Изобретение относится к способам получения изопрена из изобутилена и формальдегида.
Изопрен находит широкое применение в качестве мономера для получения каучуков, по свойствам близких натуральному, а также в органическом синтезе.
Известен ряд способов получения изопрена путем жидкофазного взаимодействия формальдегида и изобутилена или веществ, являющихся их источниками, например 4,4-диметил-1,3-диоксана и триметилкарбинола, в присутствии водного раствора кислотного катализатора, осуществляемых при повышенной температуре и давлении в одну или несколько ступеней контактирования, с отбором на последней ступени контактирования продуктов реакции и балансового количества воды в виде парового потока, с последующим охлаждением, конденсацией и разделением на водный и органический слои, с переработкой органического слоя, включающей выделение изобутилена, изопрена, фракции триметилкарбинола, фракции метилдигидропирана, фракции предшественников изопрена и высококипящего остатка, с выводом жидкого потока водного раствора катализатора на экстракцию, с последующим возвратом в зону синтеза [USP 4511751, пат. РФ 2280022, ЕР 0106323].
Во всех известных способах переработка отгоняемых продуктов синтеза включает их охлаждение и конденсацию, разделение конденсата на водный и органический слои, выделение из органического слоя последовательной ректификацией рециклового изобутилена, целевого изопрена, рецикловой фракции триметилкарбинола, фракции 4-метил-5,6-дигидро-2Н-пирана (далее метилдигидропирана), рецикловой фракции непревращенных диметилдиоксана и изоамиленовых спиртов и высококипящего остатка, выделение из водного слоя легкокипящих органических продуктов (в основном триметилкарбинола) и выводом оставшейся воды на очистку.
Для обеспечения высокой селективности процесс синтеза изопрена, как правило, осуществляется при стехиометрическом избытке изобутилена и/или триметилкарбинола. Вне зависимости от того, используется ли в процессе изобутилен или триметилкарбинол, выходящий из процесса поток содержит смесь этих веществ в соотношении, близком к равновесному [ЕР 0106323]. Таким образом, все известные способы получения изопрена предполагают выделение и рецикл триметилкарбинола.
Во всех известных способах получения изопрена путем жидкофазного взаимодействия формальдегида и изобутилена или веществ, являющихся их источниками, например 4,4-диметил-1,3-диоксана и триметилкарбинола, в присутствии водного раствора кислотного катализатора конверсия исходных реагентов не является полной. Кроме того, в продуктах синтеза содержится значительное количество веществ, отличающихся от используемого сырья, но являющихся предшественниками изопрена (4,4-диметил-1,3-диоксан, 3-метил-1-бутен-3-ол, 3-метил-3-бутен-1-ол, 3-метил-2-бутен-1-ол и др.). Выделение и рецикл непрореагировавшего сырья и продуктов, являющихся предшественниками изопрена, является неотъемлемой частью технологии процесса синтеза изопрена.
Одним из основных побочных продуктов синтеза изопрена является метилдигидропиран, который может быть выделен из продуктов синтеза и подвергнут парофазному разложению (термическому или каталитическому) в изопрен [пат. РФ 2167710, 2134679]. Разложение метилдигидропирана позволяет значительно (на 5-7%) увеличить выработку изопрена и в целом по процессу увеличить селективность его образования на 5-7% по формальдегиду и на 3-5% по изобутилену.
Проведенные нами исследования известных способов получения изопрена показали, что фракция триметилкарбинола и фракция предшественников изопрена, выделенные из продуктов синтеза ректификацией, содержат значительное количество (до 20%) примесей углеводородов, спиртов и карбонильных соединений, которые имеют близкие температуры кипения, азеотропны с целевыми продуктами выделяемых фракций и не могут быть отделены простой ректификацией.
Рецикл выделенных простой ректификацией фракции триметилкарбинола и фракции предшественников изопрена в реакционную зону синтеза изопрена приводит к накоплению в продуктах синтеза вышеуказанных примесей, которые в условиях процесса практически инертны, что в свою очередь приводит к необходимости отвода части фракций на утилизацию и соответственно к ухудшению показателей процесса по расходу сырья и энергозатратам.
Целью настоящего изобретения является устранение указанных недостатков.
Цель была достигнута путем разработки технических приемов, позволяющих отделить от целевых фракций примеси нераздельнокипящих и близкокипящих веществ и исключить их накопление в реакционной смеси и в продуктах синтеза при рецикле фракции триметилкарбинола и фракции предшественников изопрена.
Мы предлагаем способ получения изопрена путем жидкофазного взаимодействия формальдегида и изобутилена или веществ, являющихся их источниками, например 4,4-диметил-1,3-диоксана и триметилкарбинола, в присутствии водного раствора кислотного катализатора, осуществляемый при повышенной температуре и давлении в одну или несколько ступеней контактирования, с отбором на последней ступени контактирования продуктов реакции и балансового количества воды в виде парового потока, с последующим охлаждением, конденсацией и разделением на водный и органический слои, с переработкой органического слоя, включающей выделение рециклового изобутилена, целевого изопрена, рецикловых триметилкарбинола и предшественников изопрена, метилдигидропирана, направляемого на разложение, и высококипящего остатка, с выводом жидкого потока водного раствора катализатора на экстракцию, с последующим возвратом в зону синтеза, в котором выделение триметилкарбинола, метилдигидропирана и предшественников изопрена осуществляют путем азеотропной ректификации с водой.
Нами было обнаружено, что при разделении продуктов синтеза путем азеотропной ректификации с водой удается отделить примеси побочных продуктов (углеводородов, спиртов и карбонильных соединений), которые в условиях процесса инертны и при рециркуляции в зону реакции фракций триметилкарбинола и предшественников изопрена накапливаются в потоках, мешают нормальному протеканию процесса и ухудшают его показатели.
Более подробные исследования показали, что примеси побочных продуктов (углеводородов, спиртов и карбонильных соединений) либо не образуют с водой азеотропов, либо образуют азеотропы с температурами кипения вне интервала температур кипения азеотропов целевых продуктов (79-97°С), что создает возможность их отделения при азеотропной ректификации с водой.
При добавлении воды к суммарной фракции побочных продуктов, полученной после выделения изобутилена и изопрена, и отгонке фракций в виде водных азеотропов с температурами кипения Н.К. 78°С (гетерогенный), 78-87°С (гомогенный), 87-90°С (гетерогенный), 90-97°С (гетерогенный), с последующей рециркуляцией в процесс фракции 78-87°С (фракция триметилкарбинола) и фракции 90-97°С (фракция предшественников изопрена), накопления в продуктах синтеза каких-либо побочных продуктов не наблюдается, и появляется возможность осуществлять непрерывный процесс с рециклом выделенных из продуктов синтеза триметилкарбинола и предшественников изопрена, избежать ухудшения показателей синтеза за счет рецикла и накопления примесей, исключить потери триметилкарбинола и предшественников изопрена.
Добавление воды для осуществления азеотропной ректификации продуктов синтеза изопрена может осуществляться путем дробной подачи необходимого количества в каждую ректификационную колонну.
Из суммарной фракции побочных продуктов азеотропной ректификацией с водой сначала может быть отогнана широкая фракция продуктов, имеющих температуру кипения водных азеотропов до 97°С, с последующим выделением из нее целевых фракций.
Суммарная фракция побочных продуктов может быть предварительно разделена простой ректификацией на фракцию триметилкарбинола, фракцию метилдигидропирана, фракцию предшественников изопрена и высококипящий остаток, с последующей очисткой от примесей фракции триметилкарбинола и фракции предшественников изопрена азеотропной ректификацией с водой.
Количество добавляемой воды при осуществлении азеотропной ректификации должно быть достаточным для образования азеотропных смесей. Лучшие результаты достигаются при добавлении 5-15% воды по отношению к массе суммарной фракции побочных продуктов. При меньшем количестве добавляемой воды наблюдается накопление примесей в рециркулируемых фракциях триметилкарбинола и предшественников изопрена, а при большем количестве избыточная вода остается в кубовом остатке в виде отдельной плохо отделяющейся фазы, что усложняет его утилизацию.
Часть фракций, отгоняемых при азеотропной ректификации с водой, образует гетероазеотропы, которые после конденсации расслаиваются на органический и водный слои. Образующиеся водные слои могут быть использованы в качестве воды, добавляемой к исходной суммарной фракции побочных продуктов или дробно добавляемой в отдельные ректификационные колонны.
При наличии в составе производства изопрена установки парофазного термокаталитического разложения метилдигидропирана фракция предшественников изопрена может быть выделена в виде суммарной фракции с метилдигидропираном и подвергнута термокаталитическому разложению в изопрен, изобутилен и формальдегид.
Существенными отличительными признаками предлагаемого способа является выделение из продуктов синтеза триметилкарбинола, метилдигидропирана и предшественников изопрена азеотропной ректификацией с водой и рецикл в процесс триметилкарбинола и предшественников изопрена, очищенных от примесей нераздельнокипящих и близкокипящих углеводородов C8-С10 и кислородсодержащих соединений.
Предлагаемый способ позволяет выделить более концентрированную фракцию метилдигидропирана, что при ее разложении снижает энергозатраты, повышает производительность и улучшает качество изопрена.
Кроме того, возможность отделения примесей позволяет перерабатывать продукты разложения пирановой фракции совместно с продуктами синтеза изопрена.
Способ позволяет осуществлять непрерывный процесс с рециклом выделенных из продуктов синтеза триметилкарбинола и предшественников изопрена, избежать ухудшения показателей синтеза за счет рецикла и накопления примесей, исключить потери триметилкарбинола и предшественников изопрена и, как следствие, улучшить показатели процесса по расходу сырья и энергозатратам.
Промышленная осуществимость способа иллюстрируется примерами 1-5, пример 6 дан для сравнения.
Пример 1.
Принципиальная схема установки для осуществления способа согласно предлагаемому изобретению приведена на чертеже.
Установка включает реактор Р-1, сепаратор Е-2, холодильник Х-3, холодильник-конденсатор Х-4, емкость-отстойник Е-5, смеситель С-6, емкость-отстойник Е-7, колонну отгонки изобутилена К-8, колонну отгонки изопрена К-9, колонну отгонки фракции углеводородов и карбонильных соединений К-10, колонну отгонки фракции триметилкарбинола К-11, колонну отгонки фракции метилдигидропирана К-12, колонну отгонки фракции предшественников изопрена К-13, колонну отгонки органических продуктов из водного слоя синтеза К-14, технологический узел разложения пирановой фракции УРПФ.
В реактор Р-1, представляющий собой автоклав объемом 5 литров с мешалкой с герметичным электромагнитным приводом и термостатирующей рубашкой, загружают 4,5 литра 7%-ного водного раствора ортофосфорной кислоты, при включенном перемешивании в термостатирующую рубашку подают горячий теплоноситель и нагревают реактор до температуры 160°С.
В реактор подают 40%-ный формалин с расходом 256,5 г/час и концентрированный изобутилен с расходом 1747,2 г/час. Выходящий из реактора парожидкостный поток поступает в сепаратор Е-2, где разделяется на жидкую и паровую фазы.
Пары продуктов реакции и непревращенного сырья, выходящие из сепаратора Е-2, проходят холодильник-конденсатор Х-4, где охлаждаются и конденсируются, и поступают в емкость-отстойник Е-5, где расслаиваются на водный и органический слои.
Давление в реакционном узле (реактор, сепаратор, холодильник, отстойник) поддерживается на уровне 12-14 атм.
Поток жидких продуктов, представляющий собой смесь водного раствора ортофосфорной кислоты и высококипящих побочных продуктов, выводят из сепаратора Е-2 в количестве 1,6 л/час, охлаждают в холодильнике Х-3 и подают в смеситель С-6 на экстракцию органических продуктов. В качестве экстрагента в смеситель С-6 подают органический слой продуктов синтеза из емкости Е-5. Выходящая из смесителя С-6 смесь поступает в емкость-отстойник Е-7, где расслаивается на органический слой, содержащий проэкстрагированные высококипящие побочные продукты, и водный раствор ортофосфорной кислоты.
Очищенный от высококипящих органических продуктов водный раствор орто-фосфорной кислоты из емкости Е-7 насосом возвращают обратно в реактор Р-1.
Органический слой из емкости Е-7 подают на разделение. Сначала в ректификационной колонне К-8 выделяют изобутилен в количестве 1561,6 г/час, который рециркулируют в реактор Р-1, а затем в ректификационной колонне К-9 выделяют изопрен в количестве 177,4 г/час, который подают на очистку.
Кубовый продукт колонны К-9 в количестве 297,9 г/час, представляющий собой смесь побочных продуктов синтеза и содержащий 74,8% триметилкарбинола, 1,7% углеводородов C8-С10, 2,1% карбонильных соединений общей формулы С5Н10О, 4,0% метилдигидропирана, 0,3% диметилдиоксана, 1,0% изоамиленовых спиртов и 16,1% прочих продуктов, смешивают с 44,5 г/час воды и подают в блок ректификационных колонн К-10, К-11, К-12 и К-13 для разделения на фракции азеотропной ректификацией с водой. При осуществлении последовательной отгонки фракций в колонне К-10 отгоняют фракцию углеводородов C8-С10 и карбонильных соединений общей формулы С5Н10О в виде гетерогенного азеотропа с водой, имеющего температуру кипения Н.К. 78°С, в количестве 11,8 г/час, в колонне К-11 отгоняют фракцию триметилкарбинола в виде гомогенного азеотропа с водой, имеющего температуру кипения 79-81°С, в количестве 258,1 г/час, в колонне К-12 отгоняют фракцию метилдигидропирана в виде гетерогенного азеотропа с водой, имеющего температуру кипения 83-89°С, в количестве 13,4 г/час, в колонне К-13 отгоняют фракцию предшественников изопрена (смесь диметилдиоксана и изоамиленовых спиртов) в виде гетерогенного азеотропа с водой, имеющего температуру кипения 92-97°С, в количестве 4,9 г/час. Водные слои, образующиеся во флегмовых емкостях колонн К-10, К-12 и К-13 при конденсации гетерогенных азеотропов, объединяют в общий поток и направляют на смешение с потоком питания колонны К-10.
Водный слой конденсата продуктов синтеза из емкости Е-5 в необходимом количестве рециркулируют в реактор Р-1 для поддержания объема и кислотности реакционного водного слоя, а балансовое количество выводят в колонну К-14 для отгонки легкокипящих органических продуктов. В качестве дистиллата в колонне К-14 отбирают 22,5 г/час фракции, содержащей 85,9% триметилкарбинола, 13,8% воды и 0,3% других органических продуктов, а в качестве кубового продукта воду, которую выводят на очистку.
Фракцию побочных продуктов, отогнанную в колонне К-10, и кубовый остаток из колонны К-13 выводят на утилизацию.
Фракцию триметилкарбинола, отогнанную в колоннах К-11 и К-14, и фракцию предшественников изопрена, отогнанную в колонне К-13, рециркулируют в реактор Р-1.
Фракцию метилдигидропирана, отогнанную в колонне К-12, направляют на разложение в технологический узел УРПФ.
Разложение метилдигидропирана осуществляют путем контактирования его паров при температуре 400-450°С в присутствии водяного пара с алюмосиликатсодержащим катализатором, с последующим охлаждением и конденсацией продуктов разложения и разделением конденсата на водный и органический слои.
В реактор разложения подают 13,4 г/час пирановой фракции, выделенной в колонне К-12, и 40,0 г/час водяного пара и контактируют с алюмосиликатсодержащим катализатором К-97 при температуре 400-450°С. Выходящие из реактора пары продуктов разложения и воды охлаждают, конденсируют и в емкости-отстойнике расслаивают на водный и органический слои. Водный слой, содержащий 8,1% формальдегида, в количестве 43,2 г/час подают в реактор синтеза изопрена Р-1. Органический слой, содержащий 82,9% изопрена, 4,6% изобутилена и 12,5% других продуктов, в количестве 9,4 г/час подают в емкость Е-5 для последующей переработки совместно с органическим слоем синтеза изопрена.
В соответствии с балансом процесса выход изопрена составляет 76,3 мол.% по формальдегиду и 78,7 мол.% по изобутилену.
Процесс осуществляют в непрерывном режиме в течение 180 часов. Анализ состава потоков в пробах, отобранных после 48, 120 и 180 часов работы, показывает идентичные результаты, что свидетельствует об отсутствии накопления в продуктах синтеза каких-либо побочных продуктов.
Пример 2.
Процесс осуществляют как это описано в примере 1, за исключением того, что в колонне К-10 отгоняют широкую фракцию продуктов с температурой кипения водных азеотропов до 97°С, в колонне К-11 отгоняют фракцию углеводородов и карбонильных соединений с температурой кипения водных азеотропов Н.К. 78°С, в колонне К-12 отгоняют фракцию триметилкарбинола. Кубовый продукт колонны К-10 и дистиллат колонны К-11 направляют на утилизацию. Дистиллат колонны К-12 рециркулируют в реактор Р-1. Кубовый продукт колонны К-12, представляющий собой суммарную фракцию метилдигидропирана и предшественников изопрена, направляют в технологический узел УРПФ для разложения в изопрен и исходное сырье. Колонна К-13 из схемы исключена.
В соответствии с балансом процесса выход изопрена составляет 76,2 мол.% по формальдегиду и 78,9 мол.% по изобутилену.
Процесс осуществляют в непрерывном режиме в течение 180 часов. Анализ состава потоков в пробах, отобранных после 48, 120 и 180 часов работы, показывает идентичные результаты, что свидетельствует об отсутствии накопления в продуктах синтеза каких-либо побочных продуктов.
Пример 3.
Процесс осуществляют как это описано в примере 1, за исключением того, что подачу воды производят дробно в каждую колонну, с разбивкой общего количества по колоннам в следующем соотношении: в К-10 - 15%, в К-11 - 55%, в К-12 - 20% и в К-13 - 10%.
В соответствии с балансом процесса выход изопрена составляет 76,4 мол.% по формальдегиду и 78,8 мол.% по изобутилену.
Процесс осуществляют в непрерывном режиме в течение 180 часов. Анализ состава потоков в пробах, отобранных после 48, 120 и 180 часов работы, показывает идентичные результаты, что свидетельствует об отсутствии накопления в продуктах синтеза каких-либо побочных продуктов.
Пример 4.
Процесс осуществляют как это описано в примере 1, за исключением того, что в качестве источника формальдегида в реактор подают 4,4-диметил-1,3-диоксан в количестве 396,7 г/час, а вместо свежего изобутилена (185,6 г/час) в качестве сырья в процесс подают триметилкарбинол в количестве 245,2 г/час.
В соответствии с балансом процесса выход изопрена составляет 151,8 мол.% по диметилдиоксану (75,9% от теоретического) и 158,2 мол.% по триметилкарбинолу (79,1% от теоретического).
Процесс осуществляют в непрерывном режиме в течение 180 часов. Анализ состава потоков в пробах, отобранных после 48, 120 и 180 часов работы, показывает идентичные результаты, что свидетельствовало об отсутствии накопления в продуктах синтеза каких-либо побочных продуктов.
Пример 5.
Процесс осуществляют как это описано в примере 1, за исключением того, что в колонне К-10 отгоняют фракцию Н.К. 83,0°С без добавления воды, от которой в колонне К-11 с добавлением воды отгоняют фракцию Н.К. 77°С; к кубовому продукту колонны К-10 добавляют воду и последовательной азеотропной ректификацией в колоннах К-12 и К-13 выделяют соответственно фракцию метилдигидропирана и фракцию предшественников изопрена. Кубовый продукт колонны К-11 (фракцию триметилкарбинола) и фракцию предшественников изопрена рециркулируют в реактор Р-1.
В соответствии с балансом процесса выход изопрена составляет 76,1 мол.% по формальдегиду и 79,0 мол.% по изобутилену.
Процесс осуществляют в непрерывном режиме в течение 180 часов. Анализ состава потоков в пробах, отобранных после 48, 120 и 180 часов работы, показывает идентичные результаты, что свидетельствует об отсутствии накопления в продуктах синтеза каких-либо побочных продуктов.
Пример 6 (для сравнения).
Процесс осуществляют как это описано в примере 1, за исключением того, что ректификацию продуктов производят без добавления воды. При этом в соответствии с температурами кипения компонентов смеси в колонне К-10 выделяют фракцию Н.К. 78°С, в колонне К-11 - фракцию 79-83°С, в колонне К-12 - фракцию 84-119°С и в колонне К-13 - фракцию 120-140°С. Фракции 79-83°С (фракция триметилкарбинола) и 120-140°С (фракция предшественников изопрена) рециркулируют в реактор синтеза Р-1. Фракцию 84-119°С (фракция метилдигидропирана) подают на разложение в узел УРПФ. Фракции Н.К. 78°С и кубовый продукт К-13 (фракции побочных продуктов) выводят на утилизацию.
По мере осуществления процесса происходит увеличение количества отгоняемых фракций и снижение концентраций целевых продуктов.
Через 30 часов эксплуатации установки:
- количество фракции триметилкарбинола увеличивается в 1,7 раза, а содержание в ней триметилкарбинола снижается с 81,4% до 52,6%;
- количество фракции метилдигидропирана увеличивается в 3,0 раза, а содержание в ней метилдигидропирана снижается с 66,3% до 23,3%;
- количество фракции предшественников изопрена увеличивается в 2,3 раза, а содержание в ней предшественников изопрена снижается с 82,1% до 43,5%.
При дальнейшей работе установки количество отгоняемых фракций продолжает увеличиваться, и из-за ограниченных возможностей колонн по количеству отгоняемых фракций, с целью стабилизации режима работы и предотвращения дальнейшего накопления примесей в рециркулируемых фракциях, часть отгоняемых фракций отводится на утилизацию.
Через 90 часов работы установки в соответствии с балансом процесса выход изопрена составляет 71,2 мол.% по формальдегиду и 72,3 мол.% по изобутилену.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ИЗОПРЕНА | 2007 |
|
RU2332394C1 |
| СПОСОБ ВЫДЕЛЕНИЯ ИЗОПРЕНА ИЗ УГЛЕВОДОРОДНОГО СЛОЯ | 1976 |
|
SU687784A1 |
| СПОСОБ ВЫДЕЛЕНИЯ ИЗОПРЕНА | 1979 |
|
SU772074A1 |
| СПОСОБ ПРОИЗВОДСТВА ИЗОПРЕНА | 2020 |
|
RU2765441C2 |
| СПОСОБ ПОЛУЧЕНИЯ ИЗОПРЕНА | 1995 |
|
RU2099318C1 |
| СПОСОБ ПОЛУЧЕНИЯ ИЗОПРЕНА | 1995 |
|
RU2099319C1 |
| СПОСОБ ПОЛУЧЕНИЯ ИЗОПРЕНА | 2007 |
|
RU2330007C1 |
| СПОСОБ ПОЛУЧЕНИЯ ИЗОПРЕНА | 1997 |
|
RU2128637C1 |
| СПОСОБ ПОЛУЧЕНИЯ ИЗОПРЕНА | 2001 |
|
RU2197461C2 |
| СПОСОБ ПОЛУЧЕНИЯ ИЗОПРЕНА | 1999 |
|
RU2156234C1 |
Изобретение относится к способу получения изопрена путем жидкофазного взаимодействия формальдегида и изобутилена или веществ, являющихся их источниками, например 4,4-диметил-1,3-диоксана и триметилкарбинола, в присутствии водного раствора кислотного катализатора, осуществляемому при повышенной температуре и давлении с получением продуктов реакции и балансового количества воды в виде парового потока, с последующим охлаждением, конденсацией и разделением на водный и органический слои, с переработкой органического слоя, включающей выделение рециклового изобутилена, целевого изопрена, рецикловых триметилкарбинола и предшественников изопрена, фракции метилдигидропирана, фракции углеводородов C8-С10 и карбонильных соединений С5Н10О и высококипящего остатка, с переработкой водного слоя, включающей выделение органических продуктов, с выводом жидкого потока водного раствора катализатора на экстракцию, с последующим возвратом в зону синтеза, при этом выделение триметилкарбинола, метилдигидропирана и предшественников изопрена, фракции углеводородов C8-С10 и карбонильных соединений С5Н10О осуществляют путем азеотропной ректификации с добавлением воды в количестве, необходимом для образования азеотропа, с отгоняемым в колонне продуктом. Предлагаемый способ позволяет осуществить непрерывный процесс с рециклом выделенных из продуктов синтеза предшественников изопрена, избежать ухудшения показателей синтеза за счет рецикла и накопления примесей, исключить потери триметилкарбинола и предшественников изопрена и, как следствие, улучшить показатели процесса по расходу сырья и энергозатратам. 5 з.п. ф-лы, 1 ил.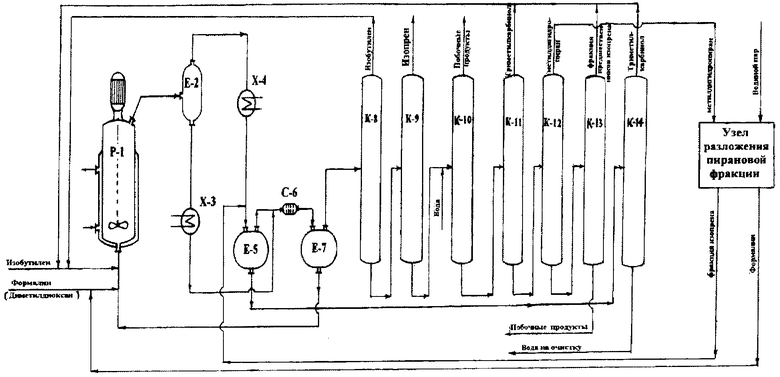
| КАТАЛИЗАТОР ДЛЯ РАСЩЕПЛЕНИЯ ВЫСОКОКИПЯЩИХ ПОБОЧНЫХ ПРОДУКТОВ СИНТЕЗА ИЗОПРЕНА | 2000 |
|
RU2167710C1 |
| СПОСОБ ПОЛУЧЕНИЯ ИЗОПРЕНА, ИЗОБУТИЛЕНА И ФОРМАЛЬДЕГИДА | 1997 |
|
RU2134679C1 |
| Приспособление к уборочным машинам для отделения початков от стеблей кукурузы | 1956 |
|
SU106323A1 |

