Данное изобретение заявляет приоритет Предварительной заявки на патент США с регистрационным номером 60/306091, поданной 17 июля 2001 года; Предварительной заявки на патент США с регистрационным номером 60/332886, поданной 6 ноября 2001 года, и Предварительной заявки на патент США с регистрационным номером 60/360361, поданной 28 февраля 2002 года, все из которых включены здесь в качестве ссылки в их полном виде.
ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Данное изобретение относится к областям клеточной и молекулярной биологии и раковой биологии. Более конкретно, данное изобретение обеспечивает способы и композиции, относящиеся к терапевтическим агентам, содержащим проапоптозную часть молекулы и клеткоспецифическую нацеливающую часть молекулы.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
В различных клинических обстоятельствах часто бывает желательной гибель отдельной клетки. Множество путей трансдукции (передачи) сигналов в клетке связаны с ее гибелью и выживанием, и доставка лимитирущего и/или решающего компонента этого пути может повлечь за собой ее разрушение. Классическим примером такого пути трансдукции сигнала является апоптоз, и многочисленные элементы апоптозных путей могли бы служить мишенью клетки в отношении смерти. Апоптоз, или программированная гибель клетки, является фундаментальным процессом, регулирующим нормальный гомеостаз ткани регуляцией баланса между клеточной пролиферацией и гибелью (Vaux et al., 1994; Jacobson et al., 1997).
Сериновая протеаза гранзим В (GrB) (Lobe et al., 1986; Schmid and Weissman, 1987; Trapani et al., 1988) интегрально участвует в апоптозной смерти клеток, индуцированной в клетках-мишенях после их подвергания действию лизосомоподобных цитоплазматических гранул (или цитолитических гранул), обнаруженных в цитотоксических Т-лимфоцитах (CTL) и природных клетках-киллерах (NK) (Henkart, 1985; Young and Cohn, 1986; Amyth and Trapani, 1995). Цитотоксические гранулы лимфоцитов содержат перфорин, образующий поры белок, и семейство сериновых протеаз, называемых гранзимами (таблица 1). Перфорин имеет некоторое структурное и функциональное сходство с белками комплемента С6, С7, С8 и С9, членами комплекса атаки мембраны комплемента (Shinkai et al., 1988). В опосредованном лимфоцитами цитолизе перфорин встраивается в мембраны клетки-мишени и, по-видимому, полимеризуется с образованием пор (Podack, 1992; Yagita et al., 1992), что опосредует доступ гранзима В к цитоплазме клетки-мишени. После вхождения внутрь клетки гранзим В индуцирует апоптоз прямой активацией каспаз и индукцией быстрой фрагментации ДНК (Shi et al., 1992).
ГРАНЗИМЫ (СЕРИНОВЫЕ ПРОТЕАЗЫ ЛИМФОЦИТОВ)
Крыса
Человек
RNKP-2, фрагментин 1
Фактор Hanukah, HTSP, гранзим 1
Крыса
Человек
Фрагментин 2, RNKP-1
HLP, гранзим 2, HSE26.1, CSPB
Крыса
RNKP-4
Человек
Гранзим 3
Триптаза
Человек
Met-аза
Гранзимы являются структурно родственными, но имеют различающееся субстратное предпочтение. Посредством его уникальной способности расщеплять после остатков аспартата, гранзим В может расщеплять многие прокаспазы in vitro и является важным инструментом в анализе созревания каспазы-3 (Darmon et al., 1995; Quan et al., 1996; Martin et al., 1996), каспазы-7 (Chinnaiyan et al., 1996; Gu et al., 1996; Fernandes-Alnemri et al., 1995), каспазы-6 (Orth et al., 1996; Fernandes-Alnemri et al., 1995), каспазы-8 (Muzio et al., 1996), каспазы-9 (Duan et al., 1996) и каспазы-10a/b (Fernandes-Alnemri et al., 1996; Vincenz and Dixit, 1997). Кроме того, он является высокотоксичным в отношении клеток-мишеней (Shi et al., 1992). До сих пор предполагалось, что гранзим В убивает клетки прямым активируемым каспазой разрушением, дополненным при определенных обстоятельствах прямым повреждением находящихся далее по ходу процесса каспазных субстратов (Andrade et al., 1998). Получив доступ к цитозолю, гранзим В быстро перемещается в ядро (Jans et al., 1996; Trapani et al., 1996) и может расщеплять поли(АДФ-рибоза)-полимеразу и антиген ядерного матрикса, иногда с использованием сайтов расщепления, отличающихся от сайтов расщепления, предпочтительных для каспаз (Andrade et al., 1998). Хотя многие прокаспазы эффективно расщепляются in vitro, индуцируемая гранзимом В активация каспазы происходит иерархическим образом в интактных клетках, начинаясь на уровне каспаз-палачей, таких как каспаза-3, за которой следует каспаза-7 (Yang et al., 1998). Это отличается от FasL-опосредованного лизиса, который основан на мембранном сигнале, генерируемом через апикальные каспазы, такие как капсаза-8 (Muzio et al., 1996; Sarin et al., 1997). Кроме того, некоторые исследования показали, что гранзим В может также индуцировать гибель клеток посредством независимого от капсаз механизма, который включает в себя прямое повреждение неядерных структур, хотя ключевые субстраты в этом пути до сих пор еще не были выяснены (Sarin et al., 1997; Trapani et al., 1998; Heibein et al., 1999; Beresford et al., 1999).
Исследования Froelich et al. предполагают, что GrB интернализуется посредством опосредованного рецепторами эндоцитоза и что роль перфорина заключается в опосредовании высвобождения гранзима В из образующихся при эндоцитозе пузырьков. Фактически, перфорин может быть заменен другими разрушающими пузырьки факторами, такими как продуцируемые аденовирусом (Froelich et al., 1996; Pinkoski et al., 1998; Browne et al., 1999).
Гранзимы обычно являются высоко гомологичными, имеющими 38-67% гомологию относительно GrB (Haddad et al., 1991), и они содержат каталитическую триаду (His-57, Asp-102 и Ser-195) сериновых протеаз семейства трипсина. Другие признаки включают в себя зрелую N-концевую Ile-Ile-Gly-Gly-последовательность, три или четыре дисульфидных мостика и консервативный мотив (PHSRPYMA), который имеется также в катепсине G нейтрофилов и химазах мастоцитов. Углеводные части молекул гранзимов являются Asn-связанными (Griffiths and Izaaz, 1993). мРНК-транскрипты гранзимов транслируются в виде пре-про-протеаз. Пре- или лидерная последовательность отщепляется сигнальной пептидазой в эндоплазматической сети. После удаления пропептидов неактивные програнзимы (зимогены) становятся активными протеазами. Пропептидные последовательности гранзимов начинаются после лидерного пептида и заканчиваются перед N-концевым Ile, необходимым для укладки (фолдинга) этой протеазы в каталитическую конформацию (Kam et al., 2000).
Среди различных апоптозных факторов, идентифицированных до сих пор, наиболее хорошо определенными регуляторами этого пути гибели клеток являются члены семейства Bcl-2. Некоторые члены семейства Bcl-2, в том числе Bcl-2, Bcl-XL, Ced-9, Bcl-w и т.д., способствуют выживанию клеток, тогда как другие члены, в том числе Вах, Bcl-Xs, Bad, Bak, Bid, Bik и Bim, как было показано, делают возможным апоптоз (Adams and Cory, 1998). К настоящему времени был предложен ряд различных гипотез, касающихся возможных биологических функций членов семейства Bcl-2. Они включают в себя образование димеров (Oltvai et al., 1993), протеазную активацию (Chinnaiyan et al., 1996), деполяризацию митохондриальных мембран (6), генерирование промежуточных активных форм кислорода (Hockenbery et al., 1993), регуляцию поступления кальция в клетки (Lam et al., 1994; Huiling et al., 1997) и образование пор (Antonsson et al., 1997; Marzo et al., 1998).
Bax, стимулирующий гибель член массой 21 кДа семейства Bcl-2, был сначала идентифицирован как белок, который коиммунопреципитировался с Bcl-2 из различных клеточных линий (Oltvai et al., 1993). Сверхэкспрессия Bax ускоряет гибель клеток в ответ на большой диапазон цитотоксических воздействий. Определение аминокислотной последовательности белка Вах показало, что она является высокогомологичной аминокислотной последовательности Bcl-2. Ген Вах состоит из шести экзонов и продуцирует альтернативные транскрипты, преобладающая форма которых кодирует мРНК 1,0 т.п.н. и названа Вахα. Подобно Bcl-2 и нескольким другим членам семейства Bcl-2, белок Вах имеет высококонсервативные области, домены ВН1, ВН2 и ВН3, и анализ гидрофобности последовательностей этих белков показывает присутствие гидрофобного трансмембранного сегмента на их С-концевых сторонах (Oltvai et al., 1993).
Вах широко экспрессируется без какой-либо видимой тканеспецифичности. Однако, после индукции апоптоза Вах перемещается в митохондрии, приводя к дисфункции митохондрий и высвобождению цитохрома с, который затем активирует пути каспаз (Hsu and Youle, 1997; Wolter et al., 1997; Gross et al., 1998). Этот процесс перемещения является быстрым и происходит на ранней стадии апоптоза (Wolter et al., 1997). Селективная сверхэкспрессия Вах при раке яичника человека переносом (трансфекцией) аденовирусного гена приводила к значительному лизису опухолевых клеток in vivo (Tai et al., 1999). Сверхэкспрессия гена Вах бинарной аденовирусной системой в культивируемых клеточных линиях из злокачественной опухоли легкого человека приводит к активации каспазы, индукции апоптоза и подавлению роста клеток. Кроме того, внутриопухолевая инъекция аденовирусного вектора, экспрессирующего ген Вах, подавляла рост ксенотрансплантатов злокачественной опухоли легкого человека, установившегося в «голых» мышах (Kagawa et al., 2000; Kagawa et al., 2000).
WO 99/45128 и Aqeilan et al., (1999) описывают химерные белки, имеющие нацеливающую на определенные клетки специфичность и апоптозиндуцирующую активность, в частности, рекомбинантный химерный белок IL-2-Вах, который специфически нацелен на IL-2-экспрессирующие клетки и индуцирует клеткоспецифический апоптоз.
WO 99/49059 относится к химерному токсину, состоящему из гонадолиберина (рилизинг-фактора гонадотропина) (GnRH) и экзотоксина А Pseudomonas (РЕ), для обнаружения связанного с опухолью эпитопа, экспрессируемого аденокарциномой человека.
WO 97/46259 относится к нацеленным химерным токсинам, содержащим нацеливающие на клетки части молекул и убивающие клетки части молекул, нацеленных на опухолевые клетки. В конкретном примере этот химерный токсин содержит гомологи гонадолиберина (рилизинг-фактора гонадотропина) и экзотоксин А Pseudomonas.
WO 97/22364 описывает нацеленное лечение аллергических реакций, посредством которого химерный цитотоксин Fc2'-3-PE40 направляется на нацеленную элиминацию клеток, экспрессирующих рецептор FceRI.
Хотя некоторые композиции химерных белков были описаны, требуются другие способы и композиции для улучшенных способов терапии, включающих в себя лизис клеток.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Данное изобретение относится к способам и композициям, участвующим в доставке химерных полипептидов, содержащих факторы пути трансдукции сигнала, которые индуцируют гибель клетки-мишени. В предпочтительном варианте этот фактор является проапоптозным фактором.
Почти все клетки содержат механизмы, ответственные за опосредование смерти клеток (апоптоза). Таким образом, в некоторых вариантах данное изобретение относится к доставке определенных проапоптозных белков, которые являются центральными медиаторами этого эффекта, во внутреннее пространство клеток-мишеней, что будет приводить к смерти клеток посредством апоптозных механизмов. Индуцирующая апоптоз часть молекулы индуцирует программированную гибель клеток после вхождения в клетку-мишень этого химерного полипептида, который доставляется для связывания с клеткой-мишенью посредством клеткоспецифической нацеливающей части молекулы. В некоторых вариантах данного изобретения и в качестве преимущества над известными в данной области способами, проапоптозные полипептиды доставляются в виде белков, а не в виде молекул нуклеиновых кислот, подлежащих трансляции для продуцирования желаемых полипептидов. В качестве дополнительного преимущества, в химерных полипептидах данного изобретения используются последовательности человека во избежание любых нежелательных иммунных реакций на чужеродный полипептид.
В дополнительных вариантах гранзим А или гранзим В является медиатором для индукции апоптоза. В конкретных вариантах, части рекомбинантный лиганд (VEGF) и/или рекомбинантное антитело (scFvMEL) сливают в виде последовательностей нуклеиновых кислот с последовательностями, которые кодируют гранзим или член семейства Bcl-2. Авторы изобретения представляют здесь данные, демонстрирующие, что химерные полипептиды, такие как гранзим В-vegf121 и гранзим В-scFvMEL, являются цитотоксичными для клеток-мишеней. При условии, что специалисту с квалификацией в данной области известно, что имеются многочисленные подобные нацеливающие на клетку и проапоптозные примеры, которые могут быть использованы взаимозаменяемо с приведенными здесь конкретными примерами, это указывает на то, что конструкции, содержащие проапоптозные белки, имеют существенный терапевтический потенциал для лечения патологических состояний и представляют новый класс терапевтических агентов с новым механизмом действия.
В варианте, в котором проапоптозные белки используют в качестве убивающей части молекулы в химерных белках, используют рекомбинантное антитело (scFvMEL), которое связывается с антигеном клеточной поверхности gp240 клеток меланомы и эффективно интернализуется. Авторы данного изобретения слили гены, кодирующие scFvMEL, с генами, кодирующими Вах, укороченный Вах1-5 и Вах 345, соответственно (названные как scFvMEL-bax, scFvMEL-Bax1-5 и scFvMEL-Bax345, соответственно). Эти гены встраивали в векторы экспрессии белка и трансформировали в бактерии. Слитые белки очищали, испытывали против клеток-мишеней в культуре, и было показано, что они являются цитотоксичными в отношении клеток-мишеней. Это предполагает, что конструкции, содержащие проапоптозный белок Вах, имеют значительный терапевтический потенциал для лечения заболеваний и представляют новый класс терапевтических агентов с новым механизмом действия.
Одним из объектов данного изобретения является химерный полипептид, содержащий клеткоспецифическую нацеливающую часть молекулы и фактор пути трансдукции сигнала.
Другим объектом данного изобретения является химерный полипептид, содержащий клеткоспецифическую нацеливающую часть молекулы и индуцирующий апоптоз фактор, где указанный индуцирующий апоптоз фактор является гранзимом. В особом варианте гранзим является гранзимом В. В другом особом варианте аминокислотная последовательность указанного гранзима В выбрана из группы, состоящей из SEQ ID NO:11, SEQ ID NO: 12, SEQ ID NO:13, SEQ ID NO:14, SEQ ID NO:15 и SEQ ID NO:16. В другом специфическом варианте аминокислотная последовательность указанного гранзима В является SEQ ID NO:60, дополнительно содержащая N-концевое удлинение SEQ ID NO:61, или SEQ ID NO:60, в которой отсутствуют первые двадцать аминокислот. В следующем специфическом варианте аминокислотная последовательность указанного гранзима В является по меньшей мере 100 смежными аминокислотами из SEQ ID NO:11, SEQ ID NO:12, SEQ ID NO:13, SEQ ID NO:14, SEQ ID NO:15, SEQ ID NO:16 или SEQ ID NO:60. В следующем специфическом варианте аминокислотная последовательность указанного гранзима В является по меньшей мере 75 смежными аминокислотами из SEQ ID NO:11, SEQ ID NO:12, SEQ ID NO:13, SEQ ID NO:14, SEQ ID NO:15, SEQ ID NO:16 или SEQ ID NO:60. В следующем специфическом варианте аминокислотная последовательность указанного гранзима В является по меньшей мере 40 смежными аминокислотами из SEQ ID NO:11, SEQ ID NO:12, SEQ ID NO:13, SEQ ID NO:14, SEQ ID NO:15, SEQ ID NO:16 или SEQ ID NO:60. В дополнительном особом варианте указанный гранзим является гранзимом А. В следующем конкретном варианте аминокислотная последовательность указанного гранзима А выбрана из группы, состоящей из SEQ ID NO:23, SEQ ID NO:24 и SEQ ID NO:25. В следующем специфическом варианте аминокислотная последовательность указанного гранзима А является по меньшей мере 100 смежными аминокислотами из SEQ ID NO:23, SEQ ID NO:24 или SEQ ID NO:25. В следующем специфическом варианте аминокислотная последовательность указанного гранзима А является по меньшей мере 75 смежными аминокислотами из SEQ ID NO:23, SEQ ID NO:24 или SEQ ID NO:25. В следующем специфическом варианте аминокислотная последовательность указанного гранзима А является по меньшей мере 40 смежными аминокислотами из SEQ ID NO:23, SEQ ID NO:24 или SEQ ID NO:25. В дополнительном специфическом варианте клеткоспецифической нацеливающей частью молекулы является цитокин, антитело, лиганд или гормон. В следующем специфическом варианте этот лиганд является VEGF. В следующем специфическом варианте VEGF является vegf121. В другом специфическом варианте антитело является одноцепочечным антителом. В следующем специфическом варианте одноцепочечное антитело является scFvMEL. В дополнительном специфическом варианте гранзим является гранзимом В, а указанная клеткоспецифическая нацеливающая часть молекулы является vegf121. В другом специфическом варианте гранзим является гранзимом В, а указанная клеткоспецифическая часть молекулы является scFvMEL. В дополнительном конкретном варианте этот полипептид дополнительно содержит линкер, такой как SEQ ID NO:50, SEQ ID NO:51 или SEQ ID NO:52. В специфическом варианте этот полипептид кодируется рекомбинантным полинуклеотидом.
Следующим объектом данного изобретения является экспрессирующая кассета, содержащая полинуклеотид, кодирующий химерный полипептид, содержащий клеткоспецифическую нацеливающую часть молекулы и индуцирующий апоптоз фактор, где указанный индуцирующий апоптоз фактор является гранзимом и где указанный полинуклеотид находится под контролем регуляторной последовательности, функциональной в клетке-хозяине. В конкретных вариантах этот гранзим является гранзимом А или гранзимом В. В специфическом варианте гранзим А кодируется полинуклеотидом SEQ ID NO:26, SEQ ID NO:27 или SEQ ID NO:28. В другом специфическом варианте гранзим В кодируется полинуклеотидом SEQ ID NO:17, SEQ ID NO:18, SEQ ID NO:19, SEQ ID NO:20, SEQ ID NO:21 или SEQ ID NO:22. В дополнительном специфическом варианте эта кассета содержится в рекомбинантном вирусном векторе, таком как аденовирусный вектор, аденоассоциированный вектор или ретровирусный вектор.
Дополнительным объектом данного изобретения является клетка-хозяин, содержащая экспрессирующую кассету, содержащую полинуклеотид, кодирующий химерный полипептид, содержащий клеткоспецифическую нацеливающую часть молекулы и индуцирующий апоптоз фактор, где указанный индуцирующий апоптоз фактор является гранзимом. В конкретных вариантах эта клетка дополнительно определяется как прокариотическая клетка-хозяин или эукариотическая клетка-хозяин.
Другим объектом данного изобретения является способ применения клетки-хозяина, содержащей экспрессирующую кассету, содержащую полинуклеотид, кодирующий химерный полипептид, содержащий клеткоспецифическую нацеливающую часть молекулы и индуцирующий апоптоз фактор, где указанный индуцирующий апоптоз фактор является гранзимом, предусматривающий культивирование этой клетки-хозяина в условиях, подходящих для экспрессии химерного полипептида.
Следующим объектом данного изобретения является способ индукции апоптоза в клетке, предусматривающий введение в указанную клетку эффективного количества химерного полипептида, содержащего клеткоспецифическую нацеливающую часть молекулы и гранзим. В конкретных вариантах этот гранзим является гранзимом А или гранзимом В. В специфических вариантах эта клетка находится in vivo и/или в человеке. Другим объектом данного изобретения является способ индукции апоптоза в клетке, предусматривающий введение в указанную клетку эффективного количества химерного полипептида, содержащего клеткоспецифическую нацеливающую часть молекулы и гранзим, где указанная клеткоспецифическая нацеливающая часть молекулы является scFvMEL, а указанный гранзим является гранзимом В. Предполагается, что клеткоспецифическая нацеливающая часть молекулы действует посредством нацеливания на специфические клетки, например, на клетки, которые экспрессируют на своей поверхности пептид или полипептид, который способен специфически связывать эту нацеливающую часть молекулы. Соединение, которое обеспечивает специфическое нацеливание на клетку, может называться мишенью. Таким образом, в некоторых вариантах данного изобретения клетки могут иметь мишень, которую узнает клеткоспецифическая нацеливающая часть молекулы.
Другим объектом данного изобретения является способ индукции апоптоза в клетке, предусматривающий введение в указанную клетку эффективного количества химерного полипептида, содержащего клеткоспецифическую нацеливающую часть молекулы и гранзим, где указанная клеткоспецифическая нацеливающая часть молекулы является vegf121, а указанный гранзим является гранзимом В.
Дополнительным объектом данного изобретения является способ индукции апоптоза в клетке, предусматривающий введение в указанную клетку эффективного количества химерного полипептида, содержащего клеткоспецифическую нацеливающую часть молекулы и проапоптозный член семейства Bcl-2. В специфическом варианте проапоптозный член семейства Bcl-2 является Вах или его фрагмент. В специфических вариантах эта клетка находится in vivo и/или в человеке. В специфическом варианте фрагмент Вах лишен по меньшей мере части полипептида, кодируемой экзоном 6 в последовательности полинуклеотида Вах.
Другим объектом данного изобретения является способ индукции апоптоза в клетке, предусматривающий введение в указанную клетку эффективного количества химерного полипептида, содержащего клеткоспецифическую нацеливающую часть молекулы и проапоптозный член семейства Bcl-2, где указанная клеткоспецифическая нацеливающая часть молекулы является scFvMEL, а указанный проапоптозный член семейства Bcl-2 является Вах или фрагментом Вах. В специфическом варианте фрагмент Вах лишен по меньшей мере части экзона 6 в последовательности полинуклеотида Вах.
Дополнительным объектом данного изобретения является способ лечения заболевания в индивидууме, предусматривающий стадии введения указанному индивидууму терапевтически эффективного количества композиции, содержащей химерный полипептид, содержащий индуцирующую апоптоз часть молекулы и клеткоспецифическую нацеливающую часть молекулы; и фармацевтический носитель. В специфическом варианте фармацевтический носитель содержит липид. В другом специфическом варианте указанным заболеванием является злокачественная опухоль, диабет, артрит или воспалительное заболевание кишечника, атеросклероз или диабетическая ретинопатия. В дополнительном конкретном варианте это заболевание является злокачественной опухолью. В другом конкретном варианте индуцирующая апоптоз часть молекулы является гранзимом. В дополнительноом конкретном варианте этот гранзим является гранзимом В или его фрагментом. В следующем конкретном варианте индуцирующей апоптоз частью молекулы является проапоптозный член семейства Bcl-2. В другом конкретном варианте проапоптозный член семейства Bcl-2 является Вах или его фрагментом. В следующем конкретном варианте фрагмент Вах лишен по меньшей мере части полипептида, кодируемой экзоном 6 в последовательности полинуклеотида Вах. В другом конкретном варианте фрагмент Вах лишен по меньшей мере части полипептида, кодируемого экзонами, выбранными из группы, состоящей из экзонов 4, 5 и 6. В следующем конкретном варианте введение выполняют внутривенной инъекцией. В другом конкретном варианте введение выполняют ингаляцией. В следующем конкретном варианте введение осуществляют внутривенно, внутрикожно, внутриартериально, внутрибрюшинно, введением в очаг повреждения, интракраниально, внутрисуставно, введением в предстательную железу, внутриплеврально, внутритрахеально, интраназально, введением в стекловидное тело, внутривлагалищно, интраректально, внутриопухолевым введением, внутримышечно, внутрибрюшинно, подкожно, субконъюнктивально, внутрипузырно, через слизистую оболочку, интраперикардиально, интраумбиликально, внутриглазно, перорально, местным введением, локальным введением, посредством ингаляции (например, арозольной ингаляции), посредством инъекции, посредством инфузии, посредством непрерывной инфузии, посредством локализованной перфузии с непосредственным омыванием клеток-мишеней, через катетер, через лаваж, в виде крема или в виде липидной композиции. В конкретном варианте этот способ дополнительно предусматривает введение указанному индивидууму противовоспалительной композиции, проведение химиотерапии, хирургического вмешательства, облучения, гормональной терапии или генотерапии.
Предполагается, что аспекты данного изобретения, обсуждаемые в контексте одного варианта данного изобретения, могут использоваться в отношении любых других вариантов изобретения.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
Следующие фигуры образуют часть данного описания и включены для дополнительной демонстрации определенных аспектов данного изобретения. Данное изобретение может быть лучше понято при ссылке на один или несколько из этих фигур в комбинации с подробным описанием конкретных представленных здесь вариантов.
Фиг.1 иллюстрирует кДНК пред-зрелого гранзима В человека из клеток Hut78. Электрофорез в 1% агарозном геле демонстрирует кДНК пред-зрелого гранзима В человека, синтезированного из клеток Hut78 при помощи ОТ-ПЦР. Дорожка 1 представляет молекулярный маркер низкомолекулярных ДНК; дорожка 2 представляет контрольную синтезированную кДНК (˜500 п.н.); дорожка 3 представляет контроль без ОТ; и дорожка 4 представляет кДНК пред-зрелого гранзима В (˜800 п.н.).
Фиг.2 показывает нуклеотидную последовательность, кодирующую пред-зрелый гранзим В человека (SEQ ID NO:54) и аминокислотную последовательность (SEQ ID NO:55).
Фиг.3 иллюстрирует конструирование слитых конструкций рЕТ32GrB-vegf121 и рЕТ32GrB-scFvMEL. Конструирование этих слитых конструкций основано на способе ПЦР. Фиг.3А показывает конструирование рЕТ32GrB-vegf121. Фиг.3В показывает конструирование рЕТ32GrB-scFvMEL. Полноразмерные гены лигировали в сайт XbaI/XhoI экспрессирующего вектора рЕТ-32а(+).
Фиг.4 демонстрирует предсказанную структуру рекомбинантных слитых белков гранзим В-vegf121 Фиг.(4А) и гранзим B-scFvMEL Фиг.(4В) в векторе рЕТ32а, экспрессируем в E. coli, и последовательности гранзим В-vegf121 (фиг.4С и 4D) (SEQ ID NO:56 для последовательности нуклеиновой кислоты и SEQ ID NO:57 для аминокислотной последовательности), гранзим В-scFvMEL (фиг.4Е и 4F) (SEQ ID NO:58 для последовательности нуклеиновой кислоты и SEQ ID NO:59 для аминокислотной последовательности). Вектор рЕТ32а(+) содержит промотор Т7 для высокопродуктивной экспрессии. Экспрессия этой нуклеиновой кислоты включает в себя последовательность, содержащую Trx.tag, за которой следуют Hic.tag, сайт расщепления тромбином и сайт расщепления энтерокиназой для конечного удаления метки (tag) очистки белка.
Фиг.5 показывает анализ электрофорезом в ДСН-ПААГ экспрессии этих слитых белков с окрашиванием Кумасси синим электрофореза в ДСН-ПААГ слитого белка гранзим В-vegf121 (фиг.5А) и слитого белка гранзим В-scFvMEL (фиг.5В) при восстанавливающих условиях. Панель А фиг.5А показывает окрашивание Кумасси синим электрофореза в ДСН-ПААГ слитого белка гранзим В-vegf121. Дорожка 1 показывает неиндуцированные лизаты общих клеток; дорожка 2 показывает индуцированные лизаты общих клеток; дорожка 3 показывает неиндуцированный растворимый материал; дорожка 4 показывает индуцированный растворимый материал; дорожка 5 показывает неиндуцированный нерастворимый материал; дорожка 6 показывает индуцированный нерастворимый материал; дорожка 7 показывает молекулярные маркеры белков. На панели В дорожка 1 показывает молекулярные маркеры белков; дорожка 2 показывает про-гранзим В-vegf121 (IMAC-элюат из Talon Resin); дорожка 3 показывает про-гранзим В-vegf121 (Imac-элюат из Nickel NTA), дорожка 4: Гранзим В-vegf121 (после разрезания rEK). На фиг.5В показано окрашивание Кумасси синим электрофореза в ДСН-ПААГ гранзим В-scFvMEL. На панели С дорожка 1 показывает молекулярные маркеры белков; дорожка 2 показывает неиндуцированные лизаты общих клеток; дорожка 3 показывает индуцированные лизаты общих клеток; дорожка 4 показывает неиндуцированный растворимый материал; дорожка 5 показывает индуцированный растворимый материал; дорожка 6 показывает неиндуцированный нерастворимый материал; дорожка 7 показывает индуцированный нерастворимый материал. На панели Д дорожка 1 показывает молекулярные маркеры белков; дорожка 2 показывает про-гранзим В-scFvMEL(IMAC-элюат из Nickel NTA); дорожка 3 показывает гранзим В-scFvMEL (после разрезания rEK).
Фиг.6 демонстрирует Вестерн-блот-анализ слитых белков гранзим В-vegf121 и гранзим В-scFvMEL.
Фиг.7 показывает активность связывания scFvMEL-части молекулы слитого белка гранзим В-scFvMEL. ELISA различных слитых белков scFvMEL выполняли на планшете, предварительно покрытом белком L.
Фиг.8 демонстрирует испытание цитотоксичности гранзим В-vegf121 против клеток log-фазы РАЕ-Flk-1 и РАЕ-Flt-1.
Фиг.9 демонстрирует испытание цитотоксичности гранзим В-scFvMEL на клетках А375-М.
Фиг.10 иллюстрирует ген Вах человека, его экзоны и домены ВН1, ВН2 и ВН3.
Фиг.11 демонстрирует клонирование кДНК Вах человека из клеток Namalwa при помощи ПЦР. Дорожка 1: маркеры низкомолекулярных ДНК, дорожки 2-6: Синтезированная контролем кДНК (˜500 п.н.), дорожки 7-8: кДНК Вах человека (˜580 п.н.) с использованием случайного праймера (дорожка 7) и с использованием олиго(dT)праймера (дорожка 8).
Фиг.12А и 12В иллюстрируют конструирование scFvMEL-bax-родственных конструкций.
Фиг.13 показывает анализ электрофорезом в ДСН-ПААГ и окрашиванием Кумасси синим экспрессии этих слитых белков.
Фиг.14 показывает экспрессию рЕН32-scFvMEL-bax и рЕТ32-Bax-scFvMEL, трансформированных в E. coli AD494(DE3)pLysS, и при IPTG-индукции.
Фиг.15 демонстрирует Вестерн-блоттинг-анализ экспрессии полноразмерных белков bax и Вах-scFvMEL. Дорожка 1: pBad/HisA (отрицательный контроль), дорожка 2: pBad/HisLacZ (положительный контроль экспрессии), дорожки 3-5: белок Bax (дорожка 3: экспрессия в среде RM+глюкоза+ампициллин, дорожка 4: экспрессия в RM+ампициллин, дорожка 5: экспрессия в среде LB+ампициллин), дорожки 6-8: белок Вах-scFvMEL (дорожка 6: экспрессия в среде RM+глюкоза+ампициллин, дорожка 7: экспрессия в RM+ампициллин, дорожка 8: экспрессия в среде LB+ампициллин).
Фиг.16А и 16В демонстрируют активность связывания scFvMEL-части слитых белков.
Фиг.17 показывает цитотоксичность слитых белков scFvMEL-bax345 и Bax345-scFvMEL на А375-М.
Фиг.18 показывает ELISA слитого белка гранзим В-Vegf121 на различных клеточных линиях (детектируемых с мышиным анти-vegf121-антителом и мышиным анти-гранзим В-антителом).
Фиг.19 демонстрирует цитотоксичность Гранзим В-VEGF121 на трансфицированных эндотелиальных клетках.
Фиг.20 показывает анализ цитотоксичности гранзим В-Vegf121 в сравнении с vegf121-rgel in vitro против клеток РАЕ/FLK-1.
Фиг.21 иллюстрирует активность каспазы на клетках РАЕ, обработанных слитым белком Гранзим В-Vegf121.
Фиг.22 демонстрирует высвобождение цитохрома с клеток РАЕ, обработанных GRB/VEGF121.
Фиг.23 показывает перемещение Вах клеток РАЕ после обработки GRB/VEGF121.
Фиг.24 иллюстрирует высвобождение цитохрома с в А375-М, обработанных GRB/scFvMEL, в сравнении с клетками SKBR3-HP.
Фиг.25 показывает, что GrB/VEGF121 индуцирует образование лесенки ДНК на клетках РАЕ/flk-1.
Фиг.26 показывает ELISA GrB/scFvMEL на положительных в отношении антигена gp240 клетках А375-М в сравнении с отрицательными в отношении антигена gp240 клетками Т-24 с детектированием с использованием мышиных mAb против GrB.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Другие цели, признаки и преимущества данного изобретения станут очевидными из следующего подробного описания. Однако, должно быть понятно, что это подробное описание и конкретные примеры, хотя они и показывают предпочтительные варианты данного изобретения, даются только в качестве иллюстрации, поскольку различные изменения и модификации в пределах сущности и объема данного изобретения будут очевидными специалистам с квалификацией в данной области из этого подробного описания.
В применении в описании здесь артикли "а" или "an" обозначают один или несколько. В применении здесь в формуле изобретения, при использовании вместе со словом "содержащие" слова "a" или "an" могут означать один или более, чем один. В применении здесь термин «другой» может означать по меньшей мере второй или более.
Термин "апоптоз" в данном контексте определяется как программированная гибель клеток; эндогенная программа смерти клетки приводит к смерти этой клетки.
Термин "цитокин" в данном контексте определяется как агент, произведенный клеткой, который влияет на поведение другой клетки. В конкретном варианте этот агент является полипептидом. Например, цитокины, произведенные лимфоцитами, часто называют лимфокинами или интерлейкинами (IL). Кроме того, цитокины действуют на специфические рецепторы цитокинов на клетках, на которые они воздействуют. В специфическом варианте, термин "цитокин" включает в себя факторы роста.
Термин "гранзим" в данном контексте определяется как фермент из гранул цитотоксических лимфоцитов, который, после вхождения в цитозоль клетки, индуцирует апоптоз и/или фрагментацию ядерной ДНК. В конкретном варианте этот гранзим является сериновой протеазой лимфоцитов. В некоторых вариантах гранзим является полноразмерным, тогда как в других вариантах гранзим является частичным.
Термин "фактор пути передачи сигнала" в данном контексте определяется как фермент, субстрат, кофактор или другой белок, который влияет на биологическую активность другого фермента, кофактора или белка. В специфическом варианте этот фактор ассоциирован с опосредованной рецепторами передачей сигнала из пространства снаружи от клеточной мембраны для модуляции реакции роста в клетке. В одном варианте реакция роста, которая модулируется, является положительной реакцией роста. В альтернативном варианте, реакция роста, которая модулируется, является отрицательной реакцией роста, такой как индукция апоптоза.
Данное изобретение относится к химерным белкам со свойствами нацеливающей на определенные клетки специфичности и деструкции клеток, такими как свойства из путей передачи сигнала, связанных прямо или опосредованно с индуцирующими апоптоз активностями. В некоторых вариантах разрушающие клетки части молекул являются индуцирующими апоптоз активностями. Химерные белки данного изобретения состоят из клеткоспецифической нацеливающей части молекулы и индуцирующей апоптоз части молекулы. Клеткоспецифическая нацеливающая часть молекулы обеспечивает клеткоспецифические связывающие свойства химерному белку, тогда как индуцирующая апоптоз часть молекулы индуцирует программированную гибель клеток после вхождения в клетку-мишень. В некоторых вариантах химерные белки данного изобретения доставляются в виде полипептидов и продуцируются рекомбинантной экспрессией слитого полинуклеотида между кодирующей последовательностью нацеливающей на клетку частью молекулы и кодирующей последовательностью индуцирующего апоптоз белка. Такие химерные белки, по-видимому, превосходят иммунотоксины, используемые в настоящее время в данной области, так как они имеют человеческое происхождение и, следовательно, имеют, как ожидается, уменьшенную иммуногенность в реципиенте-человеке. Кроме того, химерные белки убивают клетки-мишени индукцией апоптоза, который не вызывает высвобождения клеточных органелл во внеклеточную среду с образованием воспалительной реакции. Когда клетки гибнут по механизму апоптоза, они сморщиваются и конденсируются, но органеллы и плазматические мембраны сохраняют их целостность, и мертвые клетки бысто подвергаются фагоцитозу соседними клетками или макрофагами до утечки содержимого этих клеток, вызывая вследствие этого минимальную тканевую или системную реакцию.
Данное изобретение относится также к фармацевтическим композициям этих химерных белков, способам получения таких белков и способам применения их in vitro и in vivo, в частности, для элиминации специфических нежелательных клеток-мишеней и для лечения различных патологических состояний, а также для применения этих белков для диагностики заболеваний.
В данном изобретении описаны способы и композиции, касающиеся нацеленной деструкции клетки, использующие химерный полипептид. Химерный полипептид состоит по меньшей мере из двух частей молекулы: одной частью является эффективный компонент для лизиса данной клетки; второй частью молекулы является компонент доставки химерного полипептида для нацеливания убивающего компонента на представляющую интерес клетку. В некоторых вариантах данного изобретения по меньшей мере одна из этих частей молекулы, а предпочтительно обе, имеют человеческое происхождение, что элиминирует иммунную реакцию индивидуума, в которого вводят данный химерный полипептид. В одном варианте частью для лизиса клетки является компонент пути передачи сигнала, например, такой, который является лимитирующим фактором или точкой рестрикции в этом пути. Доставка через путь передачи сигнала позволяет избежать необходимости индукции более ранних стадий этого пути, и полученное введение этой точки рестрикции приводит в конечном счете к тому же самому эффекту, что и деструкция данной клетки. Специалисту с квалификацией в данной области известно, что типы агентов, которые могли бы доставляться внутриклеточно для опосредования передачи сигнала, включают в себя ферменты, такие как киназы (например, протеинкиназа В (РКВ, АКТ), которая опосредует передачу сигнала инсулина; протеинкиназа С, которая участвует в многочисленных событиях передачи сигнала; и фосфатидилинозитол-3-киназа, которая участвует в многочисленных событиях передачи сигнала); фосфатазы; протеазы (такие как каспаза-3); нуклеазы (такие как активируемая каспазой дезоксирибонуклеаза (CAD), которая является медиатором апоптоза); фосфолипазы; NCKAPI (который является апоптоз-ассоциированным белком, отрицательно регулируемым в тканях головного мозга пациентов с болезнью Альцгеймера; Suzuki et al., 2000) или кофакторы, такие как цитохром с (который участвует в передаче сигнала апоптоза) и циклический АМФ (который участвует в многочисленных путях передачи сигнала).
В особом варианте фактор пути передачи сигнала является ферментом. Этот фермент может быть гидролазой (например, дезаминазой, эстеразой, гликозидазой, липазой, пептидазой, фосфатазой, фосфодиэстеразой и протеиназой); изомеразой (например, эпимеразой, мутазой и рацемазой); лигазой или синтетазой (например, ацил-CoA-синтетазой, аминоацил-тРНК-синтетазой и карбоксилазой); лиазой (например, альдолазой, декарбоксилазой, дегидратазой и нуклеотидциклазой); оксидоредуктазой (например, дегидрогеназой, диоксигеназой, гидрогеназой, монооксигеназой, нитрогеназой, оксидазой и редуктазой); и/или трансферазой (например, ацилтрансферазой, аминотрансферазой, гликозилтрансферазой, киназой, метилтрансферазой, нуклеотидилтрансферазой, фосфорилазой и сульфотрансферазой). В специфических вариантах этот фермент классифицируется как токсин, что означает, что он является токсичным в отношении клетки, ткани или организма.
В некоторых вариантах фактор пути передачи сигнала является индуцирующим апоптоз фактором. Почти все клетки содержат механизмы, ответственные за опосредование смерти клеток (апоптоза). В конкретном варианте, и, как показано в примерах здесь, доставка белка гранзима В во внутреннее пространство клеток-мишеней приводит к смерти клеток через апоптозные механизмы. С использованием рекомбинантного лиганда (VEGF) и рекомбинантного антитела (scFvMEL), которые связываются с клеточной поверхностью опухолевых клеток и эффективно интернализуются, авторы данного изобретения сконструировали два новых связанных с гранзимом В слитых белка: GrB-vegf121 для специфического нацеливания на эндотелиальные клетки; и GrB-scFvMEL для специфического нацеливания на клетки меланомы.
Специалисту с обычной квалификацией в данной области известны клеткоспецифические нацеливающие части молекул, которые могли бы быть применимы в нацеливании химерного полипептида на представляющую интерес клетку. Например, клеткоспецифические нацеливающие части молекул могут быть антителами к маркеру (маркерам) конкретных клеток, фактору (факторам) роста, гормону (гормонам) или цитокину (цитокинам).
Специалисту с обычной квалификацией в данной области известно, что последовательности нуклеиновых кислот и аминокислотные последовательности, применимые для генерирования химерного полипептида данного изобретения, являются легко получаемыми, в частности, через общедоступные базы данных, такие как база данных GenBank Международного Центра информации по биотехнологии (NCBI) или коммерчески доступные базы данных, такие как Celera Genomics, Inc. (Rockville, MD). Например, аминокислотные последовательности гранзима В, применимые в данном исследовании, могут включать в себя, в сопровождении их номера доступа GenBank, по меньшей мере: Р10144 (SEQ ID NO:11); ХР_012328 (SEQ ID NO:12); А61021 (SEQ ID NO:13); NP_004122 (SEQ ID NO:14); САА01810 (SEQ ID NO:15); и/или ААА75490 (SEQ ID NO:16). SEQ ID NO:60 является последовательностью гранзима В человека, отражающей вариации, наблюдаемые в SEQ ID NO:11 - SEQ ID NO:16, например, при остатке 55 (Gln или Arg), при остатке 94 (Pro или Ala), такие как N-концевое удлинение, содержащее SEQ ID NO:61 (MKSLSLLHLFPLPRAKREQGGNNSSSNQGSLPEK), и/или такие как делеция остатков 1-20.
Последовательности нуклеиновых кислот гранзима В, применимые в данном изобретении, могут включать в себя по меньшей мере: ХМ_012328 (SEQ ID NO:17); BF589964 (SEQ ID NO:18); BF221604 (SEQ ID NO:19); NM_004131 (SEQ ID NO:20); А26437 (SEQ ID NO:21); и/или М28879 (SEQ ID NO:22). Аминокислотные последовательности гранзима А, применимые в данном изобретении, могут включать в себя, в сопровождении их номера доступа GenBank, по меньшей мере: Р12444 или NP_006135 (SEQ ID NO:23) или ХР_003652 (SEQ ID NO:24). SEQ ID NO:25 содержит аминокислотную последовательность гранзима А человека и отражает вариацию в SEQ ID NO:23 и SEQ ID NO:24 в остатке 121 (Thr или Met, соответственно). Последовательности нуклеиновых кислот гранзима А, применимые в данном изобретении, могут включать в себя по меньшей мере: ХМ_003652 (SEQ ID NO:26); NM_006144 (SEQ ID NO:27) и/или U40006 (SEQ ID NO:28). Специалисту с квалификацией в данной области будет известно, как найти эти и родственные им последовательности из базы данных GenBank NCBI.
I. Индуцирующие апоптоз белки
Строго регулируемая гибель клеток необходима для развития организмов с многочисленными направлениями дифференцировки и поддержания гомеостаза внутри тканей. На состояние дифференцировки индивидуальной клетки непосредственно влияет то, может ли она выполнять суицидную реакцию в ответ на стимул смерти. Были идентифицированы как положительные, так и отрицательные регуляторы программированной смерти клеток (апоптоза). Bcl-2 является репрессором программированной смерти клеток (Vaux et al., 1988), и недавно было показано, что другие гомологи Bcl-2 ингибируют апоптоз. Однако, один гомолог Bcl-2, Вах, опосредует противоположный эффект через ускорение апоптоза. В семействе Bcl-2 имеются существенная гомология, сконцентрированная в двух консервативных областях: доменах гомологии 1 и 2 Bcl-2 (ВН1 и ВН2) (Oltvai et al., 1993; Boise et al., 1993; Kozopas et al., 1993; Lin et al., 1993). Члены семейства Bcl-2 включают в себя Вах, Вс1-ХL, Mc1-1, A1 и несколько открытых рамок считывания в ДНК-вирусах. Другой консервативный домен в Вах, отличающийся от ВН1 и ВН2, назван ВН3, и он опосредует гибель клеток и функции связывания белка (Chittenden et al., 1995). Субпопуляция проапоптозных белков содержит только домен ВН3, что предполагает, что этот конкретный домен может быть доменом уникальной важности в стимуляции апоптоза (Diaz et al., 1997).
In vivo Вах гомодимеризуется и образует также гетеродимеры с BCL-2, и сверхэкспрессированный Вах преодолевает активность репрессора смерти BCL-2 (Oltvai et al., 1993). Уровни экспрессии Вах, более высокие, чем уровни экспрессии Bcl-2, в опухолях мочевого пузыря коррелируют с улучшенным прогнозом пациентов. У пациентов, опухоли которых экспрессировали больше мРНК Bcl-2, чем Вах, гораздо чаще наблюдали ранние рецидивы (Gazzaniga et al., 1996).
Недавно сообщалось, что сплайсинговый вариант Вах, Вах-альфа, экспрессировался в высоком количестве в эпителии здоровой молочной железы, тогда как только слабую экспрессию или отсутствие экспрессии обнаруживали в 39 из 40 испытанных пробах раковых тканей (Bargou et al., 1996), и в различных гистологических пробах обнаруживали понижающую регуляцию Вах-альфа. Кроме того, при трансфекции Вах-альфа в линии раковых клеток молочной железы под контролем тетрациклин-зависимой системы экспрессии Вах восстанавливал чувствительность раковых клеток в отношении как сывороточного голодания, так и запускаемого АРО-I/Fas апоптоза, значимо уменьшая рост опухоли в мышах SCID. Таким образом, было сделано предположение, что разрушение пути апоптоза может способствовать патогенезу злокачественной опухоли молочной железы по меньшей мере частично вследствие нарушения баланса между членами семейства генов Bcl-2 (Bargou et al., 1996).
Были идентифицированы дополнительные члены семейства Bcl-2 индуцирующих апоптоз белков. Bak, новый член семейства Bcl-2, экспрессируется в большом разнообразии типов клеток и связывается с гомологом Bcl-2, Bcl-х2, в дрожжах (Farrow et al., 1995; Chittenden et al., 1995). В Bak был идентифицирован домен, как необходимый, так и достаточный для активности цитотоксичности и связывания с Bcl-х1. Кроме того, в Вах и Bip1 были идентифицированы последовательности, подобные этому домену, которые являются отличающимися от ВН1 и ВН2. Этот домен является критическим для опосредования функции множественных регулирующих гибель клеток белков, которые взаимодействуют с членами семейства Bcl-2 (Chittenden et al., 1995).
Сверхэкспрессия Bak в симпатических нейронах лишенных фактора роста нервов ускоряла апоптоз и блокировала защитное действие коинфицированного Е1В 19К. Известно, что белок аденовируса Е1В 19К (19 кДа) ингибирует апоптоз, индуцированный Е1А, фактором-альфа некроза опухолей, Fas-антигеном и лишением фактора роста нервов (Farrow et al., 1995). Экспрессия Bak индуцировала быстрый и интенсивный апоптоз лишенных сыворотки фибробластов, что предполагает, что Bak непосредственно участвует в активации аппарата смерти клеток (Chittenden et al., 1995). В нормальной и неопластической толстой кишке экспрессия в слизистой оболчке иммунореактивного Bak колокализовалась с сайтами апоптоза эпителиальных клеток. Индукция апоптоза в линии клеток злокачественной опухоли толстой кишки человека НТ29 и линии клеток тонкой кишки 1ЕС 18 здоровой крысы в культуре сопровождалась увеличенной экспрессией Bak без стойких изменений в экспрессии других гомологичных Bcl-2 белков (Moss et al., 1996). Таким образом, предполагалось также, что Bak является эндогенным членом семейства Bcl-2, наилучшим образом коррелирующим с апоптозом кишечных клеток (Moss et al., 1996).
В противоположность Вах, Bak может ингибировать гибель клеток в трансформированной вирусом Эпстайна-Барр клеточной линии. Ткани с уникальным распределением мессенджер РНК Bak включают в себя ткани, содержащие долгоживущие, терминально дифференцированные типы клеток (Krajewski, et al., 1996), что предполагает, что индуцирующая гибель клеток активность является широко распространенной и что тканеспецифическая модуляция апоптоза контролируется прежде всего регуляцией молекул, которые ингибируют апоптоз (Kiefer et al., 1995).
Другой член семейства Bcl-2, Bad, обладает ключевым аминокислотным мотивом доменов ВН1 и ВН2. Bad не имеет классической С-концевой сигнальной-якорной последовательности, ответственной за интегральные мембранные положения других членов этого семейства. Bad селективно димеризуется с Bcl-ХL, а также Bcl-2, но не с Вах, Bcl-Xs-Mcl1, A1 или с самим собой. Bad обращает активность репрессора смерти Bcl-ХL, но не Bcl-2 (Yang et al., 1995; Ottilie et al., 1997; Zha et al., 1997).
Bik, другой член семейства Bcl-2, взаимодействует с усиливающими выживание клеток белками, Bcl-2 и Bcl-ХL, а также вирусными усиливающими выживание белками, BHRF1 вируса Эпстайна-Барр и E1B-19кДа аденовируса. В анализах временной трансфекции Bik способствует смерти клеток, подобно Вах и Bak, другим проапоптозным членам семейства Bcl-2. Эта проапоптозная активность Bik может быть подавлена коэкспрессией белков Bcl-2, Bcl-ХL, EBV-BHRF1 и E1B-19 кДа, что предполагает, что Bik может быть общей мишенью как для клеточных, так и для вирусных антиапоптозных белков. Хотя Bik не содержит явной гомологии относительно консервативных доменов ВН1 и ВН2, характерных для семейства Bcl-2, он имеет общий домен из 9 аминокислот (ВН3) с Вах и Bak, который может быть решающей детерминантой для стимулирующей апоптоз активности этих белков (Boyd et al., 1995; Han et al., 1996).
Семейство Bcl-2 состоит из различных пар белков-антагонистов и белков-агонистов, хотя остается невыясненным, является ли их функция взаимозависимой. С использованием моделей увеличения функции и потери функции Bcl-2 и Вах, Knudson et al. (1997) показали, что апоптоз и гипоплазия тимуса, характерные для Bcl-2-недостаточных мышей, по существу не наблюдаются у мышей, также недостаточных в отношении Вах. Единственная копия Вах усиливала апоптоз в отсутствие Bcl-2. Однако, сверхэкспрессия Bcl-2 все еще подавляла апоптоз в отсутствие Вах. Хотя существует конкуренция in vivo между Вах и Bcl-2, каждый из них способен независимо регулировать апоптоз. Было показано, что Вах образует каналы в липидных мембранах и запускает высвобождение инкапсулированного в липосомах карбоксифлуоресцеина как при нейтральном, так и кислотном рН. При физиологическом рН высвобождение могло быть блокировано Bcl-2. В более плоских липидных бислоях Вах образовывал зависимые от рН и напряжения каналы ионной проводимости. Таким образом, проапоптозные эффекты Вах могут быть индуцированы присущей ему активностью порообразования, в отношении которой Bcl-2 может быть антагонистом (Antonsson et al., 1997). Было показано, что два других члена этого семейства, Bcl-2 и Bcl-1, также образуют поры в липидных мембранах (Schnedel et al., 1997).
II. Гранзим В и апоптоз
Защитные системы хозяина против вирусов, паразитических агентов и трансформированных клеток требуют цитотоксических Т-лимфоцитов (CTL) и природных клеток-киллеров (NK) (Berke, 1995; Kagi et al., 1996), которые индуцируют апоптоз в клетках-мишенях с использованием по меньшей мере двух отдельных механизмов. В первом механизме происходит стимуляция рецепторов смерти клеточной поверхности (таких как Fas) на клетках-мишенях лигандами смерти, экспрессируемыми на поверхности эффекторной клетки (Nagata and Golstein, 1995; Ashkenazi and Dixit, 1998), которая затем приводит к активации каспазных каскадов в клетке-мишени. Во втором механизме, называемом «экзоцитозом гранул», происходит векторный перенос содержимого цитоплазматических гранул эффекторной клетки в клетку-мишень (Doherty, 1993; Shresta et al., 1995a; Shresta et al., 1995b). Перфорин и семейство гранзимов сериновых протеаз являются важными компонентами этих гранул.
Перфорин является белком массой 70 кДа, который связывается зависимым от кальция образом с фосфорилхолиновыми группами мембран (Masson and Tschopp, 1985; Young et al., 1986; Tschopp et al., 1989). После связывания перфорин встраивается в мембрану и олигомеризуется, приводя к образованию пор. Это увеличение проницаемости мембраны делает возможным вхождение других молекул, таких как гранзимы, в клетку-мишень.
Гранзимы А и В являются особенно обильными (Smyth et al., 1996) в гранулах CTL и NK-клеток. Гранзим В, который также называют фрагментином или цитотоксической протеазой Т-клеток (ССР), сходен с каспазами, имеющими свойство расщепления субстратных белков после остатка аспартата (Zunino et al., 1990; Libe et al., 1986; Odake et al., 1991; Poe et al., 1991; Shi et al., 1992). У мышей с выключенным геном гранзима В проявляется важная роль гранзима В в индукции апоптоза клетки-мишени. CTL и NK-клетки, полученные из мышей гранзим В-/-, имеют в сильной степени уменьшенную способность индуцировать апоптозную фрагментацию ДНК в клетках-мишенях (Shresta et al., 1995a; Heusel et al., 1994). Хотя более ранние дополнительные исследования показали, что очищенный гранзим В один не способствовал апоптозу при добавлении к клеткам-мишеням, совместная обработка очищенным гранзимом В и перфориновыми белками индуцировала заметную фрагментацию ДНК и признаки апоптоза в четырех линиях клеток-мишеней лимфомы (Shi et al., 1992). Таким образом, возможно, что гранзим В получает возможность вхождения в клетки-мишени через генерируемые перфорином поры, хотя это является дискуссионным. Несколько исследований показали, что гранзим В интернализуется клетками-мишенями в отсутствие добавленного перфорина (Froelich et al., 1996; Jans et al., 1996; Shi et al., 1997; Pinkoski et al., 1998; Pinkoski et al., 2000). Сообщалось, что интернализованный гранзим В находится в цитоплазме (Jans et al., 1996; Shi et al., 1997) или в новом пузырьковом компартменте (Pinkoski et al., 1998), хотя запуск апоптоза в клетках, которые содержат интернализованный гранзим В, требует дополнительного добавления перфорина к этим клеткам (Froelich et al., 1996; Jans et al., 1996; Shi et al., 1997; Pinkoski et al., 1998; Pinkoski et al., 2000). Возможно, что перфорин требуется для высвобождения гранзима В в клетку-мишень из интернализованных пузырьков. Другие исследования показали, что перфорин облегчает перемещение гранзима В в ядро и что ядерная локализация является критической для способности гранзима В вызывать апоптоз (Jans et al., 1996; Shi et al., 1997; Pinkoski et al., 1998; Pinkoski et al., 2000; Pinkoski et al., 1996; Trapani et al., 1996).
Хотя важность субклеточной локализации гранзима В остается дискуссионной, точно известно, что гранзим В обладает способностью воздействовать на каспазный путь апоптоза. Исследования in vitro показали, что гранзим В способен расщеплять прокаспазу-3, -6, -7, -8m, -9 и -10 (Darmon et al., 1996; Darmon et al., 1996; Martin et al., 1996; Quan et al., 1996; Fernandes-Alnemri et al., 1995; Orth et al., 1996; Fernandes-Alnemri et al., 1995; Chinnaiyan et al., 1996; Gu et al., 1996; Boldin et al., 1996; Muzio et al., 1996; Duan et al., 1996; Fernandes-Alnemri et al., 1996; Medema et al., 1997; Van de Craen et al., 1997; Talanian et al., 1997). Было показано, что в случае прокаспаз-3, -7 и -9, опосредуемый гранзимом В процессинг генерирует активные каспазные ферменты (Darmon et al., 1995; Quan et al., 1996; Gu et al., 1996; Duan et al., 1996). Более важно, что исследования с целыми клетками показали, что каспазы активируются в клетках-мишенях после коинкубирования с гранзимом В и перфорином (Darmon et al., 1996; Talanian et al., 1997; Shi et al., 1996). Однако, остается определить, какие каспазы являются предпочтительными субстратами in vivo для гранзима В. В любом случае, имеются основания для предположения, что гранзим В может способствовать апоптозу просто расщеплением и активацией эндогенных каспаз в клетке-мишени.
Расщепление субстратных белков каспазы PARP, ламина В и U1-70кДа наблюдают также в клетках, подвергающихся опосредованному гранзимом В/перфорином апоптозу (Medema et al., 1997; Talanian et al., 1997; Shi et al., 1996; Andrade et al., 1998). Эти события расщепления, вероятно, обусловлены каспазами, активируемыми расщеплением гранзимом В, так как расщепление всех трех белков ингибируется 100 мкМ DEVD- или VAD-содержащими пептидами, которые ингибируют каспазы, но не гранзим В (Darmon et al., 1996; Medema et al., 1997; Talanian et al., 1997; Shi et al., 1996; Andrade et al., 1998). Два дополнительных субстратных белка каспазы, DNA-PKcs и NuMA, также расщепляются в обработанных гранзимом В/перфорином клетках, но расщепление этих белков является нечувствительным к пептидным ингибиторам DEVD или VAD (Andrade et al., 1998). Кроме того, размеры протеолитических фрагментов DNA-PKcs и NuMA, генерируемых гранзимом В, отличаются от размеров фрагментов, получаемых из расщепления каспазой, что предполагает, что во время гранзим В-опосредуемого апоптоза важные клеточные субстраты расщепляются каспаза-независимым образом. Значение этих каспаза-независимых событий расщепления является невыясненным. Однако, поскольку гранзим В/перфорин-опосредованная фрагментация ДНК и апоптозная гибель существенно задерживаются 100 мкМ DEVD/VAD (Darmon et al., 1996; Talanian et al., 1997; Shi et al., 1996), это придает особое значение необходимости каспазной активации во время этой формы апоптоза.
III. Гранзим А и апоптоз
Зрелый гранзим А является сшитым дисульфидными связями гомодимером массой 50 кДа, который расщепляет субстратные белки после остатков лизина или аргинина (Odake et al., 1991; Gershenfeld et al., 1986; Masson et al., 1986), и гранзим А является наиболее обильной протеазой, обнаруживаемой в гранулах клеток CTL. Механизм действия этой протеазы существенно отличается от механизма действия гранзима В, хотя гранзим А способен индуцировать апоптоз после загрузки в клетки-мишени. Кроме того, считается, что роль гранзима А в CTL-индуцируемом апоптозе является значительно менее уловимой, чем роль гранзима В. Например, мыши, которые являются дефектными по экспрессии гранзима А (гранзим А-/- мыши), проявляют относительно нормальную CTL-опосредованную цитотоксичность (Andrade et al., 1998), хотя они неспособны к элиминации мышиного поксвируса Ectromelia (Mulbacher et al., 1996). В противоположность этому, CTL из гранзим В-/--мышей способны индуцировать гибель клеток-мишеней только после пролонгированной коинкубации (Heusel et al., 1994), и, следовательно, гранзим В имеет решающее значение для быстрого лизиса CTL. Недавние эксперименты с использованием дефектных как по гранзиму А, так и по гранзиму В мышей предполагают, что гранзим А действительно играет некоторую роль в CTL-опосредуемом лизисе. CTL из гранзим А-/-/гранзим В-/--мышей неспособны индуцировать фрагментацию ДНК клеток-мишеней, даже после пролонгированной коинкубации (Shresta et al., 1999), что свидетельствует о том, что активность гранзима А является ответственной за способность гранзим В-/- CTL индуцировать апоптоз клеток-мишеней после пролонгированного экспонирования. Таким образом, гранзим А может позволить CTL убивать клетки-мишени в условиях, когда активность гранзима В является ингибированной (например, клетки-мишени, которые экспрессируют ингибиторы гранзима В).
В исследованиях с рекомбинантными белками коинкубация гранзима А и перфорина с клетками-мишенями приводит к быстрому накоплению (в пределах 2 часов) разрывов одной цепи ДНК (Hayes et al., 1980; Beresford et al., 1999) в противоположность быстрой деградации ДНК на фрагменты олигонуклеосомной длины, наблюдаемой в клетках, обработанных гранзимом В и перфорином. Обработка гранзимом В/перфорином также приводит к ядерной конденсации (Beresford et al., 1999). Эти эффекты, которые происходят в ответ на гранзим А, являются нечувствительными к ингибиторам каспазы, что свидетельствует о том, что эти действия гранзима А являются каспаза-независимыми (Beresford et al., 1999). В соответствии с этим, обработка гранзимом А/перфорином не приводит к процессингу/активации прокаспазы-3 или расщеплению субстратных белков каспазы, PARP, ламина В или rho-GTPазы (Beresford et al., 1999). Однако, индуцируемая гранзимом В фрагментация ДНК строго зависит от активации каспаз. Как гранзим А, так и гранзим В (вместе с перфорином) также индуцируют цитолиз клеток-мишеней, причем оба случая являются каспаза-зависимыми событиями. Таким образом, существующие доказательства указывают на то, что гранзим В является первичным CTL-медиатором фрагментации ДНК клетки-мишени и апоптозной смерти и что апоптозные действия этой протеазы опосредованы прежде всего активацией каспазы. Альтернативно, гранзим А может быть молчащим или специализированным медиатором апоптоза клеток-мишеней, причем пути, инициируемые гранзимом А, являются явно отличающимися от путей, инициируемых гранзимом В.
IV. Генерирование химерных молекул
Хотя химерные белки данного изобретения могут быть получены химическими синтетическими способами или образованием химической связи между двумя частями молекулы, предпочтительно получать их слиянием кодирующей последовательности клеткоспецифической нацеливающей части молекулы и кодирующей последовательности индуцирующего апоптоз белка под контролем регуляторной последовательности, которая управляет экспрессией этого слитого полинуклеотида, в подходящей клетке-хозяине. В предпочтительных вариантах, каждый из компонентов химерного белка имеет функциональную активность в отношении их соответствующих частей, являясь клеткоспецифической нацеливающей частью молекулы и фактором пути передачи сигнала (таким как индуцирующий апоптоз белок).
Слияние двух полноразмерных кодирующих последовательностей может быть достигнуто способами, хорошо известными в области молекулярной биологии. Предпочтительно, слитый полинуклеотид содержит кодон инициации трансляции AUG на 5'-конце только первой кодирующей последовательности без инициирующего кодона второй кодирующей последовательности во избежание продуцирования двух отдельных кодируемых продуктов. Кроме того, лидерная последовательность может быть помещена на 5'-конце этого полинуклеотида для нацеливания экспрессируемого продукта на специфический сайт или компартмент в клетке-хозяине для облегчения секреции или последующей очистки после экспрессии гена. Две кодирующие последовательности могут быть слиты непосредственно без какого-либо линкера или с использованием гибкого полилинкера, такого как линкер, состоящий из пентамера Gly-Gly-Gly-Gly-Ser (SEQ ID NO:50), повторяемого 1-3 раза. Такой линкер использовали в конструировании одноцепочечных антител (scFv) встраиванием между VH и VL (Bird et al., 1988; Huston et al., 1988). Этот линкер сконструирован для обеспечения возможности точного взаимодействия между двумя бета-складками, образующими вариабельную область одноцепочечного антитела. Другие линкеры, которые могут быть использованы, включают в себя Glu-Gly-Lys-Ser-Ser-Gly-Ser-Gly-Ser-Glu-Ser-Lys-Val-Asp (SEQ ID NO:51) (Chaudhary et al., 1990) и Lys-Glu-Ser-Gly-Ser-Val-Ser-Ser-Glu-Gln-Leu-Ala-Gln-Phe-Arg-Ser-Leu-Asp (SEQ ID NO:52) (Bird et al., 1988).
А. Клеткоспецифические нацеливающие части
Химерные белки данного изобретения состоят из клеткоспецифической нацеливающей части молекулы и индуцирующей апоптоз части молекулы. Клеткоспецифическая нацеливающая часть молекулы придает этой молекуле специфическое в отношении типа клетки связывание и выбирается на основе популяции конкретных клеток, которые должны быть мишенями. Большое разнообразие белков является пригодным в качестве клеткоспецифических нацеливающих частей молекулы, в том числе, но не только, лиганды для рецепторов, такие как факторы роста, гормоны и цитокины, и антитела или их антигенсвязывающие фрагменты.
Поскольку большое количество рецепторов клеточной поверхности были идентифицированы в кроветворных клетках различных линий дифференцировки, лиганды или антитела, специфические для этих рецепторов, могут быть использованы в качестве клеткоспецифических нацеливающих молекул. Для нацеливания на IL2R+-клетки может быть использован IL2 в качестве клеткоспецифической нацеливающей части молекулы в химерном белке. Альтернативно, другие молекулы, такие как В7-1, В7-2 и CD40, могут быть использованы для специфического нацеливания на активированные Т-клетки (The Leucocyte Antigen Facts Book, 1993, Barclay et al. (tds.), Academic Press). Кроме того, В-клетки экспрессируют CD19, CD40 и рецептор IL4 и могут быть мишенями для частей молекулы, которые связывают эти рецепторы, таких как лиганд CD40, IL4, IL5, IL6 и CD28. Элиминация иммунных клеток, таких как Т-клетки и В-клетки, особенно применима в лечении аутоиммунитета, гиперчувствительности, реакций отторжения трансплантата и в лечении лимфоидных опухолей. Примерами аутоиммунных заболеваний являются рассеянный склероз, ревматоидный артрит, инсулинзависимый сахарный диабет, системная красная волчанка, склеродерма и увеит. Более конкретно, поскольку известно, что миелиновый основной белок является главной мишенью атаки иммунных клеток в рассеянном склерозе, этот белок может быть использован в качестве клеткоспецифической нацеливающей части молекулы для лечения рассеянного склероза (WO 97/19179; Becker et al., 1997).
Другие цитокины, которые могут быть использованы для нацеливания на специфические субпопуляции клеток, включают в себя интерлейкины (IL1-IL15), гранулоцитарный колониестимулирующий фактор, макрофагальный колониестимулирующий фактор, гранулоцитарно-макрофагальный колониестимулирующий фактор, ингибирующий лейкоз фактор, фактор некроза опухолей, трансформирующий фактор роста, эпидермальный фактор роста, инсулин-подобные факторы роста и/или фибробластный фактор роста (Thompson (ed.), 1994, The Cytokine Handbook, Academic Press, San Diego).
Специалисту с квалификацией в данной области известно, что существуют множество известных цитокинов, в том числе гемопоэтины (четырехспиральные пучки) (такие как Еро (эритропоэтин), IL-2 (фактор роста Т-клеток), IL-3 (образующий множество колоний CSF), IL-4 (BCGF-1, BSF-1), IL-5 (BCGF-2), IL-6, IL-4 (IFN-β2, BSF-2, BCDF), IL-7, IL-8, IL-9, IL-11, IL-13 (Р600), G-CSF, IL-15 (фактор роста Т-клеток), GM-CSF (гранулоцитарно-макрофагальный колониестимулирующий фактор), OSM (OM, онкостатин М) и LIF (фактор ингибирования лейкоза)); интерлейкины (такие как IFN-γ, IFN-α и IFN-β); суперсемейство иммуноглобулинов (таких как В7.1 (CD80) и В7.2 (В70, CD86)); TNF-семейство (например, TNF-α (кахектин), TNF-β (лимфотоксин, LT, LT-α), LT-β, CD40-лиганд (CD40L), Fas-лиганд (FasL), CD27-лиганд (CD27L), CD30-лиганд (CD30L) и 4-1BBL)); и цитокины, не относящиеся к конкретному семейству (такие как TGF-β, IL-1α, IL-1β, IL-1 RA, IL-10 (ингибитор F синтеза цитокинов), IL-12 (фактор стимуляции NK-клеток), MIF, IL-16, IL-17 (mCTLA-8) и/или IL-18 (ICIF, индуцирующий интерферон-γ фактор)).
Кроме того, некоторые молекулы клеточной поверхности экспрессируются в высокой степени в опухолевых клетках, в том числе рецепторы гормонов, такие как рецептор хорионического гонадотропина человека и рецептор рилизинг-фактора гонадотропина (Nechushtan et al., 1997). Таким образом, соответствующие гормоны могут быть использованы в качестве клеткоспецифических нацеливающих частей молекул в раковой терапии.Таким образом, в некоторых вариантах данного изобретения в химерных полипептидах не используются антитела. Однако, антитела являются чрезвычайно разносторонними и применимыми клеткоспецифическими нацеливающими частями молекул, так как они генерируются против любых представляющих интерес антигенов клеточной поверхности. Моноклональные антитела получали против рецепторов клеточной поверхности, связанных с опухолями антигенов и маркеров, специфических для лейкоцитарной линии дифференцировки, таких как CD-антигены. Гены вариабельных областей антител могут быть легко выделены из гибридомных клеток с использованием способов, хорошо известных в данной области.
На протяжении последних нескольких лет несколько моноклональных антител были одобрены для терапевтического использования, и они обеспечивали значительный клинический и коммерческий успех. Большая часть клинического применения моноклональных антител обусловлена аффинностью и специфичностью, с которыми они связываются с их мишенями, а также продолжительным существованием в кровотоке вследствие их относительно большого размера. Однако, моноклональные антитела не очень хорошо пригодны для использования в показаниях, в которых предпочтительным является короткий полупериод существования в организме или в которых их большой размер мешает им физически достигать участка потенциальной терапевтической активности.
Кроме того, антитела в их нативной форме, состоящие из двух различных полипептидных цепей, которые должны быть генерированы в приблизительно равных количествах и точно собраны, являются плохими кандидатами для терапевтических целей. Однако, может быть создан одноцепочечный полипептид, который может сохранять антигенсвязывающие свойства моноклонального антитела.
Одноцепочечные антитела (SCA) являются генетически сконструированными белками, предназначенными для расширения диапазона применения на терапевтические и диагностические применения, возможные с моноклональными антителами. SCA имеют специфичность связывания и аффинность моноклональных антител и, в их нативной форме, имеют размер одной пятой - одной шестой размера моноклонального антитела, обычно обусловливающий их очень короткие полупериоды существования в организме. SCA человека предоставляют многие преимущества в сравнении с большинством моноклональных антител, в том числе более специфическую локализацию в участках-мишенях в теле, более быстрый клиренс из тела и лучшую возможность использования перорально, интраназально, чрескожно или посредством ингаляции. Кроме этих преимуществ, полностью человеческие SCA могут быть выделены непосредственно из библиотек SCA человека без необходимости дорогостоящих и занимающих много времени процедур "гуманизации" антител. SCA легко получать посредством внутриклеточной экспрессии (внутри клеток), позволяющей их использование в применениях генотерапии, в которых молекулы SCA действуют в качестве специфических ингибиторов клеточных функций.
Вариабельные области из тяжелых и легких цепей (VH и VL) имеют длину приблизительно 110 аминокислот. Они могут быть связаны линкером из 15 аминокислот с последовательностью (SEQ ID NO:50)3, которая имеет достаточную гибкость для обеспечения возможности сборки из этих двух доменов функционального антигенсвязывающего кармана. В специфических вариантах добавление различных сигнальных последовательностей позволяет нацеливать scFv на различные органеллы внутри клетки или секретировать scFv. Добавление константной области легкой цепи (Ck) обеспечивает возможность димеризации через дисульфидные связи, что придает увеличенную стабильность и авидность. Таким образом, для одноцепочечного Fv (scFv)-SCA, хотя два домена Fv-фрагмента кодируются разными генами, была показана возможность приготовления синтетического линкера, который позволяет получать их в виде единственной белковой цепи scFv (Bird et al., 1988; Huston et al., 1988) с использованием рекомбинантных способов. Кроме того, их часто используют вследствие легкости их выделения из библиотек фагового дисплея и их способности узнавать консервативные антигены (в отношении обзора см. Adams and Schier, 1999). Например, scFv используют для нацеливания на суицидные гены в экспрессирующих карциноэмбриональный антиген (СЕА) опухолевых клетках с использованием ретровектора, представляющего анти-СЕА-scFv (Kuroki et al., 2000).
Наконец, Fc-часть тяжелой цепи антитела может быть использована для нацеливания на экспрессирующие Fc-рецептор клетки, например, применение Fc-части IgE-антитела для нацеливания на мастоциты и базофилы. Такое применение антител для нацеливания представляющего интерес полипептида или пептида при помощи нацеливаемой антителами терапии или иммунологически нацеливаемой терапии является в настоящее время одобренным и используется на существующем рынке терапевтических средств.
Таким образом, предпочтительно использовать scFv в качестве клеткоспецифической нацеливающей части молекулы в данном изобретении.
В. Индуцирующие апоптоз части молекул
Проапоптозные белки в семействе BCL2 пригодны, в частности, в качестве индуцирующих апоптоз частей молекулы в данном изобретении. Ожидается, что такие белки человека имеют уменьшенную иммуногенность в сравнении со многими иммунотоксинами, состоящими из бактериальных токсинов. Хотя Вах является применимой индуцирующей апоптоз частью молекулы в одном варианте данного изобретения, и другие члены в этом семействе являются пригодными для применения в данном изобретении и включают в себя Bak (Farrow et al., 1995; Chittenden et al., 1995; Kiefer et al., 1995), Bcl-Xs (Boise et al., 1993; Fang et al., 1994), Bad (Yang et al., 1995), Bid (Wang et al., 1996), Bik (Boyd et al., 1995), Hrk (Inohara et al., 1997) и/или Bok (Hsu et al., 1997). Нуклеотидные последовательности, кодирующие эти белки, известны в данной области, и их можно легко получить из баз данных, таких как GenBank, и, следовательно, кДНК-клоны могут быть легко получены для слияния с кодирующей последовательностью для клеткоспецифической нацеливающей части молекулы в экспрессирующем векторе.
Специфические домены конкретных членов семейства Bcl-2 исследовали в отношении их индуцирующих апоптоз активностей. Например, домен GD Bak участвует в функции апоптоза (патент США № 5656725). Кроме того, Bax и Bipla имеют общий гомологичный домен. Таким образом, любые биологически активные домены семейства Bcl-2 могут быть использованы в качестве индуцирующей апоптоз части молекулы для практики данного изобретения.
Каспазы также играют центральную роль в апоптозе и могут вполне составлять часть консенсусного центрального механизма апоптоза. Предполагается, что каспазы являются медиаторами апоптоза. С установлением того факта, что CED-3, белок, необходимый для связанной с развитием смерти клеток, имеет идентичность последовательности с цистеиновой протеазой млекопитающих интерлейкин-1-бета-превращающим ферментом (ICE), было идентифицировано семейство, состоящее из по меньшей мере 10 родственных цистеиновых протеаз. Эти белки характеризуются почти абсолютной специфичностью в отношении аспарагиновой кислоты в положении Р1. Все каспазы (ICE-подобные протеазы) содержат консервативный мотив активного сайта в виде QACKG-(где Х обозначает R, Z или G)-пентапептида. Каспазы синтезируются в виде неактивных проферментов, содержащих N-концевой пептид (продомен) вместе с одной большой и одной малой субъединицей. Кристаллические структуры как каспазы-1, так и каспазы-3 показывают, что активный фермент является гетеротетрамером, содержащим две малые и две большие субъединицы. Активация каспаз во время апоптоза приводит к расщеплению критических клеточных субстратов, в том числе поли(АДФ-рибоза)-полимеразы и ламинов, с ускорением таким образом драматических морфологических изменений апоптоза (Cohen, 1997, Biochem. J. 326:1-16). Таким образом, применение каспазы в качестве индуцирующей апоптоз части молекулы находится в пределах объема данного изобретения.
Недавно были клонированы несколько новых белков, и они были идентифицированы как факторы, необходимые для опосредования активностей белков, главным образом каспаз, участвующих в пути апоптоза. Один фактор идентифицировали как ранее известный белок транспорта электрона, цитохром с (Lin et al., 1996, Cell 86:147-157), названный Apaf-2. Кроме цитохрома с, активация каспазы-3 требует двух других цитозольных факторов Apaf-1 и Apaf-3. Apaf-1 является белком, гомологичным CED-4 С. elegans, а Apaf-3 был идентифицирован как член семейства каспаз, каспаза-9. Оба фактора связываются друг с другом через их соответствующие NH2-концевые гомологичные CED-3 домены, в присутствии цитохрома с, причем это событие приводит к активации каспазы-9. Активированная каспаза-9, в свою очередь, расщепляет и активирует каспазу-3 (Liu et al., 1996; Zou et al., 1997; Li et al., 1997). Другим белком, участвующим в апоптозном пути, является фактор фрагментации ДНК (DFF), гетеродимер из субъединиц массой 45 и 40 кДа, который функционирует далее по ходу процесса от каспазы-3, запуская фрагментацию геномной ДНК в нуклеосомных сегментах (Liu et al., 1997).
С. Получение химерных полипептидов
В соответствиии с целями данного изобретения, полинуклеотид, который кодирует химерный белок, мутантный полипептид, биологически активный фрагмент химерного белка, или его функциональный эквивалент, может быть использован для генерирования рекомбинантных молекул ДНК, которые осуществляют экспрессию этого химерного белка, химерных пептидных фрагментов, или его функционального эквивалента, в подходящих клетках-хозяевах.
Вследствие природной вырожденности генетического кода другие ДНК-последовательности, которые кодируют по существу ту же самую или функционально эквивалентную аминокислотную последовательность, могут быть использованы в практике клонирования и экспрессии химерного белка данного изобретения. Такие ДНК-последовательности включают в себя последовательности, способные гибридизоваться с химерными последовательностями или их комплементарными последовательностями в жестких условиях. В одном варианте фраза "строгие условия" в данном контексте относится к условиям гибридизации, которые (1) используют низкую ионную силу и высокую температуру для промывания, например, 0,015 М NaCl/0,0015 М цитрат натрия/0,1% ДСН при 50°С; (2) используют во время гибридизации денатурирующий агент, такой как формамид, например, 50% (об./об.) формамид, с 0,1% бычьим альбумином/0,1% Фиколлом/0,1% поливинилпирролидоном/50 мМ натрий-фосфатным буфером при рН 6,5 с 750 мМ NaCl, 75 мМ цитратом натрия при 42°С; или (3) используют 50% формамид, 5 х SSC (0,75 М NaCl, 0,075 М пирофосфат натрия, 5 х раствор Денхардта, обработанную ультразвуком ДНК спермы (50 мкг/мл), 0,1% ДСН и 10% декстрансульфат при 42°С, с промывками при 42°С в 0,2 х SSC и 0,1% ДСН.
Измененные ДНК-последовательности, которые могут быть использованы в соответствии с данным изобретением, включают в себя делеции, добавления или замены различных нуклеотидных остатков, что приводит в результате к последовательности, которая кодирует тот же самый или функционально эквивалентный продукт химерного гена. Продукт этого гена сам по себе может содержать делеции, добавления или замены аминокислотных остатков в пределах химерной последовательности, что приводит к молчащему изменению с получением функционально эквивалентного химерного белка. Такие аминокислотные замены могут быть произведены на основе сходства в полярности, заряде, растворимости, гидрофобности, гидрофильности и/или амфипатического характера включенных остатков. Например, отрицательно заряженные аминокислоты включают в себя аспарагиновую кислоту и глутаминовую кислоту; положительно заряженные аминокислоты включают в себя лизин, гистидин и аргинин; аминокислоты с незаряженными полярными головными группами, имеющие сходные величины гидрофильности, включают в себя следующие аминокислоты: глицин, аспарагин, глутамин, серин, треонин, тирозин; и аминокислоты с неполярными головными группами включают в себя аланин, валин, изолейцин, лейцин, фенилаланин, пролин, метионин, триптофан.
ДНК-последовательности данного изобретения могут быть сконструированы для изменения химерной кодирующей последовательности для различных целей, в том числе, но не только, изменений, которые модифицируют процессинг и экспрессию продукта данного гена. Например, мутации могут быть введены с использованием способов, которые хорошо известны в данной области, например, сайт-направленного мутагенеза для встраивания новых сайтов рестрикции, для изменения характера гликозилирования, фосфорилирования и т.д.
В альтернативном варианте данного изобретения кодирующая последовательность химерного белка может быть синтезирована полностью или частично с использованием химических способов, хорошо известных в данной области (см., например, Caruthers et al., 1980; Crea and Horn, 1980 и Chow and Kempe, 1981). Например, активные домены этих частей молекул могут быть синтезированы твердофазными способами, отщеплены от смолы и очищены препаративной высокоэффективной жидкостной хроматографией с последующим химическим связыванием с образованием химерного белка (например, см. Creighton, 1983, Proteins Structures And Molecular Principles, W.H. Freeman and Co., N.Y. pp. 50-60). Состав синтетических пептидов может быть подтвержден аминокислотным анализом или секвенированием (например, процедурой деградации Эдмана; см. Creighton, 1983, Proteins Structures And Molecular Principles, W.H. Freeman and Co., N.Y. pp. 34-49). Альтернативно, две молекулы химерного белка, полученные синтетическим или рекомбинантным способами, могут быть конъюгированы с использованием химических линкеров в соответствии со способами, хорошо известными в данной области (Brinkmann and Pastan, 1994).
Для экспрессии биологически активного химерного белка нуклеотидную последовательность, кодирующую химерный белок, или функциональный эквивалент, встраивают в подходящий экспрессирующий вектор, т.е. вектор, который содержит необходимые элементы для транскрипции и трансляции встроенной кодирующей последовательности. Продукты химерного гена, в также клетки-хозяева или клеточные линии, трансфицированные или трансформированные рекомбинантными химерными экспрессирующими векторами, могут быть использованы для различных целей. Они включают в себя, но не ограничиваются ими, генерирование антител (т.е. моноклональных или поликлональных антител), которые связывают с эпитопами этих белков, для облегчения их очистки.
Способы, которые хорошо известны специалистам с квалификацией в данной области, могут быть использованы для конструирования экспрессирующих векторов, содержащих кодирующую химерный белок последовательность и подходящие сигналы регуляции транскрипции/трансляции. Эти способы включают в себя in vitro-способы рекомбинантных ДНК, синтетические способы и рекомбинацию/генетическую рекомбинацию in vivo. См., например, способы, описанные в Sambrook et al., 1989, Molecular Cloning: A Laboratory Manual, Cold Spring Harbor Laboratory, N.Y. и Ausubel et al., 1989, Current Protocols in Molecular Biology, Greene Publishing Associates and Wiley Interscience, N.Y.
Различные системы хозяин-экспрессирующий вектор могут быть использованы для экспрессии кодирующей химерный белок последовательности. Они включают в себя, но не ограничиваются ими, микроорганизмы, такие как бактерии, трансформированные рекомбинантной ДНК бактериофага, содержащие плазмидную ДНК или космидную ДНК экспрессирующие векторы, содержащие кодирующую химерный белок последовательность; дрожжи, трансформированные рекомбинантными дрожжевыми экспрессирующими векторами, содержащими кодирующую химерный белок последовательность; системы клеток насекомых, инфицированные экспрессирующими рекомбинантный вирус векторами (например, бакуловирус), содержащими кодирующую химерный белок последовательность; системы клеток растений, инфицированные экспрессирующими рекомбинантный вирус векторами (например, вирус мозаичной болезни цветной капусты, CaMV; вирус мозаики табака, TMV) или трансформированные рекомбинантными плазмидными экспрессирующими векторами (например, Ti-плазмидой), содержащими кодирующую химерный белок последовательность; или системы клеток животных. Следует отметить, что, поскольку большинство индуцирующих апоптоз белков вызывают программированную гибель клеток, предпочтительно, чтобы химерный белок данного изобретения экспрессировался в прокариотических или низших эукариотических клетках. Раздел 6 показывает, что IL2-Вах может эффективно экспрессироваться в E. coli.
Экспрессирующие элементы каждой системы варьируют по их силе и специфичностям. В зависимости от используемой системы хозяин/вектор любое число подходящих элементов транскрипции и трансляции, в том числе конститутивных и индуцируемых промоторов, могут быть использованы в экспрессирующем векторе. Например, при клонировании в бактериальных системах могут быть использованы индуцируемые промоторы, такие как pL бактериофага λ, plac, ptrp, ptac (гибридный промотор ptrp-lac, промотор цитомегаловируса) и т.п.; при клонировании в системе клеток насекомых могут быть использованы такие промоторы, как промотор полиэдрина бакуловируса; при клонировании в системах клеток растений, могут быть использованы промоторы, полученные из генома клеток растений (например, промоторы белка теплового шока; промотор для малой субъединицы RUBISCO; промотор для связывающего белка хлорофилла α/β), или из вирусов растений (например, промотор РНК 35S CaMV; промотор белка оболочки TMV); для клонирования в системах клеток млекопитающих могут быть использованы промоторы, полученные из геномов клеток млекопитающих (например, промотор металлотионеина), или из вирусов млекопитающих (например, поздний промотор аденовируса; промотор 7,5 К вируса коровьей оспы); при генерировании клеточных линий, которые содержат множественные копии химерной ДНК, могут быть использованы векторы на основе SV40, BPV и EBV с подходящим селектируемым маркером.
В бактериальных системах число экспрессирующих векторов может быть предпочтительно выбрано в зависимости от применения, предназначенного для экспрессируемого химерного белка. Например, когда должны быть получены большие количества химерного белка, могут быть желательными векторы, которые управляют высокопродуктивной экспрессией белковых продуктов, которые могут быть легко очищены. Такие векторы включают в себя, но не ограничиваются ими, вектор pHL906 (Fishman et al., 1994), экспрессирующий вектор E. coli pUR278 (Ruther et al., 1983), в котором кодирующая химерный белок последовательность может быть лигирована в этот вектор в рамке считывания с кодирующей областью lacZ, так что образуется гибридный белок AS-LacZ; векторы pIN (Inouye and Inouye, 1989; Van Heeke and Schuster, 1989); и т.п.
Альтернативной системой экспрессии, которая могла бы применяться для экспрессии химерного белка, является система насекомых. В одной такой системе вирус ядерного полиэдроза Autographa californica (AcNPV) используют в качестве вектора для экспрессии чужеродных генов. Этот вирус растет в клетках Spodoptera frugiperda. Кодирующая химерный белок последовательность может быть клонирована в не-незаменимые области (например, ген полиэдрина) этого вируса и помещена под контроль промотора AcNPV (например, промотора полиэдрина). Успешное встраивание кодирующей химерный белок последовательности будет приводить к инактивации гена полиэдрина и продуцированию не заключенного в оболочку рекомбинантного вируса (т.е. вируса, не имеющего белковой оболочки, кодируемой геном полиэдрина). Затем эти рекомбинантные вирусы используют для инфицирования клеток Spodoptera frugiperda, в которых экспрессируется встроенный ген (например, см. Smith et al., 1983; патент США № 4215051).
Специфические сигналы инициации могут также требоваться для эффективной трансляции встроенной кодирующей химерный белок последовательности. Эти сигналы включают в себя кодон инициации трансляции ATG и смежные последовательности. В случаях, когда весь химерный ген, включающий в себя его собственный инициирующий кодон и смежные последовательности, встроен в подходящий экспрессирующий вектор, могут не требоваться дополнительные сигналы регуляции трансляции. Однако, в случаях, когда кодирующая химерный ген последовательность не включает в себя ее собственный инициирующий кодон, должны быть обеспечены экзогенные сигналы регуляции трансляции, в том числе инициирующая область ATG. Кроме того, инициирующий кодон должен находиться в фазе с рамкой считывания кодирующей химерный белок последовательности для гарантии трансляции полного инсерта. Эти экзогенные сигналы регуляции трансляции и инициирующие кодоны могут иметь различное происхождение и быть как природными, так и синтетическими. Эффективность экспрессии может быть усилена включением подходящих транскрипционных энхансерных элементов, терминаторов транскрипции и т.д. (см. Bittner et al., 1987).
Кроме того, может быть выбран клеточный штамм хозяина, который модулирует экспрессию встроенных последовательностей или модифицирует и процессирует продукт гена желательным специфическим образом. Такие модификации (например, гликозилирование) и процессинг (например, расщепление) белковых продуктов могут быть важными для функции белка. Присутствие консенсусных сайтов N-гликозилирования в химерном белке может требовать правильной модификации для оптимальной функции химерного белка. Различные клетки-хозяева имеют характерные и специфические механизмы для посттрансляционного процессинга и модификации белков. Подходящие клеточные линии или системы хозяев могут быть выбраны для гарантии точной модификации и процессинга химерных белков. Для этой цели могут быть использованы клетки-хозяева, которые имеют клеточный аппарат для правильного процессинга первичного транскрипта, гликозилирования и фосфорилирования химерного белка. Такие клетки-хозяева млекопитающих включают в себя, но не ограничиваются ими, СНО, VERO, BHK, HeLa, COS, MDCK, 293, W138 и т.п.
Для долгосрочного продуцирования с высоким выходом рекомбинантных химерных белков предпочтительной является стабильная экспрессия. Например, могут быть сконструированы клеточные линии, которые стабильно экспрессируют химерный белок. Клетки-хозяева могут быть трансформированы кодирующей химерной последовательностью, регулируемой подходящими регуляторными элементами экспрессии (например, промоторными, энхансерными последовательностями, терминаторами транскрипции, сайтами полиаденилирования и т.д.) и селектируемым маркером, а не экспрессирующими векторами, которые содержат вирусные сайты инициации репликации. После введения чужеродной ДНК сконструированные клетки могут расти в течение 1-2 дней в обогащенной среде и затем могут быть перенесены в селективную среду. Селектируемый маркер в рекомбинантной плазмиде придает устойчивость к отбору и позволяет клеткам стабильно интегрировать плазмиду в их хромосомы и расти с образованием очагов, которые, в свою очередь, могут быть клонированы и размножены в клеточные линии.
Могут быть использованы ряд систем отбора, включающие в себя, но не ограничивающиеся ими, применение генов тимидинкиназы вируса простого герпеса (Wigler et al., 1977), гипоксантин-гуанинфосфорибозилтрансферазы (Szybalski and Szybalski, 1962) и аденинфосфорибозилтрансферазы (Lowy et al., 1980) в клетках tk, hgprt или aprt, соответственно. Может быть также использована устойчивость к антиметаболитам в качестве основы отбора на гены dhfr, который придает устойчивость к метотрексату (Wigler et al., 1980; O'Hare et al., 1981); gpt, который придает устойчивость к микофенольной кислоте (Mulligan and Berg, 1981); neo, который придает устойчивость к аминогликозиду G-418 (Colbere-Garapin et al., 1981); и hygro, который придает устойчивость к гигромицину (Santerre et al., 1984). Были описаны другие селектируемые гены, а именно trpB, который позволяет клеткам использовать индол вместо триптофана; hisD, который позволяет клеткам использовать гистинол вместо гистидина (Hartman and Muligan, 1988); и ODC (орнитиндекарбоксилазы), который придает устойчивость к ингибитору орнитиндекарбоксилазы, 2-(дифторметил)-DL-орнитину, DFMO (McConlogue L. 1987, In: Current Communications in Molecular Biology, Cold Spring Harbor Laboratory ed.).
D. Очистка белка
Химерные белки данного изобретения могут быть очищены известными в данной области способами, такими как высокоэффективная жидкостная хроматография, ионообменная хроматография, гель-электрофорез, аффинная хроматография и т.п. Фактические условия, используемые для очистки конкретного белка, будут зависеть, отчасти, от таких факторов, как суммарный заряд, гидрофобность, гидрофильность и т.д., и будут очевидными для специалистов с квалификацией в данной области.
Для очистки аффинной хроматографией может быть использовано любое антитело, которое специфически связывает данный белок. Для получения антител различные животные-хозяева, в том числе, но не только, кролики, мыши, крысы и т.д., могут быть иммунизированы инъекцией химерного белка или его фрагмента. Этот белок может быть прикреплен к подходящему носителю, такому как бычий сывороточный альбумин (БСА), посредством боковой функциональной группы или линкеров, присоединенных к функциональной группе боковой цепи. Для увеличения иммунологической реакции могут быть использованы различные адъюванты в зависимости от вида хозяина, в том числе, но не только, адъюванты Фрейнда (полный и неполный), минеральные гели, такие как гидроксид алюминия, поверхностно-активные вещества, такие как лизолецитин, полиолы плуроников, полианионы, пептиды, масляные эмульсии, гемоцианин фиссуреллы, динитрофенол и потенциально применимые адъюванты человека, такие как BCG (бациллы Calmetter-Guerin) и Corynebacterium parvum.
Моноклональные антитела к химерному белку могут быть получены при помощи способа, который обеспечивает продуцирование молекул антитела непрерывными клеточными линиями в культуре. Эти способы включают в себя, но не ограничиваются ими, гибридомный способ, первоначально описанный Koehler and Milstein (1975), способ В-клеточных гибридом человека (Kosbor et al., 1983; Cote et al., 1983) и способ EBV-гибридом (Cote et al., 1985). Кроме того, могут быть использованы способы, разработанные для получения "химерных антител" (Morrison et al., 1984; Neuberger et al., 1984; Takeda et al., 1985) сплайсингом генов из молекулы мышиного антитела подходящей антиген-специфичности, вместе с генами из молекулы антитела человека подходящей биологической активности. Альтернативно, способы, описанные для получения одноцепочечных антител (патент США № 4946778), могут быть адаптированы для получения химерных протеинспецифических одноцепочечных антител для очистки и обнаружения химерного белка.
V. Применения химерного полипептида
После экспрессии и очистки химерного белка его идентичность и функциональные активности могут быть легко определены способами, известными в данной области. Например, антитела к двум частям молекулы этого белка могут быть использованы для идентификации этого белка в Вестерн-блот-анализе. Кроме того, этот химерный белок может быть испытан на специфическое связывание с клетками-мишенями в анализах связывания с использованием флуоресцеин-меченого или радиоактивно меченого вторичного антитела.
А. Применения in vitro и ex vivo
Химерные полипептиды данного изобретения применимы для нацеливания на специфические типы клеток в смеси клеток и для элиминации клеток-мишеней индукцией апоптоза. Химерные полипептиды данного изобретения применимы также в качестве диагностического реагента. Связывание химерного белка с клеткой-мишенью может быть легко обнаружено с использованием вторичного антитела, специфического для индуцирующей апоптоз части молекулы. В этой связи, вторичное антитело или химерный белок метят ферментом или радиоактивным изотопом для облегчения детектирования связывания химерного белка с клеткой.
В. Применения in vivo
В некоторых вариантах эффективное количество химерных полипептидов данного изобретения вводят в клетку. В других вариантах терапевтически эффективное количество химерных полипептидов данного изобретения вводят индивидууму для лечения заболевания. Термин "эффективное количество" в данном контексте определяется как количество химерных полипептидов данного изобретения, которое является необходимым для получения физиологического изменения в клетке или ткани, в которую оно введено. Термин "терапевтически эффективное количество" в данном контексте определяется как количество химерных полипептидов данного изобретения, которое элиминирует, уменьшает, задерживает или минимизирует неблагоприятные эффекты заболевания, такого как злокачественная опухоль. Специалисту с квалификацией в данной области будет понятно, что во многих случаях химерный полипептид может не привести к излечиванию, а может обеспечивать только частичную пользу. В некоторых вариантах физиологическое изменение, имеющее некоторую пользу, считается также терапевтически полезным. Таким образом, в некоторых вариантах количество химерного полипептида, которое обеспечивает физиологическое изменение, считают "эффективным количеством" или "терапевтически эффективным количеством".
Химерные белки данного изобретения могут вводиться субъекту per se или в форме фармацевтической композиции для лечения злокачественной опухоли, аутоиммунитета, отторжения трансплантата, посттравматических иммунных реакций и инфекционных заболеваний нацеливанием на вирусные антигены, такие как gp120 или ВИЧ. Более конкретно, химерные полипептиды могут быть полезны в элиминации клеток, участвующих в иммунном клеточно-опосредованном нарушении, включающем в себя лимфому; аутоиммунитет, отторжение трансплантата, реакцию трансплантат против хозяина, ишемию и инсульт. Фармацевтические композиции, содержащие белки данного изобретения, могут быть приготовлены общепринятым смешиванием, растворением, гранулированием, приготовлением драже, растиранием в порошок, эмульгированием, инкапсулированием, процессами захвата или лиофилизации. Фармацевтические композиции могут быть приготовлены общепринятым образом с использованием одного или нескольких физиологически приемлемых носителей, разбавителей, наполнителей или добавок, которые облегчают переработку этих белков в препараты, которые могут быть использованы фармацевтически. Правильное приготовление зависит от выбранного способа введения.
Для местного введения белки данного изобретения могут быть приготовлены в виде растворов, гелей, мазей, кремов, суспензий, и т.д., как хорошо известно в данной области.
Системные композиции включают в себя композиции, предназначенные для введения инъекцией, например, подкожной, внутривенной, внутримышечной, внутриоболочечной или внутрибрюшинной инъекцией, а также композиции, предназначенные для чрескожного, чресслизистого введения, введения ингаляцией, перорального или легочного введения.
Для инъекции белки данного изобретения могут быть приготовлены в водных растворах, предпочтительно в физиологически совместимых буферах, таких как раствор Хенкса, раствор Рингера или физиологический солевой раствор. Этот раствор может содержать способствующие приготовлению агенты, такие как суспендирующие, стабилизирующие и/или диспергирующие агенты.
Альтернативно, эти белки могут быть в порошкообразной форме для воссоздания с подходящим носителем, например, апирогенной водой, перед использованием.
Для трансмукозного введения в этой композиции используют усиливающие проникновение агенты, подходящие для этого барьера.Такие усиливающие проникновение агенты известны в данной области.
Для перорального введения белки могут быть легко приготовлены объединением этих белков с фармацевтически приемлемыми носителями, хорошо известными в данной области. Такие носители позволяют приготовление белков данного изобретения в виде таблеток, пилюль, драже, капсул, жидкостей, гелей, сиропов, взвесей, суспензий и т.п. для перорального введения внутрь проходящим лечение пациентом. Для пероральных композиций, таких как, например, порошки, капсулы и таблетки, подходящие эксципиенты включают в себя наполнители, такие как сахара, например, лактоза, сахароза, маннит и сорбит; препараты целлюлозы, такие как кукурузный крахмал, пшеничный крахмал, рисовый крахмал, картофельный крахмал, желатин, трагакантовая камедь, метилцеллюлоза, гидроксипропилметилцеллюлоза, натрий-карбоксиметилцеллюлоза и/или поливинилпирролидон (PVP); гранулирующие агенты и связывающие агенты. Если желательно, могут быть добавлены дезинтегрирующие агенты, такие как сшитый поливинилпирролидон, агар или альгиновая кислота или ее соль, например, альгинат натрия.
Если желательно, твердые дозированные формы могут быть покрыты сахаром или энтеросолюбильной оболочкой с использованием стандартных способов.
Для пероральных жидких препаратов, таких как, например, суспензии, эликсиры и растворы, подходящие носители, наполнители или разбавители включают в себя воду, гликоли, масла, спирты и т.д. Дополнительно могут быть добавлены улучшающие вкус и запах агенты, консерванты, красители и т.п.
Для буккального введения эти белки могут иметь форму таблеток, мазей и т.д., приготовленных общепринятым способом.
Для введения ингаляцией белки, применяемые в соответствии с данным изобретением, обычно доставляют в форме аэрозольного спрея из находящихся под давлением упаковок или распылителя, с использованием подходящего пропеллента, например, дихлордифторметана, трихлорфторметана, дихлортетрафторэтана, диоксида углерода или другого подходящяго газа. В случае находящегося под давлением аэрозоля дозированная единица может быть определена обеспечением клапана для доставки отмеренного количества. Капсулы и патроны из желатина для использования в ингаляторе или инсуффляторе могут быть приготовлены таким образом, что они содержат порошкообразную смесь белка и подходящей порошкообразной основы, такой как лактоза или крахмал.
Эти белки могут быть также приготовлены в виде ректальных или вагинальных композиций, таких как суппозитории или удерживающие клизмы, например, содержащие обычные основы суппозиториев, такие как какао-масло или другие глицериды.
Кроме композиций, описанных ранее, эти белки могут быть также приготовлены в виде депо-препарата. Такие композиции продолжительного действия могут вводиться имплантацией (например, подкожно или внутримышечно) или внутримышечной инъекцией. Так, например, эти белки могут быть приготовлены с подходящими полимерными или гидрофобными веществами (например, в виде эмульсии в приемлемом масле) или ионообменными смолами, или в виде слабо растворимых производных, например, в виде слабо растворимой соли.
Альтернативно, могут быть использованы другие фармацевтические системы доставки. Липосомы и эмульсии являются хорошо известными примерами носителей доставки, которые могут быть использованы для доставки белков данного изобретения. Некоторые органические растворители, такие как диметилсульфоксид, также могут быть использованы, хотя обычно ценой более высокой токсичности. Кроме того, эти белки могут доставляться с использованием системы пролонгированного действия, такой как полупроницаемые матриксы твердых полимеров, содержащие терапевтический агент. Были установлены различные материалы пролонгированного высвобождения, и они хорошо известны специалистам в данной области. Капсулы пролонгированного высвобождения могут, в зависимости от их химической природы, высвобождать эти белки в течение периода от нескольких недель до более 100 дней. В зависимости от химической природы и биологической стабильности химерного белка могут использоваться дополнительные стратегии для стабилизации белка.
Поскольку белки данного изобретения могут содержать заряженные боковые цепи или концы, они могут быть включены в любую из вышеописанных композиций в виде свободных кислот или оснований или в виде фармацевтически приемлемых солей. Фармацевтически приемлемыми солями являются соли, которые по существу сохраняют биологическую активность этих свободных оснований и которые получают реакцией с неорганическими кислотами. Фармацевтические соли склонны к большей растворимости в водных и других протонных растворителях, чем соответствующие формы свободных оснований.
1. Эффективные дозы
Белки данного изобретения обычно будут использоваться в количествах, эффективных для достижения предполагаемой цели. В применении для лечения или предупреждения патологического состояния белки данного изобретения вводят или наносят в терапевтически эффективном количестве. Терапевтически эффективное количество является количеством, эффективным для ослабления или предупреждения симптомов или пролонгирования выживания получающего лечение пациента. Определение терапевтически эффективного количества находится вполне в компетенции специалистов в данной области, особенно в свете обеспеченного здесь подробного описания.
Для системного введения терапевтически эффективная доза может быть определена исходно из анализов in vitro. Например, доза может быть приготовлена в моделях животных для достижения диапазона концентраций в кровотоке, который включает в себя IC50, определенную в культуре клеток. Такая информация может быть использована для более точного определения применимых доз в людях.
Первоначальные дозы могут быть также приближенно определены из данных in vivo, например, в моделях животных, с использованием способов, которые хорошо известны в данной области. Специалист с обычной квалификацией в данной области сможет легко оптимизировать введение людям на основе информации, полученной на животных.
Дозированное количество и интервал могут корректироваться индивидуально для обеспечения уровней этих белков в плазме, которые являются достаточными для поддержания терапевтического эффекта. Обычные дозы для введения пациенту инъекцией находятся в диапазоне от приблизительно 0,1 до 5 мг/кг/день, предпочтительно от приблизительно 0,5 до 1 мг/кг/день. Терапевтически эффективные уровни в сыворотке могут быть достигнуты введением множественных доз каждый день.
В случаях местного введения или селективного поглощения эффективная локальная концентрация этих белков может не соотноситься с концентрацией в плазме. Специалист с квалификацией в данной области сможет оптимизировать терапевтически эффективные локальные дозы без чрезмерного экспериментирования.
Количество вводимого белка будет, конечно, зависеть от получающего лечение субъекта, от веса субъекта, тяжести болезни, способа введения и мнения прописывающего дозу врача.
Эта терапия может повторяться периодически при детектируемых симптомах или даже в том случае, когда симптомы являются недетектируемыми. Эта терапия может предоставляться отдельно или в комбинации с другими лекарственными средствами. В случае аутоиммунных нарушений лекарственные средства, которые могут быть использованы в комбинации с IL2-Вах данного изобретения, включают в себя, но не ограничиваются ими, стероидные и нестероидные противовоспалительные агенты.
2. Токсичность
Предпочтительно, терапевтически эффективная доза описанных здесь химерных белков будет обеспечивать терапевтическую пользу без индукции существенной токсичности.
Токсичность описанных здесь белков может быть определена стандартными фармацевтическими процедурами в культурах клеток или с использованием экспериментальных животных, например, определением LD50 (дозы, летальной для 50% популяции) или LD100 (дозы, летальной для 100% популяции). Отношение доз между токсическим и терапевтическим действием является терапевтическим индексом. Предпочтительными являются белки, которые обнаруживают высокие терапевтические индексы. Данные, полученные из этих анализов в культурах клеток и исследований на животных, могут быть использованы в приготовлении диапазона доз, который не является токсичным для использования в человеке. Эта доза описанных здесь белков находится предпочтительно в пределах диапазона концентраций в кровотоке, которые включают в себя эффективную дозу с малой токсичностью или без токсичности. Эта доза может варьироваться в пределах этого диапазона в зависимости от используемой дозированной формы и используемого способа введения. Точная композиция, способ введения и доза могут быть выбраны врачом на основании состояния пациента. (См., например, Fingl et al., 1975, In: The Pharmacological Basis of Therapeutics, Ch. 1, p. 1).
VI. Биологические функциональные эквиваленты
Поскольку могут быть произведены модификации и/или изменения в структуре полинуклеотидов, кодирующих химерные полипептиды данного изобретения, и/или в самих химерных полипептидах в соответствии с данным изобретением с получением молекул, имеющих сходные или улучшенные свойства, подобные биологически функциональные эквиваленты также включены в данное изобретение.
А. Модифицированные полинуклеотиды и полипептиды
Биологический функциональный эквивалент может содержать полинуклеотид, который был сконструирован для содержания отличающихся последовательностей с одновременным сохранением способности кодировать белок "дикого типа", или стандартный белок. Это может быть выполнено до вырожденности генетического кода, т.е. присутствия множественных кодонов, которые кодируют те же самые аминокислоты. В одном примере желательным для специалиста в данной области может быть введение рестрикции последовательности узнавания фермента в полинуклеотид без нарушения способности этого полинуклеотида кодировать белок.
В другом примере полинуклеотид, кодирующий химерный полипептид, может быть биологическим функциональным эквивалентом (или может кодировать биологический функциональный эквивалент) с более существенными изменениями. Определенные аминокислоты могут быть заменены другими аминокислотами в структуре белка без ощутимой потери способности интерактивного связывания с такими структурами, как, например, антигенсвязывающие области антител, сайты связывания на молекулах субстрата, рецепторы и т.п. Так называемые "консервативные" замены не разрушают биологическую активность этого полипептида, так как это структурное изменение не является изменением, которое нарушает способность этого полипептида осуществлять его планируемую функцию. Таким образом, авторы данного изобретения предполагают, что могут быть произведены различные изменения в последовательности описанных здесь генов и белков при осуществлении все еще задач данного изобретения.
Специалисту с квалификацией в данной области, в связи с функциональными эквивалентами, понятно, что неотъемлемой в определении "биологически функциональный эквивалентный" белок и/или полинуклеотид является концепция, что существует предел числу изменений, которые могут быть произведены в определенной части молекулы с сохранением молекулы с приемлемым уровнем эквивалентной биологической активности. Таким образом, биологически функциональные эквиваленты определяются здесь как полипептиды (и полинуклеотиды), в которых могут быть заменены выбранные аминокислоты (или кодоны). Функциональная активность включает в себя способность лизиса клетки-мишени для части молекулы, являющейся фактором пути передачи сигнала, или способность клеткоспецифического нацеливания для клеткоспецифической нацеливающей части молекулы.
Обычно, чем короче длина молекулы, тем меньше изменений, которые могут быть произведены в этой молекуле при сохранении функции. Более длинные домены могут иметь промежуточное число изменений. Полноразмерный белок будет иметь наибольшую толерантность в отношении большего числа изменений. Однако, должно быть понятно, что определенные молекулы или домены, которые в высокой степени зависят от их структуры, могут выдерживать небольшое число модификаций или вообще не выдерживают модификации.
Аминокислотные замены обычно основываются на относительном сходстве боковых заместителей боковых цепей, например, их гидрофобности, гидрофильности, заряде, размере и/или т.п. Анализ размера, формы и/или типа заместителей боковых цепей аминокислот обнаруживает, что аргинин, лизин и/или гистидин, - все являются положительно заряженными остатками; что аланин, глицин и/или серин, - все имеют одинаковый размер; и/или что фенилаланин, триптофан и/или тирозин, все имеют в общем сходную форму. Таким образом, на основании этих рассмотрений аргинин, лизин и/или гистидин; аланин, глицин и/или серин; и/или фенилаланин, триптофан и/или тирозин; - определяются здесь как биологически функциональные эквиваленты.
Для выполнения более количественных изменений может учитываться индекс гидрофобности аминокислот. Каждой аминокислоте был приписан индекс гидрофобности на основании ее свойств гидрофобности и/или заряда, и эти индексы гидрофобности являются следующими: изолейцин (+4,5); валин (+4,2); лейцин (+3,8); фенилаланин (+2,8); цистеин/цистин (+2,5); метионин (+1,9); аланин (+1,8); глицин (-0,4); треонин (-0,7); серин (-0,8); триптофан (-0,9); тирозин (-1,3); пролин (-1,6); гистидин (-3,2); глутамат (-3,5); глутамин (-3,5); аспартат (-3,5); аспарагин (-3,5); лизин (-3,9) и/или аргинин (-4,5).
Важность индекса гидрофобности аминокислот в придании интерактивной биологической функции белку обычно является понимаемой в данной области (Kyte & Doolittle, 1982, включена здесь в качестве ссылки). Известно, что определенные аминокислоты могут заменять другие аминокислоты, имеющие сходный индекс и/или балл гидрофобности, и/или все еще сохранять сходную биологическую активность. При выполнении изменений на основе индекса гидрофобности предпочтительной является замена аминокислот, индексы гидрофобности которых находятся в пределах ±2, особенно предпочтительной является замена аминокислот, индексы гидрофобности которых находятся в пределах ±1, и еще более предпочтительной является замена аминокислот, индексы гидрофобности которых находятся в пределах ±0,5.
В данной области является понятным, что замена подобных аминокислот может производиться эффективно на основе гидрофильности, в частности, когда создаваемый посредством этого биологически функциональный эквивалентный белок и/или пептид предназначается для применения в иммунологических вариантах, как это имеет место в некоторых вариантах данного изобретения. Патент США 4554101, включенный здесь в качестве ссылки, устанавливает, что наивысшая локальная средняя гидрофильность белка, создаваемая гидрофильностью его смежных аминокислот, коррелирует с его иммуногенностью и/или антигенностью, т.е. с биологическим свойством данного белка.
Как подробно описано в патенте США 4544101, следующие величины гидрофильности были приписаны аминокислотным остаткам: аргинин (+3,0); лизин (+3,0); аспартат (+3,0 ± 1); глутамат (+3,0 ± 1); серин (+0,3); аспарагин (+0,2); глутамин (+0,2); глицин (0); треонин (-0,4); пролин (-0,5 ± 1); аланин (-0,5); гистидин (-0,5); цистеин (-1,0); метионин (-1,3); валин (-1,5); лейцин (-1,8); изолейцин (-1,8); тирозин (-2,3); фенилаланин (-2,5); триптофан (-3,4). В выполнении изменений на основании сходных величин гидрофильности предпочтительной является замена аминокислот, величины гидрофильности которых находятся в пределах ±2, особенно предпочтительной является замена аминокислот, величины гидрофильности которых находятся в пределах ±1, и еще более предпочтительной является замена аминокислот, величины гидрофильности которых находятся в пределах ±0,5.
В. Измененные аминокислоты
Во многих аспектах данное изобретение основывается на синтезе пептидов и полипептидов in cyto, через транскрипцию и трансляцию подходящих полинуклеотидов. Эти пептиды и полипептиды будут включать в себя двадцать «природных» аминокислот и их посттрансляционные модификации. Однако, синтез пептидов in vitro позволяет использовать модифицированные и/или необычные аминокислоты. Таблица 2 примеров, но не ограничивающая только ими, модифицированных и/или необычных аминокислот приведена здесь ниже.
Модифицированные и/или необычные аминокислоты
пиперидиновая кислота
С. Миметики
Наряду с обсуждаемыми здесь биологическими функциональными эквивалентами, авторы данного изобретения считают также, что структурно сходные соединения могут быть приготовлены для имитации ключевых частей пептидов или полипептидов данного изобретения. Такие соединения, которые могут быть названы пептидомиметиками, могут быть использованы таким же образом, что и пептиды данного изобретения, и, следовательно, являются функциональными эквивалентами.
Некоторые миметики, которые имитируют элементы вторичной и третичной структуры белка, описаны в Johnson et al., (1993). Обоснованием для применения пептидомиметиков является то, что пептидный скелет белков существует в основном для ориентации боковых цепей аминокислот таким образом, чтобы облегчить молекулярные взаимодействия, такие как взаимодействия антитела и/или антигена. Таким образом, пептидомиметик конструируют для обеспечения возможности молекулярных взаимодействий, сходных с молекулярными взаимодействиями природной молекулы.
Некоторые успешные применения концепции пептидомиметиков фокусировались на миметиках β-витков в белках, о которых известно, что они являются высокоантигенными. По-видимому, структура β-витка в полипептиде может быть предсказана компьютерными алгоритмами, обсуждаемыми здесь. После определения составляющих аминокислот витка могут быть сконструированы миметики для получения сходной пространственной ориентации этих существенных элементов боковых цепей аминокислот.
[0165] Другие подходы сосредоточивались на применении малых, содержащих много дисульфидных связей белков в качестве привлекательных структурных шаблонов для получения биологически активных конформаций, которые имитируют сайты связывания больших белков. Vita et al., (1988). Структурный мотив, который, по-видимому, является эволюционно консервативным в некоторых токсинах, является небольшим (30-40 аминокислот), стабильным и высокопермиссивным для мутации. Этот мотив состоит из бета-складки и альфа-спирали, соединенных во внутренней части тремя мостиковыми дисульфидами.
Бета II-витки имитировали успешно с использованием циклических L-пентапептидов и циклических L-пентапептидов с D-аминокислотами. Weisshoff et al., (1999). Johannesson et al. (1999) также сообщают о бициклических трипептидах с индуцирующими обращенный виток свойствами.
Способы генерирования специфических структур были описаны в данной области. Например, миметики альфа-спирали описаны в патентах США 5446128; 5710245; 5840833 и 5859184. Эти структуры делают пептид или белок более термостойким, а также увеличивают устойчивость к протеолитической деградации. Описаны шести-, семи-, одиннадцати, двенадцати, тринадцати- и четырнадцатичленные циклические структуры.
Способы генерирования конформационно ограниченных бета-витков и бета-выступов описаны, например, в патентах США 5440013; 5618914 и 5670155. Бета-витки допускают измененные боковые заместители без изменений в соответствующей конформации скелета и имеют подходящие концы для включения в пептиды с использованием стандартных процедур синтеза. Другие типы миметических витков включают в себя обращенные и гамма-витки. Миметики обращенных витков описаны в патентах США 5475085 и 5929237, а миметики гамма-витков описаны в патентах США 5672681 и 5674976.
D. Липосомное нацеливание
Связывание химерного полипептида с липосомой может улучшать биораспределение и другие свойства химерного полипептида. Например, очень успешной была опосредованная липосомами доставка нуклеиновой кислоты и экспрессия чужеродной ДНК in vitro (Nicolau and Sene, 1982; Fraley et al., 1979; Nicolau et al., 1987). Была также продемонстрирована возможность опосредованной липосомами доставки и экспрессии чужеродной ДНК в культивируемом курином эмбрионе, клетках HeLa и клетках гепатомы (Wong et al., 1980). Был осуществлен также успешный опосредованный липосомой перенос гена в крысах после внутривенной инъекции (Nicolau et al., 1987).
Предполагается, что композиция липосома/химерный полипептид может содержать дополнительные вещества для доставки к ткани. Например, в некоторых вариантах данного изобретения липид или липосома могут быть связаны с гемагглютинирующим вирусом (HVJ). Было показано, что это облегчает слияние с клеточной мембраной и способствует вхождению в клетку инкапсулированной в липосоме ДНК (Kaneda et al., 1989). В другом примере липид или липосома могут быть в комплексе или использоваться вместе с ядерными негистоновыми белками хромосом (HMG-1) (Kato et al., 1991). В дополнительных вариантах липид может быть в комплексе или использоваться вместе как с HVJ, так и с HMG-1.
Нацеленная доставка достигается добавлением лигандов без нарушения способности этих липосом доставлять большие количества химерного полипептида. Предполагается, что это позволит выполнять доставку к специфическим клеткам, тканям и органам. Специфичность нацеливания систем доставки на основе лигандов основана на распределении рецепторов лигандов на различных типах клеток. Нацеливающий лиганд может быть нековалентно или ковалентно связан с липидным комплексом и может быть конъюгирован с липосомами различными способами.
Е. Сшивающие агенты
Бифункциональные сшивающие реагенты интенсивно использовали для различных целей, в том числе для приготовления аффинных матиксов, модификации и стабилизации разнообразных структур, идентификации сайтов связывания лиганда и рецептора и структурных исследований. Гомобифункциональные реагенты, которые несут две идентичные функциональные группы, оказались высокоэффективными в индукции сшивания между идентичными и различными макромолекулами или субъединицами макромолекулы и в связывании полипептидных лигандов с их сайтами специфического связывания. Гетеробифункциональные реагенты содержат две различные функциональные группы. С использованием преимущества дифференциальных реактивностей этих двух различных функциональных групп сшивание может контролироваться как селективно, так и последовательно. Бифункциональные сшивающие реагенты могут быть подразделены в соответствии со специфичностью их функциональных групп, например, амино-, сульфгидрил-, гуанидино-, индол-, карбоксил-специфических групп. Из них особенно популярными стали реагенты, нацеленные на свободные аминогруппы, вследствие их коммерческой доступности, легкости синтеза и мягких условий реакции, при которых они могут применяться. Большая часть гетеробифункциональных сшивающих реагентов содержит первичную амино-реактивную группу и тиол-реактивную группу.
Примеры способов сшивания лигандов с липосомами описаны в патенте США 5603872 и патенте США 5401511, каждый из которых специально включен здесь в качестве ссылки в полном виде. Различные лиганды могут быть ковалентно связаны с поверхностями липосом через сшивание амино-остатков. Липосомы, в частности, многослойные пузырьки (везикулы) (MLV) или однослойные пузырьки, такие как микроэмульсифицированные липосомы (MEL) и большие однослойные липосомы (LUVET), каждый из которых содержит фосфатидилэтаноламин (РЕ), получали с использованием установленных процедур. Включение РЕ в липосомы обеспечивает активный функциональный остаток, первичную аминогруппу, на поверхности липосомы для целей сшивания. Лиганды, такие как эпидермальный фактор роста (EGF), успешно связывались с РЕ-липосомами. Лиганды связываются ковалентно с дискретными сайтами на поверхностях липосом. Число и поверхностная плотность этих сайтов будет диктоваться составом липосом и типом липосом. Поверхности липосом могут также иметь сайты нековалентного связывания. Для образования ковалентных конъюгатов лигандов и липосом сшивающие реагенты испытывали на эффективность и биосовместимость. Сшивающие реагенты включают в себя глутаровый альдегид (GAD), бифункциональный оксиран (OXR), диглицидиловый эфир этиленгликоля (EGDE) и водорастворимый карбодиимид, предпочтительно 1-этил-3-(3-диметиламинопропил)карбодиимид (EDC). Посредством химии сшивания комплексов устанавливают связь амино-остатков узнающего вещества и липосом.
В другом примере описаны гетеробифункциональные сшивающие агенты и способы применения этих сшивающих агентов (патент США 5889155, специально включенный здесь в качестве ссылки в его полном виде). Эти сшивающие реагенты объединяют нуклеофильный гидразидный остаток с электрофильным малеимидным остатком, что делает возможным связывание альдегидов со свободными тиолами, в одном примере. Этот сшивающий реагент может быть модифицирован для сшивания различных функциональных групп и, следовательно, является применимым для сшивания полипептидов и сахаров. Таблица 3 подробно описывает некоторые гетеробифункциональные сшивающие агенты, считающиеся применимыми в данном изобретении.
ГЕТЕРОБИФУНКЦИОНАЛЬНЫЕ СШИВАЮЩИЕ АГЕНТЫ
Расщепляемое сшивание
Конъюгация фермент-антитело Конъюгация гаптен-белок-носитель
Конъюгация фермент-антитело
В случаях, когда конкретный полипептид не содержит остатка, доступного для конкретного сшивающего реагента, в его природной последовательности, могут быть использованы консервативные генетические или синтетические замены аминокислот в такой первичной последовательности.
VII. Комбинированные терапии/раковые терапии
Для увеличения эффективности химерного полипептида данного изобретения или кодирующей его экспрессирующей конструкции может быть желательным комбинирование этих композиций с другими агентами, эффективными в лечении гиперпролиферативного заболевания, такими как противораковые агенты. Гиперпролиферативное заболевание включает в себя заболевания и состояния, которые связаны с любым видом аномального роста клеток или аномальной регуляции роста. В способах данного изобретения пациентом предпочтительно является человек. Различные гиперпролиферативные заболевания могут лечиться в соответствии со способами данного изобретения. Некоторыми из гиперпролиферативных заболеваний, обсуждаемых в качестве подходящих для лечения в данном изобретении, являются псориаз, ревматоидный артрит (RA), воспалительная болезнь кишечника (IBD), остеоартрит (ОА) и пре-неопластические повреждения во рту, предстательной железе, молочной железе, легком и т.д. Данное изобретение имеет важные ответвления, в частности, в отношении одного гиперпролиферативного заболевания: злокачественной опухоли.
Таким образом, в некоторых вариантах, это гиперпролиферативное заболевание определяется дополнительно как злокачественная опухоль. В других вариантах этой злокачественной опухолью является меланома, немелкоклеточная злокачественная опухоль легкого, мелкоклеточная злокачественная опухоль легкого, злокачественная опухоль легкого, гепатокарцинома, ретинобластома, астроцитома, глиобластома, злокачественная опухоль десен, злокачественная опухоль языка, лейкоз, нейробластома, злокачественная опухоль головы, злокачественная опухоль шеи, злокачественная опухоль молочной железы, злокачественная опухоль поджелудочной железы, злокачественная опухоль предстательной железы, злокачественная опухоль почки, злокачественная опухоль кости, тестикулярная злокачественная опухоль, злокачественная опухоль яичника, мезотелиома, злокачественная опухоль шейки матки, желудочно-кишечная злокачественная опухоль, лимфома, злокачественная опухоль мозга, толстой кишки, саркома или злокачественная опухоль мочевого пузыря. Злокачественная опухоль может включать в себя опухоль, состоящую из опухолевых клеток. В других вариантах гиперпролиферативным заболеванием является ревматоидный артрит, воспалительная болезнь кишечника, остеоартрит, лейомиомы, аденомы, липомы, гемангиомы, фибромы, окклюзия сосудов, рестеноз, атеросклероз, пре-неопластические повреждения (такие как аденоматозная гиперплазия и интраэпителиальная неоплазия предстательной железы), карцинома in situ, волосистая лейкоплакия полости рта или псориаз.
"Противораковый" агент способен оказывать отрицательное действие на злокачественную опухоль в субъекте, например, лизисом раковых клеток, индукцией апоптоза в раковых клетках, уменьшением размера опухоли, ингибированием роста опухоли, уменьшением кровоснабжения опухоли или раковых клеток, стимуляцией иммунной реакции против раковых клеток или опухоли, предупреждением или ингибированием прогрессирования злокачественной опухоли или увеличением продолжительности жизни субъекта со злокачественной опухолью. Более в общем виде, эти другие композиции должны обеспечиваться в комбинированном количестве, эффективном для лизиса или ингибирования пролиферации данной клетки. Этот способ может включать в себя контактирование этих клеток с экспрессирующей конструкцией и этим агентом (агентами) или множественными факторами одновременно. Это может достигаться контактированием этой клетки с единственной композицией или фармакологической готовой формой, которая включает в себя оба агента, или контактированием этой клетки с двумя отдельными композициями или готовыми формами одновременно, где одна композиция включает в себя экспрессирующую конструкцию, а другая включает в себя второй агент (агенты).
Устойчивость опухолевых клеток к агентам химиотерапии и радиотерапии представляет собой главную проблему в клинической онкологии. Одной из задач существующего исследования злокачественной опухоли является нахождение способов улучшения эффективности химиотерапии и радиотерапии комбинированием их с генотерапией. Например, ген тимидинкиназы вируса простого герпеса (HS-tK) при доставке к опухолям мозга ретровирусной векторной системой успешно индуцировал чувствительность к антивирусному агенту ганцикловиру (Culver et al., 1992). В контексте данного изобретения обсуждается, что химерные полипептиды могут быть использованы подобным образом вместе с химиотерапевтическим, радиотерапевтическим, генотерапевтическим или иммунотерапевтическим вмешательством, наряду с другими проапоптозными или регулирующими клеточный цикл агентами.
Альтернативно, эта терапия может предшествовать лечению другим агентом или следовать за лечением другим агентом с интервалами в диапазоне минут - недель. В вариантах, где другой агент и экспрессирующую конструкцию применяют к данной клетке по отдельности, следовало бы обычно гарантировать, что не будет значительного интервала между временем каждой доставки, так что этот агент и экспрессирующая конструкция все еще смогут проявить выгодным образом комбинированное действие на эту клетку. В таких случаях предполагается, что можно контактировать эту клетку с обеими формами в пределах приблизительно 12-24 часов между ними, более предпочтительно в пределах приблизительно 6-12 часов между ними. В некоторых ситуациях может быть желательным значительное удлинение этого временного периода для лечения, однако, между соответствующими введениями должен быть интервал времени от нескольких дней (2, 3, 4, 5, 6 или 7) до нескольких недель (1, 2, 3, 4, 5, 6, 7 или 8).
Могут использоваться различные приведенные ниже комбинации, в которых генотерапию обозначают как "А", а вторичный агент, такой как радио- или химиотерапия, обозначают как "В":
А/В/А В/А/В В/В/А А/А/В А/В/В В/А/А А/В/В/В В/А/В/В
B/B/B/A B/B/A/B A/A/B/B A/B/A/B A/B/B/A B/B/A/A
B/A/B/A B/A/A/B A/A/A/B B/A/A/A A/B/A/A A/A/B/A
Введение терапевтических экспрессирующих конструкций данного изобретения пациенту должно следовать обычным протоколам для введения химиотерапевтических средств с учетом токсичности вектора, если она имеется. Ожидается, что циклы лечения будут повторяться при необходимости. Ожидается также, что различные стандартные терапии, а также хирургическое вмешательство могут быть применены в комбинации с описанной терапией гиперпролиферативных клеток.
А. Химиотерапия
Терапии злокачественной опухоли включают в себя также различные комбинированные терапии с лечением как на основе химических агентов, так и на основе облучения. Комбинированные химиотерапии включают в себя, например, цисплатин (CDDP), карбоплатин, прокарбазин, меклоретамин, циклофосфамид, камптотецин, ифосфамид, мелфалан, хлорамбуцил, бусульфан, нитромочевина, дактиномицин, даунорубицин, доксорубицин, блеомицин, пликомицин, митомицин, этопозид (VP16), тамоксифен, ралоксифен, агенты связывания рецептора эстрогена, таксол, гемцитабиен, навелбин, ингибиторы фарнезилпротеинтрансферазы, трансплатину, 5-фторурацил, винкристин, винбластин и метотрексат или любой аналог или вариант предыдущих агентов.
В. Радиотерапия
Другие факторы, которые вызывают повреждение ДНК и интенсивно использовались, включают в себя то, что обычно называют γ-лучами, рентгеновскими лучами и/или нацеленной доставкой радиоактивных изотопов в опухолевые клетки. Обсуждаются также другие формы факторов повреждения ДНК, такие как микроволны и УФ-излучение. Наиболее вероятно, что все эти факторы выполняют широкий диапазон повреждений на ДНК, на предшественниках ДНК, на репликации и репарации ДНК и на сборке и поддержании хромосом. Диапазоны доз для рентгеновского облучения находятся в диапазоне суточных доз от 50 до 200 рентген в течение пролонгированных периодов времени (3-4 недели) до единственных доз 2000-6000 рентген. Диапазоны доз для радиоактивных изотопов широко варьируются и зависят от периода полураспада изотопа, силы и типа испускаемой радиации и поглощения неопластическими клетками.
Термин "контактируют" или "экспонируют" при применении к клетке используется здесь для описания способа, при помощи которого терапевтическую конструкцию и химиотерапевтический или радиотерапевтический агент доставляют к клетке-мишени или помещают в непосредственное соседство с клеткой-мишенью. Для достижения лизиса клетки или стаза оба агента доставляют к клетке в комбинированном количестве, эффективном для лизиса клетки или предотвращения ее деления.
С. Иммунотерапия
Иммунотерапевтические средства обычно основаны на применении иммунных эффекторных клеток и молекул для нацеливания на раковые клетки и разрушения раковых клеток. Иммунным эффектором может быть, например, антитело, специфическое в отношении некоторого маркера на поверхности опухолевой клетки. Антитело одно может служить в качестве эффектора терапии, или оно может рекрутировать другие клетки для фактического осуществления лизиса клеток. Антитело может быть также конъюгировано с лекарственным средством или токсином (химиотерапевтическим агентом, радионуклидом, цепью А рицина, холерным токсином, коклюшным токсином и т.д.) и служить лишь в качестве нацеливающего агента. Альтернативно, этот эффектор может быть лимфоцитом, несущим поверхностную молекулу, которая взаимодействует, прямо или опосредованно, с опухолевой клеткой-мишенью. Различные эффекторные клетки включают в себя цитотоксические Т-клетки и клетки-киллеры (NK-клетки).
Таким образом, иммунотерапия может быть использована как часть комбинированной терапии вместе с генотерапией. Ниже обсуждается общий подход для комбинированной терапии. Обычно опухолевая клетка должна нести какой-либо маркер, который доступен для нацеливания, т.е. не присутствует на большинстве других клеток. Существуют многочисленные опухолевые маркеры, и любые из них могут быть пригодны для нацеливания в контексте данного изобретения. Обычные опухолевые маркеры включают в себя карциноэмбриональный антиген, специфический для предстательной железы антиген, связанный с опухолью мочевых путей антиген, фетальный антиген, тирозиназу (р97), gp68, TAG-72, HMFG, Sialyl Lewis Antigen (сиалилированную форму CD15, лиганда для Е-, Р и L-селектинов), MucA, MucB, PLAP, рецептор эстрогена, рецептор ламинина erbB и р155.
D. Гены
В другом варианте вторичным лечением является генотерапия, в которой терапевтический полинуклеотид вводят до, после химерного полипептида данного изобретения или одновременно с химерным полипептидом данного изобретения. Доставка химерного полипептида вместе со вторым вектором, кодирующим один из следующих генных продуктов, будет оказывать комбинированное антигиперпролиферативное действие на ткани-мишени. Альтернативно, может быть использован единственный вектор, кодирующий оба гена. Многочисленные белки охватываются данным изобретением, некоторые из которых описаны ниже.
1. Индукторы клеточной пролиферации
Белки, которые индуцируют клеточную пролиферацию, дополнительно попадают в различные категории в зависимости от функции. Общим свойством всех этих белков является их способность регулировать клеточную пролиферацию. Например, форма PDGF, онкоген sis, является секретируемым фактором роста. Онкогены редко возникают из генов, кодирующих факторы роста, и в настоящее время sis является только единственным известным природно встречающимся онкогенным фактором роста. В одном варианте данного изобретения обсуждается, что антисмысловая мРНК, нацеленная на конкретный индуктор клеточной пролиферации, используется для предотвращения экспрессии этого индуктора клеточной пролиферации.
Белки FMS, ErbA, ErbB и neu являются рецепторами факторов роста. Мутации относительно этих рецепторов приводят к потере регулируемой функции. Например, точковая мутация, затрагивающая трансмембранный домен белка рецептора Neu, приводит к онкогену neu. Онкоген erbA происходит из внутриклеточного рецептора для тиреоидного гормона. Считают, что модифицированный онкогенный рецептор ErbA конкурирует с эндогенным рецептором тиреоидного гормона, вызывая неконтролируемый рост.
Самый большой класс онкогенов включает в себя белки передачи (трансдукции) сигналов (например, Src, Ab1 и Ras). Белок Src является цитоплазматической протеин-тирозинкиназой, и его превращение из протоонкогена в онкоген в некоторых случаях происходит через мутации в остатке тирозина 527. В противоположность этому, превращение ГТФазного белка ras из протоонкогена в онкоген, в одном примере, происходит из мутации валина в глицин при аминокислоте 12 в этой последовательности, с уменьшением ГТФазной активности ras.
Белки Jun, Fos и Myc являются белками, которые непосредственно проявляют их действие на ядерные функции в качестве факторов транскрипции.
2. Ингибиторы клеточной пролиферации
Опухолевые супрессорные онкогены функционируют, ингибируя избыточную клеточную пролиферацию. Инактивация этих генов разрушает их ингибиторную активность, приводя к нерегулируемой пролиферации. Опухолевые супрессоры р53, р16 и С-САМ описаны ниже.
Высокие уровни мутантного р53 были обнаружены во многих клетках, трансформированных химическим канцерогенезом, ультрафиолетовой радиацией и некоторыми вирусами. Ген р53 является частой мишенью мутационной инактивации в большом разнообразии опухолей человека, и уже документировано, что он является наиболее часто мутируемым геном в обычных злокачественных опухолях человека. Он мутируется в более, чем 50% NSCLC человека (Hollstein et al., 1991) и в широком спектре других опухолей.
Ген р53 кодирует фосфобелок из 393 аминокислот, который может образовывать комплексы с белками хозяина, такими как большой Т-антиген и Е1В. Этот белок найден в нормальных тканях и клетках, но в концентрациях, которые являются незначительными в сравнении с трансформированными клетками или опухолевой тканью.
р53 дикого типа признается как важный регулятор роста во многих типах клеток. Миссенс-мутации (мутации с изменением смысла) являются обычными для гена р53 и являются существенными для трансформирующей способности этого онкогена. Единственное генетическое изменение, вызванное точковыми мутациями, может создать канцерогенный р53. Однако, в отличие от других онкогенов известно, что точковые мутации р53 происходят в по меньшей мере 30 различных кодонах, часто создавая доминантные аллели, которые производят сдвиги в клеточном фенотипе без уменьшения до гомозиготности. Кроме того, многие из этих доминантных негативных аллелей, по-видимому, являются переносимыми в организме и переносятся в зародышевую линию. Различные мутантные аллели, по-видимому, находятся в диапазоне от минимально дисфункциональных до сильно пенетрантных, доминантных негативных аллелей (Weinberg, 1991).
Другим ингибитором клеточной пролиферации является р16. Основные переходы цикла эукариотических клеток запускаются циклин-зависимыми киназами, или CDK. Одна CDK, циклин-зависимая киназа 4 (CDK4), регулирует прогрессирование клеточного цикла через G1. Активность этого фермента может заключаться в фосфорилировании Rb в поздней G1. Активность CDK4 регулируется активирующей субъединицей, циклином D-типа, и ингибирующей субъединицей, р16INK4, которая была биохимически охарактеризована как белок, который специфически связывается с CDK4 и ингибирует CDK4 и, следовательно, может регулировать фосфорилирование Rb (Serrano et al., 1993; Serrano et al., 1995). Поскольку белок р16INK4 является ингибитором CDK4 (Serrano, 1993), делеция этого гена может увеличивать активность CDK4, приводя к гиперфосфорилированию белка Rb. Известно также, что р16 регулирует функцию CDK6.
р16INK4 принадлежит к недавно описанному классу CDK-ингибиторных белков, которые также включают в себя р16В, р19, р21WAF1 и р27KIP1. Ген р16INK4 картирован в 9р21, области хромосом, часто делетируемом во многих типах опухолей. Гомозиготные делеции и мутации гена р16INK4 являются частыми в линиях опухолевых клеток человека. Этот факт предполагает, что ген р16INK4 является геном супрессии опухолей. Однако эта интерпретация может быть оспорена наблюдением, что частота изменений гена р16INK4 является гораздо более низкой в первичных некультивируемых опухолях, чем в культивируемых клеточных линиях (Caidas et al., 1994; Cheng et al., 1994; Hussussian et al., 1994; Kamb et al., 1994; Kamb et al., 1994; Mori et al., 1994; Okamoto et al., 1994; Nobori et al., 1995; Orlow et al., 1994; Arap et al., 1995). Восстановление функции р16INK4 дикого типа трансфекцией плазмидным экспрессирующим вектором уменьшала образование колоний некоторыми линиями раковых клеток человека (Okamoto, 1994; Arap, 1995).
Другие гены, которые могут быть использованы в соответствии с данным изобретением, включают в себя Rb, APC, DCC, NF-1, NF-2, WT-1, MEN-I, MEN-II, zac1, p73, VHL, MMAC1/PTEN, DBCCR-1, FCC, rsk-3, p27, слияния p27/p16, слияния р21/р27, антитромботические гены (например, СОХ-1, TFPI), PGS, Dp, E2F, ras, myc, neu, raf, erb, fms, trk, ret, gsp, hst, abl, E1A, p300, участвующие в ангиогенезе (например, VEGF, FGF, тромбоспондин, BAI-1, GDAIF или их рецепторы) и MCC.
3. Регуляторы программированной смерти клеток
Апоптоз, или программированная гибель клеток, является основным процессом для нормального эмбрионального развития, поддержания гомеостаза в тканях взрослых и супрессии канцерогенеза (Kerr et al., 1972). Было показано, что семейство Bcl-2 белков и ICE-подобные протеазы являются важными регуляторами и эффекторами апоптоза в других системах. Белок Bcl-2, обнаруженный в связи с фолликулярной лимфомой, играет выдающуюся роль в регуляции апоптоза и усилении выживания клеток в ответ на разнообразные апоптозные стимулы (Bakhshi et al., 1985; Cleary and Sclar, 1985; Cleary et al., 1986; Tsujimoto et al., 1985; Tsujimoto and Crice, 1986). В настоящее время признается, что эволюционно консервативный белок Bcl-2 является членом семейства родственных белков, которые могут находиться в категории агонистов или антагонистов смерти клеток.
После его открытия было показано, что Bcl-2 действует, подавляя гибель клеток, запускаемую разнообразными стимулами. В настоящее время является очевидным, что существует семейство регуляторных белков Bcl-2 смерти клеток, которые имеют общие структурные гомологии и гомологии последовательностей. Было показано, что эти различные члены семейства обладают функциями, сходными с функциями Bcl-2 (например, BclXL, BclW, BclS, Mcl-1, A1, Bfl-1), или противодействуют функции Bcl-2 и способствуют смерти клеток (например, Вах, Bak, Bik, Bim, Bid, Bad, Harakiri).
Е. Хирургия
Приблизительно 60% лиц со злокачественной опухолью будут подвергаться хирургии того или иного типа, которая включает в себя превентивную, диагностическую хирургию, или хирургию для определения стадии заболевания, лечебную и паллиативную (временно облегчающую) хирургию. Лечебная хирургия является лечением злокачественной опухоли, которое может быть использовано вместе с другими терапиями, такими как лечение данного изобретения, химиотерапия, радиотерапия, гормональная терапия, генотерапия, иммунотерапия и/или альтернативные терапии.
Лечебная терапия включает в себя резекцию, в которой всю раковую ткань или часть раковой ткани физически удаляют, вырезают и/или разрушают. Резекцией опухоли называют физическое удаление по меньшей мере части опухоли. Кроме резекции опухоли, лечение посредством хирургии включает в себя лазерную хирургию, криохирургию, электрохирургию и контролируемую микроскопически хирургию (хирургию Моса). Ожидается также, что данное изобретение может быть использовано вместе с удалением неглубоких (поверхностных) раков, предраков или случайных количеств нормальной ткани.
После вырезания части или всех раковых клеток, ткани или опухоли в теле может образоваться полость. Лечение может выполняться перфузией, прямой инъекцией в эту зону или локальным нанесением на эту зону с использованием дополнительной противораковой терапии. Такое лечение может повторяться, например, каждые 1, 2, 3, 4, 5, 6 или 7 дней или каждые 1, 2, 3, 4 и 5 недель или каждые 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11 или 12 месяцев. Эти лечения могут также выполняться с варьирующими дозами.
F. Другие агенты
Предполагается, что другие агенты могут быть использованы в комбинации с данным изобретением для улучшения терапевтической активности лечения. Эти дополнительные агенты включают в себя иммуномодуляторные агенты, агенты, которые осуществляют повышающую регуляцию поверхностных рецепторов клетки и нексусов (щелевидных контактов клеток), цитостатических агентов и агентов дифференцировки, ингибиторов адгезии клеток или агентов, которые увеличивают чувствительность гиперлиферативных клеток к индукторам апоптоза. Иммуномодуляторные агенты включают в себя фактор некроза опухолей; интерферон альфа, бета и гамма; IL-2 и другие цитокины; F42K и другие аналоги цитокинов; или MIP-1, MIP-1бета, МСР-1, RANTES и другие хемокины. Далее, ожидается, что повышающая регуляция рецепторов клеточной поверхности или их лигандов, таких как Fas/Fas-лиганд, DR4 или DR5/TRAIL будет потенциировать апоптозные индуцирующие способности данного изобретения установлением аутокринного или паракринного действия на гиперпролиферативные клетки. Увеличения межклеточной передачи сигналов повышением числа нексусов (щелевидных контактов между клетками) будет увеличивать анти-гиперпролиферативные действия на соседнюю популяцию гиперпролиферативных клеток. В других вариантах могут быть использованы цитостатические агенты или агенты дифференцировки в комбинации с данным изобретением для улучшения анти-гиперпролиферативной эффективности этих обработок. Предполагается, что ингибиторы клеточной адгезии улучшают эффективность данного изобретения. Примерами ингибиторов клеточной адгезии являются ингибиторы фокальной (очаговой) адгезионной киназы (FAK) и ловастатин. Далее, предполагается, что другие агенты, которые увеличивают чувствительность гиперпролиферативной клетки к апоптозу, такие как антитело с225, могли бы использоваться в комбинации с данным изобретением для улучшения эффективности лечения.
Гормональная терапия также может быть использована вместе с данным изобретением или вместе с любой другой раковой терапией, описанной ранее. Применение гормонов может использоваться в лечении определенных раков, таких как злокачественная опухоль молочной железы, предстательной железы, яичников или шейки матки, для понижения уровня или для блокирования действий некоторых гормонов, таких как тестостерон или эстроген. Это лечение часто используют в комбинации по меньшей мере с одной другой раковой терапией в качестве возможной терапии или для уменьшения риска образования метастазов.
VIII. Фармацевтические препараты
Фармацевтические композиции данного изобретения содержат эффективное количество одного или нескольких химерных полипептидов, или химерных полипептидов и по меньшей мере одного дополнительного агента, растворенных или диспергированных в фармацевтически приемлемом носителе. Фразы "фармацевтически или фармакологически приемлемые" относятся к молекулярным частицам и композициям, которые не производят вредной, аллергической или другой неблагоприятной реакции при введении животному, такому, как, например, человек, если необходимо. Приготовление фармацевтической композиции, которая содержит по меньшей мере один химерный полипептид или дополнительный активный ингредиент, будет известно специалистам с квалификацией в данной области в свете данного описания, и примеры можно найти в Remington's Pharmaceutical Sciences, 18th Ed. Mack Printing Company, 1990, включенном здесь в качестве ссылки. Кроме того, будет понятно, что для введения животному (например, человеку) эти препараты должны удовлетворять стандартам стерильности, пирогенности, общей безопасности и чистоты, требуемым FDA Office of Biological Standards.
В применении здесь, термин "фармацевтически приемлемый носитель" включает в себя любые и все растворители, диспергирующие среды, покрытия, поверхностно-активные вещества, антиоксиданты, консерванты (например, антибактериальные агенты, противогрибковые агенты), изотонические агенты, задерживающие абсорбцию агенты, соли, консерванты, лекарственные средства, стабилизаторы лекарственных средств, гели, связывающие вещества, наполнители, дезинтегрирующие агенты, смазывающие агенты, подслащивающие агенты, улучшающие вкус и запах агенты, красители, подобные им материалы и их комбинации, как это будет известно специалисту с обычной квалификацией в данной области (см., например, Remington's Pharmaceutical Sciences, 18th Ed. Mack Printing Company, 1990, pp. 1289-1329, включенном здесь в качестве ссылки). За исключением случаев, когда любой общепринятый носитель является несовместимым с активным ингредиентом, предполагается его применение в этих терапевтических или фармацевтических композициях.
Химерный полипептид может содержать различные типы носителей в зависимости от того, вводят ли его в твердой, жидкой или аэрозольной форме, и от того, должен ли он быть стерильным для таких способов введения, как инъекция. Композиция данного изобретения может вводиться внутривенно, интрадермально, внутриартериально, внутрибрюшинно, внутрь повреждения, интракраниально, внутрисуставно, внутрь предстательной железы, интраплеврально, интратрахеально, интраназально, интравитреально (внутрь стекловидного тела), интравагинально, интраректально, локально, внутрь опухоли, внутримышечно, внутрибрюшинно, подкожно, интраконъюнктивально, внутрипузырно, через слизистую оболочку, интраперикардиально, интраумбиликально, внутриглазно, перорально, локально, ингаляцией (например, аэрозольной ингаляцией), инъекцией, инфузией, непрерывной инфузией, локализованной перфузией, омывающей клетки-мишеней непосредственно, через катетер, через лаваж, в виде кремов, в виде липидных композиций (например, липосом) или другим способом или любой комбинацией перечисленного, как будет известно специалисту с обычной квалификацией в данной области (см., например, Remington's Pharmaceutical Sciences, 18th Ed. Mack Printing Company, 1990, включенный здесь в качестве ссылки).
Фактическое дозовое количество композиции данного изобретения, вводимое пациенту-животному, может определяться физическими и физиологическими факторами, такими как вес тела, тяжесть состояния, тип заболевания, подлежащего лечению, прежние или сопутствующие терапевтические вмешательства, идиопатия пациента и способ введения. Практикующий врач, ответственный за введение, будет, в любом случае, определять концентрацию активного ингредиента (активных ингредиентов) в композиции и подходящую дозу (дозы) для индивидуального субъекта.
В некоторых вариантах фармацевтические композиции могут содержать, например, по меньшей мере приблизительно 0,1% активного соединения. В других вариантах, активное соединение может включать в себя, например, приблизительно 2% - приблизительно 75% веса этой единицы или приблизительно 25% - приблизительно 60%, и любой диапазон, произведенный в пределах этих границ. В других неограничивающих примерах доза может также содержать от приблизительно 1 микрограмм/кг/вес тела, приблизительно 5 микрограмм/кг/вес тела, приблизительно 10 микрограмм/кг/вес тела, приблизительно 50 микрограмм/кг/вес тела, приблизительно 100 микрограмм/кг/вес тела, приблизительно 200 микрограмм/кг/вес тела, приблизительно 350 микрограмм/кг/вес тела, приблизительно 500 микрограмм/кг/вес тела, приблизительно 1 миллиграмм/кг/вес тела, приблизительно 5 миллиграмм/кг/вес тела, приблизительно 10 миллиграмм/кг/вес тела, приблизительно 50 миллиграмм/кг/вес тела, приблизительно 100 миллиграмм/кг/вес тела, приблизительно 200 миллиграмм/кг/вес тела, приблизительно 350 миллиграмм/кг/вес тела, приблизительно 500 миллиграмм/кг/вес тела до приблизительно 1000 мг/кг/вес тела или более на одно введение, и любой диапазон, произведенный в пределах этих границ. В не ограничивающих изобретение примерах диапазон, произведенный из перечисленных здесь цифр, приблизительно 5 мг/кг/вес тела - приблизительно 100 мг/кг/вес тела, приблизительно 5 микрограмм/кг/вес тела - приблизительно 500 миллиграмм/кг/вес тела, и т.д., может быть введен, на основе описанных выше цифр.
В любом случае эта композиция может содержать различные антиоксиданты для замедления окисления одного или нескольких компонентов. Кроме того, предотвращение действия микроорганизмов может осуществляться консервантами, такими как различные антибактериальные и противогрибковые агенты, в том числе, но не только, парабенами (например, метилпарабенами, пропилпарабенами) хлорбутанолом, фенолом, сорбиновой кислотой, тимерозалом или их комбинациями.
Химерный полипептид может быть приготовлен в виде композиции в форме свободного основания, нейтральной или солевой форме. Фармацевтически приемлемые соли включают в себя кислотно-аддитивные соли, например, соли, образованные со свободными аминогруппами белковой композиции, или соли, которые образованы с неорганическими кислотами, такими как, например, хлористоводородная или фосфорная кислоты, или такими органическими кислотами, как уксусная, щавелевая, винная или миндальная кислота. Соли, образованные со свободными карбоксильными группами, могут быть также произведены из неорганических оснований, таких как, например, гидроксиды натрия, калия, аммония, кальция или железа (III); или таких органических оснований, как изопропиламин, триметиламин, гистидин или прокаин.
В вариантах, в которых эта композиция находится в жидкой форме, носителем может быть растворитель или дисперсионная среда, содержащая, но не ограничиваемая ими, воду, этанол, полиол (например, глицерин, пропиленгликоль, жидкий полиэтиленгликоль и т.д.), липиды (например, триглицериды, растительные масла, липосомы) и их комбинации. Подходящая текучесть может поддерживаться, например, применением покрытия, такого как лецитин; поддержанием требуемого размера частиц диспергированием в носителях, таких как, например, жидкие полиолы или липиды; применением поверхностно-активных веществ, таких как, например, гидроксипропилцеллюлоза; или комбинациями таких способов. Во многих случаях будет предпочтительно включение изотонических агентов, таких как, например, сахара, хлорид натрия или их комбинации.
В других вариантах в данном изобретении можно применять глазные капли, растворы или спреи для носа, аэрозоли или ингалируемые средства. Такие композиции обычно готовят таким образом, что они являются совместимыми с типом ткани-мишени. В не ограничивающем изобретение примере растворы для носа обычно являются водными растворами, предназначенными для введения в проходы носа, в виде капель или спреев. Растворы для носа готовят таким образом, что они во многих отношениях похожи на носовые секреции, так что сохраняется нормальное цилиарное действие. Таким образом, в предпочтительных вариантах водные растворы для носа обычно являются изотоничными или слегка забуференными для поддержания рН около 5,5 - 6,5. Кроме того, в эту композицию могут быть включены антимикробные консерванты, сходные с консервантами, применяемыми в глазных препаратах, лекарственные средства или подходящие стабилизаторы лекарственных средств, если требуется. Например, известны различные коммерческие препараты для носа, и они включают в себя лекарственные средства, такие как антибиотики и антигистамины.
В некоторых вариантах химерный полипептид готовят для введения такими способами, как введение через полость рта. В этих вариантах твердая композиция может включать в себя, например, растворы, суспензии, эмульсии, таблетки, пилюли, капсулы (например, желатиновые капсулы с твердой или мягкой оболочкой), композиции пролонгированного высвобождения, буккальные композиции, пастилки, эликсиры, суспензии, сиропы, вафли или их комбинации. Пероральные композиции могут быть непосредственно включены в состав пищевой диеты. Предпочтительные носители для перорального введения содержат инертные разбавители, ассимилируемые годные в пищу носители или их комбинации. В других аспектах данного изобретения пероральная композиция может быть приготовлена в виде сиропа или эликсира. Сироп или эликсир может содержать, например, по меньшей мере один активный агент, подслащивающий агент, консервант, улучшающий вкус и запах агент, краситель, консервант или их комбинации.
В некоторых предпочтительных вариантах пероральная композиция может содержать один или несколько связывающих агентов, наполнителей, дезинтегрирующих агентов, смазывающих веществ, улучшающих вкус и запах агентов и их комбинации. В некоторых вариантах композиция может содержать один или более из следующих компонентов: связывающий агент, например, трагакантовую камедь, аравийскую камедь, кукурузный крахмал, желатин или их комбинации; наполнитель, такой как, например, дикальцийфосфат, маннит, лактоза, крахмал, стеарат магния, натрий-сахарин, целлюлоза, карбонат магния или их комбинации; дезинтегрирующий агент, такой как, например, кукурузный крахмал, картофельный крахмал, альгиновая кислота или их комбинации; смазывающий агент, такой как, например, стеарат магния; подслащивающий агент, такой как, например, мята перечная, винтергреновое масло, вишневый ароматизатор, апельсиновый ароматизатор и т.д.; или комбинации перечисленных компонентов. Когда дозированная стандартная форма является капсулой, она может содержать, кроме веществ вышеописанного типа, носители, такие как жидкий носитель. Различные другие вещества могут присутствовать в виде покрытий или другим образом модифицировать физическую форму дозированной единицы. Например, таблетки, пилюли или капсулы могут быть покрыты шеллаком, сахаром или и тем, и другим.
Дополнительные композиции, которые пригодны для других способов введения, включают в себя суппозитории. Суппозитории являются твердыми дозированными формами различного веса и различной формы, обычно содержащие лекарственные средства, для введения в прямую кишку, влагалище или мочеиспускательный канал. После введения суппозитории размягчаются, расплавляются или растворяются в жидкостях полости. Обычно для суппозиториев общепринятые носители могут включать в себя, например, полиалкиленгликоли, триглицериды или их комбинации. В некоторых вариантах суппозитории могут быть образованы из смесей, содержащих, например, активный ингредиент в диапазоне приблизительно 0,5% - приблизительно 10% и предпочтительно приблизительно 1% - приблизительно 2%.
Стерильные инъекционные растворы готовят включением активных соединений в требуемом количестве подходящего растворителя с различными другими ингредиентами, перечисленными выше, если требуется, с последующим фильтрованием через микропористый фильтр. Обычно дисперсии готовят включением различных стерилизованных активных ингредиентов в стерильный носитель, который содержит основную дисперсионную среду и/или другие ингредиенты. В случае стерильных порошков для приготовления стерильных инъекционных растворов, суспензий или эмульсии, предпочтительными способами приготовления являются способы сушки в вакууме или лиофилизации, которые дают порошок активного ингредиента плюс любого дополнительного желательного ингредиента из их предварительно стерилизованной фильтрованием жидкой среды. Эта жидкая среда должна быть подходящим образом забуферена, если необходимо, и жидкий растворитель делают сначала изотоничным с использованием достаточного количества солевого раствора или глюкозы перед инъекцией. Предполагается также приготовление высоко концентрированных композиций для прямой инъекции, где предусматривается применение ДМСО в качестве растворителя для достижения чрезвычайно быстрого проникновения с доставкой высоких концентраций активных агентов к малой зоне.
Данная композиция должна быть стабильной в условиях приготовления и хранения и предохраненной против загрязняющего действия микроорганизмов, таких как бактерии и грибы. Должно быть понятно, что загрязнение эндотоксинами должно поддерживаться минимальным при уровне безопасности, например, меньшем, чем 0,5 нг/мг белка.
В конкретных вариантах пролонгированная абсорбция инъецируемой композиции может осуществляться применением в этих композициях агентов, задерживающих абсорбцию, таких как, например, моностеарат алюминия, желатин или их комбинации.
IX. Липидные композиции
В определенных вариантах данное изобретение использует новую композицию, содержащую один или более липидов, связанных по меньшей мере с одним химерным полипептидом. Липид является веществом, которое характерным образом нерастворимо в воде и может экстрагироваться органическим растворителем. Липиды включают в себя, например, вещества, содержащие жировые капельки, которые природно встречаются в цитоплазме, а также класс соединений, которые хорошо известны специалистам с квалификацией в данной области, которые содержат длинноцепочечные алифатические углеводороды и их производные, такие как жирные кислоты, спирты, амины, аминоспирты и альдегиды. Конечно, соединения, другие, чем специально описанные здесь, которые известны специалисту с квалификацией в области липидов, также охватываются композициями и способами данного изобретения.
Липид может быть природно встречающимся или синтетическим (т.е. сконструированным и полученным человеком). Однако, липид обычно является биологическим веществом. Биологические липиды хорошо известны в данной области и включают в себя, например, нейтральные жиры, фосфолипиды, фосфоглицериды, стероиды, терпены, лизолипиды, гликосфинголипиды, гликолипиды, сульфатиды, липиды со связанными простой эфирной связью и сложноэфирной связью жирными кислотами и полимеризованные липиды и их комбинации.
А. Типы липидов
Нейтральный жир может содержать глицерин и жирную кислоту. Типичный глицерин является трехуглеродным спиртом. Жирная кислота обычно является молекулой, содержащей углеродную цепь с кислотной частью молекулы (например, карбоновой кислотой) на конце этой цепи. Углеродная цепь жирной кислоты может быть любой длины, однако, предпочтительной является длина углеродной цепи от приблизительно 2, приблизительно 3, приблизительно 4, приблизительно 5, приблизительно 6, приблизительно 7, приблизительно 8, приблизительно 9, приблизительно 10, приблизительно 11, приблизительно 12, приблизительно 13, приблизительно 14, приблизительно 15, приблизительно 16, приблизительно 17, приблизительно 18, приблизительно 19, приблизительно 20, приблизительно 21, приблизительно 22, приблизительно 23, приблизительно 24, приблизительно 25, приблизительно 26, приблизительно 27, приблизительно 28, приблизительно 29 до приблизительно 30 или более атомов углерода и любой диапазон, произведенный в пределах этих границ. Однако, предпочтительный диапазон является диапазоном от приблизительно 14 до приблизительно 24 атомов углерода в этой являющейся цепью части жирной кислоты, причем в определенных вариантах диапазон приблизительно 16 - приблизительно 18 атомов углерода является особенно предпочтительным. В некоторых вариантах углеродная цепь жирной кислоты может содержать нечетное число атомов углерода, однако, в некоторых вариантах может быть предпочтительным четное число атомов углерода в этой цепи. Жирную кислоту, содержащую только простые связи в ее углеродной цепи, называют насыщенной, тогда как жирную кислоту, содержащую по меньшей мере одну двойную связь, называют ненасыщенной.
Конкретные жирные кислоты включают в себя, но не ограничиваются ими, линолевую кислоту, олеиновую кислоту, пальмитиновую кислоту, линоленовую кислоту, стеариновую кислоту, лауриновую кислоту, миристиновую кислоту, арахиновую кислоту, пальмитолеиновую кислоту, арахидоновую кислоту, рицинолеиновую кислоту, туберкулостеариновую кислоту, лактобацилловую кислоту. Кислотная группа одной или более жирных кислот ковалентно связана с одной или более гидроксильными группами глицерина. Таким образом, моноглицерид содержит глицерин и одну жирную кислоту, диглицерид содержит глицерин и две жирные кислоты и триглицерид содержит глицерин и три жирные кислоты.
Фосфолипид обычно содержит либо глицериновую, либо сфингозиновую часть, ионную фосфатную группу для получения амфипатического соединения и одну или более жирных кислот. Типы фосфолипидов включают в себя, например, фосфоглицериды, где фосфатная группа связана с первым углеродом глицерина диглицерида, и сфингофосфолипиды (например, сфингомиелин), где фосфатная группа этерифицирована до аминоспирта сфингозина. Другим примером сфингофосфолипида является сульфатид, который содержит ионную сульфатную группу, которая делает эту молекулу амфипатической. Фосфолипид может, конечно, содержать дополнительные химические группы, такие как, например, спирт, присоединенный к фосфатной группе. Примеры таких спиртовых групп включают в себя серин, этаноламин, холин, глицерин и инозит. Таким образом, фосфоглицериды включают в себя фосфатидилсерин, фосфатидилэтаноламин, фосфатидилхолин, фосфатидилглицерин или фосфатидилинозит. Другие фосфолипиды включают в себя фосфатидную кислоту или диацетилфосфат. В одном аспекте фосфатидилхолин содержит диолеоилфосфатидилхолин (также известный как кардиолипин), яичный фосфатидилхолин, дипальмитоилфосфалидихолин, мономиристоилфосфатидилхолин, монопальмитоилфосфатидилхолин, моностеароилфосфатидилхолин, моноолеоилфосфатидилхолин, дибутроилфосфатидилхолин, дивалероилфосфатидилхолин, дикапроилфосфатидилхолин, дигептаноилфосфатидилхолин, дикаприлоилфосфатидилхолин или дистеароилфосфатидилхолин.
Гликолипид является сфингофосфолипидом, но содержит углеводную группу, а не фосфатную группу, присоединенную к первичной гидроксильной группе сфингозина. Тип гликолипида, называемый цереброзидом, содержит одну сахарную группу (например, глюкозу или галактозу), присоединенную к первичной гидроксильной группе. Другим примером гликолипида является ганглиозид (например, моносиалоганглиозид, GM1), которые содержит приблизительно 2, приблизительно 3, приблизительно 4, приблизительно 5, приблизительно 6 - приблизительно 7 или около этого сахарных групп, которые могут быть в разветвленной цепи, присоединенной к первичной гидроксильной группе. В других вариантах гликолипид является церамидом (например, лактозилцерамидом).
Стероид является производным четырехчленной циклической системы фенантрена. Стероиды часто обладают регуляторными функциями в клетках, тканях и организмах и включают в себя, например, гормоны и родственные соединения в семействах прогестагенов (например, прогестерон), глюкокортикоидов (например, кортизол), минералокортикоидов (например, альдостерон), андрогенов (например, тестостерон) и эстрогенов (например, эстрон). Холестерин является другим примером стероида и обычно выполняет структурные, а не регуляторные функции. Витамин D является другим примером стерина и участвует в абсорбции кальция из кишечника.Терпен является липидом, содержащим одну или несколько пятиуглеродных изопреновых групп. Терпены имеют различные биологические функции и включают в себя, например, витамин А, кофермент Q и каротиноиды (например, ликопин и β-каротин).
В. Заряженные и нейтральные композиции липидов
В некоторых вариантах липидный компонент композиции является незаряженным или первично незаряженным. В одном варианте липидный компонент содержит один или несколько нейтральных липидов. В другом аспекте липидный компонент композиции может быть по существу свободным от анионных и катионных липидов, таким как некоторые фосфолипиды (например, фосфатидилхолин) и холестерин. В некоторых аспектах липидный компонент незаряженной или первично незаряженной липидной композиции содержит приблизительно 95%, приблизительно 96%, приблизительно 97%, приблизительно 98%, приблизительно 99% или 100% липидов без заряда, по существу незаряженных липидов и/или смесь липидов с равными количествами положительных и отрицательных зарядов.
В других аспектах липидная композиция может быть заряженной. Например, заряженные фосфолипиды могут быть использованы для приготовления липидной композиции в соответствии с данным изобретением и могут нести суммарный положительный заряд или суммарный отрицательный заряд. В не ограничивающем изобретение примере может быть использован диацетилфосфат для придания отрицательного заряда липидной композиции, а стеариламин может быть использован для придания положительного заряда липидной композиции.
С. Получение липидов
Липиды могут быть получены из природных источников, коммерческих источников или химически синтезированы, как это известно специалисту с обычной квалификацией в данной области. Например, фосфолипиды могут быть из природных источников, такие как яичный или соевый фосфатидилхолин, фосфатидная кислота головного мозга, фосфатидилинозит головного мозга или растений, кардиолипин сердца и растительный или бактериальный фосфатидилэтаноламин. В другом примере липиды, пригодные для применения в соответствии с данным изобретением, могут быть получены из коммерческих источников. Например, димиристилфосфатидилхолин ("DMPC") может быть получен из Sigma Chemicals Co., дицетилфосфат ("DCP") получают из К & К Laboratories (Plainview, NY); холестерин ("Chol") получают из Calbiochem-Behring; димиристилфосфатидилглицерин ("DMPG") и другие липиды могут быть получены из Avanti Polar Lipids, Inc. (Birmingham, Ala). В некоторых вариантах исходные растворы липидов в хлороформе или смеси хлороформ/метанол могут храниться при приблизительно -20°С. Предпочтительно, хлороформ используют в качестве единственного растворителя, так как он более легко выпаривается, чем метанол.
D. Структуры липидных композиций
В предпочтительном варианте данного изобретения химерный полипептид связан с липидом. Химерный полипептид, связанный с липидом, может быть диспергирован в растворе, содержащем липид, может быть растворен в липиде, эмульсифицирован в липиде, смешан с липидом, соединен с липидом, ковалентно связан с липидом, содержаться в виде суспензии в липиде, содержащемся или находящемся в комплексе с мицеллой или липосомой, или быть другим образом связан с липидом или липидной структурой. Связанная с липидом или комплексом липид/химерный полипептид композиция данного изобретения не ограничивается какой-либо конкретной структурой. Например, они могут быть просто рассеяны в растворе, возможно, с образованием агрегатов, которые не являются однородными ни по размеру, ни по форме. В другом примере они могут присутствовать в бислойной структуре, в виде мицелл, или иметь "сдувшуюся" (в состоянии "коллапса") структуру. В другом не ограничивающем изобретение примере рассматривается также комплекс липофектамин (Gibco BRL)-химерный полипептид или Superfect (Qiagen)-химерный полипептид.
В некоторых вариантах липидная композиция может содержать приблизительно 1%, приблизительно 2%, приблизительно 3%, приблизительно 4%, приблизительно 5%, приблизительно 6%, приблизительно 7%, приблизительно 8%, приблизительно 9%, приблизительно 10%, приблизительно 11%, приблизительно 12%, приблизительно 13%, приблизительно 14%, приблизительно 15%, приблизительно 16%, приблизительно 17%, приблизительно 18%, приблизительно 19%, приблизительно 20%, приблизительно 21%, приблизительно 22%, приблизительно 23%, приблизительно 24%, приблизительно 25%, приблизительно 26%, приблизительно 27%, приблизительно 28%, приблизительно 29%, приблизительно 30%, приблизительно 31%, приблизительно 32%, приблизительно 33%, приблизительно 34%, приблизительно 35%, приблизительно 36%, приблизительно 37%, приблизительно 38%, приблизительно 39%, приблизительно 40%, приблизительно 41%, приблизительно 42%, приблизительно 43%, приблизительно 44%, приблизительно 45%, приблизительно 46%, приблизительно 47%, приблизительно 48%, приблизительно 49%, приблизительно 50%, приблизительно 51%, приблизительно 52%, приблизительно 53%, приблизительно 54%, приблизительно 55%, приблизительно 56%, приблизительно 57%, приблизительно 58%, приблизительно 59%, приблизительно 60%, приблизительно 61%, приблизительно 62%, приблизительно 63%, приблизительно 64%, приблизительно 65%, приблизительно 66%, приблизительно 67%, приблизительно 68%, приблизительно 69%, приблизительно 70%, приблизительно 71%, приблизительно 72%, приблизительно 73%, приблизительно 74%, приблизительно 75%, приблизительно 76%, приблизительно 77%, приблизительно 78%, приблизительно 79%, приблизительно 80%, приблизительно 81%, приблизительно 82%, приблизительно 83%, приблизительно 84%, приблизительно 85%, приблизительно 86%, приблизительно 87%, приблизительно 88%, приблизительно 89%, приблизительно 90%, приблизительно 91%, приблизительно 92%, приблизительно 93%, приблизительно 94%, приблизительно 95%, приблизительно 96%, приблизительно 97%, приблизительно 98%, приблизительно 99%, приблизительно 100% или любой диапазон, произведенный в пределах указанных границ, конкретного липида, типа липида или нелипидного компонента, такого как лекарственное средство, белок, сахар, нуклеиновые кислоты или другой материал, описанный здесь или известный специалисту с квалификацией в данной области. В не ограничивающем изобретение примере липидная композиция может содержать приблизительно 10% - приблизительно 20% нейтральных липидов и приблизительно 33% - приблизительно 34% цереброзида и приблизительно 1% холестерина. В другом не ограничивающем изобретение примере липосома может содержать приблизительно 4% - приблизительно 12% терпенов, где приблизительно 1% этой мицеллы является ликопином, а приблизительно 3% - приблизительно 11% этой липосомы содержат другие терпены; и приблизительно 10% - приблизительно 35% фосфатидилхолина и приблизительно 1% лекарственного средства. Таким образом, предполагается, что липидные композиции данного изобретения могут содержать любой из липидов, типов липидов или других компонентов в любой комбинации или в любом процентном диапазоне.
1. Эмульсии
Липид может содержаться в эмульсии. Липидная суспензия является по существу постоянной гетерогенной жидкой смесью двух или более жидкостей, которые в норме не растворяются одна в другой при механическом перемешивании или при применении небольших количеств дополнительных веществ, известных как эмульгаторы. Способы приготовления липидных эмульсий и добавления дополнительных веществ хорошо известны в данной области (например, Modern Pharmaceutics, 1990, включенный здесь в качестве ссылки).
Например, один или более липидов добавляют к этанолу или хлороформу или любому другому подходящему органическому растворителю и перемешивают вручную или механическими способами. Затем растворитель выпаривают из этой смеси, оставляя высушенную глазурь липида. Эти липиды ресуспендируют в водной среде, такой как забуференный фосфатом солевой раствор, с получением эмульсии. Для получения более гомогенного распределения размеров эмульгированных липидов эту смесь обрабатывают ультразвуком с использованием общепринятых способов обработки ультразвуком, дополнительно эмульгируют с использованием микрофлюидизации (микропсевдоожижения) (с применением, например, микрофлюидизатора Newton, Mass.) и/или экструдируют при пониженном давлении (таком как, например, 600 фунт/кв. дюйм) с использованием экструдирующего устройства (Lipex Biomembranes, Vancouver, Canada).
2. Мицеллы
Липид может содержаться в мицелле. Мицелла является кластером или агрегатом липидных соединений, обычно в форме липидного монослоя и может быть получена с использованием любого протокола для получения мицелл, известного специалистам с квалификацией в данной области (например, Canfield et al., 1990; El-Gorab et al., 1973; Colloidal Surfactant, 1963 и Catalysis in Micellar and Macromolecular Systems, 1975, включенные здесь в качестве ссылки). Например, один или более липидов обычно готовят в виде суспензии в органическом растворителе, растворитель выпаривают, липид ресуспендируют в водной среде, обрабатывают ультразвуком и затем центрифугируют.
3. Липосомы
В конкретных вариантах липид содержит липосому. Термин "липосома" является общим термином, включающим в себя одно- или мультиламеллярные липидные пузырьки, образованные генерированием замкнутых липидных бислоев или агрегатов. Липосомы могут быть охарактеризованы как имеющие пузырьковые структуры с бислойной мембраной, обычно содержащей фосфолипид, и внутренней средой, которая обычно содержит водную композицию.
Мультиламеллярная липосома имеет множественные липидные слои, разделенные водной средой. Они образуются самопроизвольно, когда липиды, содержащие фосфолипиды, суспендируют в избытке водного раствора. Липидные компоненты подвергаются самоперегруппировке перед образованием замкнутых структур и захватывают воду и растворенные в ней вещества между липидными бислоями (Ghosh and Bachhawat, 1991). Липофильные молекулы или молекулы с липофильными областямии могут также растворяться в липидном бислое или связываться с ним.
В определенных менее предпочтительных вариантах фосфолипиды из природных источников, такие как яичный или соевый фосфатидилхолин, фосфатидная кислота головного мозга, фосфатидилинозит головного мозга или растений, кардиолипин сердца и растительный или бактериальный фосфатидилэтаноламин, предпочтительно не используют в качестве первичного фосфатида, т.е. составляющего 50% или более от общего состава фосфатидов или липосомы, вследствие нестабильности липосом и просачивания из полученных липосом.
В конкретных вариантах липид и/или химерный полипептид могут быть, например, инкапсулированы в водном внутреннем пространстве липосомы, рассеяны в липидном бислое липосомы, присоединены к липосоме через связывающую молекулу, которая связана как с липосомой, так и с химерным полипептидом, заключены в липосому, связаны в виде комплекса с липосомой и т.д.
а. Приготовление липосом
Липосома, используемая в соответствии с данным изобретением, может быть приготовлена различными способами, как будет известно специалисту с обычной квалификацией в данной области. Фосфолипиды могут образовывать разнообразные структуры, другие, чем липосомы, при диспергировании в воде в зависимости от молярного отношения липида к воде. При низких отношениях липосома является предпочтительной структурой.
Например, фосфолипид (Avanti Polar Lipids, Alabaster, AL), такой как, например, нейтральный фосфолипид диолеоилфосфатидилхолин (DOPC), растворяют в трет-бутаноле. Затем этот липид (липиды) смешивают с химерным полипептидом и/или другим компонентом (компонентами). К липидной смеси добавляют Твин 20, так что Твин 20 составляет приблизительно 5% веса композиции. К этой смеси добавляют избыток трет-бутанола, так что объем трет-бутанола равен по меньшей мере 95%. Смесь встряхивают на вортексе (встряхивателе), замораживают в бане сухой лед/ацетон и лиофилизируют в течение ночи. Лиофилизированный препарат хранят при -20°С, и он может быть использован в течение периода до трех месяцев. При необходимости лиофилизированные липосомы воссоздают (реконституируют) в 0,9% солевом растворе. Средний диаметр частиц, получаемых с использованием Твина 20 для инкапсулирования химерного полипептида, равен приблизительно 0,7 - приблизительно 1,0 мкм.
Альтернативно, липосомы могут быть получены смешиванием липидов в растворителе в контейнере, например, стеклянной грушеобразной колбе. Контейнер должен иметь объем, в десять раз больший, чем объем ожидаемой суспензии липосом. С использованием роторного испарителя растворитель удаляют при приблизительно 40°С при отрицательном давлении. Растворитель обычно удаляют в пределах приблизительно 5 минут - 2 часов, в зависимости от желаемого объема липосом. Эта композиция может быть высушена дополнительно в эксикаторе под вакуумом. Высушенные липиды обычно выбрасывают спустя приблизительно 1 неделю вследствие тенденции к ухудшению свойств со временем.
Высушенные липиды могут быть гидратированы при приблизительно 25-30 мМ фосфолипиде в стерильной, апирогенной воде встряхиванием до тех пор, пока вся липидная пленка не будет ресуспендирована. Затем водные липосомы могут быть разделены на аликвоты, каждую из которых помещают во флакон, лиофилизируют и герметизируют под вакуумом.
В других альтернативных способах липосомы могут быть приготовлены в соответствии с другими известными лабораторными процедурами (например, статьи см. Bangham et al., 1965; Gregoriadis, 1979; Deamer and Uster, 1983, Szoka and Papahadjopoulos, 1978, каждая из которых включена здесь в качестве ссылки в релевантной части). Эти способы отличаются по их соответствующим способностям захватывать водный материал и их соответствующим отношениям водного пространства к липиду.
Высушенные липиды или лиофилизированные липосомы, приготовленные, как описано выше, могут быть дегидратированы и воссозданы в растворе ингибиторного пептида и разведены до подходящей концентрации подходящим растворителем, например, DPBS. Затем эту смесь интенсивно встряхивают в вортексе (встряхивателе). Неинкапсулированные дополнительные материалы, такие как агенты, включающие в себя, но не ограничивающиеся ими, гормоны, лекарственные средства, конструкции нуклеиновых кислот и т.п. удаляют центрифугированием при 29000 х g и осадки липосом промывают. Промытые липосомы ресуспендируют при подходящей концентрации общего фосфолипида, например, около 50-200 мМ. Количество инкапсулированного дополнительного материала или активного агента может быть определено в соответствии со стандартными способами. После определения количества инкапсулированного дополнительного материала или активного агента в препарате липосом, липосомы могут быть разбавлены до подходящих концентраций и могут храниться при 4°С до использования. Фармацевтическая композиция, содержащая эти липосомы, будет обычно включать в себя стерильный, фармацевтически приемлемый носитель или разбавитель, такой как вода или солевой раствор.
Размер липосомы варьируется в зависимости от способа синтеза. Липосомы в данном изобретении могут быть различного размера. В некоторых вариантах липосомы являются небольшими, например, меньшими, чем приблизительно 100 нм, приблизительно 90 нм, приблизительно 80 нм, приблизительно 70 нм, приблизительно 60 нм или менее приблизительно 50 нм в наружном диаметре. В получении таких липосом может быть использован любой протокол, описанный здесь или известный специалисту с обычной квалификацией в данной области. Дополнительные не ограничивающие изобретение примеры получения липосом описаны в патентах США с номерами 4728578, 4728575, 4737323, 4533254, 4162282, 4310505 и 4921706; Международных заявках РСТ/US85/01161 и РСТ/US89/05040, заявке на патент Великобритании GB 2193095 А; Mayer et al., 1986; Hope et al., 1985; Mayhew et al., 1987; Mayhew et al., 1984; Cheng et al., 1987 и Liposome Technology, 1984, которые включены здесь в качестве ссылки.
Липосома, суспендированная в водном растворе, обычно находится в форме сферического пузырька, имеющего один или несколько концентрических слоев молекул липидного бислоя. Каждый слой состоит из параллельного ряда молекул, представленного формулой XY, где Х обозначает гидрофильную часть молекулы, а Y обозначает гидрофобную часть молекулы. В водной суспензии эти концентрические слои размещены таким образом, что гидрофильные части молекул стремятся оставаться в контакте с водной фазой, а гидрофобные области стремятся самоассоциироваться. Например, когда водные фазы присутствуют как в липосоме, так и без липосомы, эти липидные молекулы могут образовать бислой, известный как ламелла с аранжировкой XY-YX. Агрегаты липидов могут образовываться, когда гидрофильные и гидрофобные части более, чем одной молекулы, становятся связанными одна с другой. Размер и форма этих агрегатов будут зависеть от многочисленных различных факторов, таких как характер растворителя и присутствие других соединений в растворе.
Получение липидных композиций часто выполняют обработкой ультразвуком или последовательной экструзией липосомных смесей после I) выпаривания обращенной фазы, (II) дегидратации-регидратации, (III) детергентного диализа и (IV) гидратации тонкой пленки. В одном аспекте предполагаемым способом для получения липосом в некоторых вариантах является нагревание, обработка ультразвуком и последовательная экструзия липидов через фильтры или мембраны с уменьшающимся диаметром пор, что приводит к образованию малых, стабильных липосомных структур. Это получение дает липосомный/химерный полипептид или липосомы только подходящего и однородного размера, которые являются структурно стабильными и производят максимальную активность. Такие способы хорошо известны специалистам с квалификацией в данной области (см., например, Lang et al., 1990).
После приготовления липидные структуры могут быть использованы для инкапсулирования соединений, которые являются токсичными (например, химиотерапевтических средств) или лабильными (например, нуклеиновых кислот) в кровотоке. Физические характеристики липосом зависят от рН, ионной силы и/или присутствия двухвалентных катионов. Липосомы могут обнаруживать низкую проницаемость для ионных и/или полярных веществ, но при повышенных температурах подвергаются фазовому переходу, который заметно изменяет их проницаемость. Фазовый переход включает в себя изменение от плотноупакованной, упорядоченной структуры, известной как состояние геля, к рыхло упакованной, менее упорядоченной структуре, известной как текучее состояние. Это происходит при характерной для фазового перехода температуре и/или приводит к увеличению проницаемости для ионов, сахаров и/или лекарственных средств. Липосомное инкасулирование приводило к меньшей токсичности и более продолжительному полупериоду существования в сыворотке для таких соединений (Gabizon et al., 1990).
Липосомы взаимодействуют с клетками для доставки агентов посредством четырех различных механизмов: эндоцитоз фагоцитарными клеткми ретикулоэндотелиальной системы, такой как макрофаги и/или нейтрофилы; адсорбцию на клеточной поверхности, либо неспецифическими слабыми гидрофобными, и/либо электростатическими силами, и/либо специфическими взаимодействиями с компонентами клеточной поверхности; слияние с мембраной плазматических клеток встраиванием липидного бислоя липосомы в плазматическую мембрану, с одновременным высвобождением содержимого липосом в цитоплазму; и/или перенос липосомных липидов к клеточным и/или субклеточным мембранам, и/или наоборот, без какой-либо связи с содержимым липосом. Варьирование композиции липосом может определять, какой механизм является функциональным, хотя в одно и то же время может действовать более, чем один механизм.
Многочисленные способы лечения заболеваний используют перенос генов на основе липидов для усиления общепринятых или установления новых терапий, в частности, терапий для лечения гиперпролиферативных заболеваний. Прогресс в приготовлении липосомных композиций улучшил эффективность переноса генов in vivo (Templeton et al., 1997), и предполагается, что липосомы получают этими способами. Описаны альтернативные способы получения композиций на основе липидов (WO 99/18933).
В другой липосомной композиции амфипатический носитель, названный микроносителем разведения растворителя (SDMC), позволяет интегрировать конкретные молекулы в бислой липидного носителя (патент США 5879703). SDMC могут быть использованы для доставки липополисахаридов, полипептидов, нуклеиновых кислот и т.п. Конечно, любые другие способы получения липосом могут быть использованы квалифицированным специалистом для получения желаемой композиции липосом в данном изобретении.
b. Нацеливающие лиганды
Нацеливающий лиганд может быть либо заякорен в гидрофобной части комплекса, либо прикреплен к реакционноспособным концевым группам гидрофильной части этого комплекса. Нацеливающий лиганд может быть присоединен к липосоме через связывание с реакционноспособной группой, например, на дистальном конце этого гидрофильного полимера. Предпочтительные реакционноспособные группы включают в себя аминогруппы, карбоксильные группы, гидразидные группы и тиоловые группы. Связывание нацеливающего лиганда с гидрофильным полимером может быть выполнено стандартными способами органической химии, которые известны специалистам с квалификацией в данной области. В определенных вариантах общая концентрация нацеливающего лиганда может быть от приблизительно 0,01 до приблизительно 10 молярных %.
Нацеливающими лигандами являются любые лиганды, специфические в отношении характерного компонента области, на который нацеливает лиганд. Предпочтительные нацеливающие лиганды включают в себя белки, такие как поликлональные и моноклональные антитела, фрагменты антител или химерные антитела, ферменты или гормоны, или сахара, такие как моно-, олиго- и полисахариды (см. Heath et al., Chem. Phys. Lipids 40:347 (1986)). Например, дисиалоганглиозид GD2 является опухолевым антигеном, который был идентифицирован в опухолях нейроэктодермального происхождения, таких как нейробластома, меланома, мелкоклеточная карцинома легкого, глиома и некоторые саркомы (Mujoo et al., 1986; Schulz et al., 1984). Липосомы, содержащие моноклональные антитела против дисиалоганглиозида GD2, использовали для усиления нацеливания этих липосом на клетки, экспрессирующие этот опухолевый антиген (Montaldo et al., 1999; Pagan et al., 1999). В другом не ограничивающем изобретение примере антитела, специфические в отношении антигена злокачественной опухоли молочной железы и гинекологической злокачественной опухоли, описаны в патенте США № 5939277, включенном здесь в качестве ссылки. В дополнительном не ограничивающем изобретение примере антитела, специфические в отношении злокачественной опухоли предстательной железы, описаны в патенте США № 6107090, включенном здесь в качестве ссылки. Таким образом, ожидается, что описанные здесь антитела или известные специалисту с обычной квалификацией в данной области, могут быть использованы для нацеливания на специфические ткани и типы клеток в комбинации с композициями и способами данного изобретения. В некоторых вариантах данного изобретения обсуждаемые нацеливающие лиганды взаимодействуют с интегрином, протеогликанами, гликопротеинами, рецепторами или транспортерами. Подходящие лиганды включают в себя любые лиганды, которые являются специфическими в отношении клеток органа-мишени или в отношении структур органа-мишени, открытого для кровотока (циркуляции) в результате локальной патологии, такой как опухоли.
В некоторых вариантах данного изобретения для усиления трансдукции клеток, для увеличения трансдукции клеток-мишеней или для ограничения трансдукции нежелательных клеток антитело или нацеливающие на циклический пептид части молекулы (лиганды) связывают с липидным комплексом. Такие способы известны в данной области. Например, были описаны дополнительно липосомы, которые специфически нацеливают на клетки центральной нервной системы млекопитающих (патент США 5786214, включенный здесь в качестве ссылки). Эти липосомы состоят в основном из N-глутарилфосфатидилэтаноламина, холестерина и олеиновой кислоты, где моноклональное антитело, специфическое для нейроглии, конъюгировано с этими липосомами. Ожидается, что моноклональное антитело или фрагмент антитела могут быть использованы для нацеливания доставки на специфические клетки, ткани или органы в животном, такие как, например, головной мозг, сердце, легкое, печень и т.д.
Далее, химерный полипептид может быть доставлен к клетке-мишени через опосредованную рецептором доставку и/или нацеливающие пузырьки, содержащие липид или липосому. Они используют преимущество селективного поглощения макромолекул опосредованным рецептором эндоцитозом, который будет происходить в клетке-мишени. В связи со специфическим для типов клеток распределением различных рецепторов этот способ доставки добавляет другую степень специфичности данному изобретению.
Таким образом, в некоторых аспектах данного изобретения лиганд будет выбран для соответствия рецептору, специфически экспрессируемому на популяции клеток-мишеней. Клеткоспецифическая доставка химерного полипептида и/или нацеливающего носителя может предусматривать специфическое связывание лиганда в комбинации с липосомой. Доставляемый химерный полипептид содержится внутри липосомы, а лиганд специфического связывания функционально включен в мембрану липосомы. Таким образом, липосома будет специфически связываться с рецептором (рецепторами) клетки-мишени и доставлять содержимое к клетке. Было показано, что такие системы являются функциональными, с применением систем, в которых, например, используют эпидермальный фактор роста (EGF) в опосредованной рецептором доставке нуклеиновой кислоты к клеткам, которые проявляют повышающую регуляцию рецептора EGF.
В некоторых вариантах опосредованная рецептором доставка и/или нацеливающие носители содержат агент, специфический в отношении рецептора клетки и связывающий химерный полипептид. Другие содержат специфический в отношении рецептора клетки лиганд, с которым функционально связан доставляемый химерный полипептид. Например, для опосредованного рецептором переноса генов использовали несколько лигандов (Wu and Wu, 1987; Wagner et al., 1990; Perales et al., 1994; Myers, EPO 0273085), что подтверждает функциональность этого способа. В другом примере была описана специфическая доставка с использованием другого типа клеток млекопитающего (работа Wu and Wu, 1993; включенная здесь в качестве ссылки).
В следующих вариантах лиганд специфического связывания может содержать один или более липидов или гликопротеинов, которые направляют клеткоспецифическое связывание. Например, лактозилцерамид, галактоза-концевой асиалоганглиозид, включали в липосомы и наблюдали увеличение поглощения гена инсулина гепатоцитами (Nicolau et al., 1987). Было также показано, что асиалогликопротеин, асиалофетуин, который содержит концевые галактозильные остатки, нацеливает липосомы на печень (Spanjer and Scherphof, 1983; Hara et al., 1996). Сахара маннозил-, фукозил- или N-ацетилглюкозамин при связывании со скелетом полипептида связываются с высокой аффинностью с рецептором маннозы (патент США 5432260, специально включенный здесь в качестве ссылки в его полном виде). Ожидается, что клетко- или тканеспецифические трансформирующие конструкции данного изобретения могут специфически доставляться в клетку- или ткань-мишень подобным образом.
В другом примере лактозилцерамид и пептиды, которые нацеливают родственные LDL-рецептору белки, такие как аполипопротеин Е3 ("Apo E"), были применимы в нацеливании липосом на печень (Spanjer and Scherphof, 1983; WO 98/0748).
Фолат и рецептор фолата были описаны как применимые для клеточного нацеливания (патент США 5871727). В этом примере витамин фолат связан с этим комплексом. Рецептор фолата имеет высокую аффинность в отношении этого лиганда и сверхэкспрессируется на поверхности нескольких линий злокачественных клеток, в том числе опухолей легкого, молочной железы и головного мозга. Анти-фолат, такой как метотрексат, может быть также использован в качестве нацеливающего лиганда. Опосредованные трансферрином системы доставки нацеливают на большой диапазон реплицирующихся клеток, которые экспрессируют рецептор трансферрина (Gilliland et al., 1980).
с. Комбинации липосома/нуклеиновая кислота
В некоторых вариантах система липосома/химерный полипептид может содержать нуклеиновую кислоту, такую как, например, олигонуклеотид, полинуклеотид или конструкция нуклеиновой кислоты (например, экспрессирующий вектор). При применении бактериального промотора в конструкции ДНК, которая подлежит трансфекции в эукариотические клетки, будет также желательным включение в липосому подходящей бактериальной полимеразы.
Предполагается, что, когда композиция липосома/химерный полипептид содержит клетко- или тканеспецифическую нуклеиновую кислоту, этот способ может быть применим в данном изобретении. В некоторых вариантах невирусные композиции на основе липидов обеспечивают альтернативу вирусной генотерапии. Хотя многие исследования на культуре клеток обосновали невирусный перенос генов на основе липидов, системная доставка генов с использованием композиций на основе липидов была ограниченной. Основным недостатком невирусной доставки генов на основе липидов является токсичность катионных липидов, которые содержат невирусный носитель доставки. Эта токсичность липосом in vivo частично объясняет противоречия между результатами переноса генов in vitro и in vivo. Другим фактором, вносящим вклад в эти противоречивые данные, является различие в стабильности липосом в присутствии и в отстутствие сывороточных белков. Взаимодействие липосом и сывороточных белков оказывает разительное действие на характеристики стабильности липосом (Yang and Huang, 1997). Катионные липосомы аттрагируют и связывают отрицательно заряженные сывороточные белки. Липосомы, покрытые сывороточными белками, либо растворяются, либо поглощаются макрофагами, что приводит к их удалению из кровотока. Существующие способы липосомной доставки in vivo используют аэрозоли, подкожную, интрадермальную, внутриопухолевую или внутричерепную инъекцию во избежание проблем токсичности и стабильности, связанных с катионными липидами, находящимися в кровотоке. Взаимодействие липосом и белков плазмы является в значительной степени ответственным за несоответствие между эффективностью переноса генов in vitro (Felgner et al.,) и in vivo (Zhu et al., 1993; Philip et al., 1993; Solodin et al., 1995; Liu et al., 1995; Thierry et al., 1995; Tsukamoto et al., 1995; Aksentijevich et al., 1996).
Был описан пример способа нацеливания вирусных частиц на клетки, которые лишены единственного клеткоспецифического маркера (патент США 5849718). В этом способе, например, антитело А может иметь специфичность в отношении опухоли, но также в отношении нормальной ткани сердца и легкого, тогда как антитело В имеет специфичность в отношении опухоли, но также в отношении нормальных клеток печени. Применение антитела А или антитела В одного для доставки антипролиферативной нуклеиновой кислоты к опухоли могло бы приводить к нежелательному повреждению клеток сердца и легкого или клеток печени. Однако, антитело А и антитело В могут использоваться вместе для улучшения клеточного нацеливания. Таким образом, антитело А связывают с геном, кодирующим антипролиферативную нуклеиновую кислоту, и доставляют, через опосредованную рецептором систему поглощения, к опухоли, а также к ткани сердца и легкого. Однако, этот ген не транскрибируется в этих клетках, так как они лишены необходимого фактора транскрипции. Антитело В связывают с универсально активным геном, кодирующим фактор транскрипции, необходимый для транскрипции антипролиферативной нуклеиновой кислоты, и доставляют к опухоли и к клеткам печени. Таким образом, в клетках сердца и легкого доставляется только неактивная антипролиферативная нуклеиновая кислота, где она не транскрибируется, что приводит к отсутствию неблагоприятных эффектов. В клетках печени ген, кодирующий фактор транскрипции, доставляется и транскрибируется, но не имеет эффекта, так как не присутствует антипролиферативная нуклеиновая кислота. Однако, в опухолевых клетках оба гена доставляются и фактор транскрипции может активировать транскрипцию антипролиферативной нуклеиновой кислоты, что приводит к опухолеспецифическим токсическим эффектам.
Добавление нацеливающих лигандов для доставки генов для лечения гиперпролиферативных заболеваний обеспечивает возможность доставки генов, генные продукты которых являются более токсичными, чем в случае ненацеленных систем. Примеры более токсичных генов, которые могут быть доставлены, включают в себя проапоптозные гены, такие как Вах и Bak плюс и гены, произведенные из вирусов и других патогенов, такие как аденовирусный Е4orf4 и ген пуриннуклеозидфосфорилазы E. coli, так называемый «суицидный ген», который превращает пролекарство 6-метилпуриндезоксирибозид в токсичный пурин-6-метилпурин. Другие примеры суицидных генов, используемых в терапии с использованием пролекарств, являются ген цитозиндеаминазы E. coli и ген тимидинкиназы HSV.
Ненацеленные или нацеленные липидные комплексы могут быть также использованы для генерирования рекомбинантных или модифицированных вирусов in vivo. Например, две или более плазмиды могут быть использованы для введения ретровирусных последовательностей плюс терапевтического гена в гиперпролиферативную клетку. Ретровирусные белки, обеспеченные in trans из одной из этих плазмид, обеспечат возможность упаковки другой, несущей терапевтический ген плазмиды. Таким образом, трансдуцированные клетки станут местом для продуцирования нерепликативных ретровирусов, несущих терапевтический ген. Эти ретровирусы могут затем инфицировать соседние клетки. Промотор для терапевтического гена может быть или может не быть индуцируемым или тканеспецифическим.
Подобным образом, перенесенная нуклеиновая кислота может представлять ДНК для репликации компетентного или реплицирующегося при определенных условяих вирусного генома, такого как аденовирусный геном, который лишен всего или части аденовирусной области Е1а или Е2b или который имеет один или более тканеспецифических или индуцируемых промоторов, запускающих транскрипцию от областей Е1а или Е1b. Эта реплицирующаяся или условно реплицирующаяся нуклеиновая кислота может содержать или может не содержать дополнительный терапевтический ген, такой как опухолевый супрессорный ген или анти-онкоген.
d. Липидное введение
Фактическое дозированное количество липидной композиции (например, липосома-химерный полипептид), вводимое пациенту, может определяться физическими и физиологическими факторами, такими как вес тела, тяжесть состояния, идиопатия пациента и способ введения. С учетом этого может быть легко определена доза липидной композиции для конкретного субъекта и/или схема лечения.
Данное изобретение может вводиться внутривенно, интрадермально, внутриартериально, внутрибрюшинно, внутрь повреждения, интракраниально, внутрисуставно, внутрь предстательной железы, интраплеврально, внутритрахеально, интраназально, интравитреально (внутрь стекловидного тела), интравагинально, ректально, внутрь опухоли, внутримышечно, интраперитонеально, подкожно, внутрипузырно, через слизистую оболочку, интраперикардиально, перорально, местно, локально и/или с использованием аэрозоля, инъекции, инфузии, непрерывной инфузии, локализованной перфузии, омывающей клетки-мишени, непосредственно или через катетер и/или лаваж.
Х. Получение антител
А. Поликлональные антитела
Поликлональные антитела применимы в данном изобретении в связи со многими вариантами для химерных полипептидов. Поликлональные антитела к химерным полипептидам обычно индуцируют в животных множественными подкожными (sc) инъекциями химерного полипептида и адъюванта. Может быть полезным конъюгирование химерных полипептидов или фрагмента, содержащего аминокислотную последовательность-мишень, с белком, который является иммуногенным в видах, которые должны быть иммунизированы, например, гемоцианином фиссуреллы, сывороточным альбумином, бычьим тироглобулином или ингибитором трипсина сои, с использованием бифункционального или дериватизующего агента, например, эфира малеимидобензоилсульфосукцинимида (конъюгирование через остатки цистеина), N-гидроксисукцинимида (через остатки лизина), глутарового альдегида, янтарного ангидрида, SOCl2 или R1-N=C=NR, где R и R1 являются различными алкильными группами.
Животных иммунизируют против иммуногенных конъюгатов или производных объединением 1 мг или 1 мкг конъюгата (для кроликов или мышей, соответственно) с 3 объемами полного адъюванта Фрейнда и инъекцией этого раствора интрадермально во множественных местах. Спустя один месяц выполняют вторичную инъекцию (бустинг) 1/5-1/10 исходного количества конъюгата в полном адъюванте Фрейнда подкожной инъекцией во множественных местах. Спустя 7-14 дней у животных берут кровь и анализируют сыворотку на титр антител против химерного полипептида. Животных продолжают иммунизировать до появления плато титра. Предпочтительно, животных инъецируют конъюгатом тех же самых химерных полипептидов, но конъюгированных с отличающимся белком и/или посредством отличающегося сшивающего реагента. Конъюгаты могут быть также приготовлены в культуре рекомбинантных клеток в виде слитых белков. Для усиления иммунной реакции используют также агрегирующие агенты, такие как квасцы.
В. Моноклональные антитела
Моноклональные антитела получают из популяции по существу гомогенных антител, т.е. отдельные антитела, составляющие эту популяцию, являются идентичными, за исключением возможных природно встречающихся мутаций, которые могут присутствовать в минорных количествах. Таким образом, определение "моноклональные" указывает характер антитела как не являющегося смесью дискретных антител.
Например, моноклональные антитела против химерного полипептида данного изобретения могут быть получены с использованием гибридомного способа, впервые описанного Kohler and Milstein (1975), или могут быть получены способами рекомбинантных ДНК [Cabilly et al., патент США № 4816567].
В гибридомном способе мышь или другое подходящее животное-хозяин, например, хомячка, иммунизируют, как описано выше, для индукции лимфоцитов, которые продуцируют или способны продуцировать антитела, специфически связывающиеся с белком, используемым для иммунизации. Альтернативно, лимфоциты могут быть иммунизированы in vitro. Затем лимфоциты сливают с клетками миеломы с использованием подходящего агента слияния, такого как полиэтиленгликоль, с образованием гибридомной клетки [Goding, Monoclonal Antibodies: Principles and Practice, pp. 59-103 (Academic Press, 1986].
Полученные таким образом гибридомные клетки высевают и выращивают в подходящей культуральной среде, которая предпочтительно содержит одно или несколько веществ, которые ингибируют рост или выживание неслитых исходных миеломных клеток. Например, если исходные миеломные клетки лишены фермента гипоксантингуанин-фосфорибозилтрансферазы (HGPRT или HPRT), культуральная среда для этих гибридом обычно будет включать в себя гипоксантин, аминоптерин и тимидин (среда НАТ), т.е. вещества, предотвращающие рост HGPRT-недостаточных клеток.
Предпочтительными миеломными клетками являются клетки, которые эффективно сливаются, поддерживают стабильную экспрессию высокого уровня антитела выбранными продуцирующими антитело клетками и являются чувствительными к среде, такой как среда НАТ. Среди них предпочтительными линиями миеломных клеток являются линии миеломы мышей, такие как линии, произведенные из мышиных опухолей МОРС-21 и МРС-11, которые доступны из Salk Institute Cell Distribution Center, Calif. USA, и клетки SP-2, доступные из Американской Коллекции Типовых Культур, Rockville, Md. USA.
Культуральную среду, в которой выращиваются гибридомные клетки, анализируют на продуцирование моноклональных антител, направленных против химерных полипептидов. Предпочтительно, специфичность связывания моноклональных антител, продуцируемых гибридомными клетками, определяют иммунопреципитацией или анализом связывания in vitro, таким как радиоиммуноанализ (РИА) или твердофазный иммуноферментный анализ (ELISA).
Аффинность связывания моноклонального антитела может быть, например, определена анализом Скетчарда Munson and Pollard (1980).
После идентификации гибридомных клеток, которые продуцируют антитела желаемой специфичности, аффинности и/или активности, эти клоны могут быть субклонированы процедурами лимитирующих разведений и выращены стандартными способами. Goding, Monoclonal Antibodies: Principles and Practice, pp. 59-104 (Academic Press, 1986). Подходящие культуральные среды для этой цели включают в себя, например, модифицированную Дульбекко среду Игла или среду RPMI-1640. Кроме того, гибридомные клетки могут выращиваться in vivo в виде асцитных опухолей в животном.
Моноклональные антитела, секретируемые этими субклонами, подходящим образом выделяют из культуральной среды, жидкостей асцитов или сыворотки общепринятыми процедурами очистки иммуноглобулинов, такими как, например, хроматография на протеин А-Сефарозе, на гидроксилапатите, гель-электрофорез, диализ или аффинная хроматография.
ДНК, кодирующую моноклональные антитела данного изобретения, легко выделяют и секвенируют с использованием общепринятых процедур (например, с использованием олигонуклеотидных зондов, которые способны связываться специфически с генами, кодирующими тяжелую и легкую цепи мышиных антител). Гибридомные клетки данного изобретения служат в качестве предпочтительного источника такой ДНК. После выделения, ДНК может быть помещена в экспрессирующие векторы, которые затем трансфицируют в клетки-хозяева, такие как клетки COS обезьян, клетки яичника китайского хомячка (СНО) или миеломные клетки, которые в противном случае не продуцируют белок иммуноглобулин, для получения синтеза моноклональных антител в рекомбинантных клетках-хозяевах. ДНК может быть также модифицирована, например, путем замены кодирующей последовательности константных доменов тяжелой и легкой цепей человека вместо гомологичных мышиных последовательностей (Morrison et al., 1984) или путем ковалентного присоединения к иммуноглобулин кодирующей последовательности всей кодирующей последовательности или ее части для полипептида, не являющегося иммуноглобулином. Таким образом получают «химерные» или «гибридные» антитела, которые имеют связывающую специфичность моноклонального антитела против химерного полипептида, описанного здесь.
Обычно такими не-иммуноглобулиновыми полипептидами заменяют константные домены антитела данного изобретения или вариабельные домены одного антигенсвязывающего сайта антитела данного изобретения для создания химерного бивалентного антитела, содержащего один антигенсвязывающий сайт, имеющий специфичность в отношении химерного полипептида, и другой антигенсвязывающий сайт, имеющий специфичность в отношении отличающегося антигена.
Химерные или гибридные антитела могут быть также получены in vitro с использованием известных способов в синтетической белковой химии, в том числе способов с использованием сшивающих агентов. Например, иммунотоксины могут быть сконструированы с использованием реакции обмена дисульфидной группы или образованием тиоэфирной связи. Примеры подходящих для этой цели реагентов включают в себя иминотиолат и метил-4-меркаптобутиримидат.
Для диагностических применений антитела данного изобретения обычно метят детектируемой частью молекулы. Этой детектируемой частью молекулы может быть любая часть молекулы, которая способна производить, непосредственно или опосредованно, детектируемый сингнал. Например, детектируемой часть может быть радиоактивный изотоп, такой как 3Н, 14С, 32Р, 35S или 125I, флуоресцентное или хемилюминесцентное соединение, такое как флуоресцеинизотиоцианат, родамин или люциферин; биотин; радиоактивные изотопные метки, такие как, например, 125I, 32Р, 14С или 3Н, или фермент, такой как щелочная фосфатаза, бета-галактозидаза или пероксидаза хрена.
Может быть использован любой способ, известный в данной области для раздельного конъюгирования антитела с детектируемой частью молекулы, в том числе способы, описанные Hunter et al., (1962); David et al., (1974); Pain et al., (1981) и Nygren (1982).
Антитела данного изобретения могут быть использованы в любом известном способе анализа, таком как анализы конкурентного связывания, прямой и опосредованный сэндвич-анализ и анализы иммунопреципитации. Zola, Monoclonal Antibodies: A Manual of Techniques, pp. 147-158 (CRC Press, Inc., 1987).
Анализы конкурентного связывания основаны на способности меченого стандарта (который может быть химерным полипептидом или его иммунологически реактивным белком) конкурировать с аналитом тест-пробы (химерными полипептидами) за связывание с ограниченным количеством антитела. Количество химерных полипептидов в тест-пробе обратно пропорционально количеству стандарта, которое связывается с этими антителами. Для облегчения определения количества стандарта, которое становится связанным, эти антитела обычно делают нерастворимыми до или после конкуренции, так что стандарт или аналит, которые связываются с этими антителами, могут быть удобным образом отделены от стандарта или аналита, которые остаются несвязанными. Сэндвич-анализ включает в себя применение двух антител, каждое из которых способно связываться с отличающейся иммуногенной частью, или эпитопом, детектируемого белка. В сэндвич-анализе аналит тест-пробы связывают с первым антителом, которое иммобилизовано на твердом носителе, и после этого второе антитело связывается с этим аналитом, образуя таким образом нерастворимый состоящий из трех частей комплекс (патент США № 4376110). Второе антитело может быть само меченым детектируемой частью молекулы (прямой сэндвич-анализ) или могут быть измерены с использованием антитела против иммуноглобулина, которое является меченым детектируемой частью молекулы (непрямой сэндвич-анализ). Например, одним типом сэндвич-анализа является анализ ELISA, причем в этом случае детектируемой частью молекулы является фермент.
С. Гуманизированные антитела
Способы гуманизации антител животного, т.е. не человека, хорошо известны в данной области. Обычно гуманизированное антитело имеет один или несколько аминокислотных остатков, введенных в него, из источника, который не является человеком. Эти аминокислотные остатки не из человека часто называют "импортируемыми" остатками, которые обычно берутся из "импортируемого" вариабельного домена. Гуманизация может по существу выполняться по способу Винтера с сотрудниками (Jones et al. (1986); Riechmann et al. (1988); Verhoeyen et al. (1988)), заменой CDR или CDR-последовательностями грызунов соответствующих последовательностей антитела человека. Таким образом, такие «гуманизированные» антитела являются химерными антителами (Cabilly, supra), в которых по существу меньшая часть, чем интактный вариабельный домен человека, был заменен соответствующей последовательностью отличающегося от человека вида. На практике, гуманизированные антитела являются обычно антителами человека, в которых некоторые остатки CDR и, возможно, некоторые остатки FR заменены остатками аналогичных сайтов в антителах грызуна.
Важно, чтобы гуманизированные антитела сохраняли высокую аффинность в отношении антигена и другие благоприятные биологические свойства. Для достижения этой цели, в соответствии с предпочтительным способом, гуманизированные антитела получают способом анализа исходных последовательностей и различных концептуальных гуманизированных продуктов с использованием трехмерных моделей исходных и гуманизированных антител. Эти трехмерные модели иммуноглобулинов являются обычно доступными и известными для специалистов с квалификацией в данной области. Доступны компьютерные программы, которые иллюстрируют и отображают на дисплее возможные трехмерные конформационные структуры выбранных кандидатных последовательностей иммуноглобулинов. Обследование этих дисплеев делает возможным анализ вероятной роли этих остатков в функционировании кандидатной последовательности иммуноглобулина, т.е. анализ остатков, которые влияют на способность кандидатного иммуноглобулина связывать его антиген. Таким путем могут быть выбраны и комбинированы FR-остатки из консенсусной и импортируемой последовательности, так что достигается желаемое свойство антитела, такое как увеличенная аффинность в отношении антигена-мишени (антигенов-мишеней). Обычно остатки CDR прямо и наиболее существенным образом участвует во влиянии на связывание антигена. В отношении дополнительных подробностей см. заявку США (U.S. Application Ser. No. 07/934373 filed Aug. 21, 1992), которая является частичным продолжением заявки с регистрационным номером 07/715272, поданной 14 июня 1991 года.
D. Антитела человека
Моноклональные антитела человека могут быть получены гибридомным способом. Линии клеток миеломы человека и гетеромиеломы мышь-человек были описаны, например, Kozbor (1984) и Brodeur et al., Monoclonal Antibody Production Techniques and Applications, pp. 51-63 (Marcel Dekker, Inc. New York, 1987).
В настоящее время можно получать трансгенных животных (например, мышей), которые способны, после иммунизации продуцировать набор антител человека в отсутствие продуцирования эндогенного иммуноглобулина. Например, было описано, что гомозиготная делеция гена соединительной области (JH) тяжелой цепи в химерных и мутантных мышах эмбриональной линии приводит к полному ингибированию продуцирования эндогенных антител. Перенос ряда генов иммуноглобулинов зародышевой линии человека в таких мутантных мышей эмбриональной линии будет приводить к продуцированию антител человека после введения антигена. См., например, Jakobovits et al. (1993).
Альтернативно, может быть использована технология фагового дисплея (McCafferty et al. (1990) для получения антител и фрагментов антител человека in vitro из наборов генов вариабельного (V) домена иммуноглобулина из неиммунизированных доноров. Согласно этому способу, гены домена V антитела клонируют в рамке считывания в ген основного или минорного белка оболочки нитевидного бактериофага, такого как М13 или fd, и выставляют (отображают) в виде функциональных фрагментов антитела на поверхности фаговых частиц.
Поскольку нитевидная частица содержит одноцепочечную ДНК-копию генома фага, отборы на основе функциональных свойств этого антитела также приведут к отбору гена, кодирующего антитело, проявляющее эти свойства. Таким образом, фаг имитирует некоторые из свойств В-клеток. Фаговый дисплей может выполняться в различных форматах; в отношении обзора этих форматов см., например, Johnson and Chiswell (1993). Для фагового дисплея могут быть использованы несколько источников сегментов V-генов. Clackson et al. (1991) выделили ряд разнообразных антител против оксазолона из небольшой случайной комбинаторной библиотеки V-генов, полученной из селезенок иммунизированных мышей. Может быть сконструирован набор V-генов из иммунизированных доноров-людей, и антитела к ряду разнообразных антигенов (в том числе к собственным антигенам) могут быть выделены в основном согласно способам, описанным Marks et al. (1991) или Griffith et al. (1993). В природной иммунной реакции гены антител накапливают мутации с высокой скоростью (соматическое гипермутирование). Некоторые из введенных изменений будут придавать более высокую аффинность, и В-клетки, обнаруживающие поверхностный иммуноглобулин с высокой аффинностью, предпочтительно реплицируются и дифференцируются во время последующей стимуляции антигеном. Этот естественный процесс может быть имитирован с использованием способа, известного как «перетасовка цепей» (Marks et al., 1992). В этом способе аффинность «первичных» антител человека, полученных с использованием фагового дисплея, может быть улучшена последовательной заменой генов V-области тяжелой и легкой цепей наборами природно встречающихся вариантов (наборов) генов V-области, полученных из неиммунизированных доноров. Этот способ позволяет получать антитела и фрагменты антител с аффинностями в диапазоне нМ. Стратегия создания очень больших фаговых наборов антител (также известных как "mother-of-all libraries") была описана Waterhouse et al. (1993), а выделение человеческого антитела высокой аффинности непосредственно из такой большой фаговой библиотеки сообщается Griffith et al. (1994). Перетасовка генов может быть также использована для получения человеческих антител из антител грызунов, поскольку человеческое антитело имеет сходные аффинности и специфичности относительно исходного антитела грызуна. Согласно этому способу, который называют также "эпитопным импринтингом", ген V-домена тяжелой или легкой цепи антител грызуна, полученный с использованием способа фагового дисплея, заменяют набором генов V-домена человека с получением химер грызун-человек. Отбор на антиген приводит к выделению вариабельного домена человека, способного восстанавливать функциональный антигенсвязывающий сайт, т.е. этот эпитоп определяет (производит импринтинг) выбора партнера. При повторении этого процесса для замены остального V-домена грызуна, получают антитело человека (см. заявку на патент РСТ WO 93/06213, опубликованную 1 апреля 1993 года). В отличие от традиционной гуманизации антител грызунов пересадкой CDR, этот способ обеспечивает полностью человеческие антитела, которые не имеют каркасной области или остатков CDR происходящих из грызуна.
Е. Биспецифические антитела
Биспецифические антитела являются моноклональными, предпочтительно человеческими или гуманизированными, антителами, которые обладают специфичностями связывания в отношении по меньшей мере двух различных антигенов. В данном случае одна из специфичностей связывания является специфичностью в отношении химерного полипептида, а другая специфичность является специфичностью в отношении любого другого антигена и предпочтительно в отношении другого рецептора или субъединицы рецептора. Например, биспецифические антитела, специфически связывающие химерный полипептид и нейротрофический фактор, или два различных химерных полипептида, включены в объем данного изобретения.
F. Способы получения биспецифических антител известны в данной области.
Традиционно, рекомбинантное получение биспецифических антител основывается на коэкспрессии двух пар тяжелой цепи-легкой цепи иммуноглобулина, где эти две тяжелые цепи имеют различные специфичности (Milstein and Cuello (1983)). Вследствие случайного ассортимента тяжелых и легких цепей иммуноглобулина эти гибридомы (квадромы) образуют потенциальную смесь из 10 различных молекул антител, из которых только одна имеет правильную биспецифическую структуру. Очистка этой правильной молекулы, которую обычно выполняют при помощи стадий аффинной хроматографии, является довольно обременительной, выход продукта является низким. Подобные процедуры описаны в публикации РСТ-заявки № WO 93/08829 (опубликованной 13 мая 1993 года) и в Traunecker et al. (1991).
В соответствии с другим и более предпочтительным подходом вариабельные домены антител с требуемыми специфичностями (антигенсвязывающими сайтами антител) сливают с последовательностями константных доменов иммуноглобулина. Это слияние осуществляют с константным доменом легкой цепи иммуноглобулина, содержащим по меньшей мере часть шарнирных, CH2 и СН3 областей. Предпочтительно, чтобы первая константная область (СН1), содержащая сайт, необходимый для связывания легкой цепи, присутствовала по меньшей мере в одном из таких слияний. ДНК, кодирующие слияния тяжелых цепей иммуноглобулина и, если желательно, легкую цепь иммуноглобулина, встраивают в отдельные экспрессирующие векторы и котрансфицируют в подходящий организм-хозяин. Это обеспечивает большую гибкость в корректировке взаимных пропорций этих трех полипептидных фрагментов в вариантах, когда неравные отношения этих трех полипептидных цепей, используемых в этой конструкции, обеспечивают оптимальные выходы. Тем не менее можно встраивать кодирующие последовательности для двух или всех трех полипептидных цепей в один экспрессирующий вектор, когда экспрессия по меньшей мере двух полипептидных цепей в равных соотношениях приводит к высоким выходам или когда эти соотношения не имеют особого значения. В предпочтительном варианте этого подхода биспецифические антитела состоят из гибридной тяжелой цепи иммуноглобулина с первой специфичностью связывания в одном плече и гибридной пары тяжелая цепь-легкая цепь иммуноглобулина (обеспечивающей вторую специфичность связывания) в другом плече. Было обнаружено, что эта асимметричная структура облегчает отделение желаемого биспецифического соединения от нежелательных комбинаций цепей иммуноглобулина, так как присутствие легкой цепи иммуноглобулина только в одной половине биспецифической молекулы обеспечивает легкий способ разделения. Такой подход описан в совместно поданной заявке Ser. No.07/931811, поданной 17 августа 1992 года.
В отношении дополнительных подробностей генерирования биспецифических антител см., например, Suresh et al. (1986).
G. Гетероконъюгатные антитела
Гетроконъюгатные антитела также находятся в рамках данного изобретения. Гетероконъюгатные антитела состоят из двух ковалентно соединенных антител. Такие антитела были предложены, например, для нацеливания клеток иммунной системы на нежелательные клетки (патент США № 4676980) и для лечения ВИЧ-инфекции (Публикации заявок РСТ с номерами WO 91/00360 и WO 92/200373; ЕР 03089). Гетероконъюгатные антитела могут быть получены с использованием любых подходящих способов сшивания. Подходящие сшивающие агенты хорошо известны в данной области и описаны в патенте США № 4676980 вместе с рядом способов сшивания.
ПРИМЕРЫ
Далее дается иллюстрация предпочтительных вариантов для применения на практике данного изобретения. Однако, эти примеры не являются ограничивающими данное изобретение. В применении на практике данного изобретения возможны другие примеры и способы.
ПРИМЕР 1
МАТЕРИАЛЫ
Следующие материалы использовали для многих описанных здесь примеров. Реагенты ПЦР получали из Fisher Scientific (Pittsburgh, PA), а ферменты молекулярной биологии покупали из Boehringer Mannheim (Indianapolis, IN) или New England Biolabs (Beverly, MA). Бактериальные штаммы, бактериальные экспрессирующие плазмиды рЕТ и рекомбинантную энтерокиназу получали из Novagen (Madison, WI). Все другие химикалии были из Sigma Chemical Company (St. Louis, MO) или Fisher Scientific (Pittsburg, PA). Металл-аффинную смолу (агарозу Talon или Nichel) получали из Clontech Laboratories (Palo Alto, CA). Реагенты для культуры ткани были из Gibco BRL (Gaithersburg, MD).
Кожную Т-клеточную лимфому человека (Hut78) из Американской Коллекции Типовых Культур (АТСС, Manassas, VA) культивировали в среде RPMI 1640, содержащей 10% фетальную телячью сыворотку (ФТС). Эндотелиальные клетки аорты свиньи, трансфицированные либо рецептором flt-1 (PAE-Flt-1), либо рецептором flk-1/KDR (PAE-Flk-1), были любезным подарком от доктора J. Waltenberger, и их культивировали в питательной среде F12 (НАМ) с 10% ФТС. Клеточную линию А375М меланомы человека получали от доктора I.J. Fidler из University of Texas MD Anderson Cancer Center (Houston, TX) и культивировали в МЕМ, дополненной 10% ФТС.ПРИМЕР 2
СПОСОБЫ - КЛОНИРОВАНИЕ ГЕНА ГРАНЗИМА В ЧЕЛОВЕКА
Следующие способы использовали по меньшей мере для примера 13. РНК Hut78 выделяли с использованием системы для микровыделения РНК GlassMAX RNA Microisolation Spin Cartridge (Gibco BRL), а затем определяли количество тотальной РНК. Затем удаляли геномную ДНК инкубированием пробы с ДНКазой I в течение 15 минут при комнатной температуре. ДНКазу I инактивировали добавлением раствора ЭДТА и нагреванием в течение 15 минут при 65°С. Систему синтеза первой цепи для ОТ-ПЦР SUPERSCRIPT (Gibco BRL) использовали для синтеза первой цепи с олиго (dT). кДНК-мишень (кДНК пред-зрелого Гранзима В человека) амплифицировали с использованием праймеров: NcoIgb (5'→3'): GGTGGCGGTGGCTCCATGGAACCAATCCTGCTTCTG (SEQ ID NO:1) и CxhoIgb (5'→3'): GCCACCGCCTCCCTCGAGCTATTAGTAGCGTTTCATGGT (SEQ ID NO:2) при помощи ПЦР. Условия ПЦР включали в себя денатурацию при 95°С в течение 5 минут, ПЦР-цикл 94°С в течение 1 мин, 50°С в течение 1 мин, 72°С в течение 1 мин, для 30 циклов; стадию элонгации (удлинения) при 72°С в течение 5 мин. Проводили электрофорез в 1% агарозе для подтверждения продукта ПЦР. Затем продукт ПЦР клонировали в вектор ТА PCR 2.1 (Invitrogen; Carlsbad, CA) и обозначали как gbTA. gbTA трансформировали в компетентные клетки INVαF' и положительные клоны подвергали скринингу с использованием скрининга синих/белых колоний или способов ПЦР. ДНК для положительных клонов выделяли с использованием набора QIAprep Spin prep (Qiagen; Valencia, CA) и секвенировали для подтверждения гена гранзима В человека; этот правильный клон идентифицировали как gbTA-2 (клон № 2).
ПРИМЕР 3
СПОСОБЫ - КОНСТРУИРОВАНИЕ СЛИТЫХ ГЕНОВ ГРАНЗИМ В-VEGF121 ИЛИ ГРАНЗИМ В-scFvMEL
Следующие слитые конструкции использовали во многих описанных здесь примерах. Слитая конструкция Гранзим В-vegf121 имела формат Ek-Гранзим В-линкер G4S-Vegf121. Это конструирование было основано на способе перекрывающейся ПЦР. Вкратце, кодирующую последовательность гранзима В амплифицировали из gbTA-2 при помощи ПЦР, использующей праймеры: NgbEK (5'→3'): GGTACCGACGACGACGACAAGATCATCGGGGGACATGAG, Cgb (5'→3') (SEQ ID NO:3) и GGAGCCACCGCCACCGTAGCGTTTCATGGT (SEQ ID NO:4). Они были сконструированы для делеции сигнальной последовательности пред-зрелого гранзима В и встраивания сайта расщепления энтерокиназой на N-конце, наряду с добавлением последовательности линкера G4S к С-концу для связывания с геном vegf121. Последовательность vegf121 амплифицировали из плазмиды рЕТ22-vegf121 (любезный подарок от группы доктора Phil Thorpe, University of Texas Southwest Medical School, Dallas, TX) при помощи ПЦР с использованием праймеров: Nvegf (5'→3'): GGTGGCGGTGGCTCCGCACCCATGGCAGAA (SEQ ID NO:5) и CxhoI veg (5'→3'): AAGGCTCGTGTCGACCTCGAGTCATTACCGCCTCGGCTTGTC (SEQ ID NO:6). Последовательность scFvMEL амплифицировали из плазмиды рЕТ-scFvMEL/TNF при помощи ПЦР с использованием праймеров: Nzme2 (5'→3'): GGTGGCGGTGGCTCCACGGACATTGTGATGACCCAGTCTCAAAAATTC
(SEQ ID NO:7) и Czme2 (5'→3'): GGAGCCACCGCCACCCTCGAGCTATCATGAGGAGACGGTGAGAGTGGT (SEQ ID NO:8). Эти праймеры добавляли последовательность линкера G4S к N-концу для связывания перекрывающегося участка ПЦР с С-концом гранзима В и на С-конце встраивали сайт XhoI для облегчения следующих стадий клонирования. На С-конце добавляли два стоп-кодона непосредственно перед сайтом XhoI. Слитые гены соединяли вместе при помощи второй ПЦР с использованием праймеров NgbEK и CxhoIveg (для гранзим В-vegf121) или NgbEK и Czme2 (для гранзим В-scFvMEL). Для клонирования этих слитых генов в вектор рЕТ32а(+) с сайтом энтерокиназы при N-конце гранзима В этот фрагмент из рЕТ32а(+) амплифицировали при помощи ПЦР с использованием праймеров промотора Т7 (5'→3'): TAATACGACTCACTATAG (SEQ ID NO:9) и CpET32EK (5'→3'): CTTGTCGTCGTCGGTACCCAGATCTGG (SEQ ID NO:10). Этот праймер имеет сайт энтерокиназы на С-конце, перекрывающийся с N-концом слитого гена. С использованием ПЦР с перекрыванием слитые гены ЕК-Гранзим В-vegf121 конструировали с использованием праймеров промотора Т7 CxhoIveg, а слитые гены EK-гранзим B-scFvMEL конструировали с использованием праймера промотора Т7 и Czme2. Эти реакции ПЦР выполняли при помощи тридцати циклов 94°С в течение 1 мин, 50°С в течение 1 мин и 72°С в течение 1 мин, с реакцией удлинения при 72°С в течение 5 мин. Амплифицированные фрагменты разделяли электрофорезом в 1% агарозном геле и очищали с использованием набора для очистки ПЦР (Qiagen). Очищенные продукты ПЦР расщепляли при помощи XbaI и XhoI при 37°С в течение 3 часов и затем разделяли электрофорезом в 1% агарозном геле и очищали из этих гелей и клонировали в вектор рЕТ32а(+), названный рЕТ32GrB-vegf121 или рЕТ32GrB-scFvMEL. Смесь для лигирования трансформировали в компетентные клетки DH5α, положительные клоны подвергали скринингу при помощи ПЦР и затем секвенировали. Клон, имеющий промотор Т7, оператор lac, rbs, Trx.tag, His.tag, S-tag и сайты энтерокиназы, для получения слитых белков гранзим В-G4S-vegf121 или гранзим В-G4S-scFvMEL, без мутаций второго сайта, выбирали для трансформации в компетентные клетки AD494 (DE3)pLysS для дополнительной индукции и экспрессии.
ПРИМЕР 4
СПОСОБЫ - ИНДУКЦИЯ И ЭКСПРЕССИЯ СЛИТЫХ БЕЛКОВ ГРАНЗИМ В-VEGF121 ИЛИ ГРАНЗИМ В-scFvMEL В E. coli
Слитые конструкции примера 3 индуцировали и экспрессировали, как описано здесь, для использования во многих примерах, описанных здесь в другом месте, в том числе в примерах 17 и 18. Бактериальные колонии, трансформированные этой сконструированной плазмидой, выращивали в среде для выращивания бульона Луриа (LB), содержащей 200 мкг/мл ампициллина, 70 мкг/мл хлорамфеникола и 15 мкг/мл канамицина, при 37°С при встряхивании 240 об/мин. Затем эти культуры разбавляли 1:100 в свежей среде LB плюс антибиотики и выращивали до А600 0,5 при 37°С. После этого культуры индуцировали добавлением IPTG до конечной концентрации 0,25 мМ при 37°С в течение 1,5 час. Клетки собирали и ресуспендировали в 10 мМ Трисе (рН 8,0) и хранили в замороженном состоянии при -80°С для последующей очистки.
ПРИМЕР 5
СПОСОБЫ - ОЧИСТКА СЛИТЫХ БЕЛКОВ ГРАНЗИМ В-VEGF121 ИЛИ ГРАНЗИМ в-scFvMEL
Слитые конструкции, индуцированные и экспрессированные в примере 4, очищали, как описано здесь, для применения во многих примерах, описанных в другом месте здесь, в том числе по меньшей мере в примерах 17 и 18. Ресуспензированную культуру лизировали добавлением лизоцима до конечной концентрации 100 мкг/мл с перемешиванием в течение 30 минут при 4°С с последующей обработкой ультразвуком. Экстракты центрифугировали при 10800 g в течение 30 минут и супернатант дополнительно центрифугировали при 40000 об/мин в течение 1 часа. Супернатант, содержащий только растворимый белок, доводили до 40 мМ Триса, рН 8,0, 10 мМ имидазола и наносили на никель-NTA-агарозу, уравновешенную тем же самым буфером. После промывания никель-NTA-агарозы 500 мМ NaCl и 20 мМ имидазолом связанные белки элюировали 500 мМ NaCl и 500 мМ имидазолом. Анализы оптической плотности (280 нм) и электрофореза на ДСН-ПААГ выполняли для идентификации меченого полигистидином белка, обозначенного как Про-гранзим В-vegf121 или Про-гранзим В-scFvMEL, соответственно. Элюированный белок про-гранзим vegf121 или про-гранзим В-scFvMEL диализовали против 20 мМ Трис-HCl (рН 8,0) и 50 мМ NaCl. Часть програнзим В слитого белка гранзим В-vegf121 или слитого белка гранзим В-scFvMEL активировали добавлением рекомбинантной бычьей энтерокиназы (rEK) для удаления полигистидиновой метки в соответствии с инструкцией изготовителя (1 единица rEK расщепляла 50 мкг белка, инкубирование при комнатной температуре в течение 16 часов). rEK удаляли улавливающей ЕК агарозой. Конечный белок анализировали электрофорезом на ДСН-ПААГ и хранили при 4°С.
ПРИМЕР 6
СПОСОБЫ - ЭЛЕКТРОФОРЕЗ В ДСН-ПААГ И ВЕСТЕРН-БЛОТ-АНАЛИЗ
Следующие способы выполняли для экспериментов, описанных, например, в примере 15. Пробы белка анализировали электрофорезом на 8,5% ДСН-ПААГ при восстанавливающих условиях. Гели окрашивали Кумасси синим. Для вестерн-блот-анализа белки переносили из гелей в нитроцеллюлозные мембраны. Эти мембраны блокировали 5% обезжиренным молоком и инкубировали в течение 1 часа при комнатной температуре с мышиным моноклональным антителом против гранзима В (1,0 мкг/мл) или мышиным поликлональным антителом против vegf121 (разведение 1:2000) или кроличьим поликлональным антителом против scFvzme (разведение 1:2000). После промывания мембраны инкубировали с конъюгатом козьи антимышиные антитела/пероксидаза хрена (HRP-GAM, разведение 1:5000) или конъюгатом козьи антикроличьи антитела/пероксидаза хрена (HRP-GAR, разведение 1:5000). После дополнительного промывания мембрану проявляли с использованием системы детектирования ECL (Amersham, Piscataway, NJ) и экспонировали на рентгеновской пленке.
ПРИМЕР 7
СПОСОБЫ - АНАЛИЗЫ ФЕРМЕНТОВ
Ферментативную активность гранзима В определяли в непрерывном колориметрическом анализе с BAADT (N-α-трет-бутоксикарбонил-L-аланил-L-аланил-L-аспартил-тиобензиловым эфиром) в качестве субстрата. Анализы выполняли в 200 мкл и смеси для анализа состояли из фермента в 100 мМ HEPES, рН 7,5, 10 мМ CaCl2, 1 мМ 5,5'-дитиобис-2-нитробензойной кислоты, 0,2 мМ субстрата при 25°С. Изменение оптической плотности при OD405 измеряли на планшет-ридере Thermomax. Увеличения оптической плотности превращали в ферментативные скорости с использованием коэффициента экстинкции 13100 см-1М-1, который отличается от обычного коэффициента экстинкции 13600 см-1М-1 при 412 нм, сообщенного Ellman.
ПРИМЕР 8
СПОСОБЫ - ОБНАРУЖЕНИЕ scFvMEL-ЧАСТИ ГРАНЗИМ В-scFvMEL
Для обнаружения scFvMEL-части слитого белка Гранзим В-scFvMEL методом ELISA, использовали планшеты, покрытые белком L Reacti-Bind TM из PIERCE (Rockford, IL). Вкратце, предварительно нанесенный белок L блокировали 5% БСА и эту реакцию очищали с использованием слитого белка Гранзим В-scFvMEL или других слитых с scFvMEL белков при различных концентрациях, соответственно. После промывания эти белки инкубировали с кроличьим антителом против scFvZME, затем с HRP-GAR, затем добавляли раствор субстрата 2,2'-азино-бис-3-этилбензтиазолин-6-сульфоновой кислоты с 1 мкг/мл 30% Н2О2. Спустя 30 минут измеряли оптическую плотность при 405 нм.
ПРИМЕР 9
СПОСОБЫ - АНАЛИЗЫ ЦИТОТОКСИЧНОСТИ in vitro ПРОТИВ РАЕ-FLT-1 И PAE-FLK-1 ДЛЯ ГРАНЗИМ В-vegf121 И ПРОТИВ А375-М ДЛЯ ГРАНЗИМ В-scFvMEL
Клетки РАЕ в среде HAM F-12 с 10% ФТС или клетки А375-М в среде МЕМ с 10% ФТС высевали в 96-луночные планшеты при плотности 2,5 х 103 клеток на лунку и давали им прикрепляться в течение 24 часов при 37°С в 5% СО2. Спустя 24 часа эту среду заменяли средой, содержащей различные концентрации слитых белков гранзим В-vegf121 или гранзим В-scFvMEL. Спустя 72 часа действие гранзим В-vegf121 или гранзим В-scFvMEL на рост клеток в культуре определяли с использованием окрашивания красителем кристаллическим фиолетовым (кристаллвиолетом). Выжившие прикрепленные клетки окрашивали добавлением 100 мкл кристаллвиолета (0,5% (м/о) в этаноле). Краситель инкубировали на планшетах в течение 0,5 час, избыток красителя удаляли и планшеты промывали водой и давали им высыхать на воздухе. Оставшийся краситель солюбилизировали добавлением 150 мкл буфера Сорнсона (0,1 М цитрат натрия, рН 4,2). Планшеты считывали на микропланшет-ридере ELISA при 630 нм.
ПРИМЕР 10
ТРАНСКРИПЦИЯ И ТРАНСЛЯЦИЯ in vitro И РАСЩЕПЛЕНИЕ in vitro ПРОКАСПАЗЫ 3-ИЛИ DFF45 СЛИТЫМ С ГРАНЗИМОМ В ИЛИ ВАХ БЕЛКОМ
Экспрессирующую плазмиду, содержащую кДНК, кодирующую прокаспазу-3 или DFF54, линеаризуют расщеплением рестрикционной эндонуклеазой и генерируют 35S-меченую прокаспазу-3 или белок DFF45 с использованием in vitro-набора TNT ретикулоцитов кролика (Promega) в соответствии с инструкциями изготовителя. Вкратце, линейную плазмиду, содержащую кДНК, кодирующую прокаспазу-3 или DFF45 (1 мкг), инкубируют с реакционной смесью TNT, содержащей 25 мкл лизата ретикулоцитов кролика, 2 мкл реакционного буфера TNT, 1 мкл РНК-полимеразы Т7, 1 мМ смесь аминокислот минус цистеин, 2 мкл [35S]-цистеина или 2 мкл [35S]-метионина (10 мКи/мл) и 40 Е ингибитора РНКаз (Amershem Pharmacia Biotech, Inc.) в общем объеме 50 мкл в течение 90 мин при 30°С. Для расщепления in vitro продукты трансляции инкубируют в присутствии слитых с гранзимом В или слитых с Вах белков в 150 мМ NaCl. Реакции проводят при 30°С в конечном объеме 10 мкл в течение различных интервалов времени. Затем реакции останавливают добавлением равного объема 2 х буфера Лэммли. Затем продукты расщепления разделяют электрофорезом в 15% ДСН-ПААГ и детектируют либо иммуноблоттингом, либо получением фосфорного изображения высушенных гелей.
ПРИМЕР 11
АНАЛИЗЫ АПОПТОЗА
Морфология клеток:
Клетки А375-М или ААВ527 (для Гранзим В-scFvMEL) и клетки РАЕ-Flk1 или PAE-Flt1 (для Гранзим В-vegf121) выращивают в подходящих средах для культур клеток. Гибель клеток подвергают мониторингу с использованием ХТТ-анализа. Для визуального мониторинга опосредованного гранзимом В или опосредованного Вах апоптоза этих клеток 1 х 104 клеток засевают в каждую лунку 12-луночного предметного стекла для микроскопа. Спустя 48 часов клетки промывают один раз в ЗФР, затем обрабатывают 25 мкл бессывороточной среды, дополненной 1 мкг/мл ДТТ и слитыми с гранзимом В или слитыми с Вах белками. После инкубирования при 37°С в течение 1 часа супернатанты удаляют и заменяют 50 мкл полной среды. Спустя еще 2 часа при 37°С клетки осторожно промывают ЗФР и фиксируют смесью ацетон:метанол (1:1) в течение 2 мин при комнатной температуре. Апоптоз прикрепленных клеток визуализируют фазово-контрастной микроскопией.
Анализ фрагментации ДНК:
Для мониторинга фрагментации ДНК 5 х 105 клеток в 50 мкл бессывороточной среды дополняют 1 мкг/мл ДТТ и слитыми белками гранзима В или слитыми белками Вах. После инкубирования при 37°С в течение 1 часа их промывают один раз в ЗФР. Фрагментированную ДНК экстрагируют с использованием анализа экстракции смесью фенол/хлороформ. Вкратце, осадок клеток ресуспендируют в 25 мкл ЗФР и добавляют равный объем смеси фенол/хлороформ/изоамиловый спирт (1:1:0,1). После осторожного перемешивания и центрифугирования (10000 g в течение 2 мин) фрагментированную ДНК выделяют, обрабатывают РНКазой А в течение 1 часа при 37°С и анализируют на 2% агарозных гелях, содержащих бромид этидия.
FACS-анализ:
Клетки (5 х 105/10мл) центрифугируют при 450 х g в течение 6 мин, промывают холодным ЗФР и ресуспендируют в 300 мкл ЗФР. Эти клетки фиксируют 5 мл метанола и оставляют при -20°С по меньшей мере на 1 час. Затем клетки центрифугируют при 800 х g в течение 5 мин, ресуспендируют в 100 мкл ЗФР и разбавляют до конечного объема 1 мл ЗФР. Клетки инкубируют на льду в течение дополнительных 30 мин, центрифугируют при 800 х g в течение 5 мин и ресуспендируют в 0,5 мл ЗФР. К пробам клеток добавляют 10 мкл РНКазы (50 мкг/мл) и иодид пропидия (PI, 5 мкг/мл) и затем анализируют клеточным сортингом с возбуждением флуоресценции (FACS) на содержание ДНК как функции количества клеток.
Расщепление каспазы-3, PARP и DFF, обнаруженных в пробах клеток, обработанных слитыми белками, в сравнении с клетками, не обработанными слитыми белками, в пределах различных временных периодов или при различных дозах с использованием вестерн-блот-анализа:
Для вестерн-блоттинга пробы разделяют электрофорезом с использованием 14% ДСН-ПААГ. Белки переносят из гелей в нитроцеллюлозные мембраны. Мембраны блокируют 5% обезжиренным молоком и инкубируют в течение 1 часа при комнатной температуре с антителами против каспазы-3 или против расщепленной каспазы-3 или с антителами против PARP или против расщепленного DFF (полученными из Cell Signaling Technology). После промывания эти мембраны инкубируют с конъюгатом козьих антикроличьих или козьих антимышиных антител с пероксидазой хрена. После дополнительного промывания мембраны проявляют с использованием системы детектирования ECL Amersham и экспонируют на рентгеновской пленке.
ПРИМЕР 12
АНАЛИЗЫ НЕАПОПТОЗНЫХ ПУТЕЙ ПЕРЕДАЧИ СИГНАЛА
В следующем примере описаны эксперименты, которые выполняют для анализа путей передачи (трансдукции) сигналов, которые являются неапоптозными путями, в клетках, обработанных доставляющим носителем, содержащим протеинкиназу В, которая является критической точкой в каскаде передачи сигнала инсулина, приводящей к модуляции фермента гликогенсинтетаза-киназы-3 (GSK-3). В данном изобретении генерируют слитую конструкцию цитокина фактора роста гепатоцитов человека (HCF) и регулятора передачи сигнала протеинкиназы В (РКВ). Компонент HCF служит для связывания с гепатоцитами и для доставки активной PKB в цитоплазму клетки. От этого момента активация находящихся ниже по ходу процеса модуляторов передачи сигнала инсулина (GSK-3) будет оцениваться, как описано ниже.
Анти-GSK3-антитела получают из Transduction Laboratory. Антитело 4G10 против фосфотирозина и анти-РКВ-антитело получают из Upstate Biotechnology. Фосфоспецифическое антитело против Ser-9 GSK3 получают из Quality Controlled Biochemicals.
Культуры гепатоцитов в среде обрабатывают различными дозами HCF или слитой конструкции HCF/PKB. В различные моменты времени после добавления лекарственного средства клетки собирают. Для получения цитозольных фракций клетки промывают и собирают в охлажденный на льду забуференный фосфатом солевой раствор. Осадки клеток ресуспендируют в охлажденном на льду гипотоническом буфере (25 мМ Трис, рН 7,5, 1 мМ ЭДТА, 25 мМ NaF, 1 мМ дитиотреитол) с полной смесью ингибиторов протеаз (Roche Molrcular Biochemicals; Indianapolis, IN). Клетки лизируются после инкубирования на льду в течение 10 мин (что подтверждают микроскопическим анализом). Лизаты подвергают ультрацентрифугированию при 100000 х g в течение 30 мин при 4°С и супернатант собирают. Для иммунопреципитации клетки промывают дважды в охлажденном на льду забуференном фосфатом солевом растворе и затем лизируют в буфере IP (125 мМ NaCl, 25 мМ NaF, 25 мМ Трис, рН 7,5, 1 мМ ЭДТА, 1 мМ ЭГТА, 1% Тритон Х-100, 10 мМ глицеринфосфат, 5 мМ пирофосфат натрия, 1 мМ NaVO3, 200 нМ окадаевая кислота, 1 мМ дитиотреитол) с полной смесью ингибиторов протеаз. Анти-GSK3-антитело добавляют к осветленным лизатам и выдерживают в течение 1 часа при 4°С и затем добавляют гранулы белка G (Sigma) и выдерживают еще в течение 1 часа. Иммунопреципитаты промывают три раза буфером IP.
Для анализов киназы GSK3 иммунопреципитаты клеток, обработанных доставляющим носителем, содержащим активную слитую с протеинкиназой В конструкцию GSK3, промывают сначала один раз киназным буфером (25 мМ Тис, рН 7,5, 10 мМ MgCl2). Киназные реакции выполняют в киназном буфере со 100 мкМ [γ-32Р]АТФ и 100 мкМ пептидом 2BSP в качестве субстрата (синтезированным Biomedical Resource Center, University of California, San Francisko, CA). 2BSP основан на сайте-мишени GSK3 в eIF2B. Спустя 20 мин при 30°С реакции наносят в виде пятен на фосфоцеллюлозную бумагу Р81 (Whatman), промывают четыре раза 100 мМ фосфорной кислотой и считают в сцинтилляционном счетчике.
ПРИМЕР 13
КЛОНИРОВАНИЕ ГЕНА ГРАНЗИМА В ЧЕЛОВЕКА ИЗ HUT78
Нативный гранзим В человека является сериновой протеиназой гранул цитотоксических лимфоцитов, продуцируемых цитотоксическими Т-клетками и природными клетками-киллерами. Первоначальные попытки клонирования гена гранзима В человека из клеток HL-60, которые являются клетками промиелоцитарного лейкоза из периферической крови человека, были неуспешными. Однако, кДНК-мишень, в виде гена пред-зрелого гранзима В, была получена из клеток Hut78 кожной Т-клеточной лимфомы человека посредством выделения РНК и применения ОТ-ПЦР. Электрофорез в 1% агарозном геле показал, что кДНК пред-зрелого гранзима В человека имела размер ˜800 п.н. (фиг.1). Последовательность гена и аминокислотная последовательность (фиг.2) показали, что первые 20 аминокислот являются сигнальной последовательностью. Последовательность гранзима В человека с сигнальной последовательностью называют пред-зрелым гранзимом В. В цитотоксических клетках активный гранзим В образуется из зимогена протеолизом дипептидилпептидазой I (DPPI) (Smyth et al., 1995). Она удаляет пропептид из двух остатков (Gly19Glu20) и открывает Ile21 c образованием зрелого гранзима В с N-концевой последовательностью Ile-Ile-Gly-Gly.
ПРИМЕР 14
КОНСТРУИРОВАНИЕ СЛИТЫХ БЕЛКОВ ГРАНЗИМ В-vegf121 ИЛИ
ГРАНЗИМ В-scFvMEL
ПЦР использовали для амплификации кодирующей последовательности гранзима В от Ile21, который является первым остатком зрелого фермента, с эффективным делетированием сигнальной последовательности и домена GlyGlu. Одновременно встраивали сайт расщепления для энтерокиназы (AspAspAspLys; SEQ ID NO:53) слева и рядом с Ile21. Гранзим В присоединяли к рекомбинантному Vegf121 или scFvMEL через гибкий линкер (G4S). Затем этот слитый генный фрагмент вводили в сайты XbaI и XhoI рЕТ32а(+) с образованием экспрессирующего вектора рЕТ32CrB-vegf121 (фиг.3А) и рЕТ32GrB-scFvMEL (фиг.3В). Этот вектор содержит промотор Т7 для экспрессии высокого уровня, за которым следуют Trx.tag, His.tag, сайт расщепления тромбином и сайт расщепления энтерокиназой для конечного удаления метки (тэга) очистки белка (фиг.4А и 4В). После удаления метки (тэга) белка рекомбинантной энтерокиназой первый остаток Ile зрелого гранзима обнажается, и гранзим В-часть слитых белков гранзим В-vegf121 или гранзим В-scFvMEL активируется. Были подтверждены нуклеотидные последовательности и аминокислотные последовательности гранзим В-vegf121 (1059 п.н., 353 аминокислоты (аа)) (фиг.4С и 4D) и гранзим В-scFvMEL (1440 п.н., 480 аминокислот (аа)) (фиг.4E и 4F).
ПРИМЕР 15
ЭКСПРЕССИЯ И ОЧИСТКА СЛИТОГО БЕЛКА ГРАНЗИМ В-vegf121 ИЛИ
ГРАНЗИМ В-scFvMEL
Рекомбинантный белок гранзим В-vegf121 или гранзим В-scFvMEL экспрессировали в виде меченого полигистидином белка, обозначенного как про-гранзим В-vegf121 или про-гранзим В-scFvMEL, и затем очищали Никель-NTA-металл-аффинной хроматографией. Метку his-tag отщепляли добавлением rEK с образованием слитого белка гранзим В-vegf121 или гранзим В-scFvMEL. Один литр культуры обычно давал приблизительно 100 мкг конечного очищенного продукта гранзим В-vegf121 и 150 мкг конечного аффинного продукта гранзим В-scFvMEL.
Результаты показали индуцированную экспрессию связанных с гранзимом В слитых конструкций. Индуцированная полоса при массе ˜55 кДа для гранзим В-vegf121 и при массе ˜72 кДа для гранзим В-scFvMEL представляют, соответственно, конструкцию гранзим В-vegf121 или гранзим В-scFvMEL, содержащую метку очистки массой ˜18 кДа. Ферментативное отщепление этой метки при помощи рекомбинантной энтерокиназы (rEK) приводило к появлению полосы, мигрирующей при массе ˜38 кДа для гранзим В-vegf121 и при массе ˜53 кДа для гранзим В-scFvMEL, представляющей нативные белки. Таким образом, электрофорез в ДСН-ПААГ показал, что конечная очищенная слитая конструкция гранзим В-vegf121 мигрирует при восстанавливающих условиях в виде полосы при ожидаемой молекулярной массе 38 кДа (фиг.5А), а слитая конструкция гранзим В-scFvMEL показала полосу при ожидаемой молекулярной массе 53 кДа (фиг.5В).
Специфичность расщепленного слитого белка подтверждали Вестерн-блоттингом с использованием мышиного моноклонального антитела против гранзима В, мышиного поликлонального антитела против vegf121 или кроличьего поликлонального антитела против scFvZME (фиг.6). Эти результаты показали, что слитая конструкция гранзим В-vegf121 (фиг.6А) могла специфически связываться либо с мышиным анти-vegf121-антителом (фиг.6А (а)), либо с мышиным моноклональным антителом против гранзима В (фиг.6А (b)). Молекулярные массы про-гранзим В-vegf121 и Гранзим В-vegf121 равны приблизительно массе 55 кДа и 38 кДа, соответственно. Слитая конструкция Гранзим В-scFvMEL (фиг.6В) могла специфически связываться либо с мышиным моноклональным антителом против гранзима В (фиг.6В (с)), либо с кроличьим поликлональным антителом против scFvMEL (фиг.6В (d)). Молекулярные массы про-гранзим В-scFvMEL и Гранзим В-scFvMEL равны приблизительно массе 70 кДа и 53 кДа, соответственно.
ПРИМЕР 16
СВЯЗЫВАЮЩАЯ АКТИВНОСТЬ scFvMEL-ЧАСТИ МОЛЕКУЛЫ СЛИТОГО БЕЛКА ГРАНЗИМ В-scFvMEL
scFvMEL-часть молекулы испытывали на связывание белка L, который является иммуноглобулинсвязывающим белком, который первоначально был выделен из бактерии Peptostreptococcus magnus. Белок L имеет уникальную способность связывания через взаимодействия легкой цепи каппа без участия антигенсвязывающего сайта антитела. Это дает Белку L уникальную способность связывать одноцепочечные вариабельные фрагменты (scFv). Эти результаты показали зависимое от концентрации увеличение оптической плотности при 405 нм, что предполагает связывание scFvMEL с Белком L (фиг.7). Связывающая активность слитого белка гранзим В-scFvMEL была такой же, что и связывающая активность слитого белка scFvMEL-TNF, который может специфически связываться с антиген-положительными клетками меланомы человека и обладает цитотоксической активностью в отношении этих клеток меланомы.
ПРИМЕР 17
ЦИТОТОКСИЧЕСКИЕ ДЕЙСТВИЯ in vitro СЛИТОГО БЕЛКА
ГРАНЗИМ В-vegf121
Цитотоксичность гранзим В-vegf121 оценивали в отношении клеток РАЕ-Flk-1 log-фазы (Сверхэкспрессия рецептора flk-1/KDR) и клеток РАЕ-Flt-1 (сверхэкспрессия рецептора flt-1) в культуре, где помещали 2,5 х 103 клеток на лунку на 96-луночном планшете. 50%-ное ингибирующее рост действие было обнаружено при концентрации ˜10 нМ на клетках РАЕ-Flk-1. Однако на клетках РАЕ-Flt-1 не обнаруживали цитотоксических действий (фиг.8). Было также показано, что VEGF121 мог специфически связываться с рецептором VEGF Flk-1/KDR, но не с Flt-1. Цитотоксичность слитого белка гранзим В-vegf121 показала, что эта конструкция может специфически убивать клетки РАЕ-Flk-1, что свидетельствует о том, что Vegf121-часть молекулы этого слитого белка связывалась со сверхэкспрессирующей Flk-1 клеточной поверхностью. Затем происходила доставка гранзима В во внутреннее пространство клеток-мишеней, что приводило к цитотоксичности в отношении клеток-мишеней.
ПРИМЕР 18
ЦИТОТОКСИЧЕСКИЕ ДЕЙСТВИЯ in vitro СЛИТОГО БЕЛКА
ГРАНЗИМ В-scFvMEL
Цитотоксичность гранзим В-scFvMEL оценивали в отношении клеток А375-М меланомы человека log-фазы. Эти результаты показали, что гранзим В-scFvMEL мог убивать клетки А375-М с концентрацией IC50 приблизительно 20 нМ. При предобработке scFvMEL-3825 в концентрации 178,5 нМ в течение 6 часов с последующей обработкой слитым белком гранзим В-scFvMEL в течение 72 часов требовалась в 15 раз более высокая концентрация гранзим В-scFvMEL для проявления 50%-ной цитотоксичности в сравнении с отсутствием предобработки scFvMEL-3825 (фиг.9). В конкретном варианте это связано с тем, что антиген клеточной поверхности gp240 был уже занят scFvMEL-3825, что приводило к уменьшенному шансу связывания scFvMEL-части молекулы слитого белка гранзим В-scFvMEL с антигеном gp240, что, следовательно, ингибировало цитотоксичность гранзим В-scFvMEL на этих клетках. Эти результаты предполагали, что эта цитотоксичность, по меньшей мере частично, обусловлена взаимодействием этого антитела с его доменом клеточной поверхности.
ПРИМЕР 19
КЛОНИРОВАНИЕ ГЕНА ВАХ ЧЕЛОВЕКА
Тотальную РНК из клеток Namalwa выделяли с использованием системы для микровыделения РНК Glass MAX RNA Microisolation Spin Cartridge System (Gibco BRL). Удаление геномной ДНК выполняли добавлением ДНКазы I, не содержащей РНКаз при инкубировании при комнатной температуре в течение 15 мин. Затем ДНКазу I инактивировали добавлением раствора ЭДТА и нагреванием в течение 15 мин при 65°С. Для ОТ-ПЦР использовали систему синтеза первой цепи SUPERSCRIPT (Gibco BRL). В синтезе первой цепи использовали Олиго (dT) и затем кДНК-мишень (кДНК Вах) амплифицировали с использованием праймеров: NbaxTA (5' → 3'): GGTGATGGACGGGTCCGGGGAGCA (SEQ ID NO:29) и CbaxTA (5' → 3'): GGCCTCAGCCCATCTTCTTCCAGATGGTGA (SEQ ID NO:30) при помощи ПЦР со следующими циклами: денатурация при 95°С в течение 5 мин, 30 циклов 94°С в течение 1 мин, 50°С в течение 1 мин, 72°С в течение 1 мин и удлинение (элонгация) при 72°С в течение 5 мин. Проводили электрофорез на 1% агарозном геле для проверки продукта ПЦР. Очищенный фрагмент ПЦР клонировали в вектор ТА PCE 2.1 (Invitrogen) и полученный вектор обозначали как ВахТА. ВахТА трансформировали в компетентные клетки INVαF' и положительные клоны подвергали скринингу посредством скрининга синих/белых колоний или с использованием ПЦР-способов. ДНК для положительных клонов выделяли с использованием набора QIApre Spin prep (QIAGEN, Valencia, CA), и секвенирование подтверждало присутствие гена bax человека в правильном клоне (BaxTA-35 (клон № 35)).
ПРИМЕР 20
КОНСТРУИРОВАНИЕ РОДСТВЕННЫХ ВАХ СЛИТЫХ ГЕНОВ
Конструирование было основано на способе перекрывающейся ПЦР. Гены scFvMEL сливали с генами Вах, укороченным Вах1-5 (т.е. содержащим экзоны 1-5) или укороченным Вах345 (т.е. содержащим экзоны 3, 4 и 5) с линкером G4S в различной ориентации (обозначенные scFvMEL-bax или Bax-scFvMEL, scFvMEL-Вах1-5 или Вах1-5-scFvMEL и scFvMEL-Вах345 или Вах 345-scFvMEL, соответственно). Как показано на фиг.10, специалисту с квалификацией в данной области будет понятно, что ген Вах человека (SEQ ID NO:45), который кодирует полипептид SEQ ID NO:46, содержит шесть экзонов, с доменом ВН1 (DGNFNWGRVVA; SEQ ID NO:47) в экзоне 4, ВН2 (WIQDQGGWD; SEQ ID NO:48) в экзоне 5 и ВН3 (LKRIGDE; SEQ ID NO:49) в экзоне 3. В особом варианте химерный полипептид Вах содержит домены ВН1, ВН2 и ВН3 или их комбинации. В другом особом варианте химерный полипептид Вах состоит по существу из доменов ВН1, ВН2 и ВН3. Еще в одном особом варианте химерный полипептид Вах содержит экзоны 3, 4 и 5 или их комбинации. В дополнительном особом варианте химерный полипептид Вах состоит по существу из экзонов 3, 4 и 5.
Вкратце, кодирующую последовательность scFvMEL амплифицировали при помощи ПЦР из рЕТ32а-scFvMEL/TNF, а полноразмерный bax или укороченный Вах1-5 или укороченный Вах345 амплифицировали при помощи ПЦР из ВахТА-35. Были сконструированы различные праймеры, в которых к С-концу или N-концу добавляли линкер G4S для соединения этих слитых генов вместе при помощи второй ПЦР. Для клонирования слитых генов в векторе рЕТ32а(+) на сайтах NcoI и XhoI праймеры добавляли сайт NcoI на N-конце и два стоп-кодона, а на С-конце добавляли сайт XhoI. Первую ПЦР проводили с использованием 95°С в течение 5 мин, 30 циклов 94°С в течение 1 мин, 50°С в течение 1 мин и 72°С в течение 1 мин и затем удлинения (элонгации) при 72°С в течение 5 мин. Для конструирования scFvMEL-bax scFvMEL амплифицировали с использованием праймеров: NcoIzme (5'→3'): GGTGGCGGTGGCTCCATGGCGGACATTGTGATGACCCAGTCTCAAAAATTC (SEQ ID NO:31) и Czme (5'→3'): CGTCGGAGCCACCGCCACCGCTAGCTGAGGAGACGGTGAGAGT (SEQ ID NO:32), фрагмент Вах амплифицировали с использованием праймеров: Nbax2 (5'→3'): GGTGGCGGTGGCTCCGACGGGTCCGGGGAGCAG (SEQ ID NO:33) и Cbax (5'→3'): GGAGCCACCGCCACCCTCGAGCTATCAGCCCATCTTCTTCCAGAT (SEQ ID NO:34). Для конструирования bax-scFvMEL праймерами являются Nbax (5'→3'): GGTGGCGGTGGCTCCATGGACGGGTCCGGGGAGCAG (SEQ ID NO:35), Cbax2-1 (5'→3'): GTCCGTGGAGCCACCGCCACCGCTAGCGCCCATCTTCTTCCA (SEQ ID NO:36), Nzme2 (5'→3'): GGTGGCGGTGGCTCCACGGACATTGTGATGACCCAGTCTCAAAAATTC (SEQ ID NO:37 и Czme2 (5'→3'): GGAGCCACCGCCACCCTCGAGCTATCATGAGGAGACGGTGAGAGTGGT (SEQ ID NO:38). Для конструирования конструкции scFvMEL-Вах1-5 праймерами являются NcoIzme, Czme, Nbax2 и CxhoIbax345 (5'→3'): GGAGCCACCGCCACCCTCGAGCTATCACCAACCACCCTGGTC (SEQ ID NO:39). Для конструирования слитой конструкции Вах1-5-scFvMEL праймерами являются Nbax, Cbax345 (5'→3'): GGAGCCACCGCCACCCCAACCACCCTGGTC (SEQ ID NO:40), Nzme2 и Czme2. Для конструирования слияния scFvMEL-Вах345 праймерами являются NcoIzme, Праймер 3 (5'→3'): CCGGAGCCACCGCCACCGCTAGCTGAGGAGACTGTGA (SEQ ID NO:41), Nbax345 (5'→3'): GGTGGCGGTGGCTCCTTCATCCAGGATCGAG (SEQ ID NO:42) и CxhoIbax345. Для конструирования Вах345-scFvMEL используют NcoIВах345 (5'→3'): GGTGGCGGTGGCTCCATGGTCATCCAGGATCGAG (SEQ ID NO:43), Cbax345, Nzme2 и Czme2.
Затем проводили вторую ПЦР с использованием 30 циклов 94°С в течение 1 мин, 50°С в течение 1 мин и 72°С в течение 1 мин, затем удлинение при 72°С в течение 5 мин. Амплифицированные фрагменты разделяли электрофорезом на 1% агарозном геле, очищали с использованием набора ПЦР для очистки (Qiagen; Valencia, CA). Очищенные продукты ПЦР расщепляли при помощи NcoI и XhoI при 37°С в течение 3 часов и затем разделяли электрофорезом на 1% агарозном геле, очищали из гелей с использованием набора geneclean II (Quantium Biotechnologies, Inc., Carlsbad, CA). Очищенные фрагменты гена клонировали в вектор рЕТ32а(+), эту смесь для лигирования трансформировали в компетентные клетки DH5α, подвергали скринингу на положительные клоны с использованием ПЦР и затем секвенировали. Клоны с правильной последовательностью рамки считывания трансформировали в компетентные клетки AD494 (DE3)pLysS для дополнительной индукции и экспрессии.
Для экспрессии полноразмерного белка Вах и белка Вах-scFvMEL, последовательности Вах и Вах-scFvMEL субклонировали в вектор pBAD/His A и полученные векторы обозначали pBAD/Hisbax и pBAD/Hisbax-scFvMEL, соответственно. Вкратце, полноразмерные гены Вах и Вах-scFvMEL амплифицировали при помощи способа ПЦР. Для амплификации полноразмерного Вах в качестве матрицы использовали ВахТА-35, а праймерами были NBAXHIS (5'→3'): AAACATGCCATGGCTCACCACCACCACCACCACGACGGGTCCGGGGAGCAGCCCAGA (SEQ ID NO:44) и Cbax. Для амплификации Вах-scFvMEL в качестве матрицы использовали рЕТ32-Вах-scFvMEL (клон 2), а праймерами были NBAXHIS и Czme2. Праймер NBAXHIS был сконструирован следующим образом: сайт NcoI для клонирования, полигистидин (6 х His) для очистки и детектирования на N-конце, за которым следует инициирующий ATG; на С-конце в праймерах Cbax и Czme2 были добавлены два стоп-кодона и сайт XhoI. Очищенные фрагменты ПЦР расщепляли NcoI и XhoI и очищали с использованием набора gene clean, с последующим лигированием в те же самые расщепленные рестриктазы для вектора pBAD/His А. Смесь для лигирования трансформировали в DH5α и положительные клоны подвергали скринингу с использованием скрининга ПЦР, выделяли ДНК и эту последовательность проверяли. Положительные клоны с подтвержденной последовательностью трансформировали в компетентные клетки LMG194 для экспрессии.
ПРИМЕР 21
ИНДУКЦИЯ И ЭКСПРЕССИЯ РОДСТВЕННЫХ ВАХ СЛИТЫХ БЕЛКОВ В E. coli
Бактериальные колонии, трансформированные сконструированной плазмидой, выращивали в среде для выращивания бульоне Луриа (LB), содержащем 200 мкг/мл ампициллина, 70 мкг/мл хлорамфеникола и 15 мкг/мл канамицина, при 37°С в течение ночи при встряхивании при 24 об/мин. Затем культуры разбавляли 1:100 в свежей среде LB плюс антибиотики и выращивали до А600 = 0,6 при 37°С, после чего индуцировали добавлением IPTG до конечной концентрации 80 мкМ при 37°С в течение 2 часов. Клетки собирали, ресуспендировали в 10 мМ Трисе (рН 8,0) и хранили в замороженном виде при -80°С для дополнительной очистки.
ПРИМЕР 22
ИНДУКЦИЯ И ЭКСПРЕССИЯ ПОЛНОРАЗМЕРНЫХ ВАХ И ВАХ-scFvMEL
Бактериальные колонии, трансформированные плазмидой pBAD/Hisbax и pBAD/Hisbax-scFvMEL, выращивали в среде RM, содержащей глюкозу и 100 мкг/мл ампициллина, при 37°С при встряхивании (225 об/мин). Затем культуры разбавляли 1:100 в свежей среде RM, содержащей глюкозу и 100 мкг/мл ампициллина, и выращивали при 37°С при встряхивании до макимальной OD600, затем индуцировали добавлением 5% арабинозы при 37°С в течение 4 часов. Клетки собирали, ресуспендировали в 10 мМ Трисе (рН 8,0) и хранили в замороженном виде при -80°С для дополнительной очистки.
ПРИМЕР 23
ОЧИСТКА РОДСТВЕННЫХ ВАХ СЛИТЫХ БЕЛКОВ
Повторно приготовленную суспензию лизировали добавлением лизоцима до конечной концентрации 100 мкг/мл галтовкой в течение 30 мин при 4°С с последующей обработкой ультразвуком. Экстракты центрифугировали при 10800 g в течение 30 мин и супернатант дополнительно центрифугировали при 40000 об/мин в течение 1 часа. Супернатант, содержащий только растворимый белок, доводили до 40 мМ Триса, рН 8,0, 10 мМ имидазола и наносили на Nickel-NTA-агарозу, уравновешенную тем же самым буфером. После промывания Nickel-NTA-агарозы 500 мМ NaCl, 20 мМ имидазолом связанные белки элюировали 500 мМ NaCl, 500 мМ имидазолом. Для определения меченых полигистидином белков проводили анализ оптической плотности (280 нм) и анализ электрофорезом в ДСН-ПААГ. Конечные белки получали добавлением рекомбинантной бычьей энтерокиназы (rEK) для удаления полигистидиновой метки в соответствии с инструкциями изготовителя (1 Е rEK расщепляла 50 мкг белка при инкубировании при комнатной температуре в течение 16 часов). Затем rEK удаляли улавливающей ЕК агарозой. Конечные белки анализировали электрофорезом в ДСН-ПААГ и хранили при 4°С.
ПРИМЕР 24
ЭЛЕКТРОФОРЕЗ В ДСН-ПААГ И ВЕСТЕРН-БЛОТ-АНАЛИЗ
Пробы белков анализировали электрофорезом на 8,5% или 12% ДМН-ПААГ при восстанавливающих условиях. Гели окрашивали Кумасси синим. Для вестерн-блоттинг-анализа белки переносили из гелей в нитроцеллюлозные мембраны. Мембраны блокировали 5% обезжиренным молоком и инкубировали в течение 1 часа при комнатной температуре или в течение ночи при 4°С с кроличьим анти-scFvMEL-антителом (разведение 1:2000) или кроличьим анти-bax-антителом (разведение 1:1000). После промывания мембраны инкубировали с конъюгатом козье антикроличье антитело/пероксидаза хрена (HRP-GAR, разведение 1:5000). После дополнительного промывания мембрану проявляли с использованием системы детектирования ECL Amersham и экспонировали на рентгеновской пленке.
ПРИМЕР 25
ДЕТЕКТИРОВАНИЕ scFvMEL-ЧАСТИ МОЛЕКУЛЫ РОДСТВЕННЫХ ВАХ
СЛИТЫХ БЕЛКОВ
Для детектирования scFvMEL-части родственных Вах слитых белков использовали планшеты, покрытые белком L Reacti-Bind TM из PIERCE (Rockford, IL), или 96-луночные планшеты, содержащие прикрепленные клетки А375-М меланомы человека, на основе ELISA. Вкратце, предварительно нанесенный белок L блокировали 5% БСА и затем подвергали взаимодействию с scFvMEL-bax-родственными белками при различных концентрациях. После промывания их инкубировали с кроличьим антителом против scFvZME, затем инкубировали с HRP-GAR (разведение 1:5000) и затем добавляли раствор субстрата 2,2'-азино-бис-3-этилбензтиазолин-6-сульфоновой кислоты (ABTS) с 1 мкг/мл 30% Н2О2. Спустя 30 минут измеряли оптическую плотность при 405 нм.
ПРИМЕР 26
СПОСОБЫ - АНАЛИЗЫ ЦИТОТОКСИЧНОСТИ in vitro
Клетки А375-М меланомы человека, культивируемые в среде МЕМ с 10% ФТС, высевали в 96-луночные планшеты при плотности 2,5 х 103 клеток на лунку и давали им прикрепляться в течение 24 часов при 37°С в 5% СО2. Спустя 24 часа эту среду заменяли средой, содержащей различные концентрации различных слитых белков родственных scFvMEL-bax. Спустя 72 часа действие этих слитых белков на рост клеток в культуре определяли с использованием окрашивания красителем кристаллическим фиолетовым (кристаллвиолетом). Планшеты считывали микропланшет-ридером при 540 нм.
ПРИМЕР 27
ЦИТОТОКСИЧНОСТЬ ХИМЕРНЫХ ПОЛИПЕПТИДОВ ВАХ
Как показано на фиг.11, ген bax человека клонировали при помощи ПЦР из клеток Namalwa. Представлен электрофорез в 1% агарозном геле, демонстрирующий кДНК Вах человека, синтезированную из клеток Namalwa при помощи ОТ-ПЦР.Фиг.12А и 12В иллюстрируют конструирование scFvMEL-Вах-родственных слитых белков. Ген Вах состоит из шести экзонов, и этот ген продуцирует альтернативные транскрипты, преобладающая форма которых кодирует мРНК 1,0 т.п.н. и транскрибирует белок массой 21 кДа, который был назван Вах α. Блоки указывают экзоны, идентифицируемые цифрами. Экзон 6 является трансмембранным доменом. Гены scFvMEL сливали с Вах, укороченым Вах1-5 или Вах345 с линкером G4S в двух различных ориентациях, обозначенных scFvMEL-bax, Вах-scFvMEL, scFvMEL-Вах1-5, Вах1-5-scFvMEL, scFvMEL-Вах345 или Вах 345-scFvMEL. Слитые гены клонировали в экспрессирующий вектор рЕТ32а(+) в сайтах NcoI и XhoI. Затем эту плазмиду, содержащую слитые гены, трансформировали в AD494(DE3)pLysS E. coli для экспрессии.
Фиг.13 демонстрирует вестерн-блот-анализ, иллюстрирующий экспрессию scFvMEL-Вах-родственных слитых белков в векторе рЕТ32а(+). Электрофорез в ДСН-ПААГ и окрашивание Кумасси синим укороченных bax-родственных слитых белков имели место при восстанавливающих условиях. Эти результаты показали индуцированную экспрессию слитых белков scFvMEL и укороченного bax в экспрессирующем векторе рЕТ32а(+). Индуцированные полосы были при массе ˜62 кДа для scFvMEL-bax345 и bax345-scFvMEL и были при массе ˜65 кДа для scFvMEL-Вах1-5 и Вах1-5-scFvMEL, соответственно, которые содержали также метку для очистки ˜20 кДа.
Фиг.14 показывает экспрессию рЕТ32-scFvMEL-bax и рЕТ32-Вах-scFvMEL, трансформированных в AD494(DE3)pLysS E. coli и при индукции IPTG. Полноразмерные слитые белки bax являются очень токсичными в отношении бактерий вследствие высокогидрофобного домена-экзона 6. Бактерии, содержащие плазмиду рЕТ32-scFvMEL-Вах или рЕТ32-Вах-scFv, выращивали в среде LB, содержащей 200 мкг/мл ампициллина, 70 мкг/мл хлорамфеникола и 15 мкг/мл канамицина, до OD600 = 0,6, индуцировали добавлением IPTG до конечной концентрации 0,1 мМ, и бактерии погибали, как показано уменьшением величины OD600.
Фиг.15 демонстрирует экспрессию полноразмерных bax и Вах-scFvMEL в трансформированной вектором pBAD/HisA LMG194-E. coli. Полноразмерный ген bax и ген Вах-scFvMEL клонировали в вектор pDAD/His A в сайтах NcoI и XhoI. После инициирующего ATG следовали полигистидиновые (6 His) метки. Плазмиды, содержащие либо Вах, либо Вах-scFvMEL, трансформировали в клетки LMG194 для экспрессии и экспрессию белков Вах и Вах-scFvMEL испытывали в различных бактериальных средах для выращивания (RM, содержащей глюкозу и 100 мкг/мл ампициллина, RM, содержащей 100 мкг/мл ампициллина, LB, содержащей 100 мкг/мл ампициллина). LMG194, трансформированные pBAD/HisLacZ, использовали в качестве положительного контроля, с полосой экспрессии при массе ˜120 кДа, представляющей белок LacZ, который мог детектироваться анти-bax-антителом. LMG194, трансформированные pBAD/HisA (пустым вектором), использовали в качестве отрицательного контроля. Вестерн-блоттинг выполняли с использованием кроличьего анти-Вах-моноклонального антитела (разведение 1:1000) в качестве первичного антитела и конъюгата HRP-козье антитело против кроличьего IgG в качестве вторичного антитела. Результаты показали, что полосы при массе ˜21 кДа представляют Вах с 6Х His-меткой, а полосы при массе ˜40 кДа представляют Вах-scFvMEL с 6Х His-меткой.
На фиг.16А и 16В показана связывающая активность scFvMEL-части молекулы слитых белков. Клетки А375-М являются gp240-антиген-положительными линиями клеток меланомы человека. Белок L имеет уникальную способность связывать scFv. Слитый белок Вах345-scFvMEL мог связываться либо с А375-М (фиг.16А), либо с Белком L (фиг.16В), детектируемыми анти-scFvzme-антителом. Эта связывающая активность является той же самой, что и активность другого слитого белка scFvMEL-TNF.
Фиг.17 демонстрирует цитотоксичность scFvMEL-bax345 в сравнении с Вах345-scFvMEL на клетках А375-М. Оценивали цитотоксические действия scFvMEL-bax345 и Вах345-scFvMEL в отношении клеток А375-М меланомы человека, положительных по антигену log-фазы. Клетки А375-М помещали в 96-луночные планшеты (2,5 х 103 клеток на лунку). IC50-концентрации scFvMEL-bax345 и Вах345-scFvMEL были приблизительно 3 нМ и 10 нМ, соответственно.
ПРИМЕР 28
ELISA СЛИТОГО БЕЛКА ГРАНЗИМ В-vegf121 НА РАЗЛИЧНЫХ КЛЕТОЧНЫХ ЛИНИЯХ (ДЕТЕКТИРОВАНИЕ МЫШИНЫМ АНТИТЕЛОМ ПРОТИВ vegf121)
В ELISA-анализе связывающей активности использовали 96-луночные планшеты, содержащие прикрепленные клетки РАЕ/flk-1, PAE/flt-1, A375-M или SKBR3-HP, которые блокировали 5% БСА и затем давали им взаимодействовать с очищенным слитым белком Гранзим В-vegf121 в различных концентрациях. После промывания их инкубировали с мышиным анти-vegf121-антителом, затем с YRP-меченым козьим антителом против мышиного IgG. Затем добавляли в качестве субстрата раствор 2,2'-азино-бис-3-этилбензтиазолин-6-сульфоновой кислоты (ABTS) с 1 мкл/мл 30% Н2О2. Спустя 30 минут измеряли оптическую плотность при 405 нм.
VEGF121 может специфически связываться с рецептором flk-1/KDR на васкулярных эндотелиальных клетках (фиг.18). Этот эксперимент показал, что слитый белок Гранзим В-vegf121 специфически связывается с клетками РАЕ/flk-1, которые сверхэкспрессируют рецепторы flk-1, но связывающих активностей не наблюдали на клетках РАЕ/flt-1 (сверхэкспрессируемых рецепторах flt-1) или клетках А375-М меланомы человека и клетках SKBR3-HP злокачественной опухоли молочной железы человека. То есть, GrB/VEGF121 может специфически связываться с клетками РАЕ/flk-1, которые сверхэкспрессируют рецептор flk-1/KDR, при детектировании либо антителом против VEGF121, либо антителом против GrB.
ПРИМЕР 29
ЦИТОТОКСИЧНОСТЬ ГРАНЗИМ В/VEGF121 НА ТРАНСФИЦИРОВАННЫХ ЭНДОТЕЛИАЛЬНЫХ КЛЕТКАХ
Цитотоксичность Гранзим В-VEGF121 против клеток РАЕ/flk-1 в сравнении с клетками РАЕ/flt-1 оценивали против РАЕ/flk-1 (трансфицированных рецептором flk-1/KDR) и РАЕ/flt-1 log-фазы (трансфицированных рецептором flt-1) в культуре. Вкратце, клетки РАЕ высевали в 96-луночные планшеты при плотности 2,5 х 103 клеток на лунку. Спустя 24 часа клетки обрабатывали средой, содержащей различные концентрации Гранзим В-VEGF121. Действие на рост клеток определяли с использованием ХТТ после 72 часов. Планшеты считывали на микропланшет-ридере ELISA при 540 нм.
Результаты показали, что 50% ингибирующее рост действие (IC50) было обнаружено при концентрации ˜10 нМ на клетках РАЕ/flk-1 (фиг.19). Однако на клетках РАЕ/flt-1 не были обнаружены цитотоксические эффекты. Цитотоксичность Гранзим В-VEGF121 на клетках РАЕ/flk-1 указывает на то, что VEGF121-часть молекулы этого слитого белка специфически связана со сверхэкспрессией flk-1 на клеточной поверхности с последующей доставкой гранзима В во внутреннее пространство клеток-мишеней и цитотоксичностью в отношении этих клеток-мишеней.
ПРИМЕР 30
АНАЛИЗ ЦИТОТОКСИЧНОСТИ ГРАНЗИМ В-VEGF121 В СРАВНЕНИИ С VEGF121-RGEL in vitro ПРОТИВ PAE/FLK-1
Клетки РАЕ в среде Ham F-12 с 10% ФТС высевали в 96-луночные планшеты при плотности 2,5 х 103 клеток на лунку и давали им прикрепляться в течение 24 часов при 37°С в 5% СО2. Спустя 24 часа эту среду заменяли средой, содержащей различные концентрации Гранзим В-VEGF121 или VEGF121-rGel. Действие Гранзим В-VEGF121 или VEGF121-rGel на рост клеток определяли с использованием ХТТ после 72 часов. Планшеты считывали на микропланшет-ридере ELISA при 540 нм.
Результаты показали, что IC50 VEGF121-rGel была приблизительно 1 нМ (фиг.20). IC50 слитого белка Гранзим В-VEGF121 была в 10 раз более высокой, чем IC50 VEGF121-rGel.
ПРИМЕР 31
АКТИВНОСТЬ КАСПАЗЫ НА КЛЕТКАХ РАЕ, ОБРАБОТАННЫХ СЛИТЫМ БЕЛКОМ ГРАНЗИМ В-VEGF121
Вестерн-блоттинг активации каспазы проводили с 30 мкг лизатов целых клеток. После электрофореза в ДСН-ПААГ белки электрофоретически переносили на нитроцеллюлозные мембраны. Мембраны блокировали забуференным фосфатом солевым раствором с 0,5% Твином 20 (ЗФРТ), содержащим 5% обезжиренное молоко, и затем подвергали действию антител против каспазы-8, каспазы-3, каспазы-6, каспазы-7, расщепленной каспазы-3, PARP или расщепленного DFF45, соответственно. Мембраны промывали ЗФРТ и обрабатывали вторичными антителами, конъюгированными с пероксидазой хрена. Реакцию антиген-антитело визуализировали анализом усиленной хемилюминесценции (ECL) с использованием проявляющих реагентов ECL Amersham и экспонирования на пленке.
Эти результаты показали, что после 4 часов обработки слитым белком Гранзим В-VEGF121 расщепленную каспазу-8, расщепленную каспазу-3, расщепленный PARP и расщепленный DFF45 наблюдали на клетках РАЕ/flk-1, но не на клетках РАЕ/flt-1 (фиг.21). Однако каспаза-6 или каспаза-7 не расщеплялись Гранзим В-VEGF121, что свидетельствовало о том, что Гранзим В-VEGF121 активировал только каспазы, участвующие в каспаза-8-, каспаза-3-пути апоптоза.
ПРИМЕР 32
ОБНАРУЖЕНИЕ ГИБЕЛИ КЛЕТОК in situ (TUNEL)
Расщепление геномной ДНК во время апоптоза может давать двухцепочечные низкомолекулярные ДНК-фрагменты, а также разрывы одной цепи (ники) в высокомолекулярной ДНК. Эти разрывы цепи ДНК могут быть идентифицированы мечением свободных 3'-ОН-концов модифицированными нуклеотидами в ферментативной реакции. Способ TUNEL (опосредованное трансферазой dUTP-мечение ник-концов) использует терминальную дезоксинуклеотидилтрансферазу (TdT), которая катализирует полимеризацию нуклеотидов к свободным 3'-ОН концам ДНК зависимым от матрицы образом, для мечения разрывов цепи ДНК. Включенный флуоресцеин детектируют Fab-фрагментами анти-флуоресцеин-антитела из овцы, конъюгированными со щелочной фосфатазой (АР). После реакции с субстратом окрашенные клетки могут анализироваться под световым микроскопом.
Для обработок клеток клетки высевали на предметные стекла с 16 лунками, 2500 клеток на лунку, и инкубировали в течение ночи в условиях 37°С/5% СО2. Клетки обрабатывали слитым белком GrB-scFvMEL или GrB-vegf121 при концентрации IC50 в течение различных периодов времени (24, 48 ч и т.д.) и кратковременно промывали ЗФР.
Для TUNEL-анализа клетки фиксировали 3,7% формальдегидом при комнатной температуре в течение 20 минут, после промывания ЗФР, обрабатывали для увеличения проницаемости 0,1% Тритоном Х-100, 0,1% цитратом натрия на льду в течение 2 минут и затем промывали дважды ЗФР. Клетки инкубировали с рекционной смесью TUNEL при 37°С в течение 60 минут с последующим инкубированием с Converter-AP при 37°С в течение 30 минут и наконец давали им реагировать с раствором Fast Red (прочный красный)-субстрата при комнатной температуре в течение 10 минут. После конечной стадии промывания предметные стекла заключали в среду для заливки и анализировали под световым микроскопом.
В каждую экспериментальную схему включали положительные контроли. Фиксированные и обработанные для повышения проницаемости клетки инкубировали с 1 мг/мл ДНКазы I в течение 10 минут при 37°С для индукции разрывов цепи ДНК.
Действие GrB-scFvMEL на клетки А375-М и SKBR3-HP определяли детектированием гибели клеток in situ (TUNEL). Положительные результаты TUNEL наблюдали в отношении обработанных GrB-scFvMEL антиген-положительных клеток А375-М меланомы человека при 48 часах, но не в отношении обработанных GrB-scFvMEL антиген-отрицательных клеток SKBR3-HP злокачественной опухоли молочной железы человека. Это указывает на то, что GrB-scFvMEL мог специфически нацеливать на антиген-положительные клетки меланомы и индуцировать апоптоз клеток.
Действие GrB-vegf121 на клетках РАЕ/Flk-1 в сравнении с клетками РАЕ/Flt-1 определяли при помощи TUNEL. VEGR2/KDR сверхэкспрессировался на поверхности клеток РАЕ/Flk-1, но не РАЕ/Flt-1. VEGF121 специфически нацеливали на VEGR2/KDR, доставляя GrB в клетки РАЕ/Flk-1. Положительные результаты анализа TUNEL наблюдали в отношении обработанных GrB-vegf121 клеток РАЕ/Flk-1 при 24 часах и 48 часах, но не в отношении обработанных GrB-vegf121 клеток РАЕ/Flt-1. GrB-vegf121 индуцировал апоптоз клеток РАЕ/Flk-1.
ПРИМЕР 33
ИНТЕРНАЛИЗАЦИЯ GRB/VEGF121 В КЛЕТКИ РАЕ, НАБЛЮДАЕМАЯ ПРИ ПОМОЩИ ИММУНОФЛУОРЕСЦЕНТНОЙ МИКРОСКОПИИ
Этот пример описывает анализ интернализации GrB/VEGF121 при помощи иммунофлуоресцентной микроскопии. Клетки обрабатывали следующим образом. Клетки засевали в предметные стекла с 16 лунками (Nunc), 1 х 104 клеток на лунку, инкубировали в течение ночи в условиях 37°С/5% СО2. Клетки обрабатывали 200 нМ GrB/VEGF121 в течение 4 часов и затем промывали кратковременно ЗФР. Поверхность клеток очищали инкубированиями в течение 10 минут с глициновым буфером (500 мМ NaCl, 0,1 М глицин, рН 2,5), нейтрализовали в течение 2 мин 0,5 М Трисом, рН 7,4, и кратковременно промывали ЗФР.
Иммунофлуоресцентное окрашивание проводили следующим образом. Клетки фиксировали в 3,7% формальдегиде в течение 15 мин при комнатной температуре с последующим кратковременным промыванием ЗФР и затем обработкой для увеличения проницаемости в течение 10 мин в ЗФР, содержащем 0,2% Тритон Х-100. Затем их промывали три раза ЗФР. Пробы инкубировали с 3% БСА в течение 1 часа при комнатной температуре для блокирования сайтов неспецифического связывания и затем инкубировали с мышиным анти-гранзим В-антителом (разведение 1:100) при комнатной температуре в течение 1 часа с последующим промыванием три раза ЗФР. Пробы инкубировали со связанным с флуоресцеинизотиоцианатом (FITC) антимышиным IgG (Sigma) (разведение 1:100) при комнатной температуре в течение 1 часа и затем промывали три раза ЗФР. Стенки и перегородки осторожно удаляли. После высушивания на воздухе препарат заливали заливочной средой и анализировали под флуоресцентным микроскопом.
Результаты показали, что GrB-часть GrB/VEGF121 доставлялась в цитозоль клеток РАЕ/flk-1, но не в цитозоль клеток РАЕ/flt-1 после 4 часов обработки.
ПРИМЕР 34
GrB/VEGF121 ИНДУЦИРУЕТ АПОПТОЗ НА КЛЕТКАХ РАЕ/FLK-1, ДЕТЕКТИРУЕМЫЙ АНАЛИЗОМ TUNEL
Расщепление геномной ДНК во время апоптоза может давать двухцепочечные низкомолекулярные ДНК-фрагменты, а также разрывы одной цепи (ники) в высокомолекулярной ДНК. Эти разрывы цепи ДНК могут быть идентифицированы мечением свободных 3'-ОН-концов модифицированными нуклеотидами в ферментативной реакции. Способ TUNEL (опосредованное трансферазой dUTP-мечение ник-концов) использует терминальную дезоксинуклеотидилтрансферазу (TdT), которая катализирует полимеризацию нуклеотидов к свободным 3'-ОН концам ДНК зависимым от матрицы образом для мечения разрывов цепи ДНК. Включенный флуоресцеин детектируют Fab-фрагментами анти-флуоресцеин-антитела из овцы, конъюгированными со щелочной фосфатазой (АР). После реакции с субстратом окрашенные клетки могут анализироваться под световым микроскопом.
Обработки клеток были следующими. Клетки высевали на предметные стекла с 16 лунками, 2500 клеток на лунку, и инкубировали в течение ночи в условиях 37°С/5% СО2. Клетки обрабатывали слитым белком GrB-vegf121 при концентрации IC50 в течение различных периодов времени (24, 48 ч и т.д.) и кратковременно промывали ЗФР.
TUNEL-анализ проводили следующим образом. Клетки фиксировали 3,7% формальдегидом при комнатной температуре в течение 20 минут с последующим промыванием ЗФР и обработкой для увеличения проницаемости 0,1% Тритоном Х-100, 0,1% цитратом натрия на льду в течение 2 минут. Затем их промывали дважды ЗФР. Клетки инкубировали с рекционной смесью TUNEL при 37°С в течение 60 минут с последующим инкубированием с Converter-AP при 37°С в течение 30 минут и наконец давали им реагировать с раствором Fast Red (прочный красный)-субстрата при комнатной температуре в течение 10 минут. После конечной стадии промывания предметные стекла заключали в среду для заливки и анализировали под световым микроскопом.
Результаты показали, что VEGFR2/KDR сверхэкспрессировался на поверхности клеток РАЕ/Flk-1, но не клеток РАЕ/Flt-1. VEGF121 специфически нацеливал на VEGFR2/KDR, доставляя GrB в клетки РАЕ/Flk-1. Положительные результаты TUNEL демонстрировали для обработанных GrB/VEGF121 клеток РАЕ/Flk-1 при 24 часах и 48 часах, но не на обработанных GrB/VEGF121 клетках РАЕ/Flt-1. Таким образом,GrB/VEGF121 индуцировала апоптоз PAE/Flk-1.
ПРИМЕР 35
ВЫСВОБОЖДЕНИЕ ЦИТОХРОМА С КЛЕТОК РАЕ, ОБРАБОТАННЫХ GrB/VEGF121
Цитохром с играет важную роль в апоптозе. Этот белок локализован в пространстве между внутренней и наружной мембранами митохондрий. Стимул апоптоза запускает высвобождение цитохрома с из митохондрий в цитозоль, где он связывается с Apaf-1. Комплекс цитохром с/Apaf-1 активирует каспазу-9, которая затем активирует каспазу-3 и другие находящиеся ниже по ходу процесса каспазы.
Материалы и способы для анализа апоптоза по высвобождению цитохрома с (из Oncogene Research Products; San Diego, CA) были следующими. Клетки РАЕ/flk-1 и РАЕ/flt-1 (5 х 107) обрабатывали GrB/VEGF121 в концентрациях 0,1 нМ и 20 нМ в течение 24 часов. Клетки собирали. После промывания клеток 10 мл охлажденного на льду ЗФР клетки ресуспендировали с 0,5 мл 1 х буферной смеси для экстракции цитозоля, содержащей ДТТ и ингибиторы протеаз, и инкубировали на льду в течение 10 минут. Клетки гомогенизировали в охлаждаемом льдом стеклянном гомогенизаторе. Гомогенат переносили в микроцентрифужную пробирку на 1,5 мл и центрифугировали при 700 g в течение 10 мин при 4°С. Супернатант переносили в свежую пробирку на 1,5 мл и центрифугировали при 10000 х g в течение 30 мин при 4°С. Супернатант собирали в качестве цитозольной фракции. Осадки ресуспендировали в 0,1 мл буферной смеси для экстракции митохондрий, содержащей ДТТ и ингибиторы протеаз, встряхивали на вортексе в течение 10 секунд и хранили в качестве митохондриальной фракции. Концентрации белка определяли с использованием способа Бредфорда для определения белка Bio-Rad Laboratories, Inc. (Hercules, CA). 10 мкг каждой цитозольной и митохондриальной фракции, выделенных из необработанных и обработанных клеток, наносили на 15% ДСН-ПААГ для электрофореза с последующей стандартной процедурой Вестерн-блоттинга и зондированием антителом против цитохрома с (1 мкг/мл).
Для анализа апоптоза по высвобождению цитохрома с клетки РАЕ обрабатывали GrB/VEGF121 при различных концентрациях в течение 24 часов. Высокообогащенную митохондриальную фракцию выделяли из цитозоля. Перемещение цитохрома с из митохондрий в цитозоль во время апоптоза определяли вестерн-блоттингом с использованием антитела против цитохрома с. Фиг.22 показывает высвобождение цитохрома с на клетках РАЕ/flk-1, но не на клетках РАЕ/flt-1 после обработки GrB/VEGF121.
ПРИМЕР 36
ПЕРЕМЕЩЕНИЕ ВАХ КЛЕТОК РАЕ ПОСЛЕ ОБРАБОТКИ GrB/VEGF121
Вах, белок массой 21 кДа с высокой гомологией аминокислот с Bcl-2, вариабельно экспрессируется различными клетками. Вах как проапоптозный член семейства Bcl-2 обнаруживал некоторые структурные сходства с порообразующими белками. Таким образом, считают, что Вах может образовывать трансмембранные поры через наружную митохондриальную мембрану, что приводит к потере мембранного потенциала. Было показано, что локализация Вах меняется от цитозольной к митохондриальной после получения апоптозного стимула.
Выделение цитозольной фракции и митохондриальной фракции выполняют с использованием набора Oncogene Research Products (Cat # QIA87; San Diego, CA). Концентрации белка определяли с использованием способа Бредфорда для определения белка Bio-Rad Laboratories, Inc. (Hercules, CA). 10 мкг каждой цитозольной и митохондриальной фракции, выделенных из необработанных и обработанных клеток, наносили на 12% ДСН-ПААГ для электрофореза и затем выполняли стандартную процедуру Вестерн-блоттинга и зондировали с использованием антитела против Вах (Santa Cruz Biotechnology, Inc.; Santa Cruz, CA; разведение 1:200).
Вестерн-блоттинг-анализ выполнял экспрессию Вах на клетках РАЕ после обработки GrB/VEGF121 в течение 24 часов. Результаты показали, что локализация Вах менялась от цитозоля к митохондриям на клетках РАЕ/flk-1, но не на клетках РАЕ/flt-1 после обработки GrB/VEGF121 в концентрации 20 нМ (IC50). Фиг.23 показывает, что Вах увеличивался в митохондриях и уменьшался в цитозоле на клетках РАЕ/flk-1 после обработки GrB/VEGF121 в течение 24 часов при концентрации 20 нМ, что указывает на то, что Вах перемещался из цитозоля в митохондрии во время апоптоза.
ПРИМЕР 37
ИНТЕРНАЛИЗАЦИЯ GrB/scFvMEL В КЛЕТКИ А375-М, НАБЛЮДАЕМАЯ ПРИ ПОМОЩИ ИММУНОФЛУОРЕСЦЕНТНОЙ МИКРОСКОПИИ
Анализ интернализации GrB/scFvMEL иммунофлуоресцентной микроскопией выполняли с использованием следующих способов. Клетки засевали в предметные стекла с 16 лунками (Nunc), 1 х 104 клеток на лунку, и инкубировали в течение ночи в условиях 37°С/5% СО2. Клетки обрабатывали 100 нМ GrB/scFvMEL в течение 1 часа и 6 часов и затем промывали кратковременно ЗФР. Поверхность клеток очищали инкубированиями в течение 10 минут с глициновым буфером (500 мМ NaCl, 0,1 М глицин, рН 2,5), нейтрализовали в течение 2 мин 0,5 М Трисом, рН 7,4 и кратковременно промывали ЗФР.
Для иммунофлуоресцентного окрашивания клетки фиксировали в 3,7% формальдегиде в течение 15 мин при комнатной температуре с последующим кратковременным промыванием ЗФР и затем обработкой для увеличения проницаемости в течение 10 мин в ЗФР, содержащем 0,2% Тритон Х-100; затем эти клетки промывали три раза ЗФР. Пробы инкубировали с 3% БСА в течение 1 часа при комнатной температуре для блокирования сайтов неспецифического связывания и затем инкубировали с мышиным анти-гранзим В-антителом (разведение 1:100) при комнатной температуре в течение 1 часа с последующим промыванием три раза ЗФР. Пробы инкубировали со связанным с флуоресцеинизотиоцианатом (FITC) антимышиным IgG (Sigma) (разведение 1:100) при комнатной температуре в течение 1 часа и затем промывали три раза ЗФР. Стенки и перегородки осторожно удаляли. После высушивания на воздухе препарат заливали заливочной средой и анализировали под флуоресцентным микроскопом.
Результаты показали, что GrB-часть GrB/scFvMEL доставлялась в gp240-антиген-положительные клетки А375-М связыванием scFvMEL с антигеном gp240.
ПРИМЕР 38
GrB/scFvMEL ИНДУЦИРУЕТ АПОПТОЗ НА КЛЕТКАХ А375-М, ДЕТЕКТИРУЕМЫЙ АНАЛИЗОМ TUNEL
Клетки (3000 клеток на лунку) обрабатывали GrB/scFvMEL при концентрации IC50 в течение различных периодов времени (16 часов, 24 часа) и кратковременно промывали ЗФР. Клетки фиксировали 3,7% формальдегидом при комнатной температуре в течение 20 минут с последующим промыванием ЗФР и обрабаткой для увеличения проницаемости 0,1% Тритоном Х-100, 0,1% цитратом натрия на льду в течение 2 минут. Затем их промывали дважды ЗФР. Клетки инкубировали с рекционной смесью TUNEL при 37°С в течение 60 минут. После конечной стадии промывания клетки анализировали под флуоресцентным микроскопом.
Результаты показали, что GrB/scFvMEL индуцировал апоптоз на антиген-положительных клетках А375-М, но не на антиген-отрицательных клетках SKBR3-HP после обработки в течение 16 часов и 24 часов.
ПРИМЕР 39
ВЫСВОБОЖДЕНИЕ ЦИТОХРОМА С В ОБРАБОТАННЫХ GrB/scFvMEL КЛЕТКАХ А375-М, В СРАВНЕНИИ С КЛЕТКАМИ SKBR3-HP
Анализ апоптоза по высвобождению цитохрома выполняли, как описано (Oncogene Research Products; San Diego, CA; Cat # QIA87). Материалы и способы были следующими. Клетки А375-М меланомы человека и клетки злокачественной опухоли молочной железы человека SKBR3-HP (5 х 107) обрабатывали GrB/scFvMEL в концентрациях 5 нМ и 50 нМ в течение 24 часов. Клетки собирали. После промывания клеток 10 мл охлажденного на льду ЗФР клетки ресуспендировали с 0,5 мл 1 х буферной смеси для экстракции цитозоля, содержащей ДТТ и ингибиторы протеаз, и инкубировали на льду в течение 10 минут. Клетки гомогенизировали в охлаждаемом льдом стеклянном гомогенизаторе. Гомогенат переносили в микроцентрифужную пробирку на 1,5 мл и центрифугировали при 700 х g в течение 10 мин при 4°С. Супернатант переносили в свежую пробирку на 1,5 мл и центрифугировали при 10000 х g в течение 30 мин при 4°С. Супернатант собирали в качестве цитозольной фракции. Осадок ресуспендировали в 0,1 мл буферной смеси для экстракции митохондрий, содержащей ДТТ и ингибиторы протеаз, встряхивали на вортексе в течение 10 секунд и хранили в качестве митохондриальной фракции. Концентрации белка определяли с использованием способа Бредфорда для определения белка Bio-Rad Laboratories, Inc. (Hercules, CA). 10 мкг каждой цитозольной и митохондриальной фракции, выделенных из необработанных и обработанных клеток, наносили на 15% ДСН-ПААГ для электрофореза и затем проводили стандартную процедуру Вестерн-блоттинга и зондировали блоты антителом против цитохрома с (1 мкг/мл).
Анализ апоптоза по высвобождению цитохрома с: клетки А375-М и SKBR3-HP обрабатывали GrB/scFvMEL при различных концентрациях в течение 24 часов. Митохондриальную фракцию выделяли из цитозоля и затем высвобождение цитохрома с из митохондрий в цитозоль во время апоптоза определяли вестерн-блоттингом с использованием антитела против цитохрома с. Фиг.24 показывает высвобождение цитохрома с на клетках А375-М, но не на клетках SKBR3-HP после обработки GrB/scFvMEL.
ПРИМЕР 40
GrB/VEGF121 ИНДУЦИРУЕТ ОБРАЗОВАНИЕ ЛЕСЕНКИ (ЛЭДДЕРА) ДНК НА КЛЕТКАХ РАЕ/FLK-1
Для анализа влияния GrB/VEGF121 на клетки РАЕ/FLK-1 использовали процедуру анализа образования лесенки (лэддера) ДНК.
Клетки РАЕ (2 х 106) обрабатывали GrB/VEGF121 при концентрации IC50 в течение 24 часов. Клетки кратковременно промывали ЗФР и затем собирали трипсинизацией с последующим центрифугированием при 200 х g в течение 5 минут. Клетки ресуспендировали в 1 мл ЗФР, переносили в 10 мл охлажденного на льду 70% этанола и хранили при -20°С в течение 24 часов или более продолжительно. Фиксированные клетки центрифугировали при 800 х g в течение 5 минут и этанол тщательно удаляли. Осадки клеток ресуспендировали в 40 мкл фосфат-цитратного буфера (РСВ), состоящего из 192 частей 0,2 М Na2HPO4 и 8 частей 0,1 М лимонной кислоты (рН 7,8), и инкубировали при комнатной температуре в течение по меньшей мере 30 минут. После откручивания клеток при 1000 х g в течение 5 минут супернатант переносили в новую пробирку. К пробам добавляли 3 мкл 0,25% Нонида NP-40 и 3 мкл РНКазы (1 мг/мл) и инкубировали в течение 30 минут при 37°С с последующим добавлением 3 мкл протеиназы К (1 мг/мл) и инкубированием в течение еще 30 минут при 37°С. Проводили электрофорез на 1,5% агарозном геле для детектирования ДНК бромидом этидия под ультрафиолетовым светом.
Фиг.25 показывает, что GrB/VEGF121 индуцирует образование лесенки (лэддера) фрагментов ДНК на клетках РАЕ/flk-1, но не на клетках РАЕ/flt-1.
ПРИМЕР 41
ELISA GrB/scFvMEL НА АНТИГЕН gp240-ПОЛОЖИТЕЛЬНЫХ КЛЕТКАХ А375-М В СРАВНЕНИИ С АНТИГЕН-gp240-ОТРИЦАТЕЛЬНЫМИ КЛЕТКАМИ Т-24 С ДЕТЕКТИРОВАНИЕМ ПРИ ПОМОЩИ МОНОКЛОНАЛЬНЫХ МЫШИНЫХ АНТИТЕЛ ПРОТИВ GRB
Связывающую активность GrB/scFvMEL в отношении антиген gp240-положительных клеток А375-М меланомы человека в сравнении с антиген gp240-отрицательными клетками Т-24 мочевого пузыря человека анализировали при помощи ELISA.
96-луночные планшеты, покрытые клетками А375-М или клетками Т-24 (50000 клеток на лунку), блокировали 5% БСА и затем эти клетки реагировали с GrB/scFvMEL при различных концентрациях в течение 1 часа при комнатной температуре. После промывания пробы инкубировали с мышиным моноклональным антителом против GrB (1 мкг/мл) при комнатной температуре в течение 1 часа с последующим инкубированием с конъюгатом HRP-козье антитело против мышиного IgG. Затем добавляли раствор субстрата 2,2'-азино-бис-3-этилбензтиазолин-6-сульфоновой кислоты (ABTS) с 1 мкл/мл 30% Н2О2. Спустя 30 минут измеряли оптическую плотность при 405 нм.
Фиг.26 показывает, что GrB/scFvMEL мог специфически связываться с антиген-положительными клетками А375-М, но не с антиген-отрицательными клетками Т-24, что свидетельствует о том, что слитый белок GrB/scFvMEL имел связывающую scFvMEL-часть активность.
ССЫЛКИ
Следующие ссылки, в той степени, в какой они обеспечивают примеры процедурных или других подробностей, дополняющие примеры процедурных или других подробностей, описанные здесь, включены специально в качестве ссылок.
ПАТЕНТЫ
EPO 0273085
EPO 03089
GB 2193095A
PCT/US85/01161
PCT/US89/05040U.S.Patent No. 4,162,282
U.S.Patent No. 4,215,051
U.S.Patent No. 4,310,505
U.S. Patent No. 4,376,110
U.S. Patent No. 4,533,254
U.S. Patent No. 4,554,101
U.S. Patent No. 4,676,980
U.S. Patent No. 4,728,575
U.S. Patent No. 4,728,578
U.S. Patent No. 4,737,323
U.S. Patent No. 4,816,567
U.S. Patent No. 4,921,706
U.S. Patent No. 4,946,778
U.S. Patent No. 5,401,511
U.S. Patent No. 5,401,511
U.S. Patent No. 5,432,260
U.S. Patent No. 5,440,013
U.S. Patent No. 5,446,128
U.S. Patent No. 5,475,085
U.S. Patent No. 5,603,872
U.S. Patent No. 5,603,872
U.S. Patent No. 5,618,914
U.S. Patent No. 5,656,725
U.S. Patent No. 5,670,155
U.S. Patent No. 5,672,681
U.S. Patent No. 5,674,976
U.S. Patent No. 5,710,245U.S. Patent No. 5,786,214
U.S. Patent No. 5,840,833
U.S. Patent No. 5,849,718
U.S. Patent No. 5,859,184
U.S. Patent No. 5,871,727
U.S. Patent No. 5,879,703
U.S. Patent No. 5,889,155
U.S. Patent No. 5,889,155
U.S. Patent No. 5,929,237
U.S. Patent No. 5,939,277
U.S. Patent No. 6,107,090
U.S. Patent Serial No. 07/715,272
U.S. Patent Serial No. 07/931,811
U.S. Patent Serial No. 07/934,373
WO 91/00360
WO 92/200373
WO 93/06213
WO 93/08829
WO 97/19179
WO 97/22364
WO 97/46259
WO 98/0748
WO 99/18933
WO 99/45128
WO 99/49059
ПУБЛИКАЦИИ
Adams GP, Schier R. Generating improved single-chain Fv molecules for tumor targeting. J Immunol Methods. 1999 Dec 10; 231(l-2):249-60.
Adams J.M. and Cory S. The Bcl-2 protein family: arbiters of cell survival. Science, 281:1322-1326,1998.
Aksentijevich I, Pastan I, Lunardi-Iskandar Y, Gallo RC, Gottesman MM, Thierry AR. In vitro and in vivo liposome-mediated gene transfer leads to human MDR1 expression in mouse bone marrow progenitor cells. Hum Gene Ther. 1996 Jun 10; 7(9):llll-22.
Andrade F, Roy S, Nicholson D, Thornberry N, Rosen A, Casciola-Rosen L. Granzyme B directly and efficiently cleaves several downstream caspase substrates: implications for CTL-induced apoptosis. Immunity 1998: 8: 451-460.
Antonsson B. et al. Inhibition of Bax channel-forming activity by Bcl-2. Science. 277: 370-372, 1997.
Aqeilan R, Yarkoni S, Lorberboum-Galski H. Interleukin 2-Bax: a novel prototype of human chimeric proteins for targeted therapy. FEBS Lett. 1999 Aug 27; 457(2):271-6.
Arap et al., 1995.
Ashkenazi A, Dixit VM. Death receptors: signaling and modulation. Science 1998; 281: 1305-1308.
Ausubel et al., In: Current Protocols in Molecular Biology, John, Wiley & Sons, Inc., 1994.
Bangham, et al., "Diffusion of univalent Ions across the Lamellae of Swollen Phospholipids" J. Mol. Biol., 13:238-252,1965.Barclay et al. (eds.), The Leucocyl=te Antigen Facts Book, 1993, Academic Press.
Becker KG, Mattson DH, Powers JM, Gado AM, Biddison WE. Analysis of a sequenced cDNA library from multiple sclerosis lesions. J Neuroimmunol. 1997 Jul; 77(l):27-38.
Beresford PJ. Xia Z, Greenberg AH, Lieberman J. Granzyme A loading induces rapid cytolysis and a novel from of DNA damage independently of caspase activation. Immunity 1999; 10: 585-594.
Berke G. The CTL's kiss of death, Cell 1995; 81: 9-12. Bird, R. E. et al., Science 242, 423-426, 1988.
Boise LH, Gonzalez-Garcia M, Postema CE, Ding L, Lindsten T, Turka LA, Mao X, Nunez G, Thompson CB. bcl-x, a bcl-2-related gene that functions as a dominant regulator of apoptotic cell death. Cell. 1993 Aug 27; 74(4):597-608.
Boldin MP, Goncharov TM, Goltsev YV, Wallach D. Involvement of MACH, a novel MORTl/FADD-interacting protease, in Fas/APO-1-and TNF receptor-induced cell death. Cell 1996; 85: 803-815.
Boyd JM, Gallo GJ, Elangovan B, Houghton AB, Malstrom S, Avery BJ, Ebb RG, Subramanian T, Chittenden T, Lutz RJ, et al. Bik, a novel death-inducing protein shares a distinct sequence motif with Bcl-2 family proteins and interacts with viral and cellular survival-promoting proteins. Oncogene. 1995 Nov 2; 11 (9): 1921-8.
Brinkmann U, Di Carlo A, Vasmatzis G, Kurochkina N, Beers R, Lee B, Pastan I. Stabilization of a recombinant Fv fragment by base-loop interconnection and V(H)-V(L) permutation. J Mol Biol. 1997 Apr 25; 268(1): 107-17.Brodeur et al., Monoclonal Antibody Production Techniques & Applications, pp. 51-63 (Marcel Dekker Inc., New York, 1987).
Browne, K.A., Blink, E., Sutton, V.R., Groelich, C.J., Jans, D.A., and Trapani, J.A. Mol. Cell. Biol. 1999; 19: 8604-8615.
Canfield LM, Fritz TA, Tarara TE. Incorporation of beta-carotene into mixed micelles. Methods Enzymol. 1990; 189:418-22.
Caruthers MH, Beaucage SL, Efcavitch JW, Fisher EF, Matteucci MD, Stabinsky Y. New chemical methods for synthesizing polynucleotides. Nucleic Acids Symp Ser. 1980; (7):215-23.
Catalysis in Micellar & Macromolecular Systems, 1975.
Cheng JQ, Jhanwar SC, Klein WM, Bell DW, Lee WC, Altomare DA, Nobori T, Olopade OI, Buckler AJ, Testa JR. p16 alterations and deletion mapping of 9p21-p22 in malignant mesothelioma. Cancer Res. 1994 Nov 1; 54(21):5547-51.
Chinnaiyan A.M., Orth K., O'Rourke K., Duan, H., Poirier G.G. and Dixit, V.M. Molecular ordering of the cell death pathway. J. Biol. Chem., 271: 4573-4576, 1996.
Chinnaiyan AM, Orth K, Hanna WL, Duan HJ, Poirier GG, Froelich CJ, Dixit VM, Cytotoxic T cell-derived granzyme B activates the apoptotic protease ICE-LAP3. Curr Biol 1996; 6: 897-899.
Clackson T, Hoogenboom HR, GrifMis AD, Winter G. Making antibody fragments using phage display libraries. Nature. 1991 Aug 15; 352(6336):624-8.
Cleary ML, Sklar J. Nucleotide sequence of a t(l4; 18) chromosomal breakpoint in follicular lymphoma and demonstration of a breakpoint-cluster region near a transcriptionally active locus on chromosome 18. Proc Natl Acad Sci USA. 1985 Nov; 82(21):7439-43.
Cohen, 1997, Biochem. J. 326:1-16.
Colbere-Garapin F, Horodniceanu F, Kourilsky P, Garapin AC. A new dominant hybrid selective marker for higher eukaryotic cells. J Mol Biol. 1981 Jul 25; 150(1):1-14.
Cole SP, Vreeken EH, Roder JC. Antibody production by human X human hybridomas in serum-free medium. J Immunol Methods. 1985 Apr 22; 78(2):271-8.
Colloidal Surfactant, 1963.
Creighton, 1983, Proteins Structures & Molecular Principles, W.H. Freeman & Co., N.Y., pp. 34-60.
Darmon AJ, Ley TJ, Nicholson DW, Bleackley RC, Cleavage of CPP32 by granzyme B represents a critical role for granzyme B in the induction of target cell DNA fragmentation. J Biol Chem 1996; 271: 21709-21712.
Darmon AJ, Nicholson DW, Bleackley RC. Activation of the apoptotic protease CPP32 by cytotoxic T-cell derived granzyme B. Nature 1995; 377: 446-448.
Deamer and P. Uster, "Liposome Preparation: Methods and Mechanisms," in Liposomes (M. Ostro, ed.), Marcel Dekker, Inc., New York (1983), pp. 27-52.
Doherty PC. Cell-mediated cytotoxicity. Cell 1993; 75: 607-612.
Duan J, Orth K, Chinnaiyan AM, Poirier GG, Froelich CJ, He W-W, Dixit VM. ICE-LAP6, a novel member of the ICE/Ced-3 gene family, is activated by the cytotoxic T cell protease granzyme B. J Biol Chem 1996; 271: 16720-16724.
Ebnet K, Hausmann M, Lehmann-Grube F, Mullbacher A, Kopf M, Lamers M, Simon MM, Granzyme A-deficient mice retain potent cell-mediated cytotoxicity. EMBO J 1995; 14: 4230-4239.
El-Gorab M. Solubilization of-carotene and retinol into aqueous solutions of mixed micelles.Biochim Biophys Acta. 1973 Apr 13; 306(l):58-66.
Felgner PL, Gadek TR, Holm M, Roman R, Chan HW, Wenz M, Northrop JP, Ringold GM, Danielsen M. Lipofection: a highly efficient, lipid-mediated DNA-transfection procedure.Proc Natl Acad Sci U S A. 1987 Nov; 84(21):7413-7.
Fernandes-Alnemri T, Armstrong RC, Krebs J, Srinivasula SM, Wang L, Bullrich F, Fritz LC, Trapani JA, Tomaselli KJ, Litwack G, Ahiemri ES. In vitro activation of CPP32 and Mch3 by Mch4, a novel human apoptotic cysteine protease containing two FADD-like domains. Proc. Natl Acad SciUSA 1996; 93: 7464-7469.
Fernandes-Alnemri T, Litwack G, Alnemri S. Mch2, a new member of the apoptotic Ced-3/Ice cysteine protease gene family. Cancer Res 1995; 55: 2737-2742.
FernandeS-Alnemri T, Takahashi A, Armstrong R, Krebs J, Fritz L, Tomaselli KJ, Wang L, Yu Z, Croce CM, Salvesen G, Earnshaw WC, Litwack G, Alnemri ES. Mch3, a novel human apoptotic cysteine protease highly related to CPP32. Cancer Res 1995; 55: 6045-6052.
Fingl et al., 1975, In: The Pharmacological Basis of Therapeutics, Ch. 1, p. 1.Fraley and Fornari Kaplan, "Entrapment of a bacterial plasmid in phospholipid vesicles:potential for gene transfer," Proc. Nat'l. Acad. Sci. USA 76:3348-3352,1979.
Froelich CJ, Orth K, Turbov J, Seth P, Gottlieb R, Babior B, Shah GM, Bleackley RC, Dixit VM, Hanna W. New paradigm for lymphocyte granule-mediated cytotoxicity. Target cells bind and internalize granzyme B, but an endosomolytic agent is necessary for cytosolic delivery and subsequent apoptosis. J Biol Chem 1996; 271: 29073-29079.
Gazzaniga P, Gradilone A, Vercillo R, Gandini O, Silvestri I, Napolitano M, Albonici L, Vincenzoni A, Gallucci M, Frati L, Agliano AM. Bcl-2/bax mRNA expression ratio as prognostic factor in low-grade urinary bladder cancer. Int J Cancer. 1996 Apr 22; 69(2): 100-4.
Gershenfeld HK, Weissman IL. Cloning of cDNA for a T cell-specific serine protease from a cytotoxic T lymphocyte. Science 1986; 232: 854-858.
Ghosh and Bachhawat, "Targeting of liposomes to hepatocytes," In: Wu G. Wu C ed., Liver diseases, targeted diagnosis and therapy using specific receptors and ligands, New York: Marel Dekker, pp. 87-104,1991.
Goding, Monoclonal Antibodies: Principles & Practice, pp. 59-104 (Academic Press, 1986).
Gregoriadis G. and Davis C. "Stability of liposomes in vivo and in vitro is promoted by their cholesterol content and the presence of blood cells," Biochem Biophys Res Commun., 89(4): 1287-1293,1979.Gregoriadis, DRUG CARRIERS IN BIOLOGY AND MEDICINE, G. Gregoriadis (ed.), 1979, pp. 287-341.
Griffiths, G.M., and Isaaz, S. J. Cell Biol. 1993; 120: 885-896.
Gross A., Jockel J., Wie M.C. and Korsmeyer S.J. Enforced dimerization of Bax results in its translocation, mitochondrial dysfunction and apoptosis. EMBO J. 17: 3878-3885,1998.
Gu Y, Sarnecki C, Fleming MA, Lippke JA, Bleackley RC, Su MS-S. Processing and activation of CMH-1 by granzyme B. J Biol Chem 1996; 271: 10816-10820.
Haddad, P., Jenne, D., Tschopp, J., Clement, M.V., Mathieu-Mahul, D., and Sasportes, M. Int. Immunol. 1991; 3: 57-66.
Hayes MP, Berrebi GA, Henkart PA. Induction of target cell DNA release by the cytotoxic T lymphocyte granule protease granzyme.A. J Exp Med 1980; 170: 933-946.
Heath et al., Chem. Phys. Lipids 40:347 (1986).
Heibein, J.A., Barry, M., Motyka, B., and Bleakley, R.C. J. Immunol. 1999; 163: 4683-4693.
Henkart, P.A. Annu. Rev. Immunol. 1985; 3: 31-58.
Heusel JW, Wesselschmidt RL, Shresta S, Russell JH, Ley TJ. Cytotoxic lymphocytes require granzyme B for the rapid induction of DNA fragmentation and apoptosis in allogeneic target cells. Cell 1994; 76: 977-987.
Hockenbery D.M., Oltvai Z.N., Yin X.-M., Milliman C.L. and Korsmeyer S.J. Bcl-2 functions in an antioxidant pathway to prevent apoptosis. Cell, 75: 241-251, 1993.
Hollstein M, Sidransky D, Vogelstein B, Harris CC. p53 mutations in human cancers. Science. 1991 Jul 5; 253(5015):49-53.
Huston, J. S. et al., Proc. Natl. Acad. Sci., USA 85, 5879-5883 (1988).
Hsu Y.T. and Youle RJ. Nonionic detergents induce dimerization among members of the Bcl-2 family. J. Biol. Chem., 272:13829-13834,1997.
Huiling H., Lam M., McCormic T.S. and Distelhorst C.W. Maintenance of calcium homeostasis in the endoplasmic reticulum by Bcl-2. J. Cell Biol. 138: 1219-1228,1997.
Hussussian CJ, Struewing JP, Goldstein AM, Higgins PA, Ally DS, Sheahan MD, Clark WH Jr, Tucker MA, Dracopoli NC. Germline p16 mutations in familial melanoma. Nat Genet. 1994 Sep; 8(l):15-21.
Huston JS, Levinson D, Mudgett-Hunter M, Tai MS, Novotny J, Margolies MN, Ridge RJ, Bruccoleri RE, Haber E, Crea R, et al. Protein engineering of antibody binding sites: recovery of specific activity in an anti-digoxin single-chain Fv analogue produced in Escherichia coli. Proc Natl Acad Sci USA. 1988 Aug; 85(16):5879-83.
Inohara N, Ding L, Chen S, Nunez G. harakiri, a novel regulator of cell death, encodes a protein that activates apoptosis and interacts selectively with survival-promoting proteins Bcl-2 and Bcl-X(L). EMBO J. 1997 Apr l; 16(7):1686-94.
Jacobson M.D., Weil M, and Raff M. C. Programmed cell death in animal development. Cell, 88: 347-354,1997.Jakobovits A, Moore AL, Green LL, Vergara GJ, Maynard-Currie CE, Austin HA, Klapholz S. Germ-line transmission and expression of a human-derived yeast artificial chromosome. Nature. 1993 Mar 18; 362(6417):255-8.
Jans DA, Jans P, Briggs LJ, Sutton V, Trapani JA. Nuclear transport of granzyme B (fragmentin-2). J Biol Chem 1996; 271: 30781-30789.
Johannesson et al., 1999, "Bicyclic tripeptide mimetics with reverse turn inducing properties." J. Med. Chem. 42:601-608.
Jones PT, Dear PH, Foote J, Neuberger MS, Winter G. Replacing the complementarity-determining regions in a human antibody with those from a mouse. Nature. 1986 May 29-Jun 4; 321(6069):522-5.
Kagawa S., Gu I., Swisher S.G., Ji L., RothJ.A. Lai D., Stephens L.C. and Fang B. Antitumor effect of adenovirus-mediated Bax gene transfer on p53-sensitive and p53-resistant cancer lines. Cancer Research. 60: 1157-1161,2000.
Kagawa S., Pearson S.A., Ji L., Xu K., McDonnell T.J., Swisher S.G., Roth LA. and Fang B. A binary adeno viral vector system for expressing high levels of the proapoptotic gene bax. Gene Therapy., 7: 75-79, 2000.
Kagi D, Lederman B. Burki K, Zinkernagel RM, Hengartner H. Molecular mechanisms of lymphocyte-mediated cytoxicity and their role in immunological protection and pathogenesis in vivo. Am Rev Immunol 1996; 14:207-232.
Kam, C-M., Hudig D., and Powers, J.C. Biochimica et Biophysica Acta. 2000; 1477: 307-323.
Kamb A, Shattuck-Eidens D, Eeles R, Liu Q, Gruis NA, Ding W, Hussey C, Tran T, Miki Y, Weaver-Feldhaus J, et al. Analysis of the p16 gene (CDKN2) as a candidate for the chromosome 9p melanoma susceptibility locus. Nat Genet. 1994 Sep; 8(l):23-6.
Kaneda et al, "Increased expression of DNA cointroduced with nuclear protein in adult rat liver," Science, 243:375-378,1989.
Kaneda et al., "Introduction and expression of the human insulin gene in adult rat liver," J Biol Chem., 264(21):12126-12129, 1989.
Kato et al., "Expression of hepatitis B virus surface antigen in adult rat liver. Co-introduction of DNA and nuclear protein by a simplified liposome method," J Biol Chem., 266(6):3361-3364, 1991.
Kerr IF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br I Cancer. 1972 Aug; 26(4):239-57.
Kiefer MC, Brauer MJ, Powers VC, Wu JJ, Umansky SR, Tomei LD, Barr PI. Modulation of apoptosis by the widely distributed Bcl-2 homologue Bak. Nature. 1995 Apr 20; 374(6524):736-9.
Knudson CM, Korsmeyer SI. Bcl-2 and Bax function independently to regulate cell death. Nat Genet. 1997 Aug; 16(4):358-63.
Kozbor D, Tripputi P, Roder JC, Croce CM. A human hybrid myeloma for production of human monoclonal antibodies. I Immunol. 1984 Dec; 133(6):3001-5.
Kozopas KM, Yang T, Buchan HL, Zhou P, Craig RW. MCL1, a gene expressed in programmed myeloid cell differentiation, has sequence similarity to BCL2. Proc Natl Acad Sci USA. 1993 Aprl5; 90(8):3516-20.
Kuroki M, Arakawa F, Khare PD, Kuroki M, Liao S, Matsumoto H, Abe H, Imakiire T. Specific targeting strategies of cancer gene therapy using a single-chain variable fragment (scFv) with a high affinity for CEA. Anticancer Res. 2000 Nov-Dec; 20(6A):4067-71.
Kyte & Doolittle, 1982.
Lam M., Dubyak G., Chen L., Nunez G., Miesfeld R.L. and Distelhorst C.W. Evidence that Bcl-2 represses apoptosis by regulating endoplasmic reticulum-associated Ca2+ fluxes. Proc. Natl. Acad Sci. USA, 91: 6569-6573,1994.
Lang J, Vigo-Pelfrey C, Martin F. Liposomes composed of partially hydrogenated egg phosphatidylcholines: fatty acid composition, thermal phase behavior and oxidative stability. Chem Phys Lipids. 1990 Mar; 53(l):91-101.
Lin EY, Orlofsky A, Berger MS, Prystowsky MB. Characterization of Al, a novel hemopoietic-specific early-response gene with sequence similarity to bcl-2. J Immunol. 1993 Aug 15; 151(4):1979-88.
Liposome Technology, 1984.
Liu Y, Liggitt D, Zhong W, Tu G, Gaensler K, Debs R. Cationic liposome-mediated intravenous gene delivery. J Biol Chem. 1995 Oct 20; 270(42):24864-70.
Liu X, Kim CN, Pohl J, Wang X. Purification and characterization of an interleukin-lbeta-converting enzyme family protease that activates cysteine protease P32 (CPP32). J Biol Chem. 1996 Jun7; 271(23):13371-6.
Liu X, Zou H, Slaughter C, Wang X. DFF, a heterodimeric protein that functions downstream of caspase-3 to trigger DNA fragmentation during apoptosis. Cell. 1997 Apr 18; 89(2):175-84.
Lobe CG, Havele C, Bleackley RC. Cloning of two genes that are specifically expressed in activated cytotoxic T lymphocytes. Proc Natl Acad Sci USA 1986; 83: 1448-1452.
Lowy I, Pellicer A, Jackson JF, Sim GK, Silverstein S, Axel R. Isolation of transforming DNA: cloning the hamster aprt gene. Cell. 1980 Dec; 22(3):817-23.
Marks JD, Hoogenboom HR, Bonnert TP, McCafferty J, Griffiths AD, Winter G. By-passing immunization. Human antibodies from V-gene libraries displayed on phage. J Mol Biol. 1991 Dec 5; 222(3):581-97.
Marks JD, Griffiths AD, Malmqvist M, Clackson TP, Bye JM, Winter G. By-passing immunization: building high affinity human antibodies by chain shuffling. Biotechnology (N Y). 1992 Jul; 10(7):779-83.
Martin SJ, Amarante-Mendes GP, Shi LF, Chuang TH, Casiano CA, O'Brien GA, Fitzgerald P, Tan EM, Bokoch GM, Greenberg AH, Green DR. The cytotoxic cell protease granzyme B initiates apoptosis in a cell-free system by proteolytic processing and activation of the ICE/CED-3 family protease, CPP32, via a novel two-step mechanism. EMBO J 1996:15: 2407-2416.
Marzo I, et al. The permeability transition pore complex: a target for apoptosis regulation by caspases and bcl-2-related proteins. J. Exp. Med. 187: 1261-1271, 1998.
Masson D, Tschopp J. Isolation of a lytic pore-forming protein (perforin) from cytolytic T lymphocytes. JBiol Chem 1985; 260: 9069-9072.
Masson D, Zamai M, Tschopp J. Identification of granzyme A isolated from cytotoxic T-lymphocyte-granules as one of the proteases encoded by CTL-specific genes. FEBS Lett 1986; 208: 84-88.
Mayer LD, Hope MJ, Cullis PR. Vesicles of variable sizes produced by a rapid extrusion procedure. Biochim Biophys Acta. 1986 Jun 13; 858(l):161-8.
Mayhew E, Conroy S, King J, Lazo R, Nikolopoulus G, Siciliano A, Vail WJ. High-pressure continuous-flow system for drug entrapment in liposomes. Methods Enzymol. 198 7; 149:64-77.
McConlogue, L., 1987, In: Current communications in molecular biology, Cold Spring Harbor Lab ed.)
Medema JP, Toes REM, Scaffidi C, Zheng TS, Flavell RA, Melief CJM, Peter ME, Offringa R, Krammer PH. Cleavage of FLICE (caspase-8) by granzyme B during cytotoxic T lymphocyte-induced apoptosis. Eur J Immunol 1997; 27: 3492-3498.
Milstein C, Cuello AC. Hybrid hybridomas and their use in immunohistochemistry. Nature. 1983 Oct6-12; 305(5934):537-40.Modern Pharmaceutics, 1990.
Montaldo PG, Pagnan G, Pastorino F, Chiesa V, Raffaghello L, Kirchmeier M, Allen TM, Ponzoni M. N-(4-hydroxyphenyl) retinamide is cytotoxic to melanoma cells in vitro through induction of programmed cell death, hit J Cancer. 1999 Apr 12; 81(2):262-7.
Morrison SL, Johnson MJ, Herzenberg LA, Oi VT. Chimeric human antibody molecules: mouse antigen-binding domains with human constant region domains. Proc Natl Acad Sci U S A. 1984 Nov; 81(21):6851-5.
Mujoo K, Spiro RC, Reisfeld RA. Characterization of a unique glycoprotein antigen expressed on the surface of human neuroblastoma cells. J Biol Chem. 1986 Aug 5; 261(22):10299-305.
Mullbacher A, Ebnet K, Blanden RV, Hla RT, Stehle T, Museteanu C, Simon MM. Granzyme A is critical for recovery of mice from infection with the natural cytopathic viral pathogen, ectromelia. Proc Natl Acad Sci USA 1996; 93: 5783-5787.
Mulligan RC, Berg P. Factors governing the expression of a bacterial gene in mammalian cells. Mol Cell Biol. 1981 May; l(5):449-59.
Muzio M, Chinnaiyan AM, Kischkel FC, O'Rourke K, Shevchenko A, Ni J, Scaffidi C, Bretz JD, Zhang M, Gentz R, Mann M, Krammer PH, Peter ME, Dixit VM. FLICE, a novel FADD-homologous ICE/CED-3-like protease, is recruited to the CD95 (Fas/APO-1) death-inducing signaling complex. Cell 1996; 85: 817-827.
Muzio, M., Chinnaiyan, A.M., Kischkel, F.C., O'Rourke, K., Shevchenko, A., Scaffidi, C., Bretz, J.D., Zhang, M., Ni, J., Gentz, R., Mann, M., Krammer, P.H., Peter, M.E., and Dixit, V.M. Cell. 1996; 86: 817-821.
Nagata S, Golstein P. The Fas death factor. Science 1995; 267: 1449-1456.
Nechushtan A, Yarkoni S, Marianovsky I, Lorberboum-Galski H. Adenocarcinoma cells are targeted by the new GnRH-PE66 chimeric toxin through specific gonadotropin-releasing hormone binding sites. J Biol Chem. 1997 Apr 25; 272(17):11597-603.
Neuberger MS, Williams GT, Fox RO. Recombinant antibodies possessing novel effector functions. Nature. 1984 Dec 13-19; 312(5995):604-8.
Nicolau and Sene, "Liposome-mediated DNA transfer in eukaryotic cells: dependence of the transfer efficiency upon the type of liposomes used and the host cell cycle stage," Biochem. Biophys. Acta, 721:185-190, 1982.
Nicolau et al, "Liposomes as carriers for in vivo gene transfer and expression," Methods Enzymol., 149:157-176,1987.
Nygren H. Conjugation of horseradish peroxidase to Fab fragments with different homobifunctional and heterobifunctional cross-linking reagents. A comparative study. J Histochem Cytochem. 1982 May; 30(5):407-12.
O'Hare K, Benoist C, Breathnach R. Transformation of mouse fibroblasts to methotrexate resistance by a recombinant plasmid expressing a prokaryotic dihydrofolate reductase. Proc Natl Acad Sci USA. 1981 Mar; 78(3):1527-31.
Odake S, Kam CM, Narasimhan L, Poe M, Blake JT, Krahenbuhl O, Tschopp J, Powers JC. Human and murine cytotoxic T lymphocyte serine proteases: subsite mapping with peptide thioester substrates and inhibition of enzyme activity and cytolysis by isocoumarins. Biochem 1991; 30: 2217-2227.
Oltvai Z.N., Milliman C.L. and Korsmeyer S. J. Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell, 74: 609-619,1993.
Orth K, Chinnaiyan AM, Garg M, Froelich CJ, Dixit VM. The CED-3/ICE-like protease Mch2 is activated during apoptosis and cleaves the death substrate lamin A. J. Biol Chem 1996; 271: 16443-16446.
Ottilie S, Diaz JL, Home W, Chang J, Wang Y, Wilson G, Chang S, Weeks S, Fritz LC, Oltersdorf T. Dimerization properties of human BAD. Identification of a BH-3 domain and analysis of its binding to mutant BCL-2 and BCL-XL proteins. J Biol Chem. 1997 Dec 5; 272(49):30866-72.
Pagnan G, Montaldo PG, Pastorino F, Raffaghello L, Kirchmeier M, Allen TM, Ponzoni M. GD2-mediated melanoma cell targeting and cytotoxicity of liposome-entrapped fenretinide. Int J Cancer. 1999 Apr 12; 81(2):268-74.
Perales et al, "Gene transfer in vivo: sustained expression and regulation of genes introduced into the liver by receptor-targeted uptake," Proc. Natl. Acad. Sci. USA, 91:4086-4090, 1994.
Pinkoski MJ, Heibein JA, Barry M, Bleackley RC. Nuclear translocation of granzyme B in target cell apoptosis. Cell Death Differ 2000; 7: 17-24.
Pinkoski MJ, Hobman M, Heibein JA, Tomaselli K, Li F, Seth P, Froelich CJ, Bleackley RC. Entry and trafficking of granzyme B in target cells during granzyme B-perforin-mediated apoptosis. Blood 1998; 92: 1044-1054.
Pinkoski MJ, Winkler U, Hudig D, Bleackley RC. Biding of granzyme B in the nucleus of target cells. Recognition of an 80-kilodalton protein. J Biol Chem 1996; 271: 10225-10229.
Podack, E.R. Curr. Top. Microbiol. Immunol. 1992; 178: 175-184.
Poe M, Blake JT, Boulton DA, Gammon M, Sigal NH, Wu JK, Zweerink HJ. Human cytotoxic lymphocyte granzyme B: its purification from granules and the characterization of substrate and inhibitor specificity. J Biol Chem 1991; 266: 98-103.
Quan LT, Tewari M, O'Rourke K, Dixit VM, Snipas SJ, Poirier GG, Ray C, Pickup DJ, Salvesen GS, Proteolytic activation of the cell death protease Yama/CPP32 by granzyme B. Proc Natl Acad Sci USA 1996; 93: 1972-1976.
Remington's Pharmaceutical Sciences, 18th Ed. Mack Printing Company, 1990.
Riechmann L, Clark M, Waldmann H, Winter G. Reshaping human antibodies for therapy. Nature. 1988 Mar 24; 332(6162):323-7.
Ruther U, Muller-Hill B. Easy identification of cDNA clones. EMBO J. 1983; 2(10):1791-4. Sambrook et al., 1989.
Santerre RF, Allen ME, Hobbs JN Jr, Rao RN, Schmidt RJ. Expression of prokaryotic genes for hygromycin B and G418 resistance as dominant-selection markers in mouse L cells. Gene. 1984 Oct; 30(l-3):147-56.Sarin, A., Williams, M.S.,, Alexander-Miller, M.A., Berzofsky, J.A., Zacharchuk, C.M., and Henkart, P.A. Immunity. 1997; 6: 209-215.
Szoka and Papahadjopoulos, "Procedure for Preparation of Liposomes With Large Internal Aqueous Space... ", Proc. Natl. Acad. Sci., 75:4194-4198,1978.
Tai Y-T, Strobel T., Kufe D., and Cannistra S.A. In vivo cytotoxicity of ovarian cancer cells through tumor-selective expression of the Bax gene. Cancer Research. 59: 2121-2126,1999.
Takeda S, Naito T, Hama K, Noma T, Honjo T. Construction of chimaeric processed immunoglobulin genes containing mouse variable and human constant region sequences. Nature. 1985 Apr 4-10; 314(6010):452-4.
Talanian RV, Yang XH, Turbov J, Seth P, Ghayur T, Casiano CA, Orth K, Froelich CJ. Granule-mediated killing: pathways for granzyme B-initiated apoptosis. J Exp Med 1997; 186: 1323-1331.
Templeton NS, Lasic DD, Frederik PM, Strey HH, Roberts DD, Pavlakis GN. Improved DNA: liposome complexes for increased systemic delivery and gene expression. Nat Biotechnol. 1997 M; 15(7):647-52.
Thierry AR, Lunardi-Iskandar Y, Bryant JL, Rabinovich P, Gallo RC, Mahan LC. Systemic gene therapy: biodistribution and long-term expression of a transgene in mice. Proc Natl Acad Sci U S A. 1995 Oct 10; 92(21):9742-6.
Thompson (ed.) 1994, The Cytokine Handbook, Academic Press, San Diego.
Trapani JA, Browne KA, Smyth MJ, Jans DA. Localization of granzyme B in the nucleus. A putative role in the mechanism of cytotoxic lymphocyte-mediated apoptosis. J Biol Chem 1996; 271: 4127-4133.
Trapani, J.A., Jans, D.A., Browne, K.A., Smyth, M.J., Jans, P., and Sutton, V.R. J. Biol. Chem. 1998; 273: 27934-27938.
Trapani, J.A., Klein, J., White, P.C., and Dupont, B. Proc. Natl. Acad. Sci. U.S.A. 1988; 85: 6924-6928.
Traunecker A, Lanzavecchia A, Karjalainen K. Bispecific single chain molecules (Janusins) target cytotoxic lymphocytes on HIV infected cells. EMBO J. 1991 Dec; 10(12):3655-9.
Tschopp J, Schafer S, Masson D, Peitsch MC, Heusser C. Phosphorylcholine acts as a calcium dependent receptor molecule for lymphocyte perforin. Nature 1989; 337: 272-274.
Tsujimoto Y, Cossman J, Jaffe E, Croce CM. Involvement of the bcl-2 gene in human follicular lymphoma. Science. 1985 Jun 21; 228(4706): 1440-3.
Tsukamoto M, Ochiya T, Yoshida S, Sugimura T, Terada M. Gene transfer and expression in progeny after intravenous DNA injection into pregnant mice. Nat Genet. 1995 Mar; 9(3):243-8.
Van de Craen M, Van den brande I, Declercq W, Irmler M, Beyaert R, Tschopp J, Fiers W, Vandenabeele P. Cleavage of caspase family members by granzyme B: a comparative study in vitro. Eur J Immunol 1997; 27: 1296-1299.
Van Heeke G, Schuster SM. Expression of human asparagine synthetase in Escherichia coli. J Biol Chem. 1989 Apr 5; 264(10):5503-9.Vaux D. L., Heacher G, and Strasser A. An evolutionary perspective on apoptosis. Cell, 76: 777-779, 1994.
Verhoeyen M, Milstein C, Winter G. Reshaping human antibodies: grafting an antilysozyme activity. Science. 1988 Mar 25; 239(4847): 1534-6.
Vincenz, C., and Dixit, V.M. J. Biol. Chem. 1997; 272: 6578-6583.
Vita et al., 1998, "Novel miniproteins engineered by the transfer of active sites to small natural scaffolds." Biopolymers 47:93-100.
Wagner et al, Science, 260:1510-1513, 1990.
Wang K, Yin XM, Chao DT, Milliman CL, Korsmeyer SJ. BID: a novel BH3 domain-only death agonist. Genes Dev. 1996 Nov 15; 10(22):2859-69.
Waterhouse P, Griffiths AD, Johnson KS, Winter G. Combinatorial infection and in vivo recombination: a strategy for making large phage antibody repertoires. Nucleic Acids Res. 1993 May ll; 21(9):2265-6.
Weinberg RA. Tumor suppressor genes. Science. 1991 Nov 22; 254(5035):1138-46.
Weisshoff et al., 1999, "Mimicry of beta II'-turns of proteins in cyclic pentapeptides with one and without D-amino acids." Eur. J. Biochem. 259:776-788.
Wigler M, Silverstein S, Lee LS, Peilicer A, Cheng Y, Axel R. Transfer of purified herpes virus thymidine kinase gene to cultured mouse cells. Cell. 1977 May; ll(l):223-32.
Wigler M, Perucho M, Kurtz D, Dana S, Pellicer A, Axel R, Silverstein S. Transformation of mammalian cells with an amplifiable dominant-acting gene. Proc Natl Acad Sci USA. 1980 Jun; 77(6):3567-70.
Wolter K.G., Hsu Y-T., Smith C.L., Nechushtan A., Xi, X.-G. and Youle RJ. Movement of Bax from the cytosol to mitochondria during apoptosis. J. Cell Biol. 139: 1281-1292, 1997.
Wong et al., "Appearance of beta-lactamase activity in animal cells upon liposome-mediated gene transfer," Gene., 10(2):87-94, 1980.
Wu and Wu, "Receptor-mediated in vitro gene transfections by a soluble DNA carrier system," J. Biol. Chem., 262:4429-4432,1987.
Wu and Wu, Adv. Drug Delivery Rev., 12:159-167, 1993.
Yagita, H., Nagata, M., Kawasaki, A., Shinkai, Y., and Okumura, K. Adv. Immunol. 1992; 51:
215-242.
Yang, X., Stennicke, H.R., Wang, B., Green, D.R., Janicke, R.U., Srinivasan, A., Seth, P., Salvesen, G., and Froelich, C.J. J. Biol. Chem. 1998; 273: 34278-34283.
Young JD-E, Hengartner H, Podack ER, Cohn ZA. Purification and characterization of a cytolytic pore-forming protein from granules of cloned lymphocytes with natural killer activity. Cell 1986; 44: 849-859.
Young, J.D.E., and Cohn, Z.A. Cell. 1986; 46: 641-642.
Zamzami N., Susin S.A., Marchetti P., Hirsch T., Gomez-Monterrey L, Castedo M., and Kroemer G. Mitochondrial control of nuclear apoptosis. J. Exp. Med., 183: 1533-1544, 1996.
Zha J, Harada H, Osipov K, Jockel J, Waksman G, Korsmeyer SJ. BH3 domain of BAD is required for heterodimerization with BCL-XL and pro-apoptotic activity. J Biol Chem. 1997 Sep 26; 272(39):24101-4.
Zola, Monoclonal Antibodies: a Manual of Techniques, pp. 147-158 (CRC Press, Inc., 1987).
Zou H, Henzel WJ, Liu X, Lutschg A, Wang X. Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell. 1997 Aug 8; 90(3):405-13.
Zunino SJ, Bleackley RC, Martinez J, Hudig D. RNKP-1, a novel natural killer cell-associated serine protease gene cloned from RNK-16 cytotoxic lymphocytes. J Immunol 1990; 144: 2001-2009.
[0387] Все описанные и заявленные здесь способы и композиции могут проводиться и выполняться без чрезмерного экспериментирования в свете данного описания. Хотя композиции и способы данного изобретения описаны в виде предпочтительных вариантов, специалистам с квалификацией в данной области должно быть понятно, что могут использоваться вариации в отношении этих способов и в стадиях или в порядке стадий описанного здесь способа без отклонения от концепции, сущности и объема данного изобретения. Более конкретно, должно быть понятно, что описанные здесь агенты могут быть заменены определенными агентами, которые являются как химически, так и физиологически родственными, при получении при этом таких же или подобных результатов. Предполагается, что все такие сходные замены и модификации, очевидные специалистам с квалификацией в данной области, находятся в рамках сущности, объема и концепции данного изобретения, определяемых прилагаемой формулой изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПОЛИПЕПТИДЫ С НАПРАВЛЕННЫМ ДЕЙСТВИЕМ | 2006 |
|
RU2393874C2 |
| МОДИФИЦИРОВАННЫЕ БЕЛКИ, СКОНСТРУИРОВАННЫЕ ТОКСИНЫ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 2002 |
|
RU2305684C2 |
| СПОСОБЫ И МАТЕРИАЛЫ ДЛЯ ОБНАРУЖЕНИЯ АПОПТОЗА | 2007 |
|
RU2450273C2 |
| ПРИМЕНЕНИЕ СЛИТОГО КОНСТРУКТА ТИМП-ГФИ И СПОСОБ ЛЕЧЕНИЯ ПОВРЕЖДЕНИЙ КОЖИ ДЛЯ ПРЕДОТВРАЩЕНИЯ ИЛИ ИНГИБИРОВАНИЯ ОБРАЗОВАНИЯ РУБЦА | 2006 |
|
RU2428479C2 |
| ПЕПТИДЫ НАПРАВЛЕННОГО ДЕЙСТВИЯ НА VEGFR-1/NRP-1 | 2008 |
|
RU2488592C2 |
| ПОЛУЧЕНИЕ БИОЛОГИЧЕСКИ АКТИВНЫХ БЕЛКОВ | 2007 |
|
RU2441911C2 |
| СРЕДСТВА ДЛЯ ЛЕЧЕНИЯ ПУЗЫРЧАТКИ, СОДЕРЖАЩИЕ АНТИТЕЛА ПРОТИВ ЛИГАНДА Fas | 2009 |
|
RU2556818C2 |
| СПОСОБЫ ПРИМЕНЕНИЯ АГОНИСТОВ АРО-2L-РЕЦЕПТОРОВ И АКТИВАТОРОВ NK-КЛЕТОК | 2005 |
|
RU2395294C2 |
| АНТИТЕЛА | 2005 |
|
RU2482131C2 |
| АНТИТЕЛА К ОХ-2/СD200 И ИХ ПРИМЕНЕНИЕ | 2007 |
|
RU2520088C2 |




























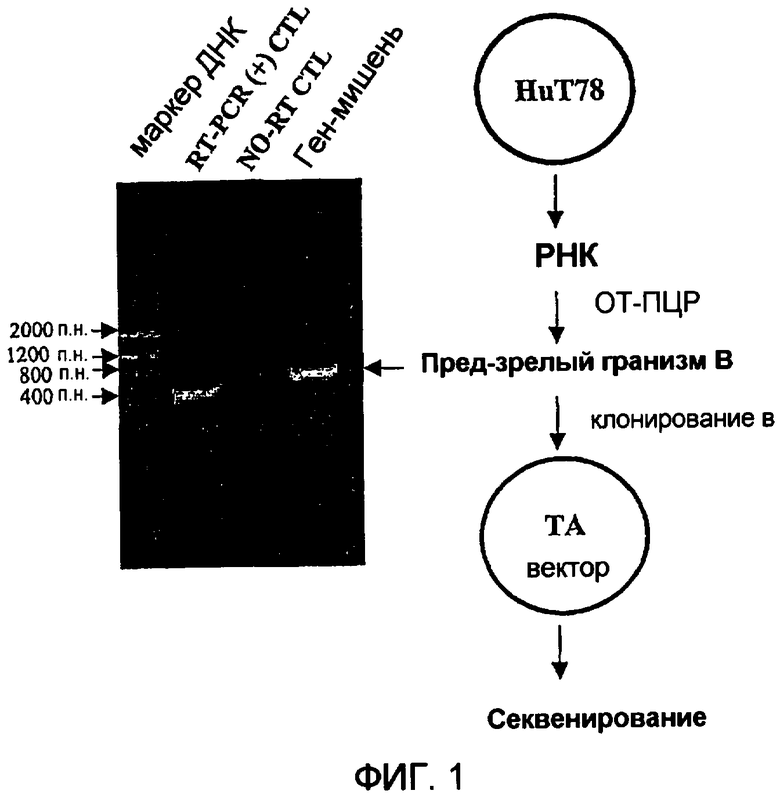

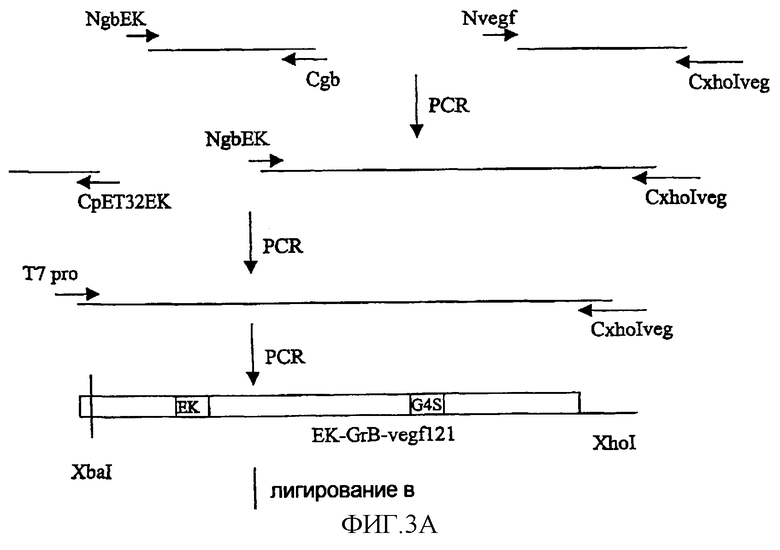




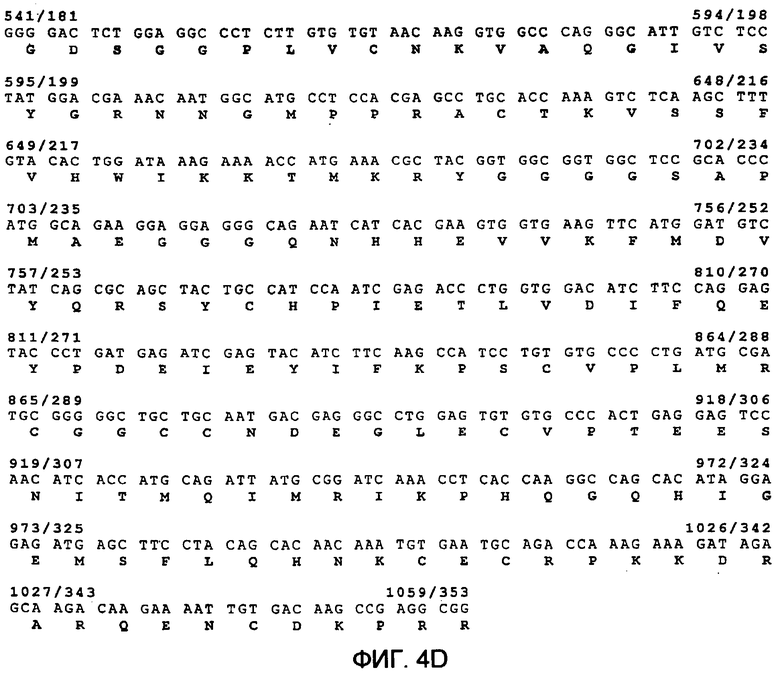

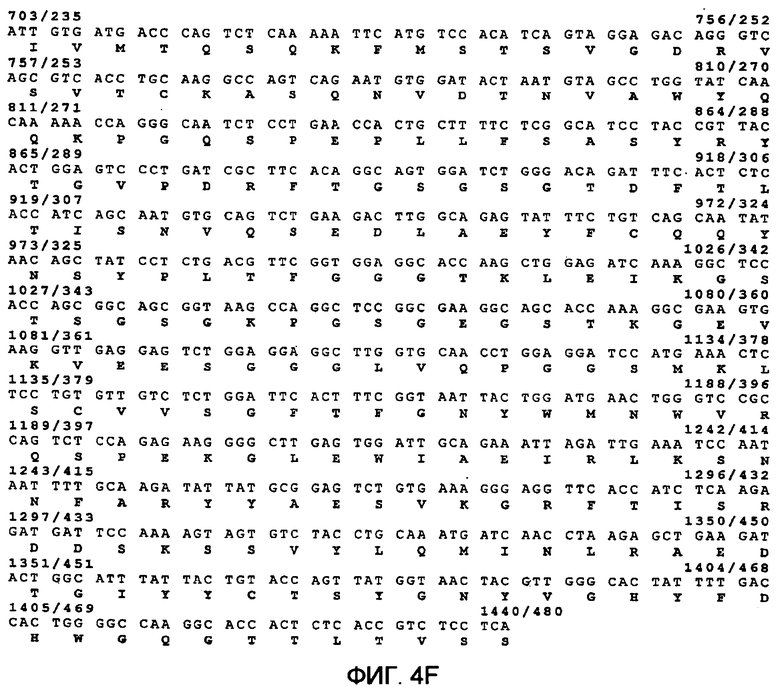
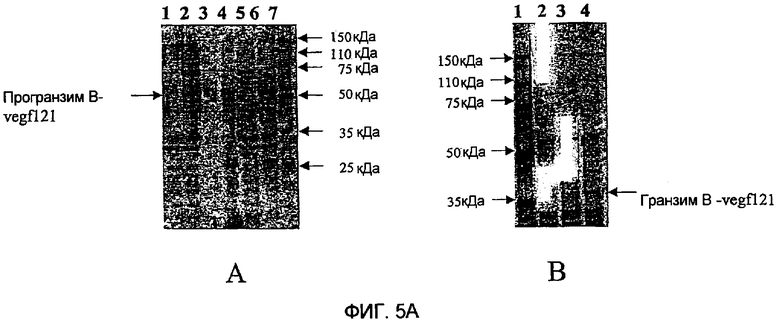
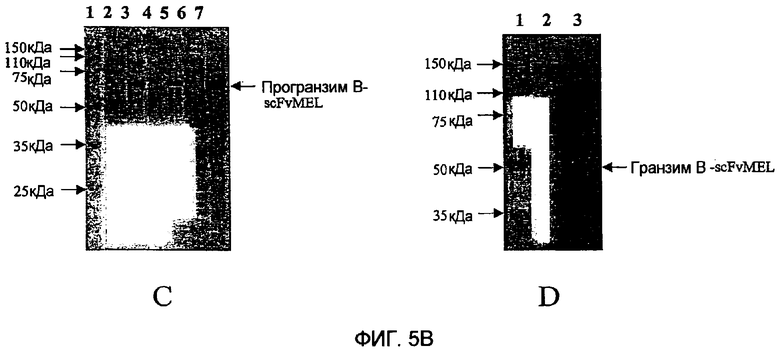
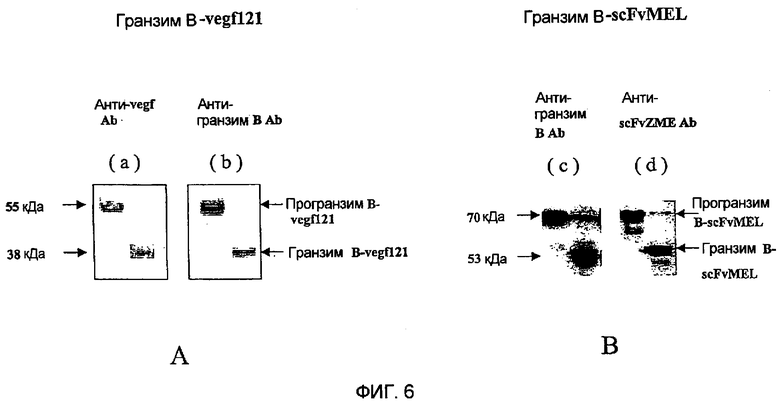
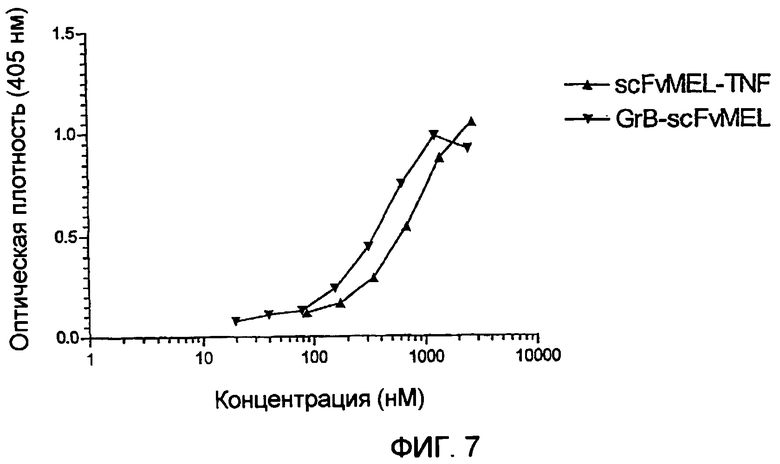
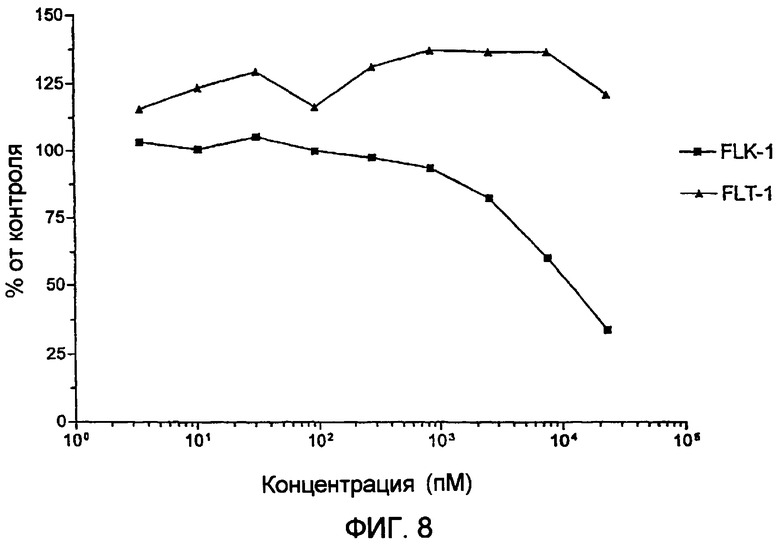
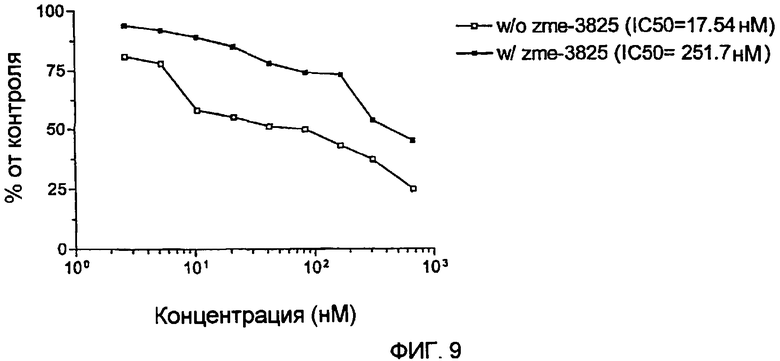
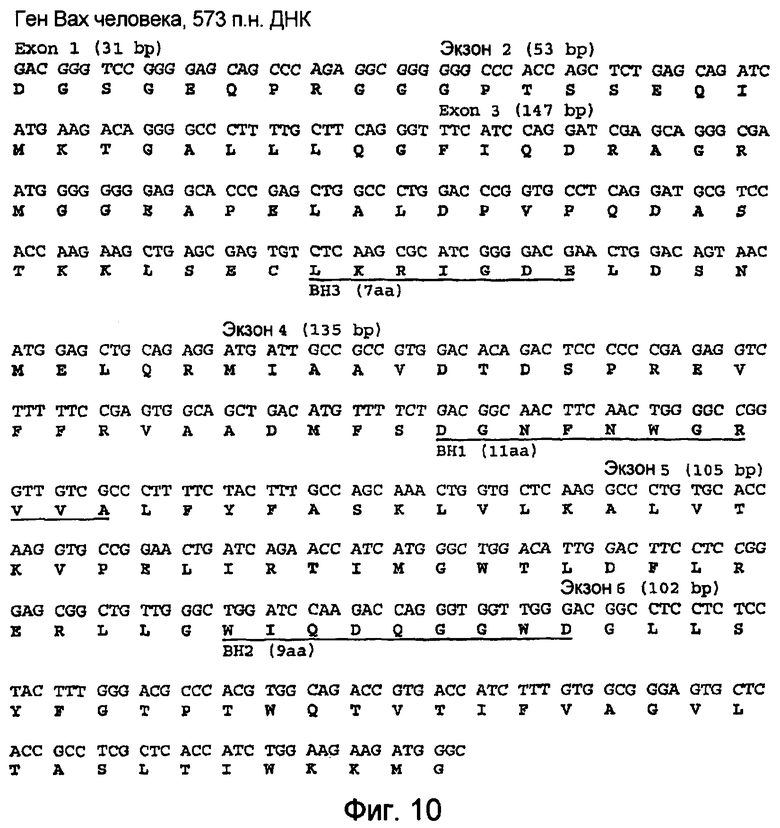
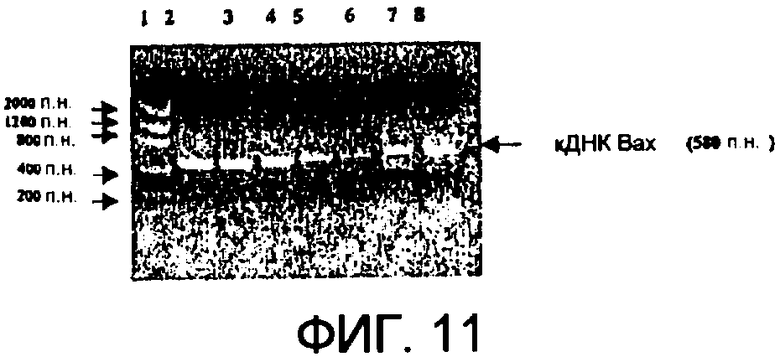



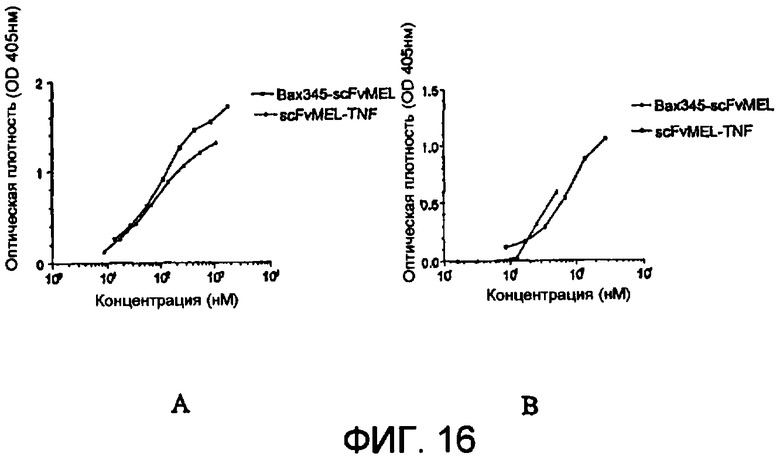

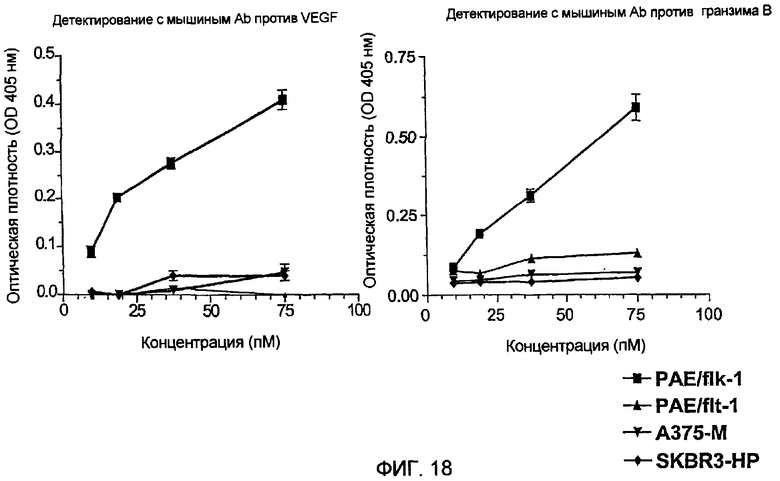
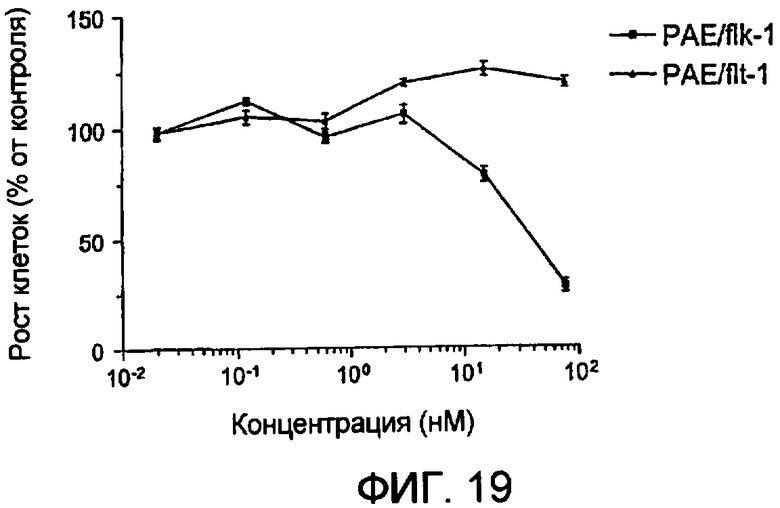
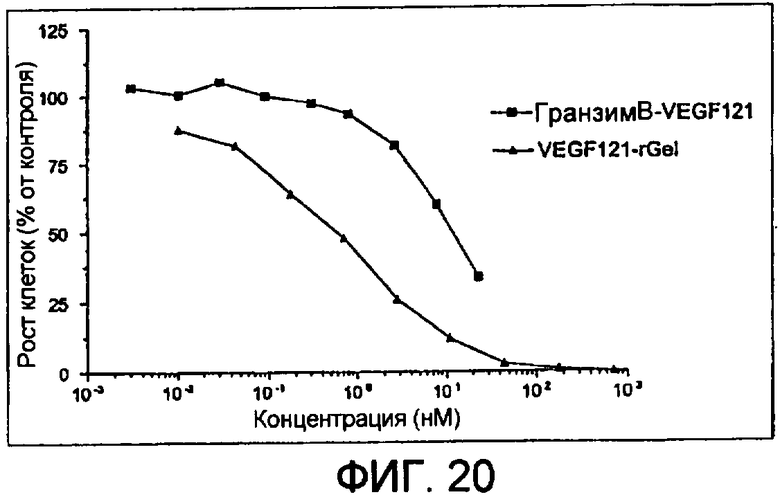
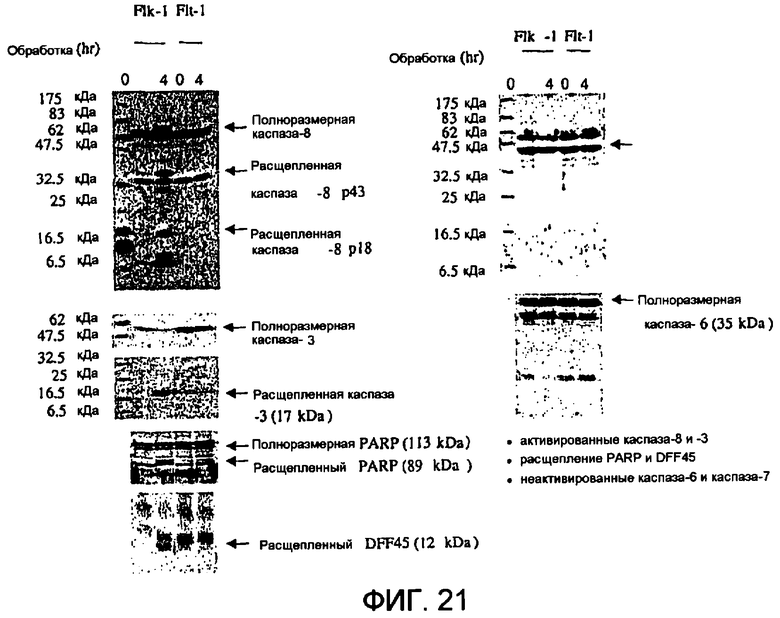



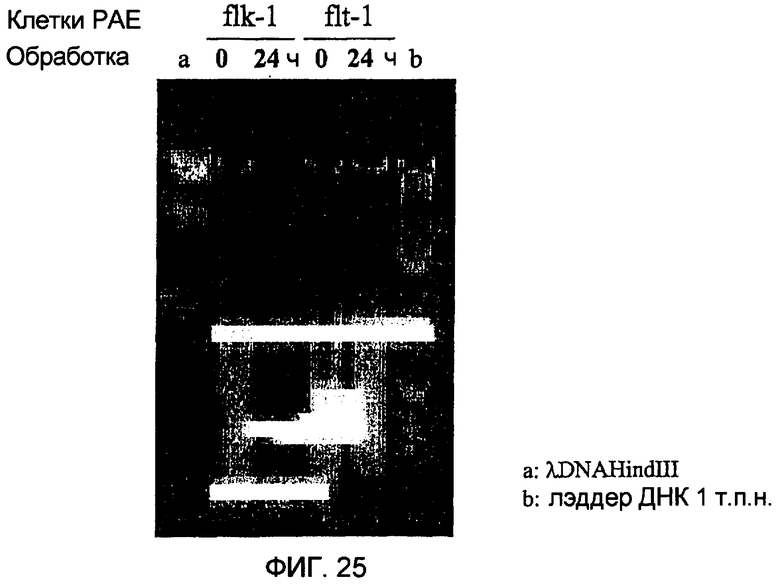

Изобретение относится к области биотехнологии и может быть использовано в терапии гиперпролиферативных нарушений. Химерный полипептид содержит клеткоспецифическую нацеливающую часть и гранзим. Полученный рекомбинантным путем полипептид используют для индуцирования апоптоза в клетке-мишени. Изобретение позволяет проводить эффективное лечение злокачественных заболеваний путем направленного лизиса опухолевых клеток. 5 н. и 19 з.п. ф-лы, 26 ил., 3 табл.
| WO 9945128 A1, 10.09.1999 | |||
| AQEILAN R | |||
| et al | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| FEBS Lett | |||
| Металлический водоудерживающий щит висячей системы | 1922 |
|
SU1999A1 |
| WILD R | |||
| et al | |||
| Ребристый каток | 1922 |
|
SU121A1 |
| Br | |||
| J | |||
| Cancer | |||
| ЩИТОВОЙ ДЛЯ ВОДОЕМОВ ЗАТВОР | 1922 |
|
SU2000A1 |

