S,4S)-4-(2,5-диметил-1Н-пиррол-1-ил)-1-изопропилциклопент-2-ен-1-карбоновой кислоты, 4, включающего следующие стадии:
(1) взаимодействие (1R,4S)-4-аминоциклопентен-2-ен-1-карбоновой кислоты с МеОН в присутствии тионилхлорида для получения метил (1R,4S)-4-аминоциклопент-2-ен-1-карбоксилата;
(2) взаимодействие упомянутого метил (1R,4S)-4-аминоциклопент-2-ен-1-карбоксилата с ацетилацетоном для получения метил (1R,4S)-4-(2,5-диметил-1Н-пиррол-1-ил)циклопент-2-ен-1-карбоксилата;
(3) взаимодействие упомянутого метил (1R,4S)-4-(2,5-диметил-1Н-пиррол-1-ил)циклопент-2-ен-1-карбоксилата с 2-иодпропаном для получения метил (1S,4S)-4-(2,5-диметил-1Н-пиррол-1-ил)-1-изопропилциклопент-2-ен-1-карбоксилата; и
(4) взаимодействие упомянутого метил-(1S,4S)-4-(2,5-диметил-1Н-пиррол-1-ил)-1-изопропилциклопент-2-ен-1-карбоксилата с NaOH и МеОН для получения (1S,4S)-4-(2,5-диметил-1Н-пиррол-1-ил)-1-изопропилциклопент-2-ен-1-карбоновой кислоты.
Синтез (1S,4S)-4-(2,5-диметил-1Н-пиррол-1-ил)-1-изопропилциклопент-2-ен-1-карбоновой кислоты, 4, представлен на схеме 3:
Схема 3
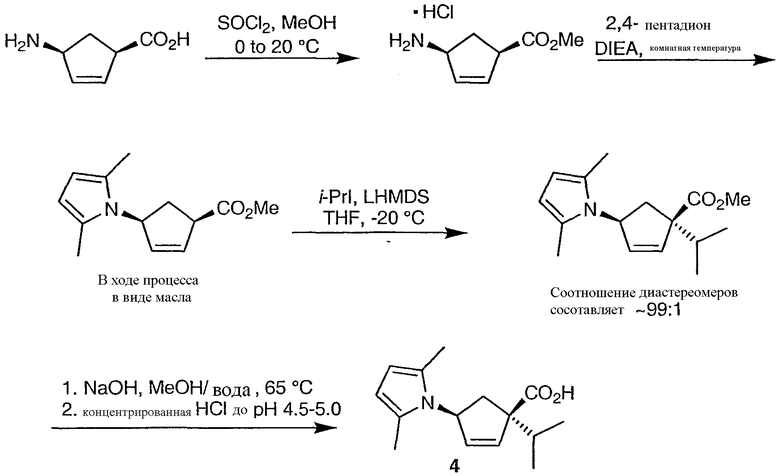
Согласно данному синтезу (1R,4S)-4-аминоциклопентен-2-ен-1-карбоновую кислоту превращают в соответствующий сложный метиловый эфир путем обработки тионилхлоридом в метаноле. Метил (1R,4S)-4-(2,5-диметил-1Н-пиррол-1-ил)циклопент-2-ен-1-карбоксилат затем синтезируют взаимодействием метил-(1R,4S)-4-аминоциклопент-2-ен-1-карбоксилата с 2,4-пентандионом в присутствии DIEA. Последующее алкилирование метил-(1R,4S)-4-(2,5-диметил-1Н-пиррол-1-ил)циклопент-2-ен-1-карбоксилата иодпропаном осуществляют, используя бис-(триметилсилил)амид лития в качестве основания. Полученный алкилированный продукт затем гидролизуют для получения (1S,4S)-4-(2,5-диметил-1Н-пиррол-1-ил)-1-изопропилциклопент-2-ен-1-карбоновой кислоты 4.
Несмотря на то, что вышеприведенное описание предполагает гидрогенизацию циклопентенового фрагмента на последней стадии для получения циклопентана, фактически, многие соединения, представленные на схемах 1 и 3, могут быть гидрогенизированы на промежуточной стадии с получением аналога циклопентана. (Благодаря тому, что пирролозащитная группа, при ее наличии, также восстанавливается до пирролидина во время такого процесса, может быть использована другая защитная группа).
Более того, согласно другому отличительному признаку данного аспекта настоящего изобретения могут быть синтезированы промежуточные соединения, имеющие защитные группы, отличные от 2,5-диметил-1Н-пиррол-1-ила. Таким образом, опытные химики стремятся получить промежуточные соединения общей формулы:
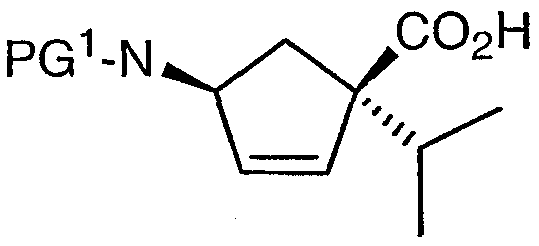
где защитная группа "PG1" включает, но не ограничивается ими, третбутоксикарбонил, бензилоксикарбонил, алкилоксикарбонил, аллилоксикарбонил, бензоил, формил, ацетил, трифторацетил, 2-нитробензолсульфонил, 4-нитробензолсульфонил, 2,4-динитробензолсульфонил, бензил, трифенилметил, имины (такие как дифенилметилен) и другие защитные группы, известные в данной области и проиллюстрированные Greene, T.; Wuts, P.G.M. Protective Groupsin Organic Synthesis, 3rd Ed., John Wiley & Sons, Inc., New York, NY 1999.
Другой аспект данного изобретения касается способа получения промежуточного 3-(трифторметил)-5,6,7,8-тетрагидро-1,6-нафтиридина, 5, включающего следующие стадии:
(1) взаимодействие 3,3,3-трифторпропановой кислоты с POCl3, DMF, NaPF6 и основанием для получения гексафторфосфата N-[3-(диметиламино)-2-(трифторметил)проп-2-енилиден]-N-метилметанамия (CF3DT);
(2) взаимодействие CF3DT с защищенным пиперидоном для получения -N-(защитная группа)-3-(трифторметил)-5,6,7,8-тетрагидро-1,6-нафтиридина (например, взаимодействие CF3DT с ВОС-пиперидоном для получения -N-(третбутоксикарбонил)-3-(трифторметил)-5,6,7,8-тетрагидро-1,6-нафтиридина); и
(3) взаимодействие упомянутого -N-(защитная группа)-3-(трифторметил)-5,6,7,8-тетрагидро-1,6-нафтиридинена (например, N-(третбутоксикарбонил)-3-(трифторметил)-5,6,7,8-тетрагидро-1,6-нафтиридин) в присутствии HCl и метанола для получения -3-(трифторметил)-5,6,7,8-тетрагидро-1,6-нафтиридина.
Синтез 3-(трифторметил)-5,6,7,8-тетрагидро-1,6-нафтиридина, 5, представлен ниже на схемах 4А и 4В:
Схема 4А
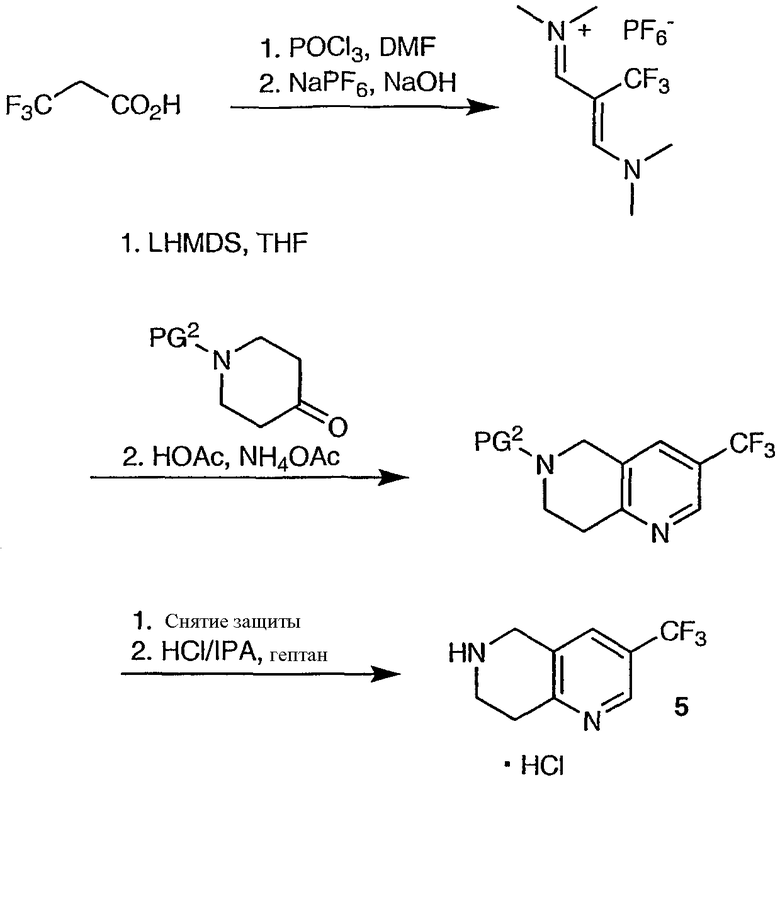
Схема 4В
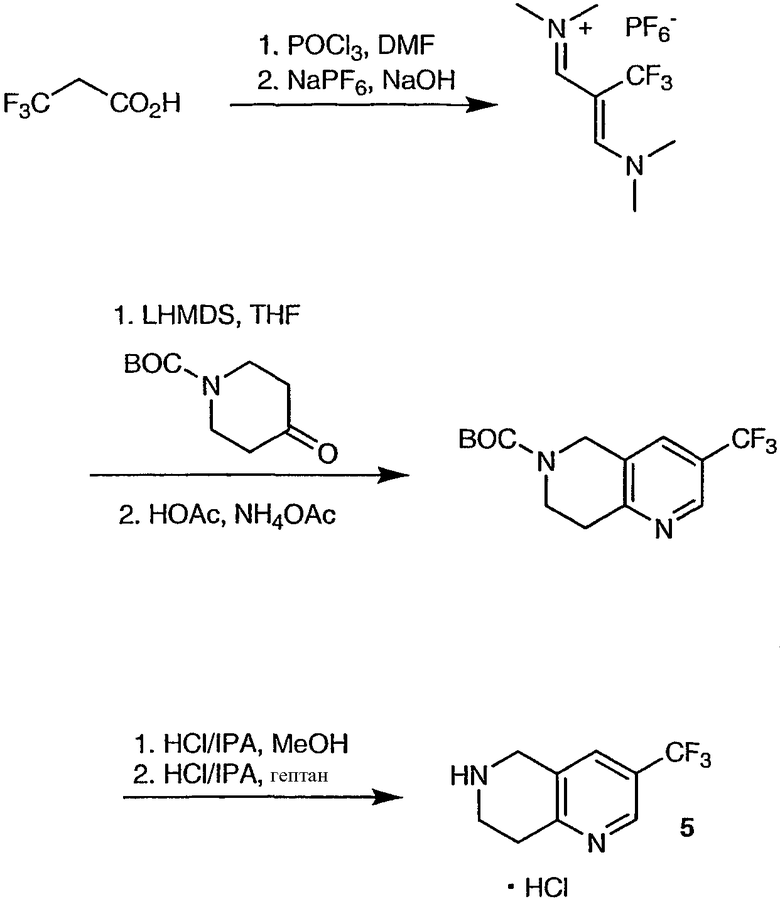
Оксихлорид фосфора (POCl3) добавляют к охлажденному DMF с такой скоростью, чтобы температура оставалась ниже 10°С (приблизительно в течение часа). Реакционную смесь нагревают до комнатной температуры, а затем добавляют 3,3,3-трифторпропановую кислоту (экзотермия приблизительно до 45°С). Реакционную смесь нагревают до 50°С и выдерживают при данной температуре в течение приблизительно 4 часов. По завершении образования винамидиния реакционной смеси дают возможность остыть до комнатной температуры. Затем реакционную смесь одновременно с 5 Н NaOH добавляют к раствору NaPF6 в воде, охлажденному до 0°С. Скорости добавления двух растворов контролируют таким образом, что температура водной суспензии остается ниже 10°С, а рН составляет от 3 до 4 (добавление осуществляют в течение приблизительно 2 часов). Завершив добавление, полученную желтую суспензию подвергают старению в течение часа при 0°С, а затем фильтруют, отделяя твердые вещества. Фильтровальный осадок промывают ледяной водой (2 раза) и сушат азотом в вакууме. Типичный выход составляет 85%.
Раствор защищенного пиперидона, например защищенного ВОС пиперидона, в ТГФ затем добавляют к охлажденному (-20°С) раствору гексаметилдисилиламида лития (LiN(TMS)2) в ТГФ, поддерживая температуру ниже 10°С для образования енолята лития (приблизительно 45 минут). После нагревания до комнатной температуры раствор енолята переносят в охлажденную (-20°С) суспензию CF3DT в ТГФ с такой скоростью, что внутренняя температура остается на уровне ниже -10°С (в течение приблизительно 45 минут). Полученную смесь подвергают старению в течение 2 часов при температуре -20°С, затем добавляют уксусную кислоту и смесь нагревают до комнатной температуры в течение 30 минут. Затем добавляют ацетат аммония и смесь нагревают до 65°С. После двух часов выдерживания при температуре 65°С реакционную смесь охлаждают до комнатной температуры. Затем добавляют воду и гептан и слои разделяют. Органический слой промывают 2 М водной лимонной кислотой, а затем анализируют на ВОС нафтиридин. Типичный выход составляет 65%.
После этого органический слой со стадии образования нафтиридина концентрируют, а растворитель заменяют метанолом. Затем добавляют HCl в IPA и смесь нагревают при 60°С до завершения снятия защиты (около часа). После охлаждения до комнатной температуры добавляют воду, а рН доводят приблизительно до 10,5. Добавляют IPAc и слои разделяют. Водный слой вновь дополнительно дважды экстрагируют изопропилацетатом (IPAc). Объединенные органические слои концентрируют, а растворитель заменяют IPA. Полученный раствор IPA фильтруют, удаляя неорганические соли, осадок на фильтре промывают IPA, а объединенный фильтрат и промывные воды вновь концентрируют до общего объема около 5 мг/л. После этого раствор свободного основания/IPA добавляют в течение 30 минут к 60°С HCl в IPA. Твердые вещества становятся заметными после добавления приблизительно 50% раствора. Закончив добавление HCl/IPA, добавляют гептан для завершения кристаллизации и суспензию охлаждают до комнатной температуры. Твердые вещества выделяют фильтрацией и промывают IPA/гептаном (3 раза по 1 мл/г), а затем сушат. Типичный выход из ВОС нафтиридина составляет 75%.
Как указано выше, могут быть использованы пиперидоны, содержащие защитные группы ("PG2"), отличные от третбутоксикарбонила. Другие защитные группы, которые могут быть полезны в синтезе нафтиридина, включают, но не ограничиваются ими, бензилоксикарбонил, алкилоксикарбонил, аллилоксикарбонил, бензоил, ацетил, формил, трифторацетил, 2-нитробензолсульфонил, 4-нитробензолсульфонил, 2,4-динитробензолсульфонил, N-бензил, трифенилметил и другие защитные группы, известные в данной области и проиллюстрированные Greene, T.; Wuts, P.G.M. Protective Groupsin Organic Synthesis, 3rd Ed., John Wiley & Sons, Inc., New York, NY 1999.
Другой аспект данного изобретения касается способа получения промежуточного (3R)-3-метокситетрагидро-4Н-пиран-4-она, 6, включающего следующие стадии:
(1) взаимодействие тетрагидро-4Н-пиран-4-она с трипропилортоформиатом и хлорбензолом для получения 4-пропокситетрагидро-2Н-пиранэтена;
(2) взаимодействие указанного 4-пропокситетрагидро-2Н-пиранэтена в присутствии ацетона, воды, простого диэфира гидрохинидин-1,4-фталазиндиила (DHQD2PHAI), дегидрата осмата калия и моногидрата 4-метилморфолин-N-оксида (NMO) для получения натриевой соли 3,4-дигидрокситетрагидро-2Н-пиран-4-сульфоновой кислоты;
(3) взаимодействие указанной натриевой соли 3,4-дигидрокситетрагидро-2Н-пиран-4-сульфоновой кислоты с метанолом и триметилортоформиатом в присутствии кислоты для получения 4,4-диметокситетрагидро-2Н-пиран-3-ола; и
(4) взаимодействие указанного 4,4-диметокситетрагидро-2Н-пиран-3-ола в присутствии ТГФ, NaOt-Bu, Me2SO4 и кислоты для получения (3R)-3-метокситетрагидро-4Н-пиран-4-она.
Синтез (3R)-3-метокситетрагидро-4Н-пиран-4-она, 6, представлен на схеме 5:
Схема 5
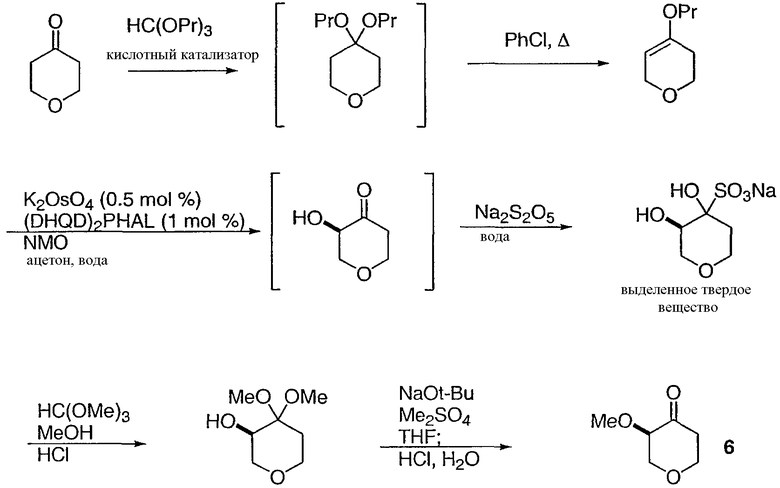
Исходный пиранон превращают в его дипропилкеталь путем обработки трипропилортоформиатом в присутствии кислотного катализатора. Затем необработанный кеталь нагревают в присутствии хлорбензола. В таких условиях удаление пропанола приводит к получению простого пропиленольного эфира. Реакцию направляют, удаляя пропанол путем дистилляции в ходе реакции. Затем необработанный простой енольный эфир окисляют в модифицированных условиях асимметричного дигидроксилирования Sharpless. В данной реакции в качестве стехиометрического окислителя используют оксид N-метилморфолина. Обычно реакция приводит к получению такого продукта, как α-гидроксикетон в количестве приблизительно от 60 до 85% энантиомерного избытка (ее). α-Гидроксикетон не выделяют непосредственно, а добавляют водный раствор Na2S2O5 для получения бисульфитного аддукта кетона. Из смеси ацетона и воды кристаллизуется рацемический бисульфитный аддукт. Рацемат удаляют фильтрацией, при этом получаемый маточный раствор обычно составляет от 95 до 99% энантиомерного избытка (ее). Ацетон удаляют в вакууме и добавляют изопропанол, получая аддукт кристаллического бисульфита с большим энантиомерным избытком. Его обрабатывают HCl и метанолом с триметилортоформиатом в качестве акцептора воды для получения диметилкеталя. Затем гидроксильную группу метилируют, используя NaOt-Bu и Me2SO4. Добавляя воду и HCl к реакционной смеси, получают целевой α-метоксипиранон с энантиомерным избытком, составляющим около 96%.
Ниже представлены значения некоторых аббревиатур, акронимов и других сокращений. Несмотря на то, что приведенные термины известны специалистам в данной области, ниже представлена таблица, суммирующая такие термины:
DIEA
IPAc
IPA
i-PrI
LHMDS
THF (ТГФ)
DMF
P.G.
BOC
NMO
TFPA
диизопропилэтиламин
изопропилацетат
изопропанол
2-иодпропан
лития гексаметилдисилазид
тетрагидрофуран
диметилформамид
защитная группа
третбутилоксикарбонил
оксид N-метилморфалина
трифторпропионовая кислота
Пример 1
((1R,3S)-3-Изопропил-3-{[3-(трифторметил)-7,8-дигидро-1,6-
нафтиридин-6(5Н)-ил]карбонил}циклопентил)[(3S,4S)-3-
метокситетрагидро-2Н-пиран-4-ил]амин
Стадия 1 - 6-{[(1S,4S)-4-(2,5-диметил-1Н-пиррол-1-ил)-1-изопропилциклопент-2-ен-1-ил]карбонил}-3-(трифторметил)-5,6,7,8-тетрагидро-1,6-нафтиридин
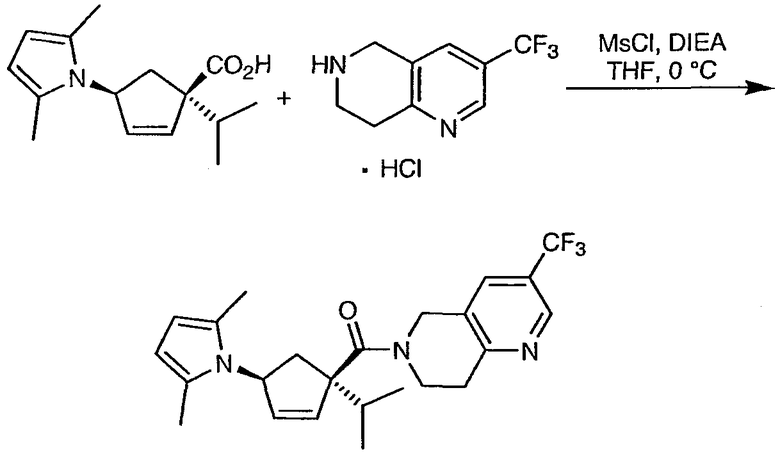
Пирролциклопентеновую кислоту (730 г, 90,4 мас.%, 2,67 ммол) и диизопропилэтиламин (0,93 л, 5,34 мол) смешивают в ТГФ (6,6 л). Полученный темный раствор (KF = 195 мг/мл) охлаждают до 0°С, затем в течение 1 минуты добавляют метансульфонилхлорид (228 мл, 2,94 мол), после чего внутренняя температура в течение нескольких минут повышается до 13°С. Охлаждающую ванну удаляют, а смесь выдерживают при комнатной температуре в течение 4 часов. Реакционный раствор охлаждают до ˜15°С и добавляют HCl соль тетрагидронафтиридина (670 г, 73,2 мас.% в виде эквивалента свободного основания, 2,42 мол). Температуру повышают до 23°С и добавляют другую порцию диизопропилэтиламина (0,93 л, 5,34 мол) с охлаждением при ˜25°С в течение 15 минут. Смесь выдерживают в течение >1 часа, а затем разбавляют 5% NaHCO3 (16 л). Продукт экстрагируют этилацетатом (EtOAc) (16 л). Органическую фазу промывают водой (10 л) и концентрируют в вакууме, замещая EtOAc метанолом до объема 10 л. Подтверждаемый анализом выход амида является количественным (1,055 кг, 2,45 мол).
Стадия 2 - (1S,4S)-4-изопропил-4-{[3-(трифторметил)-7,8-дигидро-1,6-нафтиридин-6(5Н)-ил]карбонил}циклопент-2-ен-1-амин
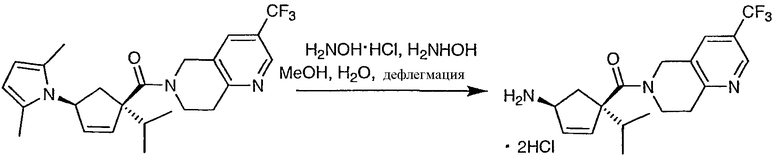
Амид (1,055 кг, 2,45 мол) в метаноле (10 л) добавляют к гидроксиламину-HCl (1 кг, 14,4 мол), 50% гидроксиламину в воде (1 л, 16,3 мол) и воде (5 л). Полученную суспензию нагревают до дефлегмации (71°С) и поддерживают при данной температуре в течение 6 часов. Реакционный раствор охлаждают до комнатной температуры, а рН доводят до 11,0 при помощи 10 Н NaOH. Реакционный раствор разбавляют водой (12 л) и продукт трижды экстрагируют хлорбензолом (14 л, 13 л и 13 л). Каждый органический слой один раз промывают этой же водой (10 л). Органические слои, содержащие продукт (согласно анализу: 0,79 кг, 2,24 мол, выход - 92%), объединяют и концентрируют. Растворитель заменяют изопропанолом (8 л). Добавляют безводный HCl в IPA (4,3 Н, 1,4 л, 6,0 мол). Смесь концентрируют до ˜2 л. Полученную суспензию нагревают до 70°С и медленно добавляют н-гептан (8 л). Суспензию охлаждают и выдерживают при комнатной температуре в течение ночи (16 часов). Твердые вещества отфильтровывают, промывают смесью 20% IPA/гептан (1,5 л) и сушат в атмосфере азота, получая 1,12 кг соли HCl (0,76 кг эквивалента свободного основания), при этом общий выход составляет 91%.
Стадия 3 -(3S,4S)-N-((1S,4S)-4-изопропил-4-{[3-(трифторметил)-7,8-дигидро-1,6-нафтиридин-6(5Н)-ил]карбонил}циклопент-2-ен-1-ил)-3-метокситетрагидро-2Н-пиран-4-амин
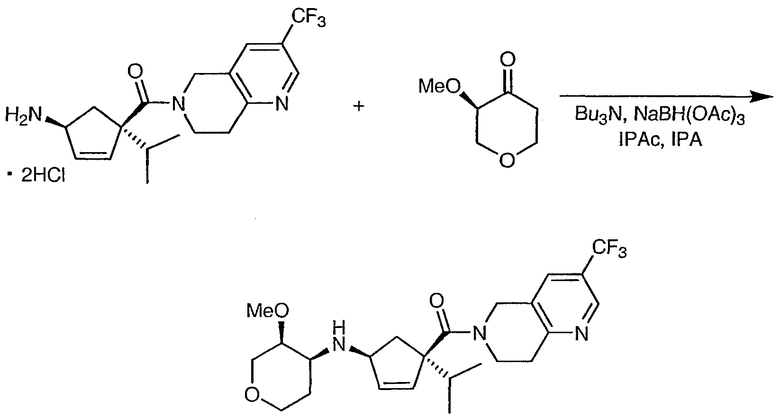
Аминдигидрохлорид (777 г, 552 г в виде свободного основания, 1,56 мол) суспендируют в IPAc (3 л). Смесь охлаждают на ледяной бане и добавляют н-Bu3N (860 мл, 3,61 мол) с последующим добавлением изопропанола (260 мл, 3,40 мол). При 5°С добавляют триацетоксиборгидрид натрия (724 г, 3,42 мол). Через час к смеси при 1°С добавляют раствор метоксипиранона в IPAc (1,76 л 160 г/л раствора, 2,17 мол). Через 6 часов смесь распределяют между насыщенным водным NaHCO3 (3 л), водой (8 л) и EtOAc (10 л). Водную фазу дополнительно экстрагируют EtOAc (15 л). Объединенные органические фазы экстрагируют насыщенным NaHCO3 (4 л), сушат над MgSO4 (600 г), а затем концентрируют. Полученное масло растворяют в CH3CN (15 л) и экстрагируют гептаном (3х4 л). Ацетонитрильная фаза содержит 654 г (1,4 мол, 90%) (3S,4S)-N-((1S,4S)-4-изопропил-4-{[3-(трифторметил)-7,8-дигидро-1,6-нафтиридин-6(5Н)-ил]карбонил}циклопент-2-ен-1-ил)-3-метокситетрагидро-2Н-пиран-4-амина. Растворитель удаляют в вакууме, получая масло, которое используют на конечной стадии гидрогенизации.
Стадия 4 - ((1R,3S)-3-изопропил-3-{[3-(трифторметил)-7,8-дигидро-1,6-нафтиридин-6(5Н)-ил]карбонил}циклопентил)[(3S,4S)-3-метокситетрагидро-2Н-пиран-4-ил]амин
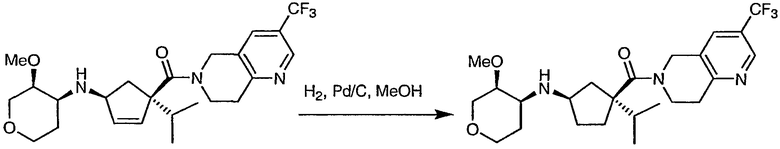
Необработанный связанный циклопентен со стадии 3 (640 г) разбавляют метанолом (3,2 л) и раствор концентрируют до масла. Разбавление метанолом (3,2 л) и концентрирование дополнительно повторяют дважды. После окончательного концентрирования масло разбавляют метанолом (6,4 л) и загружают в автоклав. Катализатор, 5% Pd/C (256 г), загружают в автоклав в виде суспензии в метаноле (1,4 л). Гидрогенизацию (40 фунтов на кв. дюйм) осуществляют в течение ночи (18 часов) при 25°С. Партию фильтруют через solka floc (глубина ˜1,5) в 8-л воронке из закаленного стекла. Автоклав промывают метанолом (5,0 л) и эту же жидкость для промывки используют для промывания осадка на фильтре. Осадок вновь промывают метанолом (1,3 л). Последовательное промывание дополнительно повторяют четырежды. Анализ при помощи ЖХ показывает, что объединенный фильтрат и промывные воды (общая масса = 17,1 кг) содержат 633 г (выход 98,5%) свободного основания. Фильтрат и промывные воды концентрируют до масла (подтверждено анализом 770 г, 1,634 мол). Масло растворяют в IPA (3,1 л) и раствор концентрируют до коричневого масла. Разбавление IPA (3,1 л) и концентрирование дополнительно повторяют дважды.
Пример 2
Сукцинат ((1R,3S)-3-изопропил-3-{[3-(трифторметил)-7,8-дигидро-1,6-нафтиридин-6(5Н)-ил]карбонил}циклопентил)[(3S,4S)-3-метокситетрагидро-2Н-пиран-4-ил]амина
Синтез 1:
Очистка от бензолсульфоната: Полученное масло из примера 1 растворяют в IPA (1,54 л) и переносят в колбу для кристаллизации. К партии добавляют промывочный IPA (2 × 385 мл). Раствор нагревают до 56°С и в этот момент добавляют бензолсульфоновую кислоту (283 г, 1,79 мол), что приводит к повышению температуры до 71°С. Раствор охлаждают до 60°С и добавляют затравку бензолсульфоната (1 г). Разбавленную суспензию выдерживают в течение 30 минут, получая густой слой затравки, после чего в течение 50 минут добавляют гептан (4,62 л). После выдерживания при температуре от 55 до 65°С в течение 2 часов, суспензию охлаждают в течение ночи до температуры окружающей среды. Суспензию фильтруют и влажный осадок промывают смесью 2:1 гептана/IPA (2 × 2,3 л). Не совсем белое твердое вещество сушат в атмосфере N2/вакуум, получая бензолсульфонат (875 г, 98,3 мас.%) в виде желтовато-коричневого твердого вещества (точно установленный выход составляет 84%). Воду (11,1 л), IPAc (32 л) и бензолсульфонат ((1R,3S)-3-изопропил-3-{[3-(трифторметил)-7,8-дигидро-1,6-нафтиридин-6(5Н)-ил]карбонил}циклопентил)[(3S,4S)-3-метокситетрагидро-2Н-пиран-4-ил]амина (1,645 кг) добавляют к раствору К2СО3 (6,58 кг) в воде (18,3 л). Смесь выдерживают в течение 15 минут. Слои разделяют и органический слой промывают водой (16 л). Раствор в IPAc свободного основания ((1R,3S)-3-изопропил-3-{[3-(трифторметил)-7,8-дигидро-1,6-нафтиридин-6(5Н)-ил]карбонил}циклопентил)[(3S,4S)-3-метокситетрагидро-2Н-пиран-4-ил]амина (60,5 кг, содержащего 1,16 кг свободного основания) концентрируют до масла. Полученное масло разбавляют этанолом (3,785 л) и вновь концентрируют до масла. Разбавление и концентрирование повторяют еще трижды.
Синтез соли янтарной кислоты: После окончательного концентрирования масло разбавляют этанолом (4,055 л). Затем раствор этанола нагревают до 65°С. Одной порцией добавляют янтарную кислоту (294 г, 2,48 мол) с последующим добавлением гептана (530 мл) в течение 10 минут. Затем раствор затравляют сукцинатом ((1R,3S)-3-изопропил-3-{[3-(трифторметил)-7,8-дигидро-1,6-нафтиридин-6(5Н)-ил]карбонил}циклопентил)[(3S,4S)-3-метокситетрагидро-2Н-пиран-4-ил]амина (5,8 г) в виде суспензии в гептане (50 мл). Полученную суспензию выдерживают в течение часа при 65°С, на протяжении которого она заметно густеет. В конце выдерживания затравочного слоя в течение 1,5 часов добавляют гептан (7,54 л). Суспензию выдерживают в течение 2 часов при 65°С, а затем ей дают возможность остыть до комнатной температуры в течение ночи (˜9 часов). Твердые вещества отфильтровывают, промывают 2:1 гептаном/этанолом (2×2,3 л, 2 мл/г свободного основания) и сушат в вакууме потоком N2 в течение 2 часов. Осадок на фильтре разбивают и дополнительно сушат в N2/вакууме в течение ˜48 часов, получая 1,328 кг сукцината (91,6%). Высушенную партию сукцината ((1R,3S)-3-изопропил-3-{[3-(трифторметил)-7,8-дигидро-1,6-нафтиридин-6(5Н)-ил]карбонил}циклопентил)[(3S,4S)-3-метокситетрагидро-2Н-пиран-4-ил]амина (1,328 г) пропускают через Comil, используя сито размером 460 мкм, получая 1,313 г.
Синтез 2
Образование сукцината: Свободное основание (3034,2 г) загружают через 1-мкм встроенный фильтр в 72-л колбу, оборудованную термопарой, подвесной мешалкой, впускным отверстием для N2/вакуума и концентратором периодического действия, при этом делают отметку на колбе на уровне 11,2 л. Объединенные концентраты дополнительно концентрируют до ˜7 л, заменяя растворитель на EtOH при постоянном объеме, используя 17 л EtOH. Раствор разбавляют при помощи EtOH до 11,2-л отметки на колбе. Вакуум выпускают, а концентратор периодического действия заменяют наливной воронкой. Добавляют 839 г янтарной кислоты и партию нагревают до 65°С. В течение 7 минут добавляют гептан (1,4 л). 5 г затравки растворяют в 100 мл гептана и добавляют к партии. Разбавленную суспензию выдерживают в течение 60 минут, получая густой слой затравки, после чего в течение 120 минут добавляют 34,9 л гептана. После выдерживания при температуре 65°С в течение 2 часов суспензию охлаждают в течение ночи до температуры окружающей среды. Анализ маточных растворов при помощи ЖХ показывает потерю 6,5 мг/мл. Суспензию фильтруют и влажный осадок промывают 2×8 л 3/1 гептана/EtOH с последующей заменой жидкости для промывки 4 л 3/1 гептана/EtOH. Не совсем белое твердое вещество сушат в атмосфере N2/вакуума на фильтровальной емкости, получая 3404 г желтовато-коричневого твердого вещества, 96,7 мас.%, при этом точно установленный выход составляет 86% с подтвержденной анализом 10,3% потерей.
Перекристаллизация сукцината (необязательная): 12,1 л EtOH загружают в визуально чистую 50-л колбу, оборудованную впускным отверстием для N2/вакуума, механической мешалкой, термопарой и впускным адаптером. Добавляют сукцинат и промывают 4,7 л EtOH. Суспензию нагревают до 65°С, после чего раствор переносят в 72-л колбу в условиях статического вакуума через встроенный 1-мкм фильтр. Завершив перенос, 50-л колбу промывают 1 л EtOH, а жидкость для промывки переносят через встроенный фильтр к партии в 72-л колбе. Фильтрованный раствор EtOH концентрируют при температуре 38-44°С и давлении 5 дюймов Hg до <12 л, при этом во время концентрирования сукцинат кристаллизуется. Суспензию разбавляют до 12,1 л, а затем нагревают до 65°С. Добавляют 1 л гептана, а затем 14 г затравки, суспендированной в 300 мл гептана. Суспензию выдерживают в течение 1 часа и 10 минут при 65°С, после чего в течение 2 часов добавляют 17,5 л гептана. Суспензию выдерживают в течение 2 часов при температуре 65°С, а затем охлаждают до температуры окружающей среды в течение ночи, ˜13 часов. Суспензию фильтруют, отделяя твердые вещества, а осадок на фильтре промывают 5,4 л жидкости для промывки суспензии из 2:1 гептана:этанола с последующей промывкой 5,4 л заменителя и 5,4 л жидкости для промывки суспензии. Осадок на фильтре сушат, пропуская через него N2 в вакууме в течение 3 часов, после чего фильтровальный осадок разбивают и дополнительно сушат N2 в вакууме в течение приблизительно 48 часов. После окончания сушки лекарственное средство в массе хранят в двойном полиэтиленовом пакете в картонном барабане до заменителя и 5,4 л жидкости для промывки суспензии. Осадок на фильтре сушат, пропуская через него N2 в вакууме в течение 3 часов, после чего фильтровальный осадок разбивают и дополнительно сушат N2 в вакууме в течение приблизительно 48 часов. После окончания сушки лекарственное средство в массе хранят в двойном полиэтиленовом пакете в картонном барабане до измельчения. В целом получают 3,018 кг сукцината (выход 93,9%).
Данные H1 и 13С ЯМР для сукцината ((1R,3S)-3-изопропил-3-{[3-(трифторметил)-7,8-дигидро-1,6-нафтиридин-6(5Н)-ил]карбонил}циклопентил)[(3S,4S)-3-метокситетрагидро-2Н-пиран-4-ил]амина:
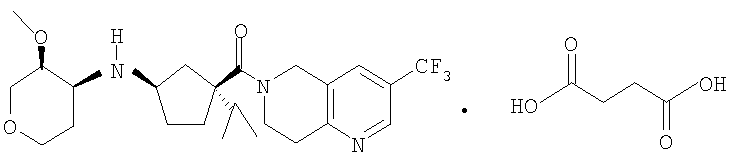
1H ЯМР (499,87 МГц, CDCl3, 47°С) δ 8,74 (д, J=1,2, 1H), 8,15 (ушир. с, 1H), 4,85-4,77 (ом, 2Н), 3,93 (дд, J=12,7, 3,6, 1H), 3,88 (м, 2Н), 3,77 (дт, J=11,5, 4,0, 1H), 3,34-3,29 (ом, 2Н), 3,26 (с, 3Н), 3,23 (дд, J=12,7, 1,6, 1H), 3,15 (м, 1H), 2,99 (м, 3Н), 2,38 (м, 1Н), 2,35 (с, 4Н), 2,23 (дд, J=12,7, 6,8, 1H), 2,08 (м, 1H), 1,84-1,78 (ом, 2Н), 1,65-1,51 (ом, 3Н), 1,24 (м, 3Н), 0,85 (д, J=6,8, 1H), 0,73 (д, J=6,8, 3Н);
13С ЯМР (125,70 МГц, CDCl3, 47°С) δ 174,73, 173,87 (2С), 159,06 (ушир.), 143,73 (кв, JCF=3,7), 131,45 (ушир.м), 129,71, 123,66 (кв, JCF=272,0), 122,90 (кв, JCF=32,6), 74,32, 64,95, 64,82, 55,76, 55,68, 55,48, 53,41, 45,62 (ушир.), 41,75 (ушир.), 38,36, 32,18, 32,05, 31,57, 30,06 (2С), 28,01, 27,25, 17,80, 17,64; Т.пл. 175°C; MC(ESI): Найдено для [С24Н34N3O3F3+Н]: 470,2631; Вычислено: 470,2631
Пример 3
(1S,4S)-4-(2,5-диметил-1H-пиррол-1-ил)-1-изопропилциклопент-2-ен-1-карбоновая кислота
Стадия 1 - Метил-(1R,4S)-4-аминоциклопент-2-ен-1-карбоксилат
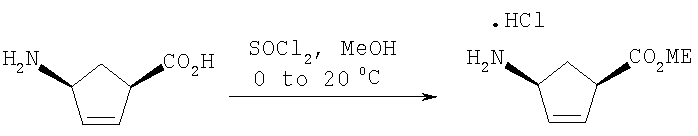
Твердую (1R,4S)-4-аминоциклопентен-2-ен-1-карбоновую кислоту (1,0 кг, 7,72 мол) растворяют в МеОН (3,0 л). Суспензию охлаждают до 0-5°С. По каплям в течение 2 часов добавляют тионилхлорид (0,576 л, 7,92 мол), поддерживая температуру <20°С. По окончании добавления тионилхлорида охолаждающую баню удаляют, а реакционную смесь выдерживают при температуре 20°С в течение 1-2 часов. Затем смесь продукта по каплям добавляют к IPAc (22,5 л) в течение 1-2 часов, после чего продукт кристаллизуется непосредственно из раствора в виде соли HCl. Партию фильтруют и сушат в вакууме в течение ночи, получая HCl-соль сложного аминоциклопентенового метилового эфира (1081 г, выход 77%).
Стадия 2 - Метил(1R,4S)-4-(2,5-диметил-1Н-пиррол-1-ил)циклопент-2-ен-1-кабоксилат

Твердую соль сложного аминоциклопентенового метилового эфира (1,076 кг, 6,059 мол) растворяют в МеОН (3 л, 2М) при температуре 20°С в атмосфере азота. Добавляют диизопропилэтиламин (DIEA, 0,78 кг, 6,059 мол) с последующим добавлением ацетонилацетона (0,711 кг, 6,241 мол). Смесь является экзотермической, повышая температуру до 32-35°С. Затем реакционную смесь выдерживают при температуре 25°С в течение 16 часов. Партию разбавляют IPAc (9-10 л) и промывают 10% NH4Cl (2×3 л) и 5% насыщенным солевым раствором (2×3 л). Партию IPAc сушат над сульфатом натрия, фильтруют и концентрируют до масла. Для промывки используют ТГФ (3 л) и партию вновь концентрируют до масла. Чувствительный к воздуху, пиррол-защищенный аминоциклопентенкарбоксилат (1189 г, выход 92%) хранят при температуре 5-7°С в атмосфере азота до осуществления стадии алкилирования.
Стадия 3 - Метил-(1S,4S)-4-(2,5-диметил-1Н-пиррол-1-ил)-1-изопропилциклопент-2-ен-1-кабоксилат
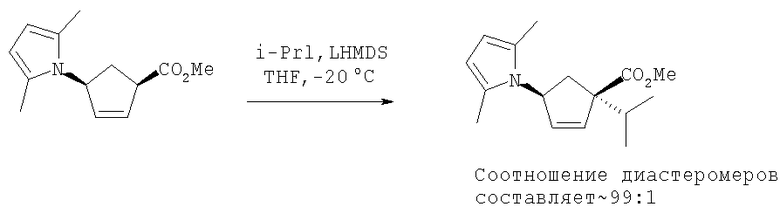
Сложный пирролметиловый эфир (1189 г), растворенный в ТГФ (1,2 л), добавляют по каплям в течение 40 минут к 1 М гексаметилдисилазиду лития (LHMDS) в ТГФ (8,65 л, 8,650 мол) при -20°С. Партию выдерживают в течение 30 минут и в течение часа добавляют 2-иодпропан. Партию выдерживают в течение часа, затем дают ей возможность нагреться до 20°С в течение 1-2 часов до завершения согласно данным ВЭЖХ (<0,5% исходного материала). Партию гасят 6% раствором NH4Cl (10 л). Загружают IPAc (20 л) и слои разделяют. Органический слой промывают 6% водным NH4Cl (10 л), 5% насыщенным солевым раствором (2×10 л) и концентрируют до масла. Чувствительный к воздуху алкилированный сложный пирролметиловый эфир (1419 г, выход 98%) хранят при температуре 5-7°С в атмосфере азота до омыления.
Стадия 4 - (1S,4S)-4-(2,5-диметил-1Н-пиррол-1-ил)-1-изопропилциклопент-2-ен-1-кабоновая кислота
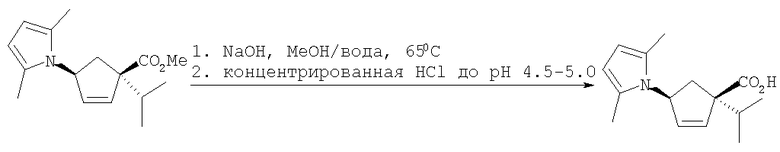
Алкилированный сложный пирролметиловый эфир (1,38 кг, 5,197 мол) растворяют в MeOH (7,7 л). Добавляют деионизированную воду (2,5 л) с последующим добавлением 10 Н NaOH (2,08 л, 20,786 мол). Затем партию нагревают до 65°С в течение 16 часов. Партию охлаждают до 10°С. Продукт кристаллизуют, доводя pH до 4,5 концентрированной HCl. Суспензию выдерживают в течение часа и к партии добавляют деионизированную воду (15 л). Суспензию выдерживают в течение 18 часов при температуре 20-25°С. Твердые вещества отфильтровывают, промывают смесью 10% МеОН/деионизированная вода и сушат в вакуумной печи (40-50°С, 25-26" Hg), получая алкилированную пирролциклопентеновую кислоту (1223 г, выход 95%).
Пример 4
3-(Трифторметил)-5,6,7,8-тетрагидро-1,6-нафтиридин
Стадия 1 - Гексафторфосфат N-[3-(диметиламино)-2-(трифторметил)проп-2-енилиден]-N-метилметанамия (CF3DT)
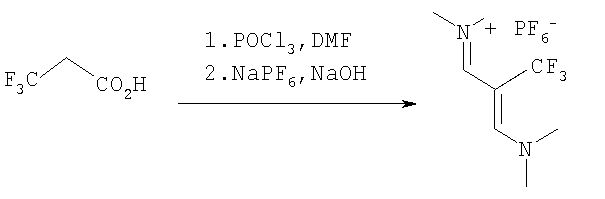
Оксихлорид фосфора (1,1 л, 11,8 мол) добавляют к диметилформамиду (2,6 л) при температуре 4°С в течение часа и реакционной смеси дают возможность нагреться до 20°С. В течение 6 минут добавляют 3,3,3-трифторпропионовую кислоту (TFPA) (771 г, 6,02 мол). Затем реакционную смесь выдерживают при температуре от 50 до 60°С в течение 4 часов, а затем дают ей возможность остыть до комнатной температуры. Реакционную смесь TFPA/POCl3/DMF загружают в 5-л наливную воронку. Реакционную колбу промывают DMF, 3×75 мл, и промывную жидкость также переносят в 5-л наливную воронку. Гексафторфосфорную кислоту (980 мл, 60% водный раствор) добавляют к воде (7,1 л) при охлаждении до температуры 4°С. Медленно добавляют гидроксид натрия (5 H, 2,0 л), поддерживая внутреннюю температуру на уровне ниже 15°С. Затем раствор охлаждают до 0°С. Гидроксид натрия (5 Н) загружают в 2-л наливную воронку и одновременно добавляют реакционную смесь TFPA/POCl3/DMF с такой скоростью, чтобы внутренняя температура оставалась на уровне ниже 5°С, а pH варьировал от 3,05 до 3,6 (приблизительно 3,2 в течение наибольшего периода добавления). Затем полученную желтую суспензию выдерживают в течение 60 минут при температуре ˜0°С. Твердые вещества отфильтровывают, промывают ледяной водой (2×4,0 л), а затем сушат потоком N2 в вакууме. В целом получают 1,785 кг (87%) CF3DT (соль винамидиния).
Стадия 2 - N-(третбутоксикарбонил)-3-(трифторметил)5,6,7,8-тетрагидро-1,6-нафтиридин
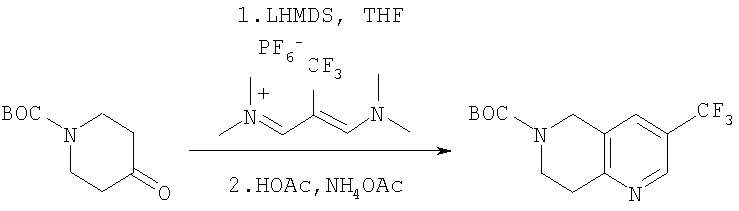
N-BOC-4-пиперидон (672 г) в ТГФ (2,72 л) загружают в раствор бис(триметилсилил)амида лития (LHMDS) (3,55 л, 1,0 М раствор в ТГФ) в ТГФ (3,7 л) при температуре -12°С в течение 45 минут. Реакционной смеси дают возможность нагреться до комнатной температуры. Смесь добавляют в течение 30 минут к суспензии CF3DT (1,17 кг) в ТГФ (5,45 л), охлажденном до -24°С. Затем реакционную смесь выдерживают в течение 2 часов при температуре ˜-20°С. Добавляют уксусную кислоту (295 мл) в течение 3 минут. Реакционную смесь нагревают до 20°С в течение 1 ч 15 мин и одной порцией добавляют ацетат аммония (741 г). Реакционную смесь нагревают до 64°С и выдерживают при данной температуре в течение 2 часов. Реакционную смесь охлаждают до комнатной температуры, а затем разбавляют водой (15,4 л) и метилциклогексаном (15,4 л). Смесь перемешивают, перемешивание прекращают и слоям дают возможность осесть. Нижний водный слой удаляют, а органический слой промывают 2 М водной лимонной кислотой (6,2 л). После перемешивания и разделения слоев анализ при помощи ЖХ показывает, что из органического слоя (общая масса = 19,8 кг) получают 617 г (64%) ВОС нафтиридина.
Стадия 3 - 3-(трифторметил)-5,6,7,8-тетрагидро-1,6-нафтиридин

Органический слой со стадии получения нафтиридина, содержащий N-(третбутоксикарбонил)-3-(трифторметил)-5,6,7,8-тетрагидро-1,6-нафтиридин (общая масса 34,2 кг, содержит 989 г ВОС нафтиридина), концентрируют до масла. Остаток разбавляют метанолом (6 л) и раствор концентрируют до масла. К остатку добавляют метанол (6 л) и полученный раствор концентрируют до 2,3 л. Раствор разбавляют метанолом до объема 7,3 л и добавляют 4,58 М HCl в IPA (3,6 л). Раствор нагревают до 55°С и выдерживают в течение часа. После охлаждения до комнатной температуры добавляют воду (5 л). Затем осторожно добавляют раствор K2CO3 (2,28 кг) в воде (5 л) (pH = 10). Смесь экстрагируют IPAc (3×10 л). Органические продукты экстрагирования содержат 601 г (91%) лишенного защиты нафтиридина. Объединенные органические слои концентрируют, остаток разбавляют IPA (6 л) и вновь концентрируют. Полученное масло разбавляют IPA (6 л) и раствор концентрируют до общего объема ˜2 л. Раствор фильтруют через воронку из закаленного стекла, а фильтрат промывают IPA (3×1 л). Объединенный фильтрат и промывную жидкость концентрируют до ˜0,5 л, а затем разбавляют IPA (1,95 л). Раствор нагревают до 60°С и в течение 40 минут добавляют HCl в IPA (790 мл, 4,33 М путем титрования). Во время добавления образование твердых веществ становится очевидным. В течение 30 минут добавляют гептан (2,75 л), после чего нагревание прекращают и суспензии дают возможность остыть до комнатной температуры в течение ночи. Добавляют дополнительное количество HCl/IPA (80 мл) и гептана (2×1,38 л). Твердые вещества отфильтровывают, промывают смесью 2:1 гептан/IPA (3×550 мл) и сушат в вакууме потоком азота, получая 678,8 г 3-(трифторметил)-5,6,7,8-тетрагидро-1,6-нафтиридина с выходом 75% в расчете на ВОС нафтиридин (479 г, 73,2 мас.% в виде свободного основания).
Пример 5
(3R)-3-Метокситетрагидро-4Н-пиран-4-он
Стадия 1 - тетрагидро-4Н-пиран-4-он. В 22-л круглодонную колбу, оборудованную механической мешалкой, термопарой и впускным отверстием для азота, загружают тетрагидро-4Н-пиран-4-он (3,00 кг, 30,0 мол) и трипропилортоформиат (5,70 кг, 30,0 мол). Насос, используемый для перекачки, промывают хлорбензолом (300 мл) и промывную жидкость добавляют к партии. Полученный раствор охлаждают на ледяной бане до 5°С. Одной порцией добавляют Amberlyst-15 (60 г), предварительно промытый деионизированной водой, а затем пропанолом и высушенный. Смесь перемешивают при температуре окружающей среды в течение 16 часов. В результате получают раствор соединения 2 (ГХ). Смесь концентрируют в вакууме (75 мм Hg). Твердые вещества удаляют фильтрацией через слой из solka floc и фильтровальный слой промывают МТВЕ (2 л). Фильтрат разбавляют МТВЕ (26 л) и экстрагируют насыщенным водным NaHCO3 (24 л), а затем водой (2×16 л). Органическую фазу концентрируют в вакууме (500 мм Hg) для удаления МТВЕ. После того как внутренняя температура достигнет 44°С, добавляют хлорбензол (3 л). Дистилляцию продолжают с использованием насадочной колонны со скоростью, поддерживающей температуру пара на уровне приблизительно от 85 до 90°С. Периодически добавляют хлорбензол с целью поддержания постоянного объема. В целом используют 15 л. Во время дистилляции температуру партии поддерживают на уровне от 122 до 125°С. Через 16 часов анализ при помощи ГХ подтверждает конверсию >9:1. Вакуум усиливают до 50 мм Hg и реакционную смесь подвергают дистилляции, получая раствор 53 мас.% соединения 3 (3,13 кг, выход 73%) и хлорбензол.
Стадия 2 - 3,4-дигидрокситетрагидро-2-Н-пиран-4-сульфоновая кислота, натриевая соль. В 50-л круглодонную колбу, оборудованную механической мешалкой, впускным отверстием для азота и термопарой, загружают ацетон (12,7 л) и воду (1,28 л). Простой диэфир гидрохинидин-1,4-фталазиндиила (DHQD2PHAL, 54,8 г, 0,070 мол), дигидрат осмата калия (12,95 г, 0,035 мол) и моногидрат N-оксида 4-метилморфолина (NMO, 1,078 кг, 7,74 мол) добавляют последовательно к растворителю и полученный раствор охлаждают до 0°С. В течение 7 часов добавляют простой пропиленольный эфир (1,85 кг 54 мас.% раствора, 7,03 мол), поддерживая реакционную температуру на уровне приблизительно 0°С. Добавляют свежеприготовленный раствор Na2S2C5 (802 г, 4,22 мол) и воду (5,63 л) с последующим добавлением ледяной уксусной кислоты (1,2 л). После выдерживания в течение 16 часов при комнатной температуре твердые вещества удаляют фильтрацией, а фильтрат концентрируют в вакууме для удаления ацетона. В течение 3,5 часов добавляют изопропанол (28 л), получая бесцветную суспензию. Твердые вещества собирают на фритте, промывают изопропанолом (6 л) и сушат в вакуумной печи, получая 958 г 92 мас.% (выход выделения составляет 57%) продукта, с 97,2% энантиомерным избытком.
Стадия 3 - 4,4-диметокситетрагидро-2Н-пиран-3-ол. В 22-л круглодонную колбу, оборудованную механической мешалкой, 5-л капельной воронкой, впускным отверстием для азота и термопарой, загружают бисульфитный аддукт (893 г 92 мас.% твердого вещества, 4,06 мол), MeOH (8,1 л) и триметилортоформиат (948 г, 8,93 мол). Полученную суспензию нагревают до 50°С и через капельную воронку в течение 40 минут добавляют 1,89 М раствор HCl в МеОН (2,48 л, 4,69 мол). Суспензию охлаждают до 7°С и в виде медленной струи добавляют 50 мас.% NaOH (340 мл). Твердые вещества собирают на фритте, а растворитель заменяют толуолом, используя в целом 12 л толуола. Партию концентрируют приблизительно до 3 л, после чего твердые вещества удаляют фильтрацией, а осадок промывают ТГФ (2 л). Получают раствор, содержащий 577 г продукта (3,56 мол в 5,33 л, выход 88%, энантиомерный избыток 97,5%).
Стадия 4 - (3R)-метокситетрагидро-4Н-пиран-4-он. В 22-л круглодонную колбу, оборудованную термопарой, впускным отверстием для азота и капельной воронкой, загружают раствор соединения 6 (3,56 мол в 5,33 л) и ТГФ (5,8 л). KF раствора показывает присутствие 1,11 мол воды. Одной порцией добавляют NaOt-Bu (494 г, 5,14 мол), получая прозрачный желтый раствор. Колбу погружают в ледяную баню и в течение 20 минут добавляют Ме2SO4 (7737 г, 5,84 мол), поддерживая внутреннюю температуру на уровне ниже 36°С. Охлаждающую баню удаляют и реакционную смесь выдерживают в течение 4 часов, получая необработанный раствор. Добавляют воду (1,5 л) с последующим добавлением 2 Н HCl (840 мл). Кажущийся pH двухфазной смеси составляет 0,6. Через 20 часов при комнатной температуре добавляют NaHCO3 (497 г) и смесь экстрагируют IPAc (4×5 л). Объединенные органические фазы концентрируют, а остаточные твердые вещества удаляют фриттой, получая раствор продукта (429 г в 2,653 кг раствора, d = 0,914, 149 г/л, выход 93%, энантиомерный избыток 97,2%).
Испытание сукцината ((1R,33)-3-изопропил-3-{[3-(трифторметил)-7,8-дигидро-1,6-нафтиридин-6(5Н)-ил]карбонил}-циклопентил)[(3S,4S)-3-метокситетрагидро-2Н-пиран-4-ил]амина в "Анализе изменения формы моноцитов цельной крови".
Как известно из предшествующего уровня техники, МСР-1 (член семейства СС-хемокинов) (белок хемоаттрактант моноцита-1), первичным рецептором которого является CCR2, продуцируется в разнообразных типах клеток в ответ на воспалительные стимулы у различных видов, включая грызунов и человека, и стимулирует хемотаксис в моноцитах и подгруппе лимфоцитов, в частности, продуцирование МСР-1 коррелирует с инфильтрацией моноцитов и макрофагов в участках воспаления. Делеция либо МСР-1, либо CCR2 гомогенной рекомбинацией у мышей дает значительное ослабление пополнения моноцитов в ответ на инъекцию тиогликолята и инфекцию Listeria monocytogenes (Lu et al., J. Exp. Med., 187, 601-608 (1998); Kurihara et al. J. Exp. Med. 186, 1757-1762 (1997); Boring et al. 1 Clin. Invest. 100, 2552-2561 (1997); Kuziel et al. Proc. Natl. Acad. Sci. 94, 12053-12058 (1997)). Кроме того, эти животные демонстрируют снижение инфильтрации моноцитов в грануломатозные поражения, индуцированные инъекцией шистосомальных или микобактериальных антигенов (Boring et al. J. Clin. Invest., 100, 2552-2561 (1997); Warmington et al. Am J.Path., 154, 1407-1416 (1999)). Эти данные подтверждают, что вызванная МСР-1 активация CCR2 играет основную роль в пополнении моноцитов в местах воспаления и что антагонизм этой активности даст значительное подавление иммунного ответа с получением терапевтической пользы при иммуновоспалительных и аутоиммунных заболеваниях.
Анализ изменения формы моноцитов цельной крови базируется на феномене, посредством которого моноциты подвергаются изменению в форме, также называемого цитоскелетной перестройкой, при связывании МСР-1 с рецепторами CCR2 моноцитов (изменение формы является предварительным условием процесса хемотаксиса, посредством которого моноцит перемещается в направлении участка воспалительного стимула). Степень изменения формы моноцита может быть соотнесена со сдвигом малоуглового рассеяния клеток в передней полусфере с использованием проточной цитометрии.
Ниже приведены данные, которые сравнивают подвергшиеся воздействию МСР-1 моноциты субъектов, получавших плацебо, с подвергшимися воздействию МСР-1 моноцитами субъектов, получавших дозы соединения (заявленного соединения, представляющего собой сукцинатную соль - антагонист CCR2), и показывают, что наблюдаются значительно сниженные доли изменения формы (соответствующие меньшему уровню рассеяния) популяции моноцитов, полученных от субъектов, обработанных испытываемым соединением. Другими словами, введение сукцинатной солевой формы, представляющей собой антагонист CCR2, ингибирует изменение формы моноцитов, вызванное воздействием МСР-1. Этот эффект обусловлен антагонистической активностью заявленной сукцинатной соли антагониста CCR2, у CCR2 рецептора при блокировании связывания МСР-1.
Данные по биологической активности сукцината ((1R,3S)-3-изопропил-3-{[3-(трифторметил)-7,8-дигидро-1,6-нафтиридин-6(5Н)-ил]карбонил}циклопентил)[(3S,4S)-3-метокситетрагидро-2Н-пиран-4-ил]амина:
Сукцинатная соль. Протокол 002 - Итоговая Таблица данных по изменению формы моноцитов
Панель А
Сукцинатная соль. Протокол 002 - Итоговая Таблица данных по изменению формы моноцитов
Панель А
Сукцинатная соль, Протокол 002 - Итоговая Таблица данных по изменению формы моноцитов
Панель В
Несмотря на то что данное изобретение было описано и проиллюстрировано со ссылкой на некоторые конкретные варианты его осуществления, специалистам в данной области понятно, что допустимы различные адаптации, изменения, модификации, замены, удаления или добавления методик и протоколов без нарушения сущности и объема данного изобретения. Например, могут быть использованы эффективные дозировки, отличные от конкретных вышеуказанных дозировок, по причине различной реактивности млекопитающих, подвергаемых лечению вышеописанными соединениями согласно данному изобретению по любому из показаний. Подобным образом, наблюдаемые конкретные фармакологические ответы могут варьироваться в соответствии и в зависимости от конкретных выбранных активных соединений либо от того, присутствуют ли в них фармакологические носители, а также от используемого вида композиции и способа введения, при этом подразумевается, что такие ожидаемые варианты или различия в результатах соответствуют целям и вариантам осуществления настоящего изобретения. Поэтому предполагается, что данное изобретение определяется объемом прилагаемой формулы изобретения и что такая формула изобретения будет интерпретироваться настолько широко, насколько это целесообразно.
| название | год | авторы | номер документа |
|---|---|---|---|
| 2-ОКСА-5-АЗАБИЦИКЛО[2.2.1]ГЕПТАН-3-ИЛЬНЫЕ ПРОИЗВОДНЫЕ | 2015 |
|
RU2697651C2 |
| N-(2-ЦИАНОГЕТЕРОЦИКЛИЛ)ПИРАЗОЛОПИРИДОНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ЯНУС-КИНАЗЫ | 2014 |
|
RU2669922C2 |
| ПРОИЗВОДНЫЕ ЭТИЛЕНДИАМИНА И СОДЕРЖАЩИЕ ИХ ИНГИБИТОР FXa И АНТИКОАГУЛЯНТ | 2001 |
|
RU2268259C2 |
| КОМБИНАЦИИ АЛЬФА-2-ДЕЛЬТА-ЛИГАНДА С СЕЛЕКТИВНЫМ ИНГИБИТОРОМ ЦИКЛООКСИГЕНАЗЫ-2 | 2003 |
|
RU2286151C2 |
| ПИРИДАЗИНОНОВЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ PARP | 2008 |
|
RU2490265C2 |
| ГЕМИНАЛЬНО-ЗАМЕЩЕННЫЕ ЦИАНОЭТИЛПИРАЗОЛОПИРИДОНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ JANUS КИНАЗ | 2014 |
|
RU2664533C2 |
| ПРОИЗВОДНЫЕ ДИАМИНОВ | 2002 |
|
RU2319699C2 |
| ПРОИЗВОДНЫЕ ДИАМИНА | 2002 |
|
RU2314303C2 |
| ЗАМЕЩЕННЫЕ ДИАМИНОКАРБОКСАМИДНЫЕ И ДИАМИНОКАРБОНИТРИЛЬНЫЕ ПРОИЗВОДНЫЕ ПИРИМИДИНОВ, ИХ КОМПОЗИЦИИ И СПОСОБЫ ЛЕЧЕНИЯ С ИХ ПОМОЩЬЮ | 2012 |
|
RU2625309C2 |
| 3-КАРБОНИЛАМИНО-5-ЦИКЛОПЕНТИЛ-1H-ПИРАЗОЛЬНЫЕ СОЕДИНЕНИЯ, ОБЛАДАЮЩИЕ АКТИВНОСТЬЮ ИНГИБИТОРОВ CDK2 | 2020 |
|
RU2797889C2 |
Данное изобретение относится к новому соединению - сукцинату ((1R,3S)-3-изопропил-3-{[3-(трифторметил)-7,8-дигидро-1,6-нафтиридин-6(5Н)-ил]карбонил}циклопентил)[(3S,4S)-3-метокситетрагидро-2Н-пиран-4-ил] амина формулы:
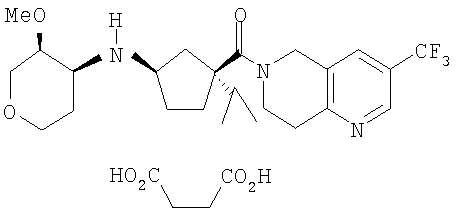
которое обладает свойством антагониста CCR-2, а также к способу модуляции активности рецептора хемокина и способу лечения, улучшения, контролирования и снижения риска воспалительного и иммунорегуляторного расстройства или заболевания и способу улучшения, контролирования и снижения риска ревматоидного артрита. 4 н.п. ф-лы, 3 табл.
| WO 02070523 A, 12.09.2002 | |||
| Топчак-трактор для канатной вспашки | 1923 |
|
SU2002A1 |
| ПРОИЗВОДНЫЕ 8-АРИЛ-1,7-НАФТИРИДИНА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ ПРОТИВОВОСПАЛИТЕЛЬНОЙ АКТИВНОСТЬЮ | 1997 |
|
RU2174123C2 |
| ПРОИЗВОДНЫЕ 1,8-БЕНЗО(B)НАФТИРИДИНА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1992 |
|
RU2047613C1 |

