ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к стабильному кристаллу 6-(3-хлор-2-фторбензил)-1-[(S)-1-гидроксиметил-2-метилпропил]-7-метокси-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты
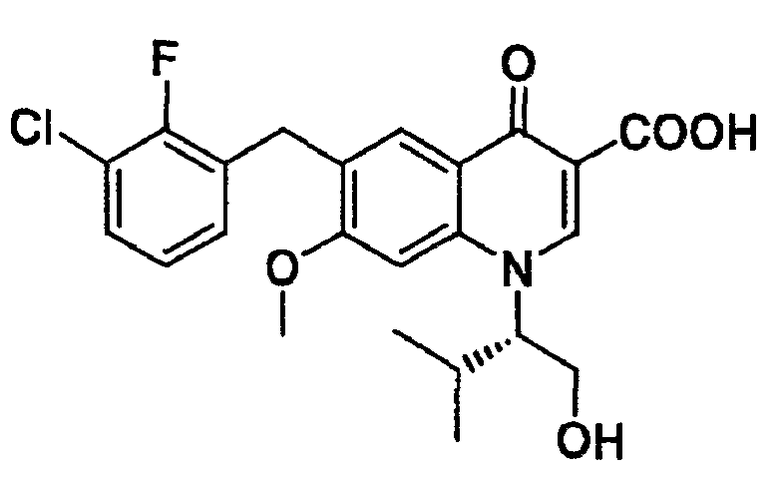
(в дальнейшем в этом документе это соединение обозначается иногда как соединение А) и смешанному кристаллу этого соединения. Настоящее изобретение также относится к фармацевтической композиции, включающей в свой состав кристалл или смешанный кристалл.
УРОВЕНЬ ТЕХНИКИ
Заявитель предоставил информацию в патенте № WO 2004/046115 (PCT/UP 2003/014773), зарегистрированном на того же самого Заявителя, о том, что вышеупомянутое соединение А обладает ингибирующим действием на интегразу, являющуюся внутренним необходимым ферментом для роста ВИЧ (вируса иммунодифицита человека), который вызывает вирусное заболевание СПИД (синдром приобретенного иммунодефицита), и проявляет анти-ВИЧ воздействие (в частности пример 4-32 и экспериментальный пример).
В общем, в том случае, если соединение применяется как фармацевтический продукт, то требуется его химическая и физическая стабильность, поскольку это обеспечивает качество и/или способствует сохранности. По этим же причинам требуется химическая и физическая стабильность не только для итоговой фармацевтической композиции, но также и для соединения, являющегося синтетическим исходным веществом.
Поэтому такое соединение предпочтительно должно быть кристаллом и особенно предпочтительно стабильным кристаллом. В том случае, если для соединения характерен кристаллический полиморфизм, то обычно выбирается наиболее стабильный кристалл.
Несмотря на то, что в случае вышеупомянутого применения описывалось соединение А, не было найдено определенного описания, имеющего отношение к кристаллической форме соединения А.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Таким образом, в настоящем изобретении изучены различные кристаллические формы соединения А. В результате установлено, что соединение А обладает кристаллическим полиморфизмом и изучен кристалл соединения А, имеющий специфическую кристаллическую форму стабильного кристалла, и на основе этого открытия сделано настоящее изобретение.
Таким образом, настоящее изобретение предоставляет следующее.
[1] кристалл (кристаллическая форма II) 6-(3-хлор-2-фторбензил)-1-[(S)-1-гидроксиметил-2-метилпропил]-7-метокси-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты, для которого в рентгенодифракционном спектре имеются следующие характеристические дифракционные пики при углах дифракции 2θ (°) 6,56, 13,20, 19,86, 20,84, 21,22, 25,22°, что измерено на рентгенодифрактометре;
[2] кристалл (кристаллическая форма III) 6-(3-хлор-2-фторбензил)-1-[(S)-1-гидроксиметил-2-метилпропил]-7-метокси-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты, для которого в рентгенодифракционном спектре имеются следующие характеристические дифракционные пики при углах дифракции 2θ (°) 8,54, 14,02, 15,68, 17,06, 17,24, 24,16, 25,74°, что измерено на рентгенодифрактометре;
[3] кристалл (кристаллическая форма III) 6-(3-хлор-2-фторбензил)-1-[(S)-1-гидроксиметил-2-метилпропил]-7-метокси-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты, имеющий экстраполированную начальную температуру при 162,1±5,0°С;
[4] кристалл, относящийся к одному из вышеупомянутых с [1] по [3], для которого степень чистоты кристалла составляет не менее чем 70%;
[5] смешанный кристалл, включающий в свой состав кристалл, вышеупомянутый как [1], и кристалл, вышеупомянутый как [2] или [3];
[6] смешанный кристалл, вышеупомянутый как [5], в котором степень чистоты кристалла составляет не менее чем 70%;
[7] фармацевтическая композиция, включающая в свой состав какой-либо из вышеупомянутых кристаллов с [1] по [4] или смешанный кристалл из вышеупомянутых [5] или [6], и фармацевтически приемлемый носитель;
[8] ингибитор интегразы, включающий в свой состав какой-либо кристалл из вышеупомянутых с [1] по [4] или смешанный кристалл из вышеупомянутых [5] или [6] в качестве активного ингредиента;
[9] антивирусное средство, включающее в свой состав какой-либо кристалл из вышеупомянутых с [1] по [4] или смешанный кристалл из вышеупомянутых [5] или [6] в качестве активного ингредиента;
[10] анти-ВИЧ средство, включающее в свой состав какой-либо кристалл из вышеупомянутых с [1] по [4] или смешанный кристалл из вышеупомянутых [5] или [6] в качестве активного ингредиента;
[11] анти-ВИЧ средство, включающее в свой состав какой-либо кристалл из вышеупомянутых с [1] по [4] или смешанный кристалл из вышеупомянутых [5] или [6] и одно или несколько видов других анти-ВИЧ активных веществ в качестве активного ингредиента; и
[12] анти-ВИЧ средство для комплексной лекарственной терапии с другими анти-ВИЧ средствами, включающее в свой состав какой-либо кристалл из вышеупомянутых с [1] по [4] или смешанный кристалл из вышеупомянутых [5] или [6] в качестве активного ингредиента.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
Фиг.1 представляет собой комплексные данные спектров рентгенодифракции, где верхняя линия показывает спектр дифракции кристаллической формы III, средняя линия показывает спектр дифракции кристаллической формы I, нижняя линия показывает спектр дифракции кристаллической формы II, на вертикальной оси показана интенсивность дифракции (cps: counts per second - подсчет за секунду, интервалы на шкале составляют 2500 cps) и на перпендикулярной оси показаны углы дифракции 2θ (°).
Фиг.2 представляет собой комплексные данные спектров рентгенодифракции, полученные после хранения образца в течение 3 дней при тестировании на стабильность кристаллической формы I. Для сравнения самая верхняя линия показывает спектр дифракции (начальные условия) для кристаллической формы II, и самая нижняя линия показывает спектр дифракции (начальные условия) для кристаллической формы I. Вторая сверху линия и последующие показывают спектры дифракции образцов после хранения в условиях #6 (60°С/75% относительная влажность, открытый контейнер, хранение в течение 3 недель), после хранения в условиях #4 (60°С, контейнер с пробкой, хранение в течение 3 недель), после хранения в условиях #5 (60°С, открытый контейнер, хранение в течение 3 недель), после хранения в условиях #3 (80°С, открытый контейнер, хранение в течение 3 недель) и после хранения в условиях #2 (80°С, контейнер с пробкой, хранение в течение 3 недель). На вертикальной оси показана интенсивность дифракции (cps: counts per second - подсчет за секунду, интервалы на шкале составляют 2500 cps) и на перпендикулярной оси показаны углы дифракции 2θ (°).
ЭФФЕКТ ИЗОБРЕТЕНИЯ
Кристалл или смешанный кристалл соединения А настоящего изобретения имеет особую вышеупомянутую кристаллическую форму и является превосходным по физической и химической стабильности, что приводит к такому преимуществу, которое заключается в возможности сохранении качества соединения А в течение длительного времени, что и обеспечивает сохранность. В дополнение ко всему они имеют преимущество в том, что выдерживают технологическую обработку в процессе производства различных фармацевтических композиций и нерасфасованных лекарственных средств, что снижает стоимость продукции.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение детально объясняется ниже.
В настоящем изобретении «кристаллическая форма II» соединения А обозначает кристалл соединения А, в рентгенодифракционном спектре которого имеются характеристические пики следующих углов дифракции 2θ (°) 6,56, 13,20, 19,86, 20,84, 21,22, 25,22°, что измерено на рентгенодифрактометре.
В настоящем изобретении «кристаллическая форма III» соединения А обозначает кристалл соединения А, в рентгенодифракционном спектре которого имеются характеристические пики следующих углов дифракции 2θ (°) 8,54, 14,02, 15,68, 17,06, 17,24, 24,16, 25,74°, что измерено на рентгенодифрактометре.
Величина дифракционных пиков при вышеупомянутых дифракционных углах 2θ (°) может иметь небольшую ошибку измерений, обусловленную погрешностями измерительных приборов или условий измерения, или им подобными факторами. Конкретно ошибка измерений может быть в пределах ±0,2, предпочтительно ±0,1, более предпочтительно ±0,06.
Кристалл соединения А настоящего изобретения также охарактеризован термическим анализом. Например, в случае, когда кристаллическая форма III соединения А настоящего изобретения подвергается дифференциальной сканирующей калориметрии (ДСК), энтальпия эндотермического пика составляет 81 Дж/г, и имеет экстраполированную начальную температуру 162,1±5,0°С, предпочтительно 162,1±3,0°С, более предпочтительно 162,1±1,0°С, где «экстраполированная начальная температура» истолковывается как определено в JIS K 7121 (метод измерения температурного перехода пластмасс), температура на пересечении экстраполированной базовой линии, построенной от положения низшей температуры до положения с максимальной температурой, с касательной линией, проведенной в точке с наибольшим тангенсом угла наклона на подъемной стороне пика плавления при низшем температурном положении кривой ДСК. Когда энтальпия и экстраполированная начальная температура эндотермического пика находятся в пределах вышеупомянутого интервала, кристалл соединения А является стабильным.
Кристалл соединения А настоящего изобретения может быть либо в кристаллической форме II либо в кристаллической форме III, либо смешанным кристаллом кристаллической формы II и кристаллической формы III. Для использования в фармацевтическом продукте соединения А и в подобных продуктах кристаллическая форма II и кристаллическая форма III предпочтительны, потому что они являются стабильными кристаллами, и кристаллическая форма III наиболее предпочтительна, потому что она является наиболее стабильным кристаллом. Кроме того, кристаллическая форма II является предпочтительной с точки зрения абсорбционной способности живыми организмами при ее назначении для приема в качестве фармацевтической композиции.
В настоящем изобретении «чистота кристалла» обозначает чистоту кристаллической формы II или кристаллической формы III соединения А. В случае смешанного кристалла из кристаллической формы II и кристаллической формы III этот термин обозначает соотношение кристалла к общему количеству вещества кристаллической формы II и кристаллической формы III. Чистота кристалла настоящего изобретения может быть определена известными методами, например, такими как рентгенодифрактометрия, термический анализ и им подобными. Для чистоты кристалла или смешанного кристалла настоящего изобретения не требуется достижения величины 100%, она может быть не менее чем 70%, предпочтительно не менее чем 80%, более предпочтительно не менее чем 90%, еще более предпочтительно не менее чем 95%, и наиболее предпочтительно не менее чем 98%. Чистота в пределах этого интервала является предпочтительной для обеспечения качества.
Кристалл или смешанный кристалл соединения А настоящего изобретения может назначаться для приема млекопитающим (человек, мышь, крыса, хомяк, кролик, кот, собака, бык, овца, обезьяна и др.) и им подобным в виде различных фармацевтических композиций, таких как анти-ВИЧ средства, ингибиторы ВИЧ-интегразы, противовирусные средства и им подобные, используемые, например, для профилактики и/или лечения СПИДа.
В случае, если кристалл или смешанный кристалл соединения А настоящего изобретения применяется как фармацевтическая композиция, он смешивается с фармацевтически приемлемыми носителями, формообразующими наполнителями, разбавителями, увеличивающими объем, разжижающими средствами, дезинтегрирующими средствами, стабилизаторами, консервирующими веществами, буферными растворами, эмульгаторами, вкусовыми ароматизирующими веществами, окрашивающими веществами, подсластителями, загустителями, нейтрализующими средствами, разбавленными кислотами и другими добавками, которые в основном известны сами по себе, это вода, растительное масло, спирт (например, этанол или бензиловый спирт и др.), полиэтиленгликоль, триацетат глицерина, желатин, углеводы (например, лактоза, крахмал и т.п.), стеарат магния, тальк, ланолин, вазелин и им подобные, формируются таблетки, пилюли, порошки, гранулы, суппозитории, инъекционные формы, глазные капли, жидкие формы, капсулы, пастилки, аэрозоли, эликсиры, суспензии, эмульсии, сиропы и им подобные с использованием стандартного способа, и назначаются для общего или местного приема перорально или парентерально.
Поскольку доза изменяется в зависимости от возраста, массы тела, симптомов, лечебного эффекта, способа приема и им подобных факторов, она обычно составляет 0,01 мг на 1 г при назначении взрослым, и вводится за один или несколько раз в день перорально или в дозированной форме для инъекций, таких как внутривенные инъекции и им подобные.
От анти-ВИЧ средства обычно требуется поддерживание его действия в течение длительного времени, чтобы оно было эффективным не только для временного подавления вирусного роста, но также и для предотвращения возобновления роста вируса. Это означает, что пролонгированный прием необходим и что высокая однократная доза может часто быть обязательной для поддержания воздействия в течение длительного периода времени, в течение ночи или подобного периода. Такой пролонгированный прием лекарства в высоких дозах повышает риск побочных эффектов.
С этой точки зрения, одним из предпочтительных способов по настоящему изобретению является такой, когда обеспечивается высокая абсорбция при пероральном приеме, и такое соединение способно поддержать свою концентрацию в крови в течение длительного времени.
Для «профилактики СПИДа» подразумевается, например, назначение приема фармацевтического средства тому пациенту, у которого выявлен положительный ВИЧ-тест, но еще не развилось состояние болезни СПИД, назначение приема фармацевтического средства тому пациенту, у которого наблюдается улучшение состояния при заболевании СПИД после лечения, но который все еще является носителем ВИЧ и для которого существует риск вторичного заболевания СПИДом, и назначение приема фармацевтического средства при возможности инфицирования.
Анти-ВИЧ композиция настоящего изобретения применяется, например, для комплексной лекарственной комбинированной терапии СПИДа. Примеры «других анти-ВИЧ активных веществ», пригодных к применению для анти-ВИЧ композиции, включают анти-ВИЧ антитела, ВИЧ вакцину, такие иммуностимуляторы, как интерферон и ему подобные, ВИЧ рибозим, ВИЧ антисмысловое лекарство, ингибитор обратной транскриптазы ВИЧ, ингибитор протеазы ВИЧ, ингибиторы связывания между вирусом и рецепторами связывания (CD4, CXCR4, CCR5 и им подобными), распознаваемыми вирусом на заражаемой клетке (клетка-хозяин), и им подобные.
Конкретные примеры ингибитора обратной транскриптазы ВИЧ включают Ретровир(R) (зидовудин), Эпивир(R) (ламивудин), Зерит(R) (санилвудин), Видекс(R) (диданозин), Хивид(R) (зальцитабин), Зиаген(R) (абакавир сульфат), Вирамун(R) (невирапин), Стокрин(R) (эфавиренз), Рескриптор(R) (делавирдин мезилат), Комбивир(R) (зидовудин+ламивудин), Тризивир(R) (абакавир сульфат+ламивидин+зидовудин), Коактинон(R) (эмивирин), Фосфоновир(R), Ковирацил(R), аловудин (3'-фтор-3'-деокситимидин), Тиовир (тиофосфономуравьиная кислота), Каправирин (5-[(3,5-дихлорфенил)тио]-4-изопропил-1-(4-пиридилметил)имидазол-2-метанол карбаминовая кислота), Тенофовир дизопроксил фумарат ((R)-[[2-(6-амино-9Н-пурин-9-ил)-1-метилтэтокси]метил]фосфоновая кислота, сложный эфир бис(изопропоксикарбонилоксиметил)а фумаровой кислоты), DPC-083 ((4S)-6-хлор-4-[(1Е)-циклопропилэтенил]-3,4-дигидро-4-трифторметил-2(1Н)-хиназолинон), DPC-961 ((4S)-6-хлор-4-(циклопропилэтинил)-3,4-дигидро-4-(трифторметил)-2(1Н)-хиназолинон), DAPD ((-)-β-D-2,6-диаминопурин диоксолан), Иммунокал, MSK-055, MSA-254, MSH-143, NV-01, TMC-120, DPC-817, GS-7340, TMC-125, SPD-754, D-A4FC, каправирин, UC-781, эмтрицитабин, аловудин, Фосфазид, UC-781, BCH-10618, DPC-083, Этравирин, BCH-13520, MIV-210, Абакавир сульфат/ламивудин, GS-7340, GW-5634, GW-695634 и им подобные, где (R) обозначает зарегистрированную торговую марку (в дальнейшем в этом документе то же самое обозначение) и наименования других фармацевтических средств являются общепринятыми наименованиями.
Конкретные примеры ингибитора протеазы ВИЧ включают Криксиван(R) (индинавир сульфат этанолат), сахинавир, Инвирас(R) (сахинавир мезилат), Норвир(R) (ритонавир), Вирасепт(R) (нелфинавир мезилат), лопинавир, Прозей(R) (ампренавир), Калетра(R) (ритонавир+лопинавир), мозенавир димезилат ([4R-(4α,5α,6β)]-1,3-бис[(3-аминофенил)метил]-гексагидро-5,6-дигидрокси-4,7-бис(фенилметил)-2Н-1,3-диазепин-2-он диметансульфонат), типранавир (3'-[(1R)-1-[(6R)-5,6-дигидро-4-гидрокси-2-оксо-6-фенилэтил-6-пропил-2Н-пиран-3-ил]пропил]-5-(трифторметил)-2-пиридинсульфонамид), лазинавир (2-метоксиэтиленамид-N-[5(S)-(трет-бутоксикарбониламино)-4(S)-гидрокси-6-фенил-2(R)-(2,3,4-триметоксибензил)гексаноил]-L-валина), KNI-272 ((R)-N-трет-бутил-3-[(2S,3S)-2-гидрокси-3-N-[(R)-2-N-(изохинолин-5-илоксиацетил)амино-3-метилтиопропаноил]амино-4-фенилбутаноил]-5,5-диметил-1,3-тиазолидин-4-карбоксамид), GW-433908, TMC-126, DPC-681, бакминстерфуллерен, МК-944A (MK944 (N-(2(R)-гидрокси-1(S)-инданил)-2(R)-фенилметил-4(S)-гидрокси-5-[4-(2-бензо[b]фуранилметил)-2(S)-(трет-бутилкарбамоил)пиперазин-1-ил]пентанамид)+индинавир сульфат), JE-2147 ([2(S)-оксо-4-фенилметил-3(S)-[(2-метил-3-окси)фенилкарбониламино]-1-оксабутил]-4-[(2-метилфенил)метиламино]карбонил-4(R)-5,5-диметил-1,3-тиазол), BMS-232632 (диметиловый эфир (3S,8S,9S,12S)-3,12-бис(1,1-диметилэтил)-8-гидрокси-4,11-диоксо-9-(фенилметил)-6-[[4-(2-пиридинил)фенил]метил]-2,5,6,10,13-пентаазатетрадекандикарбоновой кислоты), DMP-850 ((4R,5S,6S,7R)-1-(3-амино-1Н-индазол-5-илметил)-4,7-дибензил-3-бутил-5,6-дигидроксипергидро-1,3-диазепин-2-он), DMP-851, RO-0334649, Nar-DG-35, R-944, VX-385, TMC-114, Типранавир, Фозампренавир натрий, Фозампренавир кальций, Дарунавир, GM-0385, R-944, RO-033-4649, AG-1859 и им подобные.
Ингибитор интегразы ВИЧ представлен примерами S-1360, L-870810 и им подобными, ингибитор ДНК полимеразы или ингибитор синтеза ДНК представлен примерами Фоскавир(R), ACH-126443 (L-2',3'-дидегидро-дидеокси-5-фторцитидин), энтекавир ((1S,3S,4S)-9-[4-гидрокси-3-(гидроксиметил)-2-метиленциклопентил]гуанин), каланолид А ([10R(10α,11β,12α)]-11,12-дигидро-12-гидрокси-6,6,10,11-тетраметил-4-пропил-2Н,6Н,10Н-бензо[1,2-b:3,4-b':5,6-b'']трипиран-2-он), каланолид В, NSC-674447 (1,1'-азобисформамид), Искадор (экстракт viscum alubm), Рубитекан и им подобными, анти-ВИЧ антисмысловое лекарство представлено примерами HGTV-43, GEM-92 и им подобными, противовирусные антитела к ВИЧ или другие антитела представлены примерами NM-01, PRO-367, KD-247, Цитолин(R), TNX-355 (антитело к CD4), AGT-1, PRO-140 (антитело к CCR5), Анти-CTLA-4Mab и им подобными, ВИЧ вакцина или другая вакцина представлены примерами ALVAC(R), AIDSVAX(R), Ремун(R), вакцина HIVgp41, вакцина HIVgp120, вакцина HIVgp140, вакцина HIVgp160, вакцина HIVp17, вакцина HIVp24, вакцина HIVp55, Альфавакс Вектор-Система, вакцина канапирокс gp160, АнтиТат, вакцина MVA-F6 Nef, вакцина HIVrev, C4-V3 пептид, p2249f, VIR-201, HGP-30W, TBC-3B, PARTICLE-3B и им подобными, Антиферон (вакцина интерферон-α) и им подобными, интерферон или агонист интерферона представлены примерами Сумиферон(R), МультиФерон(R), интерферон-τ, Ретикулоза, человеческий лейкоцитарный интерферон-α и им подобные, антагонист CCR5 представлен примерами SCH-351125 и ему подобными, фармацевтические средства, действующие на p24 ВИЧ представлены примерами GPG-NH2 (глицил-пролил-глицинамид) и ему подобными, ингибитор ВИЧ фузии представлен примерами FP-21399 (1,4-бис[3-[(2,4-дихлорфенил)карбониламино]-2-оксо-5,8-динатрийсульфонил]нафтил-2,5-диметоксифенил-1,4-дигидразон), Т-1249, Синтетическая Полимерная Конструкция No3, пентафузид, FP-21399, PRO-542, Энфувиртид и им подобными, IL-2 агонист или антагонист представлены примерами интерлейкин-2, Имунас(R), Пролейкин(R), Мультикин(R), Онтак(R) и им подобные, антагонист TNF-б представлен примерами Таломид(R) (талидомид), Ремикад(R) (инфликсимаб), курдлан сульфат, ингибитор α-гликозидазы представлен примерами Букаст(R) и ему подобными, ингибитор пуриннуклеозидфосфорилазы представлен примерами пелдецин (2-амино-4-оксо-3Н,5Н-7-[(3-пиридил)метил]пиррол[3,2-d]пиримидин) и ему подобными, агонист или ингибитор апоптоза представлен примерами Аркин Z(R), Панавир(R), Коэнзим Q10 (2-дека(3-метил-2-бутенилен)-5,6-диметокси-3-метил-пара-бензохинон) и им подобными, ингибитор холинэстеразы представлен примерами Когнекс(R) и ему подобными, и иммуномодулятор представлен примерами Имунокс(R), Прокин(R), Мет-энкефалин (6-де-L-аргинин-7-де-L-аргинин-8-де-L-валинамид-адренорфин), WF-10 (10 кратно разбавленный раствор тетрахлордекаоксида), Пертон, PRO-542, SCH-D, UK-427857, AMD-070, AK-602 и им подобными.
Кроме того, в качестве примеров представлены Нейротропин(R), Лидакол(R), Ансер 20(R), Амплиген(R), Антикорт(R), Инактивин(R) и им подобные, PRO-2000, ген Rev M10, специфичные к ВИЧ цитотоксические Т-клетки (CTL иммунотерапия, протокол ACTG 080 терапия, CD4-ζ генотерапия), SCA связывающий белок, RBC-CD4 комплекс, Мотексафин гадолиниум, GEM-92, CNI-1493, (±)-FTC, Ушерселл, D2S, БуферГель(R), ВиваГель(R), вагинальный гель Глиминокс, лаурилсульфат натрия, 2F5, 2F5/2G12, VRX-496, Ad5gag2, BG-777, IGIV-C, BILR-255 и им подобные.
Что касается «других веществ с анти-ВИЧ активностью», применяемых для анти-ВИЧ композиций настоящего изобретения в комплексной лекарственной комбинационной терапии, предпочтительным является ингибитор обратной транскриптазы ВИЧ и ингибитор протеазы ВИЧ. Два, три или большее число фармацевтических средств могут применяться в комбинации, в которой сочетание фармацевтических средств, имеющих различные механизмы действия, представляет собой предпочтительный пример осуществления изобретения. Кроме того, предпочтителен выбор фармацевтических средств без однотипного побочного эффекта.
Конкретные примеры комбинации фармацевтических средств включают комбинирование группы средств, содержащих в своем составе эфавиренз, тенофовир, эмтрицитабин, индинавир, нелфинавир, атазанавир, ритонавир+индинавир, ритонавир+лопинавир и ритонавир+сахинавир, диданозин+ламивудин, зидовудин+диданозин, ставудин+диданозин, зидовудин+ламивудин, ставудин+ламивудин и эмтрива, и кристалла или смешанного кристалла настоящего изобретения (Guidelines for the Use of Antiretroviral Agents in HIV-Infected Adults and Adolescents. August 13, 2001). Особенно предпочтительным является совместное применение двух средств: кристалла или смешанного кристалла настоящего изобретения с эфавирензом, индинавиром, нелфинавиром, тенофовиром, эмтрицитабином, зидовудином или ламивудином, или совместное применение трех средств: кристалла или смешанного кристалла настоящего изобретения с зидовудин + ламивудин, тенофовир + ламивудин, тенофовир + зидовудин, тенофовир + эфавиренз, тенофовир + нелфинавир, тенофовир + индинавир, тенофовир + эмтрицитабин, эмтрицитабин + ламивудин, эмтрицитабин + зидовудин, эмтрицитабин + эфавиренз, эмтрицитабин + нелфинавир, эмтрицитабин + индинавир, нелфинавир + ламивудин, нелфинавир + зидовудин, нелфинавир + эфавиренз, нелфинавир + индинавир, эфавиренз + ламивудин, эфавиренз + зидовудин или эфавиренз + индинавир.
Способ получения кристалла или смешанного кристалла соединения А настоящего изобретения не связан с особыми ограничениями, и кристалл может быть получен, по существу, известным способом или способами, представленными в нижеследующих примерах и им подобными.
ПРИМЕРЫ
Несмотря на то, что способ получения кристалла соединения А настоящего изобретения объясняется нижеследующими примерами, они носят рекомендательный характер и никоим образом не ограничивают настоящего изобретения.
Справочный пример 1: Получение кристаллической формы I соединения А
Стадия 1
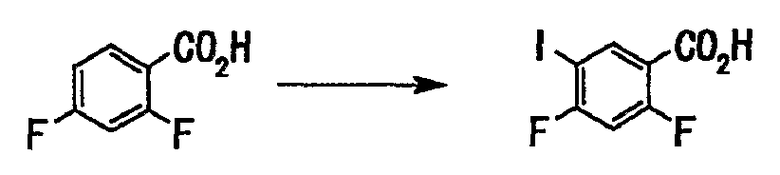
2,4-Дифторбензойную кислоту (50 г, 316 ммоль) растворяли в концентрированной серной кислоте (200 мл) и порциями добавляли N-иодсукцинимид (68 г, 300 ммоль) при температуре, не превышающей 5°С. После завершения добавления смесь перемешивали при той же температуре 4,5 часа. Реакционную смесь выливали в ледяную воду (приблизительно 600 мл), затем добавляли 10% водный раствор сульфита натрия, и смесь перемешивали. Выпавший твердый осадок отделяли фильтрованием, промывали водой и сушили в вакууме с получением неочищенных кристаллов (85 г). Неочищенные кристаллы, полученные подобным образом, объединяли (получали их общее количество 205 г) и перекристаллизовывали из 50% водного этанола (820 мл) с получением 2,4-дифтор-5-иодбензойной кислоты (148 г, выход 73%) в виде твердого вещества белого цвета.
1Н-ЯМР (CDCl3, 300 МГц) (σ) ммд: 6,94 (1Н, дд, J=10,3, 10,3 Гц), 8,46 (1Н, д, J=7,5 Гц).
Стадия 2
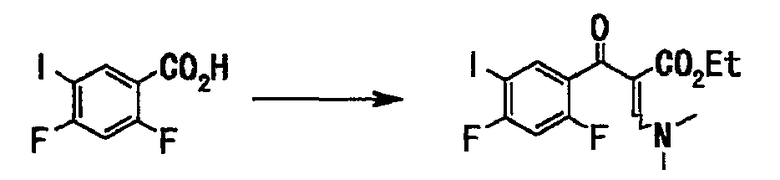
Соединение (148 г, 521 ммоль), полученное на стадии 1, растворяли в толуоле (750 мл), добавляли тионилхлорид (76 мл, 1,04 моль) и диметилформамид (каталитические количества), смесь нагревали с обратным холодильником в течение 2 часов. Нерастворившееся вещество отфильтровывали при 60°С, и фильтрат концентрировали при пониженном давлении и отгоняли азеотроп с толуолом (330 мл). Остаток растворяли в тетрагидрофуране (400 мл), и этот раствор по каплям добавляли к раствору этил-3,3-диметиламиноакрилата (82 г, 573 ммоль) и триэтиламина (87 мл, 625 ммоль) в тетрагидрофуране (400 мл), и смесь нагревали с обратным холодильником в течение 7 часов. Реакционную смесь оставляли для остывания до комнатной температуры и концентрировали при пониженном давлении. Добавляли воду (700 мл) и этилацетат (800 мл) для разделения. Органический слой последовательно промывали насыщенным водным раствором гидрокарбоната натрия (250 мл × 2), водой (300 мл) и насыщенным солевым раствором (300 мл) и сушили над сульфатом натрия. После отфильтровывания нерастворимого вещества фильтрат концентрировали при пониженном давлении с получением неочищенного вещества (210 г), этилового эфира 2-(2,4-дифтор-5-иодбензоил)-3-диметиламиноакриловой кислоты в виде твердого вещества коричневого цвета.
Стадия 3

Неочищенный продукт (210 г) со стадии 2 растворяли в тетрагидрофуране (500 мл), добавляли (S)-(+)-валинол (54 г, 521 ммоль), и смесь перемешивали при комнатной температуре 30 минут. Реакционную смесь концентрировали при пониженном давлении и остаток растворяли в диметилформамиде (600 мл). Добавляли карбонат калия (144 г, 1,04 моль) и смесь перемешивали при нагревании до 70°С в течение 2 часов. Реакционную смесь оставляли для остывания, добавляли к воде (1500 мл) и перемешивали. Выпавшее в осадок твердое вещество отделяли фильтрованием и полученное твердое вещество последовательно промывали 30% водным этанолом (500 мл) и смесью растворителей: диэтилового эфира (150 мл) и гексана (150 мл) и сушили в вакууме, получали этиловый эфир 7-фтор-1-((S)-1-гидроксиметил-2-метилпропил)-6-иод-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты (178 г, выход 76%) в виде твердого вещества бежевого цвета.
1H ЯМР (ДМСО-d6 300 МГц) (δ) ммд: 0,72 (3H, д, J=6,6 Гц), 1,10 (3H, д, J=6,6 Гц), 1,28 (3H, т, J=7,0 Гц), 2,27 (1H, ушир.), 3,77 (1H, ушир.), 3,86 (1H, ушир.), 4,23 (2H, кв, J=7,0 Гц), 4,56 (1H, ушир.), 5,12 (1H, т, J=4,9 Гц), 8,09 (1H, д, J=11,1 Гц), 8,62 (1H, д, J=7,5 Гц), 8,68 (1H, с).
МС (ESI): M+ 448.
Стадия 4

где TBDMS - трет-бутилдиметилсилильная группа.
Соединение (80 г, 179 ммоль), полученное на стадии 3, растворяли в диметилформамиде (320 мл), добавляли имидазол (16 г, 233 ммоль) и трет-бутилдиметилсилилхлорид (30 г, 197 ммоль), и смесь перемешивали при комнатной температуре в течение 1,5 часов. К реакционной смеси добавляли воду и смесь экстрагировали этилацетатом. Органический слой последовательно промывали насыщенным водным раствором хлорида аммония и насыщенным солевым раствором и сушили над сульфатом натрия. Органический слой отфильтровывали и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на силикагеле (этилацетат:гексан = 1:3 до 1:2) с получением этилового эфира 1-((S)-1-трет-бутилдиметилсилилоксиметил-2-метилпропил)-7-фтор-6-иод-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты (77 г, выход 77%) в виде бесцветного вещества в аморфной форме.
1H ЯМР (CDCl3 400 МГц) (δ) ммд: -0,07 (3H, с), -0,05 (3H, с), 0,77 (9H, с), 0,84 (3H, д, J=6,5 Гц), 1,18 (3H, д, J=6,5 Гц), 1,40 (3H, т, J=7,2 Гц), 2,35-2,50 (1H, м), 3,85-3,95 (1H, м), 3,98-4,10 (2H, м), 4,30-4,40 (2H, м), 7,26 (1H,с), 8,64 (1H, с), 8,94 (1H, д, J=7,2 Гц).
МС (ESI): M+ 562.
Стадия 5
(Приготовление раствора 3-хлор-2-фторбензилцинкбромида в тетрагидрофуране)
В токе аргона цинковый порошок (11 г, 267 ммоль) суспендировали в тетрагидрофуране (30 мл), добавляли 1,2-дибромэтан (0,15 мл, 1,8 ммоль) и триметилсилилхлорид (0,45 мл, 3,6 ммоль) при 65°С, и смесь перемешивали при нагревании в течение 30 минут. По каплям добавляли раствор 3-хлор-2-фторбензилбромида (41 г, 178 ммоль) в тетрагидрофуране (100 мл) при 65°С, и смесь перемешивали при нагревании 2 часа, и оставляли для охлаждения до комнатной температуры, получали 1М раствор 3-хлор-2-фторбензилцинкбромида в тетрагидрофуране. Этот раствор применяли на последующей основной стадии.
(Основная стадия)
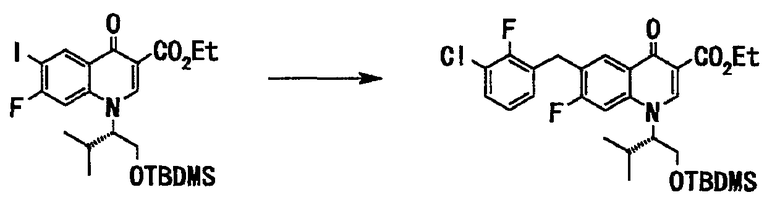
Соединение (76 г, 136 ммоль), полученное на стадии 4, растворяли в тетрагидрофуране (600 мл), и в токе аргона добавляли дибензилиденацетонпалладий (II) (3,2 г, 5,5 ммоль) и трифурилфосфин (2,6 г, 11,0 ммоль), и по каплям добавляли вышеупомянутый раствор 1М 3-хлор-2-фторбензилцинкбромид в тетрагидрофуране (178 мл, 178 ммоль) при 60°С. После завершения прикапывания смесь перемешивали с нагреванием до той же температуры в течение 2 часов. Реакционную смесь оставляли для охлаждения до комнатной температуры, добавляли насыщенный водный раствор хлорида амммония, и фильтровали смесь через целит. Фильтрат дважды экстрагировали этилацетатом. Органический слой последовательно промывали водой (дважды) и насыщенным солевым раствором и сушили над сульфатом магния. Органический слой отфильтровывали и фильтрат концентрировали при пониженном давлении, и полученный остаток очищали хроматографией на силикагеле (хлороформ:ацетон = 40:1) с получением этилового эфира 1-((S)-1-трет-бутилдиметилсилилоксиметил-2-метилпропил)-6-(3-хлор-2-фторбензил)-7-фтор-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты (68 г, выход 84%) в виде бесцветного вещества в аморфной форме.
1H ЯМР (CDCl3 400 МГц) (δ) ммд: -0,09 (3H, с), -0,05 (3H, с), 0,75 (9H, с), 0,85 (3H, д, J=6,7 Гц), 1,18 (3H, д, 6,7 Гц), 1,39 (3H, т, J=7,1 Гц), 2,45 (1H, ушир.), 3,89-3,92 (1H, м), 3,98-4,02 (1H, м), 4,07-4,12 (1H, м), 4,12 (2H, с), 4,34-4,41 (2H, м), 6,96-7,00 (1H, м), 7,03-7,05 (1H, м), 7,21-7,24 (1H, м), 7,26-7,29 (1H, м), 8,39 (1H, д, J=8,8 Гц), 8,63 (1H, с).
Стадия 6
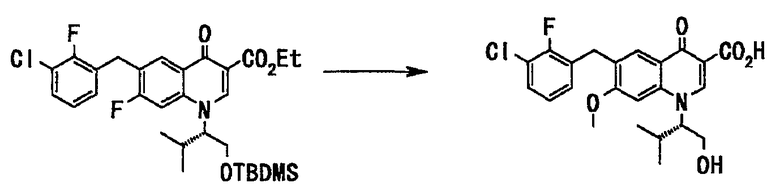
Соединение (48 г, 86 ммоль), полученное на стадии 5, растворяли в метаноле (300 мл), добавляли воду (5 мл) и 28% метанольный раствор метоксида натрия (176 мл, 862 ммоль), и смесь нагревали с обратным холодильником в течение 24 часов. Реакционную смесь оставляли для охлаждения до комнатной температуры, и смесь нейтрализовали добавлением 6 н. соляной кислоты. Метанол упаривали при пониженном давлении. К полученному раствору добавляли воду и смесь перемешивали. Выпавшее в осадок твердое вещество отделяли фильтрованием, полученное твердое вещество растворяли в этилацетате. Смесь промывали водой, сушили над сульфатом натрия. Раствор фильтровали и фильтрат концентрировали при пониженном давлении. Полученный остаток перекристаллизовывали из смеси растворителей этилацетат-гексан с получением соединения (32 г, выход 86%) в виде твердого вещества белого цвета. Полученное соединение (32 г) растворяли в бутилацетате (160 мл) при нагревании с обратным холодильником, и кристаллическая форма II была введена как затравка при 75°С. Смесь перемешивали 3,5 часа по мере того, как она остывала. Выпавший осадок отделяли фильтрованием, промывали бутилацетатом (25 мл) и сушили в вакууме с получением соединения (25 г, выход 77%) в виде твердого вещества белого цвета. Полученное соединение (4,0 г) растворяли в метаноле (40 мл) при нагревании с обратным холодильником до 50°С и добавляли по каплям в воду (40 мл) комнатной температуры. Смесь перемешивали при комнатной температуре 16 часов, отфильтровывали, и оставшееся твердое вещество промывали 66% водным метанолом, сушили в вакууме, получали кристалл соединения А (кристаллическая форма I) (3,9 г, выход 97%) в виде твердого вещества белого цвета.
Т.пл. 151-152°С
1H ЯМР (ДМСО-d6 300 МГц) (δ) ммд: 0,72 (3H, д, J=6,5 Гц), 1,16 (3H, д, J=6,5 Гц), 2,30-2,50 (1H, м), 3,70-3,90 (1H, м), 3,90-4,00 (1H, м), 4,03 (3H, с), 4,12 (2H,с), 4,80-4,90 (1H, м), 5,19 (1H, т), 7,19-7,25 (2H, м), 7,46-7,51 (2H, м), 8,04 (1H, с), 8,88 (1H, с), 15,44 (1H, с).
МС (ESI): M+ 448.
Пример 1: Получение кристаллической формы II соединения А
Стадия 1

2,4-Дифторбензойную кислоту (50 г, 316 ммоль) растворяли в концентрированной серной кислоте (200 мл), и порциями добавляли N-иодсукцинимид (68 г, 300 ммоль) при температуре, не превышающей 5°С. После завершения добавления смесь перемешивали при той же температуре 4,5 часа. Реакционную смесь выливали в ледяную воду (приблизительно 600 мл), затем добавляли 10% водный раствор сульфита натрия, и смесь перемешивали. Выпавший твердый осадок отделяли фильтрованием, промывали водой и сушили в вакууме с получением неочищенных кристаллов (85 г). Неочищенные кристаллы, полученные подобным образом, объединяли (получали их общее количество 205 г) и перекристаллизовывали из 50% водного этанола (820 мл) с получением 2,4-дифтор-5-иодбензойной кислоты (148 г, выход 73%) в виде твердого вещества белого цвета.
1H ЯМР (CDCl3 300 МГц) (σ) ммд: 6,94 (1H, дд, J=10,3, 10,3 Гц), 8,46 (1H, д, J=7,5 Гц).
Стадия 2
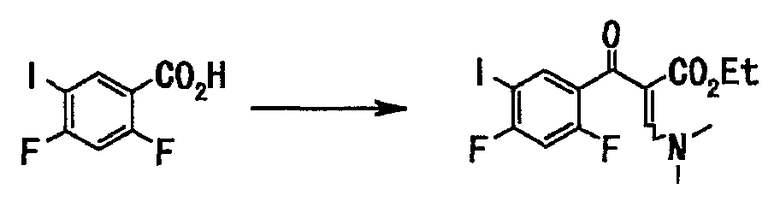
Соединение (148 г, 521 ммоль), полученное на стадии 1, растворяли в толуоле (750 мл), добавляли тионилхлорид (76 мл, 1,04 моль) и диметилформамид (каталитические количества), смесь нагревали с обратным холодильником в течение 2 часов. Нерастворившееся вещество отфильтровывали при 60°С, и фильтрат концентрировали при пониженном давлении, и отгоняли азеотроп с толуолом (330 мл). Остаток растворяли в тетрагидрофуране (400 мл), и этот раствор по каплям добавляли к раствору этил-3,3-диметиламиноакрилата (82 г, 573 ммоль) и триэтиламина (87 мл, 625 ммоль) в тетрагидрофуране (400 мл), и смесь нагревали с обратным холодильником в течение 7 часов. Реакционную смесь оставляли для остывания до комнатной температуры и концентрировали при пониженном давлении. Добавляли воду (700 мл) и этилацетат (800 мл) оставляли для разделения. Органический слой последовательно промывали дважды насыщенным водным раствором гидрокарбоната натрия (250 мл), водой (300 мл) и насыщенным солевым раствором (300 мл) и сушили над сульфатом натрия. Смесь фильтровали и фильтрат концентрировали при пониженном давлении с получением неочищенного вещества (210 г), этилового эфира 2-(2,4-дифтор-5-иодбензоил)-3-диметиламиноакриловой кислоты в виде твердого вещества коричневого цвета.
Стадия 3

Неочищенный продукт (210 г), полученный на стадии 2, растворяли в тетрагидрофуране (500 мл), добавляли (S)-(+)-валинол (54 г, 521 ммоль), и смесь перемешивали при комнатной температуре 30 минут. Реакционную смесь концентрировали при пониженном давлении, и остаток растворяли в диметилформамиде (600 мл). Добавляли карбонат калия (144 г, 1,04 моль), и смесь перемешивали при нагревании до 70°С в течение 2 часов. Реакционную смесь оставляли для охлаждения до комнатной температуры, добавляли к воде (1500 мл), и смесь перемешивали. Выпавшее в осадок твердое вещество отделяли фильтрованием. Полученное твердое вещество последовательно промывали 30% водным этанолом (500 мл) и смесью растворителей: диэтилового эфира (150 мл) и гексана (150 мл) и сушили в вакууме, получали этиловый эфир 7-фтор-1-((S)-1-гидроксиметил-2-метилпропил)-6-иод-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты (178 г, выход 76% (относительно стадии 2)) в виде твердого вещества бежевого цвета.
1H ЯМР (ДМСО-d6 300 МГц) (δ) ммд: 0,72 (3H, д, J=6,6 Гц), 1,10 (3H, д, J=6,6 Гц), 1,28 (3H, т, J=7,0 Гц), 2,27 (1H, ушир.), 3,77 (1H, ушир.), 3,86 (1H, ушир.), 4,23 (2H, кв, J=7,0 Гц), 4,56 (1H, ушир.), 5,12 (1H, т, J=4,9 Гц), 8,09 (1H, д, J=11,1 Гц), 8,62 (1H, д, J=7,5 Гц), 8,68 (1H, с).
МС (ESI): M+ 448.
Стадия 4
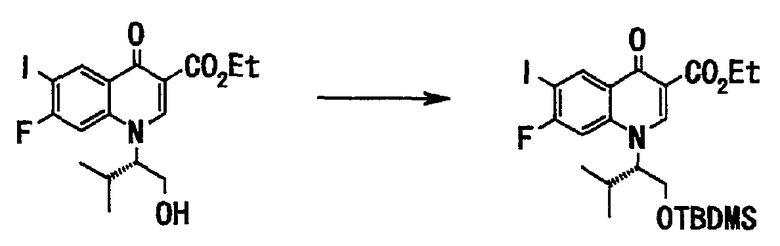
Соединение (150 г, 335 ммоль), полученное на стадии 3, растворяли в диметилформамиде (500 мл), добавляли имидазол (30 г, 436 ммоль) и трет-бутилдиметилсилилхлорид (56 г, 369 ммоль), и смесь перемешивали при комнатной температуре в течение 1,5 часов. К реакционной смеси добавляли воду и смесь экстрагировали этилацетатом. Органический слой последовательно промывали насыщенным водным раствором хлорида аммония и насыщенным солевым раствором и сушили над сульфатом натрия. Органический слой отфильтровывали и фильтрат концентрировали при пониженном давлении, и полученный остаток очищали хроматографией на силикагеле (этилацетат:гексан = 1:3 до 1:2) с получением этилового эфира 1-((S)-1-трет-бутилдиметилсилилоксиметил-2-метилпропил)-7-фтор-6-иод-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты (173 г, выход 92%) в виде бесцветного вещества в аморфной форме.
1H ЯМР (CDCl3 400 МГц) (δ) ммд: -0,07 (3H, с), -0,05 (3H, с), 0,77 (9H, с), 0,84 (3H, д, J=6,5 Гц), 1,18 (3H, д, J=6,5 Гц), 1,40 (3H, т, J=7,2 Гц), 2,35-2,50 (1H, м), 3,85-3,95 (1H, м), 3,98-4,10 (2H, м), 4,30-4,40 (2H, м), 7,26 (1H, с), 8,64 (1H, с), 8,94 (1H, д, J=7,2 Гц).
МС (ESI): M+ 562.
Стадия 5
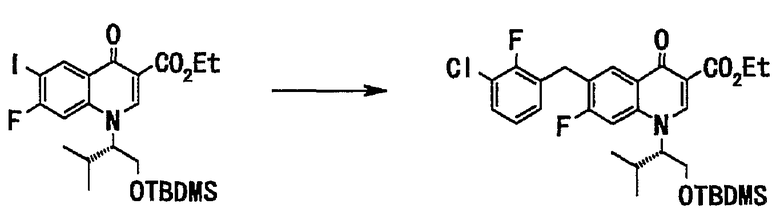
(Приготовление раствора 3-хлор-2-фторбензилцинкбромида в тетрагидрофуране)
В токе аргона цинковый порошок (11 г, 175 ммоль) суспендировали в тетрагидрофуране (30 мл), добавляли 1,2-дибромэтан (0,1 мл, 1,20 ммоль) и триметилсилилхлорид (0,29 мл, 2,4 ммоль) при 60°С, и смесь перемешивали при нагревании в течение 30 минут. По каплям добавляли раствор 3-хлор-2-фторбензилбромида (27 г, 119 ммоль) в тетрагидрофуране (60 мл) при 60°С. Смесь перемешивали при нагревании 1 час и оставляли для охлаждения до комнатной температуры, получали 1М раствор 3-хлор-2-фторбензилцинкбромида в тетрагидрофуране. Этот раствор применяли на последующей основной стадии.
(Основная стадия)
Соединение (50 г, 89 ммоль), полученное на стадии 4, растворяли в тетрагидрофуране (400 мл) и в токе аргона добавляли дихлорбис(трифенилфосфин)палладий(II) (2,1 г, 3,6 ммоль) и при 60°С по каплям добавляли раствор вышеупомянутого 1М 3-хлор-2-фторбензилцинкбромида в тетрагидрофуране. После завершения прикапывания смесь перемешивали с нагреванием до той же температуры в течение 1,5 часов. Реакционную смесь оставляли для охлаждения до комнатной температуры, добавляли 1 н. соляную кислоту, смесь экстрагировали 3 раза этилацетатом. Органический слой последовательно промывали водой и насыщенным солевым раствором и сушили над сульфатом магния. Органический слой отфильтровывали и фильтрат концентрировали при пониженном давлении, и полученный остаток очищали хроматографией на силикагеле (этилацетат:гексан = 1:2 до 1:1) с получением этилового эфира 1-((S)-1-трет-бутилдиметилсилилоксиметил-2-метилпропил)-6-(3-хлор-2-фторбензил)-7-фтор-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты (43 г, выход 83%) в виде вещества коричневого цвета в аморфной форме.
1H ЯМР (CDCl3 400 МГц) (δ) ммд: -0,09 (3H, с), -0,05 (3H, с), 0,75 (9H, с), 0,85 (3H, д, J=6,7 Гц), 1,18 (3H, д, 6,7 Гц), 1,39 (3H, т, J=7,1 Гц), 2,45 (1H, ушир.), 3,89-3,92 (1H, м), 3,98-4,02 (1H, м), 4,07-4,12 (1H, м), 4,12 (2H, с), 4,34-4,41 (2H, м), 6,96-7,00 (1H, м), 7,03-7,05 (1H, м), 7,21-7,24 (1H, м), 7,26-7,29 (1H, м), 8,39 (1H, д, J=8,8 Гц), 8,63 (1H, с).
Стадия 6
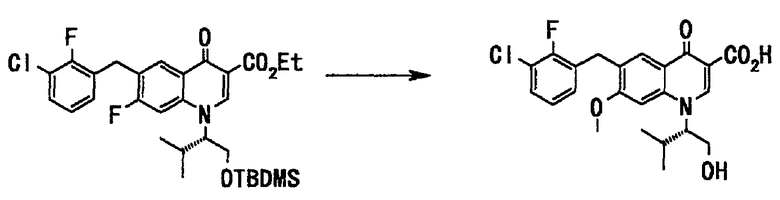
Соединение (43 г, 74 ммоль), полученное на стадии 5, растворяли в метаноле (280 мл), добавляли 28% метанольный раствор метоксида натрия (151 мл, 742 ммоль) и воду (4,3 мл), и смесь нагревали с обратным холодильником в течение 20 часов. Реакционную смесь фильтровали через целит. Фильтрат концентрировали при пониженном давлении. К остатку добавляли воду (400 мл), и смесь промывали гексаном (100 мл). Водный слой подкисляли добавлением концентрированной соляной кислоты (65 мл), и смесь экстрагировали этилацетатом. Органический слой последовательно промывали водой и насыщенным солевым раствором и сушили над сульфатом натрия. Раствор фильтровали и фильтрат концентрировали при пониженном давлении. Полученный неочищенный продукт (35 г, маслообразный, коричневого цвета) растворяли в этилацетате (49 мл) при нагревании с обратным холодильником, добавляли гексан (30 мл), оставляли для охлаждения, и смесь перемешивали в течение 18,5 часов. Выпавший твердый осадок отделяли фильтрованием, промывали смесью растворителей: этилацетат и гексан (1:1), сушили в вакууме с получением кристалла соединения А (кристаллическая форма II) (27 г, выход 82%) в виде твердого вещества белого цвета.
Т.пл. 153,7-153,9°С
1H ЯМР (ДМСО-d6 300 МГц) (δ) ммд: 0,72 (3H, д, J=6,5 Гц), 1,16 (3H, д, J=6,5 Гц), 2,30-2,50 (1H, м), 3,70-3,90 (1H, м), 3,90-4,00 (1H, м), 4,03 (3H, с), 4,12 (2H, с), 4,80-4,90 (1H, м), 5,19 (1H, т), 7,19-7,25 (2H, м), 7,46-7,51 (2H, м), 8,04 (1H, с), 8,88 (1H, с), 15,44 (1H, с).
МС (ESI): M+ 448.
Пример 2: Получение кристаллической формы II соединения А
Пример 2-1: Получение кристаллической формы II соединения А
Стадия 1

2,4-Дифторбензойную кислоту (100 г, 633 ммоль) растворяли в концентрированной серной кислоте (400 мл), и порциями добавляли N-иодсукцинимид (142 г, 601 моль) при температуре, не превышающей 5°С. После завершения добавления смесь перемешивали при той же температуре в течение 6 часов. Реакционную смесь выливали в ледяную воду (приблизительно 2400 мл), затем добавляли насыщенный водный раствор сульфита натрия и смесь перемешивали. Выпавший твердый осадок отделяли фильтрованием, промывали водой и сушили в вакууме с получением неочищенных кристаллов (188 г). Неочищенные кристаллы, полученные подобным образом, объединяли (получали их общее количество 568 г) и перекристаллизовывали из 50% водного этанола (2600 мл) с получением 2,4-дифтор-5-иодбензойной кислоты (388 г, выход 68%) в виде твердого вещества белого цвета.
1Н-ЯМР (CDCl3, 300 МГц) (σ) ммд: 6,94 (1Н, дд, J=10,3, 10,3 Гц), 8,46 (1Н, д, J=7,5 Гц).
Стадия 2

Соединение (200 г, 704 ммоль), полученное на стадии 1, растворяли в толуоле (1000 мл), добавляли тионилхлорид (103 мл, 408 ммоль) и диметилформамид (каталитические количества), смесь нагревали с обратным холодильником в течение 2 часов. Нерастворившееся вещество отфильтровывали и фильтрат концентрировали при пониженном давлении и отгоняли азеотроп с толуолом. Остаток растворяли в тетрагидрофуране (500 мл), и этот раствор по каплям добавляли к раствору этил-3,3-диметиламиноакрилата (111 г, 775 ммоль) и триэтиламина (118 мл, 845 ммоль) в тетрагидрофуране (500 мл), и смесь нагревали с обратным холодильником в течение 3 часов. Реакционную смесь оставляли для остывания до комнатной температуры и фильтровали, и фильтрат концентрировали при пониженном давлении. Добавляли воду (500 мл) и этилацетат (800 мл) для разделения. Органический слой последовательно промывали насыщенным водным раствором гидрокарбоната натрия (200 мл), водой (200 мл) и концентрированным солевым раствором, и сушили над сульфатом натрия. Органический слой фильтровали, и фильтрат концентрировали при пониженном давлении с получением неочищенного продукта (273 г), этилового эфира 2-(2,4-дифтор-5-иодбензоил)-3-диметиламиноакриловой кислоты в виде твердого вещества коричневого цвета.
Стадия 3
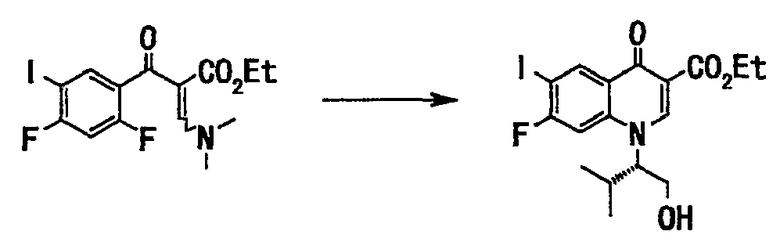
Неочищенный продукт (273 г), полученный на стадии 2, растворяли в тетрагидрофуране (650 мл), добавляли (S)-(+)-валинол (73 г, 708 ммоль) и смесь перемешивали при комнатной температуре 2 часа. Реакционную смесь концентрировали при пониженном давлении, и остаток растворяли в диметилформамиде (800 мл). Добавляли карбонат калия (195 г, 1,41 моль), и смесь перемешивали при нагревании до 70°С в течение 2,5 часов. Реакционную смесь оставляли для остывания до комнатной температуры, добавляли к воде (2000 мл) и перемешивали. Выпавшее в осадок твердое вещество отделяли фильтрованием. Полученное твердое вещество промывали суспендированием последовательно в воде и в 30% водном этаноле (650 мл) и сушили в вакууме с получением неочищенного продукта (217 г). Полученный неочищенный продукт (217 г) промывали посредством суспендирования смесью растворителей: этилацетата (650 мл) и гексана (440 мл) при нагревании с обратным холодильником. Смесь фильтровали, твердый остаток сушили в вакууме, получали этиловый эфир 7-фтор-1-((S)-1-гидроксиметил-2-метилпропил)-6-иод-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты (207 г, выход 66% (относительно стадии 2)) в виде твердого вещества бледно-коричневого цвета.
1H ЯМР (ДМСО-d6 300 МГц) (δ) ммд: 0,72 (3H, д, J=6,6 Гц), 1,10 (3H, д, J=6,6 Гц), 1,28 (3H, т, J=7,0 Гц), 2,27 (1H, ушир.), 3,77 (1H, ушир.), 3,86 (1H, ушир.), 4,23 (2H, кв, J=7,0 Гц), 4,56 (1H, ушир.), 5,12 (1H, т, J=4,9 Гц), 8,09 (1H, д, J=11,1 Гц), 8,62 (1H, д, J=7,5 Гц), 8,68 (1H, с).
МС (ESI): M+ 448.
Стадия 4
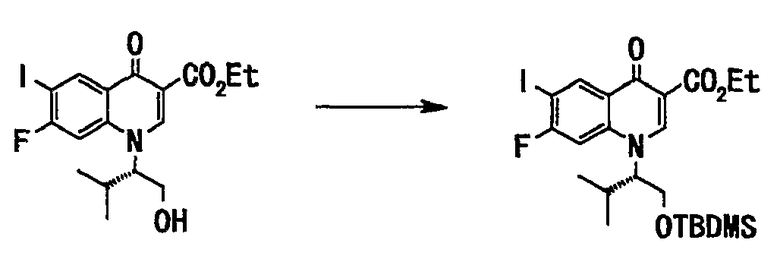
Соединение (150 г, 335 ммоль), полученное на стадии 3, растворяли в диметилформамиде (450 мл), добавляли имидазол (27 г, 397 ммоль) и трет-бутилдиметилсилилхлорид (58 г, 385 ммоль), и смесь перемешивали в течение ночи при комнатной температуре. К реакционной смеси добавляли воду (900 мл), и смесь экстрагировали этилацетатом (680 мл). Органический слой последовательно промывали водой (450 мл, 3 раза) и насыщенным солевым раствором (200 мл) и сушили над сульфатом натрия. Органический слой отфильтровывали, и фильтрат концентрировали при пониженном давлении с получением неочищенного вещества (192 г), этилового эфира 1-((S)-1-трет-бутилдиметилсилилоксиметил-2-метилпропил)-7-фтор-6-иод-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты (77 г, выход 77%) в виде вещества бледно-желтого цвета в аморфной форме.
Стадия 5
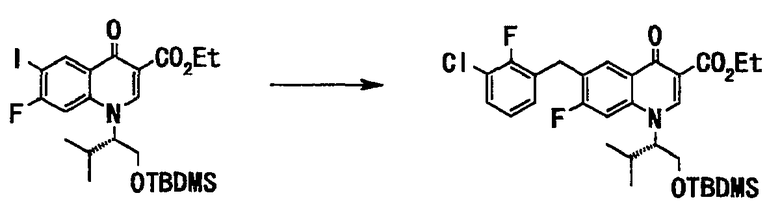
Неочищенное вещество (162 г), полученное на стадии 4, растворяли в тетрагидрофуране (160 мл) и в токе аргона добавляли дибензилиденацетонпалладий(II) (1,7 г, 2,9 ммоль) и трифурилфосфин (1,3 г, 5,8 ммоль). К этой смеси по каплям добавляли при 60°С раствор (375 мл, 375 ммоль) 1М 3-хлор-2-фторбензилцинкбромида в тетрагидрофуране, полученный таким же образом, как в примере 1, стадия 5, и после завершения прикапывания смесь перемешивали с нагреванием до той же температуры в течение 3,5 часов. Реакционную смесь оставляли для охлаждения до комнатной температуры, добавляли этилацетат (640 мл) и 10% водный раствор лимонной кислоты (400 мл) и фильтровали смесь через целит, и фильтрат расслаивался. Органический слой последовательно промывали водой (200 мл), насыщенным водным раствором гидрокарбоната натрия (400 мл) и насыщенным солевым раствором (200 мл) и сушили над сульфатом натрия. Органический слой отфильтровывали и фильтрат концентрировали при пониженном давлении с получением неочищенного вещества (186 г), этилового эфира 1-((S)-1-трет-бутилдиметилсилилоксиметил-2-метилпропил)-6-(3-хлор-2-фторбензил)-7-фтор-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты в виде маслообразного вещества коричневого цвета.
Стадия 6
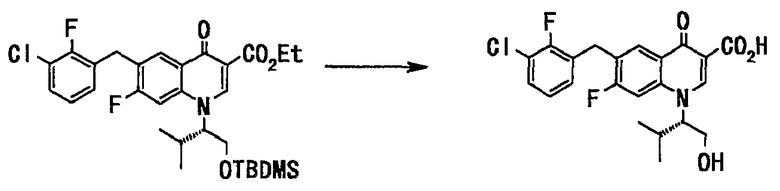
Неочищенное вещество (193 г), полученное на стадии 5, растворяли в изопропаноле (650 мл), добавляли 1 н. водный раствор гидроксида натрия (1290 мл, 1,29 моль) и смесь нагревали с обратным холодильником 2 часа. Реакционную смесь оставляли для охлаждения до комнатной температуры и фильтровали через целит. Фильтрат подкисляли добавлением концентрированной соляной кислоты и смесь перемешивали. Твердый выпавший осадок отделяли фильтрованием и сушили в вакууме с получением неочищенного вещества (132 г) в твердом виде, бледно-желтого цвета. Неочищенные вещества, полученные таким же образом, объединяли (общее количество 143 г), суспендировали в бутилацетате (430 мл) и суспензию перемешивали при нагревании с обратным холодильником 1 час. Суспензию оставляли для охлаждения до комнатной температуры и фильтровали и сушили в вакууме с получением 6-(3-хлор-2-фторбензил)-7-фтор-1((S)-1-гидроксиметил-2-метилпропил)-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты (99 г, выход 74% (относительно стадии 3)) в виде твердого вещества серого цвета.
1H ЯМР (ДМСО-d6 400 МГц) (δ) ммд: 0,71 (3H, д, J=6,5 Гц), 1,13 (3H, д, J=6,5 Гц), 2,36 (1H, ушир.), 3,77 (1H, ушир.), 3,94 (1H, ушир.), 4,25 (2H, с), 4,77 (1H, ушир.), 5,16 (1H, т, J=2,4 Гц), 7,19-7,23 (1H, м), 7,32-7,35 (1H, м), 7,48-7,52 (1H, м), 8,24-8,28 (2H, м), 9,00 (1H, с), 15,00 (1H, с).
МС (ESI): M+ 436.
Стадия 7
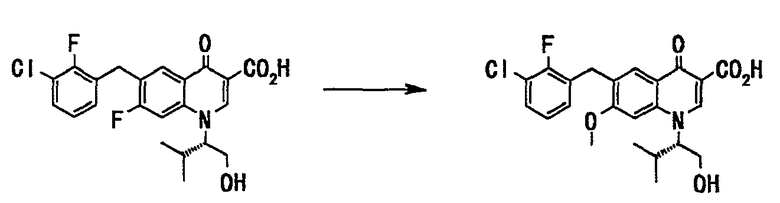
Соединение (99 г, 227 ммоль), полученное на стадии 6, растворяли в метаноле (530 мл), добавляли 28% метанольный раствор метоксида натрия (465 мл, 2,28 моль) и смесь нагревали с обратным холодильником в течение 20 часов. Реакционную смесь оставляли для охлаждения до комнатной температуры и фильтровали через целит. Фильтрат концентрировали при пониженном давлении. Остаток подкисляли путем добавления воды (200 мл) и концентрированной соляной кислоты (190 мл) и экстрагировали этилацетатом (500 мл). Органический слой дважды промывали водой (200 мл) и сушили над сульфатом натрия. Смесь фильтровали и фильтрат концентрировали при пониженном давлении с получением неочищенного вещества (108 г). Полученное неочищенное вещество (108 г) растворяли в изобутилацетате (330 мл) при нагревании и смесь перемешивали по мере того, как она охлаждалась, в течение 24 часов. Выпавший твердый осадок отделяли фильтрованием и сушили в вакууме с получением соединения А (71 г, выход 69%) в виде твердого вещества белого цвета. Неочищенные кристаллы, полученные таким же образом, объединяли (общее количество составило 233 г), растворяли в изобутилацетате (470 мл) при нагревании с обратным холодильником, и смесь перемешивалась в течение ночи, пока была оставлена для охлаждения. Выпавший твердый осадок отделяли фильтрованием и сушили в вакууме, получали кристалл соединения А (кристаллическая форма II) (206 г, выход 88%) в виде твердого вещества белого цвета.
1H ЯМР (ДМСО-d6 300 МГц) (δ) ммд: 0,72 (3H, д, J=6,5 Гц), 1,16 (3H, д, J=6,5 Гц), 2,30-2,50 (1H, м), 3,70-3,90 (1H, м), 3,90-4,00 (1H, м), 4,03 (3H, с), 4,12 (2H, с), 4,80-4,90 (1H, м), 5,19 (1H, т), 7,19-7,25 (2H, м), 7,46-7,51 (2H, м), 8,04 (1H, с), 8,88 (1H, с), 15,44 (1H, с).
МС (ESI): M+ 448.
Пример 2-2: Получение кристаллической формы II соединения А
Стадия 1
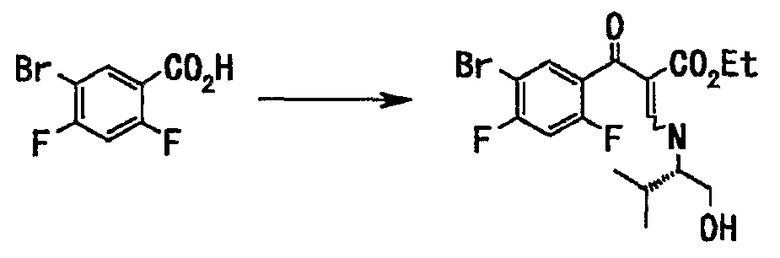
5-Бром-2,4-дифторбензойную кислоту (82,7 кг, 349 моль) растворяли в толуоле (420 л), добавляли тионил хлорид (62,3 кг, 523 моль) и диметилформамид (каталитическое количество) и смесь перемешивали при 70°С 6 часов. Реакционную смесь оставляли для охлаждения до комнатной температуры, концентрировали при пониженном давлении и снова отгоняли азеотроп с толуолом (420 л). Остаток растворяли в толуоле (220 л), этот раствор по каплям добавляли к раствору этил-3,3-диметиламиноакрилата (55,0 кг, 384 моль) и диизопропилэтиламина (58,6 кг, 523 моль) в толуоле (220 л) и смесь перемешивали при нагревании до 70°С в течение 21 часа. Реакционную смесь оставляли для охлаждения до комнатной температуры, добавляли (S)-(+)-валинол (36,0 кг, 349 моль) и смесь перемешивали при комнатной температуре 1,5 часа. К реакционной смеси добавляли воду (420 л), оставляли для расслоения, органический слой последовательно промывали 1 н. соляной кислотой (250 л, дважды), водой (420 л), 5% водным раствором гидрокарбоната натрия (250 л, дважды), водой (420 л) и 10% солевым раствором (250 л). Экстракт концентрировали при пониженном давлении и отгоняли азеотроп с диметилформамидом (420 л), получали концентрированный остаток (330 л), содержащий неочищенное вещество, этиловый эфир 2-(5-бром-2,4-дифторбензоил)-3-((S)-1-гидроксиметил-2-метилпропилметиламино)акриловой кислоты.
Стадия 2

К раствору (330 л) неочищенного вещества, полученного на стадии 1, в диметилформамиде добавляли 1,8-диазабицикло[5.4.0]ундекан (105 кг, 349 моль), и смесь перемешивали при комнатной температуре в течение 23 часов. К реакционной смеси добавляли диметилформамид (330 л) и затем воду (170 л) и после перемешивания в течение 2 часов по каплям добавляли воду (170 л). Выпавший твердый осадок отделяли фильтрованием и промывали смесью диметилформамид (170 л) - вода (170 л) и затем смешанным раствором этанол (460 л) - вода (200 л). Полученное твердое вещество сушили в вакууме, суспендировали в смеси этилацетат (330 л)-н-гептан (330 л) и промывали при суспендировании. Суспензию фильтровали и твердый остаток сушили в вакууме, получали этиловый эфир 6-бром-7-фтор-1-((S)-1-гидроксиметил-2-метилпропил)-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты (102 кг, выход 73%) в виде твердого вещества желто-белого цвета. Было подтверждено с помощью анализа высокоэффективной жидкостной хроматографией (ВЭЖХ), что это соединение эквивалентно стандартному образцу соединения.
Стадия 3

Соединение (45,0 кг, 112 моль), полученное на стадии 2, и имидазол (9,95 кг, 146 моль) суспендировали в толуоле (180 л), добавляли при 50°С раствор трет-бутилдиметилсилилхлорида (17,8 кг, 118 моль) в толуоле (45 л) и смесь перемешивали при той же температуре в течение 3 часов. К реакционной смеси добавляли толуол (230 л) и последовательно промывали водой (450 л, дважды) и 20% солевым раствором (450 л). Экстракт концентрировали при пониженном давлении и отгоняли азеотроп с тетрагидрофураном (320 л), получали концентрированный остаток (390 л), содержащий неочищенное вещество, этиловый эфир 6-бром-1-((S)-1-трет-бутилдиметилсилилоксиметил-2-метилпропил)-7-фтор-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты.
Стадия 4
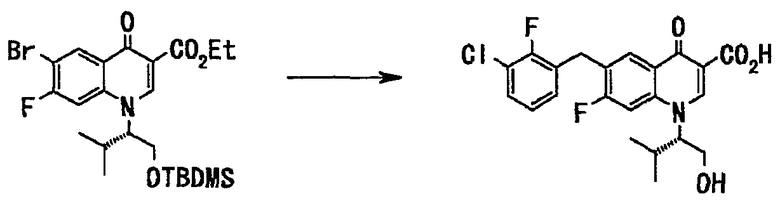
(Приготовление раствора 3-хлор-2-фторбензилцинкбромида в тетрагидрофуране)
В токе аргона цинковый порошок (18,8 кг, 287 моль) суспендировали в тетрагидрофуране (130 л), добавляли 1,2-дибромэтан (470 г, 2,50 моль) при 60°С и смесь перемешивали при той же температуре 30 минут. К этой суспензии при комнатной температуре добавляли триметилсилилхлорид (560 г, 3,10 моль) и смесь перемешивали при нагревании в течение 30 минут. По каплям добавляли раствор 3-хлор-2-фторбензилбромида (54,0 кг, 242 моль) в тетрагидрофуране (65 л) при 0°С и смесь перемешивали при 20°С 3 часа. Оставшийся цинк отфильтровывали, получали раствор 1М 3-хлор-2-фторбензилцинкбромида в тетрагидрофуране. Этот раствор применяли на последующей основной стадии.
(Основная стадия)
В токе азота растворяли трис(дибензилиденацетон)дипалладий(0) (1,96 кг, 3,36 моль) и трифенилфосфин (1,77 кг, 6,72 моль) в тетрагидрофуране (180 л) и смесь перемешивали при комнатной температуре 1 час. Добавляли по каплям при комнатной температуре раствор (390 л) неочищенного вещества в тетрагидрофуране, полученный на стадии 3, промывали тетрагидрофураном (45 л). По каплям при комнатной температуре добавляли заранее приготовленный раствор (164 кг, 157 моль) вышеупомянутого 1М 3-хлор-2-фторбензилцинкбромида в тетрагидрофуране и смесь перемешивалась при нагревании до 55°С 5 часов. Реакционную смесь оставляли для охлаждения до комнатной температуры, добавляли толуол (230 л) и 25% водный раствор хлорида аммония (230 л) и смесь перемешивали. После фильтрования смесь расслаивалась. Органический слой последовательно промывали 25% водным раствором хлорида аммония (230 л), водой (230 л), 5% водным раствором гидрокарбоната натрия (230 л, три раза) и 10% солевым раствором (230 л). Экстракт концентрировали при пониженном давлении, получали неочищенное вещество (80 л), 6-(3-хлор-2-фторбензил)-7-фтор-1-((S)-1-гидроксиметил-2-метилпропил)-4-оксо-1,4-дигидрохинолин-3-карбоновую кислоту в виде маслообразного вещества коричневого цвета.
Стадия 5
Неочищенный продукт (80 л), полученный на стадии 4, растворяли в изопропаноле (180 л), добавляли 1 н. водный раствор гидроксида натрия (180 л, 180 моль) и смесь перемешивали при нагревании до 50°С в течении 9 часов. К реакционной смеси добавляли активированный уголь (4,5 кг). Смесь перемешивали при комнатной температуре 30 мин, фильтровали через порошкообразную целлюлозу и тщательно промывали смесью изопропанол (45 л) - вода (45 л). Воду (180 л) и н-гептан (230 л) добавляли к фильтрату и после перемешивания смесь расслаивалась. Водный слой снова промывали н-гептаном (230 л). К органическому слою добавляли 4 н. соляную кислоту (45 л, 180 моль) и метилизопропилкетон (450 л), после перемешивания смесь расслаивалась. Органический слой последовательно промывали 10% солевым раствором (230 л), дважды 8,5% водным гидрокарбонатом натрия (230 л), 0,5 н. соляной кислотой (230 л) и водой (230 л). Экстракт концентрировали при пониженном давлении, три раза отгоняли азеотроп с толуолом (230 л). Остаток перемешивали при 100°С 1,5 часа, оставляли для охлаждения при комнатной температуре и перемешивали 3 часа. Выпавший твердый осадок отделяли фильтрованием, полученное твердое вещество промывали толуолом (45 л) и сушили в вакууме, получали 6-(3-хлор-2-фторбензил)-7-фтор-1-[(S)-1-гидроксиметил-2-метилпропил]-4-оксо-1,4-дигидрохинолин-3-карбоновую кислоту (42,5 кг, выход 87%) в виде твердого вещества бледно-желтого цвета. Было подтверждено с помощью анализа высокоэффективной жидкостной хроматографией (ВЭЖХ), что это соединение эквивалентно стандартному образцу.
Стадия 6

Соединение (39,2 кг, 89,9 моль), полученное на стадии 5, растворяли в метаноле (240 л), добавляли по каплям при 10°С 28% метанольный раствор метоксида натрия (173 кг, 899 моль) и смесь перемешивали с нагреванием до 70°С в течении 21 часа. К реакционной смеси добавляли активированный уголь (3,9 кг). Смесь перемешивали при комнатной температуре 1 час, фильтровали через порошкообразную целлюлозу и тщательно промывали метанолом (80 л). К фильтрату добавляли воду (29 кг, 1620 л) и смесь концентрировали при пониженном давлении. Из остатка дважды отгоняли азеотроп с изопропанолом (240 л, 120 л). К остатку добавляли 15% солевой раствор (200 л) и толуол (200 л) и после перемешивания смесь расслаивалась. Органический слой последовательно промывали 20% солевым раствором (200 л, трижды), 0,5 н. соляной кислотой (200 л), содержащей хлорид натрия (10 кг) и 20% солевым раствором (200 л). Органический слой концентрировали при пониженном давлении и отгоняли азеотроп с этилацетатом (200 л). К остатку добавляли этилацетат (320 л) и воду (200 л), после перемешивания смесь расслаивалась. Органический слой концентрировали при пониженном давлении и дважды отгоняли азеотроп с изобутилацетатом (200 л). Остаток растворяли при нагревании, фильтровали в горячем виде и тщательно промывали изобутилацетатом (20 л). Кристалл-затравку (кристаллическая форма II соединения А, 39 г) добавляли к фильтрату при 60°С и смесь перемешивали при той же температуре 1,5 часа. Смесь перемешивали при нагревании до 80°С 2 часа, оставляли для охлаждения до комнатной температуры и дополнительно перемешивали в течение 6 часов. Выпавший твердый осадок отделяли фильтрованием. Полученное твердое вещество промывали изобутилацетатом (40 л) и сушили в вакууме, получали кристалл соединения А (кристаллическая форма II) (29,0 кг, выход 72%) в виде твердого вещества белого цвета. Было подтверждено с помощью методов ВЭЖХ и рентгенодифракционного анализа (РД), что это кристаллическое соединение эквивалентно стандартному образцу кристалла (кристаллическая форма II соединения А, полученная в примере 2-1).
Пример 2-3: Получение кристаллической формы II соединения А
Кристаллическая форма II может быть также получена кристаллизацией по способам, описанным в примерах с 2-3-1 по 2-3-26.
Пример 2-3-1
Соединение А (200 мг), полученное в примере 1, растворяли в 1-бутаноле (2 мл) при нагревании с обратным холодильником. Смесь перемешивали 17 часов, давая ей охладиться. Выпавший твердый осадок отделяли фильтрованием и сушили в вакууме с получением кристаллической формы II (125 мг, выход 63%) соединения А в виде твердого вещества белого цвета.
Пример 2-3-2
Соединение А (200 мг), полученное в примере 1, растворяли в бутилацетате (2 мл) при нагревании с обратным холодильником. Смесь перемешивали 17 часов, давая ей охладиться. Выпавший твердый осадок отделяли фильтрованием и сушили в вакууме с получением кристаллической формы II (102 мг, выход 51%) соединения А в виде твердого вещества белого цвета.
Пример 2-3-3
Соединение А (200 мг), полученное в примере 1, растворяли в метилизобутилкетоне (2 мл) при нагревании с обратным холодильником. По каплям добавляли гептан (2 мл) и смесь перемешивали 6 часов, давая ей охладиться. Выпавший твердый осадок отделяли фильтрованием и сушили в вакууме с получением кристаллической формы II (168 мг, выход 84%) соединения А в виде твердого вещества белого цвета.
Пример 2-3-4
Соединение А (200 мг), полученное в примере 1, растворяли в этаноле (2 мл) при нагревании с обратным холодильником. Смесь перемешивали 17 часов, давая ей охладиться. Выпавший твердый осадок отделяли фильтрованием и сушили в вакууме с получением кристаллической формы II (56 мг, выход 28%) соединения А в виде твердого вещества белого цвета.
Пример 2-3-5
Соединение А (200 мг), полученное в примере 1, растворяли в этилацетате (2 мл) при нагревании с обратным холодильником. По каплям добавляли гептан (1,6 мл) и смесь перемешивали 6 часов, давая ей охладиться. Выпавший твердый осадок отделяли фильтрованием и сушили в вакууме с получением кристаллической формы II (166 мг, выход 83%) соединения А в виде твердого вещества белого цвета.
Пример 2-3-6
Соединение А (200 мг), полученное в примере 1, растворяли в метилэтилкетоне (2 мл) при нагревании с обратным холодильником. По каплям добавляли гептан (4 мл) и смесь перемешивали 6 часов, давая ей охладиться. Выпавший твердый осадок отделяли фильтрованием и сушили в вакууме с получением кристаллической формы II (123 мг, выход 62%) соединения А в виде твердого вещества белого цвета.
Пример 2-3-7
Соединение А (200 мг), полученное в примере 1, растворяли в 1-пропаноле (2 мл) при нагревании с обратным холодильником. Смесь перемешивали 17 часов, давая ей охладиться. Выпавший твердый осадок отделяли фильтрованием и сушили в вакууме с получением кристаллической формы II (91 мг, выход 46%) соединения А в виде твердого вещества белого цвета.
Пример 2-3-8
Соединение А (200 мг), полученное в примере 1, растворяли в изопропаноле (2 мл) при нагревании с обратным холодильником. Смесь перемешивали 17 часов, давая ей охладиться. Выпавший твердый осадок отделяли фильтрованием и сушили в вакууме с получением кристаллической формы II (88 мг, выход 44%) соединения А в виде твердого вещества белого цвета.
Пример 2-3-9
Соединение А (200 мг), полученное в примере 1, растворяли в кумоле (2 мл) при нагревании с обратным холодильником. Смесь перемешивали 17 часов, давая ей охладиться. Выпавший твердый осадок отделяли фильтрованием и сушили в вакууме с получением кристаллической формы II (188 мг, выход 94%) соединения А в виде твердого вещества белого цвета.
Пример 2-3-10
Соединение А (200 мг), полученное в примере 1, растворяли в анизоле (2 мл) при нагревании с обратным холодильником. Смесь перемешивали 17 часов, давая ей охладиться. Выпавший твердый осадок отделяли фильтрованием и сушили в вакууме с получением кристаллической формы II (107 мг, выход 54%) соединения А в виде твердого вещества белого цвета.
Пример 2-3-11
Соединение А (200 мг), полученное в примере 1, растворяли в ацетоне (2 мл) при нагревании с обратным холодильником. По каплям добавляли гептан (2 мл) и смесь перемешивали 16,5 часа, давая ей охладиться. Дополнительно добавляли гептан (4 мл) и смесь дополнительно перемешивали 24 часа. Выпавший твердый осадок отделяли фильтрованием и сушили в вакууме с получением кристаллической формы II (134 мг, выход 67%) соединения А в виде твердого вещества белого цвета.
Пример 2-3-12
Соединение А (200 мг), полученное в примере 1, растворяли в этаноле (2 мл) при нагревании с обратным холодильником. По каплям добавляли гептан (4 мл) и смесь перемешивали 19 часов, давая ей охладиться. Выпавший твердый осадок отделяли фильтрованием и сушили в вакууме с получением кристаллической формы II (129 мг, выход 65%) соединения А в виде твердого вещества белого цвета.
Пример 2-3-13
Соединение А (200 мг), полученное в примере 1, растворяли в изопропаноле (2 мл) при нагревании с обратным холодильником. По каплям добавляли гептан (4 мл) и смесь перемешивали 19 часов, давая ей охладиться. Выпавший твердый осадок отделяли фильтрованием и сушили в вакууме с получением кристаллической формы II (166 мг, выход 83%) соединения А в виде твердого вещества белого цвета.
Пример 2-3-14
Соединение А (200 мг), полученное в примере 1, растворяли в 1-пропаноле (2 мл) при нагревании с обратным холодильником. По каплям добавляли гептан (4 мл) и смесь перемешивали 19 часов, давая ей охладиться. Выпавший твердый осадок отделяли фильтрованием и сушили в вакууме с получением кристаллической формы II (158 мг, выход 79%) соединения А в виде твердого вещества белого цвета.
Пример 2-3-15
Соединение А (200 мг), полученное в примере 1, растворяли в изобутаноле (2 мл) при нагревании с обратным холодильником. Смесь перемешивали 21 час, давая ей охладиться. Выпавший твердый осадок отделяли фильтрованием и сушили в вакууме с получением кристаллической формы II (131 мг, выход 66%) соединения А в виде твердого вещества белого цвета.
Пример 2-3-16
Соединение А (200 мг), полученное в примере 1, растворяли в толуоле (2 мл) при нагревании до 100°С. Смесь перемешивали 37 часов, давая ей охладиться. Выпавший твердый осадок отделяли фильтрованием и сушили в вакууме с получением кристаллической формы II (190 мг, выход 95%) соединения А в виде твердого вещества белого цвета.
Пример 2-3-17
Соединение А (200 мг), полученное в примере 1, растворяли в метилбутилкетоне (2 мл) при нагревании до 60°С. По каплям добавляли гептан (1,8 мл) и смесь перемешивали 37 часов, давая ей охладиться. Выпавший твердый осадок отделяли фильтрованием и сушили в вакууме с получением кристаллической формы II (191 мг, выход 96%) соединения А в виде твердого вещества белого цвета.
Пример 2-3-18
Соединение А (200 мг), полученное в примере 1, растворяли в хлороформе (1 мл) при нагревании до 60°С. По каплям добавляли изопропиловый эфир (1,8 мл) и смесь перемешивали 37 часов, давая ей охладиться. Выпавший твердый осадок отделяли фильтрованием и сушили в вакууме с получением кристаллической формы II (184 мг, выход 92%) соединения А в виде твердого вещества белого цвета.
Пример 2-3-19
Соединение А (200 мг), полученное в примере 1, растворяли в тетрагидрофуране (1 мл) при нагревании до 60°С. По каплям добавляли изопропиловый эфир (2 мл) и смесь перемешивали 41 час, давая ей охладиться. Выпавший твердый осадок отделяли фильтрованием и сушили в вакууме с получением кристаллической формы II (144 мг, выход 72%) соединения А в виде твердого вещества белого цвета.
Пример 2-3-20
Соединение А (200 мг), полученное в примере 1, растворяли в изобутаноле (2 мл) при нагревании с обратным холодильником. По каплям добавляли гептан (2 мл) и смесь перемешивали 21 час, давая ей охладиться. Выпавший твердый осадок отделяли фильтрованием и сушили в вакууме с получением кристаллической формы II (160 мг, выход 80%) соединения А в виде твердого вещества белого цвета.
Пример 2-3-21
Соединение А (200 мг), полученное в примере 1, растворяли в бутаноле (2 мл) при нагревании с обратным холодильником. По каплям добавляли гептан (2 мл) и смесь перемешивали 21 час, давая ей охладиться. Выпавший твердый осадок отделяли фильтрованием и сушили в вакууме с получением кристаллической формы II (152 мг, выход 76%) соединения А в виде твердого вещества белого цвета.
Пример 2-3-22
Соединение А (200 мг), полученное в примере 1, растворяли в изобутилацетате (2 мл) при нагревании с обратным холодильником. Смесь перемешивали 21 час, давая ей охладиться. Выпавший твердый осадок отделяли фильтрованием и сушили в вакууме с получением кристаллической формы II (140 мг, выход 70%) соединения А в виде твердого вещества белого цвета.
Пример 2-3-23
Соединение А (200 мг), полученное в примере 1, растворяли в изобутилацетате (2 мл) при нагревании с обратным холодильником. По каплям добавляли гептан (2 мл) и смесь перемешивали 21 час, давая ей охладиться. Выпавший твердый осадок отделяли фильтрованием и сушили в вакууме с получением кристаллической формы II (178 мг, выход 89%) соединения А в виде твердого вещества белого цвета.
Пример 2-3-24
Соединение А (200 мг), полученное в примере 1, растворяли в бутилацетате (2 мл) при нагревании с обратным холодильником. По каплям добавляли гептан (1,5 мл) и смесь перемешивали 21 час, давая ей охладиться. Выпавший твердый осадок отделяли фильтрованием и сушили в вакууме с получением кристаллической формы II (158 мг, выход 78%) соединения А в виде твердого вещества белого цвета.
Пример 2-3-25
Соединение А (200 мг), полученное в примере 1, растворяли в анизоле (2 мл) при нагревании до 110°С. По каплям добавляли гептан (2 мл) и смесь перемешивали 21 час, давая ей охладиться. Выпавший твердый осадок отделяли фильтрованием и сушили в вакууме с получением кристаллической формы II (187 мг, выход 89%) соединения А в виде твердого вещества белого цвета.
Пример 2-3-26
Соединение А (200 мг), полученное в примере 1, растворяли в бутилацетате (2 мл) при нагревании с обратным холодильником. После быстрого охлаждения смесь перемешивали 2 часа. Выпавший твердый осадок отделяли фильтрованием и сушили в вакууме с получением кристаллической формы II (131 мг, выход 66%) соединения А в виде твердого вещества белого цвета.
Пример 2-4: Получение кристаллической формы II соединения А
Стадия 1
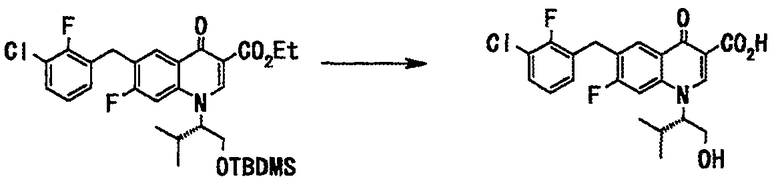
Этиловый эфир 1-((S)-1-трет-бутилдиметилсилилоксиметил-2-метилпропил)-6-(3-хлор-2-фторбензил)-7-фтор-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты (48 г, 86 ммоль), полученный в примере 1 на стадии 5, растворяли в метаноле (300 мл), добавляли воду (5 мл) и 28% метанольный раствор метоксида натрия (176 мл, 862 ммоль) и смесь нагревали с обратным холодильником 24 часа. Реакционную смесь оставляли для охлаждения до комнатной температуры, нейтрализовали 6 н. соляной кислотой, и метанол упаривали при пониженном давлении. К полученному раствору добавляли воду и после перемешивания отделяли фильтрованием выпавший твердый осадок. Полученное твердое вещество растворяли в этилацетате, промывали водой и сушили над сульфатом натрия. Смесь фильтровали и фильтрат концентрировали при пониженном давлении. Полученный остаток перекристаллизовывали из смеси этилацетат-гексан с получением соединения А (первичный кристалл 29,5 г, вторичный кристалл 2,8 г, общее количество 32,3 г, выход 86%) в виде твердого вещества белого цвета.
Т.пл. 151-152°С
1H ЯМР (ДМСО-d6 300 МГц) (δ) ммд: 0,72 (3H, д, J=6,5 Гц), 1,16 (3H, д, J=6,5 Гц), 2,30-2,50 (1H, м), 3,70-3,90 (1H, м), 3,90-4,00 (1H, м), 4,03 (3H, с), 4,12 (2H,с), 4,80-4,90 (1H, м), 5,19 (1H, т), 7,19-7,25 (2H, м), 7,46-7,51 (2H, м), 8,04 (1H, с), 8,88 (1H, с), 15,44 (1H, с).
МС (ESI): M+ 448.
Стадия 2
Соединение А (32,3 г), полученное на стадии 1, растворяли в бутилацетате (160 мл) при нагревании с обратным холодильником. Кристаллическую форму II Примера 2 добавляли как кристалл-затравку при 63°С и смесь перемешивали 3 часа, давая ей охладиться. Выпавший твердый осадок отделяли фильтрованием и сушили в вакууме с получением кристалла соединения А (кристаллическая форма II) (24,79 г, выход 77%) в виде твердого вещества белого цвета.
Пример 2-5: Получение кристаллической формы II соединения А
Стадия 1
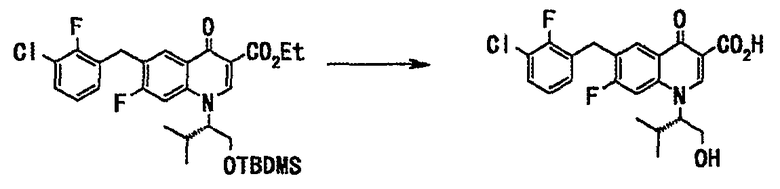
Этиловый эфир 1-((S)-1-трет-бутилдиметилсилилоксиметил-2-метилпропил)-6-(3-хлор-2-фторбензил)-7-фтор-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты (19 г, 33 ммоль), полученный в примере 1 на стадии 5, растворяли в изопропаноле (100 мл), добавляли 1 н. водный раствор гидроксида натрия (200 мл, 200 ммоль) и смесь нагревали с обратным холодильником 2,5 часа. Реакционную смесь оставляли для охлаждения до комнатной температуры и смесь фильтровали через целит. Фильтрат подкисляли добавлением концентрированной соляной кислоты и перемешивали при комнатной температуре 2 часа. Выпавший твердый осадок отделяли фильтрованием и сушили в вакууме с получением 6-(3-хлор-2-фторбензил)-7-фтор-1-((S)-1-гидроксиметил-2-метилпропил)-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты (12 г, выход 82%) в виде твердого вещества бледно-желтого цвета.
1H ЯМР (ДМСО-d6 400 МГц) (δ) ммд: 0,71 (3H, д, J=6,5 Гц), 1,13 (3H, д, J=6,5 Гц), 2,36 (1H, ушир.), 3,77 (1H, ушир.), 3,94 (1H, ушир.), 4,25 (2H, с), 4,77 (1H, ушир.), 5,16 (1H, т, J=2,4 Гц), 7,19-7,23 (1H, м), 7,32-7,35 (1H, м), 7,48-7,52 (1H, м), 8,24-8,28 (2H, м), 9,00 (1H, с), 15,00 (1H, с).
Стадия 2
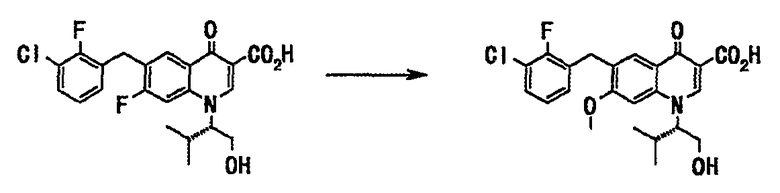
Соединение (12 г, 27 ммоль), полученное на стадии 1, растворяли в метаноле (64 мл), добавляли 28% метанольный раствор метоксида натрия (52 мл, 256 ммоль) и смесь нагревали с обратным холодильником 24 часа. Реакционную смесь оставляли для охлаждения до комнатной температуры и фильтровали через целит. Фильтрат концентрировали при пониженном давлении. Остаток подкисляли добавлением воды (360 мл) и концентрированной соляной кислоты и экстрагировали этилацетатом. Органический слой последовательно промывали водой, насыщенным солевым раствором и сушили над сульфатом натрия. Смесь фильтровали и фильтрат концентрировали при пониженном давлении с получением неочищенного вещества (13 г) в виде масла коричневого цвета. Полученное неочищенное вещество (13 г) растворяли в изобутилацетате (60 мл) при нагревании, после внесения кристалла-затравки смесь перемешивали 23 часа, давая ей охладиться. Выпавший твердый остаток отделяли фильтрованием и сушили в вакууме с получением соединения А (9,2 г, выход 75%) в виде твердого вещества белого цвета.
1H ЯМР (ДМСО-d6 300 МГц) (δ) ммд: 0,72 (3H, д, J=6,5 Гц), 1,16 (3H, д, J=6,5 Гц), 2,30-2,50 (1H, м), 3,70-3,90 (1H, м), 3,90-4,00 (1H, м), 4,03 (3H, с), 4,12 (2H, с), 4,80-4,90 (1H, м), 5,19 (1H, т), 7,19-7,25 (2H, м), 7,46-7,51 (2H, м), 8,04 (1H, с), 8,88 (1H, с), 15,44 (1H, с).
МС (ESI): M+ 448.
Стадия 3
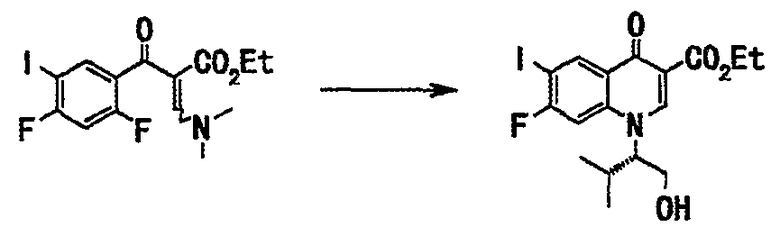
Этиловый эфир 2-(2,4-дифтор-5-иодбензоил)-3-диметиламиноакриловой кислоты (20 г), полученный в примере 1 на стадии 2, промывали суспендированием в смеси растворителей: этилацетат (60 мл) и гексан (40 мл) и нагревали с обратным холодильником. Смесь фильтровали, и оставшееся твердое вещество сушили в вакууме, получали этиловый эфир 7-фтор-1-((S)-1-гидроксиметил-2-метилпропил)-6-иод-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты (18 г, выход 94%) в виде твердого вещества бежевого цвета.
1H ЯМР (ДМСО-d6 300 МГц) (δ) ммд: 0,72 (3H, д, J=6,6 Гц), 1,10 (3H, д, J=6,6 Гц), 1,28 (3H, т, J=7,0 Гц), 2,27 (1H, ушир.), 3,77 (1H, ушир.), 3,86 (1H, ушир.), 4,23 (2H, кв, J=7,0 Гц), 4,56 (1H, ушир.), 5,12 (1H, т, J=4,9 Гц), 8,09 (1H, д, J=11,1 Гц), 8,62 (1H, д, J=7,5 Гц), 8,68 (1H, с).
МС (ESI): M+ 448.
Стадия 4

Соединение (19 г, 42 ммоль), полученное на стадии 3, растворяли в диметилформамиде (65 мл), добавляли имидазол (3,4 г, 49,9 ммоль) и трет-бутилтриметилсилилхлорид (7,2 г, 47,8 ммоль) и смесь перемешивали при комнатной температуре 1,5 часа. К реакционной смеси добавляли воду и реакционную смесь экстрагировали этилацетатом. Органический слой последовательно промывали водой, насыщенным водным раствором хлорида аммония и насыщенным солевым раствором, сушили над сульфатом натрия. Органический слой фильтровали и фильтрат концентрировали при пониженном давлении, получали неочищенное вещество (24 г), этиловый эфир 1-((S)-1-трет-бутилдиметилсилилоксиметил-2-метилпропил)-7-фтор-6-иод-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты бежевого цвета в аморфной форме.
1H ЯМР (CDCl3 400 МГц) (δ) ммд: -0,07 (3H, с), -0,05 (3H, с), 0,77 (9H, с), 0,84 (3H, д, J=6,5 Гц), 1,18 (3H, д, J=6,5 Гц), 1,40 (3H, т, J=7,2 Гц), 2,35-2,50 (1H, м), 3,85-3,95 (1H, м), 3,98-4,10 (2H, м), 4,30-4,40 (2H, м), 7,26 (1H,с), 8,64 (1H, с), 8,94 (1H, д, J=7,2 Гц).
МС (ESI): M+ 562.
Стадия 5
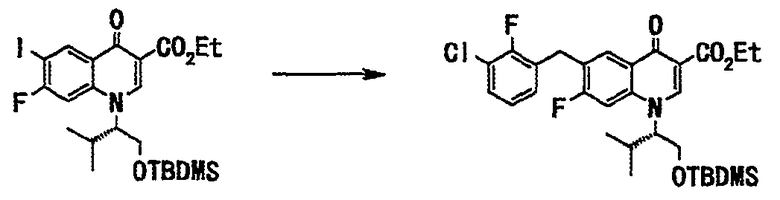
Неочищенное вещество (24 г), полученное на стадии 4, растворяли в тетрагидрофуране (200 мл) и добавляли в токе аргона дибензилиденацетонпалладий(II) (984 мг, 1,7 ммоль) и трифурилфосфин (795 мг, 3,4 ммоль) и добавляли по каплям при 60°С раствор (56 мл, 56 ммоль) 1М 3-хлор-2-фторбензилцинкбромида, полученного таким же образом, как в примере 1 на стадии 5, в тетрагидрофуране. После завершения прикапывания смесь перемешивали с нагреванием до той же температуры 2 часа. Реакционную смесь оставляли для охлаждения до комнатной температуры, добавляли насыщенный водный раствор хлорида аммония и фильтровали через целит, и фильтрат дважды экстрагировали этилацетатом. Органический слой последовательно промывали водой (дважды) и насыщенным солевым раствором и сушили над сульфатом магния. Органический слой фильтровали, фильтрат концентрировали при пониженном давлении с получением неочищенного вещества (30 г), этилового эфира 1-((S)-1-трет-бутилдиметилсилилоксиметил-2-метилпропил)-6-(3-хлор-2-фторбензил)-7-фтор-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты в виде пасты коричневого цвета.
Стадия 6
Неочищенное вещество (30 г), полученное на стадии 5, растворяли в изопропаноле (150 мл), добавляли 1 н. водный раствор гидроксида натрия (300 мл, 300 ммоль) и смесь нагревали с обратным холодильником 2,5 часа. Реакционную смесь оставляли для охлаждения до комнатной температуры и смесь фильтровали через целит. Фильтрат подкисляли добавлением концентрированной соляной кислоты и смесь перемешивали при комнатной температуре 2 часа. Выпавший твердый осадок отделяли фильтрованием, сушили в вакууме с получением неочищенного вещества (18 г) в твердом состоянии бежевого цвета. Полученное неочищенное вещество (18 г) суспендировали в бутилацетате (90 мл) и суспензию перемешивали при нагревании с обратным холодильником в течение 1 часа. Суспензию оставляли для охлаждения до комнатной температуры, фильтровали и сушили в вакууме с получением 6-(3-хлор-2-фторбензил)-7-фтор-1-((S)-1-гидроксиметил-2-метилпропил)-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты (11 г, выход 62% (относительно стадии 3)) в виде твердого вещества белого цвета.
1H ЯМР (ДМСО-d6 400 МГц) (δ) ммд: 0,71 (3H, д, J=6,5 Гц), 1,13 (3H, д, J=6,5 Гц), 2,36 (1H, ушир.), 3,77 (1H, ушир.), 3,94 (1H, ушир.), 4,25 (2H, с), 4,77 (1H, ушир.), 5,16 (1H, т, J=2,4 Гц), 7,19-7,23 (1H, м), 7,32-7,35 (1H, м), 7,48-7,52 (1H, м), 8,24-8,28 (2H, м), 9,00 (1H, с), 15,00 (1H, с).
МС (ESI): M+ 436.
Стадия 7

Соединение (11 г, 26 ммоль), полученное на стадии 6, растворяли в метаноле (60 мл), добавляли 28% метанольный раствор метоксида натрия (52 мл, 256 ммоль) и смесь нагревали с обратным холодильником 24 часа. Реакционную смесь оставляли для охлаждения до комнатной температуры и фильтровали через целит, и фильтрат концентрировали при пониженном давлении. Остаток подкисляли добавлением воды (330 мл) и концентрированной соляной кислоты и смесь экстрагировали этилацетатом. Органический слой последовательно промывали водой и насыщенным солевым раствором, сушили над сульфатом натрия. Смесь фильтровали и фильтрат концентрировали при пониженном давлении с получением неочищенного вещества (12 г) в виде масла коричневого цвета. Полученное неочищенное вещество (12 г) растворяли в изобутилацетате (60 мл) при нагревании с обратным холодильником. Добавляли кристалл-затравку (кристаллическая форма II соединения А) и смесь перемешивалась 23 часа до ее охлаждения. Выпавший твердый осадок отделяли фильтрованием и сушили в вакууме с получением соединения А (8,2 г, выход 71%) в виде твердого вещества белого цвета.
1H ЯМР (ДМСО-d6 300 МГц) (δ) ммд: 0,72 (3H, д, J=6,5 Гц), 1,16 (3H, д, J=6,5 Гц), 2,30-2,50 (1H, м), 3,70-3,90 (1H, м), 3,90-4,00 (1H, м), 4,03 (3H, с), 4,12 (2H,с), 4,80-4,90 (1H, м), 5,19 (1H, т), 7,19-7,25 (2H, м), 7,46-7,51 (2H, м), 8,04 (1H, с), 8,88 (1H, с), 15,44 (1H, с).
МС (ESI): M+ 448.
Стадия 8
Соединение А (7,66 г), полученное на стадии 7, и соединение А (9,17 г), полученное на стадии 2, растворяли в изобутилацетате (84 мл) при нагревании с обратным холодильником и смесь перемешивали 16 часов, давая ей охладиться. Выпавший твердый остаток отделяли фильтрованием и сушили в вакууме с получением кристаллической формы II (14,73 г, выход 88%) соединения А в виде твердого вещества белого цвета.
Пример 2-6: Получение кристаллической формы II соединения А
Стадия 1
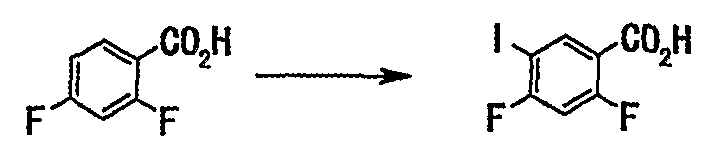
2,4-Дифторбензойную кислоту (100 г, 633 ммоль) растворяли в трифторметансульфокислоте (400 мл) и порциями добавляли N-иодсукцинимид (157 г, 696 ммоль) при температуре, не превышающей 5°С. После завершения добавления смесь перемешивали при 50°С в течение 1,5 часа. Реакционную смесь выливали в ледяную воду и перемешивали смесь 1 час. Выпавший твердый осадок отделяли фильтрованием, последовательно промывали водой и гексаном, сушили в вакууме, получали 2,4-дифтор-5-иодбензойную кислоту (179 г, выход количественный) в виде твердого вещества белого цвета.
1H ЯМР (CDCl3 300 МГц) (σ) ммд: 6,94 (1H, дд, J=10,3, 10,3 Гц), 8,46 (1H, д, J=7,5 Гц).
Стадия 2
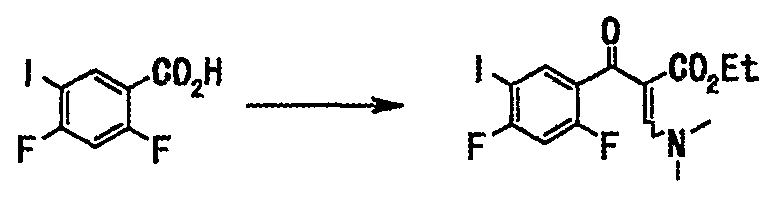
Соединение (28 г, 100 ммоль), полученное на стадии 1, растворяли в этилацетате (300 мл), добавляли оксалилхлорид (11 мл, 122 ммоль) и диметилформамид (каталитические количества) и смесь перемешивали при комнатной температуре в течение 2 часов. Фильтрат концентрировали при пониженном давлении и отгоняли азеотроп с толуолом. Остаток растворяли в тетрагидрофуране (100 мл), этот раствор по каплям добавляли к раствору этил-3,3-диметиламиноакрилата (17 г, 120 ммоль) и триэтиламина (21 мл, 150 ммоль) в тетрагидрофуране (100 мл), и смесь нагревали с обратным холодильником в течение 3 часов. Реакционную смесь оставляли для остывания и добавляли этилацетат (200 мл). Смесь последовательно промывали водой (дважды), насыщенным солевым раствором и сушили над сульфатом натрия. Смесь фильтровали и фильтрат концентрировали при пониженном давлении. Полученный остаток суспендировали при перемешивании в смеси растворителей: диэтиловый эфир (50 мл) и гексан (50 мл). Смесь фильтровали и твердый остаток сушили в вакууме с получением неочищенного продукта (26 г, выход 63%), этилового эфира 2-(2,4-дифтор-5-иодбензоил)-3-диметиламиноакриловой кислоты в виде твердого вещества желтого цвета.
Стадия 3
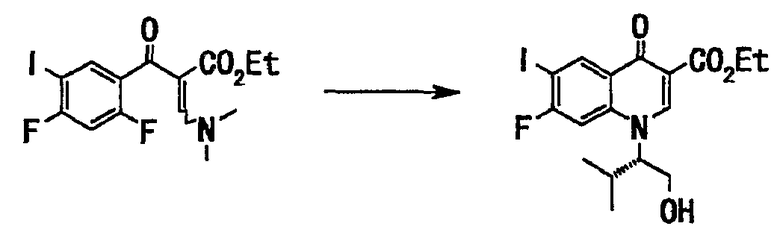
Неочищенный продукт (22 г, 55 ммоль), полученный на стадии 2, растворяли в тетрагидрофуране (110 мл), добавляли (S)-(+)-валинол (6,8 г, 65,8 ммоль) и смесь перемешивали при нагревании до 50°С 30 минут. Реакционную смесь концентрировали при пониженном давлении и остаток растворяли в этилацетате (100 мл), последовательно промывали водой и насыщенным солевым раствором и сушили над сульфатом магния. Смесь фильтровали и фильтрат концентрировали при пониженном давлении. Полученный остаток растворяли в диметилформамиде (80 мл), добавляли карбонат калия (19 г, 137 ммоль) и смесь перемешивали при нагревании до 60°С в течение 1,5 часов. Реакционную смесь оставляли для охлаждения до комнатной температуры и концентрировали при пониженном давлении. К полученному остатку добавляли воду (250 мл) и смесь перемешивали при комнатной температуре 30 мин. Выпавшее в осадок твердое вещество отделяли фильтрованием. Полученное твердое вещество последовательно промывали смесью растворителей: вода (100 мл), этилацетат (10 мл) и гексан (40 мл), и сушили в вакууме, получали этиловый эфир 7-фтор-1-((S)-1-гидроксиметил-2-метилпропил)-6-иод-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты (22 г, выход 88%) в виде твердого вещества бледно-желтого цвета.
1H ЯМР (ДМСО-d6 300 МГц) (δ) ммд: 0,72 (3H, д, J=6,6 Гц), 1,10 (3H, д, J=6,6 Гц), 1,28 (3H, т, J=7,0 Гц), 2,27 (1H, ушир.), 3,77 (1H, ушир.), 3,86 (1H, ушир.), 4,23 (2H, кв, J=7,0 Гц), 4,56 (1H, ушир.), 5,12 (1H, т, J=4,9 Гц), 8,09 (1H, д, J=11,1 Гц), 8,62 (1H, д, J=7,5 Гц), 8,68 (1H, с).
МС (ESI): M+ 448.
Стадия 4
Соединение (22 г, 48 ммоль), полученное на стадии 3, растворяли в диметилформамиде (60 мл), добавляли имидазол (3,9 г, 57,7 ммоль) и трет-бутилдиметилсилилхлорид (8,0 г, 53,0 ммоль) и смесь перемешивали при комнатной температуре в течение 1,5 часов. Реакционную смесь концентрировали при пониженном давлении и полученный остаток растворяли в этилацетате (200 мл), последовательно промывали водой (дважды) и насыщенным солевым раствором и сушили над сульфатом натрия. Смесь фильтровали и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на силикагеле (этилацетат:гексан = 3:7 до 4:6) с получением этилового эфира 1-((S)-1-трет-бутилдиметилсилилоксиметил-2-метилпропил)-7-фтор-6-иод-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты (25 г, выход 92%) в виде воскообразного вещества белого цвета.
1H ЯМР (CDCl3 400 МГц) (δ) ммд: -0,07 (3H, с), -0,05 (3H, с), 0,77 (9H, с), 0,84 (3H, д, J=6,5 Гц), 1,18 (3H, д, J=6,5 Гц), 1,40 (3H, т, J=7,2 Гц), 2,35-2,50 (1H, м), 3,85-3,95 (1H, м), 3,98-4,10 (2H, м), 4,30-4,40 (2H, м), 7,26 (1H, с), 8,64 (1H, с), 8,94 (1H, д, J=7,2 Гц).
МС (ESI): M+ 562.
Стадия 5
Соединение (25 г, 44 ммоль), полученное на стадии 4, растворяли в тетрагидрофуране (200 мл), и в токе аргона добавляли дибензилиденацетонпалладий(II) (1,0 г, 1,8 ммоль) и трифурилфосфин (824 мг, 3,5 ммоль). Раствор (58 мл, 58 ммоль) 1М 3-хлор-2-фторбензилцинкбромида, полученный таким же образом, как в примере 1 на стадии 5, в тетрагидрофуране добавляли при 60°С. После завершения прикапывания смесь нагревали с обратным холодильником в течение 3 часов. Реакционную смесь оставляли для охлаждения до комнатной температуры, добавляли этилацетат (200 мл), последовательно промывали 1 н. соляной кислотой, водой, насыщенным водным раствором гидрокарбоната натрия, водой и насыщенным солевым раствором и сушили над сульфатом магния. Смесь фильтровали и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на силикагеле (этилацетат:гексан = 4:6 до 1:1) с получением этилового эфира 1-((S)-1-трет-бутилдиметилсилилоксиметил-2-метилпропил)-6-(3-хлор-2-фторбензил)-7-фтор-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты (17 г, выход 68%) в виде маслообразного вещества бледно-желтого цвета.
1H ЯМР (CDCl3 400 МГц) (δ) ммд: -0,09 (3H, с), -0,05 (3H, с), 0,75 (9H, с), 0,85 (3H, д, J=6,7 Гц), 1,18 (3H, д, 6,7 Гц), 1,39 (3H, т, J=7,1 Гц), 2,45 (1H, ушир.), 3,89-3,92 (1H, м), 3,98-4,02 (1H, м), 4,07-4,12 (1H, м), 4,12 (2H, с), 4,34-4,41 (2H, м), 6,96-7,00 (1H, м), 7,03-7,05 (1H, м), 7,21-7,24 (1H, м), 7,26-7,29 (1H, м), 8,39 (1H, д, J=8,8 Гц), 8,63 (1H, с).
Стадия 6
Соединение (17 г, 30 ммоль), полученное на стадии 5, растворяли в метаноле (120 мл), добавляли 28% метанольный раствор метоксида натрия (62 мл, 304 ммоль) и смесь нагревали с обратным холодильником в течение 19 часов. Реакционную смесь оставляли для охлаждения до комнатной температуры, добавляли воду (200 мл), метанол упаривали при пониженном давлении. Остаток подкисляли добавлением концентрированной соляной кислоты, экстрагировали этилацетатом и сушили над сульфатом натрия. Смесь фильтровали и фильтрат концентрировали при пониженном давлении с получением неочищенного вещества (14 г) соединения А в виде масла бледно-желтого цвета.
Стадия 7
Соединение А (14,11 г), полученное на стадии 6, суспендировали в смеси растворителей этилацетат (20 мл) и гексан (20 мл) при комнатной температуре и добавляли кристалл-затравку (кристаллическая форма II соединения А) и смесь перемешивали 1 час. Суспензию фильтровали и оставшееся твердое вещество сушили в вакууме с получением соединения А (кристаллическая форма II 10,40 г, выход 77%) в виде твердого вещества белого цвета.
Пример 3: Получение кристаллической формы III соединения А
Пример 3-1: Получение кристаллической формы III соединения А
Кристаллическую форму II соединения А (10 г, 22,3 ммоль), полученную в примере 2-2, добавляли к изобутилацетату (30 мл), и кристаллическое вещество растворяли при нагревании с обратным холодильником. Раствор охлаждали до 90°С и перемешивали 5 часов, давая кристаллам выпасть в осадок. Этот раствор оставляли для дальнейшего охлаждения до комнатной температуры и осуществляли дальнейшее перемешивание в течение 12 часов. Выпавший кристаллический осадок отделяли фильтрованием. Полученный кристалл промывали изобутилацетатом (10 мл) и сушили в вакууме с получением кристалла белого цвета (9,85 г, выход 98,5%). После того, как было установлено методом рентгенодифракционного анализа, что этот кристалл отличается от кристаллической формы II, для кристалла был получен спектр рентгенодифракции (фиг.1), и этот кристалл стали считать как кристаллическая форма III.
Пример 3-2: Получение кристаллической формы III соединения А
Кристаллическую форму II соединения А (250 г, 558 ммоль), полученную в примере 2-2, добавляли к изобутилацетату (750 мл). Добавляли кристалл-затравку (12,5 г) кристаллической формы III соединения А, полученный в примере 3, при комнатной температуре, и смесь перемешивали в течение 17 часов. Выпавший кристаллический осадок отделяли фильтрованием. Полученный кристалл промывали изобутилацетатом (250 мл) и сушили в вакууме с получением целевого вещества (кристаллическая форма III, 259 г, выход 98,6%) в виде кристалла белого цвета. Было подтверждено с помощью рентгенодифракционного анализа, что этот кристалл эквивалентен стандартному образцу кристалла (кристаллическая форма III соединения А, полученная в примере 3-1).
Пример 3-3: Получение кристаллической формы III соединенияА
Соединение А (кристаллическая форма II, 10,0 г, 22,3 ммоль), полученное в примере 2-2, добавляли к изопропанолу (30 мл), и смесь нагревали с обратным холодильником для растворения кристалла. Раствор охлаждали до 70°С, добавляли кристалл-затравку (10 г) кристаллической формы III соединения А, полученный в примере 3-1, и смесь перемешивали 5 часов. Далее смесь оставляли для охлаждения до комнатной температуры, перемешивали 12 часов, и кристалл отделяли фильтрованием. Полученный кристалл промывали изопропанолом (10 мл), сушили в вакууме с получением целевого вещества (кристаллическая форма III, 9,72 г, выход 97,2%) в виде кристалла белого цвета. Было подтверждено с помощью рентгенодифракционного анализа, что этот кристалл эквивалентен стандартному образцу кристалла (кристаллическая форма III соединения А, полученная в примере 3-1).
Пример 3-4: Получение кристаллической формы III соединенияА
Кристаллическую форму II соединения А (7,00 г, 15,6 ммоль), полученная в примере 2-2, добавляли к смеси растворителей: этанол (52,5 мл) и вода (7 мл) и растворяли при нагревании. Добавляли воду (28 мл), добавляли при 70°С кристалл-затравку (10 мг) целевого вещества и смесь перемешивали 4 часа. Оставляли для охлаждения до комнатной температуры и после этого смесь охлаждали льдом и далее перемешивали в течение 2 часов и кристаллы отделяли фильтрованием. Полученные кристаллы промывали смесью растворителей: холодный этанол (8,4 мл) и вода (5,6 мл) и сушили в вакууме с получением целевого вещества в виде белых кристаллов (кристаллическая форма III, 6,77 г, выход 96,8%). Было подтверждено с помощью рентгенодифракционного анализа, что этот кристалл эквивалентен стандартному образцу (пример 3-1).
Экспериментальный пример
Характеристики свойств каждой кристаллической формы были определены нижеприведенными аналитическими тестами и для каждой кристаллической формы был осуществлен тест на стабильность с применением соответствующих показателей.
Образец
За исключением особо оговоренных случаев в качестве образцов использовались вышеупомянутые кристалл (кристаллическая форма I), полученный в справочном примере 1, кристалл (кристаллическая форма II), полученный в примере 1, и кристалл (кристаллическая форма III), полученный в примере 3-1.
Аналитический тест
1. Порошковая рентгенодифрактометрия
Этот тест имеет целью получение спектров рентгенодифракции для спецификации кристаллической формы кристаллов, полученных в справочном примере 1, примере 1 и примере 3-1. Дифракционные спектры применяются для спецификации кристаллической формы, для оценки стабильности, для определения чистоты и подобных показателей.
Образцы фиксировали в алюминиевой ячейке и измерения осуществляли с использованием рентгенодифрактометра (RINT 2000/PC Ultima+, производитель: Ridaku Corporation; источник рентгеновского излучения: Cu-Kб1 излучение; напряжение в лучевой трубке: 40 кВ; электрический ток в лучевой трубке: 40 мА; скорость сканирования: 5° в мин; минимальный шаг перемещения: 0,02°; угол дифракции: 5-40°), на основе которого были получены дифракционные спектры. Полученные дифракционные спектры представлены на фиг.1.
Как показано на фиг.1, спектры рентгенодифракции, полученные для соответствующих образцов, были различными.
Таким образом, было подтверждено, что кристаллы, полученные в справочном примере 1, примере 1 и примере 3-1, отличаются друг от друга и имеют характеристические дифракционные спектры, как это показано в спектрах рентгенодифракции. Таким образом, в настоящей спецификации они были названы как кристаллическая форма I, кристаллическая форма II и кристаллическая форма III, основываясь на их спектрах рентгенодифракции.
Для спецификации кристаллической формы может быть установлена полная характеристика дифракционного пика для каждого кристалла на основании рисунка дифракционного спектра на фиг.1.
Основные дифракционные пики и характеристические дифракционные пики, точно определенные из дифракционных спектров на фиг.1, представлены ниже.
[Кристаллическая форма I]
Основной дифракционный пик: 2θ=6,58, 14,40, 14,64, 15,24, 16,48, 19,16, 20,90, 21,14, 22,24, 24,74, 25,64, 26,12, 27,20°;
Характеристический дифракционный пик: 2θ=6,58, 14,40, 19,16, 20,90, 21,14°.
[Кристаллическая форма II]
Основной дифракционный пик: 2θ=6,56, 9,04, 13,20, 14,62, 15,24, 16,48, 19,86, 20,84, 21,22, 22,24, 25,22, 25,96, 26,12, 27,34°;
Характеристический дифракционный пик: 2θ=6,56, 13,20, 19,86, 20,84, 21,22, 25,22°.
[Кристаллическая форма III]
Основной дифракционный пик: 2θ=8,54, 14,02, 15,68, 15,90, 16,00, 17,06, 17,24, 17,84, 18,12, 19,50, 19,90, 22,26, 22,68, 23,02, 24,16, 24,76, 25,18, 25,74, 25,98, 27,50, 28,80, 30,38, 30,72, 32,54°;
Характеристический дифракционный пик: 2θ=8,54, 14,02, 15,68, 17,06, 17,24, 24,16, 25,74°.
2.Термический анализ
Этот тест имеет своей целью измерение энтальпии и экстраполированной начальной температуры эндотермического пика на кривой, полученной путем Дифференциальной Сканирующей Калориметрии (ДСК). Эти величины находятся в числе показателей стабильности вышеупомянутых кристаллической формы I, кристаллической формы II и кристаллической формы III и могут использоваться как числовой показатель для спецификации кристаллической формы.
2.1. Энтальпия и экстраполированная начальная температура кристаллической формы I и кристаллической формы II
Кристаллическая форма I и кристаллическая форма II были подвергнуты измерениям с использованием измерительной аппаратуры Дифференциальной Сканирующей Калориметрии (ДСК) (DSC8240, производитель: Rigaku Corporation), в газообразной атмосферной среде, измеряемый образец 5±1 мг, скорость роста температуры: 10°С/мин, алюминиевая открытая кювета и оксид алюминия в качестве эталона. Были определены энтальпия и экстраполированная начальная температура эндотермического пика на кривой, полученной путем ДСК.
2.2. Энтальпия и экстраполированная начальная температура кристаллической формы III
Кристаллическая форма III была подвергнута измерениям с использованием измерительной аппаратуры ДСК (DSC8240, производитель: Rigaku Corporation), в газообразной атмосферной среде, измеряемый образец 5,0±0,5 мг, скорость роста температуры: 5°С/мин, алюминиевая закрытая кювета и оксид алюминия в качестве эталона. Были определены энтальпия и экстраполированная начальная температура эндотермического пика на кривой, полученной ДСК.
Результаты представлены в таблице 1.
ДСК кривая эндотермического пика с указанием энтальпии и экстраполированной начальной температуры
Как представлено в таблице 1, кристаллическая форма III показывает наибольшую энтальпию и наивысшее значение экстраполированной начальной температуры среди трех кристаллических форм. Таким образом, было подтверждено, что кристаллическая форма III является наиболее стабильной формой.
3. Тест на чистоту
Этот тест имеет своей целью измерение чистоты соединения А. Чистоту можно использовать в качестве показателя химической стабильности.
3.1. Чистота соединения кристаллической формы I и кристаллической формы II
Каждый образец (кристаллическая форма I и кристаллическая форма II, приблизительно 10 мг) растворяли в ацетонитриле, доводя объем раствора до 10 мл, и использовали его в качестве раствора образца. Этот раствор применяли для высокоэффективной жидкостной хроматографии (ВЭЖХ) при нижеприведенных условиях. Площадь пика для раствора каждого образца измеряли автоматическим интегратором, и чистоту определяли по нижеприведенной формуле. Показатели чистоты указаны в таблицах 5 и 6, приведенных ниже.
Чистота (%) = 100 - (Asum/As) × 100
As - общая площадь пиков, полученных для раствора образца;
Asum - общая площадь пиков, отличных от главного пика, полученного для раствора образца.
Условия тестирования
Детектор: УФ - измеритель абсорбции (длина волны: 259 нм)
Колонка: CAPCELL PAK MG (внутренний диаметр 4,6 см, длина 15 см, размер частиц 5 мкм, производитель: Shiseido Co., Ltd.)
Температура колонки: постоянная температура около 40°С
Подвижная фаза А: раствор трифторуксусной кислоты (1:1000)
Подвижная фаза В: раствор трифторуксусной кислоты в ацетонитриле (1:1000)
Градиентная программа: Как представлено в нижеприведенной таблице 2, соотношение смешиваемых подвижной фазы А и подвижной фазы В изменяется для создания градиента концентраций.
3.2. Чистота соединения кристаллической формы III
Образец (кристаллическая форма III, приблизительно 50 мг) растворяли в смеси (4:1) подвижной фазы В и подвижной фазы А с получением раствора общим объемом 50 мл, который использовали в качестве раствора образца. Из этого раствора был точно отобран (1 мл) и к нему добавляли смесь (4:1) подвижной фазы В и подвижной фазы А с получением раствора с точным объемом 100 мл, который использовали в качестве стандартного раствора. Раствор образца и стандартный раствор (15 мкл) применяли для высокоэффективной жидкостной хроматографии (ВЭЖХ) при нижеприведенных условиях. Площадь пика каждого раствора измеряли автоматическим интегратором, и чистоту определяли по нижеприведенной формуле. Показатели чистоты представлены в нижеприведенной таблице 7.
Чистота (%) = 100 - (Asum/As) × 100
As - общая площадь пиков, полученных для раствора образца;
Asum - общая площадь пиков, отличных от главного пика, полученного для раствора образца.
Условия тестирования
Детектор: УФ - измеритель абсорбции (длина волны: 259 нм)
Колонка: Waters XTerra MC C18 (внутренний диаметр 4,6 см, длина 5 см, диаметр частиц 2,5 мкм, производитель: Waters)
Температура колонки: постоянная температура около 40°С
Подвижная фаза А: фосфорную кислоту добавляли к раствору гидрофосфата калия (1 → 1149) до достижения рН 7,0
Подвижная фаза В: ацетонитрил
Градиентная программа: Как представлено в нижеприведенной таблице 3, соотношение смешиваемых подвижной фазы А и подвижной фазы В изменяется для создания градиента концентраций.
4. Тест на растворимость
Этот тест имеет своей целью определение растворимости кристалла в различных тестируемых растворах при различных значениях рН. Растворимость является одним из показателей стабильности вышеупомянутых кристаллической формы I, кристаллической формы II и кристаллической формы III и может также применяться в качестве эталонного показателя способности кристалла к абсорбируемости в живых организмах.
Каждый образец (кристаллическая форма I тип I, кристаллическая форма II и кристаллическая форма III, приблизительно 10 мг) помещали в центрифужную пробирку объемом 10 мл вместе с нижеприведенным тестовым раствором (5 мл) и встряхивали с помощью шейкера (SR-1M; производитель: Tietech Co., Ltd.) в течение 14 часов. После встряхивания смесь центрифугировали (3000 об/мин, 20 мин) и супернатант фильтровали через политетрафторэтиленовый дисковый фильтр диаметром 13 мм и с размером пор 0,2 мкм (Millex-LG; производитель Millipore Corporation). Измерения проводили с помощью высокоэффективной жидкостной хроматографии (ВЭЖХ). Результаты представлены в таблице 4.
Тест на растворимость
2)Фармакопея Японии, Общий Метод Тестирования, Дезинтеграционный Тестовый Метод, 2-я жидкая среда для растворения. Образец раствора с концентрацией гидроксида натрия 0,2 моль/л (118 мл) и воду добавляли к образцу раствора с концентрацией калийдинатрий фосфата 0,2 моль/л (250 мл) до получения общего объема раствора 1000 мл. Этот раствор прозрачный и неокрашенный, и имеет рН около 6,8.
3)McIlvaine буферный раствор получали смешиванием гидрофосфата натрия и лимонной кислоты в определенном соотношении, чтобы достичь заданного значения рН.
Исходя из вышеупомянутых результатов, было установлено, что кристаллическая форма II имеет более высокую растворимость, чем кристаллическая форма III.
5. Тест на стабильность
Тест на стабильность для каждого образца осуществляли при нижеследующих консервирующих условиях. Результаты для кристаллической формы I представлены в таблице 5, результаты для кристаллической формы II представлены в таблице 6 и результаты для кристаллической формы III представлены в таблице 7.
Как показано в таблице 6 и таблице 7, для кристаллической формы II и кристаллической формы III не выявлено каких-либо отличий по результатам тестирования во всех консервирующих условиях в сравнении с начальным образцом. В противоположность этому, как показано в таблице 5, кристаллическая форма I проявляет изменения в спектре рентгенодифракции, полученном для образца после хранения при консервирующих условиях #3 (80°С, хранение в открытом контейнере 3 дня) и при консервирующих условиях #5 (60°С, хранение в открытом контейнере 3 недели), и спектр рентгенодифракции кристаллической формы I частично совпадал со спектром рентгенодифракции, полученном для кристаллической формы II. Таким образом, определили, что часть образца претерпела преобразование кристалла в кристаллическую форму II в процессе хранения. Спектры рентгенодифракции образцов, хранящихся при консервирующих условиях #1-6, для кристаллической формы I представлены на фиг.2.
Результаты теста на стабильность для кристаллической формы I
Результаты теста на стабильность для кристаллической формы II
Результаты теста на стабильность для кристаллической формы III
По результатам вышеупомянутого теста на стабильность было отмечено, что кристаллическая форма I была нестабильной, но кристаллическая форма II и кристаллическая форма III были очень стабильными при различных условиях хранения. Поэтому стало очевидным, что кристаллическая форма II и кристаллическая форма III являются предпочтительными для использования в качестве фармацевтического вещества или ему подобного.
Что касается способности к абсорбции в живых организмах, кристаллическая форма II является более предпочтительной, и кристаллическая форма III более предпочтительна, потому что она является наиболее стабильным кристаллом.
Поскольку кристаллическая форма II и кристаллическая форма III обе стабильны, их смешанный кристалл может использоваться для настоящего изобретения.
Экспериментальный пример
Нижеследующее объясняет определение способов проявления активности в ингибировании ВИЧ интегразы кристаллом или смешанным кристаллом соединения А настоящего изобретения.
(i) Конструирование содержащей интегразу рекомбинантной генетической экспрессирующейся системы
В полноразмерном первичном гене ВИЧ интегразы генетический код 185-го фенилаланина (J. Virol., 67, 425-437 (1993)) был замещен на генетический код гистидина, и полученный ген встраивали в сайты действия рестрикционных ферментов NdeI и XhoI плазмиды pET21a(+) (производитель: Novagen), в результате чего был сконструирован интегразный экспрессирующийся вектор pET21a-IN-F185H.
(ii) Получение и очистка белка интегразы
Рекомбинантная Escherichia coli BL21(DE3), трансформированная с помощью плазмиды pET21a-IN-F185H, полученной в (i), культивировалась в режиме встряхивания при 30°С в жидкой среде, содержащей ампициллин. Когда культура достигала фазу логарифмического роста, к ней добавляли изопропил-β-D-тиогалактопиранозид для промотирования экспрессии гена интегразы. Культивирование продолжали в течение 3 часов для промотирования накопления белка интегразы. Рекомбинантную E. coli отделяли в виде осадка в центрифужной пробирке при разделении центрифугированием и хранили при -80°С.
E. coli суспендировали в лизирующем буфере (20 мМ HEPES (pH 7,5), 5 мМ DTT, 10 мМ CHAPS, 10% глицерина), содержащем 1М хлорида натрия, и подвергали повторяющемуся воздействию давлением и его сбросом с целью разрушения клеток и разделяли центрифугированием при 4°С, 40 000 × g, 60 мин для извлечения водорастворимой фракции (супернатанта). Он разбавлялся в 10 раз лизирующем буфером без хлорида натрия, смешивался с SP-Сефарозой (производитель: Pharmacia Corporation) и перемешивался при 4°С 60 мин для обеспечения абсорбции белка интегразы в полимер. Полимер промывали лизирующим буфером, содержащим 100 мМ хлорида натрия, и белок интегразы элюировали лизирующим буфером, содержащим 1М хлорида натрия.
Элюированный раствор белка интегразы наносили на колонку с Superdex 75 (Pharmacia Corporation) для проведения гель-фильтрации. Белок элюировали лизирующим буфером, содержащим 1М хлорида натрия.
Полученные фракции белка интегразы собирали и хранили при -80°С.
(iii) Приготовление раствора ДНК
Нижеприведенную ДНК, синтезированную Greiner, растворяли в TE буфере (10 мМ Трис-гидрохлорида (рН 8,0), 1 мМ EDTA) и смешивали с донорской ДНК, ДНК-мишенью и их комплементарными цепями (+ или - цепи) до 1 мкМ. Смесь нагревали при 95°С 5 мин, при 80°С 10 мин, при 70°С 10 мин, при 60°С 10 мин, при 50°С 10 мин, при 40°С 10 мин и хранили при 25°С с получением двухцепочечной ДНК, которую использовали для теста.
Донорская ДНК ( - цепь содержит биотин, присоединенный к 5′нцу)
Донорская + цепь: 5'-Биотин-ACC CTT TTA GTC AGT GTG GAA AAT CTC TAG CA-3' (SEQ ID NO:1)
Донорская - цепь: 5'-ACT GCT AGA GAT TTT CCA CAC TGA CTA AAA G-3' (SEQ ID NO:2)
ДНК-мишень (и +, и - цепи содержат дигоксигенин, присоединенный к 3′нцу)
мишень + цепь: 5'-TGA CCA AGG GCT AAT TCA CT-Dig-3' (SEQ ID NO:3)
мишень - цепь: 5′GT GAA TTA GCC CTT GGT CA-Dig-3′EQ ID NO:4)
(iv) Определение активности ингибирования фермента (ВИЧинтегразы)
Донорскую ДНК разбавляли ТЕ буфером до концентрации 10 нМ, по 50 мкл этого раствора добавляли в каждую ячейку с пластиной микротитратора, поверхность которой покрыта стрептавидином (производитель: Roche), оставляли для адсорбции при 37°С на 60 мин. ДНК затем промывали фосфатным буфером (Dulbecco PBS, Sanko Junyaku Co., Ltd.), содержащим 0,1% Твина 20, и фосфатным буфером. Затем реакционную смесь (70 мкл, состав* см. ниже), тестируемое вещество (10 мкл), разбавленное реакционной смесью, и 100 мкг/мл белка интегразы (10 мкл) добавляли в каждую ячейку и проводили реакцию при 37°С в течение 60 мин.
Затем добавляли 50 нМ ДНК-мишени (10 мкл), проводили реакцию при 37°С в течение 10 мин и промывали фосфатным буфером, содержащим 0,1% Твина 20, чтобы остановить реакцию.
Затем добавляли раствор 100 мЕ/мл пероксидазы, меченной анти-дигоксигениновыми антителами (производитель: Roche, 100 мкл), и для смеси проводили реакцию при 37°С в течение 60 мин, которую завершали промывкой фосфатным буфером, содержащим 0,1% Твина 20.
Добавляли окрашенный раствор пероксидазы (производитель: Bio Rad, 100 мкл), и реакция проходила при комнатной температуре 4 мин. Цветную реакцию останавливали добавлением 1 н. серной кислоты (100 мкл). Измеряли абсорбцию при 450 нм.
Активность ингибирования ВИЧ интегразы (IC50) соединением А настоящего изобретения рассчитывали по коэффициенту ингибирования в соответствии с нижеприведенной формулой. Результаты представлены в таблице 8.
Коэффициент ингибирования (%) = [1 - (наблюдаемый опыт - холостой опыт)/(контрольный опыт - холостой опыт)] × 100
Наблюдаемый опыт: абсорбция ячейки в присутствии тестируемого соединения
Контрольный опыт: абсорбция ячейки в отсутствии тестируемого соединения
Холостой опыт: абсорбция ячейки в отсутствии тестируемого соединения, в отсутствии белка интегразы
*Состав реакционной смеси: 30 мМ морфолинпропансульфоновая кислота (MOPS), 5 мМ MgCl2, 3 мМ дитиотреитол (DTT), 0,1 мг/мл бычий сывороточный альбумин (BSA), 5% глицерин, 10% диметилсульфоксид (DMSO), 0,01% Твин 20.
Определение антивирусной активности
Эффект совместного применения кристалла или смешанного кристалла соединения А настоящего изобретения и имеющихся в наличии анти-ВИЧ средств может быть установлен следующим образом.
Например, эффект совместного применения двух средств из существующих нуклеозидных ингибиторов обратной транскриптазы (зидовудин, ламивудин, тенофовир), ингибиторов обратной транскриптазы ненуклеозидной природы (эфавиренз) или ингибиторов протеазы (индинавир, нелфинавир) и кристалла или смешанного кристалла соединения А или ему подобного определяется методом XTT с использованием CEM-SS клеток, инфицированных ВИЧ-1 IIIB.
Кроме того, определяли эффект совместного применения трех средств: кристалла или смешанного кристалла соединения А, зидовудина и ламивудина; или кристалла или смешанного кристалла соединения А, тенофовира и ламивудина и им подобных.
До тестирования совместного применения были измерены для каждого фармацевтического средства в отдельности IC50 и CC50. 5 Концентраций фармацевтического средства а и 9 концентраций фармацевтического средства b, определенные на основе этих результатов, комбинировали для определения эффекта совместного применения двух средств. Для совместного применения трех средств смешивали высококонцентрированные фармацевтическое средство b и фармацевтическое средство с и прибавляли фармацевтическое средство а в определенных концентрациях для определения эффекта.
Результаты тестирования кристалла и смешанного кристалла соединения А и комбинированного лекарства самого по себе или в их комбинации анализировали, основываясь на программах Prichard and Shipman MacSynergy II version 2.01 и Deltagraph version 1.5d.
Строили трехмерную диаграмму, изображающую % ингибирования при различных концентрациях каждого комбинируемого фармацевтического средства, полученного при тестировании три раза с доверительными пределами 95% (или 68%, 99%), и эффект совместного применения определялся на основании рассчитанных численных значений мкМ2%. Критерии определения представлены ниже:
ВОЗМОЖНОСТЬ ПРОМЫШЛЕННОГО ПРИМЕНЕНИЯ
Кристалл соединения А настоящего изобретения, который имеет вышеупомянутую специфическую кристаллическую форму, проявляет анти-ВИЧ действие настолько хорошо, насколько высока его кристаллическая стабильность. Поэтому он пригоден к использованию в качестве вещества для фармацевтической композиции, особенно фармацевтических композиций для профилактики и/или лечения СПИДа.
Это применение основывается на Японской заявке на патент № 2004-150979, содержание которой в этом документе включено в качестве ссылки. Все помещенные в этом документе ссылки, включая патенты, заявки на патент и публикации, таким образом, включены в полном объеме.


| название | год | авторы | номер документа |
|---|---|---|---|
| ВОДОРАСТВОРИМОЕ СОЕДИНЕНИЕ БЕНЗОАЗЕПИНА И СОДЕРЖАЩАЯ ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2006 |
|
RU2418804C2 |
| ТРИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ БЕНЗОПИРАНА В КАЧЕСТВЕ ПРОТИВОАРИТМИЧЕСКИХ АГЕНТОВ | 2005 |
|
RU2380370C2 |
| 3'-ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ НУКЛЕОЗИДА, СПОСОБ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ), ЛЕКАРСТВЕННОЕ СРЕДСТВО, СПОСОБ ЛЕЧЕНИЯ И ПРОФИЛАКТИКИ | 1995 |
|
RU2130029C1 |
| ПРОИЗВОДНОЕ 3'-ЭТИНИЛЦИТИДИНА | 2007 |
|
RU2441876C2 |
| ЗАМЕЩЕННЫЕ ГУАНИДИНОВЫЕ СОЕДИНЕНИЯ | 2017 |
|
RU2757279C2 |
| НОВОЕ ПРОИЗВОДНОЕ 3-АЗАБИЦИКЛО[3.1.0]ГЕКСАНА И ЕГО ПРИМЕНЕНИЕ В МЕДИЦИНСКИХ ЦЕЛЯХ | 2014 |
|
RU2701861C1 |
| ПРОЛЕКАРСТВО ПРОИЗВОДНОГО АМИНОКИСЛОТЫ | 2017 |
|
RU2739318C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, МОДЕЛИРУЮЩИЕ АКТИВНОСТЬ ХЕМОКИНОВОГО РЕЦЕПТОРА, ИХ ПРИМЕНЕНИЕ И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2001 |
|
RU2297413C2 |
| СОЛЬ ПРОИЗВОДНОГО ПИРАЗОЛОХИНОЛИНА И ЕЕ КРИСТАЛЛ | 2014 |
|
RU2655171C2 |
| ИНГИБИТОРЫ РЕПЛИКАЦИИ ВИЧ | 2011 |
|
RU2564445C2 |
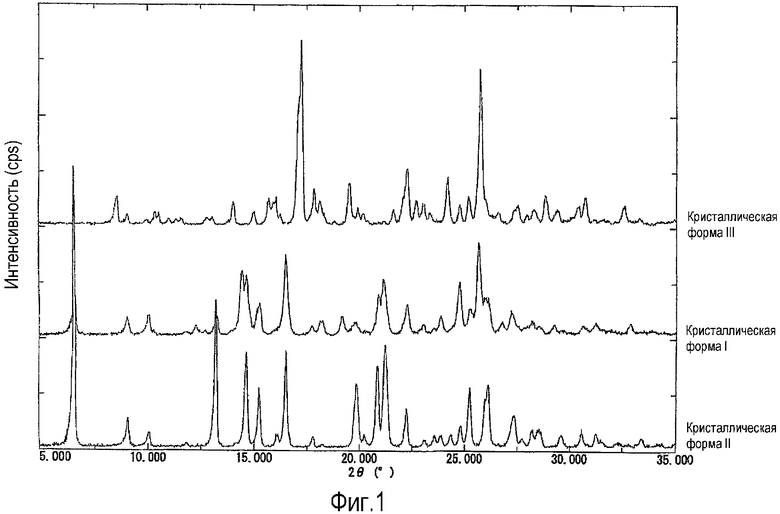
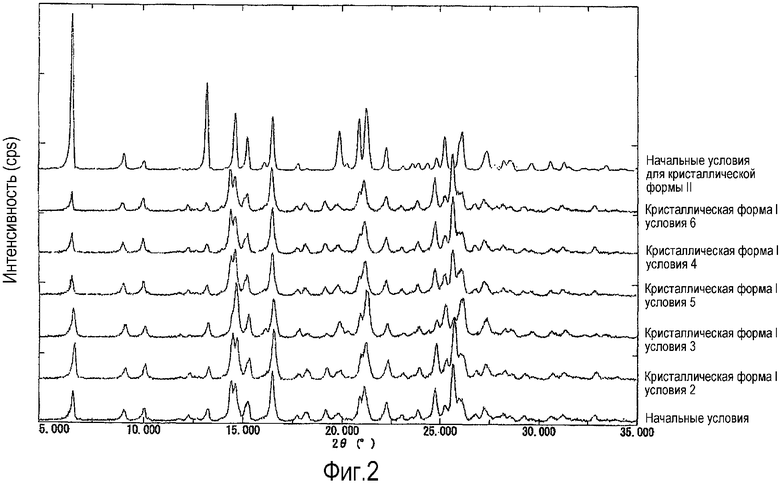
Настоящее изобретение предоставляет кристалл 6-(3-хлор-2-фторбензил)-1-[(S)-1-гидроксиметил-2-метилпропил]-7-метокси-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты, который имеет специфический рентгенодифракционный спектр с характеристическими дифракционными пиками при углах дифракции 2θ(°), что измерено на рентгенодифрактометре. Также описываются фармацевтическая композиция, обладающая активностью как ингибитор интегразы, ингибитор интегразы, антивирусное средство и анти-ВИЧ средство. Технический результат - кристалл настоящего изобретения является превосходным по физической и химической стабильности. 9 н. и 1 з.п. ф-лы, 8 табл., 2 ил.
| Дорожная спиртовая кухня | 1918 |
|
SU98A1 |
| US 3472859 A1, 14.10.1969 | |||
| СПОСОБ ПОЛУЧЕНИЯ ЭТИЛОВОГО ЭФИРА 6-ФТОР-7-ХЛОР-1,4-ДИГИДРО-4-ОКСО-3-ХИНОЛИНКАРБОНОВОЙ КИСЛОТЫ | 2002 |
|
RU2206564C1 |
| ПРОИЗВОДНЫЕ ХИНОЛИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ), ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЛЕЧЕНИЯ С ИХ ИСПОЛЬЗОВАНИЕМ | 1999 |
|
RU2213733C2 |

