Изобретение относится к способам получения изопрена из изобутилена и формальдегида.
Изопрен находит широкое применение в качестве мономера для получения каучуков по свойствам, близким к натуральному, а также в органическом синтезе.
Известен ряд способов получения изопрена конденсацией изобутилена и формальдегида или веществ, являющихся их источниками, например 4,4-диметил-1,3-диоксана и триметилкарбинола, в присутствии водного раствора кислотного катализатора, осуществляемой при повышенной температуре и повышенном давлении в одну или несколько ступеней контактирования, с отгонкой на последней ступени контактирования легкокипящей части продуктов реакции и воды, в реакционно-разделительных аппаратах различной конструкции, как правило, включающих зону подвода тепла, реакционную зону, зону сепарации и контур циркуляции [пат. РФ 1216940, пат. РФ 2085552, пат. РФ 2098398, пат. РФ 2099318, пат. РФ 2203878. пат. РФ 2280022, пат. US 4511751, пат. ЕР 0106323].
При осуществлении известных способов получения изопрена, для сохранения объема раствора катализатора постоянным, необходимо, чтобы отгонялось балансовое количество воды, которая образуется в реакции и вводится с реагентами. Кроме того, высококипящие побочные продукты также должны непрерывно или периодически выводиться из катализаторного раствора.
Наиболее близкий к предлагаемому изобретению способ заявлен в патенте ЕР 0106323 - прототип. В нем заявлено поддержание условий осуществления процесса в интервале, обеспечивающем отгонку получающегося изопрена, легкокипящих исходных и побочных продуктов и балансового количества воды. Заявлены следующие условия осуществления процесса: температура 150-220°С, давление в 1,1-2,5 раза выше давления насыщенных паров воды над раствором кислотного катализатора при температуре процесса, молярное соотношение изобутилена (и/или источника изобутилена) и формальдегида (и/или источника формальдегида) не менее 3.
В соответствии с известным способом соотношение отгоняемых легкокипящих продуктов и воды можно контролировать, регулируя давление в реакционной системе. При повышении давления в реакционной системе соотношение воды и прочих компонентов в дистилляте уменьшается, тогда как при снижении давления в реакционной системе имеет место обратная закономерность.
Принципиальным недостатком известной технологии является то, что параметры процесса определяются необходимостью обеспечить отгонку балансового количества воды, а не оптимальными условиями реакции.
В частности, реакция более селективно протекает при пониженной температуре, но в этом случае не обеспечивается отгонка балансового количества воды, раствор катализатора разбавляется водой, падает его кислотность, снижается конверсия реагентов и производительность реакционного объема. Поэтому приходится либо повышать температуру и работать при заведомо пониженной селективности, либо снижать давление, что также ведет к снижению селективности.
Известно, что селективность процесса зависит от концентрации (растворимости) изобутилена в реакционной водной фазе, которая, в свою очередь, зависит от парциального давления изобутилена в реакционной зоне. В силу этого необходимость поддержания относительно низкого давления в зоне реакции для обеспечения вывода балансового количества воды снижает селективность процесса.
Особенно актуальным это становится, когда в качестве сырья процесса используется триметилкарбинол (источник изобутилена) или его водные растворы (как правило, водный азеотроп, содержащий 12-13% воды), образующиеся при его производстве гидратацией изобутилена в составе С4-фракций. В этом случае требуется выводить повышенное количество воды, образующейся при дегидратации триметилкарбинола и вводимой с ним.
Простой расчет показывает, что при осуществлении процесса при температуре 160°С и использовании в качестве сырья 40%-ного формальдегида и 88%-ного триметилкарбинола (остальное вода) для обеспечения отгонки балансового количества воды при мольном соотношении реагентов 1:10 давление в реакционной зоне должно быть не выше 9,1 атм, а при мольном соотношении реагентов 1:5 - не выше 8,6 атм.
Аналогично при использовании в качестве сырья 4,4-диметил-1,3-диоксана и 88%-ного триметилкарбинола при мольном соотношении реагентов 1:10 давление в реакционной зоне должно быть не выше 9,7 атм, а при мольном соотношении реагентов 1:5 - не выше 9,6 атм.
Целью настоящего изобретения является устранение указанных недостатков.
Цель была достигнута путем разделения реакционной и сепарационной зон и реализации в них оптимальных для каждой зоны условий. При этом при переходе реакционной смеси из реакционной зоны в сепарационную зону осуществляется дросселирование потока.
При дросселировании потока происходит увеличение степени испарения воды, зависящее от природы присутствующих веществ и их концентраций, а также от исходных и конечных параметров дросселирования.
Нами установлено, что для каждой конкретной реакционной смеси существуют параметры, позволяющие обеспечить необходимую степень испарения воды, и определены интервалы значений этих параметров.
Мы предлагаем способ получения изопрена, который позволяет не связывать давление в реакционной зоне с парциальным давлением паров воды, соответствующим температуре в реакционной зоне (и обуславливающим отгонку балансового количества воды), и осуществлять реакцию синтеза изопрена в оптимальных для нее условиях по температуре, давлению и соотношению реагентов, при этом вывод балансового количества воды из раствора катализатора осуществлять за счет регулирования параметров зоны сепарации и регулирования количества циркулирующего раствора.
Мы предлагаем способ получения изопрена путем жидкофазного взаимодействия триметилкарбинола или его водных растворов и формальдегида или веществ, являющихся его источниками, например 4,4-диметил-1,3-диоксана, в присутствии водного раствора кислотного катализатора, осуществляемый при повышенной температуре и давлении в одну или несколько ступеней контактирования, с использованием на последней ступени контактирования реакционно-разделительного аппарата, включающего зону подвода тепла, реакционную зону и зону сепарации, с отбором продуктов реакции и воды из зоны сепарации в виде парового потока, с последующим охлаждением, конденсацией и разделением, с выводом жидкого потока водного раствора катализатора на экстракцию, с последующим возвратом в зону нагрева, в котором давление в реакционной зоне поддерживают выше, чем давление насыщенных паров воды, соответствующее температуре в реакционной зоне, а давление в зоне сепарации выдерживают ниже, чем давление насыщенных паров воды, соответствующее температуре в реакционной зоне, осуществляя дросселирование реакционного потока при переходе из реакционной зоны в зону сепарации. При этом в реакционной зоне поддерживают температуру 140-180°С и давление 8-25 атм, а в зоне сепарации давление 1,2-12 атм.
В соответствии с предлагаемым изобретением используемый реакционно-разделительный аппарат содержит зону подвода тепла, зону контактирования и зону сепарации, которые могут быть выполнены в виде единого целого либо составлять отдельные части аппарата.
Дросселирование потока из реакционной зоны в сепарационную зону может осуществляться с помощью клапана регулятора давления либо другими возможными методами создания перепада давления между зонами (установка запорно-регулирующей арматуры, трубопроводов малого сечения, создающих сопротивление потоку, и т.п.)
Подвод тепла в реакционную зону может осуществляться за счет внутренней поверхности теплообмена в зоне либо с использованием выносных теплообменников и циркуляции реакционной массы через них.
Перемешивание реакционной массы в реакционной зоне может осуществляться с использованием механического или естественного движения потоков (мешалки, циркуляционные насосы, естественное движение потоков за счет разности плотностей или разности уровней), путем установки массообменных устройств и насадок или другими возможными методами.
Используемый реакционно-разделительный аппарат может содержать две или три последовательные сепарационные зоны для предварительного разделения фаз и последующего избирательного дросселирования одной из фаз. Рабочие параметры последовательных сепарационных зон естественным образом будут отличаться.
При наличии двух зон сепарации выходящий из реакционной зоны поток дросселируют в первую зону сепарации со снижением давления на 2-20 атм, из которой паровой поток поступает на охлаждение и конденсацию, а жидкий поток дросселируют во вторую зону сепарации со снижением давления до 1,2-2,0 атм, с отбором, охлаждением и конденсацией паровой фазы и отбором, охлаждением, экстракцией и рециклом в зону нагрева жидкой фазы.
При наличии трех зон сепарации первая зона используется для предварительного более четкого разделения фаз при температуре и давлении, близких к таковым в реакционной зоне.
При осуществлении предлагаемого способа вывод балансового количества воды из водного раствора катализатора (реакционной жидкой фазы), циркулирующего по контуру: реакционная зона - зона сепарации - зона экстракции - зона нагрева - реакционная зона, дополнительно может регулироваться за счет изменения количества циркулирующей жидкой фазы. Так, при увеличении циркуляции увеличивается поток жидкой фазы, дросселируемый в зону(-ы) сепарации, и соответственно увеличивается количество дополнительно испаряемой воды. И наоборот, при снижении циркуляции количество дополнительно испаряемой воды снижается. Количество циркулирующей жидкой фазы может изменяться в интервале 0,2-6,0 частей от объема реакционной зоны.
Предлагаемый способ получения изопрена не только позволяет создать в реакционной зоне оптимальные условия для протекания химических реакций, но и несет в себе ряд других преимуществ.
Известно, что реакционная водная фаза (водный раствор кислоты) в условиях осуществления процесса (высокая температура) отличается высокой коррозионной активностью, в силу чего используемые реакционно-разделительные аппараты должны быть изготовлены из высоколегированных стойких к кислотной коррозии материалов. В предлагаемом нами способе рабочие параметры зоны сепарации отличаются более низкой температурой и давлением, что значительно снижает скорости коррозии материалов и позволяет часть аппарата (зону сепарации) изготавливать из менее легированных материалов и с меньшей металлоемкостью.
Необходимым условием осуществления процесса является очистка катализаторного раствора от высококипящих побочных продуктов, которая, как правило, осуществляется экстракцией подходящим растворителем.
Известно, что образующиеся в процессе высококипящие побочные продукты (смолы) отличаются склонностью к забивке технологического оборудования и трубопроводов, особенно при охлаждении.
При исследовании процесса в соответствии с предлагаемым изобретением нами обнаружено, что при дросселировании реакционной водной фазы в зону сепарации она охлаждается за счет испарения воды (вскипания при снижении давления ниже давления насыщенных паров). Охлаждение в режиме вскипания способствует диспергированию присутствующей гетерофазы смол и предотвращает ее отслоение и забивку оборудования при дальнейшей переработке кислотного раствора.
Существенными отличительными признаками предлагаемого способа является осуществление процесса в реакционно-разделительном аппарате, в котором давление в реакционной зоне поддерживают выше, чем давление насыщенных паров воды, соответствующее температуре в реакционной зоне, а давление в зоне сепарации выдерживают ниже, чем давление насыщенных паров воды, соответствующее температуре в реакционной зоне, осуществляя дросселирование реакционного потока при переходе из реакционной зоны в зону сепарации. При этом регулирование вывода балансового количества воды осуществляется за счет регулирования давления в зоне сепарации и количества циркулирующего водного раствора катализатора.
Способ позволяет осуществлять непрерывный процесс с выводом балансового количества воды в виде парового потока вместе с продуктами реакции, при этом поддерживая в реакционной зоне оптимальные температуру и давление, не связанные с необходимостью вывода балансового количества воды.
Промышленная осуществимость способа иллюстрируется примерами 1-5, примеры 6 и 7 даны для сравнения.
Пример 1.
Принципиальная схема установки для осуществления способа согласно предлагаемому изобретению приведена на фиг.1.
Установка включает реактор 1, сепаратор 3, холодильник 6, холодильник-конденсатор 7, емкость-отстойник 8, смеситель 9, емкость-отстойник 10, колонну отгонки изобутилена 11, колонну отгонки изопрена 12.
В реактор 1, представляющий собой автоклав объемом 5 литров с мешалкой с герметичным электромагнитным приводом и термостатирующей рубашкой, загружают 4,8 литра 7%-ного водного раствора ортофосфорной кислоты, при включенном перемешивании в термостатирующую рубашку подают горячий теплоноситель и нагревают реактор до температуры 160°С.
В реактор подают 40%-ный формалин с расходом 256,6 г/час и 88%-ный триметилкарбинол с расходом 2875,9 г/час. В реакторе поддерживают давление 12,0 атм.
Выходящий из реактора парожидкостной поток через клапан регулятора давления PC дросселируют в сепаратор 3, где поддерживают давление 4,5 атм. В сепараторе 3 поток разделяется на жидкую и паровую фазы.
Пары продуктов реакции и непревращенного сырья, выходящие из сепаратора 3, проходят холодильник-конденсатор 7, где охлаждаются и конденсируются, и поступают в емкость-отстойник 8, где расслаиваются на водный и органический слои.
Поток жидких продуктов, представляющий собой смесь водного раствора ортофосфорной кислоты и высококипящих побочных продуктов, выводят из сепаратора 3 в количестве 30,0 л/час, охлаждают в холодильнике 4 и подают в смеситель 9 на экстракцию органических продуктов. В качестве экстрагента в смеситель 9 подают органический слой продуктов синтеза из емкости 8. Выходящая из смесителя 9 смесь поступает в емкость-отстойник 10, где расслаивается на органический слой, содержащий проэкстрагированные высококипящие побочные продукты, и водный раствор ортофосфорной кислоты.
Очищенный от высококипящих органических продуктов водный раствор ортофосфорной кислоты из емкости 10 насосом возвращают обратно в реактор 1.
Органический слой из емкости 10 подают на разделение. Сначала в ректификационной колонне 11 выделяют изобутилен в количестве 1511,8 г/час, который направляют на гидратацию в триметилкарбинол (узел гидратации на схеме не показан), а затем в ректификационной колонне 12 выделяют изопрен в количестве 162,4 г/час, который подают на очистку.
Кубовый продукт колонны 12 в количестве 283,0 г/час, представляющий собой смесь побочных продуктов синтеза и непревращенного сырья, содержащий 78,2% триметилкарбинола, 4,1% метилдигидропирана, 0,3% диметилдиоксана, 1,0% изоамиленовых спиртов и 16,4% прочих продуктов, подают в узел выделения непревращенного сырья и промежуточных продуктов синтеза.
Из емкости-отстойника 8 отбирают в количестве 1175,3 г/час водный слой конденсата продуктов синтеза, содержащий 6,4% триметилкарбинола, 0,4% формальдегида и 0,2% прочих органических продуктов, и выводят на очистку перед сбросом в химзагрязненную канализацию (в узел отгонки легкокипящих органических продуктов и формальдегида).
В соответствии с балансом процесса выход изопрена на превращенное сырье составляет 73,2 мол.% по формальдегиду и 74,8 мол.% по триметилкарбинолу (изобутилену).
Процесс осуществляют в непрерывном режиме в течение 40 часов. Анализ состава потоков в пробах, отобранных после 8, 20 и 40 часов работы, показывает идентичные результаты, что свидетельствует об отсутствии накопления воды в реакционном водном слое и стабильности показателей процесса.
Пример 2.
Принципиальная схема установки для осуществления способа согласно предлагаемому изобретению приведена на фиг.2.
Установка включает реактор 1, сепаратор 3, сепаратор 4, холодильник-конденсатор 14, емкость 5, холодильник 6, холодильник-конденсатор 7, емкость-отстойник 8, смеситель 9, емкость-отстойник 10, колонну отгонки изобутилена 11, колонну отгонки изопрена 12.
В реактор 1, представляющий собой автоклав объемом 5 литров с мешалкой с герметичным электромагнитным приводом и термостатирующей рубашкой, загружают 4,8 литра 3%-ного водного раствора ортофосфорной кислоты, при включенном перемешивании в термостатирующую рубашку подают горячий теплоноситель и нагревают реактор до температуры 180°С.
В реактор подают 40%-ный формалин с расходом 256,6 г/час и 88%-ный триметилкарбинол с расходом 2875,9 г/час. В реакторе поддерживают давление 25,0 атм.
Выходящий из реактора парожидкостной поток через клапан регулятора давления PC дросселируют в сепаратор 3, где поддерживают давление 9,0 атм. В сепараторе 3 поток разделяется на жидкую и паровую фазы.
Пары продуктов реакции и непревращенного сырья, выходящие из сепаратора 3, проходят холодильник-конденсатор 7, где охлаждаются и конденсируются, и поступают в емкость-отстойник 8, где расслаиваются на водный и органический слои.
Поток жидких продуктов, представляющий собой смесь водного раствора ортофосфорной кислоты и высококипящих побочных продуктов, из сепаратора 3 через клапан регулятора уровня LC дросселируют в сепаратор 4, где поддерживают давление 2,0 атм. В сепараторе 4 происходит вскипание жидкой фазы с частичной отпаркой воды и органических продуктов. Пары воды и продуктов проходят холодильник-конденсатор 14, где охлаждаются и конденсируются, и стекают в емкость 5.
Смесь водного раствора ортофосфорной кислоты и высококипящих побочных продуктов отбирают из сепаратора 4 и насосом подают в количестве 8,0 л/час через холодильник 6 в смеситель 9 на экстракцию органических продуктов.
В качестве экстрагента в смеситель 9 подают органический слой продуктов синтеза из емкости 8. Выходящая из смесителя 9 смесь поступает в емкость-отстойник 10, где расслаивается на органический слой, содержащий проэкстрагированные высококипящие побочные продукты, и водный раствор ортофосфорной кислоты.
Очищенный от высококипящих органических продуктов водный раствор ортофосфорной кислоты из емкости 10 насосом возвращают обратно в реактор 1.
Органический слой из емкости 10 подают на разделение. Сначала в ректификационной колонне 11 выделяют изобутилен в количестве 1555,3 г/час, который направляют на гидратацию в триметилкарбинол (узел гидратации на схеме не показан), а затем в ректификационной колонне 12 выделяют изопрен в количестве 158,3 г/час, который подают на очистку.
Кубовый продукт колонны 12 в количестве 244,4 г/час, представляющий собой смесь побочных продуктов синтеза и непревращенного сырья, содержащий 73,9% триметилкарбинола, 4,9% метилдигидропирана, 0,4% диметилдиоксана, 1,2% изоамиленовых спиртов и 19,6% прочих продуктов, подают в узел выделения непревращенного сырья и промежуточных продуктов синтеза.
Из емкости-отстойника 8 и емкости 5 отбирают суммарно в количестве 1174,5 г/час водный слой конденсата паров продуктов синтеза, содержащий 5,1% триметилкарбинола, 0,5% формальдегида и 0,3% прочих органических продуктов, и выводят на очистку перед сбросом в химзагрязненную канализацию (в узел отгонки легкокипящих органических продуктов и формальдегида).
В соответствии с балансом процесса выход изопрена на превращенное сырье составляет 71,9 мол.% по формальдегиду и 73,4 мол.% по триметилкарбинолу (изобутилену).
Процесс осуществляют в непрерывном режиме в течение 40 часов. Анализ состава потоков в пробах, отобранных после 8, 20 и 40 часов работы, показывает идентичные результаты, что свидетельствует об отсутствии накопления воды в реакционном водном слое и стабильности показателей процесса.
Пример 3.
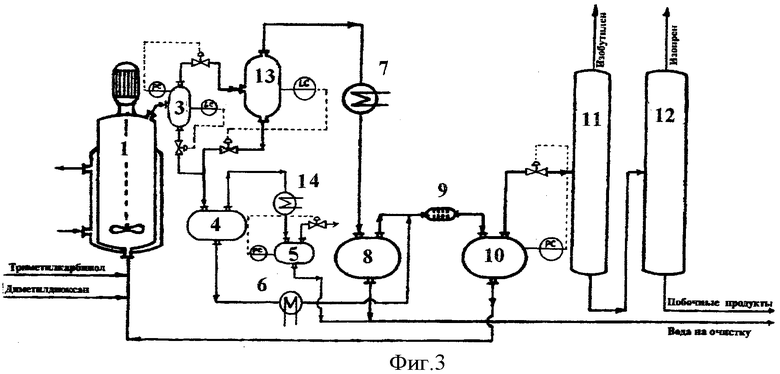
Принципиальная схема установки для осуществления способа согласно предлагаемому изобретению приведена на фиг.3.
Установка включает реактор 1, сепаратор 3, сепаратор 13, сепаратор 4, холодильник-конденсатор 14, емкость 5, холодильник 6, холодильник-конденсатор 7, емкость-отстойник 8, смеситель 9, емкость-отстойник 10, колонну отгонки изобутилена 11, колонну отгонки изопрена 12.
В реактор 1, представляющий собой автоклав объемом 5 литров с мешалкой с герметичным электромагнитным приводом и термостатирующей рубашкой, загружают 4,8 литра 9%-ного водного раствора ортофосфорной кислоты, при включенном перемешивании в термостатирующую рубашку подают горячий теплоноситель и нагревают реактор до температуры 155°С.
В реактор подают 4,4-диметил-1,3-диоксан с расходом 400,0 г/час и 88%-ный триметилкарбинол с расходом 1739,7 г/час. В реакторе поддерживают давление 12,0 атм.
Выходящий из реактора парожидкостной поток поступает в сепаратор 3, где разделяется на жидкую и паровую фазы.
Пары продуктов реакции и непревращенного сырья, а также часть неотделившихся жидких продуктов (капельный унос), выходящие из сепаратора 3, через клапан регулятора давления дросселируют в сепаратор 13, где поддерживается давление 5,3 атм. В сепараторе 13 поток дополнительно разделяется на жидкую и паровую фазы. Пары продуктов, выходящие из сепаратора 13, проходят холодильник-конденсатор 7, где охлаждаются и конденсируются, и поступают в емкость-отстойник 8, где расслаиваются на водный и органический слои.
Поток жидких продуктов, представляющий собой смесь водного раствора ортофосфорной кислоты и высококипящих побочных продуктов, из сепаратора 3 и сепаратора 13 через клапаны регуляторы уровней LC дросселируют в сепаратор 4, где поддерживают давление 1,2 атм. В сепараторе 4 происходит вскипание жидкой фазы с частичной отпаркой воды и органических продуктов. Пары воды и продуктов проходят холодильник-конденсатор 14, где охлаждаются и конденсируются, и стекают в емкость 5.
Смесь водного раствора ортофосфорной кислоты и высококипящих побочных продуктов отбирают из сепаратора 4 и насосом подают в количестве 1,0 л/час через холодильник 6 в смеситель 9 на экстракцию органических продуктов.
В качестве экстрагента в смеситель 9 подают органический слой продуктов синтеза из емкости 8. Выходящая из смесителя 9 смесь поступает в емкость-отстойник 10, где расслаивается на органический слой, содержащий проэкстрагированные высококипящие побочные продукты, и водный раствор ортофосфорной кислоты.
Очищенный от высококипящих органических продуктов водный раствор ортофосфорной кислоты из емкости 10 насосом возвращают обратно в реактор 1.
Органический слой из емкости 10 подают на разделение. Сначала в ректификационной колонне 11 выделяют изобутилен в количестве 810,5 г/час, который направляют на гидратацию в триметилкарбинол (узел гидратации на чертеже не показан), а затем в ректификационной колонне 12 выделяют изопрен в количестве 346,6 г/час, который подают на очистку.
Кубовый продукт колонны 12 в количестве 291,4 г/час, представляющий собой смесь побочных продуктов синтеза и непревращенного сырья, содержащий 59,1% триметилкарбинола, 7,7% метилдигидропирана, 2,9% диметилдиоксана, 2,2% изоамиленовых спиртов и 28,1% прочих продуктов, подают в узел выделения непревращенного сырья и промежуточных продуктов синтеза.
Из емкости-отстойника 8 и емкости 5 отбирают суммарно в количестве 691,2 г/час водный слой конденсата паров продуктов синтеза, содержащий 8,3% триметилкарбинола, 0,3% формальдегида и 0,2% прочих органических продуктов, и выводят на очистку (в узел отгонки легкокипящих органических продуктов и формальдегида) перед сбросом в химзагрязненную канализацию.
В соответствии с балансом процесса выход изопрена составляет 151,0 мол.% по диметилдиоксану, что составляет 75,5% от теоретического выхода, и 163,6 мол.% по триметилкарбинолу (изобутилену), что составляет 81,8% от теоретического выхода.
Процесс осуществляют в непрерывном режиме в течение 40 часов. Анализ состава потоков в пробах, отобранных после 8, 20 и 40 часов работы, показывает идентичные результаты, что свидетельствует об отсутствии накопления воды в реакционном водном слое и стабильности показателей процесса.
Пример 4.
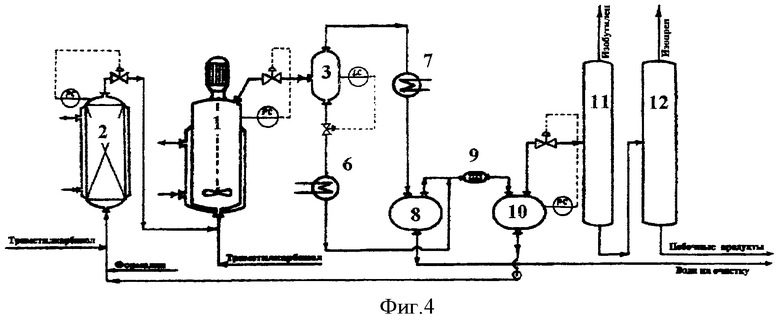
Принципиальная схема установки для осуществления способа согласно предлагаемому изобретению приведена на фиг.4.
Установка включает реакторы 1 и 2, сепаратор 3, холодильник 6, холодильник-конденсатор 7, емкость-отстойник 8, смеситель 9, емкость-отстойник 10, колонну отгонки изобутилена 11, колонну отгонки изопрена 12.
Реакторный блок для осуществления процесса в две ступени контактирования состоит из реактора синтеза предшественников изопрена 2 и реактора разложения предшественников изопрена в изопрен 1.
Реактор 2 представляет собой вертикальный аппарат колонного типа высотой 3 метра и диаметром 0,05 метра, заполненный насадкой и имеющий термостатирующую рубашку. Реактор 1 представляет собой автоклав объемом 5 литров с мешалкой с герметичным электромагнитным приводом и термостатирующей рубашкой.
В реакторный блок загружают 9,0 литров 7%-ного водного раствора ортофосфорной кислоты, в термостатирующие рубашки реакторов подают горячий теплоноситель и нагревают реактор 2 до температуры 110°С, а реактор 1 (при включенном перемешивании) до температуры 160°С.
В реактор 2 подают 40%-ный формалин с расходом 256,6 г/час и 88%-ный триметилкарбинол с расходом 860,0 г/час, и циркулирующий раствор кислоты из емкости Е-8 с расходом 3,0 л/час.В реакторе поддерживают давление 22,0 атм.
Выходящий из реактора 2 поток смешивают с дополнительным количеством триметилкарбинола, подаваемым в количестве 2015,9 г/час, и подают в реактор 1. В реакторе 1 поддерживают давление 12,0 атм.
Выходящий из реактора парожидкостной поток через клапан регулятора давления PC дросселируют в сепаратор 3, где поддерживают давление 4,5 атм. В сепараторе 3 поток разделяется на жидкую и паровую фазы.
Пары продуктов реакции и непревращенного сырья, выходящие из сепаратора 3, проходят холодильник-конденсатор 7, где охлаждаются и конденсируются, и поступают в емкость-отстойник 8, где расслаиваются на водный и органический слои.
Поток жидких продуктов, представляющий собой смесь водного раствора ортофосфорной кислоты и высококипящих побочных продуктов, выводят из сепаратора 3 в количестве 30,0 л/час, охлаждают в холодильнике 6 и подают в смеситель 9 на экстракцию органических продуктов. В качестве экстрагента в смеситель 9 подают органический слой продуктов синтеза из емкости 8. Выходящая из смесителя 9 смесь поступает в емкость-отстойник 10, где расслаивается на органический слой, содержащий проэкстрагированные высококипящие побочные продукты, и водный раствор ортофосфорной кислоты.
Очищенный от высококипящих органических продуктов водный раствор ортофосфорной кислоты из емкости 10 насосом возвращают в реактор 2 в количестве 3,0 л/час и в реактор 1 в количестве 27,0 л/час.
Органический слой из емкости 10 подают на разделение. Сначала в ректификационной колонне 11 выделяют изобутилен в количестве 1515,5 г/час, который направляют на гидратацию в триметилкарбинол (узел гидратации на чертеже не показан), а затем в ректификационной колонне 12 выделяют изопрен в количестве 164,4 г/час, который подают на очистку.
Кубовый продукт колонны 12 в количестве 273,1 г/час, представляющий собой смесь побочных продуктов синтеза и непревращенного сырья, содержащий 84,0% триметилкарбинола, 4,4% метилдигидропирана, 0,5% диметилдиоксана, 0,9% изоамиленовых спиртов и 10,2% прочих продуктов, подают в узел выделения непревращенного сырья и промежуточных продуктов синтеза.
Из емкости-отстойника 8 отбирают в количестве 1179,5 г/час водный слой конденсата продуктов синтеза, содержащий 6,9% триметилкарбинола, 0,5% формальдегида и 0,2% прочих органических продуктов, и выводят на очистку (в узел отгонки легкокипящих органических продуктов и формальдегида) перед сбросом в химзагрязненную канализацию.
В соответствии с балансом процесса выход изопрена на превращенное сырье составляет 74,9 мол.% по формальдегиду и 82,3 мол.% по изобутилену.
Процесс осуществляют в непрерывном режиме в течение 40 часов. Анализ состава потоков в пробах, отобранных после 8, 20 и 40 часов работы, показывает идентичные результаты, что свидетельствует об отсутствии накопления воды в реакционном водном слое и стабильности показателей процесса.
Пример 5.
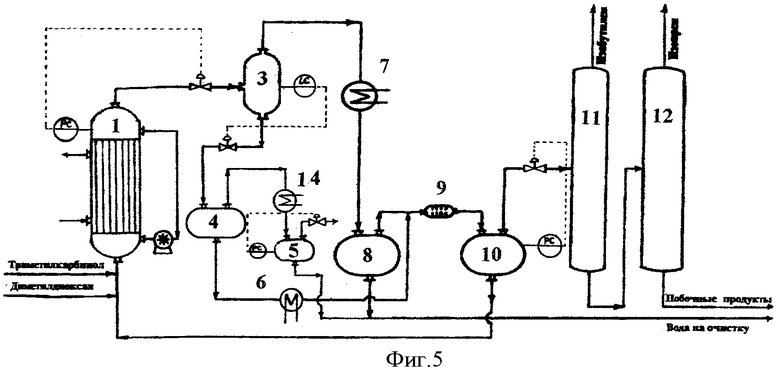
Принципиальная схема установки для осуществления способа согласно предлагаемому изобретению приведена на фиг.5.
Установка включает реактор 1, сепаратор 3, сепаратор 4, холодильник-конденсатор 14, емкость 5, холодильник 6, холодильник-конденсатор 7, емкость-отстойник 8, смеситель 9, емкость-отстойник 10, колонну отгонки изобутилена 11, колонну отгонки изопрена 12.
В реактор 1, представляющий собой трубчатый теплообменник с объемом трубного пространства 5 литров с внешней циркуляционной трубой и циркуляционным насосом, обеспечивающим циркуляцию жидкой фазы не менее 500 литров в час, загружают 4,8 литра 6%-ного водного раствора ортофосфорной кислоты, при включенном циркуляционном насосе в межтрубное пространство подают горячий теплоноситель и нагревают реактор до температуры 170°С.
В реактор подают 4,4-диметил-1,3-диоксан с расходом 400,0 г/час и 88%-ный триметилкарбинол с расходом 2875,9 г/час. В реакторе поддерживают давление 15,0 атм.
Выходящий из реактора парожидкостной поток через клапан регулятора давления PC дросселируют в сепаратор 3, где поддерживают давление 7,5 атм. В сепараторе 3 поток разделяется на жидкую и паровую фазы.
Пары продуктов реакции и непревращенного сырья, выходящие из сепаратора 3, проходят холодильник-конденсатор 7, где охлаждаются и конденсируются, и поступают в емкость-отстойник 8, где расслаиваются на водный и органический слои.
Поток жидких продуктов, представляющий собой смесь водного раствора ортофосфорной кислоты и высококипящих побочных продуктов, из сепаратора 3 через клапан регулятора уровня LC дросселируют в сепаратор 4, где поддерживают давление 1,5 атм. В сепараторе 4 происходит вскипание жидкой фазы с частичной отпаркой воды и органических продуктов. Пары воды и продуктов проходят холодильник-конденсатор 14, где охлаждаются и конденсируются, и стекают в емкость 5.
Смесь водного раствора ортофосфорной кислоты и высококипящих побочных продуктов отбирают из сепаратора 4 и насосом подают в количестве 8,0 л/час через холодильник 6 в смеситель 9 на экстракцию органических продуктов.
В качестве экстрагента в смеситель 9 подают органический слой продуктов синтеза из емкости 8. Выходящая из смесителя 9 смесь поступает в емкость-отстойник 10, где расслаивается на органический слой, содержащий проэкстрагированные высококипящие побочные продукты, и водный раствор ортофосфорной кислоты.
Очищенный от высококипящих органических продуктов водный раствор ортофосфорной кислоты из емкости 10 насосом возвращают обратно в реактор 1.
Органический слой из емкости 10 подают на разделение. Сначала в ректификационной колонне 11 выделяют изобутилен в количестве 1561,6 г/час, который направляют на гидратацию в триметилкарбинол (узел гидратации на чертеже не показан), а затем в ректификационной колонне 12 выделяют изопрен в количестве 359,7 г/час, который подают на очистку.
Кубовый продукт колонны 12 в количестве 280,3 г/час, представляющий собой смесь побочных продуктов синтеза и непревращенного сырья, содержащий 67,6% триметилкарбинола, 8,2% метилдигидропирана, 2,6% диметилдиоксана, 2,4% изоамиленовых спиртов и 19,2% прочих продуктов, подают в узел выделения непревращенного сырья и промежуточных продуктов синтеза.
Из емкости-отстойника 8 и емкости 5 отбирают суммарно в количестве 1074,3 г/час водный слой конденсата паров продуктов синтеза, содержащий 5,9% триметилкарбинола, 0,3% формальдегида и 0,2% прочих органических продуктов, и выводят на очистку перед сбросом в химзагрязненную канализацию (в узел отгонки легкокипящих органических продуктов и формальдегида).
В соответствии с балансом процесса выход изопрена составляет 156,2 мол.% по диметилдиоксану, что составляет 78,1% от теоретического выхода, и 166,8 мол.% по триметилкарбинолу (изобутилену), что составляет 83,4% от теоретического выхода.
Процесс осуществляют в непрерывном режиме в течение 40 часов. Анализ состава потоков в пробах, отобранных после 8, 20 и 40 часов работы, показывает идентичные результаты, что свидетельствует об отсутствии накопления воды в реакционном водном слое и стабильности показателей процесса.
Пример 6 (для сравнения).
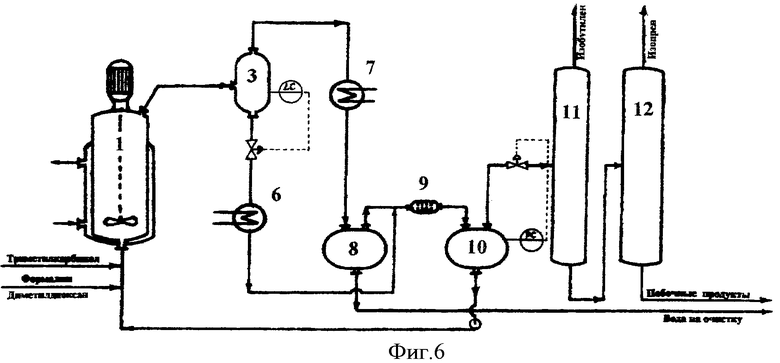
Принципиальная схема установки для осуществления способа согласно известным изобретениям приведена на фиг.6.
Установка включает реактор 1, сепаратор 3, холодильник 6, холодильник-конденсатор 7, емкость-отстойник 8, смеситель 9, емкость-отстойник 10, колонну отгонки изобутилена 11, колонну отгонки изопрена 12.
В реактор 1, представляющий собой автоклав объемом 5 литров с мешалкой с герметичным электромагнитным приводом и термостатирующей рубашкой, загружают 4,8 литра 7%-ного водного раствора ортофосфорной кислоты, при включенном перемешивании в термостатирующую рубашку подают горячий теплоноситель и нагревают реактор до температуры 160°С.
Подачи реагентов, температуру и давление в реакционной зоне выдерживают как в примере 1. В реактор подают 40%-ный формалин с расходом 256,5 г/час и 88%-ный триметилкарбинол с расходом 2875,9 г/час. Давление в реакционном узле (реактор, сепаратор, холодильник, отстойник) поддерживают на уровне 12 атм.
Выходящий из реактора парожидкостной поток поступает в сепаратор 3, где разделяется на жидкую и паровую фазы. Пары продуктов реакции и непревращенного сырья, выходящие из сепаратора 3, проходят холодильник-конденсатор 7, где охлаждаются и конденсируются, и поступают в емкость-отстойник 8, где расслаиваются на водный и органический слои.
Поток жидких продуктов, представляющий собой смесь водного раствора ортофосфорной кислоты и высококипящих побочных продуктов, выводят из сепаратора 3 в количестве 30,0 л/час, охлаждают в холодильнике 6 и подают в смеситель 9 на экстракцию органических продуктов. В качестве экстрагента в смеситель 9 подают органический слой продуктов синтеза из емкости 8. Выходящая из смесителя 9 смесь поступает в емкость-отстойник 10, где расслаивается на органический слой, содержащий проэкстрагированные высококипящие побочные продукты, и водный раствор ортофосфорной кислоты.
Очищенный от высококипящих органических продуктов водный раствор ортофосфорной кислоты из емкости 10 насосом возвращают обратно в реактор 1.
Органический слой из емкости 10 подают на разделение. Сначала в ректификационной колонне 11 выделяют изобутилен, который направляют на гидратацию в триметилкарбинол (узел гидратации на чертеже не показан), а затем в ректификационной колонне 12 выделяют изопрен, который подают на очистку.
Кубовый продукт колонны 12, представляющий собой смесь побочных продуктов синтеза и непревращенного сырья, подают в узел выделения непревращенного сырья и промежуточных продуктов синтеза.
Из емкости-отстойника 8 отбирают водный слой конденсата продуктов синтеза и выводят на очистку (в узел отгонки легкокипящих органических продуктов и формальдегида) перед сбросом в химзагрязненную канализацию.
Процесс осуществляют в непрерывном режиме. После 10 часов работы установки объем водного раствора катализатора значительно увеличивается (˜ в 1,3 раза), а кислотность его снижается, наблюдается переполнение сепаратора 3 жидкой фазой, переброс ее в систему конденсации и охлаждения паров и потери катализатора с водным слоем из емкости-отстойника 8. Процесс прекращают.
Попытки осуществления непрерывного процесса повторяют, постепенно снижая давление в реакционном узле (реактор, сепаратор, холодильник, отстойник).
Только при давлении 9,0 атм удается обеспечить балансовый вывод воды с парами продуктов и осуществить непрерывный процесс (выдержать стабильный уровень жидкой фазы в сепараторе 3).
В соответствии с балансом процесса выход изопрена на превращенное сырье составляет 71,8 мол.% по формальдегиду и 73,2 мол.% по изобутилену.
Пример 7 (для сравнения).
Процесс осуществляют как это описано в примере 6, за исключением того, что вместо формалина подают 4,4-диметил-1,3-диоксан. Подачи реагентов, температуру и давление в реакционной зоне выдерживают те же, что и в примере 3.
При осуществлении непрерывного процесса через 20 часов работы установки объем водного раствора катализатора значительно увеличивается (в 1,3 раза), а кислотность его снижается, наблюдается переполнение сепаратора 3 жидкой фазой, переброс ее в систему конденсации и охлаждения паров и потери катализатора с водным слоем из емкости-отстойника 8. Процесс прекращают.
Попытки осуществления непрерывного процесса повторяют, постепенно повышая температуру в реакторе. Только при температуре 170°С удается обеспечить балансовый вывод воды с парами продуктов и осуществить непрерывный процесс (выдержать стабильный уровень жидкой фазы в сепараторе 3).
В соответствии с балансом процесса выход изопрена составляет 144,8 мол.% по диметилдиоксану, что составляет 72,4% от теоретического выхода и 149,8 мол.% по триметилкарбинолу (изобутилену), что составляет 74,9% от теоретического выхода.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ИЗОПРЕНА | 2007 |
|
RU2332394C1 |
| СПОСОБ ПОЛУЧЕНИЯ ИЗОПРЕНА | 2007 |
|
RU2330009C1 |
| СПОСОБ ПОЛУЧЕНИЯ ИЗОПРЕНА | 2011 |
|
RU2458036C1 |
| Способ производства изопрена из изобутилена и формальдегида без выделения промежуточных продуктов | 2023 |
|
RU2828416C1 |
| СПОСОБ ПОЛУЧЕНИЯ ИЗОПРЕНА | 2001 |
|
RU2235709C2 |
| СПОСОБ ПОЛУЧЕНИЯ ИЗОПРЕНА | 2004 |
|
RU2261855C1 |
| СПОСОБ ПОЛУЧЕНИЯ ИЗОПРЕНА | 2001 |
|
RU2197461C2 |
| СПОСОБ ПОЛУЧЕНИЯ ИЗОПРЕНА | 2007 |
|
RU2341508C1 |
| СПОСОБ ПОЛУЧЕНИЯ ИЗОПРЕНА | 2004 |
|
RU2266888C1 |
| УСТАНОВКА ДЛЯ ПРОИЗВОДСТВА ИЗОПРЕНА (ВАРИАНТЫ) | 2014 |
|
RU2584259C1 |

Изобретение относится к способу получения изопрена путем жидкофазного взаимодействия триметилкарбинола или его водных растворов и формальдегида или веществ, являющихся его источниками, например 4,4-диметил-1,3-диоксана, в присутствии водного раствора кислотного катализатора, осуществляемому при повышенной температуре и давлении в одну или несколько ступеней контактирования, с использованием на последней ступени контактирования реакционно-разделительного аппарата, включающего зону подвода тепла, реакционную зону и зону сепарации, с отбором продуктов реакции и воды из зоны сепарации в виде парового потока, с последующим охлаждением, конденсацией и разделением, с выводом жидкого потока водного раствора катализатора на экстракцию, с последующим возвратом в зону нагрева, и характеризующемуся тем, что давление в реакционной зоне поддерживают выше, чем давление насыщенных паров воды, соответствующее температуре в реакционной зоне, а давление в зоне сепарации выдерживают ниже, чем давление насыщенных паров воды, соответствующее температуре в реакционной зоне, осуществляя дросселирование реакционного потока при переходе из реакционной зоны в зону сепарации. Применение данного способа позволяет проводить процесс при оптимальных условиях реакции. 4 з.п. ф-лы, 6 ил.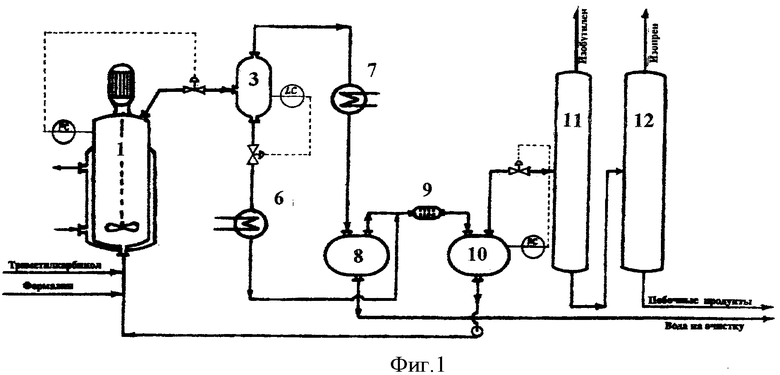
| Приспособление к уборочным машинам для отделения початков от стеблей кукурузы | 1956 |
|
SU106323A1 |
| RU 1624937 A1, 20.01.1996 | |||
| RU 508050 A1, 10.11.2001. | |||

