Изобретение относится к соединению, обладающему модуляторной активностью по отношению к ФСГ-рецептору, в частности к производному тетрагидрохинолина, к содержащей его фармацевтической композиции, а также к применению указанного соединения в медицинской терапии.
Гонадотропины играют важные роли во множестве функций организма, включая метаболизм, регулирование температуры и репродуктивный процесс. Гонадотропины воздействуют на определенные типы гонадных клеток, инициируя овариальную и яичковую дифференцировку и стероидогенез. Например, гипофизарный гонадотропин ФСГ (фолликулостимулирующий гормон) играет центральную роль в стимуляции развития фолликула и созревании, тогда как ЛГ (лютеинизирующий гормон) вызывает овуляцию (Sharp, R.M. Clin Endocrinol. 33:787-807, 1990; Dorrington and Armstrong, Recent Prog. Horm. Res. 35:301-342, 1979). В настоящее время ФСГ применяют клинически в сочетании с ЛГ или hCG для стимуляции яичников, то есть гиперстимуляции яичников для оплодотворения in vitro (IVF) и вызывания овуляции у бесплодных ановуляторных женщин (Insler, V., Int. J. Fertility 33:85-97, 1988, Navot and Rosenwaks, J. Vitro Fert. Embryo Transfer 5:3-13, 1988), а также в случаях гипогонадизма у мужчин и бесплодия у мужчин.
Гонадотропин (ФСГ) выделяется передней долей гипофиза под влиянием гонадотропинвысвобождающего гормона и эстрогенов и плацентой во время беременности. У женщин ФСГ воздействует на яичники, способствуя развитию фолликулов, и представляет собой основной гормон, регулирующий секрецию эстрогенов. У мужчин ФСГ отвечает за целостность семявыводящих каналов и влияет на клетки Сертоли для поддержания гаметогенеза. Очищенный ФСГ применяют клинически для лечения бесплодия у женщин и в некоторых случаях недостаточности сперматогенеза у мужчин. Гонадотропины, предназначенные для терапевтических целей, могут быть выделены из источников мочи человека и обладают низкой чистотой (Morse et al., Amer. J. Reproduct. Immunol. and Microbiology 17:143, 1988). Альтернативным образом их можно получить в виде рекомбинантных гонадотропинов. Рекомбинатный ФСГ человека коммерчески доступен и используется при искусственной репродукции (Olijve et al., Mol. Hum. Reprod. 2:371, 1996; Devroey et al., Lancet 339:1170, 1992).
Действия гормона ФСГ опосредуются определенным мембранным рецептором плазмы, который является членом большого семейства рецепторов, связанных с G-белком. Эти рецепторы состоят из одного полипептида с семью трансмембранными доменами и способны взаимодействовать с Gs белком, приводя, например, к активации аденилатциклазы.
ФСГ рецептор представляет собой высокоспецифичную мишень в процессе роста фолликула яичника и экспрессируется исключительно в яичнике. Блокирование данного рецептора или ингибирование передачи сигнала, которая обычно вызывается после активации ФСГ-опосредованного рецептора, нарушит развитие фолликула и, таким образом, овуляцию и фертильность. Поэтому низкомолекулярные антагонисты ФСГ могли бы создать основу для новых контрацептивов. Подобные ФСГ антагонисты могли бы привести к сниженному развитию фолликула (отсутствие овуляции), оставляя все же достаточную выработку эстрогена для избежания неблагоприятных эффектов, например, на костную массу. С другой стороны, соединения, стимулирующие активность ФСГ-рецепторов, могут служить для воспроизведения гонадотропного эффекта природного лиганда.
В настоящем изобретении описано получение низкомолекулярных аналогов гормона, селективно обладающих модуляторной активностью по отношению к ФСГ рецепторам. Соединения изобретения можно либо использовать в качестве (частичных) агонистов или (частичных) антагонистов ФСГ-рецептора.
Таким образом, в настоящее время найдено, что следующий класс тетрагидрохинолиновых соединений формулы I или их фармацевтически приемлемых солей обладают ФСГ-модуляторной активностью:
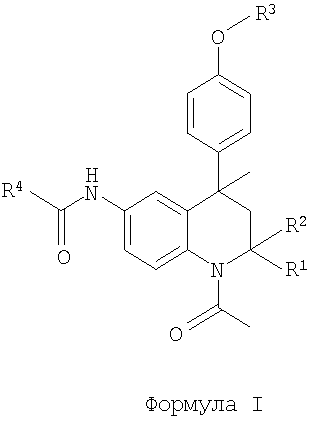
в которой R1 и R2 представляют собой Н, Me;
R3 представляет собой (2-6С)гетероциклоалкил(1-4С)алкил,
[2-5С]гетероарил(1-4С)алкил, (6С)арил(1-4С)алкил,
(1-4С)(ди)алкиламинокарбониламино(2-4С)алкил,
(2-6С)гетероциклоалкилкарбониламино(2-4С)алкил, R5-(2-4C)алкил или R5-карбонил(1-4С)алкил;
R4 представляет собой (2-5С)гетероарил, (6С)арил, (3-8С)циклоалкил, (2-6С)гетероциклоалкил или (1-6С)алкил;
R5 представляет собой (ди)(1-4С)алкиламино, (1-4С)алкокси, амино, гидрокси, (6С)ариламино, (ди)(3-4С)алкениламино, (2-5С)гетероарил(1-4С)алкиламино, (6С)арил(1-4С)алкиламино, (ди)[(1-4С)алкокси(2-4C)алкил]амино, (ди)[(1-4С)алкиламино(2-4 С)алкил]амино, (ди)[амино(2-4C)алкил]амино или (ди)[гидрокси(2-4 С)алкил]амино.
Соединения в соответствии с настоящим изобретением модулируют функцию ФСГ рецептора и могут быть использованы для тех же клинических целей, что и природный ФСГ, если они ведут себя как агонисты, с тем преимуществом, что они проявляют измененные свойства устойчивости и могут вводиться по-разному. Если они блокируют ФСГ рецептор, их можно использовать, например, в качестве контрацептивных агентов.
Таким образом, модуляторы ФСГ-рецептора настоящего изобретения можно использовать для лечения бесплодия, в целях контрацепции и для лечения гормон-зависимых нарушений, таких, как рак груди, рак простаты и эндометриоз.
Подразумевается, что следующие термины имеют указанные значения, использованные в описании и формуле изобретения, приведенных далее.
Использованный здесь термин (1-4С)алкил означает разветвленную или неразветвленную алкильную группу, содержащую 1-4 атома углерода, представляющую собой метил, этил, пропил, изопропил, бутил, втор-бутил и трет-бутил.
Использованный здесь термин (2-4С)алкил означает разветвленную или неразветвленную алкильную группу, содержащую 2-4 атома углерода, представляющую собой этил, пропил, изопропил, бутил, втор-бутил и трет-бутил.
Использованный здесь термин (1-6С)алкил означает разветвленную или неразветвленную алкильную группу, содержащую 1-6 атомов углерода, например метил, этил, пропил, изопропил, бутил, втор-бутил, трет-бутил и гексил. Предпочтительными являются (1-5С)алкильные группы, причем особенно предпочтителен (1-4С)алкил.
Использованный здесь термин (ди)(1-4С)алкиламино означает аминогруппу, монозамещенную или дизамещенную алкильными группами, каждая из которых содержит 1-4 атома углерода и имеет то же значение, что и определенное выше.
Использованный здесь термин (ди)(1-4С)алкениламино означает аминогруппу, монозамещенную или дизамещенную алкенильными группами, каждая из которых содержит 2-4 атома углерода и имеет то же значение, что и определенное выше.
Использованный здесь термин (3-8С)циклоалкил означает циклоалкильную группу, содержащую 3-8 атомов углерода, представляющую собой циклопропил, циклобутил, циклопентил, циклогексил, циклогептил и циклооктил. Предпочтительными являются (3-6С)циклоалкильные группы.
Использованный здесь термин (2-6С)гетероциклоалкил означает гетероциклоалкильную группу, содержащую 2-6 атомов углерода, предпочтительно 3-5 атомов углерода, и по меньшей мере, содержащую один гетероатом, выбранный из N, О и/или S, которая может быть связана через гетероатом, если это возможно, или через атом углерода. Предпочтительными гетероатомами являются N или О. Гетероциклоалкильная группа может быть замещена метильной или этильной группой по атому углерода или гетероатому, если это возможно. Наиболее предпочтительными гетероциклоалкильными группами являются пиперидинильная, пиперазинильная, морфолинильная, пирролидинильная и 1-метил-2-пиперидинильная.
Использованный здесь термин (1-4С)алкокси означает алкоксигруппу, содержащую 1-4 атома углерода, причем алкильная составляющая имеет то же значение, что и определенное ранее. Предпочтительными являются (1-2С)алкоксигруппы.
Использованный здесь термин (6С)арил означает фенильную группу, которая необязательно может быть замещена одним или более заместителями, выбранными из гидрокси, амино, йода, брома, хлора, фтора, нитро, трифторметила, циано, фенила, (1-4С)алкила, (1-4С)алкокси или (1-4С)(ди)алкиламино, причем алкильная, алкоксильная и (ди)алкиламиносоставляющие имеют то же значение, что и определенные ранее, например фенил, 3,5-дибромфенил, 4-бифенил, 3,5-дихлорфенил, 3-бром-6-метиламинофенил, 3-хлор-2,6-диметоксифенил и 3,5-диметилфенил.
Использованный здесь термин (2-5С)гетероарил означает замещенную или незамещенную ароматическую группу, содержащую 2-5 атомов углерода, по меньшей мере, содержащую один гетероатом, выбранный из N, О и/или S, например имидазолил, пиридил, пиримидил, тиенил или фурил. Заместители в гетероарильной группе можно выбрать из группы заместителей, приведенной для (6С)арильной группы. Гетероарильная группа может быть связана через атом углерода или гетероатом, если это возможно. Предпочтительными гетероарильными группами являются тиенильная, фурильная и пиридильная.
Использованный здесь термин (2-6С)гетероциклоалкил(1-4С)алкил означает гетероциклоалкильную группу, содержащую 2-6 атомов углерода, связанную с алкильной группой, содержащей 1-4 атома углерода, причем гетероциклоалкильная группа и алкильная группа имеют те же значения, что и определенные ранее.
Использованный здесь термин (2-6С)гетероциклоалкилкарбониламино означает гетероциклоалкильную группу, содержащую 2-6 атомов углерода, связанную с карбонильной составляющей карбониламиногруппы, причем гетероциклоалкильная группа имеет то же значение, что и определенное ранее.
Использованный здесь термин (2-6С)гетероциклоалкилкарбониламино(2-4С)алкил означает гетероциклоалкилкарбониламиногруппу, в которой гетероциклоалкильная составляющая содержит 2-6 атомов углерода, связанную через аминогруппу с алкильной группой, содержащей 2-4 атомов углерода, причем гетероциклоалкилкарбониламиногруппа и алкильная группа имеют те же значения, что и определенные ранее.
Использованный здесь термин (ди)(1-4С)алкиламинокарбонил означает (ди)алкиламино группу, алкильная группа(ы) которой содержит 1-4 атома углерода, связанную через аминогруппу с карбонильной группой, причем (ди)алкиламиногруппа имеет то же значение, что и определенное ранее.
Использованный здесь термин (3-8С)циклоалкиламинокарбонил означает циклоалкильную группу, содержащую 3-8 атомов углерода, связанную с аминосоставляющей аминокарбонильной группы, причем циклоалкильная группа имеет то же значение, что и определенное ранее.
Использованный здесь термин (ди)(1-4С) алкиламинокарбониламино означает (ди)алкиламиногруппу, алкильная группа(ы) которой содержит 1-4 атома углерода, связанную через аминогруппу с карбонильной составляющей карбониламиногруппы, обеспечивая таким образом функциональную группу мочевины, причем (ди)алкиламино группа имеет то же значение, что и определенное ранее.
Использованный здесь термин (ди)(1-4С) алкиламинокарбониламино(2-4С)алкил означает (ди)алкиламинокарбониламиногруппу, алкильная группа(ы) которой содержит 1-4 атома углерода, связанную через аминогруппу с алкильной группой, содержащей 2-4 атома углерода, причем (ди)алкиламинокарбониламиногруппа и алкильная группа имеют те же значения, что и определенные ранее.
Использованный здесь термин (2-5С)гетероарил(1-4С)алкил означает гетероарильную группу, содержащую 2-5 атомов углерода, связанную с алкильной группой, содержащей 1-4 атома углерода, причем гетероарильная группа и алкильная группа имеют те же значения, что и определенные ранее.
Использованный здесь термин (6С)арил(1-4С)алкил означает фенильную группу, необязательно замещенную одним или более заместителями, выбранными из группы заместителей, приведенных для (6С)арильной группы, связанную с алкильной группой, содержащей 1-4 атомов углерода, причем арильная группа и алкильная группа имеют те же значения, что и определенные ранее.
Использованный здесь термин (6С)ариламино означает фенильную группу, необязательно замещенную одним или более заместителями, выбранными из группы заместителей, приведенных для (6С)арильной группы, связанную с аминогруппой, причем арильная группа и алкильная группа имеют те же значения, что и определенные ранее.
Использованный здесь термин (6С)арил(1-4С)алкиламино означает фенильную группу, необязательно замещенную одним или более заместителями, выбранными из группы заместителей, приведенных для (6С)арильной группы, связанную с алкильной составляющей алкиламиногруппы, содержащей 1-4 атома углерода, причем арильная группа и алкиламиногруппа имеют те же значения, что и определенные ранее.
Использованный здесь термин (2-5С)гетероарил(1-4С)алкиламино означает гетероарильную группу, содержащую 2-5 атомов углерода, необязательно замещенную одним или более заместителями, выбранными из группы заместителей, приведенных для (6С)арильной группы, связанную с алкильной составляющей алкиламиногруппы, содержащей 1-4 атома углерода, причем гетероарильная группа и алкиламиногруппа имеют те же значения, что и определенные ранее.
Использованный здесь термин (1-4С)алкокси(2-4С)алкил означает алкоксигруппу, содержащую 1-4 атома углерода, связанную с алкильной группой, содержащей 2-4 атома углерода, причем алкоксигруппа и алкильная группа имеют те же значения, что и определенные ранее.
Использованный здесь термин (ди)([1-4С)алкокси(2-4С)алкил]амино означает аминогруппу, монозамещенную или дизамещенную (1-4С)алкокси(2-4С)алкильными группами. (1-4С)Алкокси(2-4С)алкильная группа представляет собой алкоксигруппу, содержащую 1-4 атома углерода, связанную с алкильной группой, содержащей 2-4 атома углерода и имеющую то же значение, что и определенное ранее.
Использованный здесь термин (1-4С)алкиламино(2-4С)алкил означает алкиламиногруппу, содержащую 1-4 атома углерода, связанную через аминогруппу с алкильной группой, содержащей 2-4 атома углерода, причем алкильные составляющие имеют те же значения, что и определенные ранее.
Использованный здесь термин (ди)([1-4С)алкиламино(2-4С)алкил]амино означает аминогруппу, монозамещенную или дизамещенную (1-4С)алкиламино(2-4С)алкильными группами. (1-4С)Алкиламино(2-4С)алкильная группа представляет собой алкиламиногруппу, содержащую 1-4 атома углерода, связанную через аминогруппу с алкильной группой, содержащей 2-4 атома углерода и имеющую то же значение, что и определенное ранее.
Использованный здесь термин амино(2-4С)алкил означает аминоалкильную группу, содержащую 2-4 атома углерода, причем алкильная составляющая имеет то же значение, что и определенное ранее.
Использованный здесь термин (ди)[амино(2-4С)алкил]амино означает аминогруппу, монозамещенную или дизамещенную аминоалкильными группами, содержащими 2-4 атома углерода и имеющими те же значения, что и определенные ранее.
Использованный здесь термин гидрокси(2-4С)алкил означает гидроксиалкильную группу, содержащую 2-4 атома углерода, причем алкильная составляющая имеет то же значение, что и определенное ранее.
Использованный здесь термин (ди)[гидрокси(2-4С)алкил]амино означает аминогруппу, монозамещенную или дизамещенную гидроксиалкильными группами, содержащими 2-4 атома углерода и имеющими те же значения, что и определенные ранее.
Использованный здесь термин R5-(2-4C)алкил означает R5 группу, связанную с алкильной составляющей, содержащей 2-4 атома углерода, которая имеет то же значение, что и определенное ранее.
Использованный здесь термин R5-карбонил-(1-4С)алкил означает R5 группу, связанную с карбонильной составляющей карбонилалкильной группы, причем алкильная составляющая содержит 1-4 атома углерода и имеет то же значение, что и определенное ранее.
Термин фармацевтически приемлемая соль означает такие соли, которые, в рамках медицинского заключения, подходят для использования при контакте с тканями людей и низших животных, не вызывая чрезмерной токсичности, раздражения, аллергической реакции и так далее, и соответствуют разумному соотношению выгода/риск. Фармацевтически приемлемые соли хорошо известны в данной области техники. Их можно получить при конечном выделении и очистке соединений изобретения или отдельно, вводя во взаимодействие функцию свободного основания при ее наличии с подходящей минеральной кислотой, такой, как хлористоводородная кислота, фосфорная кислота или серная кислота, или с органической кислотой, например, такой, как аскорбиновая кислота, лимонная кислота, винная кислота, молочная кислота, малеиновая кислота, малоновая кислота, фумаровая кислота, гликолевая кислота, янтарная кислота, пропионовая кислота, уксусная кислота, метансульфокислота и так далее. При наличии кислотную функцию можно ввести во взаимодействие с органическим или минеральным основанием, например гидроксидом натрия, гидроксидом калия или гидроксидом лития.
Таким образом, изобретение относится к соединениям формулы I, определенным выше.
В другом варианте осуществления в изобретении предлагаются соединения в соответствии с формулой I, в которых R1 и R2 представляют собой метил.
Изобретение относится также к соединениям формулы I, в которых R3 представляет собой (2-6С)гетероциклоалкил(1-4С)алкил, (2-5С)гетероарил(1-4С)алкил, (2-6С)гетероциклоалкилкарбониламино(2-4С)алкил, R5-(2-4C)алкил, R5-карбонил(1-4С)алкил.
В другом аспекте изобретение относится к соединениям в соответствии с формулой I, в которых R3 представляет собой (2-6С)гетероциклоалкил(1-4С)алкил, (2-5С)гетероарил(1-4С)алкил, R5-(2-4C)алкил, R5-карбонил(1-4С)алкил.
В еще одном аспекте изобретение относится к соединениям в соответствии с формулой I, в которых R3 представляет собой (2-6С)гетероциклоалкил(1-4С)алкил, (2-5С)гетероарил(1-4С)алкил или R5-(2-4C)алкил.
В еще одном аспекте изобретение относится к соединениям в соответствии с формулой I, в которых R3 представляет собой (2-6С)гетероциклоалкил(1-4С)алкил.
Согласно еще одному варианту осуществления данного изобретения гетероциклоалкильная группа в гетероциклоалкил(1-4С)алкиле в R3 в соответствии с формулой I состоит из 4, 5 или 6 атомов углерода, а гетероарильная группа в гетероарил (1-4С)алкиле в R3 состоит из 3, 4 или 5 атомов углерода.
В следующем варианте осуществления изобретение относится к соединениям в соответствии с формулой I, в которых R4 представляет собой (6С)арил.
В еще одном варианте осуществления в изобретении предлагаются соединения формулы I, в которой R5 представляет собой (ди)(1-4С)алкиламино, амино, (ди)(3-4С)алкениламино, (2-5С)гетероарил(1-4С)алкиламино, (6С)арил(1-4С)алкиламино, (ди)[(1-4С)алкокси(2-4C)алкил]амино, (ди)[(1-4С)алкиламино(2-4С)алкил]амино, (ди)[амино(2-4C)алкил]амино, (ди)[гидрокси(2-4С)алкил]амино.
В следующем варианте осуществления изобретение относится к соединениям в соответствии с формулой I, в которых R5 представляет собой (ди)(1-4С)алкиламино, (2-5С)гетероарил(1-4С)алкиламино, (ди)[(1-4С)алкокси(2-4C)алкил]амино, (ди)[(1-4С)алкиламино(2-4C)алкил]амино, (ди)[амино(2-4С)алкил]амино или (ди)[гидрокси(2-4C)алкил]амино.
В следующем варианте осуществления изобретение относится к соединениям в соответствии с формулой I, в которых R5 представляет собой (ди)(1-4С)алкиламино, амино, (ди)(3-4С)алкениламино, (2-5С)гетероарил(1-4С)алкиламино, (6С)арил(1-4С)алкиламино.
Следующий вариант осуществления изобретения составляют соединения в соответствии с формулой I, в которой R5 представляет собой (ди)(1-4С)алкиламино или амино.
В еще одном варианте осуществления изобретения предлагаются соединения в соответствии с формулой I, в которых R5 представляет собой (ди)(1-4С)алкиламино.
Еще один вариант осуществления изобретения относится к соединениям, в которых все конкретные определения групп с R1 по R5, определенные здесь выше, объединены в соединении формулы I.
Подходящие способы получения соединений данного изобретения приведены ниже.

Соединения данного изобретения формулы 1-а можно получить, исходя из описанной реакции Скраупа. Проведение данной реакции на защищенном N-трет-бутоксикарбонилом (N-Boc) 1,4-фенилендиамине (II) приводит к производному 1,2-дигидрохинолина III-а.
Похожие реакции циклоконденсации Скраупа известны в литературе: A. Knoevenagel, Chem. Ber. 54:1726, 1921; R.L.Atkins and D.E.Bliss, J. Org. Chem. 43:1975, 1978; J.V.Johnson, B.S.Rauckman, D.P.Baccanari and B.Roth, J.Med. Chem. 32:1942, 1989; W.C.Lin, S.-T.Huang and S.-T.Lin, J.Chin. Chem. Soc. 43:497, 1996; IP. Edwards, SJ. West, K.B.Marschke, D.E.Mais, M.M.Gottardis and Т.К.Jones, J.Med. Chem. 41:303, 1998.
Упомянутую выше реакцию обычно проводят при повышенной температуре в ацетоне или оксиде мезитила в присутствии йода или протонсодержащей кислоты, такой, как хлористоводородная кислота, п-толуолсульфокислота или водный йодоводород. Альтернативным образом соединения формулы III-a можно получить, вводя во взаимодействие соединение II с ацетоном в присутствии MgSO4, 4-трет-бутилкатехола и йода (L.G.Hamann, R.I.Higuchi, L.Zhi, J.P.Edwards and X.-N.Wang, J.Med. Chem, 41:623, 1998). По еще одной методике данную реакцию можно проводить в ацетоне с использованием трифлатов лантанидов (например, трифлата скандия) в качестве катализаторов. В этом случае реакцию можно проводить при комнатной температуре или при повышенной температуре при обычном нагревании или микроволновом облучении (М.Е.Theoclitou and L.A.Robinson, Tetrahedron Lett. 43:3907, 2002).
Соединения формулы III-b можно получить из N-Boc-1,4-фенилендиамина II реакцией с метилвинилкетоном. Похожие циклизации описаны в патенте Соединенных Штатов 2686182 (Badische Anilin-& Soda Fabrik Aktiengesellschaft).
Последующее 1-N-ацетилирование соединений формулы III-a-b можно осуществить в стандартных условиях. В обычном эксперименте соединения формулы III-a-b кипятят в уксусном ангидриде или вводят во взаимодействие в таком растворителе, как хлористый метилен, тетрагидрофуран, толуол или пиридин, с ацетилхлоридом в присутствии основания, такого, как N,N-диизопропилэтиламин, триэтиламин или гидрид натрия, получая производное 1-N-ацетил-4-метил-1,2-дигидрохинолина формулы IV-a-b.

Стандартное снятие защитной группы Вос в условиях, хорошо известных специалистам в данной области техники, приводит к производным 6-амино-1,2-дигидрохинолина формулы V-a-b. Данную реакцию обычно проводят в хлористом метилене в присутствии трифторуксусной кислоты.
Последующее 6-N-ацилирование соединений формулы V-a-b можно осуществить в стандартных условиях, получая соединения общей формулы VI-a-b, в которой R4 является определенным выше. Например, соединения формулы V-a-b вводят во взаимодействие в растворителе, таком, как хлористый метилен, тетрагидрофуран или толуол, с ацилгалогенидом (R4-C(O)Cl) или ангидридом кислоты (R4-С(О)-O-С(О)-R4) в присутствии основания, такого, как N,N-диизопропилэтиламин, триэтиламин, пиридин или гидрид натрия, получая 6-N-ацилированные производные 4-метил-1,2-дигидрохинолина формулы VI-a-b. Альтернативным образом ацилирование соединений общей формулы V-a-b с получением соединений общей формулы VI-a-b можно также осуществить реакцией с соответствующей карбоновой кислотой (R4-CO2H) в присутствии связывающего агента, такого, как тетрафторборат О-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония (TBTU), гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (HATU) или гексафторфосфата бромтрипирролидинфосфония (PyBrOP) и третичного основания, например N,N-диизопропилэтиламина, в растворителе, таком, как N,N-диметилформамид или хлористый метилен, при обычной или повышенной температуре.
Введение требуемой замещенной фенильной группы в 4 положение дигидрохинолинового каркаса можно осуществить алкилированием анизола по Фриделю-Крафтсу соединениями общей структуры VI-a-b, получая соединения общей формулы VII-a-b. Данную реакцию обычно проводят при повышенной температуре либо в анизоле, либо в подходящем инертном растворителе, таком, как гептан или гексан, с анизолом в качестве реагента, при катализе кислотой Льюиса (например, AlCl3, AlBr3, FeCl3 или SnCl4). Алкилирование 2,2,4-триметил-1,2-дигидрохинолинами по Фриделю-Крафтсу описано в литературе: В.A.Lugovik, L.G.Yudin and A.N.Kost, Dokl. Akad. Nauk SSSR, 170:340, 1966; В.A.Lugovik, L.G.Yudin, S.M.Vinogradova and A.N.Kost, Khim. Geterosikl. Soedin, 7:795, 1971.
Альтернативным образом N-Boc-1,4-фенилендиамин II можно ввести во взаимодействие с 2-(4-метоксифенил)пропеном и формальдегидом в ацетонитриле при обычной или повышенной температуре с последующим l-N-ацетилированием, как описано выше, получая соединение VII-b, в котором R4=О-трет-Bu. Похожие циклизации описаны в литературе: J.M.Mellor and G.D.Merriman, Tetrahedron, 51:6115, 1995. Снятие защитной группы Вос и последующее ацилирование 6-аминофункции ацилгалогенидом (R4-С(O)Cl), как описано ранее, позволяет получить соединения общей структуры VII-b, в которых R4 является определенным выше.
Расщепление ароматического метилового простого эфира в случае соединений общей формулы VII-a-b приводит к 4-(4-гидроксифенил)замещенным производным тетрагидрохинолина общей формулы VIII-a-b, устанавливая стадию функционализации ОН-группы.
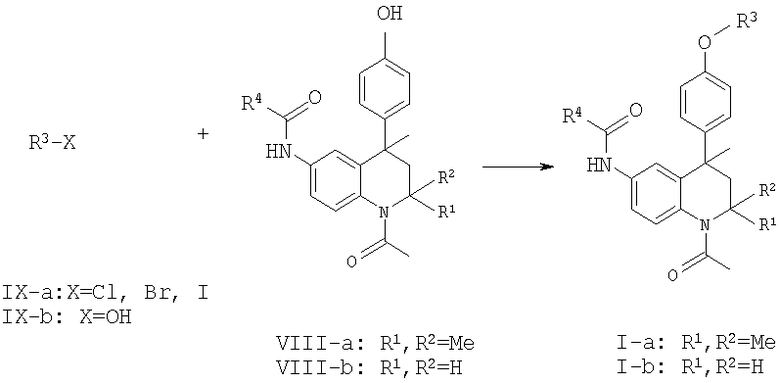
Реакции деметилирования ароматических метиловых простых эфиров хорошо известны специалисту в данной области техники. В обычном эксперименте деметилирование проводят реакцией соединения формулы VII-a-b с BBr3 в инертном растворителе, таком, как хлористый метилен, при температуре от пониженной до комнатной, получая деметилированные соединения общей формулы VIII-a-b. Альтернативным образом деметилирование можно осуществить реакцией соединения формулы VII-a-b с комплексом BF3·Ме2S при температуре окружающей среды.
Селективное O-алкилирование соединений общей формулы VIII-a-b функционализированными алкилгалогенидами общей формулы IX-a приводит к получению соединений общей формулы 1-а-b. Реакции алкилирования ароматических гидроксильных групп хорошо известны в данной области техники. Обычно раствор соединения общей формулы VIII-a-b в подходящем растворителе, таком, как 1,4-диоксан, тетрагидрофуран, хлористый метилен, ацетонитрил, ацетон или N,N-диметилформамид, обрабатывают основанием (например, N,N-диизопропиламином, триэтиламином, К2СО3, Cs2СО3 или NaOH) и соответствующим алкилирующим реагентом общей формулы IX-a, например бромистым бензилом, 3-(диметиламино)пропилхлоридом, 4-(2-хлорэтил)морфолином, 2-пиколилхлоридом или 2-хлорацетамидом. Альтернативным образом алкилирование можно осуществить при помощи известного алкилирования по Мицунобу. В этом случае раствор соединения общей формулы VIII-a-b в подходящем растворителе, таком, как 1,4-диоксан, тетрагидрофуран или хлористый метилен, обрабатывают (полимерносвязанным) трифенилфосфином, диэтил- или ди-трет-бутилазодикарбоксилатом и функционализированным спиртом общей формулы IX-b. В принципе, оба способа алкилирования можно использовать для всех R3 групп, но если R3 содержит нуклеофильную группу, такую, как вторичный амин или гидроксильная группа, может потребоваться стратегия подходящей защитной группы. Выбор защитной группы и условия снятия защиты очевидны для специалистов в данной области техники.
По другой методике получения соединений настоящего изобретения исходят из алкилирования соединений общей формулы VIII-a-b сложными эфирами общей формулы X.
Реакцию алкилирования обычно проводят в присутствии основания, такого, как N,N-диизопропилэтиламин или гидрид натрия, в подходящем растворителе, таком, как N,N-диметилформамид или тетрагидрофуран, при комнатной или повышенной температуре. Сложноэфирную функцию конечных соединений общей формулы XI-a-b, в которых А=Ме или Et, затем можно селективно восстановить в контролируемых условиях, получая соединения общей формулы XIII-а-b с использованием соответствующего восстанавливающего агента, такого, как алюмогидрид лития, при пониженной температуре, или борогидрид натрия в инертном растворителе, таком, как тетрагидрофуран. Затем свободную гидроксильную группу в соединениях общей формулы XIII-a-b можно ввести во взаимодействие с 4-толуолсульфонилхлоридом (Ts-Cl) или метансульфонилхлоридом (Ms-Cl) в инертном растворителе, таком, как 1,4-диоксан, N,N-диметилформамид или ТГФ, в присутствии подходящего основания, такого, как триэтиламин или пиридин, для получения соответствующей уходящей группы (соединения общей формулы XIV-a-b; LG=Ts или Ms соответственно). После этого нуклеофильное замещение соответствующим нуклеофилом (амином или алкоголятом) в условиях, известных специалистам в данной области техники, приводит к соединениям общей формулы I-a-b, в которых R3=R5-(2-4C)алкил и R5 являются определенными ранее.
Превращение соединений общей формулы XI-a-b, в которых А=трет-Bu, в карбоновые кислоты общей формулы XII-a-b можно осуществить путем снятия защитной трет-бутильной сложноэфирной функции. В обычном эксперименте трет-бутиловый сложный эфир общей формулы XI-a-b (А=трет-Bu) растворяют в хлористом метилене и обрабатывают сильной кислотой, такой, как трифторуксусная кислота. Полученные карбоновые кислоты общей формулы XII-a-b можно затем конденсировать с соответствующим спиртом или амином в присутствии агента сочетания, такого, как тетрафторборат О-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония (TBTU), гексафторфосфат О-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (HATU) или гексафторфосфат бромтрипирролидинофосфония (PyBrOP), и третичного основания, например N,N-диизопиропилэтиламина, в растворителе, таком, как N,N-диметилформамид или хлористый метилен, при комнатной или повышенной температуре, получая соединения общей формулы I-a-b, в которых R3=R5-карбонил(1-4С)алкил и R5 являются определенными ранее.

Некоторые соединения изобретения, которые могут находиться в форме свободного основания, можно выделить из реакционной смеси в виде фармацевтически приемлемой соли. Фармацевтически приемлемые соли можно также получить при обработке свободного основания формулы I органической или неорганической кислотой, такой, как хлористоводородная, бромистоводородная, йодистоводородная, серная кислота, фосфорная кислота, уксусная кислота, пропионовая кислота, гликолевая кислота, малеиновая кислота, малоновая кислота, метансульфокислота, фумаровая кислота, янтарная кислота, винная кислота, лимонная кислота, бензойная кислота и аскорбиновая кислота.
Соединения настоящего изобретения обладают, по меньшей мере, одним хиральным атомом углерода и, следовательно, могут быть получены в виде чистых энантиомеров или в виде смеси энантиомеров или в виде смеси диастереомеров. Способы получения чистых энантиомеров хорошо известны в данной области техники, например кристаллизация солей, полученных из оптически активных кислот и рацемической смеси, или хроматография с использованием хиральных колонок. Для диастереомеров можно использовать колонки с прямой фазой или обращенной фазой.
Соединения настоящего изобретения способны образовывать гидраты или сольваты. Специалистам в данной области техники известно, что при лиофилизации с водой заряженные соединения образуют гидратированные вещества или образуют сольваты при концентрировании в растворе с соответствующим органическим растворителем. Соединения данного изобретения включают гидраты или сольваты перечисленных соединений.
Для выбора активных соединений тестирование при 10-5 М должно приводить к активности, превышающей 20% максимальной активности при использовании ФСГ в качестве стандарта. Другим критерием может быть величина ЕС50, которая должная быть <10-5 М, предпочтительно <10-7 М.
Опытный специалист поймет, что желательные величины EC50 зависят от тестируемого соединения. Например, соединение с ЕС50, меньшей 10-5 М, обычно считают кандидатом при выборе лекарственного средства. Предпочтительно, чтобы данная величина была меньше 10-7 М. Однако соединение, которое обладает большей EC50, но селективно по отношению к конкретному рецептору, может быть даже лучшим кандидатом.
Способы определения связывания рецептора как в in vitro, так и в in vivo анализах для определения биологической активности гонадотропинов хорошо известны. Как правило, экспрессируемый рецептор приводят в контакт с тестируемым соединением и определяют связывание или стимулирование, или ингибирование функционального отклика.
Для определения функционального отклика выделенную ДНК, кодирующую ген рецептора ФСГ, предпочтительно рецептора человека, экспрессируют в подходящей клетке-хозяине. Подобная клетка может представлять собой клетку яичника китайского хомячка, но подходят также и другие клетки. Предпочтительно эти клетки имеют природу млекопитающего (Jia et al., Mol. Endocrin., 5:759-776, 1991).
Способы создания клеточных линий, экспрессирующих рекомбинантный ФСГ, хорошо известны в данной области техники (Sambrook et al., Molecular Cloning: A Laboratory Manual, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, самое последнее издание). Экспрессия рецептора достигается за счет экспрессии ДНК, кодирующей желательный белок. Методики сайт-направленного мутагенеза, лигирования дополнительных последовательностей, PCR и создание подходящих систем экспрессии, - все это в настоящее время хорошо известно в данной области техники. Части или всю ДНК, кодирующую желательный белок, можно создать искусственно с использованием стандартных твердофазных методик, предпочтительно для того, чтобы включить сайты рестрикции для простоты лигирования. Кодирующие последовательности ДНК можно снабдить подходящими контрольными элементами для транскрипции и трансляции включенной кодирующей последовательности. Как известно, в настоящее время доступны системы экспрессии, совместимые с широким кругом хозяев, включая прокариотических хозяев, таких, как бактерии, и эукариотических хозяев, таких, как дрожжи, клетки растений, клетки насекомых, клетки млекопитающих, клетки птиц и так далее.
Затем клетки, экспрессирующие рецептор, приводят в контакт с тестируемым соединением, чтобы наблюдать связывание или стимулирование, или ингибирование функционального отклика.
Альтернативным образом для определения связывания соединения можно использовать выделенные клеточные мембраны, содержащие экспрессированный рецептор.
Для определения связывания можно использовать меченные радиоактивным изотопом или флуоресцентно меченые соединения. Кроме того, можно использовать анализ на конкурентное связывание.
Другой анализ включает скрининг соединений агонистов рецептора ФСГ путем определения стимулирования опосредованного рецептором накопления цАМФ. Таким образом, подобный способ включает экспрессию рецептора на поверхность клетки-хозяина и подвергание данной клетки действию тестируемого соединения. После этого определяют количество цАМФ. Уровень цАМФ может быть понижен или повышен в зависимости от ингибирующего или стимулирующего эффекта тестируемого соединения при связывании с рецептором.
Скрининг антагонистов рецептора ФСГ включает в себя инкубацию клеток, экспрессирующих рецептор ФСГ, с тестируемым соединением в интервале концентраций в присутствии фиксированной, субмаксимально эффективной концентрации ФСГ (то есть концентрации ФСГ, вызывающей приблизительно 80% максимальной стимуляции накопления цАМФ в отсутствие тестируемого соединения). Из кривых концентрация - эффект можно вычислить величину IC50 и процент ингибирования вызванного ФСГ накопления цАМФ для каждого из тестируемых соединений. В качестве соединения сравнения можно использовать рекомбинантный ФСГ человека. В качестве альтернативы можно также применять анализы на конкурентное связывание.
Кроме прямого определения, например, уровней цАМФ в подвергнутой воздействию клетке, можно использовать клеточные линии, которые, в дополнение к трансфекции кодирующей рецептор ДНК, были также трансфицированы второй ДНК, кодирующей репортерный ген, экспрессия которого чувствительна к уровню цАМФ. Подобные репортерные гены могут быть цАМФ индуцибельными или могут быть созданы таким образом, чтобы они были связаны с новыми цАМФ элементами отклика. Вообще, экспрессию репортерных генов можно контролировать с помощью любого элемента отклика, реагирующего на изменение уровней цАМФ. Подходящими репортерными генами являются, например, гены, кодирующие β-галактозидазу, щелочную фосфатазу, люциферазу светлячков и зеленый флуоресцентный белок. Принципы таких трансактивационных анализов широко известны в данной области и описаны, например, у Stratowa, Ch., Himmler, A. and Czernilofsky, A.P. (1995) Curr. Opin. Biotechnol. 6:574.
Настоящее изобретение относится также к фармацевтическим композициям, содержащим производное тетрагидрохинолина или его фармацевтически приемлемые соли, имеющее общую формулу I, в смеси с фармацевтически приемлемыми вспомогательными веществами и необязательно другими терапевтическими агентами. Вспомогательные вещества должны быть «приемлемыми» в смысле их совместимости с прочими ингредиентами композиции и отсутствия вреда для их реципиентов.
Композиции включают, например, композиции, подходящие для перорального, сублингвального, подкожного, внутривенного, внутримышечного, местного или ректального введения и так далее, причем все это в виде единичных дозированных форм для введения.
Для перорального введения активный ингредиент может быть представлен в виде дискретных доз, таких, как таблетки, капсулы, порошки, гранулы, растворы, суспензии и так далее.
Для парентерального введения фармацевтическая композиция изобретения может быть представлена в виде контейнеров с единичной дозой или множественными дозами, например, жидкостей для инъекций в заранее определенных количествах, например, в запаянных сосудах и ампулах и может также храниться в сублимационно высушенном (лиофилизированном) состоянии с необходимостью лишь добавления перед использованием стерильного жидкого носителя, например, воды.
При смешивании с подобными фармацевтически приемлемыми вспомогательными веществами, например, как описано в стандартной ссылке: Gennaro, A.R. et al., Remington: The Science and Practice of Pharmacy (20th Edition, Lippincott Williams & Wilkins, 2000, смотри в особенности часть 5: Pharmaceutical Manufacturing), активный агент может быть сформован в твердые дозированные формы, такие, как пилюли, таблетки, или быть переработан в капсулы или суппозитории. При помощи фармацевтически приемлемых жидкостей активный агент можно применять в виде жидкой композиции, например в виде препарата для инъекции, в виде раствора, суспензии, эмульсии или в виде спрея, например назального спрея.
Для изготовления твердых дозированных форм предполагается использование дополнительных добавок, таких, как наполнители, красители, полимерные связывающие вещества и так далее. В общем, можно использовать любую фармацевтически приемлемую добавку, которая не влияет на действие активных соединений. Подходящие носители, вместе с которыми активный ингредиент данного изобретения можно вводить в виде твердой композиции, включают в себя лактозу, крахмал, производные целлюлозы и так далее или их смеси, используемые в подходящих количествах. Для парентерального введения можно использовать водные суспензии, изотонические растворы соли и стерильные растворы для инъекций, содержащие фармацевтически приемлемые диспергирующие агенты и/или увлажняющие агенты, такие, как пропиленгликоль или бутиленгликоль.
Кроме того, изобретение включает в себя фармацевтическую композицию, описанную выше, в сочетании с упаковочным материалом, подходящим для указанной композиции, при этом указанный упаковочный материал включает инструкции для использования композиции для описанного выше применения.
Производные тетрагидрохинолина данного изобретения можно также вводить в форме имплантируемых фармацевтических устройств, состоящих из ядра активного вещества, заключенного в мембрану, регулирующую скорость высвобождения. Подобные имплантаты должны применяться подкожно или местным образом, и будут высвобождать активный ингредиент с приблизительно постоянной скоростью в течение относительно длительных промежутков времени, например от недель до лет.
Способы получения имплантируемых фармацевтических устройств как таковых известны в данной области техники, например они описаны в европейском патенте 0303306 (AKZO Nobel N.V.).
Точная доза и режим введения активного ингредиента или его фармацевтической композиции будет обязательно зависеть от терапевтического эффекта, которого нужно достичь (лечение бесплодия; контрацепция), и могут изменяться для конкретного соединения, способа введения, возраста и состояния индивидуального субъекта, которому будут вводить данный медикамент.
В целом, при парентеральном введении требуются меньшие дозы, чем при других способах введения, которые в большей степени зависят от абсорбции. Однако доза для людей предпочтительно содержит 0,0001-25 мг на 1 кг веса тела. Желательную дозу можно представить в виде одной дозы или многократных поддоз, вводимых через соответствующие промежутки времени в течение дня, или, в случае реципиентов женского пола, в виде доз, которые нужно вводить через соответствующие суточные интервалы в течение менструального цикла. Доза, а также режим введения могут различаться для реципиентов женского и мужского пола.
Таким образом, соединения в соответствии с настоящим изобретением могут быть использованы в терапии.
Следующий аспект настоящего изобретения заключается в применении соединения производного тетрагидрохинолина, имеющего общую формулу I, для производства лекарственного средства, предназначенного для использования при лечении нарушений, зависимых от опосредованных рецептором ФСГ путей. Таким образом, нуждающимся в этом пациентам можно вводить подходящие количества соединений в соответствии с данным изобретением.
Следующий аспект изобретения относится к применению соединения производного тетрагидрохинолина, имеющего общую формулу I, для производства лекарственного средства, предназначенного для использования для регулирования фертильности.
В еще одном аспекте изобретение относится к применению соединения, производного тетрагидрохинолина, имеющего общую формулу I, для производства лекарственного средства, предназначенного для использования при лечении бесплодия.
В еще одном аспекте изобретение относится к применению соединения, производного тетрагидрохинолина, имеющего общую формулу I, для производства лекарственного средства, предназначенного для использования для предупреждения фертильности.
Кроме того, соединения в соответствии с изобретением можно использовать для лечения гормон-зависимых нарушений, таких, как рак груди, рак простаты и эндометриоз.
Изобретение иллюстрировано следующими примерами.
ПРИМЕРЫ
Общие примечания
В примерах использованы следующие сокращения: DMA = N,N-диметиланилин, DIPEA = N,N-диизопропилэтиламин, ТЕА = трифторуксусная кислота, D-t-BAD = ди-трет-бутилазодикарбоксилат, TBTU = тетрафторборат O-бензотриазол-1-ил-N,N,N',N'-тетраметилурония, HATU = гексафторфосфат О-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония, Fmoc = 9-флуоренилметоксикарбонил, Fmoc-Cl = 9-флуоренилметоксикарбонилхлорид, ДМФА=N,N-диметилформамид, Вос = трет-бутоксикарбонил, ТГФ = тетрагидрофуран.
Названия конечных продуктов, описанных в примерах, получены при использовании Beilstein Autonom program (версия: 2.02.119).
Если не указано иначе, все конечные продукты приведенных ниже примеров лиофилизировали из смесей вода/1,4-диоксан или смесей вода/ацетонитрил. Если соединение получали в виде соли с HCl или TFA, соответствующие кислоты добавляли в подходящих количествах к смеси растворителей перед лиофилизацией.
Для определения времен удерживания использовали следующие методы аналитической ВЭЖХ.
Метод 1: колонка 5 мкм Luna C-18(2) 150×4,6 мм; поток 1 мл/мин; детектирование 210 нм; температура колонки 40°С; растворитель А: СН3CN/Н2O=1/9 (об./об.); растворитель В: СН3CN; растворитель С: 0,1 М водная трифторуксусная кислота; градиент: растворитель от А/В/С=65/30/5 до 10/85/5 (об./об./об.) в течение 30,00 мин, затем постоянный еще в течение 10,00 мин при А/В/С=10/85/5 (об./об./об.).
Метод 2: идентичен методу 1, за исключением использованного градиента, градиент: растворитель от А/В/С=75/20/5 до 15/80/5 (об./об./об.) в течение 30,00 мин, затем постоянный еще в течение 10,00 мин при А/В/С=15/80/5 (об./об./об.).
Метод 3: колонка 3 мкм Luna C-18(2) 100×2 мм; поток 0,25 мл/мин; детектирование 210 нм; температура колонки 40°С; растворитель А: Н2О; растворитель В: СН3CN; растворитель С: 50 мМ фосфатный буфер, рН 2,1; градиент: растворитель от А/В/070/20/10 до 10/80/10 (об./об./об.) в течение 20,00 мин, затем постоянный еще в течение 10,00 мин при А/В/С=10/80/10 (об./об./об.).
Метод 4: идентичен методу 3, за исключением использованного градиента, градиент: растворитель от А/В/С=65/30/5 до 10/85/5 (об./об./об.) в течение 20,00 мин, затем постоянный еще в течение 10,00 мин при А/В/С=10/85/5 (об./об./об.).
Метод 5: идентичен методу 3, за исключением использованного градиента, градиент: растворитель от А/В=75/25 до 0/100 (об./об.) в течение 20,00 мин, затем постоянный еще в течение 10,00 мин при А/В=0/100 (об./об.).
Метод 6: идентичен методу 1, за исключением использованного градиента, градиент: растворитель от А/В/С=35/60/5 до 10/85/5 (об./об./об.) в течение 30,00 мин, затем постоянный еще в течение 10,00 мин при А/В/С=10/85/5 (об./об./об.).
Для препаративной ВЭЖХ-очистки использовали следующие методы.
Метод А: колонка=Luna С-18. Градиент: 0,1% трифторуксусная кислота в H2O/CH3CN (9/1, об./об.) /СН3CN=100/0 до 0/100 (об./об.) в течение 30-45 мин, в зависимости от простоты разделения. Детектирование: 210 нм. Соответствующие фракции собирали и концентрировали (частично) в вакууме.
Метод В: колонка=Luna С-18. Градиент: Н2О/СН3CN (9/1, об./об.)/СН3CN=80/20 до 0/100 (об./об.) в течение 30-45 мин, в зависимости от простоты разделения. Детектирование: 210 нм.
Пример 1
{1-Ацетил-4-[4-(2-диметиламиноэтокси)фенил]-2,2,4-триметил-1,2,3,4-тетрагидрохинолин-6-ил}амид бифенил-4-карбоновой кислоты
(a) трет-Бутиловый эфир (2,2,4-триметил-1,2-дигидрохинолин-6-ил)карбаминовой кислоты
Смесь N-Boc-1,4-фенилендиамина (75 г), MgSO4 (216 г), 4-трет-бутилкатехола (1,8 г) и йода (4,7 г) в безводном ацетоне (600 мл) кипятили с обратным холодильником в течение 20 ч. MgSO4 удаляли фильтрованием и концентрировали фильтрат в вакууме. Остаток хроматографировали на небольшом слое силикагеля с использованием смеси гептан/этилацетат=8/2 (об/об) в качестве элюента, получая продукт в виде коричневого масла.
Выход 41г.
(b) трет-Бутиловый эфир (1-ацетил-2,2,4-триметил-1,2-дигидрохинолин-6-ил)карбаминовой кислоты
Раствор соединения, описанного в примере 1а (41 г), в пиридине (200 мл) и CH2Cl2 (200 мл) охлаждали до 0°С. Прибавляли по каплям ацетилхлорид (21 мл) в СН2Cl2 (50 мл). По окончании прибавления смесь перемешивали в течение 3 ч при комнатной температуре. Добавляли этилацетат (2 л) и Н2O (2 л) и органический слой отделяли, сушили и концентрировали в вакууме.
Указанное в заголовке соединение получали кристаллизацией из этилацетата.
Выход 23 г.
(с) 1-Ацетил-6-амино-2,2,4-триметил-1,2-дигидрохинолин
Соединение, описанное в примере 1b (15 г), перемешивали в смеси СН2Cl2 и TFA (9/1 (об./об.), 300 мл) в течение 2 ч. Реакционную смесь охлаждали до 0°С и доводили рН до 7 при помощи 2 М водного раствора NaOH. Органический слой отделяли, промывали насыщенным раствором соли, сушили и концентрировали в вакууме, получая сырой продукт, который использовали на следующей стадии без дополнительной очистки.
Выход 10,4 г
(d) (1-Ацетил-2,2,4-триметил-1,2-дигидрохинолин-6-ил)амид бифенил-4-карбоновой кислоты
К раствору соединения, описанного в примере 1с (10 г), и DIPEA (40 мл) в СН2Cl2 (100 мл) прибавляли 4-бифенилкарбонилхлорид (9,8 г) и перемешивали полученную смесь в течение 18 ч при комнатной температуре. Добавляли воду, органический слой отделяли, сушили и концентрировали в вакууме. Продукт кристаллизовали из этилацетата.
Выход 15 г
(е) [1-Ацетил-4-(4-метоксифенил)-2,2,4-триметил-1,2,3,4-тетрагидрохинолин-6-ил]амид бифенил-4-карбоновой кислоты
К смеси соединения, описанного в примере 1d (10,0 г), и безводного анизола (50 мл) прибавляли при перемешивании треххлористый алюминий (9,7 г) и перемешивали полученную смесь при 35°С в течение 18 ч. Спустя это время добавляли воду при 0°С и экстрагировали полученную смесь этилацетатом. Органический слой отделяли, сушили и частично концентрировали в вакууме, выдерживали смесь при 0°С в течение 18 ч. Образовавшийся осадок собирали фильтрованием и сушили в вакууме, получая указанное в заголовке соединение.
Выход 7,9 г
(f) [1-Ацетил-4-(4-гидроксифенил)-2,2,4-триметил-1,2,3,4-тетрагидрохинолин-6-ил]амид бифенил-4-карбоновой кислоты
К раствору соединения, описанного в примере 1е (7,9 г), в CH2Cl2 (200 мл) при 0°С прибавляли раствор трехбромистого бора (5 мл) в CH2Cl2 (50 мл) и выдерживали полученную смесь в течение 4 ч при 0°С. Осторожно прибавляли воду (приблизительно 500 мл) и энергично перемешивали полученную смесь. Органический слой отделяли, сушили и концентрировали в вакууме. Кристаллизацией из этилацетата получали указанное в заголовке соединение.
Выход 6,1 г
(g) {1-Ацетил-4-[4-(2-диметиламиноэтокси)фенил]-2,2,4-триметил-1,2,3,4-тетрагидрохинолин-6-ил}амид бифенил-4-карбоновой кислоты
Общая методика А. К раствору соединения, описанного в примере 1f (70 мг), в ДМФА (2 мл) прибавляли Cs2СО3 (200 мг) и гидрохлорид 2-диметиламиноэтилхлорида (17 мг). Полученную смесь перемешивали в течение ночи, после чего добавляли воду и этилацетат. Органический слой отделяли, сушили и концентрировали в вакууме. Продукт очищали препаративной ВЭЖХ (метод А) и лиофилизировали из смеси СН3CN и воды, содержащий TFA, получая соответствующую соль TFA.
Выход 18 мг (соль TFA). MC-ESI: [М+Н]+=516, 6; ВЭЖХ: Rt=14,96 мин (метод 3).
Пример 2
{1-Ацетил-4-[4-(2-диметиламинопропокси)фенил]-2,2,4-триметил-1,2,3,4-тетрагидрохинолин-6-ил}амид бифенил-4-карбоновой кислоты
По общей методике А соединение, описанное в примере 1f (70 мг), алкилировали гидрохлоридом 3-диметиламинопропилхлорида (19 мг) и Cs2СО3 (200 мг) в ДМФА (2 мл). Продукт очищали препаративной ВЭЖХ (метод А) и лиофилизировали из смеси CH3CN и воды, содержащей TFA, получая соответствующую соль TFA.
Выход 58 мг (соль TFA). MC-ESI: [М+Н]+=590, 4; ВЭЖХ: Rt=15,36 мин (метод 3).
Пример 3
{1-Ацетил-2,2,4-триметил-4-[4-(3-морфолин-4-илпропокси)фенил]-1,2,3,4-тетрагидрохинолин-6-ил}амид бифенил-4-карбоновой кислоты
По общей методике А соединение, описанное в примере 1f (70 мг), алкилировали 3-морфолинпропилхлоридом (26 мг) и Cs2СО3 (200 мг) в ДМФА (2 мл). Продукт очищали препаративной ВЭЖХ (метод А) и лиофилизировали из смеси CH3CN и воды, содержащей TFA, получая соответствующую соль TFA.
Выход 56 мг (соль TFA). MC-ESI: [М+Н]+=631,6; ВЭЖХ: Rt=15,40 мин (метод 3).
Пример 4
{1-Ацетил-2,2,4-триметил-4-[4-(пиридин-2-илметокси)фенил]-1,2,3,4-тетрагидрохинолин-6-ил}амид бифенил-4-карбоновой кислоты
По общей методике А соединение, описанное в примере 1f (100 мг), алкилировали гидрохлоридом 2-пиколилхлорида (33 мг) и Cs2СО3 (325 мг) в ДМФА (5 мл). Продукт очищали препаративной ВЭЖХ (метод А) и лиофилиэировали из смеси СН3CN и воды, содержащей TFA, получая соответствующую соль TFA.
Выход 60 мг (соль TFA). MC-ESI: [М+Н]+=596,4; ВЭЖХ: Rt=19,75 мин (метод 2).
Пример 5
{1-Ацетил-2,2,4-триметил-4-[4-(1-метилпиперидин-3-илметокси)фенил]-1,2,3,4-тетрагидрохинолин-6-ил}амид бифенил-4-карбоновой кислоты
По общей методике А соединение, описанное в примере 1f (100 мг), алкилировали гидрохлоридом 3-хлорметил-1-метилпиперидина (33 мг) и Cs2СО3 (325 мг) в ДМФА (5 мл). Продукт очищали препаративной ВЭЖХ (метод А) и лиофилизировали из смеси СН3CN и воды, содержащей TFA, получая соответствующую соль TFA.
Выход 60 мг (соль TFA). MC-ESI: [M+H]+=615, 4; ВЭЖХ: Rt=16,70 мин (метод 2).
Пример 6
{1-Ацетил-4-[4-(2-диэтиламиноэтокси)фенил]-2,2,4-триметил-1,2,3,4-тетрагидрохинолин-6-ил}амид бифенил-4-карбоновой кислоты
По общей методике А соединение, описанное в примере 1f (100 мг), алкилировали гидрохлоридом 2-диэтиламиноэтилхлорида (35 мг) и Cs2СО3 (325 мг) в ДМФА (5 мл). Продукт очищали препаративной ВЭЖХ (метод А) и лиофилизировали из смеси СН3CN и воды, содержащей TFA, получая соответствующую соль TFA.
Выход 67 мг (соль TFA). MC-ESI: [M+H]+=604, 4; ВЭЖХ: Rt=16,38 мин (метод 2).
Пример 7
{1-Ацетил-2,2,4-триметил-4-[4-(пиридин-4-илметокси)фенил]-1,2,3,4-тетрагидрохинолин-6-ил}амид бифенил-4-карбоновой кислоты
По общей методике А соединение, описанное в примере 1f (100 мг), алкилировали гидрохлоридом 4-пиколилхлорида (33 мг) и Cs2СО3 (325 мг) в ДМФА (5 мл). Продукт очищали препаративной ВЭЖХ (метод А) и лиофилизировали из смеси СН3CN и воды, содержащей TFA, получая соответствующую соль TFA.
Выход 61 мг (соль TFA). MC-ESI: [M+H]+=596,4; ВЭЖХ: Rt=16,64 мин (метод 2).
Пример 8
[3-(4-{1-Ацетил-6-[(бифенил-4-карбонил)амино]-2,2,4-триметил-1,2,3,4-тетрагидрохинолин-4-ил}фенокси)пропил]амид морфолин-4-карбоновой кислоты
По общей методике А соединение, описанное в примере 1f (100 мг), алкилировали (3-хлорпропил)амидом морфолин-4-карбоновой кислоты (53 мг) и Cs2СО3 (325 мг) в ДМФА (5 мл). Продукт очищали препаративной ВЭЖХ (метод А) и лиофилизировали из смеси CH3CN и воды.
Выход 95 мг (соль TFA). MC-ESI: [M+H]+=675, 6; ВЭЖХ: Rt=18,24 мин (метод 3).
Пример 9
{1-Ацетил-4-[4-(2-азепан-1-илэтокси)фенил]-2,2,4-триметил-1,2,3,4-тетрагидрохинолин-6-ил}амид бифенил-4-карбоновой кислоты
По общей методике А соединение, описанное в примере 1f (100 мг), алкилировали гидрохлоридом 2-(гексаметиленимино)этилхлорида (42 мг) и Cs2СО3 (325 мг) в ДМФА (5 мл). Продукт очищали препаративной ВЭЖХ (метод А) и лиофилизировали из смеси CH3CN и воды, содержащей TFA, получая соответствующую соль TFA.
Выход 60 мг (соль TFA). MC-ESI: [М+Н]+=630, 6; ВЭЖХ: Rt=17,25 мин (метод 2).
Пример 10
{1-Ацетил-2,2,4-триметил-4-[4-(пиридин-3-илметокси)фенил]-1,2,3,4-тетрагидрохинолин-6-ил}амид бифенил-4-карбоновой кислоты
По общей методике А соединение, описанное в примере 1f (1,0 г), алкилировали гидрохлоридом 3-пиколоилхлорида (488 мг) и Cs2СО3 (3,2 мг) в ДМФА (10 мл). Продукт очищали препаративной ВЭЖХ (метод А) и лиофилизировали из смеси CH3CN и воды, содержащей TFA, получая соответствующую соль TFA.
Выход 884 мг (соль TFA). MC-ESI: [М+Н]+=596,4; ВЭЖХ: Rt=16,55 мин (метод 3).
Пример 11
[1-Ацетил-4-(4-карбамоилметоксифенил)-2,2,4-триметил-1,2,3,4-тетрагидрохинолин-6-ил]амид бифенил-4-карбоновой кислоты
По общей методике А соединение, описанное в примере 1f (100 мг), алкилировали 2-хлорацетамидом (24 мг) и Cs2СО3 (325 мг) в ДМФА (5 мл). Продукт очищали препаративной ВЭЖХ (метод А) и лиофилизировали из смеси СН3CN и воды, содержащей TFA, получая соответствующую соль TFA.
Выход 40 мг (соль TFA). MC-ESI: [М+Н]+=562,6; ВЭЖХ: Rt=21,63 мин (метод 2).
Пример 12
[1-Ацетил-4-(4-аллилкарбамоилметоксифенил)-2,2,4-триметил-1,2,3,4-тетрагидрохинолин-6-ил]амид бифенил-4-карбоновой кислоты
(a) трет-Бутиловый эфир (4-{1-ацетил-6-[(бифенил-4-карбонил)амино]-2,2,4-триметил-1,2,3,4-тетрагидрохинолин-4-ил}фенокси)уксусной кислоты
Смесь соединения, описанного в примере 1f (2,58 г), трет-бутилбромацетата (826 мкл), К2СО3 (2,8 г) и ацетона (100 мл) перемешивали в течение 18 ч при 50°С. Твердые вещества удаляли фильтрованием и концентрировали фильтрат в вакууме, получая продукт, который использовали на следующей стадии без дополнительной очистки.
Выход 3,2 г.
(b) (4-{1-Ацетил-6-[(бифенил-4-карбонил)амино]-2,2,4-триметил-1,2,3,4-тетрагидрохинолин-4-ил}фенокси)уксусная кислота
Соединение, описанное в примере 12а (3,2 г), перемешивали в смеси СН2Cl2 и TFA (9/1 (об./об.), 100 мл) в течение 3 ч. Добавляли толуол (100 мл) и концентрировали смесь в вакууме, получая сырой продукт, который использовали без дополнительной очистки.
Выход 3,3 г.
(c) [1-Ацетил-4-(4-аллилкарбамоилметоксифенил)-2,2,4-триметил-1,2,3,4-тетрагидрохинолин-6-ил]амид бифенил-4-карбоновой кислоты
Общая методика В. К раствору соединения, описанного в примере 12b (82 мг), аллиламина (37 мг) и DIPEA (226 мкл) в CH2Cl2 (5 мл) прибавляли TBTU (84 мг) при комнатной температуре. Если реакция не завершалась за 18 ч, добавляли еще TBTU и DIPEA.
По завершении реакции добавляли воду, органический слой отделяли, промывали насыщенным раствором соли, сушили и концентрировали в вакууме. Указанное в заголовке соединение очищали препаративной ВЭЖХ (метод А).
Выход 48 мг. MC-ESI: [М+Н]+=602,4; ВЭЖХ: Rt=18,19 мин (метод 4).
Пример 13
{1-Ацетил-4-[4-(изопропилкарбамоилметокси)фенил]-2,2,4-триметил-1,2,3,4-тетрагидрохинолин-6-ил}амид бифенил-4-карбоновой кислоты
По общей методике В соединение, описанное в примере 12b (82 мг), обрабатывали изопропиламином (38 мг), DIPEA (226 мкл), TBTU (84 мг) в СН2Cl2 (5 мл). Указанное в заголовке соединение очищали препаративной ВЭЖХ (метод А).
Выход 45 мг. MC-ESI: [M+H]+=604,6; ВЭЖХ: Rt=18,63 мин (метод 4).
Пример 14
[1-Ацетил-4-(4-диэтилкарбамоилметоксифенил]-2,2,4-триметил-1,2,3,4-тетрагидрохинолин-6-ил]амид бифенил-4-карбоновой кислоты
По общей методике В соединение, описанное в примере 12b (82 мг), обрабатывали гидрохлоридом диэтиламина (47 мг), DIPEA (226 мкл) и TBTU (84 мг) в СН2Cl2 (5 мл). Указанное в заголовке соединение очищали препаративной ВЭЖХ (метод А).
Выход 51 мг. MC-ESI: [M+H]+=618,4; ВЭЖХ: Rt=19,09 мин (метод 4).
Пример 15
[1-Ацетил-2,2,4-триметил-4-(4-{[(пиридин-4-илметил)карбамоил]метокси}фенил
ил]амид бифенил-4-карбоновой кислоты
По общей методике В соединение, описанное в примере 12b (82 мг), обрабатывали 4-пиколиламином (70 мг), DIPEA (226 мкл) и TBTU (84 мг) в СН2Cl2 (5 мл). Указанное в заголовке соединение очищали препаративной ВЭЖХ (метод А) и лиофилизировали из смеси CH3CN и воды, содержащей TFA, получая соответствующую соль TFA.
Выход 52 мг (соль TFA). MC-ESI: [М+Н]+=633,6; ВЭЖХ: Rt=11,31 мин (метод 4).
Пример 16
[1-Ацетил-4-(4-{[(фуран-2-илметил)карбамоил]метокси}фенил)-2,2,4-триметил-1,2,3,4-тетрагидрохинолин-6-ил]амид бифенил-4-карбоновой кислоты
По общей методике В соединение, описанное в примере 12b (82 мг), обрабатывали 2-фурфуриламином (63 мг), DIPEA (226 мкл) и TBTU (84 мг) в CH2Cl2 (5 мл). Указанное в заголовке соединение очищали препаративной ВЭЖХ (метод А).
Выход 50 мг. MC-ESI: [М+Н]+=642,6; ВЭЖХ: Rt=21,31 мин (метод 3).
Пример 17
[1-Ацетил-4-{4-[(2-метоксиэтилкарбамоил)метокси]фенил}-2,2,4-триметил-1,2,3,4-тетрагидрохинолин-6-ил]амид бифенил-4-карбоновой кислоты
По общей методике В соединение, описанное в примере 12b (82 мг), обрабатывали 2-метоксиэтиламином (49 мг), DIPEA (226 мкл) и TBTU (84 мг) в СН2Cl2 (5 мл). Указанное в заголовке соединение очищали препаративной ВЭЖХ (метод А).
Выход 34 мг. MC-ESI: [M+H]+=620,4; ВЭЖХ: Rt=19,70 мин (метод 3).
Пример 18
{1-Ацетил-4-[4-(бензилкарбамоилметокси)фенил]-2,2,4-триметил-1,2,3,4-тетрагидрохинолин-6-ил}амид бифенил-4-карбоновой кислоты
По общей методике В соединение, описанное в примере 12b (82 мг), обрабатывали бензиламином (49 мг), DIPEA (226 мкл) и TBTU (84 мг) в CH2Cl2 (5 мл). Указанное в заголовке соединение очищали препаративной ВЭЖХ (метод А).
Выход 53 мг. MC-ESI: [M+H]+=652,6; ВЭЖХ: Rt=22,26 мин (метод 3).
Пример 19
(1-Ацетил-4-{4-[(2-диметиламиноэтилкарбамоил)метокси]фенил}-2,2,4-триметил-1,2,3,4-тетрагидрохинолин-6-ил)амид бифенил-4-карбоновой кислоты
По общей методике В соединение, описанное в примере 12b (82 мг), обрабатывали N,N-диметилэтилендиамином (49 мг), DIPEA (226 мкл) и TBTU (84 мг) в СН2Cl2 (5 мл). Указанное в заголовке соединение очищали препаративной ВЭЖХ (метод А). В результате лиофилизации из смеси водной HCl и 1,4-диоксана получали указанное в заголовке соединение в виде HCl-соли.
Выход 11 мг (HCl-соль). MC-ESI: [M+H]+=633, 4; ВЭЖХ: Rt=13,74 мин (метод 3).
Пример 20
[1-Ацетил-2,2,4-триметил-4-(4-метилкарбамоилметоксифенил)-1,2,3,4-тетрагидрохинолин-6-ил]амид бифенил-4-карбоновой кислоты
По общей методике В соединение, описанное в примере 12b (82 мг), обрабатывали гидрохлоридом метиламина (20 мг), DIPEA (226 мкл) и TBTU (84 мг) в CH2Cl2 (5 мл).
Выход 35 мг. MC-ESI: [М+Н]+=576,4; ВЭЖХ: Rt=19,25 мин (метод 3).
Пример 21
{1-Ацетил-2,2,4-триметил-4-[4-(2-морфолин-4-ил-2-оксоэтокси)фенил]-1,2,3,4-тетрагидрохинолин-6-ил}амид бифенил-4-карбоновой кислоты
По общей методике В соединение, описанное в примере 12b (110 мг), обрабатывали морфолином (74 мг), DIPEA (296 мкл) и TBTU (109 мг) в СН2Cl2 (5 мл). Указанное в заголовке соединение очищали препаративной ВЭЖХ (метод А).
Выход 85 мг. MC-ESI: [М+Н]+=632, 4; ВЭЖХ: Rt=12,48 мин (метод 3).
Пример 22
N-{1-ацетил-2,2,4-триметил-4-[4-(3-морфолин-4-илпропокси)фенил]-1,2,3,4-тетрагидрохинолин-6-ил}-5-бром-2-метиламинобензамид
(а) 9-Флуоренилметиловый эфир (1-ацетил-2,2,4-триметил-1,2-дигидрохинолин-6-ил)карбаминовой кислоты
К раствору соединения, описанного в примере 1с (17 г), и DIPEA (40 мл) в СН2Cl2 (100 мл), добавляли FmocCl (25 г) и перемешивали полученную реакционную смесь в течение 18 ч при комнатной температуре. Добавляли этилацетат (приблизительно 200 мл) и воду (150 мл), органический слой отделяли, сушили и концентрировали в вакууме. Указанное в заголовке соединение очищали хроматографией на силикагеле с использованием СН2Cl2 в качестве элюента.
Выход 16,6 г.
(b) 9-Флуоренилметиловый эфир [1-ацетил-4-(4-метоксифенил)-2,2,4-триметил-1,2,3,4-тетрагидрохинолин-6-ил]карбаминовой кислоты
К смеси соединения, описанного в примере 22а (16,5 г), и безводного анизола (150 мл) прибавляли при перемешивании треххлористый алюминий (24,2 г) и перемешивали полученную смесь при 35°С в течение 18 ч. Спустя это время добавляли воду при 0°С и экстрагировали полученную смесь этилацетатом. Органический слой отделяли, сушили, частично концентрировали в вакууме и выдерживали смесь при 0°С в течение 18 ч. Образовавшийся осадок собирали фильтрованием и сушили в вакууме, получая указанное в заголовке соединение.
Выход 10,1 г.
(c) 9-Флуоренилметиловый эфир [1-ацетил-4-(4-гидроксифенил)-2,2,4-триметил-1,2,3,4-тетрагидрохинолин-6-ил]карбаминовой кислоты
К смеси соединения, описанного в примере 22b (10,1 г), в безводном СН2Cl2 (500 мл) прибавляли по каплям трехбромистый бор (5,05 мл) и перемешивали полученную смесь в течение 2,5 ч при комнатной температуре. Реакцию гасили ледяной водой при 0°С и добавляли СН2Cl2. Органический слой отделяли, сушили и выдерживали смесь при 4°С в течение 20 ч. Образовавшиеся твердые вещества собирали фильтрованием и сушили в вакууме, получая сырой продукт, который использовали без дополнительной очистки.
Выход 12,5 г.
(d) 1-Ацетил-6-амино-2,2,4-триметил-4-[4-(3-морфолин-4-илпропокси)фенил]-1,2,3,4-тетрагидрохинолин
Смесь соединения, описанного в примере 22с (1,0 г), Cs2СО3 (1,8 г), 4-(3-хлорпропил)морфолина (330 мг) и ДМФА (5 мл) перемешивали при 60°С в течение 18 ч. Добавляли воду и экстрагировали смесь CH2Cl2. Органический слой сушили и концентрировали в вакууме. Указанное в заголовке соединение очищали хроматографией на силикагеле с использованием СН2Cl2/2%-ного концентрированного аммиака в МеОН =1/0 ⇒ 9/1 (об./об.) в качестве элюента.
Выход 527 мг.
(e) N-{1-Ацетил-2,2,4-триметил-4-[4-(3-морфолин-4-илпропокси)фенил]-1,2,3,4-тетрагидрохинолин-6-ил}-5-бром-2-метиламинобензамид
Общая методика С. К раствору соединения, описанного в примере 22d (132 мг), 5-бром-2-метиламинобензойной кислоты (101 мг) и DIPEA (225 мкл) в СН2Cl2 (3 мл) добавляли HATU (166 мг) при комнатной температуре. Реакционную смесь перемешивали в течение 18 ч при комнатной температуре. Добавляли этилацетат (15 мл) и 2 М водную NaOH (15 мл). Органический слой отделяли и промывали 2 М водной NaOH (10 мл) и водой (15 мл), сушили и концентрировали в вакууме. Указанное в заголовке соединение очищали препаративной хроматографией (метод А).
Выход 69,8 мг. MC-ESI: [М+Н]+=663,4; ВЭЖХ: Rt=14,65 мин (метод 3).
Пример 23
N-{1-Ацетил-2,2,4-триметил-4-[4-(3-морфолин-4-илпропокси)фенил]-1,2,3,4-тетрагидрохинолин-6-ил}-3,5-дихлор-2,6-диметоксибензамид
По общей методике С соединение, описанное в примере 22d (132 мг), ацилировали 3,5-дихлор-2,6-диметоксибензойной кислотой (110 мг), DIPEA (255 мкл) и HATU (166 мг) в СН2Cl2 (3 мл). Указанное в заголовке соединение очищали препаративной ВЭЖХ (метод А).
Выход 68,3 мг. MC-ESI: [М+Н]+=684,3; ВЭЖХ: Rt=13,45 мин (метод 3).
Пример 24
[1-Ацетил-4-(4-{2-[(фуран-2-илметил)амино]этокси}фенил)-2,2,4-триметил-1,2,3,4-тетрагидрохинолин-6-ил]амид бифенил-4-карбоновой кислоты
(a) Этиловый эфир (4-{1-ацетил-6-[(бифенил-4-карбонил)амино]-2,2,4-триметил-1,2,3,4-тетрагидрохинолин-4-ил}фенокси)уксусной кислоты
Смесь соединения, описанного в примере 1f (1 г), этилбромацетата (220 мкл), К2СО3 (850 мг) и ацетона (25 мл) перемешивали в течение 6 ч при 50°С. Твердые вещества удаляли фильтрованием, а фильтрат концентрировали в вакууме, получая продукт, который использовали на следующей стадии без дополнительной очистки.
Выход 1,2 г.
(b) {1-Ацетил-4-[4-(2-гидроксиэтокси)фенил]-2,2,4-триметил-1,2,3,4-тетрагидрохинолин-6-ил}амид бифенил-4-карбоновой кислоты
К раствору соединения, описанного в примере 24а (1,2 г), в ТГФ (10 мл) при 0°С осторожно прибавляли LiAlH4 (78 мг) и перемешивали полученную смесь в течение 3 ч при комнатной температуре. Прибавляли по каплям этилацетат (50 мл), затем воду (50 мл). Водный слой отделяли и экстрагировали этилацетатом (50 мл), а объединенные органические фракции промывали насыщенным раствором соли. Органический слой сушили и концентрировали в вакууме, получая продукт, который использовали на следующей стадии без дополнительной очистки.
Выход 1 г.
(c) 2-(4-{1-Ацетил-6-[(бифенил-4-карбонил)амино]-2,2,4-триметил-1,2,3,4-тетрагидрохинолин-4-ил}фенокси)этиловый эфир метансульфокислоты
К раствору соединения, описанного в примере 24b (1 г), и DIPEA (1,7 мл) в СН2Cl2 (15 мл) прибавляли по каплям раствор метансульфонилхлорида (310 мкл) в CH2Cl2 (5 мл). Спустя 2 ч добавляли воду, органический слой отделяли, сушили и концентрировали в вакууме. Указанное в заголовке соединение очищали хроматографией на силикагеле с использованием смеси гептан/этилацетат =9/1 ⇒ 1/1 (об./об.) в качестве элюента.
Выход 870 мг.
(d) [1-Ацетил-4-(4-{2-[(фуран-2-илметил)амино]этокси}фенил)-2,2,4-триметил-1,2,3,4-тетрагидрохинолин-6-ил]амид бифенил-4-карбоновой кислоты
Общая методика D. К раствору соединения, описанного в примере 24с (87 мг), в CH3CN (5 мл) прибавляли 2-фурфуриламин (107 мг) и перемешивали полученную смесь при 70°С в течение 18 ч. Смесь концентрировали в вакууме, а продукт очищали препаративной ВЭЖХ (метод А) и лиофилизировали из смеси СН3CN и воды, содержащей TFA, получая соответствующую соль TFA.
Выход 47 мг (соль TFA). MC-ESI: [M+H]+=628,6; ВЭЖХ: Rt=11,53 мин (метод 4).
Пример 25
(1-Ацетил-4-{4-[2-(2-гидрокси-1,1-диметилэтиламино)этокси]фенил}-2,2,4-триметил-1,2,3,4-тетрагидрохинолин-6-ил)амид бифенил-4-карбоновой кислоты
По общей методике D соединение, описанное в примере 24с (87 мг), обрабатывали 2-амино-2-метилпропан-1-олом (100 мг) в СН3CN (5 мл). Указанное в заголовке соединение очищали препаративной ВЭЖХ (метод А) и лиофилизировали из смеси СН3CN и воды, содержащей TFA, получая соответствующую соль TFA.
Выход 21 мг (соль TFA). MC-ESI: [M+H]+=619,8; ВЭЖХ: Rt=10,95 мин (метод 4).
Пример 26
[1-Ацетил-2,2,4-триметил-4-(4-{2-[(пиридин-3-илметил)амино] этокси}фенил)-1,2,3,4-тетрагидрохинолин-6-ил]амид бифенил-4-карбоновой кислоты
По общей методике D соединение, описанное в примере 24с (87 мг), обрабатывали 3-аминометилпиридином (119 мг) в СН3CN (5 мл). Указанное в заголовке соединение очищали препаративной ВЭЖХ (метод А) и лиофилизировали из смеси CH3CN и воды, содержащей TFA, получая соответствующую соль TFA.
Выход 40 мг (соль TFA). MC-ESI: [M+H]+=639,4; ВЭЖХ:Rt=10,15 мин (метод 4).
Пример 27
(1-Ацетил-4-{4-[2-(2-гидроксиэтиламино)этокси]фенил}-2,2,4-триметил-1,2,3,4-тетрагидрохинолин-6-ил)амид бифенил-4-карбоновой кислоты
По общей методике D соединение, описанное в примере 24с (100 мг), обрабатывали этаноламином (100 мг) в CH3CN (5 мл). Указанное в заголовке соединение очищали препаративной ВЭЖХ (метод А) и лиофилизировали из смеси CH3CN и воды, содержащей TFA, получая соответствующую соль TFA.
Выход 50 мг (соль TFA). MC-ESI: [М+Н]+=592,6; ВЭЖХ: Rt=10,32 мин (метод 1).
Пример 28
(1-Ацетил-4-{4-[2-(2-аминоэтиламино)этокси]фенил}-2,2,4-триметил-1,2,3,4-тетрагидрохинолин-6-ил)амид бифенил-4-карбоновой кислоты
По общей методике D соединение, описанное в примере 24с (100 мг), обрабатывали этилендиамином (110 мг) в CH3CN (5 мл). Указанное в заголовке соединение очищали препаративной ВЭЖХ (метод А) и лиофилизировали из смеси CH3CN и воды, содержащей TFA, получая соответствующую соль TFA.
Выход 45 мг (соль TFA). MC-ESI: [М+Н]+=591,4; ВЭЖХ: Rt=7,04 мин (метод 1).
Пример 29
{1-Ацетил-2,2,4-триметил-4-[4-(2-пиперазин-1-илэтокси)фенил]-1,2,3,4-тетрагидрохинолин-6-ил}амид бифенил-4-карбоновой кислоты
По общей методике D соединение, описанное в примере 24с (100 мг), обрабатывали пиперазином (140 мг) в CH3CN (5 мл). Указанное в заголовке соединение очищали препаративной ВЭЖХ (метод А) и лиофилизировали из смеси СН3CN и воды, содержащей TFA, получая соответствующую соль TFA.
Выход 95 мг (соль TFA). MC-ESI: [М+Н]+=617,6; ВЭЖХ: Rt=9,54 мин (метод 1).
Пример 30
(3-{4-[1-Ацетил-6-(3,5-дихлор-2,6-диметоксибензоиламино)-2,2,4-триметил-1,2,3,4-тетрагидрохинолин-4-ил]фенокси}пропил)амид морфолин-4-карбоновой кислоты
(a) (3-{4-[1-Ацетил-6-амино-2,2,4-триметил-1,2,3,4-тетрагидрохинолин-4-ил]фенокси}пропил)амид морфолин-4-карбоновой кислоты
По методике, аналогичной описанной в примере 22d, соединение, описанное в примере 22с (1,0 г), алкилировали (с сопутствующим удалением Fmoc защитной группы) (3-хлорпропил)амидом морфолин-4-карбоновой кислоты (448 мг) с использованием Cs2СО3 (1,8 г) в ДМФА (5 мл). Указанное в заголовке соединение очищали хроматографией на силикагеле, используя СН2Cl2/2%-ный аммиак в МеОН =1/0 ⇒ 9/1 (об./об.) в качестве элюента.
Выход 894 мг.
(b) (3-{4-[1-Ацетил-6-(3,5-дихлор-2,6-диметоксибензоиламино) -2,2,4-триметил-1,2,3,4-тетрагидрохинолин-4-ил]фенокси}пропил)амид морфолин-4-карбоновой кислоты
По общей методике С соединение, описанное в примере 30а (228 мг), ацилировали 3,5-дихлор-2,6-диметоксибензойной кислотой (230 мг), DIPEA (558 мкл) и HATU (609 мг) в CH2Cl2 (5 мл). Указанное в заголовке соединение очищали препаративной ВЭЖХ (метод А).
Выход 102 мг. MC-ESI: [М+Н]+=727,4; ВЭЖХ: Rt=22,37 мин (метод 2).
Пример 31
N-{1-Ацетил-2,2,4-триметил-4-[4-(2-морфолин-4-илэтокси)фенил]-1,2,3,4-тетрагидрохинолин-6-ил}-3,5-дибромбензамид
(a) 1-Ацетил-6-амино-2,2,4-триметил-4-[4-(2-морфолин-4-илэтокси)фенил]-1,2,3,4-тетрагидрохинолин
Смесь соединения, описанного в примере 22с (1,0 г), Cs2СО3 (1,8 г), гидрохлорида N-(2-хлорэтил)морфолина (375 мг) и ДМФА (5 мл) перемешивали при 60°С в течение 18 ч. Реакция не доходила до конца, и добавляли дополнительные количества Cs2СО3 и гидрохлорида N-(2-хлорэтил)морфолина. По завершении реакции добавляли воду и экстрагировали смесь СН2Cl2. Органический слой сушили и концентрировали в вакууме. Указанное в заголовке соединение очищали хроматографией на силикагеле, используя смесь СН2Cl2/2%-ный аммиак в МеОН =1/0 ⇒ 9/1 (об./об.) в качестве элюента.
Выход 905 мг.
(b) N-{1-Ацетил-2,2,4-триметил-4-[4-(2-морфолин-4-илэтокси)фенил]-1,2,3,4-тетрагидрохинолин-6-ил}-3,5-дибромбензамид
По общей методике С соединение, описанное в примере 31а (157 мг), ацилировали 3,5-дибромбензойной кислотой (150 мг), DIPEA (313 мкл) и HATU (204 мг) в CH2Cl2 (5 мл). Указанное в заголовке соединение очищали препаративной ВЭЖХ (метод А).
Выход 71 мг (соль TFA). MC-ESI: [М+Н]+=100,2; ВЭЖХ: Rt=16,12 мин (метод 2).
Пример 32
N-{1-Ацетил-2,2,4-триметил-4-[4-(2-морфолин-4-илэтокси)фенил]-1,2,3,4-тетрагидрохинолин-6-ил}-2-хлорбензамид
По общей методике С соединение, описанное в примере 31а (150 мг), ацилировали 2-хлорбензойной кислотой (81 мг), DIPEA (299 мкл) и HATU (195 мг) в СН2Cl2 (6 мл). Указанное в заголовке соединение очищали препаративной ВЭЖХ (метод А).
Выход 162 мг (соль TFA). MC-ESI: [М+Н]+=576,4; ВЭЖХ: Rt=9,37 мин (метод 2).
Пример 33
N-{1-Ацетил-2,2,4-триметил-4-[4-(2-морфолин-4-илэтокси)фенил]-1,2,3,4-тетрагидрохинолин-6-ил}-3,5-диметилбензамид
По общей методике С соединение, описанное в примере 31а (200 мг), ацилировали 3,5-диметилбензойной кислотой (103 мг), DIPEA (399 мкл) и HATU (260 мг) в СН2Cl2 (7,5 мл). Указанное в заголовке соединение очищали препаративной ВЭЖХ (метод А).
Выход 57,5 мг (соль TFA). MC-ESI: [М+Н]+=570,4; ВЭЖХ: Rt=12,62 мин (метод 2).
Пример 34
N-{1-Ацетил-2,2,4-триметил-4-[4-(2-морфолин-4-илэтокси)фенил]-1,2,3,4-тетрагидрохинолин-6-ил}-2,5-дихлорбензамид
По общей методике С соединение, описанное в примере 31а (200 мг), ацилировали 2,5-дихлорбензойной кислотой (131 мг), DIPEA (399 мкл) и HATU (260 мг) в СН2Cl2 (7,5 мл). Указанное в заголовке соединение очищали препаративной ВЭЖХ (метод А).
Выход 130 мг (соль TFA). MC-ESI: [М+Н]+=610,2; ВЭЖХ: Rt=11,70 мин (метод 2).
Пример 35
N-{1-Ацетил-2,2,4-триметил-4-[4-(2-морфолин-4-илэтокси)фенил]-1,2,3,4-тетрагидрохинолин-6-ил}-5-метил-2-нитробензамид
По общей методике С соединение, описанное в примере 31а (157 мг), ацилировали 5-метил-2-нитробензойной кислотой (97,3 мг), DIPEA (313 мкл) и HATU (204 мг) в СН2Cl2 (5 мл). Указанное в заголовке соединение очищали препаративной ВЭЖХ (метод А).
Выход 80 мг (соль TFA). MC-ESI: [M+H]+=601,4; ВЭЖХ: Rt=9,95 мин (метод 2).
Пример 36
N-{1-Ацетил-2,2,4-триметил-4-[4-(2-морфолин-4-илэтокси)фенил]-1,2,3,4-тетрагидрохинолин-6-ил}бензамид
По общей методике С соединение, описанное в примере 31а (157 мг), ацилировали бензойной кислотой (65,6 мг), DIPEA (313 мкл) и HATU (204 мг) в CH2Cl2 (5 мл). Указанное в заголовке соединение очищали препаративной ВЭЖХ (метод А).
Выход 59 мг (соль TFA). MC-ESI: [M+H]+=524,4; ВЭЖХ: Rt=9,99 мин (метод 2).
Пример 37
N-{1-Ацетил-2,2,4-триметил-4-[4-(2-морфолин-4-илэтокси)фенил]-1,2,3,4-тетрагидрохинолин-6-ил}-4-трет-бутилбензамид
По общей методике С соединение, описанное в примере 31а (161 мг), ацилировали 4-трет-бутилбензойной кислотой (99 мг), DIPEA (322 мкл) и HATU (210 мг) в CH2Cl2 (5 мл). Указанное в заголовке соединение очищали препаративной ВЭЖХ (метод А).
Выход 80 мг (соль TFA). MC-ESI: [М+Н]+=598,2; ВЭЖХ: Rt=15,39 мин (метод 2).
Пример 38
N-{1-Ацетил-2,2,4-триметил-4-[4-(2-морфолин-4-илэтокси)фенил]-1,2,3,4-тетрагидрохинолин-6-ил}-2,3-дихлорбензамид
По общей методике С соединение, описанное в примере 31а (161 мг), ацилировали 2,3-дихлорбензойной кислотой (106 мг), DIPEA (322 мкл) и HATU (210 мг) в CH2Cl2 (5 мл). Указанное в заголовке соединение очищали препаративной ВЭЖХ (метод А).
Выход 113 мг (соль TFA). MC-ESI: [М+Н]+=610,2; ВЭЖХ: Rt=11,42 мин (метод 2).
Пример 39
N-{1-Ацетил-2,2,4-триметил-4-[4-(2-морфолин-4-илэтокси)фенил]-1,2,3,4-тетрагидрохинолин-6-ил}-4-бромбензамид
По общей методике С соединение, описанное в примере 31а (260 мг), ацилировали 4-бромбензойной кислотой (179 мг), DIPEA (517 мкл) и HATU (338 мг) в CH2Cl2 (5 мл). Указанное в заголовке соединение очищали препаративной ВЭЖХ (метод А).
Выход 127 мг (соль TFA). MC-ESI: [M+H]+=620,2; ВЭЖХ: Rt=12,24 мин (метод 2).
Пример 40
N-{1-Ацетил-2,2,4-триметил-4-[4-(2-морфолин-4-илэтокси)фенил]-1,2,3,4-тетрагидрохинолин-6-ил}-4-метокси-3-метилбензамид
По общей методике С соединение, описанное в примере 31а (260 мг), ацилировали 4-метокси-3-метилбензойной кислотой (148 мг), DIPEA (517 мкл) и HATU (338 мг) в CH2Cl2 (5 мл). Указанное в заголовке соединение очищали препаративной ВЭЖХ (метод А).
Выход 158 мг (соль TFA). MC-ESI: [М+Н]+=568,2; ВЭЖХ: Rt=11,49 мин (метод 2).
Пример 41
N-{1-Ацетил-2,2,4-триметил-4-[4-(2-морфолин-4-илэтокси)фенил]-1,2,3,4-тетрагидрохинолин-6-ил}-4-диметиламинобензамид
По общей методике С соединение, описанное в примере 31а (260 мг), ацилировали 4-диметиламинобензойной кислотой (147 мг), DIPEA (517 мкл) и HATU (338 мг) в СН2Cl2 (5 мл). Указанное в заголовке соединение очищали препаративной ВЭЖХ (метод А).
Выход 95 мг (соль TFA). MC-ESI: [М+Н]+=585,2; ВЭЖХ: Rt=9,53 мин (метод 2).
Пример 42
N-{1-Ацетил-2,2,4-триметил-4-[4-(2-морфолин-4-илэтокси)фенил]-1,2,3,4-тетрагидрохинолин-6-ил}-3-трифторметилбензамид
К раствору соединения, описанного в примере 31а (260 мг), и пиридина (500 мкл) в толуоле (4,5 мл) прибавляли 3-(трифторметил)бензоилхлорид (185 мг). Добавляли этилацетат (15 мл) и воду (15 мл). Органический слой отделяли и промывали водой (15 мл), сушили и концентрировали в вакууме. Указанное в заголовке соединение очищали препаративной ВЭЖХ (метод А).
Выход 200 мг (соль TFA). MC-ESI: [М+Н]+=610,2; ВЭЖХ: Rt=13,23 мин (метод 2).
Пример 43
N-{1-Ацетил-2,2,4-триметил-4-[4-(2-морфолин-4-илэтокси)фенил]-1,2,3,4-тетрагидрохинолин-6-ил}-3-нитробензамид
К раствору соединения, описанного в примере 31а (260 мг), и пиридина (500 мкл) в толуоле (4,5 мл) прибавляли 3-нитробензоилхлорид (165 мг). Добавляли этилацетат (15 мл) и воду (15 мл). Органический слой отделяли и промывали водой (15 мл), сушили и концентрировали в вакууме. Указанное в заголовке соединение очищали препаративной ВЭЖХ (метод А).
Выход 167 мг (соль TFA). MC-ESI: [М+Н]+=587,4; ВЭЖХ: Rt=10,28 мин (метод 2).
Пример 44
СНО-ФСГ-биоактивность in vitro
ФСГ-активность соединений проверяли на клетках яичника китайского хомячка (СНО), стабильно трансфицированных ФСГ-рецептором человека и котрансфицированных цАМФ-чувствительным элементом (CRE)/промотором, направляющим экспрессию репортерного гена люциферазы светлячков. Связывание лиганда с Gs-связанным рецептором ФСГ приведет к повышению цАМФ, которое, в свою очередь, вызовет повышенную трансактивацию люциферазной репортерной конструкции. Для проверки антагонистических свойств был добавлен рекомбинантный ФСГ в концентрации, вызывающей приблизительно 80% максимальной стимуляции накопления цАМФ в отсутствие тестируемого соединения (рек-hФСГ; 10 мЕ/мл). Сигнал люциферазы количественно оценивали с использованием счетчика люминесценции. Для тестируемых соединений рассчитывали значения EC50 (концентрация тестируемого соединения, вызывающая половину максимальной (50%) стимуляции или снижения). Для этой цели использовали компьютерную программу GraphPad PRISM, версию 3,0 (GraphPad software Inc., San Diego).
Соединения всех примеров проявляли величины ЕС50 (IC50), меньшие 10-5 М, как в агонистической, так и в антагонистической схеме анализа или и в той, и в другой. Соединения примеров 3, 4, 7, 10-13, 16, 36, 37, 39, 41 и 42 проявили величины EC50 (IC50), меньшие 10-7 М, по крайней мере, в одном из анализов.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ТЕТРАГИДРОХИНОЛИНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ, ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ РЕГУЛЯТОРОВ ФЕРТИЛЬНОСТИ | 2003 |
|
RU2328488C2 |
| ТЕТРАГИДРОХИНОЛИНОВЫЕ ПРОИЗВОДНЫЕ | 2002 |
|
RU2347570C2 |
| ПРОИЗВОДНЫЕ 4-ФЕНИЛ-5-ОКСО-1,4,5,6,7,8-ГЕКСАГИДРОХИНОЛИНА В КАЧЕСТВЕ ЛЕКАРСТВЕННЫХ СРЕДСТВ ДЛЯ ЛЕЧЕНИЯ БЕСПЛОДИЯ | 2006 |
|
RU2403249C2 |
| (ДИГИДРО)ПИРРОЛО[2,1-А]ИЗОХИНОЛИНЫ | 2009 |
|
RU2495037C2 |
| 4-ФЕНИЛ-5-ОКСО-1,4,5,6,7,8-ГЕКСАГИДРОХИНОЛИНОВЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ЛЕКАРСТВЕННЫХ СРЕДСТВ ДЛЯ ЛЕЧЕНИЯ БЕСПЛОДИЯ | 2006 |
|
RU2412170C2 |
| ПРОИЗВОДНЫЕ БОРОНОВОЙ КИСЛОТЫ | 2015 |
|
RU2717558C2 |
| ПРОИЗВОДНЫЕ 4-ФЕНИЛ-5-ОКСО-1, 4, 5, 6, 7, 8-ГЕКСАГИДРОХИНОЛИНА В КАЧЕСТВЕ ЛЕКАРСТВЕННЫХ СРЕДСТВ ДЛЯ ЛЕЧЕНИЯ БЕСПЛОДИЯ | 2006 |
|
RU2403243C2 |
| ЗАМЕЩЕННЫЕ ДИГИДРОПИРАЗОЛОНЫ ДЛЯ ЛЕЧЕНИЯ КАРДИОВАСКУЛЯРНЫХ И ГЕМАТОЛОГИЧЕСКИХ ЗАБОЛЕВАНИЙ, ИХ ПРИМЕНЕНИЕ, ЛЕКАРСТВЕННОЕ СРЕДСТВО И СПОСОБ ЛЕЧЕНИЯ И/ИЛИ ПРОФИЛАКТИКИ | 2012 |
|
RU2611012C2 |
| ИНГИБИТОР СЕРИНОВЫХ ПРОТЕАЗ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ЕГО ОСНОВЕ | 1999 |
|
RU2232760C2 |
| СОЕДИНЕНИЯ 8-МЕТИЛ-1-ФЕНИЛИМИДАЗО[1, 5-а]ПИРАЗИНА | 2011 |
|
RU2560162C2 |
Настоящее изобретение относится к производным тетрагидрохинолина, имеющим общую формулу (I), или их фармацевтически приемлемым солям, где R1 и R2 представляют собой Н, Me; R3 представляет собой (2-6С)-гетероциклоалкил(1-4С)алкил, (2-5С)гетероарил(1-4С)алкил, (6С)арил(1-4С)алкил, (2-6С)гетероциклоалкилкарбониламино(2-4С)алкил, R5-(2-4C)алкил или R5-карбонил(1-4C)алкил; R4 представляет собой (2-5С)гетероарил, (6С)арил, необязательно замещенный одним или более заместителями, выбранными из брома, хлора, нитро, фенила, (1-4С)алкила, трифторметила, (1-4С)алкокси или (1-4С)алкиламино, или (2-6С)гетероциклоалкил; R5 представляет собой (ди)(1-4С)алкиламино, (1-4С)алкокси, амино, гидрокси, (6С)ариламино, (ди)(3-4С)алкениламино, (2-5С)гетероарил(1-4С)алкиламино, (6С)арил(1-4С)алкиламино, (ди)[(1-4С)алкокси(2-4С)алкил]амино, (ди)[(1-4С)алкиламино(2-4С)алкил]амино, (ди)[амино(2-4С)алкил]амино или (ди)[гидрокси(2-4С)алкил]амино. Также изобретение относится к фармацевтической композиции на основе соединения формулы (I) и применению соединения формулы (I). Технический результат: получены новые производные тетрагидрохинолина, обладающие модулирующей активностью в отношении ФСГ рецептора. 3 н. и 7 з.п. ф-лы.
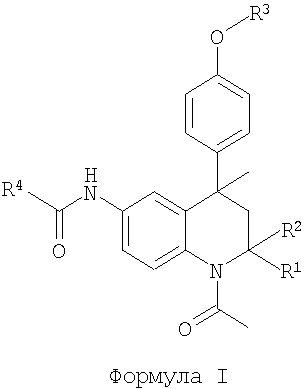
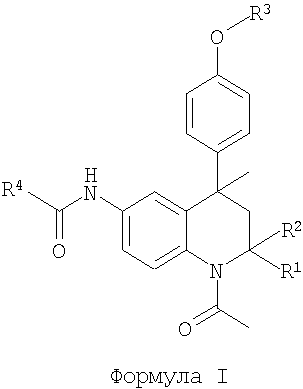
или его фармацевтически приемлемая соль, в которой
R1 и R2 представляют собой Н, Me;
R3 представляет собой (2-6С)гетероциклоалкил(1-4С)алкил, (2-5С)гетероарил(1-4С)алкил, (6С)арил(1-4С)алкил, (2-6С)гетероциклоалкилкарбониламино(2-4С)алкил, R5-(2-4C)алкил или R5-карбонил(1-4С)алкил;
R4 представляет собой (2-5С)гетероарил, (6С)арил, необязательно замещенный одним или более заместителями, выбранными из брома, хлора, нитро, фенила, (1-4С)алкила, трифторметила, (1-4С)алкокси или (1-4С)алкиламино; или (2-6С)гетероциклоалкил;
R5 представляет собой (ди)(1-4С)алкиламино, (1-4С)алкокси, амино, гидрокси, (6С)ариламино, (ди)(3-4С)алкениламино, (2-5С)гетероарил(1-4С)алкиламино, (6С)арил(1-4С)алкиламино, (ди)[(1-4С)алкокси(2-4С)алкил]амино, (ди)[(1-4С)алкиламино(2-4С)алкил]амино, (ди)[амино(2-4С)алкил]амино или (ди)[гидрокси(2-4С)алкил]амино.
| WO 0008015 А, 17.02.2000 | |||
| US 6200963 А, 13.03.2001 | |||
| Способ получения производных 1-ацил-2,3-дигидро-4 /IH/-хинолинон-4-оксима-0-сульфоновой кислоты или их солей | 1988 |
|
SU1779246A3 |
| АРИЛЦИКЛОАЛКИЛЬНЫЕ ПРОИЗВОДНЫЕ, ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ НА ИХ ОСНОВЕ | 1993 |
|
RU2125553C1 |

