Область техники
Настоящее изобретение относится к новому производному 1-α-галоген-2,2-дифтор-2-дезокси-D-рибофуранозы и способу его получения, которое является применимым в качестве промежуточного соединения при производстве гемцитабина.
Предшествующий уровень техники
Гемцитабин формулы (А), лекарственное средство для лечения немелкоклеточного рака легких (NSCLC), является синтетическим аналогом нуклеозида, имеющим нуклеооснование цитозина, стереохимически ориентированное вверх по β-направлению при атоме С-1 рибофуранозного остова.

Гемцитабин может быть получен из соединения лактола, как показано на Схеме Реакции 1 через активированное рибофуранозное промежуточное соединение, имеющее активную уходящую группу:
Схема Реакции 1

где P1 является защитной гидроксигруппой и L является уходящей группой.
Более конкретно, гемцитабин может быть получен 1а) введением реакционноспособной уходящей группы (L) к С-1 рибофуранозному кольцу соединения лактола (В) с получением активированного рибофуранозного промежуточного соединения (С), и 1b) гликозилированием соединения формулы (С) цитозином с образованием N-гликозидной связи.
В схеме реакции 1 стадия гликозилирования 1b) проходит по механизму бимолекулярного нуклеофильного замещения (SN2), и, поэтому при получении гемцитабина является важным высокая чистота α-аномера соединения (С), имеющего уходящую группу (L), ориентированную вниз. Соответственно, было предпринято много попыток разработать способ стереоселективного введения уходящей группы (L) к C-1 кольцу рибофуранозы соединения лактола (В).
Например, патенты US № 4526988 и 5453499 раскрывают активированное рибофуранозное промежуточное соединение, такое как 1-α-галогенрибофуранозы, имеющее галогенную уходящую группу, введенную к С-1 кольцу рибофуранозы. Более конкретно, патент US № 4526988 описывает способ получения производного 1-α-галогенрибофуранозы формулы (F) 2а) взаимодействием 1-гидроксигруппы соединения лактола формулы (D) с источником ацетила, таким как уксусный ангидрид, с получением 1-ацетатпроизводного формулы (Е) и 2b) взаимодействием 1-ацетатпроизводного формулы (Е) с газообразным HBr и HCl с получением 1-галогенрибофуранозы, как показано на Схеме реакции 2:
Схема Реакции 2

где R' является защитной гидроксигруппой, Ac является ацетилом и X является Br или Cl.
Однако данный способ приводит к низкому выходу требуемого α-галогенаномера из-за его низкой стереоселективности.
Патент US № 5453499 раскрывает способ получения α-обогащенной 1-галогенрибофуранозы формулы (Н), имеющей соотношение α:β от 9:1 до 10:1, взаимодействием β-сульфоната соединения формулы (G) с источником галогенида в инертном растворителе, как показано на Схеме Реакции 3:
Схема Реакции 3

где P'' является защитной гидроксигруппой, такой как бензоил, R'' является сульфонатом и Y является галогеном.
Однако 1-сульфонат соединения формулы (G), используемый в качестве исходного вещества в данном способе, полученный из соединения лактола способом, описанным в патенте US № 5401861, имеет соотношение α:β примерно 1:4 и, таким образом, суммарное соотношение стереоселективности (α:β) для 1-галогенаномера составляет примерно 3:1.
Более того, предшествующие 1-α-галогенфуранозы, имеющие защищенные 2- и 5-гидроксигруппы, например, бензоильными группами, существуют только в виде масла, что является более затруднительным для обработки и хранения, чем твердая форма, кроме того, для их выделения из смеси α- и β-изомеров требуется неэкономичный способ колоночной хроматографии. Таким образом, существуют необходимость разработки улучшенного способа получения гемцитабина с использованием α-галогенфуранозы в качестве промежуточного соединения.
Сущность изобретения
Соответственно, основной целью настоящего изобретения является обеспечение нового производного 1-α-галоген-D-рибофуранозы в твердой форме, которое может быть очищено простым методом очистки, таким как перекристаллизацией, подходящим для массового производства.
Другой целью настоящего изобретения является обеспечение высоко стереоселективного способа получения названного соединения высокой чистоты и с высоким выходом.
Также другой целью настоящего изобретения является обеспечение соединения, которое может быть использовано в качестве промежуточного в названном способе.
В соответствии с первым аспектом настоящего изобретения предусмотрено производное 1-α-галоген-2,2-дифтор-2-дезокси-D-рибофуранозы формулы (I) в твердой форме:

где
R1 является бензоилом или 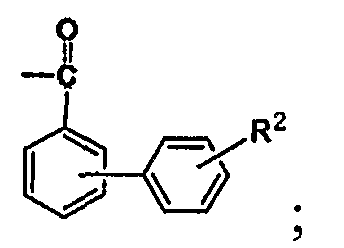
R2 является водородом, циано, галогеном, карбоалкокси, нитро, C1-2 алкокси, С1-2 алкилом или диалкиламино; и
X является Cl, Br или I.
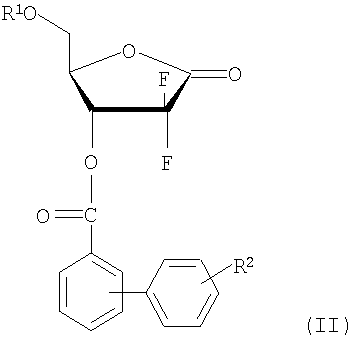
В соответствии с другим аспектом настоящего изобретения предусмотрен способ получения производного 1-α-галоген-2,2-дифтор-2-дезокси-D-рибофуранозы формулы (I), включающий стадии:
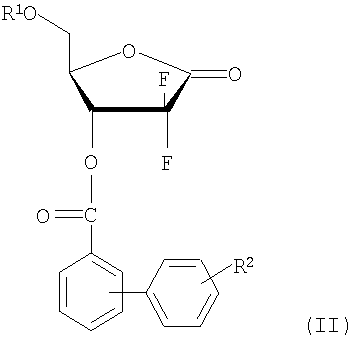
(i) восстановления соединения 1-оксорибозы формулы (II) с получением соединения лактола формулы (III);
(ii) взаимодействия соединения формулы (III) с галогенфосфатным соединением формулы (IV) в присутствии основания с получением 1-фосфатного производного фуранозы формулы (V); и
(iii) взаимодействия соединения формулы (V) с источником галогена, с последующей перекристаллизацией получившегося продукта с получением производного 1-α-галоген-2,2-дифтор-2-дезокси-D-рибофуранозы формулы (I):
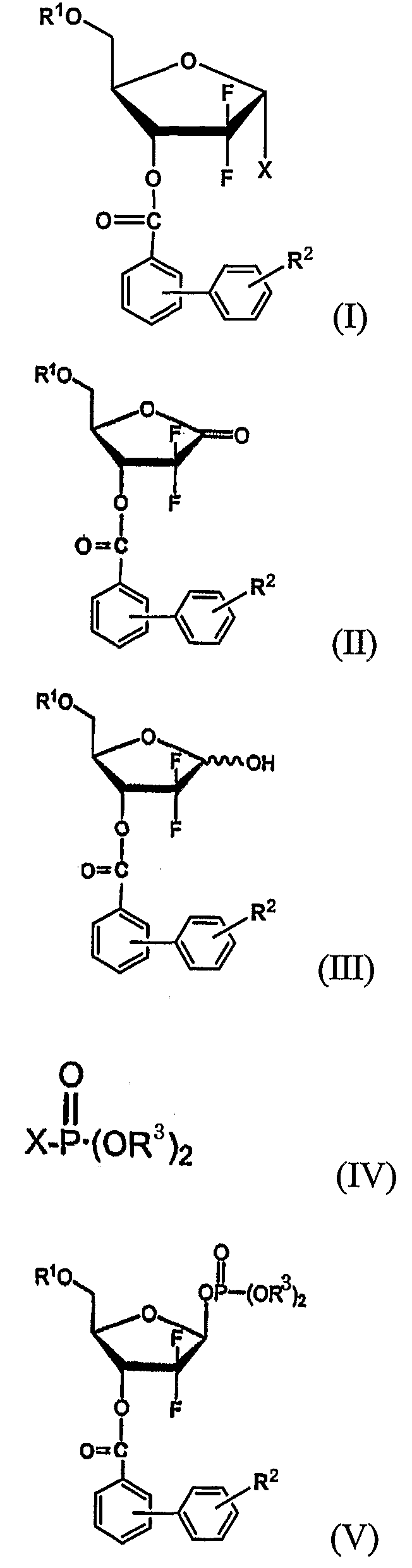
где R1, R2 и X имеют те же значения, что определены выше; и R3 является метилом, этилом или фенилом, предпочтительно фенилом.
В соответствии с еще одним аспектом настоящего изобретения, предусмотрено новое 1-фосфатное производное фуранозы формулы (V), которое может быть использовано в качестве промежуточного соединения при получении производного 1-α-галоген-D-рибофуранозы формулы (I):
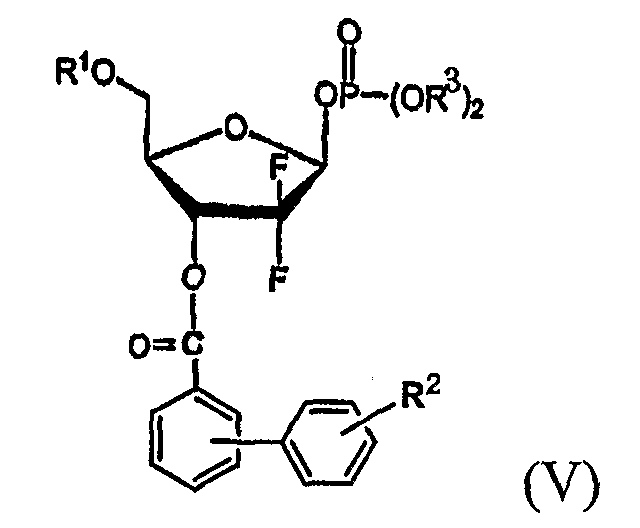
где R1, R2 и R3 имеют те же значения, что определены выше.
Подробное описание изобретения
Термин «обогащенный аномером», используемый здесь, означает смесь аномеров, имеющую содержание определенного аномера более чем 50%, предпочтительно в основном чистый аномер.
Из соединений формулы (I) настоящего изобретения предпочтительными являются те, в которых R2 является водородом.
Производное рибофуранозы формулы (I) согласно изобретению характеризуется наличием 3-гидроксигруппы, защищенной бифенилкарбонильной группой. Также производное согласно изобретению может иметь бифенилкарбонильную группу в качестве защитной 5-гидроксигруппы.
Таким образом, производное 1-α-галогенрибофуранозы согласно изобретению может быть получено в виде твердого вещества и, соответственно, оно может быть легко очищено простым методом очистки, таким как перекристаллизация, до высокой чистоты 99,5% или выше.
Также, производное 1-α-галогенрибофуранозы формулы (I) согласно изобретению может сочетаться с цитозином в обыкновенной реакции гликозилирования с получением гемцитабина, имеющего остаток цитозина при С-1 рибофуранозного кольца, ориентированного вверх β-конфигурация).
При получении гемцитабина через стадию гликозилирования с использованием производного 1-галогенрибофуранозы очень важна чистота α-галогенаномера. Если содержание β-галогенаномера увеличивается, то стереоселективность реакции гликозилирования заметно снижается, приводя к низкому выходу требуемого β-нуклеозида - гемцитабина.
Неочевидный способ получения производного 1-галогенофуранозы формулы (I) описан в Схеме Реакции 4.
Схема реакции 4
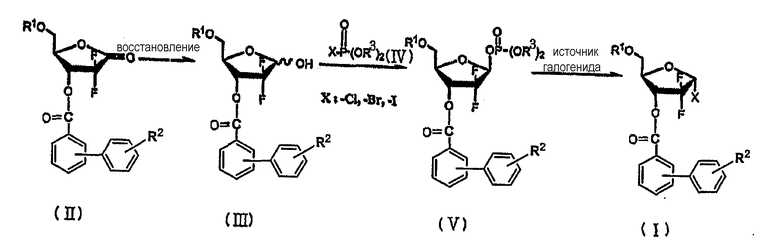
где R1, R2, R3 и X имеют те же значения, как описано выше.
В Схеме Реакции 4 производное 1-галоген-2,2-дифтор-2-дезокси-D-рибофуранозы формулы (I) может быть получено в форме, имеющей высокое содержание α-аномера 99,5% или выше, путем (i) восстановления соединения 1-оксорибозы формулы (II) в соответствии с обычными способами с получением соединения лактола формулы (III), смеси α- и β-аномеров; (ii) взаимодействия соединения формулы (III) с соединением галогенфосфата формулы (IV) в присутствии основания с получением β-обогащенной 1-фосфатфуранозы формулы (V), имеющей соотношение β/α 10 или более; и (iii) взаимодействия соединения формулы (V) с источником галогенида с получением соединения формулы (I).
Использование новых фуранозных промежуточных соединений формулы (V), имеющих фосфатную уходящую группу, является особенностью неочевидного способа получения 1-галогенрибофуранозы формулы (I), имеющей высокое содержание α-аномера.
Таким образом, на стадии (ii) получения фосфата фуранозы формулы (V) из соединения лактола формулы (III) β-аномер фосфата может быть получен с высоким соотношением β/α, больше чем 10. Также последующая стадия (iii) может быть осуществлена непрерывно без выделения промежуточного соединения с получением α-галогенфуранозы формулы (I) с высоким соотношением α/β, по меньшей мере, 10.
Более того, в соответствии с настоящим изобретением α-галогенфураноза получается в виде твердого вещества, когда бифенилкарбонильная группа принята в качестве защитной 3- и/или 5-гидроксигруппы рибофуранозного кольца, и твердая форма может быть легко очищена до высокой чистоты 99,5% или выше простым методом очистки, который делает возможным получение требуемого β-нуклеозида, имеющего высокое соотношение β/α от 4 до 14. Такое высокое соотношение β/α является заметно более высоким, чем соотношение β/α от 2 до 3, достижимое обычными способами.
Более конкретно, на стадии (i) Схемы Реакции 4 соединение лактола формулы (III) может быть получено восстановлением соединения формулы (II) восстанавливающим агентом, как описано в патенте US 4526988 и 5464826. Соединение 1-оксорибозы формулы (II), используемое в качестве исходного сырья на стадии (i), может быть получено способом, включающим стадии защиты 3-гидроксигруппы соединения формулы (VI) бифенилкарбонильной защитной группой, последующим гидролизом образовавшегося продукта в присутствии основания с получением энантиомера 3R-карбоксилата формулы (VII):
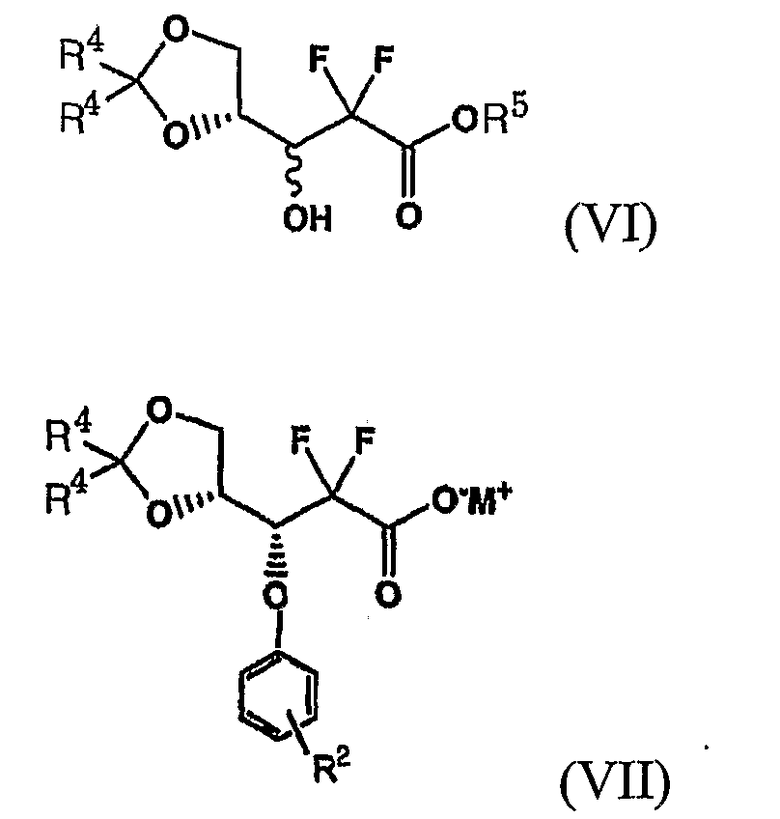
где R2 имеет то же значение, что описано выше, R4 является метилом или этилом, R5 является С1-5алкилом и M является NH4, натрием или калием.
Растворителем, пригодным для использования на стадии (i), является тетрагидрофуран, диэтиловый эфир или диоксан; и восстанавливающим агентом может быть алюмогидрид лития, ди-изо-бутилалюминогидрид или три-трет-бутоксиалюминогидрид лития, предпочтительно три-трет-бутоксиалюминогидрид лития; и восстановление может быть проведено при комнатной температуре в течение от 1 до 2 часов после добавления восстанавливающего агента при -50 до -20°С.
На данной стадии (i) восстановления соединение лактола формулы (III) получается в виде смеси α- и β-аномеров в соотношении от 1:1 до 2:1; и следующая стадия (ii) может быть проведена после выделения каждого аномера, полученного на стадии (i) или проведена без такого выделения.
На стадии (ii) 1-фосфат фуранозы формулы (V) может быть получен взаимодействием соединения формулы (III) с соединением галогенфосфата формулы (IV) в присутствии основания с получением β-обогащенного соединения формулы (V), имеющего соотношение β/α 10 или более. На данной стадии фосфатная уходящая группа может быть диметилфосфатом, диэтилфосфатом или дифенилфосфатом, предпочтительно дифенилфосфатом.
Стадия (iii) может быть проведена после выделения требуемого β-аномера, полученного на стадии (ii) перекристаллизацией с использованием таких растворителей как вода, этанол, пропанол, изопропанол, н-бутанол, этилацетат и их смеси, предпочтительно изопропанол или смесь изопропанол - вода. Также данная стадия может быть проведена с неочищенным продуктом стадии (ii) без процесса выделения.
Соединение галогенфосфата формулы (IV) может быть использовано в количестве, колеблющемся от 1,1 до 1,5 мольных эквивалентов, по отношению к лактольному соединению формулы (III). Соединение формулы (IV) является коммерчески доступным или может быть легко получено в соответствии с обычными методами, раскрытыми в Bichem. Preps., 1, 50 (1951) или J. Chem. Soc., 2921 (1949). Стадия (ii) может быть облегчена добавлением катализатора, такого как 4-диметиламинопиридин или 4-пирролидинопиридин.
Также основание, используемое для нейтрализации кислоты, полученной на стадии (ii), может быть выбрано из группы, состоящей из пиридина, триэтиламина, трибутиламина, диизопропилэтиламина и метилпиперидина, предпочтительно триэтиламина, который может быть использован в количестве, колеблющемся от 1,2 до 2,0 мольных эквивалентов, по отношению к лактольному соединению формулы (III). Растворителем, используемым на стадии (ii), может быть бензол, толуол, ацетонитрил, тетрагидрофуран, этилацетат, хлористый метилен или хлороформ, предпочтительно, толуол, и которая проводится при от -25 до 50°С в течение от 2 до 10 часов.
Кроме того, на стадии (iii) взаимодействием 1-фосфата фуранозы формулы (V) с источником галогенида с последующей перекристаллизацией получающегося продукта может быть получен высокочистый α-аномер формулы (I) 99,5% или выше (т.е., содержание β-аномера меньше, чем 0,5%).
Источник галогенида, который может быть использован на стадии (iii), включает HCl/уксусную кислоту, HBr/уксусную кислоту, HBr/пропионовую кислоту, триалкилсилилгалогенид, галогенид лития, галогенид натрия, галогенид цезия, галогенид калия, галогенид тетраалкиламмония и их смеси; из которых предпочтительными являются 30% HBr/уксусная кислота, 30% HBr/пропионовая кислота, иодид тетрабутиламмония, бромид тетрабутиламмония, триметилсилилиодид, триметилсилилбромид, триметилсилилхлорид и смесь триметилсилилхлорид - бромид лития. Такой источник галогенида используется в количестве от 5 до 30 мольных эквивалентов, предпочтительно от 10 до 20 мольных эквивалентов, по отношению к соединению формулы (V).
В случае использования 1,0 М HCl/уксусной кислоты, 30% HBr/уксусной кислоты, 30% HBr/пропионовой кислоты в качестве источника галогенида они используются в неразбавленном виде, в то время как другие источники галогенида могут быть использованы разбавленными такими растворителями, как хлористый метилен, дибромэтан, дихлорэтан, хлороформ, ТГФ, 1,4-диоксан, ацетонитрил, N,N-диметилформамид или N,N-диметилацетамид.
Стадия (iii) может быть проведена в таком растворителе, как хлористый метилен, дибромэтан, дихлорэтан или хлороформ при температуре в диапазоне от 0 до 50°С, предпочтительно от 10 до 30°С в течение от 30 минут до 24 часов.
Образующаяся 1-галогенрибофураноза является смесью α- и β-аномеров, имеющих соотношение α/β по меньшей мере 10, и требуемый α-аномер может быть выделен из смеси перекристаллизацией с использованием такого растворителя, как метанол, этанол, изопропанол, ацетонитрил, вода или их смеси, предпочтительно, изопропанол или смесь изопропанол - вода, с получением 1-α-галогенрибофуранозы с высокой чистотой 99,5% или более.
Неочевидный способ получения 1-α-галогенфуранозы формулы (I), используя 1-фосфатфуранозы формулы (V) в качестве промежуточного соединения, дает суммарный выход от 65 до 75%, который является заметно более высоким, чем достижимый обычными способами (суммарный выход примерно 45%).
Последующие Получения и Примеры даны только в целях иллюстрации и не ограничивают область изобретения.
В следующих Примерах получения и Примерах термин "-OCOBiPh" или "BiPhOCO-" относятся к 
ВЭЖХ анализы соединения формулы (V) были проведены на колонке YMC pack pro C18 RS (4,6×150 мм, 5 мкм) с использованием в качестве элюента смеси буфера и метанола (17:83, об/об); и соединения формулы (I) на колонке Capcellpak MG C18 RS (4,6×150 мм, 5 мкм) с использованием в качестве элюента смеси буфера и метанола (1:4, об/об). Буфер был получен смешением 13,8 г NaH2PO4 и 1 л дистиллированной воды и добавлением Н3PO4 до рН 2,5.
Пример получения 1: Получение D-эритро-2-дезокси-2,2-дифторопентафураноз-1-илозы-5-бензоил-3-(4-фенил)бензоата
(соединение формулы (II))
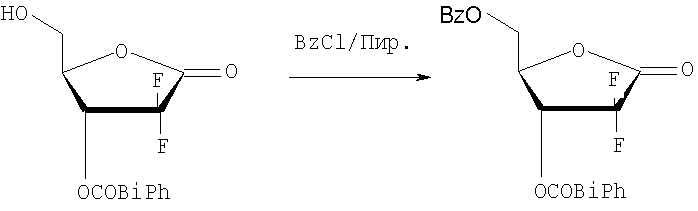
15 г D-эритро-2-дезокси-2,2-дифторопентафураноз-1-илозы-3-(4-фенил)бензоата (способ получения описан в WO 2006/009353 (Стадия 1 Способа А Примера 3 на с.14-15)) растворили в 150 мл хлористого метилена, и при перемешивании в него добавили по каплям 6,9 мл пиридина. В раствор медленно добавили 7,4 мл бензоилхлорида, растворенного в 40 мл хлористого метилена, поддерживая температуру от 5 до 10, с последующим перемешиванием в течение 7 часов при комнатной температуре. Получившуюся смесь нейтрализовали 105 мл 1N HCl и затем добавили туда воду. Органический слой отделили, последовательно промыли 100 мл насыщенного гидрокарбоната натрия и 100 мл рассола, высушили над безводным MgSO4, отфильтровали и сконцентрировали при пониженном давлении. Получившийся остаток перекристаллизовали из диэтилового эфира/гексана (5:1, об/об), получив 16,8 г названного соединения в виде белого твердого вещества (выход: 86%).
1H-ЯМР (300 МГц, CDCl3): 4,90˜4,75 (ддд, 2Н), 5,10 (дд, 1Н), 5,87 (ддд, 1H), 7,65˜7,50 (м, 5H), 7,78˜7,67 (м, 3H), 7,81 (д, 2H), 8,13 (д, 2H), 8,23 (д, 2H).
Т.пл.: 130-131°С.
Пример получения 2: Получение D-эритро-2-дезокси-2,2-дифторо-пентафураноз-1-илозы-3,5-ди(4-фенил)бензоата (соединение формулы (II))
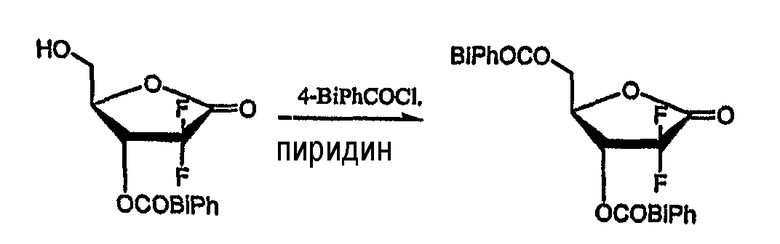
20 г D-эритро-2-дезокси-2,2-дифторопентафураноз-1-илозы-3-(4-фенил)бензоата растворили в 300 мл хлороформа, и при перемешивании в него добавили по каплям 9,5 мл пиридина. В раствор медленно добавили 10,1 мл бензоилхлорида, растворенного в 55 мл хлороформа с последующим перемешиванием в течение 6 часов при комнатной температуре. Получившуюся смесь нейтрализовали 140 мл 1N HCl и последовательно промыли 150 мл воды, 150 мл насыщенного гидрокарбоната натрия и 150 мл рассола. Органический слой отделили, высушили над безводным MgSO4, отфильтровали и сконцентрировали при пониженном давлении. Получившийся остаток перекристаллизовали из этилацетата/гексана (3:1 об/об), получив 21,8 г названного соединения в виде белого твердого вещества (выход: 72%).
1H-ЯМР (300 МГц, CDCl3): 4,72˜4,79 (м, 2H), 5,03 (кв, 1H), 5,84˜5,76 (м, 1H), 7,48˜7,44 (м, 6H), 7,72˜7,60 (м, 8H), 8,15˜8,07 (м, 4H).
Т.пл.: 137-139°С.
Пример 1: Получение 1-α-бромо-2-дезокси-2,2-дифторо-D-рибофуранозил-5-бензоил-3-(4-фенил)бензоата (соединение формулы (I); R1=бензоил и R2=4-фенил)
Стадия 1) Получение 2-дезокси-2,2-дифторо-D-рибофуранозил-3-бензоил-5-(4-фенил)бензоата (соединение формулы (III))

13,5 г три-трет-бутоксиалюминогидрида лития растворили в 160 мл ТГФ и перемешивали в течение 30 минут при комнатной температуре с последующим охлаждением до -40°С. В раствор добавили соединение, полученное в примере получения 1, растворенное в 80 мл ТГФ, смесь медленно нагрели до комнатной температуры и оставили реагировать при этой температуре в течение 2 часов. По завершении реакции в реакционную смесь добавили по каплям 220 мл 1N HCl для разложения избытка три-трет-бутоксиалюминогидрида лития. Органический (ТГФ) и водный слои разделили, и водный слой проэкстрагировали 220 мл диэтилового эфира. Эфирный экстракт объединили с ТГФ слоем и последовательно промыли 220 мл воды, 220 мл насыщенного гидрокарбоната натрия и 220 мл рассола. Органический слой отделили, высушили над безводным MgSO4, отфильтровали и сконцентрировали при пониженном давлении. Получившийся остаток очистили флэш-хроматографией, получив 18,3 г названного соединения в виде бледно-желтого сиропа (выход: 91%).
1H-ЯМР (300 МГц, CDCl3): 3,89˜3,91 (д, 1H), 4,61˜4,81 (м, 2H), 5,31˜5,92 (м, 2H), 7,26˜7,70 (м, 10H), 8,05˜8,16 (м, 4H).
Стадия 2) Получение 2-дезокси-2,2-дифторо-D-рибофуранозил-3-бензоил-5-(4-фенил)бензоил-1β-дифенилфосфата (соединение формулы (V))
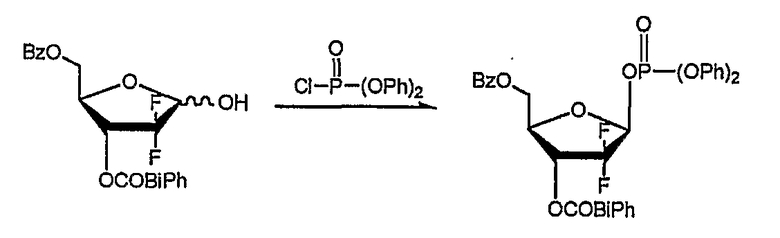
18,3 г соединения, полученного на стадии 1, растворили в 146 мл толуола и добавили к нему 6,7 мл триэтиламина. В раствор по каплям добавили 12,4 мл дифенилхлорфосфата, растворенного в 37 мл толуола, с последующим перемешиванием в течение 4 часов при комнатной температуре. По завершении реакции оставшийся триэтиламин нейтрализовали добавлением 48 мл 1N HCl, слои толуола и воды отделили и водный слой проэкстрагировали 48 мл диэтилового эфира. Эфирный экстракт объединили со слоем толуола и последовательно промыли водой, насыщенным гидрокарбонатом натрия и насыщенным рассолом. Органический слой отделили, высушили над безводным MgSO4, отфильтровали и сконцентрировали при пониженном давлении с получением смеси α- и β-фосфатов в виде твердого вещества. Смесь исследовали 1H ЯМР и обнаружили, что соотношение α-фосфат:β-фосфат составило 1:10,6. β-фосфат селективно перекристаллизовали из изопропанол/вода (3:1 об/об), получив 26,5 г названного соединения в виде белого твердого вещества (выход: 87%).
1H-ЯМР (300 МГц, CDCl3): 4,56-4,25 (м, 3H), 5,80 (м, 1H), 5,95 (т, 1H), 7,44-6,98 (м, 16H), 7,51 (д, 2H), 7,57 (д, 2H), 7,89 (д, 2H), 8,01 (д, 2H).
Т. пл.: 101-103°С.
ВЭЖХ чистота (% пика): α-фосфат аномер 1,76%, β-фосфат аномер 98,24%.
Стадия 3) Получение 1-α-бромо-2-дезокси-2,2-дифторо-D-рибофуранозил-3-бензоил-5-(4-фенил)бензоата (соединение формулы (I))

22,8 г соединения, полученного на стадии 2, добавили к 80,5 мл 30% HBr/уксусной кислоте с последующим перемешиванием в течение 6 часов при комнатной температуре. По завершении реакции образовавшуюся смесь разбавили 400 мл хлористого метилена и вылили на 500 мл лед/вода. Органический слой отделили, последовательно промыли водой, насыщенным гидрокарбонатом натрия и рассолом, высушили над безводным MgSO4 и сконцентрировали при пониженном давлении с получением смеси α- и β-бромоаномеров в виде твердого вещества. Смесь исследовали 1H ЯМР и обнаружили, что соотношение α-бромо:β-бромо составило 10,7:1. α-бромосоединение селективно перекристаллизовали из изопропанола, получив 17,0 г названного соединения в виде белого твердого вещества (выход: 82%).
1H-ЯМР (300 МГц, CDCl3): 8,19 (д, 2H), 8,06 (д, 2H), 7,73 (д, 2H), 7,63 (д, 2H), 7,64-7,41 (м, 6H), 6,56 (д, 1H), 5,60 (дд, 1H).
Т. пл.: 111-112°С.
ВЭЖХ чистота (% пика): α-бромоаномер 99,74%, β-бромоаномер 0,26%.
Пример 2: Получение 1-α-бромо-2-дезокси-2,2-дифторо-D-рибофуранозил-3,5-ди(4-фенил)бензоата (соединение формулы (I); R1=4-бифенилкарбонил и R2=4-фенил).
Стадия 1) Получение 2-дезокси-2,2-дифторо-D-рибофуранозил-3,5-ди(4-фенил)бензоата (соединение формулы (III))
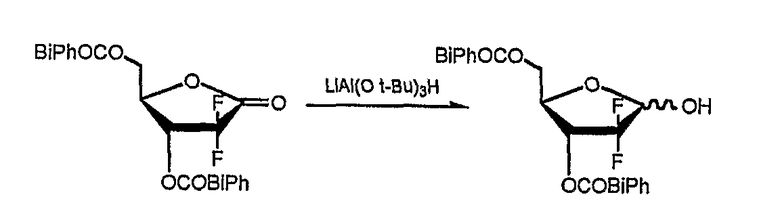
8,66 г три-трет-бутоксиалюминогидрида лития растворили в 120 мл ТГФ и перемешивали в течение 30 минут при комнатной температуре с последующим охлаждением до -40°С. В раствор добавили соединение, полученное в Примере Получения 2, растворенное в 100 мл ТГФ, и перемешивали в течение 1 часа при комнатной температуре. По завершении реакции в реакционную смесь добавили по каплям 142 мл 1N HCl для разложения избытка три-трет-бутоксиалюминогидрида лития, ТГФ и водный слои разделили, и водный слой проэкстрагировали 150 мл диэтилового эфира. Эфирный экстракт объединили с ТГФ слоем и последовательно промыли водой, насыщенным гидрокарбонатом натрия и рассолом. Органический слой отделили, высушили над безводным MgSO4, и сконцентрировали при пониженном давлении. Остаток перекристаллизовали из толуола, получив 13,4 г названного соединения в виде белого твердого вещества (выход: 89%).
1H-ЯМР (300 МГц, CDCl3): 3,45 (с, 1H), 3,8 (с), 4,85˜4,50 (м, 3H), 5,8˜5,4 (м, 2H), 7,49˜7,43 (м, 6H), 7,71˜7,61 (м, 8H), 8,18˜8,12 (м, 4H).
Т. пл.: 156-158°С.
Стадия 2) Получение 2-дезокси-2,2-дифторо-D-рибофуранозил-3,5-ди(4-фенил)бензоил-1β-дифенилфосфата (соединение формулы (V))

13 г соединения, полученного на стадии 1, растворили в смеси 130 мл толуола и 100 мл хлористого метилена, и добавили к нему 5,1 мл триэтиламина. К образующейся смеси по каплям добавили 7,6 мл дифенилхлорфосфата и перемешивали в течение 5 часов при комнатной температуре. По завершении реакции растворитель удалили при пониженном давлении, получившееся твердое вещество растворили в 130 мл хлористого метилена и добавили к нему 65 мл 1N HCl. Органический слой отделили, последовательно промыли водой, насыщенным гидрокарбонатом натрия и рассолом, высушили над безводным MgSO4 и сконцентрировали при пониженном давлении с получением смеси α- и β-фосфатов в виде твердого вещества. Смесь исследовали 1H ЯМР и обнаружили, что соотношение α-фосфат:β-фосфат составило 1:10,8. β-фосфат селективно перекристаллизовали из изопропанола, получив 15,6 г названного соединения в виде белого твердого вещества (выход: 83%).
1H-ЯМР (300 МГц, CDCl3): 4,70-4,40 (м, 3H), 5,90 (м, 1H), 6,08 (т, 1H), 7,70˜7,08 (м, 24H), 8,15˜8,04 (дд, 4H).
Т. пл.: 145-147°С.
ВЭЖХ чистота (% пика): α-фосфат аномер 1,29%, β-фосфат аномер 98,71%.
Стадия 3) Получение 1-α-бромо-2-дезокси-2,2-дифторо-D-рибофуранозил-3,5-ди-(4-фенил)бензоата (соединение формулы (I))

13 г соединения, полученного на стадии 2, растворили в 83,2 мл 30% HBr/уксусной кислоте и перемешивали в течение 7 часов при комнатной температуре. В раствор добавили 50 мл смеси лед/вода и отфильтровали образовавшееся твердое вещество. Отфильтрованное твердое вещество являлось смесью α- и β-бромоаномеров, и 1H ЯМР анализ показал, что соотношение составило α-бромо:β-бромо 10,9:1. α-бромосоединение селективно перекристаллизовали из этанола, получив 8,45 г названного соединения в виде белого твердого вещества (выход: 83%).
1H-ЯМР (300 МГц, CDCl3): 4,89˜4,22 (м, 3H), 5,62 (дд, 1H), 6,55 (д, 1H), 7,73˜7,42 (м, 14H), 8,63˜8,11 (дд, 4H).
Т. пл.: 151-153°С.
ВЭЖХ чистота (% пика): α-бромоаномер 99,67%, β-бромоаномер 0,33%.
Пример 3: Получение 1-α-бромо-2-дезокси-2,2-дифторо-D-рибофуранозил-3-бензоил-5-(4-фенил)бензоата (получение in situ)
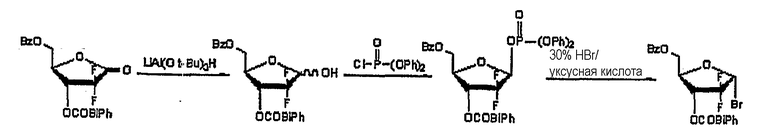
6,5 г три-трет-бутоксиалюминогидрида лития растворили в 100 мл ТГФ и перемешивали в течение 30 минут при комнатной температуре, и охладили до -40°С. В раствор по каплям добавили 10 г соединения, полученного в Примере Получения 1, растворенного в 50 мл ТГФ, и перемешивали в течение 2 часов при комнатной температуре. По завершении реакции в реакционную смесь добавили по каплям 120 мл 1N HCl для разложения избытка три-трет-бутоксиалюминогидрида лития, ТГФ и водный слои разделили, и водный слой проэкстрагировали 150 мл диэтилового эфира. Эфирный экстракт объединили с ТГФ слоем и последовательно промыли водой, насыщенным гидрокарбонатом натрия и рассолом. Органический слой отделили, высушили над безводным MgSO4, отфильтровали и сконцентрировали при пониженном давлении, с получением 10,5 г остатка в виде сиропа.
Образовавшийся остаток растворили в 100 мл толуола и добавили к нему 4,0 мл триэтиламина. К образовавшейся смеси по каплям добавили 6,4 мл дифенилхлорфосфата, растворенного в 30 мл толуола, с последующим перемешиванием в течение 4 часов при комнатной температуре. По завершении реакции для нейтрализации оставшегося триэтиламина к смеси добавили 30 мл 1N HCl, слои толуола и воды разделили, и водный слой проэкстрагировали 30 мл диэтилового эфира. Эфирный экстракт объединили со слоем толуола и последовательно промыли водой, насыщенным гидрокарбонатом натрия и рассолом. Органический слой отделили, высушили над безводным MgSO4, отфильтровали и сконцентрировали при пониженном давлении, получив 14,9 г смеси α- и β-фосфатов в виде сиропа. Смесь исследовали 1H ЯМР и найдено, что соотношение α-фосфат:β-фосфат составило 1:10,3.
Затем к смеси фосфатов добавили 57,2 мл 30% HBr/уксусной кислоты и перемешивали в течение 7 часов при комнатной температуре. По завершении реакции смесь разбавили 280 мл хлористого метилена, вылили на лед/воду и отделили слой хлористого метилена. Слой хлористого метилена последовательно промыли смесью лед/вода, насыщенным гидрокарбонатом натрия и рассолом. Органический слой отделили, высушили над безводным MgSO4, отфильтровали и сконцентрировали при пониженном давлении с получением смеси α- и β-изомеров в виде твердого вещества. Смесь исследовали 1H ЯМР и обнаружили, что соотношение α-бромо:β-бромо составило 10,5:1. α-бромосоединение селективно перекристаллизовали из изопропанола, получив 8,0 г названного соединения в виде белого твердого вещества (выход: 70%).
Данные 1H ЯМР и т. пл. были теми же, что найденные на стадии 4 Примера 1.
ВЭЖХ чистота (% пика): α-бромоаномер 99,51%, β-бромоаномер 0,48%.
Пример 4: Получение 1-α-иодо-2-дезокси-2,2-дифторо-D-рибофуранозил-3-бензоил-5-(4-фенил)бензоата

5,6 мл триметилсилилиодида добавили к 40 мл хлористого метилена, к ним добавили 1,8 г соединения, полученного на стадии 2 Примера 1, и смесь перемешивали в течение 0,5 часа при комнатной температуре. При охлаждении на ледяной бане смесь по каплям добавили к 100 мл насыщенного гидрокарбоната натрия и перемешивали в течение 0,5 часа. Слой хлористого метилена отделили, высушили над безводным MgSO4 и сконцентрировали при пониженном давлении, получив смесь α- и β-изомеров в виде твердого вещества. Смесь исследовали 1H ЯМР и обнаружили, что соотношение α-иодо:β-иодо составило 14,2:1. α-иодосоединение селективно перекристаллизовали из изопропанола, получив 1,36 г названного соединения в виде белого твердого вещества (выход: 92%).
1H-ЯМР (300 МГц, CDCl3): 8,24 (д, 2H), 8,06 (д, 2H), 7,74 (д, 2H), 7,66 (д, 2H), 7,64-7,43 (м, 6H), 6,93 (д, 1H), 5,60 (дд, 1H), 4,86˜4,68 (м, 3H).
ВЭЖХ чистота (% пика): α-иодоаномер 99,81%, β-иодоаномер 0,18%.
Сравнительный Пример 1: Получение 1-α-иодо-2,2-дифтор-2-дезокси-D-рибофуранозил-3,5-дибензоата
Названное соединение было получено в соответствии со способом, раскрытым в патенте U.S. № 5453499, как описано ниже.
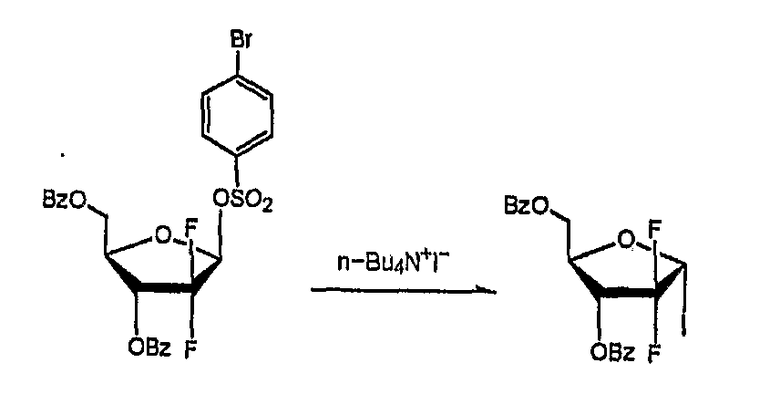
К 1 г 2,2-дифтор-2-дезокси-D-рибофуранозил-3,5-дибензоил-1-β-(п-бромбензол)сульфоната добавили 80 мл тетрагидрофурана и 80 мл иодида тетрабутиламмония, и смесь кипятили с обратным холодильником в течение 3,5 часов. Образовавшаяся смесь включала смесь α-иодо и β-иодо, анализ 1H ЯМР показал, что соотношение α-иодо:β-иодо составило 10:1.
Для того чтобы выделить α-иодосоединение смесь охладили и разбавили дихлорметаном и водой. Органический слой отделили, последовательно промыли 1N HCl, карбонатом натрия, рассолом и водой, высушили над безводным MgSO4 и сконцентрировали при пониженном давлении с получением остатка в виде сиропа. Образовавшийся осадок очистили флэш-хроматографией на силикагеле (толуол/гексан (2:1, об/об)), получив 302 мг названного соединения (выход: 45%).
1H-ЯМР (300 МГц, CDCl3): 8,12 (м, 4H), 7,72˜7,4 (м, 6H), 6,92 (д, 1H), 5,60 (дд, 1H), 4,91 ˜4,62 (м, 3H).
Хотя изобретение было описано относительно конкретных вышеназванных воплощений, следует осознавать, что специалистом в данной области могут быть сделаны различные модификации и изменения, которые также попадают в рамки изобретения, как определено в прилагаемой формуле изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ 2'-ДЕЗОКСИ-2', 2'-ДИФТОРЦИТИДИНА | 2005 |
|
RU2360919C2 |
| СПОСОБ ПОЛУЧЕНИЯ ОБОГАЩЕННЫХ БЕТА-АНОМЕРОМ НУКЛЕОЗИДОВ | 1993 |
|
RU2131880C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНОГО D-ЭРИТРО-2,2-ДИФТОРО-2-ДЕЗОКСИ-1-ОКСОРИБОЗЫ | 2005 |
|
RU2337917C1 |
| СПОСОБ ПОЛУЧЕНИЯ КАПЕЦИТАБИНА И ИСПОЛЬЗУЕМОГО ПРИ ЭТОМ ОБОГАЩЕННОГО β-АНОМЕРОМ ТРИАЛКИЛКАРБОНАТНОГО СОЕДИНЕНИЯ | 2008 |
|
RU2439064C1 |
| НОВЫЕ НУКЛЕОЗИДЫ, ИМЕЮЩИЕ БИЦИКЛИЧЕСКУЮ САХАРНУЮ ГРУППИРОВКУ, И СОДЕРЖАЩИЕ ИХ ОЛИГОНУКЛЕОТИДЫ | 1999 |
|
RU2211223C2 |
| СПОСОБ ПОЛУЧЕНИЯ 2'-ФТОР-2'-АЛКИЛЗАМЕЩЕННЫХ ИЛИ ДРУГИХ ЗАМЕЩЕННЫХ РИБОФУРАНОЗИЛПИРИМИДИНОВ И ПУРИНОВ И ИХ ПРОИЗВОДНЫХ | 2005 |
|
RU2433124C2 |
| СИНТЕЗ β-L-2'-ДЕЗОКСИНУКЛЕОЗИДОВ | 2004 |
|
RU2361875C2 |
| СПОСОБ ПОЛУЧЕНИЯ И ОЧИСТКИ ГЕМЦИТАБИНА ГИДРОХЛОРИДА | 2006 |
|
RU2345087C2 |
| МОНОЦИКЛИЧЕСКИЕ L-НУКЛЕОЗИДЫ, ИХ АНАЛОГИ И ПРИМЕНЕНИЯ | 1997 |
|
RU2188828C2 |
| ПРОИЗВОДНЫЕ 5,6-ДИХЛОРБЕНЗИМИДАЗОЛА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКИЙ СОСТАВ И СПОСОБ ЛЕЧЕНИЯ И ПРОФИЛАКТИКИ ВИРУСНЫХ ИНФЕКЦИЙ | 1995 |
|
RU2145963C1 |
Предложены производные 1-α-галоген-2,2-дифтор-2-дезокси-D-рибофуранозы общей формулы (I) в твердой форме, где R1 - бензоил или  R2 - водород; и Х - Cl, Br или I; которые могут быть использованы в качестве промежуточных в стереоселективном способе получения гемцитабина. Кроме того, предложен стереоселективный способ получения соединений общей формулы (I), включающий стадии: (i) восстановления 1-оксорибозы формулы (II) с получением лактола формулы (III); (ii) взаимодействия соединения формулы (III) с галогенфосфатным соединением формулы (IV) в присутствии основания с получением 1-фосфатфуранозного производного формулы (V); и (iii) взаимодействия соединения формулы (V) (также предложенного в качестве нового) с источником галогена, с последующей перекристаллизацией получившегося продукта; где R1, R2 и Х имеют указанные выше значения, а R3 является фенилом. 3 н. и 8 з.п. ф-лы.
R2 - водород; и Х - Cl, Br или I; которые могут быть использованы в качестве промежуточных в стереоселективном способе получения гемцитабина. Кроме того, предложен стереоселективный способ получения соединений общей формулы (I), включающий стадии: (i) восстановления 1-оксорибозы формулы (II) с получением лактола формулы (III); (ii) взаимодействия соединения формулы (III) с галогенфосфатным соединением формулы (IV) в присутствии основания с получением 1-фосфатфуранозного производного формулы (V); и (iii) взаимодействия соединения формулы (V) (также предложенного в качестве нового) с источником галогена, с последующей перекристаллизацией получившегося продукта; где R1, R2 и Х имеют указанные выше значения, а R3 является фенилом. 3 н. и 8 з.п. ф-лы.

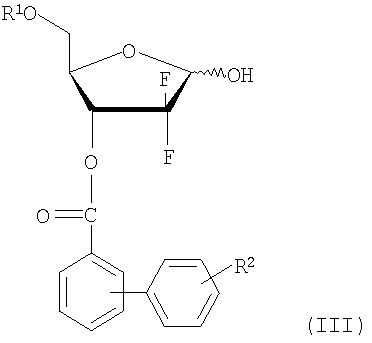
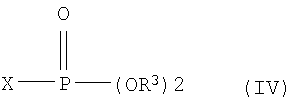
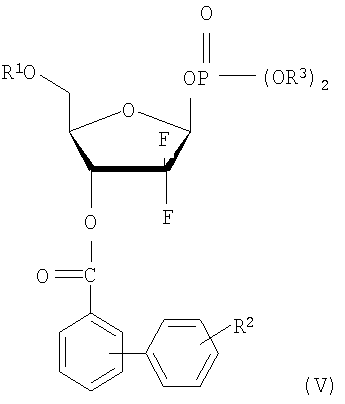

где R1 является бензоилом или 
R2 является водородом; и
Х является Cl, Br или I.
(i) восстановления соединения 1-оксорибозы формулы (II) с получением соединения лактола формулы (III);
(ii) взаимодействия соединения формулы (III) с галогенфосфатным соединением формулы (IV) в присутствии основания с получением 1-фосфатфуранозного производного формулы (V); и
(iii) взаимодействия соединения формулы (V) с источником галогена, с последующей перекристаллизацией получившегося продукта с получением производного 1-α-галоген-2,2-дифтор-2-дезокси-D-рибофуранозы формулы (I)


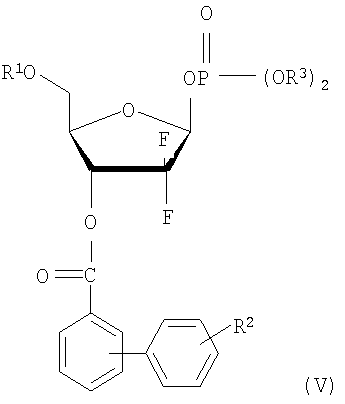
где R1, R2 и X имеют те же значения, что определенные в п.1; и R3 является фенилом.
где R1 является бензоилом или
R2 является водородом; и
R3 является фенилом.
| US 5945547 А, 31.08.1999 | |||
| US 5453499 А, 26.09.1995 | |||
| US 5744597 А, 28.04.1998 | |||
| US 5401861 А, 28.03.1995 | |||
| СПОСОБ ПОЛУЧЕНИЯ ОБОГАЩЕННЫХ БЕТА-АНОМЕРОМ НУКЛЕОЗИДОВ | 1993 |
|
RU2131880C1 |

