Перекрестные ссылки на родственные заявки
Данная патентная заявка претендует на приоритет патентных заявок Индии №183/СНЕ/2006 и 874/СНЕ/2006, зарегистрированных 6 февраля 2006 г. и 18 мая 2006 г. соответственно, которые целиком включены сюда в качестве ссылок, и временной патентной заявки США №60/804836, зарегистрированной 15 июня 2006 г., которая целиком включена сюда в качестве ссылки.
Уровень техники
Настоящее изобретение относится к способу получения гемцитабина гидрохлорида. Более конкретно, изобретение относится к способу, позволяющему получить гемцитабина гидрохлорид, не содержащий примесей, связанных с процессом его производства.
Гемцитабина гидрохлорид известен под химическим названием 2'-дезокси-2',2'-дифторцитидина моногидрохлорид (β-аномер) (здесь и далее обозначается принятым названием гемцитабина гидрохлорид) и имеет структурную формулу I
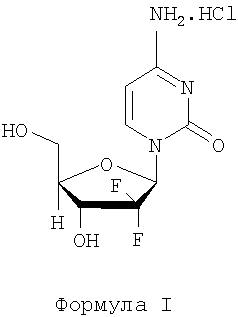
Гемцитабина гидрохлорид является нуклеозидным аналогом, проявляющим противоопухолевую активность, и доступен на рынке под торговой маркой GEMZAR® в форме препарата для инъекций. Флакон Gemzar содержит количество гемцитабина гидрохлорида, эквивалентное 200 мг или 1 г свободного основания.
Способы увеличения количества β-аномера известны специалистам. Например, патент США №5945547 раскрывает способ очистки гемцитабина гидрохлорида в отношении его аномерной чистоты. Способ включает растворение 1:1 α/β аномерной смеси в горячей воде с последующим прибавлением ацетона при нагревании до кипения с обратным холодильником, охлаждение раствора до температуры примерно от -10 до 50°С. Осадок гемцитабина гидрохлорида собирают и подвергают дальнейшей очистке путем повторения описанного выше процесса до получения гемцитабина гидрохлорида с чистотой примерно 99%.
Способы получения гемцитабина также хорошо известны специалистам. Например, патент США №4808614 раскрывает гемцитабина гидрохлорид, композиции, содержащие гемцитабина гидрохлорид, и их применение для лечения инфекций вируса герпеса. Он также описывает способ получения гемцитабина гидрохлорида, изображенный на схеме I.
Схема I:
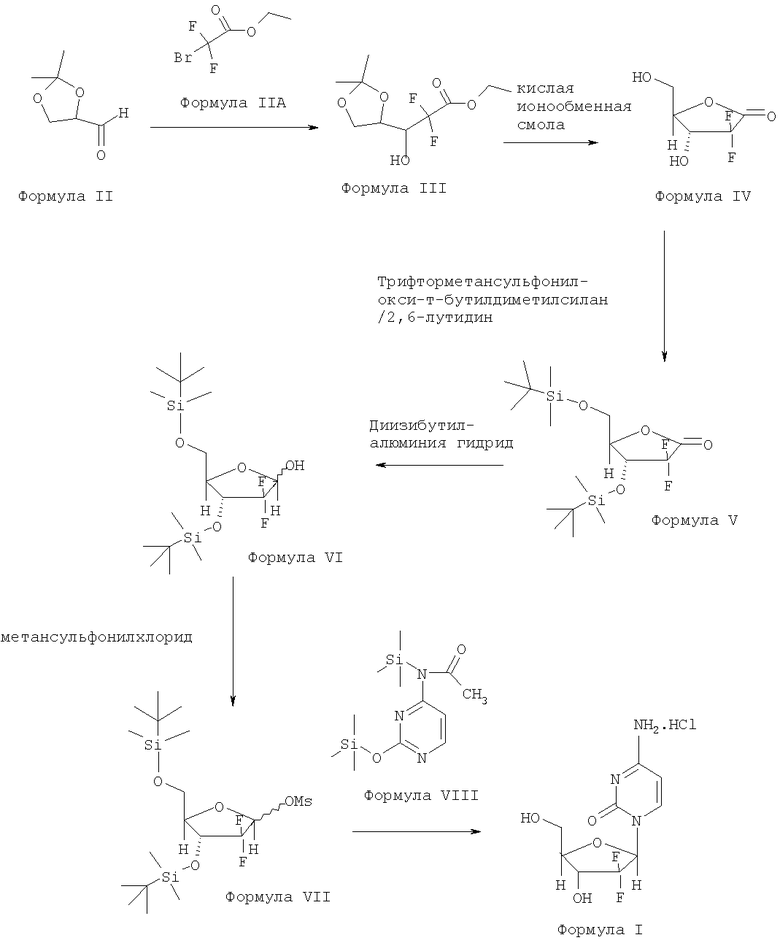
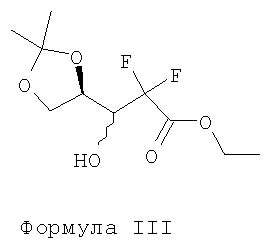
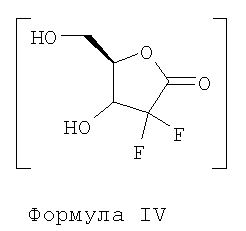
Вышеупомянутый способ раскрывает использование гидролитических реагентов, таких как слабокислые ионообменные смолы, для превращения соединения Формулы III в соединение Формулы IV.
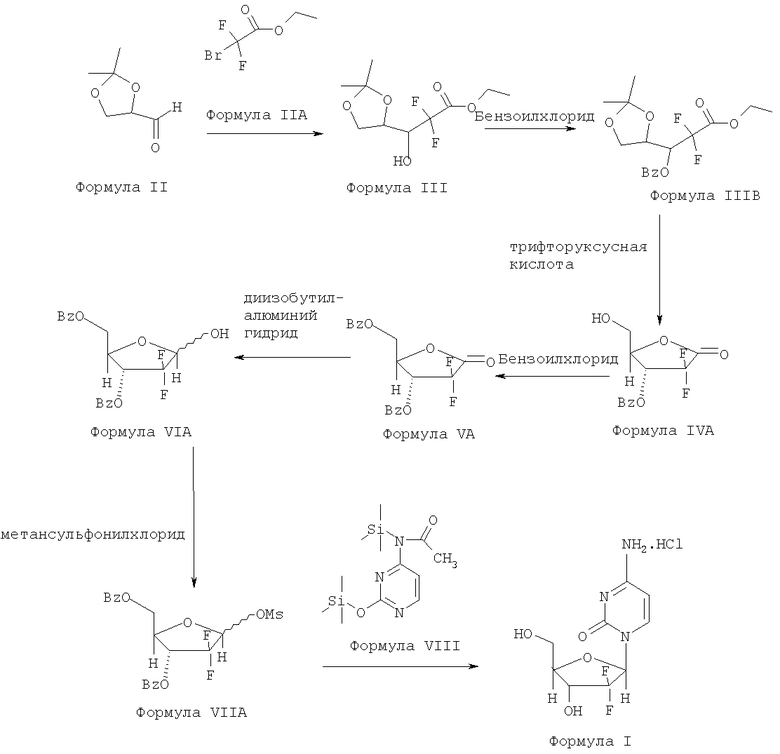
Патент США №5223608 раскрывает способ получения гемцитабина гидрохлорида путем использования гидролитических реагентов, таких как сильные кислоты, для получения 2-дезокси-2,2-дифтор-D-эритропентофураноз-1-улоза-3,5-дибензоата Формулы VA.
Этот способ представлен схемой II.
Схема II:
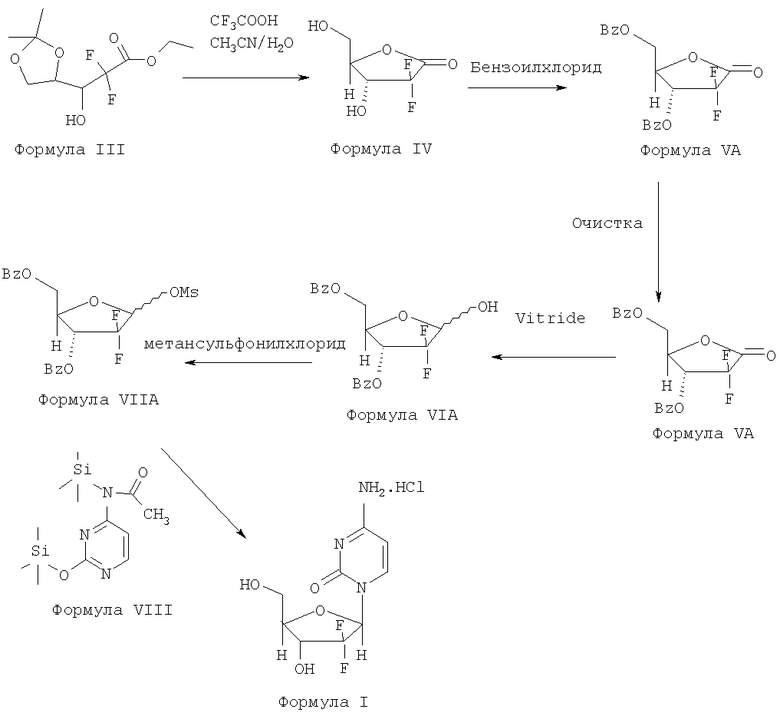
Публикация заявки РСТ №2005/095430 А1 раскрывает способ получения гемцитабина гидрохлорида. Эта заявка описывает использование гидролитических реагентов, таких как сильные кислоты, для получения 2-дезокси-2,2-дифтор-D-эритропентофураноз-1-улоза-3,5-дибензоата Формулы VA и также описывает очистку соединения Формулы VA. Эта заявка также описывает способ очистки гемцитабина гидрохлорида путем растворения 95% обогащенного β-аномера гемцитабина гидрохлорида в воде и выделения путем использования растворителей, таких как изопропиловый спирт или ацетонитрил или ацетон.
Способ представлен схемой III.
Схема III:
Это основные методы получения гемцитабина гидрохлорида. Указанные выше подходы используют слабокислые ионообменные смолы и сильные кислоты в качестве агентов гидролиза в момент образования незащищенного или защищенного лактона. Оба типа гидролизующих агентов имеют недостаток, связанный с образованием лактонового кольца. Могут образовываться нежелательные продукты реакции в виде примесей и лактон часто возвращается к своему прекурсору с открытой цепью из-за чувствительности к сильным кислотам и смолам.
Настоящее изобретение относится к способу получения промежуточных продуктов и усовершенствованию методики очистки с целью получения требуемого β-аномера гемцитабина гидрохлорида, по существу не содержащего α-аномера.
В соответствии с настоящим изобретением предлагается удобный способ получения гемцитабина гидрохлорида и его промежуточных продуктов с требуемой чистотой и выходом путем использования лучших методов синтеза, которые являются простыми, экологически безопасными, экономически эффективными, надежными и пригодными для использования в промышленных масштабах.
Сущность изобретения
Настоящее изобретение предлагает способ очистки гемцитабина гидрохлорида с целью получения гемцитабина гидрохлорида, обогащенного его β-аномером.
Согласно варианту осуществления способ очистки гемцитабина гидрохлорида, обогащенного его β-аномером, включает стадии:
a) получения раствора гемцитабина гидрохлорида в воде при соотношении воды и гемцитабина гидрохлорида от примерно 3:1 до примерно 12:1 (мас./об.);
b) обработки раствора активированным углем в количестве от примерно 0,1 до 10 вес.% от количества гемцитабина гидрохлорида в растворе;
c) удаления активированного угля из раствора с образованием фильтрата;
d) повышения концентрации гемцитабина гидрохлорида в фильтрате до соотношения фильтрата и гемцитабина гидрохлорида от примерно 1:1 до примерно 1:5 (мас./об.), эффективного для осаждения гемцитабина гидрохлорида, и
e) выделения осажденного гемцитабина гидрохлорида.
В другом аспекте настоящее изобретение относится к способу получения гемцитабина гидрохлорида и его промежуточных продуктов.
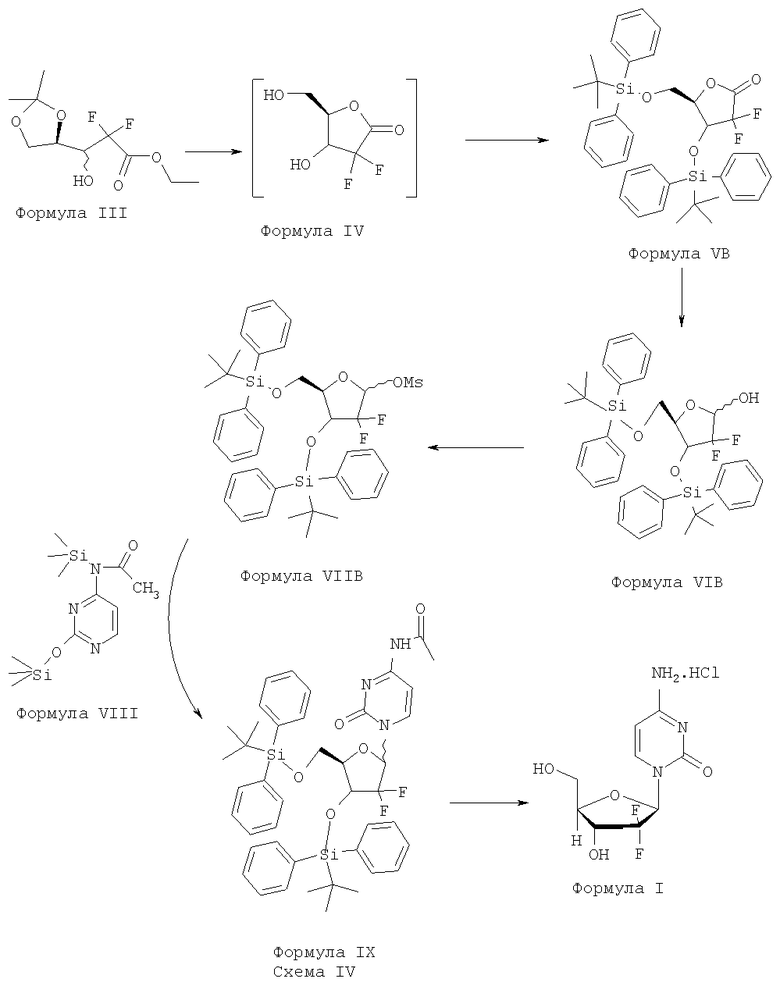
Согласно варианту осуществления способ получения гемцитабина гидрохлорида Формулы I включает:
i) превращение соединения Формулы III в соединение Формулы IV с использованием реагентов дециклизации в присутствии пригодного органического растворителя с последующей азеотропной перегонкой;
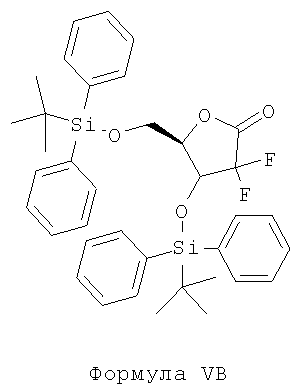
ii) защиту гидроксильных групп соединения Формулы IV с помощью пригодных реагентов защиты гидроксильных групп в присутствии пригодного органического растворителя при желательной температуре с образованием соединения Формулы VB;
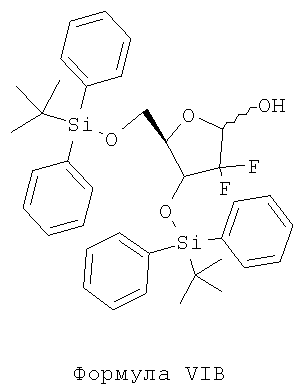
iii) восстановление соединения Формулы VB с помощью пригодного восстановителя в присутствии пригодного органического растворителя с образованием соединения Формулы VIB;
iv) защиту соединения Формулы VIB с помощью алкил- или арилсульфонилхлоридов, в присутствии пригодного основания и пригодного органического растворителя с образованием соединения Формулы VIIB;
v) конденсацию соединения Формулы VIIB с соединением Формулы VIII в присутствии пригодного органического растворителя и пригодных оснований с образованием соединения Формулы IX и
vi) удаление защитных групп соединения Формулы IX с помощью пригодного реагента с последующим проведением реакции с хлористоводородной кислотой в присутствии пригодного органического растворителя с образованием гемцитабина гидрохлорида Формулы I.
Еще один аспект настоящего изобретения предусматривает кристаллический гемцитабина гидрохлорид, охарактеризованный его регтгеновской порошковой дифрактограммой (РПД), кривой диффренциальной сканирующей калориметрии (ДСК) и/или спектром инфракрасного поглощения (ИК).
В еще одном аспекте изобретение предусматривает гемцитабина гидрохлорид, по существу не содержащий остаточных растворителей.
Краткое описание чертежей
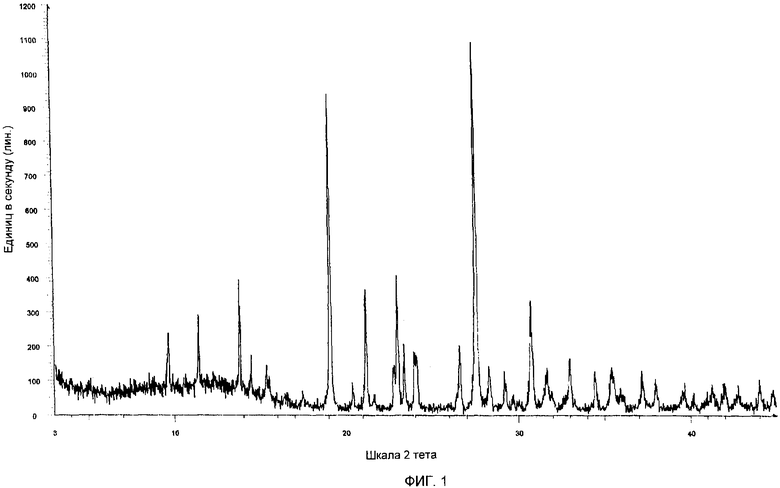
Фиг.1 представляет собой рентгеновскую порошковую дифрактограмму гемцитабина гидрохлорида, полученного, как описано в примере 8.
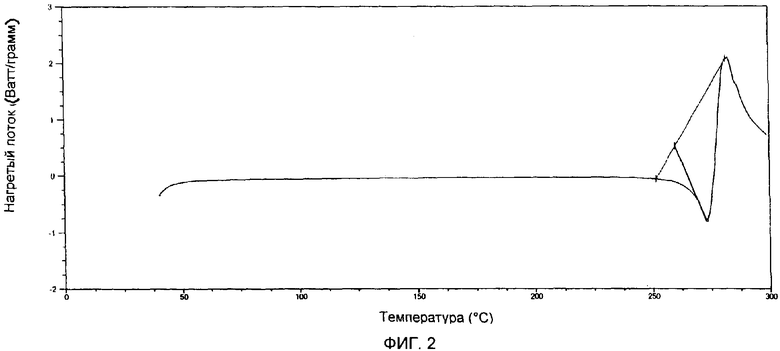
Фиг.2 представляет собой результаты дифференциальной сканирующей калориметрии образца гемцитабина гидрохлорида, полученного в соответствии с примером 8.
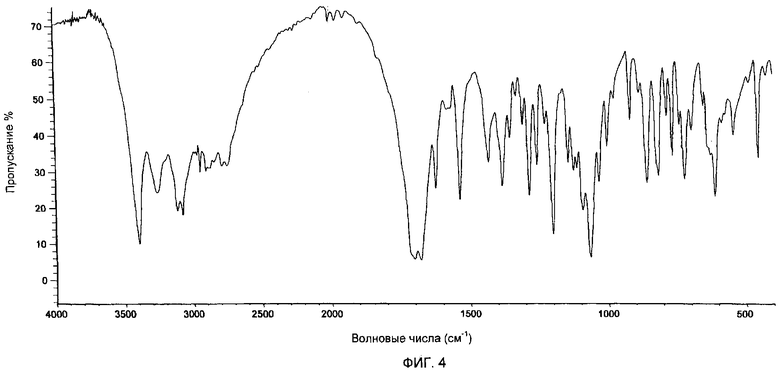
Фиг.3 представляет собой ИК-спектр образца гемцитабина гидрохлорида, полученного в соответствии с Примером 8.
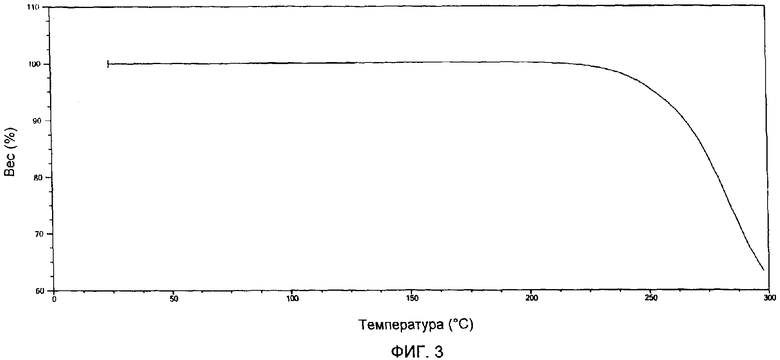
Фиг.4 представляет собой термогравиметрическую кривую образца гемцитабина гидрохлорида, полученного в соответствии с Примером 8.
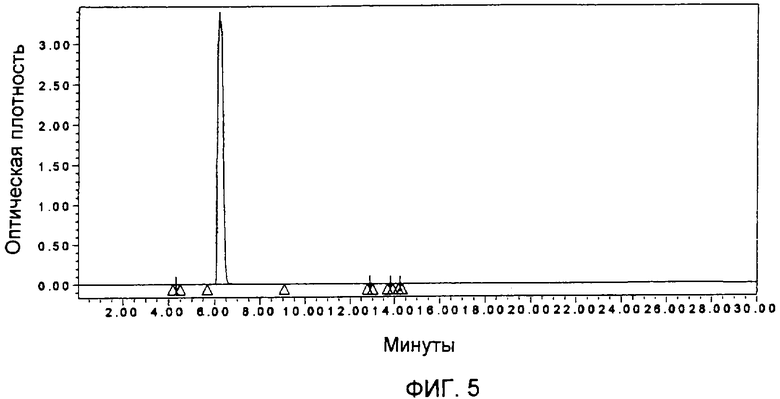
Фиг.5 представляет собой ВЭЖХ-хроматограмму (хроматограмму, полученную методом высокоэффективной жидкостной хроматографии) образца гемцитабина гидрохлорида, полученного в соответствии с Примером 8.
Подробное описание изобретения
Патент США №5945547 описывает способ, использующий ацетон/воду. Авторы настоящего изобретения неожиданно обнаружили, что использование воды без ацетона приводит к улучшению аномерной чистоты.
Таким образом, настоящее изобретение предусматривает способ очистки гемцитабина гидрохлорида с образованием гемцитабина гидрохлорида, обогащенного его β-аномером.
Согласно варианту осуществления способа очистки гемцитабина гидрохлорида, обогащенного его β-аномером:
a) берут раствор гемцитабина гидрохлорида в воде при соотношении воды и гемцитабина гидрохлорида от примерно 3:1 до примерно 12:1 (мас./об.);
b) обрабатывают раствор активированным углем в количестве от примерно 0,1 до 10 вес.% от количества гемцитабина гидрохлорида в растворе;
c) удаляют активированный уголь из раствора с образованием фильтрата;
d) повышают концентрацию гемцитабина гидрохлорида в фильтрате до соотношения фильтрата и гемцитабина гидрохлорида от примерно 1:1 до примерно 1:5 (мас./об.), эффективного для осаждения гемцитабина гидрохлорида, и
e) выделяют осажденный гемцитабина гидрохлорид.
Стадия (а) включает обеспечение раствора гемцитабина гидрохлорида в растворителе, состоящем по существу из воды.
Раствор гемцитабина гидрохлорида может быть получен путем его растворения в воде. Для получения раствора пригодна любая форма гемцитабина гидрохлорида, такая как любая кристаллическая или аморфная форма гемцитабина гидрохлорида.
Гемцитабина гидрохлорид, предназначенный для растворения, может быть получен методами, известными специалистам, или описанными выше способами.
Концентрация смеси аномерных солей в воде не является критической, при условии, что используется количество воды, достаточное для обеспечения полного растворения. Во избежание избыточных потерь продукта в процессе кристаллизации и выделения поддерживается небольшое количество используемой воды. Количество воды, используемой для выделения бета-аномера, составляет от примерно 3 до примерно 12-кратного (мас./об.) по отношению к гемцитабина гидрохлориду.
Раствор может быть приготовлен при температуре в интервале от примерно 25 до 100°С. В зависимости от взятого количества растворителя растворение может быть проведено в интервале температур 25-100°С или растворение может потребовать использования повышенных температур в диапазоне примерно от 50 до 100°С.
Раствор может быть необязательно обработан активированным углем для улучшения окраски соединения с последующей фильтрацией через среду, такую как через слой кальцинированной в присутствии спекающего агента диатомовой земли (Hyflow) для удаления углерода.
Предпочтительное количество углерода угля, используемого для выделения бета-аномера с улучшенной окраской, составляет примерно от 0,1 до примерно 10 мас./мас. углерода угля на грамм смеси α/β аномеров.
Обработка углем может проводиться при температуре растворения или после охлаждения раствора до более низкой температуры.
Раствор может быть необязательно профильтрован путем пропускания через бумагу, стекловолокно или другой мембранный материал или осветляющее средство, такое как целит. В зависимости от используемого оборудования, концентрации и температуры раствора фильтрующий аппарат может нуждаться в предварительном подогреве во избежание преждевременной кристаллизации.
Концентрация раствора может составлять от примерно 1 г/мл до примерно 20 г/мл растворителя или в интервале от 1 г/мл до 5 г/мл.
Стадия (b) включает увеличение концентрации гемцитабина гидрохлорида в указанном растворе для того, чтобы вызвать осаждение указанного β-аномера.
Концентрирование может осуществляться путем испарения, перегонки при атмосферном давлении или перегонки под вакуумом.
Перегонка растворителя может проводиться под вакуумом от примерно 100 мм рт.ст. до примерно 600 мм рт.ст. при температуре от примерно 40°С до примерно 70°С. Могут быть использованы любые температуры и значения вакуума, при условии, что концентрирование происходит без увеличения уровней содержания примесей.
Концентрирование раствора может производиться до состояния, в котором начинается выпадение осадка гемцитабина гидрохлорида из раствора с превращением раствора в суспензию. В общем, концентрирование прекращают, когда отношение растворителя к гемцитабину гидрохлориду становится равным от примерно 1:1 до примерно 1:5.
Реакционная масса может далее выдерживаться при температуре ниже температуры концентрирования, например ниже температуры от примерно 40°С до примерно 45°С, в течение периода времени, необходимого для более полного выделения продукта. Точная температура охлаждения и время, необходимое для полной кристаллизации, могут быть легко определены специалистом в данной области техники и будут также зависеть от таких параметров, как концентрация и температура раствора или суспензии.
Стадия (с) включает выделение указанного β-аномера.
Выделение твердого вещества может быть проведено обычными методами, такими как фильтрация, декантирование, центрифугирование и т.п., или путем фильтрации в инертной атмосфере с использованием таких газов, как, например, азот и т.п.
Влажный фильтровальный осадок, полученный на стадии (с), может быть необязательно дополнительно высушен. Высушивание может быть удобно проведено в лотковой сушилке, вакуумной печи, сушильном шкафу, сушилке с псевдоожиженным слоем, вращающейся распылительной сушилке, распылительной сушилке и т.п. Высушивание может проводиться при температуре от примерно 35°С до примерно 70°С. Высушивание может проводиться в течение любого желательного периода времени от примерно 1 до 20 часов.
В конкретном варианте осуществления изобретения описанный выше способ по изобретению может быть адаптирован для использования в качестве основы непрерывного процесса кристаллизации. Чистоту продукта, полученного на стадии (с), проверяют для определения процентного содержания примесей альфа-аномера. Если содержание примесей не снизилось до требуемого уровня ниже 0,1% по результатам анализа методом ВЭЖХ, то стадии (а)-(с) повторяют для влажного материала, полученного на стадии (с). Если на стадии (с) достигается требуемая чистота, то цикл останавливают.
Таким образом, создается цикл операций, который может повторяться бесконечно, что позволяет адаптировать способ по изобретению к непрерывному процессу с очевидными сопутствующими преимуществами при использовании в коммерческих масштабах.
Очищенный гемцитабина гидрохлорид, полученный выше, содержит менее 0,1% или менее 0,01% любой из примесей цитозина и α-аномера.
Гемцитабина гидрохлорид, полученный описанным выше способом, анализировали методом ВЭЖХ в соответствии с методикой, описанной в Фармакопее США (USP) 29; NF 24; 2006; page No.990-991, как указано в таблице.
В одном аспекте настоящее изобретение предусматривает способ получения гемцитабина гидрохлорида и его промежуточных продуктов.
Согласно варианту осуществления способ получения гемцитабина гидрохлорида и его промежуточных продуктов включает стадии:
i) превращения соединения Формулы III в соединение Формулы IV с использованием реагентов дециклизации в присутствии пригодного органического растворителя с последующей азеотропной перегонкой;
ii) защиты гидроксильных групп соединения Формулы IV с помощью пригодных реагентов защиты гидроксильных групп в присутствии пригодного органического растворителя при желательной температуре с образованием соединения Формулы VB;
iii) восстановления соединения Формулы VB с помощью пригодного восстановителя в присутствии пригодного органического растворителя с образованием соединения Формулы VIB;
iv) защиты соединения Формулы VIB с использованием алкил- или арилсульфонилхлоридов в присутствии пригодного основания и пригодного органического растворителя с образованием соединения Формулы VIIB;
v) конденсации соединения Формулы VIIB с соединением Формулы VIII в присутствии пригодного органического растворителя и пригодных оснований с образованием соединения Формулы IX и
vi) удаления защитных групп соединения Формулы IX с использованием пригодного реагента с последующим проведением реакции с хлористоводородной кислотой в присутствии пригодного органического растворителя с образованием гемцитабина гидрохлорида Формулы I.
Стадия i) включает превращение соединения Формулы III в соединение Формулы IV с использованием реагентов дециклизации в присутствии пригодного органического растворителя с последующей азеотропной перегонкой.
Пригодный температурный интервал для проведения реакции составляет от примерно 20°С до примерно 70°С.
Дополнительно реакционная масса подвергается азеотропной перегонке после прибавления йода и спиртового растворителя. Перегонка может быть проведена при температуре от примерно 50°С до примерно 150°С.
Пригодные растворители, которые могут быть использованы для реакции, включают, без ограничений, спиртовые растворители, такие как метанол, этанол, н-пропанол, изопропанол, н-бутанол и т.п.; углеводороды, такие как н-гексан, циклогексан, н-гептан, толуол, хлорбензол, 1,2-дихлорбензол, ксилолы и т.п.
Стадия ii) включает защиту гидроксильных групп соединения Формулы IV с помощью пригодных реагентов защиты гидроксильных групп в присутствии пригодного органического растворителя при желательной температуре с образованием соединения Формулы VB.
Согласно варианту осуществления защитная группа, используемая для защиты гидроксильной группы соединения Формулы IV, является трет-бутилдифенилсилилхлоридом, с образованием соединения Формулы VB.
Пригодные защитные группы гидроксилов, которые могут быть использованы в приведенной выше реакции, включают, без ограничений, силильные гидроксизащитные группы, такие как трет-бутилдифенилсилил, триметилсилилхлорид, изопропилдиметилсилил, метилдиизопропилсилил, триизопропилсилил и т.п., формил, 2-хлорацетил, бензил, дифенилметил, трифенилметил, 4-нитробензил, феноксикарбонил, т-бутил, метоксиметил, фенилоксиацетил, изобутирил, этоксикарбонил, бензилоксикарбонил и т.п.
Пригодные основания, которые могут быть использованы, включают, без ограничений, органические основания, такие как пиридин, триэтиламин, имидазол, 2,6-лутидин, 2,3-лутидин, 3,5-лутидин и т.п.
Пригодные растворители, которые могут быть использованы в описанной выше реакции, включают, без ограничений, простые эфиры, такие как тетрагидрофуран, 1,4-диоксан, диэтиловый эфир и 1,2-диметоксиэтан и т.п.; углеводороды, такие как н-гексан, циклогексан, н-гептан, толуол, хлорбензол, 1,2-дихлорбензол, ксилолы и т.п.; апротонные полярные растворители, такие как N,N-диметилформамид (ДМФ), диметилсульфоксид, диметилацетамид, ацетонитрил и т.п.; кетоны, такие как ацетон, метилизобутилкетон, метил-трет-бутилкетон и т.п., или их смеси.
Пригодные температуры для проведения реакции могут находиться в интервале от примерно 5 до 50°С.
Стадия iii) включает восстановление соединения Формулы VB с помощью пригодного восстановителя в присутствии пригодного органического растворителя с образованием соединения Формулы VIB.
Пригодные восстановители, которые могут быть использованы, включают, без ограничений, натрий-бис(2-метоксиэтокси)алюминийгидрид (Vitride), борогидрид натрия (NaBH4), гидрид лития-алюминия (LiAIH4), диизобутилалюминийгидрид (DIBAL-H).
Пригодные растворители, которые могут быть использованы для указанной выше реакции, включают, без ограничений, спирты, такие как метанол, этанол, н-пропанол, изопропиловый спирт, н-бутанол и т.п.; простые эфиры, такие как тетрагидрофуран, 1,4-диоксан, диэтиловый эфир и 1,2-диметоксиэтан и т.п.; углеводороды, такие как н-гексан, циклогексан, н-гептан, толуол, хлорбензол, 1,2-дихлорбензол, ксилолы и т.п., или их смеси.
Пригодные для проведения реакции температуры могут находиться в интервале от примерно -50 до 100°С.
Стадия iv) предусматривает защиту соединения Формулы VI В с использованием алкил- или арилсульфонилхлоридов в присутствии пригодного основания и пригодного органического растворителя с образованием соединения Формулы VIIB.
Согласно варианту осуществления настоящего изобретения защитная группа, используемая для защиты гидроксильной группы соединения Формулы VIB, представляет собой метансульфонилхлорид, с образованием соединения Формулы VIIB.
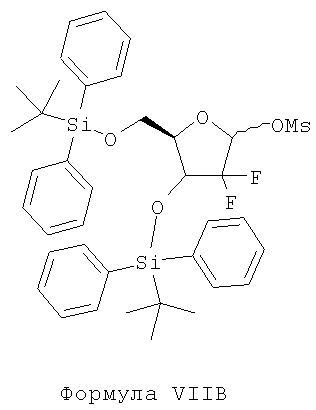
Пригодные защитные группы, которые могут быть использованы для защиты гидроксильных групп, включают, без ограничений, алкил- и арилсульфонилхлориды, такие как метансульфонилхлорид, бензолсульфонилхлорид и т.п.
Пригодные растворители, которые могут быть использованы для вышеуказанной реакции, включают, без ограничений, простые эфиры, такие как тетрагидрофуран, 1,4-диоксан, диэтиловый эфир и 1,2-диметоксиэтан и т.п.; углеводороды, такие как н-гексан, циклогексан, н-гептан, толуол, хлорбензол, 1,2-дихлорбензол, ксилолы и т.п.; апротонные полярные растворители, такие как N,N-диметилформамид (ДМФ), диметилсульфоксид, диметилацетамид, ацетонитрил и т.п.; сложные эфиры, такие как этилацетат, изопропилацетат и т.п.; кетоны, такие как ацетон, метилизобутилкетон, метил-трет-бутилкетон и т.п., или их смеси.
Пригодные температуры для проведения реакции могут находиться в интервале от примерно 0 до 50°С.
Стадия v) включает конденсацию соединения Формулы VII В с соединением Формулы VIII в присутствии пригодного органического растворителя и пригодного основания с образованием соединения Формулы IX.
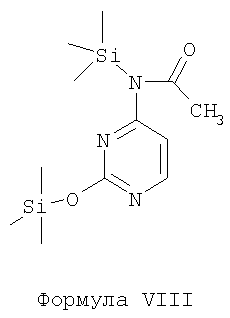
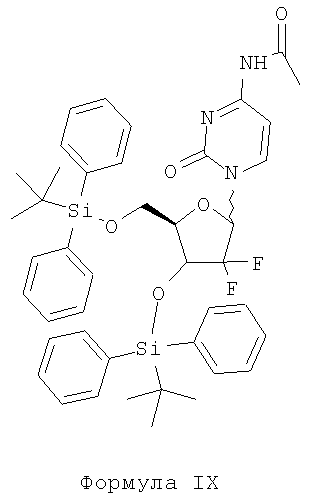
Пригодные растворители, которые могут быть использованы в описанной выше реакции, включают, без ограничений, простые эфиры, такие как тетрагидрофуран, 1,4-диоксан, диэтиловый эфир, 1,2-диметоксиэтан и т.п.; углеводороды, такие как н-гексан, циклогексан, н-гептан, толуол, хлорбензол, 1,2-дихлорбензол, ксилолы и т.п.; сложные эфиры, такие как этилацетат, изопропилацетат и т.п.; кетоны, такие как ацетон, метилизобутилкетон, метил-трет-бутилкетон и т.п., или их смеси.
Пригодные основания, которые могут быть использованы, включают, без ограничений, органические основания, такие как пиридин, триэтиламин, гексаметилдисилазан, триметилсилилтрифлат и т.п.
Пригодные температуры для проведения реакции могут находиться в интервале от примерно 20 до 100°С.
Стадия vi) включает удаление защитных групп соединения Формулы IX с использованием пригодного реагента с последующим проведением реакции с хлористоводородной кислотой в присутствии пригодного органического растворителя с образованием гемцитабина гидрохлорида Формулы I.
Пригодные реагенты для удаления защитных групп, которые могут быть использованы, могут быть выбраны из трет-бутиламмонийфторида, аммиака, ацетилхлорида, разбавленной хлористоводородной кислоты и т.п.
Растворители, используемые в описанных выше реакциях, включают, без ограничений, нерастворители, такие как вода; спирты, такие как метанол, этанол, этанола гидрохлорид, н-пропанол, изопропанол, изопропиловый спирт, н-бутанол и т.п.; галогенированные растворители, такие как дихлорметан, дихлорэтан, хлороформ и т.п.; простые эфиры, такие как тетрагидрофуран, 1,4-диоксан, диэтиловый эфир и 1,2-диметоксиэтан и т.п.; углеводороды, такие как н-гексан, циклогексан, н-гептан, толуол, хлорбензол, 1,2-дихлорбензол, ксилолы и т.п.; апротонные полярные растворители, такие как N,N-диметилформамид (ДМФ), диметилсульфоксид, диметилацетамид, ацетонитрил и т.п.; сложные эфиры, такие как этилацетат, изопропилацетат и т.п.; кетоны, такие как ацетон, метилизобутилкетон, метил-трет-бутилкетон и т.п., или их смеси.
Необязательно, одна или несколько из последовательных стадий (i)-(iv) проводятся без выделения соединений промежуточных продуктов. В одном варианте осуществления изобретения стадия (i) проводится без выделения промежуточного продукта, с последующим выделением соединения Формулы VB.
Пригодные температуры для проведения реакции могут находиться в интервале от примерно -10 до 70°С.
Необязательно, влажный фильтровальный осадок, полученный на разных стадиях процесса, может быть затем высушен. Высушивание может удобно проводиться в лотковой сушилке, вакуумной печи, сушильном шкуфу, сушилке с псевдоожиженным слоем, вращающейся распылительной сушилке, распылительной сушилке и т.п. Высушивание может быть проведено при температуре от примерно 35°С до примерно 70°С. Высушивание может быть проведено в течение любого желательного периода времени от примерно 1 до 20 часов.
Процесс в целом может быть представлен схемой IV:
Еще один аспект настоящего изобретения предусматривает кристаллический гемцитабина гидрохлорид, охарактеризованный его рентгеновской порошковой дифрактограммой (РПД), кривой диффренциальной сканирующей калориметрии (ДСК) и/или спектром инфракрасного поглощения (ИК).
Кристаллический гемцитабина гидрохлорид, полученный по настоящему изобретению, характеризуется его РПД-рентгенограммой. Все данные РПД, описанные тут, были получены с использованием Cu Кα излучения, имеющего длину волны 1,541 Å, и были получены с помощью рентгеновского дифрактометра Bruker Axe D8 Advance Powder X-ray Diffractometer.
Кристаллический гемцитабина гидрохлорид характеризуется дифрактограммой РПД, имеющей характеристические пики при значениях 2θ примерно 9.57, 11.35, 13.74, 14.38, 15.29, 19.09, 20.36, 21.15, 22.755, 22.98, 23.36, 24.02, 26.58, 27.56, 28.29, 29.19, 30.74, 31.66, 34.45, 35.42, 37.1, 37.96, 41.99, 44.023±0,2 градуса.
Анализ методом дифференциальной сканирующей калориметрии был проведен на приборе модели DSC Q1000 фирмы ТА Instruments при скорости нагрева 5°С/минуту с модуляцией времени, равной 60 секундам, и модуляцией температуры ±1°С. Начальная температура составляла 0°С и конечная температура 200°С.
Кристаллический гемцитабина гидрохлорид имеет характеристическую кривую диффренциальной сканирующей калориметрии, по существу совпадающую с приведенной на фиг.2 и имеет эндотермический пик при 259-274°С.
Инфракрасные (ИК) спектры гемцитабина гидрохлорида были записаны с помощью спектрофотометра модели Perkin Elmer System Spectrum 1, в диапазоне от 450 до 4000 см-1, с разрешением 4 см-1, в таблетке бромида калия, при концентрации исследуемого соединения 1 мас.%.
Кристаллический гемцитабина гидрохлорид характеризуется ИК-спектром, имеющим характеристические пики при примерно 3392, 3259, 3117, 3078, 1679, 1535, 1283, 1199, 1065, 856, 814±1 см-1.
В еще одном аспекте изобретение предусматривает гемцитабина гидрохлорид, по существу не содержащий остаточных растворителей.
Гемцитабина гидрохлорид, полученный с использованием способа по настоящему изобретению, имеет величину содержания остаточного растворителя, соответствующую пределам, установленным нормативами Международной конференции по гармонизации технических требований к регистрации фармацевтических средств, предназначенных для использования людьми (International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use, "ICH"). Нормативные значения уровней растворителя зависят от типа растворителя, но составляют не выше примерно 5000 млн-1, или примерно 4000 млн-1, или примерно 3000 млн-1.
Гемцитабина гидрохлорид, полученный по данному изобретению, содержит менее чем примерно 100 млн-1 или менее чем примерно 500 млн-1 ацетона, менее чем примерно 100 млн-1 или менее чем примерно 500 млн-1 изопропанола, менее чем примерно 100 млн-1 или менее чем примерно 500 млн-1 дихлорметана.
Некоторые специфические аспекты и варианты осуществления настоящего изобретения будут объяснены более подробно со ссылкой на следующие примеры, которые приведены только для иллюстрации и не должны рассматриваться как каким-либо образом огрничивающие объем изобретения.
ПРИМЕРЫ
Пример 1
Способ получения 3,5-бис(трет-бутилдифенилсилилокси)-2-дезокси-2,2-дифтор-1-оксорибозы (Формула VB)
Загружают 100,3 г этил-2,2-дифтор-3-гидрокси-3-(2,2-диметилдиоксолан-4-ил)пропионата Формулы III, 2000 мл метанола и 10,02 г йода в чистую и сухую круглодонную колбу и перемешивают в течение ночи. Раствор 22,5 г тиосульфата натрия в 225 мл воды прибавляют к указанной выше реакционной массе при температуре примерно 25-35°С в течение примерно 30 минут с последующей перегонкой 1000 мл метанола из реакционной смеси при примерно 80-95°С. Одновременно 500 мл толуола прибавляют к полученной реакционной массе и 500 мл растворителя отгоняют под атмосферным давлением при 95°С. Описанную выше стадию повторяют 5 раз при 120°С и применяют вакуум от 650 до 750 мм для полного удаления толуола, получая соединение 2-дезокси-2,2-дифтор-1-оксорибозу Формулы IV.
Полученный остаток растворяют в 1325 мл N,N-диметилформамида (ДМФ) под атмосферой азота при примерно 25-35°С. Загружают 80,5 г имидазола и 303 мл трет-бутилдифенилсилилхлорида (TBDPSi-CI) в полученную, как указано выше, реакционную смесь при 25-35°С, а затем перемешивают при температуре примерно 25-35°С в течение ночи. Проверяют полноту протекания реакции с помощью тонкослойной хроматографии и охлаждают до примерно 10-15°С. Прибавляют 2600 мл воды при 10°С с одновременным перемешиванием с последующей экстракцией 2×500 мл дихлорметана. Органический и водный слои разделяют, после чего органический слой промывают 500 мл воды, а затем органический слой полностью перегоняют при 50°С под вакуумом. Полученный сырой продукт очищают хроматографией на колонке с использованием системы растворителей петролейного эфира и этилацетата, получая 100 г чистого названного в заголовке соединения.
МС: 662,4 (M+NH4) атомных единиц массы (а.m.u.). 1H ЯМР: значения δ 80,8 млн-1 (9Н), 1,1 млн-1 (9Н), 4,5 млн-1 (2Н), 3,4 млн-1 (1Н), 3,7 млн-1 (9Н).
Пример 2
Способ получения 3,5-бис(трет-бутилдифенилсилилокси)-2-дезокси-2,2-дифторрибозы (формула VIB)
Загружают в круглодонную колбу 100 г 3,5-бис(трет-бутилдифенилсилилокси)-2-дезокси-2,2-дифтор-1-оксорибозы Формулы VB и 1000 мл тетрагидрофурана (ТГФ) под атмосферой азота при примерно 25-35°С с одновременным перемешиванием. Реакционную смесь охлаждают до температуры от -45 до -50°С, после чего прибавляют 75,4 мл натрий-бис(2-метоксиэтокси)алюминийгидрида (Vitride) в течение примерно 5-10 минут с последующим перемешиванием под давлением в течение примерно 10-15 минут. Проверяют полноту протекания реакции с помощью тонкослойной хроматографии. После завершения реакции к полученной, как указано выше, реакционной массе прибавляют 200 мл насыщенного раствора хлорида аммония при примерно -45°С с последующим разделением двух слоев. Полученный водный слой экстрагируют 3×500 мл этилацетата, после чего разделяют органический и водный слои. Органический слой полностью перегоняют под вакуумом, получая 103,5 г указанного в заголовке соединения.
МС: 664,4 (M+NH4) а.е.м. (a.m.u.). 1H ЯМР: значения δ 0,9 млн-1 (9Н), 1,1 (9Н), 5,1 млн-1 (1Н).
Пример 3
Способ получения 3,5-бис(трет-бутилдифенилсилилокси)-1-метансульфонилокси-2-дезокси-2,2-дифторрибозы (формула VIIB)
Загружают в круглодонную колбу 103 г 3,5-бис(трет-бутилдифенилсилилокси)-2-дезокси-2,2-дифторрибозы Формулы VIB, 1030 мл дихлорметана и 55,5 мл триэтиламина под атмосферой азота с одновременным перемешиванием. Реакционную массу охлаждают до 0-5°С, после чего к указанной выше реакционной массе прибавляют 18,6 мл метансульфонилхлорида при 0°С в течение 10 минут. Реакционной массе дают нагреться до температуры 25-35°С и перемешивают под давлением в течение примерно 3-4 часов до израсходования исходного материала. Полноту протекания реакции проверяют с помощью тонкослойной хроматографии. К полученной, как указано выше, реакционной массе прибавляют 300 мл воды и два слоя разделяют, после чего водный слой снова экстрагируют 300 мл дихлорметана. Полученные органические слои полностью перегоняют под вакуумом, получая 111,0 г указанного в заголовке соединения.
МС: 742,2 (M+NH4) а.е.м. (a.m.u.). 1H ЯМР: значения δ 5,9 млн-1 (1Н), 0,8 (9Н), 1,1 млн-1 (9Н), 3,1 млн-1 (3Н).
Пример 4
Способ получения 1-[2'-дезокси-2',2'-дифтор-3',5'-рибофураноза-3,5-бис(трет-бутилдифенилсилилокси)-N-ацетилцитозина (формула IX)
Загружают в круглодонную колбу 92,98 г н-ацетилцитозина и 1925 мл толуола под атмосферой азота. Прибавляют в указанную колбу также 204,6 мл гексаметилдисилазана (HMDS) в течение примерно 5 минут при 25-35°С с последующей перегонкой 20% растворителя под вакуумом при примерно 65°С и дают реакционной массе остыть до примерно 50°С. К указанной выше реакционной смеси прибавляют 41 мл триметилсилилхлорида, после чего реакционную массу нагревают до кипения с обратным холодильником и поддерживают реакционную массу при температуре кипения с обратным холодильником в течение примерно 2-3 часов под давлением. Отгоняют из реакционной смеси 585 мл толуола, после чего прибавляют 1375 мл толуола и реакционную массу охлаждают до 25-35°С. К указанной выше реакционной массе прибавляют 102,3 мл триметилсилилтрифлата (ТМС-трифлат) в течение периода 5-10 минут и реакционную массу перемешивают в течение примерно 20-30 минут. Раствор мезилированного соединения Формулы VIIB в 550 мл толуола прибавляют к полученной выше силилированной реакционной массе при 25-35°С, после чего реакционную массу нагревают до кипения с обратным холодильником и выдерживают реакционную массу в течение примерно 15 часов под давлением. Реакционную массу охлаждают до примерно 25-35°С с одновременным перемешиванием, после чего прибавляют 40 мл метанола в течение примерно 10 минут. Из полученной реакционной массы отгоняют 50% растворителя от общего объема при примерно 50°С под вакуумом, после чего охлаждают реакционную массу до 0-5°С в течение примерно 15-20 минут. Полученную смолистую реакционную массу декантируют и органический слой промывают 700 мл насыщенного раствора NaHCO3 и разделяют на два слоя. Органический слой перегоняют под вакуумом, получая 134 г указанного в заголовке соединения.
Пример 5
Способ получения гемцитабина гидрохлорида формулы I
Загружают в круглодонную колбу 133 г 1-[2'-дезокси-2',2'-дифтор-3',5'-рибофураноза-3,5-бис(трет-бутилдифенилсилилокси)-N-ацетилцитозина Формулы IX, полученного в соответствии с примером 4, под атмосферой азота. Прибавляют 7,5 л метанола и реакционный раствор охлаждают до 0-5°С, после чего прибавляют к реакционному раствору 1330 мл аммиака. Полученный раствор концентрируют при 50°С до минимального объема и реакционную массу экстрагируют 12 литрами этилацетата, после чего концентрируют органический слой при 50°С. Прибавляют к полученной, как описано выше, реакционной массе 1180 мл тетрагидрофурана (ТГФ) и 79,8 мл TBAF (тетра-н-бутиламмонийфторида) при 25-30°С и перемешивают в течение примерно 1 часа. Полноту протекания реакции контролируют с помощью тонкослойной хроматографии и затем концентрируют реакционную массу под вакуумом при 50°С. Прибавляют к указанной реакционной массе деминерализованную воду и затем водный слой промывают дихлорметаном. Полученный водный слой концентрируют при 55°С, получая 72% α-аномера и 24% β-аномера. К полученной выше сырой массе прибавляют 190 мл изопропилового спирта и затем реакционную массу нагревают до 60°С, после чего прибавляют 25 мл концентрированной хлористоводородной кислоты. Полученную реакционную массу охлаждают до 10-15°С и перемешивают в течение примерно 3 часов при 10°С. Полученное твердое вещество фильтруют, получая 69,9% α-аномера и 28,48% β-аномера, и полученный фильтрат концентрируют при 45°С. К полученному сырому фильтрату прибавляют ацетон и перемешивают в течение примерно одной ночи. Полученное твердое вещество фильтруют, получая 2% α-аномера и 71% β-аномера. Полученное твердое вещество растворяют в деминерализованной воде (qty) и прибавляют материал darco (уголь), после чего перемешивают в течение примерно 30-45 минут. Полученную реакционную массу фильтруют и полученный фильтрат концентрируют до минимального количества, получая 700 мг названного в заголовке соединения. Чистота по результатам ВЭЖХ: 95% чистый β-аномер.
Пример 6
Очистка гемцитабина гидрохлорида
Помещают в реактор 26,4 л (3 объема) деминерализованной воды и нагревают до температуры примерно 80-85°С. В указанный выше реактор прибавляют 6,6 кг необработанного гемцитабина гидрохлорида, имеющего примерно 1:1 соотношение смеси α/β аномерных солей по результатам ВЭЖХ с одновременным перемешиванием до образования прозрачного раствора. Реакционный раствор охлаждают до примерно 20-25°С, затем фильтруют выделившееся твердое вещество под атмосферой азота и твердое вещество промывают 5 л ацетона. Полученное твердое вещество высушивают под разрежением в течение примерно 1 часа под давлением азота, получая 2,7 кг гемцитабина гидрохлорида. Выделение обогащенного β-аномером соединения может проводиться, например, при температуре от 0 до 10°С. Чистота по данным ВЭЖХ: 98,1%. % α-аномера: 1,7%.
Пример 7
Очистка гемцитабина гидрохлорида
Помещают в реактор 860 мл очищенной воды вместе с 86 г необработанного гемцитабина гидрохлорида, имеющего соотношение α/β аномерной смеси примерно 1:1. Смесь нагревают до температуры примерно 35-40°С с одновременным перемешиванием до образования прозрачного раствора. Прибавляют к реакционной массе 0,1 объема угля при 35-40°С при перемешивании в течение примерно 30 минут. Реакционную массу фильтруют через Hyflow, после чего промывают 3×30 мл деминерализованной воды при комнатной температуре. Полученный фильтрат концентрируют до 3 объемов реакционной массы при 45-50°С под вакуумом 600 мм рт.ст. и затем охлаждают до 15-20°С с одновременным перемешиванием в течение примерно 30 минут при 15-20°С. Реакционную массу фильтруют, а потом промывают 50 мл ацетона при 15-20°С и высушивают под разрежением в течение 30 минут. Полученную твердую массу высушивают при температуре примерно 35-40°С в течение примерно 4 часов под вакуумом 600-620 мм рт.ст., получая 31 г гемцитабина гидрохлорида. Чистота по данным ВЭЖХ: 95,6%, % α-аномера 3,8%.
Пример 8
Очистка гемцитабина гидрохлорида
Помещают в реактор 342 мл (12 объемов) очищенной воды вместе с 28,5 г гемцитабина гидрохлорида, имеющего чистоту 95,6% и нагревают до температуры примерно 35-40°С с одновременным перемешиванием до образования прозрачного раствора. Прибавляют к реакционной массе 0,1 объема угля при 35-40°С при перемешивании в течение примерно 30 минут. Реакционную массу фильтруют через Hyflow и затем промывают 3×25 деминерализованной воды при комнатной температуре. Полученный фильтрат концентрируют до 2 объемов реакционной массы при 45-50°С под вакуумом 600-620 мм рт.ст. и затем охлаждают до 25-30°С. Реакционную массу фильтруют, а затем промывают 31 мл ацетона при 25-30°С. Описанный выше процесс повторяют один раз и, наконец, полученную твердую массу высушивают при температуре 35-40°С в течение примерно 4 часов под вакуумом 600-620 мм рт.ст., получая 24 г гемцитабина гидрохлорида. Чистота по данным ВЭЖХ: 99,96%. % α-аномера 0,01%. Оптическое вращение: [α]D 20 (10 мг/мл водного раствора): +47,1°.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ 2'-ДЕЗОКСИ-2', 2'-ДИФТОРЦИТИДИНА | 2005 |
|
RU2360919C2 |
| СПОСОБ ПОЛУЧЕНИЯ ОБОГАЩЕННЫХ БЕТА-АНОМЕРОМ НУКЛЕОЗИДОВ | 1993 |
|
RU2131880C1 |
| ПРОИЗВОДНЫЕ 1-α-ГАЛОГЕН-2,2-ДИФТОР-2-ДЕЗОКСИ-D-РИБОФУРАНОЗЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2005 |
|
RU2346948C2 |
| СПОСОБ ПОЛУЧЕНИЯ И ОЧИСТКИ ГЕМЦИТАБИНА ГИДРОХЛОРИДА | 2007 |
|
RU2355400C2 |
| Способ получения нуклеозида или его фармацевтически приемлемых солей | 1984 |
|
SU1442076A3 |
| СПОСОБ ПОЛУЧЕНИЯ ГЕМЦИТАБИН ГИДРОХЛОРИДА | 1995 |
|
RU2154648C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНОГО D-ЭРИТРО-2,2-ДИФТОРО-2-ДЕЗОКСИ-1-ОКСОРИБОЗЫ | 2005 |
|
RU2337917C1 |
| СПОСОБ ПОЛУЧЕНИЯ 2'-ДЕЗОКСИКСИЛОТИМИДИНА, ПРОИЗВОДНЫЕ D-КСИЛОФУРАНОЗЫ, ПРОИЗВОДНЫЕ КСИЛОТИМИДИНА | 1994 |
|
RU2108339C1 |
| ПОЛИМЕРНОЕ ПРОИЗВОДНОЕ АНТИМЕТАБОЛИТА ЦИТИДИНА | 2006 |
|
RU2404980C2 |
| НОВЫЙ ПИРИМИДИНОВЫЙ НУКЛЕОЗИД ИЛИ ЕГО СОЛЬ | 2006 |
|
RU2395517C2 |
Настоящее изобретение относится к способу очистки гемцитабина гидрохлорида, предполагающему обогащение гемцитабина гидрохлорида его β-аномером, согласно которому берут раствор гемцитабина гидрохлорида в воде при соотношении воды и гемцитабина гидрохлорида от 3:1 до 12:1 (мас./об.); обрабатывают раствор активированным углем, причем активированный уголь берут в количестве от 0,1 до 10 вес.% от количества гемцитабина гидрохлорида в растворе; удаляют активированный уголь из раствора с образованием профильтрованного раствора; повышают концентрацию гемцитабина гидрохлорида в профильтрованном растворе до соотношения профильтрованного раствора и гемцитабина гидрохлорида от 1:1 до 1:5 (мас./об.), эффективного для осаждения гемцитабина гидрохлорида; выделяют осажденный гемцитабина гидрохлорид, и, если содержание примесей в осажденном гемцитабине гидрохлориде не снизилось до требуемого уровня, то стадии (а)-(е) повторяют. Данное изобретение относится также к способу получения гемцитабина гидрохлорида с использованием вышеуказанной методики очистки. 2 н. и 3 з.п. ф-лы, 1 табл., 5 ил.
a) проводят реакцию 2-дезокси-2,2-дифтор-1-оксорибозы Формулы IV с т-бутилдифенилсилилхлоридом с образованием 3,5-бис(т-бутилдифенилсилилокси)-2-дезокси-2,2-дифторрибозы Формулы VB;
b) восстанавливают 3,5-бис(т-бутилдифенилсилилокси)-2-дезокси-2,2- дифторрибозу Формулы VB, используя натрий-бис(2-метоксиэтокси) алюминий-гидрид, для получения 3,5-бис(т-бутилдифенилсилилокси)-2-дезокси-2,2-дифторрибозы Формулы VIB;
c) проводят защиту гидроксильной группы в 3,5-бис(т-бутилдифенилсилилокси)-2-дезокси-2,2-дифторрибозы Формулы VIB, используя метансульфонилхлорид, с образованием 3,5-бис(трет-бутилдифенилсилилокси)-1-метансульфонилокси-2-дезокси-2,2-дифторрибозы Формулы VIIB;
d) проводят конденсацию 3,5-бис(трет-бутилдифенилсилилокси)-1-метансульфонилокси-2-дезокси-2,2-дифторрибозу Формулы VIIB дисилильным соединением Формулы VIII с образованием 1-[2'-дезокси-2',2,-дифтор-3',5'-рибофураноза-3,5-бис(трет-бутилдифенилсилилокси)-N-ацетилцитозина Формулы IX;
e) снимают защиту 1-[2'-дезокси-2',2'-дифтор-3',5'-рибофураноза-3,5-бис(трет-бутилдифенилсилилокси)-N-ацетилцитозина Формулы IX, используя трет-бутиламмонийфторид, с последующей реакцией с соляной кислотой, для получения гемцитабина гидрохлорида; и
f) проводят очистку гемцитабина гидрохлорида способом по п.1.
| US 5945547 А, 31.08.1999 | |||
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| СПОСОБ ПОЛУЧЕНИЯ ГЕМЦИТАБИН ГИДРОХЛОРИДА | 1995 |
|
RU2154648C2 |

