Перекрестная ссылка на родственные заявки
По данной заявке испрашивается приоритет по заявке Соединенных Штатов No. 60/310313, поданной 6 августа 2001 г., которая тем самым включена здесь во всей ее полноте в качестве ссылки.
Область техники, к которой относится изобретение
Настоящее изобретение относится к новым депсипептидным соединениям. Изобретение также относится к фармацевтическим композициям на основе указанных соединений и способам применения данных соединений в качестве антибактериальных агентов. Изобретение также относится к способам получения указанных новых депсипептидных соединений и промежуточных продуктов, применяемых для получения данных соединений.
Известный уровень техники
Быстрое увеличение случаев грамположительных инфекций, включая вызываемые устойчивыми бактериями, заново возбудило интерес к разработке новых классов антибиотиков. Классом соединений, которые показали себя как потенциальные антибиотические агенты, являются циклические депсипептиды. Примечательным членом циклических депсипептидов являются липопептиды A-21978C, описанные, например, в патентах Соединенных Штатов RE 32333; RE 32445; RE 32311; RE 32310; 4482487; 4537717 и 5912226 и в международных патентных заявках WO 01/44272; WO 01/44274 и WO 01/44271. Кроме того, класс A54145 соединений, описанных в патентах Соединенных Штатов 4994270; 5039789 и 5028590, также, как показано, обладает антибиотической активностью.
Даптомицин, также известный как LY 146032, включает н-деканоильную боковую цепь, соединенную с N-концевым триптофаном цепи из трех аминокислот, соединенной с циклическим пептидом из 10 аминокислот. Даптомицин обладает сильной антибактериальной активностью in vitro и in vivo против клинически значимых грамположительных бактерий, которые вызывают серьезные и угрожающие жизни заболевания. Данные бактерии включают устойчивые патогены, такие как устойчивые к ванкомицину энтерококки (VRE), устойчивые к метициллину Staphylococcus aureus (MRSA), восприимчивые к промежуточному продукту гликопептида Staphylococcus aureus (GISA), устойчивые к ванкомицину Staphylococcus aureus (VRSA), негативные в отношении коагулазы стафилококки (CNS) и устойчивые к пенициллину Streptococcus pneumoniae (PRSP), для которых существует очень мало терапевтических альтернатив. Смотри, например, Tally et al., 1999, Exp. Opin. Invest. Drugs 8:1223-1238.
Несмотря на перспективы, которые связаны с существующими антибактериальными агентами, продолжает сохраняться потребность в новых антибиотиках. Многие патогены повторно экспонировались с обычно применяемыми антибиотиками. Данная экспозиция привела к отбору вариантов антибактериальных штаммов, устойчивых к широкому спектру антибиотиков. Потеря силы и эффективности антибиотика, определяемые возникновением механизмов устойчивости, приводит к неэффективности антибиотика и, следовательно, может вести к некоторым угрожающим жизни инфекциям, которые практически не поддаются лечению. Как только на рынке появляются новые антибиотики, у патогенов может развиться устойчивость или переходная устойчивость к данным новым лекарствам, эффективно создавая потребность в потоке новых антибактериальных агентов для борьбы с данными возникающими штаммами. Кроме того, соединения, которые проявляют бактерицидную активность, имеют преимущества по сравнению с имеющимися в настоящее время бактериостатическими соединениями. Таким образом, следует ожидать, что новые антибактериальные агенты будут пригодны для воздействия не только на «природные» патогены, но также и на патогены с переходной устойчивостью и устойчивые патогены, потому что патоген никогда не был экспонирован с новым антибактериальным агентом. Новые антибактериальные агенты могут характеризоваться различной эффективностью против различных типов патогенов.
Краткое изложение существа изобретения
В настоящем изобретении предложены новые соединения, которые обладают антибактериальной активностью в отношении широкого спектра бактерий, включая устойчивые к лекарствам бактерии, и способы получения таких соединений.
В настоящем изобретении предлагаются в одном аспекте соединения формулы
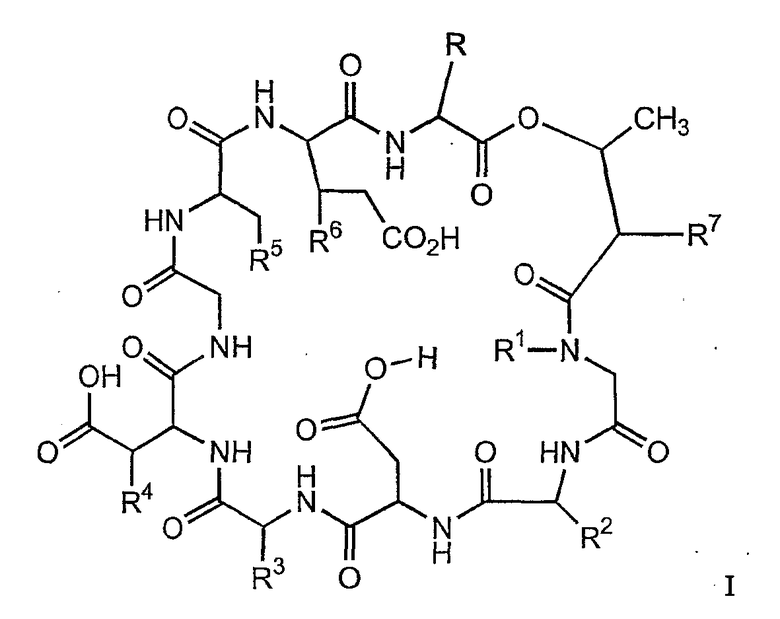
и их соли;
где
(a) R представляет собой 2-бутил, изопропил или 2-(2'-аминофенацил);
(b) каждый из R1 и R6 независимо представляет собой гидридо или метил;
(c) R2 представляет собой метил или -CH2CH2CH2R8;
(d) R3 представляет собой метил или -CH2CH2CH2CH2R9;
(e) R4 представляет собой гидридо или метокси;
(f) R5 представляет собой гидрокси или карбоксиамино;
(g) каждый из R7, R8 и R9 независимо представляет собой амино, монозамещенный амино, дизамещенный амино, ациламино, уреидо, гуанидино, карбамоил, сульфонамино, тиоациламино, тиоуреидо, иминоамино или фосфонамино;
(h) при условии, что
(1) когда R2 представляет собой -CH2CH2CH2R8, R7 является отличным от
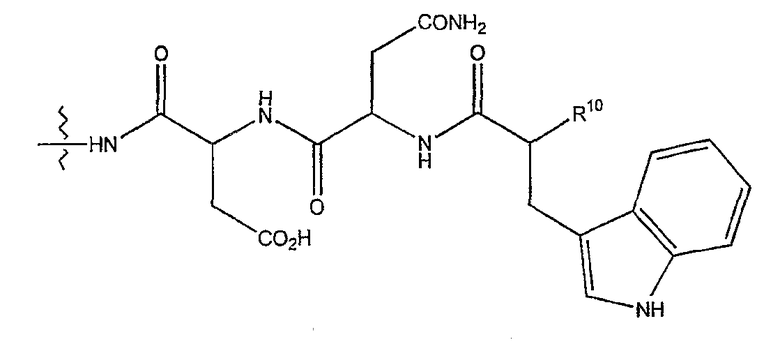
где R10 представляет собой амино, монозамещенный амино, дизамещенный амино, ациламино, уреидо, гуанидино, карбамоил, сульфонамино, тиоациламино, иминоамино или фосфонамино;
(2) когда R2 представляет собой метил, R7 является отличным от
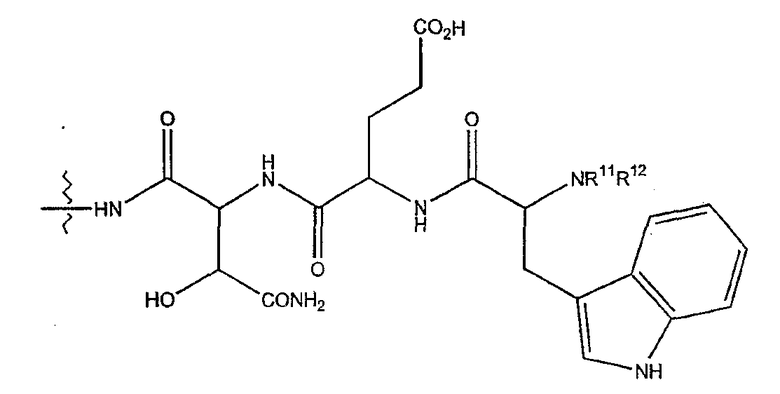
где каждый из R11 и R12 представляет собой гидридо, C6-C18 незамещенный алканоил, C8-C18 незамещенный алкеноил, C8-C18 незамещенный алкил или C8-C18 выбранный замещенный алкил; или альтернативно R11 и R12 вместе представляют собой C8-C18 алкилиденил.
В другом аспекте настоящее изобретение относится к фармацевтическим композициям, содержащим соединения формулы I, и к способам их применения.
В дополнительном аспекте настоящее изобретение относится к способу получения соединений формулы I.
В еще одном аспекте настоящее изобретение относится к соединениям, пригодным в качестве промежуточных продуктов для получения соединений формулы I.
Подробное описание изобретения
Определения
Термин «активирующая группа» обозначает группу, которая при присоединении к карбонильной группе активирует карбонильную группу для воздействия на нее нуклеофильного амина, что ведет к потере активирующей группы и образованию амидной связи. Примерами активирующих групп являются арилокси, ацилокси, имидазолил
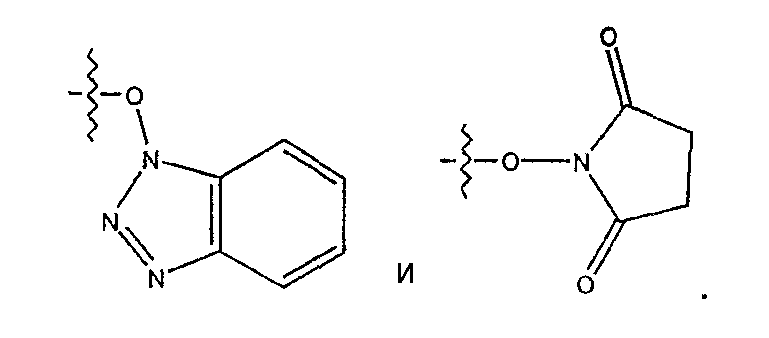
Предпочтительными активирующими группами являются арилоксигруппы. Наиболее предпочтительная активирующая группа представляет собой пентафторфенокси.
Термин «ацил» обозначает карбонильный радикал, присоединенный к алкилу, алкенилу, алкинилу, циклоалкилу, гетероциклилу, арильной или гетероарильной группе, причем примеры включают без ограничения такие радикалы, как ацетил и бензоил. Термин ацил подразделяется на (1) «незамещенный алканоил», который определяют как карбонильный радикал, присоединенный к незамещенной алкильной группе, и (2) «незамещенный алкеноил», который определяют как карбонильный радикал, присоединенный к незамещенной алкенильной группе.
Термин «ациламино» обозначает азотный радикал, присоединенный к ацильной группе.
Термин «ацилокси» обозначает кислородный радикал, присоединенный к ацильной группе.
Термин «алкенил» определяют как линейные или разветвленные радикалы, имеющие от двух до приблизительно двадцати атомов углерода, предпочтительно, от трех до приблизительно десяти атомов углерода, и содержащие, по меньшей мере, одну углерод-углеродную двойную связь. Один или несколько атомов водорода могут также быть заменены на заместитель, выбранный из ацила, ациламино, ацилокси, алкенила, алкокси, алкила, алкинила, амино, арила, арилокси, карбамоила, карбоалкокси, карбокси, карбоксиамидо, карбоксиамино, циано, дизамещенного амино, формила, гуанидино, галогена, гетероарила, гетероциклила, гидрокси, иминоамино, монозамещенного амино, нитро, оксо, фосфонамино, сульфинила, сульфонамино, сульфонила, тио, тиоациламино, тиоуреидо или уреидо. Двойная(ые) связь(и) ненасыщенной углеводородной цепи могут иметь либо цис, либо транс конфигурацию. Примеры алкенильных групп включают без ограничения этиленил или фенилэтиленил. Термин алкенил подразделяется на «незамещенный алкенил», который определяют как алкенильную группу, которая не имеет заместителей.
Термин «алкокси» обозначает кислородный радикал, замещенный алкильной, циклоалкильной или гетероциклильной группой. Примеры включают без ограничения метокси, трет-бутокси, бензилокси и циклогексилокси.
Термин «алкил» определяют как линейный или разветвленный радикал, имеющий от одного до приблизительно двадцати атомов углерода, если не указано иначе. Термин «низший алкил» определяют как алкильную группу, содержащую 1-4 атомов углерода. Один или несколько атомов водорода могут быть заменены на заместители, выбранные из ацила, ациламино, ацилокси, алкенила, алкокси, алкила, алкинила, амино, арила, арилокси, карбамоила, карбоалкокси, карбокси, карбоксиамидо, карбоксиамино, циано, дизамещенного амино, формила, гуанидино, галогена, гетероарила, гетероциклила, гидрокси, иминоамино, монозамещенного амино, нитро, оксо, фосфонамино, сульфинила, сульфонамино, сульфонила, тио, тиоациламино, тиоуреидо или уреидо. Примеры алкильных групп включают без ограничения метил, бутил, трет-бутил, изопропил, трифторметил, нонил, ундецил, октил, додецил, метоксиметил, 2-(2'-аминофенацил), 3-индолилметил, бензил и карбоксиметил. Термин алкил подразделяется на (1) «незамещенный алкил», который определяют как алкильную группу, которая не имеет заместителей, (2) «замещенный алкил», которым обозначают алкильный радикал, в котором один или несколько атомов водорода заменены на заместитель, выбранный из ацила, ациламино, ацилокси, алкенила, алкокси, алкила, алкинила, амино, арила, арилокси, карбамоила, карбоалкокси, карбокси, карбоксиамидо, карбоксиамино, циано, дизамещенного амино, формила, гуанидино, галогена, гетероарила, гетероциклила, гидрокси, иминоамино, монозамещенного амино, нитро, оксо, фосфонамино, сульфинила, сульфонамино, сульфонила, тио, тиоациламино, тиоуреидо или уреидо, и (3) «выбранный замещенный алкил», которым обозначают алкильный радикал, в котором (a) один протон заменен на группу, выбранную из гидрокси, карбокси, C1-C8 алкокси или (b) от одного до трех протонов заменены галогеном.
Термин "алкилиденил" определяется как карбоновый радикал формулы
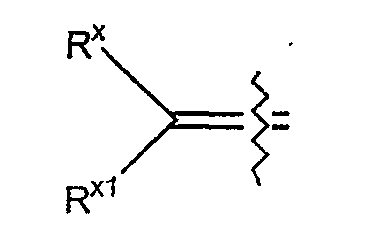
где Rx и Rx1 независимо выбраны из гидридо или C7-C17 незамещенного алкила, где суммарное количество атомов углерода от Rx и Rx1 не превышает 17.
Термин «алкинил» обозначает линейные или разветвленные радикалы, содержащие от двух до приблизительно десяти атомов углерода и содержащие, по меньшей мере, одну углерод-углеродную тройную связь. Один или несколько атомов водорода могут быть также заменены на заместителя, выбранный из ацила, ациламино, ацилокси, алкенила, алкокси, алкила, алкинила, амино, арила, арилокси, карбамоила, карбоалкокси, карбокси, карбоксиамидо, карбоксиамино, циано, дизамещенного амино, формила, гуанидино, галогена, гетероарила, гетероциклила, гидрокси, иминоамино, монозамещенного амино, нитро, оксо, фосфонамино, сульфинила, сульфонамино, сульфонила, тио, тиоациламино, тиоуреидо или уреидо. Пример алкинильной группы включает без ограничения пропинил.
Термин "амино" определяется как NH2 радикал.
Термин "аминокислотный остаток" обозначает соединение формулы
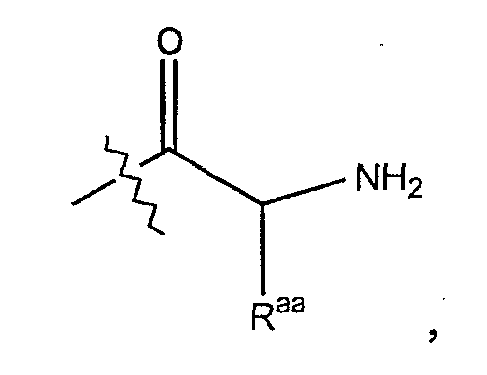
где Raa представляет собой боковую цепь аминокислоты.
Термин "боковая цепь аминокислоты" обозначает любую боковую цепь (R группу) существующей в природе или синтетической аминокислоты. Например, 3-индолилметил можно было бы также назвать боковой цепью триптофана.
Термин "2-(2'-аминофенацил)" относится к радикалу формулы
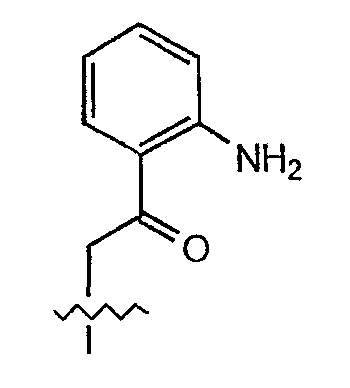
Термин "аминозащитная группа" относится к любому химическому соединению, которое может быть использовано для того, чтобы предотвратить химическую реакцию аминогруппы молекулы, при том, что в другом участке молекулы происходит химическое изменение. Специалистам в данной области техники известны многочисленные аминозащитные группы, и примеры можно найти в "Protective Groups in Organic Synthesis" by Theodora W. Greene, John Wiley and Sons, New York, 1981. Примеры аминозащитных групп включают фталимидо, трихлорацетил, STA-основание, бензилоксикарбонил, трет-бутоксикарбонил, трет-амилоксикарбонил, изоборнилоксикарбонил, адамантилоксикарбонил, хлорбензилоксикарбонил, нитробензилоксикарбонил и тому подобное. Предпочтительными аминозащитными группами являются "карбаматные аминозащитные группы", которые определяются как аминозащитные группы, которые при связывании с аминогруппой образуют карбамат. Предпочтительными аминокарбаматными защитными группами являются аллилоксикарбонильная (alloc), карбобензилокси (CBZ) и трет-бутоксикарбонильная защитные группы.
Термин "арил" или "арильное кольцо" обозначает ароматический радикал в одинарной или конденсированной карбоциклической кольцевой системе, содержащий от пяти до четырнадцати кольцевых членов. В предпочтительном осуществлении кольцевая система содержит от шести до десяти членов кольца. Один или несколько атомов водорода могут также быть заменены на заместитель, выбранный из ацила, ациламино, ацилокси, алкенила, алкокси, алкила, алкинила, амино, арила, арилокси, азидо, карбамоила, карбоалкокси, карбокси, карбоксиамидо, карбоксиамино, циано, дизамещенного амино, формила, гуанидино, галогена, гетероарила, гетероциклила, гидрокси, иминоамино, монозамещенного амино, нитро, оксо, фосфонамино, сульфинила, сульфонамино, сульфонила, тио, тиоациламино, тиоуреидо или уреидо. Примеры арильных групп включают без ограничения фенил, нафтил, бифенил, терфенил.
Термин "арилокси" обозначает содержащие окси-содержащие радикалы, замещенные арильной или гетероарильной группой. Примеры включают без ограничения фенокси.
Термин "карбамоил" обозначает азотный радикал формулы
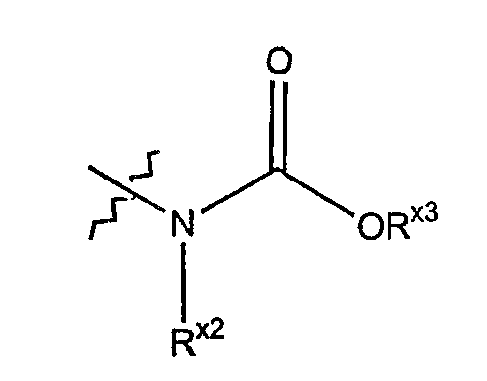
где Rx2 выбран из гидридо, алкила, арила, циклоалкила, гетероарила или гетероциклила, а Rx3 выбран из алкила, арила, циклоалкила, гетероарила или гетероциклила.
Термин "карбоалкокси" определяется как карбонильный радикал, присоединенный к алкокси или арилоксигруппе.
Термин "карбокси" обозначает радикал COOH.
Термин "карбоксиамино" обозначает радикал CONH2.
Термин "карбоксиамидо" обозначает карбонильный радикал, присоединенный к монозамещенной или дизамещенной аминогруппе.
Термин "α-карбоксиаминокислотная боковая цепь" обозначает углеродный радикал формулы
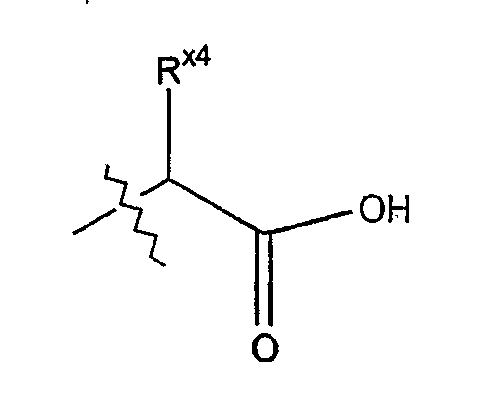
где Rx4 определяется как боковая цепь аминокислоты.
Термин "карбоксиметил" обозначает радикал CH2CO2H.
Термин "циклоалкил" или "циклоалкильное кольцо" обозначает насыщенное или частично ненасыщенное карбоциклическое кольцо в одинарной или конденсированной карбоциклической кольцевой системе, содержащей от трех до двенадцати кольцевых членов. В предпочтительном осуществлении циклоалкил представляет собой кольцевую систему, содержащую от трех до семи членов в кольце. Один или несколько атомов водорода могут также быть заменены на заместитель, выбранный из ацила, ациламино, ацилокси, алкенила, алкокси, алкила, алкинила, амино, арила, арилокси, карбамоила, карбоалкокси, карбокси, карбоксиамидо, карбоксиамино, циано, дизамещенного амино, формила, гуанидино, галогена, гетероарила, гетероциклила, гидрокси, иминоамино, монозамещенного амино, нитро, оксо, фосфонамино, сульфинила, сульфонамино, сульфонила, тио, тиоациламино, тиоуреидо или уреидо. Примеры циклоалкильной группы включают без ограничения циклопропил, циклобутил, циклогексил и циклогептил.
Термин "дизамещенный амино" обозначает аминорадикал, содержащий две замещающие группы, независимо выбранные из алкила, циклоалкила, гетероциклила, арила или гетероарила. Предпочтительными дизамещенными аминорадикалами являются "низшие дизамещенные амино" радикалы, где замещающие группы являются низшими алкилами. Также предпочтительными дизамещенными аминорадикалами являются аминорадикалы, в которых один заместитель является низшей алкильной группой, а другой заместитель является боковой цепью α-карбоксиаминокислоты.
Группа "Fmoc" представляет собой 9-флуоренилметоксикарбонильную группу.
Термин "гуанидино" обозначает азотный радикал формулы
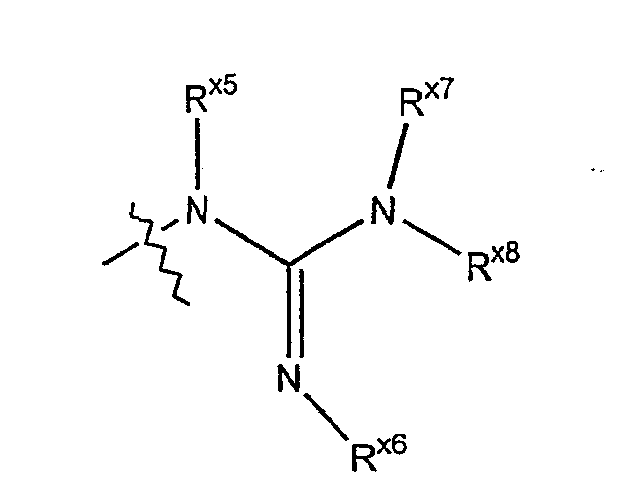
где каждый из Rx5, Rx7 и Rx8 независимо выбраны из гидридо, алкильной, арильной, циклоалкильной, гетероарильной или гетероциклильной группы; и Rx6 выбран из алкильной, арильной, циклоалкильной, гетероарильной или гетероциклильной группы. Термин "галоген" относится к радикалу брома, хлора, фтора или йода.
"Гетероарил" или "гетероарильное кольцо" обозначает ароматический радикал, который содержит от одного до четырех гетероатомов или гетерогрупп, выбранных из O, N, S или SO, в одинарной или конденсированной гетероциклической кольцевой системе, содержащей от пяти до пятнадцати кольцевых членов. В предпочтительном осуществлении гетероарильная кольцевая система содержит от шести до десяти кольцевых членов. Один или несколько атомов водорода могут также быть заменены на заместитель, выбранный из ацила, ациламино, ацилокси, алкенила, алкокси, алкила, алкинила, амино, арила, арилокси, карбамоила, карбоалкокси, карбокси, карбоксиамидо, карбоксиамино, циано, дизамещенного амино, формила, гуанидино, галогена, гетероарила, гетероциклила, гидрокси, иминоамино, монозамещенного амино, нитро, оксо, фосфонамино, сульфинила, сульфонамино, сульфонила, тио, тиоациламино, тиоуреидо или уреидо. Примеры гетероарильных групп включают без ограничения пиридинильную, тиазолильную, тиадиазолильную, изохинолинильную, пиразолильную, оксазолильную, оксадиазоильную, триазолильную и пирролильную группы.
Термин "гетероциклильный", "гетероциклический" или "гетероциклильное кольцо" обозначает насыщенное или частично ненасыщенное кольцо, содержащее от одного до четырех гетероатомов или гетерогрупп, выбранных из O, N, NH, N(низший алкил), S, SO или SO2 в одинарной или конденсированной гетероциклической кольцевой системе, содержащей от трех до двенадцати кольцевых членов. В предпочтительном осуществлении гетероциклил представляет собой кольцевую систему, содержащую от трех до семи кольцевых членов. Один или несколько атомов водорода могут также быть заменены на заместитель, выбранный из ацила, ациламино, ацилокси, алкенила, алкокси, алкила, алкинила, амино, арила, арилокси, карбамоила, карбоалкокси, карбокси, карбоксиамидо, карбоксиамино, циано, дизамещенного амино, формила, гуанидино, галогена, гетероарила, гетероциклила, гидрокси, иминоамино, монозамещенного амино, нитро, оксо, фосфонамино, сульфинила, сульфонамино, сульфонила, тио, тиоациламино, тиоуреидо или уреидо. Примеры гетероциклической группы включают без ограничения морфолинил, пиперидинил и пирролидинил.
Термин "гидридо" обозначает одиночный атом водорода (H).
Термин "иминоамино" обозначает азотный радикал формулы:
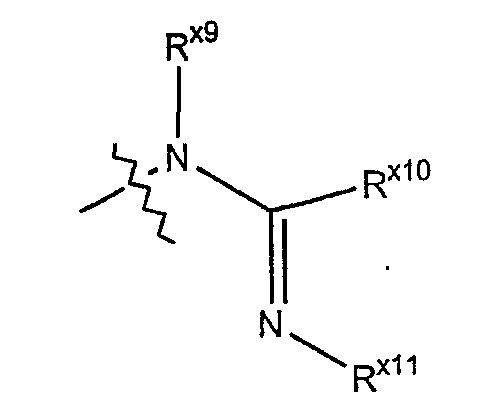
где Rx9 и Rx11 независимо выбраны из гидридо, алкильной, циклоалкильной, арильной, гетероарильной или гетероциклильной группы; и Rx10 выбран из алкильной, циклоалкильной, арильной, гетероарильной или гетероциклильной группы.
Термин "модифицирующий агент" определяется как (a) нуклеофильный акцептор или (b) альдегид или кетон, который взаимодействует с амином в условиях восстановления с образованием алкилированного амина.
Термин "монозамещенный амино" обозначает аминорадикал, содержащий гидридогруппу и замещающую группу, выбранную из алкила, циклоалкила, гетероциклила, арила или гетероарила. Предпочтительными монозамещенными аминорадикалами являются "низшие монозамещенные амино" радикалы, где замещающая группа представляет собой низшую алкильную группу. Более предпочтительными монозамещенными аминорадикалами являются аминорадикалы, содержащие боковую цепь α-карбоксиаминокислоты.
Термин "нуклеофильный акцептор" обозначает соединение, которое подвержено нуклеофильной атаке первичного или вторичного амина. Примеры нуклеофильных акцепторов включают без ограничения изоцианаты, изотиоцианаты, активированные сложные эфиры, хлорангидриды кислоты, сульфонилхлориды, активированные сульфонамиды, активированные гетероциклы, активированные гетероарилы, хлорформиаты, цианоформиаты, тиоэфиры, фосфорилхлориды, фосфорамидаты, имидаты и лактоны.
Термин "фосфонамино" обозначает азотный радикал формулы:
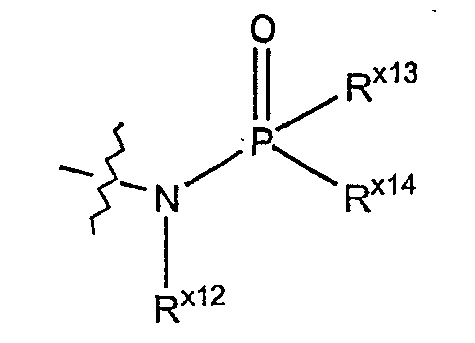
где Rx12 выбран из гидридо, алкила, арила, циклоалкила, гетероарила или гетероциклила; где каждый из Rx13 и Rx14 независимо выбран из алкила, алкокси, арила, арилокси, циклоалкила, гетероарила и гетероциклила.
Термин "сульфинил" обозначает четырехвалентный радикал серы, замещенный заместителем оксо и вторым заместителем, выбранным из группы, включающей алкильную, циклоалкильную, гетероциклильную, арильную или гетероарильную группы.
Термин сульфонамино обозначает аминорадикал формулы:
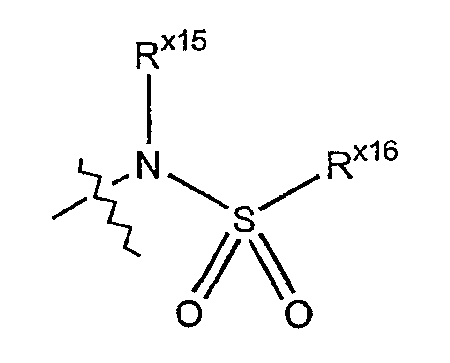
где Rx15 выбран из гидридо, алкильной, циклоалкильной, арильной, гетероарильной или гетероциклильной группы; а Rx16 выбран из алкильной, циклоалкильной, арильной, гетероарильной или гетероциклильной группы.
Термин "сульфонил" обозначает радикал шестивалентной серы, замещенный двумя заместителями оксо, а третий заместитель выбран из алкила, циклоалкила, гетероциклила, арила или гетероарила.
Термин "тио" обозначает радикал, содержащий замещающую группу, независимо выбранную из гидридо, алкила, циклоалкила, гетероциклила, арила и гетероарила, присоединенную к двухвалентному атому серы, такой как метилтио и фенилтио.
Термин "тиоациламино" обозначает аминорадикал формулы
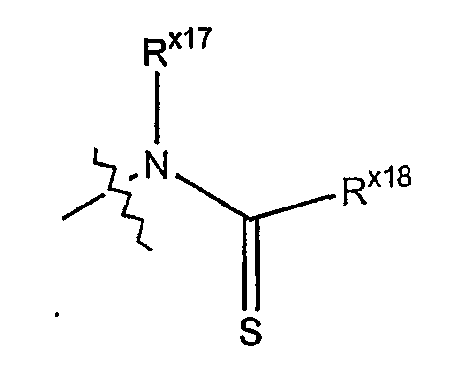
где Rx17 выбран из гидридо, алкильной, арильной, циклоалкильной, гетероарильной или гетероциклильной группы; и где Rx18 выбран из алкильной, арильной, циклоалкильной, гетероарильной или гетероциклильной группы.
Термин "тиоуреидо" обозначает радикал серы формулы:
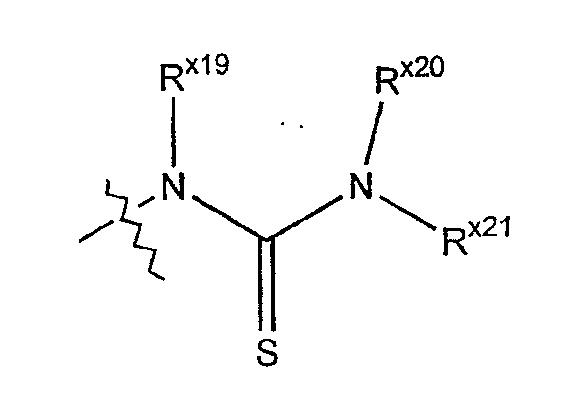
где каждый из Rx19 и Rx20 независимо выбран из гидридо, алкильной, арильной, циклоалкильной, гетероарильной или гетероциклильной группы; а Rx21 выбран из алкильной, арильной, циклоалкильной, гетероарильной или гетероциклильной группы.
Тритильная группа представляет собой трифенилметильную группу.
Термин "уреидо" обозначает азотный радикал формулы
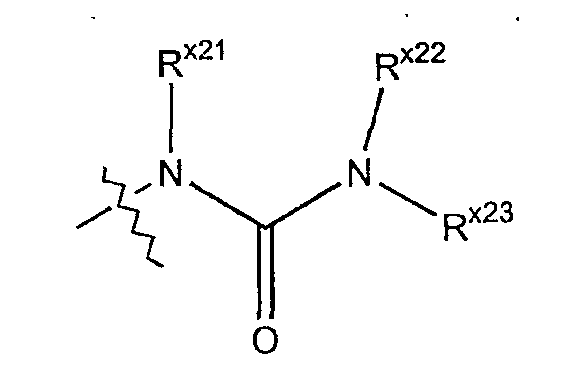
где каждый из Rx21 и Rx22 независимо выбран из гидридо, алкильной, арильной, циклоалкильной, гетероарильной или гетероциклильной группы; а Rx23 выбран из алкильной, арильной, циклоалкильной, гетероарильной или гетероциклильной группы.
Соли соединений по изобретению включают аддитивные соли кислот и аддитивные соли оснований. В предпочтительном осуществлении соль является фармацевтически приемлемой солью соединения формулы I. Термин "фармацевтически приемлемые соли" охватывает соли и используется для обозначения солей щелочных металлов и обозначения аддитивных солей свободных кислот или свободных оснований. Природа соли не является ограничивающим фактором в том случае, если она является фармацевтически приемлемой. Подходящие фармацевтически приемлемые аддитивные соли кислот соединений по изобретению могут быть получены из неорганической кислоты или органической кислоты. Примеры таких неорганических кислот включают без ограничения хлористоводородную, бромистоводородную, йодистоводородную, азотную, карбоновую, серную и фосфорную кислоты. Подходящие органические кислоты могут быть выбраны из классов алифатических, циклоалифатических, ароматических, арилалифатических, гетероциклических, карбоксильных и сульфоновых органических кислот, примеры которых включают без ограничения муравьиную, уксусную, пропионовую, янтарную, гликолевую, глюконовую, малеиновую, эмбоновую (памоевую) кислоты, метансульфокислоту, этансульфокислоту, 2-гидроксиэтансульфокислоту, пантотеновую кислоту, бензолсульфокислоту, толуолсульфокислоту, сульфаниловую, мезиловую кислоты, циклогексиламиносульфокислоту, стеариновую, альгиновую, β-гидроксимасляную, малоновую, галактоновую и галактуроновую кислоты. Подходящие фармацевтически приемлемые аддитивные соли оснований соединений по изобретению включают, но не ограничиваются этим, соли металлов, производные алюминия, кальция, лития, магния, калия, натрия и цинка, или органические соли, производные N,N'-дибензилэтилендиамина, хлорпрокаина, холина, диэтаноламина, этилендиамина, N-метилглюкамина, лизина и прокаина. Все данные соли могут быть получены с помощью обычных способов из соответствующего соединения по изобретению путем обработки, например, соединения по изобретению подходящей кислотой или основанием. Соединения по изобретению могут содержать один или несколько асимметричных атомов углерода и, таким образом, могут существовать в виде оптических изомеров, а также в виде их рацемических или нерацемических смесей. Соединения по изобретению могут применяться в настоящем изобретении в виде одного изомера или в виде смеси стереохимических изомерных форм. Диастереоизомеры, т.е. не накладываемые стереохимические изомеры, могут быть разделены обычными способами, такими как хроматография, перегонка, кристаллизация или сублимация. Оптические изомеры могут быть получены путем разделения рацемических смесей обычными способами, например, путем образования диастереоизомерных солей при обработке оптически активной кислотой или основанием. Примеры подходящих кислот включают без ограничения винную, диацетилвинную, дибензоилвинную, дитолуилвинную и камфорсульфокислоту. Смесь диастереомеров может быть разделена с помощью кристаллизации с последующим отделением оптически активных оснований от оптически активных солей. Альтернативный способ разделения оптических изомеров включает применение хиральной хроматографической колонки, оптимально выбранной для максимального разделения энантиомеров. Еще один способ включает синтез ковалентных диастереоизомерных молекул путем взаимодействия соединений по изобретению с оптически чистой кислотой в активной форме или оптически чистым изоцианатом. Синтезированные диастереоизомеры могут быть разделены традиционными способами, такими как хроматография, перегонка, кристаллизация или сублимация, и затем гидролизованы с получением энантиомерно чистого соединения. Оптически активные соединения по изобретению могут также быть получены с применением оптически активных исходных материалов. Данные изомеры могут находиться в виде свободной кислоты, свободного основания, сложного эфира или соли.
Изобретение также относится к выделенным соединениям. Выделенное соединение относится к соединению, которое представляет, по меньшей мере, 10%, предпочтительно, по меньшей мере, 20%, более предпочтительно, по меньшей мере, 50% и наиболее предпочтительно, по меньшей мере, 80% соединения, присутствующего в смеси. В предпочтительном осуществлении соединение, его фармацевтически приемлемая соль или фармацевтическая композиция, включающая соединение, проявляют определяемую (т.е. статистически значимую) антимикробную активность при исследовании с помощью обычных биологических методов, таких как описанные здесь.
Депсипептидные соединения
В одном аспекте изобретение относится к соединению формулы I
и его солям;
где (a) R представляет собой 2-бутил, изопропил или 2-(2'-аминофенацил);
(b) каждый из R1 и R6 независимо представляет собой гидридо или метил;
(c) R2 представляет собой метил или -CH2CH2CH2R8;
(d) R3 представляет собой метил или -CH2CH2CH2CH2R9;
(e) R4 представляет собой гидридо или метокси;
(f) R5 представляет собой гидрокси или карбоксиамино;
(g) каждый из R7, R8 и R9 независимо представляет собой амино, монозамещенный амино, дизамещенный амино, ациламино, уреидо, гуанидино, карбамоил, сульфонамино, тиоациламино, тиоуреидо, иминоамино или фосфонамино;
(h) при условии, что,
(1) когда R2 представляет собой -CH2CH2CH2R8, R7 является отличным от
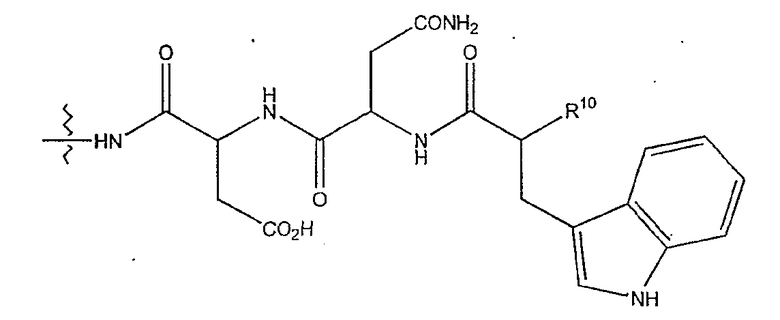
где R10 представляет собой амино, монозамещенный амино, дизамещенный амино, ациламино, уреидо, гуанидино, карбамоил, сульфонамино, тиоациламино, тиоуреидо, иминоамино и фосфонамино;
(2) когда R2 представляет собой метил, R7 является отличным от
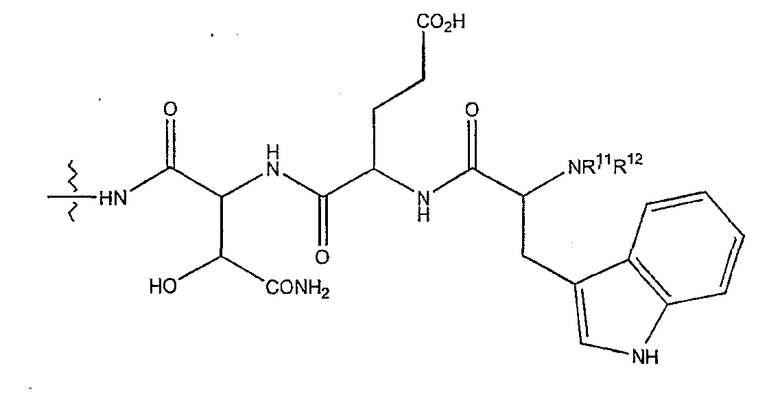
где каждый из R11 и R12 представляет собой гидридо, C6-C18 незамещенный алканоил, C8-C18 незамещенный алкеноил, C8-C18 незамещенный алкил или C8-C18 выбранный замещенный алкил; или альтернативно R11 и R12 вместе представляют собой C8-C18 алкилиденил.
Предпочтительно, R7 представляет собой
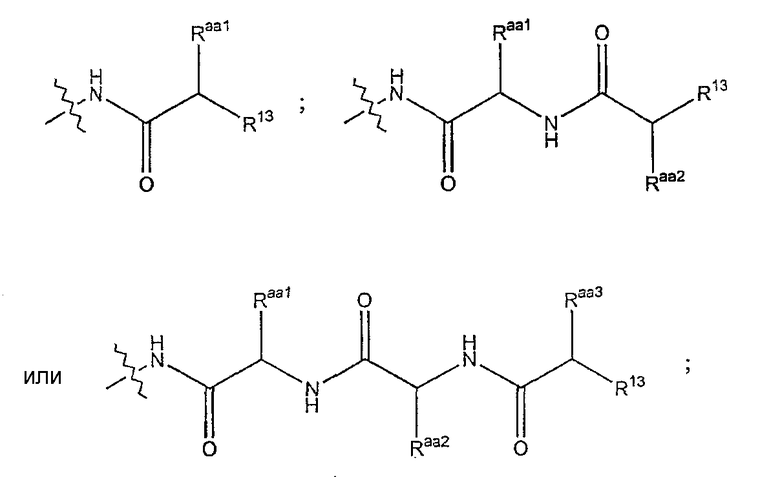
где каждый из Raa, Raa2 и Raa3 независимо представляет собой боковую цепь аминокислоты и где R13 представляет собой амино, монозамещенный амино, дизамещенный амино, ациламино, уреидо, гуанидино, карбамоил, сульфонамино, тиоациламино, тиоуреидо, иминоамино или фосфонамино.
В одном осуществлении изобретения R представляет собой 2-(2'-аминофенацил); каждый из R1 и R4 представляет собой гидридо; R2 представляет собой -CH2CH2CH2R8; каждый из R3 и R6 представляет собой метил; а R5 представляет собой гидроксил. В данном осуществлении предлагается соединение формулы II.
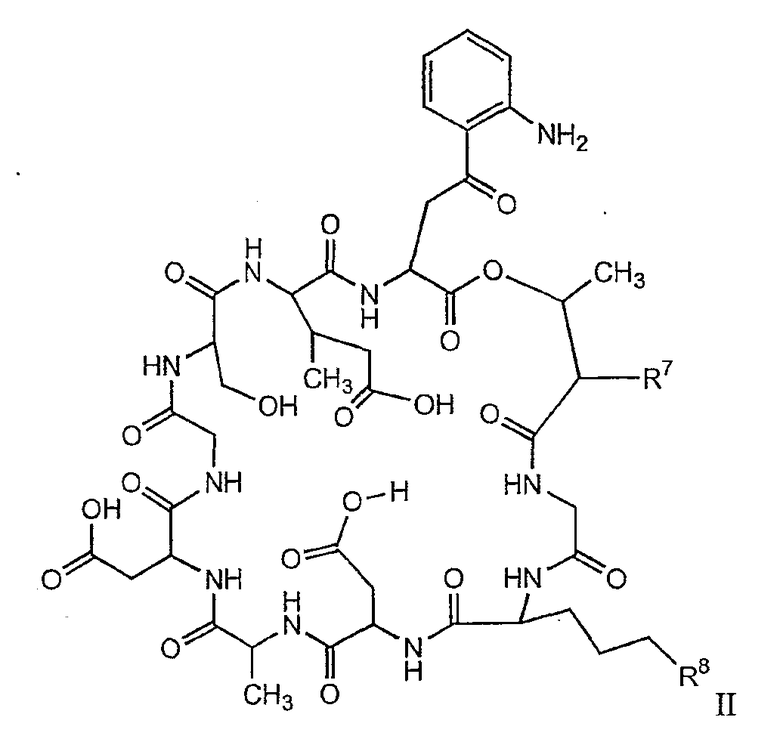
В другом осуществлении изобретения R представляет собой изопропил или 2-бутил; каждый из R1 и R2 представляет собой метил; R3 представляет собой - CH2CH2CH2CH2R9; каждый из R4 представляет собой метокси, а R5 представляет собой карбоксиамино. В данном осуществлении предлагается соединение формулы III.
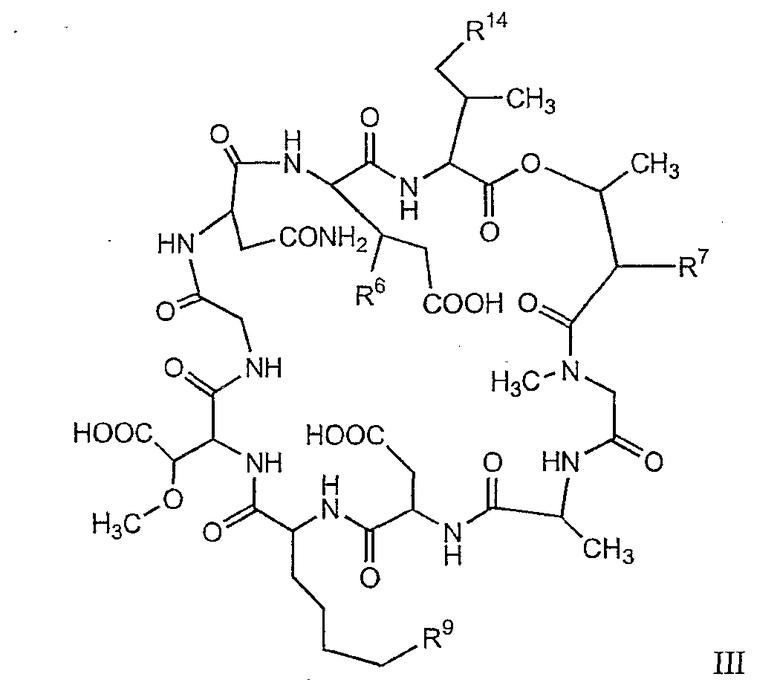
где R14 представляет собой гидридо или метил.
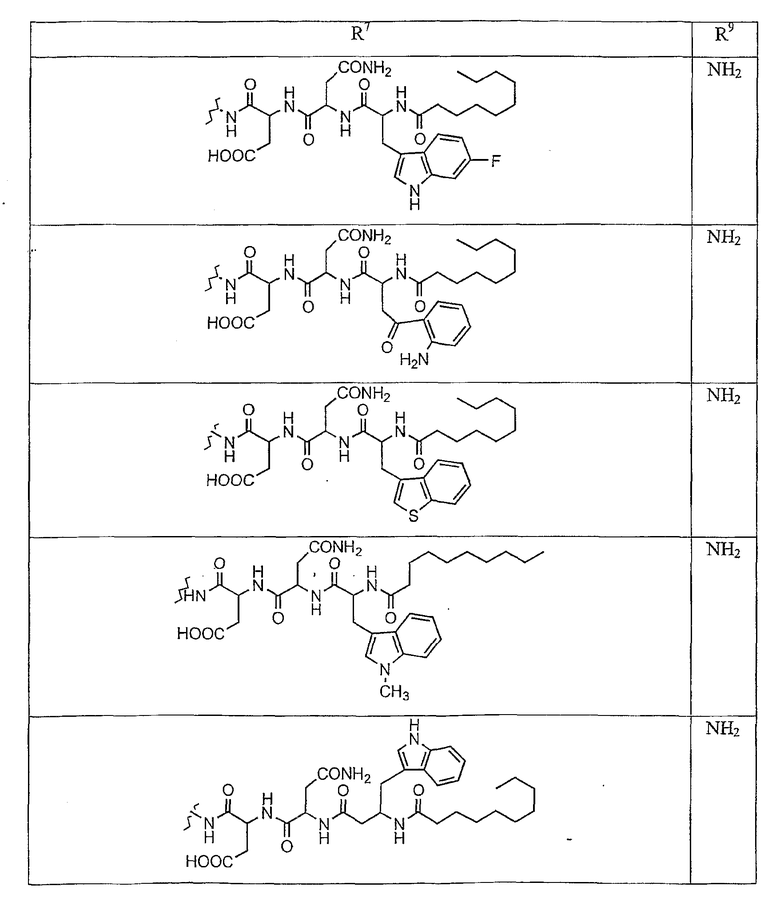
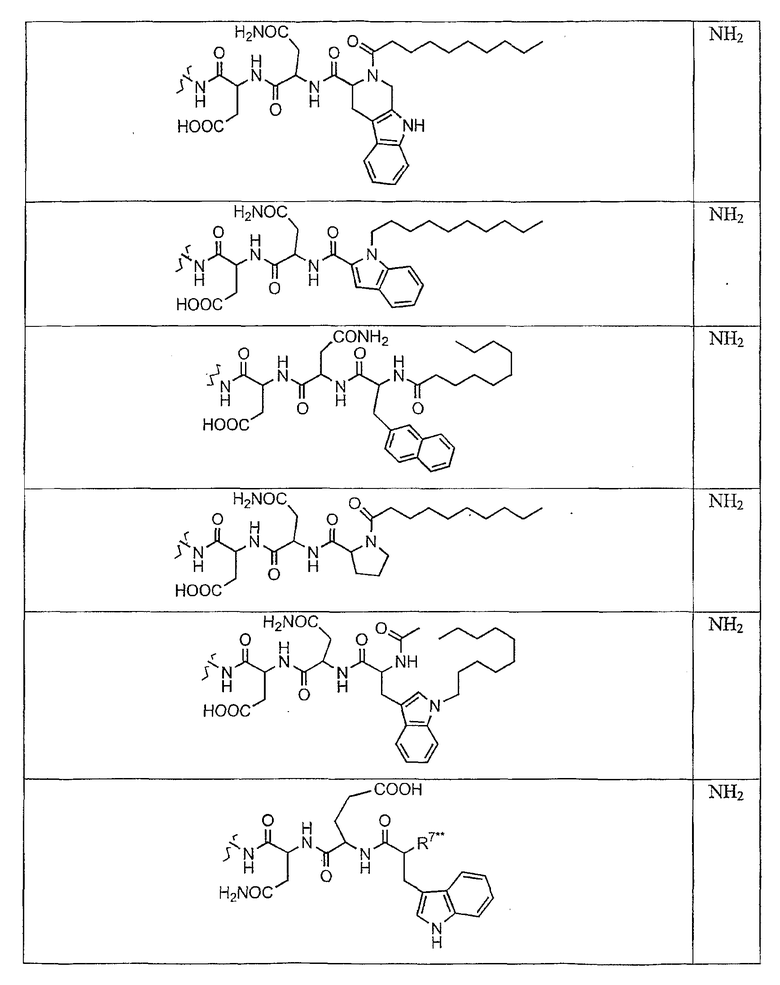
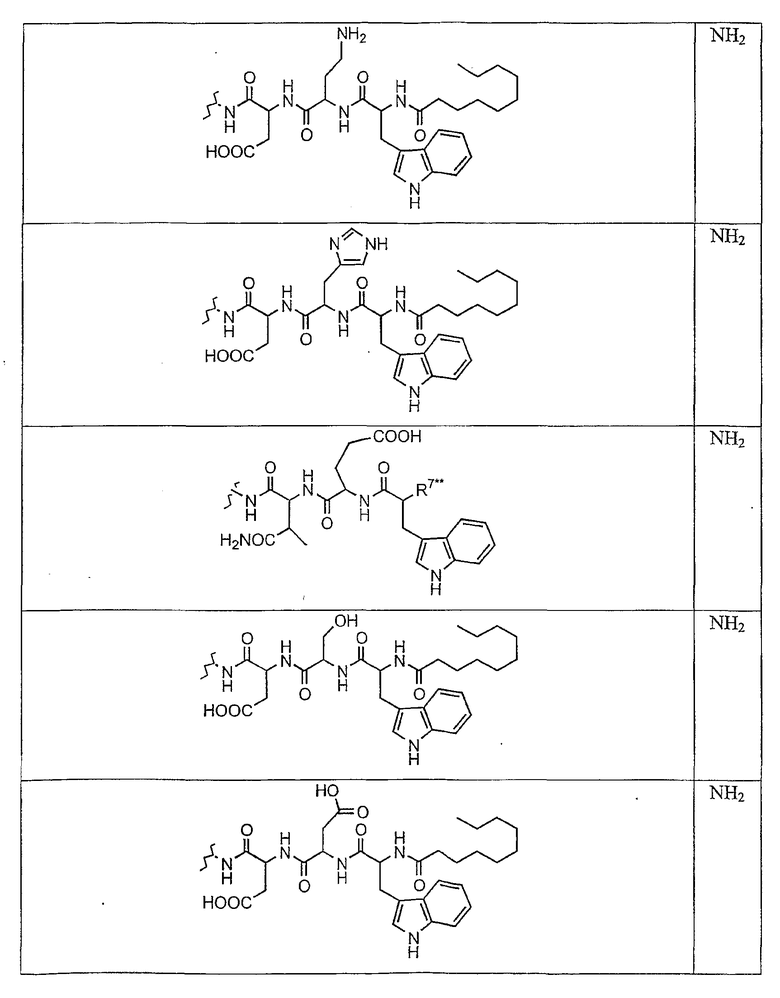
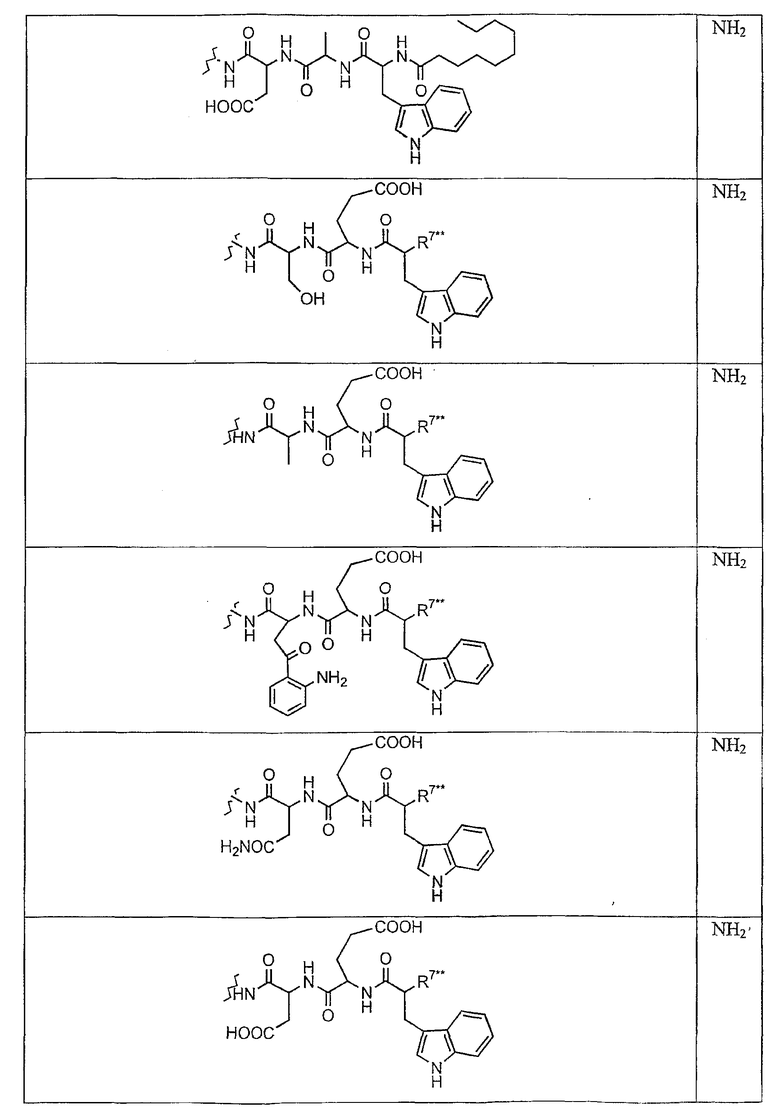
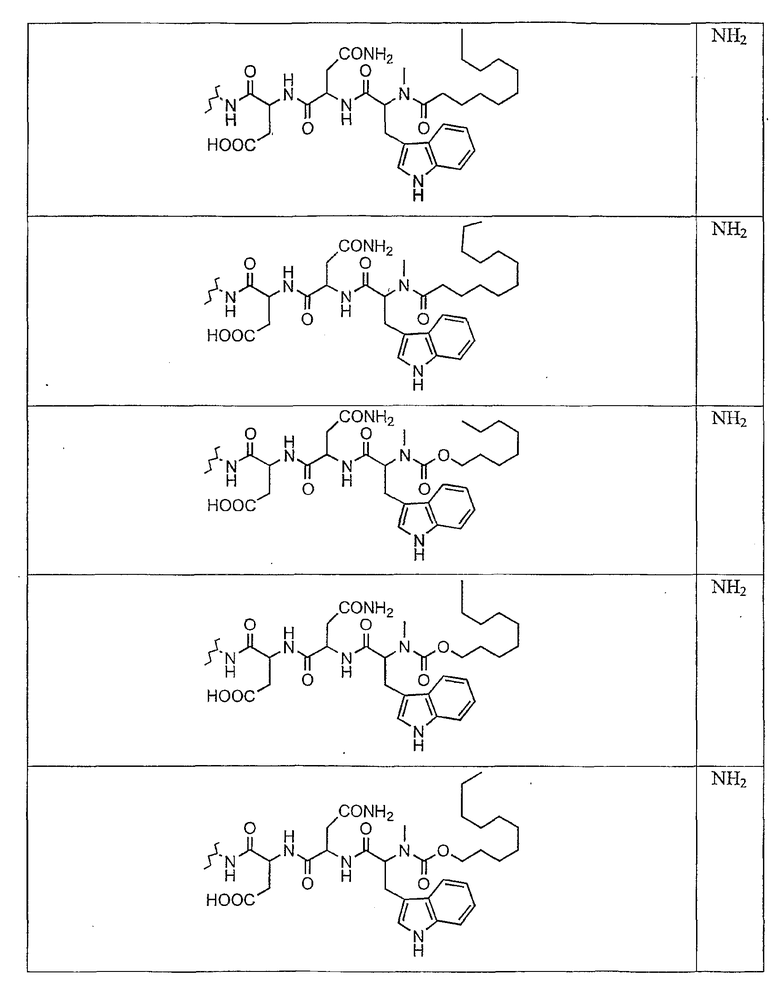
В таблице I представлены типичные соединения формулы II.
Таблица I
Соединения формулы II
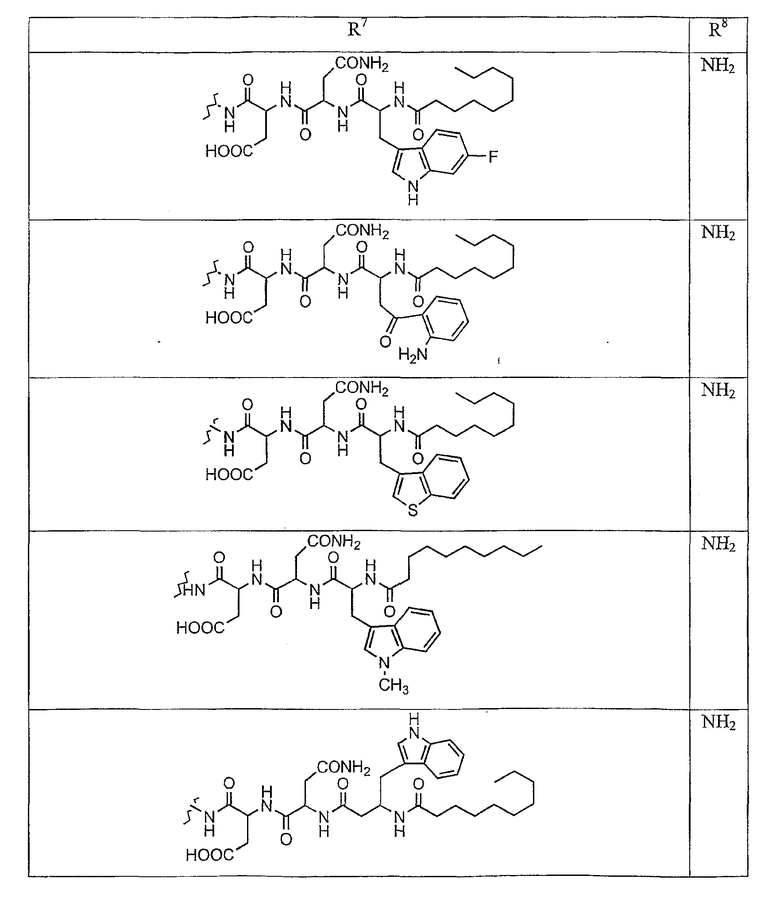
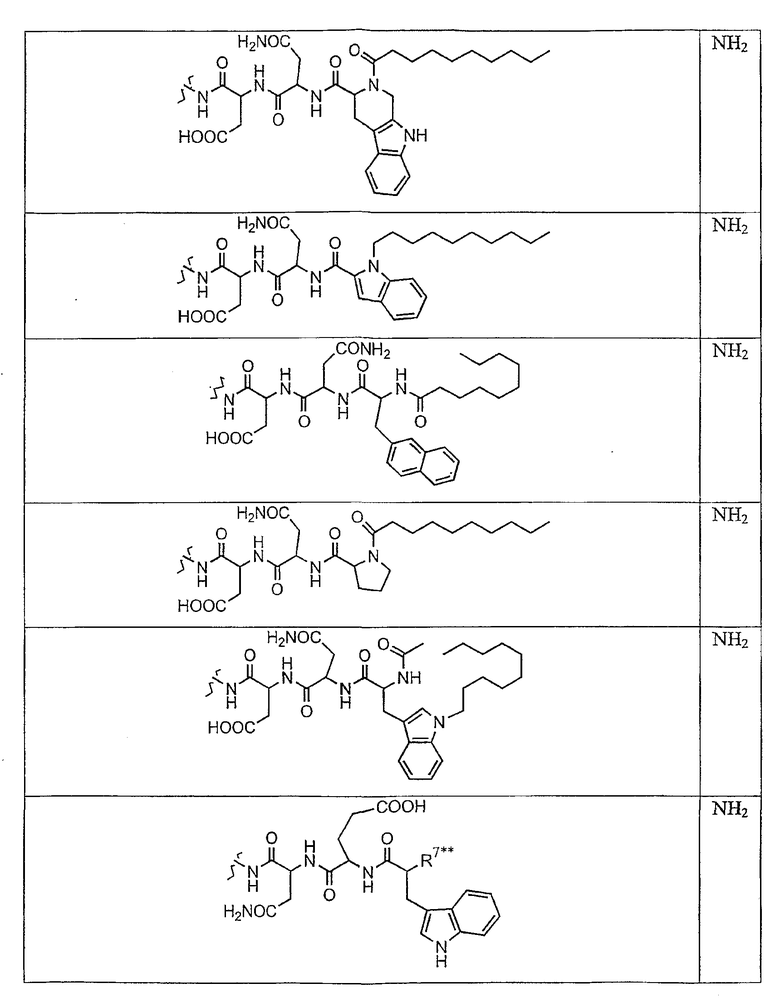
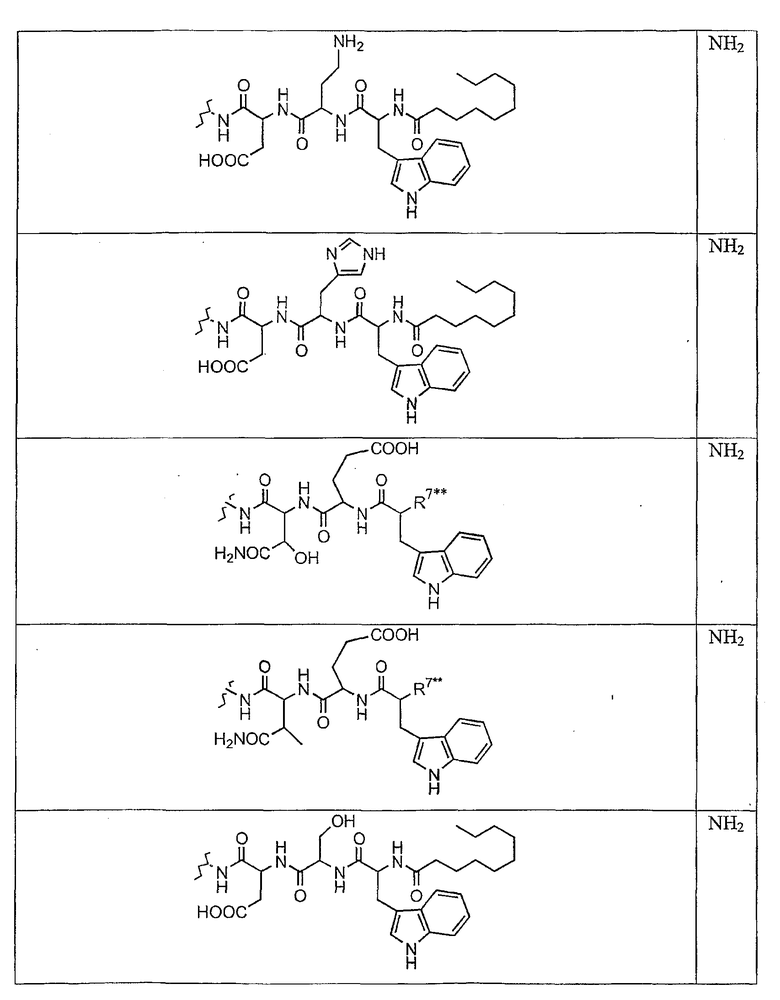
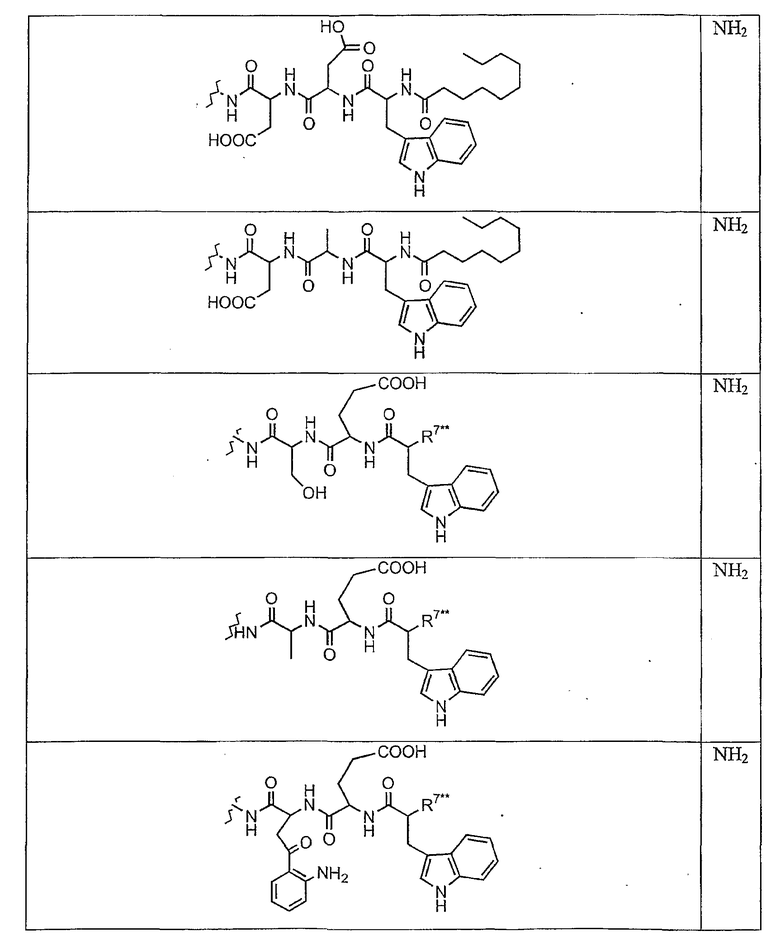
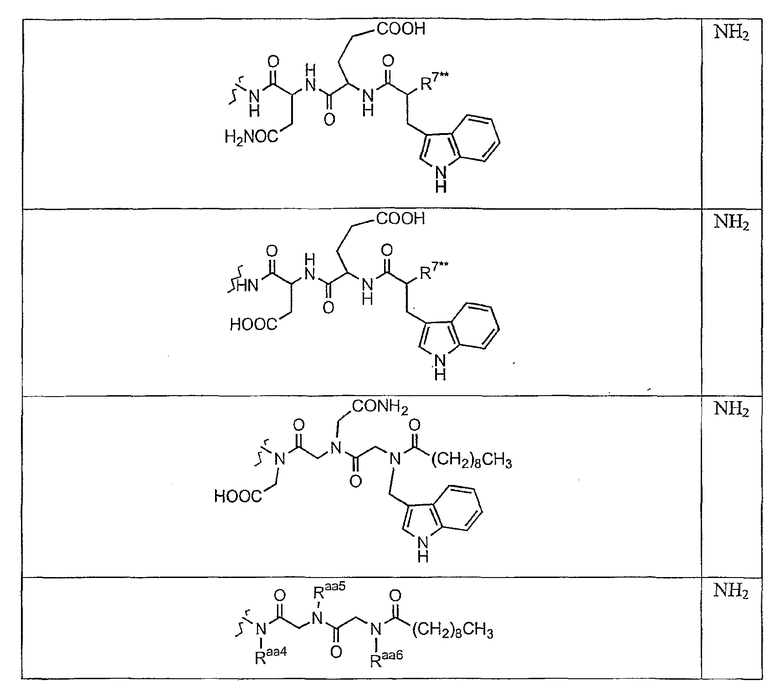
где R7** представляет собой амино, монозамещенный амино, дизамещенный амино, ациламино, уреидо, гуанидино, карбамоил, сульфонамино, тиоациламино, тиоуреидо, иминоамино или фосфонамино и каждый из Raa4, Raa5 и Raa6 независимо представляет собой боковую цепь аминокислоты.
В таблице II представлены типичные соединения формулы III.
Таблица II
Соединения формулы III
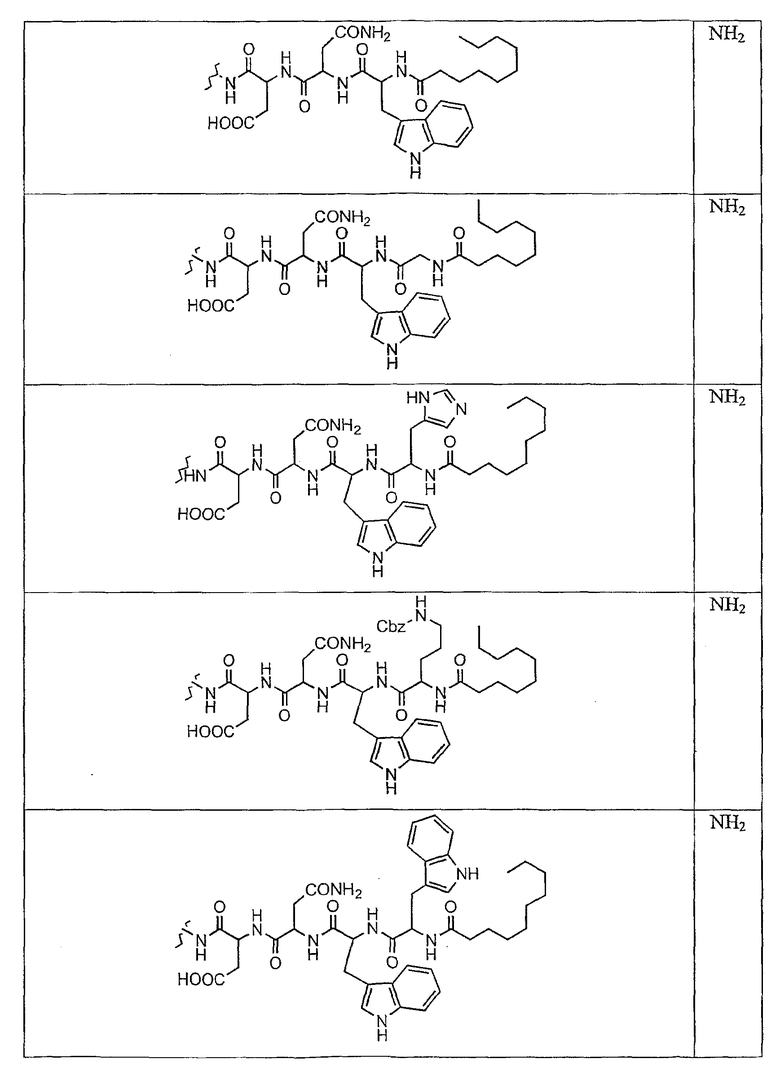
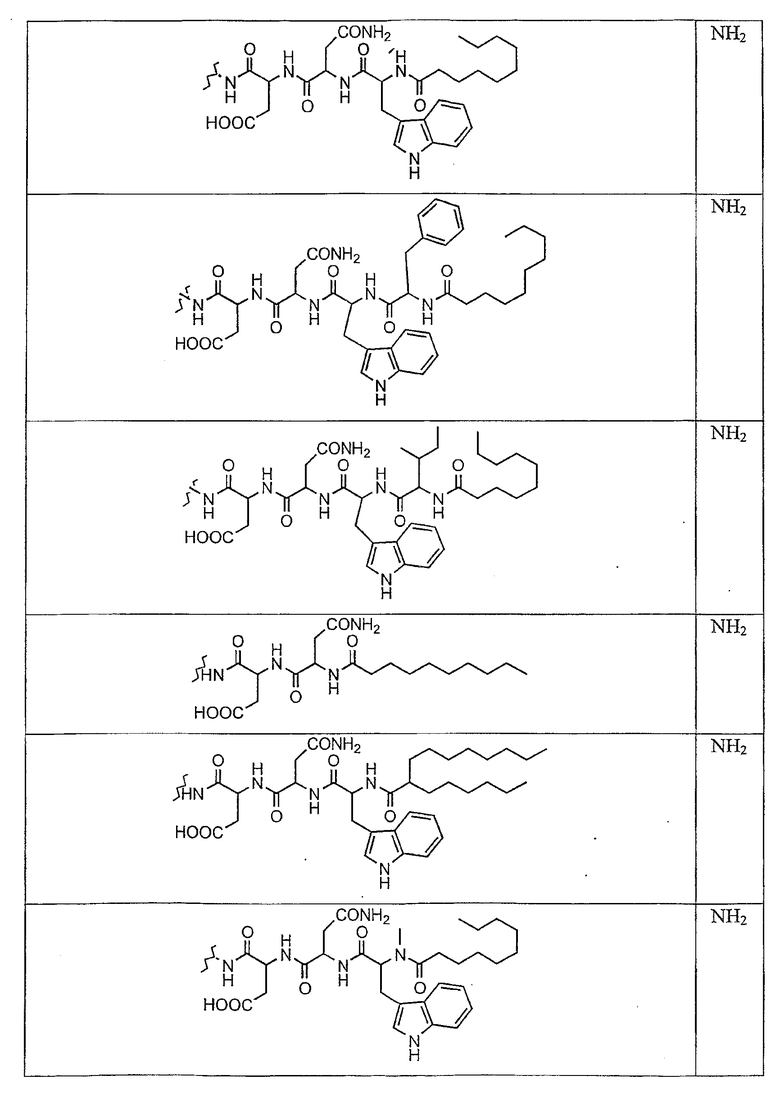
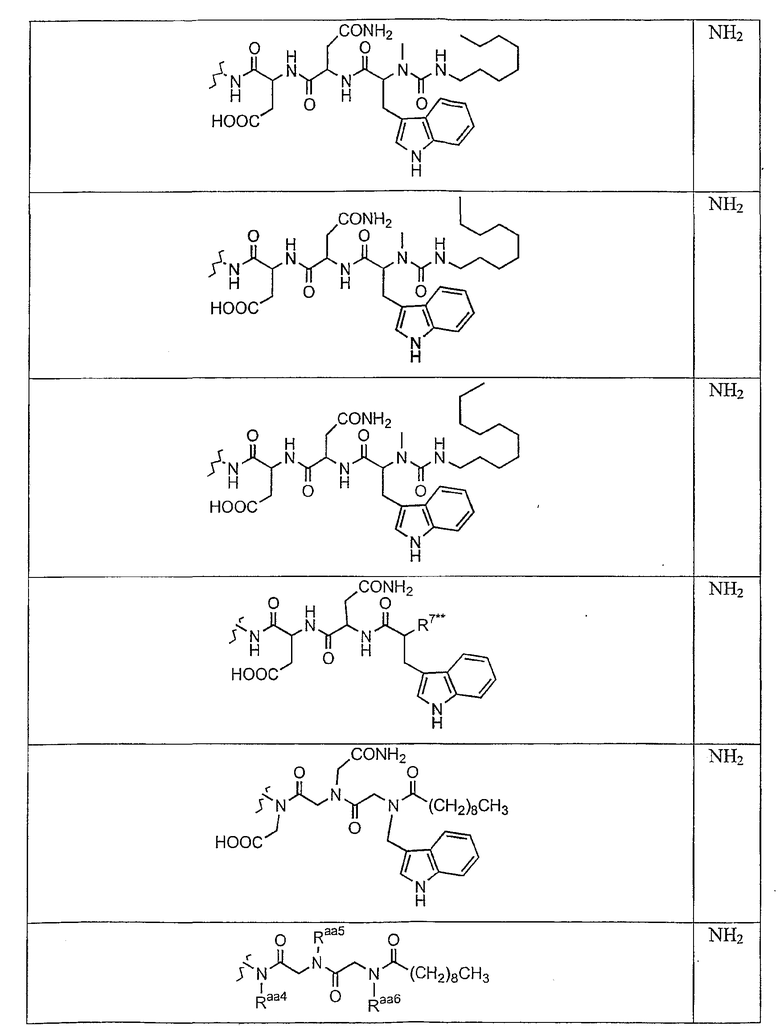
где R7** представляет собой амино, монозамещенный амино, дизамещенный амино, ациламино, уреидо, гуанидино, карбамоил, сульфонамино, тиоациламино, тиоуреидо, иминоамино или фосфонамино и каждый из Raa4, Raa5 и Raa6 независимо представляет собой боковую цепь аминокислоты.
Промежуточные продукты
В настоящем изобретении также предлагаются соединения формулы IV, которые особенно пригодны в качестве промежуточных продуктов для получения соединений формулы I.
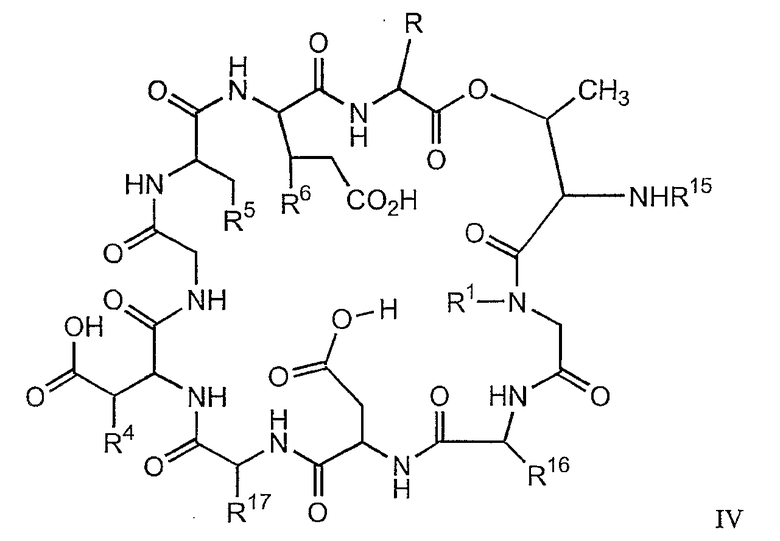
и их соли;
где
(a) R представляет собой 2-бутил, изопропил или 2-(2'-аминофенацил);
(b) каждый из R1 и R6 независимо представляет собой гидридо или метил;
(c) R4 представляет собой гидридо или метокси;
(d) R5 представляет собой гидрокси или карбоксиамино;
(e) R15 представляет собой гидридо,
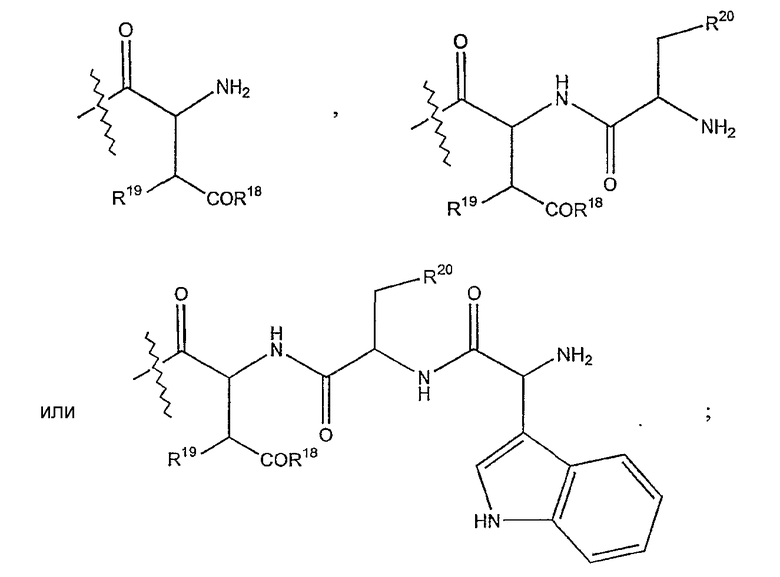
где R18 представляет собой амино или гидрокси; R19 представляет собой гидридо или гидрокси; а R20 представляет собой карбоксиамино или карбоксиметил
(f) R16 представляет собой метил или -CH2CH2CH2R21;
(g) R17 представляет собой метил или -CH2CH2CH2CH2R22;
где каждый из R21 и R22 независимо представляет собой амино, монозамещенный амино, дизамещенный амино, ациламино, уреидо, гуанидино, карбамоил, сульфонамино, тиоациламино, тиоуреидо, иминоамино или фосфонамино.
В предпочтительном осуществлении изобретения каждый из R21 и R22 представляет собой -NHR23, где R23 представляет собой аминозащитную группу. В более предпочтительном осуществлении изобретения R23 представляет собой карбаматную аминозащитную группу, выбранную из аллилоксикарбонила, карбобензилоксикарбонила и трет-бутоксикарбонила. В наиболее предпочтительном осуществлении R23 представляет собой аллилоксикабонил.
В более предпочтительном осуществлении в настоящем изобретении предложены промежуточные продукты формул V, VI, VII, VIII, IX и X, которые особенно пригодны в качестве промежуточных продуктов для получения соединений формулы I.
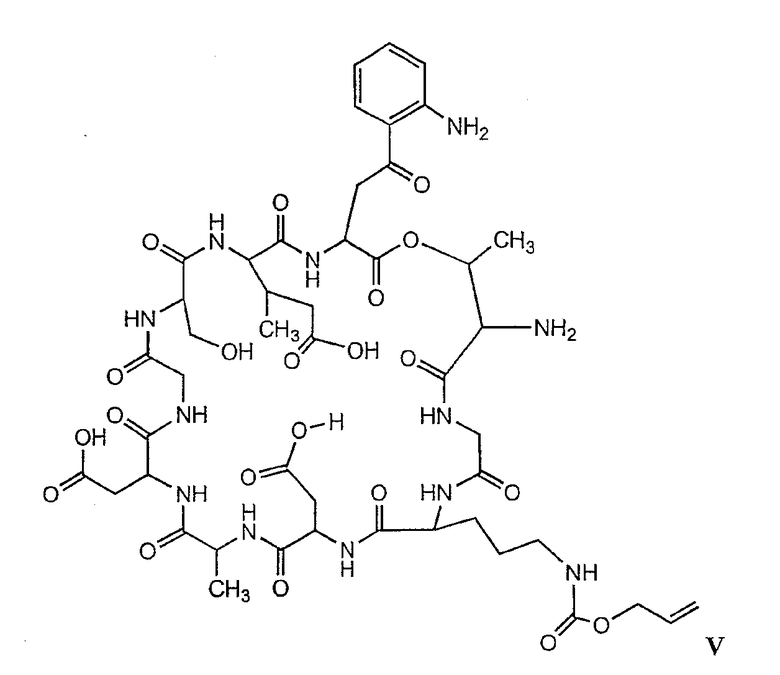
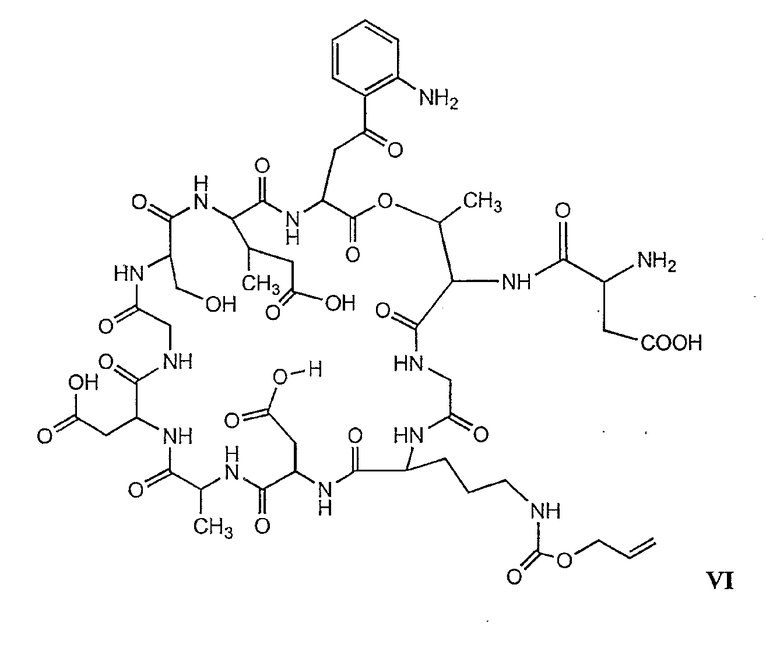
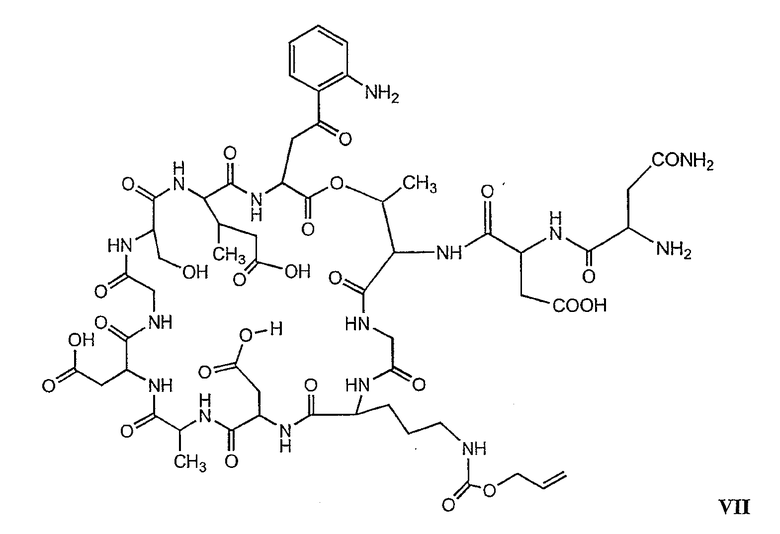
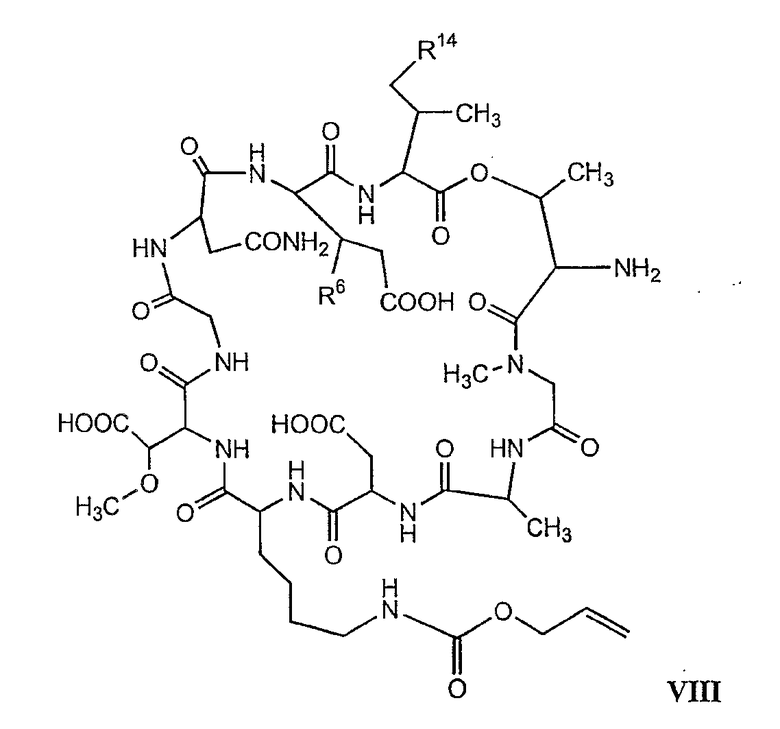
где R6 и R14 представляют собой определенное ранее.
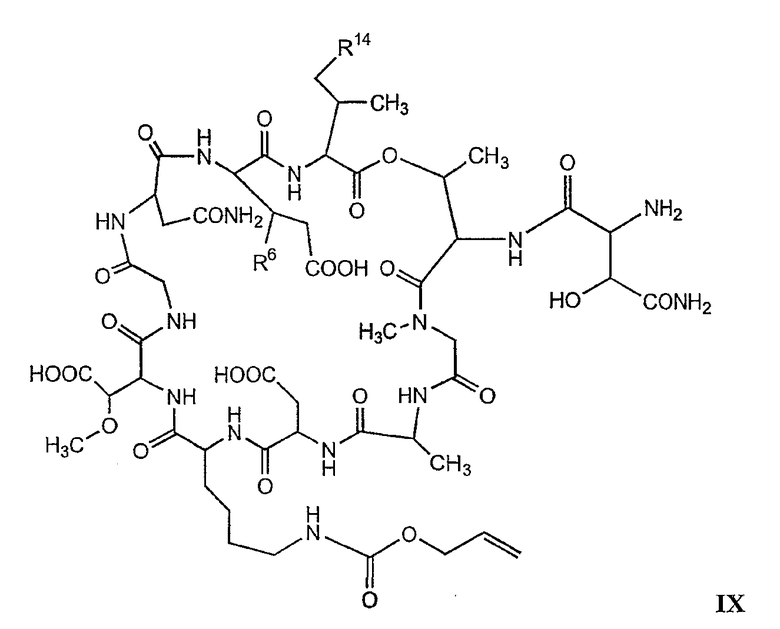
где R6 и R14 представляют собой определенное ранее.
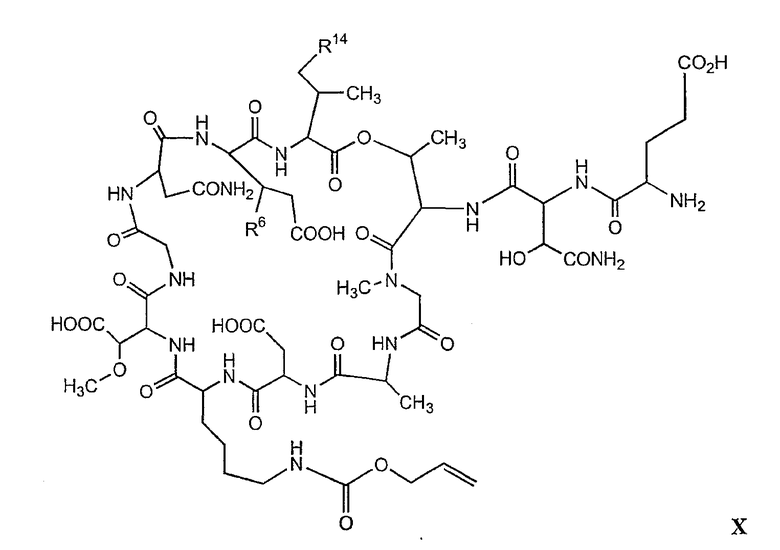
где R6 и R14 представляют собой определенное ранее.
Фармацевтические композиции и способы их применения
В настоящем изобретении предложены фармацевтические композиции или составы, включающие соединения формулы I или их соли.
Соединения по настоящему изобретению, предпочтительно, соединения формулы I, или их фармацевтически приемлемые соли могут быть включены в состав для перорального, внутривенного, внутримышечного, подкожного или парентерального введения для терапевтического или профилактического лечения заболеваний, в частности, бактериальных инфекций. Для перорального или парентерального введения соединения по настоящему изобретению могут быть смешаны с обычно используемыми фармацевтическими носителями и наполнителями и могут применяться в виде таблеток, капсул, эликсиров, суспензий, сиропов, облаток и тому подобного. Композиции, включающие соединение данного изобретения, должны содержать от приблизительно 0,1 до приблизительно 99% по массе активного соединения, и более обычно от приблизительно 10 до приблизительно 30%.
Описанные здесь фармацевтические препараты получают в соответствии со стандартными процедурами и их вводят в дозах, которые выбраны для снижения, предотвращения или уничтожения инфекции (смотри, например, Remington's Pharmaceutical Sciences, Mack Publishing Company, Easton, PA and Goodman and Gliman's The Pharmaceutical Basis of Therapeutics, Pergamon Press, New York, NY, содержание которых включено здесь в качестве ссылки для общего описания способов введения различных антимикробных агентов для лечения человека). Композиции по настоящему изобретению, предпочтительно, соединения формулы I, могут быть доставлены с применением систем доставки с контролируемым (например, капсулы) или с поддерживаемым (например, биодеградируемые матриксы) высвобождением. Типичные системы доставки с отставленным высвобождением для доставки лекарств, которые пригодны для введения композиций изобретения, предпочтительно формулы I, описаны в патентах США № 4452775 (владелец патента Kent), 5239660 (Leonard), 3854480 (Zaffaroni).
Фармацевтически приемлемые композициия по настоящему изобретению содержат одно или несколько соединений по изобретению, предпочтительно, соединения формулы I, в сочетании с одним или более нетоксичными фармацевтически приемлемыми носителями и/или разбавителями, и/или адъювантами, и/или наполнителями, обобщенно обозначаемые здесь как вещества "носителя", и, если это желательно, с другими активными ингредиентами. Композиции могут содержать обычные носители и наполнители, такие как кукурузный крахмал или желатин, лактоза, сахароза, микрокристаллическая целлюлоза, каолин, маннит, дикальцийфосфат, хлорид натрия и альгиновая кислота. Композиции могут содержать кроскармеллозу натрия, микрокристаллическую целлюлозу, кукурузный крахмал, натриевую соль гликолата крахмала и альгиновую кислоту.
Связующими агентами, которые могут входить в состав таблеток, являются камедь, метилцеллюлоза, натриевая соль карбоксиметилцеллюлозы, поливинилпирролидон (Povidone), гидроксипропилметилцеллюлоза, сахароза, крахмал и этилцеллюлоза.
Смазывающие агенты, которые могут использоваться, включают стеарат магния или другие стеараты металлов, стеариновую кислоту, силиконовую жидкость, тальк, воски, масла и коллоидный диоксид кремния.
Могут также использоваться ароматизаторы, такие как перечная мята, масло зимолюбки, вишневая отдушка или тому подобное. Может быть также желательным добавление окрашивающего агента для получения более эстетичной по виду дозированной формы или для помощи в идентификации продукта.
Для перорального применения особенно пригодны твердые составы, такие как таблетки и капсулы. Могут быть также применены препараты с поддерживаемым высвобождением или препараты, покрытые для всасывания в кишечнике. Для применения в педиатрии и гериатрии особенно пригодными являются суспензии, сиропы и жевательные таблетки. Для перорального введения фармацевтические композиции могут быть в виде, например, таблетки, капсулы, суспензии или жидкости. Фармацевтическую композицию предпочтительно изготавливают в виде стандартной лекарственной формы, содержащей терапевтически эффективное количество активного ингредиента. Примерами таких стандартных лекарственных форм являются таблетки и капсулы. Для терапевтических целей таблетки и капсулы могут содержать, в дополнение к активному ингредиенту, обычные носители, такие как связующие агенты, например, аравийскую камедь, желатин, поливинилпирролидон, сорбит или трагакант; наполнители, например, фосфат кальция, глицин, лактозу, маисовый крахмал, сорбит или сахарозу; смазывающие агенты, например, стеарат магния, полиэтиленгликоль, диоксид кремния или тальк; разрыхляющие агенты, например, картофельный крахмал, ароматизирующие или окрашивающие агенты, или приемлемые увлажняющие агенты. Пероральные жидкие препараты обычно находятся в виде водных или масляных растворов, суспензий, эмульсий, сиропов или эликсиров, и могут содержать традиционные добавки, такие как суспендирующие агенты, эмульгирующие агенты, неводные агенты, консерванты, окрашивающие агенты и ароматизирующие агенты. Примеры добавок для жидких препаратов включают камедь, миндальное масло, этиловый спирт, фракционированное кокосовое масло, желатин, глюкозный сироп, глицерин, гидрогенизированные пищевые жиры, лецитин, метилцеллюлозу, метил или пропил пара-гидроксибензоат, пропиленгликоль, сорбит или сорбиновую кислоту.
Для внутривенного (в/в) применения соединение по настоящему изобретению может быть растворено или суспендировано в любой из обычно применяемых для внутривенного введения жидкостях и введено путем инфузии. Внутривенные жидкости включают без ограничения физиологический раствор или раствор Рингера. Внутривенное введение может быть осуществлено с помощью без ограничения шприца, минипомпы или внутривенной системы.
Составы для парентерального введения могут находиться в виде водных или неводных изотонических стерильных растворов или суспензий для инъекции. Данные растворы или суспензии могут быть получены из стерильных порошков или гранул, содержащих один или несколько носителей, упоминавшихся для применения в составах для перорального введения. Соединения могут быть растворены в полиэтиленгликоле, пропиленгликоле, этаноле, кукурузном масле, бензиловом спирте, хлориде натрия и/или различных буферах.
Для внутримышечных препаратов стерильный состав соединения по настоящему изобретению или подходящая растворимая солевая форма соединения, например, гидрохлоридная соль, могут быть растворены и введены в фармацевтическом разбавителе, таком как вода для инъекций (WFI), физиологический раствор или 5% глюкоза. Подходящая нерастворимая форма соединения может быть получена и введена в виде суспензии на водной основе или на фармацевтически приемлемой масляной основе, например, в эфире жирной кислоты с длинной цепью, таком как этилолеат.
Доза внутривенного, внутримышечного или парентерального состава соединения по настоящему изобретению может быть введена в виде болюса или путем медленной инфузии. Болюс представляет собой стандартную лекарственную форму, которую вводят менее чем за 30 минут. В предпочтительном осуществлении болюс вводят менее чем за 15 или менее, чем за 10 минут. В более предпочтительном осуществлении болюс вводят менее чем за 5 минут. В еще более предпочтительном осуществлении болюс вводят в течение одной минуты или менее. Инфузия представляет собой дозу, которую вводят на протяжении 30 минут или более. В предпочтительном осуществлении инфузия продолжается один час или более. В другом осуществлении инфузия по существу является постоянной.
Для местного применения соединения по настоящему изобретению, предпочтительно, соединения формулы I, могут быть также получены в формах, подходящих для нанесения на кожу или на мембраны слизистых носа и горла, и могут быть в виде кремов, мазей, жидких спреев или составов для ингаляции, лепешек или смазывающих средств для горла. Такие местные составы могут дополнительно включать химические соединения, такие как диметилсульфоксид (ДМСО), для облегчения проникновения активного ингредиента через поверхность. Для нанесения в глаза или уши соединения по настоящему изобретению, предпочтительно, соединения формулы I могут быть представлены в жидкой или полужидкой форме, изготовленной на гидрофобной или гидрофильной основе, в виде мазей, кремов, лосьонов, смазывающих средств или порошков.
Для ректального введения соединения по настоящему изобретению, предпочтительно соединения формулы I, можно вводить в виде суппозиториев, на основе смесей с обычно используемыми носителями, такими как масло какао, воск или другой глицерид.
Альтернативно, соединения по настоящему изобретению, предпочтительно, соединения формулы I, могут быть в виде порошка для распределения его в подходящем фармацевтически приемлемом носителе во время использования. В другом осуществлении стандартная лекарственная форма соединения может быть раствором соединения или, предпочтительно, его соли в подходящем разбавителе в стерильных запаянных ампулах или стерильных шприцах. Концентрация соединения в стандартной лекарственной форме может варьироваться, например, от приблизительно 1 процента до приблизительно 50 процентов, в зависимости от используемого соединения и его растворимости и желаемой врачом дозы. Если композиции содержат единичные дозы, каждая единичная доза, предпочтительно, содержит 1-500 мг активного вещества. Для лечения взрослого человека применяемая доза, предпочтительно, варьирует от 5 мг до 10 г в день, в зависимости от пути и частоты введения.
В другом аспекте в данном изобретении предложен способ ингибирования роста микроорганизмов, предпочтительно, бактерий, включающий контактирование указанных организмов с соединением по настоящему изобретению в условиях, которые обеспечивают контакт соединения с указанным организмом и с указанным микроорганизмом. Такие условия известны специалисту в данной области техники и проиллюстрированы в примерах. Данный способ включает контактирование микробной клетки с терапевтически эффективным количеством соединения(ний) по изобретению, предпочтительно, соединения(ний) формулы I, in vivo или in vitro.
В соответствии с данным аспектом изобретения новые описанные здесь композиции помещают в фармацевтически приемлемый носитель и они доставляются субъекту-реципиенту (предпочтительно, человеку) в соответствии с известными способами доставки лекарств. Обычно в способах по изобретению для доставки композиций по изобретению in vivo применяются известные в данной области техники методы осуществления доставки агента, по существу, только с одним изменением, заключающемся в том, что при осуществлении этих известных в данной области техники методов в качестве лекарств используют соединения по настоящему изобретению, предпочтительно, соединений формулы I. Более того, в способах применения заявленной композиции для обработки клеток в культуре, например, для уничтожения или снижения уровня бактериального загрязнения клеточной культуры, используют известные в данной области техники способы для обработки клеточных культур антибактериальным(ми) агентом(тами) по существу только с одним изменением способа, заключающимся в том, что при осуществлении этих известных в данной области методик используют соединения по настоящему изобретению, предпочтительно, соединения формулы I.
В одном осуществлении в данном изобретении предложен способ лечения инфекции, в частности, вызываемой грамположительными бактериями, у субъекта с помощью терапевтически эффективного количества соединения по изобретению. Примеры методов для доставки антибактериального агента описаны в патенте США No. 5041567 и патентной заявке PCT номер EP94/02552 (публикация WO 95/05384), полное содержание этих документов включено здесь в качестве ссылок. Используемая здесь фраза «терапевтически эффективное количество» обозначает количество соединения по настоящему изобретению, которое предотвращает возникновение, облегчает симптомы или останавливает развитие бактериальной инфекции. Термин «лечение» определяется как введение субъекту терапевтически эффективного количества соединения по изобретению, как для предотвращения возникновения инфекции, так и для контролирования или уничтожения инфекции. Описываемый здесь термин «субъект» определяется как млекопитающее, растение или клеточная культура. В предпочтительном осуществлении субъектом является человек или другое больное животное, нуждающееся в антибактериальном лечении.
Способ включает введение субъекту эффективной дозы соединения по настоящему изобретению. Эффективная доза обычно составляет от приблизительно 0,1 до приблизительно 100 мг/кг соединения по изобретению или его фармацевтически приемлемой соли. Предпочтительная доза составляет от приблизительно 0,1 до приблизительно 50 мг/кг соединения по изобретению или его фармацевтически приемлемой соли. Более предпочтительная доза составляет от приблизительно 1 до 25 мг/кг соединения по изобретению или его фармацевтически приемлемой соли. Эффективная доза для клеточной культуры обычно составляет от приблизительно 0,1 до 1000 мкг/мл, более предпочтительно от 0,1 до 200 мкг/мл.
Композиции, содержащие соединения по изобретению, могут быть введены в виде единичной суточной дозы или в виде разбитых доз в день. Для лечения может потребоваться введение в течение продолжительных периодов времени, например, в течение нескольких дней или от двух до четырех недель. Количество на вводимую дозу или суммарное вводимое количество должно зависеть от таких факторов, как природа и тяжесть инфекции, возраст и общее состояние здоровья больного, толерантность больного к соединению и микроорганизм или микроорганизмы, вовлеченные в инфекцию. Способ введения больному даптомицина, другого члена депсипептидного класса соединений, описан в патенте США с серийным № 09/406568, поданным 24 сентября 1999 г., по которому испрашивается приоритет по предварительным заявкам США №№ 60/101828, поданной 25 сентября 1998 г., и 60/125750, поданной 24 марта 1999 г., содержание которых включено здесь в качестве ссылки.
Соединения по настоящему изобретению могут также быть введены больному или животному при кормлении или с пищей. При введении в виде части от суммарного количества потребляемой пищи количество применяемого соединения может составлять менее 1% от массы продуктов и, предпочтительно, не более 0,5% по массе. Продукты для животных могут быть обычными продуктами, к которым может быть добавлено соединение, или оно может быть добавлено к заранее приготовленной смеси.
В настоящем изобретении также предложены способы введения соединения формулы I или его фармацевтической композиции нуждающемуся в нем субъекту в количестве, которое эффективно для снижения или уничтожения бактериальной инфекции. Соединение может быть введено перорально, парентерально, с помощью ингаляции, местно, ректально, интраназально, защечно, вагинально или с помощью имплантируемого резервуара, наружной помпы или катетера. Соединение может быть адаптировано для офтальмологического или аэрозольного применения. Соединения по настоящему изобретению могут быть введены в виде аэрозоля для лечения пневмонии или других легочных инфекций. Предпочтительным средством для аэрозольной доставки является безводный ингалятор или ингалятор сухого порошка. Соединения формулы I или их фармацевтическая композиция могут также быть инъецированы или введены непосредственно в абсцесс, желудочек или сустав. Парентеральное введение включает подкожную, внутривенную, внутримышечную, внутрисуставную, интрасиновиальную, полостную, подоболочечную, внутрипеченочную, в область повреждения и внутричерепную инъекцию или инфузию. В предпочтительном осуществлении соединения по настоящему изобретению вводят внутривенно, подкожно или перорально. В предпочтительном осуществлении для введения соединения формулы I в клеточную культуру соединение может быть включено в питательную среду.
Способ по настоящему изобретению может быть применен для лечения субъекта, имеющего бактериальную инфекцию, у которого инфекция обусловлена или осложнена любым типом бактерий, в частности, грамположительными бактериями. В одном осуществлении соединение по настоящему изобретению или его фармацевтическую композицию вводят больному в соответствии со способами изобретения. В предпочтительном осуществлении бактериальная инфекция может быть вызвана или осложнена грамположительными бактериями. Данные грамположительные бактерии включают, но не ограничиваются этим, чувствительные к метициллину и устойчивые к метициллину стафилококки (включая Staphylococcus aureus, S. epidermidis, S. haemolyticus, S. hominis, S. saprophyticus и коагулаза-негативные стафилококки), восприимчивые к промежуточному продукту гликопептида S. aureus (GISA), устойчивые к ванкомицину Staphylococcus aureus (VRSA), восприимчивые к пенициллину и устойчивые к пенициллину с стрептококки (включая Streptococcus pneumoniae, S. pyogenes, S. agalactiae, S. avium, S. bovis, S. lactis, S. sangius и стрептококки группы C, стрептококки группы G и стрептококки Streptococci Group C, Streptococci Group G и зеленящие стрептококки), энтерококки (включая восприимчивые к ванкомицину и устойчивые к ванкомицину штаммы, такие Enterococcus faecalis и E. Faecium), Clostridium difficile, C. clostridiiforme, C. innocuum, C. perfringens, C. ramosum, Haemophilus influenzae, Listeria monocytogenes, Corynebacterium jeikeium, Bifidobacterium spp., Eubacterium aerofaciens, E. lentum, Lactobacillus acidophilus, L. casei, L. plantarum, Lactococcus spp., Leuconostoc spp., Pediococcus, Peptostreptococcus anaerobius, P. asaccarolyticus, P. magnus, P. micros, P. prevotii, P. productus, Propionibacterium acnes, Actinomyces spp., Moraxella spp. (включая M. catarrhalis) и Escherichia spp. (включая E. coli).
В предпочтительном осуществлении антибактериальная активность соединений формулы I в отношении классических «устойчивых» штаммов сравнима с таковой в отношении «восприимчивых» штаммов в экспериментах in vitro. В другом предпочтительном осуществлении величина минимальной ингибиторной концентрации (MIC) соединений в соответствии с данным изобретением в отношении восприимчивых штаммов является обычно такой же или ниже таковой ванкомицина. Таким образом, в предпочтительном осуществлении соединение по данному изобретению или его фармацевтическую композицию вводят в соответствии со способами данного изобретения больному, у которого обнаружена бактериальная инфекция, которая является устойчивой к другим соединениям, включая ванкомицин или даптомицин. Кроме того, в отличие от гликопептидных антибиотиков депсипептидные соединения, такие как описанные в настоящем изобретении, проявляют быструю, дозозависимую бактерицидную активность против грамположительных организмов. Таким образом, в предпочтительном осуществлении соединение в соответствии с настоящим изобретением или его фармацевтическую композицию вводят в соответствии со способами данного изобретения больному, нуждающемуся в быстро действующей терапии антибиотиками.
Способ по настоящему изобретению может быть применен для любой бактериальной инфекции любого органа или ткани организма. В предпочтительном осуществлении бактериальная инфекция вызывается грамположительными бактериями. Данные органы или ткань включают, без ограничения, скелетную мышцу, кожу, кровоток, почки, сердце, легкие и кости. Способ изобретения может быть применен для лечения без ограничения инфекций кожи и мягких тканей, бактериемий и инфекций мочевого тракта. Способ по изобретению может быть применен для лечения передающихся контактным путем респираторных инфекций, включая, без ограничения, отит среднего уха, синусит, хронический бронхит и пневмонию, включая пневмонию, вызываемую устойчивыми к лекарствам S. Pneumoniae или H. Influenzae. Способ по изобретению может быть также применен для лечения смешанных инфекций, которые включают различные типы грамположительных бактерий, или которые включают как грамположительные, так и грамотрицательные бактерии. Данные типы инфекций включают внутрибрюшинные инфекции и акушерско/гинекологические инфекции. Способ изобретения также может быть применен для лечения инфекции, включающей, без ограничения, эндокардит, нефрит, септический артрит, внутрибрюшинный сепсис, инфекции костей и суставов и остеомиелит. В предпочтительном осуществлении любое из описанных выше заболеваний можно лечить с применением соединений в соответствии с данным изобретением или их фармацевтических композиций.
Способ по настоящему изобретению может быть также использован на практике наряду с одновременным введением одного или нескольких других антимикробных агентов, таких как антибактериальные агенты (антибиотики) или противогрибковые агенты. В одном аспекте способ может быть применен на практике путем введения более чем одного соединения в соответствии с данным изобретением. В другом осуществлении способ может быть применен на практике путем введения соединения в соответствии с данным изобретением с липопептидным соединением, таким как даптомицин или липопептидными соединениями, описанными, например, в международных патентных заявках WO 01/44272; WO 01/44274 и WO 01/44271.
Антибактериальные агенты и их классы, которые могут быть введены совместно с соединением по изобретению, включают, без ограничения, пенициллины и родственные им лекарства, карбапенемы, цефалоспорины и родственные им лекарства, аминогликозиды, бацитрацин, грамицидин, мупироцин, хлорамфеникол, тиамфеникол, фузидат натрия, линкомицин, клиндамицин, макролиды, новобиоцин, полимиксины, рифамицины, спектиномицин, тетрациклины, ванкомицин, теикопланин, стрептограмины, противофолатные агенты, включая сульфонамиды, триметоприм и их сочетания, а также пириметамин, синтетические антибактериальные агенты, включая нитрофураны, манделат метенамина и гиппурат метенамина, нитроимидазолы, хинолоны, фторхинолоны, изониазид, этамбутол, пиразинамид, пара-аминосалициловую кислоту (PAS), циклосерин, капреомицин, этионамид, протионамид, тиацетазон, виомицин, эверниномицин, гликопептид, глицилциклин, кетолиды, оксазолидинон; имипенен, амикацин, нетилмицин, фосфомицин, гентамицин, цефриаксон, зирацин, LY 333328, CL 331002, HMR 3647, Zyvox®, Synercid®, азтреонам и метронидазол, эпироприм, OCA-983, GV-143253, санфетринем натрия, CS-834, биапенем, A-99058.1, A-165600, A-179796, KA 159, динемицин А, DX8739, DU 6681; цефлупренам, ER 35786, цефоселес, санфетринемцелексетил, HGP-31, цефпиром, HMR-3647, RU-59863, мерсацидин, KP 736, рифалазил; AM 1732, MEN 10700, ленапенем, BO 2502A, NE-1530, PR 39, K130, OPC 20000, OPC 2045, венеприм, PD 138312, PD 140248, CP 111905, сулопенем, ритипенамакоксил, RO-65-5788, циклотиалидин, Sch-40832, SEP-132613, микакокидин A, SB-275833, SR-15402, SUN A0026, TOC 39, карумонам, цефозопран, цефетаметпивоксил и T 3811.
Противогрибковые агенты, которые могут быть введены совместно с соединением по изобретению, включают, без ограничения, каспофунген, вориконазол, сертаконазол, IB-367, FK-463, LY-303366, Sch-56592, ситафлоксазин, DB-289, полиены, такие как амфотерицин, нистатин, примарицин; азолы, такие как флуконазол, итраконазол и кетоконазол; аллиламины, такие как нафтифин и тербинафин; и антиметаболиты, такие как флуцитозин. Другие противогрибковые агенты включают без ограничения описанные Fostel et al., Drug Descovery Today 5:25-32 (2000), включенной здесь в качестве ссылки. Fostel et al. раскрывают противогрибковые соединения, включающие коринекандин, Mer-WF3010, фузакандины, артрихитин/LL 15G256, сордарины, циспентацин, азоксибациллин, ауреобасидин и кафрефунгин.
Соединение в соответствии с данным изобретением можно вводить согласно данному способу до тех пор, пока бактериальная инфекция не будет уничтожена или ослаблена. В одном осуществлении соединение формулы I вводят в течение периода времени от 2 дней до 6 месяцев. В предпочтительном осуществлении соединение формулы I вводят в течение от 7 до 56 дней. В более предпочтительном осуществлении соединение формулы I вводят в течение от 7 до 28 дней. В еще более предпочтительном осуществлении соединение формулы I вводят в течение от 7 до 14 дней. Соединение формулы I можно вводить в течение более длительного или более короткого периода времени, если это желательно.
Получение новых депсипептидов
1. Полусинтетический способ
Способ получения соединений формулы I, в которых, по меньшей мере, один из R2 и R3 отличен от метила и каждый из R8 и R9 независимо представляет собой NH2.
Способ A
Для соединений формулы I, в которых, по меньшей мере, один из R2 и R3 отличен от метила и каждый из R8 и R9 независимо представляет собой NH2, способ в соответствии с одним аспектом изобретения включает стадии:
(a) получения депсипептидного производного формулы XI
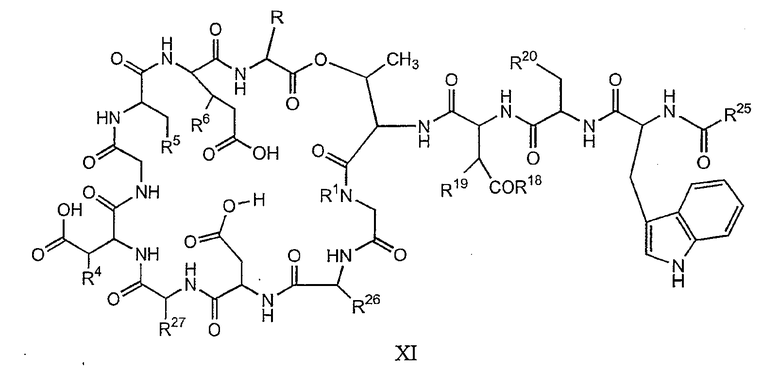
где R, R1, R4, R5, R6, R18, R19 и R20 описаны выше; R25 представляет собой алкильную группу; R26 представляет собой метил или -CH2CH2CH2NH2; и R27 представляет собой метил или -CH2CH2CH2CH2NH2; или его соли(b) защиты свободных(ой) аминогрупп(ы) соединения формулы XI защитной группой для получения защищенного депсипептидного соединения;
(c) обработки защищенного депсипептидного соединения, полученного на стадии (b), дезацилирующим агентом для получения соединения с концевым аминосоединением;
(d) удаления триптофанового аминокислотного остатка из соединения с концевым аминосоединением, полученного на стадии (c), для получения дезтриптофанового соединения;
(e) удаления концевого аминокислотного остатка из дезтриптофанового соединения, полученного на стадии (d), для получения дездипептидного соединения;
(f) удаления концевого аминокислотного остатка из дездипептидного соединения, полученного на стадии (e), для получения депсипептидного соединения;
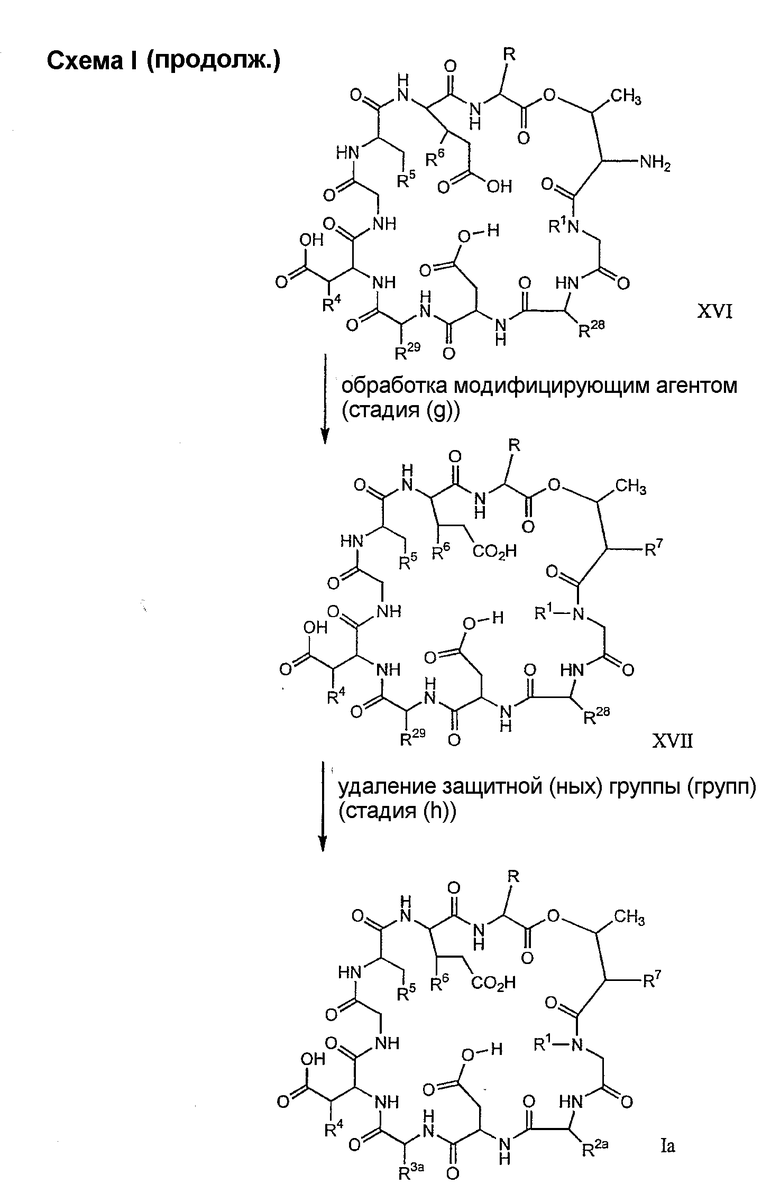
(g) обработки депсипептидного базового соединения со стадии (f) модифицирующим агентом; и
(h) удаления защитной группы из защищенного соединения формулы I для получения соединения формулы Ia.
Процедура A иллюстрирована на схеме I.
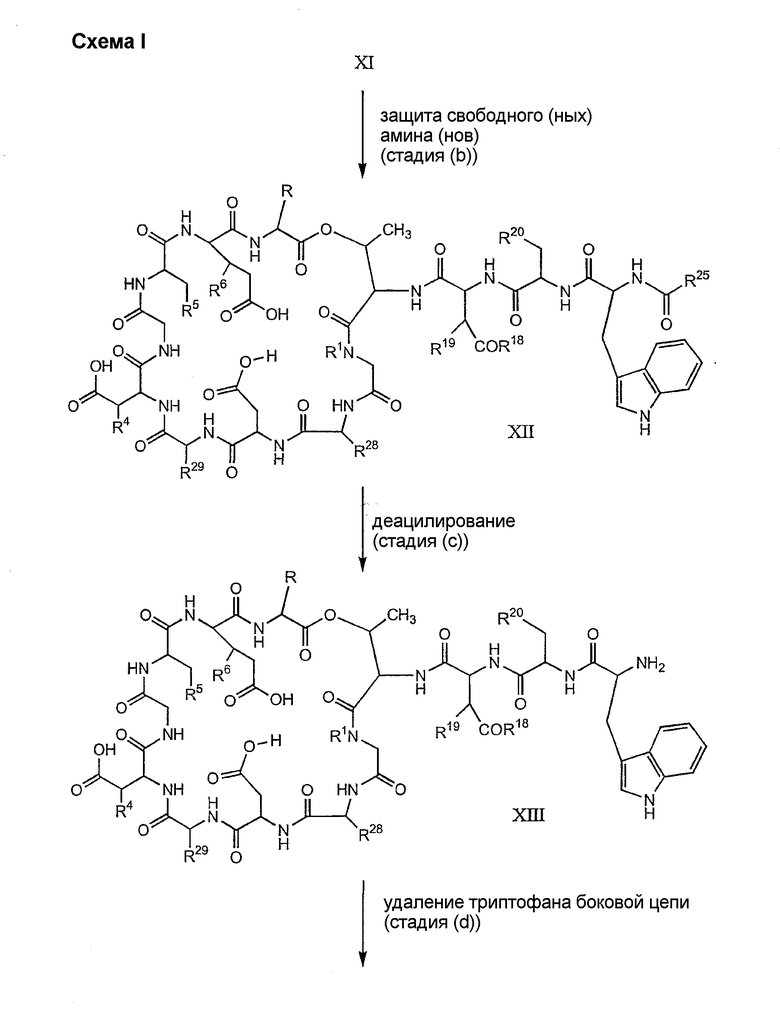
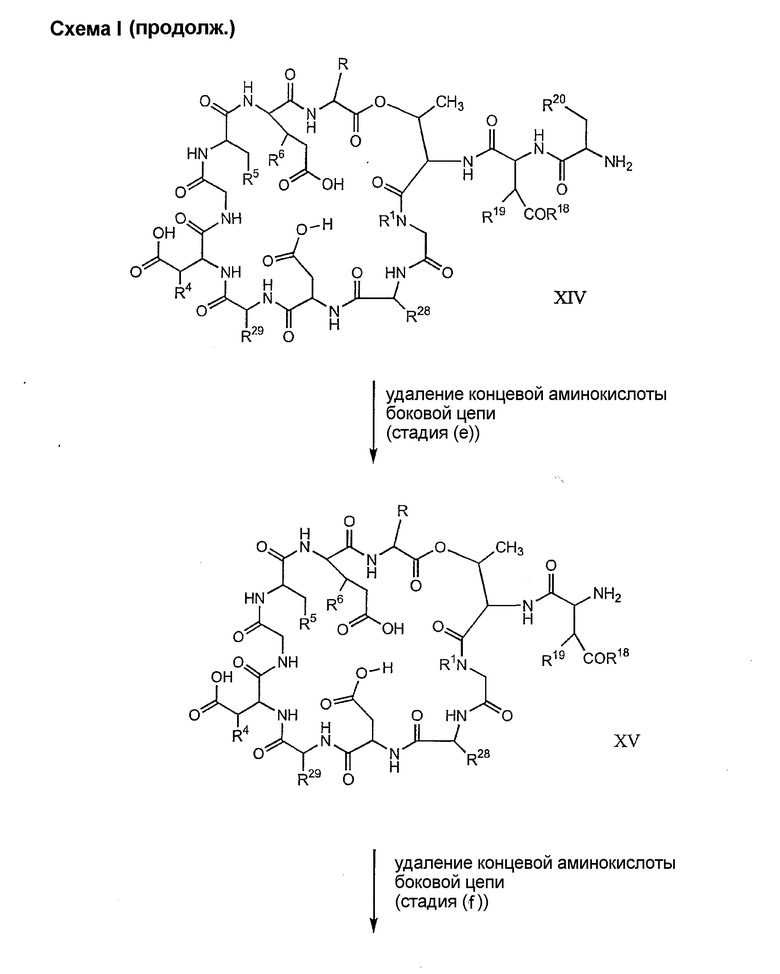
Полусинтетический способ в соответствии с одним аспектом изобретения включает получение соединения формулы XI (стадия a)). Соединения формулы XI могут быть получены с помощью способов, описанных в патентах США RE 32333; RE 32455; RE 32311; 4482487; 4537717; 4800157; 4874843; 4885243; 5912226; 4994270; 5039789 и 5028590; заявках на международные патенты с номерами регистрации WO01/44272, WO01/44274, WO01/44271, WO01/53330 и WO02/059322, каждый из которых включен здесь в качестве ссылки.
В предпочтительном осуществлении соединение формулы XI является таким, в котором каждый из R1 и R4 представляет собой гидридо; каждый из R3 и R6 представляет собой метил; R5 представляет собой гидроксил; R25 представляет собой 7-метилнонил, 9-метилдецил, 9-метилундецил, нонил, децил или их смеси, а R26 представляет собой - CH2CH2CH2NH2.
В другом предпочтительном осуществлении соединение формулы XI является таким, в котором R представляет собой изопропил или 2-бутил; каждый из R1 и R2 представляет собой метил; R4 представляет собой метокси; R5 представляет собой карбоксиамино; R25 представляет собой 8-метилнонаноил, н-деканоил или 8-метилдеканоил; а R27 представляет собой -CH2CH2CH2CH2NH2.
Свободный амин соединения формулы XI обрабатывают защитной группой с получением защищенного депсипептидного соединения формулы XII (стадия (b)), где R, R1, R4, R5, R6, R18, R19, R20 и R25 описаны выше; R28 представляет собой метил или -CH2CH2CH2HP; R29 представляет собой метил или -CH2CH2CH2CH2NHP; где P представляет собой аминозащитную группу, или его соли.
Примеры аминозащитных групп и способы защиты аминов с помощью указанных групп можно найти в Protective Groups in Organic Synthesis by Theodora W.Greene, (New York: John Wiley and Sons, Inc.), 1981, здесь и далее "Greene", включенный здесь в качестве ссылки. Предпочтительными аминозащитными группами являются карбаматные аминозащитные группы. Более предпочтительными аминогруппами являются аллилоксикарбонильная (alloc), карбобензилокси (CBZ) и трет-бутоксикарбонильная защитные группы. Наиболее предпочтительной аминозащитной карбаматной группой является аллилоксикарбонил. Способы защиты амина даптомицина, A54145 и родственных липопептидов можно найти в патентах Соединенных Штатов RE 32310; RE 32311; 4482487; 4524135; 4537717; 5039789 и 5028590; заявках на международные патенты с номерами регистрации WO01/44272, WO01/44274 и WO01/44271.
Защищенное депсипептидное соединение затем обрабатывают дезацилирующим агентом с образованием соединения с концевой аминокислотой формулы XIII (стадия (c)). Дезацилирующими агентами, пригодными для изобретения, являются ферментативные дезацилирующие агенты. Фермент, который пригоден для дезацилирования соединения формулы XII, продуцируется определенным микроорганизмом семейства Actinoplanaceae. Некоторые из данных известных видов и множества данного семейства включают Actinoplanes philippinensis, Actinoplanes armeniacus, Actinoplanes utahensis, Actinoplanes missouriensis, Spirillospora albida, Streptosporiangium roseum, Streptosporangium vulgare, Streptosporangium roseum var hollandensi, Streptosporangium album, Streptosporangium viridialbum, Amorphosporangium auranticolor, Ampullariella regularis, Ampullariella campanulata, Ampullariella lobata, Ampullariella digitata, Pilimelia terevasa, Pimelia anulata, Planomonspora parontospora, Planomonspora venezuelensis, Planobispora longispora, Planobispora rosea, Dactylosporangium aurantiacum и Dactylosporangium thailandende. Все природные и искусственные варианты и мутанты, которые получаются из Actinoplanacea и которые продуцируют фермент, могут быть использованы в данном изобретении.
Предпочтительными источниками дезацилирующего фермента являются Actinoplanes utahensi: NRRL 12052; Actinoplanes missouriensis NRRL 12053; Actinoplanes sp.: NRRL 8122; Actinoplanes sp.: NRRL 12065; Streptosporsngium roseum var hollandensis: NRRL 12064; Actinoplanes utahenis ATCC 14539 и Actinoplanes missouriensis ATCC 14538. Более предпочтительным источником дезацилирующего фермента является вид Actinoplanes utahensi. Наиболее предпочтительным источником дезацилирующего фермента является рекомбинантный Streptomyces lividans, который экспрессирует дезацилирующий фермент Actinoplanes utahensis, как описано в J. Ind. Microbiol. Biotechnol. 2000, 24(3) 173-180. Данный фермент известен также как эхинокандин B деацилаза или ECB деацилаза.
Подходящие способы ферментативного деацилирования соединений формулы XII можно найти в патентах США 4524135; 4537717; 4482487; RE 32310; RE 32311, 5039789 и 5028590; заявках на международные патенты с номерами регистрации WO01/44272, WO01/44274 и WO01/44271, каждый из которых включен здесь в качестве ссылки.
Удаление триптофанового аминокислотного остатка из соединения с концевой аминокислотой формулы XIII ведет к образованию соединения формулы XIV (стадия (d)). Способы удаления триптофанового аминокислотного остатка известны специалистам в данной области техники. Предпочтительный способ удаления триптофанового аминокислотного остатка соответствует условиям деградации по Эдману.
Деградация по Эдману является хорошо обоснованной реакцией, известной специалистам в данной области техники (смотри, например, P. Edman, 1950, Acta Chem. Scan. 4:283-93 и P. Edman, 1956, Acta Chem Scan 10:761-768). В данной реакции концевая NH2 группа взаимодействует с изотиоцианатом с образованием тиомочевинного производного пептида. После обработки кислотой или основанием тиомочевинный пептид подвергается реакции циклизации с получением тиогидантоила и укороченного пептида (смотри схему II).
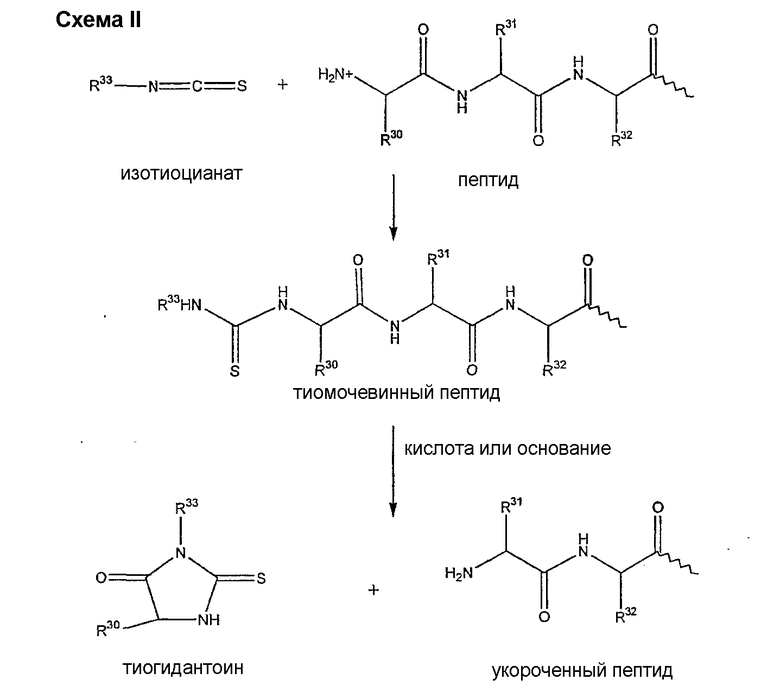
где каждый из R30, R31 и R32 представляет собой аминокислоту боковой цепи; а R33 представляет собой арильную или алкильную группу.
Деградацию по Эдману можно проводить при разнообразных условиях. На первом этапе последовательности деградации по Эдману изотиоцианат взаимодействует с амином при нейтральных или умеренно-щелочных (pH<9,5) условиях в таких растворителях, как тетрагидрофуран, N,N'-диметилформамид, дихлорметан, диоксан или этанол. Может быть применено множество изотиоцианатов (смотри K. K. Han et al. Biochemie 1977, 59:557-576).
Последующие циклизация и отщепление могут быть осуществлены в различных условиях. Обычно применяют безводную трифторуксусную кислоту, гептафтормасляную кислоту (смотри, например, W.F. Brandt et al., 1976, Z. Physiol. Chem. 357:1505-1508) или концентрированную соляную кислоту (смотри, например, G.E. Tarr, 1977, Methods in Enzymology, 47:335-337). Могут быть также применены умеренно-щелочные условия, такие как триэтиламин или N,N-диметилаллиамин/уксусная кислота (pH˜9) (смотри G.C. Barrett et al., 1985, Tetrahedron Letters 26(36):4375-4378). В качестве обзора для данной реакции смотри K.K. Han, 1985, Int. J. Biochem 17(4):429-445.
В предпочтительном осуществлении тиомочевинный пептид (соединение формулы XVIII), образованный при взаимодействии тиоизоцианата с соединением формулы XIII, обрабатывают в кислых условиях с получением соединения формулы XIV. В более предпочтительном осуществлении изобретения соединение формулы XVIII обрабатывают трифторуксусной кислотой с получением соединения формулы XIV (схема III).
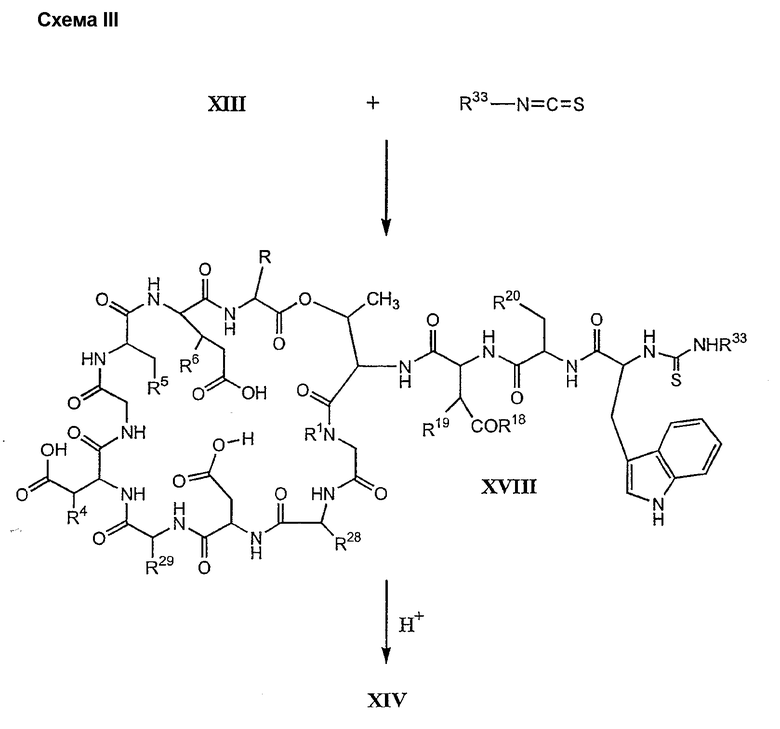
где каждый из R, R1, R4, R5, R6, R18, R19, R20, R28, R29 и R33 описан выше.
В предпочтительном осуществлении R33 представляет собой фенил, н-децил, нонил или октил. В более предпочтительном осуществлении R33 представляет собой н-децил.
Удаление концевого аминокислотного остатка из соединения формулы XIV ведет к образованию дездипептидного соединения формулы XV (стадия (e)). Способы удаления концевого аминокислотного остатка известны специалистам в данной области техники. Предпочтительный способ удаления концевого аминокислотного остатка соответствует условиям деградации по Эдману (смотри выше).
В предпочтительном осуществлении тиомочевинный пептид (соединение формулы XIX), образовавшийся при взаимодействии тиоизоцианата с соединением формулы XIV, обрабатывают в кислых условиях с получением дездипептидного соединения формулы XV. В предпочтительном осуществлении изобретения соединение формулы XIX обрабатывают трифторуксусной кислотой с получением соединения формулы XV (схема IV).
Схема IV
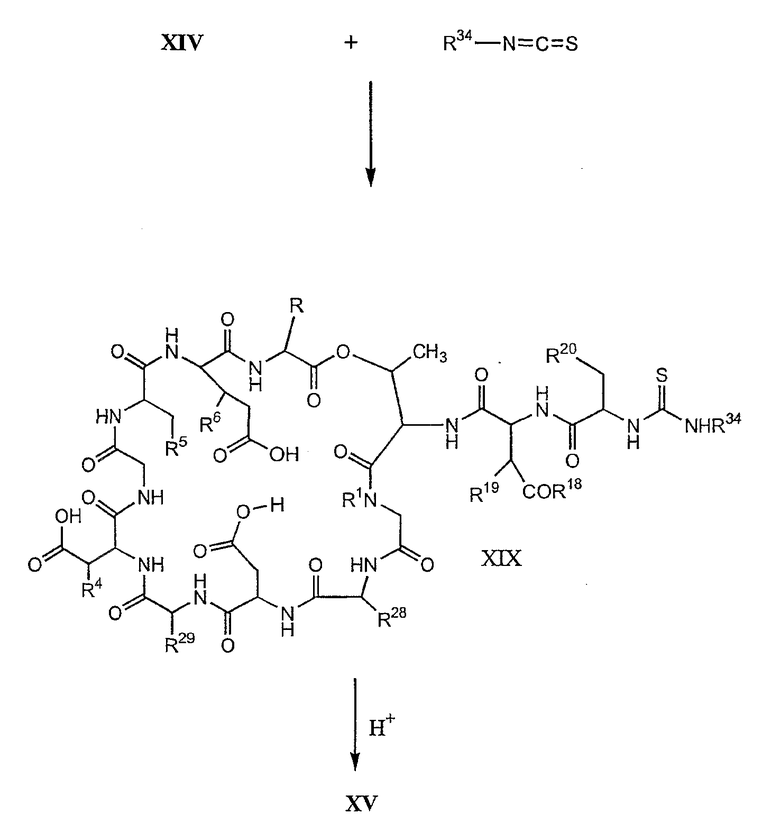
где каждый из R, R1, R4, R5, R6, R18, R19, R20, R28 и R29 описан выше; а R34 представляет собой алкил или арил.
Удаление концевого аминокислотного остатка из соединения формулы XV ведет к образованию депсипептидного базового соединения формулы XVI (стадия (f)). Способы удаления концевого аминокислотного остатка известны специалистам в данной области техники. Предпочтительный способ удаления концевого аминокислотного остатка соответствует условиям деградации по Эдману (смотри выше).
В предпочтительном осуществлении тиомочевинный пептид (соединение формулы XX), образовавшийся при взаимодействии тиоизоцианата с соединением формулы XV, обрабатывают в кислых условиях с получением депсипептидного базового соединения формулы XVI. В предпочтительном осуществлении изобретения соединение формулы XX обрабатывают трифторуксусной кислотой с получением соединения формулы XVI (схема IV A).
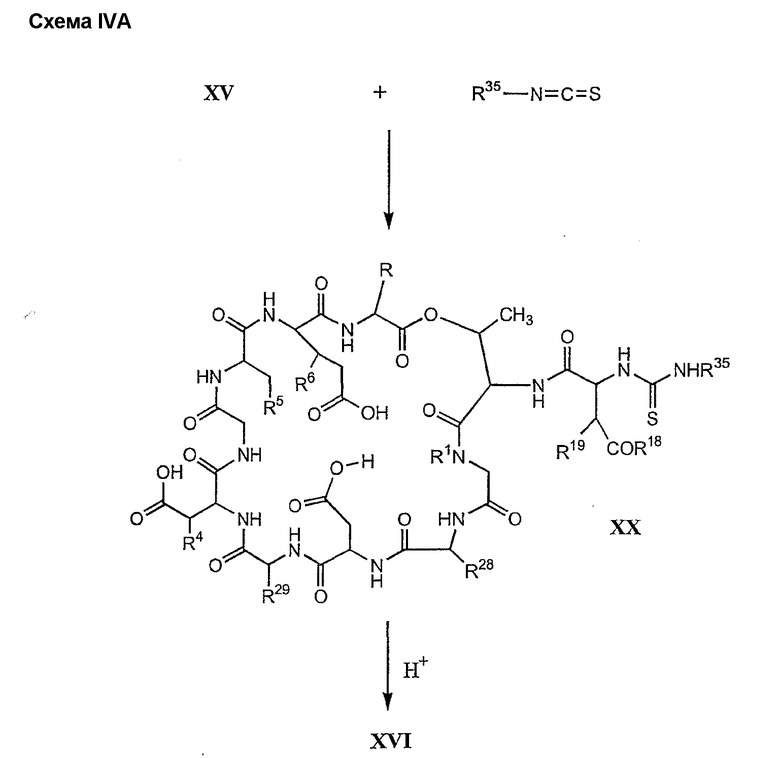
где каждый из R, R1, R4, R5, R6, R18, R19, R20, R28 и R29 описан выше; а R35 представляет собой алкил или арил.
Обработка депсипептидного базового соединения формулы XVI модифицирующим агентом ведет к образованию защищенного соединения формулы I (соединения формулы XVII, стадия (f)). Взаимодействие амина с модифицирующими агентами, как определено здесь, является хорошо известным специалистам в данной области техники. Например, обработка соединения формулы XVI изоцианатом дает соединения формулы XVII, в которых R7 представляет собой уреидо. Подобным образом, обработка соединения формулы XVI активированным сложным эфиром, лактоном или хлорангидридом кислоты дает соединения формулы XVII, в которых R7 представляет собой ациламино. Обработка соединения формулы XVI сульфонилхлоридом или активированным сульфонамидом дает соединение формулы XVII, в котором R7 представляет собой сульфонамино. Обработка соединения формулы XVI активированным гетероциклом дает соединение формулы XVII, в котором R7 представляет собой гетероциклический амино. Обработка соединения формулы XVI активированным гетероарилом дает соединение формулы XVII, в котором R7 представляет собой гетероарильный амино. Обработка соединения формулы XVI карбонатом, хлорформиатом или цианоформиатом дает соединения формулы XVII, в которых R7 представляет собой карбамат. Обработка соединения формулы XVI тиоэфиром дает соединения формулы XVII, в которых R7 представляет собой тиоациламино. Обработка соединения формулы XVI фосфорилхлоридом или фосфорамидатом дает соединения формулы XVII, в которых R7 представляет собой фосфонамино. Обработка соединения формулы XVI имидатом дает соединения формулы XVII, в которых R7 представляет собой иминоамино. Обработка соединения формулы XVI тиоизоцианатом дает соединения формулы XVII, в которых R7 представляет собой тиоуреидо. Обработка соединения формулы XVI альдегидом или кетоном в восстанавливающих условиях дает соединения формулы XVII, в которых R7 представляет собой монозамещенную амино или дизамещенную аминогруппу. Обработка соединения формулы XVI имидатом дает соединения формулы XVII, в которых R7 представляет собой иминоамино. Обработка соединения формулы XVI гуанидинирующим агентом, таким как
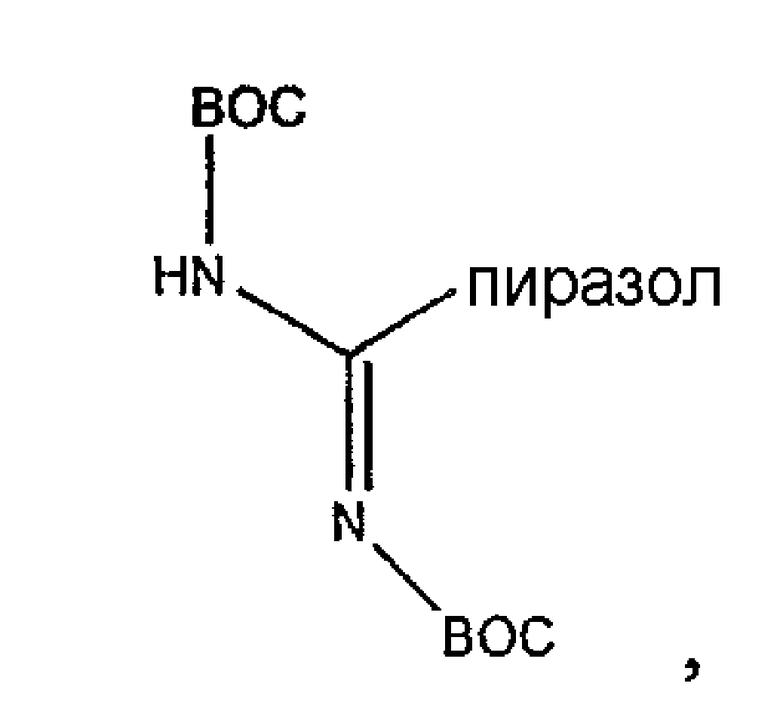
дает соединения формулы XVII, в которых R7 представляет собой гуанидино.
Специалистам в данной области техники должно быть понятным, что, если модифицирующий агент содержит заместители, которые несовместимы с условиями реакции, при которых образуется соединение формулы XVII, указанные заместители должны быть защищены перед применением в реакции. Подходящие защитные группы и способы их получения можно найти у Greene (смотри выше).
Взаимодействие аминов сложных молекул, таких как даптомицин и родственные депсипептиды, можно найти в патентах Соединенных Штатов 4399067; 4482487; и 4537717; 5039789; и 5028590; и Международных патентных заявках с номерами регистрации WO01/44272, WO01/44274 и WO01/44271.
В предпочтительном осуществлении реакции модифицирующий агент представляет собой активированный сложный эфир. В более предпочтительном осуществлении реакции модифицирующий агент представляет собой
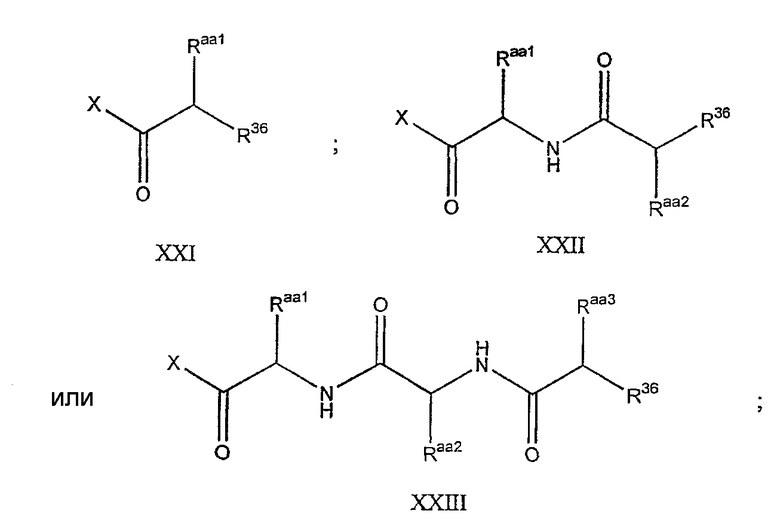
где каждый из Raa1, Raa2 и Raa3 представляет собой боковую цепь аминокислоты или защищенную форму боковой цепи аминокислоты; R36 представляет собой амино, монозамещенный амино, дизамещенный амино, ациламино, уреидо, гуанидино, карбамоил, сульфонамино, тиоациламино, тиоуреидо, иминоамино или фосфонамино; а × представляет собой активирующую группу. В даже более предпочтительном осуществлении × представляет собой арилоксигруппу. В еще более предпочтительном осуществлении × представляет собой пентафторфенокси.
Соединения формул XXI, XXII и XXIII могут быть получены из соответствующего пептида или аминокислоты обработкой активирующими агентами, такими как ангидриды, хлорформиаты, пентафторфенол/дициклогексилкарбодиимид, N',N'-карбонилдиимидазол, гидроксибензотриазол или N-гидроксисукцинимид. Пептиды могут быть получены с помощью любой стандартной процедуры для пептидов. В качестве обзора некоторых процедур образования пептидов смотри Vogel's Textbook of Practical Organic Chemistry, 5th Ed., eds. B.S. Furniss, A.J. Hannaford; P.W.G. Smith; A.R. Tatchell (New York: John Wiley and Sons, Inc.), 1089, p. 750-763 и Introduction to Organic Chemistry, 2nd Ed. by A. Streitwieser, Jr. and C.H. Heathcock (New York: MacMillan Publishing Co., Inc.), p. 954-962. Другие способы, которые пригодны для получения пептидов по настоящему изобретению, включают твердофазный синтез. Конкретные примеры таких методов подробно описаны в примерах (смотри ниже).
Удаление защитной группы (групп) из защищенного соединения формулы I (соединения формулы XVII) приводит к образованию соединения формулы Ia (стадия h), в котором R1, R4, R5, R6 и R7 описаны выше; R2a представляет собой метил или -CH2CH2CH2NH2, а R3a представляет собой метил или -CH2CH2CH2CH2NH2. Удаление аминозащитной группы может быть осуществлено методами, описанными в Greene (смотри выше). Специалистам в данной области техники понятно, что выбор аминозащитной группы, применяемой на первой стадии способа, определяет реагенты и методы, используемые для удаления указанной аминозащитной группы.
Когда модифицирующий агент содержит одну или более защитную группу (группы), указанная защитная группа (группы) также должна быть удалена. Выбор защитной группы (групп), применяемой на заместителе (заместителях) модифицирующего агента, определяет реагенты и методы, используемые для удаления указанной защитной группы (групп). Когда защитная группа (группы), применяемые на заместителе (заместителях) модифицирующего агента, и защитная группа, применяемая на стадии (b), являются совместимыми, защитные группы могут быть удалены в одну стадию. Однако, когда защитная группа (группы) являются несовместимыми, для удаления всех защитных групп может потребоваться несколько стадий.
Способ получения соединений формулы I, где, по меньшей мере, один из R2 и R3 отличен от метила и каждый из R8 и R9 отличен от NH2.
Способ B
Для соединений формулы I, в которых, по меньшей мере, один из R2 и R3 отличен от метила и каждый из R8 и R9 отличен от NH2, способ в соответствии с другим аспектом изобретения включает дополнительные стадии:
(i) обработку соединения формулы I со свободным амином со стадии (h) способа A с модифицирующим агентом с получением соединения формулы I.
Обработка соединения формулы I свободным амином формулы Ia модифицирующим агентом хорошо известна специалистам в данной области техники и описана для стадии (g) способа A (смотри выше).
Описание взаимодействия свободных аминов сложных молекул, таких как даптомицин и родственных депсипептидов, можно найти в патентах США 4399067, 4482487; и 4537717; и заявках на международные патенты WO01/44272, WO01/44274 и WO01/44271.
Специалистам в данной области техники понятно, что, если R7 или модифицирующий агент со стадии (i) содержит заместители, которые несовместимы с условиями реакции, в которой образуется соединение формулы I, то указанные заместители должны быть защищены перед стадией (i). Подходящие защитные группы и способы их получения можно найти в Greene (смотри выше).
Когда R7 и модифицирующий агент стадии (i) содержат защитную группу (группы), указанная защитная группа (группы) может быть удалена. Выбор защитной группы (групп) для заместителя (заместителей) R7 и модифицирующего агента должен определять реагенты и способы, используемые для удаления указанной защитной группы (групп). Когда защитная группа (группы), используемые на заместителе (заместителях) R7 и модифицирующего агента совместимы, защитные группы могут быть удалены в одну стадию. Однако, когда защитная группа (группы) являются несовместимыми, для удаления всех защитных групп может потребоваться несколько стадий.
Способ C
Альтернативный способ для получения соединений формулы I, в которых, по меньшей мере, один из R2 и R3 отличен от метила и каждый из R8 и R9 отличен от NH2, включает следующие стадии:
(a) получение депсипептидного производного формулы XI
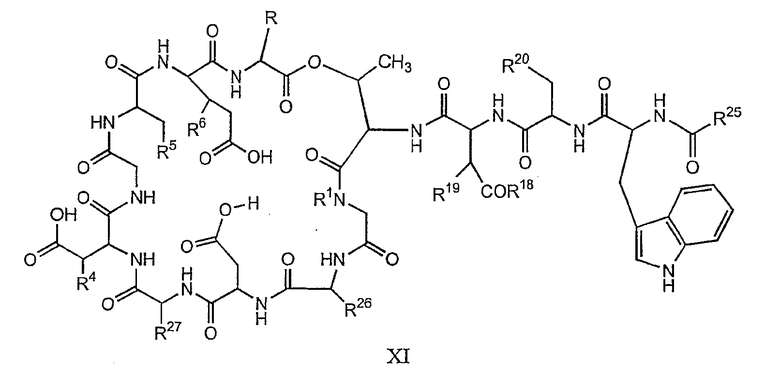
где R, R1, R4, R5, R6, R18, R19, R20, R25, R26 и R27 описаны выше;
(b) обработку свободной аминогруппы (групп) соединения формулы XI модифицирующим агентом с получением блокированного депсипептидного соединения, где указанный модифицирующий агент выбран таким образом, что образованное блокированное депсипептидное соединение стабильно на стадиях (c), (d), (e), (f) и (g);
(c) обработку блокированного депсипептидного соединения, полученного на стадии (b), дезацилирующим агентом с получением соединения с концевым аминосоединением;
(d) удаление триптофанового аминокислотного остатка из соединения с концевым аминосоединением, полученного на стадии (c), с получением дезтриптофанового соединения;
(e) удаление концевого аминокислотного остатка из дезтриптофанового соединения, полученного на стадии (d), с получением дездипептидного соединения;
(f) удаление концевого аминокислотного остатка из дездипептидного соединения, полученного на стадии (e), с получением депсипептидного соединения;
(g) обработку депсипептидного базового соединения со стадии (f) модифицирующим агентом.
Способ C проиллюстрирован схемой V

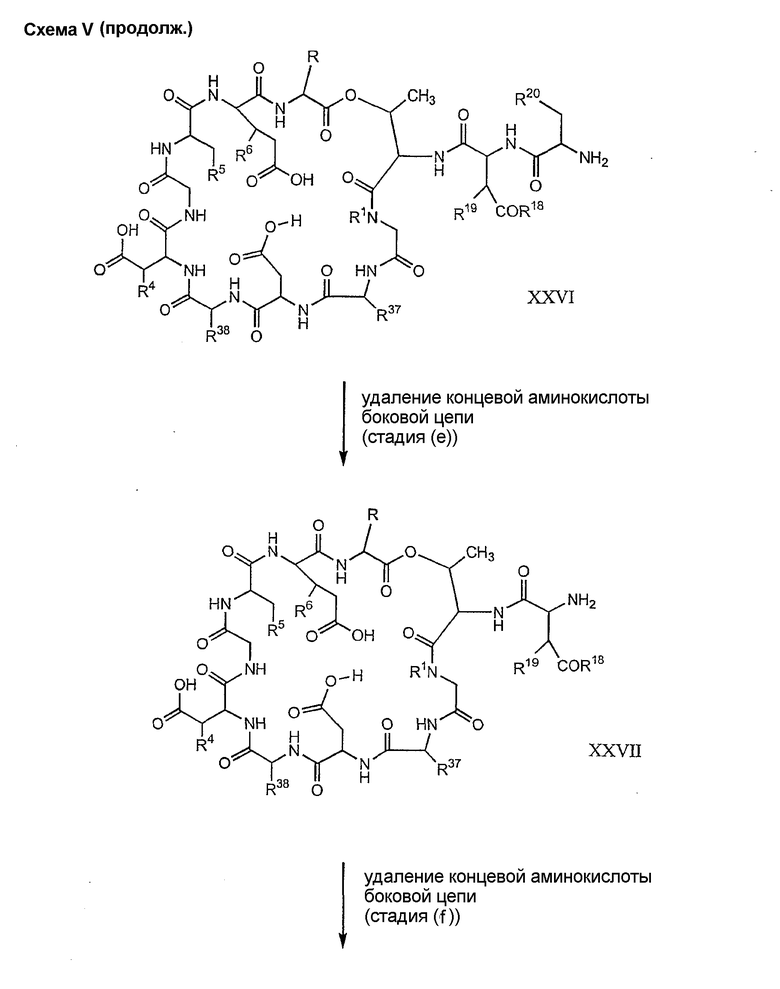
Обработка соединения формулы XII модифицирующим агентом ведет к образованию защищенного соединения формулы I (соединения формулы XXIV, стадия (b)), где R37 представляет собой метил или -CH2CH2CH2R8; и R38 представляет собой метил или -CH2CH2CH2CH2R9; прилагаемые так, что, по меньшей мере, один из R37 и R38 должен отличаться от метила, и R8 и R9 должны отличаться от амино. Образование защищенного соединения формулы I осуществляется так, как описано ранее для стадии (g) способа A (смотри выше).
Дезацилирование соединения XXIV ведет к образованию концевого аминосоединения XXV. Подходящими агентами для дезацилирования соединений формулы XXVI являются ферментативные дезацилирующие агенты (смотри выше).
Удаление триптофанового аминокислотного остатка из соединения с концевым аминосоединением формулы XXV ведет к образованию соединения формулы XXVI (стадия (d)). Способы удаления триптофанового аминокислотного остатка известны специалистам в данной области техники. Предпочтительный способ удаления триптофанового аминокислотного остатка соответствует условиям деградации по Эдману (смотри выше).
Удаление концевого аминокислотного остатка из соединения формулы XXVI ведет к образованию дездипептидного соединения формулы XXVII (стадия (e)). Способы удаления концевого аминокислотного остатка известны специалистам в данной области техники. Предпочтительный способ удаления концевого аминокислотного остатка соответствует условиям деградации по Эдману (смотри выше).
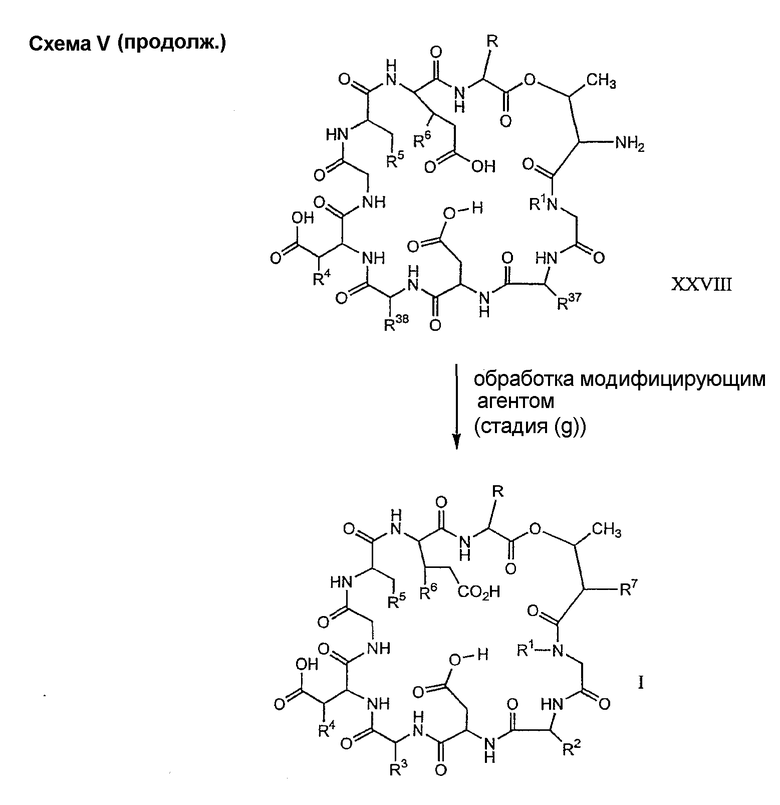
Удаление концевого аминокислотного остатка из соединения формулы XXVII ведет к образованию депсипептидного базового соединения формулы XXVIII (стадия (f)). Способы удаления концевого аминокислотного остатка известны специалистам в данной области техники. Предпочтительный способ удаления концевого аминокислотного остатка соответствует условиям деградации по Эдману (смотри выше).
Обработка депсипептидного соединения формулы XXVIII модифицирующим агентом хорошо известна специалистам в данной области техники и описана для стадии (g) способа A (смотри выше).
Специалистам в данной области техники понятно, что, если модифицирующий агент содержит заместители, которые несовместимы с условиями реакции, при которых образуется соединение формулы I, указанные заместители должны быть защищены перед взаимодействием соединения формулы XXVIII. Подходящие защитные группы и способы их получения можно найти у Greene (смотри выше).
Когда это желательно, защитные группы могут быть удалены. Когда защитные(ая) группы (группа) являются(ется) совместимыми, защитные группы могут быть удалены в одну стадию. Однако когда защитные(ая) группы (группа) являются(ется) несовместимыми, для удаления всех защитных групп может потребоваться несколько стадий.
Способ D
Соединения формулы I и соединения формулы Ia, в которых R7 представляет собой
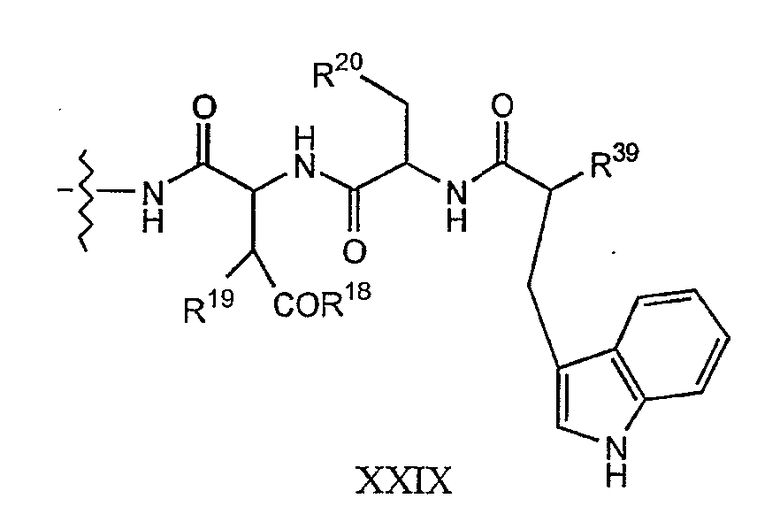
где каждый из R18, R19 и R20 соответствует описанному ранее, и R39 представляет собой амино, монозамещенный амино, дизамещенный амино, ациламино, уреидо, гуанидино, карбамоил, сульфонамино, тиоациламино, тиоуреидо, иминоамино или фосфонамино; могут быть получены из соединения XIII, как описано на схеме VI, или через соединение XXV, как описано на схеме VII.
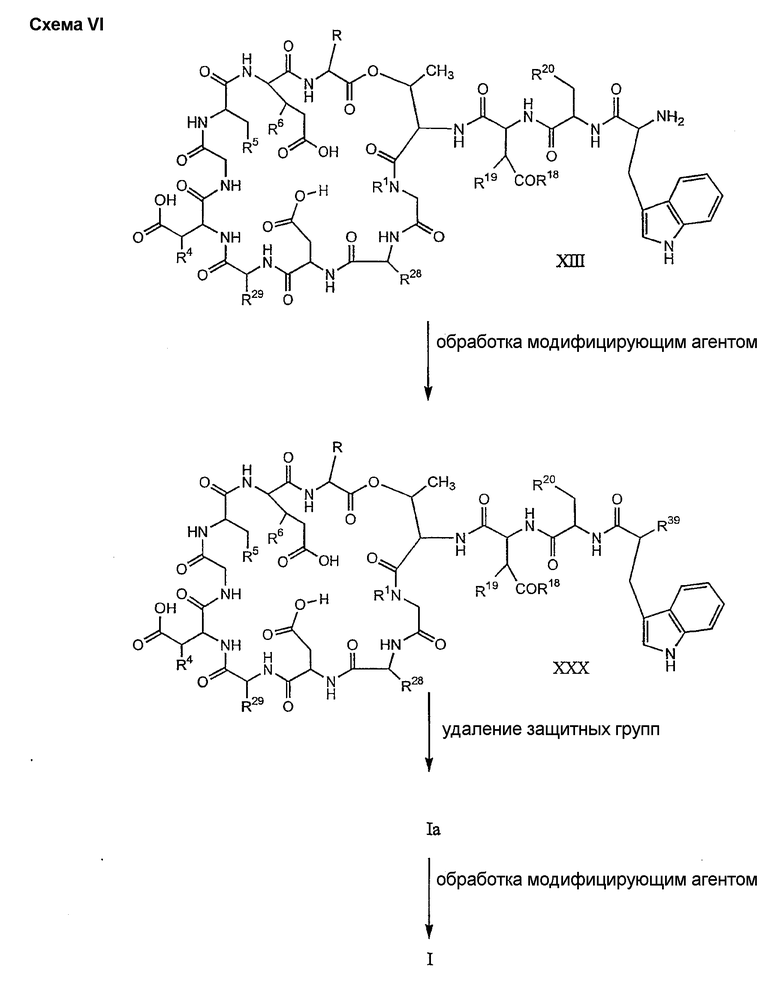
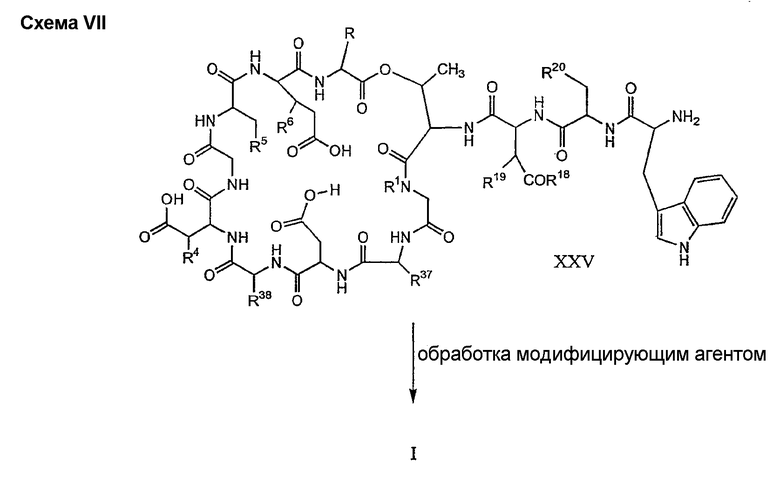
Способ E
Сходным образом, соединения формулы I и соединения формулы Ia, в которых R7 представляет собой
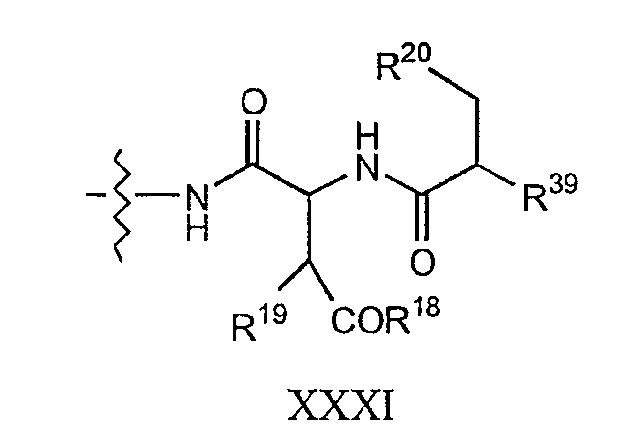
где каждый из R18, R19 и R20 соответствует описанному ранее, и R39 представляет собой амино, монозамещенный амино, дизамещенный амино, ациламино, уреидо, гуанидино, карбамоил, сульфонамино, тиоациламино, тиоуреидо, иминоамино или фосфонамино; могут быть получены из соединения XIV, как описано на схеме VIII, или через соединение XXVI, как описано на схеме IX.
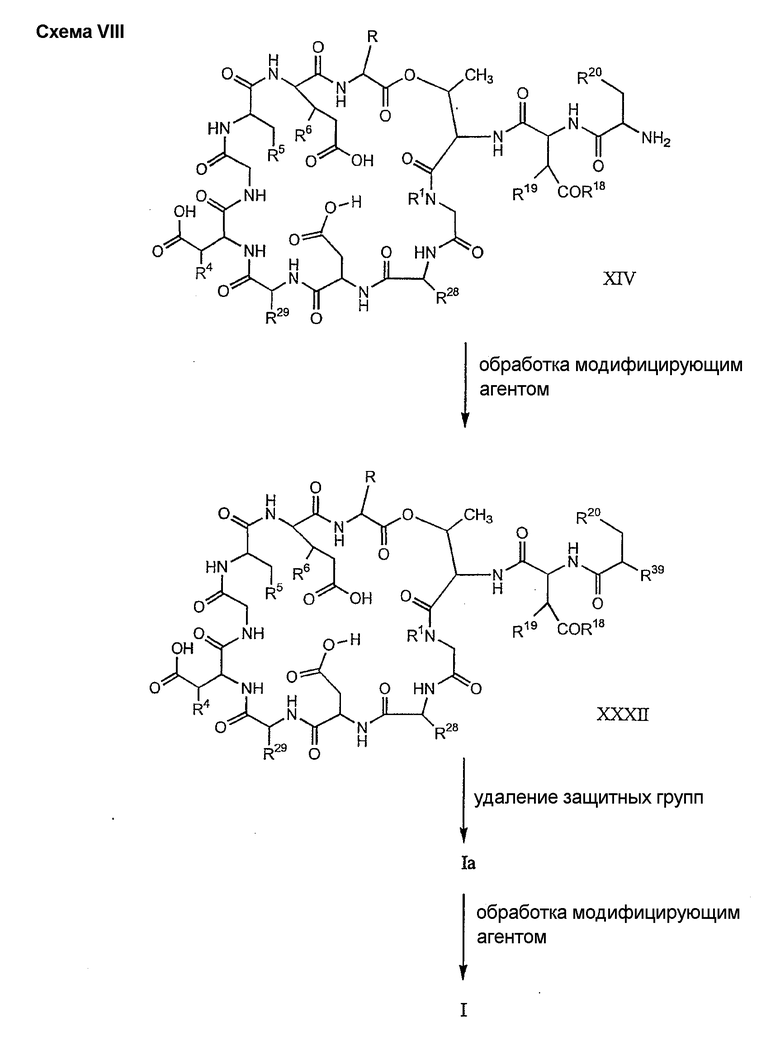
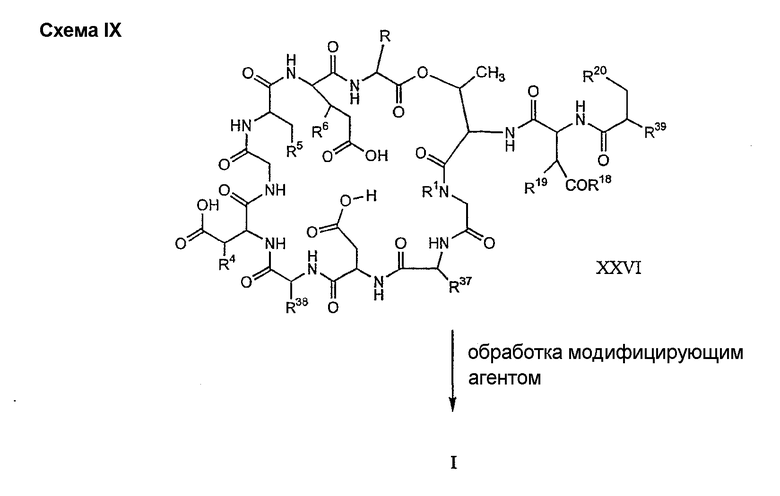
Способ F
Сходным образом, соединения формулы I и соединения формулы Ia, в которых R7 представляет собой
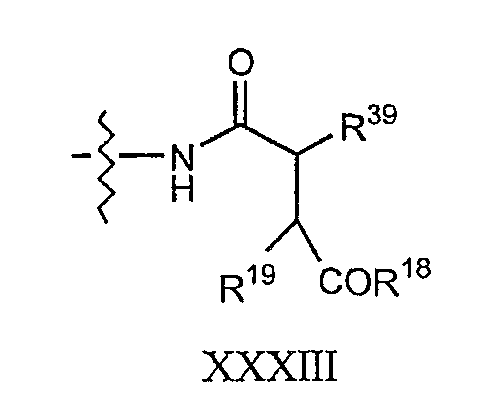
где каждый из R18 и R19 соответствует описанному ранее, и R39 представляет собой амино, монозамещенный амино, дизамещенный амино, ациламино, уреидо, гуанидино, карбамоил, сульфонамино, тиоациламино, тиоуреидо, иминоамино или фосфонамино; могут быть получены из соединения XV, как описано на схеме X, или через соединение XXVII, как описано на схеме XI.
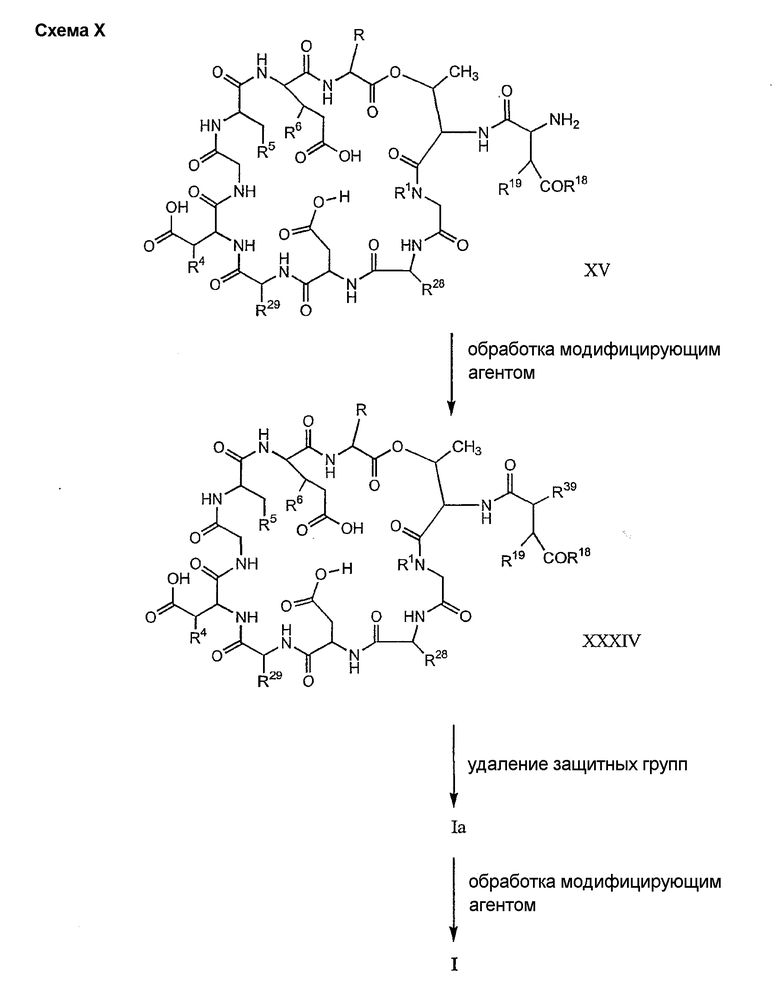
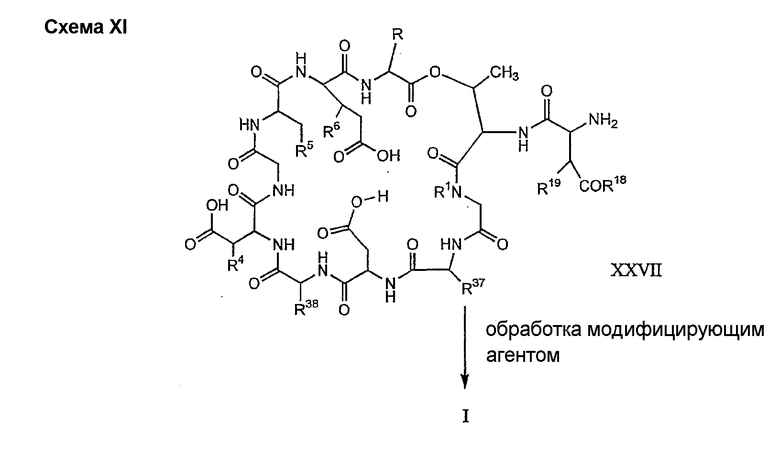
2. Синтетический способ
Твердофазный синтез депсипептидных соединений
В альтернативном осуществлении изобретения депсипептидные соединения формулы I могут быть синтезированы на твердой основе (схема XII, схема XIII и схема XV).
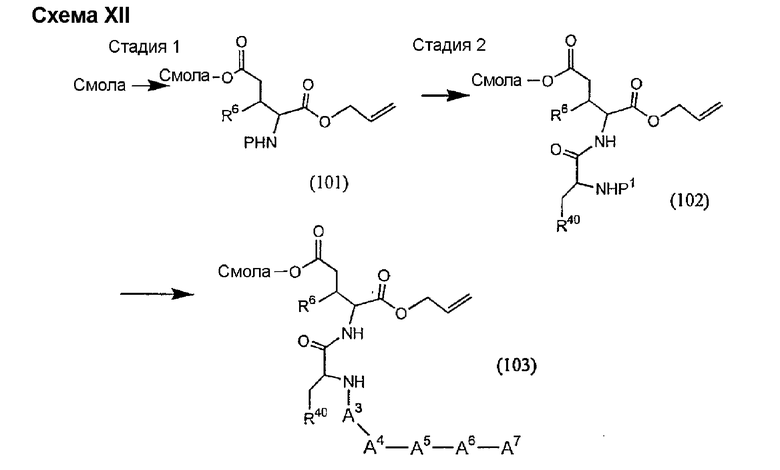
На стадии 1 N-защищенный O-аллиловый сложный эфир β-метилглутаминовой кислоты или защищенный O-аллиловый сложный эфир глутаминовой кислоты присоединяют к смоле с получением соединения 101, где R6 описан выше. В данной реакции может быть применена смола или твердая основа, такая как, но не ограничиваясь этим, Wang, HMPA, Safety Catch, Rink Acid, 2-хлортритилхлоридная смола, тритилхлоридная смола, 4-метилтритилхлоридная смола, 4-метокситритилхлоридная смола или смола PAM.
Снятие защиты с соединения 101 с последующим соединением свободного амино с защищенным серином или защищенным аспарагином дает соединение 102, где R40 представляет собой -OP2 или -CONHP3; и каждый из P, P1, P2 и P3 независимо представляет собой защитную группу. Данный способ присоединения к пептиду, т.е. снятие защиты с альфа-аминогруппы с последующим соединением с защищенной аминокислотой, повторяют до присоединения к смоле желаемого количества аминокислот. На схеме XII было соединено в общей сложности семь аминокислот с получением соединения 103, где A3 представляет собой  ; A4 представляет собой
; A4 представляет собой  ,
,
где P4 представляет собой защитную группу, и R4 описан выше; A5 представляет собой  ,
,
где R44 представляет собой метил или -CH2CH2CH2CH2NP5,
где P5 представляет собой аминозащитную группу; A6 представляет собой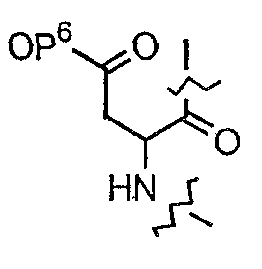 ,
,
где P6 представляет собой защитную группу; и A7 представляет собой  ,
,
где R45 представляет собой метил или -CH2CH2CH2NP7, где P7 представляет собой аминозащитную группу. Второй пептид присоединяют к смоле сходным образом, как показано на схеме XIII.
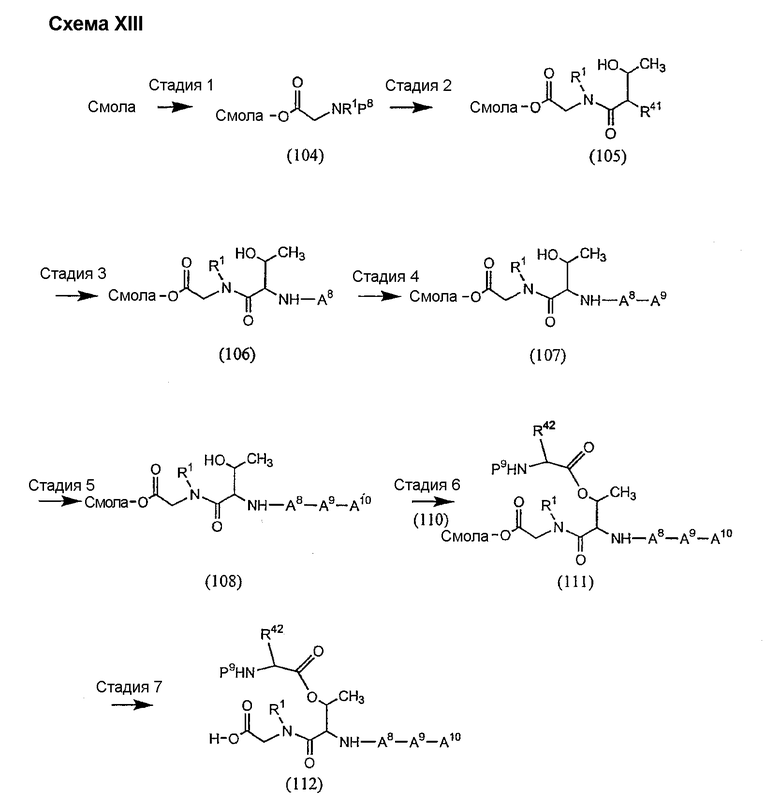
На стадии 1 к смоле присоединяют N-замещенный глицин или саркозин с получением соединения 104, где R1 описан выше, и P8 является аминозащитной группой. Выбор смолы, применяемой на стадии 1, зависит от природы аминокислоты, присоединяемой на стадиях 2-6. Если боковые цепи аминокислоты содержат защитные группы, смолу следует выбирать так, чтобы защитные группы оставались интактными при отделении смолы от пептида на стадии 7. Смолы, которые могут быть отщеплены при сохранении защитных групп пептидов, включают, но не ограничиваются этим, Safety Catch, Rink Acid, 2-хлортритилхлоридную смолу, тритилхлоридную смолу, 4-метилтритилхлоридную смолу, 4-метокситритилхлоридную смолу или смолу PAM.
Снятие защиты у защищенного амино соединения 104 сопровождается соединением свободного амино с
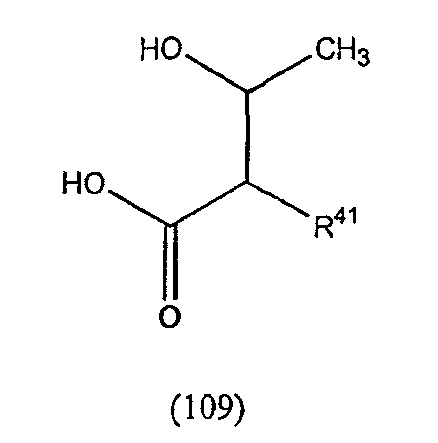
с получением соединения 105, где R41 представляет собой амино, монозамещенный амино, дизамещенный амино, ациламино, уреидо, гуанидино, карбамоил, сульфонамино, тиоациламино, тиоуреидо, иминоамино или фосфонамино.
Когда R41 представляет собой амино, данный способ пептидного связывания, т.е. снятие защиты с альфа-аминогруппы с последующим соединением с замещенной аминокислотой, повторяют до достижения желаемого количества присоединенных к смоле аминокислот. На схеме XIII было соединено пять аминокислот с получением соединения 108, где A8 представляет собой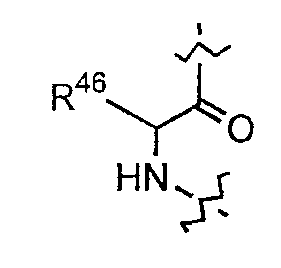 ,
,
где R46 представляет собой гидридо, боковую цепь аминокислоты или боковую цепь защищенной аминокислоты;
A9 представляет собой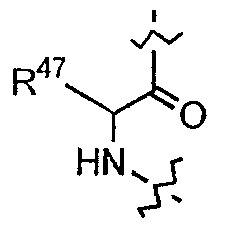 ,
,
где R47 представляет собой гидридо, боковую цепь аминокислоты или боковую цепь защищенной аминокислоты;
A10 представляет собой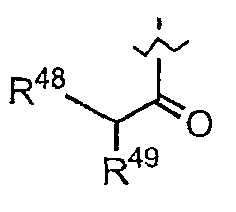 ,
,
где R48 представляет собой гидридо, боковую цепь аминокислоты или боковую цепь защищенной аминокислоты, и R49 представляет собой амино, монозамещенный амино, дизамещенный амино, ациламино, уреидо, гуанидино, карбамоил, сульфонамино, тиоациламино, тиоуреидо, иминоамино или фосфонамино.
Соединение 108 соединяют с
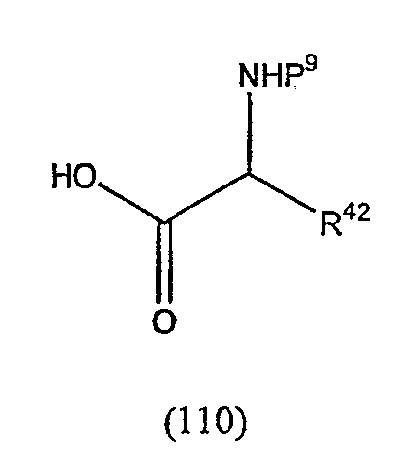
с получением соединения 111, где P9 представляет собой защитную группу, и R42 представляет собой 2-бутил, изопропил или
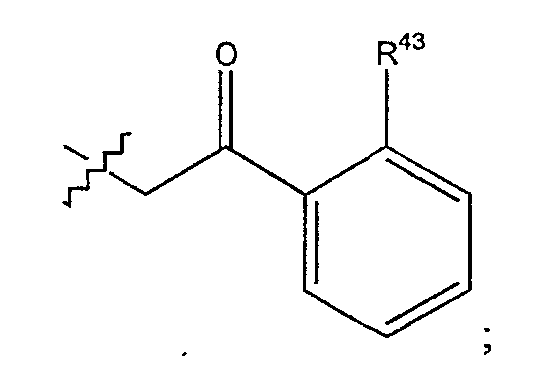
где R43 представляет собой группу, способную к превращению в аминогруппу. Например, R43 может быть азидо, защищенным амино фтальимидо или нитро.
Пептид 111 затем отделяют от смолы с получением соединения 112.
Соединение пептидных фрагментов 103 и 112 изображено на схеме XIV
Пептидные фрагменты 103 и 112 соединяют с получением связанного со смолой пептида 113. Снятие защиты защитной группы P9 и O-аллилового сложного эфира с последующей кристаллизацией дает связанный со смолой депсипептид 114. Отщепление депсипептида от смолы с последующим снятием защиты любых оставшихся защитных групп дает соединения формулы I (схема XVI).
Когда желательны соединения формулы I, в которых нет экзоциклических аминокислот, вместо соединения 108 может быть использовано соединение 105 (схема XV). Соединение 105 соединяют с соединением 110 с получением соединения 115; после отделения от смолы получают соединение 116; после соединения с соединением 103 получают соединение 117; затем снимают защиту и циклизуют с получением связанного со смолой депсипептида (118) и отщепляют от смолы, как описано ранее на схемах XIII и XIV (смотри выше).
Когда желательны соединения формулы I, в которых имеется одна экзоциклическая аминокислота, вместо соединения 108 может быть использовано соединение 106 (схема XVII). Соединение 106 соединяют с соединением 110 с получением соединения 119; после отделения от смолы получают соединение 120; после соединения с соединением 103 получают соединение 121; затем снимают защиту и циклизуют с получением связанного со смолой депсипептида (122) и отщепляют от смолы, как описано ранее на схемах XIII и XIV (смотри выше).
Когда желательны соединения формулы I, в которых имеются две экзоциклические аминокислоты, вместо соединения 108 может быть использовано соединение 107 (схема XVIII). Соединение 107 соединяют с соединением 110 с получением соединения 123; после отделения от смолы получают соединение 124; после соединения с соединением 103 получают соединение 125; затем снимают защиту и циклизуют с получением связанного со смолой депсипептида (126) и отщепляют от смолы, как описано ранее на схемах XIII и XIV (смотри выше).
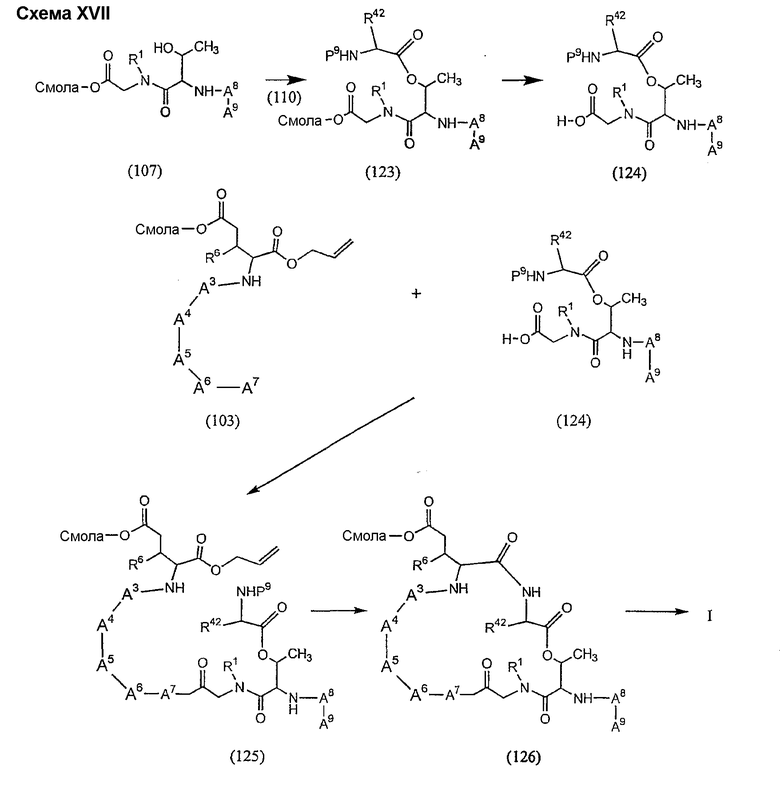
После описанных выше схем синтеза (схемы XII-XII) понятно, что и аминогруппа аминокислоты, и функциональные группы боковой цепи аминокислоты должны быть ортогонально защищены перед их присоединением к наращиваемой пептидной цепи. Подходящими защитными группами могут быть любые аминозащитные группы, пригодные для пептидного синтеза. Такие пары защитных групп хорошо известны. Смотри, например, "Synthesis Notes" в каталоге Novabiochem и Peptide Synthesis Handbook (1999), страницы S1-S93 и цитированные в нем ссылки.
Специалистам в данной области техники также должно быть понятно, что выбор защитной группы для функциональных групп боковой цепи аминокислоты должен приводить или не приводить к отщеплению защитной группы одновременно с конечным отщеплением пептида от смолы, что должно давать функциональную природную кислоту или ее защищенное производное, соответственно. Когда защитные группы не отщепляются одновременно с отщеплением депсипептида от смолы, может потребоваться дополнительное снятие защиты.
3. Биосинтетический способ
Соединения по настоящему изобретению могут быть также получены рекомбинантными способами. В данном способе модифицированную нерибосомальную пептидсинтетазу даптомицина вводят в клетку, способную продуцировать соединение формулы I, и клетку культивируют с образованием соединения формулы I. О нерибосомальных пептидсинтетазах даптомицина, а также о модификациях данных синтетаз сообщалось (смотри Международную патентную заявку с номером регистрации 02/059322).
Для того чтобы данное изобретение могло быть понято более полно, представлены следующие примеры. Данные примеры предназначены для иллюстративной цели и их не следует толковать как ограничивающие объем изобретения каким-либо образом.
Пример 1: Синтез пептидной смолы соединения I:
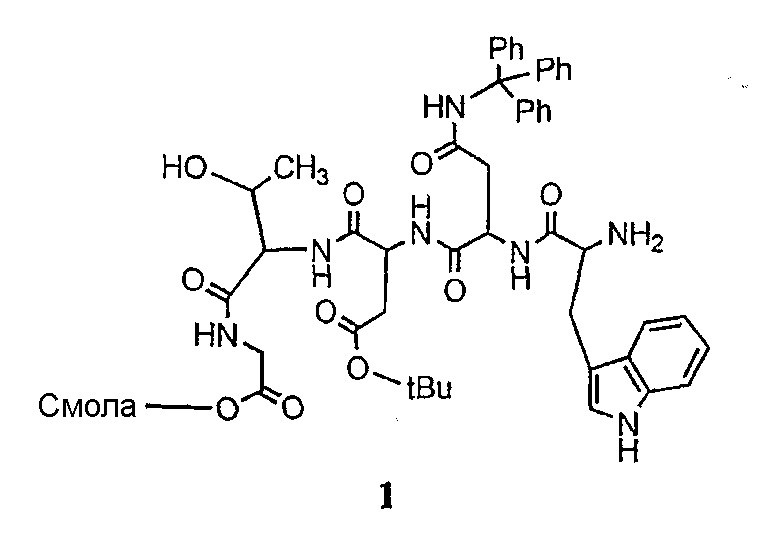
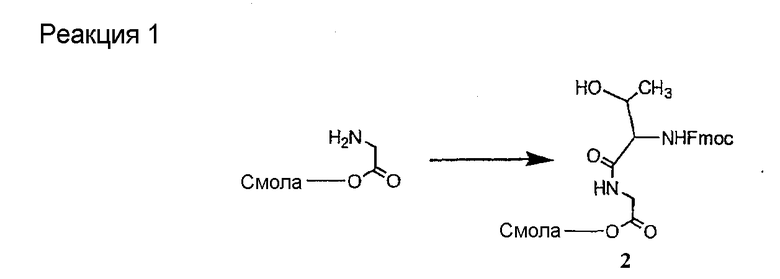
Имеющиеся в продаже Nα-(9-флуоренилметоксикарбонил)-L-треонин (2 мл 0,5 молярного раствора в N-метилпиролидине), 1,3-диизопропилкарбодиимид (2 мл 0,5 молярного раствора в N-метилпиролидине) и 1-гидроксибензотриазол (2 мл 0,5 молярного раствора в N-метилпиролидине) добавляли к имеющемуся в продаже глицин 2-хлортритильной смоле (334 мг). Смесь перемешивали в течение одного часа, фильтровали и несколько шариков тестировали на наличие свободного амина с применением стандартного теста Кайзера (смотри E. Kaiser, et al (1970) Anal. Biochem., 34, 595; и Advanced Chemtech Handbook of Combinatorial, Organic and Peptide Chemistry 2003-2004 стр. 208). Тест Кайзера дал синюю окраску, так что указанные выше условия присоединения были воспроизведены. После фильтрации несущую продукт смолу промывали N-метилпиролидином (3 × 6 мл), метанолом (3 × 6 мл) и вновь N-метилпиролидином (3 × 6 мл) с получением соединения 2.
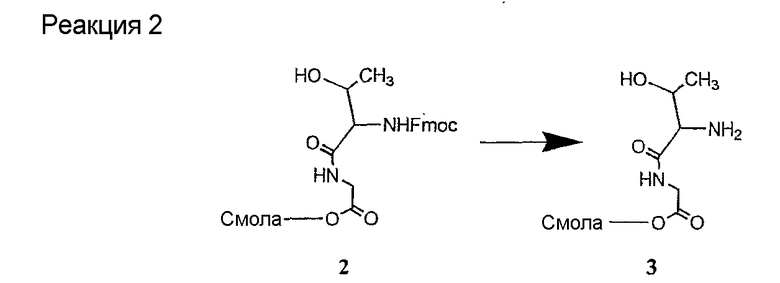
Соединение 2 встряхивали в 20% пиперидине в N-метилпиролидине (6 мл) в течение 30 минут. Смолу фильтровали и вновь суспендировали в 20% пиперидине в N-метилпиролидине (6 мл) и встряхивали в течение 30 минут. Реакционную смесь фильтровали, после чего твердое вещество промывали N-метилпиролидином (3 × 6 мл), метанолом (3 × 6 мл) и вновь N-метилпиролидином (3 × 6 мл) с получением соединения 3.
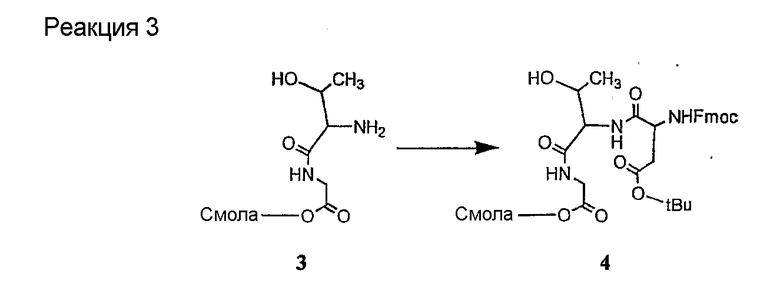
Имеющиеся в продаже β-трет-бутиловый эфир Nα-(9-флуоренилметоксикарбонил)-L-аспарагиновой кислоты (2 мл 0,5 молярного раствора в N-метилпиролидине), 1,3-диизопропилкарбодиимид (2 мл 0,5 молярного раствора в N-метилпиролидине) и 1-гидроксибензотриазол (2 мл 0,5 молярного раствора в N-метилпиролидине) добавляли к соединению 3. Смесь перемешивали в течение одного часа, фильтровали и присоединение повторяли. Реакционную смесь фильтровали и затем твердое вещество промывали N-метилпиролидином (3 × 6 мл), метанолом (3 × 6 мл) и вновь N-метилпиролидином (3 × 6 мл) с получением соединения.
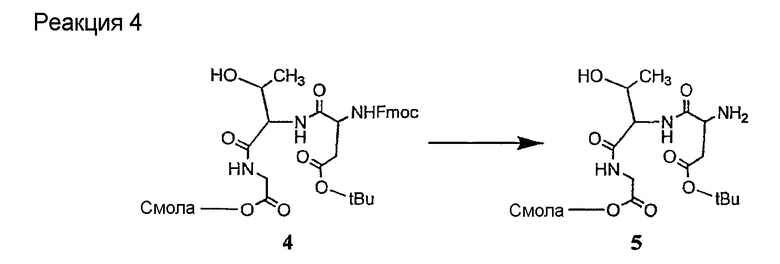
Соединение 4 встряхивали в 20% пиперидине в N-метилпиролидине (6 мл) в течение 30 минут. Смолу фильтровали и вновь суспендировали в 20% пиперидине в N-метилпиролидине (6 мл) и встряхивали в течение 30 минут. Реакционную смесь фильтровали и затем промывали N-метилпиролидином (3 × 6 мл), метанолом (3 × 6 мл) и вновь N-метилпиролидином (3 × 6 мл) с получением соединения 5.
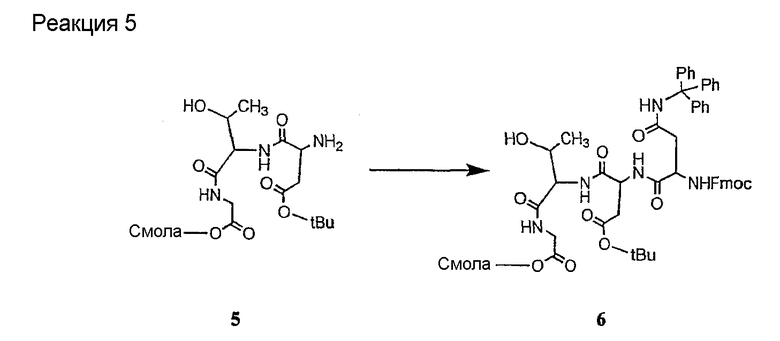
Имеющиеся в продаже Nα-(9-флуоренилметоксикарбонил)-L-аспарагин δ-N-тритил (2 мл 0,5 молярного раствора в N-метилпиролидине), 1,3-диизопропилкарбодиимид (2 мл 0,5 молярного раствора в N-метилпиролидине) и 1-гидроксибензотриазол (2 мл 0,5 молярного раствора в N-метилпиролидине) добавляли к смоле 5. Реакционную смесь перемешивали в течение одного часа, фильтровали и присоединение повторяли. Реакционную смесь фильтровали и затем твердое вещество промывали N-метилпиролидином (3 × 6 мл), метанолом (3 × 6 мл) и вновь N-метилпиролидином (3 × 6 мл) с получением соединения 6.
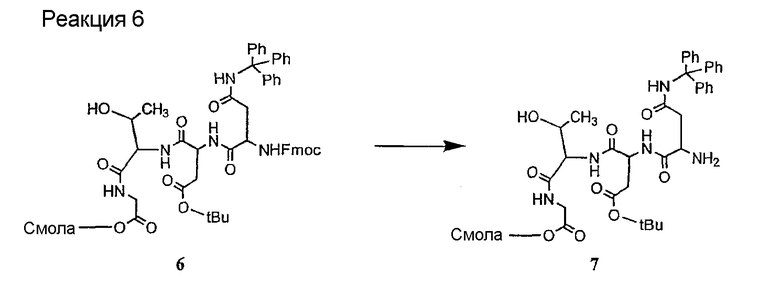
Соединение 6 встряхивали в 20% пиперидине в N-метилпиролидине (6 мл) в течение 30 минут. Смолу фильтровали и вновь суспендировали в 20% пиперидине в N-метилпиролидине (6 мл) и встряхивали в течение 30 минут. Реакционную смесь фильтровали и затем твердое вещество промывали N-метилпиролидином (3 × 6 мл), метанолом (3 × 6 мл) и вновь N-метилпиролидином (3 × 6 мл) с получением соединения 7.
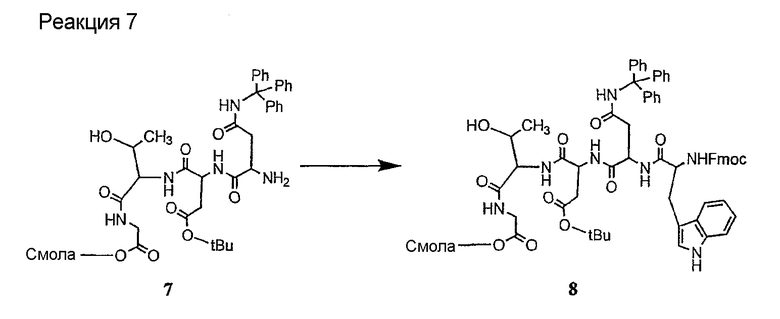
Имеющиеся в продаже Nα-(9-флуоренилметоксикарбонил)-L-триптофан (2 мл 0,5 молярного раствора в N-метилпиролидине), 1,3-диизопропилкарбодиимид (2 мл 0,5 молярного раствора в N-метилпиролидине) и 1-гидроксибензотриазол (2 мл 0,5 молярного раствора в N-метилпиролидине) добавляли к смоле. Реакционную смесь перемешивали в течение одного часа, затем фильтровали и присоединение повторяли. Реакционную смесь фильтровали и затем твердое вещество промывали N-метилпиролидином (3 × 6 мл), метанолом (3 × 6 мл) и вновь N-метилпиролидином (3 × 6 мл) с получением соединения 8.
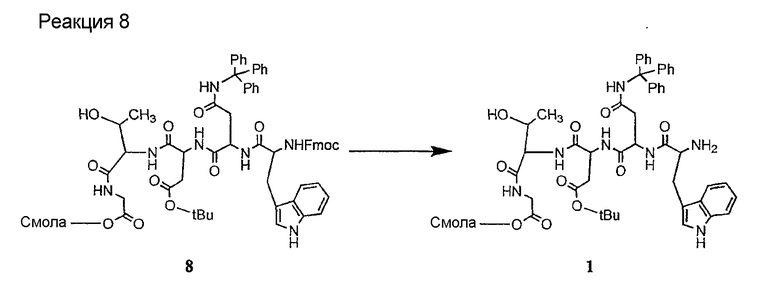
Соединение 8 встряхивали в 20% пиперидине в N-метилпиролидине (6 мл) в течение 30 минут. Смолу фильтровали и вновь суспендировали в 20% пиперидине в N-метилпиролидине (6 мл) и встряхивали в течение 30 минут. Реакционную смесь фильтровали и затем твердое вещество промывали N-метилпиролидином (3 × 6 мл), метанолом (3 × 6 мл) и вновь N-метилпиролидином (3 × 6 мл) с получением пептидного соединения смолы 1.
Пример 2: Синтез пептидной смолы соединения 9:
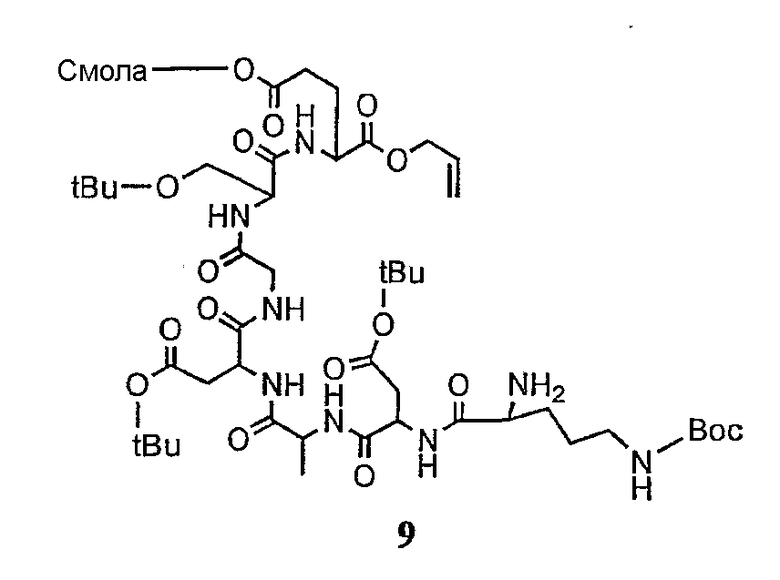
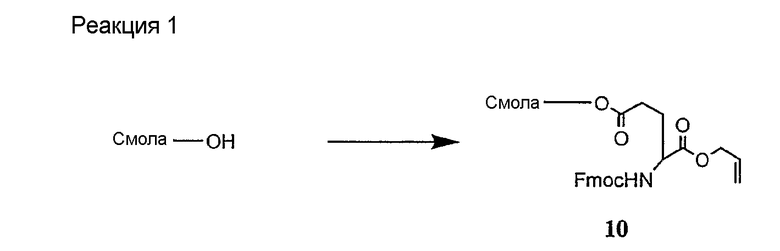
К суспензии имеющейся в продаже 4-гидроксиметилфенокси смолы (смолы Ванга) (5 г, 0,4 ммоль/г) в дихлорметане (60 мл) добавляли 1,3-диизопропилкарбодиимид (0,940 мл), 4-диметиламинопиридин (24 мг в N-метилпиролидине (1 мл)) и имеющийся в продаже α-аллиловый эфир Nα-(9-флуоренилметоксикарбонил)-L-глутаминовой кислоты (2,46 г в N-метилпиролидине (9 мл)). Реакционную смесь перемешивали в течение 16 часов, фильтровали и твердое вещество промывали N-метилпиролидином и дихлорметаном и сушили с получением соединения 10.
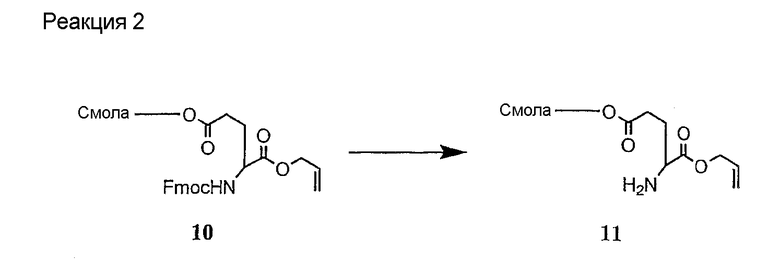
Соединение 10 (526 мг) встряхивали в 20% пиперидине в N-метилпиролидине (6 мл) в течение 30 минут. Смолу фильтровали и вновь суспендировали в 20% пиперидине в N-метилпиролидине (6 мл) и встряхивали в течение 30 минут. Реакционную смесь фильтровали и затем твердое вещество промывали N-метилпиролидином (3 × 6 мл), метанолом (3 × 6 мл) и вновь N-метилпиролидином (3 × 6 мл) с получением соединения 11.
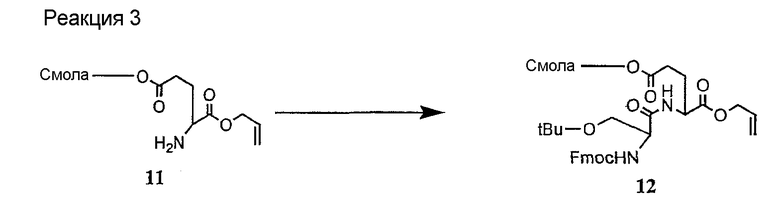
Имеющиеся в продаже трет-бутиловый эфир Nα-(9-флуоренилметоксикарбонил)-L-серина (2 мл 0,5 молярного раствора в N-метилпиролидине), 1,3-диизопропилкарбодиимид (2 мл 0,5 молярного раствора в N-метилпиролидине) и 1-гидроксибензотриазол (2 мл 0,5 молярного раствора в N-метилпиролидине) добавляли к смоле 11. Реакционную смесь перемешивали в течение одного часа, затем фильтровали и присоединение повторяли. Реакционную смесь фильтровали и затем твердое вещество промывали N-метилпиролидином (3 × 6 мл), метанолом (3 × 6 мл) и вновь N-метилпиролидином (3 × 6 мл) с получением соединения 12.
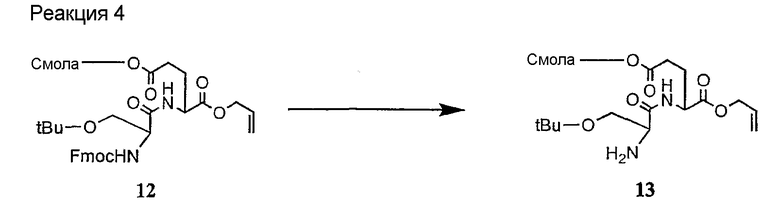
Соединение 12 встряхивали в 20% пиперидине в N-метилпиролидине (6 мл) в течение 30 минут. Смолу фильтровали и вновь суспендировали в 20% пиперидине в N-метилпиролидине (6 мл) и встряхивали в течение 30 минут. Реакционную смесь фильтровали и затем твердое вещество промывали N-метилпиролидином (3 × 6 мл), метанолом (3 × 6 мл) и вновь N-метилпиролидином (3 × 6 мл) с получением соединения 13.
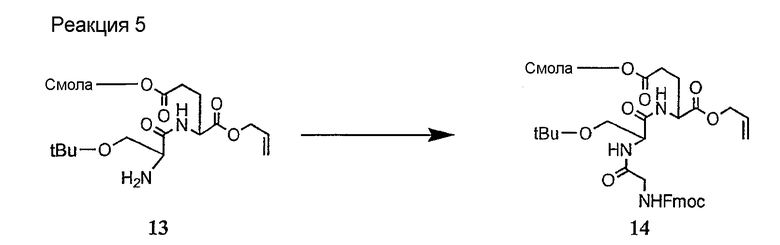
Имеющиеся в продаже Nα-(9-флуоренилметоксикарбонил)-L-глицин (2 мл 0,5 молярного раствора в N-метилпиролидине), 1,3-диизопропилкарбодиимид (2 мл 0,5 молярного раствора в N-метилпиролидине) и 1-гидроксибензотриазол (2 мл 0,5 молярного раствора в N-метилпиролидине) добавляли к смоле 13. Реакционную смесь перемешивали в течение одного часа, затем фильтровали и присоединение повторяли. Реакционную смесь фильтровали и затем твердое вещество промывали N-метилпиролидином (3 × 6 мл), метанолом (3 × 6 мл) и вновь N-метилпиролидином (3 × 6 мл) с получением соединения 14.
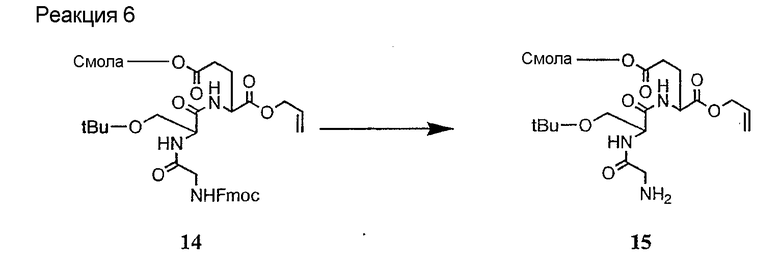
Соединение 14 встряхивали в 20% пиперидине в N-метилпиролидине (6 мл) в течение 30 минут. Смолу фильтровали и вновь суспендировали в 20% пиперидине в N-метилпиролидине (6 мл) и встряхивали в течение 30 минут. Реакционную смесь фильтровали и затем твердое вещество промывали N-метилпиролидином (3 × 6 мл), метанолом (3 × 6 мл) и вновь N-метилпиролидином (3 × 6 мл) с получением соединения 15.
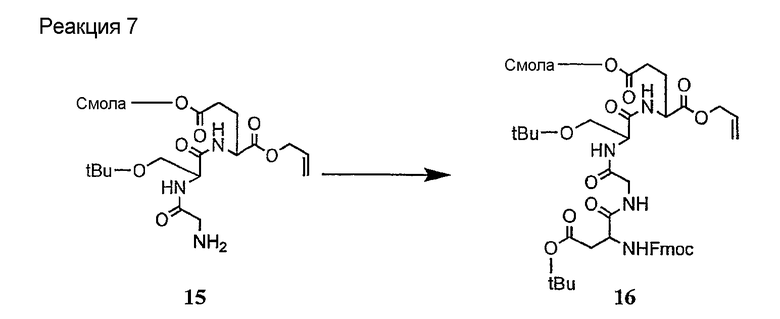
Имеющиеся в продаже β-трет-бутиловый эфир Nα-(9-флуоренилметоксикарбонил)-L-аспарагиновой кислоты (2 мл 0,5 молярного раствора в N-метилпиролидине), 1,3-диизопропилкарбодиимид (2 мл 0,5 молярного раствора в N-метилпиролидине) и 1-гидроксибензотриазол (2 мл 0,5 молярного раствора в N-метилпиролидине) добавляли к смоле 15. Реакционную смесь перемешивали в течение одного часа, затем фильтровали и присоединение повторяли. Реакционную смесь фильтровали и затем твердое вещество промывали N-метилпиролидином (3 × 6 мл), метанолом (3 × 6 мл) и вновь N-метилпиролидином (3 × 6 мл) с получением соединения 16.
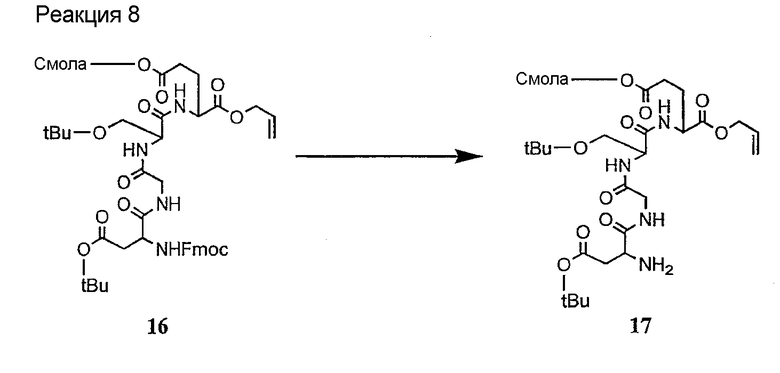
Соединение 16 встряхивали в 20% пиперидине в N-метилпиролидине (6 мл) в течение 30 минут. Смолу фильтровали и вновь суспендировали в 20% пиперидине в N-метилпиролидине (6 мл) и встряхивали в течение 30 минут. Реакционную смесь фильтровали и затем твердое вещество промывали N-метилпиролидином (3 × 6 мл), метанолом (3 × 6 мл) и вновь N-метилпиролидином (3 × 6 мл) с получением соединения 17.
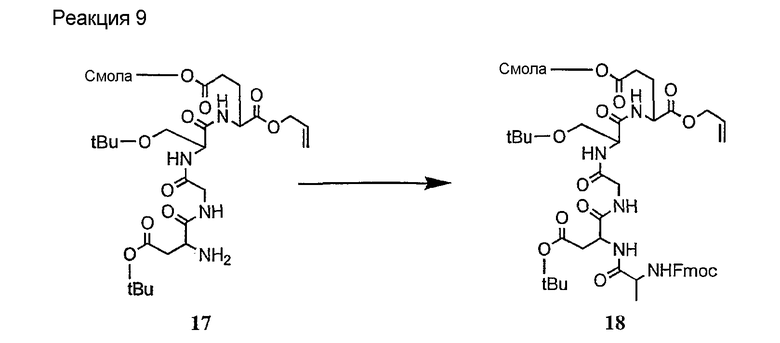
Имеющиеся в продаже Nα-(9-флуоренилметоксикарбонил)-L-аланин (2 мл 0,5 молярного раствора в N-метилпиролидине), 1,3-диизопропилкарбодиимид (2 мл 0,5 молярного раствора в N-метилпиролидине) и 1-гидроксибензотриазол (2 мл 0,5 молярного раствора в N-метилпиролидине) добавляли к смоле 17. Реакционную смесь перемешивали в течение одного часа, затем фильтровали и присоединение повторяли. Реакционную смесь фильтровали, и затем твердое вещество промывали N-метилпиролидином (3 × 6 мл), метанолом (3 × 6 мл) и вновь N-метилпиролидином (3 × 6 мл) с получением соединения 18.
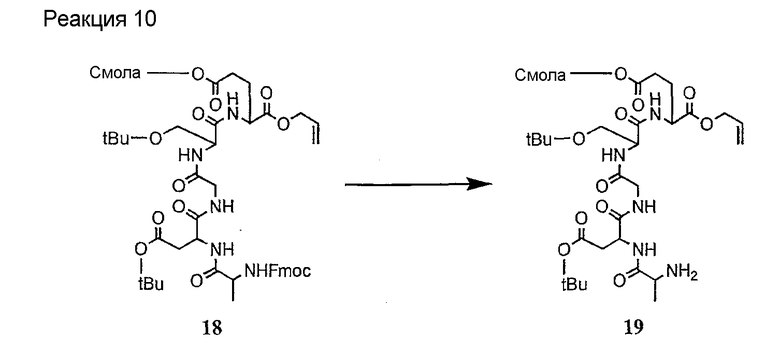
Соединение 18 встряхивали в 20% пиперидине в N-метилпиролидине (6 мл) в течение 30 минут. Смолу фильтровали и вновь суспендировали в 20% пиперидине в N-метилпиролидине (6 мл) и встряхивали в течение 30 минут. Реакционную смесь фильтровали и затем твердое вещество промывали N-метилпиролидином (3 × 6 мл), метанолом (3 × 6 мл) и вновь N-метилпиролидином (3 × 6 мл) с получением соединения 19.
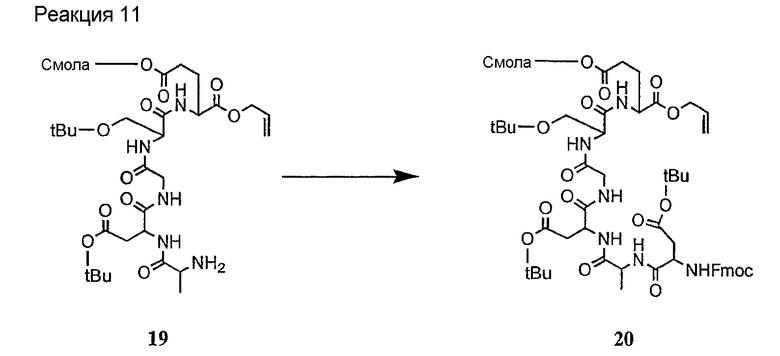
Имеющиеся в продаже β-трет-бутиловый эфир Nα-(9-флуоренилметоксикарбонил)-L-аспарагиновой кислоты (2 мл 0,5 молярного раствора в N-метилпиролидине), 1,3-диизопропилкарбодиимид (2 мл 0,5 молярного раствора в N-метилпиролидине) и 1-гидроксибензотриазол (2 мл 0,5 молярного раствора в N-метилпиролидине) добавляли к смоле 19. Реакционную смесь перемешивали в течение одного часа, фильтровали и присоединение повторяли. Реакционную смесь фильтровали и затем твердое вещество промывали N-метилпиролидином (3 × 6 мл), метанолом (3 × 6 мл) и вновь N-метилпиролидином (3 × 6 мл) с получением соединения 20.
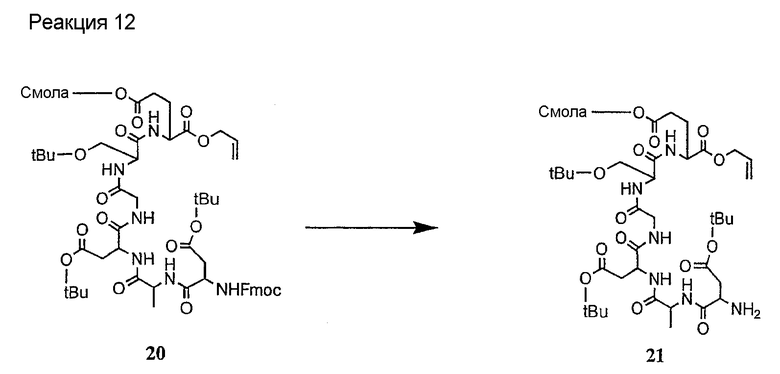
Соединение 20 встряхивали в 20% пиперидине в N-метилпиролидине (6 мл) в течение 30 минут. Смолу фильтровали и вновь суспендировали в 20% пиперидине в N-метилпиролидине (6 мл) и встряхивали в течение 30 минут. Реакционную смесь фильтровали и затем твердое вещество промывали N-метилпиролидином (3 × 6 мл), метанолом (3 × 6 мл) и вновь N-метилпиролидином (3 × 6 мл) с получением соединения 21.
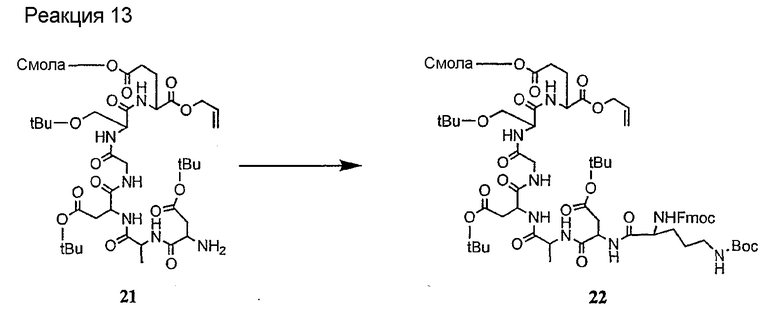
Имеющиеся в продаже Nα-(9-флуоренилметоксикарбонил)-Nδ-(трет-бутоксикарбонил)-L-орнитин (2 мл 0,5 молярного раствора в N-метилпиролидине), 1,3-диизопропилкарбодиимид (2 мл 0,5 молярного раствора в N-метилпиролидине) и 1-гидроксибензотриазол (2 мл 0,5 молярного раствора в N-метилпиролидине) добавляли к смоле 21. Реакционную смесь перемешивали в течение одного часа, затем фильтровали и присоединение повторяли. Реакционную смесь фильтровали и затем твердое вещество промывали N-метилпиролидином (3 × 6 мл), метанолом (3 × 6 мл) и вновь N-метилпиролидином (3 × 6 мл) с получением соединения 22.
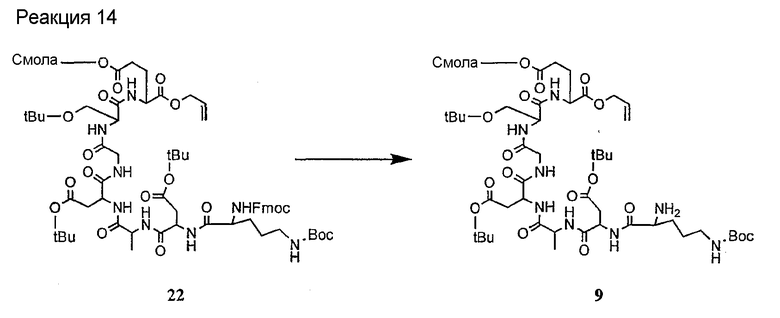
Соединение 22 встряхивали в 20% пиперидине в N-метилпиролидине (6 мл) в течение 30 минут. Смолу фильтровали и вновь суспендировали в 20% пиперидине в N-метилпиролидине (6 мл) и встряхивали в течение 30 минут. Реакционную смесь фильтровали и затем твердое вещество промывали N-метилпиролидином (3 × 6 мл), метанолом (3 × 6 мл) и вновь N-метилпиролидином (3 × 6 мл) с получением соединения 9.
Пример 3: Синтез пептидной смолы соединения 23:
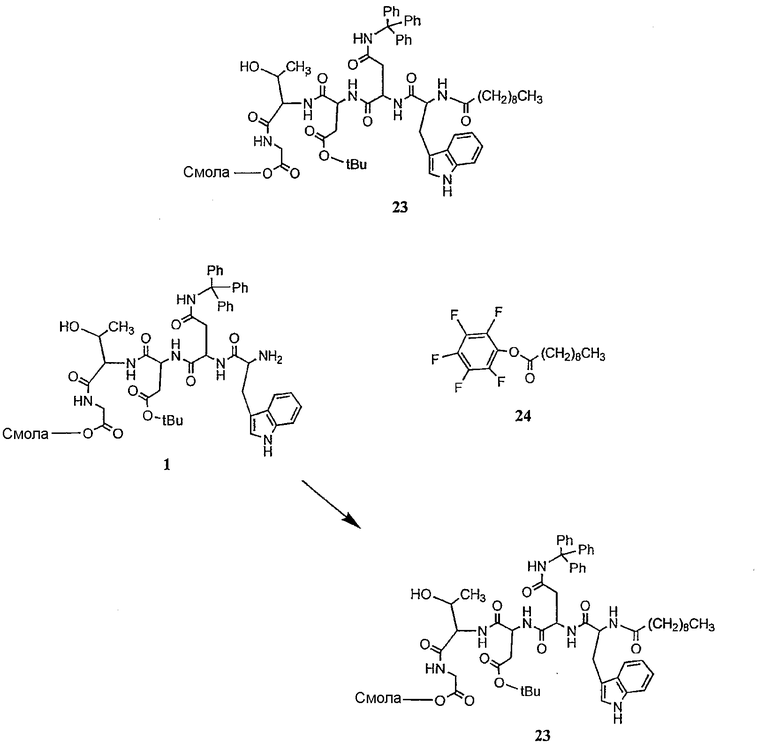
Пептидную смолу 1 (2 г) добавляли к раствору пентафторфенилового эфира декановой кислоты 24 (440 мг, получение соединения 24 смотри пример 6, реакция 1) в дихлорметане. Смесь перемешивали в течение 17 часов, фильтровали и с помощью теста Кайзера (смотри выше) было определено, что реакция прошла не полностью. Декановую кислоту (517 мг), 1-гидроксибензотриазол (446 мг) и 1,3-диизопропилкарбодиимид (438 мкл) растворяли в N-метилпиролидине (8 мл) и перемешивали в течение одного часа. Затем к смеси добавляли смолу и перемешивали в течение 8 часов, фильтровали и промывали N-метилпиролидином (3 × 6 мл), метанолом (3 × 6 мл) и вновь N-метилпиролидином (3 × 6 мл). С помощью теста Кайзера было обнаружено, что реакция прошла полностью с получением связанного со смолой липопептида 23.
Пример 4: Синтез соединения 29:
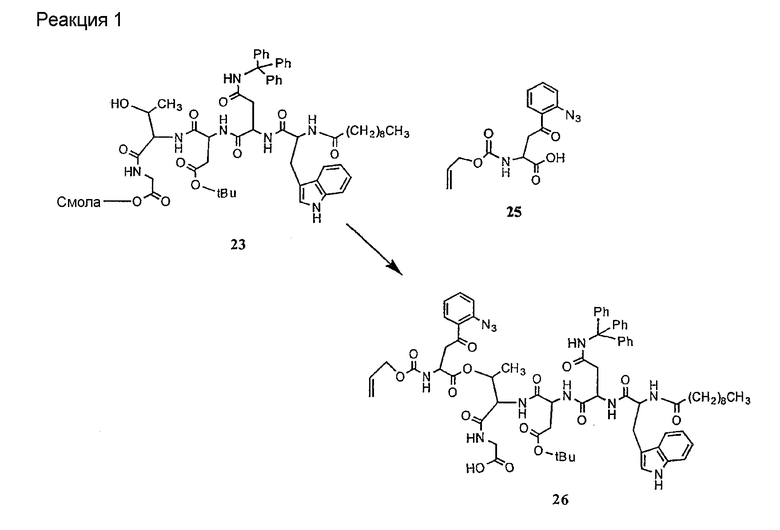
L-2-N-(Аллилоксикарбонил)-4-(2-азидофенил)-4-оксомасляную кислоту 25 (636 мг, смотри пример 15 (смотри ниже)), 4-диметиламинопиридин (25 мг) и N-метил-2-хлорпиридиний йодид (511 мг) тщательно промывали струей аргона и затем суспендировали в дихлорметане (10 мл). Добавляли триэтиламин (560 мкл) и реакционную смесь перемешивали с получением однородного раствора. К раствору добавляли липопептидную смолу 23 (667 мг) и сосуд вновь продували аргоном и перемешивали в течение 17 часов. Отбирали 20-мг образец смолы для проверки завершения реакции (20 мг смолы в дихлорметане (0,6 мл), обрабатывали 2,2,2-трифторэтанолом (0,2 мл) и уксусной кислотой (0,2 мл) и перемешивали в течение 3 часов. Реакционную смесь фильтровали и растворитель выпаривали с получением остатка. Анализ с помощью жидкостной хроматографии/масс-спектрометрии показал, что реакция не прошла полностью. Присоединение было оценено как неполное, и поэтому смолу сушили при пониженном давлении в течение 5 дней, и описанное выше присоединение повторяли еще в течение 17 часов. Реакционную смесь фильтровали и твердое вещество тщательно промывали дихлорметаном. Твердое вещество затем суспендировали в дихлорметане (6 мл), 2,2,2-трифторэтаноле (2 мл), уксусной кислоте (2 мл) и перемешивали в течение 5 часов. Реакционную смесь фильтровали и упаривание фильтрата дало сырой желаемый пептид 26 (44 мг). Сырой продукт очищали ВЭЖХ с обращенной фазой (C18 10 мкм колонка Jupiter 250 × 21,2 мм) с элюированием градиентом смеси от 20% ацетонитрил - 0,5% муравьиная кислота: 80% вода - 0,5% муравьиная кислота до 80% ацетонитрил - 0,5% муравьиная кислота: 20% вода - 0,5% муравьиная кислота в течение 25 минут. Содержащие продукт фракции лиофилизовали с получением чистого продукта 26 (10,6 мг).
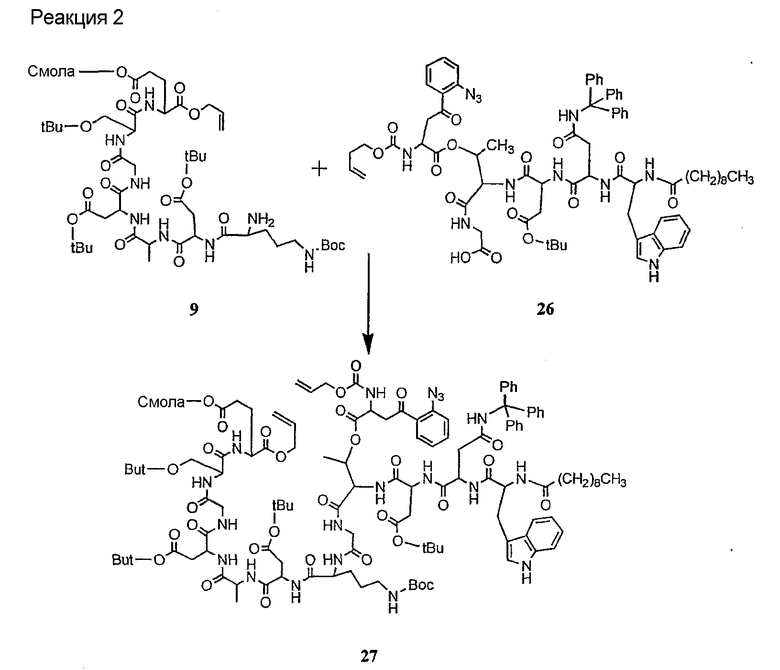
К раствору соединения 26 (10,6 мг) в N-метилпиролидине (0,7 мл) добавляли гидроксибензотриазол (5 мг), 1,3-диизопропилкарбодиимид (6 мкл) и пептидную смолу 9 (12,3 мг) и затем перемешивали в течение 22 часов. Смолу фильтровали и полноту присоединения оценивали с помощью теста Кайзера, что дало связанный со смолой липопептид 27.
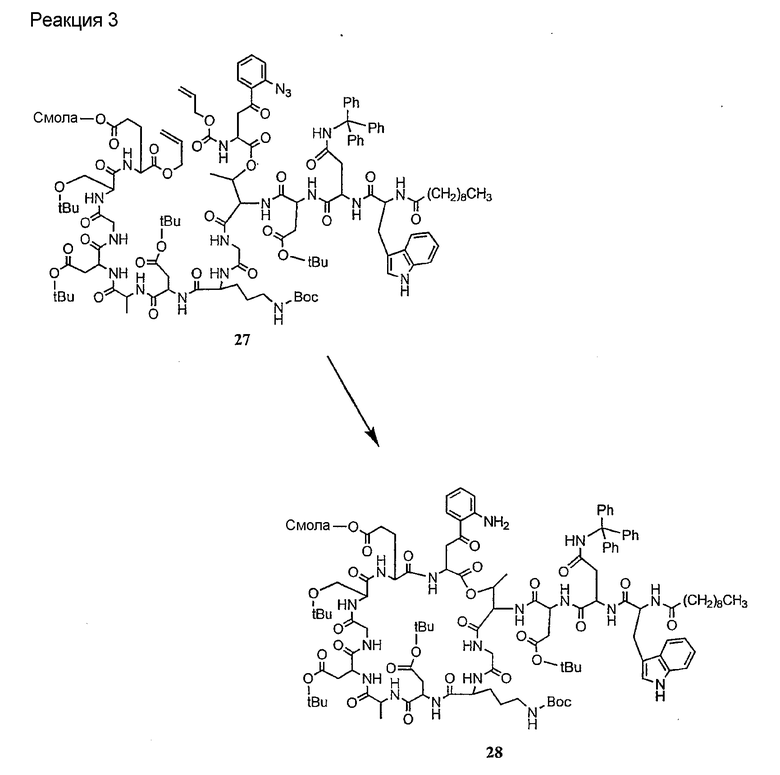
Высушенную смолу 27 помещали в атмосфере аргона и обрабатывали раствором тетракис-(трифенилфосфин)палладия(0) (19 мг) в дихлорметане (1,47 мл), уксусной кислоте (74 мкл) и N-метилморфолине (37 мкл). Смесь перемешивали в течение 4 часов при температуре окружающей среды, фильтровали и твердое вещество промывали два раза N-метилморфолином, два раза метанолом и вновь два раза N-метилморфолином. К смоле добавляли 1-гидроксибензотриазол (0,5 мл 0,5 молярного раствора в N-метилморфолине) и 1,3-диизопропилкарбодиимид (0,5 мл 0,5 молярного раствора в N-метилморфолине). Реакционную смесь перемешивали в течение 17 часов, фильтровали и тщательно промывали N-метилморфолином с получением связанного со смолой циклизованного депсипептида 28.
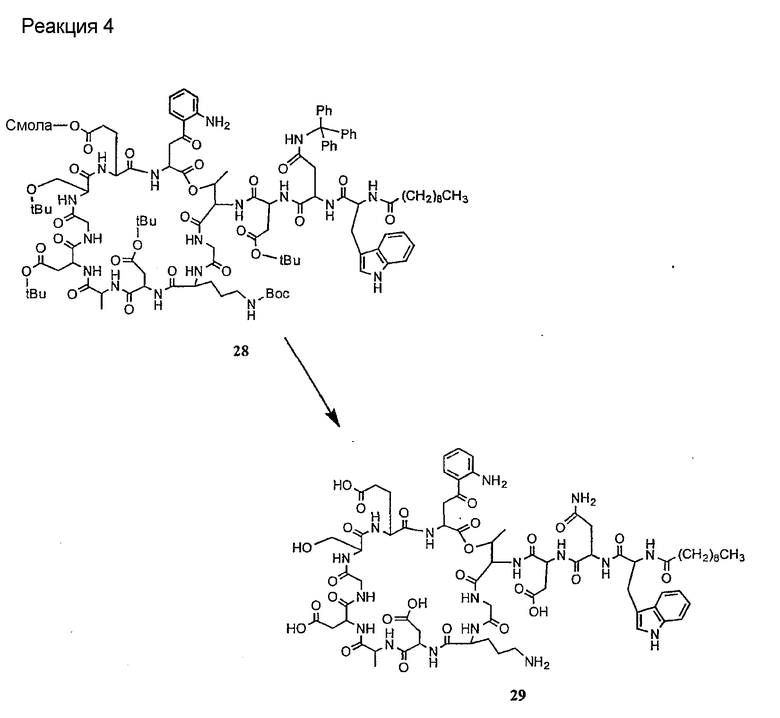
Высушенную смолу 28 суспендировали в дихлорметане (4 мл), трифторуксусной кислоте (6 мл), этандитиоле (250 мкл) и триизопропилсилане (250 мкл) и реакционную смесь перемешивали в течение 3 часов при температуре окружающей среды. Смолу фильтровали и объединенные фильтраты упаривали при пониженном давлении. Сырой продукт затем подвергали распределению между диэтиловым эфиром (6 мл) и водой (3 мл). Водный слой лиофилизовали с получением сырого продукта. Сырой продукт очищали ВЭЖХ с обращенной фазой (C18 10 мкм колонка Jupiter 250 × 21,2 мм) с элюированием градиентом смеси от 20% ацетонитрил - 0,5% муравьиная кислота: 80% вода - 0,5% муравьиная кислота до 80% ацетонитрил - 0,5% муравьиная кислота: 20% вода - 0,5% муравьиная кислота в течение 25 минут. Содержащие продукт фракции объединяли и лиофилизовали с получением чистого продукта 29 (1,0 мг).
Пример 5: Синтез соединения 33:
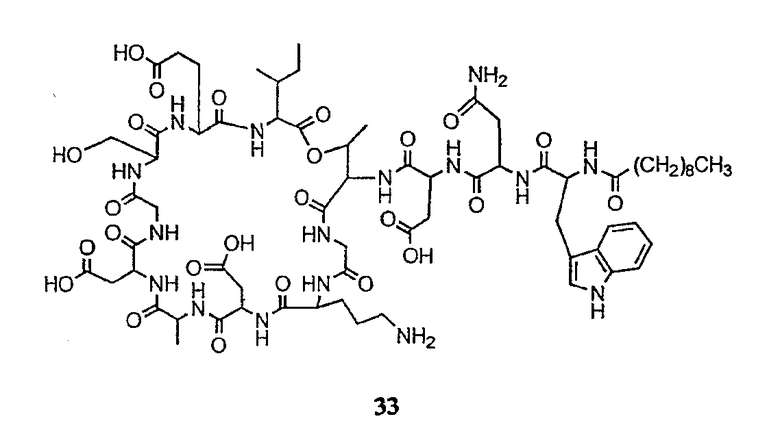
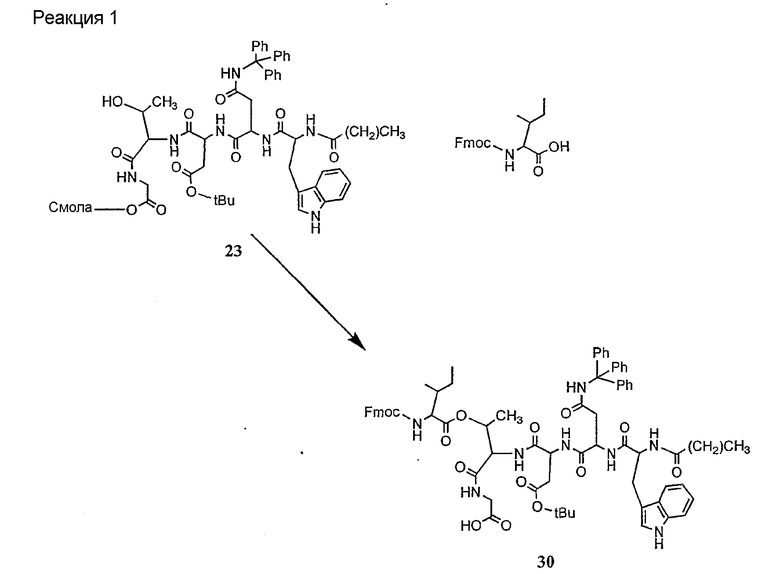
Имеющиеся в продаже Nα-(9-флуоренилметоксикарбонил)-L-изолейцин (95 мг), 4-диметиламинопиридин (6 мг) и N-метил-2-хлорпиридиний йодид (69 мг) тщательно промывали струей аргона и затем суспендировали в дихлорметане (2,7 мл). Добавляли триэтиламин (76 мкл) и реакционную смесь перемешивали с получением однородного раствора. К раствору добавляли липопептидную смолу 23 (200 мг) и сосуд вновь продували аргоном и затем перемешивали в течение 14 часов. Полученную смолу затем фильтровали и промывали дихлорметаном. Твердое вещество суспендировали в дихлорметане (6 мл), 2,2,2-трифторэтаноле (2 мл) и уксусной кислоте (2 мл) и перемешивали в течение 3 часов. Смолу фильтровали и упаривание фильтрата дало желаемый пептид 30 (54 мг) в виде белого твердого вещества.
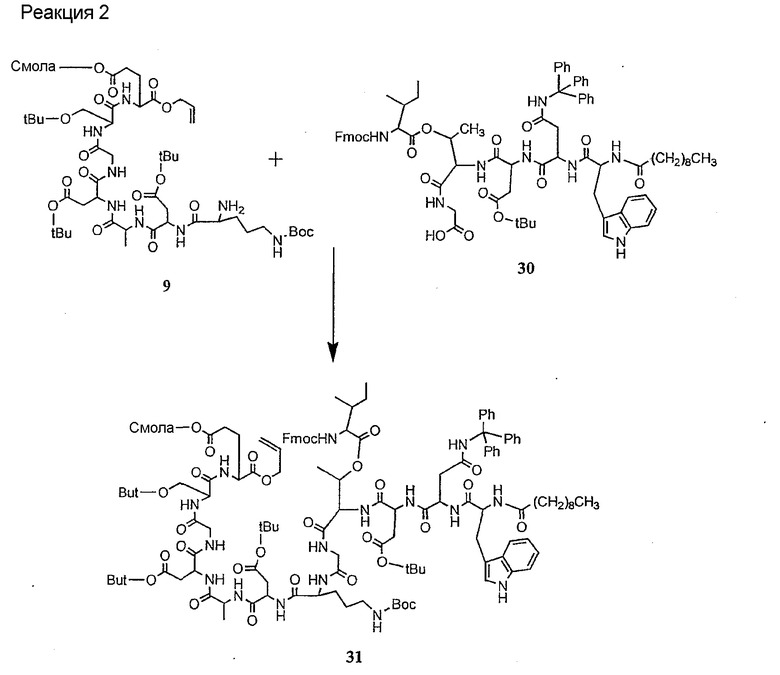
1-Гидроксибензотриазол (26 мг), 1,3-диизопропилкарбодиимид (30 мкл) и пептидную смолу 9 (64 мг) добавляли к раствору депсипептида 30 (54 мг) в N-метилморфолине (3,8 мл) и полученную смесь перемешивали в течение 22 часов. Смолу фильтровали и с помощью теста Кайзера присоединение было оценено как полное с получением связанного со смолой депсипептида 31.
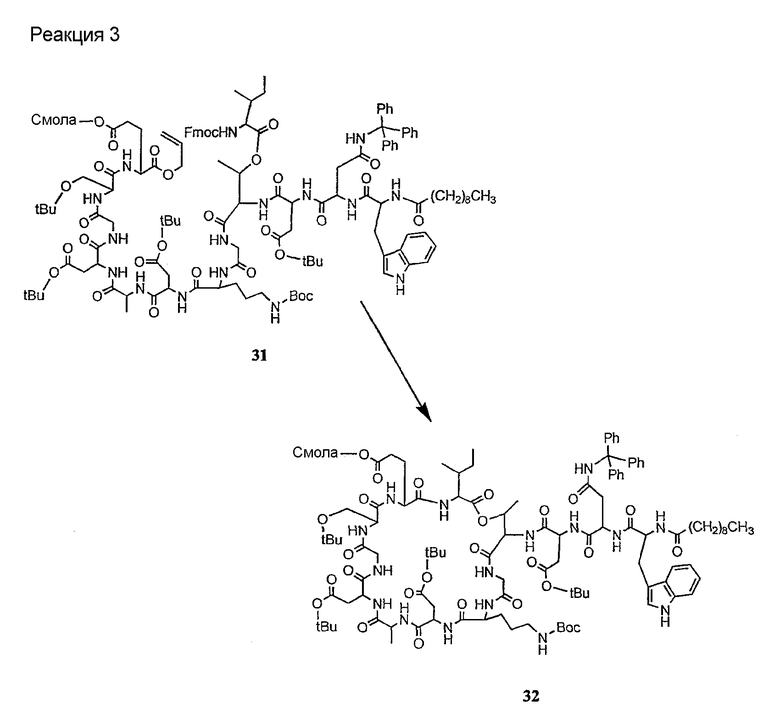
Высушенную смолу 31 помещали в атмосфере аргона и обрабатывали раствором тетракис-(трифенилфосфин)палладия(0) (48 мг в дихлорметане (7,63 мл)), уксусной кислоте (0,38 мл) и N-метилморфолине (0,19 мл). Смесь перемешивали в течение 4 часов при температуре окружающей среды, фильтровали и твердое вещество промывали два раза N-метилморфолином, два раза метанолом и вновь два раза N-метилморфолином. Твердую смолу суспендировали в 20% пиперидине в N-метилморфолине (7 мл) в течение 105 минут, фильтровали и твердое вещество тщательно промывали N-метилморфолином. К смоле добавляли 1-гидроксибензотриазол (0,3 мл 0,5 молярного раствора в N-метилморфолине) и 1,3-диизопропилкарбодиимид (0,3 мл 0,5 молярного раствора в N-метилморфолине). Реакционную смесь перемешивали в течение 17 часов, фильтровали и осадок тщательно промывали N-метилморфолином с получением связанного со смолой циклизованного депсипептида 32.
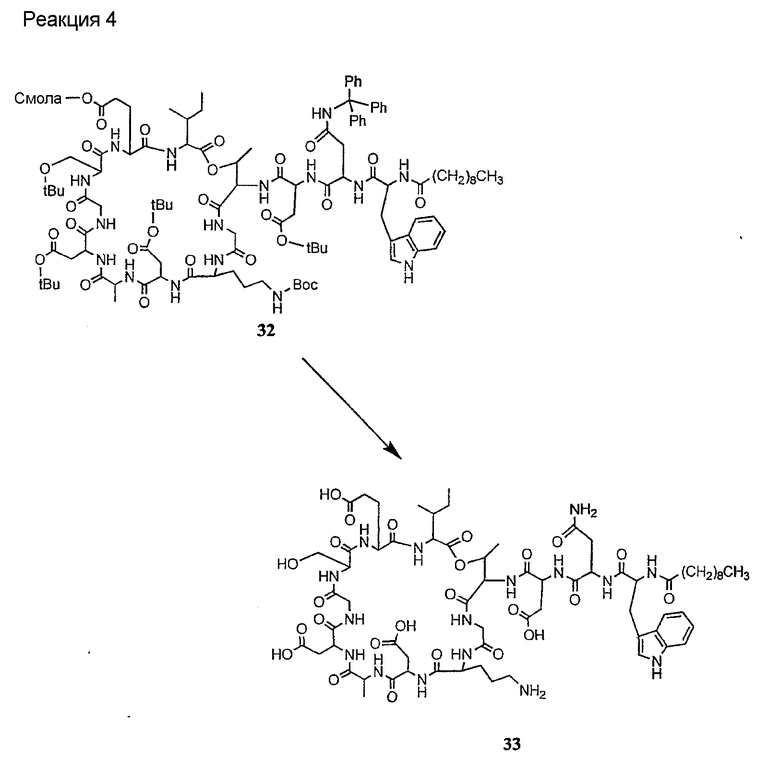
Высушенную смолу 32 суспендировали в дихлорметане (4 мл), трифторуксусной кислоте (6 мл), этандитиоле (250 мкл) и триизопропилсилане (250 мкл) и перемешивали в течение 3 часов при температуре окружающей среды. Реакционную смесь фильтровали и промывали дихлорметаном (2 × 2 мл) и объединенные фильтраты упаривали при пониженном давлении. Сырой продукт затем подвергали распределению между диэтиловым эфиром (6 мл) и водой (3 мл). Водный слой отделяли и лиофилизовали с получением сырого продукта 33 (21,5 мг). Сырой продукт затем очищали ВЭЖХ с обращенной фазой (C18 10 мкм колонка Jupiter 250 × 21,2 мм) с элюированием градиентом смеси от 20% ацетонитрил - 0,5% муравьиная кислота: 80% вода - 0,5% муравьиная кислота до 80% ацетонитрил - 0,5% муравьиная кислота: 20% вода - 0,5% муравьиная кислота в течение 25 минут. Содержащие продукт фракции объединяли и лиофилизовали с получением чистого продукта 33 (1,8 мг).
Пример 6: Синтез соединения 34:
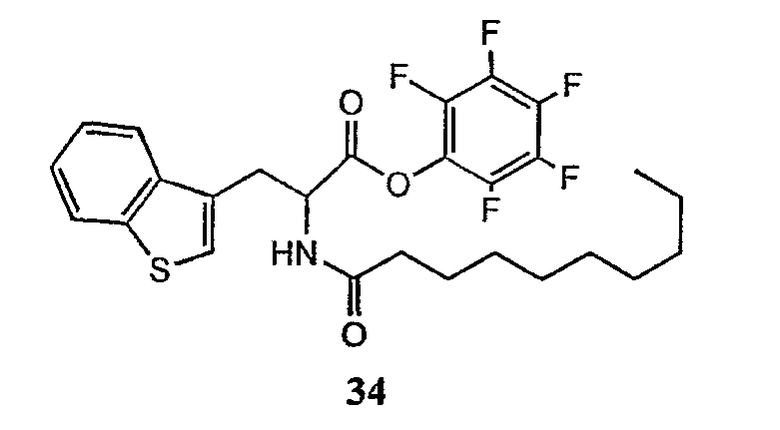
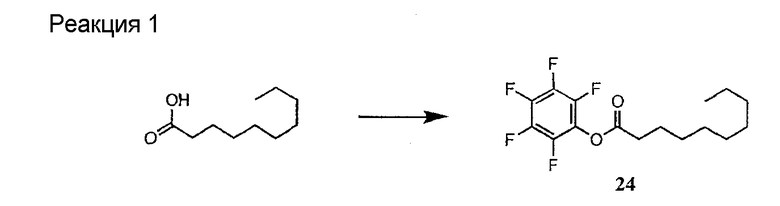
К раствору имеющейся в продаже декановой кислоты (13,78 г), в тетрагидрофуране (40 мл) добавляли 1,3-диизопропилкарбодиимид (13,76 мл). Через 5 минут добавляли раствор пентафторфенола (16,20 г) в тетрагидрофуране (40 мл) и реакционную смесь перемешивали в течение 72 часов. Полученную смесь фильтровали; остаток промывали тетрагидрофураном (40 мл) и тетрагидрофурановые фильтраты объединяли. Растворитель выпаривали и остаток очищали на силикагеле с применением смеси 9:1 гексан:этилацетат в качестве элюента с получением желаемого продукта 24 в виде масла (23,49 г).
Раствор имеющегося в продаже Boc-L-3-бензотиенилаланина 35 (1,0 г) в безводном метаноле (20 мл) охлаждали до 0°С на бане со льдом. По каплям добавляли тионилхлорид (1,63 г) и реакционной смеси давали нагреться до температуры окружающей среды в течение 16 часов. Выпаривание растворителя дало сырой продукт, который подвергали распределению между этилацетатом и водным бикарбонатом калия. Органический слой промывали водным бикарбонатом калия и насыщенным хлоридом натрия, сушили безводным сульфатом натрия и упаривали с получением продукта 36 (0,66 г).
К раствору метилового эфира бензотиенилаланина 36 (0,66 г) в тетрагидрофуране (10 мл) добавляли раствор деканоилпентафторфенолового эфира 24 (1,04 г) в тетрагидрофуране (10 мл). Полученный раствор перемешивали в течение 24 часов. Реакционную смесь выливали в этилацетат и промывали последовательно 1 н. хлористоводородной кислотой, 10% водным карбонатом калия и насыщенным хлоридом натрия. Объединенный органический слой затем сушили безводным сульфатом натрия и упаривали с получением продукта 37 (1,40 г), который использовали в качестве сырого вещества в следующей реакции.
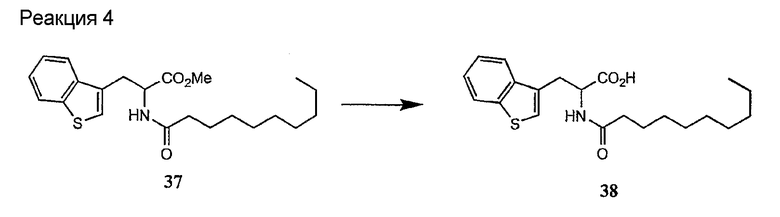
Моногидрат гидроксида лития (0,59 г) добавляли к раствору соединения 37 (1,40 г), метанола (15 мл) и воды (3 мл) и реакционную смесь перемешивали в течение одного часа. Метанол удаляли выпариванием и водный раствор подкисляли до pH 1 с помощью 1 н. хлористоводородной кислоты и экстрагировали этилацетатом. Органические слои объединяли и сушили безводным сульфатом натрия и упаривали с получением продукта 38.
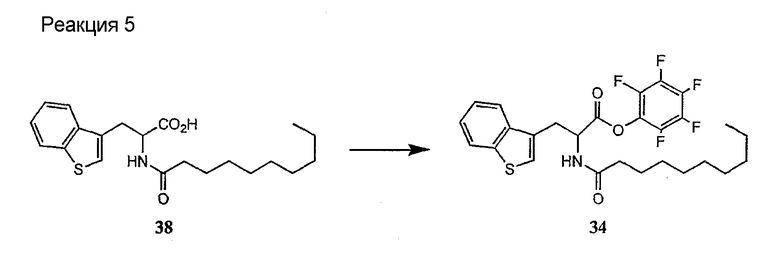
1,3-Диизопропилкарбодиимид (121 мг) добавляли к раствору соединения 38 (200 мг) и пентафторфенола (108 мг) в тетрагидрофуране (5 мл) и реакционную смесь перемешивали в течение одного часа. Полученную смесь фильтровали, растворитель выпаривали и остаток очищали на силикагеле. Применение смеси 9:1 гексан:этилацетат в качестве элюента дало загрязнение с большей подвижностью, а смесь 4:1 гексан:этилацетат в качестве элюента дала продукт 34 (80 мг).
Пример 7: Синтез соединения 39:
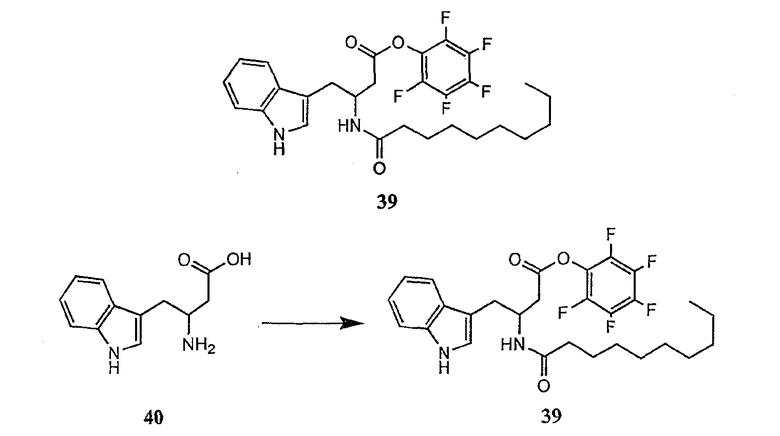
Соединение 39 получали из имеющегося в продаже соединения 40 при описанных выше условиях следующим образом: Соединение 39 превращали в соответствующий метиловый эфир при условиях, описанных в примере 6, реакция 2. Метиловый эфир ацилировали при условиях, описанных в примере 6, реакция 3. Ацилированное соединение затем сапонифицировали при условиях реакции, описанных в примере 6, реакция 4. Сапонифицированное вещество затем превращали в соединение 39 при условиях, описанных в примере 6, реакция 5.
Пример 8: Синтез соединения 41:
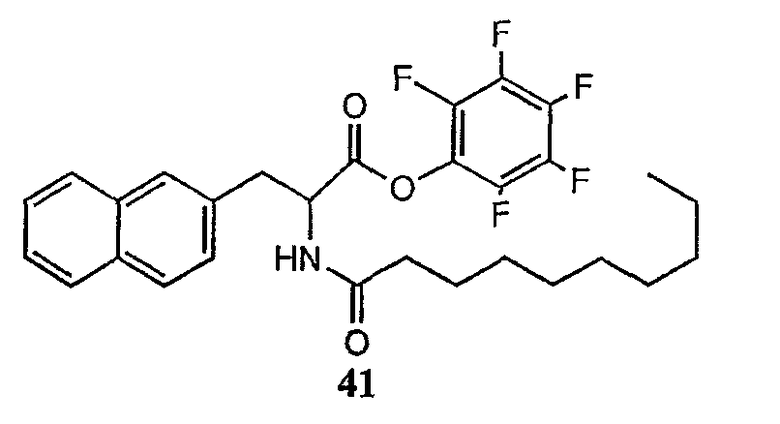
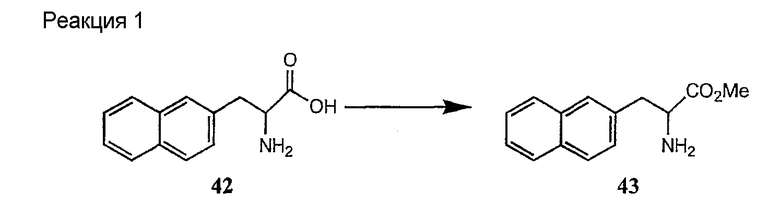
Имеющееся в продаже соединение 42 превращали в соединение 43 при условиях, описанных в примере 6, реакция 2.
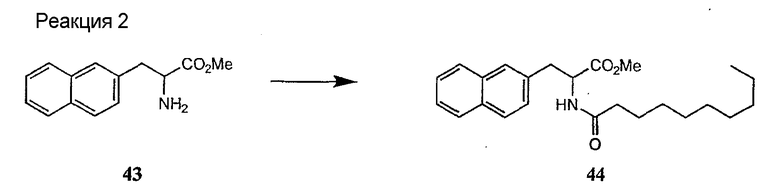
Деканоилхлорид (0,51 мл) добавляли к раствору метилового эфира аминокислоты 43 (440 мг) и триэтиламина (0,43 мл), охлажденному до 0°С на бане со льдом. Реакционной смеси давали нагреться до температуры окружающей среды в течение трех часов. Реакционную смесь затем выливали в этилацетат и последовательно промывали водой и насыщенным хлоридом натрия. Органические слои затем сушили безводным сульфатом натрия и упаривали с получением продукта 44.
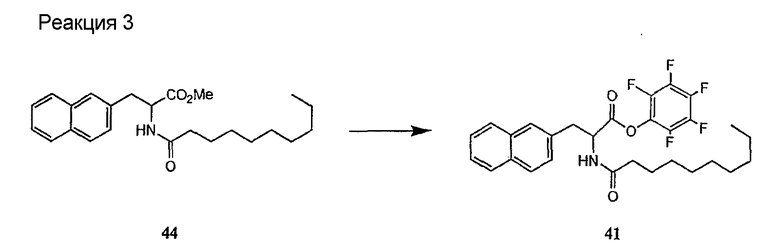
Соединение 44 превращали в соединение 41 при условиях, описанных ниже. Соединение 44 сапонифицировали при условиях реакции, описанных в примере 6, реакция 4. Сапонифицированное вещество затем превращали в соединение 41 при условиях, описанных в примере 6, реакция 5.
Пример 9: Синтез соединения 45:
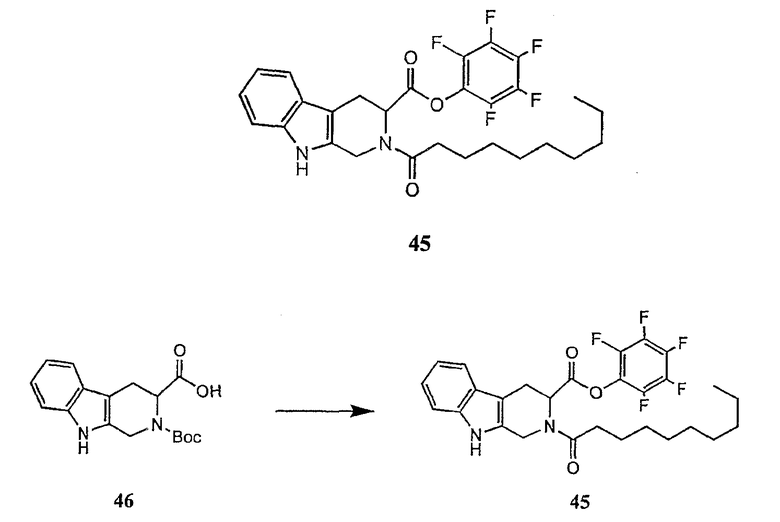
Соединение 45 получали из имеющегося в продаже соединения 46 при описанных выше условиях следующим образом: Соединение 46 превращали в соответствующий метиловый эфир при условиях, описанных в примере 6, реакция 2. Метиловый эфир ацилировали при условиях, описанных в примере 8, реакция 2. Ацилированное соединение затем сапонифицировали при условиях реакции, описанных в примере 6, реакция 4. Сапонифицированное вещество затем превращали в соединение 45 при условиях, описанных в примере 6, реакция 5.
Пример 10: Синтез соединения 47:
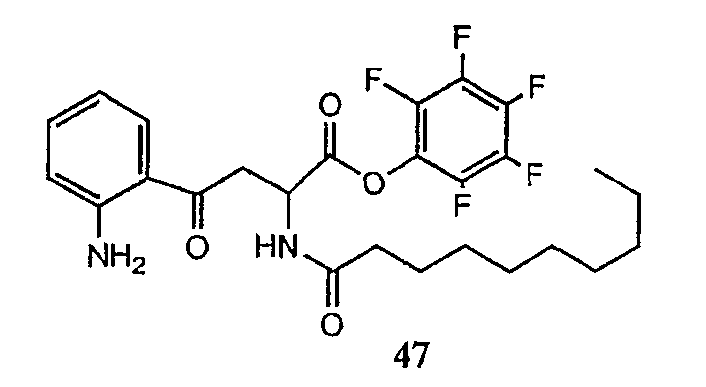
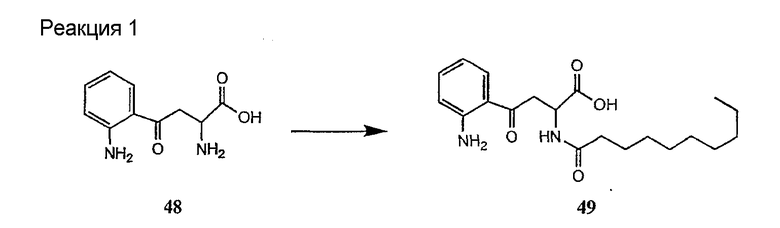
Имеющееся в продаже соединение 48 обрабатывали при условиях, описанных выше в примере 6, реакция 3, с получением соединения 49. Обработка соединения 49, как описано в примере 6, реакция 5, дала соединение 47.
Пример 11: Синтез соединения 50:
Имеющееся в продаже соединение 51 обрабатывали при условиях, описанных выше в примере 6, реакция 3, с единственной модификацией, заключавшейся в применении растворителя N,N'-диметилформамида вместо растворителя тетрагидрофурана, с получением соединения 52. Обработка соединения 52, как описано выше в примере 6, реакция 5, с единственной модификацией, заключавшейся в применении растворителя N,N'-диметилформамида вместо растворителя тетрагидрофурана, дала соединение 50.
Пример 12: Синтез соединения 53:
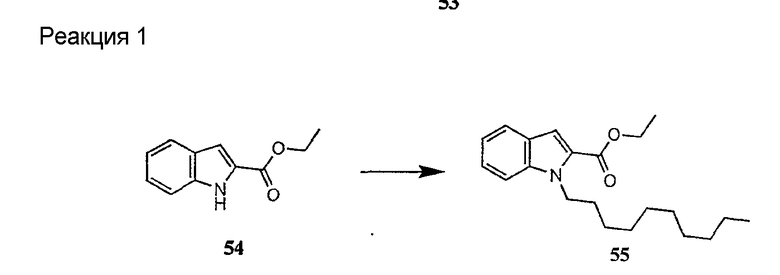
Раствор имеющегося в продаже этилиндол-2-карбоксилата 54 (2 г), йоддекана (2,25 мл) и карбоната калия (2,92 г) в безводном диметилформамиде (30 мл) перемешивали в течение 24 часов. Реакционную смесь выливали в этилацетат (200 мл) и промывали водой (300 мл). Органический слой затем сушили безводным сульфатом магния и упаривали с получением продукта 55 (2,604 г).
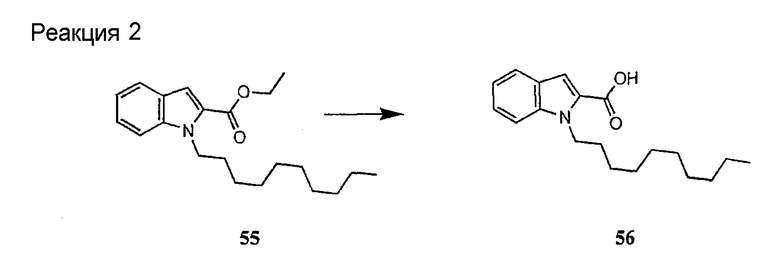
Гидроксид лития/вода (1,65 г) в воде (16 мл) добавляли к раствору соединения 55 (2,6 г) в тетрагидрофуране (16 мл) и перемешивали в течение 4 дней. Добавляли этилацетат (20 мл) и воду (10 мл), pH водного слоя доводили до 1 и экстрагировали этилацетатом (3 × 5 мл). Органические слои объединяли, сушили и упаривали с получением масла. Гидролиз был неполным, в связи с чем масло растворяли в смеси метанол/вода 2:1 и добавляли гидроксид калия (0,88 г). Реакционную смесь перемешивали в течение 15 часов. Добавляли этилацетат (20 мл) и воду (10 мл), слои разделяли, pH водного слоя доводили до 1 и экстрагировали этилацетатом (3 × 5 мл). Органические слои объединяли, сушили и упаривали с получением сырого продукта в виде масла, который очищали на силикагеле с применением смеси 10:1 гексан:этилацетат в качестве элюента с получением продукта 56 (0,35 г).
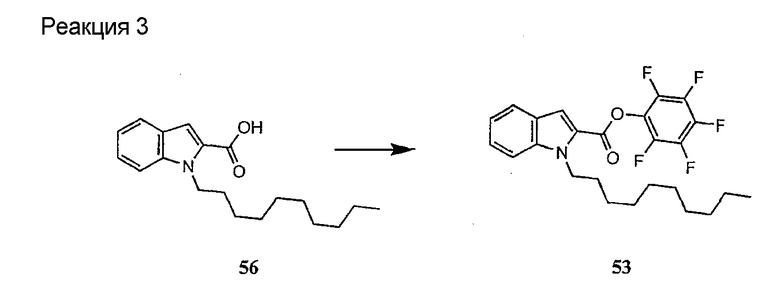
1,3-Диизопропилкарбодиимид (206 мг) добавляли к раствору соединения 56 (340 мг) и пентафторфенола (227 мг) в дихлорметане (5 мл) и реакционную смесь перемешивали в течение 15 часов. Смесь гасили гексаном, фильтровали и растворитель выпаривали. Очистка остатка на силикагеле с применением смеси 20:1 гексан:этилацетат в качестве элюента дала продукт 53 (490 мг).
Пример 13: Синтез соединения 57:
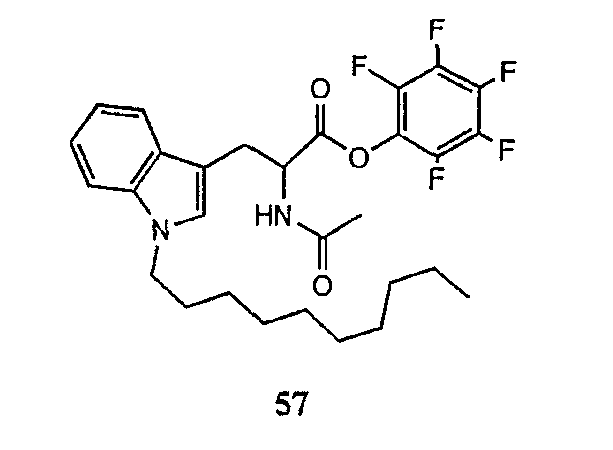
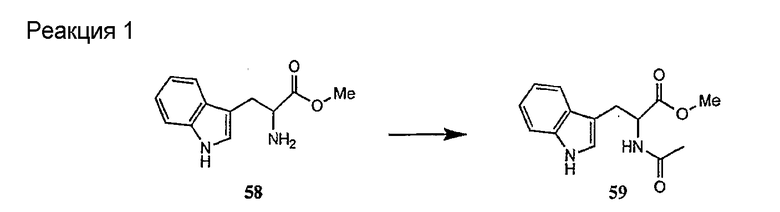
К имеющемуся в продаже раствору метилового эфира триптофана 58 (2,54 г) и триэтиламина (2,9 мл) в дихлорметане (10 мл) добавляли 4-диметиламинопиридин (0,12 г) и уксусный ангидрид (1,03 мл). Через 18 часов реакционную смесь гасили 1 н. хлористоводородной кислотой (10 мл) и водный слой экстрагировали этилацетатом (3 × 10 мл). Объединенные органические слои промывали насыщенным бикарбонатом натрия и насыщенным хлоридом натрия, затем сушили безводным сульфатом натрия и упаривали с получением сырого продукта. Очистка на силикагеле с применением смеси 2:1 гексан:этилацетат дала продукт 59 (1,99 г).
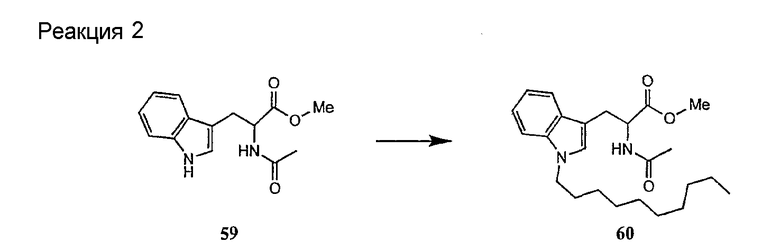
Соединение 59 алкилировали при условиях, описанных выше в примере 8, реакция 2, с заменой гидроксида калия на карбонат калия с получением соединения 60.
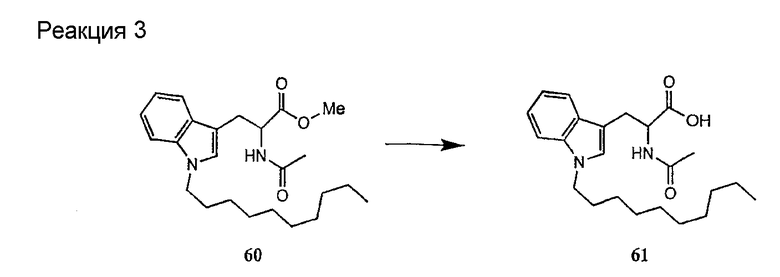
Гидролиз соединения 60 с применением процедуры, описанной в примере 6, реакция 4, дал соединение 61.
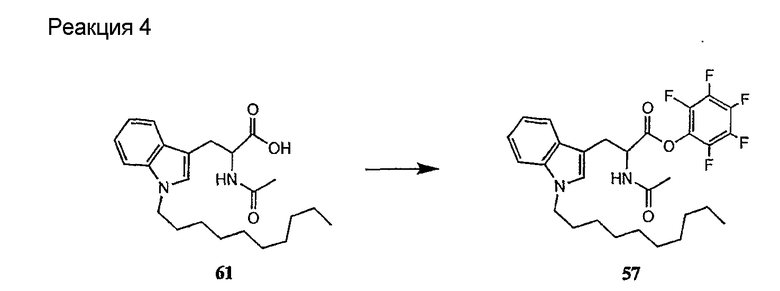
Соединение 61 превращали в соединение 57 с применением условий, описанных в примере 6, реакция 4.
Пример 14: Синтез соединения 62:
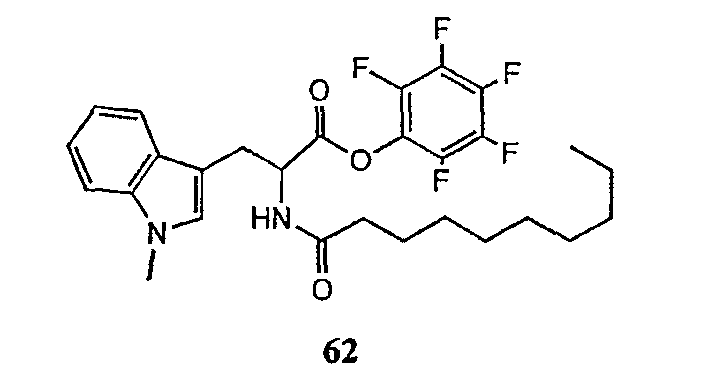
Имеющийся в продаже метиловый эфир триптофана 58 превращали в соединение 63 посредством условий, описанных в примере 8, реакция 2.
Соединение 63 превращали в соединение 64 с применением условий, описанных в примере 12, реакция 1, с применением метилйодида вместо йоддекана.
Соединение 64 гидролизовали с применением условий, описанных в примере 13, реакция 3, с получением соединения 65.
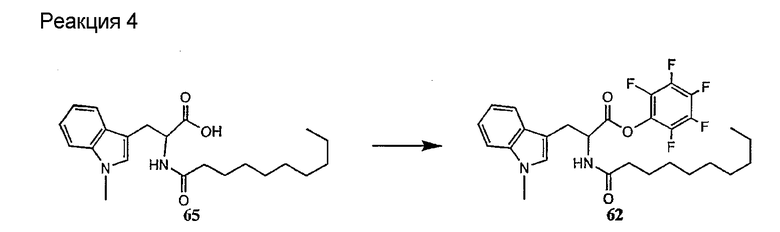
Соединение 65 превращали в соединение 62 с применением условий, описанных в примере 13, реакция 4.
Пример 15: Синтез соединения 25:
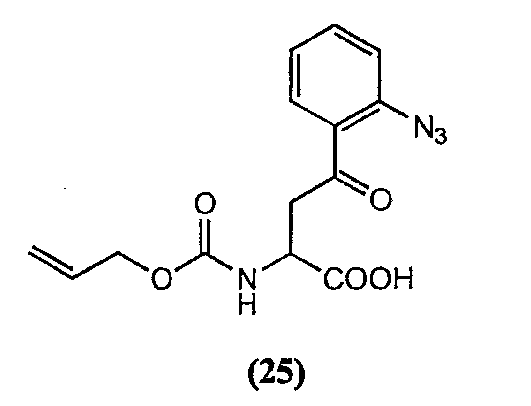
К суспензии имеющейся в продаже L-2-амино-4-(2-аминофенил)-4-оксомасляной кислоты A (3,93 г) и триэтиламина (5,79 мл) в тетрагидрофуране (181 мл) добавляли воду в количестве, достаточном для получения гомогенного раствора. Раствор затем охлаждали до 0°С на бане со льдом и по каплям добавляли диаллилпирокарбонат (3,36 мл). Реакционной смеси давали нагреться до температуры окружающей среды в течение 16 часов и затем концентрировали до одной трети от исходного объема. Смесь выливали в дихлорметан (350 мл), промывали 1 н. хлористоводородной кислотой (3 × 100 мл) и насыщенным хлоридом натрия (1 × 100 мл). Объединенные водные слои вновь экстрагировали этилацетатом (4 × 100 мл) и объединенную органическую фракцию сушили безводным сульфатом натрия. Выпаривание растворителя дало продукт B в виде желтого твердого вещества (5,14 г).
К раствору соединения B (5,00 г) в воде (130 мл) и концентрированной хлористоводородной кислоты (43 мл) при -2°С по каплям добавляли раствор нитрита натрия (1,30 г) в воде (3 мл), так, что температура оставалась ниже 0°С. Реакционную смесь перемешивали при -4°С в течение 135 минут и добавляли раствор азида натрия (3,34 г) в воде (3 мл). Реакционной смеси давали нагреться до температуры окружающей среды в течение 16 часов. Смесь затем выливали в воду (130 мл) и водную фазу экстрагировали дихлорметаном (4 × 80 мл). Объединенный органический слой затем сушили безводным сульфатом натрия. Выпаривание растворителя дало продукт 25 в виде оранжевой пены (5,14 г).
Пример 16: Синтез соединения 77:
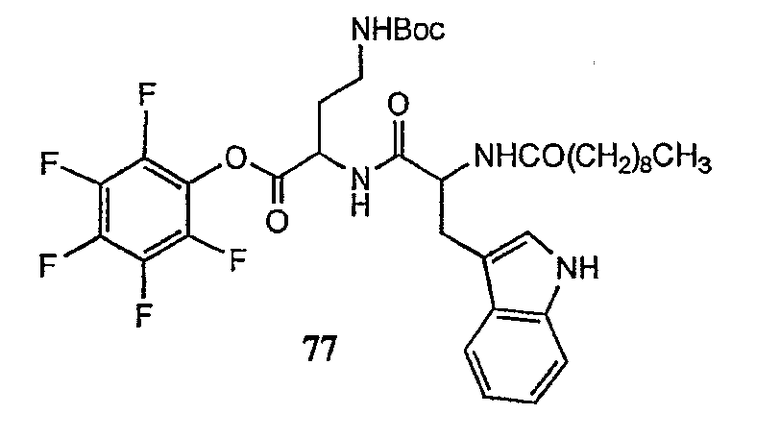
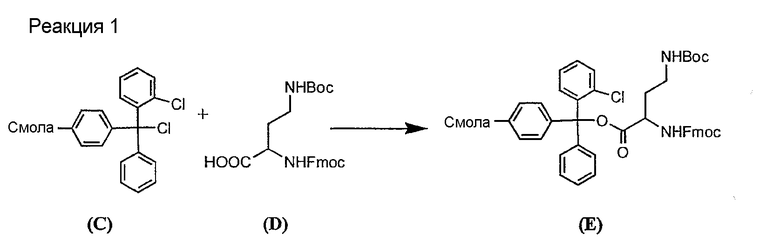
К суспензии имеющейся в продаже 2-хлортритиловой смолы C (1 г) и имеющейся в продаже 2-N-(9-флуоренилметоксикарбонил)-4-N(трет-бутоксикарбонил)-L-2,4-диаминомасляной кислоты в дихлорметане D (10 мл) добавляли диизопропилэтиламин (1,65 мл). Смесь перемешивали в течение 3 часов, фильтровали и твердое вещество промывали дихлорметаном (10 мл), затем сушили с получением соединения E.
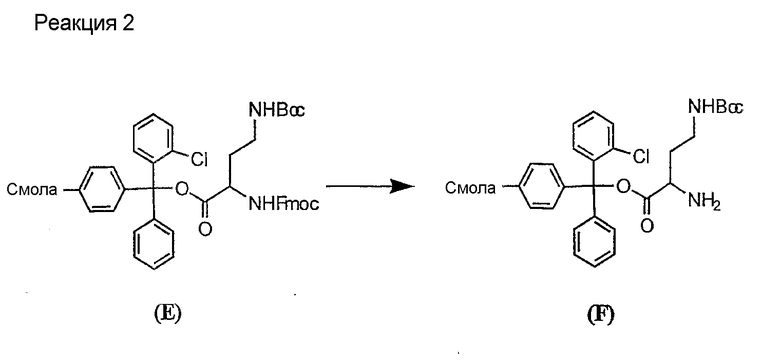
Соединение E встряхивали в 20% пиперидине в N-метилпиролидине (20 мл) в течение 60 минут. Реакционную смесь фильтровали, после чего твердое вещество промывали N-метилпиролидином (20 мл) с получением соединения F.
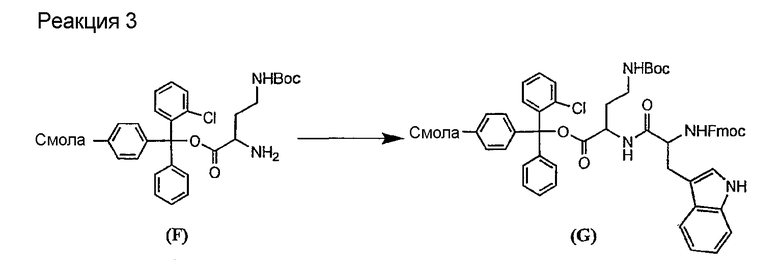
Диизопропилэтиламин (0,5 мл) добавляли к раствору имеющегося в продаже Nα-(9-флуоренилметоксикарбонил)-L-триптофана (1,23 г), TBTU (0,92 г) и N,N-диметилформамида (10 мл) и полученную смесь перемешивали в течение 10 минут. Затем раствор добавляли к смоле F, перемешивали в течение одного часа, фильтровали и твердое вещество промывали N-метилпиролидином (10 мл) с получением соединения G.
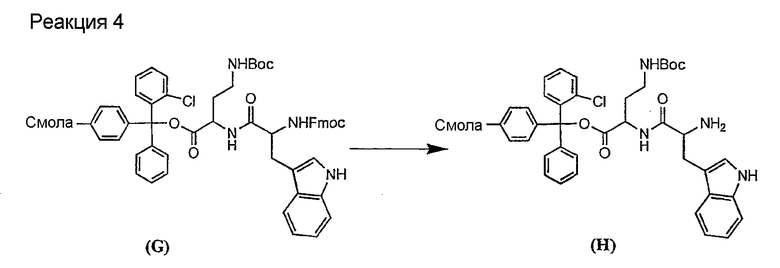
Соединение G встряхивали в 20% пиперидине в N-метилпиролидине (20 мл) в течение 60 минут. Реакционную смесь фильтровали, после чего твердое вещество промывали N-метилпиролидином (20 мл) с получением соединения H.
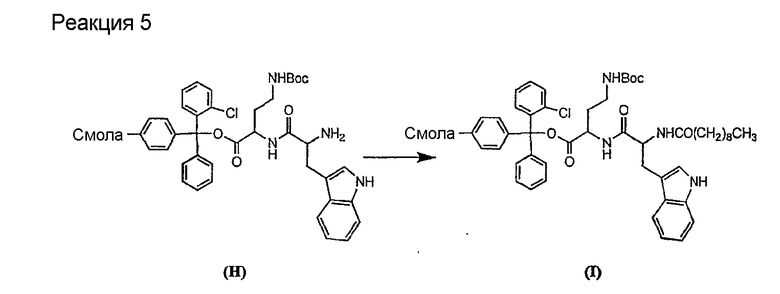
Диизопропилэтиламин (0,5 мл) добавляли к раствору имеющейся в продаже декановой кислоты (0,50 г), TBTU (0,92 г) и N,N-диметилформамида (15 мл) и полученную смесь перемешивали в течение 10 минут. Затем раствор добавляли к смоле H, перемешивали в течение одного часа, фильтровали и твердое вещество промывали N-метилпиролидином (10 мл) с получением соединения I.
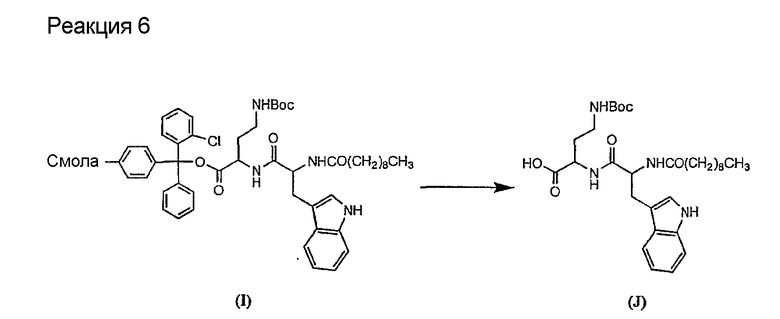
Соединение I обрабатывали смесью дихлорметан:2,2,2-трифторэтанол:уксусная кислота (3:1:1) и полученную смесь перемешивали в течение 3 часов. Реакционную смесь затем фильтровали и фильтрат упаривали с получением соединения J в виде белого твердого вещества (180 мг).
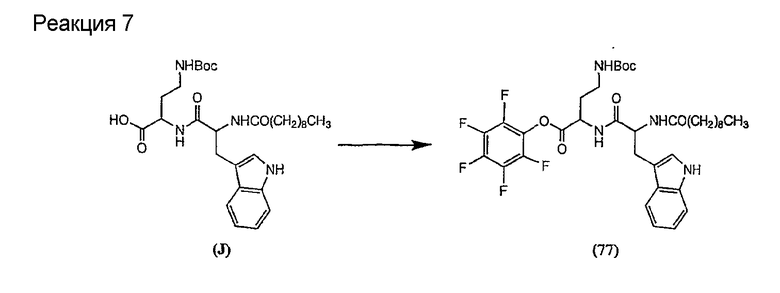
Соединение J превращали в соединение 77 с применением условий, описанных в примере 6, реакция 4.
Пример 17: Синтез защищенного Alloc даптомицина:
соединения 67:
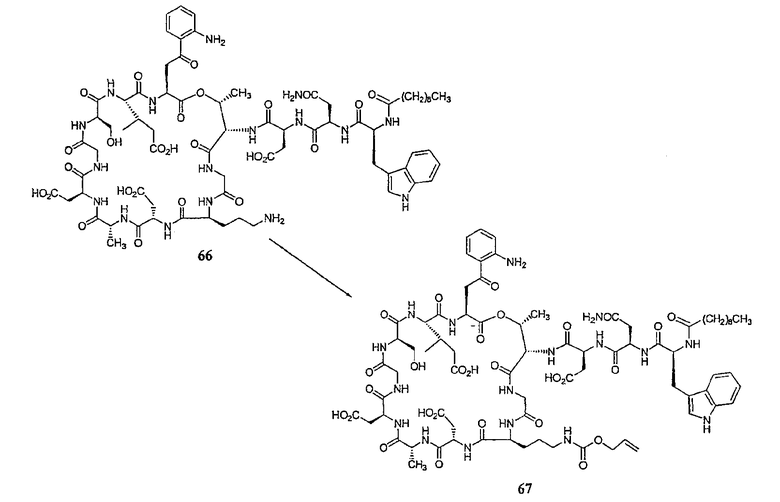
К раствору даптомицина 66 (10 г) в безводном N,N'-диметилформамиде (40 мл) при 0°С добавляли аллил-1-бензотриазолилкарбонат (13,5 г). Реакционной смеси давали нагреться до комнатной температуры и перемешивали в течение 18 часов. Смесь разбавляли водой (200 мл), после чего наносили на смолу Bondesil 40 мкМ C8 (400 г), которую предварительно промывали метанолом (1 л) и водой (1 л). Смолу промывали водой (1 л), и продукт элюировали метанолом (1 л). Выпаривание метанола дало соединение 67 в виде желтого твердого вещества (1 г).
Пример 18: Получение соединения 68:
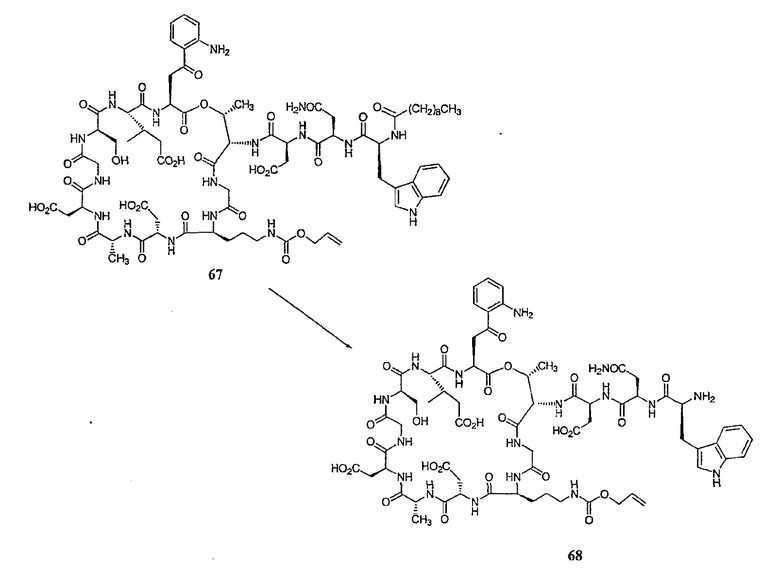
Препарат фермента деацилазы получали из рекомбинантного Streptomyces lividans, который экспрессирует фермент деацилазу Actinoplanes utahensis. Фермент в водном этиленгликоле (10 мл) добавляли к раствору соединения 67 (15 г в воде, 1,9 л) при pH 8. Реакционную смесь перемешивали при комнатной температуре в течение 18 часов и pH доводили до 8 с помощью 1 М гидроксида натрия. Реакционную смесь выливали на смолу Bondesil 40 мкМ C8 (400 г), которую предварительно промывали метанолом (1 л) и водой (1 л). Продукт элюировали 20% ацетонитрилом в воде (1 л) и лиофилизовали с получением соединения 68 в виде желтого твердого вещества (9,1 г).
Пример. 19: Деградация по Эдману для удаления триптофанового препарата соединения 70:
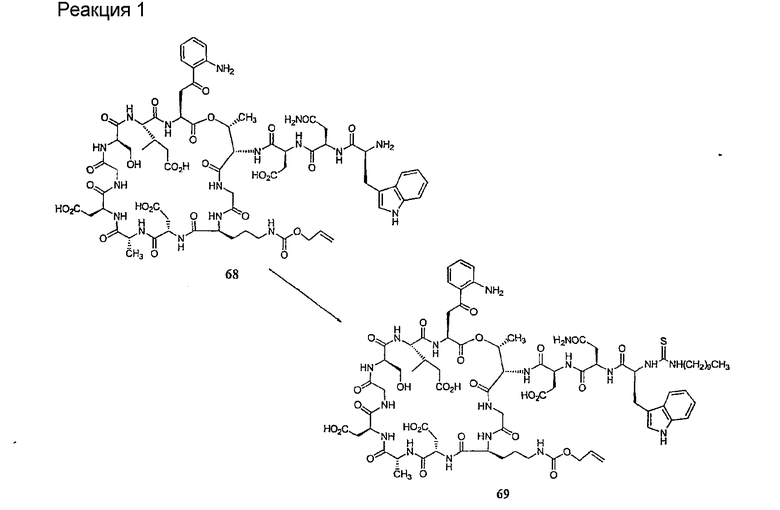
К суспензии соединения 68 (9,1 г) в безводном N,N'-диметилформамиде (15 мл) добавляли н-децилизотиоцианат (1,2 мл). Реакционную смесь перемешивали при комнатной температуре в течение 18 часов. Реакционную смесь выливали на смолу Bondesil 40 мкМ C8 (400 г), которую предварительно промывали метанолом (1 л) и водой (1 л). Продукт элюировали метанолом (800 мл) после предварительной промывки водой (800 мл) и затем 20% ацетонитрилом в воде (800 мл). Выпаривание метанола дало соединение 69 в виде желтого твердого вещества (7,3 г).
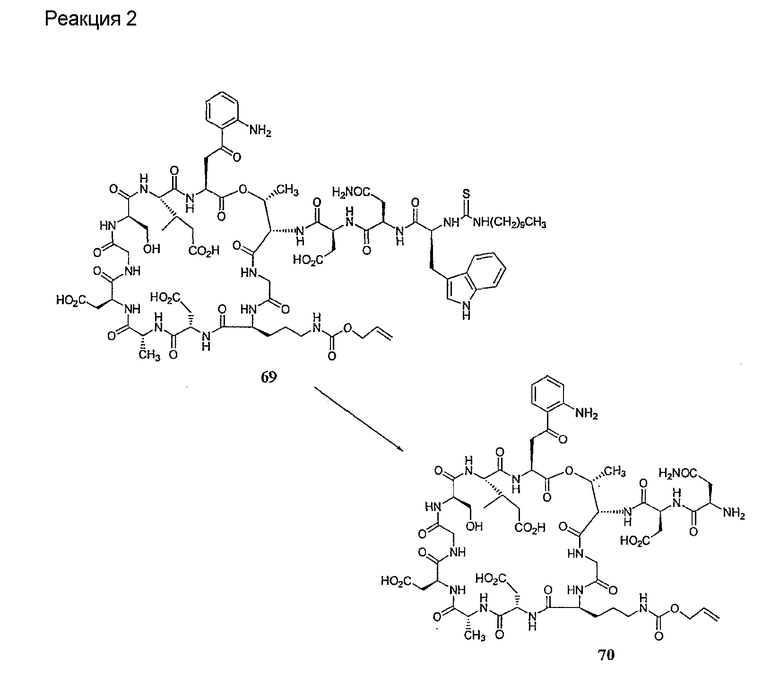
Соединение 69 (7,3 г) перемешивали при комнатной температуре в 25% трифторуксусной кислоте в безводном дихлорметане (30 мл) в течение 2 часов перед упариванием досуха. Остаток растворяли в воде (50 мл) и выливали на смолу Bondesil 40 мкМ C8 (400 г), которую предварительно промывали метанолом (1 л) и водой (1 л). Продукт элюировали градиентом от 20 до 40% ацетонитрила в воде и лиофилизовали с получением соединения 70 в виде желтого твердого вещества (1,05 г).
Пример. 20: Деградация по Эдману для удаления аспарагинового препарата дезаспарагинового соединения 72:
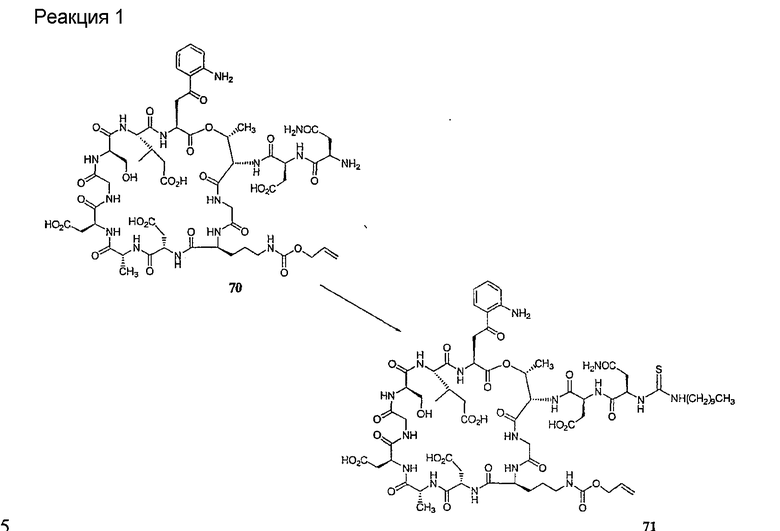
К суспензии соединения 70 (0,57 г) в безводном N,N'-диметилформамиде (5 мл) добавляли н-децилизотиоцианат (0,16 мл). Реакционную смесь перемешивали при комнатной температуре в течение 18 часов перед упариванием досуха. Остаток растирали с диэтиловым эфиром (5 мл) с получением соединения 71 в виде желтого твердого вещества (0,54 г).
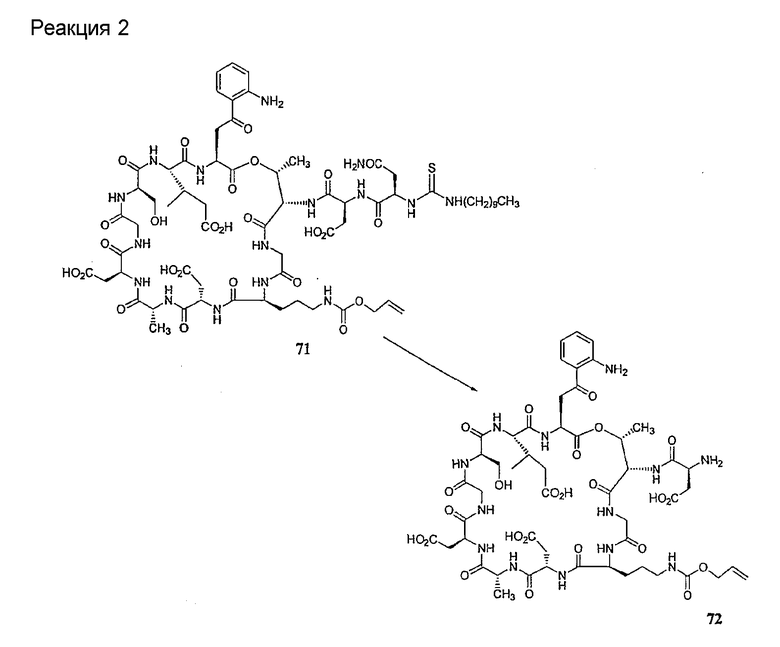
Соединение 71 (0,54 г) перемешивали в 50% трифторуксусной кислоте в безводном дихлорметане (4 мл) в течение 2 часов перед упариванием досуха. Остаток растворяли в воде (25 мл) и выливали на смолу Bondesil 40 мкМ C8 (50 г), которую предварительно промывали метанолом (100 мл) и водой (100 мл). Продукт элюировали 20% ацетонитрилом в воде после предварительной промывки водой (100 мл). Элюент выпаривали досуха с получением соединения 72 в виде желтого твердого вещества (0,40 г).
Пример. 21: Деградация по Эдману для удаления препарата аспарагиновой кислоты базового соединения 74:
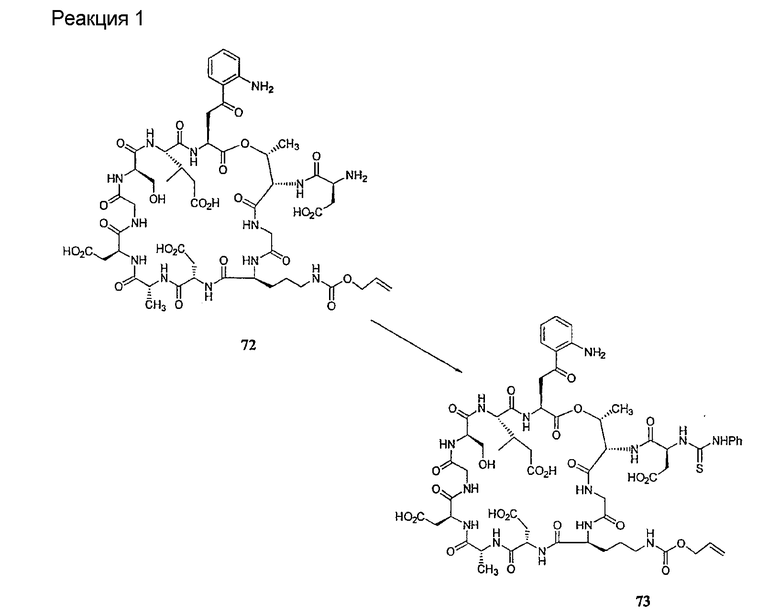
К суспензии соединения 72 (1,0 ммоль) в безводном N,N'-диметилформамиде (5 мл) добавляли фенилизотиоцианат (1,0 ммоль). Реакционную смесь перемешивали при температуре окружающей среды в течение 18 часов и затем упаривали досуха. Остаток растирали с диэтиловым эфиром (5 мл) с получением продукта 73.
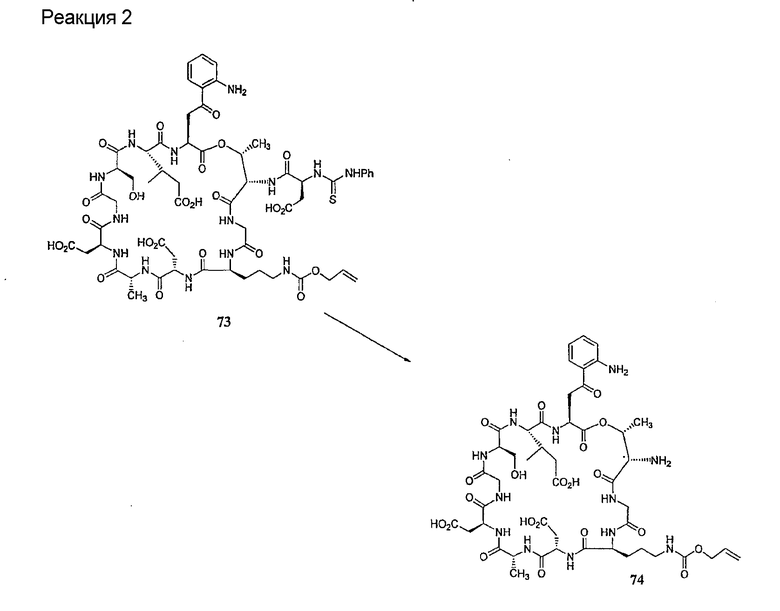
Соединение 73 перемешивали в дихлорметане (2 мл) и трифторуксусной кислоте (2 мл) в течение 2 часов перед упариванием досуха с получением продукта в виде соли трифторуксусной кислоты 74.
Пример 22: Получение соединения 76:
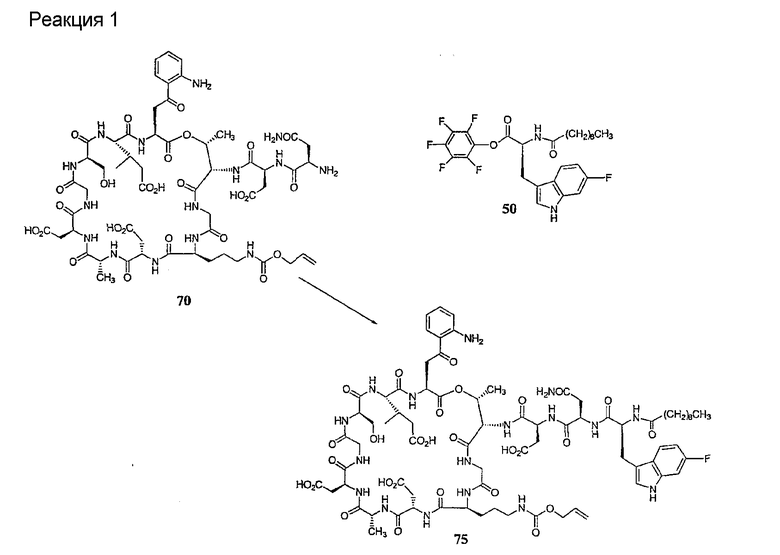
Соединение 50 (397 мг) добавляли к раствору деацилированного дезтриптофанового даптомицина 70 (500 мг) и диизопропилэтиламина (2-3 капли) в N,N'-диметилформамиде (5 мл). Реакцию отслеживали с помощью аналитической ВЭЖХ с обращенной фазой (элюирование градиентом смеси от 5% до 95% ацетонитрил - 0,1% муравьиная кислота в воде - 0,1% муравьиная кислота с применением luna C18 5 мкМ (150 × 3 мм) и полный расход соединения 70 наблюдался после перемешивания в течение 16 часов. Растворитель удаляли выпариванием и сырой продукт 75 (631 мг) применяли далее на следующей стадии.
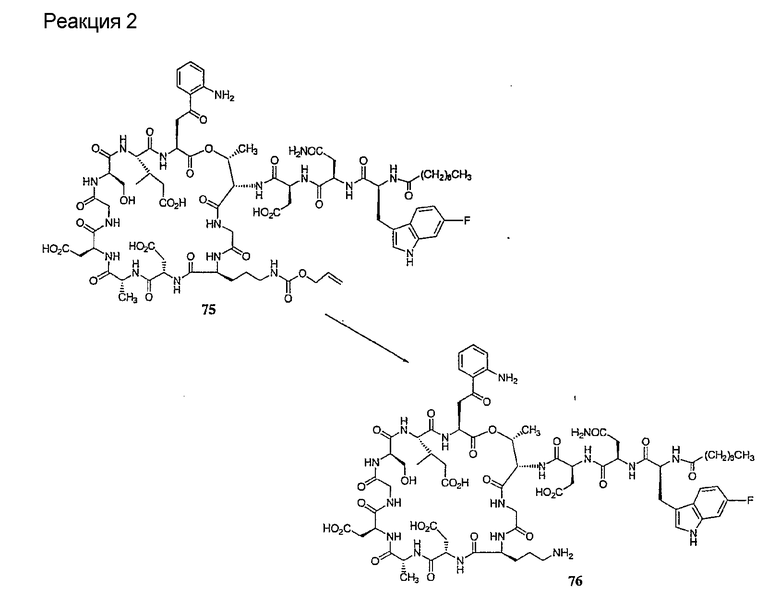
Тетракис-(трифенилфосфин)палладия(0) (631 мг) добавляли к раствору соединения 75 (631 мг), N-метилморфолина (631 мкл) в диоксане (10 мл) и 1 н. хлористоводородной кислоте (6,31 мл). После перемешивания в течение 16 часов реакционную смесь фильтровали и очищали ВЭЖХ с обращенной фазой с применением колонки C18 10 мкМ Jupiter 250 × 21,2 мм с элюированием градиентом смеси ацетонитрил:вода:муравьиная кислота от 30:70:0,1 до 90:10:0,1 в течение 25 минут. Упаривание содержащих продукт фракций дало продукт 76 (44 мг).
Пример 23: Получение соединения 79:
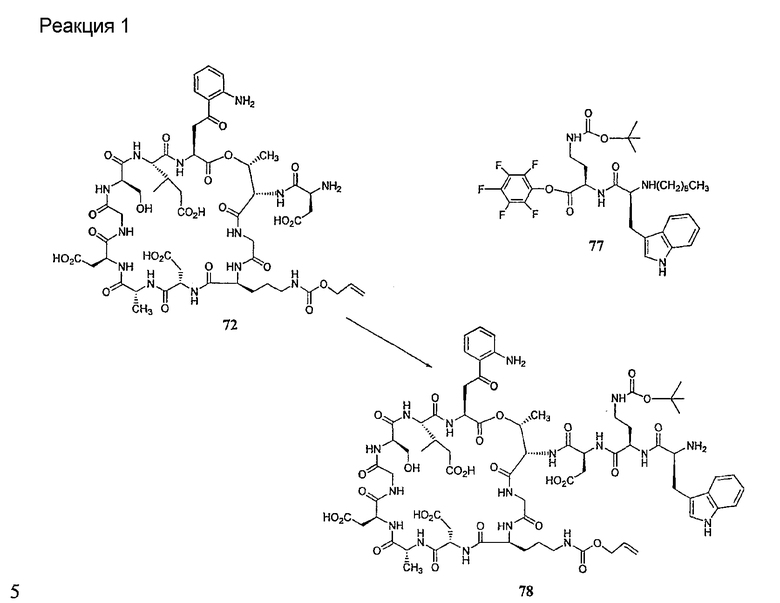
Соединение 77 (1,2 ммоль, смотри пример 16 (смотри выше)) добавляли к раствору деацилированного дезтриптофан-дезаспарагинового даптомицина 72 (1,0 ммоль) и диизопропилэтиламина (2-3 капли) в безводном диметилформамиде (5 мл). Реакционную смесь перемешивали при температуре окружающей среды до расходования всего исходного вещества по данным аналитической ВЭЖХ с обращенной фазой (элюирование градиентом смеси от 5% до 95% ацетонитрил - 0,1% муравьиная кислота в воде - 0,1% муравьиная кислота с применением luna C18 5 мкМ 150 × 3 мм). Смесь упаривали досуха и остаток растирали с эфиром (5 мл) с получением желаемого продукта 78.
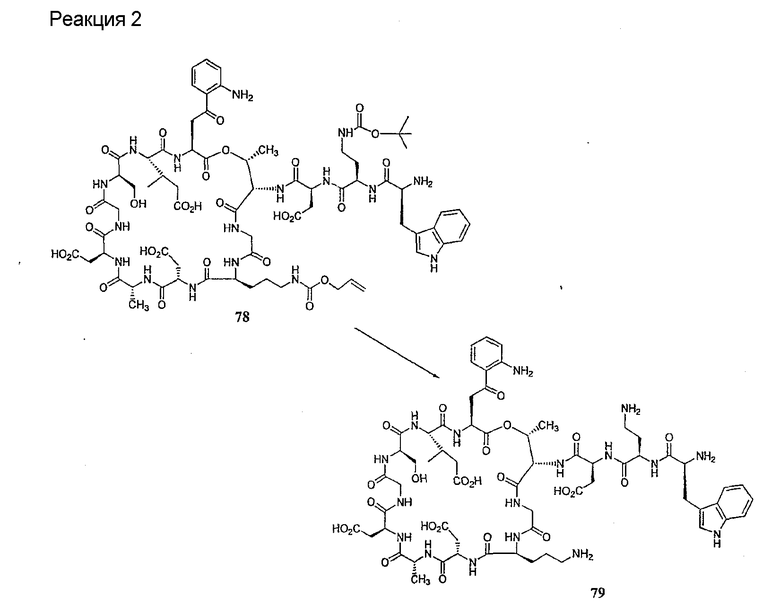
К раствору соединения 78 (1 ммоль) в 0,5 М хлористоводородной кислоте (1 мл) и диоксане (3 мл) добавляли N-метилморфолин (0,1 мл) и тетракис-(трифенилфосфин)палладия(0) (100 мг). Реакционную смесь перемешивали в атмосфере аргона в течение 24 часов, фильтровали и концентрировали с получением сырого полупродукта. Остаток растворяли в безводном дихлорметане (4 мл). Добавляли триизопропилсилан (0,1 мл) и трифторуксусную кислоту (1 мл) и реакционную смесь перемешивали в течение 2 часов перед упариванием досуха. Остаток очищали препаративной ВЭЖХ с применением колонки C8 10 мкм Jupiter 250 × 21,2 мм с элюированием градиентом 30-80% ацетонитрила в водной 0,1% муравьиной кислоте в качестве элюента. Выпаривание растворителя из содержащих продукт фракций дало желаемый продукт 79.
Пример 24
Биологическая активность
Соединения согласно формуле I тестировали на противомикробную активность с набором организмов в соответствии со стандартными процедурами, описанными Национальным комитетом клинических лабораторных стандартов (документ NCCLS M7-A5, Vol. 20, No. 2, 2000), за исключением того, что все тестирование проводили при 37°С. Соединения растворяли в 100% диметилсульфоксиде и разводили до конечной реакционной концентрации (0,1 мкг/мл-100 мкг/мл) в среде роста микробов. Во всех случаях конечная концентрация диметилсульфоксида при инкубации с клетками была ниже или равна 1%. Для расчетов минимальной ингибирующей концентрации (MIC) в лунки планшета для микротитрования, содержащие 5x104 бактериальных клеток в конечном объеме 100 мкл среды (бульона Мюллера-Хинтона с добавкой 50 мг/л Ca2+) добавляли 2-кратные разведения соединений. С помощью имеющегося в продаже планшетного ридера измеряли оптическую плотность (OD) бактериальных клеток, которая служит мерой роста и пролиферации бактерий. MIC определяли как наименьшую концентрацию соединения, ингибирующую рост тестируемого организма. Величины MIC (в мкг/мл) типичных соединений настоящего изобретения перечислены в таблице III.
Таблица III
Биологическая активность соединений формулы I
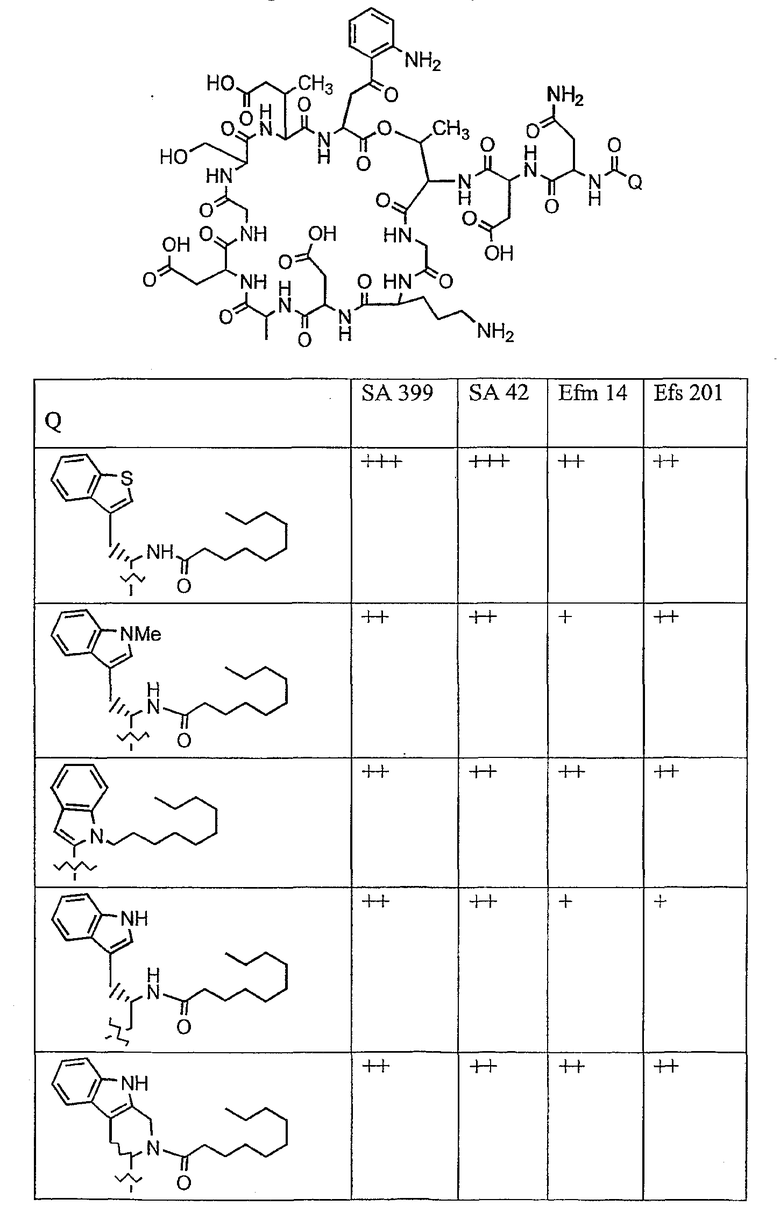
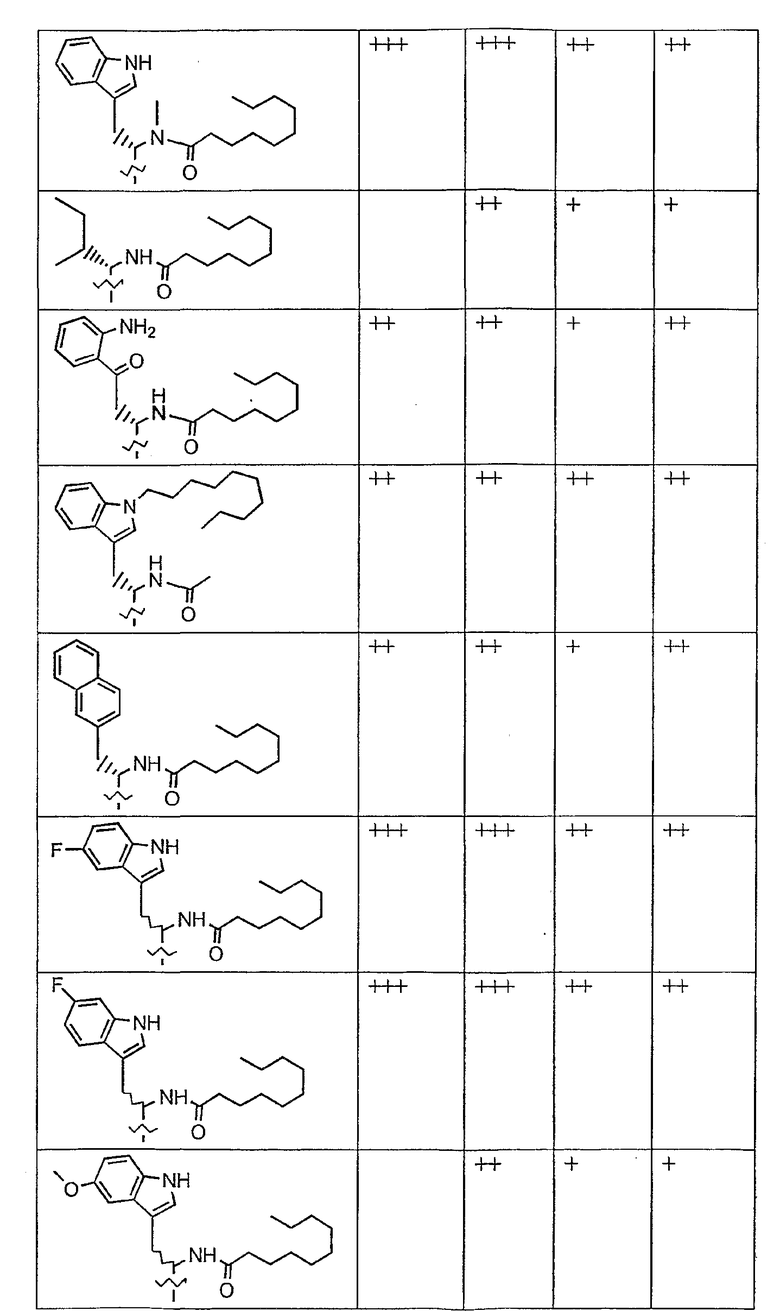
SA399 представляет собой устойчивый к метициллину Staphylococcus aureus.
SA42 представляет собой Staphylococcus aureus дикого типа.
EF14 представляет собой Enterococcus faecium дикого типа.
EF201 представляет собой Enterococcus faecalis дикого типа.
Где "+++" обозначает, что соединение имеет MIC (мкг/мл) 1 мкг/мл или ниже или ED50 1 мг/кг или ниже;
"++" обозначает, что соединение имеет MIC (мкг/мл) или ED50 более 1 мкг/мл или 1 мг/кг, соответственно, но меньшие или равные 10 мкг/мл или 10 мг/кг для ED50, соответственно; и
"+" обозначает, что соединение имеет MIC (мкг/мл) выше 10 мкг/мл или ED50 выше 10 мг/кг.
Пример 25
Активность in vivo
Тест защиты мышей является производственным стандартом для измерения эффективности тестируемого соединения in vivo [примеры данной модели смотри в J. J. Clement, et al., Antimicrobial Agents and Chemotherapy, 38 (5), 1071-1078, (1994)]. Как иллюстрировано ниже, данный тест применяют для демонстрации эффективности соединений настоящего изобретения против бактерий in vivo.
Противобактериальную активность in vivo устанавливают путем инфицирования самок мышей CD-1 (Charles River Lab, MA) массой 19-23 г внутрибрюшинно устойчивым к метициллину инокулятом S. aureus (MRSA). Инокулят получают из устойчивого к метициллину S. aureus (ATCC 43300). Инокулят MRSA культивируют в бульоне Мюллера-Хинтона (MH) при 37°С в течение 18 часов. Оптическую плотность при 600 нм (OD600) определяют для разведения 1:10 ночной культуры. Бактерии (8 × 108 КОЕ) добавляют к 20 мл забуференного фосфатом физиологического раствора (Sigma P-0261), содержащего 5% свиного желудочного муцина (Sigma M-2378). Всех животных инъецируют 0,5 мл инокулята, эквивалентного 2 × 107 cfu/мышь, что представляет собой дозу, вызывающую 100% гибель животных без лечения.
Тестируемое соединение растворяют в 10,0 мл 50 мМ фосфатного буфера с получением раствора 1 мг/мл (pH 7,0). Раствор последовательно разводят носителем в 4 раза (1,5 мл до 6,0 мл) с получением растворов 0,25, 0,063 и 0,016 мг/мл. Все растворы фильтруют с помощью фильтра для шприца 2 m Nalgene. Сразу после бактериальной инокуляции группе 1 животных подкожно (п/к) инъецируют буфер (без тестируемого соединения), а группы со 2 по 5 получают тестируемое соединение п/к в дозе 10,0, 2,5, 0,63 и 0,16 мг/кг, соответственно. Группа 6 животных получает тестируемое соединение п/к в дозе 10 мг/кг (или наибольшую терапевтическую дозу данного соединения) лишь для отслеживания острой токсичности. Данные инъекции повторяют еще раз для соответствующих групп через 4 часа после инокуляции. Объем инъекции каждый раз составляет 10 мл на килограмм массы тела. 50% защитную дозу (PD50) рассчитывают на основе количества выживших мышей через 7 дней после инокуляции.
Все публикации и патентные заявки, цитированные в данном описании, включены здесь в качестве ссылки, как если бы для каждой публикации или патентной заявки было конкретно и отдельно указано, что она включена в качестве ссылки. Хотя указанное выше изобретение было описано довольно подробно в виде иллюстраций и примеров с целью облегчения понимания, в свете указаний данного изобретения специалисту в данной области техники должно быть вполне очевидно, что в него могут быть внесены определенные изменения и модификации без выхода за пределы духа и объема прилагаемой формулы изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПЕПТИДНЫЕ АНТИБИОТИКИ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 2005 |
|
RU2428429C2 |
| ПРОИЗВОДНЫЕ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ N-АДАМАНТИЛМЕТИЛА В КАЧЕСТВЕ ФАРМАЦЕВТИЧЕСКИХ КОМПОЗИЦИЙ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 2002 |
|
RU2300525C2 |
| НЕПЕПТИДНЫЕ АНТАГОНИСТЫ GnRH | 2003 |
|
RU2329251C2 |
| ПРОИЗВОДНЫЕ ПУРИНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЛЕЧЕНИЯ | 1997 |
|
RU2191778C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ТИАЗОЛИДИНОНА ИЛИ ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМОЙ СОЛИ | 1990 |
|
RU2036915C1 |
| БЕНЗОИЛАМИНОГЕТЕРОЦИКЛИЛЬНЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ АКТИВАТОРОВ ГЛЮКОКИНАЗЫ (GLK) | 2007 |
|
RU2440992C2 |
| СОЕДИНЕНИЯ, ПОЛЕЗНЫЕ В КАЧЕСТВЕ АНТИПРОЛИФЕРАТИВНЫХ АГЕНТОВ И ИНГИБИТОРОВ GARFT, СПОСОБ ИНГИБИРОВАНИЯ И ПРОЛИФЕРАЦИИ И ПРОИЗВОДНЫЕ ЭФИРА ТИОФЕНКАРБОНОВОЙ КИСЛОТЫ | 1995 |
|
RU2152945C2 |
| БИАРИЛЗАМЕЩЕННЫЕ ТРИАЗОЛЫ КАК БЛОКАТОРЫ НАТРИЕВЫХ КАНАЛОВ | 2004 |
|
RU2356897C2 |
| СОЕДИНЕНИЯ ФОСФОНОВЫХ КИСЛОТ В КАЧЕСТВЕ ИНГИБИТОРОВ СЕРИНОВОЙ ПРОТЕИНАЗЫ | 2002 |
|
RU2311421C2 |
| ЗАМЕЩЕННЫЕ ДИАЛКИЛ (ОКСИДО)-Λ-СУЛЬФАНИЛИДЕН НИКОТИНАМИД ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ | 2014 |
|
RU2711749C2 |
Настоящее изобретение относится к новым депсипептидным соединениям, а также относится к фармацевтическим композициям данных соединений и применению данных соединений в качестве антибактериальных соединений. Изобретение также относится к способам получения данных новых депсипептидных соединений и промежуточных продуктов, применяемых при получении данных соединений. 9 н. и 22 з.п. ф-лы, 3 табл.
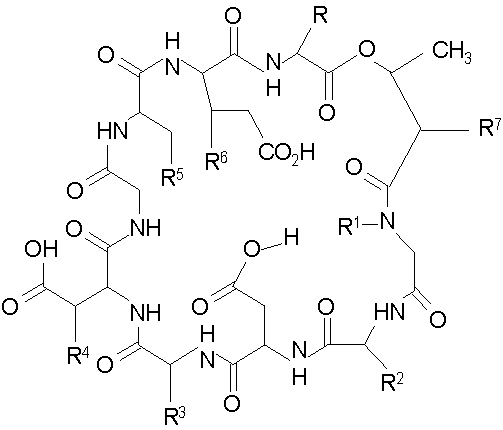
или его фармацевтически приемлемая соль,
где R представляет собой 2-бутил, изопропил или 2-(2'-аминофенацил);
каждый из R1 и R6 независимо представляет собой водород или метил;
R2 представляет собой -CH2CH2CH2R8 где R8 представляет собой амино или карбамоил;
R3 представляет собой метил;
R4 представляет собой водород;
R5 представляет собой гидрокси;
R7 выбран из группы состоящей из
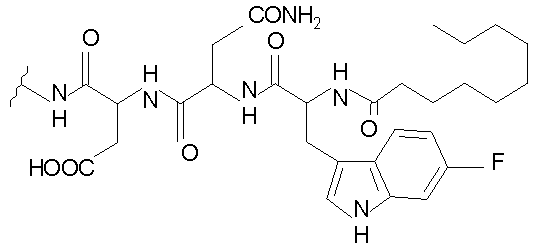
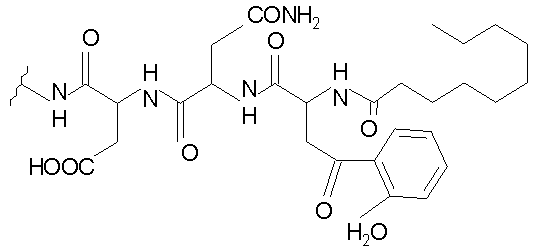
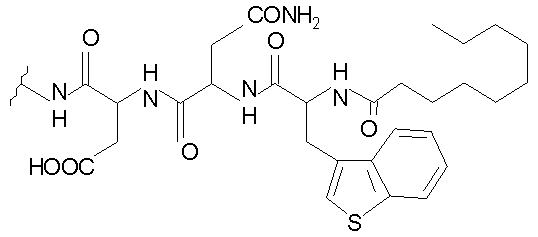
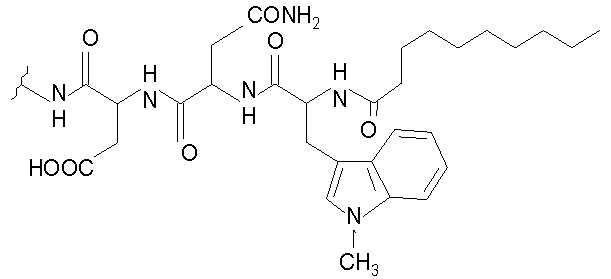
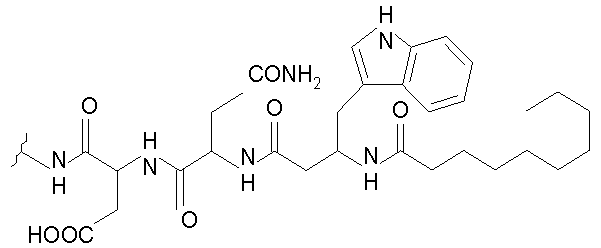
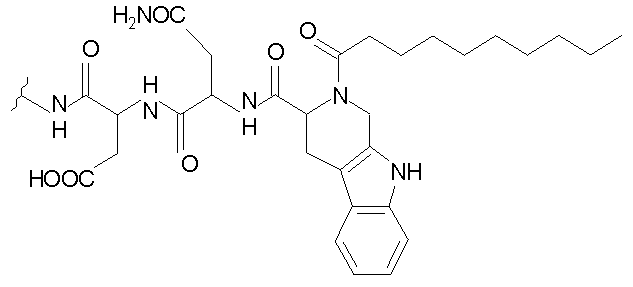
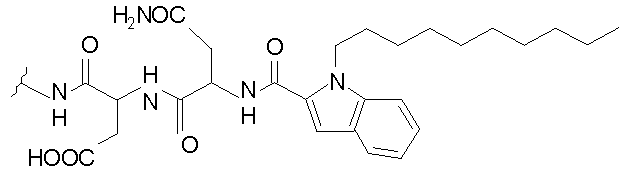
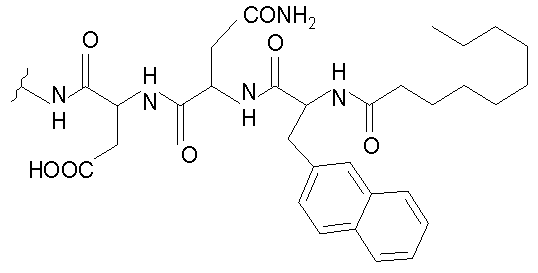
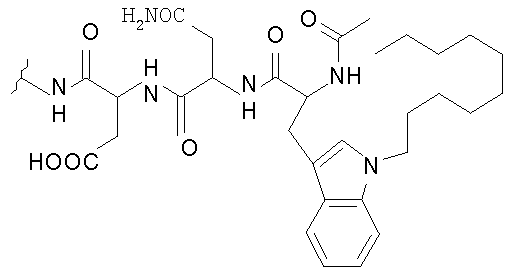
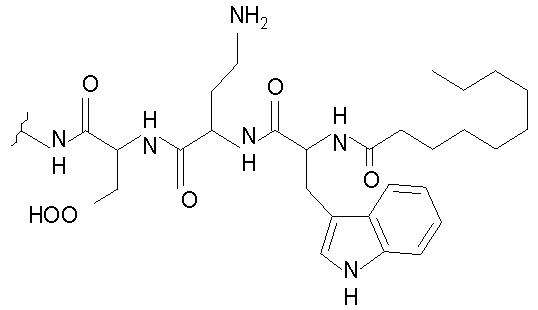
или R7 представляет собой
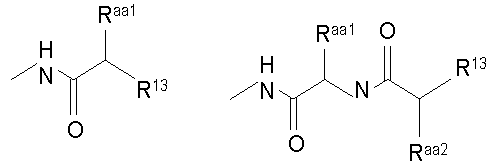
где
Raa1 представляет собой СН2СООН;
Raa2 представляет собой СН2CONH2;
R13 представляет собой аминогруппу, тиоуреидогруппу формулы
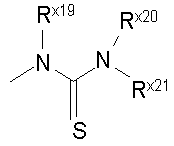
где
Rx19 и Rx20 каждый независимо представляет водород; Rx21 представляет собой (C1-С10)алкил или (C6-С10)арил.
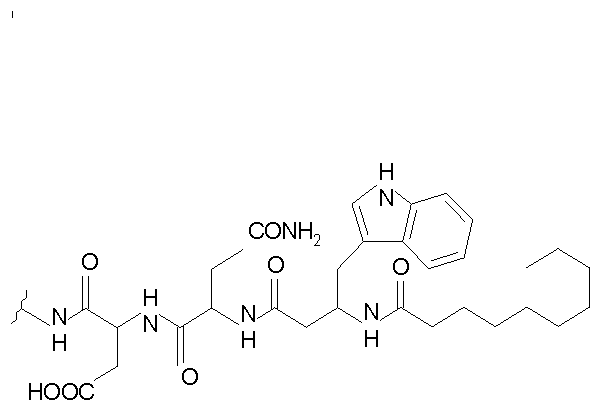
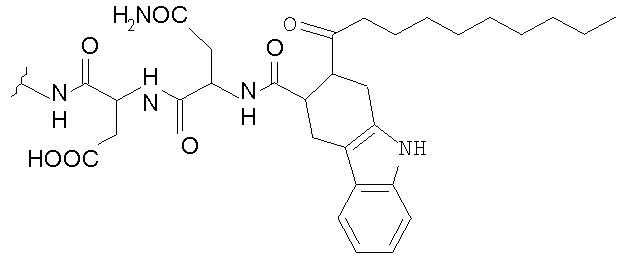
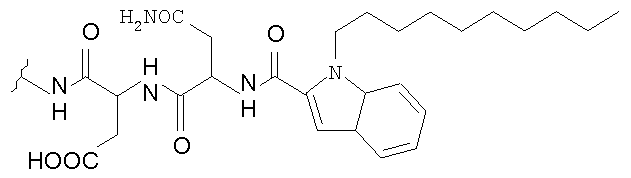
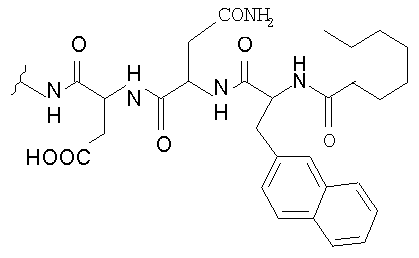
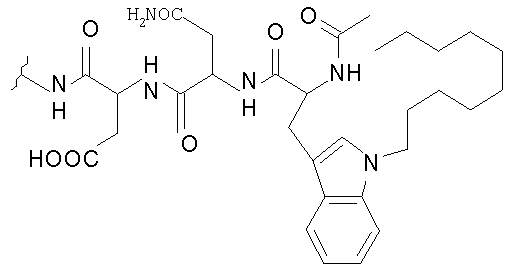
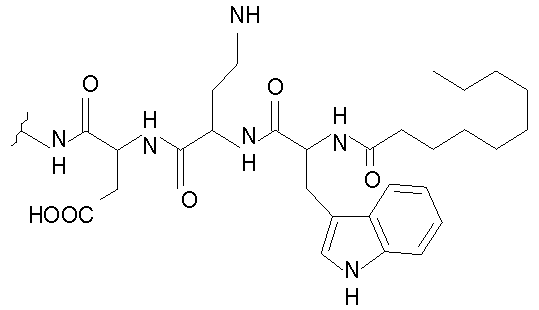
где
Raa1 представляет собой CH2COOH;
Raa2 представляет собой СН2CONH2;
R13 представляет собой аминогруппу, тиоуреидогруппу формулы
где
Rx19 и Rx20 каждый независимо представляет водород; Rx21 представляет собой (C1-С10)алкил или (C6-С10)арил.
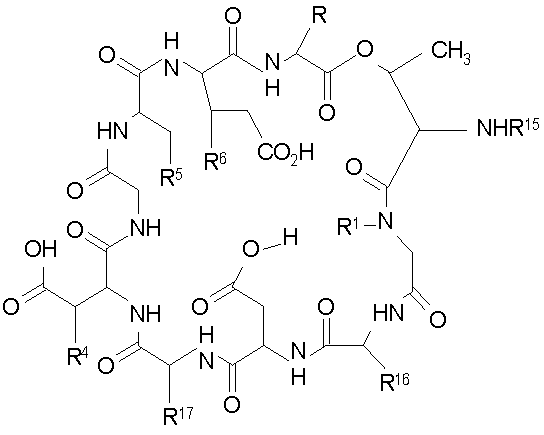
или его фармацевтически приемлемая соль, где
(a) R представляет собой 2-бутил, изопропил или 2-(2'-аминофенацил);
(b) каждый из R1 и R6 независимо представляет собой водород или метил;
(c) R4 представляет собой водород;
(d) R5 представляет собой гидрокси;
(e) R15 представляет собой водород,
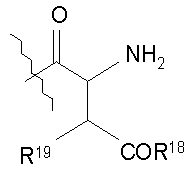 или
или 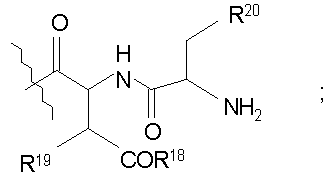
где R18 представляет собой амино или гидрокси; R19 представляет собой водород и R20 представляет собой карбоксиамино;
(f) R16 представляет собой метил или -CH2CH2CH2R21;
(d) R17 представляет собой метил;
где R21 независимо представляет собой амино или карбамоил.
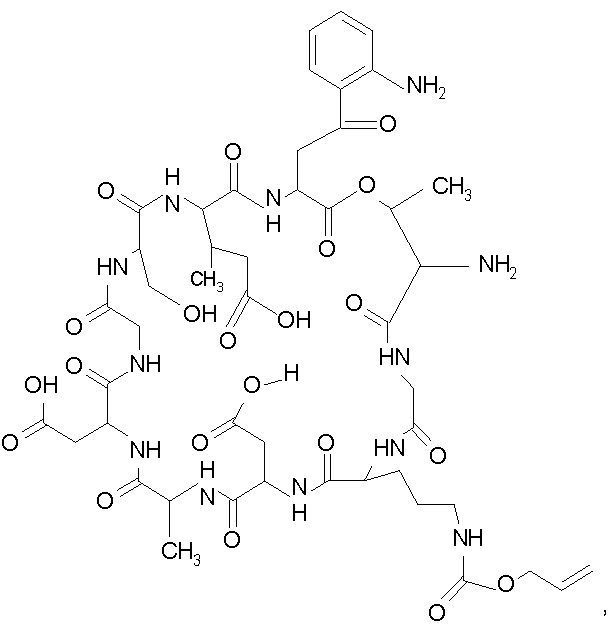
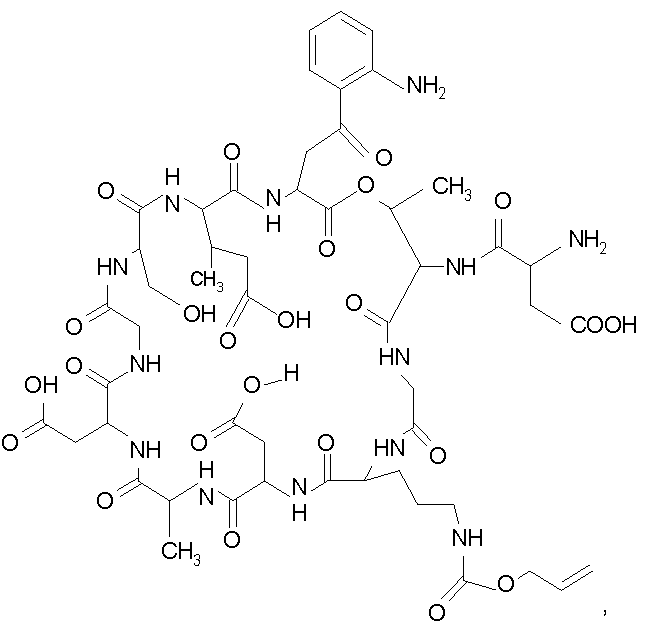
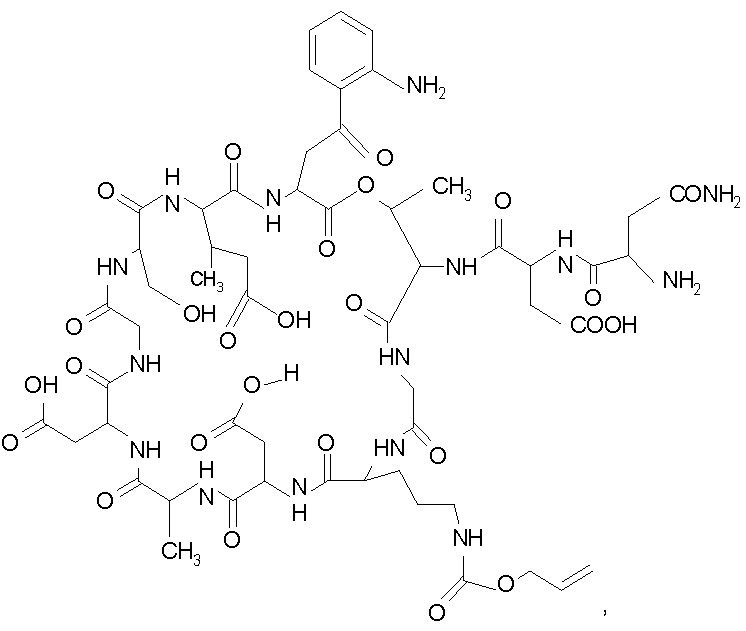
где каждый из R6 и R14 представляет собой независимо водород или метил.
где
Rаа1 представляет собой CH2COOH;
Rаа2 представляет собой CH2CONH2;
R13 представляет собой аминогруппу, тиоуреидогруппу.
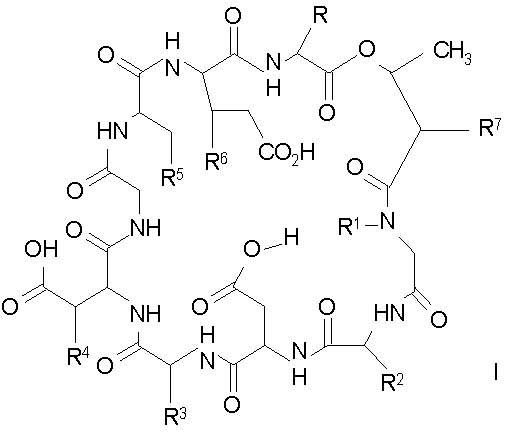
или его соль,
где R представляет собой 2-бутил, изопропил или 2-(2'-аминофенацил);
каждый из R1 и R6 независимо представляет собой водород или метил;
R2 представляет собой -СН2СН2СН2R8 где R8 представляет собой амино или карбамоил;
R3 представляет собой метил;
R4 представляет собой водород;
R5 представляет собой гидрокси;
R7 выбран из группы, состоящей из

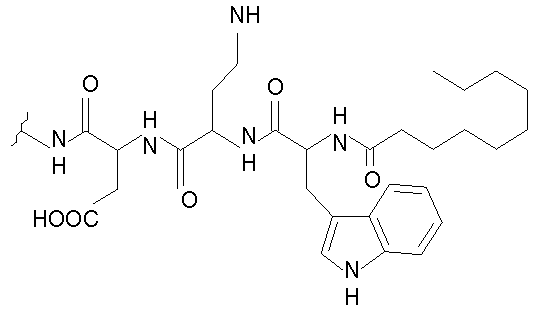
или R7 представляет собой
где Rаа1 представляет собой СН2СООН;
Rаа2 представляет собой CH2CONH2;
R13 представляет собой аминогруппу, тиоуреидогруппу формулы
где Rx19 и Rх20 каждый независимо представляет водород; Rх21 представляет собой (C1-С10)алкил или (C6-С10)арил,
включающий удаление аминозащитной группы из соединения формулы XVII
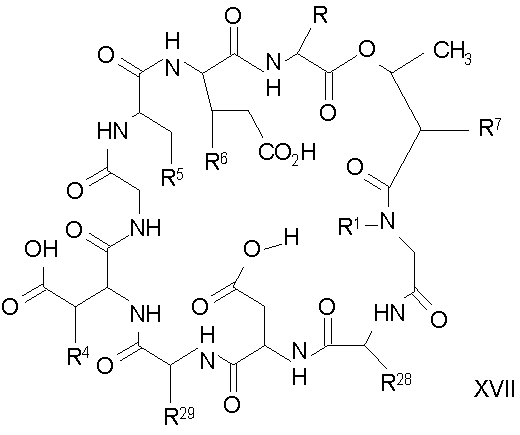
или его фармацевтически приемлемой соли,
где R28 представляет собой -CH2CH2CH2NHP; R29 представляет собой метил и Р представляет собой аминозащитную группу.
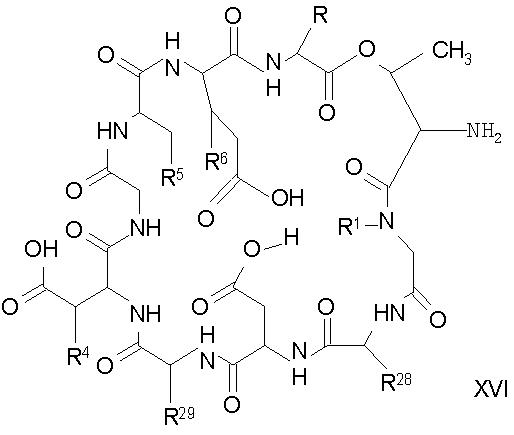
или его фармацевтически приемлемой соли, модифицирующим агентом с получением соединения формулы XVII или его соли.
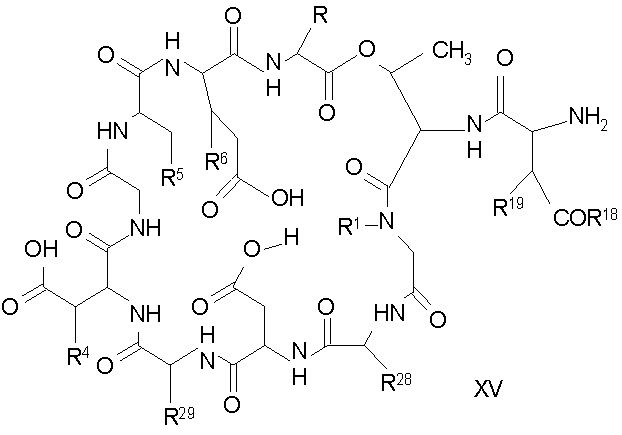
или его фармацевтически приемлемой соли,
где R18 представляет собой гидрокси; R19 представляет собой водород;
с получением соединения формулы XVI или его фармацевтически приемлемой соли.
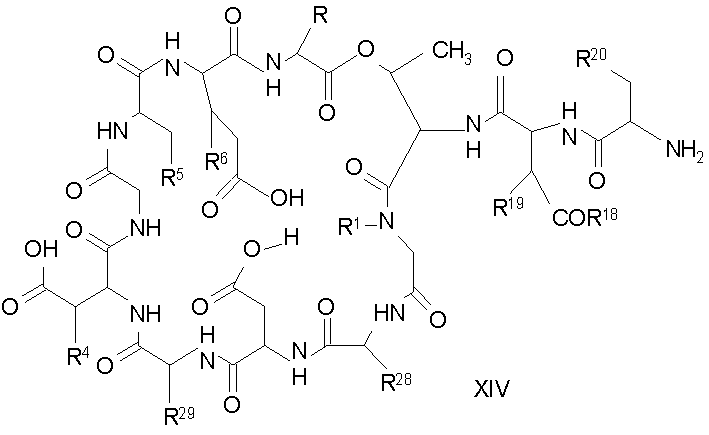
или его фармацевтически приемлемой соли,
где R20 представляет собой карбоксиамино;
с получением соединения формулы XV или его соли.
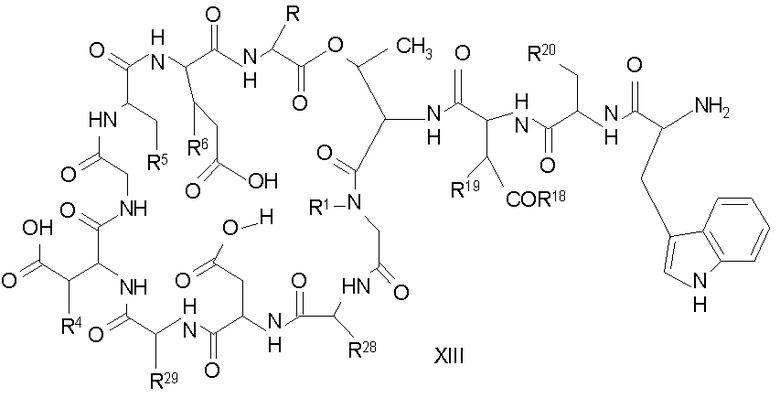
или его фармацевтически приемлемой соли с получением соединения формулы XIV или его соли.
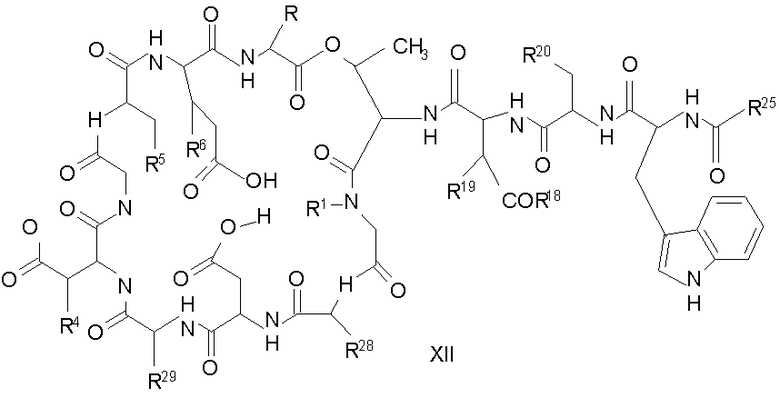
или его фармацевтически приемлемой соли,
где R25 представляет собой алкильную группу,
с получением соединения формулы XIII или его фармацевтически приемлемой соли.
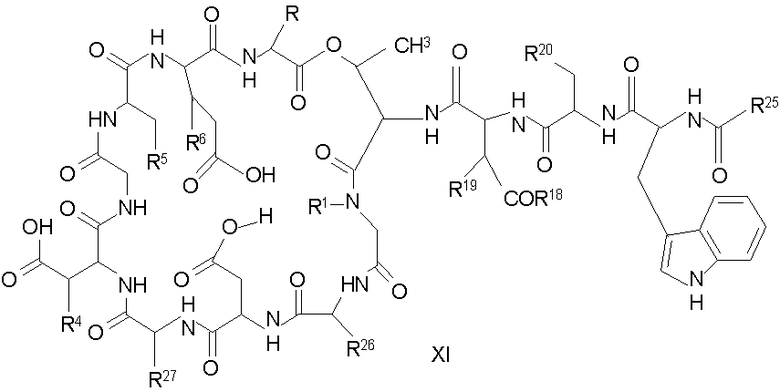
или его фармацевтически приемлемой соли,
где R26 представляет собой метил или -CH2CH2CH2NH2; a R27 представляет собой метил;
с получением соединения формулы XII или его фармацевтически приемлемой соли.
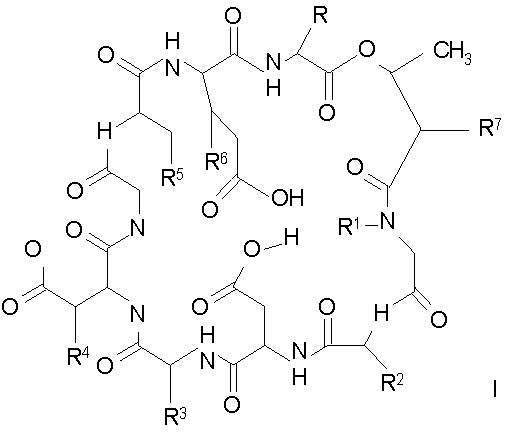
или его фармацевтически приемлемой соли,
где R представляет собой 2-бутил, изопропил или 2-(2'-аминофенацил);
каждый из R1 и R6 независимо представляет собой водород или метил;
R2 представляет собой -CH2CH2CH2R8, где R8 представляет собой амино или карбамоил;
R3 представляет собой метил;
R4 представляет собой водород;
R5 представляет собой гидрокси;
R7 выбран из группы состоящей из
или R7 представляет собой
где Raa1 представляет собой СН2СООН;
Rаа2 представляет собой CH2CONH2;
R13 представляет собой аминогруппу, тиоуреидогруппу формулы
Rх19 и Rх20 каждый независимо представляет водород; Rх21 представляет собой (C1-С10)алкил или (C6-C10) арил,
включающий обработку соединения формулы Ia
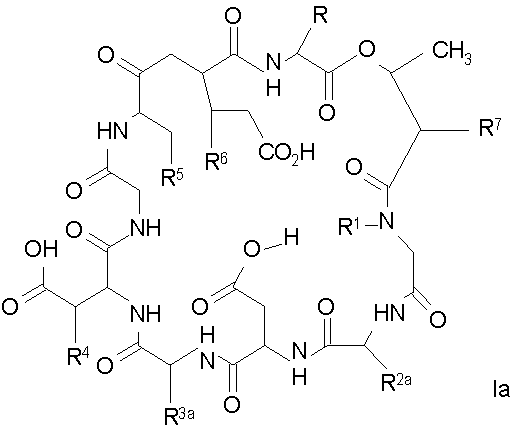
или его фармацевтически приемлемой соли,
где R2а представляет собой -СН2СН2СН2NH2; и R3а представляет собой метил; модифицирующим агентом.
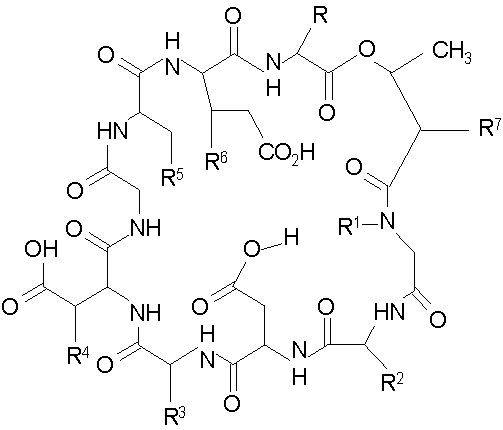
или его фармацевтически приемлемой соли,
где R представляет собой 2-бутил, изопропил или 2-(2'-аминофенацил);
каждый из R1 и R6 независимо представляет собой водород или метил;
R2 представляет собой -CH2CH2CH2R8, где R8 представляет собой амино или карбамоил;
R3 представляет собой метил;
R4 представляет собой водород;
R5 представляет собой гидрокси;
R7 выбран из группы, состоящей из

или R7 представляет собой
где Raa1 представляет собой СН2СООН;
Rаа2 представляет собой CH2CONH2;
R13 представляет собой аминогруппу, тиоуреидогруппу формулы
где Rх9 и Rх20 каждый независимо представляет водород; Rх21 представляет собой (C1-С10)алкил или (C6-C10)арил,
включающий обработку соединения формулы XXVIII
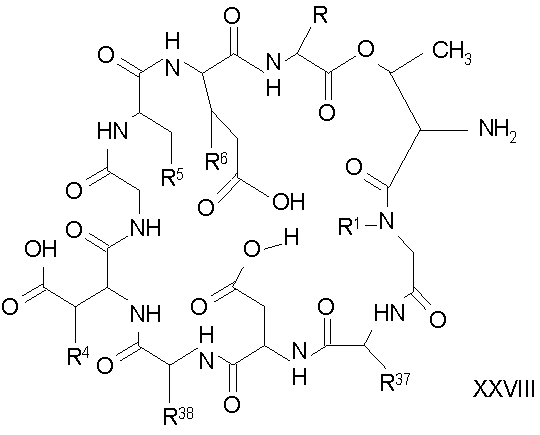
или его фармацевтически приемлемой соли,
где R37 представляет собой -CH2CH2CH2R8 и R38 представляет собой метил, модифицирующим агентом.
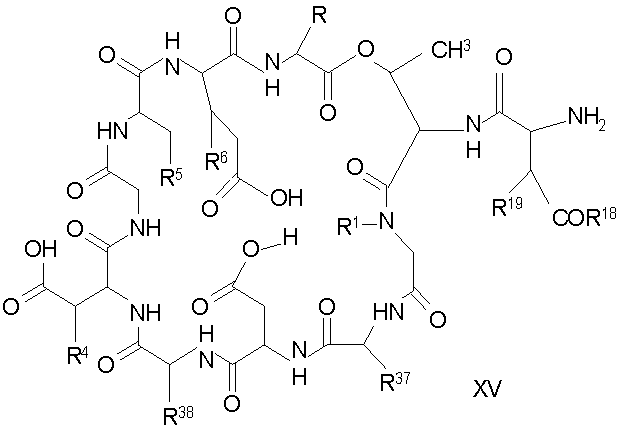
или его фармацевтически приемлемой соли,
где R18 представляет собой гидрокси и R19 представляет собой водород,
с получением соединения формулы XXVIII или его фармацевтически приемлемой соли.
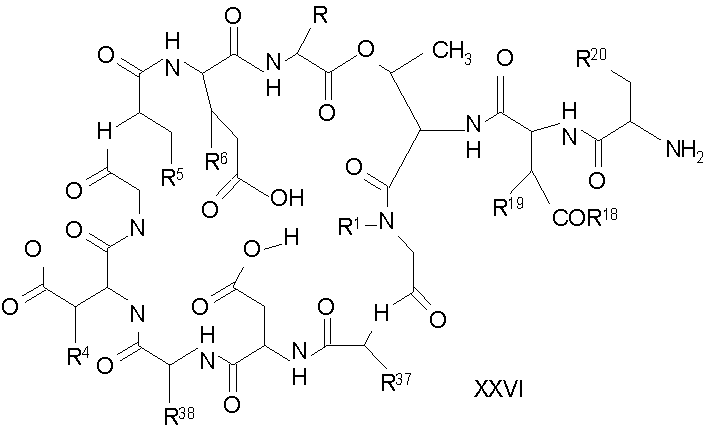
или его фармацевтически приемлемой соли,
где R20 представляет собой карбоксиамино,
с получением соединения формулы XXVII или его фармацевтически приемлемой соли.
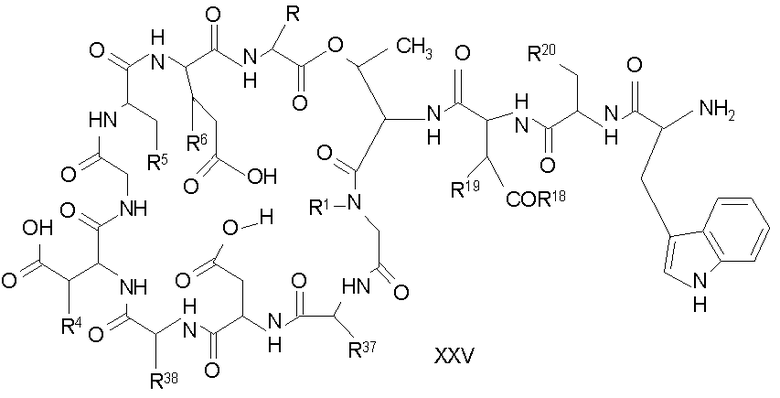
или его фармацевтически приемлемой соли с получением соединения формулы XXVI или его фармацевтически приемлемой соли.

или его фармацевтически приемлемой соли,
где R25 представляет собой алкильную группу, с получением соединения формулы XXV или его фармацевтически приемлемой соли.
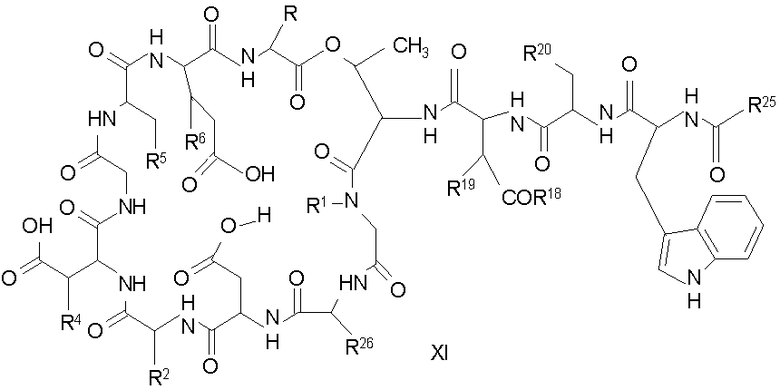
или его фармацевтически приемлемой соли,
где R26 представляет собой метил или -CH2CH2CH2NH2 и R27 представляет собой метил, модифицирующим агентом с получением соединения формулы XXV или его фармацевтически приемлемой соли.
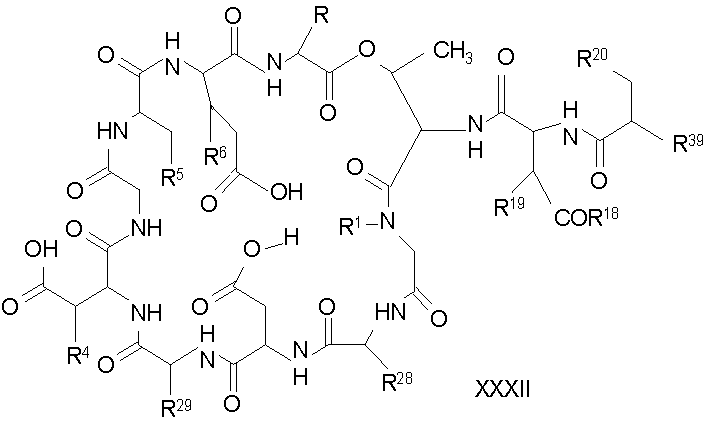
или его фармацевтически приемлемой соли,
где (a) R представляет собой 2-бутил, изопропил или 2-(2'-аминофенацил);
(b) каждый из R1 и R6 независимо представляет собой водород или метил;
(c) R4 представляет собой водород;
(d) R5 представляет собой гидрокси или карбоксиамино;
(e) R18 представляет собой амино или гидрокси;
(f) R19 представляет собой водород;
(g) R20 представляет собой карбоксиамино;
(h) R28 представляет собой -CH2CH2CH2NHP;
(i) R29 представляет собой метил;
(j) P представляет собой аминозащитную группу;
(l) при условии, что,
(1) когда R2 представляет собой -CH2CH2CH2R8, R7 является отличным от
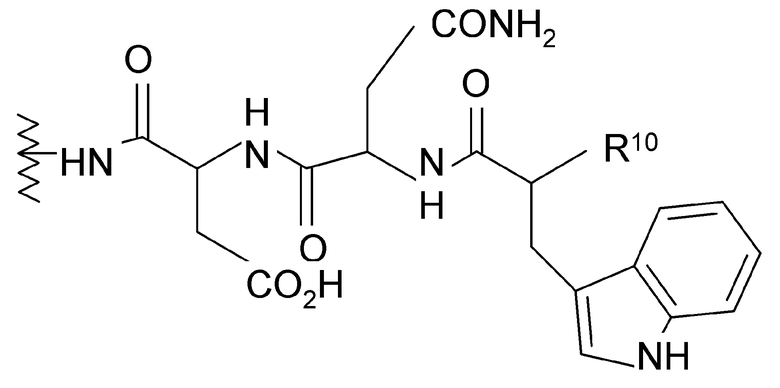
где R10 представляет собой амино, монозамещенный амино, дизамещенный амино, ациламино, уреидо, гуанидино, карбамоил, сульфонамино, тиоациламино, иминоамино или фосфонамино;
(2) когда R2 представляет собой метил, R7 является отличным от
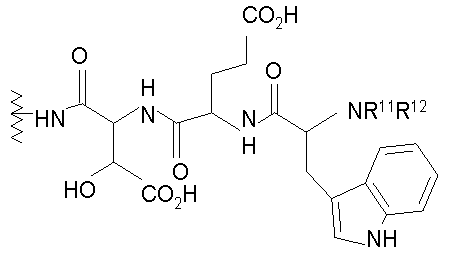
где каждый из R11 и R12 представляет собой водород, C6-C18 незамещенный алканоил, C8-C18 незамещенный алкеноил, C8-C18 незамещенный алкил или C8-C18 выбранный замещенный алкил; или альтернативно R11 и R12 совместно представляют собой С8-С18 алкилиденил;
включающий обработку соединения формулы XIV
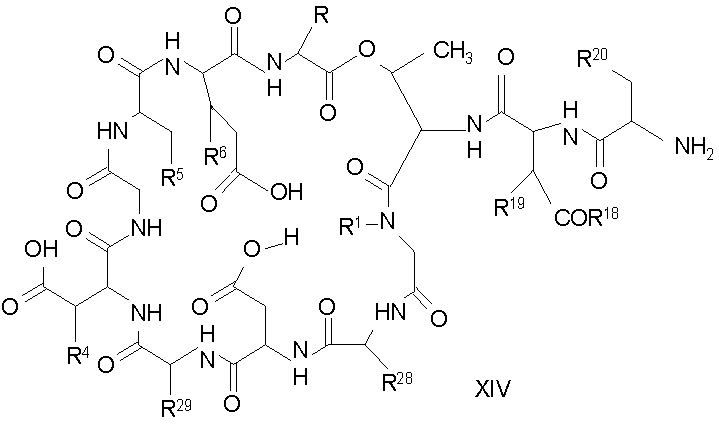
или его фармацевтически приемлемой соли, модифицирующим агентом.
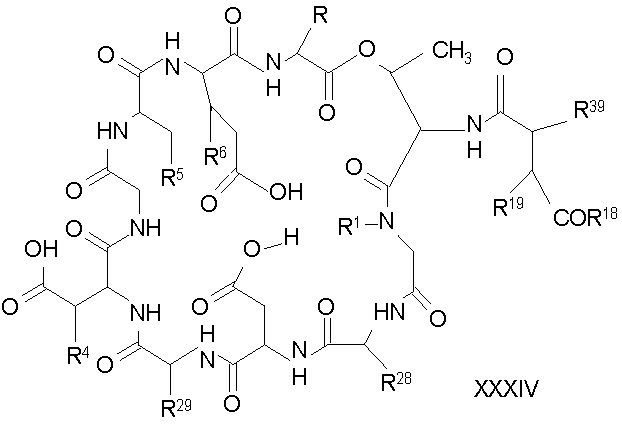
или его фармацевтически приемлемой соли;
где R представляет собой 2-бутил, изопропил или 2-(2'-аминофенацил);
каждый из R1 и R6 независимо представляет собой водород или метил;
R4 представляет собой водород;
R5 представляет собой гидрокси или карбоксиамино;
R18 представляет собой амино или гидрокси;
R19 представляет собой водород;
R20 представляет собой карбоксиамино;
R28 представляет собой -CH2CH2CH2NHP;
R29 представляет собой метил;
Р представляет собой аминозащитную группу;
R39 независимо представляет собой амино, тиоуреидо;
(k) при условии, что, (1) когда R2 представляет собой -CH2CH2CH2R8, R7 является отличным от
где R10 представляет собой амино, монозамещенный амино, дизамещенный амино, ациламино, уреидо, гуанидино, карбамоил, сульфонамино, тиоациламино, иминоамино или фосфонамино;
(2) когда R2 представляет собой метил, R7 является отличным от
где каждый из R11 и R12 представляет собой водород, C6-C18 незамещенный алканоил, C8-C18 незамещенный алкеноил, C8-C18 незамещенный алкил или C8-C18 выбранный замещенный алкил; или альтернативно R11 и R12 совместно представляют собой C8-C18 алкилиденил,
включающий обработку соединения формулы XV
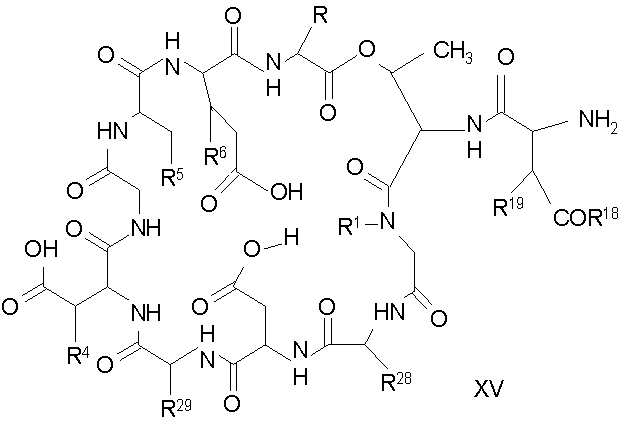
или его фармацевтически приемлемой соли, модифицирующим агентом.
| Способ получения циклопептида | 1968 |
|
SU535902A3 |
| ЦИКЛИЧЕСКИЕ ПЕПТИДЫ ИЛИ ИХ АДДИТИВНЫЕ СОЛИ КИСЛОТЫ, КОМПОЗИЦИЯ, ПРОЯВЛЯЮЩАЯ ПРОТИВОГРИБКОВУЮ И АНТИПНЕВМОЦИСТНУЮ АКТИВНОСТЬ, СПОСОБ ЛЕЧЕНИЯ ГРИБКОВЫХ ИНФЕКЦИЙ | 1994 |
|
RU2122548C1 |
| US 4537717 A, 27.08.1985 | |||
| US 5039789 A, 13.08.1991 | |||
| US 5912226 A, 15.06.1999. | |||

