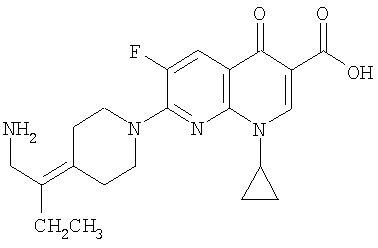
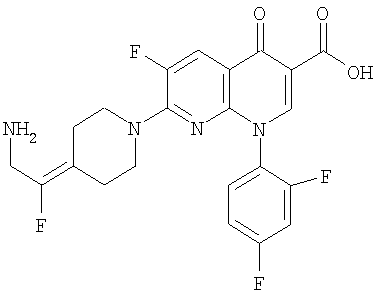
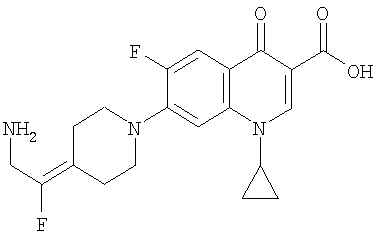
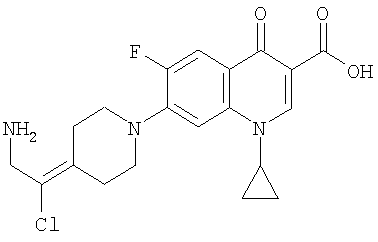
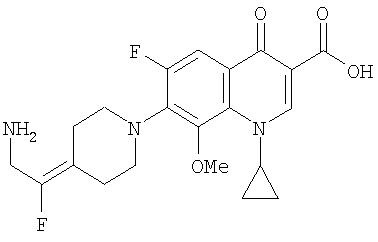
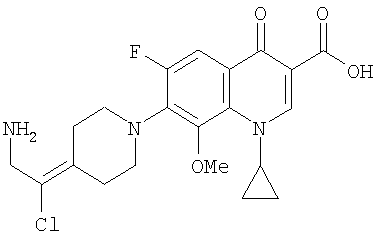
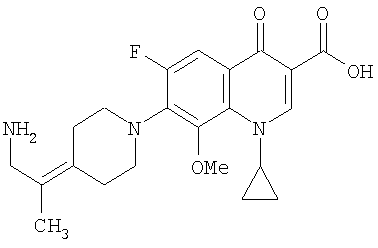
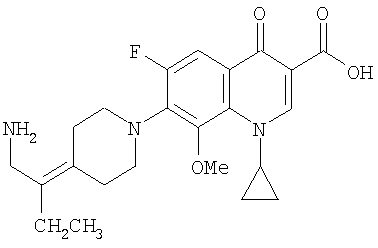
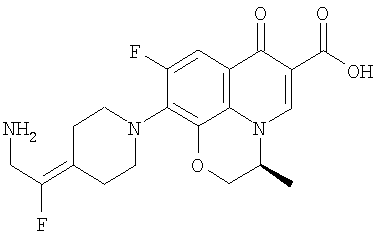
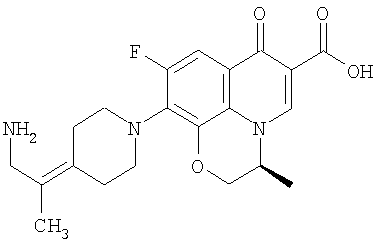
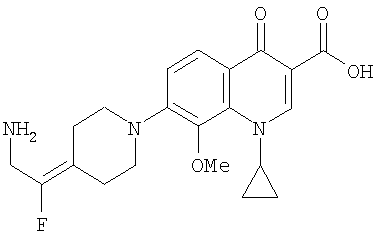
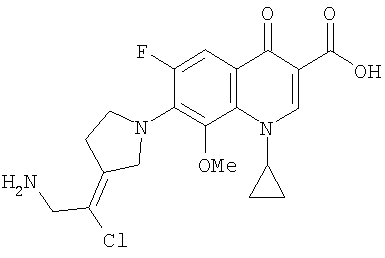
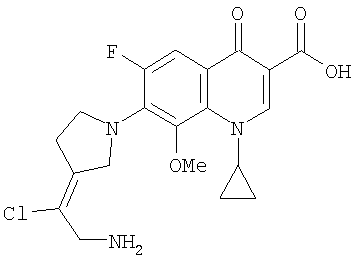
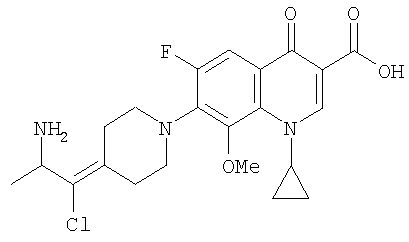
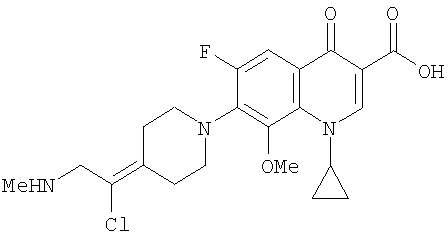
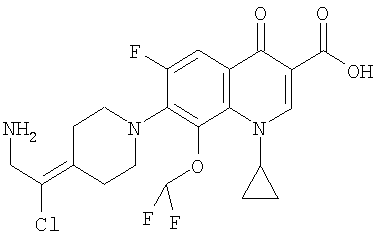
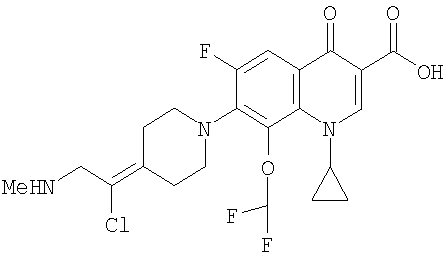
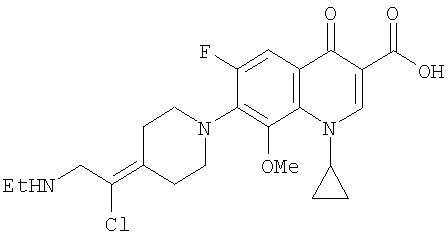
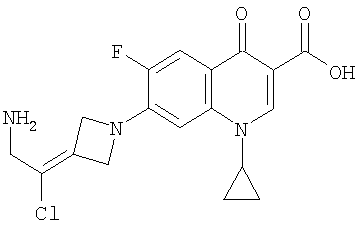
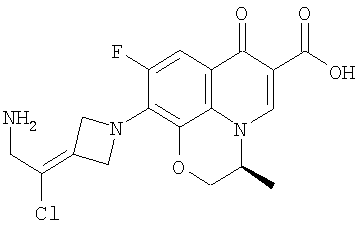
Перекрестная ссылка на родственную заявку
Настоящая заявка, согласно разделу 35 Кодекса законов США § 119(е), претендует на преимущество по отношению к предварительной заявке с порядковым номером 60/504924, поданной 22 сентября 2003 года, которая полностью включена в данное описание в качестве ссылки.
Область техники, к которой относится изобретение
Объект изобретения относится к новым противомикробным соединениям, композициям их содержащим и их применениям.
Предпосылки создания изобретения
В химической и медицинской литературе описываются соединения, которые называют противомикробными, то есть, способными противодействовать или подавлять рост или размножение микроорганизмов, таких как бактерии. Например, такие антибактериальные агенты описаны в Antibiotics, Chemotherapeutics and Antibacterial Agents for Disease Control (M. Greyson, изд., 1982); E. Gale и др., The Molecular Basis of Antibiotic Action, 2-ое изд. (1981); Recent Research Developments in Antimicrobial Agents & Chemotherapy (S.G. Pandalai, изд., 2001); Quinolone Antimicrobial Agents (Jonn S Wolfson., David C Hooper, изд., 1989) и F. O'Grady, H.P. Lambert, R.G. Finch, D. Greenwood, Martin Dedicoat, "Antibiotic and Chemotherapy, 7-ое изд." (1997).
Механизмы действия этих антибактериальных агентов различны. Однако, в целом, полагают, что они действуют в одном или более направлениях: ингибированием синтеза клеточной оболочки или ее восстановлением; изменением проницаемости клеточной оболочки; ингибированием синтеза белка или ингибированием синтеза нуклеиновых кислот. Например, бета-лактамные антибактериальные агенты действуют через ингибирование основных пенициллинсвязывающих белков (РВР) в бактериях, которые отвечают за синтез клеточной оболочки. В качестве другого примера, хинолоны действуют, по меньшей мере, отчасти путем ингибирования синтеза ДНК, таким образом предохраняя клетку от репликации.
Фармакологические характеристики противомикробных агентов и их пригодность для любого данного клинического применения отличаются. Например, классы противомикробных агентов (и представители в пределах класса) могут отличаться по 1) их относительной эффективности против различных типов микроорганизмов; 2) их чувствительности к развитию устойчивости микроорганизмов; 3) их фармакологическим характеристикам, таким как биодоступность и биораспределение. Следовательно, при выборе подходящего противомикробного агента для данной клинической ситуации требуется изучение многих факторов, включая тип организма, вовлеченного в патологический процесс, желательный способ введения, локализацию инфекции, которую нужно лечить, и другие факторы.
Однако множество попыток получить улучшенные противомикробные агенты дало неоднозначные результаты. Действительно, получено немного противомикробных агентов, которые являются клинически приемлемыми с точки зрения спектра их противомикробной активности, аннулирования устойчивости микроорганизмов и фармакологии. Таким образом, остается необходимость в широком спектре противомикробных агентов, которые эффективны против устойчивых микроорганизмов.
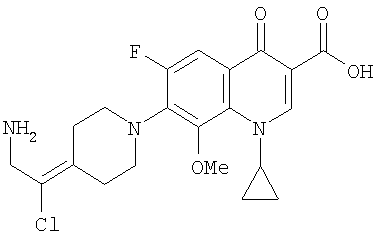
Согласно уровню техники известно, что некоторые 1,4-дигидрохинолоновые, нафтиридиновые или родственные гетероциклические составляющие обладают противомикробной активностью и описаны в следующих ссылках: R. Albrecht Prog. Drug Research, том 21, с. 9 (1977); J. Wolfson и др., "The Fluoroquinolones: Structures, Mechanisms of Action and Resistance, and Spectra of Activity in Vitro", Antimicrob. Agents and Chemother., том 28, с. 581 (1985); G. Klopman и др., Antimicrob. Agents and Chemother., том 31, с.1831 (1987); M.P. Wentland и др., Ann. Rep. Med. Chem., том 20, с. 145 (1986); J.B. Cornett и др., Ann. Rep. Med. Chem., том 21, с. 139 (1986); P.B. Fernandes и др., Ann. Rep. Med. Chem., том 22, с. 117 (1987); A. Koga и др., "Structure-Activity Relationships of Antibacterial 6,7- and 7,8-Disubstituted 1-alkyl-1,4-dihydro-4-oxoquinoline-3-carboxylic Acids", J. Med. Chem., том 23, сс. 1358-1363 (1980); J.M. Domagala и др., J. Med. Chem., том 31, с. 991 (1988); T. Rosen и др., J. Med. Chem., том 31, с. 1598 (1988); B. Ledoussal и др., "Non 6-Fluoro Substituted Quinolone Antibacterials: Structure and Activity", J. Med. Chem., том 35, cc. 198-200 (1992); патент США 6329391; A.M. Emmerson и др., "The quinolones: Decades of development and use", J. Antimicrob. Chemother., том 51, сс. 13-20 (2003); J. Ruiz "Mechanisms of resistance to quinolones: target alterations, decreased accumulation and DNA gyrase protection", J. Antimicrob. Chemother., том 51, сс. 1109-1117 (2003); Y. Kuramoto и др., "A Novel Antibacterial 8-Chloroquinolone with a Distorted Orientation of the N1-(5-Amino-2,4-difluorophenyl) Group", J. Med. Chem., том 46, cc. 1905-1917 (2003); публикация патента Японии 06263754; публикация Европейского патента 487030; публикация Международной заявки на патент WO 0248138; публикация Международной заявки на патент WO 9914214; публикация патента США 2002/0049192; публикация Международной заявки на патент WO 02085886; публикация Европейского патента 572259; публикация Международной заявки на патент WO 0136408; патент США 5677456; публикация Европейского патента 362759; патент США 5688791; патент США 4894458; публикация Европейского патента 677522; патент США 4822801; патент США 5256662; патент США 5017581; публикация Европейского патента 304087; публикация Международной заявки на патент WO 0136408; публикация Международной заявки на патент WO 02085886; публикация патента Японии 01090184; публикация Международной заявки на патент WO 9209579; публикация Международной заявки на патент WO 0185728; публикация Европейского патента 343524; публикация патента Японии 10130241; публикация Европейского патента 413455; публикация Международной заявки на патент WO 0209758; публикация Международной заявки на патент WO 0350107; публикация Международной заявки на патент WO 9415933; публикация Международной заявки на патент WO 9222550; публикация патента Японии 07300472; публикация Международной заявки на патент WO 0314108; публикация Международной заявки на патент WO 0071541; публикация Международной заявки на патент WO 0031062 и патент США 5869670.
В WO 03050107 описывается ряд дигидрохинолоновых, нафтиридиновых и родственных гетероциклических антибактериальных агентов. Особый интерес представляет раскрытие соединений формулы:
где R8 и R8' означают водород, алкил, замещенный алкил, алкиламино или аралкил; R9 означает водород, алкил, алкиламино, диалкиламино, арил, аралкил или тригалогеналкил; и Х означает гидрокси, алкокси, ацилокси, аминогруппу или замещенную аминогруппу.
В публикации Европейского патента 362759 раскрываются 1,4-дигидрохинолоновые и нафтиридиновые антибактериальные агенты формулы:
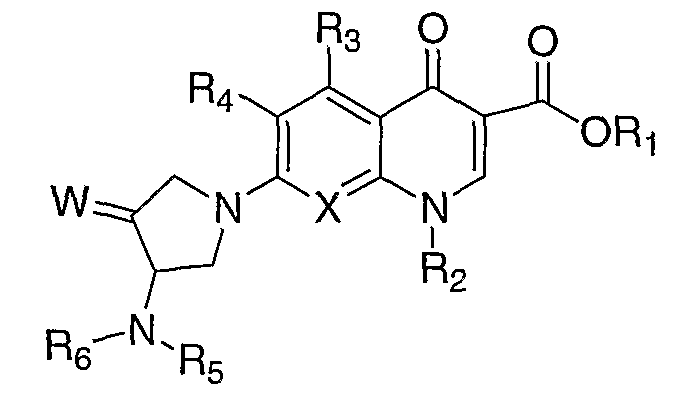
где W означает С1-3-алкилиден и R5 и R6 означают водород или алкил.
В публикации Международной заявки на патент WO 99/14214 и патенте США 6329391 раскрываются хинолоновые антибактериальные агенты с С7-пиперидинильными, С7-азетидинильными или С7-пирролидинильными заместителями формулы:
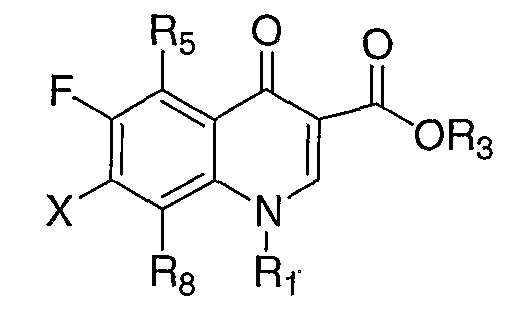
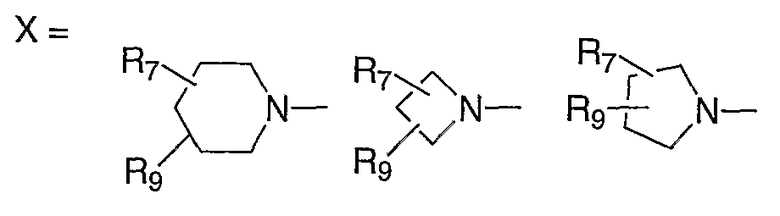
Особый интерес представляют такие соединения, где R7 означает аминогруппу, аминоалкил или замещенный аминоалкил и R9 выбирают из группы, состоящей из водорода, С1-С4-алканила, С2-С6-алкенила, С2-С6-алкинила или конденсированного или спироциклического С3-С6-алкильного кольца. Для соединений с замещенным пиперидином в положении 7 хинолонкарбоновой кислоты, предпочтительными заместителями являются 3-амино-4-метил, 3-амино-4,4-диметил, 3-амино-4-спироциклопропил, 3-амино-6-циклопропил, 3-аминометил, 4-аминометил и 3-метиламино. Для соединений с замещенным пирролидином в положении 7 цикла хинолонкарбоновой кислоты, предпочтительные заместители включают 3-(1-аминоэтил), 3-аминометил, 4-(1-аминоэтил)-2,2-диметил и 2-аминометил. В случае соединений с азетидиновым заместителем в положении 7 хинолонкарбоновой кислоты, соединения, имеющие заместители, такие как 3-амино, 3-аминометил и 3-(1-амино-1-метил)этил, относятся к числу предпочтительных примеров.
В публикации Европейского патента 241206А2 раскрываются соединения формулы:
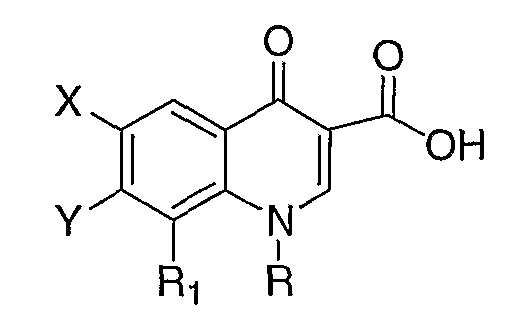
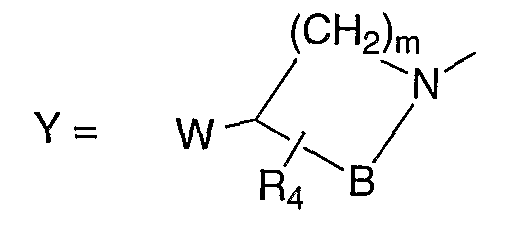
где В означает -СН2-, -(СН2)2- или (СН2)3-; R4 означает водород, С1-С3-алкил, гидрокси или С1-С3-алкокси; W означает гидрокси, С1-С3-алкокси или группу формулы R5R6N-(CH2)n-, в которой n имеет значение 0 или 1; и R5 и R6 являются одинаковыми или различными и каждый представляет собой атом водорода, С1-С3-алкильную группу или аралкильную группу, и m имеет значение 1 или 2. Каждый символ определен в описании вышеуказанной публикации. Для пиперидинового заместителя в положении 7 хинолонкарбоновой кислоты, соединения, имеющие заместители 4-амино-3-метил, 4-метиламино-3-метил, 4-гидрокси-3-метил, включены там в качестве предпочтительных примеров.
В публикации Европейского патента 0394553В1 раскрываются противовирусные соединения формулы:
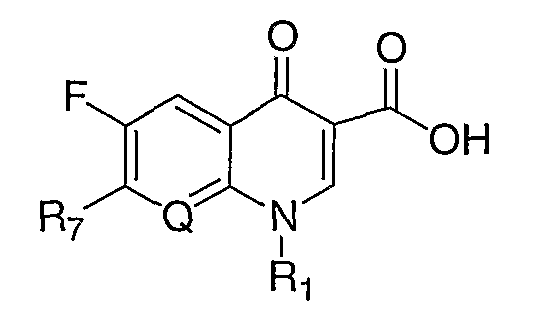
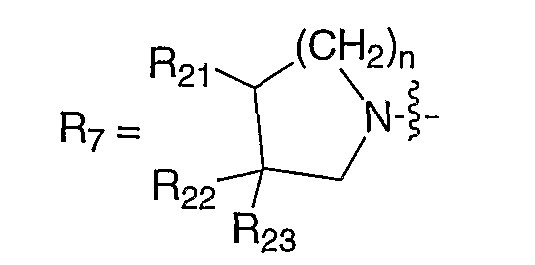
где R21, R22 и R23, каждый, независимо, означают атом водорода, атом галогена, аминогруппу, С1-С6-алкил, С1-С8-алкокси или С1-С8-аминоалкил и два из них можно комбинировать друг с другом для образования спиро-кольца; и n имеет значение 1 или 2.
В публикации Европейского патента 0572259А1 раскрываются противовирусные соединения формулы:
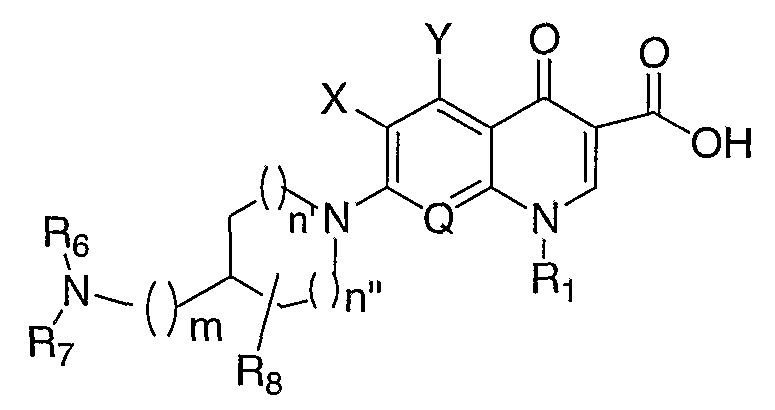
где R6 и R7 могут быть одинаковыми или различными и каждый представляет собой атом водорода или низшую алкильную группу; m имеет значение 0 или 1; n' имеет значение 1 или 2; n" имеет значение 1, 2, 3 или 4; и R8 означает атом водорода, низшую алкильную группу, гидроксильную группу или низшую алкоксильную группу.
В публикации Международной заявки на патент WO 9324479 раскрываются соединения формулы:
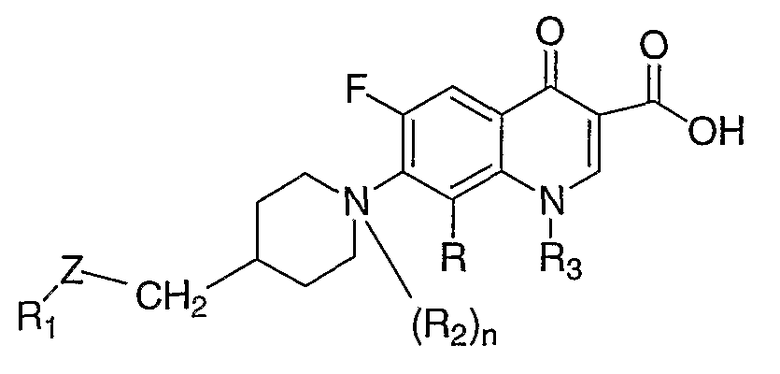
где Z означает амино-радикал; R1 означает водород, необязательно гидроксилированный низший алкильный радикал, ацильный радикал карбоновой кислоты, алкилкарбоновой кислоты или арилсульфоновой кислоты, или ариламинокарбонильный радикал; R2 означает атом кислорода; и n имеет значение 0 или 1.
Примеры бактериальных инфекций, устойчивых к терапии антибиотиками, ранее описаны; в настоящее время они представляют значительную опасность для здоровья людей в развивающемся мире. Развитие устойчивости микроорганизмов (возможно, как результат интенсивного применения антибактериальных агентов в течение длительного времени) вызывает возрастающее беспокойство медиков. «Устойчивость» можно определять как существование организмов, в рамках популяции данных видов микроорганизмов, которые являются менее чувствительными к действию данного противомикробного агента. Эта устойчивость касается отношения окружающей среды в клиниках и частных больницах, где относительно высокая степень инфицирования и интенсивное применение антибактериальных агентов. См., например, W. Sanders, Jr. и др., "Inducible Beta-lactamases: Clinical and Epidemiologic Implications for the Use of Newer Cephalosporins", Review of Infectious Diseases, с. 830 (1988).
Известно, что болезнетворные бактерии могут приобретать устойчивость посредством некоторых особых механизмов, включая инактивацию антибиотика бактериальными ферментами (как, например, β-лактамазы, гидролизующую пенициллин и цефалоспорин); удаление антибиотика при использовании насосов, вызывающих отток; изменение мишени антибиотика мутацией и генетической рекомбинацией (например, устойчивость к пенициллину у Neiserria gonorrhoeae); и приобретение легко переносимого гена от внешнего источника для создания устойчивой мишени (например, устойчивость к метициллину у Staphylococcus aureus). Имеются некоторые грамположительные патогены, такие как устойчивый к ванкомицину Enterococcus faecium, которые устойчивы фактически ко всем коммерчески доступным антибиотикам.
Следовательно, существующие антибактериальные агенты ограничены в преодолении устойчивости. Таким образом, полезно получение новых антибактериальных агентов, которые можно применять против устойчивых микроорганизмов.
Краткое изложение сущности изобретения
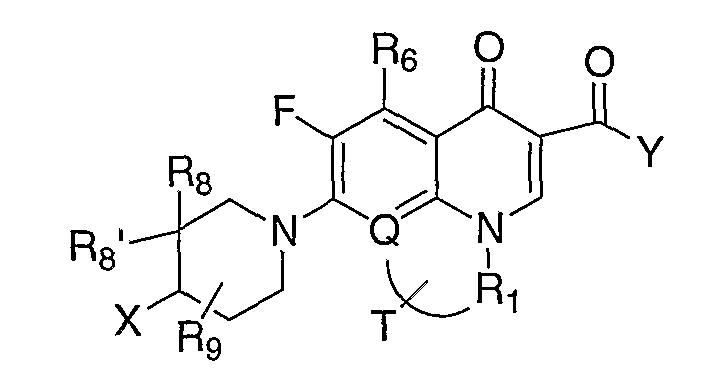
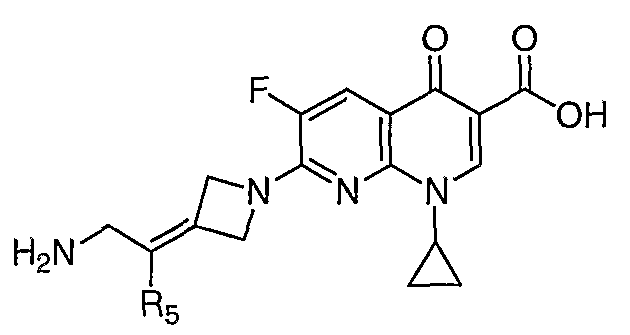
Заявители получили новый ряд хинолонов и близких к ним соединений, которые эффективны против устойчивых микроорганизмов и обладают значительными преимуществами по активности относительно известных из уровня техники. В частности, изобретение относится к соединениям, имеющим структуру формулы (I):
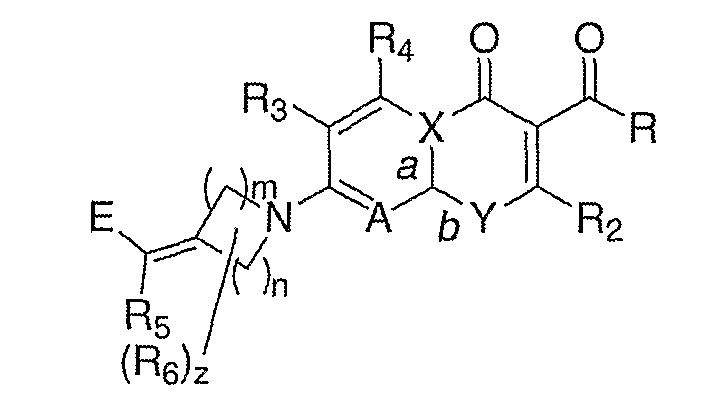
Формула I
где:
n означает целое число от 1 до 3;
m означает целое число от 1 до 3;
z означает целое число от 0 до 3;
R выбирают из водорода, гидрокси или алкокси;
R2 означает водород;
R3 и R4 независимо выбирают из группы, состоящей из водорода, галогена, аминогруппы, гидрокси, алкокси, алкилтиогруппы, алкила, алкенила и алкинила;
R5 выбирают из группы, состоящей из водорода, галогена, алкила, арила, алкокси и алкилтиогруппы;
R6 независимо выбирают из группы, состоящей из алкила, гидрокси, алкокси, алкилтиогруппы, алкенила, алкинила, арила, алкоксииминогруппы и галогена; или R5 и R6 связаны с образованием 4-7-членного карбоцикла, где каждый атом углерода в кольце может быть необязательно замещен R12, где R12 выбирают из группы, состоящей из галогена, аминогруппы, гидрокси, алкокси, алкилтиогруппы, алкила, алкенила, алкинила, оксогруппы, алкоксииминогруппы и гидроксииминогруппы;
Е выбирают из группы, состоящей из:
1) 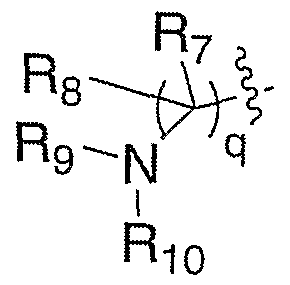
где
q означает целое число от 1 до 3;
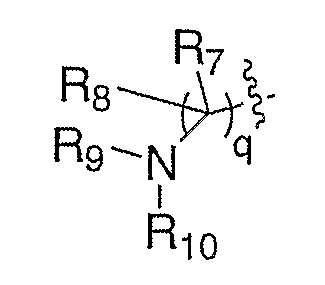
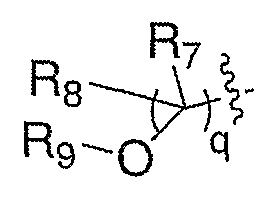
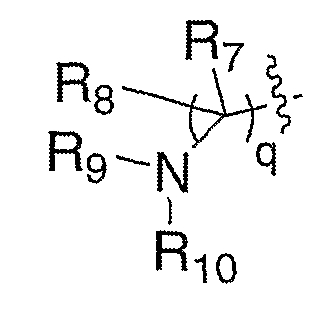
R7 и R8, каждый, независимо выбирают из водорода и алкила; или R7 и R8 связаны с образованием 3-6-членного карбоцикла, или либо, R7 либо R8 независимо могут быть связаны либо с R9, либо с R10 с образованием гетероцикла, содержащего атом азота, к которому присоединены R9 или R10, где R9 и R10, каждый, независимо выбирают из группы, состоящей из водорода, алкила, ацила, алкоксикарбонила или сульфонила, или, альтернативно, R9 и R10 связаны с образованием гетероцикла, содержащего атом азота, к которому они присоединены;
2) 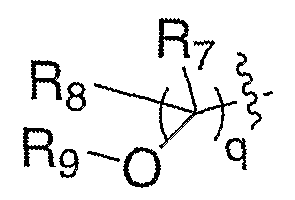
где
q означает, как описано выше;
R7 и R8, каждый, независимо выбирают из водорода и алкила; или R7 и R8 связаны с образованием 3-6-членного карбоцикла; и R9 выбирают из группы, состоящей из водорода, алкила, ацила, алкоксикарбонила или сульфонила; и
3) алкенила;
А выбирают из N и C(R11), где R11 выбирают из группы, состоящей из водорода, алкила, галогена, гидрокси, алкокси, алкилтиогруппы и цианогруппы;
Х выбирают из С и N, где, если Х означает С, а означает двойную связь и b означает простую связь, и, если Х означает N, а означает простую связь и b означает двойную связь; и
Y выбирают из N(R1) и С(R1), при условии, что, когда Y означает N(R1), Х означает С и, когда Y означает С(R1), Х означает N, где R1 выбирают из С3-С6-циклоалкила, С4-С6-гетероциклоалкила, алкила, алкена, 6-членного арила и 6-членного гетероарила; при условии, что
если А означает C(R11), Х означает С и Y означает N(R1), тогда R11 и R1 могут быть связаны с образованием 6-членного гетероцикла, или
если А означает C(R11), Х означает С и Y означает N(R1), тогда R2 и R1 могут быть связаны с образованием моноциклического или бициклического гетероцикла, или
если А означает C(R11), Х означает С и Y означает N(R1), тогда R2 и R могут быть связаны с образованием 5-членного гетероцикла; или
их оптическим изомерам, диастереомерам или энантиомерам; их фармацевтически приемлемым солям, гидратам или пролекарствам.
Кроме того, способы применения соединений согласно данному изобретению в качестве исходных материалов также рассматриваются в данном изобретении.
Обнаружено, что соединения согласно данному изобретению и композиции, содержащие эти соединения, являются эффективными противомикробными агентами против широкого диапазона патогенных микроорганизмов с преимуществами в отношении активности против устойчивых микроорганизмов.
Соответственно, настоящее изобретение также относится к способу лечения млекопитающего, имеющего состояние, вызванное бактериальной инфекцией или которому способствует бактериальная инфекция, включающему введение вышеуказанному млекопитающему терапевтически эффективного количества соединения формулы (I).
Настоящее изобретение, далее, относится к способу профилактики страдания млекопитающего, вызываемого бактериальной инфекцией и которому способствует бактериальная инфекция, включающему введение вышеуказанному млекопитающему профилактически эффективной дозы фармацевтической композиции, содержащей соединение формулы (I).
Подробное описание
Объект изобретения относится к соединениям формулы (I):
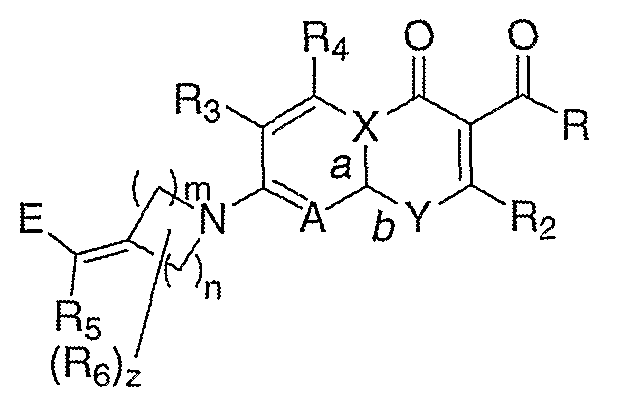
Формула I
где:
a, b, n, m, z, R, R2, R3, R4, R5, R6, A, E, X и Y имеют значение, как описано выше в разделе «Краткое изложение сущности изобретения».
По отношению к вышеуказанному описанию, некоторые определения имеют следующие значения.
Если специально не оговорено согласно стандартной номенклатуре, используемой в данном описании, концевая часть указанной боковой цепи описывается первой, с последующей соседней функциональностью в направлении места присоединения.
Если специально не оговорено, термины «алкил», «алкенил» и «алкинил», или используемые отдельно или как часть замещающей группы, включают линейные или разветвленные цепи, имеющие 1-8 атомов углерода, или любое число в этом диапазоне. Термин «алкил» относится к углеводородам с линейной или разветвленной цепью. Термин «алкенил» относится к углеводородам с линейной или разветвленной цепью, по меньшей мере, с одной углерод-углеродной двойной связью. Термин «алкинил» относится к углеводородам с линейной или разветвленной цепью, по меньшей мере, с одной углерод-углеродной тройной связью. Например, алкильные радикалы включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, н-пентил, 3-(2-метил)бутил, 2-пентил, 2-метилбутил, неопентил, н-гексил, 2-гексил и 2-метилпентил. «Алкокси»-радикалы представляют собой включающие кислород части простых эфиров, образованных из вышеописанных алкильных групп с линейной или разветвленной цепью. «Циклоалкильные» группы содержат 3-8 атомов углерода в кольце и, предпочтительно, 5-7 атомов углерода в кольце. Алкильные, алкенильные, алкинильные, циклоалкильные группы и алкоксигруппы могут быть независимо замещенными одним или более членами групп, включающими, но не ограничиваясь этим, гидроксииминогруппу, галоген, алкил, алкенил, алкинил, циклоалкил, алкокси, оксогруппу, алкоксииминоарил, гетероарил, гетероциклическую группу, CN, нитрогруппу, -OCOR13, -OR13, -SR13, -SOR13, -SO2R13, -COOR13, -NR13R14, -CONR13R14, -OCONR13R14, -NHCOR13, -NHCOOR13 и -NHCONR13R14, где R13 и R14 независимо выбирают из группы, состоящей из водорода, алкила, алкенила, алкинила, циклоалкила, арила, гетероарила, гетероциклической группы, аралкила, гетероаралкила и гетероциклоалкила, или, альтернативно, R14 и R15 могут быть связаны с образованием гетероцикла, содержащего атом азота, к которому они присоединены.
Термин «ацил», как здесь используется, или используют отдельно или как часть замещающей группы, означает органический радикал, имеющий 2-6 атомов углерода (линейная или разветвленная цепь), органической кислоты. Термин «Ас», как здесь используется, или используемый отдельно или как часть замещающей группы, означает ацетил.
Термин «галоген» означает фтор, хлор, бром или иод. (Моно-, ди-, три- и пер-)галогеналкил означает алкил, замещенный независимой заменой атомов водорода галогеном.
«Арил» или «Ar», или используемый отдельно или как часть замещающей группы, представляет собой карбоциклический ароматический радикал, включающей, но не ограничиваясь этим, фенил, 1- или 2-нафтил и тому подобное. Карбоциклический ароматический радикал может быть замещен независимой заменой 1-3 атомов водорода арилом, гетероарилом, галогеном, ОН, CN, меркапто, нитро, амино, С1-С8-алкилом, С2-С8-алкенилом, С1-С8-алкоксилом, С1-С8-алкилтио, С1-С8-алкиламино, ди(С1-С8-алкил)амино, (моно-, ди-, три- и пер-)галогеналкилом, формилом, карбокси, алкоксикарбонилом, С1-С8-алкил-СО-О-, С1-С8-алкил-СО-NH- или карбоксамидом. Иллюстративные арильные радикалы включают, например, фенил, нафтил, бифенил, фторфенил, дифторфенил, бензил, бензоилоксифенил, карбоэтоксифенил, ацетилфенил, этоксифенил, феноксифенил, гидроксифенил, карбоксифенил, трифторметилфенил, метоксиэтилфенил, ацетамидофенил, толил, ксилил, диметилкарбамилфенил и тому подобные. «Ph» или «PH» означает фенил. «Bz» означает бензоил.
Или используемый отдельно или как часть замещающей группы, термин «гетероарил» относится к циклическому, полностью ненасыщенному радикалу, имеющему 5-10 атомов в кольце, где один атом кольца выбирают из S, O и N; 0-2 атома кольца представляют собой дополнительные гетероатомы, независимо выбираемые из S, O и N; и остальные атомы кольца представляют собой атомы углерода. Радикал может быть связан с остальной частью молекулы посредством любого из атомов кольца. Типичные гетероарильные группы включают, например, пиридинил, пиразинил, пиримидинил, пиридазинил, пирролил, пиразолил, имидазолил, тиазолил, оксазолил, изоксазолил, тиадиазолил, триазолил, триазинил, оксадиазолил, тиенил, фуранил, хинолинил, изохинолинил, индолил, изотиазолил, N-оксопиридил, 1,1-диоксотиенил, бензотиазолил, бензоксазолил, бензотиенил, хинолинил-N-оксид, бензимидазолил, бензопиранил, бензизотиазолил, бензизоксазолил, бензодиазинил, бензофуразанил, индазолил, индолизинил, бензофурил, циннолинил, хиноксалинил, пирролопиридинил, фуропиридинил (такой как фуро[2,3-с]пиридинил, фуро[3,2-b]пиридинил или фуро[2,3-b]пиридинил), имидазопиридинил (такой как имидазо[4,5-b]пиридинил или имидазо[4,5-с]пиридинил), нафтиридинил, фталазинил, пуринил, пиридопиридил, хиназолинил, тиенофурил, тиенопиридил и тиенотиенил. Гетероарильная группа может быть замещена путем независимой замены 1-3 атомов водорода в ней с помощью арила, гетероарила, галогена, ОН, CN, меркапто, нитро, амино, С1-С8-алкила, С1-С8-алкоксила, С1-С8-алкилтио, С1-С8-алкиламино, ди(С1-С8-алкил)амино, (моно-, ди-, три- и пер-)галогеналкила, формила, карбокси, алкоксикарбонила, С1-С8-алкил-СО-О-, С1-С8-алкил-СО-NH- или карбоксамида.
Гетероарил может быть замещен моно-оксогруппой с получением, например, 4-оксо-1Н-хинолина.
Термины «гетероцикл», «гетероциклический» и «гетероцикло» относятся к необязательно замещенной, полностью насыщенной, частично насыщенной или неароматической циклической группе, которая означает, например, 4-7-членную моноциклическую, 7-11-членную бициклическую или 10-15-членную трициклическую систему, которая имеет, по меньшей мере, один гетероатом в кольце, содержащем, по меньшей мере, один атом углерода. Каждое кольцо гетероциклической группы, содержащей гетероатом, может иметь 1, 2 или 3 гетероатома, выбираемых из атомов азота, атомов кислорода и атомов серы, где гетероатомы азота и серы также могут быть необязательно окисленными. Атомы азота могут быть необязательно четвертичными. Гетероциклическая группа может быть присоединена к любому гетероатому или атому углерода. Гетероциклическая группа может быть замещена независимой заменой 1-3 атомов водорода арилом, гетероарилом, галогеном, ОН, CN, меркапто, нитро, амино, С1-С8-алкилом, С1-С8-алкоксилом, С1-С8-алкилтио, С1-С8-алкиламино, ди(С1-С8-алкил)амино, (моно-, ди-, три- и пер-)галогеналкилом, формилом, карбокси, алкоксикарбонилом, С1-С8-алкил-СО-О-, С1-С8-алкил-СО-NH- или карбоксамидом.
Типичные моноциклические гетероциклические группы включают пирролидинил, оксетанил, пиразолинил, имидазолинил, имидазолидинил, оксазолинил, оксазолидинил, изоксазолинил, тиазолидинил, изотиазолидинил, тетрагидрофурил, пиперидинил, пиперазинил, 2-оксопиперазинил, 2-оксопиперидинил, 2-оксопирролидинил, 4-пиперидонил, тетрагидропиранил, тетрагидротиопиранил, тетрагидротиопиранилсульфон, морфолинил, тиоморфолинил, тиоморфолинилсульфоксид, тиоморфолинилсульфон, 1,3-диоксолан, диоксанил, тиетанил, тииранил, 2-оксазепинил, азепинил и тому подобные. Типичные бициклические гетероциклические группы включают хинуклидинил, тетрагидроизохинолинил, дигидроизоиндолил, дигидрохиназолинил (такой как 3,4-дигидро-4-оксохиназолинил), дигидробензофурил, дигидробензотиенил, бензотиопиранил, дигидробензотиопиранил, дигидробензотиопиранилсульфон, бензопиранил, дигидробензопиранил, индолинил, хромонил, кумаринил, изохроманил, изоиндолинил, пиперонил, тетрагидрохинолинил и тому подобные.
Термин «карбоциклический» относится к насыщенному или ненасыщенному, неароматическому, моноциклическому, углеводородному кольцу с 3-7 атомами углерода.
Замещенный арил, замещенный гетероарил и замещенный гетероцикл также могут быть замещены другим замещенным арилом, другим замещенным гетероарилом или другим замещенным гетероциклом с получением, например, 4-пиразол-1-илфенила или 4-пиридин-2-илфенила.
Указанные количества атомов углерода (например, С1-С8 или С1-8) независимо относятся к количеству атомов углерода в алкильном или циклоалкильном остатке или в алкильной части большего заместителя, в котором алкил указывается по типу приставки к корню.
Если специально не оговорено, подразумевают, что определение любого заместителя или переменного в определенном положении в молекуле является независимыми от его определений где-либо в другом месте в данной молекуле. Подразумевают, что заместители и схемы замещения соединений согласно данному изобретению могут быть выбраны обычными специалистами в данной области для получения соединений, которые химически стабильны и которые можно легко синтезировать с помощью методик, известных в данной области, а также изложенных в данном контексте способов.
Термин «гидроксизащитная группа» относится к группам, известным в данной области для подобной цели. Как правило, используемые гидроксизащитные группы описываются, например, в руководстве T.H. Greene и P.G.M. Wuts, Protective Groups in Organic Synthesis, 2-ое изд., John Wiley & Sons, Нью Йорк (1991), которое включено в данный контекст путем ссылки. Иллюстративные гидроксизащитные группы включают, но не ограничиваясь этим, тетрагидропиранил, бензил, метилтиометил, этилтиометил, пивалоил, фенилсульфонил, трифенилметил, тризамещенный силил, такой как триметилсилил, триэтилсилил, трибутилсилил, триизопропилсилил, трет-бутилдиметилсилил, три-трет-бутилсилил, метилдифенилсилил, этилдифенилсилил, трет-бутилдифенилсилил; ацил и ароил, такие как ацетил, бензоил, пивалоилбензоил, 4-метоксибензоил, 4-нитробензоил и арилацил.
Когда соединения согласно данному изобретению имеют, по меньшей мере, один стереогенный центр, они могут, следовательно, существовать в виде энантиомеров. Когда соединения имеют два или более стереогенных центра, они могут дополнительно существовать в виде диастереомеров. Кроме того, некоторые из кристаллических форм соединений могут существовать в виде полиморфов и подразумевают, что они как таковые включены в настоящее изобретение. Кроме того, некоторые из соединений могут образовывать сольваты с водой (то есть, гидраты) или обычными органическими растворителями и подразумевают, что такие сольваты также входят в рамки объема данного изобретения.
Некоторые из соединений согласно настоящему изобретению могут иметь транс- и цис-изомеры. Кроме того, когда способы получения соединений согласно данному изобретению приводят к смеси стереоизомеров, эти изомеры можно разделять с помощью таких обычных способов, как препаративная хроматография. Соединения можно получать в виде отдельного стереоизомера или в рацемической форме, в виде смеси из нескольких возможных стереоизомеров. Нерацемические формы можно получать или синтезом или разделением. Соединения, например, можно разделять на их энантиомеры такими стандартными способами, как образование диастереомерных пар путем солеобразования. Соединения также можно разделять с помощью ковалентного связывания с хиральным вспомогательным веществом с последующим хроматографическим разделением и/или кристаллографическим разделением и удалением хирального вспомогательного вещества. Альтернативно, соединения могут быть разделены, с использованием хиральной хроматографии.
Фраза «фармацевтически приемлемая соль» означает одну или более солей свободного основания или свободной кислоты, которые обладают желаемой фармакологической активностью свободного основания или свободной кислоты, соответственно, и которые ни биологически, ни иным образом, не являются нежелательными. Эти соли можно получать из неорганических или органических кислот. Примерами неорганических кислот являются соляная кислота, азотная кислота, бромводородная кислота, серная кислота или фосфорная кислота. Примерами органических кислот являются уксусная кислота, пропионовая кислота, гликолевая кислота, молочная кислота, пировиноградная кислота, малоновая кислота, янтарная кислота, яблочная кислота, малеиновая кислота, фумаровая кислота, винная кислота, лимонная кислота, бензойная кислота, коричная кислота, миндальная кислота, метансульфоновая кислота, этансульфоновая кислота, п-толуолсульфоновая кислота, бензолсульфоновая кислота, салициловая кислота и тому подобные. Кроме того, подходящими солями являются таковые неорганические или органические основания, такие как КОН, NaOH, Ca(OH)2, Al(OH)3, пиперидин, морфолин, этиламин, триэтиламин и тому подобные.
В рамки объема данного изобретения входят гидратированные формы соединений, содержащие различные количества воды, например, гидратные, полугидратные и полуторагидратные формы. Настоящее изобретение также включает, в рамках данного объема, пролекарства соединений согласно данному изобретению. Вообще, такие пролекарства являются функциональными производными соединений, которые легко in vivo превращаются в необходимое соединение. Таким образом, в способах лечения согласно настоящему изобретению термин «введение» включает в себя лечение различных описанных заболеваний с помощью конкретно раскрытого соединения или с помощью соединения, которое конкретно не раскрыто, но которое in vivo превращается в точно определенное соединение после введения пациенту. Обычные методики подбора и получения подходящих пролекарственных производных раскрыты, например, в "Design of Prodrugs", изд. H. Bundgaard, Elsevier, 1985.
Термин «субъект» включает, без ограничения, любое животное или искусственно модифицированное животное. В качестве отдельного воплощения, субъектом является человек.
Термин «устойчивый к лекарственному средству» или «устойчивость к лекарственному средству» относится к характеристикам микроорганизма в отношении выживания в присутствии доступного в настоящее время противомикробного агента, такого как антибиотик в его обычной эффективной концентрации.
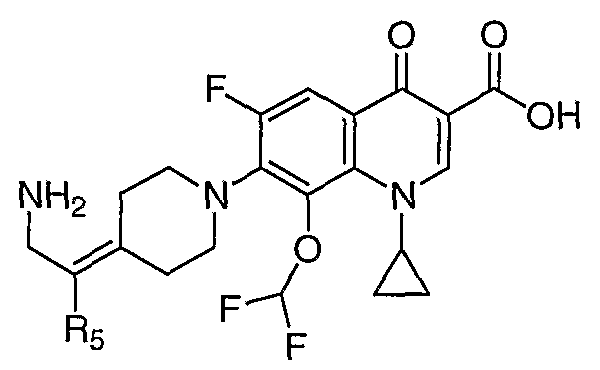
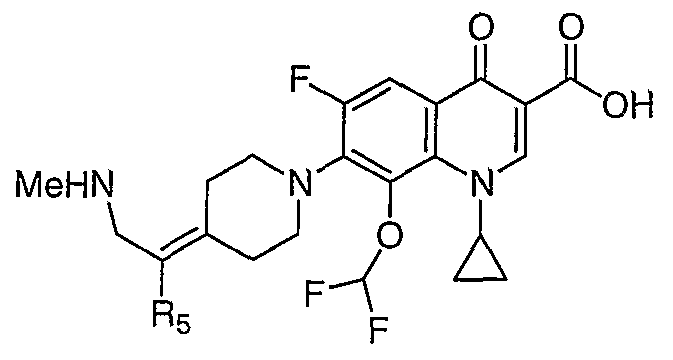
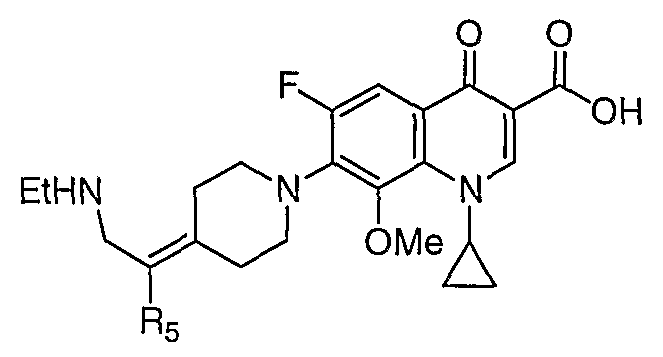
Таблица 1 содержит не исчерпывающий перечень предпочтительных соединений формулы (I).
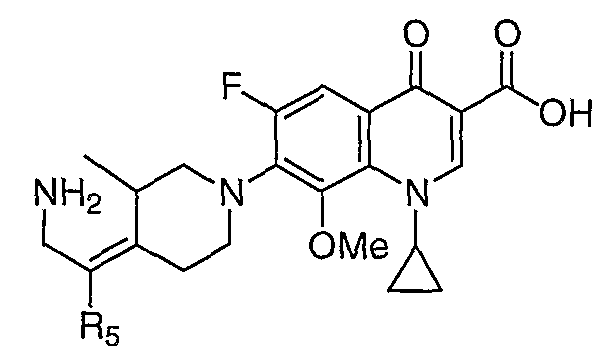
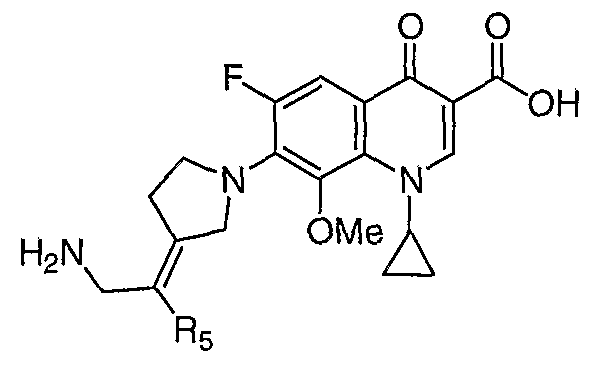
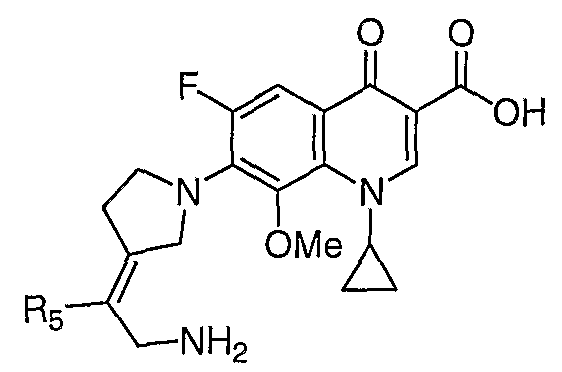
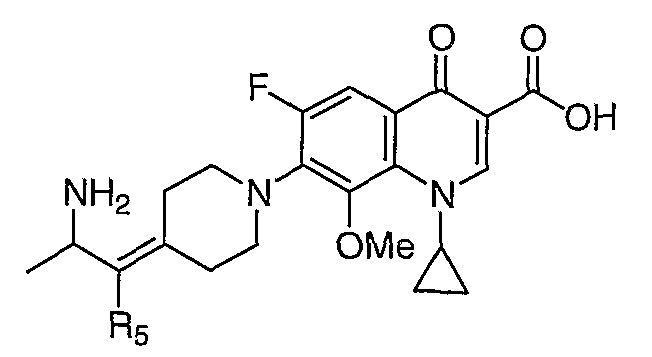
Общая реакционная схема получения соединения
При получении соединений согласно данному изобретению порядок стадий синтеза можно изменять для увеличения выхода желаемого продукта. Кроме того, квалифицированному специалисту также известен разумный выбор реакций, растворителей и температур, являющихся важной составляющей успешного синтеза. Хотя определение оптимальных условий и т.д. является общепринятой практикой, должно быть понятно, что множество соединений можно получать, руководствуясь нижеприводимыми схемами.
Исходные вещества, используемые при получении соединений согласно данному изобретению, являются известными, получаемыми опубликованными синтетическими способами или коммерчески доступными.
Известно, что квалифицированный специалист в области органической химии может без труда осуществлять стандартные манипуляции с органическими соединениями без дальнейшей инструкции; то есть, в компетенцию и практику квалифицированного специалиста входит осуществление подобных манипуляций. Они включают, но не ограничиваясь этим, восстановление карбонильных соединений в их соответствующие спирты, окисление, ацилирование, такие замещения в ароматическом ряду, как электрофильное, нуклеофильное, получение простых эфиров, этерификацию, омыление и тому подобное. Примеры таких манипуляций рассматриваются в стандартных руководствах, таких как March, Advanced Organic Chemistry (Wiley), Carey and Sundberg, Advanced Organic Chemistry (том 2), Feiser & Feiser, Reagents for Organic Synthesis (16 томов), L. Paquette, Encyclopedia of Reagents for Organic Synthesis (8 томов), Frost & Fleming, Comprehensive Organic Synthesis (9 томов) и тому подобное.
Квалифицированному специалисту понятно, что некоторые реакции наилучшим образом протекают, когда другие функциональные группы в молекуле маскированы или защищены, избегая таким образом нежелательных побочных реакций и/или для увеличения выхода реакции. Квалифицированный специалист часто применяет защитные группы для увеличения выходов или для избежания побочных реакций. Примеры таких манипуляций можно найти, например, в руководстве T. Greene, Protecting Groups in Organic Synthesis.
Общие методики получения гетероциклов, пригодные для получения соединений согласно данному изобретению, раскрыты в следующих ссылках, которые все включены в данный контекст в качестве ссылки (включая перечисленные статьи со ссылками): патент США 6329391, публикация Европейского патента 342849, публикация Международной заявки на патент WO 9711068, публикация Европейского патента 195316, публикация Европейского патента 1031569, патент США 6025370, публикация Европейского патента 153828, публикация Европейского патента 191451, публикация Европейского патента 153163, публикация Европейского патента 230053, публикация Европейского патента 976749, публикация Международной заявки на патент WO 0118005, публикация Международной заявки на патент WO 9407873, патент США 4777253, публикация Европейского патента 421668, публикация Международной заявки на патент WO 0248138, публикация Европейского патента 230295, публикация Международной заявки на патент WO 9914214, публикация патента США 2002/0049223, публикация Международной заявки на патент WO 9921849, публикация Международной заявки на патент WO 9729102, публикация Международной заявки на патент WO 0334980, публикация Международной заявки на патент WO 0209758, публикация Международной заявки на патент WO 9619472, публикация патента Германии DE 3142854, публикация Международной заявки на патент WO 0328665, публикация Европейского патента 47005, публикация Международной заявки на патент WO 0311450 и публикация Европейского патента 688772.
Соединения по данному изобретению можно получать разными путями. Разнообразные методологии получения соединений согласно данному изобретению показаны на схеме I, ниже, где L означает удаляемую группу, такую как фтор или хлор.
Схема I
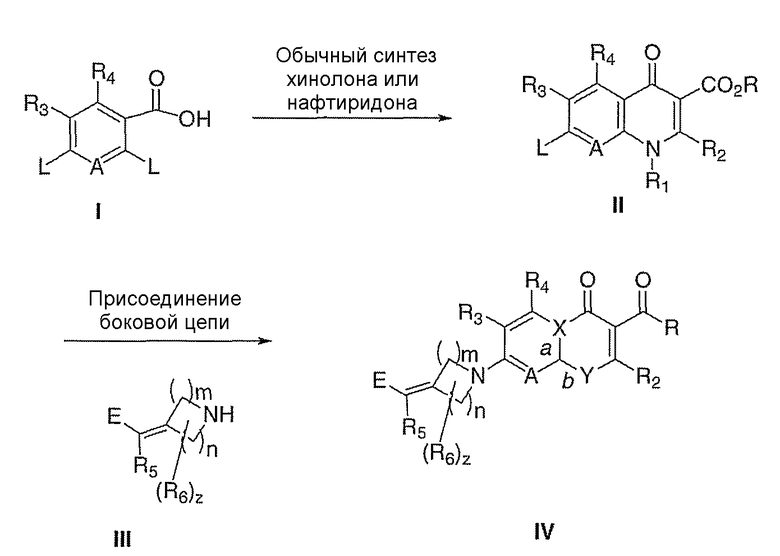
В случае, когда Е означает:
и, по меньшей мере, один из R9 и R10 означает водород, и может быть необходима защита концевого азота для осуществления селективного превращения в желаемый продукт (схема II). В таком случае, обычные аминозащитные группы, известные специалисту в данной области, такие как трет-бутилоксикарбонил (Вос), бензилоксикарбонил (Cbz), бензил (Bn), 9-флуоренилметоксикарбонил (Fmoc), аллилоксикарбонил (Alloc), 2-триметилсилилэтоксикарбонил (Теос), N-формил, N-ацетил, N-бензоил или фталимид, можно использовать для маскирования концевого амина, как в соединении V. После присоединения боковой цепи защитную группу можно удалять в стандартных условиях, известных специалисту в данной области, чтобы получить желаемый продукт VII. Продукт VII можно далее превращать, например, путем алкилирования, в другие соединения VIII согласно данному изобретению.
Схема II
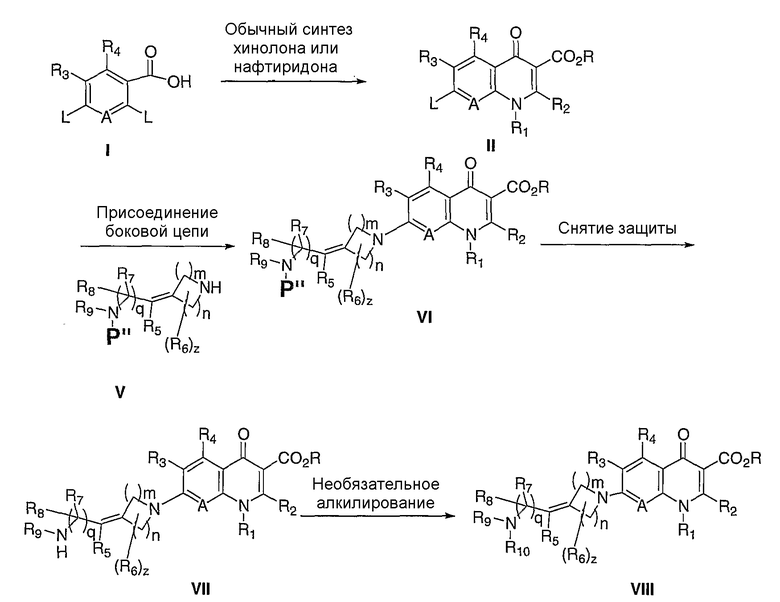
Р" = защитная группа
Методологии получения соединений согласно данному изобретению, где Х означает N и Y означает C(R1), показаны на схеме III, ниже.
Схема III
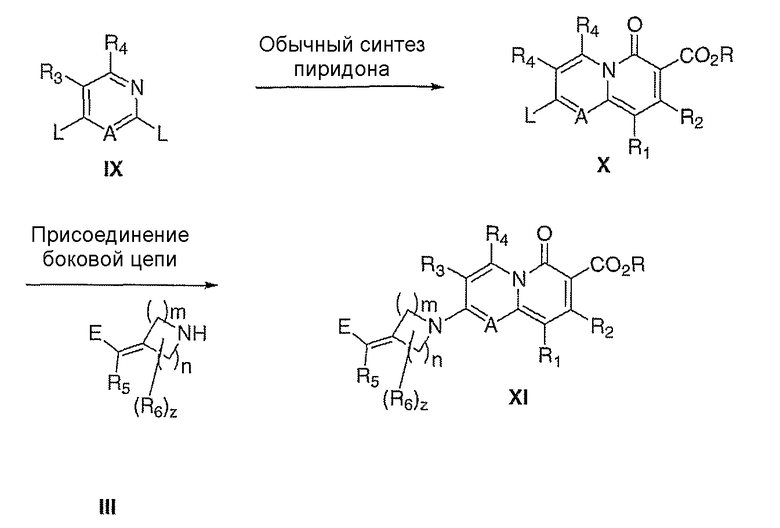
Как раньше, где Е означает:
и, по меньшей мере, один из R9 и R10 означает водород, может быть необходима защита концевого азота для осуществления селективного превращения в желаемый продукт (схема IV). В таком случае, обычные аминозащитные группы, известные специалисту в данной области, такие как трет-бутилоксикарбонил (Вос), бензилоксикарбонил (Cbz), бензил (Bn), 9-флуоренилметоксикарбонил (Fmoc), аллилоксикарбонил (Alloc), 2-триметилсилилэтоксикарбонил (Теос), N-формил, N-ацетил, N-бензоил или фталимид, можно использовать для маскировки концевого амина, как в соединении V. После присоединения боковой цепи защитную группу можно удалять в стандартных условиях, известных специалисту в данной области, чтобы получить желаемый продукт XIII. Продукт XIII можно далее превращать, например, путем алкилирования, в другие соединения XIV согласно данному изобретению.
Схема IV
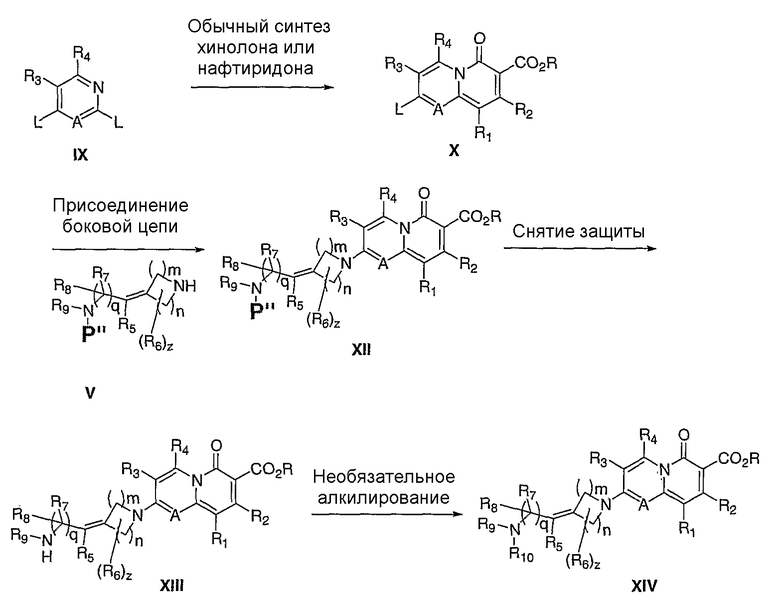
Р" = защитная группа
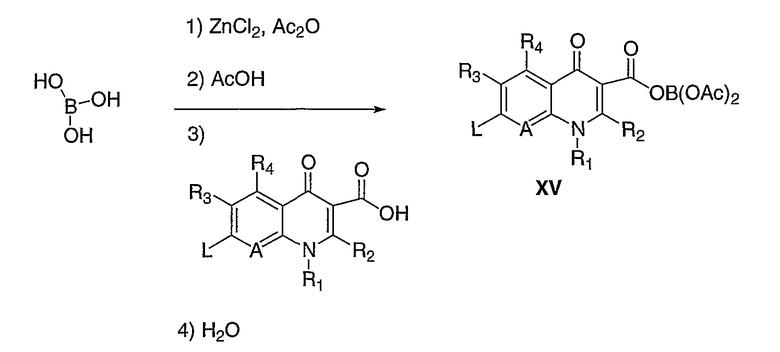
Иногда, амины с боковой цепью недостаточно реакционноспособны для эффективного присоединения к гетероциклу (II или X) в условиях, проиллюстрированных на схемах I-IV, особенно, когда А означает С(R11), где R11 означает алкокси. Гетероцикл можно активировать для нуклеофильной атаки добавлением кислоты Льюиса, такой как, но не ограничиваясь этим, трифторид бора, триацетоксиборат и литийхлорид. Предпочтительный способ активации раскрыт в патенте США 5157117. Хинолоны обрабатывают триацетоксиборатом, получаемым in situ, в таких растворителях, как уксусная кислота или пропионовая кислота, но не ограничиваясь этим, и нагревают в течение 1-24 часов при температуре от 60°С до 120°С. Диацилхинолинилборат (XV) выделяют фильтрацией, потом удаляют растворитель. Схема V иллюстрирует этот предпочтительный способ активации.
Схема V
Получение предшественника - амина с боковой цепью III
Схема VI иллюстрирует синтез амина с боковой цепью III, где Е означает:
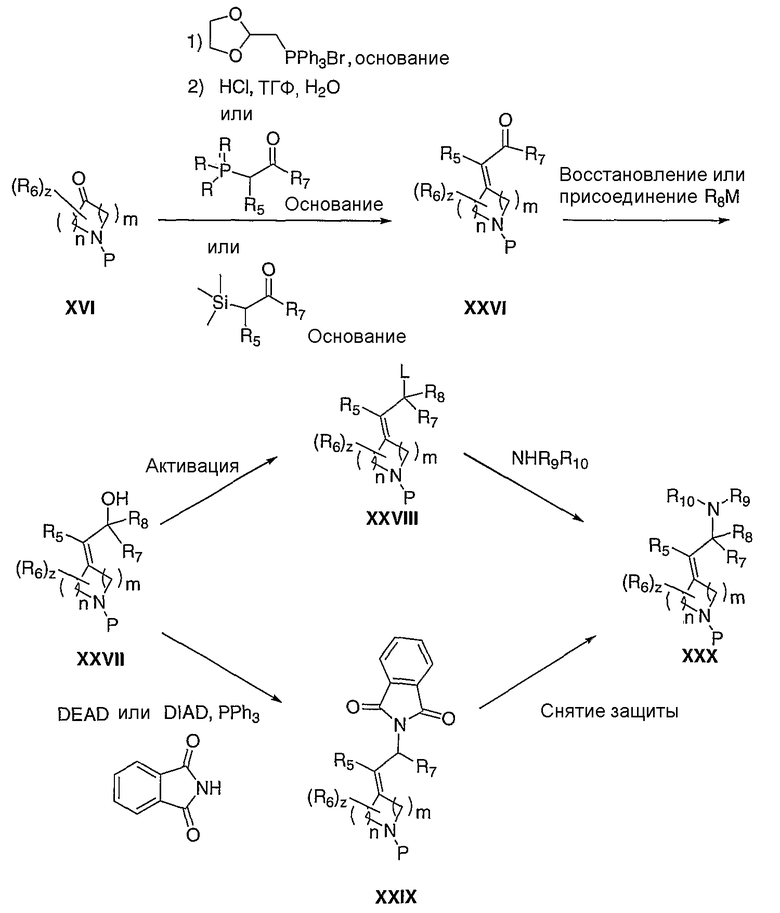
R7 и R8 означают водород, и q означает 1. Тризамещенные или тетразамещенные алкилидены ХХ можно получать олефинированием по Петерсону, Виттигу или Wadsworth-Horner-Emmons'у соответственно замещенного кетона (XVI) в таких растворителях, как, но не ограничиваясь этим, тетрагидрофуран, диметилсульфоксид или метиленхлорид, в течение 1-24 часов при температуре от -78°С до 120°С в присутствии такого основания, как, но не ограничиваясь этим, н-бутиллитий, гидрид натрия или карбонат калия. Полученный сложный эфир (XVII) можно восстанавливать с помощью таких восстановителей, как, но не ограничиваясь этим, диизобутилалюминийгидрид, литийтриэтилборгидрид или боргидрид натрия, в таких растворителях, как, но не ограничиваясь этим, толуол, метиленхлорид или тетрагидрофуран, в течение 1-24 часов при температуре от 0°С до 120°С, с получением соответствующего спирта XVIII, где q=1. Превращение спирта XVIII в соединение с удаляемой группой XIX, такой как, но не ограничиваясь этим, хлорид, бромид, мезилат или тозилат, в стандартных условиях и замещение удаляемой группы с помощью соответственно замещенного амина, в таких растворителях, как, но не ограничиваясь этим, диметилформамид, диметилсульфоксид или тетрагидрофуран, в течение 1-24 часов при температуре от 0°С до 120°С, приводит к превращению спирта XVIII в амин ХХ. Удаление защитной группы, Р, в стандартных условиях, известных специалисту в данной области, приводит к получению амина III, где Е означает:
где R7 и R8 означают водород, и q означает 1. Альтернативно, прямое замещение спирта XVIII можно проводить с помощью реакции Mitsunobu с фталимидом и диалкилазодикарбоксилатом, приводящей к получению XXI. Снятие защиты фталимида (XXI) с помощью гидразина в таких растворителях, как метанол или этанол, приводит к получению амина (ХХ), где R9 и R10 означают водород. Защитную группу, Р, можно удалять из XXI в стандартных условиях, известных специалисту в данной области, с получением амина V, где R7 и R8 означают водород, и R9 и Р" вместе с азотом, к которому они присоединены, образуют фталимидную группу.
Схема VI
где L означает удаляемую группу; Р означает защитную группу.
Схема XXII иллюстрирует превращение спиртов формулы XVIII в соединения формулы III, где Е означает алкенил (LVIII). Кроме того, на схеме показан синтез соединений формулы III, где Е означает:
где R7 и R8 означают водород и R9 означает ацил, алкоксикарбонил или сульфонил (LX). Окисление спирта XVIII любым из целого ряда таких подходящих окислителей, как периодинан Десс-Мартина, реактив Корей-Кима или реактив Свема, приводит к получению соответствующего альдегида (LVI). Альдегид можно подвергать промотируемой основанием реакции олефинирования, такой как, но не ограничиваясь этим, реакция Виттига, с получением LVII, где Rc означает водород или алкил. Удаление защитной группы, Р, из LVII в стандартных условиях, известных специалисту в данной области, приводит к получению амина III, где Е означает алкенил (LVIII). Схема ХХ также иллюстрирует превращение спиртов формулы XVIII в соединения формулы III, где Е означает:
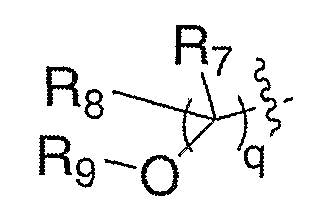
где R7 и R8 означают водород и R9 означает ацил, алкоксикарбонил или сульфонил (LX).
Взаимодействие спирта XVIII с ацилирующим агентом в присутствии амина в качестве такого основания, как пиридин, в таких инертных растворителях, как дихлорметан, тетрагидрофуран или толуол, при температуре от -20°С до 60°С в течение 1-48 часов, приводит к получению соединений формулы III, где Е означает:
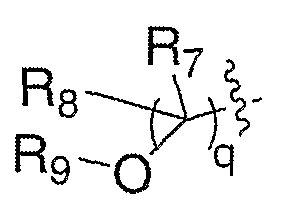
где R7 и R8 означают водород и R9 означает ацил (LIX). Ацилирующие агенты включают галогенангидриды кислот, ангидриды кислот и кислоты в присутствии таких активирующих агентов, как дициклогексилкарбодиимид, EDCI, BOP-CI, BOP, PyBOP и тому подобные. Спирты формулы XVIII можно превращать в соединения формулы III, где Е означает:
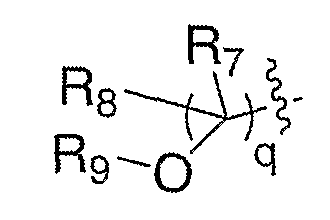
где R7 и R8 означают водород и R9 означает алкоксикарбонил (LIX), путем взаимодействия с карбонилирующим агентом в присутствии такого амина в качестве основания, как пиридин, в таких инертных растворителях, как дихлорметан, тетрагидрофуран или толуол, при температуре от -20°С до 60°С в течение 1-48 часов. Карбонилирующие агенты включают хлорформиаты, фторформиаты, азидоформиаты и пирокарбонаты. Спирты формулы XVIII можно превращать в соединения формулы III, где Е означает:
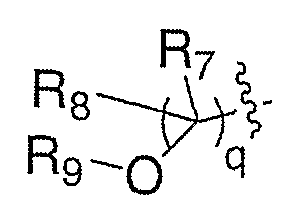
где R7 и R8 означают водород и R9 означает сульфонил (LIX), путем взаимодействия с сульфонилхлоридом или ангидридом сульфокислоты, в присутствии такого амина в качестве основания, как пиридин, в таких инертных растворителях, как дихлорметан, тетрагидрофуран или толуол, при температуре от -20°С до 60°С в течение 1-48 часов. Удаление защитной группы, Р, из LIX в стандартных условиях, известных специалисту в данной области, приводит к получению амина III, где Е означает:
где R7 и R8 означают водород и R9 означает ацил, алкоксикарбонил или сульфонил (LX).
Схема XXII
где Р означает защитную группу.
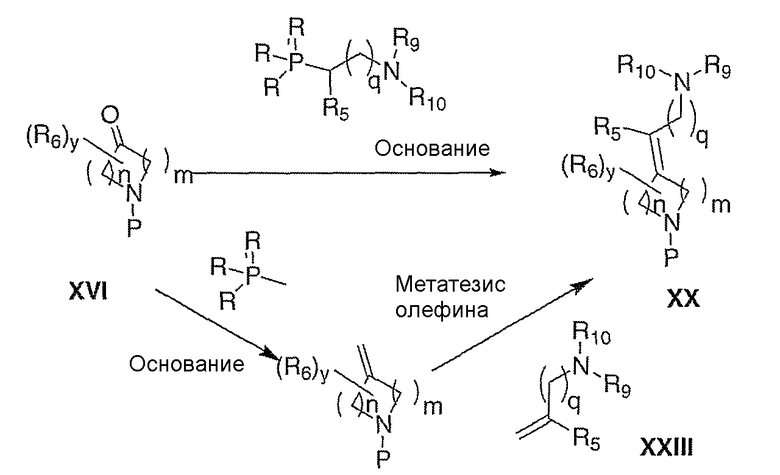
Схема VII иллюстрирует прямое превращение кетона XVI в олефин ХХ, используя промотируемые основанием такие реакции олефинирования, как, но не ограничиваясь этим, реакция Виттига, Wadsworth-Horner-Emmons'а или Петерсона. Альтернативно, амин ХХ можно получать с помощью метатезиса олефина из соединения с концевой олефиновой связью XXII, используя соответственно замещенный амин XXIII. Удаление защитной группы, Р, из ХХ в стандартных условиях, известных специалисту в данной области, приводит к получению амина III, где Е означает:
и R7 и R8 означают водород.
Схема VII
где Р означает защитную группу.
Схема VIII иллюстрирует гидроксилирование XXIV диоксидом селена, приводящее к получению аллилового спирта XXV. Превращение осуществляют в таких растворителях, как, но не ограничиваясь этим, метиленхлорид, толуол или тетрагидрофуран, при температуре от 25°С до 150°С, необязательно в присутствии такого соокислителя, как трет-бутилгидропероксид. Удаление защитной группы, Р, из XXV в стандартных условиях, известных специалисту в данной области, приводит к получению амина III, где Е означает:
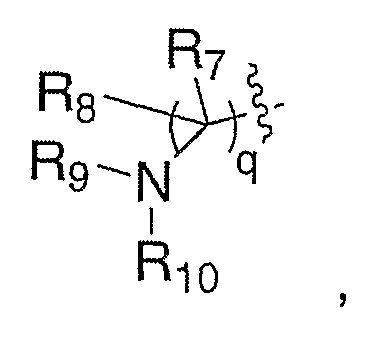
и один из R6 означает гидрокси.
Схема VIII
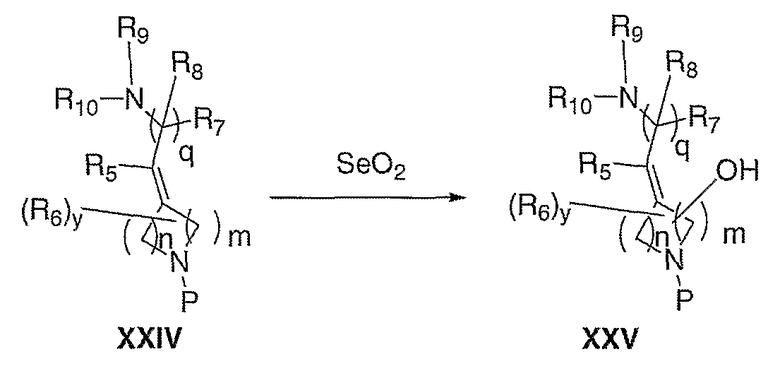
где Р означает защитную группу.
Схема IX иллюстрирует получение α,β-ненасыщенного карбонильного соединения XXVI, где R7 имеет значение, как ранее определено, используя методики олефинирования Петерсона, Виттига или Wadsworth-Horner-Emmons'а, из соответственно замещенного кетона (XVI) в таких растворителях, как, но не ограничиваясь этим, тетрагидрофуран, диметилсульфоксид или метиленхлорид, в течение 1-24 часов при температуре от -78°С до 120°С, в присутствии таких оснований, как, но не ограничиваясь этим, н-бутиллитий, гидрид натрия или карбонат калия. Полученное карбонильное соединение (XXVI) можно восстанавливать с помощью таких восстановителей, как, но не ограничиваясь этим, диизобутилалюминийгидрид, литийтриэтилборгидрид или боргидрид натрия, в таких растворителях, как, но не ограничиваясь этим, толуол, метиленхлорид или тетрагидрофуран, в течение 1-24 часов при температуре от 0°С до 120°С, что приводит к получению соответствующего спирта XXVII. Альтернативно, карбонильное соединение можно подвергать нуклеофильному присоединению соответственно таких замещенных металлоорганических агентов (R8M, где М означает металл), как литийорганическое соединение или реактив Гриньяра, что приводит к получению соответствующего спирта XXVII, где R8 означает алкил. Подходящие растворители для последнего превращения включают диэтиловый эфир, тетрагидрофуран или толуол, при температуре от -78°С до 20°С от 30 минут до 48 часов. В случае, где один из R7 или R8 означает водород, превращение спиртовой функциональной группы XXVII в такие удаляемые группы, как, но не ограничиваясь этим, бромид, мезилат или тозилат, как в XXVIII, в стандартных условиях и замещение удаляемой группы соответственно замещенным амином в таких растворителях, как, но не ограничиваясь этим, диметилформамид, диметилсульфоксид или тетрагидрофуран, в течение 1-24 часов при температуре от 0°С до 120°С, приводит к превращению спирта XXVII в амин ХХХ. Удаление защитной группы, Р, из ХХХ в стандартных условиях, известных специалисту в данной области, приводит к получению амина III, где Е означает:
и один из R7 и R8 означает водород. Альтернативно, в случае где один из R7 или R8 означает водород, прямое замещение спирта XXVII можно выполнять по реакции Mitsunobu с фталимидом и диалкилазодикарбоксилатом, с последующим снятием защиты фталимида с помощью гидразина, в таких растворителях, как метанол или этанол, что приводит к получению амина ХХХ. Защитную группу, Р, можно удалять из XXIX в стандартных условиях, известных специалисту в данной области, с получением амина V, где R8 означает водород и R9 и Р" вместе с атомом азота, к которому они присоединены, образуют фталимидную группу.
Схема IX
где L означает удаляемую группу,
Р означает защитную группу.
Схема Х изображает получение XXXVI, где R5 означает галоген. Алкилидены XXXI, где R5 означает водород, можно галогенировать такими соответствующими галогенирующими агентами, как, но не ограничиваясь этим, 1-бром-2,5-пирролидиндион, 1,1,1-трис(ацетилокси)-1,1-дигидро-2-бензиодоксол-3(1Н)-он и тетраалкиламмонийбромид или тионилхлорид, с получением XXXII. Алкилиден XXXII можно восстанавливать с помощью таких восстановителей, как, но не ограничиваясь этим, диизобутилалюминийгидрид, литийтриэтилборгидрид или боргидрид натрия, в таких растворителях, как, но не ограничиваясь этим, толуол, метиленхлорид или тетрагидрофуран, в течение 1-24 часов при температуре от 0°С до 120°С, что приводит к получению соответствующего спирта XXXIII. Альтернативно, карбонильное соединение можно подвергать нуклеофильному присоединению таких соответственно замещенных металлоорганических агентов, как литийорганическое соединение или реактив Гриньяра, что приводит к получению соответствующего спирта XXXIII, где R8 означает алкил. Подходящие растворители для последнего превращения включают диэтиловый эфир, тетрагидрофуран или толуол, при температуре от -78°С до 20°С от 30 минут до 48 часов. В случае, где один из R7 или R8 означает водород, превращение функциональной спиртовой группы XXXIII в такие удаляемые группы, как, но не ограничиваясь этим, бромид, мезилат или тозилат, как в XXXIV, в стандартных условиях и замещение удаляемых групп соответственно замещенным амином в таких растворителях, как, но не ограничиваясь этим, диметилформамид, диметилсульфоксид или тетрагидрофуран, в течение 1-24 часов при температуре от 0°С до 120°С, приводит к превращению XXXIV в амин XXXVI. Удаление защитной группы, Р, из XXXVI в стандартных условиях, известных специалисту в данной области, приводит к получению амина III, где Е означает:
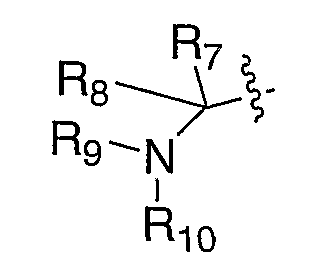
и один из R7 и R8 означает водород. Альтернативно, в случае, где один из R7 или R8 означает водород, прямое замещение спирта XXXIII можно выполнять по реакции Mitsunobu с фталимидом и диалкилазодикарбоксилатом, с последующим снятием защиты фталимида с помощью гидразина, в таких растворителях, как метанол или этанол, что приводит к получению амина XXXVI. Защитную группу, Р, можно удалять из XXXV в стандартных условиях, известных специалисту в данной области, с получением амина V, где R8 означает водород и R9 и Р" вместе с атомом азота, к которому они присоединены, образуют фталимидную группу.
Схема Х
Схема XI иллюстрирует синтез амина с боковой цепью III, где Е означает:
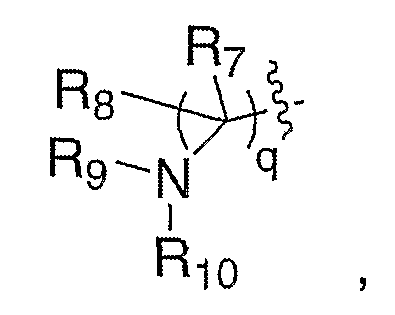
где R7 и R8 означают водород и R5 означает замещенный или с разветвленной цепью алкил. На схеме XI галогенированное карбонильное соединение XXXVII, где Rа означает водород или алкил, можно получать так же, как галогенированное карбонильное соединение XXXII. Карбонильное соединение XXXVII, где Rа означает водород или алкил, можно восстанавливать с помощью таких восстановителей, как, но не ограничиваясь этим, диизобутилалюминийгидрид, литийтриэтилборгидрид или боргидрид натрия в таких растворителях, как, но не ограничиваясь этим, толуол, метиленхлорид или тетрагидрофуран, в течение 1-24 часов при температуре от 0°С до 120°С, что приводит к получению соответствующего спирта XXXVIII, где Rа означает водород или алкил, один из Rb означает водород, а другой Rb означает гидроксил. Альтернативно, карбонильное соединение XXXVII, где Rа означает алкил, можно подвергать нуклеофильному присоединению соответственно таких замещенных металлоорганических агентов, как литийорганическое соединение или реактив Гриньяра, что приводит к получению соответствующего спирта XXXVIII, где Rа означает алкил, один из Rb означает алкил, а другой Rb означает гидроксил. Наконец, карбонильное соединение XXXVII, где Rа означает водород или алкил, или спирт XXXVIII, где Rа означает водород или алкил, один из Rb означает водород, а другой Rb означает гидроксил, можно фторировать, используя такие нуклеофильные фторирующие реагенты, как, но не ограничиваясь этим, (N-этилэтанаминато)сератрифторид (DAST) или бис(2-метоксиэтил)аминосератрифторид (Deoxofluor), в таком подходящем растворителе, как метиленхлорид, в течение 1-24 часов при температуре от 0°С до 60°С, что приводит к получению XXXVIII, где, в случае, когда карбонильное соединение XXXVII представляет собой субстрат, Rа означает водород или алкил и Rb означает фтор, и где, в случае, когда спирт XXXVIII представляет собой субстрат, Rа означает водород или алкил, один из Rb означает водород, а другой Rb означает фтор. Галогенированный алкилиден XXXVIII можно карбонилировать в присутствии таких катализаторов на основе переходных металлов, как, но не ограничиваясь этим, палладийацетат, дикарбонилбис(трифенилфосфин)никель или тетракис(трифенилфосфин)палладий, в атмосфере монооксида углерода, в присутствии другой добавки, такой как метанол, необязательно в качестве растворителя, или в таких растворителях, как, но не ограничиваясь этим, диметилсульфоксид или тетрагидрофуран, в течение 1-24 часов при температуре от 0°С до 120°С, что приводит к получению сложного эфира XXXIX. XXXIX можно восстанавливать с помощью таких восстановителей, как, но не ограничиваясь этим, диизобутилалюминийгидрид, литийтриэтилборгидрид или боргидрид натрия, в таких растворителях, как, но не ограничиваясь этим, толуол, метиленхлорид или тетрагидрофуран, в течение 1-24 часов при температуре от 0°С до 120°С, что приводит к получению соответствующего спирта XL, где q=1. Превращение спирта XL в соединение с такими удаляемыми группами, как, но не ограничиваясь этим, бромид, мезилат или тозилат, в стандартных условиях и замещение удаляемой группы соответственно замещенным амином в таких растворителях, как, но не ограничиваясь этим, диметилформамид, диметилсульфоксид или тетрагидрофуран, в течение 1-24 часов при температуре от 0°С до 120°С, приводит к превращению спирта XL в амин XLIII. Удаление защитной группы, Р, из XLIII в стандартных условиях, известных специалисту в данной области, приводит к получению амина III, где Е означает:

где R7 и R8 означают водород, и R5 означает CRaRaRb. Альтернативно, прямое замещение спирта XL можно осуществлять по реакции Mitsunobu с фталимидом и диалкилазодикарбоксилатом, с получением XLII. Снятие защиты фталимида (XLII) с помощью гидразина в таких растворителях, как метанол или этанол, приводит к получению амина XLIII. Защитную группу, Р, можно удалять из XLII в стандартных условиях, известных специалисту в данной области, с получением амина V, где R7 и R8 означают водород, R9 и Р" вместе с азотом, к которому они присоединены, образуют фталимидную группу и R5 означает CRaRaRb.
Схема XI
Схема XII иллюстрирует синтез амина с боковой цепью III, где Е означает:
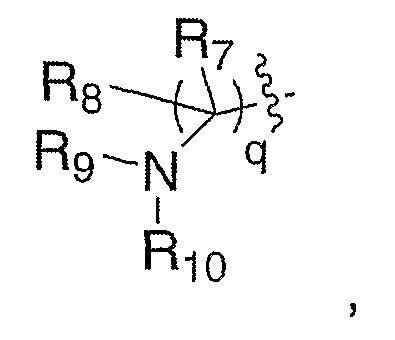
где один из R7 или R8 означает водород и другой означает алкил, R5 означает замещенный или с разветвленной цепью алкил и q означает 1. Соединение XXXVIII, полученное, как описано выше, можно карбонилировать в присутствии катализаторов на основе таких переходных металлов, как, но не ограничиваясь этим, палладийацетат, дикарбонилбис(трифенилфосфин)никель или тетракис(трифенилфосфин)палладий, в атмосфере монооксида углерода, в присутствии металлоорганических реагентов R7M, где R7 имеет значение, как ранее описано, и включает такие реагенты, как трибутилоловогидрид или алкилиндий (Organic Letters, 2003, 5(7), 1103-1106), в таких растворителях, как, но не ограничиваясь этим, метанол, диметилсульфоксид или тетрагидрофуран, в течение 1-24 часов при температуре от 0°С до 120°С, что приводит к получению XLIV, где R7 имеет значение, как ранее описано. Карбонильное соединение XLIV можно восстанавливать с помощью таких восстановителей, как, но не ограничиваясь этим, диизобутилалюминийгидрид, литийтриэтилборгидрид или боргидрид натрия в таких растворителях, как, но не ограничиваясь этим, толуол, метиленхлорид или тетрагидрофуран, в течение 1-24 часов при температуре от 0°С до 120°С, что приводит к получению соответствующего спирта XLV. Альтернативно, карбонильное соединение можно подвергать нуклеофильному присоединению соответственно таких замещенных металлоорганических реагентов, как литийорганическое соединение или реактив Гриньяра, что приводит к получению соответствующего спирта XLV, где R8 означает алкил. Подходящие растворители для последнего превращения включают диэтиловый эфир, тетрагидрофуран или толуол, при температуре от -78°С до 20°С от 30 минут до 48 часов. В случае, где один из R7 или R8 означает водород, превращение функциональной спиртовой группы XLV в такие удаляемые группы, как, но не ограничиваясь этим, бромид, мезилат или тозилат, как в XLVI, в стандартных условиях и замещение удаляемой группы соответственно замещенным амином в таких растворителях, как, но не ограничиваясь этим, диметилформамид, диметилсульфоксид или тетрагидрофуран, в течение 1-24 часов при температуре от 0°С до 120°С, приводит к превращению спирта XLV в амин XLVIII. Удаление защитной группы, Р, из XLVIII в стандартных условиях, известных специалисту в данной области, приводит к получению амина III, где Е означает:
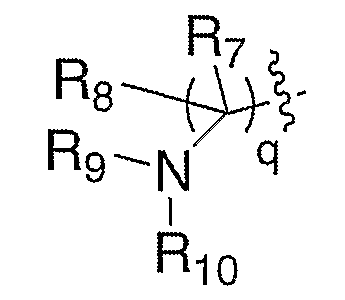
где один из R7 и R8 означает водород и другой означает алкил, R5 означает замещенный или с разветвленной цепью алкил и q означает 1. Альтернативно, в случае, где один из R7 или R8 означает водород, прямое замещение спирта XLV можно проводить по реакции Mitsunobu с фталимидом и диалкилазодикарбоксилатом, с последующим снятием защиты фталимида с помощью гидразина, в таких растворителях, как метанол или этанол, что приводит к получению XLVIII. Защитную группу, Р, можно удалять из XLVIII в стандартных условиях, известных специалисту в данной области, с получением амина V, где один из R7 и R8 означает водород и другой означает алкил, R9 и Р" вместе с азотом, к которому они присоединены, образуют фталимидную группу, R5 означает замещенный или с разветвленной цепью алкил и q означает 1.
Схема XII
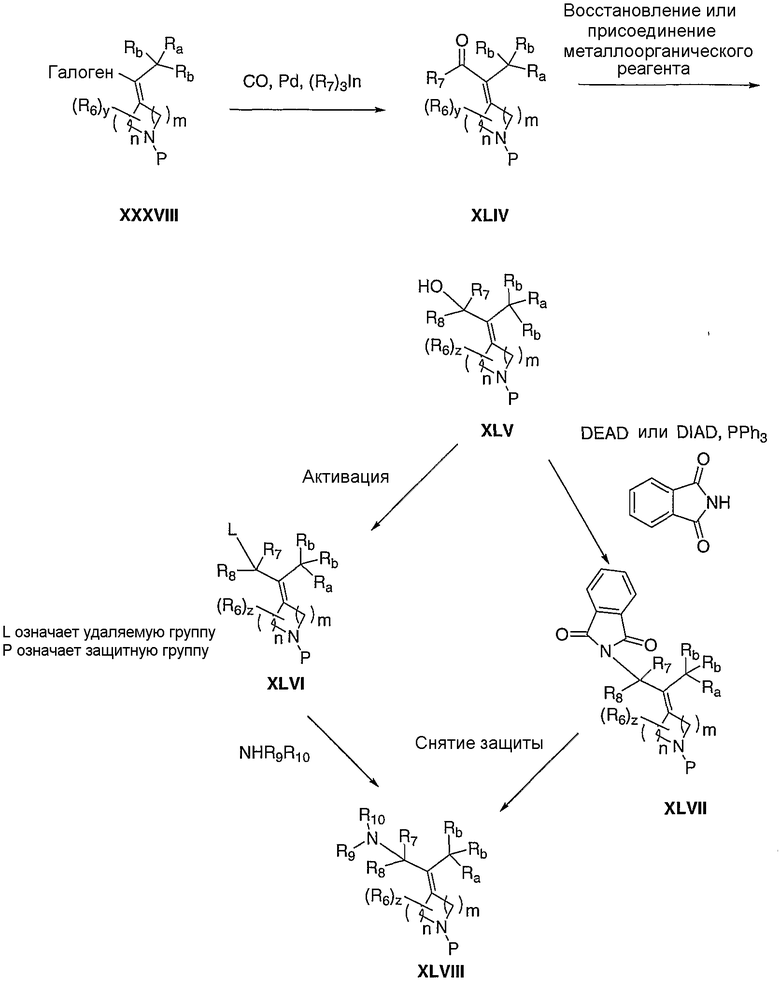
Схема XIII иллюстрирует превращение кетона XVIa в олефин LIII, используя протокол промотируемого основанием аннелирования винилсилана Stork-Jung по Робинсону (Tetrahedron Letters, 2001, 42, 9123). Конденсация кетона XVIa с аллилиодидом XLIX, где Rc означает алкильную группу и P' означает гидроксизащитную группу (Tetrahedron Letters, 2001, 42, 9123), приводит к получению алкилированного кетона L. Эпоксидирование кетона L такими эпоксидирующими агентами, как, но не ограничиваясь этим, диметилдиоксиран или м-хлорпербензойная кислота, что приводит к получению оксирана LI. Протодесилилирование LI такими агентами, как, но не ограничиваясь этим, тетра-н-бутиламмонийфторид или пиридинийполи(фтороводород) и водная кислота, с сопровождающим раскрытием эпоксидного кольца, приводит к получению кетона LII. Аннелирование LII можно осуществлять обработкой LII таким основанием, как, но не ограничиваясь этим, метилат натрия, что приводит к получению LIII. α,β-Ненасыщенный кетон LIII можно восстанавливать с помощью таких восстановителей, как, но не ограничиваясь этим, диизобутилалюминийгидрид, литийтриэтилборгидрид или боргидрид натрия, в таких растворителях, как, но не ограничиваясь этим, толуол, метиленхлорид или тетрагидрофуран, в течение 1-24 часов при температуре от 0°С до 120°С, с последующим удалением гидроксизащитной группы, что приводит к получению соответствующего спирта LIV, где один из R12 означает водород, а другой R12 означает гидрокси. Альтернативно, LIII можно подвергать нуклеофильному присоединению соответственно таких замещенных металлоорганических реагентов, как литийорганическое соединение или реактив Гриньяра, с последующим удалением гидроксизащитной группы, что приводит к получению соответствующего спирта LIV, где один из R12 означает алкил, а другой R12 означает гидрокси. Подходящие растворители для последнего превращения включают диэтиловый эфир, тетрагидрофуран или толуол, при температуре от -78°С до 20°С от 30 минут до 48 часов. Наконец, карбонильное соединение LIII можно фторировать, используя такие нуклеофильные фторирующие реагенты, как, но не ограничиваясь этим, (N-этилэтанаминато)сератрифторид (DAST) или бис(2-метоксиэтил)аминосератрифторид (Deoxofluor), в таких подходящих растворителях, как метиленхлорид, в течение 1-24 часов при температуре от 0°С до 60°С, с последующим удалением гидроксизащитной группы, что приводит к получению спирта LIV, где R12 означает фтор.
Схема XIII
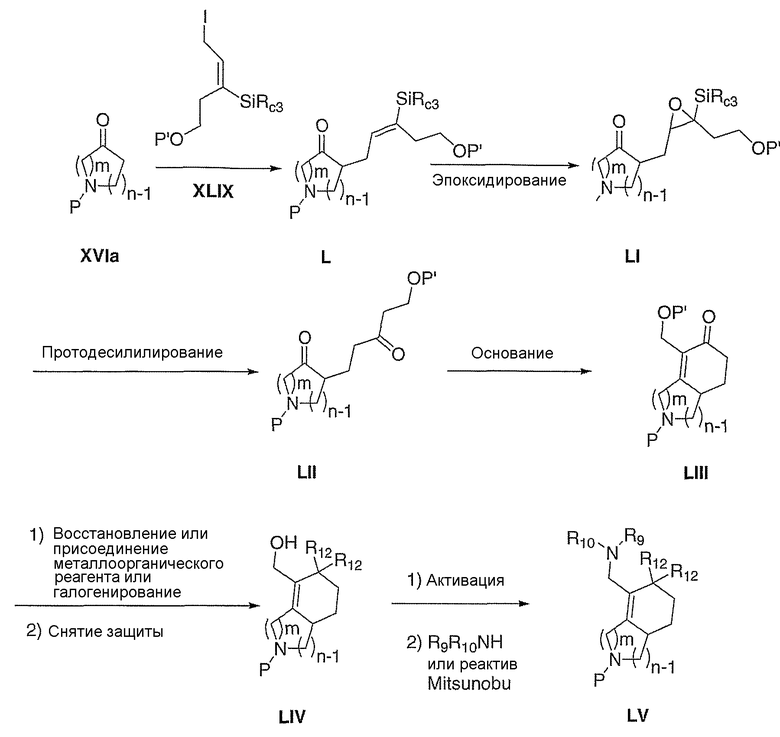
где Р означает защитную группу.
Спирт LIV можно превращать в соединение с такими удаляемыми группами, как, но не ограничиваясь этим, бромид, мезилат или тозилат, в стандартных условиях. Замещение удаляемой группы соответственно замещенным амином в таких растворителях, как, но не ограничиваясь этим, диметилформамид, диметилсульфоксид или тетрагидрофуран, в течение 1-24 часов при температуре от 0°С до 120°С, приводит к превращению LIV в амин LV. Удаление защитной группы, Р, из LV в стандартных условиях, известных специалисту в данной области, приводит к получению соответствующего вторичного амина III, где Е означает:
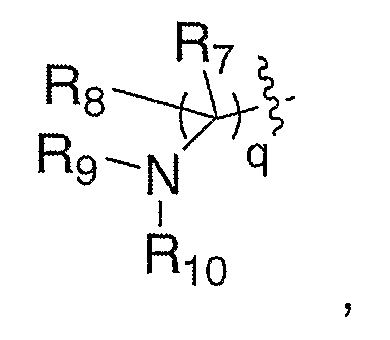
где R7 и R8 означают водород, и R5 и R6 связаны с образованием 6-членного карбоцикла, и q означает 1.
Альтернативно, прямое замещение гидроксильной группы в спирте LIV можно проводить по реакции Mitsunobu с фталимидом и диалкилазодикарбоксилатом, с последующим снятием защиты фталимида с помощью гидразина в таких растворителях, как метанол или этанол, что приводит к получению амина LV, где R9 и R10 означают водород.
Экспериментальный раздел
Получение предшественников - гетероциклов
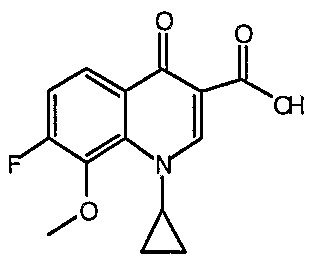
Все гетероциклы, такие как 1-циклопропил-1,4-дигидро-6,7-дифтор-8-метокси-4-оксохинолин-3-карбоновая кислота, 7-хлор-1-циклопропил-6-фтор-4-оксо-1,4-дигидронафтиридин-3-карбоновая кислота, 9,10-дифтор-2,3-дигидро-3-метил-7-оксо-7Н-пиридо[1,2,3-de]-1,4-бензоксазин-6-карбоновая кислота, 1-циклопропил-1,4-дигидро-6,7-дифтор-4-оксохинолин-3-карбоновая кислота, 7-хлор-1-(2,4-дифторфенил)-6-фтор-4-оксо-1,4-дигидронафтиридин-3-карбоновая кислота и 1-циклопропил-1,4-дигидро-7-фтор-8-метокси-4-оксохинолин-3-карбоновая кислота получают по методикам, описанным в литературе (см. приведенное выше обсуждение общих методик для получения гетероциклов) или получают из коммерческих источников.
Получение предшественника А - получение диацилхинолинилборатов
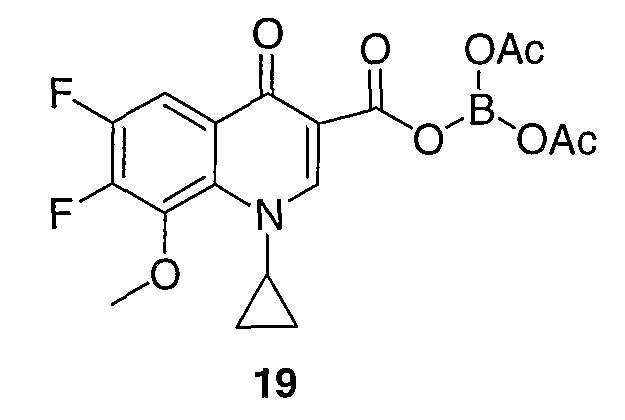
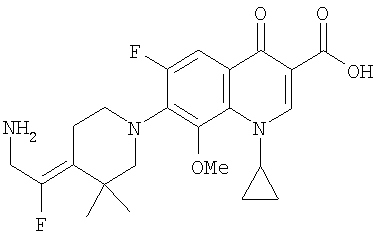
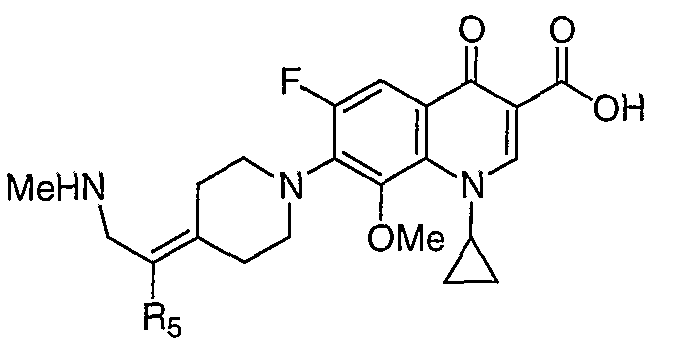
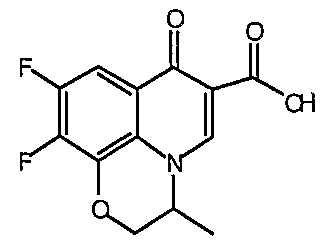
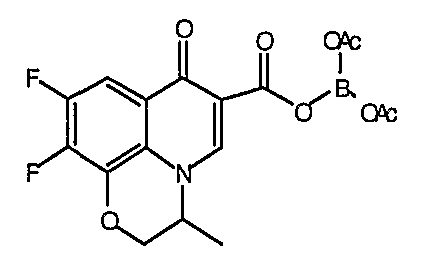
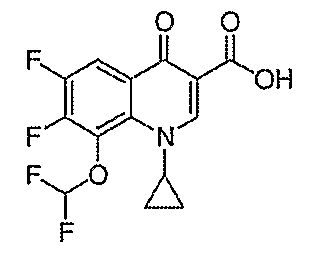
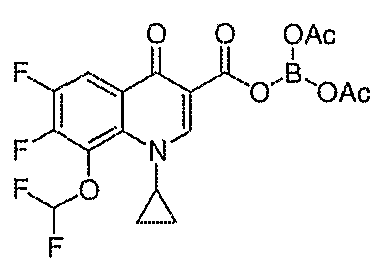
Соединение 19 (формула XV: L = F, A = C-OMe, R1 = циклопропил, R2 = H, R3 = F, R4 = H)
Диацилхинолинилбораты получают по методике, описанной в патенте США 5157117. Смесь борной кислоты (2,4 г, 38,7 ммоль), уксусного ангидрида (13,8 мл, 146 ммоль) и хлорида цинка (52 мг, 0,38 ммоль) выдерживают при 110°С в течение 1,5 часов, обрабатывают уксусной кислотой (51 мл) и перемешивают в течение часа при 110°С. Полученную смесь оставляют охлаждаться до 60°С, обрабатывают 1-циклопропил-1,4-дигидро-6,7-дифтор-8-метокси-4-оксохинолин-3-карбоновой (18) (7,3 г, 25,9 ммоль) и уксусной кислотами (26 мл). Полученный раствор выдерживают при 60°С в течение 5 часов, охлаждают до комнатной температуры и концентрируют в вакууме. Остаток обрабатывают водой (50 мл) и твердое вещество собирают фильтрацией. Полученное твердое вещество промывают водой (3×50 мл) и сушат с получением указанного в заголовке соединения в виде твердого вещества белого цвета, которое используют, как таковое, в ближайшей реакции.
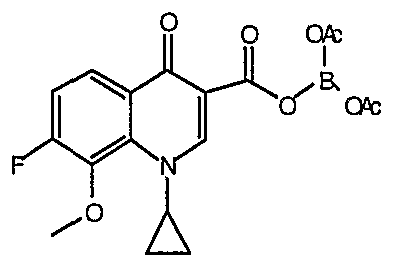
Такую же методику, как указано выше, применяют для превращения каждой из соответствующих гетероциклических карбоновых кислот, перечисленных в таблице 2, в соответствующее диацилборатное производное (соединения 17, 21, 23 и 83).
Получение предшественника В - боковой цепи III
Схема XIV
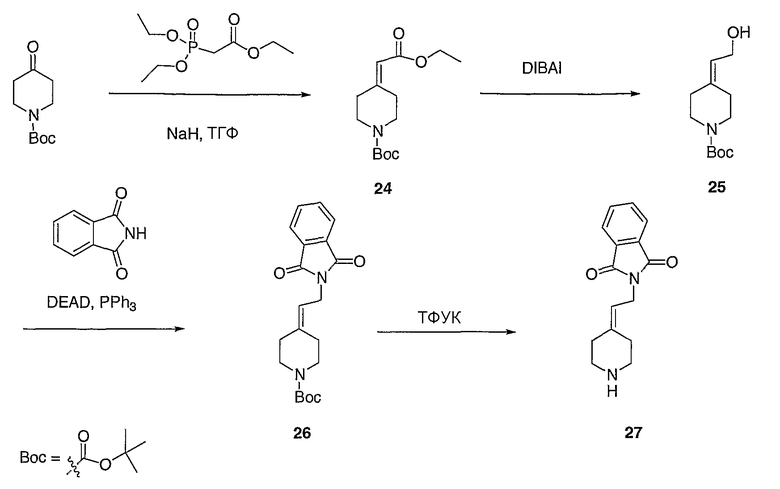
Соединение 27 схемы XIV:
трет-Бутил-4-(2-этокси-2-оксоэтилиден)пиперидинил-1-карбоксилат (24) получают по методике, описанной Sato и др., Heterocycles, 2001, 54, 747.
трет-Бутил-4-(2-гидроксиэтилиден)пиперидинил-1-карбоксилат (25) получают по методике, описанной Sato и др., Heterocycles, 2001, 54, 747.
трет-Бутил-4-[2-(1,3-дигидро-1,3-диоксо-2Н-изоиндол-2-ил)этилиден]пиперидинил-1-карбоксилат (26) получают по адаптированной методике из Synthesis, 1995, 756. Раствор соединения 25 (250 мг, 1,10 ммоль), фталимида (208 мг, 1,40 ммоль) и трифенилфосфина (366 мг, 1,40 ммоль) в безводном ТГФ (10 мл) обрабатывают диэтилазодикарбоксилатом (0,25 мл, 1,40 ммоль), добавляя его с помощью шприца в отсутствие света в атмосфере азота. Спустя 5 часов реакционную смесь обрабатывают водой (10 мл), разбавляют этилацетатом (50 мл), промывают 10%-ным водным раствором бикарбоната натрия (2×25 мл) и сушат (MgSO4). Очистку проводят флэш-хроматографией (0-30% этилацетат/гексаны) с получением указанного в заголовке соединения (389 мг, выход 78%) в виде пенообразного вещества белого цвета. MC=357 (М+Н).
4-[2-(1,3-Дигидро-1,3-диоксо-2Н-изоиндол-2-ил)этилиден]-1-пиперидинтрифторацетат (27). Раствор соединения 26 (380 мг, 1,03 ммоль) растворяют в CH2Cl2 (50 мл) и обрабатывают трифторуксусной кислотой (1 мл) при комнатной температуре. Спустя 1 час реакционную смесь концентрируют в вакууме с получением указанного в заголовке соединения 27 (363 мг, выход 100%) в виде масла. MC=257 (М+Н).
1-(трет-Бутоксикарбонил)-4-пиперидинон приводят во взаимодействие с каждым из соответствующих фосфоноацетатов, перечисленных в таблице 3, и продукты подвергают аналогичным операциям, как описано в случае синтеза соединения 27, с получением соответствующих спиртов (соединения 28-30, 84) и происходящих от них аминов (соединения 31-33, 85).
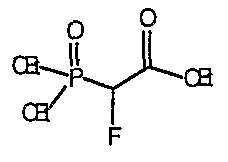
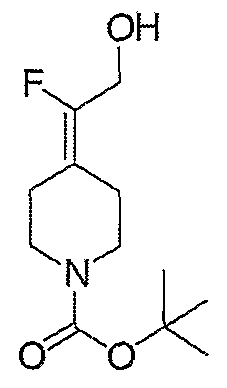
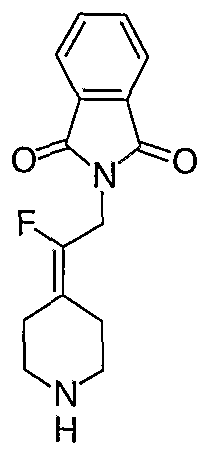
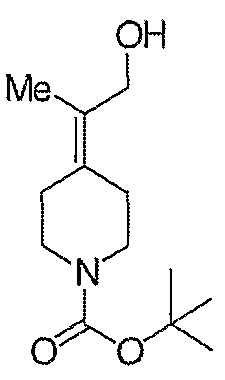
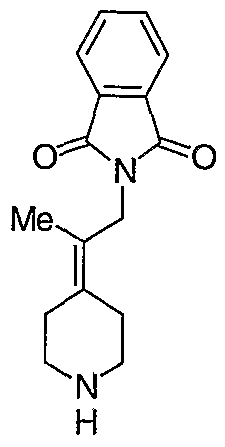
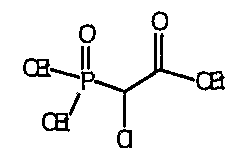
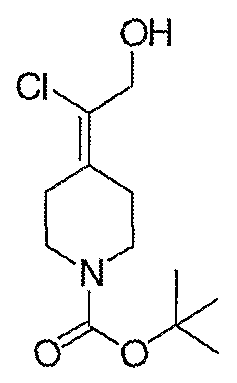
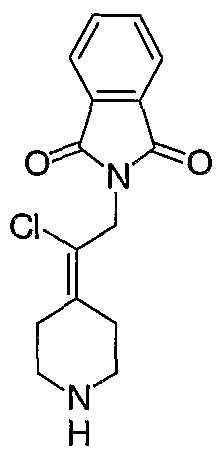
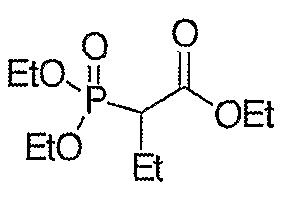
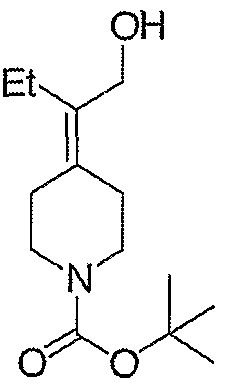
Схема XXIII
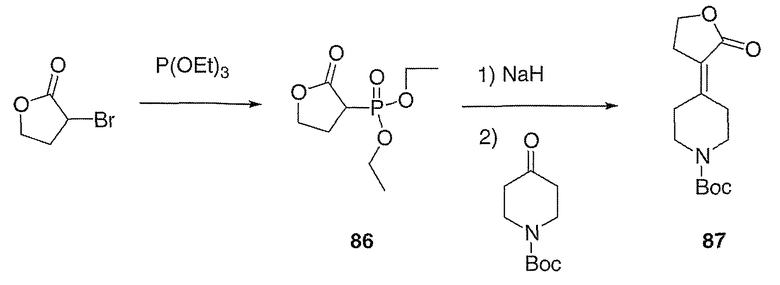
Диэтиловый эфир (2-оксотетрагидрофуран-3-ил)фосфоновой кислоты (86; схема XXIII) получают по методике, описанной Murphy и др., Chemical Communications, 1996, 6, 737-8.
трет-Бутиловый эфир 4-(2-оксодигидрофуран-3-илиден)пиперидин-1-карбоновой кислоты (87; схемаXXIII) получают по методике, аналогичной таковой, описанной Sato и др., Heterocycles, 2001, 54, 747; МС=267 (М+Н).
Схема XXIV
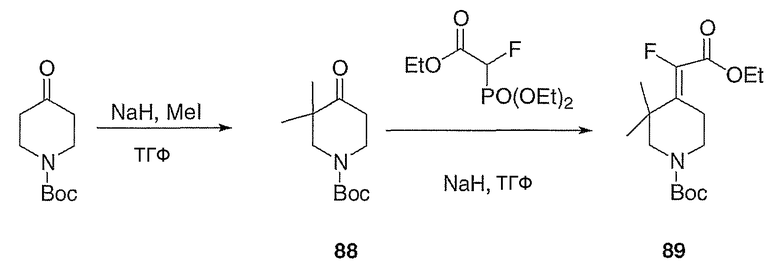
трет-Бутиловый эфир 3,3-диметил-4-оксопиперидин-1-карбоновой кислоты (88; схема XXIV) получают по методике, описанной Vice и др., J. Org. Chem., 2001, 66, 2487-2492.
трет-Бутиловый эфир 4-(2-этокси-1-фтор-2-оксоэтилиден)-3,3-диметилпиперидин-1-карбоновой кислоты (89, схема XXIV) получают по методике, аналогичной таковой, описанной Sato и др., Heterocycles, 2001, 54, 747.
Схема XXV
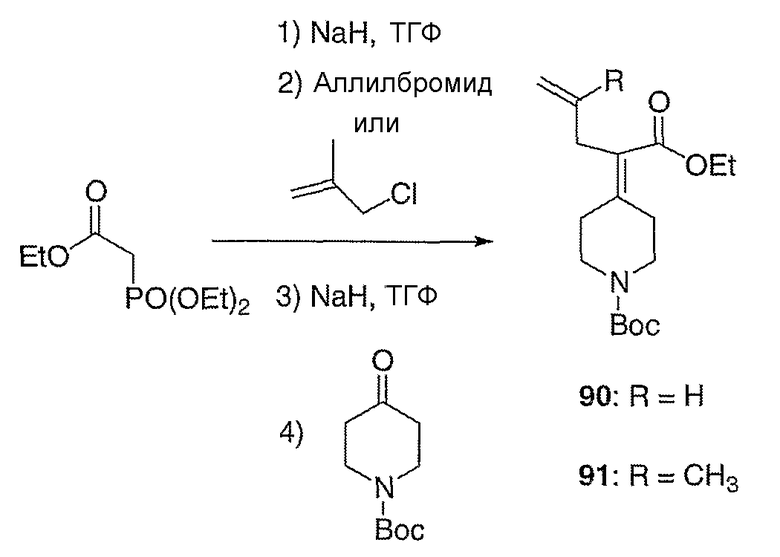
трет-Бутиловый эфир 4-(1-этоксикарбонилбут-3-енилиден)-пиперидин-1-карбоновой кислоты (90, схема XXV). Суспензию гидрида натрия (1,50 г, 37,6 ммоль) в ТГФ (100 мл) при 0°С в атмосфере азота осторожно обрабатывают триэтилфосфоноацетатом (8,12 мл, 37,6 ммоль), вводя его с помощью шприца. Спустя 30 мин реакционную смесь обрабатывают аллилбромидом (3,3 мл, 37,6 ммоль) и оставляют нагреваться до 25°С в течение 12 часов. Полученную смесь вторично охлаждают до 0°С, обрабатывают гидридом натрия (1,50 г, 37,6 ммоль) и полученную суспензию перемешивают в течение 30 мин при 0°С. Раствор 1-(трет-бутоксикарбонил)-4-пиперидинона (5,0 г, 25 ммоль) в ТГФ (50 мл) добавляют с помощью канюли в течение 10 мин и полученный раствор оставляют нагреваться до 25°С в течение 12 часов. Реакцию гасят добавлением 15%-ного водного раствора бикарбоната натрия (50 мл) и полученную смесь разбавляют этилацетатом (100 мл), промывают 15%-ным водным раствором бикарбоната натрия (2×100 мл) и концентрируют в вакууме. Очистку проводят с помощью хроматографии (0-50% EtOAc/гексаны) с получением указанного в заголовке соединения (1,93 г, выход 25%) в виде масла желтого цвета; MC=310 (М+Н).
трет-Бутиловый эфир 4-(1-этоксикарбонил-3-метилбут-3-енилиден)пиперидин-1-карбоновой кислоты (91, схема XXV) получают по методике, описанной для соединения 90, за исключением того, что метилаллилхлорид используют вместо аллилбромида.
Схема XXVI
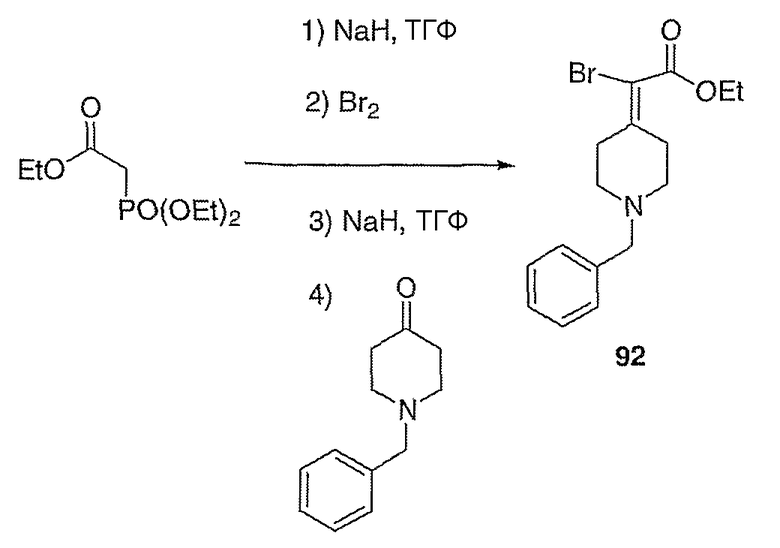
Этиловый эфир (1-бензилпиперидин-4-илиден)бромуксусной кислоты (92; схема XXVI). Суспензию гидрида натрия (1,50 г, 37,6 ммоль) в ТГФ (100 мл) при 0°С в атмосфере азота осторожно обрабатывают триэтилфосфоноацетатом (8,12 мл, 37,6 ммоль), вводя его с помощью шприца. Спустя 30 мин реакционную смесь обрабатывают бромом (1,95 мл, 37,6 ммоль) с помощью капельной воронки в течение 10 минут и полученную смесь перемешивают в течение 3 часов. Реакционную смесь обрабатывают гидридом натрия (1,50 г, 37,6 ммоль) и полученную суспензию перемешивают в течение 30 мин при 0°С. Раствор 1-бензилпиперидин-4-она (5,0 г, 25 ммоль) в ТГФ (50 мл) добавляют с помощью канюли в течение 10 мин и полученный раствор оставляют нагреваться до 25°С в течение 12 часов. Реакцию гасят добавлением 15%-ного водного раствора бикарбоната натрия (50 мл) и полученную смесь разбавляют этилацетатом (100 мл), промывают 15%-ным водным раствором бикарбоната натрия (2×100 мл) и концентрируют в вакууме. Очистку проводят с помощью хроматографии (0-50% EtOAc/гексаны) с получением указанного в заголовке соединения (6,35 г, выход 74%) в виде масла красно-оранжевого цвета; MC=339 (М+Н).
Спирты, перечисленные в таблице 6, получают подобным образом, как описано для трет-бутил-4-(2-гидроксиэтилиден)пиперидинил-1-карбоксилата (25), за исключением того, что соответствующий этилиденкарбоксилат используют вместо трет-бутил-4-(2-этокси-2-оксоэтилиден)пиперидинил-1-карбоксилата (24).
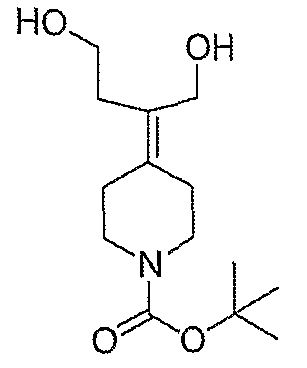
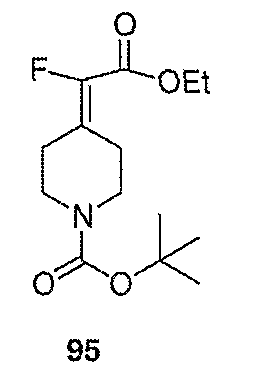
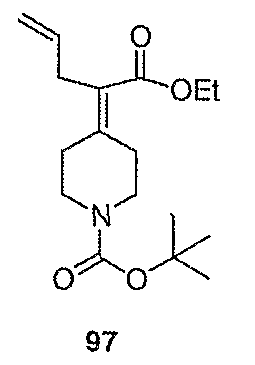
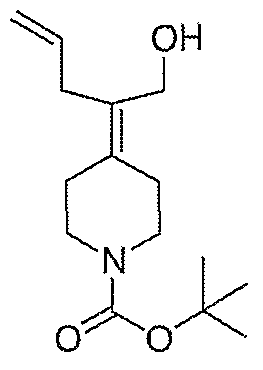
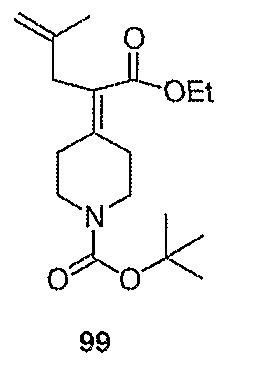
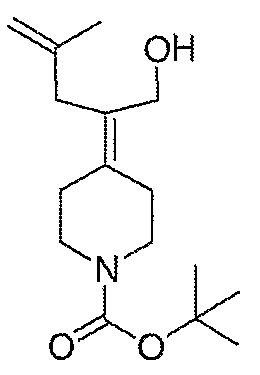
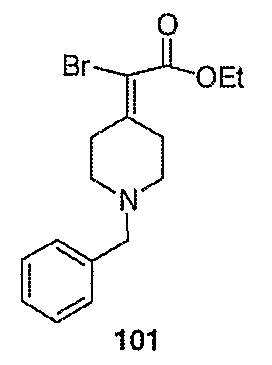
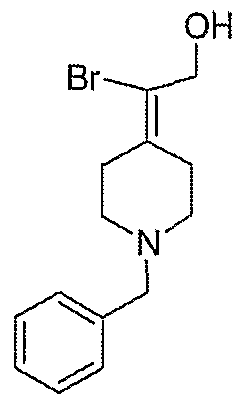
Схема XXVII
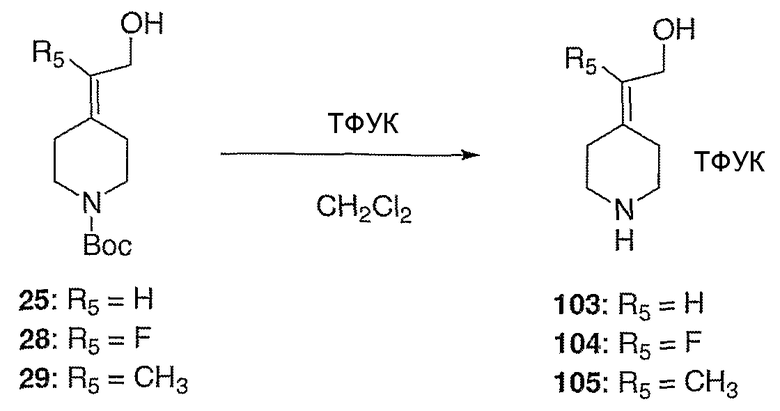
2-Пиперидин-4-илиденэтанолтрифторацетат (103; схема XXVII). Раствор соединения 25 (191 мг, 0,5 ммоль) растворяют в СН2Cl2 (10 мл) и обрабатывают трифторуксусной кислотой (0,5 мл) при комнатной температуре. Спустя 1 час реакционную смесь концентрируют в вакууме с получением указанного в заголовке соединения (64 мг, выход 100%) в виде масла. MC=129 (М+Н).
2-Пиперидин-4-илиденпропан-1-ол-трифторацетат (105; схема XXVII) получают по методике, описанной для соединения 103, за исключением того, что используют соединение 29. MC=142 (М+Н).
2-Фтор-2-пиперидин-4-илиденэтанолтрифторацетат (104; схема XXVII) получают по методике, описанной для соединения 103, за исключением того, что используют соединение 28. MC=146 (М+Н).
Схема XXVIII
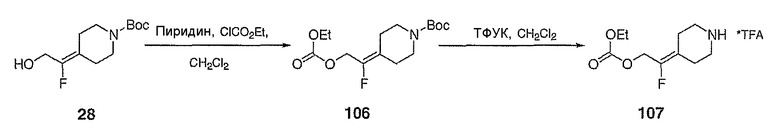
трет-Бутил-4-(2-этоксикарбонилокси-1-фторэтилиден)пиперидин-1-карбоксилат (106; схема XXVIII). К спирту 28 (0,5064 г, 2,064 ммоль) в СН2Cl2 (10 мл) при комнатной температуре добавляют пиридин (0,23 мл, 2,8 ммоль) и затем этилхлорформиат (0,22 мл, 2,2 ммоль). После перемешивания в течение ночи добавляют насыщенный водный раствор NH4Cl (10 мл) и смесь экстрагируют с помощью СН2Cl2 (5×10 мл), сушат над Na2SO4, концентрируют и хроматографируют на диоксиде кремния (20% EtOAc/гексан в качестве элюента) с получением указанного в заголовке соединения 106 (0,4546 г, выход 69%) в виде прозрачного масла. MC=318 (М+Н).
4-(2-Этоксикарбонилокси-1-фторэтилиден)пиперидин (107; схема XXVIII). К соединению 106 (0,1787 г, 0,5631 ммоль) в СН2Cl2 (3 мл) добавляют трифторуксусную кислоту (0,56 мл, 7,3 ммоль) и смесь перемешивают в течение 3 часов, после чего все летучие вещества удаляют в вакууме с получением сырого указанного в заголовке соединения, которое используют без дальнейшей очистки. MC=218 (М+Н).
Схема XXIX
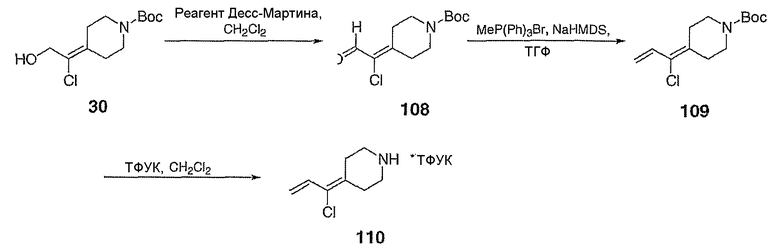
трет-Бутил-4-(1-хлор-2-оксоэтилиден)пиперидин-1-карбоксилат (108; схема XXIX). К спирту 30 (6,01 г, 23,0 ммоль) в СН2Cl2 при комнатной температуре и на открытом воздухе добавляют реагент Десс-Мартина (21,17 г, 49,9 ммоль) и реакционную смесь перемешивают в течение ночи, после чего смесь промывают насыщенным водным раствором Na2S2O3 (60 мл) и насыщенным водным раствором NaHCO3 (3×30 мл). Органический слой сушат над Na2SO4, концентрируют и хроматографируют на диоксиде кремния (выход 25%, EtOAc/гексан в качестве элюента) с получением указанного в заголовке соединения 108 (5,22 г, выход 88%) в виде кристаллического твердого вещества белого цвета. MC=260 (М+Н).
трет-Бутил-4-(1-хлор-2-пропенилиден)пиперидин-1-карбоксилат (109; схема XXIX). Метилтрифенилфосфонийбромид (5,51 г, 15,4 ммоль) в ТГФ (40 мл) при 0°С обрабатывают бис(триметилсилил)амидом натрия (15,4 мл, 1,0М в ТГФ) и перемешивают в течение 20 мин, после чего добавляют с помощью канюли соединение 108 (2,05 г, 7,89 ммоль) в ТГФ (15 мл) и смесь перемешивают 3 часа, нагревая до комнатной температуры. Смесь гасят добавлением насыщенного водного раствора NH4Cl (20 мл) и водный слой экстрагируют EtOAc (6×20 мл). Комбинированные органические слои сушат над Na2SO4, концентрируют и хроматографируют на диоксиде кремния (градиентное элюирование с 0-10% МеОН/СН2Cl2) с получением указанного в заголовке соединения 109 (1,94 г, выход 96%) в виде кристаллического твердого вещества белого цвета. MC=258 (М+Н).
4-(1-Хлор-2-пропенилиден)пиперидинтрифторацетат (110; схема XXIX). К соединению 109 (0,1415 г, 0,5489 ммоль) в СН2Cl2 (5 мл) добавляют трифторуксусную кислоту (0,55 мл, 7,1 ммоль) и смесь перемешивают в течение 3 часов, после чего все летучие вещества удаляют в вакууме. Таким образом, полученное сырое указанное в заголовке соединение используют без дальнейшей очистки. MC=158 (М+Н).
Защищенные амины, перечисленные в таблице 7, получают подобным образом, как описано для трет-бутил-4-[2-(1,3-дигидро-1,3-диоксо-2Н-изоиндол-2-ил)этилиден]пиперидинил-1-карбоксилата (26), за исключением того, что соответствующий спирт используют вместо трет-бутил-4-(2-гидроксиэтилиден)пиперидинил-1-карбоксилата (25).
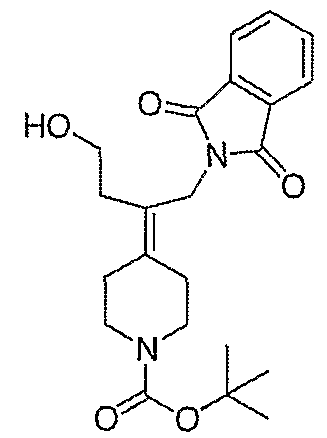
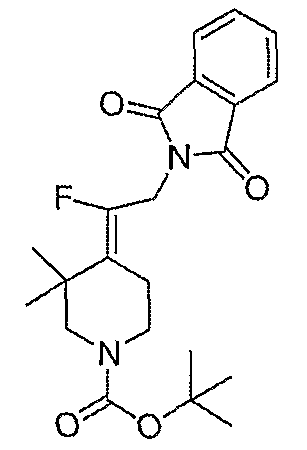
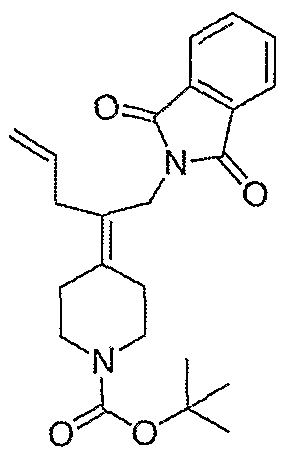
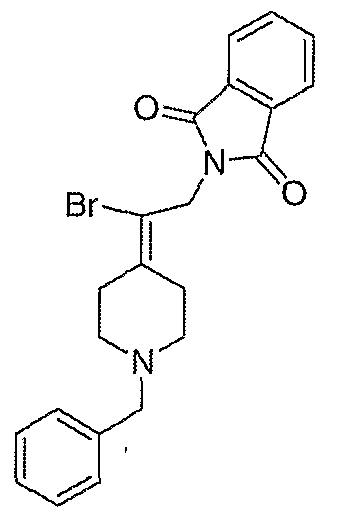
Амины, перечисленные в таблице 8, получают подобным образом, как описано для 4-[2-(1,3-дигидро-1,3-диоксо-2Н-изоиндол-2-ил)этилиден]-1-пиперидинтрифторацетата (27), за исключением того, что соответствующий защищенный амин используют вместо трет-бутил-4-[2-(1,3-дигидро-1,3-диоксо-2Н-изоиндол-2-ил)этилиден]пиперидинил-1-карбоксилата (26).
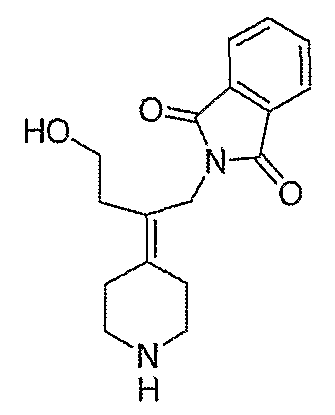
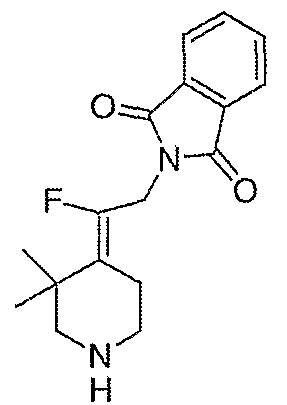
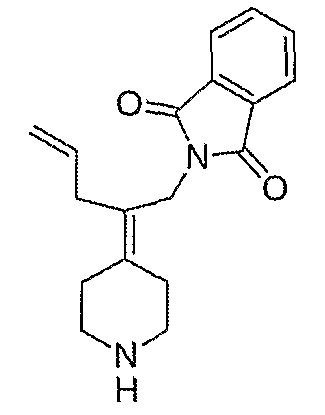
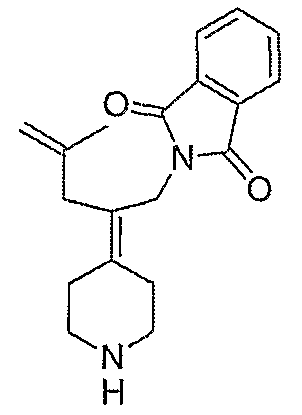
Схема ХХХ
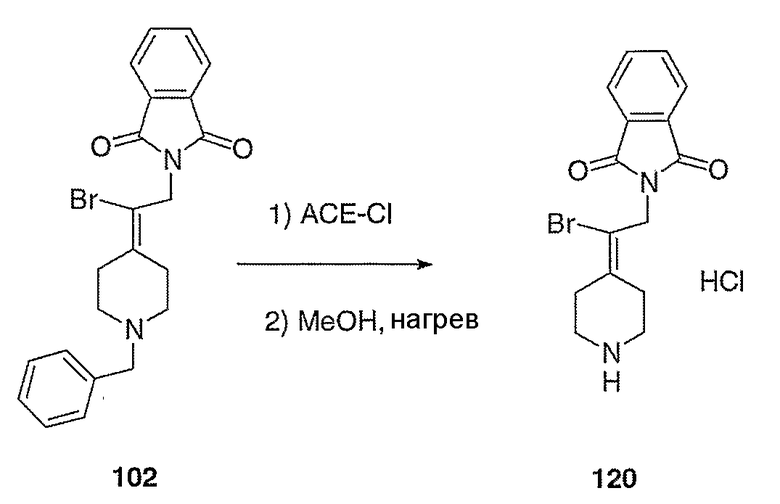
2-(2-Бром-2-пиперидин-4-илиденилэтил)изоиндол-1,3-дионгидрохлорид (120; схема ХХХ). Смесь соединения 102 (0,50 г, 1,17 ммоль) и 1-хлорэтилхлорформиата (0,7 мл, 6,2 ммоль) в дихлорэтане (10 мл) выдерживают при температуре кипения с обратным холодильником в течение 2 часов. Полученный раствор оставляют охлаждаться до комнатной температуры и концентрируют в вакууме. Остаток растворяют в метаноле (50 мл) и выдерживают при температуре кипения с обратным холодильником в течение 2 часов. Реакционную смесь оставляют охлаждаться до комнатной температуры и концентрируют в вакууме с получением твердого вещества белого цвета. Остаток промывают диэтиловым эфиром (2 раза) и сушат с получением указанного в заголовке соединения (432 мг, выход 100%) в виде масла оранжевого цвета. MC=336 (М+Н).
Соединения Z-37 и E-37 схемы XV:
(Е/Z)-Этилхлор(1-бензил-3-пирролидинилиден)ацетат (34). Получают по методике, как при синтезе соединения 24, за исключением того, что 1-бензилпирролидин-3-он используют вместо 1-(трет-бутоксикарбонил)-4-пиперидинона и используют триэтил-2-хлорфосфоноацетат вместо триэтилфосфоноацетата. MC=280 (М+Н).
(Е/Z)-2-(1-Бензил-3-пирролидинилиден)-2-хлорэтанол (35). Получают по методике, как при синтезе соединения 25, за исключением того, что соединение 34 используют вместо соединения 24. MC=283 (М+Н).
Схема XV
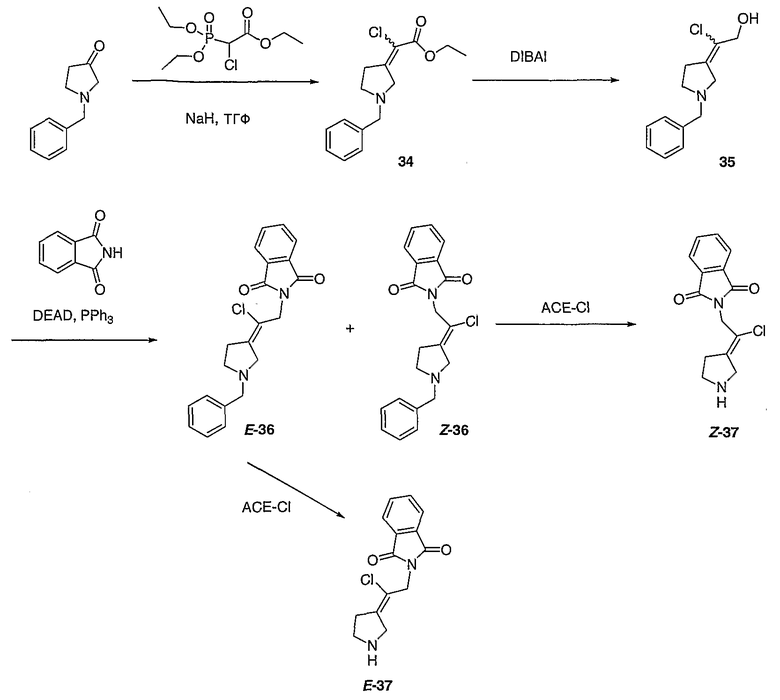
(Е/Z)-2-[2-(1-Бензил-3-пирролидинилиден)-2-хлорэтил]-1Н-изоиндол-1,3(2Н)дион (Е-36 и Z-36). Получают по методике, как при синтезе соединения 26, за исключением того, что соединение 35 (1,58 г) используют вместо соединения 25. Е/Z-Изомеры разделяют с помощью жидкостной хроматографии среднего давления (MPLC) (0-45% этилацетат/гексаны) с получением Z-36 (430 мг, MC=367 (М+Н)) в виде масла красноватого цвета и Е-36 (420 мг, MC=367 (М+Н)) в виде масла красноватого цвета.
(Е)-2-[2-Хлор-2-(3-пирролидинилиден)этил]-1Н-изоиндол-1,3(2Н)-дионгидрохлорид (Е-37). Смесь соединения Е-36 (0,430 г, 1,45 ммоль) и 1-хлорэтилхлорформиата (0,7 мл, 6,2 ммоль) в дихлорэтане (10 мл) выдерживают при температуре кипения с обратным холодильником в течение 2 часов. Полученный раствор оставляют охлаждаться до комнатной температуры и концентрируют в вакууме. Остаток растворяют в метаноле (50 мл) и выдерживают при температуре кипения с обратным холодильником в течение 2 часов. Реакционную смесь оставляют охлаждаться до комнатной температуры и концентрируют в вакууме с получением твердого вещества белого цвета. Остаток промывают диэтиловым эфиром (2 раза) и сушат с получением соединения Е-37 (200 мг, выход 50%) в виде масла коричневого цвета. MC=277 (М+Н).
(Z)-2-[2-Хлор-2-(3-пирролидинилиден)этил]-1Н-изоиндол-1,3(2Н)-дионгидрохлорид (Z-37). Получают по методике, как при синтезе соединения Е-37, за исключением того, что Z-36 используют вместо Е-36. MC=277 (М+Н).
Схема XVI
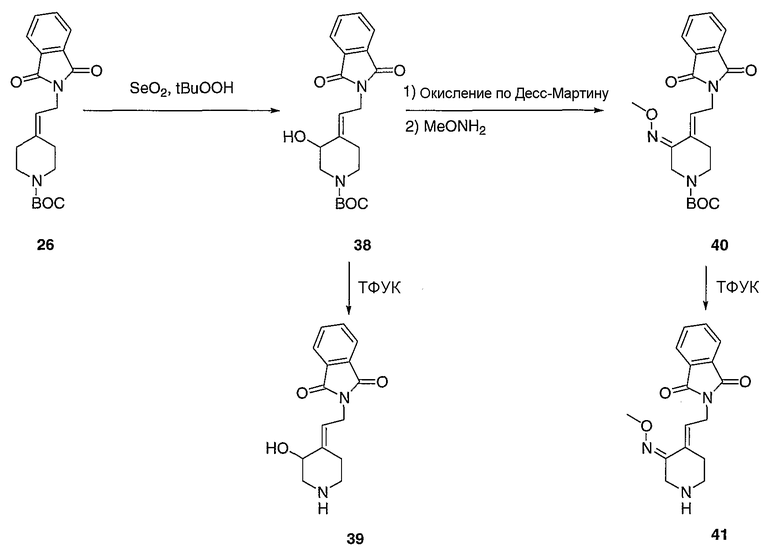
Соединения 39 и 41 схемы XVI:
трет-Бутил-(Е)-4-[2-(1,3-диоксо-1,3-дигидро-2Н-изоиндол-2-ил)этилиден]-3-гидроксипиперидинил-1-карбоксилат (38). Суспензию SeO2 (0,5 г, 6,06 ммоль) в СН2Cl2 (5 мл) при 0°С обрабатывают трет-бутилгидропероксидом (2,5 мл, 9,09 ммоль, 5-6 М, 10%-ный раствор в ундекане) путем введения его с помощью шприца. Спустя 20 мин реакционную смесь обрабатывают раствором этилидена 26 (1,44 г, 4,04 ммоль) в СН2Cl2 (15 мл) и полученную смесь перемешивают в течение 12 часов при комнатной температуре. Реакцию осторожно гасят добавлением 15%-ного водного раствора тиосульфата натрия (15 мл) и реакционную смесь разбавляют СН2Cl2 (25 мл). Слои разделяют, и органический слой промывают 15%-ным водным раствором тиосульфата натрия (15 мл), сушат (MgSO4), отфильтровывают и концентрируют в вакууме. Очистку проводят флэш-хроматографией (силикагель, 0-75% этилацетат/гексаны) с получением указанного в заголовке соединения 38 (0,51 г, выход 33%) в виде твердого вещества белого цвета. MC=373 (М+Н).
(Е)-4-[2-(1,3-Диоксо-1,3-дигидро-2Н-изоиндол-2-ил)этилиден]-3-гидроксипиперидин (39). Получают с помощью подобной методики, как при синтезе соединения 27, за исключением того, что соединение 38 используют вместо соединения 26. MC=273 (М+Н).
(Е)-трет-Бутил-4-[2-(1,3-диоксо-1,3-дигидро-2Н-изоиндол-2-ил)этилиден]-3-метоксииминопиперидинил-1-карбоксилат (40). Раствор соединения 38 (0,51 г, 1,37 ммоль) в СН2Cl2 (15 мл) при 25°С обрабатывают периодинаном Десс-Мартина (0,254 г, 0,60 ммоль). Спустя 1 час реакционную смесь разбавляют СН2Cl2 (25 мл), промывают 10%-ным водным раствором NaHCO3 (3×25 мл), сушат (MgSO4), отфильтровывают и концентрируют в вакууме. Остаток используют на ближайшей стадии без дальнейшей очистки. Раствор остатка растворяют в пиридине (6 мл), обрабатывают метоксиамингидрохлоридом (0,835 г, 6,0 ммоль) в метаноле (36 мл) при 25°С. Спустя 2 мин реакционную смесь кипятят с обратным холодильником в течение 5 часов, разбавляют этилацетатом (25 мл), промывают 10%-ным водным раствором NaHCO3 (3×25 мл), сушат (MgSO4), отфильтровывают и концентрируют в вакууме с получением соединения 40 (230 мг, выход 42%) в виде остатка оранжевого цвета. Остаток используют на ближайшей стадии без дальнейшей очистки. MC=400 (М+Н).
(Е)-4-[2-(1,3-Диоксо-1,3-дигидро-2Н-изоиндол-2-ил)этилиден]-3-метоксииминопиперидин (41). Получают с помощью подобной методики, как при синтезе соединения 27, за исключением того, что соединение 40 используют вместо соединения 26. MC=300 (М+Н).
Схема XVII
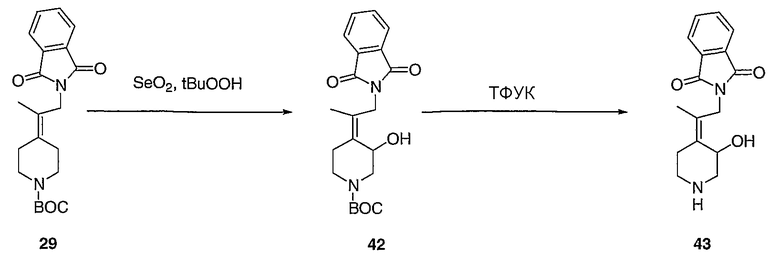
Соединение 43 схемы XVII:
(Z)-трет-Бутил-4-[2-(1,3-диоксо-1,3-дигидро-2Н-изоиндол-2-ил)-1-метилэтилиден]-3-гидроксипиперидинил-1-карбоксилат (42). Суспензию SeO2 (1,3 г, 11,4 ммоль) в СН2Cl2 (15 мл) при 0°С обрабатывают трет-бутилгидропероксидом (4 мл, 22 ммоль, 5-6 М, 10%-ный раствор в ундекане) путем введения его с помощью шприца. Спустя 20 мин реакционную смесь обрабатывают раствором этилидена 29 (3,4 г, 9,1 ммоль) в СН2Cl2 (15 мл) и полученную смесь перемешивают в течение 12 часов при комнатной температуре. Реакцию осторожно гасят добавлением 15%-ного водного раствора тиосульфата натрия (15 мл) и реакционную смесь разбавляют СН2Cl2 (25 мл). Слои разделяют и органический слой промывают 15%-ным водным раствором тиосульфата натрия (15 мл), сушат (MgSO4), отфильтровывают и концентрируют в вакууме. Очистку проводят флэш-хроматографией (силикагель, 0-75% этилацетат/гексаны) с получением указанного в заголовке соединения 42 (1,2 г, выход 34%) в виде твердого вещества белого цвета. MC=387 (М+Н).
(Z)-4-[2-(1,3-Диоксо-1,3-дигидро-2Н-изоиндол-2-ил)-1-метилэтилиден]-3-гидроксипиперидин (43). Получают с помощью подобной методики, как при синтезе соединения 27, за исключением того, что соединение 42 используют вместо соединения 26. MC=287 (М+Н).
Схема XVIII
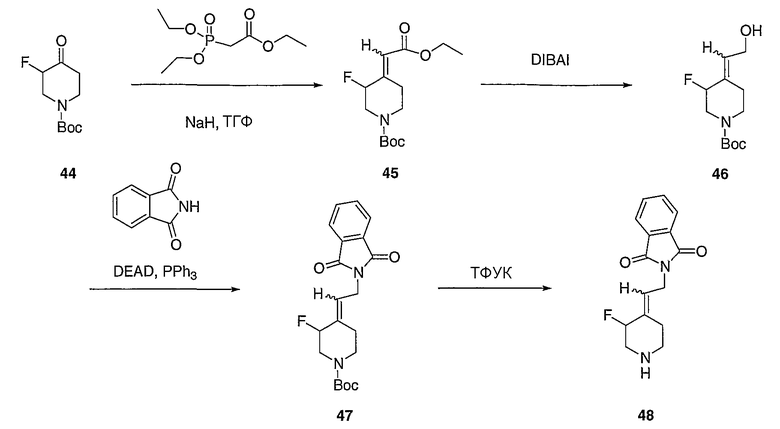
Соединение 48 схемы XVIII:
трет-Бутил-3-фтор-4-оксопиперидинил-1-карбоксилат (44) получают в соответствии с патентом США 5837715.
(Е/Z)-трет-Бутил-4-(2-этокси-2-оксоэтилиден)-3-фторпиперидинил-1-карбоксилат (45). Получают с помощью подобной методики, как при синтезе соединения 24, за исключением того, что соединение 44 используют вместо 1-(трет-бутоксикарбонил)-4-пиперидинона. MC=288 (М+Н).
(Е/Z)-трет-Бутил-4-(2-гидроксиэтилиден)-3-фторпиперидинил-1-карбоксилат (46). Получают с помощью подобной методики, как при синтезе соединения 25, за исключением того, что соединение 45 используют вместо соединения 24. MC=246 (М+Н).
(Е/Z)-трет-Бутил-4-[2-(1,3-дигидро-1,3-диоксо-2Н-изоиндол-2- ил)этилиден]-3-фторпиперидинил-1-карбоксилат (47). Получают с помощью подобной методики, как при синтезе соединения 26, за исключением того, что соединение 46 используют вместо соединения 25. MC=375 (М+Н).
(Е/Z)-4-[2-(1,3-Дигидро-1,3-диоксо-2Н-изоиндол-2-ил)этилиден]-3-фторпиперидинтрифторацетат (48). Получают с помощью подобной методики, как при синтезе соединения 27, за исключением того, что соединение 47 используют вместо соединения 26. MC=275 (М+Н).
Схема XIX
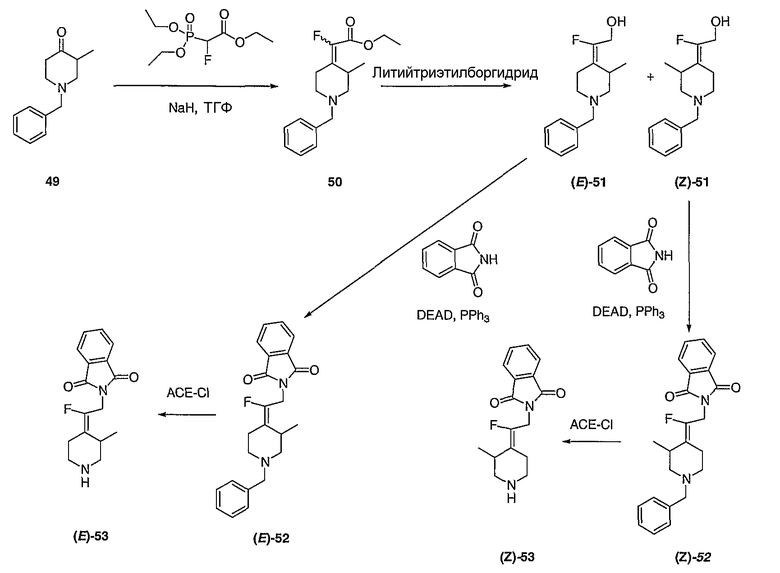
Соединения Z-53 и Е-53 схемы XIX:
Этил-1-[3-метил-1-(фенилметил)-4-пиперидинилиденил]-1-фторацетат (50). Получают с помощью подобной методики, как при синтезе соединения 24, за исключением того, что соединение 49 используют вместо 1-(трет-бутоксикарбонил)-4-пиперидинона и триэтил-2-фторфосфоноацетат используют вместо триэтилфосфоноацетата. MC=292 (М+Н).
2-[3-Метил-1-(фенилметил)-4-пиперидинилиденил]-2-фторэтанол (Е-51 и Z-51). Раствор соединения 50 (2,68 г, 9,19 ммоль) в тетрагидрофуране (50 мл) при 0°С обрабатывают раствором Супер-гидрида™ (23 мл, 23 ммоль, 1,0М в тетрагидрофуране, Aldrich) в атмосфере азота. Спустя 1 час реакционную смесь осторожно обрабатывают метанолом (10 мл), разбавляют этилацетатом (50 мл), промывают 10%-ным водным раствором NaHCO3 (3×50 мл), сушат (MgSO4) и концентрируют в вакууме. Очистку проводят путем MPLC (силикагель, 0-50% этилацетат/гексаны) с получением Z-изомера 51 (0,84 г, выход 37%) (MC=250 (М+Н)) в виде бесцветного масла и Е-изомера 51 (0,97 г, выход 42%) (MC=250 (М+Н)) в виде бесцветного масла.
(Z)-2-[2-(3-Метил-1-(фенилметил)-4-пиперидинилиденил)-2-фторэтил]-1Н-изоиндол-1,3(2Н)-дион (Z-52). Получают с помощью подобной методики, как при синтезе соединения 26, за исключением того, что соединение Z-51 используют вместо соединения 25. MC=379 (М+Н).
(Е)-2-[2-(3-Метил-1-(фенилметил)-4-пиперидинилиденил)-2-фторэтил]-1Н-изоиндол-1,3(2Н)-дион (Е-52). Получают с помощью подобной методики, как при синтезе соединения 26, за исключением того, что соединение Е-51 используют вместо соединения 25. MC=379 (М+Н).
(Z)-2-[2-Фтор-2-(3-метил-4-пиперидинилиденил)этил]-1Н-изоиндол-1,3(2Н)-дионгидрохлорид (Z-53). Смесь соединения Z-52 (0,550 г, 1,45 ммоль) и 1-хлорэтилхлорформиата (0,63 мл, 5,8 ммоль) в дихлорэтане (15 мл) кипятят с обратным холодильником в течение 2 часов. Полученный раствор оставляют охлаждаться до комнатной температуры и концентрируют в вакууме. Остаток растворяют в метаноле (50 мл) и выдерживают при температуре кипения с обратным холодильником в течение 2 часов. Реакционную смесь оставляют охлаждаться до комнатной температуры и концентрируют в вакууме с получением твердого вещества белого цвета. Остаток промывают диэтиловым эфиром (2 раза) и сушат с получением соединения Z-53 (260 мг, выход 55%) в виде твердого вещества белого цвета. MC=289 (М+Н).
(Е)-2-[2-Фтор-2-(3-метил-4-пиперидинилиденил)этил]-1Н-изоиндол-1,3(2Н)-дионгидрохлорид (Е-53). Получают с помощью подобной методики, как при синтезе соединения Z-52, за исключением того, что Е-52 используют вместо Z-52. MC=289 (М+Н).
Схема ХХ
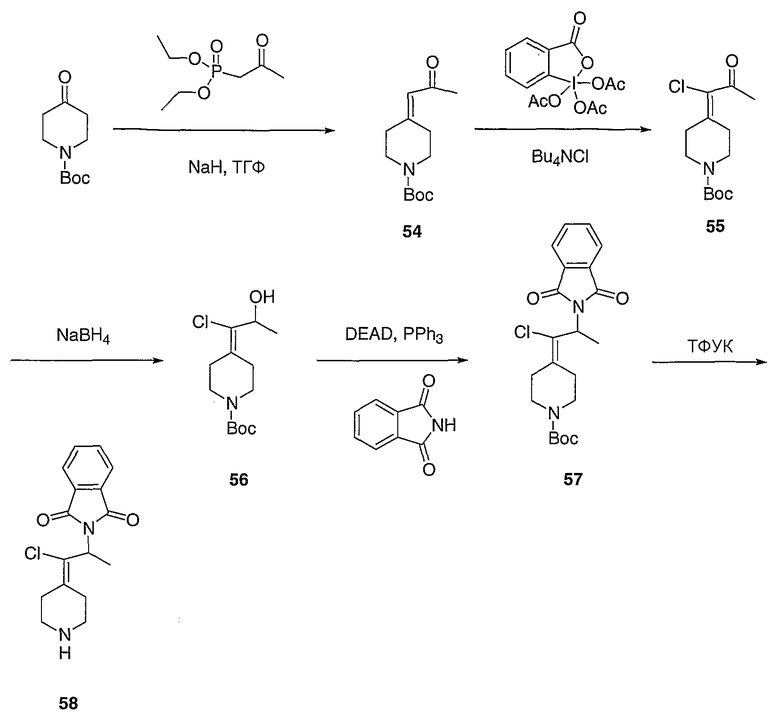
трет-Бутиловый эфир 4-(2-оксопропилиден)пиперидин-1-карбоновой кислоты (54). Получают с помощью подобной методики, описанной в публикации Международной заявки на патент WO 0285901.
трет-Бутиловый эфир 4-(1-хлор-2-оксопропилиден)пиперидин-1-карбоновой кислоты (55). Суспензию тетрабутиламмонийхлорида (11,1 г, 40,1 ммоль) в СН2Cl2 (50 мл) при 25°С обрабатывают 1,1,1-трис(ацетилокси)-1,1-дигидро-1,2-бензиодоксол-3(1Н)-оном (17,0 г, 40,1 ммоль) и полученный раствор светло-желтого цвета перемешивают в течение 10 мин. Реакционную смесь обрабатывают раствором соединения 54 в СН2Cl2 (50 мл) и полученный раствор перемешивают в течение 3 часов. Раствор светло-желтого цвета осторожно вливают в 10%-ный водный раствор бикарбоната натрия (100 мл), разбавляют СН2Cl2 (50мл), вызывая выпадение осадка, отфильтровывают и осадок отбрасывают. Полученный прозрачный раствор промывают 10%-ным водным раствором бикарбоната натрия (1×100 мл), насыщенным раствором соли (1×100 мл), сушат (MgSO4) и концентрируют в вакууме. Очистку проводят путем MPLC (0-40% этилацетат/гексаны) с получением соединения 55 (1,24 г, выход 34%) в виде бесцветного масла. MC=274 (М+Н).
трет-Бутиловый эфир 4-(1-хлор-2-гидроксипропилиден)-пиперидин-1-карбоновой кислоты (56). Раствор соединения 55 (1,24 г, 4,53 ммоль) в этаноле (25 мл) обрабатывают боргидридом натрия (102 мг, 2,72 ммоль) при 25°С. Спустя 1 час реакционную смесь концентрируют в вакууме, разбавляют этилацетатом (40 мл), осторожно обрабатывают 5%-ным водным раствором соляной кислоты (1×25 мл), слои разделяют и сушат (MgSO4). Полученный раствор концентрируют в вакууме с получением соединения 56 (902,1 мг, выход 72%) в виде бесцветного остатка, который используют без дальнейшей очистки. MC=298 (М+Na).
трет-Бутиловый эфир 4-[1-хлор-2-(1,3-диоксо-1,3-дигидроизоиндол-2-ил)пропилиден]пиперидин-1-карбоновой кислоты (57). Получают с помощью подобной методики, как при синтезе соединения 26, за исключением того, что соединение 56 используют вместо соединения 25. MC=427 (М+Na).
2-[(2-Хлор-1-метил-2-(4-пиперидинилиден)этил]-1Н-изоиндол-1,3(2Н)-дион (58). Получают с помощью подобной методики, как при синтезе соединения 27, за исключением того, что соединение 56 используют вместо соединения 26. MC=305 (М+Н).
Схема XXXI
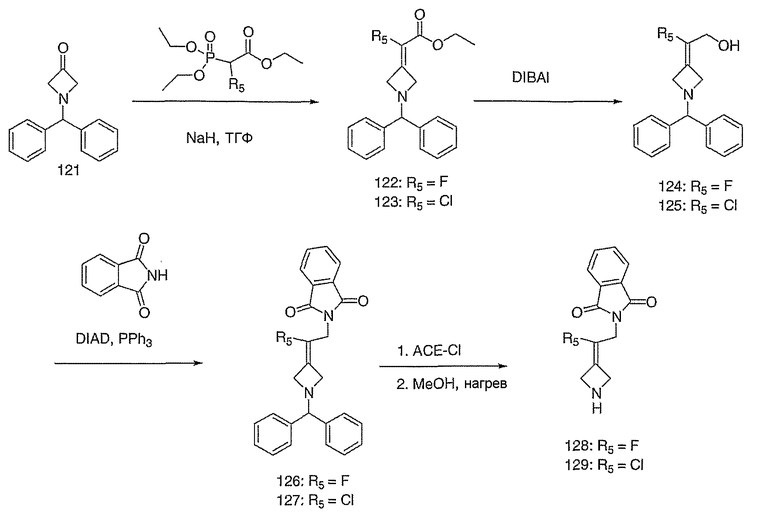
Этил-1-[1-дифенилметилазетидин-3-илиден]-1-фторацетат (122; R5=F). Триэтил-2-фтор-2-фосфоноацетат (0,63 мл, 3,10 ммоль) добавляют к NaH (60%-ная дисперсия в масле, 115 мг, 2,87 ммоль) в безводном ТГФ (6 мл) при 0°С. После перемешивания в течение 15 минут, добавляют раствор кетона 121 (562 мг, 2,37 ммоль) в безводном ТГФ (6 мл). Реакционную смесь нагревают до комнатной температуры и перемешивают в течение ночи. Реакционную смесь разбавляют этилацетатом (100 мл), промывают насыщенным раствором NaHCO3 (2×100 мл), сушат (MgSO4), отфильтровывают и концентрируют в вакууме. Сырое вещество хроматографируют (100% СН2Cl2) с получением сложного эфира 122 (R5=F) в виде масла желтого цвета (392 мг, выход 66%). MC=326 (М+Н).
Этил-1-[1-дифенилметилазетидин-3-илиден]-1-хлорацетат (123; R5=Cl). Получают по методике, описанной выше, за исключением того, что в реакции используют триэтил-2-хлор-2-фосфоноацетат вместо триэтил-2-фтор-2-фосфоноацетата. Сложный эфир 123 (R5=Cl) выделяют в виде твердого вещества белого цвета (выход 77%). MC=342, 344 (М+Н).
2-(1-Дифенилметилазетидин-3-илиден)-2-фторэтанол (124; R5=F). Диизобутилалюминийгидрид (DIBAL) (1 М в толуоле, 4,2 мл, 4,2 ммоль) добавляют к раствору сложного эфира 122 (R5=F) (510 мг, 1,56 ммоль) в толуоле (8 мл) при -78°С в течение нескольких минут. Реакционную смесь перемешивают в течение 5 часов и затем гасят медленным добавлением раствора метанола в толуоле. Реакционную смесь разбавляют этилацетатом (100 мл), промывают раствором NaOH (1 н., 2×50 мл), водой (50 мл), сушат (MgSO4) и концентрируют в вакууме с получением спирта 124 (R5=F, 289 мг, выход 65%) в виде твердого вещества бледно-желтого цвета после растирания в порошок с простым эфиром/гексанами. MC=284 (М+Н).
2-(1-Дифенилметилазетидин-3-илиден)-2-хлорэтанол (125; R5=Cl). Получают по методике, описанной выше, за исключением того, что сложный эфир 123 (R5=Cl) используют вместо сложного эфира 122 (R5=F). Спирт 125 (R5=Cl) выделяют с помощью хроматографии (20% этилацетат/гексаны) в виде твердого вещества белого цвета (выход 53%). MC=300, 302 (М+Н).
2-[2-(1-Дифенилметилазетидин-3-илиден)-2-фторэтил]изоиндол-1,3-дион (126; R5=F). Диизопропилазодикарбоксилат (DIAD) (0,89 мл, 4,489 ммоль) добавляют к раствору спирта 124 (R5=F) (1,00 г, 3,533 ммоль), трифенилфосфина (1,14 г, 4,34 ммоль) и фталимида (0,648 г, 4,527 ммоль) в безводном ТГФ (30 мл) при 0°С. Реакционную смесь нагревают до комнатной температуры и перемешивают в течение 36 часов. Летучие вещества выпаривают и остаток хроматографируют на силикагеле (5% этилацетат/гексаны) с получением фталимида 126 (R5=F) (952 мг, выход 65%) в виде твердого вещества белого цвета. MC=413 (М+Н).
2-[2-(1-Дифенилметилазетидин-3-илиден)-2-хлорэтил]изоиндол-1,3-дион (127; R5=Cl). Получают по методике, описанной выше, за исключением того, что спирт 125 (R5=Cl) используют вместо спирта 124 (R5=F) в реакции Mitsunobu. Фталимид 127 (R5=Cl) выделяют с помощью хроматографии (15% этилацетат/гексаны) в виде твердого вещества белого цвета (выход 68%). MC=429, 431 (М+Н).
2-(2-Азетидин-3-илиден-2-фторэтил)изоиндол-1,3-дионгидрохлорид (128; R5=F). Фталимид 126 (R5=F) (350 мг, 0,8491 ммоль) и АСЕ-Cl (0,50 мл, 4,65 ммоль) в 1,2-дихлорэтане (20 мл) выдерживают при температуре кипения с обратным холодильником в атмосфере азота в течение 24 часов. После охлаждения, летучие вещества выпаривают и к полученному остатку добавляют метанол (25 мл). Кипятят с обратным холодильником в течение 3 часов, после чего метанол выпаривают с получением соединения 128 (R5=F) в виде порошка бежевого цвета (230 мг, выход 96%). MC=247 (М+Н).
2-(2-Азетидин-3-илиден-2-хлорэтил)изоиндол-1,3-дионгидрохлорид (129; R5=Cl). Получают по методике, описанной выше, за исключением того, что фталимид 127 (R5=Cl) используют в реакции вместо фталимида 126 (R5=F). Соединение выделяют в виде порошка белого цвета (выход 86%). МС=263, 265 (М+Н).
Схема XXXII
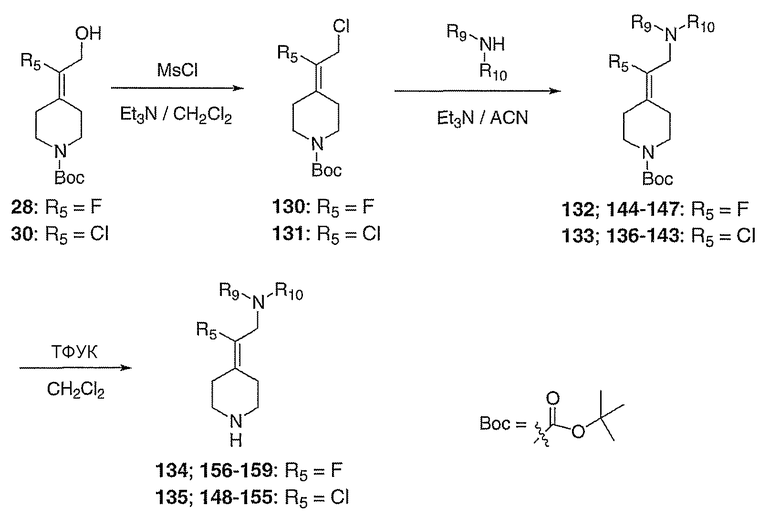
трет-Бутил-4-(1,2-дихлорэтилиден)пиперидинил-1-карбоксилат (131; R5=Cl). Раствор соединения 30 (R5=Cl) (4,24 г, 16,20 ммоль) и триэтиламина (6,8 мл, 48,60 ммоль) в СН2Cl2 (120 мл) обрабатывают метансульфонилхлоридом (1,9 мл, 24,30 ммоль) при 0°С, затем нагревают до комнатной температуры и перемешивают в течение ночи. Полученную смесь гасят добавлением насыщенного водного раствора NaHCO3 (100 мл) и продукт экстрагируют СН2Cl2. Очистку проводят флэш-хроматографией (0-20% этилацетат/гексаны) с получением указанного в заголовке соединения (3,1 г, выход 68%) в виде твердого вещества белого цвета.
трет-Бутил-4-(2-хлор-1-фторэтилиден)пиперидинил-1-карбоксилат (130; R5=F). Получают по методике, описанной выше, за исключением того, что спирт 28 (R5=F) используют вместо спирта 30 (R5=Cl).
трет-Бутил-4-[2-(N-бензил-N-метиламино)-1-хлорэтилиден]пиперидинил-1-карбоксилат (133; R5=Cl, R9=метил, R10=бензил). Раствор соединения 131 (R5=Cl) (600 мг, 2,14 ммоль) и триэтиламина (1,5 мл, 10,71 ммоль) в ацетонитриле (18 мл) обрабатывают N-бензилметиламином (0,45 мл, 3,43 ммоль) при комнатной температуре и перемешивают в течение ночи. Полученную смесь концентрируют в вакууме и остаток разбавляют этилацетатом (20 мл), промывают водой (2×10 мл) и сушат (MgSO4). Очистку проводят флэш-хроматографией (0-15% этилацетат/гексаны) с получением указанного в заголовке соединения (690 мг, выход 88%) в виде твердого вещества белого цвета. МС=365 (М+Н).
трет-Бутил-4-[2-(N-бензил-N-метиламино)-1-фторэтилиден]пиперидинил-1-карбоксилат (132; R5=F, R9=метил, R10=бензил). Получают по методике, описанной выше, за исключением того, что хлорид 130 (R5=F) используют вместо хлорида 131 (R5=Cl). МС=349 (М+Н).
N-Бензил-N-метил-(2-хлор-2-пиперидин-4-илиден)этиламин (135; R5=Cl). Раствор соединения 133 (R5=Cl) (690 мг, 1,89 ммоль) растворяют в СН2Cl2 (15 мл) и обрабатывают трифторуксусной кислотой (1,5 мл) при комнатной температуре. Спустя 5 часов реакционную смесь концентрируют в вакууме с получением указанного в заголовке соединения (количественный выход) в виде масла, которое используют на ближайшей стадии без дальнейшей очистки. МС=265 (М+Н).
N-Бензил-N-метил-(2-фтор-2-пиперидин-4-илиден)этиламин (134; R5=F). Получают по методике, описанной выше, за исключением того, что амин 132 (R5=F) используют вместо амина 133 (R5=Cl). МС=249 (М+Н).
В таблице 9 перечислены Вос-защищенные амины (136-147) и происходящие от них амины (148-159), полученные с помощью аналогичных методик, подробно описанных выше.
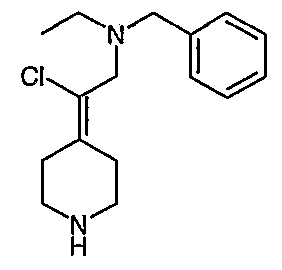
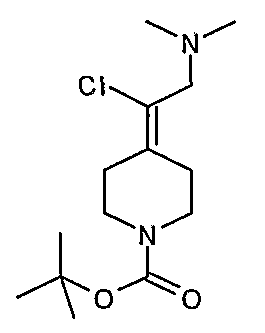
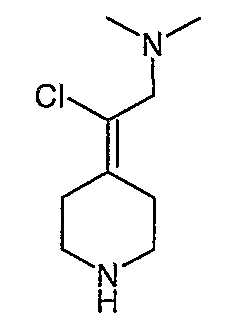
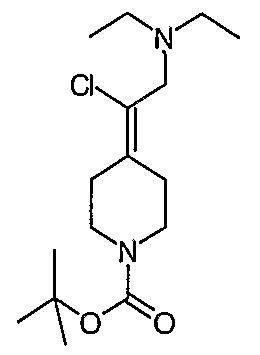
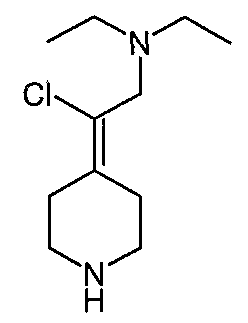
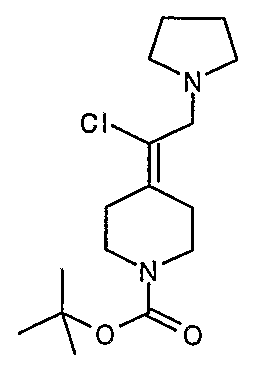
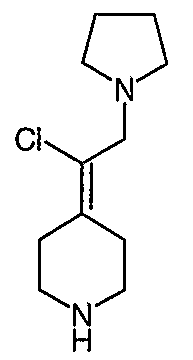
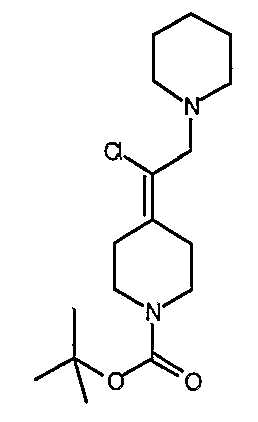
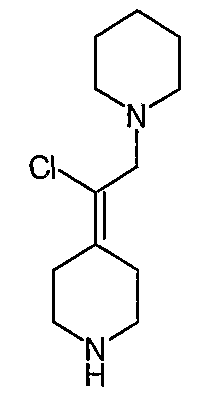
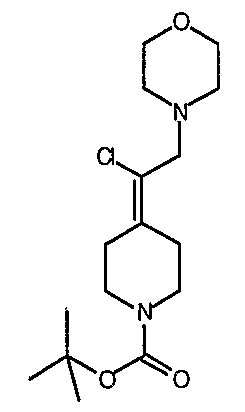
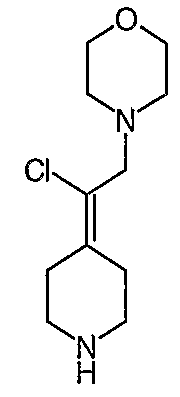
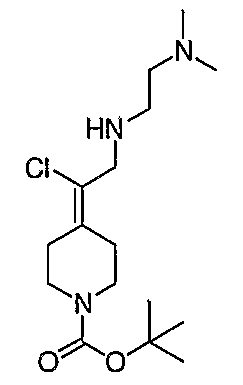
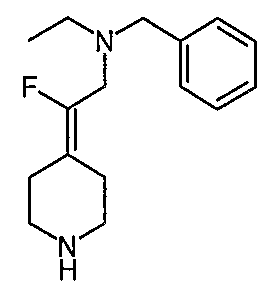
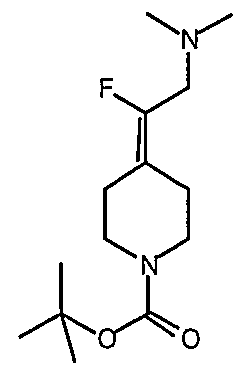
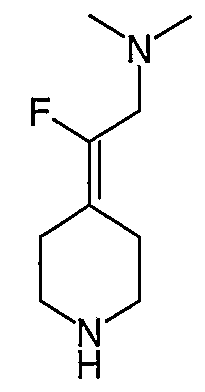
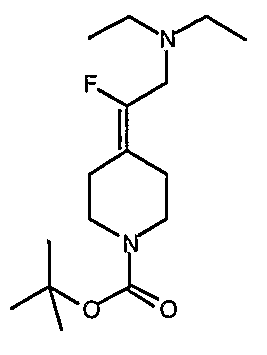
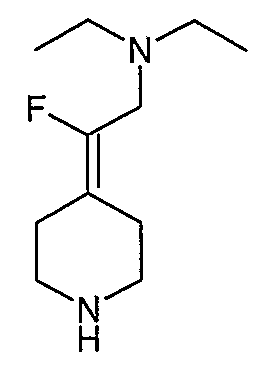
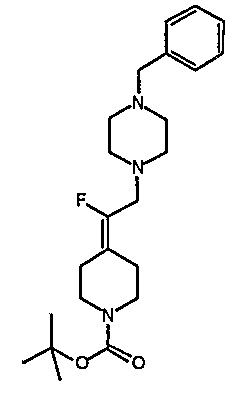
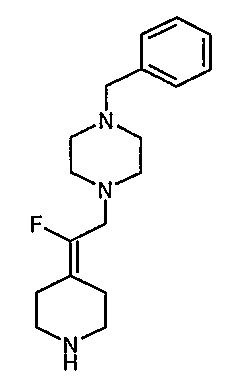
Получение конечного продукта
7-[4-(2-Амино-1-фторэтилиден)пиперидин-1-ил]-1-циклопропил-6-фтор-4-оксо-1,4-дигидронафтиридин-3-карбоновая кислота (1). Раствор амина 31 (612 мг, 1,57 ммоль) и триэтиламина (0,7 мл, 5,0 ммоль) в ацетонитриле (4 мл) обрабатывают 7-хлор-1-циклопропил-6-фтор-4-оксо-1,4-дигидронафтпиридин-3-карбоновой кислотой (222 мг, 0,787 ммоль) в атмосфере азота и реакционную смесь перемешивают в течение 12 часов. Полученную смесь концентрируют в вакууме, и остаток промывают водой (3×10 мл). Остаток оставляют высыхать в течение 15 мин. Твердое вещество собирают, повторно суспендируют в метаноле (5 мл) и реакционную смесь обрабатывают гидразином (1 мл). Спустя 5 мин реакционную смесь нагревают до температуры кипения с обратным холодильником и полученную смесь перемешивают в течение 1 часа. Реакционную смесь концентрируют в вакууме, разбавляют водой и твердые вещества собирают фильтрацией. Полученный продукт белого цвета промывают водой (3×20 мл), оставляют высыхать в течение ночи, получая указанное в заголовке соединение 1 (40,4 мг, выход 13%). МС=391 (М+Н).
Трифторацетат 7-[4-(2-амино-1-фторэтилиден)пиперидин-1-ил]-1-циклопропил-6-фтор-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты (5). Раствор амина 31 (311 мг, 0,80 ммоль) и триэтиламина (0,55 мл, 4,0 ммоль) в ацетонитриле (4 мл) обрабатывают диацетилхинолинилборатом 17 (300 мг, 0,60 ммоль) в атмосфере азота. Спустя 5 мин реакционную смесь нагревают до температуры кипения с обратным холодильником и перемешивают в течение 12 часов. Полученную смесь оставляют охлаждаться до комнатной температуры, концентрируют в вакууме и остаток промывают водой (3×10 мл). Остаток растворяют в тетрагидрофуране (3 мл) и обрабатывают 10%-ным водным раствором соляной кислоты (5 мл) при комнатной температуре. Спустя 30 мин, реакционную смесь концентрируют в вакууме, разбавляют водой (10 мин) и твердое вещество собирают фильтрацией. Твердый остаток промывают водой (3×5 мл) и оставляют высыхать в течение 15 мин. Твердое вещество собирают и повторно суспендируют в метаноле (5 мл) и реакционную смесь обрабатывают гидразином (1 мл). Спустя 5 мин, реакционную смесь нагревают до температуры кипения с обратным холодильником и полученную смесь перемешивают в течение 1 часа. Реакционную смесь концентрируют в вакууме и остаток очищают ВЭЖХ (колонка с обращенной фазой С-18, 0-55% ацетонитрил/вода, содержащая 0,1% трифторуксусной кислоты) с получением соли трифторуксусной кислоты 5 (61,3 мг, выход 20%) в виде твердого вещества светло-желтого цвета. МС=390 (М+Н).
7-[3-(2-Амино-1-фторэтилиден)азетидин-1-ил]-1-циклопропил-6-фтор-4-оксо-1,4-дигидро[1,8]нафтиридин-3-карбоновая кислота (80). 7-Хлор-1-циклопропил-6-фтор-4-оксо-1,4-дигидронафтиридин-3-карбоновую кислоту (57 мг, 0,2016 ммоль), амин 128 (R5=F) (67 мг, 0,2389 ммоль) и триэтиламин (0,5 мл) в ацетонитриле (10 мл) выдерживают при температуре кипения с обратным холодильником в течение ночи. После охлаждения, летучие вещества выпаривают и остаток суспендируют в воде (25 мл). Полученное твердое вещество собирают фильтрацией и сушат. К твердому веществу добавляют этанол (5 мл), затем гидразин (0,01 мл, 0,3138 ммоль). Реакционную смесь выдерживают при температуре кипения с обратным холодильником в течение 1 часа, после чего летучие вещества выпаривают. К остатку добавляют воду (15 мл) и полученное твердое вещество собирают фильтрацией, промывают дополнительно водой и сушат с получением соединения 87 (49,1 мг, выход 69%) в виде порошка не совсем белого цвета. МС=363 (М+Н).
7-[3-(2-Амино-1-фторэтилиден)азетидин-1-ил]-1-циклопропил-8-дифторметокси-6-фтор-4-оксо-1,4-дигидрохинолин-3-карбоновая кислота (160). Раствор амина 128 (R5=F) (83 мг, 0,2923 ммоль), диацетилхинолинилбората 83 (111 мг, 0,2413 ммоль) и триэтиламина (0,5 мл) в ацетонитриле (10 мл) выдерживают при температуре кипения с обратным холодильником в течение ночи. Летучие вещества выпаривают и затем к остатку добавляют ТГФ (5 мл) и 10%-ный водный раствор HCl (4 мл). Эту смесь перемешивают приблизительно 1 час. Полученное твердое вещество собирают фильтрацией, промывают водой и сушат. К твердому веществу добавляют этанол (4 мл) и гидразин (0,01 мл) и реакционную смесь выдерживают при температуре кипения с обратным холодильником в течение 1,5 часов. Этанол выпаривают в вакууме и к оставшемуся веществу добавляют воду (20 мл). Твердое вещество собирают и сушат с получением соединения 160 в виде твердого вещества желтого цвета (выход 20%). МС=428 (М+Н).
Трифторацетат 7-{4-[2-(N-бензил-N-метиламино)-1-хлорэтилиден]пиперидин-1-ил}-1-циклопропил-6-фтор-8-метокси-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты (161). Раствор амина 135 (1,89 ммоль) и триэтиламина (1,2 мл, 8,59 ммоль) в ацетонитриле (15 мл) обрабатывают диацетилхинолинилборатом 19 (727 мг, 1,72 ммоль) в атмосфере азота. Спустя 5 мин реакционную смесь нагревают до температуры кипения с обратным холодильником и реакционную смесь перемешивают в течение 24 часов. Полученную смесь оставляют охлаждаться до комнатной температуры и затем концентрируют в вакууме. Остаток растворяют в тетрагидрофуране (5 мл), обрабатывают 10%-ным водным раствором соляной кислоты (5 мл) при комнатной температуре и перемешивают в течение ночи. Полученную смесь концентрируют в вакууме и остаток очищают ВЭЖХ (колонка с обращенной фазой С-18, 30-90% ацетонитрил/вода, содержащая 0,1% трифторуксусной кислоты) с получением соли трифторуксусной кислоты 161 (632 мг, выход 56%) в виде твердого вещества желтого цвета. МС=540 (М+Н).
7-{4-[2-(N-Бензил-N-метиламино)-1-хлорэтилиден]пиперидин-1-ил}-1-циклопропил-6-фтор-4-оксо-1,4-дигидро[1,8]нафтиридин-3-карбоновая кислота (162). Раствор амина 135 (0,48 ммоль) и триэтиламина (0,28 мл, 2,0 ммоль) в ацетонитриле (7 мл) обрабатывают 7-хлор-1-циклопропил-6-фтор-4-оксо-1,4-дигидро[1,8]нафтиридин-3-карбоновой кислотой (113 мг, 0,40 ммоль) в атмосфере азота. Спустя 5 мин реакционную смесь нагревают до температуры кипения с обратным холодильником и реакционную смесь перемешивают в течение 24 часов. Полученную смесь оставляют охлаждаться до комнатной температуры, концентрируют в вакууме и остаток разбавляют водой. Продукт собирают фильтрацией и затем промывают водой и небольшим количеством метанола с получением указанного в заголовке соединения (178 мг, выход 87%) в виде твердого вещества белого цвета. МС=511 (М+Н).
7-[4-(2-Гидроксиэтилиден)пиперидин-1-ил]-1-циклопропил-6-фтор-4-оксо-1,4-дигидронафтиридин-3-карбоновая кислота (163). Раствор амина 103 (256 мг, 1,06 ммоль) и триэтиламина (0,5 мл, 3,55 ммоль) в ацетонитриле (4 мл) обрабатывают 7-хлор-1-циклопропил-6-фтор-4-оксо-1,4-дигидронафтиридин-3-карбоновой кислотой (200 мг, 0,71 ммоль) в атмосфере азота и реакционную смесь перемешивают в течение 16 часов. Полученную смесь концентрируют в вакууме, и остаток промывают водой (3×10 мл) и оставляют сушиться в течение ночи, получая указанное в заголовке соединение 163 (105 мг, выход 40%). МС=374 (М+Н).
7-[4-(Гидроксиэтилиден)пиперидин-1-ил]-1-циклопропил-6-фтор-4-оксо-1,4-дигидрохинолин-3-карбоновая кислота (164). Раствор амина 103 (146 мг, 0,61 ммоль) и триэтиламина (0,55 мл, 4,0 ммоль) в ацетонитриле (4 мл) обрабатывают диацетилхинолинилборатом 17 (125 мг, 0,60 ммоль) в атмосфере азота. Спустя 5 мин реакционную смесь нагревают до температуры кипения с обратным холодильником и реакционную смесь перемешивают в течение 12 часов. Полученную смесь оставляют охлаждаться до комнатной температуры, концентрируют в вакууме и остаток промывают водой (3×10 мл). Остаток растворяют в тетрагидрофуране (3 мл) и обрабатывают 10%-ным водным раствором соляной кислоты (5 мл) при комнатной температуре. Спустя 30 мин реакционную смесь концентрируют в вакууме, разбавляют водой (10 мин) и твердое вещество собирают фильтрацией. Твердый остаток промывают водой (3×5 мл) и оставляют сушиться в течение 15 мин. Твердое вещество собирают с получением соединения 157 (5,1 мг, выход 2,2%) в виде твердого вещества светло-желтого чвета. МС=373 (М+Н).
В таблице 4 перечислены дополнительные соединения согласно настоящему изобретению, полученные с помощью экспериментальных методик, подробно описанных выше. В случае нафтиридинов 2-4, 59-63, 69 и 173-176, для их получения используют экспериментальную методику, аналогичную таковой для получения соединения 1. Для нафтиридинов 171, 172 и 185 применяют аналогичную методику, как для получения соединения 163. Для нафтиридинов 81, 183 и 184 применяют аналогичную методику, как для получения соединения 80. Для нафтиридинов 165-170, 177-182 и 186 применяют методику, подобную таковой для получения соединения 162. Для хинолонов 6-15, 64-66, 70, 71, 73, 78, 187, 188, 201-204, 206-208 и 210 применяют аналогичную методику, как для получения соединения 5. Для хинолонов 76, 77, 189, 205 и 209 применяют аналогичную методику, как для получения соединения 160. Для хинолонов 190-200 применяют аналогичную методику, как для получения соединения 161. Для хинолона 211 применяют подобную методику, как для получения соединения 163.
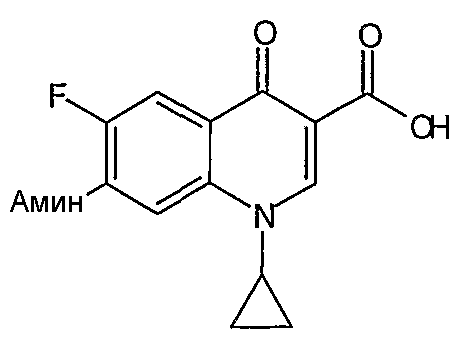
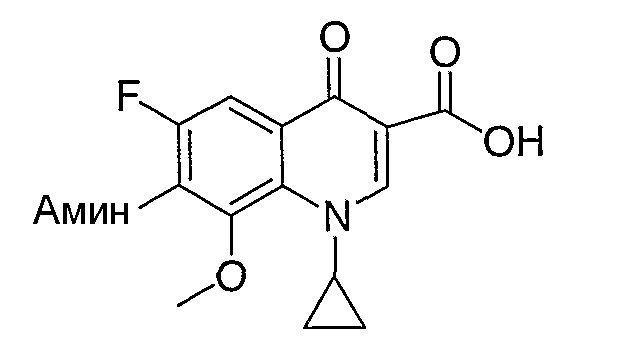
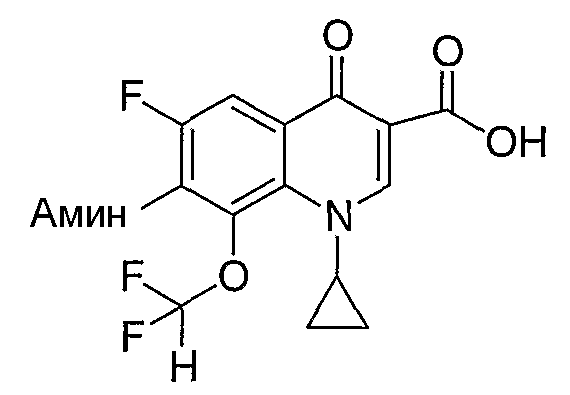
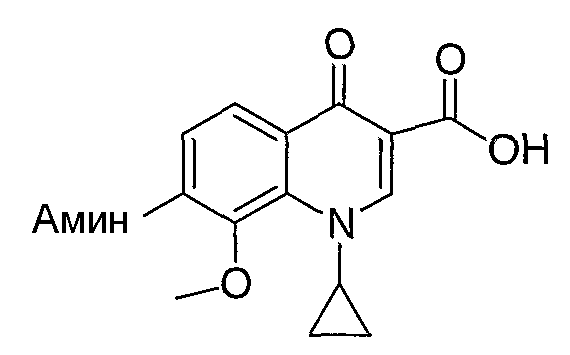
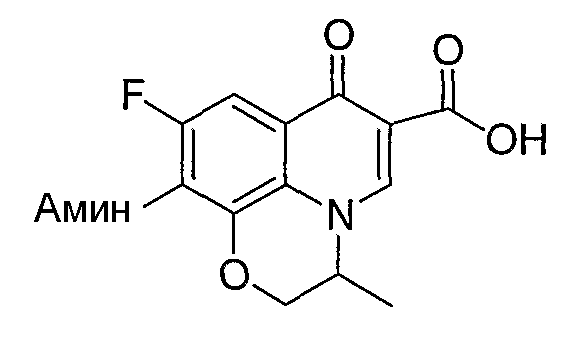
Схема XXXIII
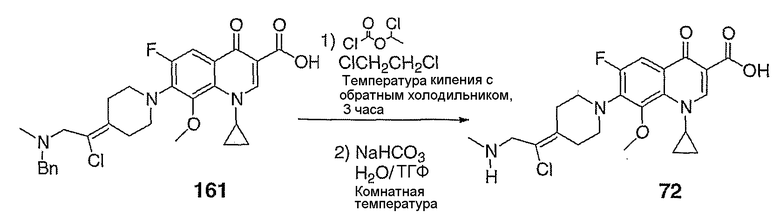
Трифторацетат 7-[4-(1-хлор-2-метиламиноэтилиден)пиперидин-1-ил]-1-циклопропил-6-фтор-8-метокси-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты (72). Раствор соединения 154 (160 мг, 0,24 ммоль) растворяют в 1,2-дихлорэтане (4 мл) и обрабатывают 1-хлорэтилхлорформиатом (0,8 мл, 7,3 ммоль) в атмосфере азота. Спустя 5 мин реакционную смесь нагревают до температуры кипения с обратным холодильником и реакционную смесь перемешивают в течение 3 часов. Полученную смесь оставляют охлаждаться до комнатной температуры и затем концентрируют в вакууме. Остаток растворяют в тетрагидрофуране (5 мл), устанавливают рН>7 добавлением NaHCO3 и воды при комнатной температуре и перемешивают в течение ночи. Полученную смесь концентрируют в вакууме и остаток очищают ВЭЖХ (колонка с обращенной фазой С-18, 35-90% ацетонитрил/вода, содержащая 0,1% трифторуксусной кислоты) с получением соли трифторуксусной кислоты 72 (37 мг, выход 27%) в виде твердого вещества желтого цвета. МС=450 (М+Н).
В таблице 10 перечислены конечные продукты (74, 75, 79, 213-220), полученные с помощью методик, аналогичных таковым, указанным выше.
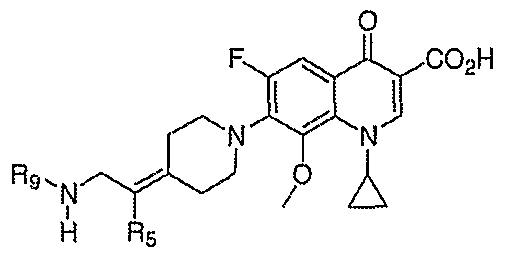
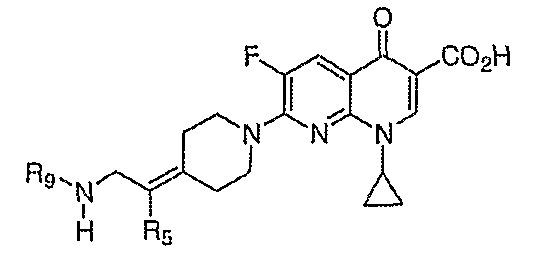
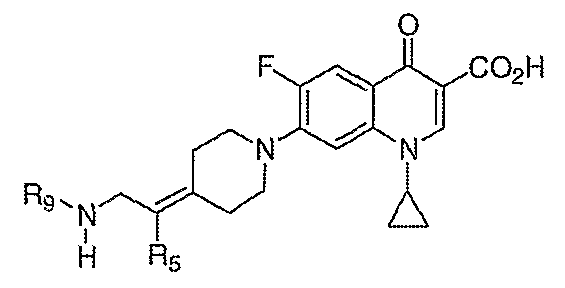
Схема XXXIV
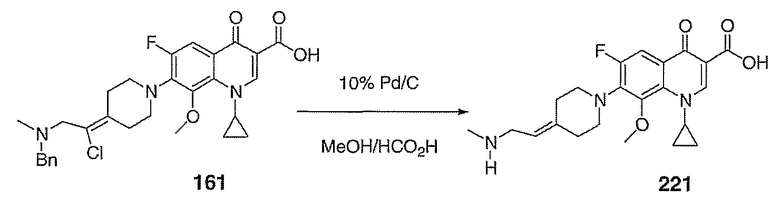
Трифторацетат 1-циклопропил-6-фтор-8-метокси-7-[4-(2-метиламиноэтилиден)пиперидин-1-ил]-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты (221). Раствор соединения 161 (70 мг, 0,11 ммоль) в смеси метанол/муравьиная кислота (об./об.=20/1) (14 мл) обрабатывают 10% Pd/C (35 мг, 7,3 ммоль) в атмосфере азота при комнатной температуре и перемешивают в течение 3 часов. Полученную смесь отфильтровывают и концентрируют в вакууме. Остаток очищают ВЭЖХ (колонка с обращенной фазой С-18, 35-90% ацетонитрил/вода, содержащая 0,1% трифторуксусной кислоты) с получением соли трифторуксусной кислоты 221 (8,3 мг, выход 15%) в виде твердого вещества желтого цвета. МС=416 (М+Н).
Схема XXI
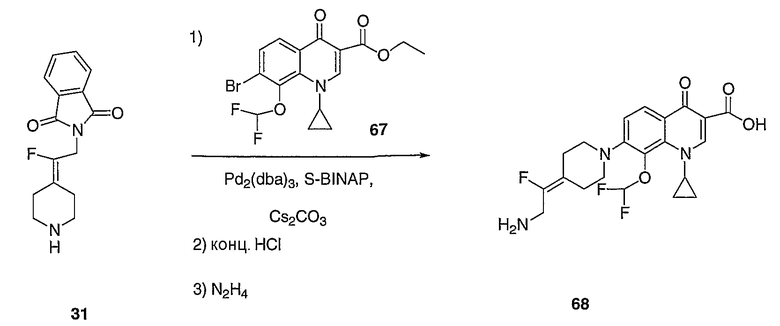
7-[4-(2-Амино-1-фторэтилиден)пиперидин-1-ил]-1-циклопропил-8-дифторметокси-4-оксо-1,4-дигидрохинолин-3-карбоновая кислота (68). Раствор амина 31 (534 мг, 1,94 ммоль), хинолона 67 (587 мг, 1,46 ммоль) (полученный, как описано выше в ЕР 1031569), карбоната цезия (717 мг, 2,2 ммоль), (1S)-[1,1'-бинафталин]-2,2'-диилбис[дифенилфосфина](137 мг, 0,22 ммоль) в толуоле (75 мл) обрабатывают Pd2(dba)3 (66 мг, 0,072 ммоль) и реакционную смесь выдерживают при температуре кипения с обратным холодильником. Спустя 12 часов реакционную смесь оставляют охлаждаться до комнатной температуры, концентрируют в вакууме и остаток промывают водой (3×10 мл). Очистку проводят с помощью MPLC (0-100% этилацетат/гексаны) с получением остатка желтого цвета. Остаток растворяют в концентрированной соляной кислоте (5 мл) и выдерживают при температуре кипения с обратным холодильником. Спустя 3 часа реакционную смесь концентрируют в вакууме, разбавляют водой (10 мл) и твердое вещество собирают фильтрацией. Твердый остаток промывают водой (3×5 мл) и оставляют сушиться в течение 15 мин. Твердое вещество собирают и повторно суспендируют в метаноле (5 мл) и реакционную смесь обрабатывают гидразином (1 мл). После 5 мин реакционную смесь нагревают до температуры кипения с обратным холодильником и полученную смесь перемешивают в течение 1 часа. Реакционную смесь концентрируют в вакууме и очищают ВЭЖХ (колонка с обращенной фазой С-18, 0-55% ацетонитрил/вода, содержащая 0,1% трифторуксусной кислоты) с получением соли трифторуксусной кислоты указанного в заголовке соединения 68 (75 мг, выход 12%) в виде твердого вещества светло-желтого цвета. МС=438 (М+Н).
Схема XXXV
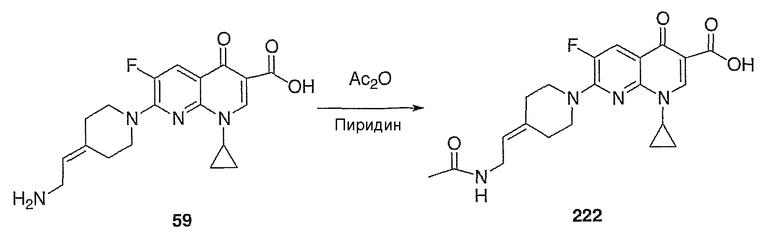
7-[4-(2-Ацетиламиноэтилиден)пиперидин-1-ил]-1-циклопропил-6-фтор-4-оксо-1,4-дигидро[1,8]нафтиридин-3-карбоновая кислота (222). Смесь соединения 59 (25 мг, 0,067 ммоль) и уксусного ангидрида (94 мкл, 0,100 ммоль) в пиридине (1 мл) перемешивают в течение 12 часов при 25°С. Полученную смесь концентрируют в вакууме и остаток промывают водой (3×10 мл) и оставляют сушиться в течение ночи, получая указанное в заголовке соединение 222 (15 мг, выход 54%). МС=415 (М+Н).
Биологическая активность
Соединения, описанные в настоящем изобретении, обладают антибактериальной активностью вследствие их новой структуры и пригодны в качестве антибактериальных средств для лечения бактериальных инфекций у людей и животных.
Минимальная ингибирующая концентрация (MIC) является индикатором in vitro антибактериальной активности, широко применяемым в данной области. In vitro противомикробную активность соединений определяют по методу микроразведений бульона с последующим тестированием согласно Национальному Комитету по клиническим лабораторным стандартам (NCCLS). Этот метод описан в документе NCCLS М7-А4, том 17, № 2, "Methods for Dilution Antimicrobial Susceptibility Test for Bacteria that Grow Aerobically-Fourth Edition", который включен в данный контекст в качестве ссылки.
Согласно этому методу, двукратные серийные разведения лекарственного средства в бульоне Mueller-Hinton с установленным содержанием катионов добавляют в лунки планшетов для микроразведений. Тест-организмы получают путем установления мутности активно растущих на бульоне культур так, чтобы конечная концентрация тест-организма после его добавления в лунки составляла приблизительно 5×104 колониеобразующих единиц (КОЕ) на лунку.
После инокуляции для микроразведений планшетов, планшеты инкубируют при 35°С в течение 16-20 часов и затем считывают. MIC представляет собой наименьшую концентрацию тестируемого соединения, которая полностью ингибирует рост тест-организма. Величину роста в лунках, содержащих тестируемое соединение, сравнивают с величиной роста в контрольных лунках роста (без тестируемого соединения), используемых в каждом планшете. Как представлено в таблице 5, соединения согласно настоящему изобретению тестировали против множества патогенных бактерий, получая диапазон активностей в зависимости от тестируемого организма.
(А: Staphylococcus aureus OC4172; штаммы В, С и D представляют собой фторхинолон-резистентные, клинические изоляты Streptococcus pneumoniae, которые содержат различные совокупности замещений аминокислот в QRDR-области; Е: Streptococcus pneumoniae АТСС 49619).
| название | год | авторы | номер документа |
|---|---|---|---|
| БЕТА-КАРБОЛИНОВЫЕ СОЕДИНЕНИЯ, ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ НА ИХ ОСНОВЕ И СПОСОБЫ СВЯЗЫВАНИЯ, ДОСТИЖЕНИЯ АГОНИСТИЧЕСКОГО/АНТАГОНИСТИЧЕСКОГО ЭФФЕКТА | 1999 |
|
RU2233841C2 |
| СОЕДИНЕНИЕ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1999 |
|
RU2237060C2 |
| ПРОИЗВОДНЫЕ БИПИПЕРИДИНА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2002 |
|
RU2296751C2 |
| ВОДОРАСТВОРИМЫЕ АНАЛОГИ СС-1065 И ИХ КОНЪЮГАТЫ | 2007 |
|
RU2489423C2 |
| ПРОИЗВОДНЫЕ АРИЛ- И ГЕТЕРОАРИЛПИПЕРИДИНКАРБОКСИЛАТОВ, ИХ ПОЛУЧЕНИЕ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ФЕРМЕНТА FAAH | 2005 |
|
RU2376305C2 |
| ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ ПУРИНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЙ ПРЕПАРАТ | 1997 |
|
RU2228335C2 |
| СОЕДИНЕНИЯ АЛКИЛСУЛЬФИНИЛ-ЗАМЕЩЕННЫХ ТИАЗОЛИДОВ | 2011 |
|
RU2605745C2 |
| НОВЫЕ АРОМАТИЧЕСКИЕ ФТОРГЛИКОЗИДНЫЕ ПРОИЗВОДНЫЕ, СОДЕРЖАЩИЕ ЭТИ СОЕДИНЕНИЯ ЛЕКАРСТВЕННЫЕ СРЕДСТВА, И ИХ ПРИМЕНЕНИЕ | 2003 |
|
RU2340622C2 |
| СЕРУСОДЕРЖАЩИЕ ФОСФОНОВЫЕ КИСЛОТЫ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ ИЛИ ЭФИРЫ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1993 |
|
RU2136691C1 |
| ЛИГАНДЫ МЕЛАНОКОРТИНОВЫХ РЕЦЕПТОРОВ | 2001 |
|
RU2246501C2 |
Описываются соединения, имеющие структуру формулы (I), где n и m означают целое число от 1 до 3; z означает целое число от 0 до 1; R выбирают из водорода, гидрокси или алкокси; R2 означает водород; R3 и R4 независимо выбирают из группы, состоящей из водорода, галогена; R5 выбирают из группы, состоящей из галогена, алкила; R6 представляет собой алкил; Е выбирают из группы, указанной в п.1 формулы изобретения, А выбирают из N и C(R11), X выбирают из С, а означает двойную связь и b означает простую связь; и Y выбирают из N(R1), при условии, что, когда Y означает N(R1), X означает С, где R1 выбирают из С3-С6-циклоалкила, фенила; при условии, что если А означает C(R11), X означает С и Y означает N(R1), тогда R11 и R1 могут быть связаны с образованием морфолинила. Соединения, описанные в настоящем изобретении, обладают антибактериальной активностью и пригодны в качестве антибактериальных средств для лечения бактериальных инфекций у людей и животных. 12 з.п. ф-лы, 10 табл.
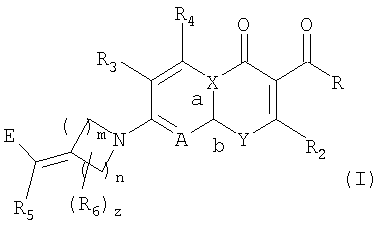
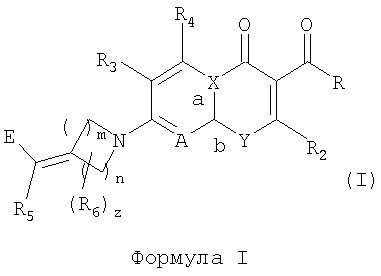
где n означает целое число от 1 до 3;
m означает целое число от 1 до 3;
z означает целое число от 0 до 1;
R выбирают из водорода, гидрокси или алкокси;
R2 означает водород;
R3 и R4 независимо выбирают из группы, состоящей из водорода, галогена;
R5 выбирают из группы, состоящей из галогена, алкила;
R6 представляет собой алкил;
Е выбирают из группы, состоящей из:
1) 
где q означает целое число от 1 до 3;
R7 и R8, каждый, независимо выбирают из водорода и алкила; или R7 и R8 связаны с образованием бензила;
R9 и R10, каждый, независимо выбирают из группы, состоящей из водорода, алкила, ацила;
2) 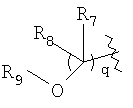
где q означает, как описано выше;
R7 и R8, каждый, независимо представляет собой водород; и R9 представляет собой водород; и
А выбирают из N и C(R11), где R11 представляет собой алкокси;
X выбирают из С, а означает двойную связь и b означает простую связь; и
Y выбирают из N(R1), при условии, что, когда Y означает N(R1), X означает С, где R1 выбирают из С3-С6-циклоалкила, фенила; при условии, что
если А означает C(R11), X означает С и Y означает N(R1), тогда R11 и R1 могут быть связаны с образованием морфолинила,
или его оптический изомер; его фармацевтически приемлемая соль.