Область техники, к которой относится изобретение
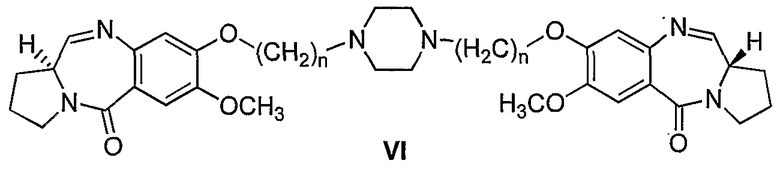
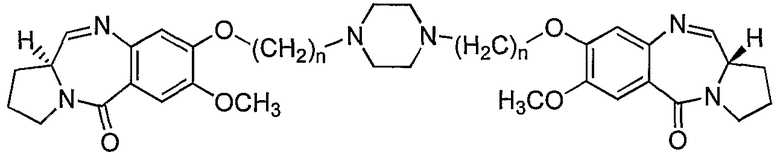
Настоящее изобретение относится к новым пирроло[2,1-с][1,4]бензодиазепинам, полезным в качестве потенциальных противоопухолевых средств. Это изобретение относится к способу получения новых пирроло[2,1-с][1,4]бензодиазепинов, полезных в качестве противоопухолевых средств. Более конкретно, изобретение предлагает способ получения 1,1'-{[(бисалкан-1,N-диил)пиперазин]диокси}бис[(11aS)-7-метокси-1,2,3,11а-тетрагидро-5Н-пирроло[2,1-c][1,4]бензодиазепин-5-онов] с изменяющейся длиной алифатической цепи, а также описывает противораковую (противоопухолевую) активность. Структурная формула новых пирроло[2,1-с][1,4]бензодиазепинов является следующей, где n=2-10.
Предшествующий уровень техники
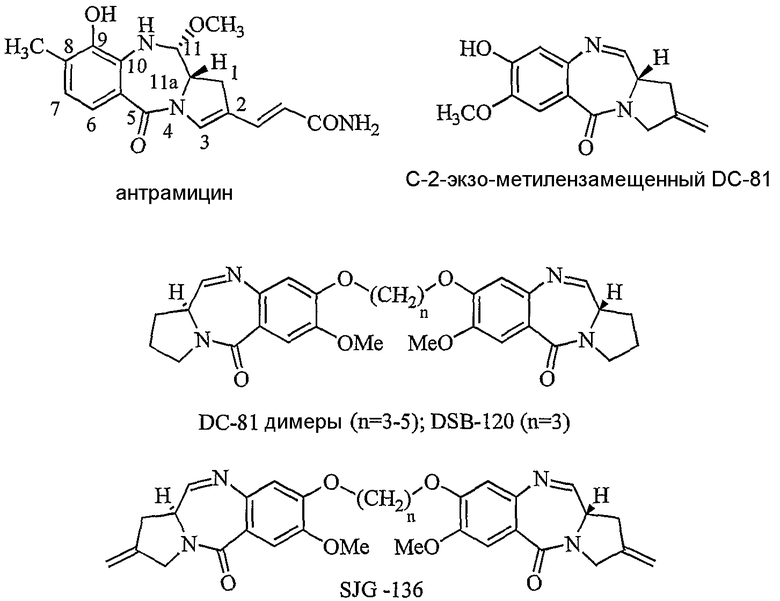
Пирроло[2,1-с][1,4]бензодиазепиновые противоопухолевые антибиотики общеизвестны как соединения класса антрамицина. В последние несколько лет проявляется растущий интерес к разработке новых пирроло[2,1-с][1,4]бензодиазепинов (ПБД). Эти антибиотики ковалентно взаимодействуют с ДНК, образуя N2-аддукты гуанина, которые лежат внутри малой бороздки двухцепочечной ДНК, посредством аминальной связи, неустойчивой к действию кислот, с электрофильным имином в положении N10-C11. (S. Kunimoto et al., J. Antibiot., 1980, 33, 665; K.W. Kohn and C.L. Speous, J. Mol. Biol., 1970, 51, 551; L.H. Hurley et al., Biochem. Biophys. Acta., 1977, 475, 521; D.J. Kaplan and L.H. Hurley, Biochemistry, 1981, 20, 7572). Молекулы имеют правостороннюю закрученность, которая позволяет им следовать кривизне малой бороздки В-формы двухцепочечной ДНК, охватывая три пары оснований. Недавно были разработаны димеры ПБД, содержащие две С2-экзо-метилензамещенные субъединицы DC-81, связанные в положении C-8 инертным пропандиоксидным линкером (S.J. Gregson et al., J. Med. Chem., 2001, 44, 737). Недавней разработкой стала связка двух единиц ПБД в положениях С-8 с образованием бифункциональных алкилирующих реагентов, способных сшивать ДНК. (D.E. Thruston et al., J. Org. Chem., 1996, 61, 8141-8147). Недавно были синтезированы несшитые смешанные имин-амидные димеры ПБД, имеющие значительную способность связывать ДНК и сильную противоопухолевую активность (A. Kamal et al., US Pat. 636233, 26.03.2002; A. Kamal et al., J. Med. Chem., 2002, 45, 4679).
Природные пирроло[2,1-с][1,4]бензодиазепины принадлежат к группе противоопухолевых антибиотиков, производимых видом Streptomyces. Недавно появилось много побудительных причин для синтеза ПБД систем, так как они могут узнавать и связывать определенные последовательности ДНК. Примеры природных ПБД включают антрамицин, DC-81, томаимицин, сибиромицин и неотрамицин.
 .
.
Однако клинической эффективности этих антибиотиков мешают несколько ограничений, таких как плохая растворимость в воде, кардиотоксичность, развитие устойчивости к лекарственному средству и метаболическая инактивация.
Цели изобретения
Основной целью настоящего изобретения является предложение новых пирроло[2,1-с][1,4]бензодиазепинов, полезных в качестве противоопухолевых средств.
Другой целью настоящего изобретения является предложение фармацевтических композиций, содержащих новые пирроло[2,1-с][1,4]бензодиазепины, полезные в качестве противораковых средств.
Еще одной целью настоящего изобретения является предложение способа получения новых пирроло[2,1-с][1,4]бензодиазепинов.
Сущность изобретения
Таким образом, настоящее изобретение обеспечивает новые пирроло[2,1-с][1,4]бензодиазепины формулы VI, где n=2-10, и способ их получения.
Подробное описание изобретения
Соответственно, настоящее изобретение обеспечивает аналоги 1,1'-{[(бисалкан-1,N-диил)пиперазин]диокси}бис[(11aS)-7-метокси-1,2,3,11а-тетрагидро-5Н-пирроло[2,1-c][1,4]бензодиазепин-5-она] формулы (VI)
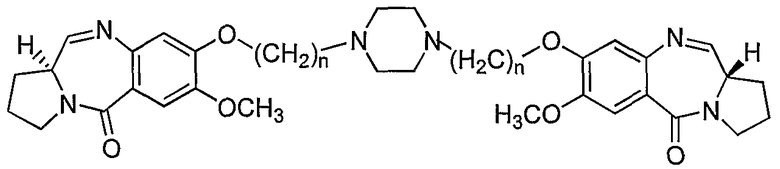
где n=2-10.
Другой вариант осуществления изобретения касается новых пирролобензодиазепинов структурной формулы, представленной ниже, (n=2)
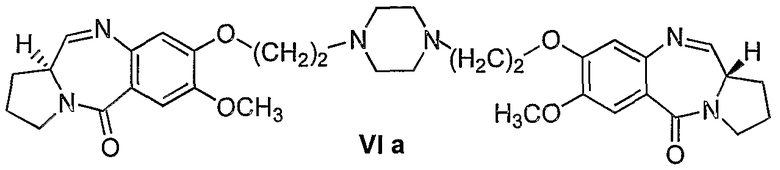
Еще один вариант осуществления изобретения касается новых пирролобензодиазепинов структурной формулы, представленной ниже, (n=3)
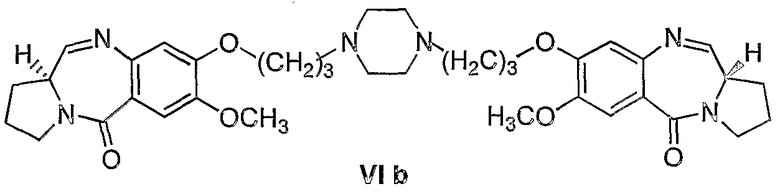
Еще один вариант осуществления изобретения касается новых пирролобензодиазепинов структурной формулы, представленной ниже, (n=4)
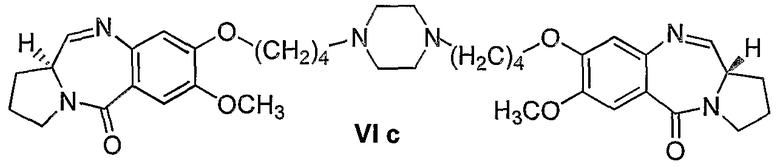
Еще один вариант осуществления изобретения касается новых пирролобензодиазепинов структурной формулы, представленной ниже, (n=5)
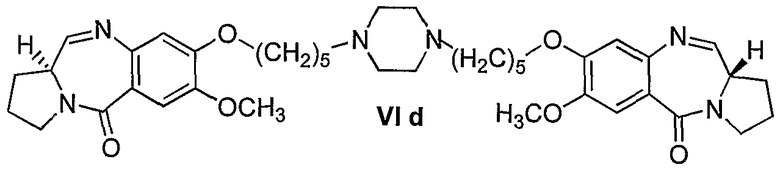
Еще один вариант осуществления изобретения касается новых пирролобензодиазепинов структурной формулы, представленной ниже, (n=6)
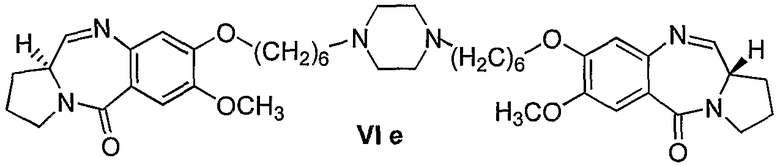
Еще один вариант осуществления изобретения касается новых пирролобензодиазепинов структурной формулы, представленной ниже, (n=7)
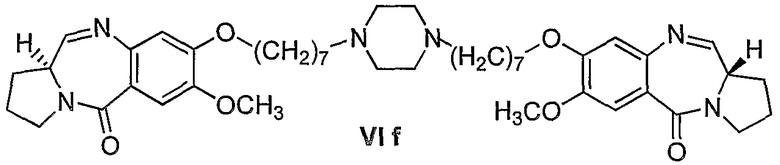
Еще один вариант осуществления изобретения касается новых пирролобензодиазепинов структурной формулы, представленной ниже, (n=8)
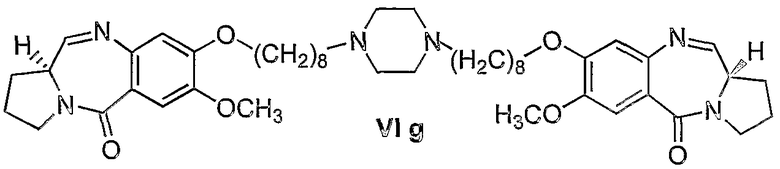
Еще один вариант осуществления изобретения касается новых пирролобензодиазепинов структурной формулы, представленной ниже, (n=9)
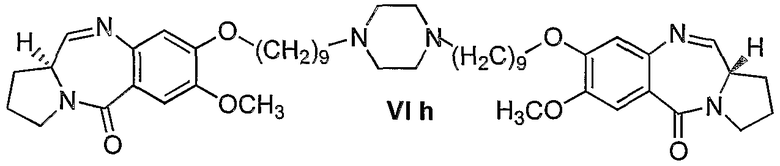
Еще один вариант осуществления изобретения касается новых пирролобензодиазепинов структурной формулы, представленной ниже, (n=10)
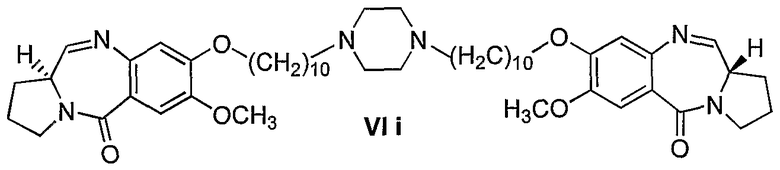
В одном варианте осуществления изобретение относится к способу получения аналогов 1,1'-{[(бисалкан-1,N-диил)пиперазин]диокси}бис[(11aS)-7-метокси-1,2,3,11а-тетрагидро-5Н-пирроло[2,1-c][1,4]бензодиазепин-5-она] формулы (VI), причем способ включает стадии:
a) взаимодействие соединения формулы (I) с 1,2-дибромэтаном в смешивающемся с водой органическом растворителе в присутствии основания при температуре кипения с обратным холодильником в течение 20-48 часов,
b) выливание реакционной смеси стадии (a) в воду, экстракцию этилацетатом, отделение этилацетатного слоя и отбрасывание водного слоя,
c) упаривание этилацетатного слоя стадии (b), чтобы получить остаток, который далее очищают, чтобы получить чистое соединение формулы (II),
d) выдерживание раствора соединения формулы (II) в кетонном растворителе в присутствии основания при температуре кипения с обратным холодильником в течение 20-48 часов,
e) выливание реакционной смеси стадии (d) в воду, экстракцию этилацетатом, отделение этилацетатного слоя, упаривание этилацетатного слоя, чтобы получить остаток, очистку остатка, чтобы получить соединение формулы (IV),
f) растворение соединения формулы (IV) в спирте, добавление дигидрата хлорида олова(II) и кипячение с обратным холодильником в течение 0,5-1,5 часа,
g) доведение рН реакционной смеси стадии (f) до 8,0 раствором бикарбоната щелочного металла,
h) экстракцию раствора с рН 8,0 стадии (g) этилацетатом, отделение этилацетатного экстракта, сушку этилацетатного экстракта над безводным сульфатом натрия, фильтрование и упаривание этилацетатного раствора, чтобы получить неочищенное соединение формулы (V),
i) растворение соединения формулы (V) стадии (h) в смеси ацетонитрил-вода, добавление хлорида ртути, оксида ртути и перемешивание в течение 6-12 часов при температуре окружающей среды,
j) упаривание органического слоя стадии (i), разбавление остатка этилацетатом, добавление насыщенного раствора бикарбоната при комнатной температуре, фильтрование через слой целита, промывание слоя этилацетатом, чтобы получить прозрачный фильтрат; и
k) упаривание фильтрата стадии (j), чтобы получить остаток, который очищают, пропуская через колонку силикагеля, с получением чистого соединения формулы (VI).
В другом варианте осуществления изобретения используемое основание выбирают из группы, состоящей из карбоната лития, карбоната натрия, карбоната калия или карбоната цезия.
В другом варианте осуществления изобретения используемый кетонный растворитель выбирают из группы, состоящей из ацетона, метилэтилкетона и метилизобутилкетона.
В другом варианте осуществления изобретения используемый спирт выбирают из группы, состоящей из метанола, этанола и изопропанола, предпочтительно метанола.
Еще один вариант изобретения предлагает фармацевтическую композицию, полезную в качестве противоопухолевого средства, причем композиция содержит эффективное количество одного или нескольких аналогов 1,1'-{[(бисалкан-1,N-диил)пиперазин]диокси}бис[(11aS)-7-метокси-1,2,3,11а-тетрагидро-5Н-пирроло[2,1-c][1,4]бензодиазепин-5-она] формулы (VI).
В еще одном варианте осуществления изобретения композиция необязательно содержит фармацевтически приемлемые добавки.
В еще одном варианте осуществления изобретения композицию вводят млекопитающим, включая людей.
В еще одном варианте осуществления изобретения композиция может быть введена перорально, системно или любыми другими известными способами.
Способ получения пирроло[2,1-е][1,4]бензодиазепинов формулы VI представлен на чертеже, сопровождающем описание, где n=2-10; способ включает: взаимодействие диэтилтиоацеталя (2S)-N-[4-гидрокси-5-метокси-2-нитробензоил]-2-карбоксальдегида формулы I с дибромалканами в апротонном смешивающемся с водой органическом растворителе, таком как ацетон, ТГФ и ДМФА, в присутствии слабого неорганического основания, такого как К2СО3, CsCO3 и ВаСО3, при температуре до температуры кипения с обратным холодильником в течение до 48 часов, взаимодействие диэтилтиоацеталя (2S)-N-[4-(n-бромалкокси)-5-метокси-2-нитробензоил]пирролидин-2-карбоксальдегида формулы II с пиперазином формулы III в присутствии слабых неорганических оснований, таких как K2СО3, CsCO3 и ВаСО3, и в присутствии апротонных смешивающихся с водой органических растворителей при температуре до температуры кипения с обратным холодильником в течение до 48 часов, выделение диэтилтиоацеталя 1,1'-{[(бисалкан-1,N-диил)пиперазин]диокси}бис[(11aS)-7-метокси-2-нитробензоилпирролидин-2-карбоксальдегида] формулы IV, где n=2-10, обычными методами, восстановление нитросоединения формулы IV SnCl2·2H2O в присутствии органического растворителя при температуре до температуры кипения с обратным холодильником, выделение диэтилтиоацеталя 1,1'-{[(бисалкан-1,N-диил)пиперазин]диокси}бис[(11aS)-7-метокси-2-аминобензоилпирролидин-2-карбоксальдегида] формулы V, где n=2-10, обычными методами, взаимодействие аминосоединения формулы V с известными реагентами, удаляющими защиту, с получением новых пирроло[2,1-е][1,4]бензодиазепинов формулы VI, где n имеет значения, указанные выше.
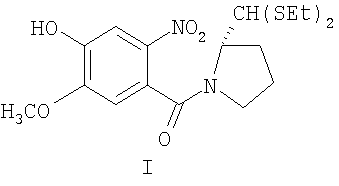
Предшественник, диэтилтиоацеталь (2S)-N-(4-гидрокси-2-метокси-2-нитробензоил)пирролидин-2-карбоксальдегида формулы I (промежуточные соединения DC-81), получают методом, описанным в литературе (D.E. Thurston et al., Synthesis, 1990, 81).
Некоторые типичные соединения формулы VI настоящего изобретения приведены ниже.
1) 1,1'-{[(бисэтан-1,N-диил)пиперазин]диокси}бис[(11aS)-7-метокси-1,2,3,11а-тетрагидро-5Н-пирроло[2,1-c][1,4]бензодиазепин-5-он].
2) 1,1'-{[(биспропан-1,N-диил)пиперазин]диокси}бис[(11aS)-7-метокси-1,2,3,11а-тетрагидро-5Н-пирроло[2,1-c][1,4]бензодиазепин-5-он].
3) 1,1'-{[(бисбутан-1,N-диил)пиперазин]диокси}бис[(11aS)-7-метокси-1,2,3,11а-тетрагидро-5Н-пирроло[2,1-c][1,4]бензодиазепин-5-он].
Эти новые аналоги димеров пирроло[2,1-c][1,4]бензодиазепина, связанных через пиперазин в положениях С-8, показали перспективную противораковую активность в различных линиях клеток. Синтезированные молекулы имеют очень большое биологическое значение из-за возможного последствия селективного ДНК связывания. Это привело к разработке и синтезу новых представителей того же класса, как показано на схеме 1, который включает:
1. Образование простой эфирной связи в положении С-8 промежуточных соединений DC-81 с пиперазином.
2. Кипячение реакционной смеси в течение 24-48 часов.
3. Синтез С-8-связанных димерных иминов ПБД с активностью противоопухолевых антибиотиков.
4. Очистку колоночной хроматографией с использованием различных растворителей, таких как этилацетат, гексан, дихлорметан и метанол.
Способ получения новых несшитых пирроло[2,1-c][1,4]бензодиазепинов раскрыт и заявлен в совместно рассматриваемой заявке заявителей.
Следующие примеры приведены для иллюстрации и, следовательно, не должен быть истолкованы как ограничение объема изобретения.
Краткое описание чертежа, который представляет схематическую диаграмму получения соединения общей формулы VI(a-i).
Пример 1
Раствор диэтилтиоацеталя (2S)-N-(4-гидрокси-5-метокси-2-нитробензоил)пирролидин-2-карбоксальдегида формулы I (800 мг, 2 ммоль), 1,2-дибромэтана (940 мг, 2,5 ммоль) и К2СО3 (828 мг, 3 ммоль) в сухом ацетоне (40 мл) кипятили с обратным холодильником 48 часов. После завершения реакции, о чем свидетельствует ТСХ (EtOAc-гексан (7:3)), реакционную смесь выливали в воду и затем экстрагировали этилацетатом. Упаривание органического слоя дало неочищенный продукт, который далее очищали колоночной хроматографией на силикагеле, элюируя смесью EtOAc-гексан (1:1), и получали чистый диэтилтиоацеталь (2S)-N-[4-(2-бромэтокси)-5-метокси-2-нитробензоил]пирролидин-2-карбоксальдегида формулы II.
1Н-ЯМР: (CDCl3) δ 1,20-1,4 (м, 6Н), 1,75-2,2 (м, 4Н), 2,6-2,9 (м, 4Н), 3,20-3,33 (м, 2Н) 3,67 (т, 2Н), 3,95 (с, 3Н), 4,37 (т, 2Н), 4,62-4,78 (м, 1Н), 4,85 (д, 1Н), 6,82 (с, 1Н), 7,67 (с, 1Н).
Раствор диэтилтиоацеталя (2S)-N-[4-(3-бромэтокси)-5-метокси-2-нитробензоил]пирролидин-2-карбоксальдегида формулы II (507 мг, 1 ммоль), пиперазина (0,043 мг, 0,5 ммоль) формулы III и К2СО3 (414 мг, 3 ммоль) в сухом ацетоне (20 мл) кипятили с обратным холодильником 48 часов. После завершения реакции, о чем свидетельствует ТСХ (EtOAc), реакционную смесь выливали в воду и затем экстрагировали этилацетатом. Упаривание органического слоя дало неочищенный продукт, который далее очищали колоночной хроматографией на силикагеле, элюируя смесью EtOAc-гексан (9:1), и получали чистый диэтилтиоацеталь 1,1'-{[(бисэтан-1,N-диил)пиперазин]диокси}бис[(11aS)-7-метокси-2-нитробензоилпирролидин-2-карбоксальдегида].
1Н-ЯМР: (CDCl3) δ 1,29-1,41 (м, 12H), 1,7-2,39 (м, 8H), 2,60-2,90 (м, 20H), 3,17-3,3 (м, 4H), 3,92 (с, 6H), 4,2 (т, 4H), 4,60-4,70 (м, 2H), 4,81 (д, 2H), 6,8 (с, 2H), 7,7 (с, 2H).
Масс-спектрометрия с бомбардировкой быстрыми атомами (FAB MS): 939 (М+Н)+.
Диэтилтиоацеталь 1,1'-{[(бисэтан-1,N-диил)пиперазин]диокси}бис[(11aS)-7-метокси-2-нитробензоилпирролидин-2-карбоксальдегида] формулы IV (939 мг, 1,0 ммоль) растворяли в метаноле (10 мл), добавляли SnCl2·2H2O (1,124 г, 5,0 ммоль) и кипятили с обратным холодильником 1,5 часа. Реакционную смесь затем осторожно доводили до рН 8 насыщенным раствором NaHCO3 и экстрагировали этилацетатом (3×20 мл). Объединенную органическую фазу сушили над Na2SO4, упаривали в вакууме и получали неочищенный диэтилтиоацеталь 1,1'-{[(бисэтан-1,N-диил)пиперазин]диокси}бис[(11aS)-7-метокси-2-аминобензоилпирролидин-2-карбоксальдегида].
Раствор диэтилтиоацеталя 1,1'-{[(бисэтан-1,N-диил)пиперазин]диокси}бис[(11aS)-7-метокси-2-аминобензоилпирролидин-2-карбоксальдегида] формулы V (879 мг, 1 ммоль), HgCl2 (794 мг, 2,93 ммоль), HgO (686 мг, 3,18 ммоль) в CH3CN/H2O (3:1, 15 мл) перемешивали при комнатной температуре 12 часов до тех пор, пока ТСХ (EtOAc) не покажет полную потерю исходного материала. Затем органический слой упаривают в вакууме и остаток разбавляют этилацетатом. К образующемуся раствору медленно добавляли насыщенный NaHCO3 при комнатной температуре и смесь фильтровали через целит и промывали этилацетатом. Фильтрат упаривали в вакууме и получали неочищенный 1,1'-{[(бисэтан-1,N-диил)пиперазин]диокси}бис[(11aS)-7-метокси-1,2,3,11а-тетрагидро-5Н-пирроло[2,1-c][1,4]бензодиазепин-5-он] формулы VIa, который далее очищали колоночной хроматографией на силикагеле, элюируя сначала этилацетатом, чтобы удалить следы солей ртути, затем смесью CHCl3-метанол (9:1).
1Н-ЯМР: (CDCl3) δ 1,92-2,42 (м, 8H), 2,60-2,95 (м, 12H), 3,2-3,88 (м, 6H), 3,92 (с, 6H), 4,14-4,28 (м, 4H), 6,76 (с, 2H), 7,5 (с, 2H), 7,66 (д, 2H).
FAB MS: 631 (М+Н)+.
Пример 2
Раствор диэтилтиоацеталя (2S)-N-(4-гидрокси-5-метокси-2-нитробензоил)пирролидин-2-карбоксальдегида формулы I (400 мг, 1 ммоль), 1,3-дибромпропана (502 мг, 2,5 ммоль) и К2СО3 (414 мг, 3 ммоль) в сухом ацетоне (20 мл) кипятили с обратным холодильником 48 часов. После завершения реакции, о чем свидетельствует ТСХ (EtOAc-гексан (7:3)), реакционную смесь выливали в воду и затем экстрагировали этилацетатом. Упаривание органического слоя дало неочищенный продукт, который далее очищали колоночной хроматографией на силикагеле, элюируя смесью EtOAc-гексан (1:1), и получали чистый диэтилтиоацеталь (2S)-N-[4-(4-бромпропокси)-5-метокси-2-нитробензоил]пирролидин-2-карбоксальдегида формулы II.
1Н-ЯМР: (CDCl3) δ 1,25-1,4 (м, 6H), 1,85-2,35 (м, 4H), 2,38-2,5 (м, 2H), 2,6-2,9 (м, 4H), 3,18-3,33 (м, 2H), 3,64 (т, 2H), 3,97 (с, 3H), 4,29 (т, 2H), 4,67-4,78 (м, 1H), 4,83 (д, 1H), 6,78 (с, 1H), 7,7 (с, 1H).
Раствор диэтилтиоацеталя (2S)-N-[4-(4-бромпропокси)-5-метокси-2-нитробензоил]пирролидин-2-карбоксальдегида формулы II (520 мг, 1 ммоль), пиперазина (0,043 мг, 1 ммоль) формулы III и К2СО3 (414 мг, 3 ммоль) в сухом ацетоне (20 мл) кипятили с обратным холодильником 48 часов. После завершения реакции, о чем свидетельствует ТСХ (EtOAc), реакционную смесь выливали в воду и затем экстрагировали этилацетатом. Упаривание органического слоя дало неочищенный продукт, который далее очищали колоночной хроматографией на силикагеле, элюируя смесью EtOAc-гексан (9:1), и получали чистый диэтилтиоацеталь 1,1'-{[(биспропан-1,N-диил)пиперазин]диокси}бис[(11aS)-7-метокси-2-нитробензоилпирролидин-2-карбоксальдегида] формулы IV.
1Н-ЯМР: (CDCl3) δ 1,3-1,42 (м, 12H), 1,9-2,32 (м, 8H), 2,47-2,6 (м, 4H), 2,7-2,9 (м, 24H), 3,2-3,3 (м, 4H), 3,95 (с, 6H), 4,1-4,2 (т, 4H), 4,62-4,75 (м, 2H), 4,82 (д, 2H), 6,75 (с, 2H), 7,67 (с, 2H).
FAB MS: 967 (М+Н)+.
Диэтилтиоацеталь 1,1'-{[(биспропан-1,N-диил)пиперазин]диокси}бис[(11aS)-7-метокси-2-нитробензоилпирролидин-2-карбоксальдегида] формулы IV (966 мг, 1,0 ммоль) растворяли в метаноле (10 мл), добавляли SnCl2·2H2O (1,124 г, 5,0 ммоль) и кипятили с обратным холодильником 1,5 часа. Реакционную смесь затем осторожно доводили до рН 8 насыщенным раствором NaHCO3 и экстрагировали этилацетатом (3×20 мл). Объединенную органическую фазу сушили над Na2SO4, упаривали в вакууме и получали неочищенный диэтилтиоацеталь 1,1'-{[(биспропан-1,N-диил)пиперазин]диокси}бис[(11aS)-7-метокси-2-аминобензоилпирролидин-2-карбоксальдегида] формулы V.
Раствор диэтилтиоацеталя 1,1'-{[(биспропан-1,N-диил)пиперазин]диокси}бис[(11aS)-7-метокси-2-аминобензоилпирролидин-2-карбоксальдегида] формулы V (907 мг, 1 ммоль), HgCl2 (794 мг, 2,93 ммоль), HgO (687 мг, 3,18 ммоль) в CH3CN/H2O (3:1, 15 мл) перемешивали при комнатной температуре 12 часов до тех пор, пока ТСХ (EtOAc) не покажет полную потерю исходного материала. Затем органический слой упаривают в вакууме и остаток разбавляют EtOAc. К образующемуся раствору медленно добавляли насыщенный NaHCO3 при комнатной температуре и смесь фильтровали через целит и промывали этилацетатом. Фильтрат упаривали в вакууме и получали неочищенный 1,1'-{[(биспропан-1,N-диил)пиперазин]диокси}бис[(11aS)-7-метокси-1,2,3,11а-тетрагидро-5Н-пирроло[2,1-c][1,4]бензодиазепин-5-он] формулы VIb, который далее очищали колоночной хроматографией на силикагеле, элюируя сначала этилацетатом, чтобы удалить следы солей ртути, затем смесью CHCl3-метанол (9:1).
1Н-ЯМР: (CDCl3) δ 1,92-2,37 (м, 8H), 2,57-2,8 (м, 16H), 3,32-3,75 (м, 6H), 3,95 (с, 6H), 4,12-4,45 (м, 4H), 6,85 (с, 2H), 7,52 (с, 2H), 7,82 (д, 2H).
FAB MS: 659 (М+Н)+.
Пример 3
Раствор диэтилтиоацеталя (2S)-N-[4-гидрокси-5-метокси-2-нитробензоил]пирролидин-2-карбоксальдегида формулы I (400 мг, 1 ммоль), 1,4-дибромбутана (540 мг, 2,5 ммоль) и К2СО3 (414 мг, 3 ммоль) в сухом ацетоне (20 мл) кипятили с обратным холодильником 48 часов. После завершения реакции, о чем свидетельствует ТСХ (EtOAc-гексан (7:3)), реакционную смесь выливали в воду и затем экстрагировали этилацетатом. Упаривание органического слоя дало неочищенный продукт, который далее очищали колоночной хроматографией на силикагеле, элюируя смесью EtOAc-гексан (1:1), и получали чистый диэтилтиоацеталь (2S)-N-[4-(5-бромбутанокси)-5-метокси-2-нитробензоил]пирролидин-2-карбоксальдегида формулы II.
1Н-ЯМР: (CDCl3) δ 1,3-1,45 (м, 6H), 1,88-2,38 (м, 4H), 2,69-2,88 (м, 8H), 3,20-3,33 (м, 2H), 3,51 (т, 2H), 3,97 (с, 3H), 4,16 (т, 2H), 4,63-4,76 (м, 1H), 4,86 (д, 1H), 6,79 (с, 1H), 7,67 (с, 1H).
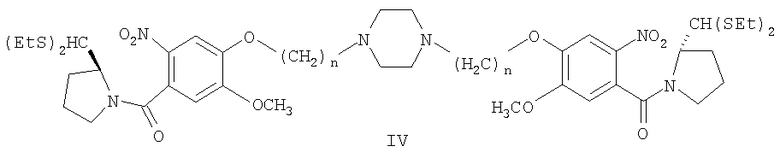
Раствор диэтилтиоацеталя (2S)-N-[4-(5-бромбутанокси)-5-метокси-2-нитробензоил]пирролидин-2-карбоксальдегида формулы II (53 мг, 1 ммоль), пиперазина (0,043 мг, 1 ммоль) формулы III и К2СО3 (414 мг, 3 ммоль) в сухом ацетоне (20 мл) кипятили с обратным холодильником 48 часов. После завершения реакции, о чем свидетельствует ТСХ (EtOAc), реакционную смесь выливали в воду и затем экстрагировали этилацетатом. Упаривание органического слоя дало неочищенный продукт, который далее очищали колоночной хроматографией на силикагеле, элюируя смесью EtOAc-гексан (9:1), и получали чистый диэтилтиоацеталь 1,1'-{[(бисбутан-1,N-диил)пиперазин]диокси}бис[(11aS)-7-метокси-2-нитробензоилпирролидин-2-карбоксальдегида] формулы IV.
1Н-ЯМР: (CDCl3) δ 1,30-1,43 (м, 12H), 2,74-2,35 (м, 12H), 2,51-2,66 (м, 16H), 3,20-3,3 (м, 4H), 3,97 (с, 6H), 4,12 (т, 4H), 4,64-4,76 (м, 2H), 4,87 (д, 2H), 6,84 (с, 2H), 7,66 (с, 2H).
FAB MS: 995 (М+Н)+.
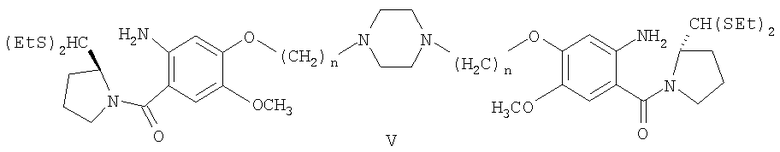
Диэтилтиоацеталь 1,1'-{[(бисбутан-1,N-диил)пиперазин]диокси}бис[(11aS)-7-метокси-2-нитробензоилпирролидин-2-карбоксальдегида] формулы IV (730 мг, 1,0 ммоль) растворяли в метаноле (10 мл), добавляли SnCl2·2H2O (1,124 г, 5,0 ммоль) и кипятили с обратным холодильником 1,5 часа. Реакционную смесь затем осторожно доводили до рН 8 насыщенным раствором NaHCO3 и экстрагировали этилацетатом (3×20 мл). Объединенную органическую фазу сушили над Na2SO4, упаривали в вакууме и получали неочищенный диэтилтиоацеталь 1,1'-{[(бисбутан-1,N-диил)пиперазин]диокси}бис[(11aS)-7-метокси-2-аминобензоилпирролидин-2-карбоксальдегида] формулы V.
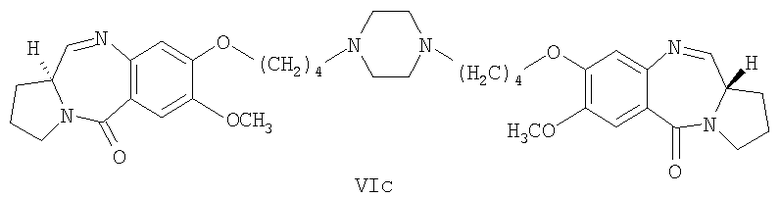
Раствор диэтилтиоацеталя 1,1'-{[(бисбутан-1,N-диил)пиперазин]диокси}бис[(11aS)-7-метокси-2-аминобензоилпирролидин-2-карбоксальдегида] формулы V (935 мг, 1 ммоль), HgCl2 (794 мг, 2,93 ммоль HgO (687 мг, 3,18 ммоль) в CH3CN/H2O (3:1, 15 мл) перемешивали при комнатной температуре 12 часов до тех пор, пока ТСХ (EtOAc) не показывала полное исчезновение исходного материала. Затем органический слой упаривали в вакууме и остаток разбавляли этилацетатом. К образующемуся раствору медленно добавляли насыщенный NaHCO3 при комнатной температуре и смесь фильтровали через целит и промывали этилацетатом. Фильтрат упаривали в вакууме и получали неочищенный 1,1'-{[(бисбутан-1,N-диил)пиперазин]диокси}бис[(11aS)-7-метокси-1,2,3,11а-тетрагидро-5Н-пирроло[2,1-c][1,4]бензодиазепин-5-он] формулы VIc, который далее очищали колоночной хроматографией на силикагеле, элюируя сначала этилацетатом, чтобы удалить следы солей ртути, затем смесью CHCl3-метанол (9:1).
1Н-ЯМР: (CDCl3) δ 1,78-2,24 (м, 8H), 2,30-2,75 (м, 20H), 3,4-3,7 (м, 6H), 3,92 (с, 6H), 4,1-4,23 (м, 4H), 6,73 (с, 2H), 7,48 (с, 2H), 7,60 (д, 2H).
FAB MS: 687 (М+Н)+.
Биологическая активность: изучение in vitro биологической активности проведено в Национальном Институте Рака (США).
Цитотоксичность: Соединения VIa-d оценивали in vitro по отношению к шестидесяти опухолевым клеткам человека, происходящим из девяти типов рака (лейкоз, меланома, немелкоклеточный рак легких, рак толстой кишки, ЦНС, яичников, простаты и молочной железы). Для каждого соединения измеряли кривые зависимости доза-эффект по каждой линии клеток минимум для пяти концентраций при десятикратном разбавлении. Использовали протокол 48-часового непрерывного действия лекарства и анализ белка сульфородамин В (SRB) для оценки жизнеспособности или роста клеток. Вычисляли концентрации, вызывающие 50% ингибирование роста клеток (ИР50), полное ингибирование роста клеток (ПИР, 0% роста) и 50% гибель клеток (ЛК50, -50% рост), в сравнении с контролем. Среднеграфические значения средней точки log10ПИР и log10ЛК50, а также log10ИР50 перечислены в таблице 1. Как демонстрируют среднеграфические примеры, соединение VIc проявляет интересный профиль активности и селективности для разных линий клеток. Среднеграфические значения средней точки log10ПИР и log10ЛК50 показали картину, подобную картине для среднеграфических значений средней точки log10ИР50.
Среднеграфические значения средней точки log10ИР50, log10ПИР и log10ЛК50 данных по in vitro цитотоксичности соединений VIa-d для линий опухолевых клеток человека
Противораковая in vitro активность четырех представителей соединений VI дана в таблице 2. Сравнение данных таблицы 2 выявляет важное значение алканового линкера. По мере того как алкановый линкер увеличивается с двух до четырех атомов углерода цитотоксическая активность умеренно растет. Четырехуглеродный линкер соединения VIc придает подходящее соответствие малой бороздке двойной спирали ДНК, и это соединение показывает немного более высокую активность в ряду соединенений VIa-d.
Значения log10ЛК50 (концентрация в мол/л,
вызывающая 50% летальность) для соединений VIa-d
| название | год | авторы | номер документа |
|---|---|---|---|
| ПИРРОЛО [2.1-C][1.4] БЕНЗОДИАЗЕПИНЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2003 |
|
RU2314309C2 |
| ИМИДАЗОДИАЗЕПИНЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ЛЕКАРСТВЕННОЕ СРЕДСТВО | 1995 |
|
RU2139873C1 |
| НОВЫЕ КОНЪЮГАТЫ АНТИТЕЛ И ИХ ПРИМЕНЕНИЯ | 2014 |
|
RU2684468C2 |
| КОНЪЮГАТ АНТИТЕЛО-ПРОИЗВОДНОЕ ПИРРОЛОБЕНЗОДИАЗЕПИНА | 2018 |
|
RU2820928C2 |
| СОЕДИНЕНИЯ ДЛЯ ЛЕЧЕНИЯ PD-L1 ЗАБОЛЕВАНИЙ | 2020 |
|
RU2838028C2 |
| ПРОИЗВОДНЫЕ МАННОЗЫ ДЛЯ ЛЕЧЕНИЯ БАКТЕРИАЛЬНЫХ ИНФЕКЦИЙ | 2013 |
|
RU2667060C2 |
| ПРОЛЕКАРСТВА СОПРЯЖЕННО-БИЦИКЛИЧЕСКИХ АНТАГОНИСТОВ C5aR | 2019 |
|
RU2794327C2 |
| ТРИАРИЛЬНЫЕ СОЕДИНЕНИЯ ДЛЯ ЛЕЧЕНИЯ PD-L1 ЗАБОЛЕВАНИЙ | 2020 |
|
RU2840345C2 |
| СОЕДИНЕНИЯ 8-МЕТИЛ-1-ФЕНИЛИМИДАЗО[1, 5-а]ПИРАЗИНА | 2011 |
|
RU2560162C2 |
| ПРОИЗВОДНЫЕ ХИНАЗОЛИНА | 2005 |
|
RU2414468C2 |
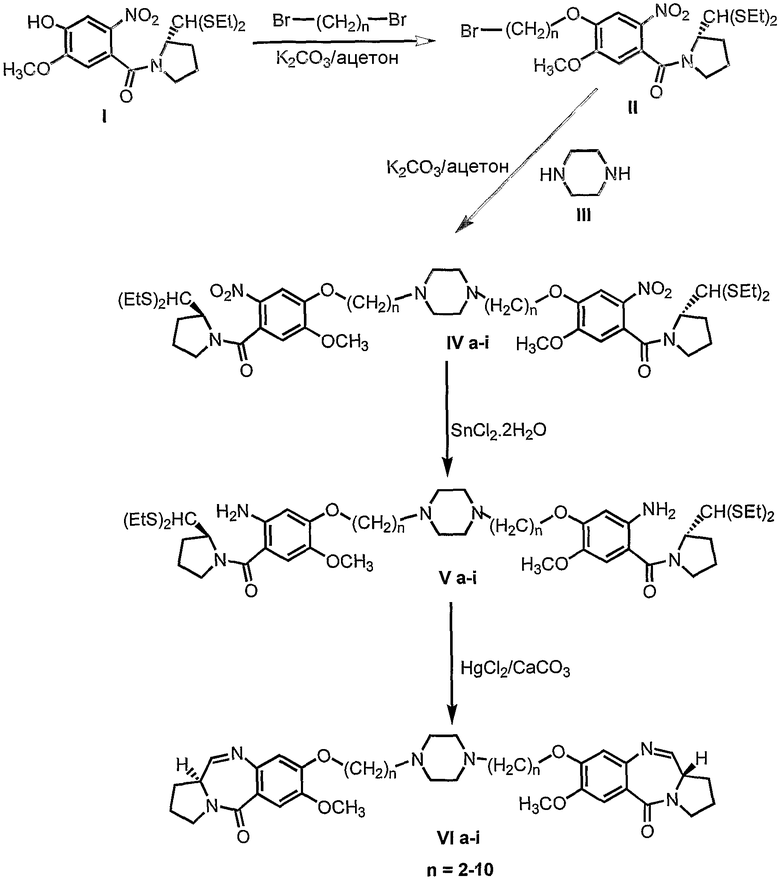
Описываются новые производные пирроло[2,1-с][1,4]бензодиазепина общей формулы
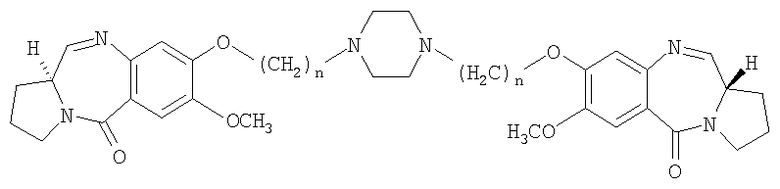
где n=2-10. Фармацевтическая композиция, их содержащая, и способ их получения. Соединения обладают противоопухолевой активностью и могут применяться в медицине. 6 н. и 12 з.п. ф-лы, 2 табл., 1 ил.
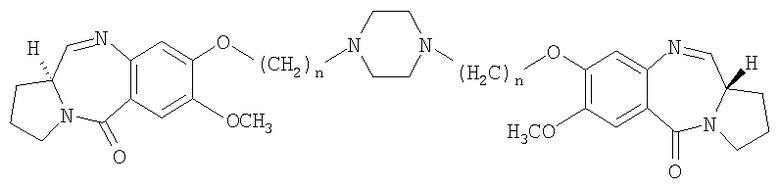
где n=2-10.
а) взаимодействие диэтилтиоацеталя (2S)-N-(4-гидрокси-5-метокси-2-нитробензоил)пирролидин-2-карбоксальдегида формулы I формулы (I)
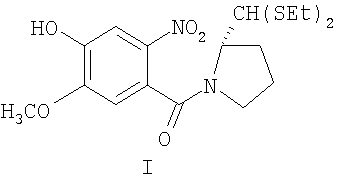
с дибром(С2-С10)алканом в смешивающемся с водой органическом растворителе в присутствии основания при температуре кипения в течение 20-48 ч,
b) выливание реакционной смеси стадии (а) в воду, экстракцию этилацетатом, отделение этилацетатного слоя и отбрасывание водного слоя,
c) упаривание этилацетатного слоя стадии (b) для получения остатка, который затем очищают с получением чистого диэтилтиоацеталя (2S)-N-[4-(n-бромалкокси)-5-метокси-2-нитробензоил]пирролидин-2-карбоксальдегида формулы (II),
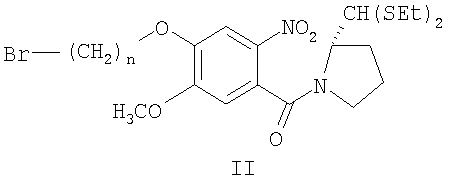
где n=2-10;
d) выдерживание раствора соединения формулы (II) и
пиперазина (III) в кетонном растворителе в присутствии основания при температуре кипения в течение 20-48 ч,
e) выливание реакционной смеси стадии (d) в воду, экстракцию этилацетатом, отделение этилацетатного слоя, упаривание
этилацетатного слоя с получением остатка, очистку остатка для получения диэтилтиоацеталя 1,1'-{[(бисалкан-1,N-диил)пиперазин]диокси}бис[(11аS)-7-метокси-2-нитробензоилпирролидин-2-карбоксальдегида] формулы (IV),
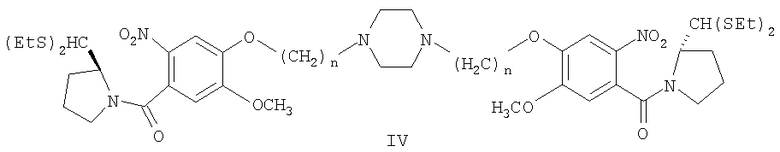
где n=2-10;
f) растворение соединения формулы (IV) в спирте, добавление дигидрата хлорида олова (II) и кипячение в течение 0,5-1,5 ч,
g) доведение рН реакционной смеси стадии (f) до 8,0 раствором бикарбоната щелочного металла,
h) экстракцию раствора с рН 8,0 стадии (g) этилацетатом, отделение этилацетатного экстракта, сушку этилацетатного экстракта над безводным сульфатом натрия, фильтрование и упаривание этилацетатного раствора с получением неочищенного диэтилтиоацеталя 1,1'-{[(бисалкан-1,N-диил)пиперазин]диокси}бис[(11аS)-7-метокси-2-аминобензоилпирролидин-2-карбоксальдегида формулы (V),
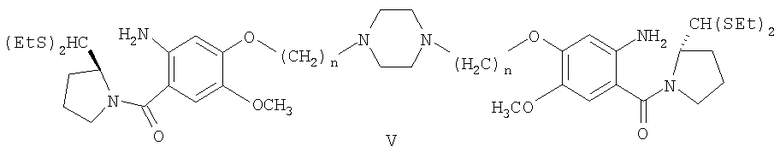
где n=2-10;
i) растворение соединения формулы (V) стадии (h) в смеси ацетонитрил-вода, добавление хлорида ртути, оксида ртути и перемешивание в течение 6-12 ч при температуре окружающей среды,
j) упаривание органического слоя стадии (i), разбавление остатка этилацетатом, добавление насыщенного раствора бикарбоната при комнатной температуре, фильтрование через слой целита, промывание слоя этилацетатом для получения прозрачного фильтрата; и
k) упаривание фильтрата стадии (j) для получения остатка, который очищают, пропуская через колонку силикагеля, с получением чистого соединения формулы (VI) по п.1.
а) взаимодействие соединения формулы (I)
с 1,4-дибромбутаном в смешивающемся с водой органическом растворителе в присутствии основания при температуре кипения в течение 20-48 ч,
b) выливание реакционной смеси стадии (а) в воду, экстракцию этилацетатом, отделение этилацетатного слоя и отбрасывание водного слоя,
c) упаривание этилацетатного слоя стадии (b) для получения остатка, который далее очищают для получения чистого соединения формулы (II),
где n=4;
d) выдерживание раствора соединения формулы (II) и пиперазина в кетонном растворителе в присутствии основания при температуре кипения в течение 20-48 ч,
e) выливание реакционной смеси стадии (d) в воду, экстракцию этилацетатом, отделение этилацетатного слоя, упаривание этилацетатного слоя с получением остатка, очистку остатка с получением соединения формулы (IV),
где n=4;
f) растворение соединения формулы (IV) в спирте, добавление дигидрата хлорида олова (II) и кипячение в течение 0,5-1,5 ч,
g) доведение рН реакционной смеси стадии (f) до 8,0 раствором бикарбоната щелочного металла,
n) экстракцию раствора с рН 8,0 стадии (g) этилацетатом, отделение этилацетатного экстракта, сушку этилацетатного экстракта над безводным сульфатом натрия, фильтрование и упаривание этилацетатного раствора для получения неочищенного соединения формулы (V),
где n=4;
i) растворение соединения формулы (V) стадии (h) в смеси ацетонитрил-вода, добавление хлорида ртути, оксида ртути и перемешивание в течение 6-12 ч при температуре окружающей среды,
j) упаривание органического слоя стадии (i), разбавление остатка этилацетатом, добавление насыщенного раствора бикарбоната при комнатной температуре, фильтрование через слой целита, промывание слоя этилацетатом для получения прозрачного фильтрата; и
k) упаривание фильтрата стадии (j) для получения остатка, который очищают, пропуская через колонку силикагеля, с получением чистого соединения формулы (VIc) по п.10.
| Домовый номерной фонарь, служащий одновременно для указания названия улицы и номера дома и для освещения прилежащего участка улицы | 1917 |
|
SU93A1 |
| KAMAL at al | |||
| Топка с несколькими решетками для твердого топлива | 1918 |
|
SU8A1 |
| Chem | |||
| Lett | |||
| Способ гальванического снятия позолоты с серебряных изделий без заметного изменения их формы | 1923 |
|
SU12A1 |
| ТРИЦИКЛИЧЕСКИЕ ДИАЗЕПИНОВЫЕ АНТАГОНИСТЫ ВАЗОПРЕССИНА И АНТАГОНИСТЫ ОКСИТОЦИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ЛЕЧЕНИЯ | 1994 |
|
RU2126006C1 |
| ТРИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЛЕЧЕНИЯ | 1998 |
|
RU2213094C2 |

