Настоящее изобретение относится к 2,3,4-замещенным 5,6,7,8-тетрагидро[1]бензотиено[2,3-b]пиридиновым соединениям и их фармацевтически приемлемым солям и к их применению в качестве ингибиторов репликации вируса иммунодефицита человека (ВИЧ). Соединения по настоящему изобретению полезны для прямого или опосредованного ингибирования активности одного или более белков ВИЧ и для лечения заболеваний или состояний, опосредованных ВИЧ, таких как, например, синдром приобретенного иммунодефицита (СПИД). Без всякой связи с какой-либо конкретной теорией, полагают, что соединения по настоящему изобретению ингибируют репликацию ВИЧ в результате прямого или опосредованного ингибирования взаимодействия между ферментом ВИЧ-интеграза и эндогенным фактором роста из эпителия хрусталика (LEDGF). Обсуждение этого механизма как возможной мишени для терапии ВИЧ смотри в Llano, M etal. Science, 314, 461-464 (2006).
В WO 2010/130842 раскрыты производные тиено[2,3-b]пиридина, обладающие противовирусной активностью, более конкретно свойствами ингибирования репликации ВИЧ.
Несмотря на то, что в данной области уже осуществлено большое количество исследований, все еще существует острая потребность в сильнодействующих ингибиторах ВИЧ в данной области. Поэтому задачей настоящего изобретения является удовлетворение этой насущной потребности путем идентификации эффективных фармацевтически активных ингредиентов, которые обладают активностью против ВИЧ, являются менее токсичными, более стабильными (т.е. химически стабильными и метаболически стабильными), эффективными против вирусов, резистентных к доступным в настоящее время лекарственным средствам, и/или которые более устойчивы к мутациям вируса, чем существующие противовирусные лекарственные средства, и которые полезны, либо сами по себе, либо в комбинации с другими активными ингредиентами, для лечения ретровирусных инфекций, в частности лентивирусных инфекций, и, более конкретно, ВИЧ-инфекций, у млекопитающих и, более конкретно, у людей. Специалистам в данной области известно также, что физико-химические свойства известных лекарственных средств, а также их ADME-Tox (введение, распределение, метаболизм, выведение и токсикология) свойств могут ограничивать или не допускать их применение в лечении заболеваний. Следовательно, проблемы с существующими лекарственными средствами, которые предпочтительно должны быть преодолены, могут быть выбраны из плохих или неадекватных физико-химических или ADME-Tox свойств, таких как растворимость, LogP, ингибирование CYP (цитохром P450), печеночная стабильность и стабильность в плазме. В частности, преимуществом будет создание соединения с фармакокинетическими (PK) свойствами, которые делают его пригодным для введения дозы один раз в сутки, т.е. обеспечивающего правильные характеристики баланса всасывания, метаболизма и выведения для достижения профиля воздействия, согласующегося с введением дозы один раз в сутки. Задача настоящего изобретения также состоит в том, чтобы дополнить существующие противовирусные лекарственные средства таким образом, чтобы результирующая комбинация лекарственных средств имела более высокую активность или более высокую устойчивость к мутации вируса, чем каждое из индивидуальных соединений. Опять же, преимуществом будет то, что такая комбинация будет обеспечивать режим, подходящий для введения дозы один раз в сутки.
В первом аспекте настоящего изобретения предложено соединение, выбранное из
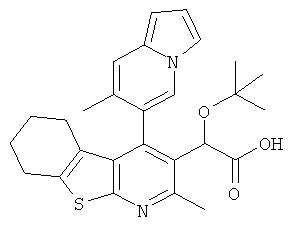 ,
, 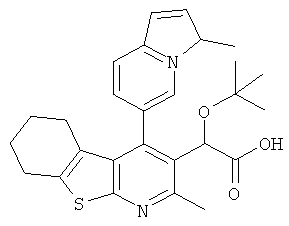 ,
, 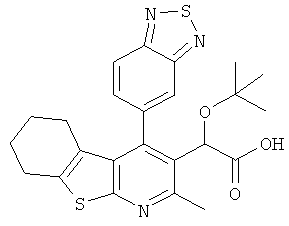 ,
, 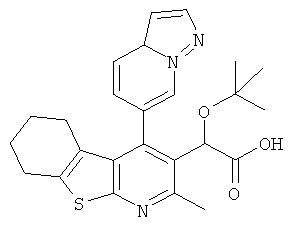
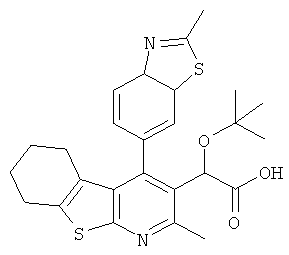 ,
, 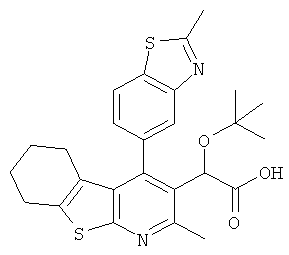 ,
, 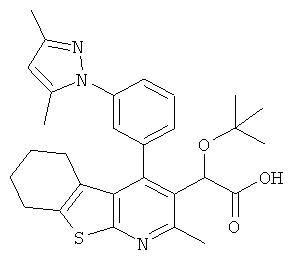 ,
, 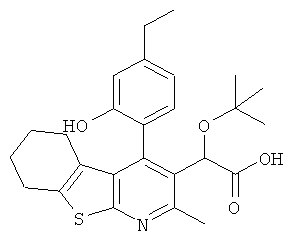 ,
,
 ,
, 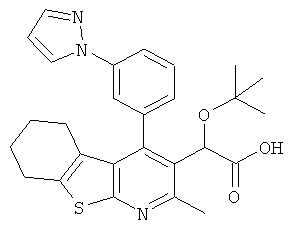 ,
, 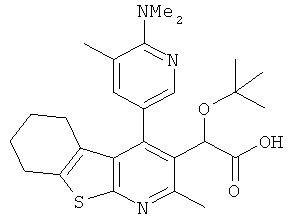 ,
, 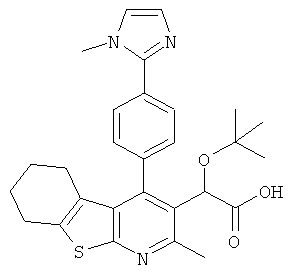 ,
,
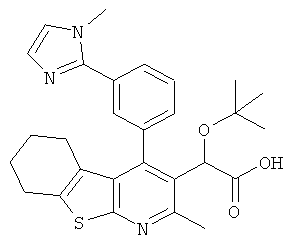 ,
, 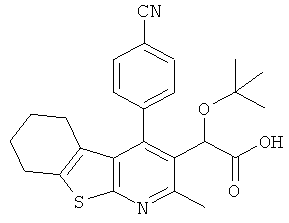 ,
, 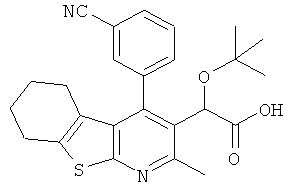 ,
, 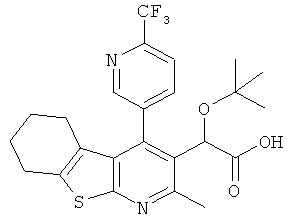 ,
,
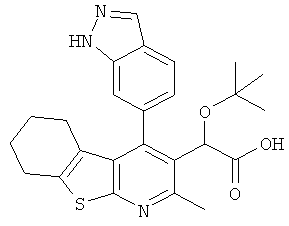 ,
, 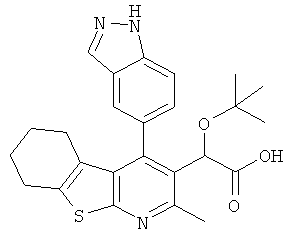 ,
, 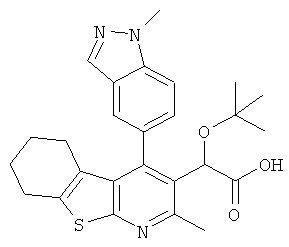 ,
, 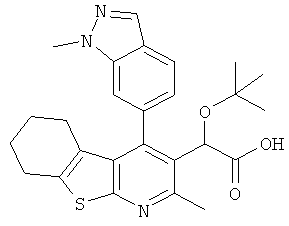 ,
,
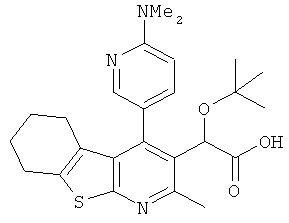 ,
, 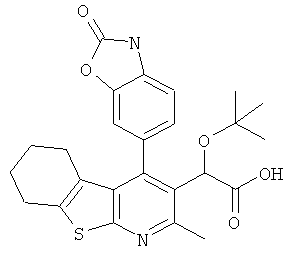 ,
, 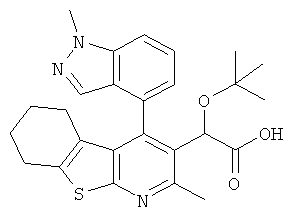 ,
, 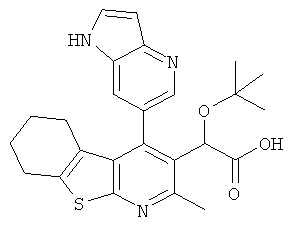 ,
,
 ,
, 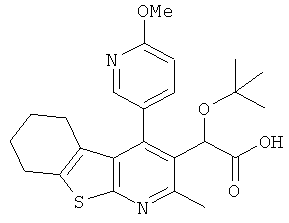 ,
,  ,
,  ,
,
 ,
,  ,
, 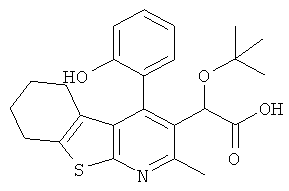 ,
, 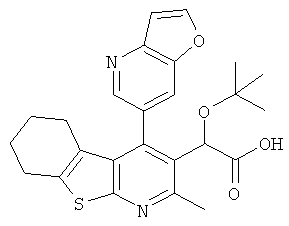 ,
,
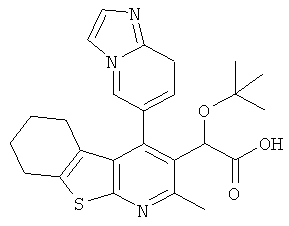 ,
, 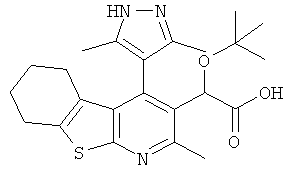 ,
, 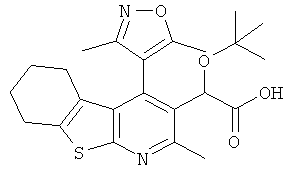 ,
, 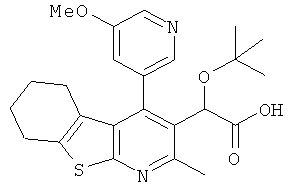 ,
,
 ,
, 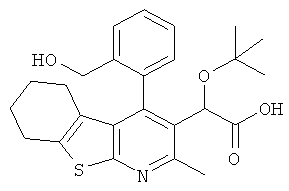 ,
, 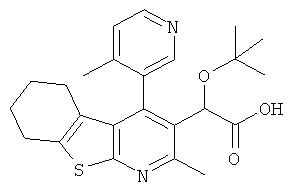 ,
, 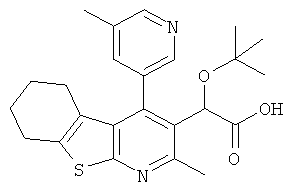 ,
,
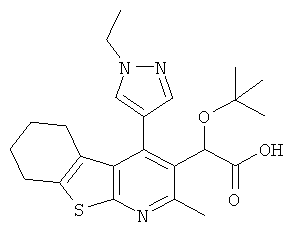 ,
, 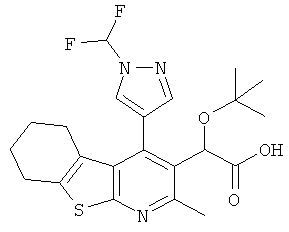 ,
, 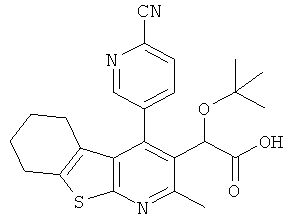 ,
, 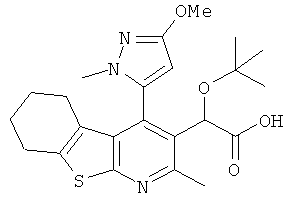 ,
,
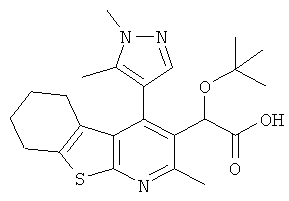 ,
, 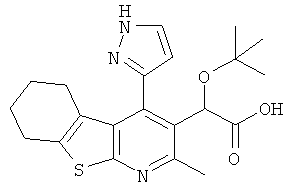 ,
, 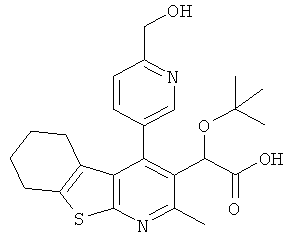 ,
,  ,
,
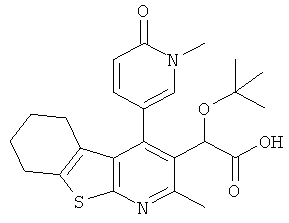 ,
, 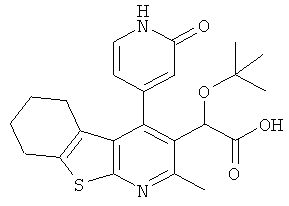 ,
, 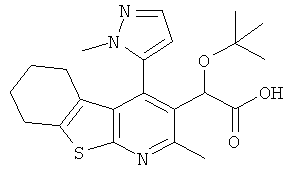 ,
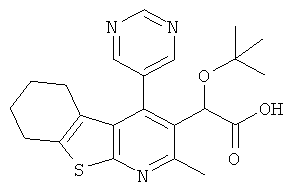
, 
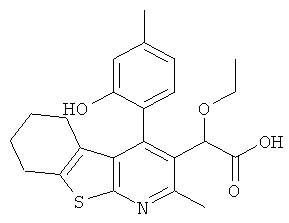 ,
, 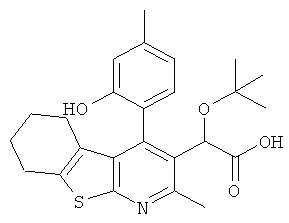 ,
, 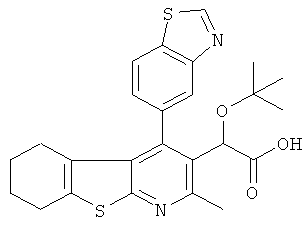 ,
, 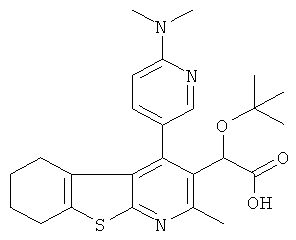 ,
,
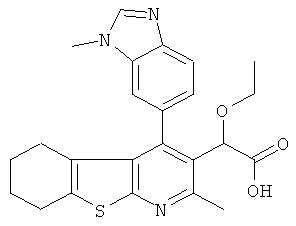 ,
, 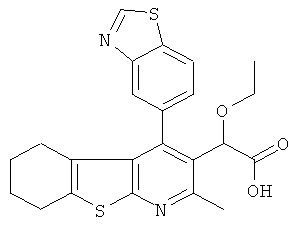 ,
, 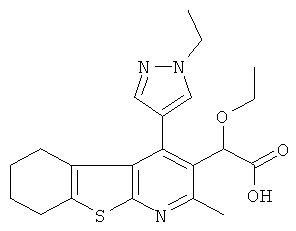 ,
, 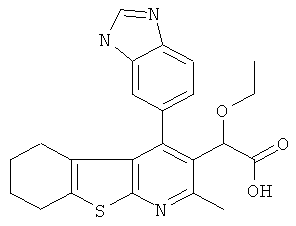 ,
,
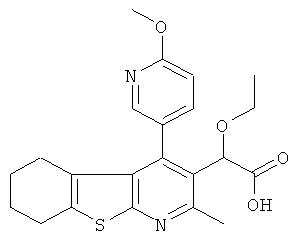 ,
, 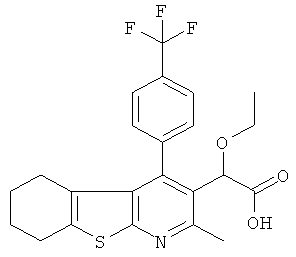 ,
, 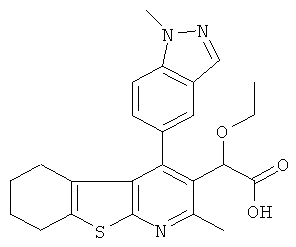 ,
, 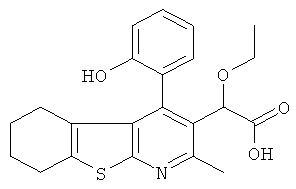 ,
,
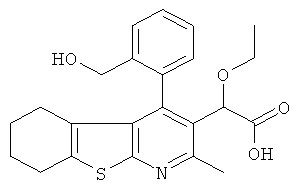 ,
, 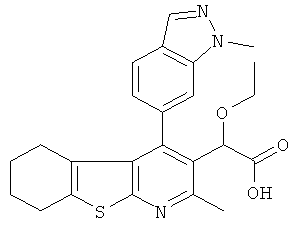 ,
, 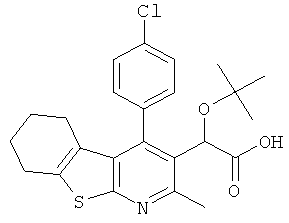 ,
, 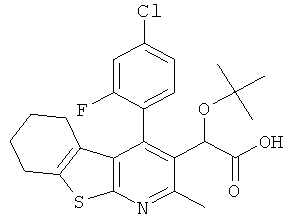 ,
,
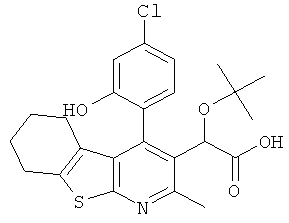 ,
, 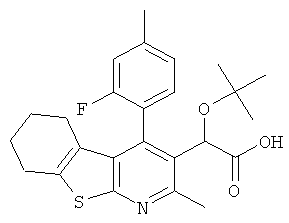 ,
, 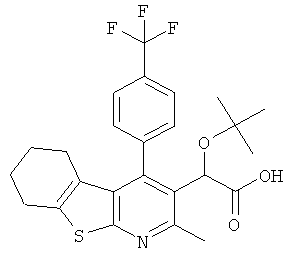 ,
, 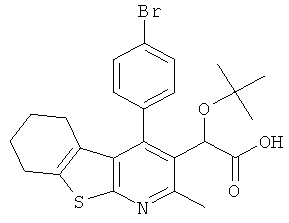 ,
,
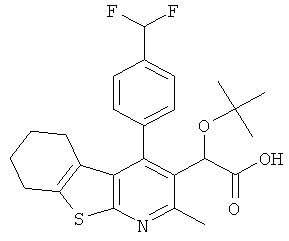 и
и 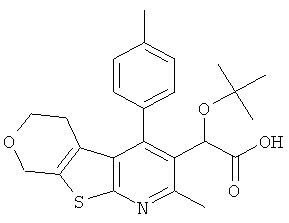
или его фармацевтически приемлемая соль.
В другом воплощении первого аспекта настоящего изобретения предложено соединение, выбранное из
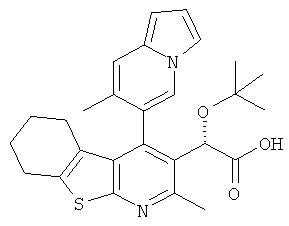 ,
, 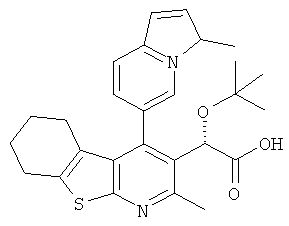 ,
, 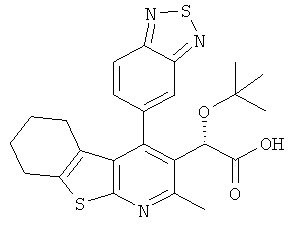 ,
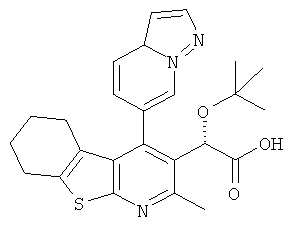
, 
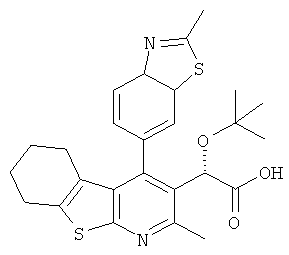 ,
,  ,
, 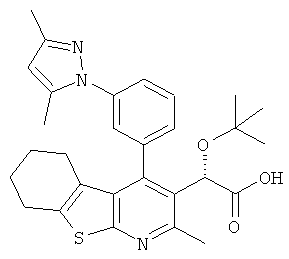 ,
, 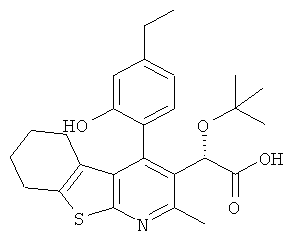 ,
,
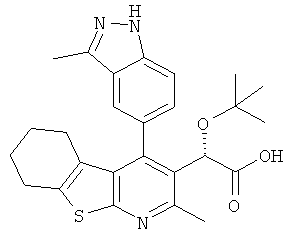 ,
, 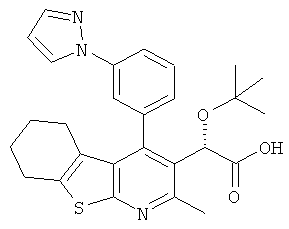 ,
,  ,
, 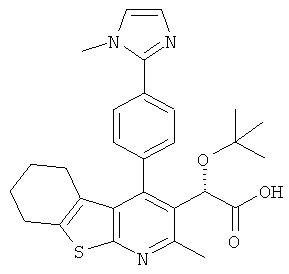 ,
,
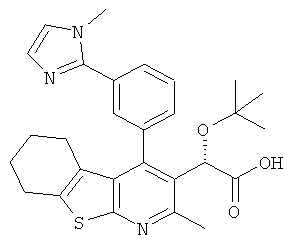 ,
, 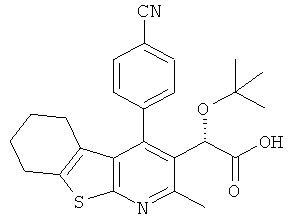 ,
, 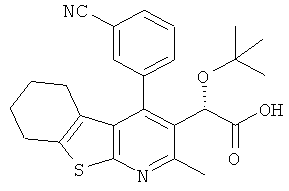 ,
, 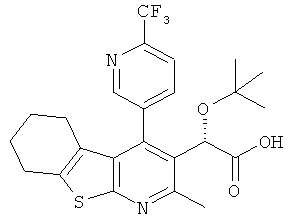 ,
,
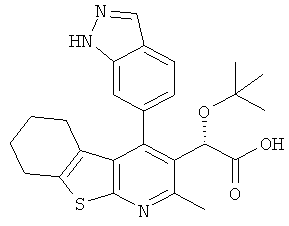 ,
, 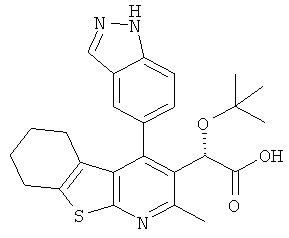 ,
, 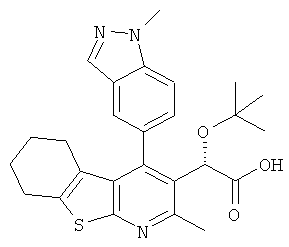 ,
, 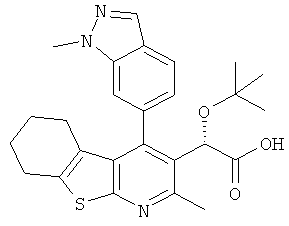 ,
,
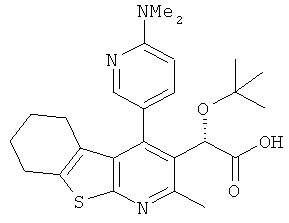 ,
, 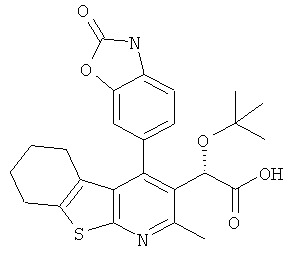 ,
, 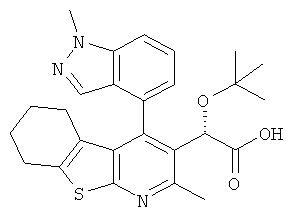 ,
, 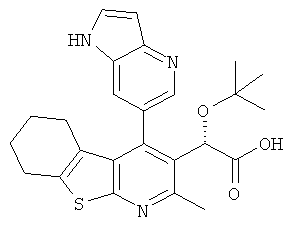 ,
,
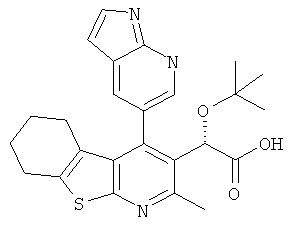 ,
, 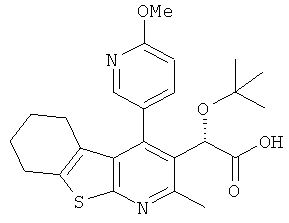 ,
, 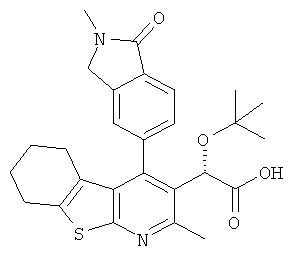 ,
, 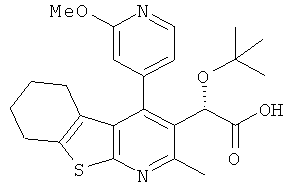 ,
,
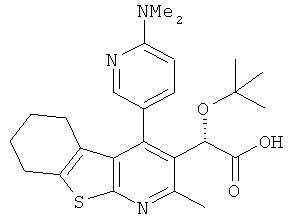 ,
, 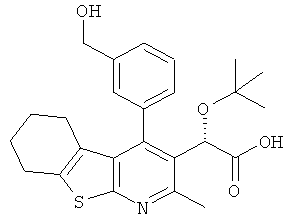 ,
, 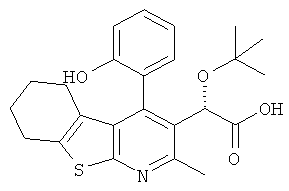 ,
, 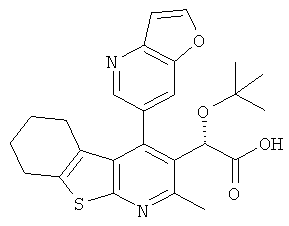 ,
,
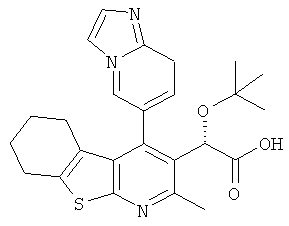 ,
, 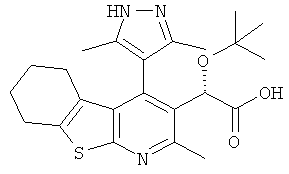 ,
, 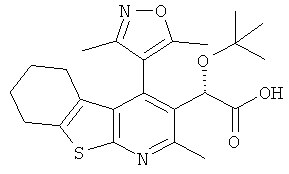 ,
, 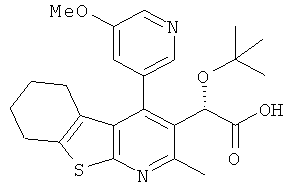 ,
,
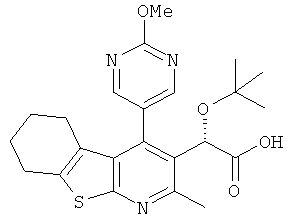 ,
, 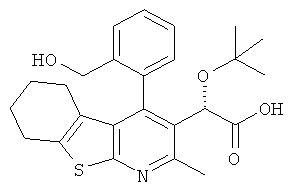 ,
, 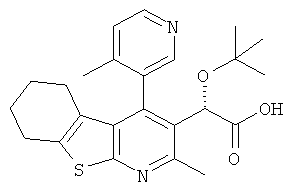 ,
, 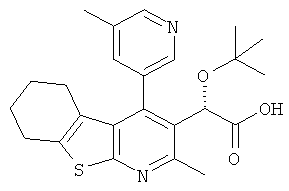 ,
,
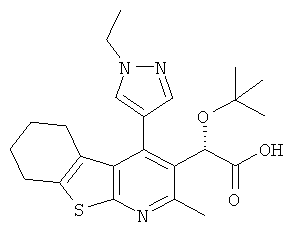 ,
, 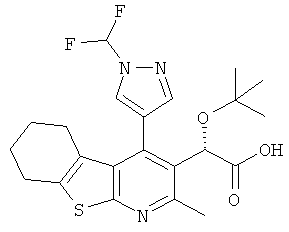 ,
, 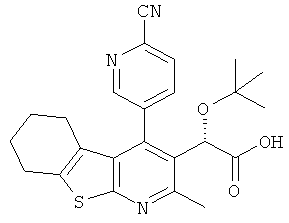 ,
, 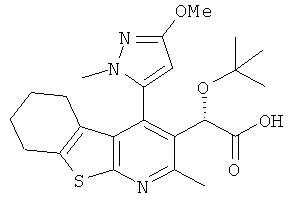 ,
,
 ,
, 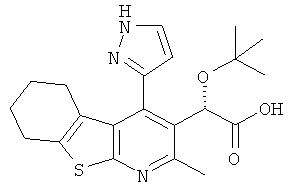 ,
, 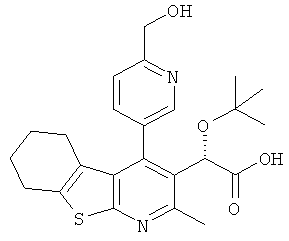 ,
, 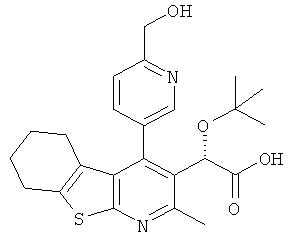 ,
,
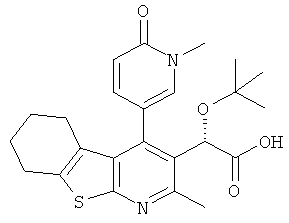 ,
,  ,
,  ,
, 
 ,
, 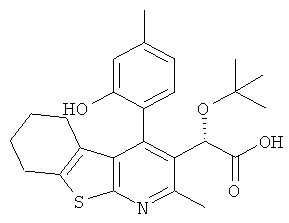 ,
, 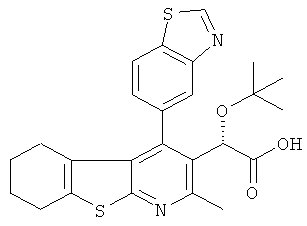 ,
, 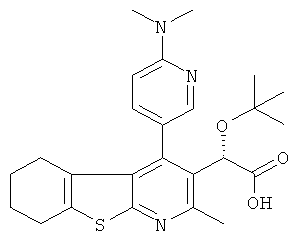 ,
,
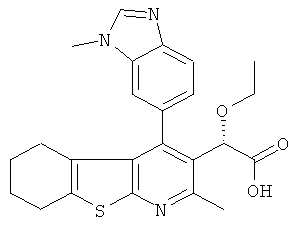 ,
,  ,
, 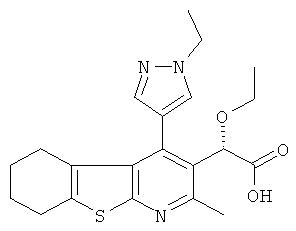 ,
, 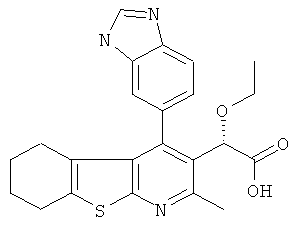 ,
,
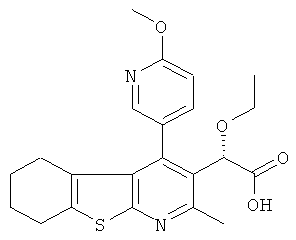 ,
, 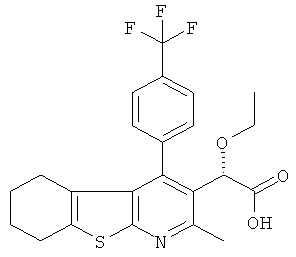 ,
, 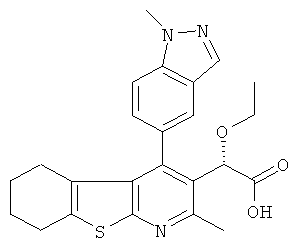 ,
, 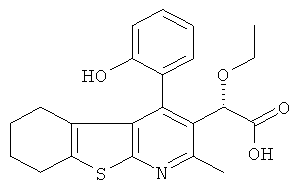 ,
,
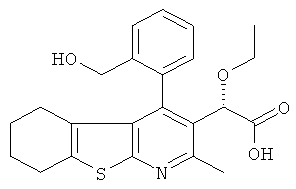 ,
, 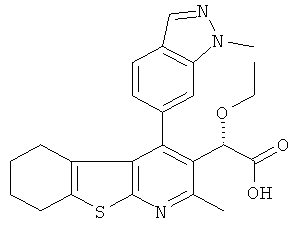 ,
, 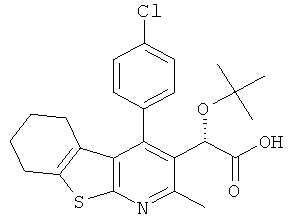 ,
, 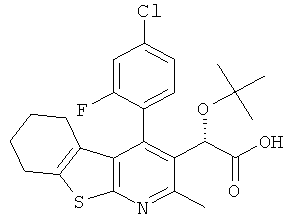 ,
,
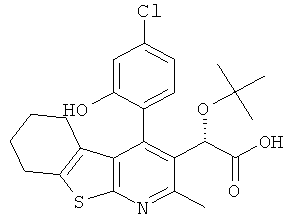 ,
, 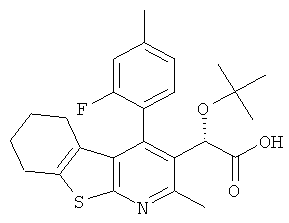 ,
, 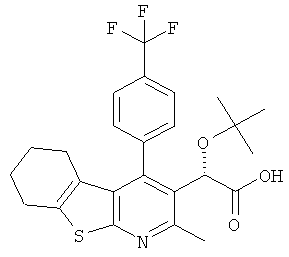 ,
, 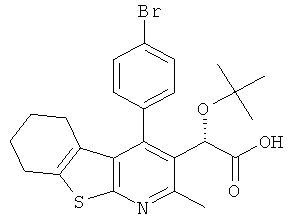 ,
,
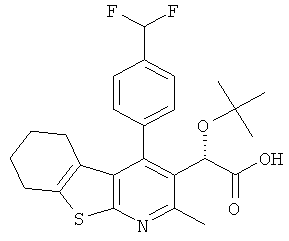 и
и 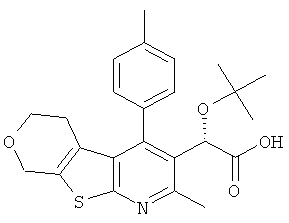
или его фармацевтически приемлемая соль.
В еще одном воплощении первого аспекта настоящего изобретения предложено соединение формулы (I)
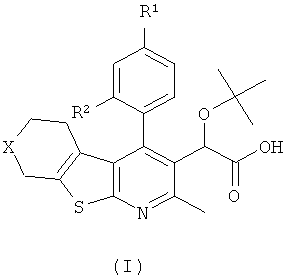
где:
R1 представляет собой CH3, CH2CH3, Cl, Br, CHF3 или CF3;
R2 представляет собой H, OH или F;
X представляет собой CH2 или O;
при условии, что когда R1 представляет собой CH3, и R2 представляет собой H, тогда X представляет собой O;
или его фармацевтически приемлемая соль.
В еще одном воплощении первого аспекта настоящего изобретения предложено соединение, выбранное из
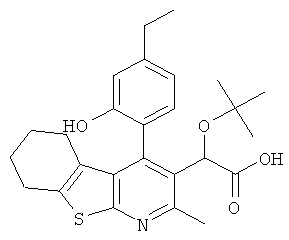 ,
, 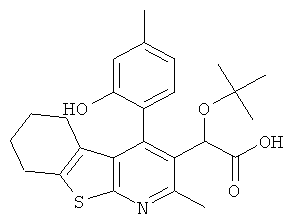 , , ,
, , ,
, 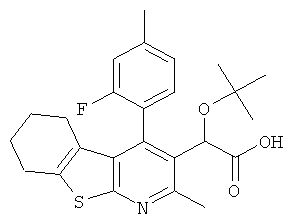 , , ,
, , ,
и ,
или его фармацевтически приемлемая соль.
В еще одном воплощении первого аспекта настоящего изобретения предложено соединение формулы (Ia)
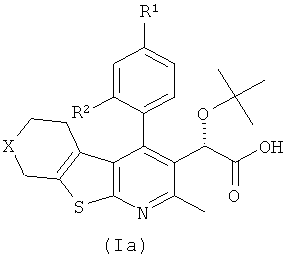
где:
R1 представляет собой CH3, CH2CH3, Cl, Br, CHF2 или CF3;
R2 представляет собой H, OH или F;
X представляет собой CH2 или O;
при условии, что когда R1 представляет собой CH3, и R2 представляет собой H, тогда X представляет собой О;
или его фармацевтически приемлемая соль.
В еще одном воплощении первого аспекта настоящего изобретения предложено соединение, выбранное из
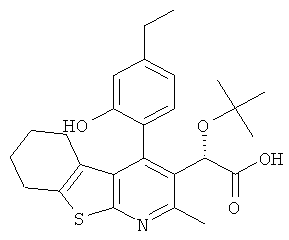 ,
, 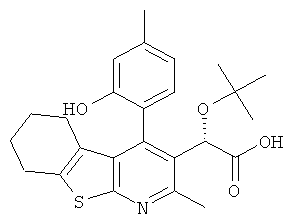 ,
, 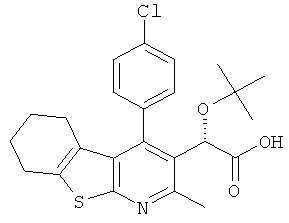 ,
, 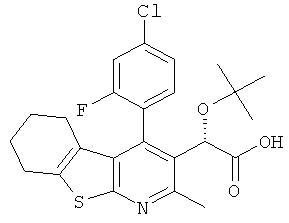 ,
,
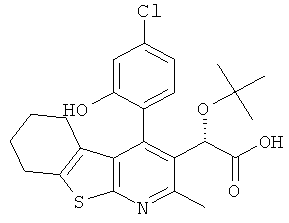 ,
, 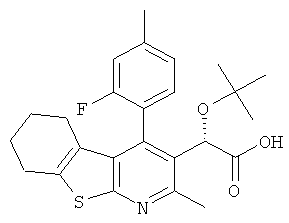 ,
, 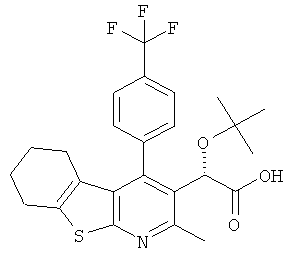 ,
, 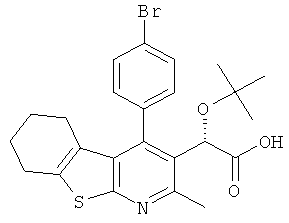 ,
,
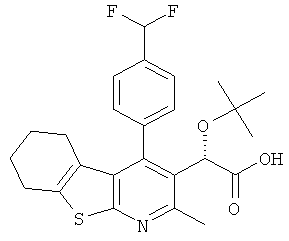 и
и 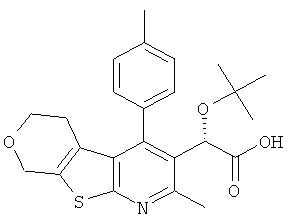
или его фармацевтически приемлемая соль.
В еще одном воплощении первого аспекта настоящего изобретения предложено соединение, выбранное из
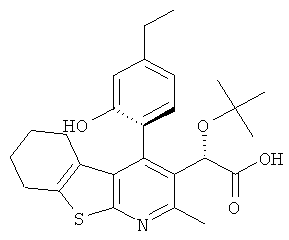 ,
, 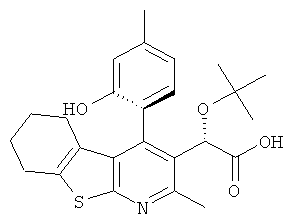 и
и 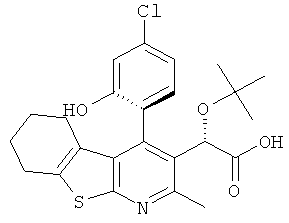
или его фармацевтически приемлемая соль.
Фармацевтически приемлемые соли соединений, описанных выше, включают соли присоединения кислоты и соли с основаниями.
Подходящие соли присоединения кислоты могут быть образованы с кислотами, которые образуют нетоксичные соли. Примеры могут охватывать соли ацетат, адипат, аспартат, бензоат, безилат, бикарбонат/карбонат, бисульфат/сульфат, борат, камзилат, цитрат, цикламат, эдисилат, эзилат, формиат, фумарат, глуцептат, глюконат, глюкуронат, гексафторфосфат, гибензат, гидрохлорид/хлорид, гидробромид/бромид, гидройодид/йодид, изетионат, лактат, малат, малеат, малонат, мезилат, метилсульфат, нафтилат, напсилат, никотинат, нитрат, оротат, оксалат, пальмитат, памоат, фосфат/гидрофосфат/дигидрофосфат, пироглутамат, сахарат, стеарат, сукцинат, таннат, тартрат, тозилат, трифторацетат и ксинофоат.
Подходящие соли с основаниями могут быть образованы с основаниями, которые образуют нетоксичные соли. Примеры могут охватывать соли алюминия, соли с аргинином, с бензатином, соли кальция, соли с холином, с диэтиламином, с диоламином, с глицином, с лизином, соли магния, соли с меглумином, с оламином, соли калия, натрия, соли с трометамином и соли цинка.
Также могут быть образованы гемисоли с кислотами и основаниями, например, соль гемисульфат и гемикальциевая соль. Обзор по подходящим солям смотри в "Handbook of Pharmaceutical Salts: Properties, Selection, and Use" by Stahl and Wermuth (Wiley-VCH, Weinheim, Germany, 2002).
Соединения по изобретению могут существовать в континууме твердых состояний в диапазоне от полностью аморфного состояния до полностью кристаллического состояния. Термин "аморфный" относится к состоянию, в котором у вещества отсутствует дальний порядок на молекулярном уровне, и, в зависимости от температуры, вещество может проявлять физические свойства твердого вещества или жидкости. Обычно такие вещества не дают характерных картин дифракции рентгеновских лучей и, хотя они и проявляют свойства твердого вещества, их более формально описывают как жидкость. При нагревании происходит изменение свойств от свойств твердого вещества до свойств жидкости, которое характеризуется изменением состояния, обычно второго порядка ("стеклование"). Термин "кристаллический" относится к твердой фазе, в которой вещество имеет регулярную упорядоченную внутреннюю структуру на молекулярном уровне и дает картину дифракции рентгеновских лучей с характерными пиками. Такие вещества при достаточном нагревании также будут проявлять свойства жидкости, но переход от твердого состояния в жидкое состояние характеризуется фазовым превращением, обычно первого порядка ("точка плавления").
Соединения по изобретению могут существовать как в несольватированной форме, так и в сольватированной форме. Термин "сольват" использован здесь для описания молекулярного комплекса, содержащего соединение по изобретению и стехиометрическое количество одной или более молекул фармацевтически приемлемого растворителя, например этанола. Термин "гидрат" используется, когда указанным растворителем является вода. Признанной в настоящее время системой классификации органических гидратов является система, которая определяет гидраты изолированных сайтов, канальные гидраты или гидраты, координированные с ионом металла (смотри "Polymorphism in Pharmaceutical Solids" by K.R. Morris (Ed. H.G. Brittain, Marcel Dekker, 1995). Гидратами изолированных сайтов являются гидраты, в которых молекулы воды изолированы от прямого контакта друг с другом находящимися между ними органическими молекулами. В канальных гидратах молекулы воды расположены в каналах решетки, где они соседствуют с другими молекулами воды. В гидратах, координированных с ионами металла, молекулы воды связаны с ионом металла. Если растворитель или вода связан(а) прочно, то комплекс будет иметь четко определенную стехиометрию независимо от влажности. Однако если растворитель или вода связан(а) слабо, как в канальных сольватах и гигроскопических соединениях, то содержание воды/растворителя будет зависеть от влажности и условий сушки. В таких случаях нормой будет отсутствие стехиометрии.
В объем изобретения также входят многокомпонентные комплексы (иные, чем соли и сольваты), в которых лекарственное средство и по меньшей мере один другой компонент присутствуют в стехиометрических или нестехиометрических количествах. Комплексы этого типа включают клатраты (комплексы включения лекарственное средство-хозяин) и сокристаллы. Последние обычно определяют как кристаллические комплексы нейтральных молекулярных составляющих, которые связаны вместе посредством нековалентных взаимодействий, но которые также могут представлять собой комплекс нейтральной молекулы с солью. Сокристаллы могут быть получены в результате кристаллизации из расплава, перекристаллизации из растворителей или совместного физического измельчения компонентов (смотри O. Almarsson and M.J. Zaworotko, Chem. Commun., 17, 1889-1896 (2004)). Общую информацию по таким многокомпонентным комплексам смотри в Haleblian, J. Pharm. Sci, 64 (8), 1269-1288 (August 1975).
Соединения по изобретению также могут существовать в мезоморфном состоянии (мезофаза или жидкий кристалл) под воздействием соответствующих условий. Мезоморфное состояние является промежуточным состоянием между истинным кристаллическим состоянием и истинным жидким состоянием (либо расплав, либо раствор). Мезоморфизм, возникающий в результате изменения температуры, описывают как "термотропный", а мезоморфизм, являющийся результатом добавления второго компонента, такого как вода или другой растворитель, описывают как "лиотропный". Соединения, способные образовывать лиотропные мезофазы, описывают как "амфифильные", и они состоят из молекул, которые обладают ионной (например -COO-Na+ -COO-K+ или -SO3 -Na+) или неионной (например -N-N+(CH3)3) полярной концевой группой. Дополнительную информацию смотри в N.H. Hartshorne and A. Stuart, "Crystals and the Polarizing Microscope", 4th Edition (Edward Arnold, 1970).
Далее все ссылки на соединение включают ссылки на их соли, сольваты, полиморфы, кристаллические габитусы, многокомпонентные комплексы и жидкие кристаллы и на их сольваты, полиморфы, кристаллические габитусы, многокомпонентные комплексы и жидкие кристаллы солей.
Соединения по настоящему изобретению имеют хиральный центр у карбоксильной группы. Поэтому соединения, которые не проявляют также атропизомерию (подробнее описана ниже), могут существовать в виде двух стереоизомеров (т.е. энантиомеров). Например:
симметричный 4-заместитель
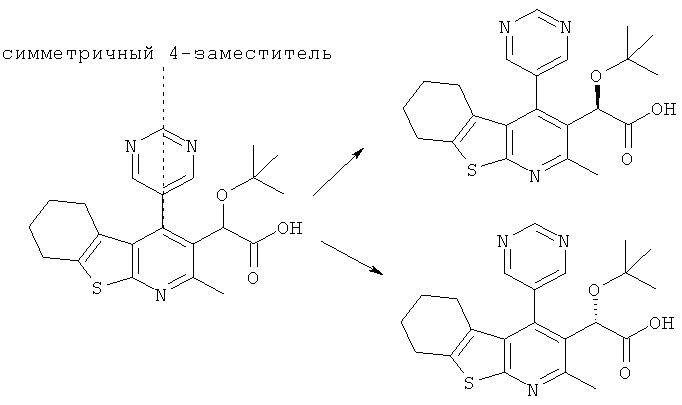
Когда 4-заместитель является несимметричным относительно плоскости связи в положении 4, тогда может также возникать атропизомерия. Это происходит из-за того, что ароматическое кольцо 4-заместителя и пиридиновая часть 5,6,7,8-тетрагидро[1]бензотиено[2,3-b]пиридина лежат более или менее перпендикулярно друг к другу, и вращение относительно связи в положении 4 2,3,4-замещенного 5,6,7,8-тетрагидро[1]бензотиено[2,3-b]пиридинового соединения по настоящему изобретению может быть ограниченным. Такие соединения могут, следовательно, существовать в виде четырех стереоизомеров (т.е. диастереоизомеров). Например:
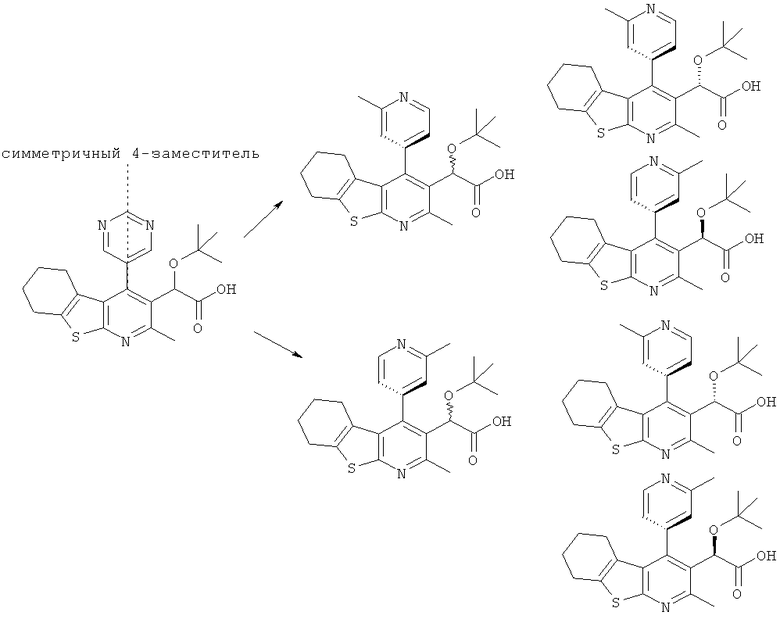
Соединения формулы (I) также содержат ароматические группировки, такие как имидазольные кольца, в которых может иметь место таутомерная изомерия ("таутомерия"). Таутомерия может принимать форму протонной таутомерии (например в имидазольных кольцах), а также валентной таутомерии (например в других ароматических группировках). Отсюда следует, что единственное соединение может проявлять более чем один тип изомерии.
В объем заявленных соединений по настоящему изобретению входят все стереоизомеры и таутомерные формы соединений, включая соединения, проявляющие более чем один тип изомерии, и смеси одного или более из них.
Если соединение или формула изображено(а) с указанной стереохимией по одному или более хиральным центрам, то подразумевается, что соединение или формула имеет стереоизомерный избыток по меньшей мере 80% (например по меньшей мере 90% конкретно указанного изомера и самое большое 10% других возможных изомеров), предпочтительно по меньшей мере 90%, более предпочтительно по меньшей мере 94% и наиболее предпочтительно по меньшей мере 99%.
Общепринятые методы получения/выделения индивидуальных энантиомеров/диастереоизомеров включают хиральный синтез из подходящего оптически чистого предшественника или разделение рацемата (или рацемата соли или производного) с использованием, например, хиральной жидкостной хроматографии высокого давления (ЖХВД). Альтернативно, рацемат (или рацемический предшественник) может быть подвергнут взаимодействию с подходящим оптически активным соединением, например спиртом, или, в случае, когда соединение формулы I содержит кислотную или основную группировку, с кислотой или основанием, например с винной кислотой или 1-фенилэтиламином. Полученная диастереомерная смесь может быть разделена хроматографией и/или фракционной кристаллизацией, и один или оба диастереоизомера могут быть превращены в соответствующий(ие) чистый(ые) энантиомер(ы) способами, известными специалисту.
Хиральные соединения по изобретению (и их хиральные предшественники) могут быть получены в энантитомерно обогащенной форме с использованием хроматографии, обычно ЖХВД, на смоле с асимметрической стационарной фазой и с подвижной фазой, состоящей из углеводорода, обычно гептана или гексана, содержащего от 0 до 50 об.% изопропанола, обычно от 2 до 20%, и от 0 до 5 об.% алкиламина, обычно 0,1% диэтиламина. Концентрирование элюента приводит к получению обогащенной смеси. Альтернативно, разделение может быть осуществлено с использованием SFC (сверхкритическая флюидная хроматография) на смоле с асимметрической стационарной фазой и с подвижной фазой, состоящей из градиента CO2, растворенного в метаноле.
Смеси стереоизомеров могут быть разделены обычными методами, известными специалистам в данной области (смотри, например, "Stereochemistry of Organic Compound" by E L Eliel (Wiley, New York, 1994)).
Абсолютная конфигурация единственного стереоизомера соединения по настоящему изобретению может быть определена путем выяснения структуры кристалла кристаллического комплекса фермент-соединение с использованием методов, известных специалистам в данной области.
Соединения по изобретению могут быть синтезированы описанными ниже общими способами.
Схема 1
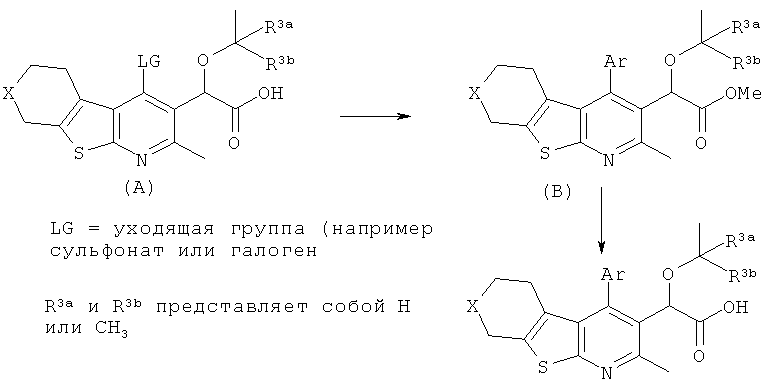
Реакция сочетания соединения формулы (A) с подходящим арильным (Ar) предшественником, осуществляемая по известным методикам (аминирование, реакция сочетания Сузуки, реакция сочетания Негиши, реакция сочетания Штилле и т.п.), приводит к образованию соединений формулы (B), которые могут быть превращены в целевые соединения по изобретению в стандартных условиях гидролиза.
Схема 2
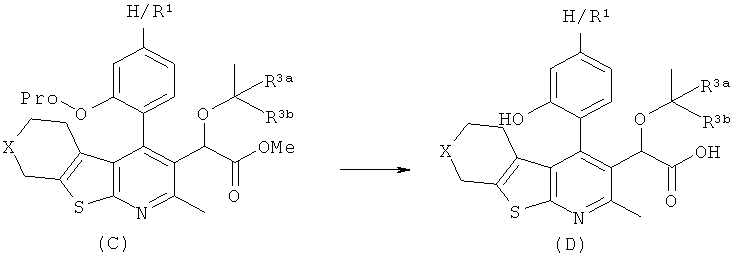
Pro = уходящая группа (например аллил) R3a и R3b представляют собой H или CH3
Соединения формулы (D) могут быть также получены путем удаления фенольной защитной группы, например аллильной, бензильной, в стандартных условиях удаления защитных групп либо до, либо после, либо одновременно с гидролизом эфирной группы соединений (C).
Во втором аспекте настоящего изобретения предложена фармацевтическая композиция, содержащая соединение по настоящему изобретению или его фармацевтически приемлемую соль или сольват, вместе с фармацевтически приемлемым эксципиентом.
Термин "эксципиент" используется в данном документе для описания любого ингредиента, не являющегося соединением по изобретению. Выбор эксципиента в большой степени будет зависеть от таких факторов, как конкретный способ введения, влияние эксципиента на растворимость и стабильность и природа лекарственной формы.
Фармацевтические композиции, подходящие для доставки соединений по настоящему изобретению, и способы их приготовления будут понятны специалистам в данной области. Такие композиции и способы их приготовления можно найти, например, в "Remington's Pharmaceutical Sciences", 19th Edition (Mack Publishing Company, 1995).
Соединения по изобретению можно вводить перорально. Пероральное введение может включать проглатывание, когда соединение поступает в желудочно-кишечный тракт, или можно использовать трансбуккальное или сублингвальное введение, в результате которого соединение поступает в кровоток прямо из ротовой полости. Композиции, подходящие для перорального введения, включают как твердые, так и жидкие композиции.
Твердые формы композиций включают таблетки, капсулы (содержащие твердые частицы, жидкости или порошки), пастилки (включая пастилки с жидким наполнителем), жевательные резинки, мульти- и нано-частицы, гели, твердые растворы, липосомные препараты, пленки, овули и спреи.
Жидкие формы композиций включают суспензии, растворы, сиропы и эликсиры. Такие формы могут быть использованы в качестве наполнителей в мягких или твердых капсулах и обычно содержат носитель, например воду, этанол, полиэтиленгликоль, пропиленгликоль, метил целлюлозу или подходящее масло, и один или более эмульгаторов и/или суспендирующих агентов. Жидкие композиции могут быть также приготовлены путем разведения твердого вещества, например из саше.
Соединения по изобретению можно также использовать в быстрорастворимых, быстрораспадающихся лекарственных формах, таких как формы, описанные в Expert Opinion in Therapeutic Patents, 11 (6), 981-986, by Liang and Chen (2001).
В таблеточных лекарственных формах в зависимости от дозы содержание лекарственного средства может составлять от 1 масс.% до 80 масс.% от массы лекарственной формы, более типично от 5 масс.% до 60 масс.% от массы лекарственной формы.
В дополнение к лекарственному средству таблетки обычно содержат разрыхлитель. Примеры разрыхлителей включают крахмал-гликолят натрия, натрий-карбоксиметилцеллюлозу, кальций-карбоксиметилцеллюлозу, натрий-кроскармелозу, кросповидон, поливинилпирролидон, метилцеллюлозу, микрокристаллическую целлюлозу, замещенную низшим алкилом гидроксипропилцеллюлозу, крахмал, прежелатинизированный крахмал и альгинат натрия. Как правило, содержание разрыхлителя будет составлять от 1 масс.% до 25 масс.%, предпочтительно от 5 масс.% до 20 масс.% от массы лекарственной формы.
Связывающие вещества обычно используют для придания когезионных свойств композициям в форме таблетки. Подходящие связывающие вещества включают микрокристаллическую целлюлозу, желатин, сахара, полиэтиленгликоль, натуральные и синтетические камеди, поливинилпирролидон, прежелатинизированный крахмал, гидроксипропилцеллюлозу и гидроксипропилметилцеллюлозу.
Таблетки могут также содержать разбавители, такие как лактоза (моногидрат, подвергнутый распылительной сушке моногидрат, безводный и т.п.), маннит, ксилит, декстроза, сахароза, сорбит, микрокристаллическая целлюлоза, крахмал и дигидрат двухосновного фосфата кальция.
Таблетки возможно могут также содержать поверхностно-активные вещества, такие как лаурилсульфат натрия и полисорбат 80, и скользящие вещества, такие как диоксид кремния и тальк. Содержание поверхностно-активных веществ, если они присутствуют, может составлять от 0,2 масс.% до 5 масс.% от массы таблетки, и содержание скользящих веществ может составлять от 0,2 масс.% до 1 масс.% от массы таблетки.
Таблетки также обычно содержат смазывающие вещества, такие как стеарат магния, стеарат кальция, стеарат цинка, стеарилфумарат натрия и смеси стеарата магния с лаурилсульфатом натрия. Содержание смазывающих веществ составляет от 0,25 масс.% до 10 масс.%, предпочтительно от 0,5 масс.% до 3 масс.% от массы таблетки.
Другие возможные ингредиенты включают антиоксиданты, красители, корригенты, консерванты и агенты, маскирующие вкус.
Иллюстративная таблетка содержит вплоть до примерно 80% лекарственного средства, от примерно 10 масс.% до примерно 90 масс.% связывающего вещества, от примерно 0 масс.% до примерно 85 масс.% разбавителя, от примерно 2 масс.% до примерно 10 масс.% разрыхлителя и от примерно 0,25 масс.% до примерно 10 масс.% смазывающего вещества.
Таблеточные смеси можно прессовать непосредственно или с помощью валика для формования таблеток. Таблеточные смеси или порции смесей перед таблетированием могут быть альтернативно подвергнуты влажному, сухому гранулированию или гранулированию из расплава, застыванию расплава или экструзии. Конечный препарат может содержать один или более слоев и может быть покрыт оболочкой или может не иметь оболочки; он может даже быть инкапсулированным.
Приготовление таблеток обсуждается в "Pharmaceutical Dosage Forms: Tablets", Vol.1, by H. Lieberman and L Lachman (Marcel Dekker, New York, 1980).
Годные к употреблению пероральные пленки обычно представляют собой лекарственные формы в виде гибких водорастворимых или набухающих в воде тонких пленок, которые могут быть быстрорастворимыми или мукоадгезивными и обычно содержат соединение формулы (I), пленкообразующий полимер, связывающее вещество, растворитель, увлажнитель, пластификатор, стабилизатор или эмульгатор, модификатор вязкости и растворитель. Некоторые компоненты композиции могут осуществлять более чем одну функцию. Пленкообразующий полимер может быть выбран из натуральных полисахаридов, белков или синтетических гидроколлоидов и обычно присутствует в количестве в пределах от 0,01 до 99 масс.%, более типично в пределах от 30 до 80 масс.%. Другие возможные ингредиенты включают антиоксиданты, красители, корригенты и усилители вкуса, консерванты, агенты, стимулирующие слюноотделение, охлаждающие агенты, сорастворители (включая масла), смягчающие вещества, агенты, создающие объем, антивспениватели, поверхностно-активные вещества и агенты, маскирующие вкус. Пленки в соответствии с изобретением обычно изготавливают путем испарительной сушки тонких водных пленок, нанесенных на отслаивающуюся подложку или бумагу. Эту процедуру можно выполнять в термостате или в туннельной сушилке, обычно в комбинированном устройстве-сушилке для нанесения покрытий, или путем сублимационной сушки или вакуумирования.
Твердые формы композиций для перорального введения могут быть приготовлены с возможностью немедленного и/или модифицированного высвобождения. Формы композиций с модифицированным высвобождением включают формы с замедленным, длительным, импульсным, контролируемым, направленным и программированным высвобождением.
Подходящие для целей данного изобретения формы композиций с модифицированным высвобождением описаны в патенте США №6106864. Подробности других подходящих технологий высвобождения, таких как высокоэнергетические дисперсии и осмотические и покрытые оболочкой частицы можно найти в "Pharmaceutical Technology On-line", 25(2), 1-14, by Verma et al (2001). Применение жевательной резинки для достижения контролируемого высвобождения описано в WO 00/35298.
Соединения по изобретению можно также вводить прямо в кровоток, в мышцу или во внутренний орган. Подходящие способы парентерального введения включают внутривенное, внутриартериальное, интраперитонеальное, интратекальное, интравентрикулярное, интрауретральное, интрастернальное, интракраниальное, внутримышечное и подкожное введение. Подходящие устройства для парентерального введения включают игольные (включая микроигольные) шприцы, безыгольные инъекторы и оборудование для инфузий.
Парентеральные композиции обычно представляют собой водные растворы, которые могут содержать эксципиенты, такие как соли, углеводы и буферные агенты (предпочтительно до pH от 3 до 9), но для некоторых применений их предпочтительно готовят в виде стерильного неводного раствора или в виде высушенной формы для использования с подходящим разбавителем, таким как стерильная апирогенная вода.
Приготовление парентеральных композиций в стерильных условиях, например путем лиофилизации, без труда может быть осуществлено с использованием стандартных фармацевтических методов, известных специалистам в данной области.
Растворимость соединения по настоящему изобретению, используемого в приготовлении парентеральных растворов, может быть увеличена за счет использования соответствующих методов приготовления композиций, таких как введение в композицию увеличивающих растворимость агентов.
Композиции для парентерального введения могут быть приготовлены с возможностью немедленного и/или модифицированного высвобождения. Композиции модифицированного высвобождения включают композиции замедленного, длительного, импульсного, контролируемого, направленного и программированного высвобождения. Так, соединение по изобретению может быть приготовлено в виде твердого вещества, полутвердого вещества или тиксотропной жидкости для введения в виде имплантированного депо, обеспечивающего модифицированное высвобождение активного соединения. Примеры форм таких композиций включают стенты, покрытые лекарственным средством и микросферы из сополимера dl-молочной кислоты и гликолевой кислоты (PGLA).
Соединения по изобретению можно также вводить местно на кожу или слизистую оболочку, т.е. дермально или трансдермально. Типичные формы композиций для этой цели включают гели, гидрогели, лосьоны, растворы, кремы, мази, присыпки, повязки, пенки, пленки, пластыри, облатки, имплантаты, губки, фибры, бандажи и микроэмульсии. Липосомы также могут быть использованы. Типичными носителями являются спирт, вода, минеральное масло, жидкий вазелин, белый вазелин, глицерин, полиэтиленгликоль и пропиленгликоль. В композицию могут быть включены усилители проницаемости (смотри, например, J Pharm Sci, 88 (10), 955-958, by Finnin and Morgan (October 1999).
Другие способы местного введения включают доставку посредством электропорации, ионтофореза, фонофореза, сонофореза и микроигольной или безыгольной (например Powderject™, Bioject™ и т.д.) инъекции.
Композиции для местного введения могут быть приготовлены с возможностью немедленного и/или модифицированного высвобождения. Композиции с модифицированным высвобождением включают композиции с замедленным, длительным, импульсным, контролируемым, направленным и программированным высвобождением.
Соединения по изобретению можно вводить ректально или вагинально, например в форме суппозитория, пессария или клизмы. Масло какао является традиционной суппозиторной основой, но могут быть использованы различные альтернативные основы, которые являются подходящими.
Композиции для ректального/вагинального введения могут быть приготовлены с возможностью немедленного и/или модифицированного высвобождения. Композиции с модифицированным высвобождением включают композиции с замедленным, длительным, импульсным, контролируемым, направленным и программированным высвобождением.
Соединения по изобретению могут быть объединены с растворимыми макромолекулярными веществами, такими как циклодекстрин и подходящие его производные или полиэтиленгликоль-содержащие полимеры, для улучшения их растворимости, скорости растворения, маскировки вкуса, биодоступности и/или стабильность для использования в любых вышеупомянутых способах введения.
Было установлено, что комплексы лекарственное средство - циклодекстрин, например, полезны для большинства лекарственных форм и путей введения. Можно использовать как комплексы включения, так и комплексы невключения. В качестве альтернативы прямому комплексообразованию с лекарственным средством, циклодекстрин можно использовать в качестве вспомогательной добавки, т.е. в качестве носителя, разбавителя или солюбилизатора. Чаще всего используемыми для этих целей являются альфа-, бета- и гамма-циклодекстрины, примеры которых можно найти в публикациях международных патентных заявок WO 91/11172, WO 94/02518 и WO 98/55148.
В третьем аспекте настоящего изобретения предложено соединение по настоящему изобретению или его фармацевтически приемлемая соль для применения в качестве лекарственного средства.
Конкретным воплощением этого аспекта изобретения является соединение по настоящему изобретению или его фармацевтически приемлемая соль для применения в лечении ВИЧ-инфекции.
В четвертом аспекте настоящего изобретения предложено применение соединения по настоящему изобретению или его фармацевтически приемлемой соли для изготовления лекарственного средства для лечения ВИЧ-инфекции.
В пятом аспекте настоящего изобретения предложен способ лечения млекопитающего, включая человека, с целью лечения ВИЧ-инфекции, включающий введение указанному млекопитающему эффективного количества соединения по настоящему изобретению или его фармацевтически приемлемой соли или сольвата.
Используемый в данном документе термин "лечение" охватывает как превентивное, так и куративное лечение заболевания или расстройства. Он также охватывает замедление, прерывание, контролирование или остановку прогрессирования заболевания или расстройства. Он также охватывает предупреждение, излечивание, замедление, прерывание, контролирование или прекращение симптомов заболевания или расстройства.
Соединение по настоящему изобретению можно вводить в комбинации с одним или более дополнительными агентами для лечения млекопитающего, например человека, страдающего ВИЧ-инфекцией или любым другим заболеванием или состоянием, которое связано с ВИЧ-инфекцией. Агенты, которые могут быть использованы в комбинации с соединением по настоящему изобретению, включают, но не ограничены ими, агенты, полезные в качестве ингибиторов протеазы ВИЧ, ингибиторов обратной транскриптазы БИЧ, ненуклеозидных ингибиторов обратной транскриптазы ВИЧ, ингибиторов интегразы ВИЧ, ингибиторов CCR5, ингибиторов слияния ВИЧ или других ингибиторов проникновения ВИЧ, ингибиторов созревания, агентов, которые действуют, нарушая мультимеризацию капсида ВИЧ или вирусную коровую устойчивость, соединений, направленно воздействующих на белки организма, необходимые для вирусной репликации или ускользания от иммунологического надзора (таких как, без ограничения, PSIP1), соединения, полезные в качестве иммуномодуляторов, соединения, которые ингибируют ВИЧ по неизвестным механизмам, соединения, полезные для лечения вирусов герпес, соединения, полезные в качестве противоинфекционных средств, и другие, которые описаны
ниже.
Соединения, полезные в качестве ингибиторов протеазы ВИЧ, которые можно использовать в комбинации с соединением по настоящему изобретению, включают, но не ограничены ими, 141 W94 (ампренавир), CGP-73547, CGP-61755, DMP-450 (мозенавир), нелфинавир, ритонавир, саквинавир (инвираза), лопинавир, ТМС-126, атазанавир, палинавир, GS-3333, KN 1-413, KNI-272, LG-71350, CGP-61755, PD 173606, PD 177298, PD 178390, PD 178392, U-140690, ABT-378, DMP-450, AG-1776, MK-944, VX-478, индинавир, типранавир, ТМС-114 (дарунавир), DPC-681, DPC-684, фосампренавир-кальций (Lexiva), бензолсульфонамидные производные, описанные в WO 03053435, R-944, Ro-03-34649, VX-385 (бреканавир), GS-224338, OPT-TL3, PL-100, SM-309515, AG-148, DG-35-VIII, DMP-850, GW-5950X, KNI-1039, L-756423, LB-71262, LP-130, RS-344, SE-063, UIC-94-003, Vb-19038, A-77003, BMS-182193, BMS-186318, SM-309515, JE-2147, GS-9005, телинавир (SC-52151), BILA-2185 BS, DG-17, PPL-100, A-80987, GS-8374, DMP-323, U-103017, CGP-57813 и CGP-53437.
Соединения, полезные в качестве ингибиторов обратной транскриптазы ВИЧ, которые можно использовать в комбинации с соединением по настоящему изобретению, включают, но не ограничены ими, абакавир, эмтрицитабин (FTC), GS-840 (адефовир), ламивудин, адефовир дипивоксил, бета-фтор-ddA, залцитабин, диданозин, ставудин, зидовудин, тенофовир, тенофовира дисопроксилфумарат, амдоксовир, SPD-754 (априцитабин), SPD-756, рацивир, реверсет (DPC-817), MIV-210 (FLG), 6eTa-L-Fd4C (ACH-126443, элвуцитабин), MIV-310 (аловудин, FLT), dOTC, DAPD, энтекавир, GS-7340, стампидин, D-d4FC (декселвуцитабин), фосфазид, фозивудин дитоксил и фосалвудин дитоксил.
Соединения, полезные в качестве ненуклеозидных ингибиторов обратной транскриптазы ВИЧ, которые можно использовать в комбинации с соединением по настоящему изобретению, включают, но не ограничены ими, эфавиренц, HBY-097, невирапин, дапивирин (ТМС-120), ТМС-125, этравирин, делавирдин, DPC-083, DPC-961, ТМС-120, каправирин, GW-678248, GW-695634, каланолид, рилпивирин (ТМС-278), ловирид, эмивирин (МКС-442), DPC-963, MIV-150, BiLR 355 BS, VRX-840773, лерсивирин (UK-453061), RDEA806 и трициклические пиримидиноновые производные, которые описаны в WO 03062238.
Соединения, полезные в качестве ингибиторов CCR5, которые можно использовать в комбинации с соединением по настоящему изобретению, включают, но не ограничены ими, ТАК-779, SC-351125, SCH-D, UK-427857 (маравирок), PRO-140 и GW-873140 (аплавирок, Ono-4128, AK-602), SCH-417690 (вицивирок, SCH-D), INCB-9471, INCB-15050, TBR-220 (ТАК-220), CCR5 mAb004. Другие соединения, полезные в качестве ингибиторов CCR5, которые можно использовать в комбинации с соединением по настоящему изобретению, включают, но не ограничены ими, (N-{(1S)-3-[3-изопропил-5-метил-4Н-1,2,4-триазол-4-ил]-экзо-8-азабицикло[3.2.1]окт-8-ил}-1-фенилпропил)-4,4-дифторциклогексанкарбоксамид), метил-1-эндо-{8-[(3S)-3-(ацетиламино)-3-(3-фторфенил)пропил]-8-азабицикло[3.2.1]окт-3-ил}-2-метил-4,5,6,7-тетрагидро-1H-имидазо[4,5-c]пиридин-5-карбоксилат и N-{(1S)-3-[3-эндо-(5-изобутиоил-2-метил-4,5,6,7-тетрагидро-1H-имидазо[4,5-c]пиридин-1-ил)-8-азабицикло[3.2.1]окт-8-ил]-1-(3-фторфенил)пропил}ацетамид).
Соединения, полезные в качестве ингибиторов интегразы ВИЧ, которые можно использовать в комбинации с соединением по настоящему изобретению, включают, но не ограничены ими, ралтегравир, элвитегравир (GS-9137, JTK-303), GSK-364735, MK-2048, BMS-707035, S-1360 (GW-810781), L-870810, L-870812, AR-177, ВА-011, производные 1,5-нафтиридин-3-карбоксамида, раскрытые в WO 03062204, соединения, раскрытые в WO 03047564, соединения, раскрытые в WO 03049690, производные 5-гидроксипиримидин-4-карбоксамида, раскрытые в WO 03035076, и L-000810810.
Ингибиторы слияния для лечения ВИЧ, которые можно использовать в комбинации с соединением по настоящему изобретению, включают, но ими не ограничены, энфувиртид (T-20), T-1249, AMD-3100, силфувиртид, FB-006M, TRI-1144, PRO-2000 и конденсированные трициклические соединения, раскрытые в JP 2003171381.
Ингибиторы созревания для лечения ВИЧ, которые можно использовать в комбинации с соединением по настоящему изобретению, включают, но не ограничены ими, бевиримат и вивекон.
ВИЧ фиксированные комбинации лекарственных средств для лечения ВИЧ, которые можно использовать в комбинации с соединением по настоящему изобретению включают, но не ограничены ими, комбивир, атрипла, тризивир, трувада, калетра и эпзиком.
Ингибиторы CXCR4 для лечения ВИЧ, которые можно использовать в комбинации с соединением по настоящему изобретению включают, но не ограничены им, AMD-070.
Ингибиторы проникновения для лечения ВИЧ, которые можно использовать в комбинации с соединением по настоящему изобретению, включают, но не ограничены им, SP-01A.
Ингибиторы Gp 120 для лечения ВИЧ, которые можно использовать в комбинации с соединением по настоящему изобретению, включают, но не ограничены ими, BMS-488043 и BMS-378806.
Ингибиторы G6PD и NADH-оксидазы для лечения ВИЧ, которые можно использовать в комбинации с соединением по настоящему изобретению, включают, но не ограничены им, иммунитин.
Другие соединения, которые являются полезными ингибиторами ВИЧ и которые можно использовать в комбинации с соединением по настоящему изобретению, включают, но не ограничены ими, Soluble CD4, PRO-542, ибализумаб (TNX-355) и соединения, раскрытые в JP 2003119137.
Соединения, полезные в лечении или контролировании инфекции вирусами, иными, чем ВИЧ, которые можно использовать в комбинации с соединением по настоящему изобретению, включают, но не ограничены ими, ацикловир, фомивирсен, пенцикловир, НРМРС, оксетаноцин G, AL-721, цидофовир, цитомегаловирусный иммуноглобулин, цитовен, фомивганцикловир, фамцикловир, фосканет-натрий, Isis 2922, KNI-272, валацикловир, виразол рибавирин, валганцикловир, МЕ-609, PCL-016, DES6, ODN-93, ODN-112, VGV-1, амплиген, HRG-214, цитолин, VGX-410, KD-247, AMZ-0026, CYT-99007A-221, DEBIO-025, BAY 50-4798, MDX-010 (ипилимумаб), PBS-119, ALG-889, PA-1050040 (PA-040) и филибувир (PF-00868554).
Соединения, которые действуют в качестве иммуномодуляторов и которые можно использовать в комбинации с соединением по настоящему изобретению, включают, но не ограничены ими, AD-439, AD-519, интерферон альфа, AS-101, бропиримин, ацеманнан, CL246.738, EL10, FP-21399, интерферон гамма, гранулоцитарно-макрофагальный колониестимулирующий фактор, IL-2, внутривенный иммуноглобулин, IMREG-1, IMREG-2, имутиолдиэтилдитиокарбамат, интерферон альфа-2, метионин-энкефалин, МТР-РЕ, гранулоцитарный колониестимулирующий фактор, ремун, rCD4, рекомбинантный растворимый человеческий CD4, интерферон альфа-2, SK&F106528, растворимый T4 ихимопентин, фактор некроза опухоли (TNF), тукарезол, рекомбинантный человеческий интерферон бета и интерферон альфа n-3.
Противоинфекционные средства, которые можно использовать в комбинации с соединением по настоящему изобретению, включают, но не ограничены ими, атоваквон, азитромицин, кларитромицин, триметоприм, тровафлоксацин, пириметамин, даунорубицин, клиндамицин с примаквином, пастилл, орнидил, эфлорнитин пентамидин, рифабутин, спирамицин, тнтраконазол-R51211, триметрексат, даунорубицин, хлороквин, рекомбинантный человеческий эритропоэтин, рекомбинантный человеческий гормон роста, мегестрола ацетат, тестером и общее энтеральное питание.
Противогрибковые средства, которые можно использовать в комбинации с соединением по настоящему изобретению, включают, но не ограничены ими, анидулафунгин, C31G, каспофунгин, DB-289, флуконазол, итраконазол, кетоконазол, микафунгин, посаконазол и вориконазол.
Другие соединения, которые можно использовать в комбинации с соединением по настоящему изобретению, включают, но не ограничены ими, ацеманнан, ансамицин, LM 427, AR177, BMS-232623, BMS-234475, CI-1012, курдлана сульфат, декстрана сульфат, STOCRINE EL10, гиперицин, лобукавир, новапрен, октапептидная последовательность пептида Т, тринатрия фосфонрформиат, пробукол и RBC-CD4.
Кроме того, соединение по настоящему изобретению можно использовать в комбинации с антипролиферативными агентами для лечения таких состояний, как саркома Капоши. Такие агенты включают, но не ограничены ими, ингибиторы металломатриксных протеаз, А-007, бевацизумаб, BMS-275291, галофугинон, интерлейкин-12, ритуксимаб, паклитексел, порфимер-натрий, ребимастат и COL-3.
Такую комбинацию можно вводить, когда соединение по настоящему изобретению присутствует в той же фармацевтической композиции, что и дополнительный(ые) агент(ы), описанный(ые) выше. Альтернативно, такую комбинацию можно вводить, когда соединение по настоящему изобретению присутствует в фармацевтической композиции, отдельной от фармацевтической композиции, в которой находится(ятся) дополнительный(ые) агент(ы). Если соединение по настоящему изобретению вводят отдельно от дополнительного(ых) агента(ов), такое введение может происходить параллельно или последовательно с подходящим интервалом времени между их введением.
Дополнительно, соединение по настоящему изобретению можно вводить в комбинации с одним или более дополнительными агентами, которые оказывают эффект увеличения воздействия соединения по изобретению на млекопитающее. Термин "воздействие", используемый в данном документе, относится к концентрации соединения по изобретению в плазме крови млекопитающего, измеренной за некоторый период времени. Воздействие соединения на млекопитающего может быть измерено путем введения соединения по изобретению млекопитающему в подходящей форме, взятия образцов плазмы крови в предопределенные точки времени и измерения количества соединения по изобретению в плазме с использованием подходящего аналитического метода, такого как жидкостная хроматография или жидкостная хроматография/масс-спектроскопия. Количество соединения по изобретению, присутствующего в плазме крови в определенные моменты времени, определяют, и данные по концентрации и времени для всех образцов наносят на график для построения кривой. Вычисляют площадь под кривой и получают воздействие соединения на млекопитающее. Термины "воздействие", "площадь по кривой" и "площадь под кривой концентрация/время" имеют одинаковое значение и могут быть использованы взаимозаменяемым образом.
Среди агентов, которые могут быть использованы для увеличения воздействия соединения по настоящему изобретению на млекопитающее, находятся агенты, которые могут действовать в качестве ингибиторов по меньшей мере одной изоформы цитохрома Р450 (CYP450). Изоформы CYP450, которые преимущественно могут быть ингибированы, включают, но не ограничены ими, CYP1A2, CYP2D6, CYP2C9, CYP2C19 и CYP3A4. Подходящие агенты, которые могут быть использованы для ингибирования CYP3A4, включают, но не ограничены ими, ритонавир, делавирдин, N-(3,4-дифторбензил)-2-{[(4-метоксипиридин-3-ил)амино]сульфонил}-N-метилбензамид и N-(1-(5-(4-фторбензил)-3-(пиридин-4-ил)-1H-пиразол-1-карбонил)пиперидин-4-ил)метансульфонамид.
Такую комбинацию можно вводить, когда соединение по настоящему изобретению присутствует в той же композиции, что и дополнительный(ые) агент(ы), описанный(ые) выше. Альтернативно, такую комбинацию можно вводить, когда соединение по настоящему изобретению присутствует в фармацевтической композиции, отдельной от фармацевтической композиции, в которой находится(ятся) дополнительный(ые) агент(ы). Если соединение по настоящему изобретению вводят отдельно от дополнительного(ых) агента(ов), то такое введение может происходить параллельно или последовательно с подходящим интервалом времени между их введением.
Ввиду того, что может быть желательно вводить комбинацию активных соединений, например в целях лечения конкретного заболевания или состояния, в объеме настоящего изобретения предусмотрено, что две или более фармацевтических композиций, по меньшей мере одна из которых содержит соединение по изобретению, могут быть объединены в форме набора, подходящего для совместного введения композиций.
Так, набор по изобретению содержит две или более отдельных фармацевтических композиций, по меньшей мере одна из которых содержит соединение по настоящему изобретению, и средства для раздельного хранения указанных композиций, такие как контейнер, секционированный флакон или секционированный пакет из фольги. Примером такого набора является известная блистерная упаковка, используемая для упаковки таблеток, капсул и т.п.
Набор по изобретению является, в частности, особенно подходящим для введения разных лекарственных форм, например пероральных и парентеральных, для введения отдельных композиций в разных диапазонах доз, или для титрования отдельных композиций одного относительно друг друга. Чтобы способствовать соблюдению пациентами режима приема, набор обычно содержит инструкции по введению и может быть снабжен так называемой памяткой.
Описанные ниже методики иллюстрируют способы, подходящие для получения соединений по настоящему изобретению.
Общие методы
ЖХ7МС (жидкостная хроматография/масс-спектрометрия) (2 мин, кислотная) A: 0,1% муравьиная кислота в воде, B: 0,1% муравьиная кислота в ацетонитриле; колонка: Agilent Extend C18 фаза 30×3 мм с размером частиц 3 микрона; градиент: 90-0% А за 1,8 мин, выдержка 0,55 мин, снова уравновешивание 0,15 мин; скорость потока 1,6 мл/мин; УФ: 210 нм-450 нм DAD (детектор на диодной матрице); температура: 50°C.
ЖХ/МС (5 мин, кислотная) A: 0,1% муравьиная кислота в воде В: 0,1% муравьиная кислота в ацетонитриле; колонка: Agilent Extend C18 фаза 50×3 мм с размером частиц 3 микрона; градиент: 95-0% А за 3,5 мин, выдержка 1 мин, снова уравновешивание 0,4 мин; скорость потока 1,2 мл/мин; УФ: 210 нм-450 нм DAD; температура: 50°C
ЖХ/МС (12 мин, кислотная) A: 0,1% муравьиная кислота в воде B: 0,1% муравьиная кислота в ацетонитриле; колонка: Agilent SB C18 фаза 50×3 мм с размером частиц 3 микрона; градиент: 95% А выдержка 1 мин, 95-0% А за 8 мин, выдержка 2,5 мин, снова уравновешивание 0,50 мин; скорость потока 1,2 мл/мин; УФ: 210 нм-450 нм DAD; температура: 50°C
ЖХ/МС (5 мин, основная) A: метанол, В: 10 мМ бикарбонат аммония в воде @ pH10; колонка: XBridge C18 2,1×30 мм с размером частиц 5 микрон; градиент: 95-5% А за 2,9 мин, выдержка 0,9 мин, снова уравновешивание 0,1 мин; скорость потока 0,5 мл/мин; УФ: 215-350 нм DAD; температура 25°C
1H ЯМР регистрировали на Varian Gemini 400 МГц при 30°C.
Подготовительный пример 1: Этил-2-амино-4,5,6,7-тетрагидро-1-бензотиофен-3-карбоксилат
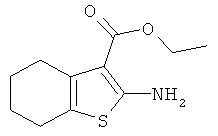
К раствору этилцианоацетата (427 мл, 4 моль) в этаноле (4 л) добавляли серу (153,88 г, 4,80 моль), морфолин (422 мл, 4,80 моль) и циклогексанон (497 мл, 4,80 моль), и полученный раствор перемешивали при 50°C в течение 18 часов. Реакционную смесь охлаждали до комнатной температуры и фильтровали для удаления твердого вещества. Остаток на фильтре промывали холодным этанолом и затем сушили с получением указанного в заголовке соединения в виде бледно-желтого твердого вещества, 608,4 г. Маточные жидкости охлаждали в ледяной бане, и полученный осадок собирали фильтрованием. Твердое вещество очищали флэш-хроматографией на сухой колонке, элюируя этилацетатом в гептане (20-30%), с получением дополнительно 38,62 г указанного в заголовке соединения. Оба твердых вещества объединяли с получением 647,02 г указанного в заголовке соединения выход 72%. 1H ЯМР (400 МГц, CDCl3) δ м.д. (миллионные доли) 1.34 (t, 3H), 1.73-1.75 (m, 4H), 2.46-2.49 (m, 2H), 2.63-2.69 (m, 2H), 4.25 (q, 2H), 5.92 (brs, 2H).
Подготовительный пример 2: Этил-3-этоксибут-2-еноат
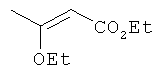
К раствору этилацетоацетата (2,7 кг, 20,74 моль) в этаноле (4 л) добавляли конц. N2SO4 (4 мл) при 25°C в атмосфере азота. Смесь нагревали до 50°C, затем по каплям добавляли триэтилортоформиат (3073,6 г, 20,74 моль). Смесь перемешивали при 50°C в течение 16 ч. Смесь концентрировали при пониженном давлении с получением указанного в заголовке соединения (2,8 кг, 85%) в виде желтого масла. 1H ЯМР (400 МГц, CDCl3): 8 4.9 (s, 1H), 4.1 (m, 2H), 3.7 (m, 2H), 2.2 (s, 3H), 1.2 (m, 3H), 1.1 (m, 3H).
Подготовительный пример 3: Этил-(4-гидрокси-2-метил-5,6,7,8-тетрагидро[1]бензотиено[2,3-b]пиридин-3-ил)карбоксилат
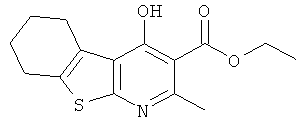
В реакционном сосуде емкостью 10 л к перемешиваемому раствору этил-2-амино-4,5,6,7-тетрагидробензо[b]тиофен-3-карбоксилата (Подготовительный пример 1) и этил-3-этоксибут-2-еноата (Подготовительный пример 2) в толуоле (6,0 л) добавляли пиридиний-пара-толуолсульфоновую кислоту (39,7 г, 158 ммоль) при комнатной температуре в атмосфере аргона. Полученную смесь нагревали до температуры образования флегмы и перемешивали при температуре образования флегмы в течение 6 часов. Смесь охлаждали до 40°C и перемешивали при 40°C в течение 16 часов. Смесь нагревали до 50°C (до растворения осадка), и раствор выпускали из реакционного сосуда. В реакционный сосуд загружали 21%-ный (масс./масс.) раствор этоксида натрия в этаноле (1,1 л, 3,48 моль) и этанол (2,0 л), и полученную смесь нагревали до 50°C. Реакционную смесь затем вливали назад в реакционный сосуд в течение 5 минут. Полученную смесь нагревали до температуры образования флегмы и перемешивали при температуре образования флегмы в течение 2 часов. Смесь охлаждали до 30°C и разделяли на 2 приблизительно равные порции. Каждую порцию обрабатывали Celite® (приблизительно 2 л) и концентрировали при пониженном давлении. Полученное твердое вещество суспендировали в воде (2,0 л, 40°C) и энергично перемешивали до тех пор, пока все твердое вещество не перешло в текучее состояние. Твердое вещество собирали фильтрованием через слой Celite®, и остаток на фильтре промывали водой (2,5 л, 40°C). Фильтрат промывали диэтиловым эфиром (2×1,3 л) и затем охлаждали до 15°C. Осторожно добавляли 6 М соляную кислоту до достижения pH 4. Полученный осадок собирали фильтрованием, и остаток на фильтре промывали разбавленной соляной кислотой (500 мл). Остаток на фильтре сушили в сушильном шкафу при 50°C в течение 2 суток. В результате получили указанное в заголовке соединение в виде желтого твердого вещества (584 г, 64%).
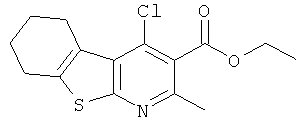
Подготовительный пример 4: Этил-4-хлор-2-метил-5,6,7,8-тетрагидро[1]бензотиено[2,3-b]пиридин-3-карбоксилат
Этил-4-гидрокси-2-метил-5,6,7,8-тетрагидро[1]бензотиено[2,3-b]пиридин-3-карбоксилат (Подготовительный пример 3, 150 г, 515 ммоль) порциями добавляли к оксихлориду фосфора (450 мл, 4,92 моль), и полученную смесь перемешивали при 100°C в течение 1 часа и затем охлаждали до комнатной температуры, Летучие вещества удаляли в вакууме, и остаток осторожно вливали в энергично перемешиваемую смесь лед/вода. Этилацетат (500 мл) добавляли в эту смесь. pH смеси доводили до значения 8 добавлением ЮМ водного раствора гидроксида натрия. Слои разделяли, и водный слой дополнительно экстрагировали этилацетатом (3×500 мл). Объединенные органические экстракты промывали рассолом (300 мл), сушили над MgSO4 и концентрировали в вакууме. Полученное масло вливали в кристаллизатор, где оно затвердевало при стоянии с образованием указанного в заголовке соединения, 140,69 г, выход 88%. 1H ЯМР (400 МГц, CDCl3) δ м.д. 1.42 (t, 3H), 1.86-1.89 (m, 4H), 2.59 (s, 3H), 2.82-2.84 (m, 2H), 3.08-3.11 (m, 2H), 4.46 (q, 2H).
Подготовительный пример 5: Этил-(4-йод-2-метил-5.6.7.8-тетрагидро[1]бензотиено[2.3b]пиридин-3-ил)карбоксилат
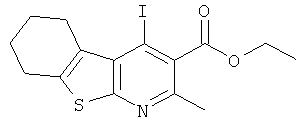
Йодид натрия (994,0 г, 6,63 моль) добавляли в течение 5 минут к перемешиваемому раствору ацетилхлорида (177 мл, 2,48 моль) и этил-(4-хлор-2-метил-5,6,7,8-тетрагидро[1]бензотиено[2,3-b]пиридин-3-ил)карбоксилата
(Подготовительный пример 4,257 г, 0,83 моль) в ацетонитриле (2,0 л) при комнатной температуре. Полученную смесь нагревали до температуры образования флегмы и перемешивали в течение 30 часов. Смесь охлаждали до комнатной температуры и выдерживали в течение 72 часов. Твердое вещество собирали фильтрованием и промывали холодным ацетонитрилом (500 мл).
Твердое вещество распределяли между дихлорметаном (1,5 л) и водой (0,75 л). Водный слой подщелачивали до pH 9 добавлением 2 М раствора гидроксида натрия, и два слоя разделяли. Органический слой промывали 2 М раствором тиосульфата натрия (800 мл), рассолом (800 мл), сушили (Na2SO4), фильтровали и концентрировали при пониженном давлении с получением указанного в заголовке соединения (260,0 г, 78%) в виде бежевого твердого вещества. Это вещество, как было определено 1H ЯМР спектроскопией, представляет собой смесь 13:1 этил-(4-йод-2-метил-5,6,7,8-тетрагидро[1]бензотиено[2,3-b]пиридин-3-ил)карбоксилата и этил-(4-хлор-2-метил-5,6,7,8-тетрагидро[1]бензотиено[2,3-b]пиридин-3-ил)карбоксилата соответственно.
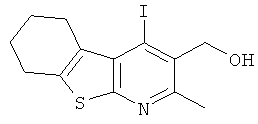
Подготовительный пример 6; (4-Йод-2-метил-5,6,7,8-тетрагидро[1]бензотиено[2,3-b]пиридин-3-ил)метанол
К раствору этил-(4-йод-2-метил-5,6,7,8-тетрагидро[1]бензотиено[2,3-b]пиридин-3-ил)карбоксилата (Подготовительный пример 5, 260,2 г, 0,65 моль) в CH2Cl2 (2,0 л) добавляли 25% (масс./масс.) гидрид диизобутилалюминия (2,0 л, 1,97 моль) при 5°C в течение 1,5 часов в атмосфере аргона. Полученной смеси давали возможность нагреться до комнатной температуры и перемешивали в течение 16 часов. Полученную смесь разбавляли дихлорметаном (1,0 л), охлаждали до 0°C и очень осторожно добавляли 2 М соляную кислоту (200 мл). Затем добавляли 6 М соляную кислоту до достижения pH 2. Полученную смесь энергично перемешивали в течение 1 часа. Осадок собирали фильтрованием, и остаток на фильтре промывали дихлорметаном (300 мл) и водой (300 мл). Твердое вещество суспендировали в дихлорметане (250 мл) и 1 М растворе гидроксида натрия (400 мл) и энергично перемешивали до тех пор, пока твердое вещество не перешло в текучее состояние. Твердое вещество собирали фильтрованием, промывая остаток на фильтре водой (200 мл). Остаток на фильтре концентрировали, удаляя последовательно пропан-2-ол (500 мл), метанол (500 мл) и дихлорметан (500 мл) при пониженном давлении, с получением указанного в заголовке соединения в виде твердого вещества кремового цвета (212,3 г, 91%).
Подготовительный пример 7: (4-Йод-2-метил-5,6,7,8-тетрагидро[1]бензотиено[2,3-b]пиридин-3-ил)карбальдегид
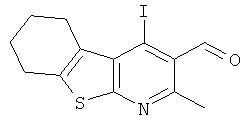
К суспензии (4-йод-2-метил-5,6,7,8-тетрагидро[1]бензотиено[2,3-b]пиридин-3-ил)метанола (Подготовительный пример 6, 150,0 г, 0,42 моль) в триэтиламине (174 мл, 1,25 моль) и диметилсульфоксиде (1,1 л) порциями при 5°C добавляли комплекс триоксид серы-пиридин. Полученную смесь перемешивали при комнатной температуре в течение 16 часов. Смесь вливали в энергично перемешиваемую ледяную воду (1,0 л). Твердое вещество собирали фильтрованием, и остаток на фильтре промывали водой (750 мл). Полученное твердое вещество растворяли в смеси дихлорметан/этилацетат (2,0 л, 9:1) (это требовало нагревания), и раствор сушили (MgSO4), фильтровали и концентрировали при пониженном давлении с получением указанного в заголовке соединения в виде белого твердого вещества (135,0 г, 91%). 1H ЯМР (400 МГц, CDCl3) δ 10.36 (s, 1H), 3.32-3.20 (m, 2H), 2.89-2.79 (m, 2H), 2.76 (s, 3H) 1.95-1.83 (m, 4H).
Подготовительный пример 8: 2-(4-Йод-2-метил-5,6,7,8-тетрагидро[1]бензотиено[2,3-b]пиридин-3-ил)-2-[(триметилсилил)окси]ацетонитрил
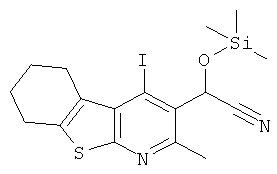
К суспензии 4-йод-2-метил-5,6,7,8-тетрагидро[1]бензотиено[2,3-b]пиридин-3-карбальдегида (Подготовительный пример 7, 115,31 г, 322,8 ммоль) в дихлорметане (1,75 л) добавляли йодид цинка (41,1 г, 128,8 ммоль), и полученную смесь охлаждали до 5°C. По каплям добавляли триметилсилилцианид (129 мл, 968,4 ммоль) добавляли в течение 10 минут, и смеси давали возможность нагреться до комнатной температуры в течение 2 часов. Добавляли воду (750 мл) и дихлорметан (1 л), и после перемешивания в течение 1 часа слои разделяли. Водный слой экстрагировали дихлорметаном (500 мл), и объединенные органические слои промывали водой (700 мл) и рассолом (700 мл), сушили над Na2SO4 и концентрировали в вакууме с получением указанного в заголовке соединения, 145,61 г, выход 99%. 1H ЯМР (400 МГц, CDCl3) δ м.д. 0.22 (s, 9H), 1.82-1.93 (m, 4H), 2.56-2.86 (m, 2H), 2.92 (s, 3H), 3.18-3.25 (m, 2H), 6.48 (s, 1H).
Подготовительный пример 9: Метил-2-[4-йод-2-метил-5,6,7,8-тетрагидро[1]бензотиено[2,3-b]пиридин-3-ил]-2-гидроксиацетат
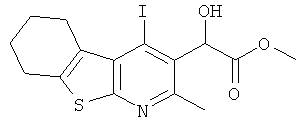
К перемешиваемой суспензии 2-(4-йод-2-метил-5,6,7,8-тетрагидро[1]бензотиено[2,3-b]пиридин-3-ил)-2-триметилсилоксиацетонитрила (Подготовительный пример 8, 168,0 г, 369,10 ммоль) в метаноле (2,5 л) добавляли концентрированную серную кислоту (410 мл, 7,38 моль) при 5°C. Смесь нагревали до 70°C и перемешивали в течение 36 часов. После охлаждения смеси до комнатной температуры смесь разбавляли водой (1,0 л) и этилацетатом (2,0 л) и затем охлаждали до 5°C. pH водного слоя доводили до значения pH 8 осторожным добавлением 6 М водного раствора гидроксида натрия. Это привело к осаждению твердого вещества. Твердое вещество собирали фильтрованием, и остаток на фильтре промывали теплой водой (500 мл) и этилацетатом (200 мл). Фильтрат сохраняли для дальнейшей обработки. Остаток на фильтре суспендировали в метаноле (500 мл) и энергично перемешивали до тех пор, пока твердое вещество не перешло в текучее состояние. Твердое вещество собирали фильтрованием, и остаток на фильтре промывали метанолом (2×200 мл) и диэтиловым эфиром (2×200 мл). Полученное твердое вещество сушили при 50°C в течение 16 часов. Это привело к получению указанного в заголовке соединения (66,5 г, 43%) в виде белого твердого вещества. Слои сохраненных фильтратов разделяли, и органический слой концентрировали при пониженном давлении. Полученный остаток растирали с метанолом (100 мл), этилацетатом (100 мл) и диэтиловым эфиром (100 мл). Полученное твердое вещество собирали фильтрованием и сушили при 50°C в течение 16 часов. Это привело к получению дополнительной порции указанного в заголовке соединения (38,3 г, 25%) в виде белого твердого вещества.
Подготовительный пример 10: Метил-трет-бутокси(4-йод-2-метил-5,6,7,8-тетрагидро[1]бензотиено[2,3-b]пиридин-3-ил)ацетат
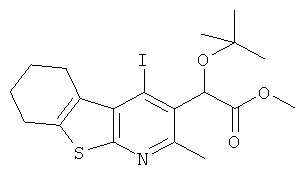
Суспензию метил-гидрокси(4-йод-2-метил-5,6,7,8-тетрагидро[1]бензотиено[2,3-b]пиридин-3-ил)ацетата (Подготовительный пример 9, 10,0 г, 23,97 ммоль) в трет-бутилацетате (250 мл) энергично перемешивали в течение 5 минут, после чего по каплям добавляли перхлорную кислоту (70%, 6,15 мл, 71,90 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 20 минут. Смесь нейтрализовали добавлением насыщенного водного раствора гидрокарбоната натрия (60 мл) и экстрагировали этилацетатом (250 мл). Слои разделяли, и водный слой дополнительно экстрагировали этилацетатом (250 мл). Объединенные органические слои промывали рассолом (250 мл), сушили над MgSO4 и концентрировали в вакууме с получением неочищенного продукта. Остаток очищали колоночной флэш-хроматографией, элюируя смесью этилацетат/гептан (10-40%), с получением указанного в заголовке соединения, 4,26 г, выход 38%. 1H ЯМР (400 МГц, CDCl3) δ м.д. 1.23 (s, 9H), 1.81-1.92 (m, 4H), 2.66 (s, 3H), 2.79-2.89 (m, 2H), 3.21-3.27 (m, 2H), 3.68 (s, 3H), 5.95 (s, 1H).
Подготовительный пример 11: Этил-2-трет-бутокси(4-йод-2-метил-5,6,7,8-тетрагидроГПбензотиено[2,3-b]пиридин-3-ил)ацетат
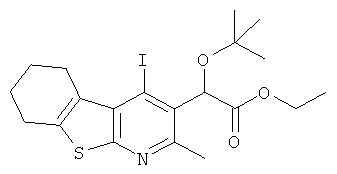
Этил-2-[4-йод-2-метил-5,6,7,8-тетрагидро[1]бензотиено[2,3-b]пиридин-3-ил]-2-гидроксиацетат (3 г, 7 ммоль) переносили в 20 мл дихлорметана и трет-бутилацетат (20 мл, 170 ммоль), и смесь охлаждали до 3°C в бане лед/вода. Затем по каплям добавляли концентрированную серную кислоту (1,14 мл, 21 ммоль), и смеси давали возможность нагреться до комнатной температуры в течение 3 часов. Реакцию гасили добавлением 100 мл 1М NaOH и 100 мл воды, и органический слой отделяли. Органическую фазу промывали рассолом (100 мл), сушили над MgSO4 и упаривали при пониженном давлении. Остаток очищали флэш-хроматографией, используя градиент EtOAc в гептане в качестве элюента (от 0:100 до 30:70) с получением указанного в заголовке соединения в виде белого твердого вещества (1,22 г, 36%). 1H ЯМР (400МГц, CDCl3) δ 1.20 (t, 3H), 1.23 (s, 9H), 1.81-1.94 (m, 4H), 2.66 (s, 3H), 2.80-2.87 (m, 2H), 3.22-3.31 (m, 2H), 4.10-4.22 (m, 2H), 5.90 (s, 1H).
Подготовительный пример 12: трет-Бутокси(4-йод-2-метил-5,6,7,8-тетрагидро[1]бензотиено[2.3-b]пиридин-3-ил)уксусная кислота
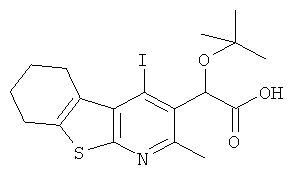
К раствору метил-трет-бутокси(4-йод-2-метил-5,6,7,8-тетрагидро[1]бензотиено[2,3-b]пиридин-3-ил)ацетата (Подготовительный пример 10, 63,95 г, 135 ммоль) в тетрагидрофуране (1 л) и денатурате (1 л) по каплям добавляли водный гидроксид натрия (1 M, 811 мл, 811 ммоль). Полученную смесь перемешивали при 60°C в течение 90 минут и затем охлаждали до комнатной температуры в течение одной ночи. Летучие растворители удаляли в вакууме, и оставшийся водный остаток разбавляли водой (400 мл) и экстрагировали трет-бутил-метиловым эфиром (600 мл). Органический слой промывали водой (400 мл) и затем охлаждали на льду. Добавляли 2 М соляную кислоту до pH 5, и полученное твердое вещество собирали фильтрованием, промывая водой. Твердое вещество растворяли в 2-метил-тетрагидрофуране (300 мл) и перемешивали в течение 10 минут. Появился водный слой, и его отделяли. Водный слой экстрагировали 2-метил-тетрагидрофураном (2×300 мл). Объединенные органические слои сушили над MgSO4 и концентрировали в вакууме с получением указанного в заголовке соединения в виде бледно-желтого твердого вещества, 54,35 г, выход 88%. 1H ЯМР (400 МГц, CDCl3) δ м.д. 1.27 (s, 9H), 1.81-1.93 (m, 4H), 2.65 (s, 3H), 2.76-2.88 (m, 2H), 3.20-3.27 (m, 2H), 6.13 (s, 1H), 9.66 (brs, 1H).
Подготовительный пример 13: (4R)-4-Бензил-3-[(2R)-2-трет-бутокси-2-(4-йод-2-метил-5,6,7,8-тетрагидро[1]бензотиено[2,3-b]пиридин-3-ил)ацетил1-1,3-оксазолидин-2-он (13A) и (4R)-4-бензил-3-[(2S)-2-трет-бутокси-2-(4-йод-2-метил-5,6,7,8-тетрагидро[1]бензотиено[2,3-b]пиридин-3-ил)ацетил1-1,3-оксазолидин-2-он (13B)
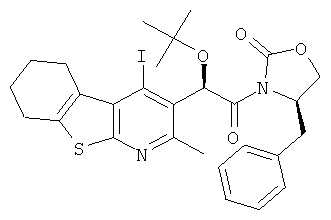
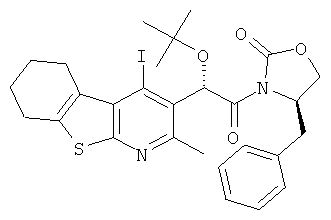
К раствору трет-бутокси(4-йод-2-метил-5,6,7,8-тетрагидро[1]бензотиено[2,3-b]пиридин-3-ил)уксусной кислоты (Подготовительный пример 12, 54,35 г, 118 ммоль) в тетрагидрофуране (1 л) добавляли гексафторфоссрат [(бензотриазол-1-илокси)-диметиламино-метилен]-диметил-аммония (67,31 г, 177 ммоль) и этил-ди-изопропил-амин (61,8 мл, 355 ммоль), и полученную реакционную смесь перемешивали при 40°C в течение 2 часов. Через 90 минут (R)-4-бензил-2-оксазолидинон (41,93 г, 237 ммоль) растворяли в тетрагидрофуране (THF) (500 мл) и обрабатывали гидридом натрия (60% в минеральном масле) и перемешивали в течение 30 минут при комнатной температуре. После этого две смеси объединяли и перемешивали при 40°C в течение 8 часов. Выпавшее в осадок вещество собирали фильтрованием. Твердое вещество распределяли между этилацетатом (500 мл) и насыщенным водным раствором гидрокарбоната натрия (500 мл). Слои разделяли, и водный слой дополнительно экстрагировали этилацетатом (2×500 мл). Объединенные органические слои промывали рассолом (500 мл), сушили над MgSO4 и концентрировали в вакууме с получением коричневого масла, 1,2 г. Первоначальный THF-фильтрат промывали насыщенным водным раствором гидрокарбоната натрия (500 мл) с добавлением этилацетата (1 л) для разделения слоев. Водный слой промывали этилацетатом (2×250 мл). Объединенные органические экстракты промывали рассолом (500 мл), сушили над MgSO4 и концентрировали в вакууме. Неочищенный остаток объединяли с коричневым маслом после обработки твердого вещества и очищали флэш-хроматографией на сухой колонке, элюируя с градиентом гептана и этилацетата (от 0% до 20%). Фракции, содержащие продукт, концентрировали в вакууме с получением бледно-желтого полутвердого вещества, 72 г.Остаток дополнительно очищали колоночной хроматографией, элюируя с градиентом гептана и этилацетата (от 0% до 10%) для разделения диастереомеров. Оба подвергали кристаллизации из денатурата. Верхнюю зону выделяли в виде белого твердого вещества, 23,75 г, выход 32% (13A). Нижнюю зону выделяли в виде белого твердого вещества, 21,36 г, выход 29% (13B). 13A: 1H ЯМР (400 МГц, CDCl3) δ м.д. 1.25 (s, 9H), 1.80-1.92 (m, 4H), 2.85 (s, 3H), 2.79-2.90 (m, 2H), 2.94 (dd, 1H), 3.15-3.30 (m, 2H), 4.16 (dd, 1H), 4.26 (t, 1H), 4.71-4.80 (m, 1H), 6.90 (s, 1H), 7.23-7.36 (m, 4H). 13B: 1H ЯМР (400 МГц, CDCl3) δ м.д. 1.17 (s, 9H), 1.82-1.95 (m, 4H), 2.84 (s, 3H), 2.78-2.87 (m, 3H), 3.18-3.31 (m, 2H), 4.17-4.22 (m, 1H), 4.30 (t, 1H), 4.70-4.78 (m, 1H), 6.93 (s, 1H), 7.17-7.33 (m, 4H).
Подготовительный пример 14: (2R)-трет-Бутокси(4-йод-2-метил-5.6.7.8-тетрагидро[1]бензотиено[2.3-b]пиридин-3-ил)уксусная кислота
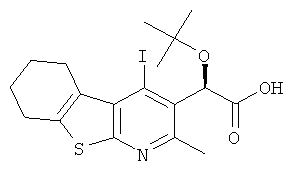
К раствору (4R)-4-бензил-3-[(2R)-2-трет-бутокси-2-(4-йод-2-метил-5,6,7,8-тетрагидро[1]бензотиено[2,3-b]пиридин-3-ил)ацетил]-1,3-оксазолидин-2-она (Подготовительный пример 13A, 23,75 г, 38,40 ммоль) в тетрагидрофуране (450 мл) и воде (150 мл) при 0°C по каплям добавляли раствор гидрата гидроксида лития (3,35 г, 80,64 ммоль) и перекись водорода (27%-ный водный раствор, 18,3 мл, 161,27 ммоль). Полученный раствор перемешивали при 0°C в течение 15 минут, затем в течение 2 часов при комнатной температуре. Добавляли насыщенный раствор сульфита натрия (500 мл), затем добавляли воду (500 мл), и смесь перемешивали в течение 15 минут. Смесь подкисляли до pH 4 добавлением 6 М соляной кислоты. Смесь экстрагировали дихлорметаном (4×500 мл). Объединенные органические слои сушили над MgSO4 и концентрировали в вакууме. Полученное белое твердое вещество подвергали кристаллизации из денатурата (150 мл) при нагревании при температуре образования флегмы в течение 5 минут. Смесь охлаждали до комнатной температуры в течение ночи. Полученное твердое вещество собирали фильтрованием, промывали холодным денатуратом и сушили в вакууме с получением указанного в заголовке соединения в виде белого твердого вещества, 12,01 г, выход 68%. 1H ЯМР (400 МГц, CDCl3) δ м.д. 1.27 (s, 9H), 1.81-1.93 (m, 4H), 2.65 (s, 3H), 2.75-2.88 (m, 2H), 3.18-3.28 (m, 2H), 6.13 (s, 1H), 9.66 (brs, 1H).
Подготовительный пример 15: (2S)-трет-Бутокси(4-йод-2-метил-5,6,7,8-тетрагидро[1]бензотиено[2,3-b]пиридин-3-ил)уксусная кислота
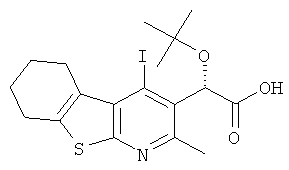
К раствору (4R)-4-бензил-3-[(2S)-2-трет-бутокси-2-(4-йод-2-метил-5,6,7,8-тетрагидро[1]бензотиено[2,3-b]пиридин-3-ил)ацетил]-1,3-оксазолидин-2-она
(Подготовительный пример 13 В, 21,36 г, 34,53 ммоль) в тетрагидрофуране (400 мл) и воде (130 мл) при 0°C по каплям добавляли раствор гидрата гидроксида лития (3,04 г, 72,52 ммоль) и перекись водорода (27%-ный водный раствор, 16,4 мл, 145,04 ммоль). Полученный раствор перемешивали при 0°C в течение 15 минут, затем в течение 2 часов при комнатной температуре. Добавляли насыщенный раствор сульфита натрия (500 мл), затем добавляли воду (500 мл), и смесь перемешивали в течение 15 минут. Смесь подкисляли до pH 4 добавлением 6 М соляной кислоты. Смесь экстрагировали дихлорметаном (4×500 мл). Объединенные органические слои сушили над MgSO4 и концентрировали в вакууме. Полученное белое твердое вещество подвергали кристаллизации из денатурата (150 мл) с нагреванием при температуре образования флегмы в течение 5 минут. Смесь охлаждали до комнатной температуры в течение ночи. Полученное твердое вещество собирали фильтрованием и промывали холодным денатуратом и сушили в вакууме с получением указанного в заголовке соединения в виде белого твердого вещества, 10,25 г, выход 65%. 1H ЯМР (400 МГц, CDCl3) δ м.д. 1.27 (s, 9H), 1.81-1.93 (m, 4H), 2.65 (s, 3H), 2.75-2.88 (m, 2H), 3.18-3.28 (m, 2H), 6.13 (s, 1H).
Подготовительный пример 16: Метил-(2S)-трет-бутокси(4-йод-2-метил-5,6,7,8-тетрагидроГПбензотиено[2,3-b]пиридин-3-ил)ацетат
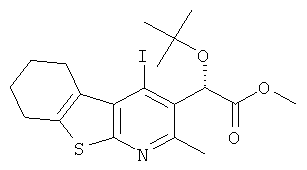
К раствору (2S)-трет-бутокси(4-йод-2-метил-5,6,7,8-тетрагидро[1]бензотиено[2,3-b]пиридин-3-ил)уксусной кислоты (Подготовительный пример 15, 1,0 г, 2,178 ммоль) в дихлорметане (34 мл) добавляли гексафторфосфат [(бензотриазол-1-илокси)-диметиламино-метилен]-диметил-аммония (1,24 г, 3,266 ммоль) и этил-ди-изопропил-амин (600 мкл, 3,266 ммоль), и полученную реакционную смесь перемешивали при 30°C в течение 2 часов. Добавляли метанол (17 мл), и реакционную смесь перемешивали при 30°C в течение 18 часов. Реакционную смесь охлаждали до комнатной температуры и разбавляли дихлорметаном (50 мл). Раствор промывали насыщенным водным раствором гидрокарбоната натрия (50 мл), водой (50 мл) и рассолом (50 мл). Органический слой сушили над MgSO4 и концентрировали в вакууме. Остаток очищали флэш-хроматографией, элюируя 5% этилацетатом в гептане с получением указанного в заголовке соединения в виде белого твердого вещества, 963 мг, выход 93%. 1H ЯМР (400 МГц, CDCl3) δ м.д. 1.22 (s, 9H), 1.85-1.88 (m, 4H), 2.65 (s, 3H), 2.82-2.86 (m, 2H), 3.23-3.26 (m, 2H), 3.68 (s, 3H), 5.94 (s, 1H).
Подготовительный пример 17: (4-Хлор-2-гидроксифенил)бороновая кислота
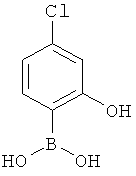
К раствору (4-хлор-2-метоксифенил)бороновой кислоты (1,0 г, 5,36 ммоль) в дихлорметане (5 мл) при 0°C добавляли трибромид бора (1 М в дихлорметане, 10 мл, 10 ммоль). Реакционную смесь перемешивали при 0°C в течение 1 часа, затем оставляли нагреваться до комнатной температуры и перемешивали в течение 18 часов. Реакцию осторожно гасили водой. Полученный осадок собирали фильтрованием с получением белого твердого вещества, 260 мг.
Слои разделяли, и органический слой сушили над Na2SO4 и концентрировали в вакууме с получением белого твердого вещества, 300 мг. Две порции объединяли с получением указанного в заголовке соединения в виде белого твердого вещества, 560 мг, выход 61%. Это вещество использовали в Подготовительном примере 18 без очистки.
Подготовительный пример 18: 5-Хлор-2-(4,4,5,5-тетраметил-1.3.2-диоксаборолан-2-ил)фенол
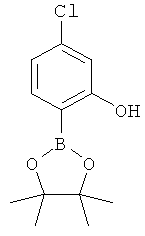
К раствору (4-хлор-2-гидроксифенил)бороновой кислоты (Подготовительный пример 15, 200 мг, 1,16 ммоль) в дихлорметане (15 мл) добавляли пинакон (274 мг, 2,32 ммоль), и полученный раствор перемешивали при комнатной температуре в течение 18 часов. Реакционную смесь промывали водой (10 мл), сушили над Na2SO4 и концентрировали в вакууме с получением указанного в заголовке соединения в виде бледно-желтого твердого вещества, 280 мг, выход 95%. 1H ЯМР (400 МГц, CDCl3) δ м.д. 1.35 (s, 12H), 6.86-6.88 (m, 2H), 7.51 (d, 1H), 7.89 (s, 1H).
Подготовительный пример 19: 2-(2-Фтор-4-метилфенил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан
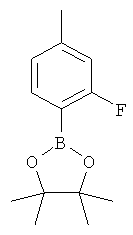
К раствору (2-фтор-4-метилфенил)бороновой кислоты (500 мг, 3,24 ммоль) в диэтиловом эфире (20 мл) добавляли пинакон (360 мг, 3,24 ммоль) и моногидрат 4-толуолсульфоновой кислоты (30 мг, 162 мкмоль), и полученный раствор перемешивали при комнатной температуре в течение 18 часов.
Реакционную смесь промывали насыщенным водным раствором гидрокарбоната натрия (20 мл), сушили над MgSO4 и концентрировали в вакууме с получением указанного в заголовке соединения в виде белого твердого вещества, 740 мг, выход 97%. 1H ЯМР (400 МГц, CDCl3) δ м.д. 1.33 (s, 12H), 2.34 (s, 3H), 6.82 (d, 1H), 6.93 (d, 1H), 7.60 (d, 1H).
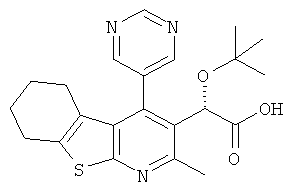
Пример 1: трет-Бутокси(2-метил-4-пиримидин-5-ил-5,6,7,8-тетрагидро[1]бензотиено[2,3-b]пиридин-3-ил)уксусная кислота
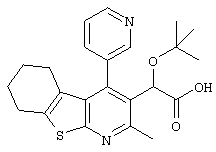
Метил-трет-бутокси[4-йод-2-метил-5,6,7,8-тетрагидро[1]бензотиено[2,3-b]пиридин-3-ил]ацетат (Подготовительный пример 10) может быть подвергнут взаимодействию с пиримидин-бороновой кислотой в присутствии карбоната калия и тетракис(трифенилфосфин)палладия(0) с получением метилового эфира вышеуказанного соединения. Гидролиз водным гидроксидом лития может быть использован для получения конечного продукта.
Противовирусная активность > 20 мкМ (n=2) (S8737E)
Пример 2: (2S)-трет-бутокси(2-метил-4-пиримидин-5-ил-5,6,7,8-тетрагидро[1]бензотиено[2,3-b]пиридин-3-ил)уксусная кислота
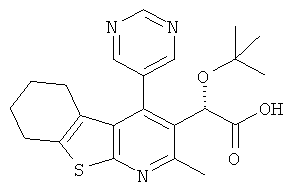
Образец продукта из Примера 1 может быть растворен в смеси метанола и дихлорметана (1:1), нанесен на колонку Chiralpak 1C (250×20 мм внутренний диаметр) и элюирован метанолом/OC2 (60:40) при температуре окружающей среды и скорости потока 60 г/мин. Фракции, содержащие единственный энантиомер, могут быть объединены и упарены при пониженном давлении с образованием продукта-энантиомера. Хиральная чистота может быть оценена хиральной ЖХВД с использованием колонки Chiralpak 1C, элюируя смесью гексан/изопропанол.
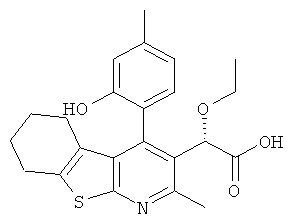
Пример 3: Этокси[4-(2-гидрокси-4-метилФенил)-2-метил-5,6,7,8-тетрагидро[1]бензотиено[2,3-b]пиридин-3-ил]уксусная кислота
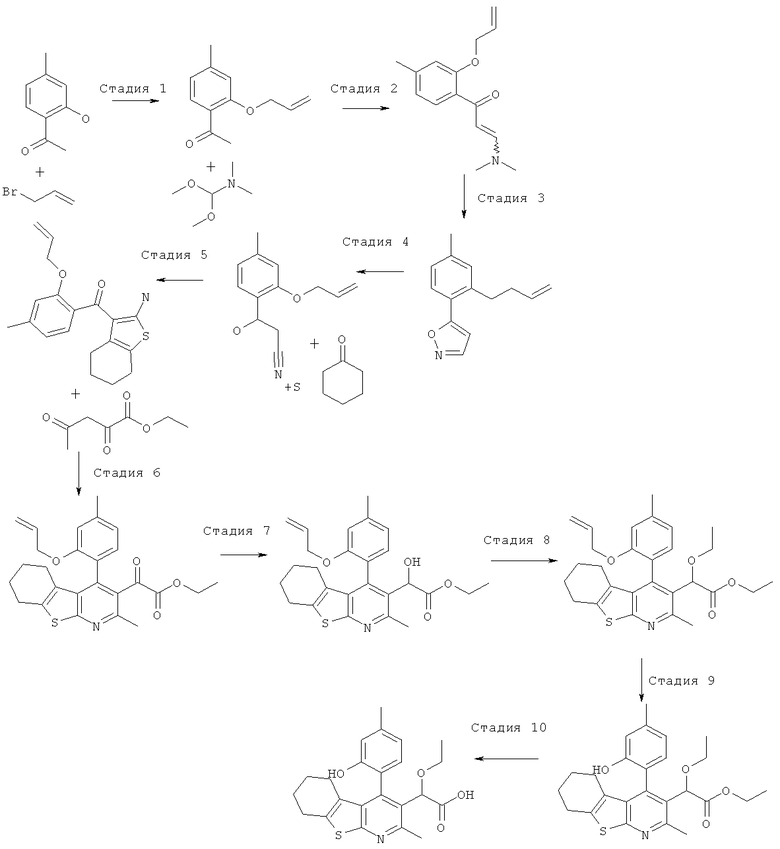
Стадия 1
К раствору 1-(2-гидрокси-4-метил-фенил)-этанона (10 г, 67 ммоль) в диметилформамиде (100 мл) добавляли карбонат калия (18,4 г, 2 экв., 133 ммоль), затем добавляли аллилбромид (8,06 г, 5,75 мл, 1 экв., 67 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Остаток распределяли между этилацетатом (200 мл) и водой (800 мл), органическую фазу отделяли, промывали рассолом (50 мл), сушили (MgSO4), фильтровали и концентрировали в вакууме. После охлаждения концентрата до комнатной температуры 1-(2-аллилокси-4-метил-фенил)-этанон кристаллизовался в виде бесцветных пластинок, 12,40 г (выход 98%). ЖХ/МС (2 мин, кислотная) 1,20 мин, УФ-чистота 72-100%, ЭРИ+/АД+ (электрораспылительная ионизация с регистрацией положительных ионов/ионизация при атмосферном давлении с регистрацией положительных ионов) 191. 1H ЯМР (CDCl3) чистота > 95% 7.68 (d, J=8.01 Гц, 1H), 6.80-6.83 (m, 1H), 6.76 (s, 1H), 6.10 (m, 1H), 5.44 (dq, J=17.5, 1.5 Гц, 1H), 5.33 (dq, J=11.0 Гц, 1.5 Гц, 1H), 4.61-4.66 (m, 2H), 2.63 (s, 3H), 2.37 (s, 3H).
Стадия 2
К 1-(2-аллилокси-4-метил-фенил)-этанону (13,9 г, 73 ммоль) добавляли диметилформамид диметилацеталь (56 мл, 422 моль, 5,8 экв.), и реакционную смесь нагревали до температуры образования флегмы в течение ночи. Реакционную смесь затем концентрировали в вакууме с получением 1-(2-аллилокси-4-метил-фенил)-3-диметиламино-пропенона (17,9 г) в виде оранжевого масла, которое без очистки переносили на следующую стадию.
Стадия 3
К перемешиваемому раствору неочищенного 1-(2-аллилокси-4-метил-фенил)-3-диметиламино-пропенона (14,1 г, 57,5 ммоль) в метаноле (70 мл) добавляли гидрохлорид гидроксиламина (4,4 г, 63 ммоль, 1,1 экв.), и эту реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Бесцветные игольчатые кристаллы отфильтровывали и анализировали, и установили, что они представляют собой 5-(2-аллилокси-4-метил-фенил)-изоксазол, 7,3 г (выход 58%). ЖХ/МС (2 мин, кислотная) 1,32 мин, УФ-чистота 64-91%, ЭРИ+/АД+216. 1H ЯМР (CDCl3) чистота>95% 7.88 (d, J=8.01 Гц, 1H), 6.88-6.92 (m, 1H), 6.82 (s, 1H), 34 6.77 (m, 1H), 6.05-6.20 (m, 1H), 5.46 (dq, J=17.0, 1.5 Гц, 1H), 5.35 (dq, J=10.5, 1.5 Гц, 1H), 4.67 (m, 2
Стадия 4
К перемешиваемой суспензии 5-(2-аллилокси-4-метил-фенил)-изоксазола (7,3 г, 33,8 ммоль) в этаноле (40 мл) добавляли этоксид натрия (21%-ный раствор в этаноле, 40 мл, 110 ммоль, 3,2 экв.), и эту реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь подкисляли до pH 2 соляной кислотой (2 н., водный раствор), и твердое вещество отфильтровывали и сушили на воздухе в течение 1 часа. Не совсем белое твердое вещество, 4,8 г (выход 66%), анализировали, и было установлено, что оно представляет собой чистый 3-(2-аллилокси-4-метил-фенил)-3-оксо-пропионитрил. ЖХ/МС (2 мин, кислотная) 1,12 мин, УФ-чистота 51-100%, ЭРИ+/АД+216, ЭРИ-/АД - (электрораспылительная ионизация с регистрацией отрицательных ионов/ионизация при атмосферном давлении с регистрацией отрицательных ионов) 214. 1H ЯМР (CDCl3) чистота > 95% 7.80 (d, J=8.0 Гц, 1H), 6.88 (dd, J=8.0, 1.0 Гц, 1H), 6.79 (s, 1H), 6.13 (m, 1H), 5.37-5.50 (m, 2H), 4.67-4.70 (m, 2H), 4.08 (s, 2H), 2.41 (s, 3H).
Стадия 5
К перемешиваемому раствору 3-(2-аллилокси-4-метил-фенил)-3-оксо-пропионитрила (1 г, 4,6 ммоль) в этаноле (20 мл) добавляли циклогексанон (684 мг, 722 мкл, 7 ммоль, 1,5 экв.) и серу (224 мг, 7 ммоль, 1,5 экв.), затем добавляли морфолин (607 мг, 610 мкл, 7 ммоль, 1,5 экв.), и эту реакционную смесь перемешивали в течение ночи при 40°C. Реакционную смесь концентрировали в вакууме. Остаток очищали, используя ISCO Companion с силикагелевым (40 г) картриджем Redisep и градиент гептана и этилацетата (от 0% до 40%). Фракции, содержащие целевой продукт, объединяли и концентрировали в вакууме с получением (2-аллилокси-4-метил-фенил)-(2-амино-4,5,6,7-тетрагидро-бензо[b]тиофен-3-ил)-метанона в виде желтой смолы, 1,1 г (выход 72%). ЖХ/МС (2 мин, кислотная) 1,45 мин, УФ-чистота 58-100%, ЭРИ+/АД+ 328. 1H ЯМР (CDCl3) чистота > 95% 7.08 (d, J=7.5 Гц, 1H) 6.91 (br. s., 2H) 6.78 (dq, J=7.5, 0.5 Гц, 1H) 6.69 (s, 1H) 5.92 (m, J=17.0, 10.5, 5.0, 5.0 Гц, 1H) 5.13-5.28 (m, 2H) 4.52 (dt, J=5.0, 2.0 Гц, 2H) 2.47 (tt, J=6.0, 2.0 Гц, 2H) 2.36 (s, 3H) 1.78 (tt, J=6.0, 2.0 Гц, 2H) 1.64-1.71 (m, 2H) 1.44-1.51 (m, 2H).
Стадия 6
К перемешиваемому раствору (2-аллилокси-4-метил-фенил)-(2-амино-4,5,6,7-тетрагидро-бензо[b]тиофен-3-ил)-метанона (882 мг, 2,69 ммоль) в этаноле (30 мл) добавляли этил-2,4-диоксопентаноат (426 мг, 378 мкл, 1 экв.), затем добавляли ацетилхлорид (846 мг, 766 мкл, 4 экв.), и эту реакционную смесь нагревали до 50°C в течение 1 часа. Реакционную смесь затем концентрировали в вакууме с получением гидрохлорида этилового эфира [4-(2-аллилокси-4-метил-фенил)-2-метил-5,6,7,8-тетрагидро-бензо[4,5]тиено[2,3-b]пиридин-3-ил]-оксо-уксусной кислоты в виде бледно-желтого масла, 1 г (выход 86%). ЖХ/МС.(2 мин, кислотная) 1,70 мин, УФ-чистота 54-100%, ЭРИ+/АД+450. 1H ЯМР (CDCl3) чистота > 90% 6.95 (d, J=7.0 Гц, 1H) 6.87 (d, J=7.0 Гц, 1H) 6.76 (s, 1H) 5.82 (m, 1H) 5.09-5.20 (m, 2H) 4.39-4.51 (m, 2H) 3.84-3.94 (m, 2H) 2.95 (s, 3H) 2.87-2.93 (m, 2H) 2.43 (s, 3H) 1.92-2.08 (m, 2H) 1.78-1.89 (m, 2H) 1.55-1.70 (m, 2H) 1.12 (t, J=7.1 Гц, 3H).
Стадия 7
К перемешиваемому раствору гидрохлорида этилового эфира [4-(2-аллилокси-4-метил-фенил)-2-метил-5,6,7,8-тетрагидро-бензо[4,5]тиено[2,3-b]пиридин-3-ил]-оксо-уксусной кислоты (1,1 г, 2,56 ммоль) в этаноле (20 мл) добавляли боргидрид натрия (145 мг, 1,5 экв., 3,84 ммоль), и эту реакционную смесь перемешивали при комнатной температуре в течение 5 минут.Реакционную смесь концентрировали в вакууме, и остаток распределяли между этилацетатом (20 мл) и соляной кислотой (водный раствор, 1 н., 30 мл). Органическую фазу отделяли, промывали рассолом (10 мл), сушили (MgSO4), фильтровали и концентрировали в вакууме с получением неочищенного этилового эфира [4-(2-аллилокси-4-метил-фенил)-2-метил-5,6,7,8-тетрагидро-бензо[4,5]тиено[2,3-b]пиридин-3-ил]-гидрокси-уксусной кислоты в виде бледно-оранжевой смолы, 1,1 г (выход 91%). ЖХ/МС (2 мин, кислотная) 1,53 мин, УФ-чистота 52-81%, ЭРИ+/АД+ 452. 1H ЯМР (CDCl3) чистота > 80% 7.02 (d, J=7.4 Гц, 1H) 6.81-6.85 (m, 1H) 6.79 (s, 1H) 5.86 (m, 1H) 5.18 (s, 1H) 5.08-5.17 (m, 2H) 4.47-4.51 (m, 2H) 4.08-4.22 (m, 2H) 2.81 (t, J=6.2 Гц, 2H) 2.63 (s, 3H) 2.43 (s, 3H) 1.71-1.87 (m, 4H) 1.53-1.64 (m, 2H) 1.17-1.21 (m, 3H).
Стадия 8
К перемешиваемому раствору этилового эфира [4-(2-аллилокси-4-метил-фенил)-2-метил-5,6,7,8-тетрагидро-бензо[4,5]тиено[2,3-b]пиридин-3-ил]-гидрокси-уксусной кислоты (100 мг, 221 мкмоль, 1 экв.) в MeCN (5 мл) добавляли оксид серебра (102 мг, 442 мкмоль, 2 экв.), затем добавляли йодэтан (172 мг, 89 мкл, 5 экв.), и эту реакционную смесь перемешивали в течение 16 часов при 60°C. Добавляли дополнительную порцию йодэтана (1 мл, 50 экв.) и оксида серебра(1) (102 мг, 2 экв.), и продолжали нагревание реакционной смеси при 60°C в течение 16 часов. Реакционную смесь разбавляли MeCN (5 мл) и фильтровали. Фильтрат затем концентрировали в вакууме. Остаток очищали, используя ISCO Companion с силикагелевым (12 г) картриджем Redisep и градиент гептана и этилацетата (от 0% до 30%). Фракции, содержащие этиловый эфир [4-(2-аллилокси-4-метил-фенил)-2-метил-5,6,7,8-тетрагидро-бензо[4,5]тиено[2,3-b]пиридин-3-ил]-этокси-уксусной кислоты объединяли и концентрировали в вакууме с получением целевого продукта в виде бледно-оранжевого масла, 22 мг (выход 22%). ЖХ/МС (12 мин, кислотная) 7,55 мин, УФ-чистота 100%, ЭРИ+/ХИАД+480.
Стадия 9
К перемешиваемому раствору этилового эфира [4-(2-аллилокси-4-метил-фенил)-2-метил-5,6,7,8-тетрагидро-бензо[4,5]тиено[2,3-b]пиридин-3-ил]-этокси-уксусной кислоты (22 мг, 46 мкмоль) в дихлорметане (3 мл) добавляли 1,3-диметилбарбитуровую кислоту (38 мг, 240 мкмоль, 5 экв.), и реакционный сосуд вакуумировали и заполняли азотом. Добавляли тетракис(трифенилрфосфин)палладий (1,2 мг, 2 мол. %), и реакционную смесь нагревали до температуры образования флегмы в течение 16 часов. Реакционную смесь концентрировали в вакууме. Остаток очищали, используя ISCO Companion с силикагелевым (4 г) картриджем Redisep и градиент гептана и этилацетата (от 0% до 40%). Фракции, содержащие целевой продукт, объединяли и концентрировали в вакууме с получением этилового эфира этокси-[4-(2-гидрокси-4-метил-фенил)-2-метил-5,6,7,8-тетрагидро-бензо[4,5]тиено[2,3-b]пиридин-3-ил]-уксусной кислоты в виде бледно-желтого масла, 20 мг (выход 95%). ЖХ/МС (12 мин, кислотная) 6,40 мин, УФ-чистота 58-69%, ЭРИ+/ХИАД+ (электрораспылительная ионизация с регистрацией положительных ионов/химическая ионизация при атмосферном давлении с регистрацией положительных ионов) 440, ЭРИ-/ХИАД- (электрораспылительная ионизация с регистрацией отрицательных ионов/химическая ионизация при атмосферном давлении с регистрацией отрицательных ионов) 438; 6,66 мин, УФ-чистота 27-31%, ЭРИ+/ХИАД+440, ЭРИ-/ХИАД-438.
Стадия 10
К перемешиваемому раствору этилового эфира этокси-[4-(2-гидрокси-4-метил-фенил)-2-метил-5,6,7,8-тетрагидро-бензо[4,5]тиено[2,3-b]пиридин-3-ил]-уксусной кислоты (20 мг, 45 мкмоль) в этаноле (1 мл) и тетрагидрофуране (1 мл) добавляли гидроксид натрия (2 н. водный раствор, 0,5 мл, 20 экв.), и эту реакционную смесь перемешивали при 60°C в течение 5 часов. Реакционную смесь концентрировали в вакууме до удаления всех органических растворителей, водный остаток затем подкисляли соляной кислотой (2 н. водный раствор) до рН 2. Бледно-желтое твердое вещество, 4 мг (выход 16%) отфильтровывали и анализировали, и было установлено, что оно содержит этокси-[4-(2-гидрокси-4-метил-фенил)-2-метил-5,6,7,8-тетрагидро-бензо[4,5]тиено[2,3-b]пиридин-3-ил]-уксусную кислоту. ЖХ/МС (12 мин, кислотная) 5,04 мин, УФ-чистота 47-43%, ЭРИ+/ХИАД+412, ЭРИ-/ХИАД-410; 5,80 мин, УФ-чистота 12-13%, ЭРИ+/ХИАД+412, ЭРИ-/ХИАД-410.
Пример 4: 2-трет-бутокси(4-йод-2-метил-5,6,7,8-тетрагидро[1]бензотиено[2,3-b]пиридин-3-ил)уксусная кислота
5-Бензотиазол-бороновую кислоту (150 мкмоль) и этил-2-mpem-бутокси(4-йод-2-метил-5,6,7,8-тетрагидро[1]бензотиено[2,3-b]пиридин-3-ил)ацетат (Подготовительный пример 11, 1 мл 0,1М раствора в диоксане, 100 мкмоль) добавляли в реакционный сосуд. Затем добавляли 1М раствор карбоната цезия (200 мкл, 200 мкмоль) в воде, затем добавляли 5 мкмоль Pd(dppf)Cl2, и все вместе перемешивали при 100°C в течение 16 ч, после чего охлаждали до комнатной температуры, и раствор упаривали при пониженном давлении с получением желтого остатка. В каждый сосуд добавляли 2М раствор гидроксида лития в воде (200 мкл, 400 мкмоль), затем добавляли 1 мл THF, и смесь встряхивали при комнатной температуре в течение 16 ч. pH раствора доводили до нейтрального значения, используя 1М водный раствор соляной кислоты, после чего смесь упаривали досуха при пониженном давлении, и остаток затем очищали препаративной ЖХВД на колонке C18, используя смесь ацетонитрила и воды в качестве подвижной фазы. После упаривания соответствующих фракций при пониженном давлении получили указанное в заголовке соединение (11 мг, 23%) в виде белого твердого вещества. ЭРИ/ХИАД(+): 467 (M+H).
Соединения указанных ниже Примеров синтезировали согласно способу, описанному в Пример 4, используя соответствующую арилбороновую кислоту.
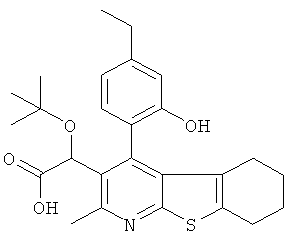
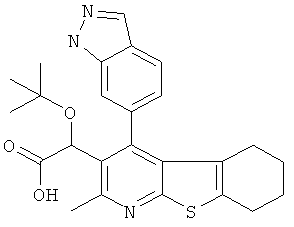
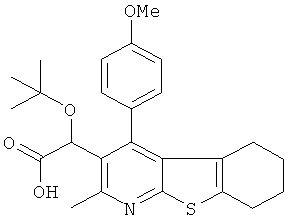
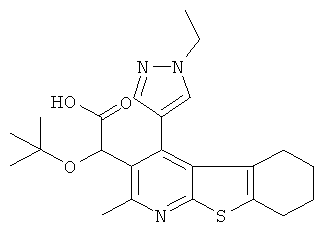
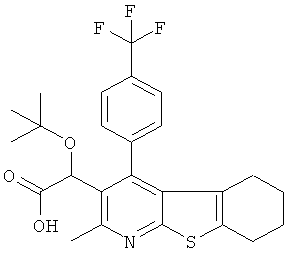
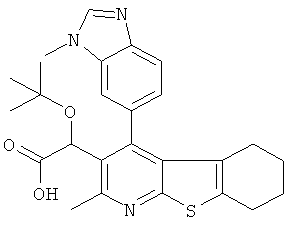
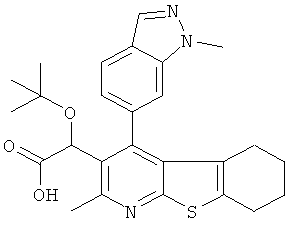
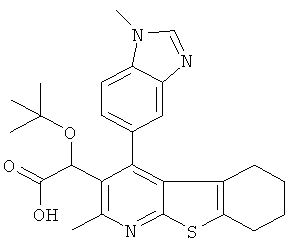
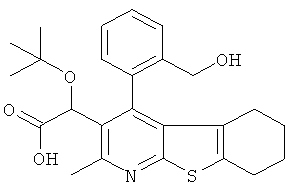
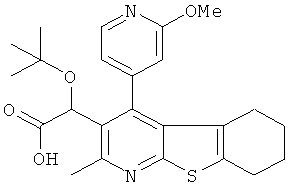
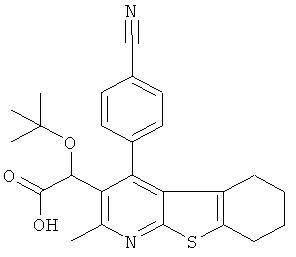
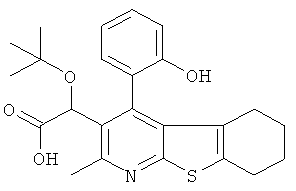
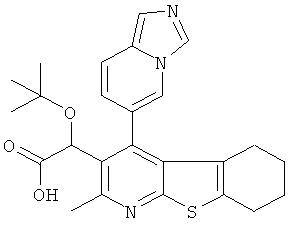
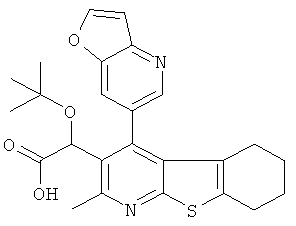
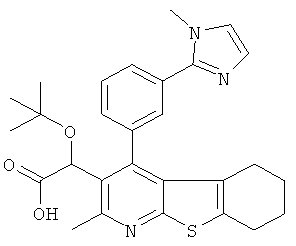
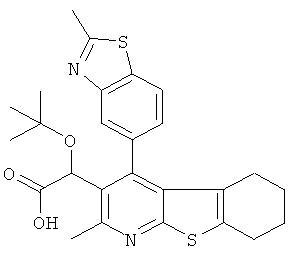
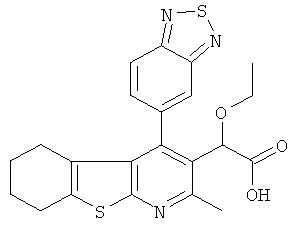
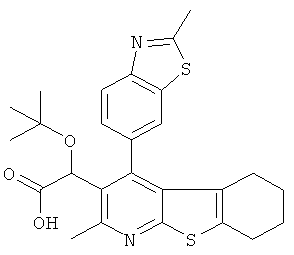
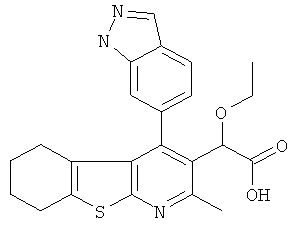
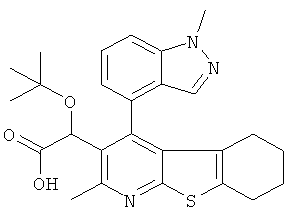
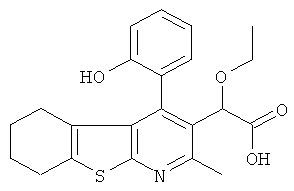
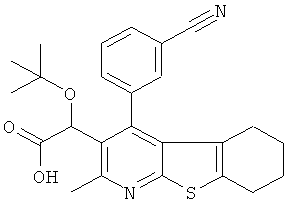
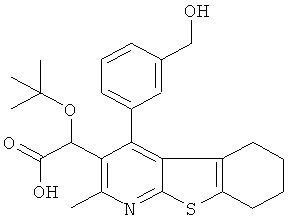

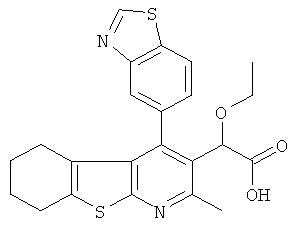
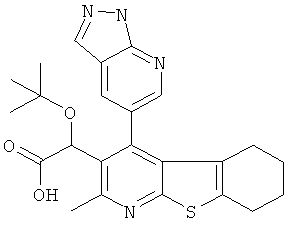
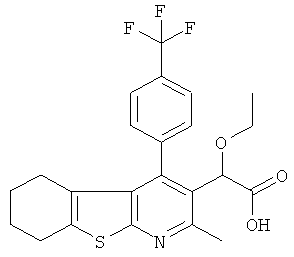
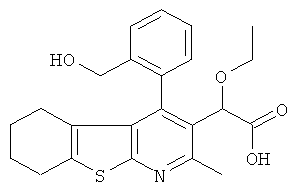
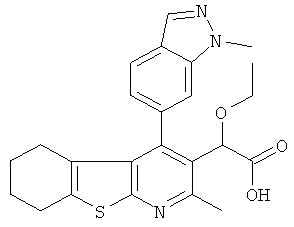
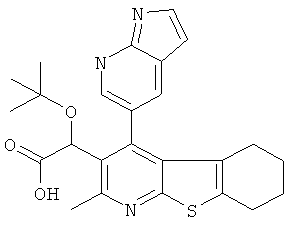
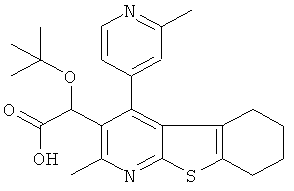
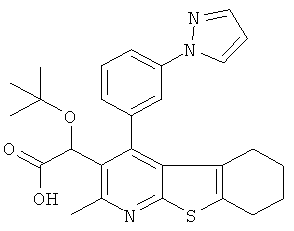
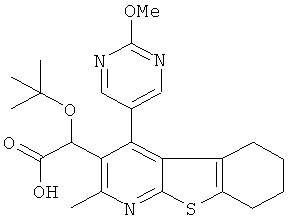
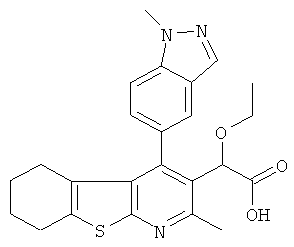
Пример 44: (23)-трет-бутокси[4-(4-хлорфенил)-2-метил-5,6,7,8-тетрагидро[1]бензотиено[2,3-b]пиридин-3-ил]уксусная кислота
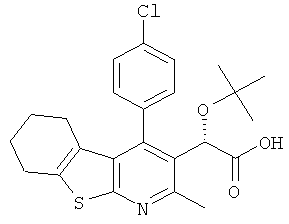
Стадия 1: Метил-(2S)-трет-бутокси[4-(4-хлорфенил)-2-метил-5,6,7,8-тетрагидро[1]бензотиено[2,3-b]пиридин-3-илацетат
К раствору метил-(28)-трет-бутокси(4-йод-2-метил-5,6,7,8-тетрагидро[1]бензотиено[2,3-b]пиридин-3-ил)ацетата (Подготовительный пример 16, 100 мг, 212 мкмоль) в диоксане (2 мл) добавляли 4-хлорбензол-бороновую кислоту (66 мг, 422 мкмоль), этил-ди-изопропил-амин (120 мкл, 636 мкмоль), воду (500 мкл) и тетракис(трифенилрфосфин)палладий (25 мг, 20 мкмоль). Реакционную смесь дегазировали и перемешивали при 100°C в герметично закрытой пробирке. Реакционную смесь охлаждали до комнатной температуры и разбавляли этилацетатом (10 мл). Смесь пропускали через слой Celite. Органический фильтрат промывали водой (10 мл) и рассолом (10 мл), сушили над MgSO4 и концентрировали в вакууме с получением неочищенного продукта. Остаток очищали флэш-хроматографией, элюируя этилацетатом в гептане (0-6%), с получением указанного в заголовке соединения в виде белого полутвердого вещества, 60 мг, выход 62%. 1H ЯМР (400 МГц, CDCl3) δ м.д. 0.97 (s, 9H), 1.45-1.48 (m, 1H), 1.62-1.73 (m, 3H), 1.78-1.82 (m, 2H), 2.71 (s, 3H), 2.78-2.81 (m, 2H), 3.66 (s, 3H), 4.99 (s, 1H), 7.18-7.21 (m, 1H), 7.39-7.42 (m, 3H).
Стадия 2; (23)-трет-бутокси[4-(4-хлорфенил)-2-метил-5,6,7,8-тетрагидро[1]бензотиено[2,3-b]пиридин-3-ил]уксусная кислота
К раствору метил-(2S)-трет-бутокси[4-(4-хлорфенил)-2-метил-5,6,7,8-тетрагидро[1]бензотиено[2,3-b]пиридин-3-ил]ацетата (60 мг, 131 мкмоль) в тетрагидрофуране (2 мл) и денатурате (2 мл) добавляли водный раствор гидроксида натрия (1 M, 790 мкл, 790 мкмоль). Полученный раствор перемешивали при 60°C в течение 3 часов. Летучие растворители удаляли в вакууме, и остаток разбавляли водой (10 мл). В смесь добавляли дихлорметан (20 мл), и затем подкисляли ее до pH 5 добавлением 2 М водного раствора соляной кислоты. Органический слой отделяли и промывали водой (10 мл) и рассолом (10 мл), сушили над MgSO4 и концентрировали в вакууме. Остаток подвергали перекристаллизации из 2-пропанола, и полученное твердое вещество собирали фильтрованием с получением указанного в заголовке соединения в виде белого твердого вещества, 11,2 мг, выход 19%. 1H ЯМР (400 МГц, CDCl3) δ м.д. 1.02 (s, 9H), 1.45-1.48 (m, 1H), 1.67-1.73 (m, 3H), 1.78-1.84 (m, 2H), 2.69 (s, 3H), 2.79-2.81 (m, 2H), 5.11 (s, 1H), 7.18-7.22 (m, 1H), 7.42-7.46 (m, 2H), 7.59-7.61 (m, 1H).
Противовирусная активность = 0,036 мкМ (n=6) (S8737E).
HTRF анализ взаимодействия = 576 нМ (n=10) (S9118)
Пример 45: (23)-трет-бутокси[4-(4-хлор-2-фторфенил)-2-метил-5,6,7,8-тетрагидроГПбензотиено[2,3-b]пиридин-3-ил1уксусная кислота
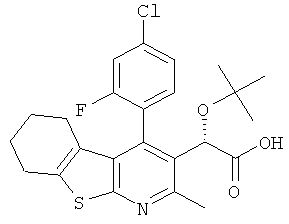
Стадия 1: Метил-(2S)-трет-бутокси[4-(4-хлор-2-срторсренил)-2-метил-5,6,7,8-тетрагидроГПбензотиено[2,3-b]пиридин-3-ил]ацетат
Указанное в заголовке соединение было получено из метил-(2S)-трет-бутокси(4-йод-2-метил-5,6,7,8-тетрагидро[1]бензотиено[2,3-b]пиридин-3-ил)ацетата (Подготовительный пример 16, 100 мг, 212 мкмоль) и (4-хлор-2-фторфенил)-бороновой кислоты (74 мг, 424 мкмоль) способом, описанным в Примере 44, Стадия 1, с выходом 57 мг, 56%. Полученное вещество представляло собой смесь атропизомеров в соотношении 3:1. 1H ЯМР (400 МГц, 43 CDCl3) δ м.д. 1.01 (s, 9Н), 1.48-1.55 (m, 1Н), 1.69-1.75 (m, 3H), 1.95-2.03 (m, 1H), 2.72 (s, 3H), 2.80-2.86 (m, 3H), 3.66 (s, 3H), 4,98 (s, 1H), 7,19-7,21 (m, 1H), 7,25-7,27 (m, 1H), 7,35-7,37 (m, 1H).
Стадия 2: (2S)-трет-бутокси[4-(4-хлор-2-фторфенил-2-метил-5,6,7,8-тетрагидро[1]бензотиено[2,3-b]пиридин-3-ил1уксусная кислота
Указанное в заголовке соединение было получено из метил-(2S)-трет-бутокси[4-(4-хлор-2-фторфенил)-2-метил-5,6,7,8-тетрагидро[1]бензотиено[2,3-b]пиридин-3-ил]ацетата (57 мг, 119 мкмоль) способом, описанным в Примере 44, Стадия 2. В результате очистки колоночной флэш-хроматографией, элюируя 5% метанолом в дихлорметане, получили указанное в заголовке соединение, 24 мг, с выходом 24%. Вещество представляет собой единственный атропизомер. Второй атропизомер не был выделен. 1H ЯМР (400 МГц, CDCl3) δ м.д. 1.04 (s, 9H), 1.45-1.51 (m, 1H), 1.62-1.76 (m, 3H), 1.82-1.84 (m, 1H), 2.05-2.10 (m, 1H), 2.69 (s, 3H), 2.80-2.84 (m, 2H), 5.08 (s, 1H), 7.22-7.28 (m, 2H), 7.59-7.61 (m, 1H).
Противовирусная активность = 0,023 мкМ (n=6) (S8737E).
HTRF анализ взаимодействия = 565 нМ (n=10) (S9118)
Пример 46: (2S)-трет-бутокси[4-(4-хлор-2-гидроксифенил)-2-метил-5,6,7,8-тетрагидро[1]бензотиено[2,3-b]пиридин-3-ил]уксусная кислота

Стадия 1: Метил-(2S)-трет-бутоксих4-(4-хлор-2-гидроксифенил)-2-метил-5,6,7,8-тетрагидро[1]бензотиено[2,3-b]пиридин-3-ил]ацетат
К раствору метил-(2S)-трет-бутокси(4-йод-2-метил-5,6,7,8-тетрагидро[1]бензотиено[2,3-b]пиридин-3-ил)ацетата (Подготовительный пример 16, 200 мг, 422 мкмоль) в диоксане (4 мл) добавляли (4-хлор-2-гидроксифенил)-бороновую кислоту (Подготовительный пример 17, 191 мг, 761 мкмоль), дихлор-[1,1'-бис(ди-трет-бутилфосфино)]ферроцен-палладий(II)™ (Pd-118) (Johnson-Matthey, 27 мг, 10 мол. %, 42,2 мкмоль), фосфат калия (180 мг, 844 мкмоль) и воду (1 мл). Полученную смесь перемешивали при 105°C в герметично закрытой пробирке в течение 18 часов. Добавляли дополнительную порцию Pd-118 (10 мг, 3,7 мол. %, 15,6 мкмоль), и смесь перемешивали при 105°C в течение 72 часов. Реакционную смесь охлаждали до комнатной температуры и фильтровали через короткую колонку с диоксидом кремния, промывая этилацетатом. Растворитель удаляли в вакууме. Неочищенный продукт очищали колоночной хроматографией, элюируя смесью этилацетат/гептан (0-10%), с получением указанного в заголовке соединения в виде коричневого твердого вещества, 37,5 мг, выход 19%. Полученное вещество представляло собой смесь атропизомеров в соотношении примерно 2:1, и его переносили на стадию гидролиза 2 без дополнительной очистки.
Стадия 2: (2S)-трет-бутокси[4-(4-хлор-2-гидроксифенил)-2-метил-5,6,7,8-тетрагидроГПбензотиено[2,3-b]пиридин-3-ил]уксусная кислота
Указанное в заголовке соединение было получено из метил-(2S)-трет-бугокси[4-(4-хлор-2-гидроксифенил)-2-метил-5,6,7,8-тетрагидро[1]бензотиено[2,3-b]пиридин-3-ил]ацетата (70 мг, 148 мкмоль) способом, описанным в Примере 44, Стадия 2. В результате очистки колоночной флэш-хроматографией, элюируя этилацетатом, затем 20% метанолом в этилацетате, получены выделенные атропизомеры 46А: 26 мг, выход 24%, и 46B: 7 мг, выход 10%. 46A: 1H ЯМР (400 МГц, CD3OD) δ м.д. 1.02 (s, 9Н), 1.50-1.53 (m, 1H), 1.65-1.98 (m, 4H), 2.18-2.21 (m, 1H), 2.64 (s, 3H), 2.79-2.81 (m, 2H), 5.13 (s, 1H), 6.91 (s, 1Н), 6.97 (d, 1H), 7.27 (d, 1H). 46B: 1H ЯМР(400 МГц, CD2OD) δ м.д. 1.00 (s, 9H), 1.49-1.51 (m, 1H), 1.65-1.85 (m, 4H), 2.20-2.26 (m, 1H), 2.69 (s, 3H), 2.79-2.81 (m, 2H), 5.05 (s, 1H), 6.88 (s, 1H), 6.92 (d, 1H), 7.40 (d, 1H).
Атропизомер 46A: Противовирусная активность = 0,014 мкМ (n=2) (S8737E)
HTRF анализ взаимодействия = 558 нМ (n=6) (S9118)
Атропизомер 46B: Противовирусная активность = 0,040 мкМ (n=2) (S8737E).
HTRF анализ взаимодействия = 798 нМ (n=2) (S9118)
Пример 47: (2S)-трет-бутокси[4-(2-фтор-4-метилфенил)-2-метил-5,8,7,8-тетрагидро[1]бензотиено[2,3-b]пиридин-3-ил]уксусная кислота
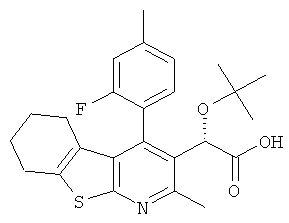
Стадия 1: Метил-(2S)-трет-бутокси[4-(2-фтор-4-метилфенил)-2-метил-5,6,7,8-тетрагидро[1]бензотиено[2,3-b]пиридин-3-ил]ацетат
Указанное в заголовке соединение было получено из метил-(28)-трет-бутокси(4-йод-2-метил-5,6,7,8-тетрагидро[1]бензотиено[2,3-Ь]пиридин-3-ил)ацетата (Подготовительный пример 16, 100 мг, 212 мкмоль) и 2-(2-фтор-4-метилфенил)-4,4,5,5-тетраметил-1,3,2-диоксаборолана (Подготовительный пример 19, 99 мг, 424 мкмоль) способом, описанным в Примере 44, Стадия 1, выход 100 мг, 96%. Полученное вещество представляло собой смесь атропизомеров в соотношении 2:1.
1H ЯМР (400 МГц, CDCl3) δ м.д. 0.99 (s, 5.8H), 1.05 (s, 3.2H), 1.26-2.08 (m, 6H), 2.45 (s, 3H), 2.71 (s, 1.8H), 2.76-2.80 (m, 1.2H), 3.58 (s, 1.2H), 3.66 (s, 1.8H), 5.06 (s, 0.66H), 5.07 (s, 0.33H), 6.94-7.02 (m, 2.4H), 7.23-7.25 (m, 0.6H).
Стадия 2: (2S)-трет-бутокси[4-(2-фтор-4-метилфенил)-2-метил-5.6.7.8-тетрагидро[1]бензотиено[2,3-b]пиридин-3-ил1уксусная кислота
Указанное в заголовке соединение было получено из метил-(2S)-трет-бутокси[4-(2-фтор-4-метилфенил)-2-метил-5,6,7,8-тетрагидро[1]бензотиено[2,3-b]пиридин-3-ил]ацетата (100 мг, 220 мкмоль) способом, описанным в Примере 44, Стадия 2. В результате очистки колоночной флэш-хроматографией, элюируя 5% метанолом в дихлорметане, разделения атропизомеров не произошло, и смесь все еще состояла из двух атропизомеров в соотношении 2:1, 40 мг, 41%. 1H ЯМР (400 МГц, CDCl3) δ м.д. 0.95 (s, 5.8Н), 1.05 (s, 3.2H), 1.26-2.08 (m, 6H), 2.44 (s, 1.2H), 2.45 (s, 1.8H), 2.68 (s, 1.8H), 2.75 (s, 1.2H), 2.76-2.81 (m, 2H), 5.05 (s, 0.66H), 5.11 (s, 0.33H), 7,02-7.13 (m, 2.4H), 7.45-7.49 (m, 0.6H).
Противовирусная активность = 0,071 мкМ (n=2) (S8737E).
HTRF анализ взаимодействия = 657 нМ (n=2) (S9118)
Пример 48: трет-Бутокси[4-(2-гидрокси-4-метилсЬенил)-2-метил-5,6,7,8-тетрагидро[1]бензотиено[2,3-b]пиридин-3-ил]уксусная кислота
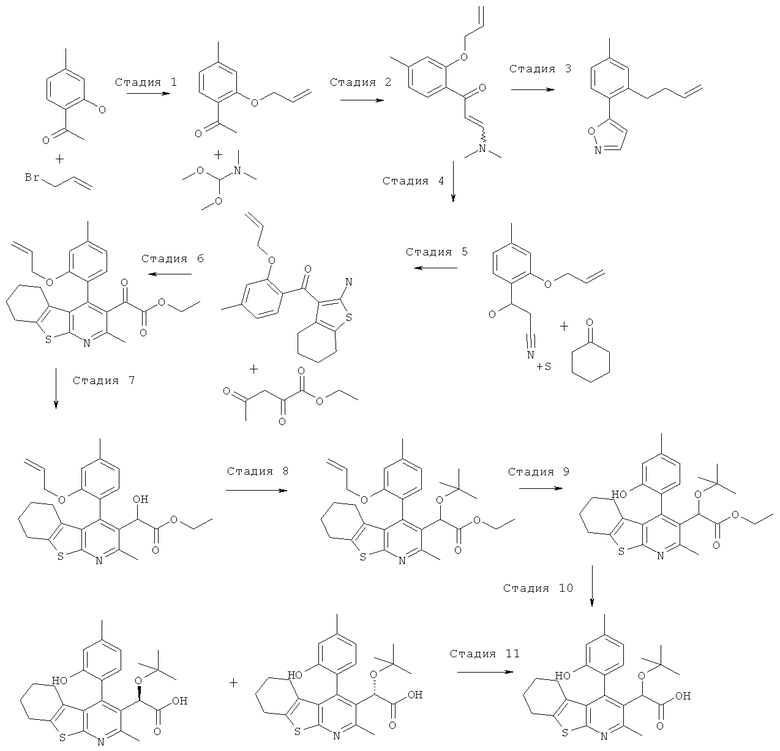
Стадия 1: 1-[2-(аллилокси)-4-метилфенил]этанон К раствору 1-(2-гидрокси-4-метил-фенил)-этанона (10 г, 67 ммоль) в диметилформамиде (100 мл) добавляли карбонат калия (18,4 г, 133 ммоль), затем добавляли аллилбромид (5,75 мл, 67 ммоль). Эту реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Остаток распределяли между этилацетатом (200 мл) и водой (800 мл), органическую фазу отделяли, промывали рассолом (50 мл), сушили над MgSO4 и концентрировали в вакууме. После охлаждения концентрата до комнатной температуры указанное в заголовке соединение кристаллизовалось в виде бесцветных пластинок, 12,40 г, выход 98%. ЖХ/МС (2 мин, кислотная) 1,20 мин, УФ-чистота 72-100%, ЭРИ+/АД+191. 1H ЯМР (400 МГц, CDCl3) δ м.д. 2.37 (s, 3H), 2.63 (s, 3H), 4.61-4.66 (m, 2H), 5.33 (dq, 1H), 5.44 (dq, 1H), 6.10 (m, 1H), 6.76 (s, 1H), 6.80-6.83 (m, 1H), 7.68 (d, 1H).
Стадия 2: (2E)-1-[2-(аллилокси)-4-метилфенил1-3-(диметиламино)проп-2-ен-1-он
К 1-[2-(аллилокси)-4-метилфенил]этанону (Стадия 1, 13,9 г, 73 ммоль) добавляли диметилформамид диметилацеталь (56 мл, 422 ммоль), и эту реакционную смесь нагревали до температуры образования флегмы в течение 18 часов. Реакционную смесь затем концентрировали в вакууме с получением указанного в заголовке соединения в виде оранжевого масла, 17,9 г, которое без очистки переносили на следующую реакционную стадию.
Стадия 3: 5-[2-(аллилокси)-4-метилфенил]изоксазол
К перемешиваемому раствору неочищенного (2E)-1-[2-(аллилокси)-4-метилфенил]-3-(диметиламино)проп-2-ен-1-она (Стадия 2, 14,1 г, 57,5 ммоль) в метаноле (70 мл) добавляли гидрохлорид гидроксиламина (4,4 г, 63 ммоль), и эту реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Бесцветные игольчатые кристаллы отфильтровывали и анализировали, и было установлено, что это вещество представляет собой указанное в заголовке соединение, 7,3 г, выход 58%. ЖХ/МС (2 мин, кислотная) 1,32 мин, УФ-чистота 64-91% УФ, ЭРИ+/АД+216. 1H ЯМР (400 МГц, CDCl3) δ м.д. 2.40 (s, 3H), 4.67 (m, 2H), 5.35 (dq, 1H), 5.46 (dq, 1H), 6.05-6.20 (m, 1H), 6.77 (m, 1H), 6.82 (s, 1H), 6.88-6.92 (m, 1H), 7.88 (d, 1H).
Стадия 4: 3-[2-(аллилокси)-4-метилфенил]-3-оксопропаннитрил
К перемешиваемой суспензии 5-[2-(аллилокси)-4-метилфенил]изоксазола (Стадия 3, 7,3 г, 33,8 ммоль) в этаноле (40 мл) добавляли этоксид натрия (21% раствор в этаноле, 40 мл, 110 ммоль), и эту реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь подкисляли до pH 2 соляной кислотой (2 н. водный раствор), и твердое вещество отфильтровывали и сушили на воздухе в течение 1 часа с получением указанного в заголовке соединения в виде не совсем белого твердого вещества, 4,8 г, выход 66%. ЖХ/МС (2 мин, кислотная) 1,12 мин, УФ-чистота 51-100%, ЭРИ+/АД+216, ЭРИ-/АД-214. 1H ЯМР (400 МГц, CDCl3) δ м.д. 2.41 (s, 3H), 4.08 (s, 2H), 4.67-4.70 (m, 2H), 5.37-5.50 (m, 2H), 6.13 (m, 1H), 6.79 (s, 1H), 6.88 (dd, 1H), 7.80 (d, 1H).
Стадия 5: [2-(аллилокси)-4-метилфенил1(2-амино-4,5,6,7-тетрагидро-1-бензотиен-3-ил)метанон
К перемешиваемому раствору 3-[2-(аллилокси)-4-метилфенил]-3-оксопропаннитрила (Стадия 4, 1 г, 4,6 ммоль) в этаноле (20 мл) добавляли циклогексанон (722 мкл, 7 ммоль) и серу (224 мг, 7 ммоль), затем добавляли морфолин (610 мкл, 7 ммоль), и эту реакционную смесь перемешивали в течение ночи при 40°C. Реакционную смесь концентрировали в вакууме. Остаток очищали, используя ISCO Companion с силикагелевым (40 г) картриджем Redisep и градиент гептана и этилацетата (от 0% до 40%). Фракции, содержащие целевой продукт, объединяли и концентрировали в вакууме с получением указанного в заголовке соединения в виде желтой смолы, 1,1 г, выход 72%. ЖХ/МС (2 мин, кислотная) 1,45 мин, УФ-чистота 58-100%, ЭРИ+/АД+328. 1H ЯМР (400 МГц, CDCl3) δ м.д. 1.44-1.51 (m, 2H), 1.64-1.71 (m, 2H), 1.78 (tt, 2Н), 2.36 (s, 3H), 2.47 (tt, 2H), 4.52 (dt, 2H), 5.13-5.28 (m, 2H), 5.92 (m, 1H), 6.69 (s, 1H), 6.78 (dq, 1H), 6.91 (br s, 2H), 7.08 (d, 1H).
Стадия 6: гидрохлоридная соль этил-{4-[2-(аллилокси)-4-метилфенил]-2-метил-5,6,7,8-тетрагидро[1]бензотиено[2,3-b]пиридин-3-ил)(оксо)ацетата
К перемешиваемому раствору [2-(аллилокси)-4-метилфенил](2-амино-4,5,6,7-тетрагидро-1-бензотиен-3-ил)метанона (Стадия 5, 882 мг, 2,69 ммоль) в этаноле (30 мл) добавляли этил-2,4-диоксопентаноат (378 мкл, 2,69 ммоль), затем добавляли ацетилхлорид (766 мкл, 10,8 ммоль), и эту реакционную смесь нагревали до 50°C в течение 1 часа. Реакционную смесь затем концентрировали в вакууме с получением смеси диастереоизомеров указанного в заголовке соединения в форме гидрохлоридной соли в виде бледно-желтого масла, 1 г, выход 86%. ЖХ/МС (2 мин, кислотная) 1,70 мин, УФ-чистота 54-100%, ЭРИ+/АД+450. 1H ЯМР(400 МГц, CDCl3) δ м.д. 1.12 (t, 3H), 1.55-1.70 (m, 2H), 1.78-1.89 (m, 2H), 1.92-2.08 (m, 2H), 2.43 (s, 3H), 2.87-2.93 (m, 2H), 2.95 (s, 3H), 3.84-3.94 (m, 2H), 4.39-4.51 (m, 2H), 5.09-5.20 (m, 2H), 5.82 (m, 1H), 6.76 (s, 1H), 6.87 (d, 1H),6.95(d, 1H).
Стадия 7: Этил-{4-[2-(аллилокси)-4-метилфенил1-2-метил-5,6,7,8-тетрагидро[1]бензотиено[2,3-b]пиридин-3-ил}(гидрокси)ацетат
К перемешиваемому раствору гидрохлоридной соли этил-{4-[2-(аллилокси)-4-метилфенил]-2-метил-5,6,7,8-тетрагидро[1]бензотиено[2,3-b]пиридин-3-ил}(оксо)ацетата (Стадия 6, 1,1 г, 2,56 ммоль) в этаноле (20 мл) добавляли боргидрид натрия (145 мг, 3,84 ммоль), и эту реакционную смесь перемешивали при комнатной температуре в течение 5 минут. Реакционную смесь концентрировали в вакууме, и остаток распределяли между этилацетатом (20 мл) и соляной кислотой (1 н. водный раствор, 30 мл). Органический слой отделяли, промывали рассолом (10 мл), сушили над MgSO4, фильтровали и концентрировали в вакууме с получением смеси диастереоизомеров указанного в заголовке соединения в виде бледно-оранжевой смолы, 1,1 г, выход 91%. ЖХ/МС (2 мин, кислотная) 1,53 мин, УФ-чистота 52-81%, ЭРИ+/АД+452. 1H ЯМР (400 МГц, CDCl3 δ м.д. 1.17-1.21 (m, 3H), 1.53-1.64 (m, 2H), 1.71-1.87 (m, 4H), 2.43 (s, 3H), 2.63 (s, 3H), 2.81 (t, 2H), 4.08-4.22 (m, 2H), 4.47-4.51 (m, 2H), 5.08-5.17 (m, 2H), 5.18 (s, 1H), 5.86 (m, 1H), 6.79 (s, 1H), 6,81-6.85 (m, 1H), 7.02 (d, 1H).
Стадия 8: Этил-{4-[2-(аллилокси)-4-метилфенил]-2-метил-5,6,7,8-тетрагидро[1]бензотиено[2,3-b]пиридин-3-ил}(трет-бутокси)ацетат
К перемешиваемому раствору этил-{4-[2-(аллилокси)-4-метилфенил]-2-метил-5,6,7,8-тетрагидро[1]бензотиено[2,3-b]пиридин-3-ил}(гидрокси)ацетата (Стадия 7, 720 мг, 1,59 ммоль) в дихлорметане (2.5 мл) добавляли трет-бутилацетат (2,5 мл), затем добавляли концентрированную серную кислоту (244 мкл, 4,78 ммоль), и эту реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь гасили добавлением 1 М водного раствора гидроксида натрия до тех пор, пока pH не достиг значения pH 5. Летучие растворители удаляли в вакууме, и остаточный водный слой экстрагировали этилацетатом (30 мл). Органический слой промывали рассолом (10 мл), сушили над MgSO4 и концентрировали в вакууме. Остаток очищали, используя ISCO Companion с силикагелевым (12 г) картриджем Redisep и градиент гептана и этилацетата (от 0% до 40%). Фракции, содержащие продукт, концентрировали в вакууме с получением смеси диастереоизомеров указанного в заголовке соединения в виде бесцветного масла, 370 мг, выход 45%. Было показано, что это вещество представляет собой смесь диастереомеров в соотношении приблизительно 80:20.
Основной диастереомер (примерно 80%)
1H ЯМР (400 МГц, CDCl3) δ м.д. 0.95 (s, 9H) 1.19 (t, 3H) 1.53-1.82 (m, 4H) 1.89-1.99 (m, 2H) 2.42 (s, 3H) 2.70 (s, 3H) 2.80 (m, 2H) 4.11 (m, 2H) 4.26-4.45 (m, 2H) 4.94-5.04 (m, 2H) 5.05 (s, 1H) 5.70-5.80 (m, 1H) 6.71 (s, 1H) 6.80 (d, 1H) 7.15 (d, 1H).
Минорный диастереомер (примерно 20%)
1H ЯМР (400 МГц, CDCl3) δ м.д. 1.00 (s, 9H) 1.13 (t, 3H) 1.53-1.82 (m, 4H) 1.89-1.99 (m, 2H) 2.42 (s, 3H) 2.70 (s, 3H) 2.80 (m, 2H) 4.11 (m, 2H) 4.26-4.45 (m, 2H) 4.94-5.04 (m, 2H) 5.05 (s, 1H) 5.70-5.80 (m, 1H) 6.73 (s, 1H) 6.78 (d, 1H) 6.93 (d, 1H).
Стадия 9: Этил-трет-бутокси[4-(2-гидрокси-4-метилфенил)-2-метил-5,6,7,8-тетрагидроГПбензотиено[2,3-b]пиридин-3-ил]ацетат
К перемешиваемому раствору этил-{4-[2-(аллилокси)-4-метилфенил]-2-метил-5,6,7,8-тетрагидро[1]бензотиено[2,3-b]пиридин-3-ил}(трет-бутокси)ацетата (Стадия 8, 370 мг, 729 мкмоль) в дихлорметане (10 мл) добавляли 1,3-диметилбарбитуровую кислоту (569 мг, 3,64 ммоль), и реакционный сосуд вакуумировали и заполняли азотом. Добавляли тетракис(трифенилрфосфин)палладий (17 мг, 15 мкмоль), и реакционную смесь нагревали до температуры образования флегмы в течение 16 часов. Добавляли дополнительную порцию тетракис(трифенилрфосфин)палладия (17 мг, 15 мкмоль), и реакционную смесь нагревали до температуры образования флегмы в течение еще 4 часов. Реакционную смесь концентрировали в вакууме и адсорбировали на диоксид кремния. Остаток очищали, используя ISCO Companion с силикагелевым (12 г) картриджем Redisep и градиент гептана и этилацетата (от 0% до 20%). Фракции, содержащие продукт, концентрировали в вакууме с получением указанного в заголовке соединения в виде пары диастереоизомеров в виде бесцветного масла, 214 мг, выход 63%. Другая пара минорных диастереоизомеров совместно элюировалась с непрореагировавшим исходным веществом, и не была выделена. ЖХ/МС (12 мин, кислотная) 6,89 мин, УФ-чистота 100%, ЭРИ+/ХИАД+468, ЭРИ-/ХИАД-466 1H ЯМР (400 МГц, CDCl3) δ м.д. 1.01 (s, 9Н) 1.20 (t, 3H) 1.62-1.85 (m, 4H) 2.02-2.14 (m, 2H) 2.41 (s, 3H) 2.74 (s, 3H) 2.78-2.85 (m, 2H) 4.08-4.18 (m, 2H) 5.14 (s, 1H) 6.78 (s, 1H) 6.82 (d, 1H) 7.17 (d, 1H).
Стадия 10: трет-Бутокси[4-(2-гидрокси-4-метилфенил)-2-метил-5,6,7,8-тетрагидро[1]бензотиено[2,3-b]пиридин-3-ил]уксусная кислота
К перемешиваемому раствору основной диастереоизомерной пары этил-трет-бутокси[4-(2-гидрокси-4-метилфенил)-2-метил-5,6,7,8-тетрагидро[1]бензотиено[2,3-b]пиридин-3-ил]ацетата (Стадия 9, 214 мг, 458 мкмоль) в этаноле (5 мл) и тетра гидрофура не (5 мл) добавляли раствор гидроксида натрия (2 н. водный раствор, 2 мл, 2,75 ммоль), и эту реакционную смесь перемешивали при 60°C в течение 18 часов. Реакционную смесь концентрировали в вакууме до удаления всех органических растворителей, водный остаток затем подкисляли соляной кислотой (2 н. водный раствор) до pH 2. Выпавшее в осадок твердое вещество собирали фильтрованием и промывали трет-бутил-метиловым эфиром (10 мл) и сушили в вакууме с получением смеси диастереоизомеров указанного в заголовке соединения в виде белого твердого вещества, 76 мг, выход 38%. ЖХ/МС (12 мин, кислотная) 5,51 мин, УФ-чистота 81%, ЭРИ+/ХИАД+440, ЭРИ-/ХИАД-438. 1H ЯМР (400 МГц, CDCl3) δ м.д. 1.04 (s, 9H) 1.66-1.88 (m, 4H) 2.09-2.21 (m, 2H) 2.40 (s, 3H) 2.73 (s, 3H) 2.82 (t, 2H) 5.33 (s, 1H) 6.78 (s, 1H) 6.85 (d, 1H) 7.36 (d, 1H).
Стадия 11: Хиральное разделение
Диастереомерную пару, выделенную на Стадии 10, разделяли с использованием колонки Chiralpak 1C, элюируя смесью 70:30 гептан:1РА (изопропиловый спирт) при 18 мл в минуту и при времени прогона 9 минут.Первый энантиомер элюировался через 3,56 минут, а второй энантиомер элюировался через 4,54 минут, под мониторингом УФ.
Первый элюировавшийся энантиомер был определен как (2R)-трет-бутокси[4-(2-гидрокси-4-метилфенил)-2-метил-5,6,7,8-тетрагидро[1]бензотиено[2,3-b]пиридин-3-ил]уксусная кислота.
Противовирусная активность = 0,089 мкМ (n=2) (S8737E).
HTRF анализ взаимодействия = 4120 нМ (n=2) (S9118)
Второй элюировавшийся энантиомер был определен как (2S)-трет-бутокси[4-(2-гидрокси-4-метилфенил)-2-метил-5,6,7,8-тетрагидро[1]бензотиено[2,3-b]пиридин-3-ил]уксусная кислота с атропизомерной конфигурацией, показанной ниже (подтверждено рентгеноструктурным анализом):
Противовирусная активность = 0,013 мкМ (n=2) (S8737E).
HTRF анализ взаимодействия = 576 нМ (n=2) (S9118)
Пример 9: Получение (S)-2-(трет-бутокси)-2-(4-(4-(диФторметил)(ренил)-2-метил-5,6,7,8-тетрагидробензо[4,5]тиено[2,3-b]пиридин-3-ил)уксусной кислоты
Стадия 1: (S)-метил-2-(трет-бутокси)-2-(4-(4-(дифторметил)фенил)-2-метил-5,6,7,8-тетрагидробензо[4,5]тиено[2,3-b]пиридин-3-ил)ацетат
2-(4-(Дифторметил)фенил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан (1,04 г, 4,09 ммоль), фосфат калия (1,35 г, 6,36 ммоль), воду (750 мкл) и дихлор[1,1'-бисс(ди-трет-бутилфосфино)]ферроценпалладий(II)™ (101 мг, 155 мкмоль) добавляли к перемешиваемому раствору (З)-метил 2-(трет-бутокси)-2-(4-йод-2-метил-5,6,7,8-тетрагидробензо[4,5]тиено[2,3-b]пиридин-3-ил)ацетата (750 мг, 1,58 ммоль) в диоксане (11 мл) в реакционной пробирке. Реакционную смесь дегазировали аргоном в течение 2 минут, герметично закрывали и затем перемешивали при 100°C в течение 16 часов. Реакционную смесь охлаждали до комнатной температуры, разбавляли этилацетатом (30 мл) и водой (30 мл) и затем пропускали через слой целита. Слои фильтрата разделяли, и водный слой экстрагировали этилацетатом (3×30 мл). Объединенные органические слои сушили (Na2SO4) и концентрировали в вакууме с получением неочищенного продукта. Остаток очищали колоночной флэш-хроматографией, элюируя этилацетатом в гептане (10%), с получением указанного в заголовке соединения (639 мг, 85%) в виде желтого масла. 1H ЯМР (400 МГц, CDCl3) δ=0.96 (s, 9H), 1.85-1.35 (m, 6H), 2.72 (s, 3H), 2.85-2.75 (m, 2H), 3.67 (s, 3H), 4.95 (s, 1H), 6.76 (t, 1H), 6.45 (d, 1H), 7.61-7.51 (m, 3H). ЖХ/МС (время прогона = 5 минут, основная): Rt=3,37 минут: m/z474,23 [M+H+].
Стадия 2: (S)-2-(трет-бутокси)-2-(4-(4-(дифторметил)фенил)-2-метил-5,6,7,8-тетрагидробензо[4,5]тиено[2,3-b]пиридин-3-ил)уксусная кислота
2 М водный раствор гидроксида натрия (1,93 мл, 3,86 ммоль) добавляли к раствору (S)-метил-2-(трет-бутокси)-2-(4-(4-(дифторметил)фенил)-2-метил-5,6,7,8-тетрагидробензо[4,5]тиено[2,3-b]пиридин-3-ил)ацетата (183 мг, 0,39 ммоль) в смеси 1:1 тетрагидрофуран/денатурат (4 мл) при комнатной температуре. Полученную смесь перемешивали при комнатной температуре в течение 64 ч. Реакционную смесь распределяли между этилацетатом (20 мл) и водой (10 мл). Водный раствор доводили до pH 4 добавлением 2 М водного раствора соляной кислоты. Слои разделяли, и водный слой экстрагировали этилацетатом (2×10 мл). Объединенные органические слои сушили (MgSO4) и концентрировали в вакууме с получением неочищенного продукта в виде бледно-оранжевого твердого вещества. Это вещество очищали колоночной флэш-хроматографией на диоксиде кремния, элюируя метанолом в дихлорметане (3-5%), с получением указанного в заголовке соединения (111 мг, 63%) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ=1.01 (s, 9H), 1.90-1.35 (m, 6H), 2.70 (s, 3H), 2.85-2.78 (m, 2H), 5.07 (brs, 1H), 6.75 (t, 1H), 7.34-7.41 (brm, 1H), 7.61 (d, 2H), 7.80-7.70 (brm, 1H). ЖХ/МС (время прогона = 5 минут, основная): r(=3,04 минут; m/z 460,22 [M+H+]. Противовирусная активность = 0,081 мкМ (n=6) (S8737E). HTRF анализ взаимодействия = 11010 нМ (n=6) (S9118) Пример 50: Получение (S)-2-(трет-бутокси)-2-(2-метил-4-(4-(трифторметил)Фенил)-5,6,7,8-тетрагидробензо[4,5]тиено[2,3-b]пиридин-3-ил)уксусной кислоты
Стадия 1: (S)-метил-2-(тоет-бутокси)-2-(2-метил-4-(4-(трифторметил)фенил)-5,6,7,8-тетрагидробензо[4,5]тиено[2,3-b]пиридин-3-ил)ацетат
2-(4-(Трифторметил)фенил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан (115 г, 0,42 ммоль), N-этил-N-изопропилпропан-2-амин (120 мкл, 0,64 ммоль) и воду (500 мкл) добавляли к перемешиваемому раствору (S)-метил-2-(трет-бутокси)-2-(4-йод-2-метил-5,6,7,8-тетрагидробензо[4,5]тиено[2,3-b]пиридин-3-ил)ацетата (100 мг, 0,21 ммоль) в диоксане (2 мл) в реакционной пробирке. Реакционную смесь дегазировали аргоном в течение 2 минут, затем добавляли тетракис(трифенилфосфин)палладий(0) (25 мг, 21 мкмоль), и сосуд герметично закрывали и нагревали при 100°C в течение 16 часов. Реакционную смесь охлаждали до комнатной температуры и разбавляли этилацетатом (30 мл) и водой (30 мл), и смесь пропускали через слой целита. Слои фильтрата разделяли, и органический слой промывали рассолом (30 мл), сушили (MgSO4) и концентрировали в вакууме с получением неочищенного продукта в виде коричневой смолы. Остаток очищали колоночной флэш-хроматографией, элюируя этилацетатом в гептане (10%), с получением указанного в заголовке соединения (75 мг, 72%) в виде желтого масла. 1H ЯМР (400 МГц, CDCl3) δ=0.96 (s, 9H), 1.85-1.35 (m, 6H), 2.72 (s, 3H), 2.85-2.75 (m, 2H), 3.69 (s, 3H), 4.91 (s, 1H), 7.40 (d, 1H), 7.61 (d, 1H), 7.71 (m, 2H). ЖХ/МС (время прогона = 5 минут, основная): Rt=3,64 минут; m/z 492,06 [M+H+].
Стадия 2: (S)-2-(тоет-бутокси)-2-(2-метил-4-(4-(трифторметил)фенил)-5,6,7,8-тетрагидробензо[4,5]тиено[2,3-b]пиридин-3-ил)уксусная кислота
1 М водный раствор гидроксида натрия (1,6 мл, 1,6 ммоль) добавляли к раствору (S)-метил-2-(трет-бутокси)-2-(2-метил-4-(4-(трифторметил)фенил)-5,6,7,8-тетрагидробензо[4,5]тиено[2,3-b]пиридин-3-ил)ацетата (75 мг, 0,15 ммоль) в смеси 1:1 тетрагидрофуран/денатурат (4 мл) при комнатной температуре. Полученную смесь нагревали при 60°C в течение 1,5 ч. Полученный раствор концентрировали в вакууме, и остаток растворяли в воде (10 мл). Водный раствор доводили до pH 4 добавлением 2 М водного раствора соляной кислоты, и полученную суспензию экстрагировали дихлорметаном (20 мл). Органический слой промывали рассолом, сушили (MgSO4) и концентрировали в вакууме с получением неочищенного продукта в виде бледно-желтого твердого вещества. Это вещество очищали колоночной флэш-хроматографией на диоксиде кремния, элюируя метанолом в дихлорметане (5%), с получением указанного в заголовке соединения (40,3 мг, 56%) в виде бледно-желтого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ=1.01 (s, 9H), 1.89-1.30 (m, 6H), 2.71 (s, 3H), 2.90-2.75 (m, 2H), 5.02 (s, 1H), 7.48-7.34 (m, 1H), 7.90-7.64 (m, ЗН). ЖХ/МС (время прогона = 5 минут, основная): Rt=3,30 минут; m/z 478,01 [M+H+].
Противовирусная активность = 0,055 мкМ (n=4) (S8737E).
HTRF анализ взаимодействия = 1500 нМ (n=4) (S9118)
Пример 51: Получение (S)-2-(4-(4-бромфенил)-2-метил-5,6,7,8-тетрагидробензо[4,5]тиено[2,3-b]пиридин-3-ил)-2-(трет-бутокси)уксусной кислоты
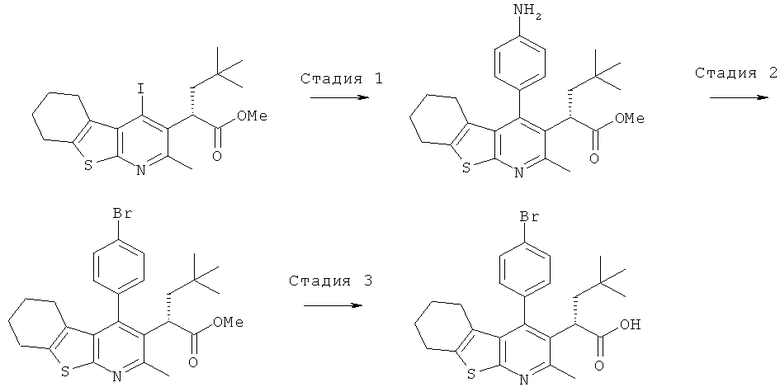
Стадия 1: (S)-метил-2-(4-(4-аминосренил-2-метил-5,6,7,8-тетрагидробензо[4,5]тиено[2,3-b]пиридин-3-ил)-2-(трет-бутокси)ацетат
4-(4,4,5,5-Тетраметил-1,3,2-диоксаборолан-2-ил)анилин (140 мг, 0,63 ммоль), гидрокарбонат натрия (178 мг, 2,1 ммоль) и воду (200 мкл) добавляли к перемешиваемому раствору (б)-метил-2-(трет-бутокси)-2-(4-йод-2-метил-5,6,7,8-тетрагидробензо[4,5]тиено[2,3-b]пиридин-3-ил)ацетата (200 мг, 0,42 ммоль) в N,N-диметилацетамиде (4 мл) в реакционной пробирке. Реакционную смесь дегазировали аргоном в течение 2 минут, затем добавляли бис(три-трет-бутилфосфин)палладий(0) (22 мг, 42 мкмоль), и сосуд герметично закрывали и нагревали при 100°C в течение 16 часов. Реакционную смесь охлаждали до комнатной температуры и распределяли между дихлорметаном (30 мл) и водой (30 мл). Слои разделяли, и органический слой сушили (MgSO4) и концентрировали в вакууме с получением неочищенного продукта в виде коричневой смолы. Остаток очищали колоночной флэш-хроматографией на силикагеле, элюируя этилацетатом в гептане (25%), с получением указанного в заголовке соединения (114 мг, 67%) в виде белой пены. 1H ЯМР (400 МГц, CDCl3) δ=0.97 (s, 9H), 1.95-1.30 (m, 6H), 2.68 (s, 3H), 2.85-2.77 (m, 2H), 3.65 (s, 3H), 3.80 (brs, 2H), 5.19 (s, 1H), 6.75-6.68 (m, 2H), 6.99 (d, 1H), 7.19 (d,1H).
Стадия 2: (S)-метил-2-(4-(4-бромфенил)-2-метил-5,6,7,8-тетрагидробензо[4,5]тиено[2,3-b]пиридин-3-ил)-2-(трет-бутокси)ацетат
Раствор бромида меди(II) (73 мг, 0,33 ммоль) и mpem-бутил нитрита (60 мкл, 0,43 ммоль) в ацетонитриле (2 мл) добавляли к перемешиваемому раствору (S)-метил-2-(4-(4-аминофенил)-2-метил-5,6,7,8-тетрагидробензо[4,5]тиено[2,3-b]пиридин-3-ил)-2-(трет-бутокси)ацетата (114 мг, 0,26 ммоль) в ацетонитриле (1 мл) при комнатной температуре, и полученную смесь перемешивали при комнатной температуре в течение 16 часов. Смесь распределяли между 2 М водным раствором соляной кислоты (5 мл) и добавляли дихлорметан (10 мл). Слои разделяли, и органический слой промывали водой (10 мл), рассолом (10 мл), сушили (MgSO4) и концентрировали в вакууме с получением неочищенного продукта. Этот продукт очищали колоночной флэш-хроматографией на диоксиде кремния, элюируя этилацетатом в гептане (10%), с получением указанного в заголовке соединения (83 мг, 63%) в виде коричневого масла. 1H ЯМР (400 МГц, CDCl3) δ=0.96 (s, 9H), 1.85-1.40 (m, 6H), 2.71 (S, 3H), 2.81-2.75 (m, 2H), 3.65 (s, 3H), 4.99 (s, 1H), 7.12 (s, 1H), 7.31 (d, 1H), 7.60-7.50 (m, 2H). ЖХ/МС (время прогона = 5 минут, основная): Rt=3,57 минут; m/z 504,12 [M+H+].
Стадия 3: (S)-2-(4-(4-бромфенил)-2-метил-5,6,7,8-тетрагидробензо[4,5]тиено[2,3-b]пиридин-3-ил)-2-(трет-бутокси)уксусная кислота
1 М водный раствор гидроксида натрия (1,6 мл, 1.6 ммоль) добавляли к раствору (S)-метил-2-(4-(4-бромфенил)-2-метил-5,6,7,8-тетрагидробензо[4,5]тиено[2,3-b]пиридин-3-ил)-2-(трет-бутокси)ацетата (83 мг, 0,16 ммоль) в смеси 1:1 тетрагидрофуран/денатурат (4 мл) при комнатной температуре. Полученную смесь нагревали до 60°C и перемешивали при 60°C в течение 2 часов. Полученный раствор концентрировали в вакууме, и остаток растворяли в воде (10 мл). Водный раствор доводили до pH 5 добавлением 2 М водного раствора соляной кислоты, и полученную суспензию экстрагировали дихлорметаном (20 мл). Органический слой промывали рассолом, сушили (MgSO4) и концентрировали в вакууме с получением неочищенного продукта в виде бледно-желтого твердого вещества. Это вещество очищали растиранием с денатуратом (5 мл). Полученное твердое вещество собирали фильтрованием с получением указанного в заголовке соединения (27 мг, 34%) в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ=1.02 (s, 9H), 2.00-1.40 (m, 6H), 2.69 (s, 3H), 2.83-2.70 (m, 2H), 5.11 (s, 1H), 7.25-7.06 (m, 1H), 7.63-7.42 (m, 3H). ЖХ/МС (время прогона = 5 минут, основная): Rt=3,19 минут; m/z 490,00 [M+H+].
Противовирусная активность = 0,080 мкМ (n=2) (S8737E).
HTRF анализ взаимодействия = 1160 нМ (n=2) (S9118)
Пример 52: трет-Бутокси-(2-метил-4-пара-толил-5,8-дигидро-6H-7-окса-9-тиа-1-аза-флуорен-3-ил)-уксусная кислота
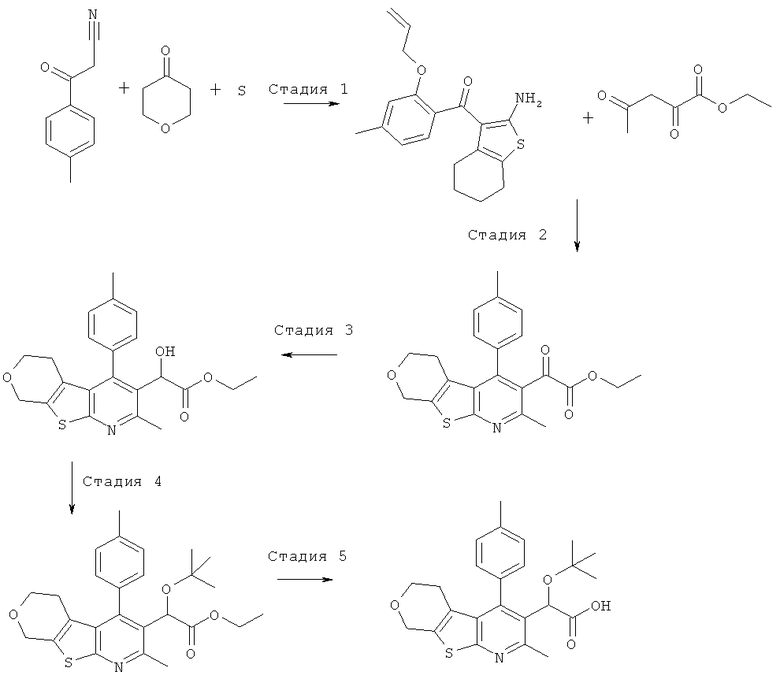
Стадия 1: (2-Амино-4,7-дигидро-5H-тиено[2,3-c]пиран-3-ил)-пара-толил-метанон
4-Пиранон (2,31 мл, 24,97 ммоль) и серу (801 мг, 24,97 ммоль) добавляли к раствору 3-оксо-3-пара-толил-пропионитрила (3,79 г, 23,8 ммоль) в этаноле (40 мл), затем добавляли морфолин (2,18 мл, 24,97 ммоль), и эту реакционную смесь нагревали при 40°C в течение 16 часов. Реакционную смесь концентрировали в вакууме, и остаток очищали флэш-хроматографией на диоксиде кремния, элюируя смесью 0-20% этилацетата/гептаны, с получением (2-амино-4,7-дигидро-5H-тиено[2,3-c]пиран-3-ил)-пара-толил-метанона в виде желтого твердого вещества (2,2 г, выход 36%). ЖХ/МС (2 мин, кислотная) 1,07 мин ЭРИ+/АД+274(MH+). 1H ЯМР (400 МГц, CDCl3) δ м.д. 2.00-2.03 (m, 2H), 2.42 (s, 3H), 3.80-3.84 (m, 2H), 4.80 (s, 2H), 6.60 (br s, 2H), 7.20 (d, 2H), 7.41 (d, 2H).
Стадия 2: Этиловый эфир (2-метил-4-пара-толил-5,8-дигидро-6H-7-окса-9-тиа-1-аза-флуорен-3-ил)-оксо-уксусной кислоты
Этиловый эфир 2,4-диоксо-пентановой кислоты (1,2 мл, 8,6 ммоль) добавляли к раствору 2-амино-4,7-дигидро-5Н-тиено[2,3-с]пиран-3-ил)-пара-толил-метанона (2,3 г, 8,6 ммоль) в этаноле (50 мл), затем добавляли ацетилхлорид (2,43 мл, 34,3 ммоль), и эту реакционную смесь подогревали до 50°C в течение 1 часа. Реакционную смесь затем концентрировали в вакууме и адсорбировали на диоксид кремния. Остаток очищали флэш-хроматографией на диоксиде кремния, элюируя 0-40% этилацетатом в гептанах, с получением указанного в заголовке соединения в виде красного твердого вещества (675 мг, 19%). ЖХ/МС (2 мин, кислотная) 2,78 мин ЭРИ+/АД+396 (MH+).
Стадия 3: Этиловый эфир гидрокси-(2-метил-4-пара-толил-5,8-дигидро-6H-7-окса-9-тиа-1-аза-флуорен-3-ил)-уксусной кислоты
Боргидрид натрия (97 мг, 2,56 ммоль) добавляли к перемешиваемому раствору этилового эфира гидрокси-(2-метил-4-пара-толил-5,8-дигидро-6H-7-окса-9-тиа-1-аза-флуорен-3-ил)-уксусной кислоты (675 мг, 1,71 ммоль) в этаноле (20 мл). Добавляли 2 М HCl в воде (3 мл), и затем реакционную смесь концентрировали в вакууме. Остаток распределяли между водой (50 мл) и этилацетатом (30 мл). Слои разделяли, и органическую фазу сушили (MgSO4) и концентрировали в вакууме с получением указанного в заголовке соединения в виде кремового твердого вещества (543 мг, 80%), которое использовали без дополнительной очистки. ЖХ/МС (2 мин, кислотная) 1,20 мин ЭРИ+/АД+398. 1H ЯМР (400 МГц, CDCl3) δ м.д. 1.20 (t, 3H), 1.82-1.88 (m, 2H), 2.41 (s, 3H), 2.61 (s, 3H), 3.68-3.72 (m, 2H), 4.10-4.21 (m, 2H), 4.81 (s, 2H), 7.16-7.26 (m, 4H).
Стадия 4: Этиловый эфир трет-бутокси-(2-метил-4-пара-толил-5,8-дигидро-6H-7-окса-9-тиа-1-аза-флуорен-3-ил-уксусной кислоты
трет-Бутилацетат (0,8 мл) добавляли к раствору этилового эфира гидрокси-(2-метил-4-пара-толил-5,8-дигидро-6H-7-окса-9-тиа-1-аза-флуорен-3-ил)-уксусной кислоты (100 мг, 0,252 ммоль) в дихлорметане (0,8 мл), затем добавляли концентрированную серную кислоту (40 мкл), и эту реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Добавляли раствор бикарбоната натрия (10% водный, 2 мл), и реакционную смесь упаривали в вакууме, пока не осталась только водная фаза. Водную фазу экстрагировали этилацетатом (2 мл), и органический слой промывали рассолом (1 мл), сушили и концентрировали в вакууме. Остаток очищали флэш-хроматографией на диоксиде кремния, элюируя смесью 0-30% этилацетата/гептаны, с получением бесцветного твердого вещества (66 мг, 45%). ЖХ/МС (12 мин, кислотная) 7,25 мин, ЭРИ+/АД+454 (МН+) 1H ЯМР (400 МГц, CDCl3) δ м.д. 0.98 (s, 9Н), 1.20 (t, 3H), 1.55-1.65 (m, 2H), 2.43 (s, 3H), 2.77 (s, ЗН), 3.57-3.61 (m, 1H), 3.78-3.82 (m, 1H), 4.06-4.20 (m, 2H), 4.79-4.82 (m, 2H), 7.16 (d, 2H), 7.32 (d, 2H).
Стадия 5: трет-Бутокси-(2-метил-4-пара-толил-5,8-дигидро-6H-7-окса-9-тиа-1-аза-Флуорен-3-ил)-уксусная кислота
Водный гидроксид натрия (2 н., 2 мл, 4 ммоль) добавляли к перемешиваемому раствору этилового эфира трет-бутокси-(2-метил-4-лара-толил-5,8-дигидро-6H-7-окса-9-тиа-1-аза-флуорен-3-ил)-уксусной кислоты (66 мг, 0,15 ммоль) в этаноле (2 мл) и тетрагидрофуране (2 мл), и эту реакционную смесь перемешивали в течение 2 часов при 60°C. Реакционную смесь концентрировали в вакууме, пока не осталась только водная фаза. pH доводили до значения 2 добавлением 2 н. водного раствора HCl. Полученное твердое вещество собирали фильтрованием и очищали препаративной ЖХВД с получением указанного в заголовке соединения. ЖХ/МС 3,63 мин, ЭРИ+/АД+425,1 (MH+).
Колонка: Gemini-NX 3 мкм С18 110А, температура окружающей среды; детектирование: УФ 225 нм - МС Скорость потока: 1,5 мл/мин; подвижная фаза: A: H2O+0,1% муравьиной кислоты, В: MeCN+0,1% муравьиной кислоты. Градиент (время/мин, %B) - (0,5), (3,95), (4,95), (4,1,5), (5,5).
Противовирусная активность = 0,189 мкМ (n=2) (S8737E).
HTRF анализ взаимодействия = 2750 нМ (n=2) (S9118)
Оценка анти-ВИЧ активности соединений по изобретению Противовирусный анализ на основе МТ-2 (S8737E)
Этот анализ разработан для определения эффектов небольших молекул на репликацию ВИЧ-1 в лимфобластоидной клеточной линии, МТ2, и он дает возможность определять противовирусный эффект соединений, действующих на любой стадии цикла репликации ВИЧ-1. Данный анализ, наряду со связанным с ним анализом цитотоксичности, был подробно описан в 2005 году (Cao et al., Antimicrobial Agents and Chemotherapy 2005; 49 (9), p.3833-3841).
Клетки МТ2 инфицируют вирусом ВИЧ-1 (штамм NL4.3; Adachi et al., Journal of Virology 1986) и трансфицируют в планшеты для анализа, содержащие последовательные разведения тестируемых соединений. Планшеты для анализа инкубируют в течение 3 суток (МТ2), чтобы обеспечить прохождение нескольких раундов вирусной репликации/инфицирования. В конце этого срока надосадочную жидкость переносят в новые планшеты, содержащие клетки JC53BL.
Клетки JC53BL экспрессируют рецепторы CXCR4, CCR5 и CD4 и ВИЧ-1-LTR-β-Gal. В нормальных культуральных условиях экспрессируются недетектируемые уровни β-галактозидазы, но в присутствии ВИЧ-1 вирусный белок Tat способен активировать ВИЧ-1-LTR в клетках JC53BL, приводя к увеличению экспрессии фермента β-Gal. Экспрессия может быть измерена с использованием анализа "FluorAce β-Galactosidase reporter". Уровни β-Gal прямо пропорциональны уровням Tat (вплоть до порогового значения), что дает возможность выполнения количественного определения вирусов. Соединения, которые ингибируют репликацию вируса, будут давать пониженный сигнал, и можно построить кривую доза-ответ для каждого соединения. Ее затем используют для определения IC50, показателя силы действия соединения, для каждого соединения.
Анализ на противовирусную активность на основе МТ-2 требует отдельной оценки цитотоксичности соединений. Ее осуществляют с использованием 3-суточного анализа на цитотоксичность в отношении МТ-2 следующим образом:
3-суточный анализ на цитотоксичность в MT2 (S8738E)
Анализ разработан для тестирования в отношении того, обладают или не обладают соединения цитотоксической активностью в клетках MT2, путем измерения жизнеспособности этих клеток в присутствии соединений. Анализ проводят путем добавления в планшеты для анализа MT2, содержащие последовательные разведения соединений, предназначенных для скрининга. После инкубирования в течение 3 суток жизнеспособность клеток, оставшихся в планшетах, анализируют с использованием коммерчески доступного реагента CellTiter-Glo (Promega Ltd). Полученные данные затем используют для вычисления концентрации соединения, необходимой для того, чтобы вызвать 50%-ную цитотоксичность (CC50).
Все соединения, примеры которых приведены, имели CC50>20 мкМ.
Ссылки:
Adachi, A., Gendelman, H., Koenig, S., Folks, Т., Willey, R., Rabson, A. and Martin, M (1986) Production of acquired immunodeficiency syndrome-associated retrovirus in human и nonhuman cells transfected with an infectious molecular clone, J. Virol., 59,284-291.
Cao J, Isaacson J, Patick AK, Blair WS. (2005) High-throughput human immunodeficiency virus type 1 (HIV-1) full replication assay that includes HIV-1 Vif as an antiviral target. Antimicrobial Agent and Chemotherapy; 49 (9), p 3833-3841.
Анализ для измерения ингибиторной активности в отношении взаимодействия LEDGF-интеграза соединений по изобретению
HTRF анализ взаимодействия (S9118)
Гомогенный флуоресцентный с временным разрешением (HTRF) анализ осуществляют по аналогии с ранее описанными HTRF анализами взаимодействия белок-белок, которые рассмотрены в Mathis, Clin. Chem., 2005. Процедуру анализа выполняют следующим образом: реакции проводят в конечном объеме 20 мкл в 384-луночных черных микротитрационных планшетах малого объема (Greiner). Конечный реакционный буфер содержит 29 мМ фосфатного буфера (pH 7), 10 мМ буфера HEPES (pH 7,4), 68,5 мМ NaCl, 1,4 мМ KCl, 400 мМ KF, 0,05% (масс./об.) плюрониловой кислоты (P104, Sigma Aldrich) и 1% (об./об.) DMSO (диметилсульфоксид). Интегразу с His6-меткой (конечная концентрация 78 нМ) инкубируют с манноза-связывающим белком, слитым с А325 С-концевым интеграза-связывающим доменом LEDGF в присутствии соединения в течение 2 часов при комнатной температуре. Оба эти белковые реагенты предоставляет проф. Zeger Debyser из Katholieke Universiteit Leuven, Leuven, Belgium. Соединения добавляют в концентрациях, варьирующих в широких пределах от 0,1 вплоть до 100 мкМ. После этого добавляют 8,3 нМ конъюгированного с криптатом европия анти-MBP моноклонального антитела и 17 нМ анти-His антитела, конъюгированного с акцепторным флуорофором d2. После инкубирования в течение 2 часов при комнатной температуре планшеты считывают на микропланшет-ридере EnVision™ (Perkin Elmer), используя длину волны возбуждения 320 нм. Соотношение флуоресценции, испускаемой при 665 нм и 620 нм, используют для оценки степени ингибирования взаимодействия белок-белок.
Ссылки: Mathis G., Probing molecular interactions with homogeneous techniques based on rare earth cryptates and fluorescence energy transfer. Clin. Chem.41 (9), 1391-7(1995).
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНОЕ НИТРОИМИДАЗОЛА ПРОТИВ ТУБЕРКУЛЕЗА ЛЕГКИХ | 2016 |
|
RU2675622C1 |
| ПРОИЗВОДНЫЕ ТЕТРАГИДРОХИНОЛИНА, ДЕМОНСТРИРУЮЩИЕ ЗАЩИТНОЕ ОТ ВИЧ-ИНФЕКЦИИ ДЕЙСТВИЕ | 2005 |
|
RU2350604C2 |
| БИЦИКЛИЧЕСКИЕ ЛАКТАМЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2716136C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ИХ ПРИМЕНЕНИЕ | 2001 |
|
RU2289581C2 |
| АМИНОПИРИДИНСОДЕРЖАЩИЕ ИНГИБИТОРЫ ТИРОЗИНКИНАЗЫ СЕЛЕЗЕНКИ (SYK) | 2012 |
|
RU2612217C2 |
| ЗАМЕЩЕННЫЕ ПИРИМИДИНТИОАЛКИЛЬНЫЕ ИЛИ АЛКИЛЭФИРНЫЕ СОЕДИНЕНИЯ И СПОСОБ ИНГИБИРОВАНИЯ ОБРАТНОЙ ТРАНСКРИПТАЗЫ ВИРУСОВ | 1996 |
|
RU2167155C2 |
| БИЦИКЛИЧЕСКИЕ ЛАКТАМЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2827714C1 |
| ПРОИЗВОДНЫЕ ПИПЕРИДИНОВ | 2011 |
|
RU2554353C2 |
| ИНГИБИТОРЫ ИНТЕГРИНА AVB6 | 2018 |
|
RU2769702C2 |
| СОЕДИНЕНИЯ ГЕТЕРОАРИЛА В КАЧЕСТВЕ ИНГИБИТОРОВ ТКБ И ИХ ПРИМЕНЕНИЕ | 2015 |
|
RU2742122C2 |
Изобретение относится к соединениям формулы (I)
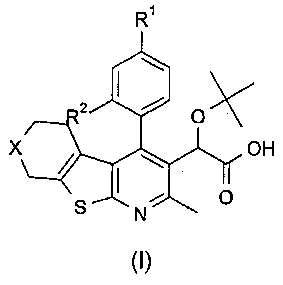
где: R1 представляет собой СН3, СН2СН3, Cl, Br, CHF2 или CF3; R2 представляет собой Н, ОН или F; X представляет собой СН2 или О при условии, что когда R1 представляет собой СН3 и R2 представляет собой Н, тогда X представляет собой О; и к применению таких соединений в качестве ингибиторов репликации ВИЧ.6 н. и 9 з.п. ф-лы, 52 пр.
1. Соединение формулы (I)
где:
R1 представляет собой СН3, СН2СН3, Cl, Br, CHF2 или CF3;
R2 представляет собой Н, ОН или F; X представляет собой СН2 или О;
при условии, что когда R1 представляет собой СН3 и R2 представляет собой Н, тогда X представляет собой О;
или его фармацевтически приемлемая соль.
2. Соединение по п. 1, выбранное из:
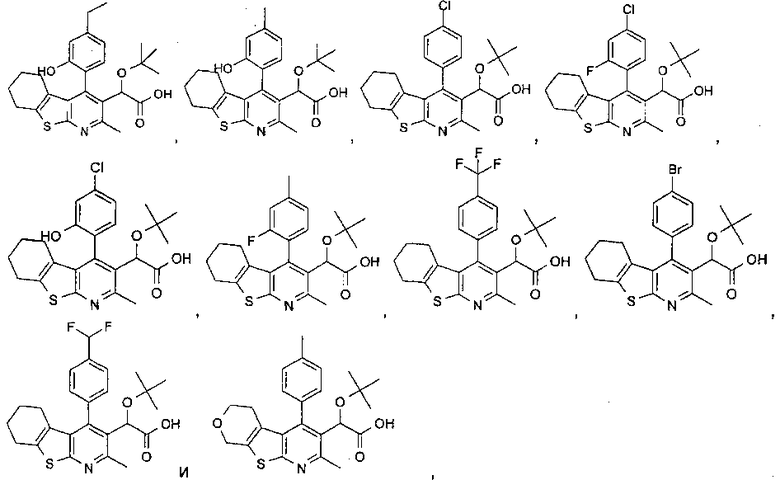
или его фармацевтически приемлемая соль.
3. Соединение по п. 1 формулы (Ia)
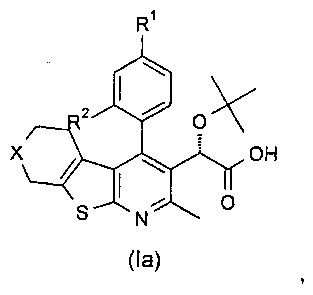
где:
R1 представляет собой СН3, СН2СН3, Cl, Br, CHF2 или CF3;
R2 представляет собой Н, ОН или F; X представляет собой СН2 или О;
при условии, что когда R1 представляет собой СН3 и R2 представляет собой Н, тогда X представляет собой О;
или его фармацевтически приемлемая соль.
4. Соединение по п. 1, выбранное из:
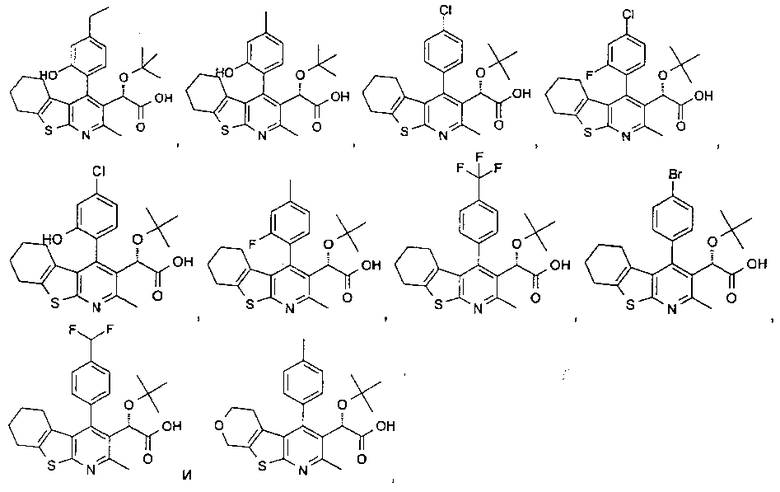
или его фармацевтически приемлемая соль.
5. Соединение по п. 1, выбранное из:
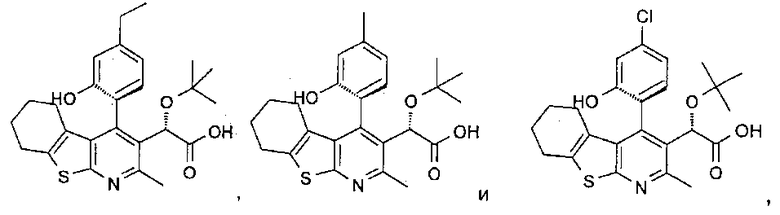
или его фармацевтически приемлемая соль.
6. Соединение, выбранное из:
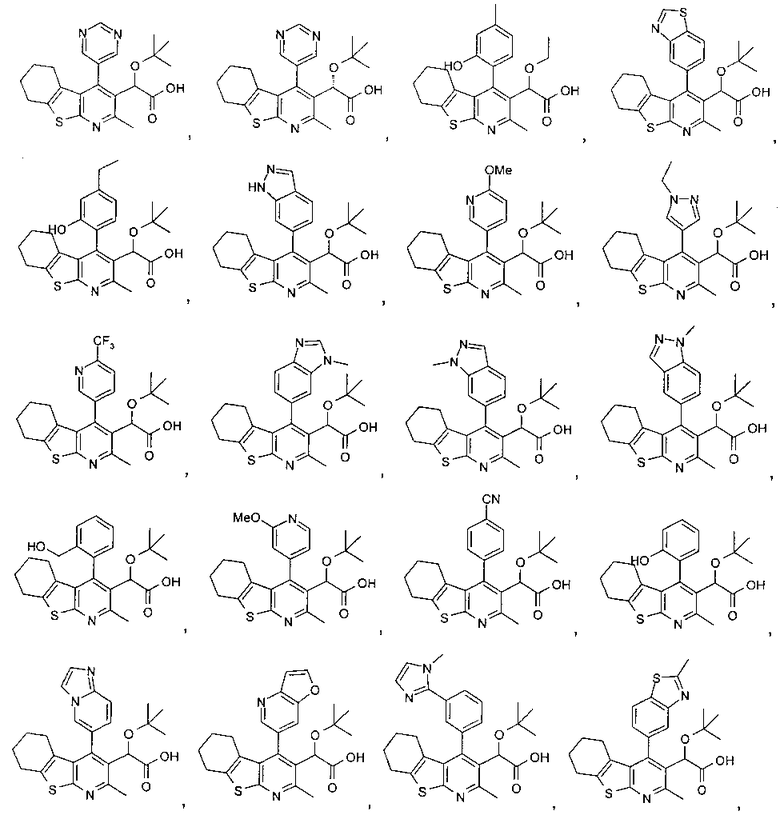
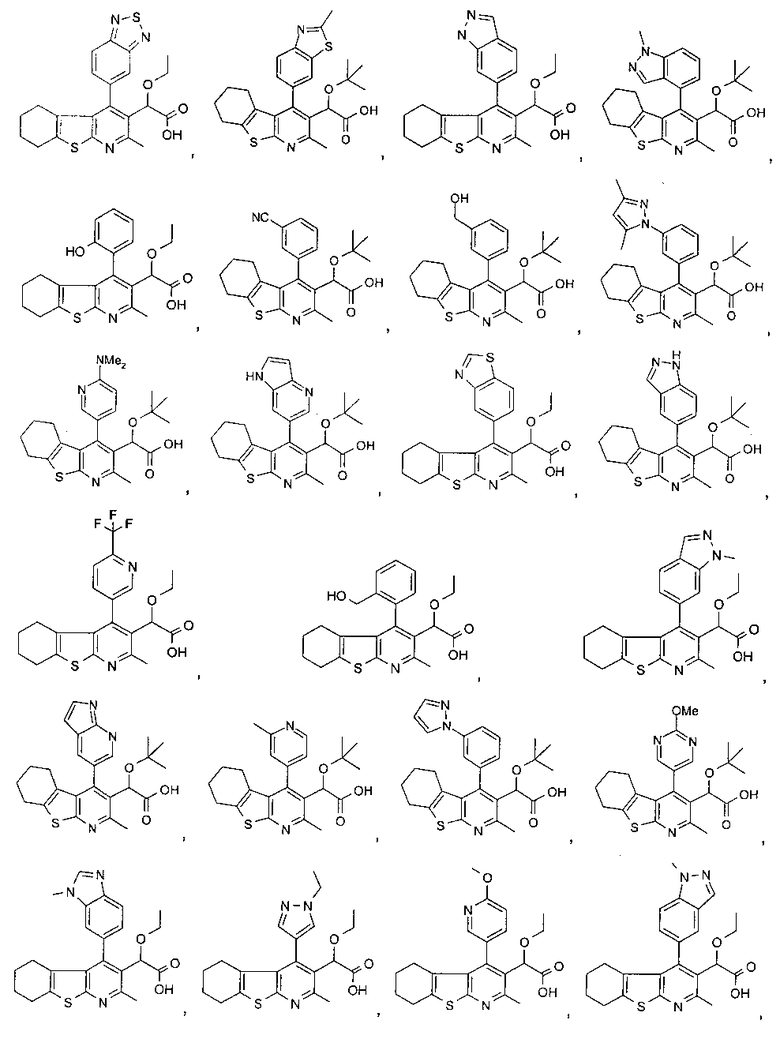
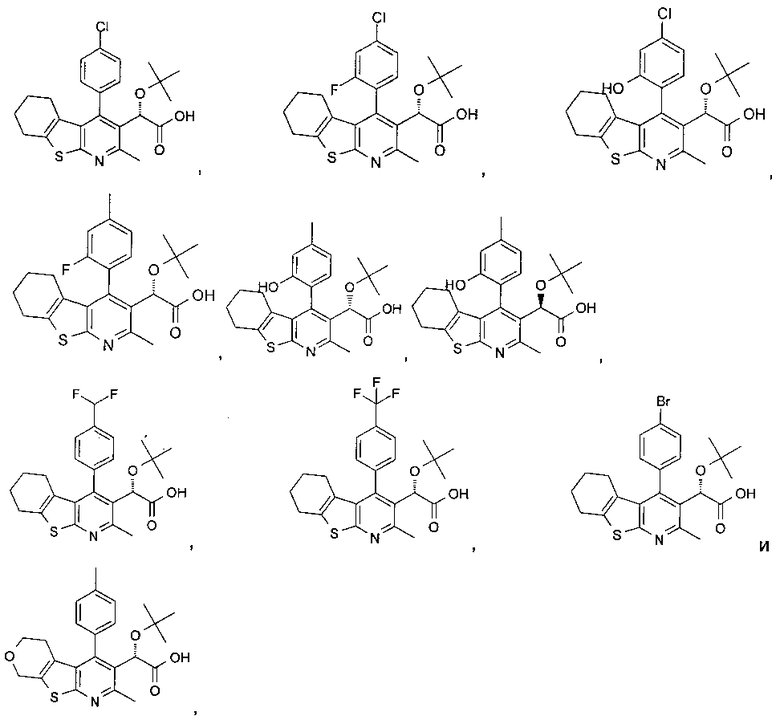
или его фармацевтически приемлемая соль.
7. Соединение по п. 6, выбранное из:
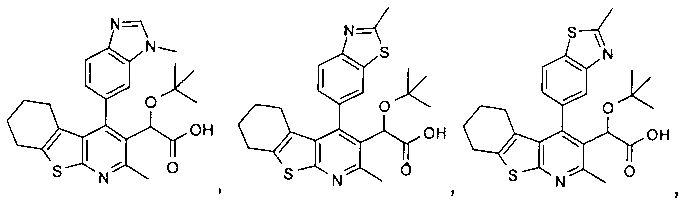
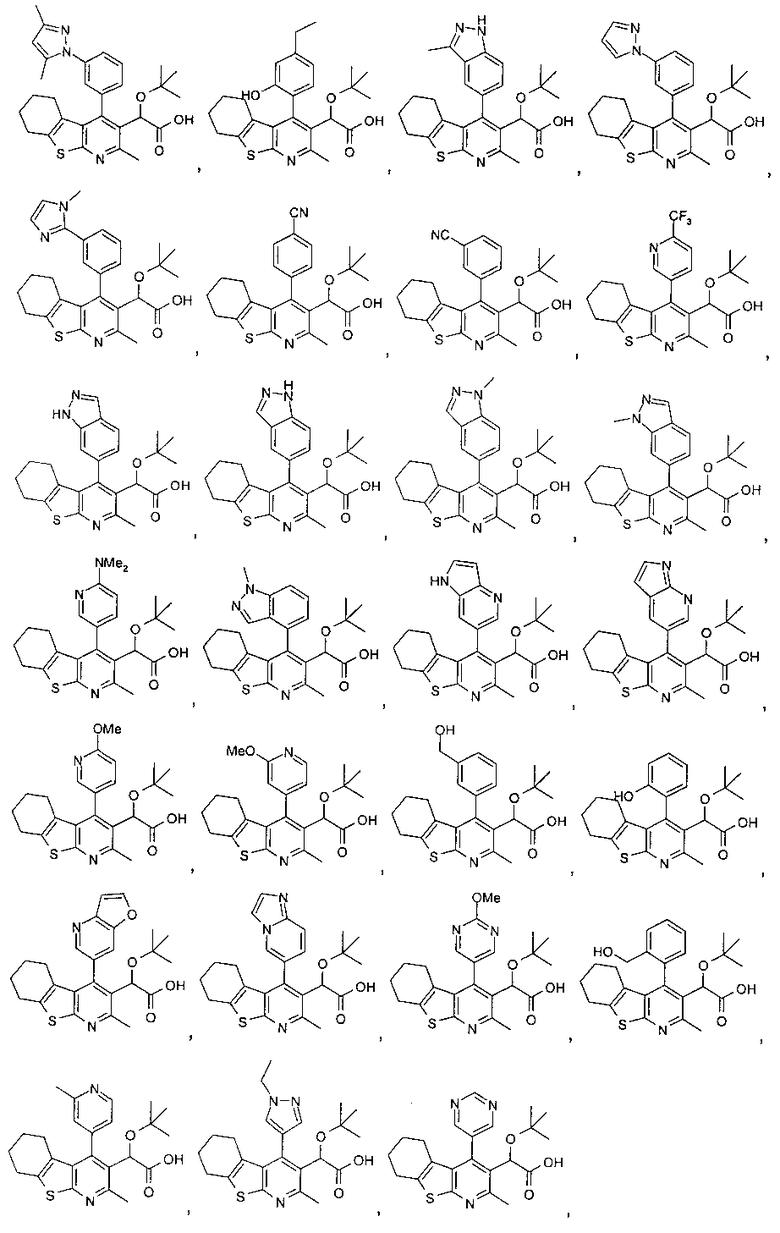
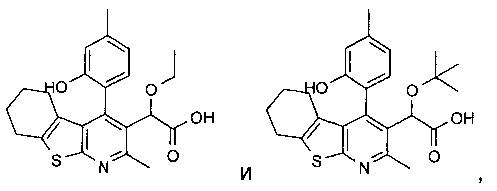
или его фармацевтически приемлемая соль.
8. Соединение по п. 6, выбранное из:
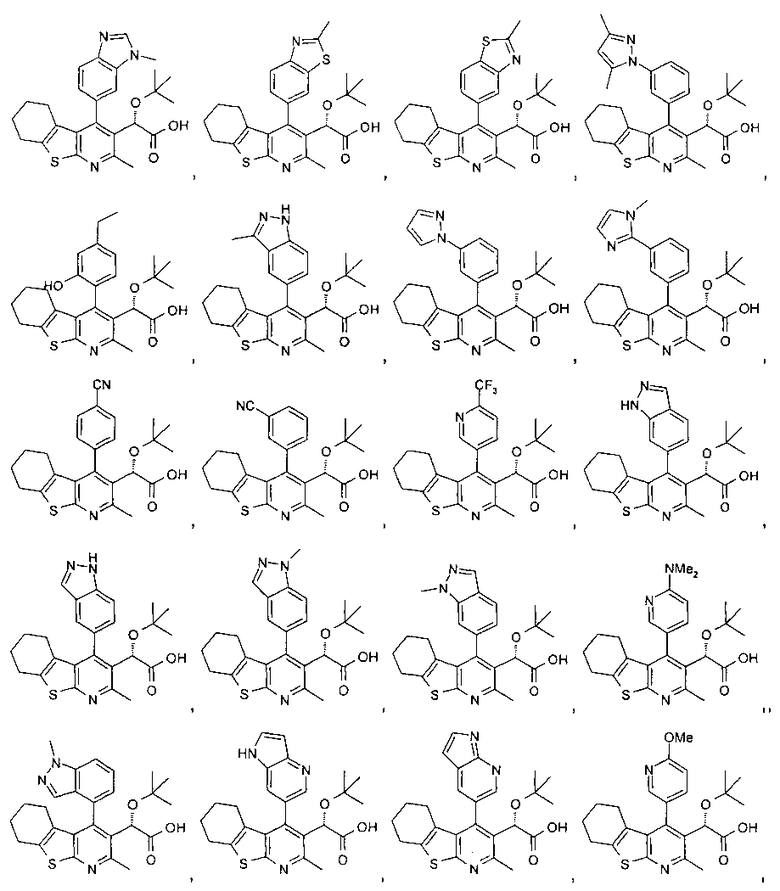
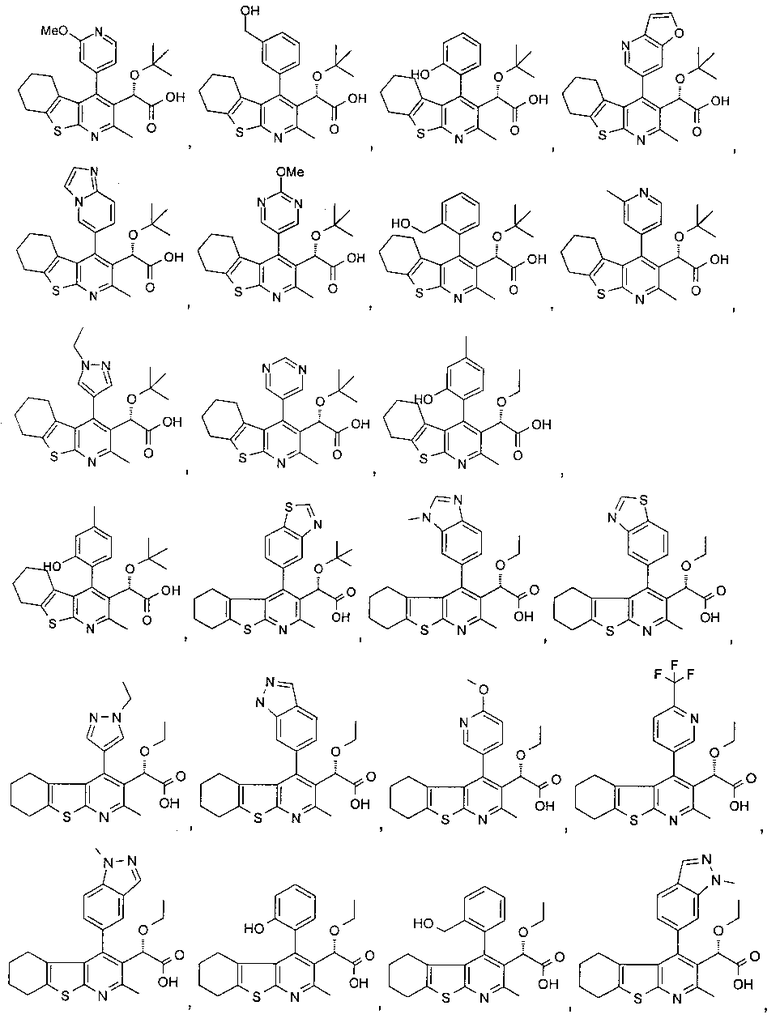
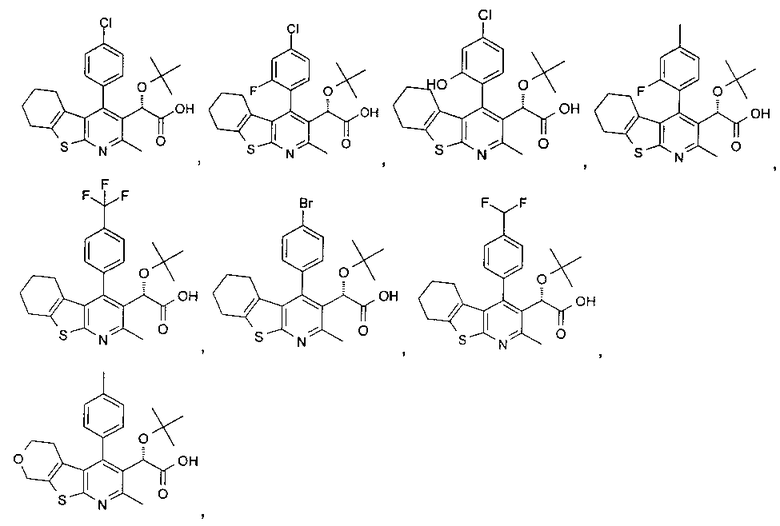
или его фармацевтически приемлемая соль.
9. Соединение по п. 6, выбранное из:
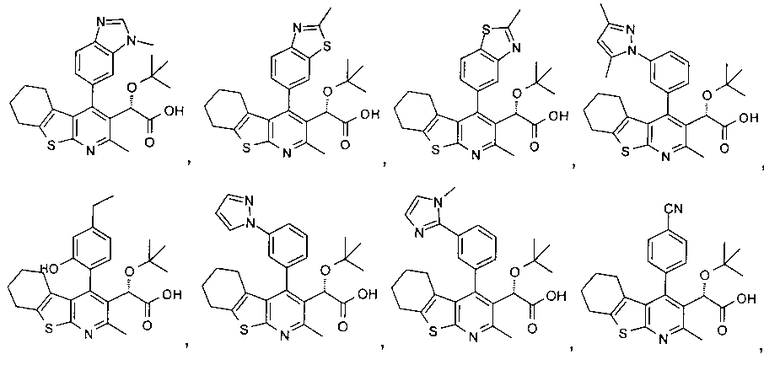
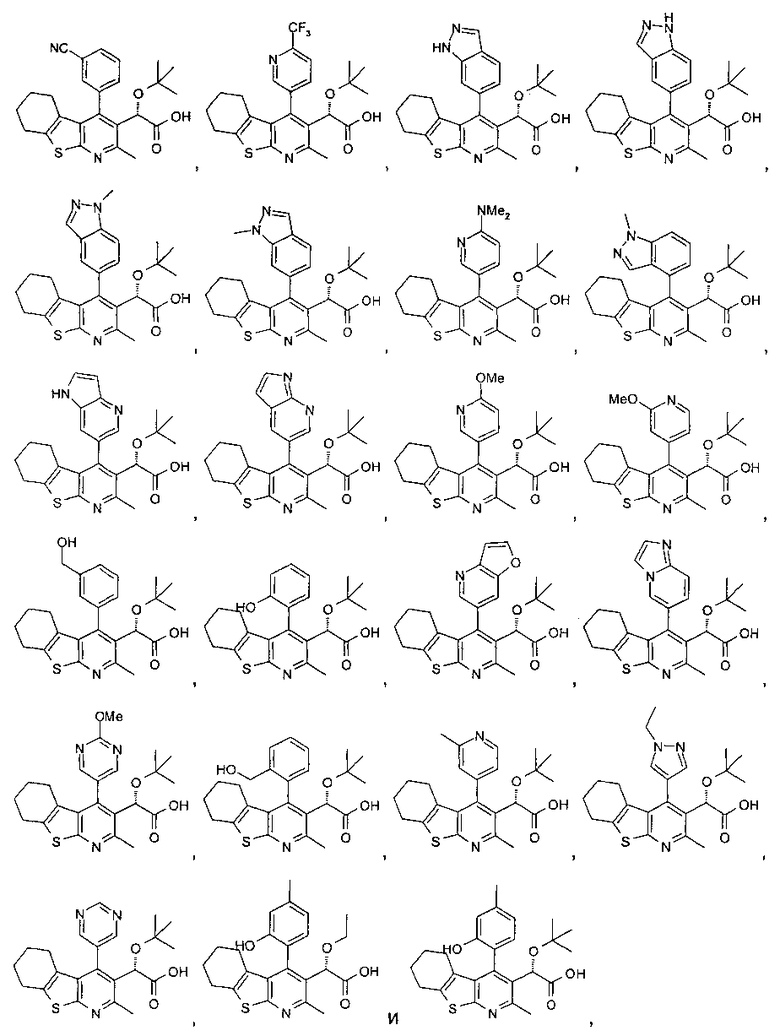
или его фармацевтически приемлемая соль.
10. Фармацевтическая композиция, обладающая ингибирующей активностью в отношении репликации вируса иммунодефицита человека (ВИЧ), содержащая соединение по любому из пп. 1-9 вместе с фармацевтически приемлемым эксципиентом.
11. Соединение по любому из пп. 1-9 для применения в качестве лекарственного средства для лечения ВИЧ-инфекции.
12. Соединение по любому из пп. 1-9 для применения в лечении ВИЧ-инфекции.
13. Применение соединения по любому из пп. 1-9 для изготовления лекарственного средства для лечения ВИЧ-инфекции.
14. Способ лечения млекопитающего, включая человека, с целью лечения ВИЧ-инфекции, включающий введение указанному млекопитающему эффективного количества соединения по любому из пп. 1-9.
15. Продукт, содержащий соединение по любому из пп. 1-9 и один или более других фармакологически активных агентов, в виде комбинированного препарата для одновременного, раздельного или последовательного применения в терапии ВИЧ-инфекции.
| WO 2005076861,A2, 25.08.2005 | |||
| WO 2010130842,A1, 18.11.2010 | |||
| RU 2008137929, A, 27.03.2010 | |||
| ПРОИЗВОДНЫЕ БЕНЗОТИОФЕНА, БЕНЗОФУРАНА, ИНДОЛТИАЗЕПИНОНА, ОКСАЗЕПИНОНА И ДИАЗЕПИНОНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ ИНГИБИРУЮЩЕЙ КЛЕТОЧНУЮ АДГЕЗИЮ ИЛИ ВИЧ-АКТИВНОСТЬЮ, СПОСОБ ТОРМОЖЕНИЯ АДГЕЗИИ ЛЕЙКОЦИТОВ К ЭНДОТЕЛИАЛЬНЫМ КЛЕТКАМ ПРИ ЛЕЧЕНИИ ВЫЗВАННЫХ ЕЮ БОЛЕЗНЕЙ, СПОСОБ ЛЕЧЕНИЯ МЛЕКОПИТАЮЩИХ, ЗАРАЖЕННЫХ ВИЧ | 1995 |
|
RU2144033C1 |

