Данное изобретение относится к соединению, обладающему модуляторной активностью в отношении FSH рецептора, в частности, к производному тетрагидрохинолина, фармацевтической композиции, содержащей данное соединения, а также к применению указанного соединения в терапии.
Гонадотропины выполняют функции, важные для различных процессов в организме, включая метаболизм, регуляцию температуры и репродуктивный процесс. Гонадотропины воздействуют на специфические клетки гонадного типа, инициируя овариальную и тестикулярную дифференциацию и стероидогенез. Например, гипофизарный гонадотропный FSH (фолликулостимулирующий гормон) играет ключевую роль в стимулировании развития и созревания фолликул, в то время как LH (лютеинизирующий гормон) индуцирует овуляцию (Sharp, R.M. Clin Endocrinol. 33:787-807, 1990; Dorrington and Armstrong, Recent Prog. Horm. Res. 35:301-342, 1979). В настоящее время FSH клинически применяется в сочетании с LH или hCG для овариального стимулирования, то есть овариального гиперстимулирования для in vitro оплодотворения (in vitro fertilization - IVF), и индукции овуляции у женщин, страдающих бесплодием вследствие отсутствия овуляции (Insler, V., Int. J. Fertility 33:85-97, 1988, Navot and Rosenwaks, J. Vitro Fert. Enbryo Transfer 5:3-13, 1988), а также для лечения мужского гипогонадизма и мужского бесплодия.
Гонадотропный FSH высвобождается из передней доли гипофиза под влиянием гонадотропин-высвобождающего гормона и эстрогенов и из плаценты во время беременности. В женском организме FSH воздействует на яичники, ускоряя развитие фолликул, и является основным гормоном, регулирующим секрецию эстрогенов. В мужском организме FSH является ответственным за целостность семенных канальцев и воздействует на клетки Сертоли для поддержки гаметогенеза. Очищенный FSH используется для лечения женского бесплодия и некоторых типов нарушений сперматогенеза у мужчин. Гонадотропины, предназначенные для терапевтических целей, могут быть выделены из мочи человека и имеют низкий уровень чистоты (Morse et al., Amer. J. Reproduct. Immunol. and Microbiology 17:143, 1988). Альтернативно, они могут быть получены в виде рекомбинантных гонадотропинов. Рекомбинантный FSH человека является коммерчески доступным и используется для вспомогательной репродукции. (Olijve et al. Mol. Hum. Reprod. 2:371, 1996; Devroey et al. Lancet 339:1170, 1992).
Действия FSH гормона опосредуются специфическим плазменным мембранным рецептором, который является представителем большого семейства рецепторов, связываемых G-белком. Эти рецепторы состоят из единственного полипептида с семью трансмембранными доменами и способны взаимодействовать с G-белком, что приводит, например, к активации аденилатциклазы.
FSH рецептор является высоко специфической мишенью в процессе роста овариального фолликула и экспрессирован только в яичнике. Блокирование данного рецептора или ингибирование передачи сигнала, который обычно индуцируется после активации FSH-опосредуемого рецептора, будет нарушать развитие фолликула и, как следствие, приводить к нарушению овуляции и фертильности. Поэтому FSH антагонисты с низкой молекулярной массой составили основу новых контрацептивных средств. Такие FSH антагонисты способны вызывать задержку развития фолликулы (отсутствие овуляции) с сохранением выработки эстрогена на уровне, достаточном для предотвращения неблагоприятных эффектов, например, на костную массу. С другой стороны, соединения, которые стимулируют активность FSH рецептора, могут имитировать гонадотропное действие на природный лиганд.
Данное изобретение описывает получение низкомолекулярных аналогов гормона, которые обладают селективной модуляторной активностью в отношении FSH рецептора. Соединения данного изобретения могут использоваться в качестве либо (частичных) агонистов, либо (частичных) антагонистов FSH рецептора.
Итак, в настоящее время установлено, что представленный далее класс производных тетрагидрохинолина формулы I или их фармацевтически приемлемые соли обладают модуляторной активностью в отношении FSH.
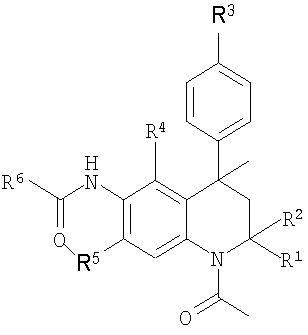
Формула I
где
R1 и R2 представляют собой Н или Ме;
R3 представляет собой Н, гидрокси, (1-4С)алкокси, (ди)(1-4С)алкиламино(2-4С)алкокси или (2-6)гетероциклоалкил(2-4С)алкокси;
R4 представляет собой Н, ОН, (1-4С)алкокси или R7;
R5 представляет собой Н, ОН, (1-4С)алкокси или R7;
при условии, что если R4 представляет собой Н, то R5 отличен от Н, ОН или (1-4С)алкокси, и если R5 представляет собой Н, то R4 отличен от Н, ОН или (1-4С)алкокси;
R6 представляет собой (2-5С)гетероарил, (6С)арил, (3-8С)циклоалкил, (2-6С)гетероциклоалкил или (1-6С)алкил;
R7 представляет собой амино, (ди)(1-4С)алкиламино, (6С)арилкарбониламино, (6С)арилкарбонилокси, (2-5С)гетероарилкарбониламино, (2-5С)гетероарилкарбонилокси, R8-(2-4С)алкиламино, R8-(2-4С)алкокси, R9-метиламино или R9-метокси;
R8 представляет собой гидрокси, амино, (1-4С)алкокси, (ди)(1-4С)алкиламино, (2-6С)гетероциклоалкил, (2-6С)гетероциклоалкилкарбониламино, (ди)(1-4С)алкиламинокарбониламино или (1-4С)алкоксикарбониламино; и
R9 представляет собой аминокарбонил, (ди)(1-4С)алкиламинокарбонил, (2-5С)гетероарил или (6С)арил.
R4 и R5 могут быть независимо выбраны из каждой из групп, описанных выше, и не должны быть одинаковыми.
Соединения согласно данному изобретению модулируют функцию FSH рецептора и могут использоваться для таких же клинических задач, что и природный FSH, если они ведут себя подобно агонистам, с тем преимуществом, что они проявляют свойства измененной стабильности и могут вводиться иначе. Если они блокируют FSH рецептор, то могут использоваться, например, в качестве контрацептивного средства.
Следовательно, модуляторы FSH рецептора данного изобретения могут использоваться для лечения бесплодия, для контрацепции и для лечения гормон-зависимых расстройств, таких как рак молочной железы, рак предстательной железы и эндометриоз.
Представленные далее термины, которые используются в описании и формуле данного изобретения, как подразумевается, имеют следующие значения.
Термин «(1-4С)алкил», используемый в данном описании, означает разветвленную или неразветвленную алкильную группу, содержащую 1-4 атома углерода, например, метил, этил, пропил, изопропил, бутил, втор-бутил и трет-бутил.
Термин «(1-6С)алкил», используемый в данном описании, означает разветвленную или неразветвленную алкильную группу, содержащую 1-6 атомов углерода, например, метил, этил, пропил, изопропил, бутил, втор-бутил, трет-бутил и гексил. (1-5С)Алкильные группы являются предпочтительными, причем наиболее предпочтителен (1-4С)алкил.
Термин «(3-8С)циклоалкил» означает циклоалкильную группу, содержащую 3-8 атомов углерода, например, циклопропил, циклобутил, циклопентил, циклогексил, циклогептил и циклооктил. Предпочтительными являются (3-6С)циклоалкильные группы.
Термин «(2-6С)гетероциклоалкил» означает гетероциклоалкильную группу, содержащую 2-6 атомов углерода, предпочтительно 3-5 атомов углерода, и включающую, по меньшей мере, один гетероатом, выбранный из атомов N, O и/или S, которая может присоединяться через гетероатом, если это выполнимо, или через атом углерода. Предпочтительными гетероатомами являются N или О. Наиболее предпочтительными являются пиперидил, пиперазинил, морфолинил и пирролидинил.
Термин «(1-4С)алкокси» означает алкоксигруппу, содержащую 1-4 атома углерода, причем алкильный фрагмент принимает значение, определенное выше. (1-2С)Алкоксигруппы являются предпочтительными.
Термин «(2-4С)алкокси» означает алкоксигруппу, содержащую 2-4 атома углерода, причем алкильный фрагмент принимает значение, определенное выше.
Термин «(ди)(1-4С)алкиламино» в данном описании означает аминогруппу, монозамещенную или дизамещенную алкильными группами, каждая из которых содержит 1-4 атома углерода и принимает значение, определенные выше.
Термин «(6С)арил» в данном описании означает фенильную группу, которая может быть необязательно замещенной одним или несколькими заместителями, выбранными из гидрокси, амино, йода, брома, хлора, фтора, нитро, трифторметила, циано, фенила, (1-4С)алкила, (1-4С)алкокси, (1-4С)(ди)алкиламино, алкила, алкокси и (ди)алкиламиногрупп, значения которых определены выше, например фенила, 3,5-дибромфенила, 4-бифенила, 3,5-дихлорфенила, 3-бром-6-метиламинофенила, 3-хлор-2,6-диметоксифенила и 3,5-диметилфенила.
Термин «(2-5С)гетероарил» означает замещенную или незамещенную ароматическую группу, содержащую 2-5 атомов углерода и, по меньшей мере, один гетероатом, выбранный из N, O и/или S, например, имидазолил, пиридил, пиримидил, тиенил или фурил. Заместители на гетероарильной группе могут выбираться из группы заместителей, указанных для (6С)арильной группы. Гетероарильная группа может присоединяться через атом углерода или гетероатом, если это возможно. Предпочтительными гетероарильными группами являются тиенил, фурил и пиридил.
Термин «ди(1-4С)алкиламино(2-4С)алкокси» в данном описании означает (ди)алкиламиногруппу, в которой алкильный фрагмент или алкильные фрагменты, каждый из которых содержит 1-4 атома углерода, присоединен(ы) через аминогруппу к алкильному фрагменту алкоксигруппы, содержащей 2-4 атома углерода, где (ди)алкиламиногруппа и алкоксигруппа принимают значения, определенные выше.
Термин «(2-6С)гетероциклоалкил(2-4С)алкокси» в данном описании означает гетероциклоалкильную группу, содержащую 2-6 атомов углерода и присоединенную к алкильному фрагменту алкоксигруппы, содержащей 2-4 атома углерода, где алкоксигруппа и гетероциклоалкильная группа принимают значения, определенные выше.
Термин «(6С)арилкарбониламино» в данном описании означает фенильную группу, необязательно замещенную одним или несколькими заместителями, выбранными из группы заместителей, указанных для (6С)арильной группы, которая присоединена к карбонильному фрагменту карбониламиногруппы, где (6С)арильный фрагмент принимает значения, определенные выше.
Термин «(6С)арилкарбонилокси» в данном описании означает фенильную группу, необязательно замещенную одним или несколькими заместителями, выбранными из группы заместителей, указанных для (6С)арильной группы, которая присоединена к карбонильному фрагменту карбонилоксигруппы, где (6С)арильный фрагмент принимает значения, определенные выше.
Термин «(2-5С)гетероарилкарбониламино» в данном описании означает гетероарильную группу, содержащую 2-5 атомов углерода, необязательно замещенную одним или несколькими заместителями, выбранными из группы заместителей, указанных для (6С)арильной группы, и присоединенную к карбонильному фрагменту карбониламиногруппы. Гетероарильный фрагмент в гетероарилкарбониламиногруппе принимает значения, определенные выше.
Термин «(2-5С)гетероарилкарбонилокси» в данном описании означает гетероарильную группу, содержащую 2-5 атомов углерода, необязательно замещенную одним или несколькими заместителями, выбранными из группы заместителей, указанных для (6С)арильной группы, и присоединенную к карбонильному фрагменту карбонилоксигруппы. Гетероарильный фрагмент в гетероарилкарбонилоксигруппе принимает значения, определенные выше.
Термин «(2-6С)гетероциклоалкилкарбониламино» в данном описании означает гетероциклоалкильную группу, содержащую 2-6 атомов углерода и присоединенную к карбонильному фрагменту карбониламиногруппы, где гетероциклоалкильная группа принимает значения, определенные выше.
Термин «(ди)(1-4С)алкиламинокарбонил» в данном описании означает (ди)алкиламиногруппу, в которой алкильная(ые) группа(ы) содержит(ат) 1-4 атома углерода и которая присоединена к карбонильной группе через аминогруппу, где (ди)алкиламиногруппа принимает значения, определенные выше.
Термин «(ди)(1-4С)алкиламинокарбониламино» в данном описании означает (ди)алкиламиногруппу, в которой алкильная(ые) группа(ы) содержит(ат) 1-4 атома углерода и которая присоединена через аминогруппу к карбонильному фрагменту карбониламиногруппы, обеспечивая, таким образом, функциональность мочевины, где (ди)алкиламиногруппа принимает значения, определенные выше.
Термин «(1-4С)алкоксикарбониламино» в данном описании означает алкоксигруппу, содержащую 1-4 атома углерода, присоединенную к карбонильному фрагменту карбониламиногруппы, обеспечивая, таким образом, карбаматную функциональность, где алкоксигруппа принимает значения, определенные выше.
Термин «R8-(2-4C)алкиламино» в данном описании означает группу R8, присоединенную к алкильному фрагменту (2-4С)алкиламиногруппы, значения которой определенные выше.
Термин «R8-(2-4С)алкокси» в данном описании означает группу R8, присоединенную к алкильному фрагменту (2-4С)алкоксигруппы, значения которой определены выше.
Термин «R9-метиламино» в данном описании означает группу R9, присоединенную к метильному фрагменту метиламиногруппы.
Термин «R9-метокси» в данном описании означает группу R9, присоединенную к метильному фрагменту метоксигруппы.
Термин «фармацевтически приемлемая соль» относится к таким солям, которые в области медицинского применения подходят для использования в контакте с тканями людей и низших животных без избыточной токсичности, раздражения, аллергической реакции и т.п. и характеризуются разумным соотношением польза/риск. Фармацевтически приемлемые соли хорошо известны в данной области техники. Они могут быть получены в процессе конечного выделения и очистки соединений данного изобретения или отдельно взаимодействием функциональной группы свободного основания, если она присутствует, с подходящей минеральной кислотой, такой как хлористоводородная кислота, фосфорная кислота или серная кислота, или с органической кислотой, такой как, например, аскорбиновая кислота, лимонная кислота, винная кислота, молочная кислота, малеиновая кислота, малоновая кислота, фумаровая кислота, гликолевая кислота, янтарная кислота, пропионовая кислота, уксусная кислота, метансульфоновая кислота и т.п. Функциональная группа кислоты, если таковая присутствует, может подвергаться взаимодействию с органическим или минеральным основанием, таким как гидроксид натрия, гидроксид калия или гидроксид лития.
Таким образом, изобретение относится к соединениям формулы I, которые определены выше.
В другом примере осуществления данного изобретения оно относится к соединениям формулы I, где R3 представляет собой Н, гидрокси или (1-4С)алкокси.
Изобретение также относится к соединениям формулы I, где R4 представляет собой Н, ОН или (1-4С)алкокси.
В еще одном варианте осуществления данного изобретения оно предоставляет соединения формулы I, где R5 представляет собой ОН, (1-4С)алкокси или R7.
В еще одном варианте осуществления данного изобретения оно предоставляет соединения формулы I, где R6 представляет собой (2-5С)гетероарил, (6С)арил, (3-8С)циклоалкил или (1-6С)алкил.
В еще одном аспекте данное изобретение относится к соединениям формулы I, где R6 представляет собой (2-5С)гетероарил или (6С)арил.
В еще одном аспекте гетероарильная группа в R6 состоит из 4 или 5 атомов углерода.
Изобретение также относится к соединениям формулы I, где R7 представляет собой (ди)(1-4С)алкиламино, (2-5С)гетероарилкарбониламино, (2-5С)гетероарилкарбонилокси, R8-(2-4C)алкокси, R9-метиламино или R9-метокси.
Еще одним аспектом данного изобретения являются соединения согласно формуле I, где R7 представляет собой (ди)(1-4С)алкиламино, (2-5С)гетерарилкарбонилокси, R8-(2-4C)алкокси, R9-метиламино или R9-метокси.
В еще одном аспекте данное изобретение относится к соединениям формулы I, где R7 представляет собой (ди)(1-4С)алкиламино, R8-(2-4С)алкокси, R9-метиламино или R9-метокси.
В еще одном аспекте данное изобретение относится к соединениям формулы I, где R8-(2-4С)алкокси в группе R7 представляет собой R8-этокси.
В еще одном аспекте данное изобретение относится к соединениям формулы I, где R8-(2-4С)алкиламино в группе R7 представляет собой R8-этиламино.
В еще одном аспекте данное изобретение предоставляет соединения формулы I, где R8 представляет собой амино, (ди)(1-4С)алкиламино, (2-6С)гетероциклоалкил, (2-6С)гетероциклоалкилкарбониламино или (1-4С)алкоксикарбониламино.
В еще одном примере осуществления данного изобретения оно предоставляет соединения формулы I, где R8 представляет собой амино, (ди)(1-4С)алкиламино, (2-6С)гетероциклоалкил или (1-4С)алкоксикарбониламино.
В еще одном примере осуществления данного изобретения оно предоставляет соединения формулы I, где R8 представляет собой амино, (ди)(1-4С)алкиламино, (2-6С)гетероциклоалкил или (2-6С)гетероциклоалкилкарбониламино.
Изобретение относится также к соединениям согласно формуле I, где R8 представляет собой амино, (ди)(1-4С)алкиламино или (2-6С)гетероциклоалкил.
В еще одном аспекте данного изобретения R8 в соединениях формулы I представляет собой (ди)(1-4С)алкиламино или (2-6С)гетероциклоалкил.
В еще одном аспекте данное изобретение относится к соединениям согласно формуле I, где гетероциклоалкильная группа в R8 состоит из 4 или 5 атомов С.
Согласно еще одному примеру осуществления данного изобретения R9 в соединениях согласно формуле I представляет собой аминокарбонил, (2-5С)гетероарил или (6С)арил.
Согласно еще одному примеру осуществления данного изобретения гетероарильная группа в R9 соединений согласно формуле I состоит из 3, 4 или 5 атомов С.
Еще один аспект данного изобретения относится к соединениям, в которых все конкретные значения групп R1-R9, определенные в описании выше, объединены в соединении формулы I.
Далее представлены приемлемые способы получения соединений данного изобретения.

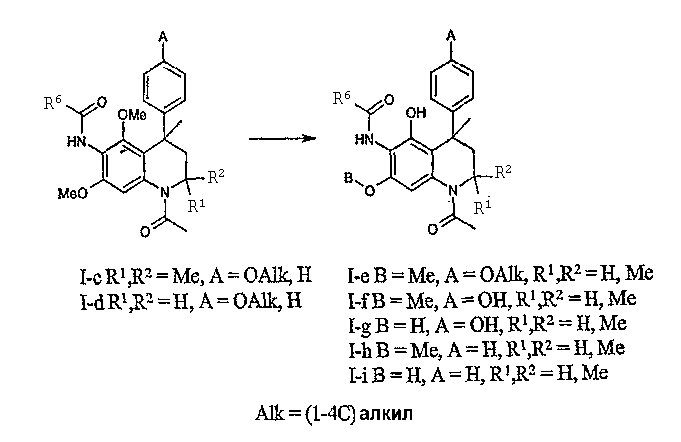
Соединения данного изобретения, в которых R4 и R5 представляют собой (1-4С)алкокси, R1 и R2 представляют собой метил и R6 принимает значения, определенные выше, могут быть получены исходя из подходящих замещенных анилинов общей формулы II в соответствии с реакцией Скраупа (Skraup), подробно описанной в литературе, с получением производных 2,2,4-триметил-1,2-дигидрохинолина формулы IIIa.
Аналогичные реакции циклоконденсации Скраупа описаны в литературе: A. Knoevenagel, Chem. Ber. 54:1726, 1921; R.L. Atkins and D.E. Bliss, J. Org. Chem. 43:1975, 1978; J.V. Johnson, B.S. Rauckman, D.P. Baccanari, B. Roth, J.Med. Chem. 32:1942, 1989; W.C. Lin, S.-T. Huang, S.-T. Lin, J. Chin. Chem. Soc. 43:497, 1996; J.P. Edwards, S.J. West, K.B. Marschke, D.E. Mais, M.M. Gottardis and T.K. Jones, J. Med. Chem. 41:303, 1998.
Описанная выше реакция обычно проводится при повышенной температуре в ацетоне или мезитилоксиде в присутствии йода или протонной кислоты, такой как хлористоводородная кислота, п-толуолсульфокислота или водный йодистый водород. Альтернативно 1,2-дигидро-2,2,4-триметилхинолины формулы III-a могут быть получены взаимодействием соответствующего анилина формулы II с ацетоном в присутствии MgSO4, 4-трет-бутилкатехола и йода (L.G. Hamann, R.I. Higuchi, L. Zhi, J.P. Edwards and X.-N. Wang, J. Med. Chem., 41:623, 1998). В соответствии с другой методикой реакция может проводиться в ацетоне с использованием трифлатов лантанидов (например, трифлата скандия) в качестве катализаторов. В этом случае реакция может проводиться при комнатной температуре или при повышенных температурах с использованием обычного нагрева или микроволнового облучения (M.E. Theoclitou, L.A. Robinson, Tetrahedron Lett. 43:3907, 2002).
Исходные вещества могут быть легко получены непосредственно из коммерческих источников или легко синтезированы квалифицированными специалистами в данной области техники.
Соединения формулы III-b могут быть получены из анилинов общей формулы II взаимодействием с метилвинилкетоном. Аналогичные реакции циклизации описаны в патенте США № 2686182 (Badische Anilin- & Soda-Fabric Aktiengesellschaft).
Последующее 1-N-ацетилирование соединений формулы III-a-b, где R1 и R2 принимают значения, описанные выше, может проводиться с использованием стандартных условий. В обычном эксперименте соединения формулы III кипятят с обратным холодильником в уксусном ангидриде или подвергают взаимодействию в растворителе, таком как дихлорметан, тетрагидрофуран, толуол или пиридин, с хлорангидридом уксусной кислоты в присутствии основания, такого как N,N-диизопропилэтиламин, триэтиламин или гидрид натрия, с получением N-ацетилированных 4-метил-1,2-дигидрохинолин-производных формулы IV-a-b.
Аналогичные реакции N-ацилирования дигидрохинолиновой основы описаны в литературе: G. Reddelien, A. Thurm, Chem. Ber. 65:1511, 1932; Zh. V. Shmyreva, Kh. S. Shikhaliev, E.B. Shpanig, Izv. Vyssh. Uchebn. Zaved., Khim. Khim. Teknol. 31:45, 1988; Zh. V. Shmyreva, Kh. S. Shikhaliev, L.P. Zalukaev, Y.A. Ivanov, Y.S. Ryabokobylko, I.E. Pokrovskaya, Zh. Obshch. Khim. 59:1391, 1989.
Введение необходимой (замещенной) фенильной группы в положение 4 дигидрохинолиновой основы может проводиться посредством алкилирования бензола или подходящего замещенного бензола соединениями общей структуры IV-a-b в соответствии с реакцией Фриделя-Крафтса. Данная реакция обычно проводится при повышенных температурах в чистом бензоле или в подходящем замещенном бензоле, либо в подходящем инертном растворителе, таком как гептан или гексан, с бензолом, или подходящим замещенным бензолом в качестве реагента, в присутствии катализатора - кислоты Льюиса (например, AlCl3, AlBr3, FeCl3 или SnCl4). Реакции алкилирования Фриделя-Крафтса с 2,2,4-триметил-1,2-дигидрохинолинами описаны в литературе: B.A. Lugovik, L.G. Yudin, A.N. Kost, Dokl. Akad. Nauk SSSR, 170:340, 1966; B.A. Lugovik, L.G. Yudin, S.M. Vinogradova, A.N. Kost, Khim. Geterosikl. Soedin., 7:795, 1971.
Алтернативно анилины общей структуры II могут подвергаться взаимодействию с подходящим замещенным производным 1-метилстирола и формальдегидом в ацетонитриле при температуре окружающей среды или при повышенной температуре для получения соединений общей формулы V-b. Аналогичные реакции циклизации описаны в литературе: J.M. Mellor, G.D. Merriman, Tetrahedron, 51:6115, 1995.
Соединения общей структуры V-a-b после этого могут подвергаться региоселективному нитрованию в положение 6 тетрагидрохинолиновой основы для получения соединений общей структуры VI-a-b. Данная реакция обычно проводится при температурах в интервале от -10°С до комнатной температуры в дихлорметане с использованием смеси азотной кислоты и уксусного ангидрида в качестве нитрующего агента. Альтернативно азотная кислота может добавляться в раствор соединений общей структуры V-a-b в ледяной уксусной кислоте или в смеси уксусной кислоты и дихлорметана. Аналогичные реакции региоселективного нитрования тетрагидрохинолинов описаны в литературе: B. Golankiewicz, Pol. J. Chem., 54:355, 1980; Zh. V. Shmyreva, Kh. S. Shikhaliev, L.P. Zalukaev, Y.A. Ivanov, Y.S. Ryabokobylko, I.E. Pokrovskaya, Zh. Obshch. Khim. 59:1391, 1989.
Восстановление нитрогруппы соединений общей структуры VI-a-b может проводиться с использованием различных способов, хорошо известных в данной области техники для восстановления ароматических нитросоединений, таких как каталитическое гидрирование с использованием в качестве катализаторов переходных металлов, обработка сульфидами, обработка железом или другими металлами и (слабой) кислотой, обработка дихлоридом в кислотных условиях и т.п. В частности, восстановление нитрогруппы соединений общей формулы VI-a-b может осуществляться обработкой порошкообразным цинком и уксусной кислотой в ТГФ или 1,4-диоксане при температуре в интервале от 0 до 100°С.
Последующее 6-N-ацилирование соединений формулы VII-a-b может проводиться в стандартных условиях, хорошо известных квалифицированным специалистам в данной области техники, для получения соединений общей структуры I-a-b. Например, соединения формулы VII подвергаются взаимодействию в растворителе, таком как дихлорметан, тетрагидрофуран, толуол или пиридин, с ацилгалогенидом (R6-C(O)-Cl) или ангидридом кислоты (R6-C(O)-O-C(O)-R6) в присутствии основания
такого как N,N-диизопропиленэтиламин, триэтиламин, пиридин или гидрид натрия, для получения 6-N-ацилированных производных 1,2,3,4-тетрагидрохинолина формулы I-a-b. Альтернативно ацилирование соединений общей формулы VII-a-b для получения соединений общей формулы I-a-b может проводиться взаимодействием с подходящей карбоновой кислотой (R6-CO2H) в присутствии связывающего реагента, такого как тетрафторборат О-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония (TBTU), гексафторфосфат О-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (HATU) или гексафторфосфат бромтрипирролидинфосфония (PyBrOP) и третичное основание, например, N,N-диизопропилэтиламин, в растворителе, таком как N,N-диметилформамид или дихлорметан, при температуре окружающей среды или при повышенной температуре.
Соединения данного изобретения, в которых R3 = H, OH или (1-4С)алкокси, R4 = ОН, R5 = OH или (1-4С)алкокси и R1, R2 и R6 принимают значения, определенные выше, могут быть получены реакциями деметилирования соединений общей формулы I-c-d. Реакции деметилирования простых ароматических метиловых эфиров хорошо известны специалистам в данной области техники. Обычно деметилирование осуществляется при взаимодействии соединения формулы I-c-d c BBr3 в инертном растворителе, таком как дихлорметан, при температуре в интервале от пониженной до температуры окружающей среды для получения деметилированных соединений общей формулы I-e-i. Альтернативно деметилирование может осуществляться при взаимодействии соединений формулы I-c-d с BF3·Me2S комплексом при температуре окружающей среды. Степень деметилирования может отчасти контролироваться осторожным регулированием температуры реакции и количеством деметилирующего реагента. Обычно получают смеси моно-, ди- и, если это возможно, тригидрокси-производных общей формулы I-e-i, которые могут разделяться хроматографически. Реакция деметилирования обычно протекает со средней степенью селективности с предпочтительным деметилированием в положение 5 тетрагидрохинолиновой основы. Скорость реакции деметилирования (деалкилирования) соединений общей формулы I-c-d соответствует соотношению 5-OMe>4-(п-ОАлк-фенил)>7-OMe.
Соединения данного изобретения, в которых R3 представляет собой Н или (функционализированную) алкоксигруппу и R4 и/или R5 представляют собой (функционализированные) алкоксигруппы или ацилоксигруппы, могут быть получены реакциями реалкилирования или ацилирования гидроксильных групп соединений общей формулы I-e-i (функционализированными) алкилгалогенидами (например, хлорэтилпирролидином) или ацилгалогенидами (например 2-фуроилхлоридом или метилхлорформиатом), соответственно, в стандартных условиях.
Соединения данного изобретения, в которых R4 = H и R5 присоединены к тетрагидрохинолиновой основе через атом азота и R1, R2 и R6 принимают значения, определенные выше, могут быть получены из N-Boc-1,4-фенилендиамина (VIII). Последовательность реакций (а) Скраупа, (b) ацилирования и (с) алкилирования бензола или замещенного бензола в соответствии с реакцией Фриделя-Крафтса, как описано выше, приводит к получению соединений общей формулы Х-а. Следует отметить, что защитная Вос-группа отщепляется в условиях реакции Фриделя-Крафтса.
Альтернативно N-Boc-1,4-фенилендиамин может подвергаться обработке метилвинилкетоном с последующим ацилированием и реакцией Фриделя-Крафтса, как описано выше, для получения соединений общей формулы X-b.
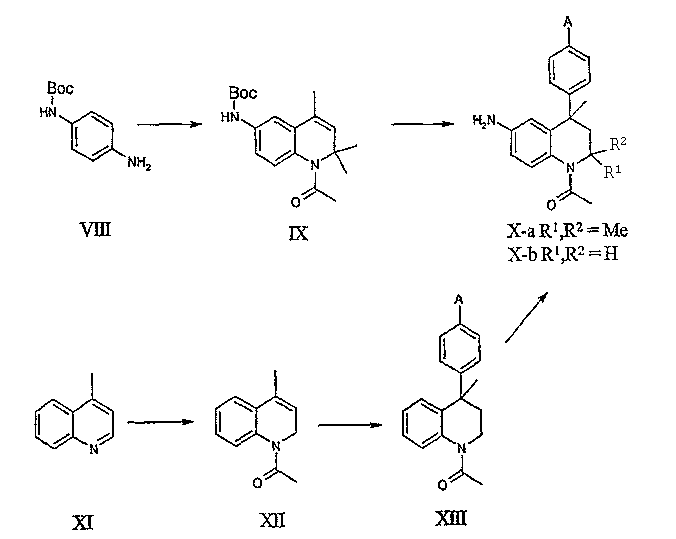
В еще одной методике соединения общей формулы X-b могут быть получены исходя из частичного восстановления 4-метилхинолина (XI) с BH3·THF комплексом и бис(2-метоксиэтокси)алюмодигидридом натрия для получения 4-метил-1,2-дигидрохинолина и последующим ацетилированием, как описано выше, для получения соединения XII. Реакции восстановления аналогичных хинолинов с получением 1,2-дигидрохинолинов описаны в литературе: см., например, D. Roberts, J.A. Joule, J. Org. Chem. 62:568, 1997; R.F. Heier, L.A. Dolak, J.N. Duncan, D.K. Hyslop, M.F. Lipton, I.J. Martin, M.A. Mauragis, M.F. Piercey, N.F. Nichols, P.J.K.D. Schreur, M.W. Smith, M.W. Moon, J. Med. Chem. 40: 639, 1997. Реакция Фриделя-Крафтса соединения XII с бензолом или подходящим замещенным бензолом приводит к получению соединений общей формулы XIII, которые могут подвергаться превращению в соединения общей формулы X-b региоселективным нитрованием в положение 6 и восстановлением до соответствующего 6-амино-производного с использованием описанных выше условий. Реакции региоселективного нитрования аналогичных структур были описаны в литературе, например, в публикации Zh. V. Shmyreva et al., J. Gen. Chem. USSR (англ. перевод) 59:1234, 1989.
Соединения общей формулы X-a-b после этого могут подвергаться реакции введения защитной 9-флуоренилметилоксикарбонильной группы (Fmoc-группа), см., например: T.W. Greene and P.M.Wuts, Protective groups in organic synthesis (3rd ed., John Wiley & Sons, Inc., 1999, см., в частности, стр. 506). Указанная выше защита обычно вводится с использованием FmocCl в ТГФ с пиридином в качестве основания.
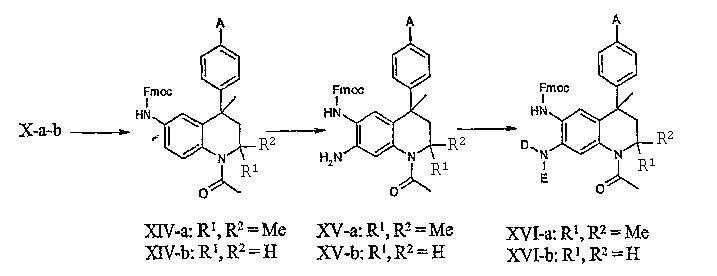
Региоселективное нитрование в положение 7 тетрагидрохинолиновой основы соединений общей формулы XIV-a-b с последующим восстановлением нитрогруппы (см. выше) приводит к получению производных 7-амино-1,2,3,4-тетрагидрохинолина общей формулы XV-a-b. Реакции региоселективного нитрования аналогичных основ с аналогичным замещением описаны в литературе; см., например: S.H. Reich. M.A. Fuhly, D. Nguyen, M.J. Pino et al., J. Med. Chem. 35:847, 1992; A. Ivobe, M. Uchida, K. Kamata, Y. Hotei, H. Kusama, H. Harada, Chem. Pharm. Bull. 49:822, 2001. Условия нитрования аналогичны условиям, описанным выше.

Восстановительное алкилирование аминогруппы в положении 7 тетрагидрохинолин-производных общей формулы XV-a-b с использованием соответствующих замещенных альдегидов и подходящего восстановителя (например, цианоборгидрида натрия или триацетоксиборгидрида натрия) в подходящем растворителе, таком как метанол или N,N-диметилформамид, приводит к получению соединений общей формулы XVIa-b. Обычно формальдегид приводит к преобладающему получению 7-диметиламинотетрагидрохинолиновых производных (D = E = Me), в то время как применение других альдегидов приводит к преимущественному получению моноалкилированных соединений общей формулы XVIa-b (D = H, E = (функционализированный)алкил). Реакции восстановительного алкилирования ароматических аминов хорошо знакомы специалистам в данной области.
Стандартное отщепление защитной Fmoc группы с использованием пиперидина в дихлорметане приводит к получению 6-аминотетрагидрохинолин-производных общей формулы XVII-a-b, которые могут подвергаться селективному алкилированию в положение 6, как описано выше для соединений данного изобретения общей формулы I-j-k.
В соответствии с другой методикой аминогруппа в положении 7 тетрагидрохинолин-производных общей формулы XV-a-b может подвергаться ацилированию (гетеро)арилкарбоновыми кислотами (G-CO2H) или ацилхлоридами (G-C(O)-Cl), как описано выше. На последующих стадиях стратегия удаления защиты-ацилирования (удаление защитной 6-N-Fmoc группы и ацилирование полученной 6-NH2 группы), описанная выше, приводит к получению соединений данного изобретения общей формулы I-l-m.

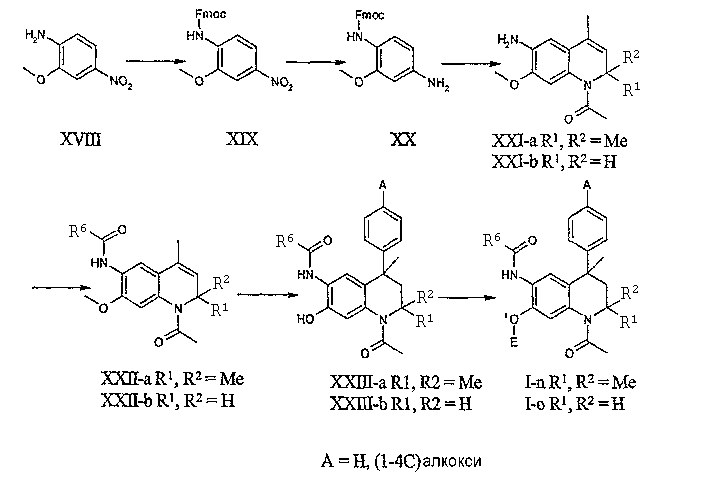
Соединения данного изобретения, в которых R4 = H, R5 присоединен к тетрагидрохинолиновой основе через атом кислорода и R1, R2 и R6 принимают значения, определенные выше, могут быть получены, исходя из 2-метокси-4-нитроанилина (XVII). Последовательность реакций (a) введения защитной Fmoc-группы для получения XIX, (b) восстановления нитрогруппы с получением ХХ, (с) региоселективная реакция Скраупа, (d) ацетилирование и (е) удаление защитной Fmoc-группы, как описано выше, приводит к получению соединений общей формулы XXI-a.
Соединения общей формулы XXI-b могут быть получены обработкой соединения ХХ метилвинилкетоном в условиях, описанных выше, для превращения соединений формулы II в соединения III-b с последующим 1-N-ацетилированием и удалением защитной Fmoc-группы, как описано выше.
Последующее превращение соединений общей формулы XXI в соединения XXIII может осуществляться ацилированием 6-аминогруппы с использованием подходящего ацилирующего агента, например, ацилхлорида R6-C(O)-Cl, и последующей реакцией Фриделя-Крафтса с бензолом или подходящим производным бензола в условиях, описанных выше. При проведении реакции Фриделя-Крафтса в присутствии кислоты Льюиса имеет место сопутствующее деметилирование 7-Оме функциональной группы в соединениях общей формулы XII. Полученная таким образом свободная 7-ОН группа в соединениях общей формулы XXIII может подвергаться повторному алкилированию или ацилированию (функционализированными) алкилгалогенидами (например, хлорэтилпирролидином) или ацилгалогенидами (например, 2-фуроилхлоридом или метилхлорформиатом), соответственно, в стандартных условиях для получения соединений общей формулы I-n-o (E = функционализированный алкил, ацил или карбамат).
Соединения данного изобретения, в которых R4 и R5 присоединены к тетрагидрохинолиновой основе через атом азота, могут быть получены из соединений общей формулы XXIV, где PG представляет собой защитную группу атома азота, например, Вос, ацетил, метилкарбамат или Fmoc, посредством описанных выше реакций, например, реакции Скраупа или циклоконденсации с метилвинилкетоном, 1-N-ацетилирования, расщепления защитной группы, N-алкилирования, реакции Фриделя-Крафтса, нитрования, восстановления нитрогруппы и ацилирования (см. выше).

Взаимодействие соединений общей формулы XXV с ацетоном или мезитилоксидом в соответствии с реакцией Скраупа может приводить к двум различным региоизомерным продуктам общей формулы XXVI-a и XXVII-a соответственно. Превращение соединений общей формулы XXV с использованием метилвинилкетона в описанных выше условиях может приводить к региоизомерным продуктам общей формулы XXVI-b или XXVII-b соответственно. Обычно такие региоизомерные дигидрохинолины могут разделяться хроматографическими методами (силикагель, ВЭЖХ) или кристаллизацией и могут впоследствии подвергаться превращению в соединения данного изобретения описанными выше способами.
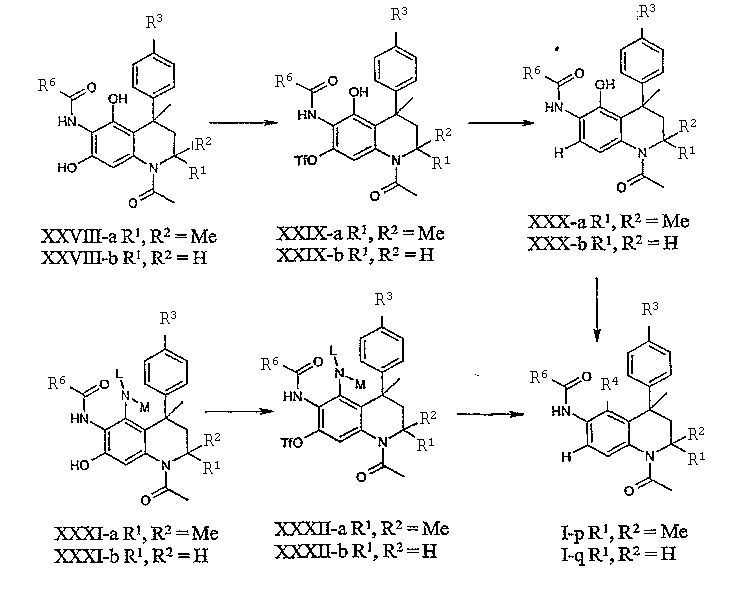
Соединения данного изобретения, в которых R5 = Н, могут быть получены восстановительным 7-деоксигенерированием соединений общей формулы XXVIII или XXXI (L и/или М представляет собой подходящий (замещенный) алкил, ацил, алкилоксикарбонил или алкиламинокарбнил) через селективное 7-О-трифлатирование и последующее восстановление группы 7-OTf (Tf = трифторметилсульфонил). Целевые соединения общей формулы XXXI доступны из производных общей формулы XXVII с использованием описанных выше условий. Реакция (региоселективного) трифлатирования может осуществляться в контролируемых условиях с использованием Tf2N-фенила и N,N-диизопропилэтиламина в ДМФА при комнатной температуре. Обычно имеет место предпочтительное трифлатирование 7-ОН группы. Последующее восстановление может осуществляться с использованием смеси трифетилфосфина, триэтиламина, муравьиной кислоты и ацетата палладия (II), как описано в литературе (см., например, K.A. Parker, O. Ding, Tetrahedron 56:10249, 2000). Последующее превращение полученных таким образом соединений общей формулы XXX или XXXII с использованием описанных выше условий приводит к получению соединений общей формулы I-p-q, в которых R5 = H.
Некоторые соединения данного изобретения, которые могут находиться в форме свободного основания, могут выделяться из реакционной смеси в форме фармацевтически приемлемой соли. Фармацевтически приемлемые соли также могут быть получены обработкой свободного основания формулы I органической или неорганической кислотой, такой как хлористоводородная кислота, бромистоводородная кислота, йодистоводородная кислота, серная кислота, фосфорная кислота, уксусная кислота, пропионовая кислота, гликолевая кислота, малеиновая кислота, малоновая кислота, метансульфоновая кислота, фумаровая кислота, янтарная кислота, винная кислота, лимонная кислота, бензойная кислота и аскорбиновая кислота.
Соединения данного изобретения обладают, по меньшей мере, одним хиральным атомом углерода и, следовательно, могут быть получены в виде чистых энантиомеров, в виде смеси энантиомеров или в виде смеси диастереомеров. Способы получения чистых энантиомеров хорошо известны в данной области техники, например кристаллизация солей, которые получены из оптически активных кислот и рацемической смеси, или хроматография с использованием хиральных колонок. Для диастереомеров могут использоваться колоночная хроматография с прямой или обращенной фазой.
Соединения данного изобретения могут образовывать гидраты или сольваты. Специалистам в данной области известно, что заряженные соединения образуют гидратированные соединения при лиофилизации с водой или образуют сольваты при концентрировании в растворе подходящего органического растворителя. Соединения данного изобретения включают гидраты или сольваты указанных соединений.
Для скрининга активных соединений испытание при 10-5 М должно привести к активности, составляющей более 20% максимальной активности при использовании FSH в качестве контроля. Другим критерием может служить значение ЕС50, которое должно составлять <10-5 М, предпочтительно <10-7 М.
Специалисту в данной области будет понятно, что желательное значение ЕС50 зависит от используемого соединения. Например, соединение со значением ЕС50 менее 10-5 М обычно рассматривается в качестве возможного кандидата для получения лекарственного средства. Предпочтительно данное значение составляет менее 10-7 М. Однако соединение, которое обладает более высоким значением ЕС50, но селективно в отношении конкретного рецептора, может быть даже более перспективным соединением.
Способы определения рецепторного связывания гонадотропинов in vitro и in vivo для определения биологической активности хорошо известны специалистам в данной области техники. Обычно экспрессированный рецептор подвергается контактированию с соединением, подлежащим испытанию, и количественно определяется связывание, стимулирование или ингибирование функционального ответа.
Для определения функционального ответа изолированная ДНК, кодирующая FSH рецепторный ген предпочтительно рецептора человека, экспрессируется в подходящие клетки-хозяева. Такой клеткой может быть клетка яичника китайского хомячка, но приемлемы и другие клетки. Предпочтительно клетки являются клетками млекопитающих (Jia et al., Mol. Endocrin., 5:759-776, 1991).
Способы создания рекомбинантного FSH, экспрессирующего клеточные линии, хорошо известны в данной области техники (Sambrook et al., Molecular Cloning: a Laboratory Manual, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, latest edition). Экспрессия рецептора достигается экспрессией ДНК, кодирующей желаемый белок. Методы сайт-направленного мутагенеза, лигирования дополнительных последовательностей, PCR и создания подходящих экспрессионных систем сегодня являются хорошо известными в данной области. Части ДНК или вся ДНК, кодирующая желательный белок, могут быть получены синтетически с использование стандартных твердофазных методов предпочтительно для включения сайтов рестрикции для облегчения лигирования. Подходящие контрольные элементы для транскрипции и трансляции включенной кодирующей последовательности могут предоставляться для ДНК кодирующих последовательностей. Как хорошо известно, системы экспрессии, доступные в настоящее время, совместимы с широким спектром организмов-хозяев, включая как прокариотические хозяева, такие как бактерии, так и эукариотические хозяева, такие как дрожжи, растительные клетки, клетки насекомых, клетки млекопитающих, клетки птиц и т.д.
Клетки, экспрессирующие рецептор, затем контактируют с испытываемым соединением для наблюдения связывания, стимулирования или ингибирования функционального ответа.
Альтернативно для количественного определения связывания вещества могут использоваться изолированные клеточные мембраны, содержащие экспрессированный рецептор.
Для количественного определения связывания могут использоваться соединения с радиоактивной или флуоресцентной меткой. Может осуществляться также определение конкурентного связывания.
Другой опыт включает скрининг соединений, представляющих собой агонист FSH рецептора, определением стимулирования рецептор-опосредуемого аккумулирования сАМР. Таким образом, этот способ включает экспрессию рецептора на поверхности клетки-хозяина и экспозицию клетки испытываемым соединением. После этого определяют количество сАМР. Уровень содержания сАМР может снижаться или повышаться в зависимости от ингибирующего или стимулирующего действия испытываемого соединения на связывание с рецептором.
Скрининг антагонистов FSH рецепторов включает выдерживание FSH рецептор-экспрессирующих клеток в испытываемом соединении, взятом в некотором интервале концентраций, в присутствии FSH, взятого при фиксированной, близкой к максимально эффективной концентрации (т.е. концентрации FSH, которая индуцирует приблизительно 80% максимального стимулирования аккумулирования сАМР в отсутствие испытываемого соединения). Значение IC50 и процент ингибирования FSH-индуцированного аккумулирования сАМР может определяться для каждого из испытываемого соединений из графиков зависимости концентрация-эффект.
Помимо прямого количественного определения, например, уровней содержания сАМР в подвергающейся воздействию клетке, могут использоваться клеточные линии, которые в дополнение к трансфекции рецептор-кодирующей ДНК трансфицированы второй кодирующей ДНК репортерного гена, экспрессия которого реагирует на уровень содержания сАМР. Такие репортерные гены могли бы индуцироваться сАМР или могли бы создаваться так, чтобы они соединялись с новыми элементами, чувствительными к сАМР. Обычно экспрессия репортерного гена может контролироваться любым чувствительным элементом, реагирующим на изменение уровней содержания сАМР. Подходящими репортерными генами являются, например, гены, кодирующие β-галактозидазу, алкалинфосфатазу, люциферазу светлячка и зеленый флуоресцентный белок. Принципы таких опытов трансактивации хорошо известны в данной области и описаны, например, в публикации Stratowa, Ch., Himmler, A., Czernilofsky, A.P., (1995) Curr. Opin. Biotechnol. 6:574. В качестве контрольного соединения может использоваться рекомбинантный FSH человека. Альтернативно могут проводиться конкурентные эксперименты.
Данное изобретение относится также к фармацевтической композиции, содержащей производное тетрагидрохинолина общей формулы I или его фармацевтически приемлемые соли в смеси с фармацевтически приемлемыми вспомогательными добавками и, необязательно, с другими терапевтическими средствами. Термин «приемлемый» в отношении вспомогательных добавок означает, что вспомогательные добавки должны быть совместимыми с другими ингредиентами композиции и не оказывать неблагоприятного воздействия на принимающих композицию реципиентов.
Композиции включают, например, композиции, приемлемые для перорального, сублингвального, подкожного, внутривенного, внутримышечного, местного или ректального введения и т.п. в единичных дозированных формах.
Для перорального введения активный ингредиент может быть представлен в виде отдельных единиц, таких как таблетки, капсулы, порошки, гранулы, растворы, суспензии и т.п.
Для парентерального введения фармацевтическая композиция данного изобретения может предоставляться в контейнерах, содержащих единичную дозу или многократные дозы, например, в виде жидкостей для инъекций в предопределенных количествах в запаянных пузырьках или ампулах, и может также храниться в высушенном сублимацией (лиофилизированном) состоянии, требующем лишь добавления стерильного жидкого носителя, например воды, перед применением.
Активный ингредиент, смешанный с такими фармацевтически приемлемыми вспомогательными добавками, например, как описано в традиционно используемой ссылке Gennaro, A.R. et al., Remington: The Science and Practice of Pharmacy (20th Edition., Lippincott Williams & Wilkins, 2000, см., в частности, Part 5: Pharmaceutical Manufacturing), может прессоваться в твердые дозированные единицы, такие как пилюли, таблетки, или перерабатываться в капсулы или свечи. При использовании фармацевтически приемлемых жидкостей активный ингредиент может применяться в виде жидкой композиции, например, в виде препарата для инъекции, в форме раствора, суспензии, эмульсии, или в виде спрея, например, назального спрея.
Для получения твердых форм дозированных единиц предполагается применение традиционных добавок, таких как наполнители, красители, полимерные связующие вещества и т.п. Обычно может использоваться любая фармацевтически приемлемая добавка, которая не влияет на функцию активных ингредиентов. Подходящие носители, с которыми может вводиться активный ингредиент данного изобретения в виде твердых композиций, включают лактозу, крахмал, производные целлюлозы и т.п. или их смеси, используемые в подходящих количествах. Для парентерального введения могут применяться водные суспензии, изотонические солевые растворы и стерильные растворы для инъекций, содержащие фармацевтически приемлемые дисперсанты и/или смачивающие агенты, такие как пропиленгликоль или бутиленгликоль.
Изобретение включает также фармацевтическую композицию, описанную выше, в сочетании с упаковкой, подходящей для указанной композиции, причем указанная упаковка снабжена инструкцией для применения композиции, которая определена выше.
Производные тетрагидрохинолина данного изобретения могут также вводиться в форме имплантируемых фармацевтических средств, состоящих из ядра активного ингредиента, покрытого мембраной, регулирующей скорость высвобождения активного ингредиента. Такие имплантаты должны применяться подкожно или местно и будут высвобождать активный ингредиент с приблизительно постоянной скоростью в течение относительно длительных периодов времени, составляющих, например, от нескольких недель до нескольких лет. Способы получения имплантируемых фармацевтических средств как таковые известны в данной области и описаны, например, в европейской заявке на патент 303306 (AKZO Nobel N.V.).
Точная дозировка и схема введения активного ингредиента или его фармацевтической композиции будут обязательно зависеть от терапевтического эффекта, который необходимо достигнуть (лечение бесплодия; контрацепция) и могут изменяться в зависимости от конкретного соединения, способа введения и возраста и состояния отдельного субъекта, которому должно вводиться лекарственное средство.
Обычно для парентерального введения требуются меньшие дозировки, чем при введении другими способами, которые в большей степени зависят от абсорбции. Однако дозировка для людей предпочтительно содержит 0,0001-25 мг на кг веса тела. Желательная доза может вводиться в разовой дозе или делиться на несколько небольших доз, вводимых с подходящими интервалами в течение дня, или в случае реципиента-женщины в дозах, подлежащих введению с подходящими суточными интервалами в течение менструального цикла. Дозировка также как и схема введения для мужчин и женщин могут быть разными.
Таким образом, соединения данного изобретения могут использоваться в терапевтическом лечении.
Дополнительный аспект данного изобретения относится к применению производного тетрагидрохинолина общей формулы I для производства лекарственного средства, предназначенного для применения при лечении расстройств, чувствительных к метаболическим путям, опосредуемым FSH-рецептором. Таким образом, пациентам, нуждающимся в этом, могут вводиться подходящие количества соединений согласно данному изобретению.
В другом аспекте изобретение относится к применению производного тетрагидрохинолина общей формулы I для производства лекарственного средства, предназначенного для применения для контроля фертильности.
В еще одном аспекте изобретение относится к применению производного тетрагидрохинолина общей формулы I для производства лекарственного средства, предназначенного для применения при лечении бесплодия.
В еще одном аспекте данное изобретение относится к применению производного тетрагидрохинолина общей формулы I для производства лекарственного средства, предназначенного для применения для предупреждения фертильности.
Соединения данного изобретения могут также применяться для лечения гормон-зависимых расстройств, таких как рак молочной железы, рак предстательной железы и эндометриоз.
Далее изобретение иллюстрируется следующими примерами.
Примеры
Общие пояснения:
В примерах используются следующие сокращения: ДМА = N,N-диметиланилин, DIPEA = N,N-диизопропилэтиламин, ТФУК = трифторуксусная кислота, DtBAD = ди-трет-бутилазодикарбоксилат, HATU = гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N'N'-тетраметилурония, Fmoc = 9-флуоренилметоксикарбонил, Fmoc-Cl = 9-флуоренилметоксикарбонилхлорид, ДМФА = N,N-диметилформамид, DMAP = 4-диметиламинопиридин, ТГФ = тетрагидрофуран.
Если особо не оговорено другое условие, все целевые продукты примеров, приведенных ниже, лиофилизованы из смесей вода/1,4-диоксан или вода/ацетонитрил. Если соединение получено в виде HCl- или ТФУ-соли, соответствующие кислоты были добавлены в подходящих количествах к смеси растворителей перед лиофилизацией.
Названия целевых продуктов, описанных в примере, составлены с использованием программы Beilstein Autonom program (version: 2.02.119).
Для определения времени удерживания использовались следующие аналитические методы ВЭЖХ:
Метод 1. Колонка: 5 мкм Luna C-18(2) 150×4,6 мм; скорость потока: 1 мл/мин; обнаружение: 210 нм; температура колонки: 40°С; растворитель А: CH3CN/H2О = 1/9 (об./об.); растворитель В: CH3CN; растворитель С: 0,1М водная трифторуксусная кислота; элюирование с градиентом: А/В/С от 65/30/5 до 10/85/5 (об./об./об.) в течение 30 мин, затем в течение дополнительных 10,00 мин элюирование постоянным элюентом А/В/С = 10/85/5 (об./об./об.).
Метод 2 идентичен методу 1 за исключением элюирования с градиентом: элюент А/В/С от 75/20/5 до 15/80/5 (об./об./об.) в течение 30 минут, затем в течение дополнительных 10,00 мин элюирование постоянным элюентом А/В/С 15/80/5 (об./об./об.).
Метод 3 идентичен методу 1 за исключением элюирования с градиентом: А/В/С от 35/60/5 до 10/85/5 (об./об./об.) в течение 30,00 мин, затем в течение дополнительных 10,00 минут элюирование постоянным элюентом А/В/С = 10/85/5 (об./об./об.).
Метод 4. Колонка: 3 мкм Luna C-18(2) 100×2 мм; скорость потока: 0,25 мл/мин; обнаружение: 210 нм; температура колонки: 40°С; растворитель А: Н2О; растворитель В: СН3CN; элюирование с градиентом: А/В от 75/25 до 0/100 (об./об.) в течение 20 минут, затем в течение дополнительных 10,00 мин элюирование постоянным элюентом А/В = 0/100 (об./об.).
Метод 5. Колонка: 3 мкм Luna C-18(2) 100×2 мм; скорость потока: 0,25 мл/мин; обнаружение: 210 нм; температура колонки: 40°С; растворитель А: Н2О; растворитель В: СН3CN; растворитель С: 50 мМ фосфатный буфер, рН 2,1; элюирование с градиентом: А/В/С от 75/20/10 до 10/80/100 (об./об.) в течение 20 минут, затем в течение дополнительных 10,00 мин элюирование постоянным элюентом А/В/С = 10/80/10 (об./об./об.).
Метод 6 идентичен методу 5 за исключением элюирования с градиентом: А/В/С от 65/30/5 до 10/85/5 (об./об./об.) в течение 20,00 мин, затем в течение 10,00 мин элюирование постоянным элюентом А/В/С = 10/85/5 (об./об./об.).
Для очистки продуктов препаративной ВЭЖХ используются следующие методы:
Метод А. Колонка: Luna C-18; элюирование с градиентом: 0,1% трифторуксусная кислота в Н2О/CH3CN (9/1 об./об.)/СН3CN от 80/20 до 0/100 (об./об.) в течение 30-45 мин в зависимости от легкости разделения; обнаружение: 210 нм.
Метод В. Колонка: Luna C-18; элюирование с градиентом: Н2О/CH3CN (9/1 об./об.)/СН3CN от 80/20 до 0/100 (об./об.) в течение 30-45 мин в зависимости от легкости разделения; обнаружение: 210 нм.
Пример 1
N-[1-Ацетил-5,7-диметокси-4-(4-метоксифенил)-2,2,4-триметил-1,2,3,4-тетрагидрохинолин-6-ил]-3-хлор-2,6-диметоксибензамид
(а) 5,7-Диметокси-2,2,4-триметил-1,2-дигидрохинолин
Раствор 3,5-диметоксианилина (50 г) в ацетоне (800 мл) по каплям добавляют к смеси MgSO4 (100 г) и Sc(OTf)3 (8,0 г) в 1 л ацетона при комнатной температуре. Спустя 5 часов добавляют другую порцию Sc(OTf)3 (3,2 г) и реакционную смесь перемешивают до тех пор, пока не останется исходного вещества. Раствор фильтруют, ацетон частично выпаривают в вакууме, вызывая кристаллизацию указанного в заголовке соединения, которое отфильтровывают, получая после сушки в вакууме 22 г продукта. Оставшуюся маточную жидкость концентрируют в вакууме и остаток очищают хроматографией на силикагеле (элюирование с градиентом: гептан/этилацетат от 1/0 до 0/1 (об./об.), получая еще 19,4 г указанного в заголовке соединения.
Выход: 42 г.
(b) 1-Ацетил-5,7-диметокси-2,2,4-триметил-1,2-дигидрохинолин
Смесь соединения, описанного в примере 1а (42 г), и уксусного ангидрида (100 мл) перемешивают при 100°С в течение 20 часов. Реакционную смесь выливают в 500 мл ледяной воды при перемешивании. Образующийся твердый осадок отфильтровывают и сушат в вакууме при 40°С в течение 2 дней. Полученное твердое вещество коричневого цвета используют на следующей стадии без дополнительной очистки.
Выход 45 г.
(с) 1-Ацетил-5,7-диметокси-4-(4-метоксифенил)-2,2,4-триметил-1,2,3,4-тетрагидрохинолин
Смесь соединения, описанного в примере 1b (30 г), и AlCl3 (44 г) в анизоле (500 мл) перемешивают при 50°С в течение 18 часов. Реакционную смесь охлаждают (0°С), гасят водой и добавляют этилацетат. Смесь перемешивают в течение ночи. Органический слой отделяют, сушат над MgSO4, фильтруют и концентрируют в вакууме. Остаток хроматографируют на силикагеле (элюирование: гептан/этилацетат = 8/2 (об./об.)).
Выход: 15 г.
(d) 1-Ацетил-5,7-диметокси-4-(4-метоксифенил)-2,2,4-триметил-6-нитро-1,2,3,4-тетрагидрохинолин
Раствор уксусного ангидрида (450 мкл) в дымящей азотной кислоте (22,5 мл) по каплям добавляют к раствору соединения, описанного в примере 1с (15 г) в CH2Cl2 (500 мл) при 0°С. После завершения добавления реакционную смесь перемешивают при комнатной температуре в течение 3 часов. Добавляют воду, органический слой отделяют, сушат над MgSO4, фильтруют и концентрируют в вакууме. Остаток кристаллизуют из этанола, получая указанное в заголовке соединение в виде твердого кристаллического вещества.
Выход: 10 г.
(е) 1-Ацетил-6-амино-5,7-диметокси-4-(4-метоксифенил)-2,2,4-триметил-1,2,3,4-тетрагидрохинолин
Раствор соединения, описанного в примере 1d (11,75 г), и уксусной кислоты (15,5 мл) в ТГФ (600 мл) охлаждают до 0°С. Порошкообразный цинк (36 г) добавляют порциями и ледяную баню удаляют. Температура быстро возрастает до 30°С, после чего реакционной смеси дают охладиться до комнатной температуры. Избыток цинка удаляют фильтрованием и к фильтрату добавляют СН2Cl2 и насыщенный водный раствор Na2CO3. Органический слой отделяют, сушат над MgSO4, фильтруют и концентрируют в вакууме. Продукт используют на следующей стадии без дополнительной очистки.
Выход: 10,9 г.
(f) N-[1-Ацетил-5,7-диметокси-4-(4-метоксифенил)-2,2,4-триметил-1,2,3,4-тетрагидрохинолин-6-ил]-3-хлор-2,6-диметоксибензамид
Общая методика А: К раствору соединения, описанного в примере 1е (100 мг), 3-хлор-2,6-диметоксибензойной кислоты (60 мг) и DIPEA (132 мкл) в CH2Cl2 (2 мл) добавляют HATU (143 мг) при комнатной температуре. Если реакция не завершится по истечении 18 часов, добавляют дополнительное количество HATU и DIPEA. После завершения реакции добавляют насыщенный водный раствор NaHCO3, органический слой отделяют, сушат (MgSO4) и концентрируют в вакууме. Указанное в заголовке соединение очищают препаративной ВЭЖХ (метод А).
Выход: 87 мг; МС-ESI: [M+H]+ = 597,4.
ВЭЖХ: Rt=17,98 мин (метод 1).
Пример 2
[1-Ацетил-5,7-диметокси-4-(4-метоксифенил)-2,2,4-триметил-1,2,3,4-тетрагидрохинолин-6-ил]амид 4,5-диметилфуран-2-карбоновой кислоты
Общая методика В: К раствору соединения, описанного в примере 1е (800 мг), 4,5-диметилфуран-2-карбоновой кислоты (308 мг) и ДМА (768 мкл) в ДМФА (10 мл) при комнатной температуре добавляют HATU (1,1 г). Если реакция не завершится по истечении 18 часов, реакционную смесь нагревают до 50°С. После завершения реакции добавляют воду и этилацетат, органический слой отделяют, сушат (MgSO4) и концентрируют в вакууме. Указанное в заголовке соединение очищают хроматографией на силикагеле (элюирование с градиентом: гептан/этилацетат от 1/0 до 0/1 (об./об.)).
Выход: 444 мг; МС-ESI: [M+H]+ = 521,4.
ВЭЖХ: Rt=16,96 мин (метод 1).
Пример 3
[1-Ацетил-5,7-диметокси-4-(4-метоксифенил)-2,2,4-триметил-1,2,3,4-тетрагидрохинолин-6-ил]амид 5-бромтиофен-2-карбоновой кислоты
В соответствии с общей методикой В соединение, описанное в примере 1е (800 мг), ацилируют 5-бромтиофен-2-карбоновой кислотой (456 мг), ДМА (768 мкл) и HATU (1,1 г) в CH2Cl2 (10 мл). Указанное в заголовке соединение очищают хроматографией на силикагеле (элюирование с градиентом: гептан/этилацетат от 1/0 до 0/1 (об./об.)).
Выход: 1,0 г; МС-ESI: [M+H]+ = 589,2; ВЭЖХ: Rt=18,90 мин (метод 2).
Пример 4
[1-Ацетил-5,7-диметокси-4-(4-метоксифенил)-2,2,4-триметил-1,2,3,4-тетрагидрохинолин-6-ил]амид бифенил-4-карбоновой кислоты
Общая методика С: К раствору соединения, описанного в примере 1е (800 мг), и 4-бифенилкарбонилхлорида (475 мг) в CH2Cl2 (10 мл) при комнатной температуре добавляют ДМА (768 мкл). Реакционную смесь перемешивают до тех пор, пока не останется исходного вещества, после чего добавляют воду. Органический слой отделяют, сушат (MgSO4) и концентрируют в вакууме. Указанное в заголовке соединение очищают хроматографией на силикагеле (элюирование с градиентом: гептан/этилацетат от 1/0 до 0/1 (об./об.)).
Выход: 678 мг; МС-ESI: [M+H]+ = 579,4; ВЭЖХ: Rt=26,19 мин (метод 2).
Пример 5
[1-Ацетил-5-гидрокси-7-метокси-4-(4-метоксифенил)-2,2,4-триметил-1,2,3,4-тетрагидрохинолин-6-ил]амид фуран-2-карбоновой кислоты
(а) [1-Ацетил-5,7-диметокси-4-(4-метоксифенил)-2,2,4-триметил-1,2,3,4-тетрагидрохинолин-6-ил]амид фуран-2-карбоновой кислоты
В соответствии с общей методикой С соединение, описанное в примере 1е (800 мг), ацилируют 2-фуроилхлоридом (217 мкл) и ДМА (768 мкл) в CH2Cl2 (10 мл). Указанное в заголовке соединение очищают хроматографией на силикагеле (элюирование с градиентом: гептан/этилацетат от 1/0 до 0/1 (об./об.)).
Выход: 896 мг.
(b) [1-Ацетил-5-гидрокси-7-метокси-4-(4-метоксифенил)-2,2,4-триметил-1,2,3,4-тетрагидрохинолин-6-ил]амид фуран-2-карбоновой кислоты
Общая методика D: Раствор соединения, описанного в примере 5а (50 мг), в CH2Cl2 (4 мл) охлаждают до -78°С в атмосфере N2. По каплям добавляют трибромид бора (28 мкл) и после завершения добавления реакционной смеси дают медленно нагреться до комнатной температуры. Реакцию гасят водой и добавляют CH2Cl2. Органический слой отделяют, сушат (MgSO4) и концентрируют в вакууме. Указанное в заголовке соединение очищают препаративной ВЭЖХ (метод А). В описанных выше условиях обычно образуются смеси соединений с различной степенью деметилирования, которые могут разделяться препаративной ВЭЖХ.
Выход: 9,1 мг; МС-ESI: [M+H]+ = 479,4; ВЭЖХ: Rt=23,40 мин (метод 2).
Пример 6
N-[1-Ацетил-5-гидрокси-7-метокси-4-(4-метоксифенил)-2,2,4-триметил-1,2,3,4-тетрагидрохинолин-6-ил]-3,5-дихлорбензамид
(а) N-[1-Ацетил-5,7-диметокси-4-(4-метоксифенил)-2,2,4-триметил-1,2,3,4-тетрагидрохинолин-6-ил]-3,5-дихлорбензамид
В соответствии с общей методикой С соединение, описанное в примере 1е (800 мг), ацилируют 3,5-дихлорбензоилхлоридом (460 мг) и ДМА (768 мкл) в CH2Cl2 (10 мл). Указанное в заголовке соединение очищают хроматографией на силикагеле (элюирование с градиентом: гептан/этилацетат от 1/0 до 0/1 (об./об.)).
Выход: 1,03 г.
(b) N-[1-Ацетил-5-гидрокси-7-метокси-4-(4-метоксифенил)-2,2,4-триметил-1,2,3,4-тетрагидрохинолин-6-ил]-3,5-дихлорбензамид
В соответствии с общей методикой D соединение, описанное в примере 6а (50 мг), обрабатывают трибромидом бора (24 мкл) в CH2Cl2 (4 мл). Указанное в заголовке соединение очищают препаративной ВЭЖХ (метод А).
Выход: 9,6 мг; МС-ESI: [M+H]+ = 557,2; ВЭЖХ: Rt=23,40 мин (метод 2).
Пример 7
[1-Ацетил-5-гидрокси-7-метокси-4-(4-метоксифенил)-2,2,4-триметил-1,2,3,4-тетрагидрохинолин-6-ил]амид 5-хлортиофен-2-карбоновой кислоты
(а) [1-Ацетил-5,7-диметокси-4-(4-метоксифенил)-2,2,4-триметил-1,2,3,4-тетрагидрохинолин-6-ил]амид 5-хлортиофен-2-карбоновой кислоты
В соответствии с общей методикой В соединение, описанное в примере 1е (800 мг), ацилируют 5-хлортиофен-2-карбоновой кислотой (456 мг), ДМА (768 мкл) и HATU (1,1 г) в CH2Cl2. Указанное в заголовке соединение очищают хроматографией на силикагеле (элюирование с градиентом: гептан/этилацетат от 1/0 до 0/1 (об./об.)).
Выход: 1,0 г.
(b) [1-Ацетил-5-гидрокси-7-метокси-4-(4-метоксифенил)-2,2,4-триметил-1,2,3,4-тетрагидрохинолин-6-ил]амид 5-хлортиофен-2-карбоновой кислоты
В соответствии с общей методикой D соединение, описанное в примере 7а (200 мг), обрабатывают трибромидом бора (350 мкл) в CH2Cl2 (25 мл), поддерживая температуру на уровне не выше -30°С. Указанное в заголовке соединение очищают хроматографией на силикагеле (элюирование с градиентом: гептан/этилацетат от 1/0 до 0/1 (об./об.)), затем препаративной ВЭЖХ (метод А).
Выход: 35 мг; МС-ESI: [M+H]+ = 529,2; ВЭЖХ: Rt=28,24 мин (метод 2).
Пример 8
[1-Ацетил-5-гидрокси-7-метокси-4-(4-метоксифенил)-2,2,4-триметил-1,2,3,4-тетрагидрохинолин-6-ил]амид бифенил-4-карбоновой кислоты
В соответствии с общей методикой D соединение, описанное в примере 4 (50 мг), обрабатывают трибромидом бора (100 мкл) в CH2Cl2 (4 мл), поддерживая температуру на уровне не выше 0°С. Реакционная смесь содержит также продукт, описанный в примере 10. Указанное в заголовке соединение очищают хроматографией на силикагеле (элюирование с градиентом: гептан/этилацетат от 1/0 до 0/1 (об./об.))
Выход: 43 мг; МС-ESI: [M+H]+ = 565,4; ВЭЖХ: Rt=32,53 мин (метод 2).
Пример 9
[1-Ацетил-5,7-дигидрокси-4-(4-гидроксифенил)-2,2,4-триметил-1,2,3,4-тетрагидрохинолин-6-ил]амид бифенил-4-карбоновой кислоты
В соответствии с общей методикой D соединение, описанное в примере 4 (50 мг), обрабатывают трибромидом бора (100 мкл) в CH2Cl2 (4 мл), поддерживая температуру на уровне не выше 15°С. Указанное в заголовке соединение очищают хроматографией на силикагеле (элюирование с градиентом: гептан/этилацетат от 1/0 до 0/1 (об./об.)).
Выход: 33 мг; МС-ESI: [M+H]+ = 537,4; ВЭЖХ: Rt=24,16 мин (метод 2).
Пример 10
[1-Ацетил-5-гидрокси-4-(4-гидроксифенил)-7-метокси-2,2,4-триметил-1,2,3,4-тетрагидрохинолин-6-ил]амид бифенил-4-карбоновой кислоты
В соответствии с общей методикой D соединение, описанное в примере 4 (400 мг), обрабатывают трибромидом бора (800 мкл) в CH2Cl2 (25 мкл), поддерживая температуру на уровне не выше 0°С. Указанное в заголовке соединение (а = бипродукт, как описано в примере 8) очищают хроматографией на силикагеле (элюирование с градиентом: гептан/этилацетат от 1/0 до 0/1 (об./об.)).
Выход: 50 мг; МС-ESI: [M+H]+ = 551,4; ВЭЖХ: Rt=27,58 мин (метод 2).
Пример 11
[1-Ацетил-5-гидрокси-7-метокси-4-(4-метоксифенил)-2,2,4-триметил-1,2,3,4-тетрагидрохинолин-6-ил]амид 4,5-диметилфуран-2-карбоновой кислоты
В соответствии с общей методикой D соединение, описанное в примере 2 (200 мг), обрабатывают трибромидом бора (336 мкл) в CH2Cl2 (25 мл), поддерживая температуру на уровне не выше -78°С. Указанное в заголовке соединение очищают препаративной ВЭЖХ (метод А).
Выход: 51 мг; МС-ESI: [M+H]+ = 507,4; ВЭЖХ: Rt=24,32 мин (метод 1).
Пример 12
N-[1-Ацетил-5-гидрокси-4-(4-гидроксифенил)-7-метокси-2,2,4-триметил-1,2,3,4-тетрагидрохинолин-6-ил]-3,5-дихлорбензамид
В соответствии с общей методикой D соединение, описанное в примере 6а (75 мг), обрабатывают трибромидом бора (38 мкл) в CH2Cl2 (5 мл). Указанное в заголовке соединение очищают препаративной ВЭЖХ (метод А).
Выход: 11 мг; МС-ESI: [M+H]+ = 543,4; ВЭЖХ: Rt=25,66 мин (метод 2).
Пример 13
N-[1-Ацетил-5-гидрокси-7-метокси-4-(4-метоксифенил)-2,2,4-триметил-1,2,3,4-тетрагидрохинолин-6-ил]-3,5-диметилбензамид
(а) N-[1-Ацетил-5,7-диметокси-4-(4-метоксифенил)-2,2,4-триметил-1,2,3,4-тетрагидрохинолин-6-ил]-3,5-диметилбензамид
В соответствии с общей методикой В соединение, описанное в примере 1е (800 мг), ацилируют 3,5-диметилбензойной кислотой (330 мг), ДМА (768 мкл) и HATU (1,1 г) в CH2Cl2 (10 мл). Указанное в заголовке соединение очищают хроматографией на силикагеле (элюирование с градиентом: гептан/этилацетат от 1/0 до 0/1 (об./об.)).
Выход 1,18 г.
(b) N-[1-Ацетил-5-гидрокси-7-метокси-4-(4-метоксифенил)-2,2,4-триметил-1,2,3,4-тетрагидрохинолин-6-ил]-3,5-диметилбензамид
В соответствии с общей методикой D соединение, описанное в примере 13а (300 мг), обрабатывают трибромидом бора (513 мкл) в CH2Cl2 (25 мл), поддерживая температуру не уровне не выше -40°С. Указанное в заголовке соединение очищают препаративной ВЭЖХ (метод А).
Выход: 41 мг; МС-ESI: [M+H]+ = 517,4; ВЭЖХ: Rt=13,89 мин (метод 3).
Пример 14
N-[1-Ацетил-5-гидрокси-7-метокси-4-(4-метоксифенил)-2,2,4-триметил-1,2,3,4-тетрагидрохинолин-6-ил]-3,5-дибромбензамид
(а) N-[1-Ацетил-5,7-диметокси-4-(4-метоксифенил)-2,2,4-триметил-1,2,3,4-тетрагидрохинолин-6-ил]-3,5-дибромбензамид
В соответствии с общей методикой В соединение, описанное в примере 1е (800 мг), ацилируют 3,5-дибромбензойной кислотой (616 мг), ДМА (768 мкл) и HATU (1,1 г) в ДМФА (10 мл). Указанное в заголовке соединение очищают хроматографией на силикагеле (элюирование с градиентом: гептан/этилацетат от 1/0 до 0/1 (об./об.)).
Выход: 900 мг.
(b) N-[1-Ацетил-5-гидрокси-7-метокси-4-(4-метоксифенил)-2,2,4-триметил-1,2,3,4-тетрагидрохинолин-6-ил]-3,5-дибромбензамид
В соответствии с общей методикой D соединение, описанное в примере 14а (300 мг), обрабатывают трибромидом бора (639 мкл) в CH2Cl2 (25 мл), поддерживая температуру на уровне не выше -60°С. Указанное в заголовке соединение очищают препаративной ВЭЖХ (метод В).
Выход: 28 мг; МС-ESI: [M+H]+ = 647,2; ВЭЖХ: Rt=16,29 мин (метод 3).
Пример 15
[1-Ацетил-2,2,4-триметил-7-(2-морфолин-4-илэтокси)-4-фенил-1,2,3,4-тетрагидрохинолин-6-ил]амид бифенил-4-карбоновой кислоты
(а) 1-Fmoc-2-метокси-4-нитроанилин
Раствор 2-метокси-4-нитроанилина (3,0 г) и пиридина (1,6 мл) в ТГФ (30 мл) охлаждают до 0°С. Небольшими порциями добавляют Fmoc (5,07 г), после завершения добавления ледяную баню удаляют и смесь перемешивают в течение 5 часов. ТГФ удаляют в вакууме, и остаток растворяют в CH2Cl2 (175 мл). Добавляют метанол (˜100 мл) и CH2Cl2 частично удаляют в вакууме до тех пор, пока не образуется осадок. Смесь оставляют на 1 час, после чего кристаллы отфильтровывают и сушат в вакууме, получая указанное в заголовке соединение.
Выход: 6,32 г; МС-ESI: [M+H]+ = 391,2.
(b) 9-флуоренилметил-N-(2-метокси-4-аминофенил)карбамат
Общая методика Е: Смесь соединения, описанного в примере 15а (6,07 г), уксусной кислоты (8,9 мл) и ТГФ (150 мл) охлаждают до 0°С. Несколькими порциями добавляют порошкообразный цинк (20,4 г) и ледяную баню удаляют. После того как температура медленно достигнет 10°С, она быстро возрастает до 45°С. Реакционной смеси дают медленно охладиться до комнатной температуры, избыток цинка удаляют фильтрованием и добавляют CH2Cl2 (˜500 мл). Смесь промывают насыщенным водным раствором NaHCO3 (3×200 мл) и насыщенным раствором соли (1×200 мл). Органический слой отделяют, сушат (MgSO4), фильтруют и концентрируют в вакууме до образования осадка. Смесь оставляют на ночь при температуре 0°С, после чего кристаллы отфильтровывают и сушат в вакууме, получая указанное в заголовке соединение.
Выход: 4,45 г.
(с) 9Н-флуорен-9-илметиловый эфир (7-метокси-2,2,4-триметил-1,2-дигидрохинолин-6-ил)карбаминовой кислоты
Смесь соединения, описанного в примере 15b (4,45 г), I2 (157 мг), MgSO4 (7,4 г), 4-трет-бутилкатехола (61 мг) и ацетона (˜350 мл) кипятят с обратным холодильником в течение 5 часов. MgSO4 удаляют фильтрованием и фильтрат концентрируют в вакууме. Указанное в заголовке соединение получают хроматографией на силикагеле (элюирование с градиентом: гептан/этилацетат от 9/1 до 7/3 (об./об.)).
Выход: 4,24 г.
(d) 9Н-флуорен-9-илметиловый эфир (1-ацетил-7-метокси-2,2,4-триметил-1,2-дигидрохинолин-6-ил)карбаминовой кислоты
К раствору соединения, описанного в примере 15с (4,24 г), в пиридине (25 мл) и CH2Cl2 (25 мл) добавляют небольшое количество DMAP (˜20 мг). Затем медленно добавляют ацетилхлорид (2,0 мл) в CH2Cl2 (20 мл). После завершения добавления смесь разбавляют CH2Cl2 (˜100 мл) и промывают водой (3×100 мл), 0,1М HCl (3×100 мл), 0,5М водн. HCl (1×100 мл) и насыщенным раствором соли (1×100 мл). Органический слой сушат (MgSO4) и концентрируют в вакууме. Указанное в заголовке соединение получают хроматографией на силикагеле (элюирование с градиентом: гептан/этилацетат от 9/1 до 7/3 (об./об.)).
Выход: 3,91 г.
(е) 1-Ацетил-6-амино-7-метокси-2,2,4-триметил-1,2-дигидрохинолин
Пиперидин (8,0 мл) добавляют к раствору соединения, описанного в примере 15d (3,91 г), в CH2Cl2 (80 мл). Через 1,5 часа реакционную смесь концентрируют в вакууме и указанное в заголовке соединение получают хроматографией на силикагеле (элюирование с градиентом: гептан/этилацетат от 9/1 до 7/3 (об./об.)).
Выход: 2,2 г.
(f) (1-Ацетил-7-метокси-2,2,4-триметил-1,2-дигидрохинолин-6-ил)амид бифенил-4-карбоновой кислоты
Общая методика F: К смеси соединения, описанного в примере 15е (2,2 г), толуола (45 мл) и пиридина (5 мл) добавляют 4-бифенилкарбонилхлорид (2,21 г). Если реакция не завершится по истечении 3 часов при комнатной температуре, вводят дополнительное количество 4-бифенилкарбонилхлорида (2,0 г). Перемешивание продолжают в течение 30 минут, после чего реакционную смесь концентрируют в вакууме. Остаток переносят в этилацетат (˜100 мл) и промывают насыщенным водным раствором NaHCO3 (100 мл), 1М водн. HCl (3×100 мл) и насыщенным раствором соли (100 мл). Органический слой сушат (MgSO4) и концентрируют в вакууме. К остатку добавляют CH2Cl2 (˜50 мл), твердые вещества удаляют фильтрованием и больше не используют. Фильтрат концентрируют в вакууме и указанное в заголовке соединение получают хроматографией на силикагеле (элюирование с градиентом: гептан/этилацетат от 1/0 до 1/1 (об./об.)).
Выход: 3,1 г.
(g) (1-Ацетил-7-гидрокси-2,2,4-триметил-1,2,3,4-тетрагидрохинолин-6-ил)амид бифенил-4-карбоновой кислоты
Общая методика G: К раствору соединения, описанного в примере 15f (3,1 г), в бензоле (100 мл) добавляют трихлорид алюминия (5,6 г), и реакционную смесь перемешивают при комнатной температуре в течение 20 часов. Реакцию гасят Н2О (˜100 мл) и значение рН смеси доводят до 8 добавляя 2М водн. NaOH при энергичном перемешивании. Добавляют этилацетат (˜300 мл) и органический слой промывают Н2О (2×150 мл) и насыщенным раствором соли (1×150 мл), сушат (MgSO4) и концентрируют в вакууме, получая продукт, который используют на следующей стадии без дополнительной очистки.
Выход: 3,5 г.
(h) [1-Ацетил-2,2,4-триметил-7-(2-морфолин-4-илэтокси)-4-фенил-1,2,3,4-тетрагидрохинолин-6-ил]амид бифенил-4-карбоновой кислоты
Общая методика Н: Смесь соединения, описанного в примере 15g (70 мг), гидрохлорида N-(2-хлорэтил)морфолина (31 мг), Cs2CO3 и ДМФА (3 мл) перемешивают при 50°С до тех пор, пока не останется исходного вещества. Реакционную смесь разбавляют этилацетатом (15 мл) и добавляют воду (˜15 мл). Органический слой промывают водой (3×15 мл), отделяют, сушат (MgSO4), фильтруют и концентрируют в вакууме. Указанное в заголовке соединение получают в виде HCl соли лиофилизацией из смеси 1,4-диоксана и Н2О, содержащей HCl.
Выход: 63 мг (HCl соль); МС-ESI: [M+H]+ = 618,6; ВЭЖХ: Rt=19,49 мин (метод 4).
Пример 16
(1-Ацетил-7-диметилкарбамоилметокси-2,2,4-триметил-4-фенил-1,2,3,4-тетрагидрохинолин-6-ил]амид бифенил-4-карбоновой кислоты
В соответствии с общей методикой Н соединение, описанное в примере 15g (79 мг), алкилируют 2-хлор-N,N-диметилацетамидом (23 мг) и CsCO3 (255 мг) в ДМФА (2 мл). Указанное в заголовке соединение очищают кристаллизацией из CH3CN.
Выход: 15 мг; МС-ESI: [M+H]+ = 590,6; ВЭЖХ: Rt=23,58 мин (метод 5).
Пример 17
[1-Ацетил-2,2,4-триметил-4-фенил-7-(3-пиперидин-1-илпропокси)-1,2,3,4-тетрагидрохинолин-6-ил]амид бифенил-4-карбоновой кислоты
В соответствии с общей методикой Н соединение, описанное в примере 15g (79 мг), алкилируют гидрохлоридом N-(3-хлорпропил)пиперидина (37,4 мг) и Cs2CO3 (255 мг) в ДМФА (2 мл). Указанное в заголовке соединение очищают кристаллизацией из CH3CN.
Выход: 83 мг (HCl соль); МС-ESI: [M+H]+ = 630,8; ВЭЖХ: Rt=15,49 мин (метод 5).
Пример 18
[1-Ацетил-2,2,4-триметил-4-фенил-7-(пиридин-2-илметокси)-1,2,3,4-тетрагидрохинолин-6-ил)амид бифенил-4-карбоновой кислоты
В соответствии с общей методикой H соединение, описанное в примере 15g (79 мг), алкилируют гидрохлоридом 2-пиколилхлорида (31 мг) и Cs2CO3 (255 мг) в ДМФА (2 мл). Указанное в заголовке соединение очищают кристаллизацией из CH3CN.
Выход: 32 мг (HCl соль); МС-ESI: [M+H]+ = 596,6; ВЭЖХ: Rt=22,41 мин (метод 6).
Пример 19
[1-Ацетил-2,2,4-триметил-4-фенил-7-(пиридин-3-илметокси)-1,2,3,4-тетрагидрохинолин-6-ил)амид бифенил-4-карбоновой кислоты
В соответствии с общей методикой H соединение, описанное в примере 15g (79 мг), алкилируют гидрохлоридом 3-пиколилхлорида (31 мг) и Cs2CO3 (255 мг) в ДМФА (2 мл). Указанное в заголовке соединение очищают кристаллизацией из CH3CN.
Выход: 36 мг (HCl соль); МС-ESI: [M+H]+ = 596,6; ВЭЖХ: Rt=19,70 мин (метод 6).
Пример 20
[1-Ацетил-2,2,4-триметил-4-фенил-7-(пиридин-4-илметокси)-1,2,3,4-тетрагидрохинолин-6-ил)амид бифенил-4-карбоновой кислоты
В соответствии с общей методикой H соединение, описанное в примере 15g (79 мг), алкилируют гидрохлоридом 4-пиколилхлорида (31 мг) и Cs2CO3 (255 мг) в ДМФА (2 мл). Указанное в заголовке соединение очищают кристаллизацией из CH3CN.
Выход: 31 мг (HCl соль); МС-ESI: [M+H]+ = 596,4; ВЭЖХ: Rt=17,09 мин (метод 6).
Пример 21
[1-Ацетил-7-(2-метилэтокси)-2,2,4-триметил-4-фенил-1,2,3,4-тетрагидрохинолин-6-ил]амид бифенил-4-карбоновой кислоты
В соответствии с общей методикой H соединение, описанное в примере 15g (79 мг), алкилируют гидрохлоридом 2-диметиламиноэтилхлорида (27 мг) и Cs2CO3 (255 мг) в ДМФА (2 мл). Указанное в заголовке соединение очищают кристаллизацией из CH3CN.
Выход: 55 мг (HCl соль); МС-ESI: [M+H]+ = 576,6; ВЭЖХ: Rt=14,94 мин (метод 5).
Пример 22
(1-Ацетил-7-карбамоилметокси-2,2,4-триметил-4-фенил-1,2,3,4-тетрагидрохинолин-6-ил]амид бифенил-4-карбоновой кислоты
В соответствии с общей методикой H соединение, описанное в примере 15g (79 мг), алкилируют 2-хлорацетамидом (18 мг) и Cs2CO3 (255 мг) в ДМФА (2 мл). Указанное в заголовке соединение очищают препаративной ВЭЖХ (метод А).
Выход: 60,2 мг; МС-ESI: [M+H]+ = 562,4; ВЭЖХ: Rt=20,47 мин (метод 5).
Пример 23
(3-{1-Ацетил-6-[(бифенил-4-карбонил)амино]-2,2,4-триметил-4-фенил-1,2,3,4-тетрагидрохинолин-7-илокси}пропил)амид морфолин-4-карбоновой кислоты
В соответствии с общей методикой H соединение, описанное в примере 15g (79 мг), алкилируют морфолин-4-карбоновой кислотой (40 мг) и Cs2CO3 (255 мг) в ДМФА (2 мл). Указанное в заголовке соединение очищают препаративной ВЭЖХ (метод А).
Выход: 52,4 мг; МС-ESI: [M+H]+ = 675,6; ВЭЖХ: Rt=22,31 мин (метод 5).
Пример 24
1-Ацетил-6-(3,5-дибромбензоиламино)-2,2,4-триметил-4-фенил-1,2,3,4-тетрагидрохинолин-7-иловый эфир фуран-2-карбоновой кислоты
(а) N-(1-Ацетил-7-метокси-2,2,4-триметил-1,2-дигидрохинолин-6-ил)-3,5-дибромбензамид
В соответствии с общей методикой F соединение, описанное в примере 15e (1,0 мг), ацилируют 3,5-дибромбензоилхлоридом (1,72 г) в толуоле (9 мл) и пиридине (1 мл). Указанное в заголовке соединение очищают хроматографией на силикагеле (элюирование: гептан/этилацетат = 8/2 (об./об.)).
Выход: 1,3 мг.
(b) N-(1-Ацетил-7-гидрокси-2,2,4-триметил-1,2,3,4-тетрагидрохинолин-6-ил)-3,5-дибромбензамид
В соответствии с общей методикой G соединение, описанное в примере 24а (1,3 г), перемешивают с AlCl3 (1,0 г) в бензоле (50 мл). Полученный продукт используют на следующей стадии без дополнительной очистки.
Выход: 1,39 г.
(с) 1-Ацетил-6-(3,5-дибромбензоиламино)-2,2,4-триметил-4-фенил-1,2,3,4-тетрагидрохинолин-7-иловый эфир фуран-2-карбоновой кислоты
Смесь соединения, описанного в примере 24b (100 мг), фуроилхлорида (16 мкл) и DIPEA (60 мкл) в CH2Cl2 (5 мл) перемешивают при комнатной температуре до тех пор, пока не останется исходных веществ. Добавляют воду, органический слой отделяют, промывают насыщенным раствором соли, сушат (MgSO4) и концентрируют в вакууме. Указанное в заголовке соединение очищают препаративной ВЭЖХ (метод А).
Выход: 47 мг; МС-ESI: [M+H]+ = 681,2; ВЭЖХ: Rt=31,6 мин (метод 2).
Пример 25
N-[1-Ацетил-7-(2-аминоэтокси)-2,2,4-триметил-4-фенил-1,2,3,4-тетрагидрохинолин-6-ил]-3,5-дибромбензамид
Общая методика I: Смесь соединения, описанного в примере 24b (100 мг), трет-бутил-N-(2-гидроксиэтил)карбамата (29 мг), DtBAD (79 мг), DIPEA (60 мкл) и избытка полимер-связанного трифенилфосфина в CH2Cl2 (5 мл) перемешивают при комнатной температуре до тех пор, пока не останется исходного вещества. Реакционную смесь фильтруют и промывают водой и насыщенным раствором соли. Органический слой отделяют, сушат (MgSO4) и концентрируют в вакууме. Сырой продукт переносят в CH3CN (˜1 мл) и добавляют несколько капель ТФУК для способствования отщеплению трет-бутилкарбамата. Указанное в заголовке соединение очищают препаративной ВЭЖХ (метод А).
Выход: 17 мг (ТФУ соль); МС-ESI: [M+H]+ = 630,2; ВЭЖХ: Rt=15,6 мин (метод 2).
Пример 26
Трет-Бутиловый эфир {2-[1-ацетил-6-(3,5-диметилбензоиламино)-2,2,4-триметил-4-фенил-1,2,3,4-тетрагидрохинолин-7-илокси]этил}карбаминовой кислоты
(а) N-(1-ацетил-7-метокси-2,2,4-триметил-1,2-дигидрохинолин-6-ил)-3,5-диметилбензамид
В соответствии с общей методикой F соединение, описанное в примере 15е (1,0 г), ацилируют 3,5-диметилбензоилхлоридом (0,97 г) в толуоле (9 мл) и пиридине (1 мл). Указанное в заголовке соединение очищают хроматографией на силикагеле (элюирование: гептан/этилацетат = 8/2 (об./об.)).
Выход: 1,1 г.
(b) N-(1-Ацетил-7-гидрокси-2,2,4-триметил-4-фенил-1,2,3,4-тетагидрохинолин-6-ил)-3,5-диметилбензамид
В соответствии с общей методикой G соединение, описанное в примере 26a (1,1 г), перемешивают с AlCl3 (1,0 г) в бензоле (50 мл). Полученный продукт используют на следующей стадии без дополнительной очистки.
Выход: 1,3 г.
(с) Трет-Бутиловый эфир {2-[1-ацетил-6-(3,5-диметилбензоиламино)-2,2,4-триметил-4-фенил-1,2,3,4-тетрагидрохинолин-7-илокси]этил}карбаминовой кислоты
В соответствии с общей методикой I соединение, описанное в примере 26b (100 мг), алкилируют трет-бутил-N-(2-гидроксэтил)карбаматом (37 мл), DtBAD (101 мг), DIPEA (77 мкл) и избытком полимер-связанного трифенилфосфина в CH2Cl2 (5 мл). В данном случае группа трет-бутилкарбамата не подвергается расщеплению, что приводит после очистки препаративной ВЭЖХ (метод А) и лиофизации к получению указанного в заголовке продукта.
Выход: 38 мг; МС-ESI: [M+H]+ = 600,4; ВЭЖХ: Rt=33,1 мин (метод 2).
Пример 27
N-[1-Ацетил-7-(фуран-2-илметокси)-2,2,4-триметил-4-фенил-1,2,3,4-тетрагидрохинолин-6-ил]-3,5-диметилбензамид
В соответствии с общей методикой I соединение, описанное в примере 26b (100 мг), алкилируют 2-(гидроксиметил)фураном (21 мкл), DtBAD (101 мг), DIPEA (77 мкл) и избытком полимер-связанного трифенилфосфина в CH2Cl2 (5 мл). Указанное в заголовке соединение очищают препаративной ВЭЖХ (метод А) и затем лиофилизацией.
Выход: 16 мг; МС-ESI: [M+H]+ = 537,4; ВЭЖХ: Rt=33,8 мин (метод 2).
Пример 28
N-[1-Ацетил-2,2,4-триметил-4-фенил-7-(пиридин-4-илметокси)-1,2,3,4-тетрагидрохинолин-6-ил]-3,5-дихлорбензамид
(а) N-(1-Ацетил-7-метокси-2,2,4-триметил-1,2-дигидрохинолин-6-ил)-3,5-дихлорбензамид
В соответствии с общей методикой F соединение, описанное в примере 15e (1,0 г), ацилируют 3,5-дихлорбензоилхлоридом (1,2 г) в толуоле (9 мл) и пиридине (1 мл). Указанное в заголовке соединение очищают хроматографией на силикагеле (элюирование: гептан/этилацетат 8/2 (об./об.)).
Выход: 1,47 г.
(b) N-(1-Ацетил-7-гидрокси-2,2,4-триметил-4-фенил-1,2,3,4-тетрагидрохинолин-6-ил)-3,5-дихлорбензамид
В соответствии с общей методикой G соединение, описанное в примере 28a (1,47 г), перемешивают с AlCl3 (1,5 г) в бензоле (75 мл). Полученный продукт используют на следующей стадии без дополнительной очистки.
Выход: 1,51 г.
(с) N-[1-Ацетил-2,2,4-триметил-4-фенил-7-(пиридин-4-илметокси)-1,2,3,4-тетрагидрохинолин-6-ил]-3,5-дихлорбензамид
В соответствии с общей методикой H соединение, описанное в примере 28b (100 мг), алкилируют гидрохлоридом 4-пиколилхлорида (36 мг) и Cs2CO3 (255 мг) в смеси ДМФА (1 мл) и CH2Cl2 (4 мл). Указанное в заголовке соединение получают в виде соответствующей ТФУ соли после препаративной ВЭЖХ (метод А) и последующей лиофилизации.
Выход: 35 мг (ТФУ соль); МС-ESI: [M+H]+ = 588,4; ВЭЖХ: Rt=18,0 мин (метод 2).
Пример 29
N-[1-Ацетил-2,2,4-триметил-4-фенил-7-(2-пирролидин-1-илэтокси)-1,2,3,4-тетрагидрохинолин-6-ил]-3,5-диметилбензамид
Общая методика J: Смесь соединения, описанного в примере 26b (100 мг), гидрохлорида 2-хлорэтилпирролидина (41 мг) и DIPEA (77 мкл) в CH2Cl2 (5 мл) перемешивают при комнатной температуре до тех пор, пока не останется исходного вещества. Добавляют воду, органический слой отделяют, промывают насыщенным раствором соли, сушат (MgSO4) и концентрируют в вакууме. Очистка препаративной ВЭЖХ с последующей лиофилизацией приводит к получению указанного в заголовке соединения в виде соответствующей ТФУ соли.
Выход: 104 мг (ТФУ соль); МС-ESI: [M+H]+ = 554,4; ВЭЖХ: Rt=15,2 мин (метод 2).
Пример 30
N-[1-Ацетил-2,2,4-триметил-7-(5-метилизоксазол-3-илметокси)-4-фенил-1,2,3,4-тетрагидрохинолин-6-ил]-3,5-диметилбензамид
В соответствии с общей методикой J соединение, описанное в примере 26b (100 мг), алкилируют (хлорметил)-5-метилизоксазолом (32 мг) и DIPEA (77 мкл) в CH2Cl2 (5 мл). Очистка препаративной ВЭЖХ (метод А) с последующей лиофилизацией приводит к получению указанного в заголовке соединения.
Выход: 41 мг; МС-ESI: [M+H]+ = 552,4; ВЭЖХ: Rt=31,3 мин (метод 2).
Пример 31
N-[1-Ацетил-7-(2-диэтиламиноэтокси)-2,2,4-триметил-4-фенил-1,2,3,4-тетрагидрохинолин-6-ил]-3,5-диметилбензамид, ТФУ соль
В соответствии с общей методикой J соединение, описанное в примере 26b (100 мг), алкилируют гидрохлоридом N,N-диэтиламиноэтилхлорида (42 мг) и DIPEA (77 мкл) в CH2Cl2 (5 мл). Очистка препаративной ВЭЖХ (метод А) с последующей лиофилизацией приводит к получению указанного в заголовке соединения в виде соответствующей ТФУ соли.
Выход: 43 мг (ТФУ соль); МС-ESI: [M+H]+ = 556,4; ВЭЖХ: Rt=15,2 мин (метод 2).
Пример 32
N-[1-Ацетил-2,2,4-триметил-4-фенил-7-(пиридин-4-илметокси)-1,2,3,4-тетрагидрохинолин-6-ил]-5-бром-2-метиламинобензамид
(а) N-(1-Ацетил-7-метокси-2,2,4-триметил-1,2-дигидрохинолин-6-ил)-5-бром-2-метиламинобензамид
В соответствии с общей методикой F соединение, описанное в примере 15e (1,0 г), ацилируют 5-бром-2-метиламинобензоилхлоридом (1,43 г) в толуоле (9 мл) и пиридине (1 мл). Указанное в заголовке соединение получают хроматографией на силикагеле (элюирование: гептан/этилацетат 8/2 (об./об.)).
Выход: 595 мг.
(b) N-(1-Ацетил-7-гидрокси-2,2,4-триметил-4-фенил-1,2,3,4-тетрагидрохинолин-6-ил)-5-бром-2-метиламинобензамид
В соответствии с общей методикой G соединение, описанное в примере 32a (595 мг), перемешивают с AlCl3 (0,75 г) в бензоле (50 мл). Полученный продукт используют на следующей стадии без дополнительной очистки.
Выход: 437 мг.
(с) N-[1-Ацетил-2,2,4-триметил-4-фенил-7-(пиридин-4-илметокси)-1,2,3,4-тетрагидрохинолин-6-ил]-5-бром-2-метиламинобензамид
В соответствии с общей методикой H соединение, описанное в примере 32b (44 мг), алкилируют гидрохлоридом 4-пиколилхлорида (15 мг) и Cs2CO3 (˜100 мг) в смеси ДМФА (1 мл) и CH2Cl2 (4 мл). Указанное в заголовке соединение получают в виде соответствующей ТФУ соли после препаративной ВЭЖХ (метод В).
Выход: 18 мг (ТФУ соль); МС-ESI: [M+H]+ = 629,4; ВЭЖХ: Rt=18,1 мин (метод 2).
Пример 33
(1-Ацетил-7-диметиламино-2,2,4-триметил-4-фенил-1,2,3,4-тетрагидрохинолин-6-ил)амид фуран-2-карбоновой кислоты
(а) Трет-Бутиловый эфир (2,2,4-триметил-1,2-дигидрохинолин-6-ил)карбаминовой кислоты
Смесь N-Вос-1,4-фенилендиамина (75 г), MgSO4 (216 г), 4-трет-бутилкатехола (1,8 г) и йода (4,7 г) в безводном ацетоне (600 мл) кипятят с обратным холодильником в течение 20 часов. MgSO4 удаляют фильтрованием и фильтрат концентрируют в вакууме. Остаток хроматографируют на силикагеле (элюирование: гептан/этилацетат = 8/2 (об./об.)), получая продукт в виде коричневого масла.
Выход 41 г.
(b) Трет-бутиловый эфир (1-ацетил-2,2,4-триметил-1,2-дигидрохинолин-6-ил)карбаминовой кислоты
Раствор соединения, описанного в примере 33а (41 г), в пиридине (200 мл) и CH2Cl2 (200 мл) охлаждают до 0°С. По каплям добавляют ацетилхлорид (21 мл) в CH2Cl2 (50 мл). После завершения добавления смесь перемешивают в течение 3 часов при комнатной температуре. Добавляют этилацетат (2 л) и Н2О (2 л) и органический слой отделяют, сушат (MgSO4) и концентрируют в вакууме. Указанное в заголовке соединение получают кристаллизацией из этилацетата.
Выход: 23 г.
(с) 1-Ацетил-6-амино-2,2,4-триметил-4-фенил-1,2,3,4-тетрагидрохинолин
В соответствии с общей методикой G соединение, описанное в примере 33b (33,3 г), перемешивают с AlCl3 (40,4 г) в бензоле (700 мл). Полученный продукт очищают хроматографией на силикагеле (элюирование: гептан/этилацетат = 8/2 (об./об.)).
Выход: 22,4 г.
(d) 9-Флуоренилметиловый эфир (1-ацетил-2,2,4-триметил-4-фенил-1,2,3,4-тетрагидрохинолин-6-ил)карбаминовой кислоты
К раствору соединения, описанного в примере 33с (22,4 г), в ТГФ (300 мл) добавляют пиридин (6,4 мл) и полученную смесь охлаждают до 0°С. По каплям добавляют раствор FmocCl (20,7 г) в ТГФ (100 мл), и после завершения добавления смесь перемешивают в течение 1 часа при комнатной температуре. Реакционную смесь концентрируют в вакууме и добавляют этилацетат (800 мл) и 0,3М водн. HCl (500 мл). Органический слой отделяют, промывают 0,3М HCl (2Ч500 мл), Н2О (500 мл) и насыщенным раствором соли (500 мл), сушат (MgSO4) и концентрируют в вакууме. Продукт используют на следующей стадии без дополнительной очистки.
Выход: 43 г.
(е) 9-Флуоренилметиловый эфир (1-ацетил-2,2,4-триметил-7-нитро-4-фенил-1,2,3,4-тетрагидрохинолин-6-ил)карбаминовой кислоты
Дымящую азотную кислоту (3,07 мл) по каплям в течение 10 минут добавляют к смеси соединения, описанного в примере 33d (43 г), и уксусной кислоты (230 мл) в CH2Cl2 (230 мл). Реакционную смесь перемешивают до полного превращения исходного вещества, после чего добавляют Н2О (150 мл). Водный слой отделяют и экстрагируют CH2Cl2 (150 мл). Объединенные органические слои промывают насыщенным водным раствором NaHCO3 (3×200 мл) и насыщенным раствором соли (200 мл), затем сушат над MgSO4 и фильтруют. Добавляют метанол (˜200 мл) и CH2Cl2 осторожно удаляют в вакууме, после чего смесь оставляют на ночь при комнатной температуре. Ярко-желтые кристаллы отфильтровывают и сушат (MgSO4) в вакууме.
Выход: 29,3 г.
(f) 9-Флуоренилметиловый эфир (1-ацетил-7-амино-2,2,4-триметил-4-фенил-1,2,3,4-тетрагидрохинолин-6-ил)карбаминовой кислоты
В соответствии с общей методикой Е соединение, описанное в примере 33е (20 г), восстанавливают с использованием порошкообразного цинка (45 г) и уксусной кислоты (20 мл) в ТГФ (˜600 мл), получая продукт, который используют на следующей стадии без дополнительной очистки.
Выход: 21 г.
(g) 9-Флуоренилметиловый эфир (1-ацетил-7-диметиламино-2,2,4-триметил-4-фенил-1,2,3,4-тетрагидрохинолин-6-ил)карбаминовой кислоты
Водный раствор формальдегида (37%, 3,8 мл) добавляют к раствору соединения, описанного в примере 33f (12 г), уксусной кислоты (15,7 мл) и цианоборгидрида натрия (2,9 г) в метаноле (200 мл), что приводит к экзотермической реакции и образованию белого осадка. Для способствования перемешиванию добавляют дополнительное количество МеОН. Смесь перемешивают в течение 15 минут, осадок отфильтровывают и промывают смесью МеОН/Н2О = 1/1 (об./об.). Фильтрат частично концентрируют для получения большего количества твердого вещества, которое также собирают. Объединенные твердые продукты перекристаллизовывают из CH2Cl2/МеОН, получая диметилированное соединение.
Выход: 9,7 г.
(h) 1-Ацетил-6-амино-7-диметиламино-2,2,4-триметил-4-фенил-1,2,3,4-тетрагидрохинолин
Пиперидин (7,7 мл) добавляют к раствору соединения, описанного в примере 33g (4,5 г), в CH2Cl2 (70 мл). Через 24 часа реакционную смесь разбавляют CH2Cl2 (100 мл) и промывают 0,5М водн. HCl (2×150 мл), водой (100 мл) и насыщенным раствором соли (1 мл). Органический слой сушат (MgSO4) и разбавляют до общего объема 200 мл. Полученный раствор указанного в заголовке соединения (˜13,8 мг/мл) используют в последующем синтезе.
(i) (1-Ацетил-7-диметиламино-2,2,4-триметил-4-фенил-1,2,3,4-тетрагидрохинолин-6-ил)амид фуран-2-карбоновой кислоты
Триэтиламин (38 мкл) и 2-фуроилхлорид (27 мкл) добавляют к раствору соединения, описанного в примере 33h (96,6 мг), в CH2Cl2 (10 мл) и полученную смесь перемешивают до полного превращения исходного вещества. Указанное в заголовке соединение очищают хроматографией на силикагеле (элюирование с градиентом: гептан/этилацетат от 1/0 до 0/1 (об./об.)).
Выход: 5,5 мг; МС-ESI: [M+H]+ = 446,2; ВЭЖХ: Rt=19,02 мин (метод 2).
Пример 34
(1-Ацетил-7-диметиламино-2,2,4-триметил-4-фенил-1,2,3,4-тетрагидрохинолин-6-ил)амид 5-метилтиофен-2-карбоновой кислоты
В соответствии с общей методикой А соединение, описанное в примере 33h (96,6 мг), ацилируют 5-метилтиофен-2-карбоновой кислотой (39,1 мг), HATU (157 мг) и DIPEA (239 мкл) в CH2Cl2 (10 мл). Указанное в заголовке соединение очищают препаративной ВЭЖХ (метод В).
Выход: 35,5 мг; МС-ESI: [M+H]+ = 476,2; ВЭЖХ: Rt=21,26 мин (метод 2).
Пример 35
(1-Ацетил-7-диметиламино-2,2,4-триметил-4-фенил-1,2,3,4-тетрагидрохинолин-6-ил)амид бифенил-4-карбоновой кислоты
В соответствии с общей методикой А соединение, описанное в примере 33h (96,6 мг), ацилируют 4-бифенилкарбоновой кислотой (54,4 мг), HATU (157 мг) и DIPEA (239 мкл) в CH2Cl2 (10 мл). Указанное в заголовке соединение очищают препаративной ВЭЖХ (метод В).
Выход: 31,5 мг; МС-ESI: [M+H]+ = 532,4; ВЭЖХ: Rt=24,92 мин (метод 2).
Пример 36
N-(1-Ацетил-7-диметиламино-2,2,4-триметил-4-фенил-1,2,3,4-тетрагидрохинолин-6-ил)-3,5-дибромбензамид
В соответствии с общей методикой А соединение, описанное в примере 33h (96,6 мг), ацилируют 3,5-дибромбензойной кислотой (77 мг), HATU (157 мг) и DIPEA (239 мкл) в CH2Cl2 (10 мл). Указанное в заголовке соединение очищают кристаллизацией из смеси CH2Cl2/CH3CN.
Выход: 24,3 мг; МС-ESI: [M+H]+ = 614,2; ВЭЖХ: Rt=27,71 мин (метод 2).
Пример 37
(1-Ацетил-7-диметиламино-2,2,4-триметил-4-фенил-1,2,3,4-тетрагидрохинолин-6-ил)амид циклопентанкарбоновой кислоты
В соответствии с общей методикой А соединение, описанное в примере 33h (137 мг), ацилируют циклопентанкарбоновой кислотой (128 мкл), HATU (224 мг) и DIPEA (400 мкл) в CH2Cl2 (10 мл). Указанное в заголовке соединение очищают хроматографией на силикагеле (элюирование с градиентом: гептан/этилацетат от 1/0 до 0/1 (об./об.)).
Выход: 148 мг; МС-ESI: [M+H]+ = 448,4; ВЭЖХ: Rt=12,93 мин (метод 1).
Пример 38
N-(1-Ацетил-7-диметиламино-2,2,4-триметил-4-фенил-1,2,3,4-тетрагидрохинолин-6-ил)изобутирамид
В соответствии с общей методикой А соединение, описанное в примере 33h (137 мг), ацилируют изомасляной кислотой (110 мкл), HATU (224 мг) и DIPEA (400 мкл) в CH2Cl2 (10 мл). Указанное в заголовке соединение очищают хроматографией на силикагеле (элюирование с градиентом: гептан/этилацетат от 1/0 до 0/1 (об./об.)).
Выход: 43 мг; МС-ESI: [M+H]+ = 422,4; ВЭЖХ: Rt=9,99 мин (метод 1).
Пример 39
(1-Ацетил-7-фуран-2-илкарбониламино-2,2,4-триметил-4-фенил-1,2,3,4-тетрагидрохинолин-6-ил)амид фуран-2-карбоновой кислоты
К раствору соединения, описанного в примере 33f (150 мг), и триэтиламина (43 мкл) в CH2Cl2 (1 мл) добавляют 2-фуроилхлорид (30 мкл). После полного превращения исходного вещества добавляют 1М водную HCl, органический слой отделяют, добавляют пиперидин (1 мл) и полученную смесь перемешивают в течение ночи. Реакционную смесь промывают 1М водной HCl, органический слой отделяют, сушат (MgSO4) и концентрируют в вакууме. Указанное в заголовке соединение очищают хроматографией на силикагеле (элюирование с градиентом: гептан/этилацетат от 1/0 до 0/1 (об./об.)) и затем препаративной ВЭЖХ (метод А).
Выход: 18 мг; МС-ESI: [M+H]+ = 512,4; ВЭЖХ: Rt=19,92 мин (метод 2).
Пример 40
(1-Ацетил-2,2,4-триметил-4-фенил-7-пропиламино-1,2,3,4-тетрагидрохинолин-6-ил)амид 5-метилтиофен-2-карбоновой кислоты
(а) 1-Ацетил-6-амино-2,2,4-триметил-4-фенил-7-пропиламино-1,2,3,4-тетрагидрохинолин
Общая методика К: К смеси соединения, описанного в примере 33f (750 мг), уксусной кислоты (953 мкл), цианоборгидрида натрия (135 мг) и МеОН (10 мл) добавляют пропиональдегид (94,2 мкл). Смесь перемешивают в течение 18 часов, добавляют воду и осадок отфильтровывают. Осадок переносят в CH2Cl2 (10 мл), добавляют пиперидин (1 мл) и полученную смесь перемешивают в течение 18 часов. Реакционную смесь промывают 1М водной HCl, органический слой отделяют и разбавляют до общего объема 50 мл. Полученный раствор используют на следующих стадиях синтеза.
(b) (1-Ацетил-2,2,4-триметил-4-фенил-7-пропиламино-1,2,3,4-тетрагидрохинолин-6-ил)амид 5-метилтиофен-2-карбоновой кислоты
В соответствии с общей методикой А соединение, описанное в примере 40а (100 мг), ацилируют 5-метилтиофен-2-карбоновой кислотой (39,1 мг), HATU (157 мг) и DIPEA (239 мкл) в CH2Cl2 (10 мл). Указанное в заголовке соединение очищают препаративной ВЭЖХ и лиофилизуют.
Выход: 18,3 мг; МС-ESI: [M+H]+ = 490,4; ВЭЖХ: Rt=23,96 мин (метод 2).
Пример 41
(1-Ацетил-7-этиламино-2,2,4-триметил-4-фенил-1,2,3,4-тетрагидрохинолин-6-ил)амид бифенил-4-карбоновой кислоты
(а) 1-Ацетил-6-амино-7-этиламино-2,2,4-триметил-4-фенил-1,2,3,4-тетрагидрохинолин
В соответствии с общей методикой К соединение, описанное в примере 33f (750 мг), алкилируют ацетальдегидом (73,3 мкл) и удаляют защитную группу пиперидином (1 мл), получая после обработки и разбавления раствор указанного в заголовке соединения в CH2Cl2.
(b) (1-Ацетил-7-этиламино-2,2,4-триметил-4-фенил-1,2,3,4-тетрагидрохинолин-6-ил)амид бифенил-4-карбоновой кислоты
В соответствии с общей методикой А соединение, описанное в примере 41а (100 мг), ацилируют 4-бифенилкарбоновой кислотой (54,4 мг), HATU (157 мг) и DIPEA (239 мкл) в CH2Cl2 (10 мл). Указанное в заголовке соединение очищают препаративной ВЭЖХ и лиофилизуют.
Выход: 9,8 мг; МС-ESI: [M+H]+ = 532,4; ВЭЖХ: Rt=25,55 мин (метод 2).
Пример 42
{1-Ацетил-2,2,4-триметил-4-фенил-7-[(пиридин-4-илметил)амино]-1,2,3,4-тетрагидрохинолин-6-ил}амид 5-метилтиофен-2-карбоновой кислоты
(а) 1-Ацетил-6-амино-2,2,4-триметил-4-фенил-7-[(пиридин-4-илметил)амино]-1,2,3,4-тетрагидрохинолин
В соответствии с общей методикой К соединение, описанное в примере 33f (750 мг), алкилируют 4-пиридинкарбоксальдегидом (125 мкл) и удаляют защитную группу пиперидином (1 мл), получая после обработки и разбавления раствор указанного в заголовке соединения в CH2Cl2.
(b) {1-Ацетил-2,2,4-триметил-4-фенил-7-[(пиридин-4-илметил)амино]-1,2,3,4-тетрагидрохинолин-6-ил}амид 5-метилтиофен-2-карбоновой кислоты
В соответствии с общей методикой А соединение, описанное в примере 42а (114 мг), ацилируют 5-метилтиофен-2-карбоновой кислотой (39,1 мг), HATU (157 мг) и DIPEA (239 мкл) в CH2Cl2 (10 мл). Указанное в заголовке соединение очищают препаративной ВЭЖХ и лиофилизуют.
Выход: 51 мг; МС-ESI: [M+H]+ = 539,4; ВЭЖХ: Rt=13,19 мин (метод 2).
Пример 43
{1-Ацетил-2,2,4-триметил-4-фенил-7-[(пиридин-3-илметил)амино]-1,2,3,4-тетрагидрохинолин-6-ил}амид 5-метилтиофен-2-карбоновой кислоты
(а) 1-Ацетил-6-амино-2,2,4-триметил-4-фенил-7-[(пиридин-3-илметил)амино]-1,2,3,4-тетрагидрохинолин
В соответствии с общей методикой К соединение, описанное в примере 33f (750 мг), алкилируют 3-пиридинкарбоксальдегидом (125 мкл) и удаляют защитную группу пиперидином (1 мл), получая после обработки и разбавления раствор указанного в заголовке соединения в CH2Cl2.
(b) {1-Ацетил-2,2,4-триметил-4-фенил-7-[(пиридин-3-илметил)амино]-1,2,3,4-тетрагидрохинолин-6-ил}амин 5-метилтиофен-2-карбоновой кислоты
В соответствии с общей методикой А соединение, описанное в примере 43а (114 мг), ацилируют 5-метилтиофен-2-карбоновой кислотой (39,1 мг), HATU (157 мг) и DIPEA (239 мкл) в CH2Cl2 (10 мл). Указанное в заголовке соединение очищают препаративной ВЭЖХ и лиофилизуют.
Выход: 44 мг; МС-ESI: [M+H]+ = 539,4; ВЭЖХ: Rt=13,45 мин (метод 2).
Пример 44
N-(1-Ацетил-7-изобутиламино-2,2,4-триметил-4-фенил-1,2,3,4-тетрагидрохинолин-6-ил)-3,5-дибромбензамид
(а) 1-Ацетил-6-амино-7-изобутиламино-2,2,4-триметил-4-фенил-1,2,3,4-тетрагидрохинолин
В соответствии с общей методикой К соединение, описанное в примере 33f (750 мг), алкилируют изобутилальдегидом (119 мкл) и удаляют защитную группу пиперидином (1 мл), получая после обработки и разбавления раствор указанного в заголовке соединения в CH2Cl2.
(b) N-(1-Ацетил-7-изобутиламино-2,2,4-триметил-4-фенил-1,2,3,4-тетрагидрохинолин-6-ил)-3,5-дибромбензамид
В соответствии с общей методикой А соединение, описанное в примере 44а (114 мг), ацилируют 3,5-дибромбензойной кислотой (77 мг), HATU (157 мг) и DIPEA (239 мкл) в CH2Cl2 (10 мл). Указанное в заголовке соединение очищают препаративной ВЭЖХ и лиофилизуют.
Выход: 54 мг; МС-ESI: [M+H]+ = 642,4; ВЭЖХ: Rt=29,47 мин (метод 2).
Пример 45
(1-Ацетил-7-бензиламино-2,2,4-триметил-4-фенил-1,2,3,4-тетрагидрохинолин-6-ил)амид бифенил-4-карбоновой кислоты
(а) 1-Ацетил-6-амино-7-бензиламино-2,2,4-триметил-4-фенил-1,2,3,4-тетрагидрохинолин
В соответствии с общей методикой К соединение, описанное в примере 33f (750 мг), алкилируют бензальдегидом (133 мкл) и удаляют защитную группу пиперидином (1 мл), получая после обработки и разбавления раствор указанного в заголовке соединения в CH2Cl2.
(b) (1-Ацетил-7-бензиламино-2,2,4-триметил-4-фенил-1,2,3,4-тетрагидрохинолин-6-ил)амид бифенил-4-карбоновой кислоты
В соответствии с общей методикой А соединение, описанное в примере 45a (113 мг), ацилируют 4-бифенилкарбоновой кислотой (54,4 мг), HATU (157 мг) и DIPEA (239 мкл) в CH2Cl2 (10 мл). Указанное в заголовке соединение очищают препаративной ВЭЖХ и лиофилизуют.
Выход: 114 мг; МС-ESI: [M+H]+ = 594,4; ВЭЖХ: Rt=26,46 мин (метод 2).
Пример 46
(1-Ацетил-7-бензиламино-2,2,4-триметил-4-фенил-1,2,3,4-тетрагидрохинолин-6-ил)амид 5-метилтиофен-2-карбоновой кислоты
В соответствии с общей методикой А соединение, описанное в примере 45а (113 мг), ацилируют 5-метилтиофен-2-карбоновой кислотой (39,1 мг), HATU (157 мг) и DIPEA (239 мкл) в CH2Cl2 (10 мл). Указанное в заголовке соединение очищают препаративной ВЭЖХ и лиофилизуют.
Выход: 107 мг; МС-ESI: [M+H]+ = 538,4; ВЭЖХ: Rt=18,59 мин (метод 2).
Пример 47
N-{1-Ацетил-2,2,4-триметил-4-фенил-7-[(пиридин-3-илметил)амино]-1,2,3,4-тетрагидрохинолин-6-ил}-3,5-дибромбензамид
В соответствии с общей методикой А соединение, описанное в примере 43а (114 мг), ацилируют 3,5-дибромбензойной кислотой (77 мг), HATU (157 мг) и DIPEA (239 мкл) в CH2Cl2 (10 мл). Указанное в заголовке соединение очищают препаративной ВЭЖХ и лиофилизуют.
Выход: 41 мг; МС-ESI: [M+H]+ = 677,2; ВЭЖХ: Rt=14,88 мин (метод 2).
Пример 48
N-(1-Ацетил-7-диметиламино-4-метил-4-фенил-1,2,3,4-тетрагидрохинолин-6-ил)-3,5-дибромбензамид
(а) 4-Метил-1,2-дигидрохинолин
Раствор лепидина (10,0 г) в ТГФ охлаждают до -78°С, после чего добавляют раствор BH3·THF в ТГФ (1М, 70 мл). Через 2 часа добавляют раствор бис(2-метоксиэтокси)алюмодигидрид натрия в толуоле (3,5М, 40 мл) и реакционную смесь перемешивают в течение дополнительных 2 часов. Добавляют воду и смесь разбавляют этилацетатом. Органический слой отделяют, сушат (MgSO4) и частично концентрируют в вакууме, что приводит к кристаллизации указанного в заголовке соединения. Кристаллы отфильтровывают, получая после сушки в вакууме 3,5 г продукта. Оставшуюся маточную жидкость концентрируют в вакууме и остаток очищают хроматографией на силикагеле (элюирование с градиентом: гептан/этилацетат от 0/1 до 1/0 (об./об.)), получая дополнительные 4,4 г указанного в заголовке соединения.
Выход: 7,9 г.
(b) 1-Ацетил-4-метил-1,2-дигидрохинолин
В соответствии с общей методикой С соединение, описанное в примере 48а (7,9 г), ацилируют ацетилхлоридом (11,8 мл) и DMA (34 мл) в CH2Cl2 (50 мл) при 0°С. Указанное в заголовке соединение очищают хроматографией на силикагеле (элюирование: гептан/этилацетат = 6/4).
Выход: 8,8 г.
(с) 1-Ацетил-4-метил-4-фенил-1,2,3,4-тетрагидрохинолин
В соответствии с общей методикой G соединение, описанное в примере 48b (8,8 г), перемешивают с AlCl3 (18,8 г) в бензоле (250 мл). Полученный продукт используют на следующей стадии без дополнительной очистки.
Выход: 12,0 г.
(d) 1-Ацетил-4-метил-6-нитро-4-фенил-1,2,3,4-тетрагидрохинолин
К раствору соединения, описанного в примере 48с (5,0 г), и уксусного ангидрида (189 мкл) в CH2Cl2 (50 мл) по каплям добавляют дымящую HNO3 (9,4 мл). После завершения реакции добавляют воду, органический слой промывают насыщенным раствором соли, отделяют и концентрируют в вакууме. Указанное в заголовке соединение очищают хроматографией на силикагеле (элюирование: гептан/этилацетат = 6/4 (об./об.)).
Выход: 3,86 г.
(е) 1-Ацетил-6-амино-4-метил-4-фенил-1,2,3,4-тетрагидрохинолин
В соответствии с общей методикой Е соединение, описанное в примере 48d (3,86 г), восстанавливают порошкообразным цинком (16 г) и уксусной кислотой (7 мл) в ТГФ (˜250 мг), получая продукт, который используют на следующей стадии без дополнительной очистки.
Выход: 2,2 г.
(f) 9-Флуоренилметиловый эфир (1-ацетил-4-метил-4-фенил-1,2,3,4-тетрагидрохинолин-6-ил)карбаминовой кислоты
К раствору соединения, описанного в примере 48е (1,0 г), в ТГФ (10 мл) добавляют пиридин (314 мкл) и полученную смесь охлаждают до 0°С. Добавляют FmocCl (1,01 г) и смесь перемешивают в течение 18 часов при комнатной температуре, после чего реакционную смесь концентрируют в вакууме. Указанное в заголовке соединение очищают хроматографией на силикагеле (элюирование: гептан/этилацетат = 6/4 (об./об.)).
Выход: 950 мг.
(g) 9-Флуоренилметиловый эфир (1-ацетил-4-метил-7-нитро-4-фенил-1,2,3,4-тетрагидрохинолин-6-ил)карбаминовой кислоты
К раствору соединения, описанного в примере 48f (850 мг), и уксусного ангидрида (17 мкл) в CH2Cl2 (5 мл) по каплям добавляют дымящую HNO3 (842 мкл). После завершения реакции добавляют воду, органический слой промывают насыщенным раствором соли, отделяют и концентрируют в вакууме. Указанное в заголовке соединение очищают хроматографией на силикагеле (элюирование с градиентом: гептан/этилацетат от 9/1 до 0/1 (об./об.)).
Выход: 714 мг.
(h) 9-Флуоренилметиловый эфир (1-ацетил-7-амино-4-метил-4-фенил-1,2,3,4-тетрагидрохинолин-6-ил)карбаминовой кислоты
В соответствии с общей методикой Е соединение, описанное в примере 48g (2,37 г), восстанавливают порошкообразным цинком (5,6 г) и уксусной кислотой (2,4 мл) в ТГФ (˜50 мг), получая продукт, который используют на следующей стадии без дополнительной очистки.
Выход: 2,66 г.
(i) 9-Флуоренилметиловый эфир (1-ацетил-7-диметиламино-4-метил-4-фенил-1,2,3,4-тетрагидрохинолин-6-ил)карбаминовой кислоты
Водный раствор формальдегида (37%, 650 мкл) добавляют к раствору соединения, описанного в примере 48h (2,66 г), уксусной кислоты (3,1 мл) и цианоборгидрида натрия (232 мг) в метаноле (50 мл), и полученную смесь перемешивают в течение 18 часов. Реакционную смесь концентрируют в вакууме, остаток переносят в этилацетат и промывают водой и насыщенным раствором соли. Органический слой отделяют, сушат (MgSO4) и концентрируют в вакууме. Указанное в заголовке соединение очищают хроматографией на силикагеле (элюирование с градиентом: гептан/этилацетат от 9/1 до 0/1 (об./об.)).
Выход: 1,0 г.
(j) 1-Ацетил-6-амино-7-диметиламино-4-метил-4-фенил-1,2,3,4-тетрагидрохинолин
Пиперидин (1,8 мл) добавляют к раствору соединения, описанного в примере 48i (1 г), в CH2Cl2 (20 мл) и смесь перемешивают до тех пор, пока не останется исходного вещества. Реакционную смесь концентрируют в вакууме, и указанное в заголовке соединение очищают хроматографией на силикагеле (элюирование с градиентом: гептан/этилацетат от 9/1 до 0/1 (об./об.)).
Выход: 370 мг.
(k) N-(1-Ацетил-7-диметиламино-4-метил-4-фенил-1,2,3,4-тетрагидрохинолин-6-ил)-3,5-дибромбензамид
В соответствии с общей методикой А соединение, описанное в примере 48j (74 мг), ацилируют 3,5-дибромбензойной кислотой (70 мг), HATU (131 мг) и DIPEA (120 мкл) в CH2Cl2 (3 мл). Указанное в заголовке соединение очищают препаративной ВЭЖХ и лиофилизуют.
Выход: 82 мг; МС-ESI: [M+H]+ = 586,2; ВЭЖХ: Rt=22,40 мин (метод 2).
Пример 49
(1-Ацетил-7-диметиламино-4-метил-4-фенил-1,2,3,4-тетрагидрохинолин-6-ил)амид 5-бромтиофен-2-карбоновой кислоты
В соответствии с общей методикой А соединение, описанное в примере 48j (74 мг), ацилируют 5-бромтиофен-2-карбоновой кислотой (52 мг), HATU (131 мг) и DIPEA (120 мкл) в CH2Cl2 (3 мл). Указанное в заголовке соединение очищают препаративной ВЭЖХ и лиофилизуют.
Выход: 69 мг; МС-ESI: [M+H]+ = 514,2; ВЭЖХ: Rt=17,01 мин (метод 2).
Пример 50
(1-Ацетил-7-диметиламино-4-метил-4-фенил-1,2,3,4-тетрагидрохинолин-6-ил)амид 5-хлортиофен-2-карбоновой кислоты
В соответствии с общей методикой А соединение, описанное в примере 48j (74 мг), ацилируют 5-хлортиофен-2-карбоновой кислотой (52 мг), HATU (131 мг) и DIPEA (120 мкл) в CH2Cl2 (3 мл). Указанное в заголовке соединение очищают препаративной ВЭЖХ и лиофилизуют.
Выход: 81 мг; МС-ESI: [M+H]+ = 468,2; ВЭЖХ: Rt=17,49 мин (метод 2).
Пример 51
Биологическая активность CHO-FSH in vitro
Активность соединений в отношении FSH испытывают на клетках яичника китайского хомячка (Chinese Hamster Ovary - CHO), стабильно трансфицированных рецептором FSH человека и совместно трансфицированных с сАМР чувствительным элементом (cAMP responsive element - CRE)/промотором, направляющим экспрессию репортерного гена люциферазы светлячка. Связывание лиганда с Gs-связанным FSH рецептором будет приводить к повышению содержания сАМР, который в свою очередь будет вызывать повышение трансактивации репортерной конструкции люциферазы. Для испытания свойств антагониста рекомбинантный FSH добавляют в концентрации, которая индуцирует приблизительно 80% максимального стимулирования аккумуляции сАМР в отсутствие испытываемого соединения (rec-hFSH, 10 мU/мл). Сигнал люциферазы количественно определяют с использованием люминесцентного счетчика. Для испытываемых соединений значения ЕС50 (концентрация испытываемого соединения, вызывающая полумаксимальное (50%) стимулирование или снижение) вычисляют. Для этого используют программу GraphPad PRISM, version 3.0 (GraphPad software Inc., San Diego).
Соединения всех примеров демонстрируют значения ЕС50 (IC50) менее 10-5 М в опытах по определению свойств агонистов или антагонистов или обоих свойств. Соединения примеров 5-8, 10-14, 16, 18-20, 33-35, 37, 38, 41 и 45-50 продемонстрировали значение ЕС50 менее 10-7 М, по меньшей мере, в одном из опытов определения биологической активности.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ТЕТРАГИДРОХИНОЛИНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ, ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ РЕГУЛЯТОРОВ ФЕРТИЛЬНОСТИ | 2003 |
|
RU2328487C2 |
| ТЕТРАГИДРОХИНОЛИНОВЫЕ ПРОИЗВОДНЫЕ | 2002 |
|
RU2347570C2 |
| (ДИГИДРО)ПИРРОЛО[2,1-А]ИЗОХИНОЛИНЫ | 2009 |
|
RU2495037C2 |
| БИЦИКЛИЧЕСКИЕ ГЕТЕРОАРОМАТИЧЕСКИЕ СОЕДИНЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И ПРИМЕНЕНИЕ | 2001 |
|
RU2271360C2 |
| 4-ФЕНИЛ-5-ОКСО-1,4,5,6,7,8-ГЕКСАГИДРОХИНОЛИНОВЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ЛЕКАРСТВЕННЫХ СРЕДСТВ ДЛЯ ЛЕЧЕНИЯ БЕСПЛОДИЯ | 2006 |
|
RU2412170C2 |
| НОВЫЕ ИЗОИНДОЛИНОВЫЕ ИЛИ ИЗОХИНОЛИНОВЫЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2014 |
|
RU2689305C2 |
| СОЕДИНЕНИЯ И СПОСОБЫ ПРИМЕНЕНИЯ | 2009 |
|
RU2561109C2 |
| ПРОИЗВОДНЫЕ ТИЕНО[2,3-D]ПИРИМИДИНА И ИХ ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ НАРУШЕНИЙ ФЕРТИЛЬНОСТИ | 2002 |
|
RU2298011C2 |
| БИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ МОЧЕВИНЫ, ТИОМОЧЕВИНЫ, ГУАНИДИНА И ЦИАНОГУАНИДИНА, ПРИГОДНЫЕ ДЛЯ ЛЕЧЕНИЯ БОЛИ | 2013 |
|
RU2664541C2 |
| БИЦИКЛИЧЕСКИЕ ГЕТЕРОАРОМАТИЧЕСКИЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ LH АГОНИСТОВ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2000 |
|
RU2248979C2 |
Данное изобретение относится к производным тетрагидрохинолина общей формулы (I)
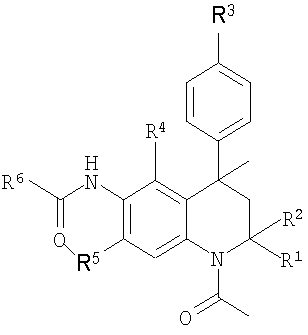
или их фармацевтически приемлемым солям, где R1 и R2 представляют собой Н или Me; R3 представляет собой Н, гидрокси или (1-4С)алкокси; R4 представляет собой Н, ОН, (1-4С)алкокси; R5 представляет собой ОН, (1-4С)алкокси или R7; при условии, что если R4 представляет собой Н, то R5 отличен от ОН или (1-4С)алкокси; R6 представляет собой (2-5С)гетероарил, необязательно замещенный одним или несколькими заместителями, выбранными из (1-4С)алкила, брома или хлора; (6С)арил, необязательно замещенный одним или несколькими заместителями, выбранными из (1-4С)алкила, (1-4С)алкокси, брома, хлора, фенила или (1-4С)(ди)алкиламино; (3-8С)циклоалкил, (2-6С)гетероциклоалкил или (1-6С)-алкил; R7 представляет собой амино, (ди)(1-4С)алкиламино, (6С)арилкарбониламино, (2-5С)гетероарилкарбониламино, (2-5С)гетероарил-карбонилокси, R8-(2-4С)алкокси, R9-метиламино или R9-метокси; R8 представляет собой амино, (1-4С)алкокси, (ди)(1-4С)-алкиламино, (2-6С)-гетероциклоалкил, (2-6С)гетероциклоалкилкарбониламино или (1-4С)-алкоксикарбониламино; и R9 представляет собой аминокарбонил, (ди)(1-4С)алкиламинокарбонил, (2-5С)гетероарил или (6С)арил. Также изобретение относится к фармацевтической композиции, содержащей указанные производные, и применению данных производных для регуляции фертильности. Технический результат: получены новые производные тетрагидрохинолина, обладающие модулирующей активностью в отношении FSH рецептора. 4 н. и 11 з.п. ф-лы.
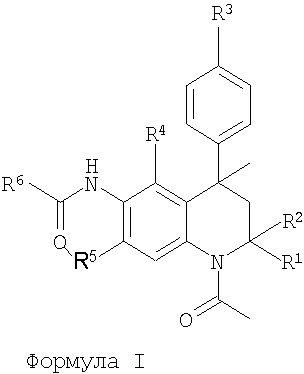
или его фармацевтически приемлемая соль, где
R1 и R2 представляют собой Н или Me;
R3 представляет собой Н, гидрокси или (1-4С)алкокси;
R4 представляет собой Н, ОН, (1-4С)алкокси;
R5 представляет собой ОН, (1-4С)алкокси или R7;
при условии, что если R4 представляет собой Н, то R5 отличен от ОН или (1-4С)алкокси;
R6 представляет собой (2-5С)гетероарил, необязательно замещенный одним или несколькими заместителями, выбранными из (1-4С)алкила, брома или хлора;
(6С)арил, необязательно замещенный одним или несколькими заместителями, выбранными из (1-4С)алкила, (1-4С)алкокси, брома, хлора, фенила или (1-4С)(ди)алкиламино;
(3-8С)циклоалкил, (2-6С)гетероциклоалкил или (1-6С)алкил;
R7 представляет собой амино, (ди)(1-4С)алкиламино, (6С)арилкарбониламино, (2-5С)гетероарилкарбониламино, (2-5С)гетероарилкарбонилокси, R8-(2-4С)алкокси, R9-метиламино или R9-метокси;
R8 представляет собой амино, (1-4С)алкокси, (ди)(1-4С)алкиламино, (2-6С)гетероциклоалкил, (2-6С)гетероциклоалкилкарбониламино или (1-4С)алкоксикарбониламино; и
R9 представляет собой аминокарбонил, (ди)(1-4С)алкиламинокарбонил, (2-5С)гетероарил или (6С)арил.
(6С)арил, необязательно замещенный одним или несколькими заместителями, выбранными из (1-4С)алкила, (1-4С)алкокси, брома, хлора, фенила или (1-4С)(ди)алкиламино;
(3-8С)циклоалкил или (1-6С)алкил.
(6С)арил, необязательно замещенный одним или несколькими заместителями, выбранными из (1-4С)алкила, (1-4С)алкокси, брома, хлора, фенила или (1-4С)(ди)алкиламино.
| WO 0008015 А, 17.02.2000 | |||
| US 6200963 B1, 13.03.2001 | |||
| Способ получения производных 1-ацил-2,3-дигидро-4 /IH/-хинолинон-4-оксима-0-сульфоновой кислоты или их солей | 1988 |
|
SU1779246A3 |
| АРИЛЦИКЛОАЛКИЛЬНЫЕ ПРОИЗВОДНЫЕ, ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ НА ИХ ОСНОВЕ | 1993 |
|
RU2125553C1 |

